searching genomes for noncoding rna cs374 leticia britos 10/03/06
Post on 20-Dec-2015
214 views
TRANSCRIPT


Searching genomes for Searching genomes for noncoding RNA noncoding RNA
CS374
Leticia Britos
10/03/06

DNA to RNA, and genes
G
A
G
U
C
A
G
C
DNA, ~3x109 long in humansContains ~ 22,000 genes
RNA: carries the “message” for “translating”, or “expressing” one gene
transcription translation
folding
1
2easy
3

“Structural genes encode proteins and regulatory genes produce non-coding RNA”
F. Jacob and J. Monod (1961)

Where are the genes?Where are the genes?
Gene Finding

atg
tga
ggtgag
ggtgag
ggtgag
cagatg
cagttgcaggcc
ggtgag
Where are the genes?Where are the genes?
Gene Finding
In humans:
~22,000 genes~1.5% of human DNA

An expanding universe of noncoding RNA
• rRNArRNA (structure/function of ribosomes)• tRNAtRNA (translation)• snRNAsnRNA (RNA splicing, telomere maintenance)• snoRNAsnoRNA (chemical modification of rRNA)• miRNAmiRNA (translational regulation)• gRNAgRNA (mRNA editing)• tmRNAtmRNA (degradation of defective proteins)• riboswitches riboswitches (translational and transcriptional regulation)• ribozymesribozymes (autocatalytic RNA)• RNAi RNAi (gene regulation by dsRNA)

Exciting times for the RNA world (and for Stanford)

atg
tga
ggtgag
ggtgag
ggtgag
caggtg
cagatg
cagttgcaggcc
ggtgag
How to find ncRNAs?

How to find ncRNAs?

Riboswitches
5’ 3’
promoter
5’ UTR exons 3’ UTR
introns
coding
5’ 3’
promoter
5’ UTR exons 3’ UTR
introns
coding
noncoding
5’ 3’
promoter
5’ UTR exons 3’ UTR
introns
coding

Sequence conservation is not enough
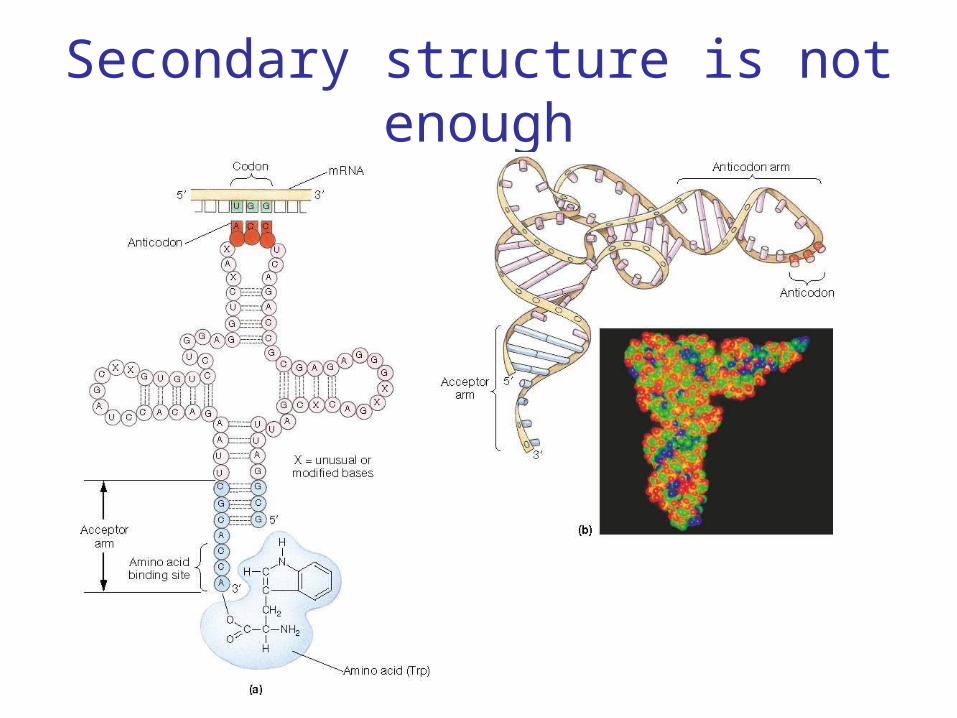
Secondary structure is not enough

