serum cystatin c and emphysema: results from the national health and nutrition examination survey...
TRANSCRIPT

Serum Cystatin C and Emphysema: Results from the NationalHealth and Nutrition Examination Survey (NHANES)
Haala K. Rokadia • Shikhar Agarwal
Received: 7 July 2011 / Accepted: 10 January 2012 / Published online: 28 January 2012
� Springer Science+Business Media, LLC 2012
Abstract
Background Cystatin C (CysC) is a potent nonorgan-
specific cysteine protease inhibitor and may contribute to
elastolysis and tissue destruction by a mechanism of pro-
tease–antiprotease imbalance. Given the prevalence of
CysC in the serum of smokers and its role in tissue
destruction, we aimed to evaluate the association between
CysC and emphysema.
Methods Pooled cross-sectional data from the National
Health and Nutrition Examination Survey 1999–2002 were
used. Emphysema and chronic bronchitis were defined by a
self-reported history ascertained using standardized ques-
tionnaires. Active smokers were defined as self-reported
current smokers or measured serum cotinine C10 ng/mL.
Nonactive smokers with a serum cotinine level [0.05 ng/
mL were defined as environmental tobacco smoke (ETS)-
exposed.
Results The prevalence (95% CI) of emphysema was
1.3% (range = 0.9–1.8%). The mean (SE) CysC level in
the emphysema group was significantly higher than in
normal controls [1,139 (22) vs. 883 (8) lg/L; p = 0.001].
Upon stratification of the study population by C-reactive
protein (CRP) concentrations, we demonstrated a progres-
sive increase in the mean serum CysC level with serially
increasing CRP concentrations. Active smokers with
emphysema had 115.4 (46.5) lg/L higher mean (SE) CysC
levels than the normal controls (p \ 0.001). Upon adjusted
analysis, we observed that nonactive smokers with signif-
icant ETS exposure had 31.2 (15.2) lg/L higher mean (SE)
serum CysC levels as compared to ETS unexposed non-
active smokers (p = 0.04).
Conclusion In a large representative noninstitutionalized
US population, we demonstrated an association between
emphysema and serum CysC. Active smokers with
emphysema had significantly higher CysC levels. These
findings suggest that CysC may play a role in the patho-
genesis of smoking-related emphysema.
Keywords Cystatin C � Emphysema � Chronic bronchitis �Chronic obstructive pulmonary disease � Smoking �National
Health and Nutrition Examination Survey
Introduction
Chronic obstructive pulmonary disease (COPD) is char-
acterized by significant airflow limitation and is an
important cause of lung-related morbidity and mortality.
COPD represents a group of chronic inflammatory dis-
eases, including chronic bronchitis and emphysema. The
histological hallmark of COPD is the presence of inflam-
matory cells, including neutrophils and alveolar macro-
phages, in the microvasculature, interstitium, and alveoli.
Several animal and cell culture-based studies have dem-
onstrated the role of serine and cysteine proteases in the
development of COPD [1–3]. In addition, several in vitro
studies have suggested that inhibition of these proteases
may attenuate the contribution of proteases to the patho-
genesis of COPD [4]. Furthermore, proteolytic enzymes
have been demonstrated to play a role in fibrotic lung
remodeling by direct action on transforming growth factor
[5, 6].
H. K. Rokadia
Department of Internal Medicine, Cleveland Clinic, Cleveland,
OH 44195, USA
S. Agarwal (&)
Department of Cardiovascular Medicine, Cleveland Clinic,
9500 Euclid Avenue, Mail Code J2, Cleveland, OH 44195, USA
e-mail: [email protected]
123
Lung (2012) 190:283–290
DOI 10.1007/s00408-012-9374-z

Cathepsins are a family of cysteine proteases that are
responsible for macrophage-mediated extracellular matrix
degeneration [7]. Cathepsins B, H, L, and S have been
implicated in the pathogenesis of COPD. These cathepsins
are ubiquitous proteins secreted by inflammatory cells,
including neutrophils and alveolar macrophages. The
activity of these potent proteolytic enzymes is typically
inhibited by the cystatin family of proteins. Cystatin C
(CysC) is the most widespread and potent inhibitor of
cathepsins B and L, which are involved primarily in lung
tissue destruction [3, 8]. CysC binds to cathepsins in a
competitive fashion using noncovalent binding and results
in formation of inactive protease–cystatin complexes.
CysC is a nonorgan-specific cysteine protease inhibitor
that is secreted into the bloodstream by inflammatory cells,
including alveolar macrophages [9]. Although cathepsins
are measurable in the serum, the serum levels may not be
representative of the parenchymal cathepsin activity due to
primarily a local tissue action of these proteins [10, 11]. In
contrast, cystatins are secreted into the bloodstream and the
levels of these proteins may indirectly yet reliably predict
cathepsin activity.
CysC has been extensively studied in kidney disease and
cardiovascular disease and has been shown to be valuable
in assessing renal function and predicting cardiovascular
mortality, particularly in the elderly [12, 13]. Elevated
serum CysC has been associated with smoking, increased
age, male gender, C-reactive protein (CRP), and cardio-
vascular risk factors such as hypertension, low high-density
lipoprotein cholesterol and elevated body mass index [14].
