s5 ch22 toxic_effectsofpesticides
TRANSCRIPT

UNIT 5
TOXIC AGENTS
2996R_ch22_761-810 4/16/01 4:37 PM Page 761
Copy
right
ed M
ater
ial
Copyright © 2001 by The McGraw-Hill Companies Retrieved from: www.knovel.com

INTRODUCTION
The U.S. Environmental Protection Agency (U.S. EPA) defines apesticide as any substance or mixture of substances intended forpreventing, destroying, repelling, or mitigating any pest. A pesti-cide may also be described as any physical, chemical, or biologi-cal agent that will kill an undesirable plant or animal pest. Theterm pest includes harmful, destructive, or troublesome animals,plants, or microorganisms. Pesticide is a generic name for a vari-ety of agents that are classified more specifically on the basis ofthe pattern of use and organism killed. In addition to the majoragricultural classes that encompass insecticides, herbicides, andfungicides, one finds pest-control agents grouped as acaricides, lar-vacides, miticides, molluscides, pediculicides, rodenticides, scabi-cides, plus attractants (pheromones), defoliants, desiccants, plantgrowth regulators, and repellants.
HISTORICAL DEVELOPMENT
Over the centuries, humans have developed many ingeniousmethods in their attempts to control the invertebrates, vertebrates,
and microorganisms that constantly threatened the supply of foodand fiber as well as posing a threat to health. The historical liter-ature is replete with descriptions of plant diseases and insectplagues and the measures taken to control them. Sulfur was usedas a fumigant by the Chinese before 1000 B.C. and as a fungicidein the 1800s in Europe against powdery mildew on fruit; it is stillthe major pesticide used in California today. In sixteenth-centuryJapan, poor-quality rendered whale oil was mixed with vinegar andsprayed on paddies and fields to prevent the development of insectlarvae by weakening the cuticle. The Chinese applied moderateamounts of arsenic-containing compounds as insecticides in thesixteenth century. As early as 1690, water extracts of tobacco leaves(Nicotiana tabacum) were sprayed on plants as insecticides, andnux vomica, the seed of Strychnos nux-vomica (strychnine), wasintroduced to kill rodents. In the mid-1800s, the pulverized root ofDerris eliptica, containing rotenone, was used as an insecticide, aswas pyrethrum extracted from the flowers of the chrysanthemum(Chrysanthemum cineariaefolum). In the late 1800s, arsenic triox-ide was used as a weed killer, particularly for dandelions. Bordeauxmixture—copper sulfate, lime [Ca(OH)2], and water—was intro-duced in 1882 to combat vine downy mildew (Plasmopara viti-
INTRODUCTION
HISTORICAL DEVELOPMENT
REGULATORY MANDATE
Exposure
INSECTICIDES
Organochlorine CompoundsSigns and Symptoms of PoisoningSite and Mechanism of Toxic ActionsBiotransformation, Distribution, and StorageTreatment of Poisoning
Anticholinesterase AgentsSigns and Symptoms of PoisoningMechanism of Toxic ActionBiotransformation, Distribution, and StorageTreatment of Poisoning
Pyrethroid EstersSigns and Symptoms of PoisoningSite and Mechanism of ToxicityBiotransformation, Distribution, and StorageTreatment of Poisoning
AvermectinsMechanism of Action
Newer Chemical InsecticidesNitromethylenesChloronicotinylPhenylpyrazoles
BOTANICAL INSECTICIDES
NicotineRotenoids
HERBICIDES
Chlorophenoxy CompoundsBipyridyl DerivativesChloroacetanilidesPhosphonomethyl Amino Acids
GlyphosateGlufosinate
FUNGICIDES
HexachlorobenzenePentachlorophenolPhthalimidesDithiocarbamates
FUMIGANTS
PhosphineEthylene Dibromide/Dibromochloropropane
RODENTICIDES
Zinc PhosphideFluoroacetic Acid and DerivativesA-Naphthyl ThioureaAnticoagulants
CONCLUSIONS
CHAPTER 22
TOXIC EFFECTS OF PESTICIDES
Donald J. Ecobichon
763
2996R_ch22_761-810 4/16/01 4:37 PM Page 763
Copy
right
ed M
ater
ial
Copyright © 2001 by The McGraw-Hill Companies Retrieved from: www.knovel.com

764 UNIT 5 TOXIC AGENTS
cola), a disease introduced into France from the United States whenphylloxera-resistant vine rootstocks were imported. Sulfuric acid,at a concentration of 10% v/v, was used in the early 1900s to de-stroy dicotyledonous weeds that would absorb the acid, whereascereal grains and substitute plants, having a smooth and waxymonocotyledon, were protected. Paris Green (copper arsenite) wasintroduced to control the Colorado beetle in the late 1800s; cal-cium arsenate replaced Paris Green and lead arsenate was a majorcornerstone in the agriculturalist’s armamentarium against insectpests in the early 1900s. By the 1900s, the widespread use of ar-senical pesticides caused considerable public concern becausesome treated fruits and vegetables were found to have toxicresidues. Although some of these early pesticides caused only min-imal harm to the humans exposed, other agents were exceedinglytoxic, and the medical literature of the era is sprinkled with anec-dotal reports of poisonings. Looking back over the early years ofpesticide development, before the 1930s, it is somewhat surprisingto realize just how few pesticides were available (Cremlyn, 1978).