Noncoding RNA signals in the genome are not as strong as the signals for protein
coding genes
Look for structure in evolutionary conserved sequences

Identify new instances of a given ncRNA family in a genome

Existing algorithms
• CMSearch
• RSEARCH
• ERPIN

Example: finding 5S RNAs in a 1.6Mb genome
• RSEARCH: 6.5 h
• FastR: 103 s

FastR

What is a Database filter?
A computational procedure that takes a DB as input and outputs a subset of it.
filter
The object being searched for remains in the DB after filtering (sensitivity)
The filtered DB is significantly smaller
The filtering operation is fast (efficiency)

Problem
• Given an RNA sequence with known structure, find homologous sequences in a RNA DB
AGAGCGUAUCGAUUUAGAGAGCUAUAGCUAGAGAGGAGA
UUAUAGCGCGCAUAUAGGACAAACAGUCUCUAUGGGGAC
AUUCCGGGAACAUAGUAUAGGCGACGGAUUAGCUAGCCAA
AUCGCGCUAUAGCUAGCGAGGACAGCUAUAGCUAGCGAG
AUAUCGGGCUGUGGACACUAUACGAUCGAAUCUAGCUAU
AUCGCGCUAUAGCUAGCGAGGACAGCUAUAGCUAGCGAG
AUAUCGGGCUGUGGACACUAUACGAUCGAAUCUAGCUAU
QUERY DB
AUCGCGCUAUAGCUAGCGAGGACAGCUAUAGCUAGCGAG

• Stage 2: alignalign the selected sequences in the DB with the query and determine the best alignments
Solution
filterfilter
• Stage 1: filterfilter the DB

Filtering
• Sequence alone is not sufficient
• Structure alone is not sufficient

Filter using both sequence and structural features

Structural features: (k,w)-stacks(k,w)-stacks
AUUCCGGGAACAUAGUAUAGGCGACGGAUUAGCUAGCCAA
3 6 25 28
a a’
a a’
a a’
a a’

AU
UC
CG
GG
AA
CA
UA
GU
AU
AG
GC
GA
CG
GA
UU
AG
CU
AG
CC
AA
Definition of a (k,w)-stack(k,w)-stack
AUUCCGGGAACAUAGUAUAGGCGACGGAUUAGCUAGCCAA
d = 18
A pair of substrings of at least length k, that are at most w bases apart
a
a’
Is a,a’ :(4,18)-stack?(4,20)-stack?(4,9)-stack?(3, 20)-stack?

Use of (k,w)-stacks as filters in the search for ncRNAs
If we use a (7,70)-stack filter, we eliminate 90% of the DB from consideration