It has been suggested that CysC may more accurately
estimate glomerular filtration rate (GFR) than creatinine,
particularly in the setting of the elderly and those with mild
renal insufficiency [15, 16]. Furthermore, it has been pos-
tulated that CysC plays a role in pathogenesis of malig-
nancy and metastasis [17–21]. Cathepsins B and L and
their potent inhibitor CysC have been studied in the path-
ogenesis of multiple pulmonary processes, including
bronchiectasis [22], lung malignancy [23], and pleural
effusions [24].
It is suggested that a cathepsin–cystatin imbalance
contributes to tissue destruction by under-regulated prote-
ase activity [8, 25, 26]. It may be speculated that this
overactivity of proteases in the lung parenchyma upregu-
lates the formation of CysC in an attempt to attenuate lung
tissue destruction. Given the evidence of a role of CysC in
tissue destruction and its prevalence in smokers, there
might be a role of CysC in emphysema. There is a striking
paucity of data relating CysC and emphysema. With this
background, we aim to evaluate the association between
CysC and emphysema in a large representative noninsti-
tutionalized US population. Figure 1 is a causal diagram
demonstrating the relationship between CysC and
emphysema. In this figure we have considered several
different etiopathogenic pathways that may result in ele-
vation of CysC in subjects with emphysema.
Methods
Study Population
This study analyzed pooled data from the 1999–2000 and
2001–2002 National Health and Nutrition Examination
Surveys (NHANES) [27]. NHANES is an on-going cross-
sectional survey of the civilian, noninstitutionalized US
population designed to provide a representative sample
from which to make national estimates on health and
nutritional status.
Exposure and Outcome Assessment
Cigarette smoking status was determined using serum
cotinine measurements along with the questionnaire items,
including ‘‘Have you smoked at least 100 cigarettes in your
entire life?’’ and ‘‘Do you now smoke cigarettes?’’ In the
NHANES, serum cotinine was measured using isotope
dilution, high-performance liquid chromatography/atmo-
spheric pressure chemical ionization tandem mass spec-
trometry. Active smokers were defined as self-reported
cigarette smokers or those with a measured serum cotinine
C10 ng/mL. All nonactive smokers by self-report were
classified as former smokers and never smokers based on
survey question responses. All nonactive smokers were
Fig. 1 Causal diagram representing the relationship between cystatin
C, smoking, and emphysema. The possible pathways that may result in
elevation of serum cystatin C are demonstrated using dashed arrows.
This diagram demonstrates that cystatin C may be a partial mediator
in the association between cigarette smoking and emphysema. HTNhypertension, DM diabetes mellitus, CKD chronic kidney disease,
CAD coronary artery disease, CHF congestive heart failure
284 Lung (2012) 190:283–290
123

subcategorized into those with elevated cotinine levels
([0.05 ng/mL) and those with nonelevated cotinine levels.
Nonactive smokers with elevated cotinine levels were
defined as environmental tobacco smoke (ETS)-exposed.
CysC was assayed from stored serum samples from
NHANES 1999–2000 and 2001–2002 from all participants
aged 60 years or more and 25% random sampling of par-
ticipants aged 12–59 years. Samples were assayed using an
automated particle-enhanced nephelometric assay (N Latex
Cystatin C; Dade Behring, Deerfield, IL). The assay,
ranging from 0.23 to 7.25 mg/dl, is currently the most
precise automated assay across the clinical concentration
range.
The study outcomes of emphysema or chronic bronchitis
were determined historically using standardized question-
naires. Patients that reported only a history of asthma or
symptoms of asthma were not included.
Measurement of Confounders and Mediators
CysC has been associated with cardiovascular disease and
chronic kidney disease (CKD) in several studies [12–16].
Due to clustering of chronic diseases like cardiovascular
disease and CKD with COPD by virtue of common risk
factors, we adjusted for the presence of cardiovascular
disease, congestive heart failure, stroke, CKD, and tradi-
tional cardiovascular risk factors, including age, gender,
race, hypertension, hyperlipidemia, body mass index, and
diabetes. Since CRP has been implicated in the causation
of cardiovascular disease, we included it in our regression
modeling in order to serve two independent purposes. First,
it provided conservative estimates of association between
CysC and emphysema. Second, it helped elucidate if there
was a possibility of a noninflammatory mediated pathway
in the pathogenesis of emphysema.
Hypertension was defined as systolic blood pressure
[140 mmHg, diastolic blood pressure [90 mmHg, self-
reported diagnosis of hypertension by a physician, or self-
reported use of antihypertensive medication. Hyperlipidemia
was defined as a total blood cholesterol [240 mg/dl, self-
reported diagnosis of hyperlipidemia by a physician, or self-
reported use of cholesterol-lowering medication. Diabetes
was defined as self-reported diagnosis of diabetes by a phy-
sician or use of insulin or oral hypoglycemic medication.
GFR was estimated using the modification of diet in
renal disease study formula [28, 29] based on age, serum
creatinine, race, and gender. Serum creatinine was stan-
dardized across surveys based on previously published
calibration equations [30]. CKD was defined as an esti-
mated GFR \60 mL/min/1.73 m2. Body mass index was
calculated from self-reported current height and weight.
CRP was measured on stored venipuncture samples by
latex-enhanced nephelometry with results reported within
the range of 0.01–18.5 mg/dl.
Statistical Analysis
Statistical analysis was performed using Stata v10.0 (Stata
Corp., College Station, TX, USA). Data from NHANES
1999–2000 and 2001–2002 surveys were pooled using
standard methods and, subsequently, 4 year combined
weights were calculated. Survey statistics traditionally used
to analyze complex semirandom survey designs were
employed to analyze these data. Multivariate linear
regression analysis was performed with CysC as the out-
come measure in the regression models to obtain adjusted-
effect estimates and their 95% confidence intervals (CI)
after accounting for the above-mentioned confounders.