The 1930s ushered in the era of modern synthetic chemistry,including the development of a variety of agents such as alkyl thio-cyanate insecticides, dithiiocarbamate fungicides, ethylene dibro-mide, methyl bromide, and ethylene oxide (Cremlyn, 1978). Bythe beginning of World War II, there were a number of pesticides,including dichlorodiphenyltrichloroethane (DDT), dinitrocresol,4-chloro-2-methyloxyacetic acid (MCPA), and 2.4-dichlorophe-noxyacetic acid (2,4-D) under experimental investigation, much ofthis activity being kept under wraps during the war (Kirby, 1980).In the postwar era, there was rapid development in the agrochem-ical field, with a plethora of insecticides, fungicides, herbicides,and other chemical agents being introduced. In no other field ofchemistry has there been such a diversity of structures arising fromthe application of the principles of chemistry to the mechanism(s)of action in pests to develop selectivity and specificity in agentstoward certain species while reducing toxicity to other forms oflife.
Despite the modern-day development of second- and third-generation derivatives of the early chemical pesticides, all pesti-cides possess an inherent degree of toxicity to some living organ-ism, otherwise they would be of no practical use. Unfortunately,the target-species selectivity of pesticides is not as well developedas might be hoped for, and nontarget species frequently are affectedbecause they have physiologic and/or biochemical systems similarto those of the target organisms. There is no such thing as a com-pletely safe pesticide. There are, however, pesticides that can beused safely and/or that present a low level of risk to human healthwhen applied with proper attention to the label’s instructions.Despite the current controversy over pesticide use and the presenceof low levels of residues in food, groundwater, and air, these agentsare integral components of our crop- and health-protection pro-grams. As long as pesticides continue to be used, accidental and/orintentional poisoning of wildlife, domestic stock, and humans canbe anticipated and will require treatment.
On a worldwide basis, intoxications attributed to pesticideshave been estimated to be as high 3 million cases of acute, severepoisoning annually, with as many or more unreported cases andsome 220,000 deaths (WHO, 1990). These estimates suffer frominadequate reporting of data for developing countries, but they maynot be too far off the mark. From estimations based on Californiadata, a total of some 25,000 cases of pesticide-related illness oc-cur annually among agricultural workers in that state, with nationalestimate for the United States as a whole being on the order 80,000
cases per year (Coye et al., 1986). Results from California, a statethat uses a vast amount of chemical pesticides, revealed that 1087occupationally related exposures occurred in 1978. A breakdownof these poisonings by job category, as shown in Fig. 22-1, revealedthat ground applicators were at greatest risk, whereas aerial appli-cators and workers involved in mosquito-abatement programs hadthe least pesticide-related illness (Kilgore, 1980, 1988). Of 1211cases of pesticide-related illness reported to California physiciansin 1986, a total of 1065 were occupational in nature (Edmiston andMaddy, 1987). However, in other countries, the incidence of poi-soning is very low; for example, in the United Kingdom, fewerthan 20 agricultural incidents with organophosphates are reportedeach year (Weir et al., 1992). Such data are not representative ofthe rest of the agricultural world.