Structural features: nestednested (k,w,l)-stacks
AUUCCGGGAACAUAGUAUAGGCGACGGAUUAGCUAGCCAA
3 6 25 2812 1614 18
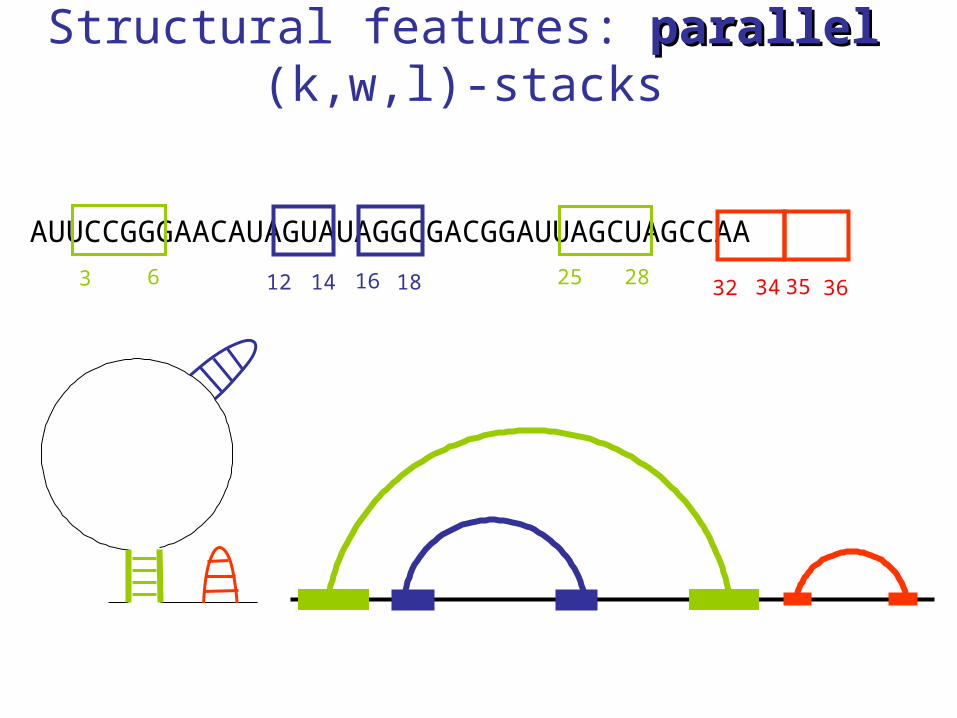
Structural features: parallelparallel (k,w,l)-stacks
AUUCCGGGAACAUAGUAUAGGCGACGGAUUAGCUAGCCAA
3 6 25 2812 1614 18 32 3534 36

Structural features: multiloopmultiloop (k,w,l)-stacks

Filtering criteria
nested stacks
Parallel stacks
Multiloop stacks

Filtering algorithm
1. Build a hash table hash table of kmers in the DB
AUUCCGGGAACAUAUUCUAGGCGACGGAUUAGAAUGCCAA
kmer indexAUUC
k=4
1

Filtering algorithm
1. Build a hash table hash table of kmers in the DB
AUUCCGGGAACAUAUUCUAGGCGACGGAUUAGAAUGCCAA
kmer indexAUUC
k=4
1UUCC 2

Filtering algorithm
1. Build a hash table hash table of kmers in the DB
AUUCCGGGAACAUAUUCUAGGCGACGGAUUAGAAUGCCAA
kmer indexAUUC
k=4
UUCG
1UUCC 2
3

Filtering algorithm
1. Build a hash table hash table of kmers in the DB
AUUCCGGGAACAUAUUCUAGGCGACGGAUUAGAAUGCCAA
kmer indexAUUC
k=4
UUCGCCGG
1UUCC 2
34

Filtering algorithm
1. Build a hash table hash table of kmers in the DB
AUUCCGGGAACAUAUUCUAGGCGACGGAUUAGAAUGCCAA
kmer indexAUUC
k=4
UUCGCCGG
1UUCC 2
34
14

Filtering algorithm
2. Identify (k,w)-stacks
AUUCCGGGAACAUAUUCUAGGCGACGGAUUAGAAUGCCAA
kmer indexAUUC
k=4
UUCGCCGG
1UUCC 2
34
14
reverse complement
GAAU n

Filtering algorithm
2. Identify (k,w)-stacks
AUUCCGGGAACAUAUUCUAGGCGACGGAUUAGAAUGCCAA
kmer indexAUUC
k=4
UUCGCCGG
1UUCC 2
34
14
reverse complement
GAAU n
d w?

Filtering algorithm
3. Compute complex stacks using DP
nested
parallel
multiloop

Result of stage 1 (filtering)AGAGCGUAUCGAUUUAGAGAGCUAUAGCUAGAGAGGAGA
UUAUAGCGCGCAUAUAGGACAAACAGUCUCUAUGGGGAC
AUUCCGGGAACAUAGUAUAGGCGACGGAUUAGCUAGCCA
AUCGCGCUAUAGCUAGCGAGGACAGCUAUAGCUAGCGAG
AUAUCGGGCUGUGGACACUAUACGAUCGAAUCUAGCUAU
AUCGCGCUAUAGCUAGCGAGGACAGCUAUAGCUAGCGAG
AUAUCGGGCUGUGGACACUAUACGAUCGAAUCUAGCUAU