Results
The prevalence (95% CI) of emphysema and chronic bron-
chitis in the US population was estimated as 1.3%
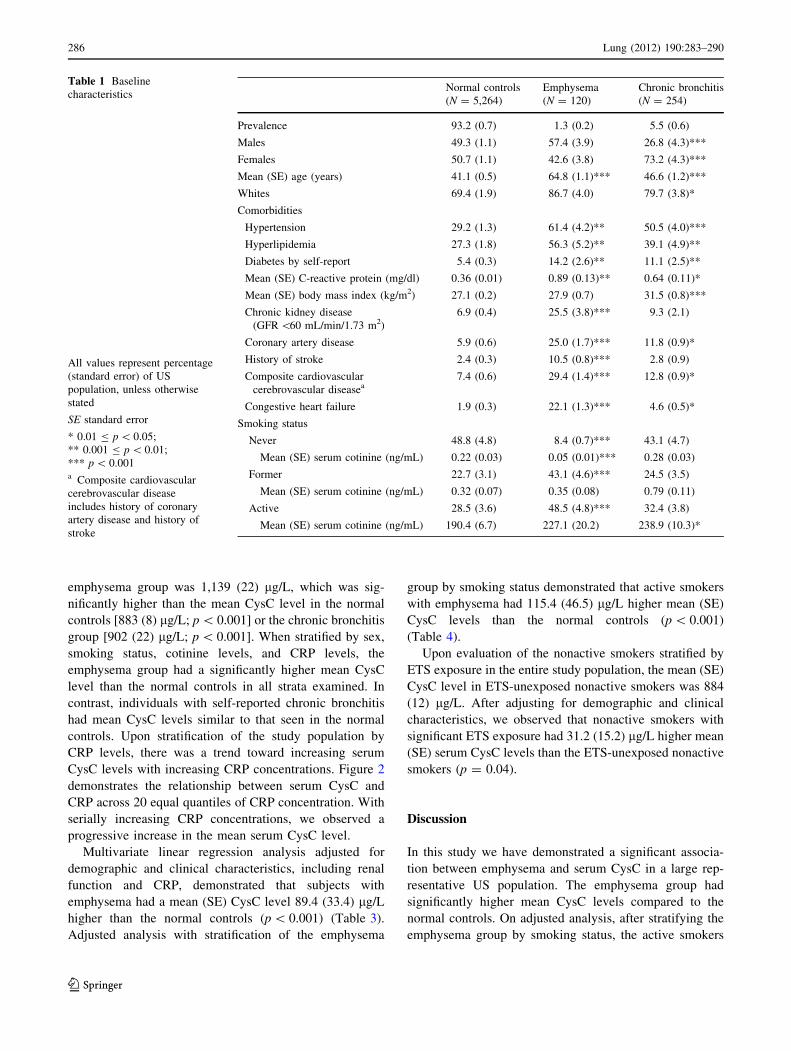
(0.9–1.8%) and 5.5% (4.2–6.7%), respectively. Table 1
gives the demographics and clinical characteristics of the
study population. We observed that the subjects with self-
reported emphysema were significantly older than the nor-
mal controls (p \ 0.001). The chronic bronchitis group had
a female (p \ 0.001) and white (p = 0.03) predominance.
We observed that the emphysema group had a significantly
higher prevalence of coronary artery disease, cerebrovas-
cular disease, and CKD compared to the other groups
(p \ 0.001 for all comparisons). In addition to cardiovas-
cular disease, the emphysema group had a significantly
higher prevalence of traditional cardiovascular risk factors,
specifically hypertension (61.4%), hyperlipidemia (56.3%),
and diabetes (14.2%). In comparison to the control popula-
tion, the mean CRP was significantly higher in the emphy-
sema (p = 0.003) and chronic bronchitis (p = 0.02) groups.
There were significant differences noted in the distri-
bution of smoking characteristics in the study groups. In
comparison to the normal controls, we observed a signifi-
cantly higher prevalence of active smoking (48.5% vs.
28.5%; p \ 0.001) and former smoking (43.1% vs. 22.7%;
p \ 0.001) in the emphysema group. Only 8.4% of subjects
with self-reported emphysema were never smokers com-
pared to 48.8% of normal controls (p \ 0.001). Of the
never and former smokers (nonactive smokers), the pro-
portion (SE) of individuals with significant ETS exposure
was 48.3% (2.3%), 53.9% (3.1%), and 49.2% (2.4%)
among the normal control group, emphysema group, and
chronic bronchitis group, respectively.
Table 2 gives the distribution of mean CysC levels in
the study population. The mean (SE) CysC level in the
Lung (2012) 190:283–290 285
123
emphysema group was 1,139 (22) lg/L, which was sig-
nificantly higher than the mean CysC level in the normal
controls [883 (8) lg/L; p \ 0.001] or the chronic bronchitis
group [902 (22) lg/L; p \ 0.001]. When stratified by sex,
smoking status, cotinine levels, and CRP levels, the
emphysema group had a significantly higher mean CysC
level than the normal controls in all strata examined. In
contrast, individuals with self-reported chronic bronchitis
had mean CysC levels similar to that seen in the normal
controls. Upon stratification of the study population by
CRP levels, there was a trend toward increasing serum
CysC levels with increasing CRP concentrations. Figure 2
demonstrates the relationship between serum CysC and
CRP across 20 equal quantiles of CRP concentration. With
serially increasing CRP concentrations, we observed a
progressive increase in the mean serum CysC level.