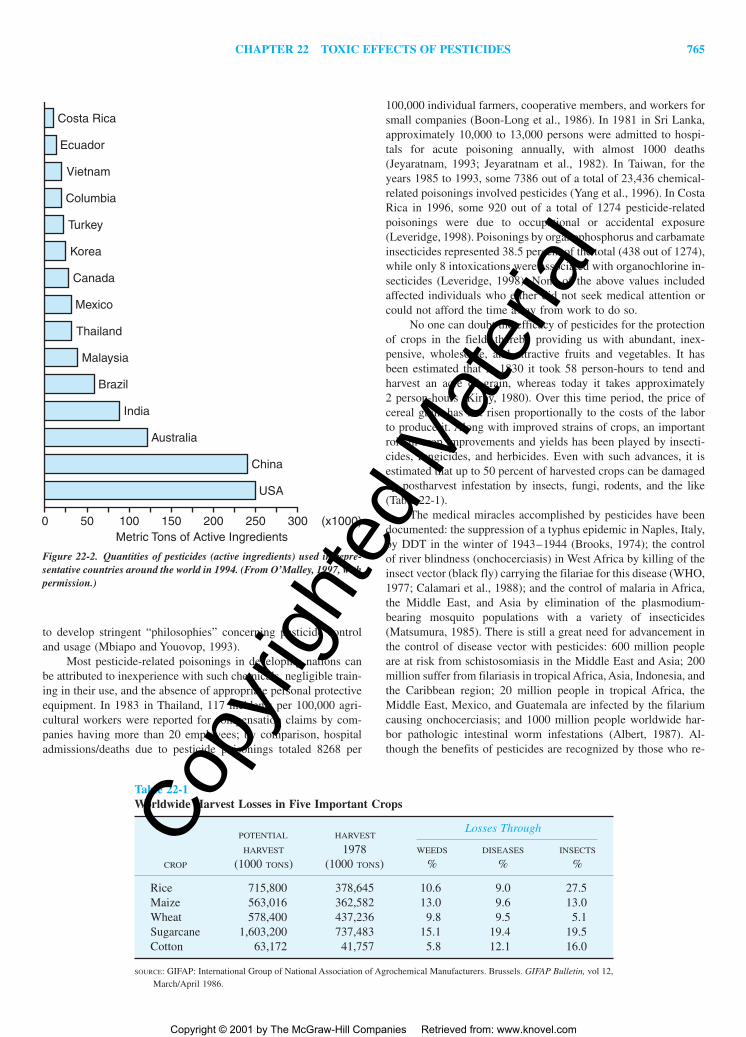
The incidence of poisoning is 13-fold higher in developingcountries than in highly industrialized nations, which consume 85percent of world’s pesticide production (Forget, 1989). A recentlypublished proceedings gives a good overview of the situation indeveloping nations, where there are few regulations controlling theimportation, registration, and sale of pesticides (Forget, 1993).Many countries in Central and South America, Africa, and South-east Asia are becoming “breadbaskets” for countries of more tem-perate climate, being sources of fresh fruits, vegetables, cut flow-ers, and so on in the off-season, since they are capable of growingtwo or three crops of export produce each year. Figure 22-2 showsthe quantities of pesticidal active ingredients used in a number ofcountries in 1994 (UN FAO, 1994). A more complete listing canbe found in O’Malley (1997). In developed countries, more herbi-cides are used than any other class of pesticides, whereas in trop-ical, developing nations, there is a predominant use of insecticides.As other developing nations explore the global export produce mar-ket, pesticide consumption—now around 3000 to 10,000 metrictons—will increase dramatically. Most developing nations have yet
250200150100500Number of Human Pesticide Exposure Illnesses
Mosquito Abatement
Aerial Applicators
Fumigators
Firemen, Police
Manufacturing and Pest Control
Agricultural Workers and Others
Field Workers
Structural
Mixers and Loaders
Warehouse and Indoors
Gardeners and Nurserymen
Ground Applicators
Figure 22-1. Frequency of pesticide poisoning related to occupation andpotential for exposure. [From the records of the California Departmentof Public Health (Kilgore, 1988)].
2996R_ch22_761-810 4/17/01 11:47 AM Page 764
Copy
right
ed M
ater
ial
Copyright © 2001 by The McGraw-Hill Companies Retrieved from: www.knovel.com
CHAPTER 22 TOXIC EFFECTS OF PESTICIDES 765
to develop stringent “philosophies” concerning pesticide controland usage (Mbiapo and Youovop, 1993).
Most pesticide-related poisonings in developing nations canbe attributed to inexperience with such chemicals, negligible train-ing in their use, and the absence of appropriate personal protectiveequipment. In 1983 in Thailand, 117 incidents per 100,000 agri-cultural workers were reported for compensation claims by com-panies having more than 20 employees; by comparison, hospitaladmissions/deaths due to pesticide poisonings totaled 8268 per
100,000 individual farmers, cooperative members, and workers forsmall companies (Boon-Long et al., 1986). In 1981 in Sri Lanka,approximately 10,000 to 13,000 persons were admitted to hospi-tals for acute poisoning annually, with almost 1000 deaths(Jeyaratnam, 1993; Jeyaratnam et al., 1982). In Taiwan, for theyears 1985 to 1993, some 7386 out of a total of 23,436 chemical-related poisonings involved pesticides (Yang et al., 1996). In CostaRica in 1996, some 920 out of a total of 1274 pesticide-relatedpoisonings were due to occupational or accidental exposure(Leveridge, 1998). Poisonings by organophosphorus and carbamateinsecticides represented 38.5 percent of the total (438 out of 1274),while only 8 intoxications were associated with organochlorine in-secticides (Leveridge, 1998). None of the above values includedaffected individuals who either did not seek medical attention orcould not afford the time away from work to do so.