• Stage 2: alignalign the selected sequences in the DB with the query and determine the best alignments
Solution
filterfilter
• Stage 1: filterfilter the DB

Possible ways to align RNAs
1. sequence to sequence
2. structure to structure
3. sequence to structure

RNA sequence structure alignment
AGAGCGUAUCGAUUUAGAGAGCUAUAGCUAGAGAGGAGA
QueryQuery
s [1,……………………………………………………..m]
UUAUAGCGCGCAUAUAGGACAAACAGUCUCUAUGGGGAC
t [1,……………………………………………………..n]
DB (filtered)DB (filtered)
S (set of base pairings)

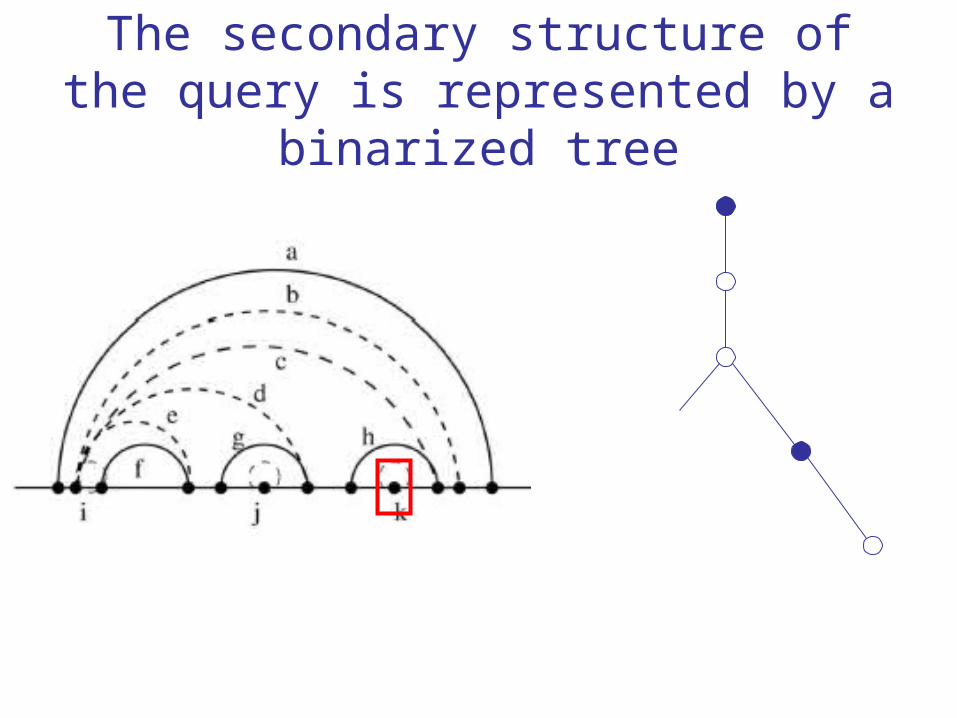
The secondary structure of the query is represented by a binarized tree
ji j -1i +1
i - jRule 1: when i and j are
paired

The secondary structure of the query is represented by a binarized tree
ji j -1
Rule 2: when j is unpaired

Rule 3: when j is paired
but not to the left-most base
The secondary structure of the query is represented by a binarized tree
jk -1i k

The secondary structure of the query is represented by a binarized tree
ji

The secondary structure of the query is represented by a binarized tree

The secondary structure of the query is represented by a binarized tree

The secondary structure of the query is represented by a binarized tree

The secondary structure of the query is represented by a binarized tree
The secondary structure of the query is represented by a binarized tree

The secondary structure of the query is represented by a binarized tree

The secondary structure of the query is represented by a binarized tree

The secondary structure of the query is represented by a binarized tree

The secondary structure of the query is represented by a binarized tree

The secondary structure of the query is represented by a binarized tree

The secondary structure of the query is represented by a binarized tree

The secondary structure of the query is represented by a binarized tree

The secondary structure of the query is represented by a binarized tree

The secondary structure of the query is represented by a binarized tree

The secondary structure of the query is represented by a binarized tree
Final binary tree