Multivariate linear regression analysis adjusted for
demographic and clinical characteristics, including renal
function and CRP, demonstrated that subjects with
emphysema had a mean (SE) CysC level 89.4 (33.4) lg/L
higher than the normal controls (p \ 0.001) (Table 3).
Adjusted analysis with stratification of the emphysema
group by smoking status demonstrated that active smokers
with emphysema had 115.4 (46.5) lg/L higher mean (SE)
CysC levels than the normal controls (p \ 0.001)
(Table 4).
Upon evaluation of the nonactive smokers stratified by
ETS exposure in the entire study population, the mean (SE)
CysC level in ETS-unexposed nonactive smokers was 884
(12) lg/L. After adjusting for demographic and clinical
characteristics, we observed that nonactive smokers with
significant ETS exposure had 31.2 (15.2) lg/L higher mean
(SE) serum CysC levels than the ETS-unexposed nonactive
smokers (p = 0.04).
Discussion
In this study we have demonstrated a significant associa-
tion between emphysema and serum CysC in a large rep-
resentative US population. The emphysema group had
significantly higher mean CysC levels compared to the
normal controls. On adjusted analysis, after stratifying the
emphysema group by smoking status, the active smokers
Table 1 Baseline
characteristics
All values represent percentage
(standard error) of US
population, unless otherwise
stated
SE standard error
* 0.01 B p \ 0.05;
** 0.001 B p \ 0.01;
*** p \ 0.001a Composite cardiovascular
cerebrovascular disease
includes history of coronary
artery disease and history of
stroke
Normal controls
(N = 5,264)
Emphysema
(N = 120)
Chronic bronchitis
(N = 254)
Prevalence 93.2 (0.7) 1.3 (0.2) 5.5 (0.6)
Males 49.3 (1.1) 57.4 (3.9) 26.8 (4.3)***
Females 50.7 (1.1) 42.6 (3.8) 73.2 (4.3)***
Mean (SE) age (years) 41.1 (0.5) 64.8 (1.1)*** 46.6 (1.2)***
Whites 69.4 (1.9) 86.7 (4.0) 79.7 (3.8)*
Comorbidities
Hypertension 29.2 (1.3) 61.4 (4.2)** 50.5 (4.0)***
Hyperlipidemia 27.3 (1.8) 56.3 (5.2)** 39.1 (4.9)**
Diabetes by self-report 5.4 (0.3) 14.2 (2.6)** 11.1 (2.5)**
Mean (SE) C-reactive protein (mg/dl) 0.36 (0.01) 0.89 (0.13)** 0.64 (0.11)*
Mean (SE) body mass index (kg/m2) 27.1 (0.2) 27.9 (0.7) 31.5 (0.8)***
Chronic kidney disease
(GFR \60 mL/min/1.73 m2)
6.9 (0.4) 25.5 (3.8)*** 9.3 (2.1)
Coronary artery disease 5.9 (0.6) 25.0 (1.7)*** 11.8 (0.9)*
History of stroke 2.4 (0.3) 10.5 (0.8)*** 2.8 (0.9)
Composite cardiovascular
cerebrovascular diseasea7.4 (0.6) 29.4 (1.4)*** 12.8 (0.9)*
Congestive heart failure 1.9 (0.3) 22.1 (1.3)*** 4.6 (0.5)*
Smoking status
Never 48.8 (4.8) 8.4 (0.7)*** 43.1 (4.7)
Mean (SE) serum cotinine (ng/mL) 0.22 (0.03) 0.05 (0.01)*** 0.28 (0.03)
Former 22.7 (3.1) 43.1 (4.6)*** 24.5 (3.5)
Mean (SE) serum cotinine (ng/mL) 0.32 (0.07) 0.35 (0.08) 0.79 (0.11)
Active 28.5 (3.6) 48.5 (4.8)*** 32.4 (3.8)
Mean (SE) serum cotinine (ng/mL) 190.4 (6.7) 227.1 (20.2) 238.9 (10.3)*
286 Lung (2012) 190:283–290
123

with emphysema were observed to have higher CysC levels
compared to the nonsmokers. In addition, there was a
higher mean CysC level observed in the nonactive smokers
with significant ETS exposure in comparison to the normal
controls, suggesting that any active or second-hand smoke
exposure might contribute to increased serum CysC levels.
Cathepsins belong to the family of cysteine proteases
that catalyze the splitting of large proteins into amino acids
and play a significant role in pathogen killing and regula-
tion of inflammatory processes [31]. The activity of
cathepsins is closely regulated by a group of compounds
called cysteine protease inhibitors, which include stefins
and cystatins. The cystatin superfamily consists of three
types of compounds: type 1 cystatins (A and B) act intra-
cellularly, type 2 cystatins (C, D, E/M, F, G, and S) act
extracellularly, and type 3 cystatins are kininogens, which
are intravascular proteins [32]. Type 2 cystatins belong to a
heterogeneous group of ubiquitous proteins. CysC is the
most widespread type 2 cystatin and is secreted by virtually
all organs and is present in all body fluids. CysC inhibits
the action of cathepsins by noncovalent, reversible, com-
petitive binding and formation of inactive protease–cysta-
tin complexes. Tipping the balance in favor of proteases
may result in increased tissue destruction. It may be
speculated that increased tissue destruction resulting from
enhanced protease activity might lead to a compensatory
increase in the concentration of cystatins. The increase in
the concentration of cystatins as a maker of tissue
destruction has been utilized in several diseases.
Table 2 Mean (SE) cystatin C (lg/L) levels in study population
Normal
controls
Emphysema Chronic
bronchitis
Total 883 (8) 1139 (22)*** 902 (22)
Males 904 (8) 1167 (24)** 919 (23)
Females 863 (9) 1102 (25)*** 895 (20)
Smoking status
Never 870 (12) 1068 (21)** 904 (21)
Former 921 (13) 1217 (28)** 897 (24)
Active 907 (16) 1082 (28)* 902 (26)
Cotinine level (ng/mL)
\0.05 881 (12) 1138 (20)*** 895 (25)
0.05–10 867 (10) 1251 (26)* 908 (30)
C10 905 (15) 1082 (24)* 902 (22)
CRP level (mg/dL)
\0.1 826 (7) 975 (29)* 834 (22)
0.1–0.24 870 (9) 998 (28)** 877 (28)
0.25–0.49 919 (14) 1349 (32)** 911 (29)
C0.50 966 (16) 1200 (22)* 939 (26)
CRP C-reactive protein, SE standard error
* 0.01 B p \ 0.05; ** 0.001 B p \ 0.01; *** p \ 0.001
Fig. 2 Relationship between serum cystatin C and serum CRP. This
figure demonstrates the mean cystatin C level across 20 equal
quantiles of serum CRP concentrations in the entire study population.
The shaded square represents the mean cystatin C level and the
vertical lines represent the 95% confidence interval for each
respective quantile of CRP concentration
Table 3 Multivariate linear regression analysis with cystatin C
(lg/L) as outcome
Coefficient 95% confidence
interval
p value
Normal controls Reference
Emphysema 89.4 23.9 to 154.9 \0.001
Chronic bronchitis -11.8 -43.5 to 20.0 0.5
Age (years) 2.3 1.6 to 3.0 \0.001
Male sex 50.0 35.1 to 65.0 \0.001
Whites Reference
Blacks -17.2 -41.7 to 7.3 0.2
Mexican American -51.1 -71.4 to -30.7 \0.001
Other -38.6 -67.5 to -9.8 \0.001
Hypertension 20.1 1.7 to 38.4 \0.001
Hyperlipidemia -11.2 -32.2 to 9.8 0.3
Diabetes 15.7 -30.5 to 61.9 0.5
Cardiovascular disease 47.3 19.0 to 75.5 \0.001
GFR [90 mL/min/
1.73 m2Reference
GFR 60–90 mL/min/
1.73 m262.1 39.3 to 84.9 \0.001
GFR 30–59 mL/min/
1.73 m2371.7 327.4 to 416.0 \0.001
GFR \30 mL/min/
1.73 m23,155.9 2,369.6 to
3,942.3
\0.001
Logarithm transformed
CRP (mg/dL)
46.6 27.3 to 65.9 \0.001
BMI (kg/m2) 2.0 0.3 to 3.6 \0.001
Constant 664.6 589.4 to 739.8 \0.001
GFR glomerular filtration rate, CRP C-reactive protein, BMI body
mass index
Lung (2012) 190:283–290 287
123

In the current literature, CysC has been studied exten-
sively in kidney disease, cardiovascular disease, and solid
organ malignancy [12–21]. CysC has been studied as a
more sensitive marker for renal dysfunction than serum
creatinine [15, 16]. The proposed mechanism of CysC in
estimation of glomerular filtration arises from its low
molecular weight which allows it to be freely filtered by the
glomerulus and reabsorbed, but not secreted, by tubular
cells [33]. CysC has a proposed role in atherosclerosis and
may serve as an earlier marker for renal insufficiency
associated with adverse outcomes [34]. In addition, CysC
has been shown to be associated with increased cardio-
vascular and all-cause mortality [35].
Higher levels and activity of cathepsins and lower levels
of CysC have been shown to be present in higher-grade
malignancies and metastatic disease [20]. The suggested
mechanism of action is that cathepsins are secreted to
degrade the basement membrane and extracellular matrix,
in effect facilitating metastasis. With lower levels of CysC,
the inhibition of this function is insufficient [18–20].
There are multiple protease–antiprotease mechanisms
that have been implicated in the pathogenesis of pulmonary
disease. More widely known are the underregulated neu-
trophil elastase, a serine protease, and matrix metallopro-
teinase activity contributing to the pathogenesis of
emphysema [31]. Finlay et al. [36] demonstrated increased
levels of the two major matrix metalloproteinases, colla-
genase and gelatinase, in the bronchoalveolar lavage fluid
of emphysematous patients compared to smoking controls
and suggested that the presence of collagenase was a better
indicator of emphysema in smoking patients. CysC is the
most potent inhibitor of cathepsin B and L, which have
been implicated in pulmonary diseases [3, 8, 37, 38]. Both
these cysteine proteases and their inhibitors are secreted by
alveolar macrophages [24].
The protease–antiprotease activity of cathepsins and
CysC has been studied in multiple pulmonary diseases.