The secondary structure of the query is represented by a binarized tree
Binary treeBinary tree

Alignment algorithm

Alignment algorithm
Optimal alignment between the query (with structure v) and substring (i-j) of the DB
black node
white node / one child
white node / two children
Optimal alignment between the query (with structure v) and substring (i-j) of the DB
Optimal alignment between the query (with structure v) and substring (i-j) of the DB

Optimal alignment
i
jv
A [i,j,v]

Alignment algorithm
white node / one child
white node / two children
black node

Alignment algorithm
white node / one child
white node / two children
black node
ji j -1i +1
i - j
j -1i +1
i - j
alignment score for pairing and structure

Alignment algorithm
black node
white node / one child
white node / two children
ji j -1
i - j
ji
j -1
i - j
j-1
alignment score for pairing

Alignment algorithm
black node
white node / one child
white node / two children
jk -1i k
sliding k

Validation
Known instances of ncRNAs (tRNA, 5S rRNA, ribozymes, riboswitches) are
inserted in a random sequence (1Mb)
Filtering and alignment algorithms are applied (with k, l, w, %GC)

Validation: filtering
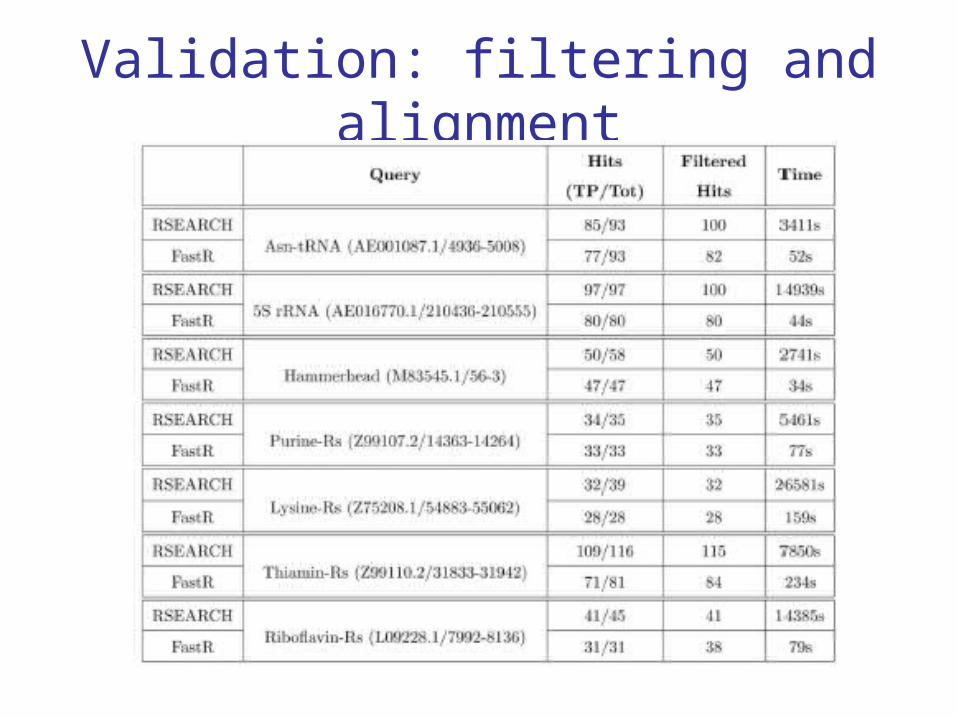
Validation: filtering and alignment

Results: riboswitches
5’ 3’
promoter
5’ UTR exons 3’ UTR
introns
coding
non-coding

Riboswitches

Riboswitches

Riboswitch families

Results: new riboswitches

Results: new riboswitches

RealReal motivation of bioinformaticists:
“We design novel filters and show that they dominatedominate the HMM filters of Weiberg and Ruzzo…”
world domination