Smoking activates neutrophils and increases alveolar
macrophage activity in the lung [39]. Upon alveolar mac-
rophage activation, both proteases and antiproteases are
released [40, 41]. Warfel [25] demonstrated elevated CysC
and cathepsin B levels in the culture medium of alveolar
macrophages of smokers relative to nonsmokers. Abboud
[8] measured cathepsin L and CysC levels in bronchoal-
veolar lavage samples of smokers and found elevated
levels in smokers with emphysema compared to smokers
without emphysema. This suggestion that smokers that
develop clinically significant emphysema have higher
CysC levels compared to their nonemphysematous smok-
ing counterparts is also corroborated in our study. Lesser
[3] showed that intratracheal instillation of cathepsin B in
hamsters can induce emphysema. In a baboon model of
bronchopulmonary dysplasia, a neonatal disease of alter-
nating areas of emphysema and atelectasis, elevated
cathepsin activity was found without a concurrent elevation
in CysC activity, leading to a net increased cysteine pro-
tease activity [26]. These studies suggest that underregu-
lated cathepsin activity may lead to clinically relevant
pulmonary disease. Levels of total and unbound serum
cathepsin B and L were elevated in patients with pul-
monary malignancy [17, 19, 21, 42].
The finding of elevated CysC levels in smokers was
corroborated in our study, specifically showing elevated
CysC in emphysema patients who were active smokers. As
shown in Fig. 1, several mechanistic associations might
explain this observed relationship. CysC might be elevated
as a direct response to cigarette smoking or to the devel-
opment of emphysema or even to nonspecific lung tissue
destruction. The other, more plausible mechanism of CysC
elevation might be cathepsins–CysC imbalance in the
process of tissue destruction. Data suggest that elevated
cathepsin B and elevated CysC might still represent an
imbalance in favor of protease activity; despite elevated
Table 4 Multivariate linear regression analysis with stratification of
the emphysema group by smoking status
Coefficient 95% confidence
interval
p value
Normal controls Reference
Nonsmoker -37.2 -106.4 to 31.9 0.3
Former smoker 86.6 -43.4 to 216.7 0.2
Active smoker 115.4 24.2 to 206.6 \0.001
Age (years) 2.3 1.6 to 3.1 \0.001
Male sex 50.6 34.5 to 66.7 \0.001
Whites Reference
Blacks -18.2 -43.3 to 6.9 0.1
Mexican American -51.6 -71.5 to -31.6 \0.001
Other -32.8 -56.5 to -9.2 \0.001
Hypertension 21.0 2.6 to 39.5 \0.001
Hyperlipidemia -16.1 -39.8 to 7.6 0.2
Diabetes 1.2 -44.3 to 46.8 1.0
Cardiovascular disease 54.1 18.9 to 89.3 \0.001
GFR [90 mL/min/
1.73 m2Reference
GFR 60–90 mL/min/
1.73 m261.6 38.8 to 84.4 \0.001
GFR 30–59 mL/min/
1.73 m2372.5 324.0 to 421.1 \0.001
GFR \30 mL/min/
1.73 m23337.5 2594.6 to 4080.5 \0.001
Logarithm transformed
CRP (mg/dL)
43.5 23.7 to 63.3 \0.001
BMI (kg/m2) 2.5 0.7 to 4.3 \0.001
Constant 648.3 570.8 to 725.8 \0.001
GFR glomerular filtration rate, CRP C-reactive protein, BMI body
mass index
288 Lung (2012) 190:283–290
123

CysC levels, the cathepsin activity level might still be
active [3, 25]. Alternatively, the elevated levels of CysC
might reflect the body’s lagging response to counteract the
effect of increased concentration of the cathepsins induced
by smoking. However, the degree of elevation in CysC
might not be sufficient to inhibit the simultaneous
increased cathepsin levels. It has been demonstrated in
animal studies that the capacity of CysC to neutralize
excessive local proteolytic activity of the cathepsins might
be limited in the lung [26]. CysC might not achieve suf-
ficient levels in the microenvironment of the alveolar
macrophage where cathepsins are released by lysosomes
[43]. In addition, CysC has affinity to primarily cathepsin
B; other cathepsins might be less inhibited by CysC and
might contribute to tissue destruction.
Figure 2 demonstrates a dose-dependent trend of
increasing CysC levels with increasing serum CRP. As
demonstrated in Fig. 1, activation of inflammatory cells
might lead to activation of cathepsins with a consequent
increase in CysC. However, further analysis revealed that
the significant elevation in CysC levels tended to persist
despite an adjustment for serum CRP, suggesting that there
may be a non-CRP-mediated role of cathepsins and/or CysC
in the pathogenesis of emphysema. It is important to note
that by suggesting a non-CRP-mediated role of CysC, we do
not discount a concomitant CRP-mediated role of CysC. As
smoking and elevated CRP levels contribute to the inflam-
matory phenomenon of alveolar macrophage activation and
ultimately emphysema, there might be an additional role of
CRP within the inflammatory pathway and should be fur-
ther investigated in future studies. Furthermore, the non-
CRP-mediated role of CysC should be further elucidated to
clarify any preventative or therapeutic potential.
Limitations
Our data set was a semirandom sample that was based on
self-report and was susceptible to reporter’s bias. With this
cross-sectional study, an evaluation of causality could not
be made. In addition, as our study population was a non-
institutionalized civilian population, there was a potential
of survival bias as sicker patients with more severe risk
factors were likely not included in this study. However,
despite evaluating a potentially less sick group, our results
have likely underestimated the association between CysC
and emphysema.
We performed an analysis on nonsmokers with signifi-
cant ETS exposure based on reported nonsmokers with
serum cotinine levels [0.05 ng/mL. This categorization
might not capture all subjects with significant ETS expo-
sure, depending on the extent of exposure and the time
interval between the exposure and serum cotinine level
measurement.
A major limitation of our study was the use of self-
reported outcomes and the lack of spirometric or imaging
confirmation of emphysema. Due to this limitation, an
analysis of the outcomes stratified by severity of disease
was not possible. Given the association between CysC and
emphysema that we found in this study, further study into
this relationship, including correlation to radiographic
imaging and pulmonary function testing, would be of
interest.
As comorbidities, including cardiovascular disease,
renal disease, and smoking, may affect serum CysC levels,
these confounding factors were identified and controlled
for in our analysis. We performed a rigorous multivariate
linear regression analysis that controlled for demographic
information and clinical comorbidities, including renal
disease by stages of CKD, cardiovascular disease, and
cardiac risk factors. Importantly, despite controlling for
CRP level and stratifying by smoking history and despite a
robust analysis to control for confounding factors that
might affect CysC levels, we continued to find a significant
association between serum CysC and emphysema. We
acknowledge the possibility of residual confounding due to
unmeasured factors which might affect our analysis.
Conclusions
Our study demonstrated a significant association between
serum CysC levels and emphysema in a large representative
noninstitutionalized US population. Active smokers with
emphysema were observed to have significantly higher
levels of CysC in comparison to the normal controls. In
addition, never smokers with significant ETS exposure had
significantly higher CysC compared to those without a
significant ETS exposure. Due to a significant association of
CRP and CysC in our study population, it might be specu-
lated that elevation of CysC occurs secondary to inflam-
matory processes in the lung. However, persistence of
significant association between emphysema and CysC
despite adjustment for serum CRP might indicate a nonin-
flammatory pathway in the etiopathogenesis of emphysema.
Conflict of interest The authors have no conflicts of interest to
disclose.
References
1. Jannoff A, Sloan B, Weinbaum G et al (1977) Experimental
emphysema induced with purified human neutrophil elastase:
tissue localization of the instilled protease. Am Rev Respir Dis
115:461–478
2. Owen CA, Campbell EJ (1999) The cell biology of leukocyte-
mediated proteolysis. J Leukoc Biol 65:137–150
Lung (2012) 190:283–290 289
123

3. Lesser M, Padilla ML, Cardozo C (1992) Induction of emphy-
sema in hamsters by intratracheal instillation of cathepsin B. Am
Rev Respir Dis 145:661–668
4. Chughtai B, O’Riordan TG (2004) Potential role of inhibitors of
neutrophil elastase in treating diseases of the airway. J Aerosol
Med 17:289–298
5. Chua F, Laurent GJ (2006) Neutrophil elastase: mediator of
extracellular matrix destruction and accumulation. Proc Am
Thorac Soc 3:424–427
6. Lungarella G, Cavarra E, Lucattelli M, Martorana PA (2008) The
dual role of neutrophil elastase in lung destruction and repair. Int
J Biochem Cell Biol 40:1287–1296
7. Owen CA (2008) Roles for proteinases in the pathogenesis of
chronic obstructive pulmonary disease. Int J Chronic Obstr Pulm
Dis 3:253–268
8. Abboud RT, Vimalanathan S (2008) Pathogenesis of COPD. Part
1. The role of protease–antiprotease imbalance in emphysema. Int
J Tuberc Lung Dis 12:361–367
9. Kos J, Lah TT (1998) Cysteine proteinases and their endogenous
inhibitors: target proteins for prognosis, diagnosis, and therapy in
cancer. Oncol Rep 5:1349–1361
10. Chapman HA, Stone OL (1984) Comparison of live human
neutrophils and alveolar macrophage elastolytic activity in vitro.
Relative resistance of macrophage elastolytic activity to serum
and alveolar proteinase inhibitors. J Clin Invest 74:1693–1700
11. Abbott DE, Margaryan NV, Jeruss JS et al (2010) Reevaluating
cathepsin D as a biomarker for breast cancer: serum activity
levels versus histopathology. Cancer Biol Ther 9:23–30
12. Meng L, Yang Y, Qi LT, Wang XJ, Xu GB, Zhang BW (2011)
Elevated serum cystatin C is an independent predictor of car-
diovascular events in people with relatively normal renal func-
tion. J Nephrol. doi:10.5301/jn.5000020
13. Bevc S, Hojs R, Ekart R, Gorenjak M, Puklavec L (2011) Simple
cystatin C formula compared to sophisticated CKD-EPI formulas
for estimation of glomerular filtration rate in the elderly. Ther
Apher Dial 15:261–268
14. Kottgen A, Selvin E, Stevens LA et al (2008) Serum cystatin C in
the United States: the third National Health and Nutrition
Examination Survey (NHANES III). Am J Kidney Dis 51:
385–394
15. Kyhse-Andersen J, Schmidt C, Nordin G et al (1994) Serum
cystatin C, determined by a rapid, automated particle-enhanced
turbidimetric method, is a better marker than serum creatinine for
glomerular filtration rate. Clin Chem 40:1921–1926
16. Dharnidharka VR, Kwon C, Stevens G (2002) Serum cystatin C
is superior to serum creatinine as a marker of kidney function: a
meta-analysis. Am J Kidney Dis 40:221–226
17. Zore I, Krasovec M, Cimerman N et al (2001) Cathepsin
B/Cystatin C complex levels in sera from patients with lung and
colorectal cancer. Biol Chem 382:805–810
18. Poteryaeva ON, Falameyeva OV, Korolenko TA et al (2000)
Cysteine proteinase inhibitor level in tumor and normal tissues in
control and cured mice. Drugs Exp Clin Res 26:301–306
19. Krepela E, Prochazka J, Karova B, Cermak J, Roubkova H (1998)
Cysteine proteases and cysteine protease inhibitors in non-small
cell lung cancer. Neoplasma 45:318–331
20. Kos J, Werle B, Lah T, Brunner N (2000) Cysteine proteinases
and their inhibitors in extracellular fluids: markers for diagnosis
and prognosis in cancer. Int J Biol Markers 15:84–89
21. Werle B, Schanzenbacher U, Lah TT et al (2006) Cystatins in
non-small cell lung cancer: tissue levels, localization and relation
to prognosis. Oncol Rep 16:647–655
22. Buttle DJ, Burnett D, Abrahamson M (1990) Levels of neutrophil
elastase and cathepsin B activities, and cystatins in human spu-
tum: relationship to inflammation. Scand J Clin Lab Invest
50:509–516
23. Luthgens K, Ebert W, Trefz G, Gabrijelcic D, Turk V, Lah T
(1993) Cathepsin B and cysteine proteinase inhibitors in bron-
choalveolar lavage fluid of lung cancer patients. Cancer Detect
Prev 17:387–397
24. Werle B, Sauckel K, Nathanson CM et al (2003) Cystatins C,
E/M, and F in human pleural fluids of patients with neoplastic and
inflammatory lung disorders. Biol Chem 382:281–287
25. Warfel AH, Cardozo C, Yoo OH, Zucker-Franklin D (1991)
Cystatin C and cathepsin B production by alveolar macrophages
from smokers and nonsmokers. J Leukoc Biol 49:41–47
26. Altiok O, Yasumatsu R, Bingol-Karakoc G et al (2006) Imbal-
ance between cysteine proteases and inhibitors in a baboon model
of bronchopulmonary dysplasia. Am J Respir Crit Care Med
173:318–326
27. Centers for Disease Control and Prevention (2010) NHANES–
National Health and Nutrition Examination Survey homepage.
Available at http://www.cdc.gov/nchs/nhanes.htm/. Accessed 3
August 2010
28. Levey AS, Coresh J, Balk E et al (2003) National Kidney
Foundation practice guidelines for chronic kidney disease: eval-
uation classification and stratification. Ann Intern Med 139:
137–147
29. Coresh J, Selvin E, Stevens LA et al (2007) Prevalence of chronic
kidney disease in the United States. JAMA 298:2038–2047
30. Selvin S, Manzi J, Stevens LA et al (2007) Calibration of serum
creatinine in the National Health and Nutrition Examination
Surveys (NHANES) 1988–1994, 1999–2004. Am J Kidney Dis
50:918–926
31. Korkmaz B, Horwitz MS, Jenne DE, Gauthier F (2010) Neutro-
phil elastase, proteinase 3, and cathepsin G as therapeutic targets
in human disease. Pharmacol Rev 62:726–759
32. Abrahamson M (1994) Cystatins. Methods Enzymol 244:
685–700
33. Salgado JV, Neves FA, Bastos MG et al (2010) Monitoring renal
function: measured and estimated glomerular filtration rates—a
review. Braz J Med Biol Res 43:528–536
34. Lafarge JC, Naour N, Clement K, Guerre-Millo M (2010)
Cathepsins and cystatin C in atherosclerosis and obesity. Bio-
chimie 92:1580–1586
35. Shlipak MG, Marnak MJ, Katz R et al (2005) Cystatin C and the
risk of death and cardiovascular events among elderly persons.
N Engl J Med 352:2049–2060
36. Finlay GA, Russell KJ, McMahon KJ et al (1997) Elevated levels
of matrix metalloproteinases in bronchoalveolar lavage fluid of
emphysematous patients. Thorax 52:502–506
37. Henskens YM, Veerman EC, Amerongen AV (1996) Cystatins in
health and diseases. Biol Chem Hoppe Seyler 377:71–86
38. Abrahamson M, Alvarez-Fernandez M, Nathanson CM (2003)
Cystatins. Biochem Soc Symp 70:179–199
39. Finkelstein R, Fraser RS, Ghezzo H, Cosio MG (1995) Alveolar
inflammation and its relation to emphysema in smokers. Am J
Respir Crit Care Med 152:1666–1672
40. Takeyabu K, Betsuyaku T, Nishimura M et al (1998) Cysteine
proteinases and cystatin C in bronchoalveolar lavage fluid from
subjects with subclinical emphysema. Eur Respir J 12:1033–1039
41. Chapman HA, Reilly JJ, Yee R, Grubb A (1990) Identification of
cystatin C, a cysteine proteinase inhibitor, as a major secretory
product of human alveolar macrophages in vitro. Am Rev Respir
Dis 141:698–705
42. Sloane BF, Rozhin J, Robinson D, Honn KV (1990) Role for
cathepsin B and cystatins in tumor growth and progression. Biol
Chem Hoppe Seyler 371:193–198
43. Woischnik M, Bauer A, Aboutaam R et al (2008) Cathepsin H
and napsin A are active in the alveoli and increased in alveolar
proteinosis. Eur Respir J 31:1197–1204
290 Lung (2012) 190:283–290
123