No one can doubt the efficacy of pesticides for the protectionof crops in the field, thereby providing us with abundant, inex-pensive, wholesome, and attractive fruits and vegetables. It hasbeen estimated that in 1830 it took 58 person-hours to tend andharvest an acre of grain, whereas today it takes approximately 2 person-hours (Kirby, 1980). Over this time period, the price ofcereal grain has not risen proportionally to the costs of the laborto produce it. Along with improved strains of crops, an importantrole in crop improvements and yields has been played by insecti-cides, fungicides, and herbicides. Even with such advances, it isestimated that up to 50 percent of harvested crops can be damagedby postharvest infestation by insects, fungi, rodents, and the like(Table 22-1).
The medical miracles accomplished by pesticides have beendocumented: the suppression of a typhus epidemic in Naples, Italy,by DDT in the winter of 1943–1944 (Brooks, 1974); the controlof river blindness (onchocerciasis) in West Africa by killing of theinsect vector (black fly) carrying the filariae for this disease (WHO,1977; Calamari et al., 1988); and the control of malaria in Africa,the Middle East, and Asia by elimination of the plasmodium-bearing mosquito populations with a variety of insecticides(Matsumura, 1985). There is still a great need for advancement inthe control of disease vector with pesticides: 600 million peopleare at risk from schistosomiasis in the Middle East and Asia; 200million suffer from filariasis in tropical Africa, Asia, Indonesia, andthe Caribbean region; 20 million people in tropical Africa, theMiddle East, Mexico, and Guatemala are infected by the filariumcausing onchocerciasis; and 1000 million people worldwide har-bor pathologic intestinal worm infestations (Albert, 1987). Al-though the benefits of pesticides are recognized by those who re-
0 50 100 150 200 250 300 (x1000)Metric Tons of Active Ingredients
USA
China
Australia
India
Brazil
Malaysia
Thailand
Mexico
Canada
Korea
Turkey
Columbia
Vietnam
Ecuador
Costa Rica
Figure 22-2. Quantities of pesticides (active ingredients) used in repre-sentative countries around the world in 1994. (From O’Malley, 1997, withpermission.)
Table 22-1Worldwide Harvest Losses in Five Important Crops
Losses Through
HARVEST 1978 WEEDS DISEASES INSECTS
CROP (1000 TONS) (1000 TONS) % % %
Rice 715,800 378,645 10.6 9.0 27.5Maize 563,016 362,582 13.0 9.6 13.0Wheat 578,400 437,236 9.8 9.5 5.1Sugarcane 1,603,200 737,483 15.1 19.4 19.5Cotton 63,172 41,757 5.8 12.1 16.0
SOURCE: GIFAP: International Group of National Association of Agrochemical Manufacturers. Brussels. GIFAP Bulletin, vol 12,March/April 1986.
POTENTIAL HARVEST
2996R_ch22_761-810 4/16/01 4:37 PM Page 765
Copy
right
ed M
ater
ial
Copyright © 2001 by The McGraw-Hill Companies Retrieved from: www.knovel.com

766 UNIT 5 TOXIC AGENTS
quire them, certain parts of the world are experiencing an envi-ronmentalist- and media-evoked backlash toward all pesticide usebecause of the carelessness, misuse, and/or abuse of some agentsby a relatively few individuals in a limited number of well-publi-cized incidents. Without bearing any direct responsibility for plan-ning or involvement in health care or food or fiber production, someenvironmental and consumer advocacy groups propose a total banon pesticide use. Between the two extremes of overwhelming useand total ban lies a position advocating the careful and rational useof the beneficial chemicals.
REGULATORY MANDATE
The widespread use and misuse of the early, toxic pesticides cre-ated an awareness of the potential health hazards posed by themand the need to protect the consumer from residues in foods. It wasnot until 1906 that the Wiley or Sherman Act was passed, creatingthe first Federal Food and Drugs Act. This was replaced by theFederal Food, Drug and Cosmetic Act (FDCA) in 1938, with spe-cific pesticide amendments being passed in 1954 and 1958; theserequired that pesticide tolerances be established for all agriculturalcommodities. The 1958 amendment contained the famous Delaneyclause (Section 409), which states that “no additive shall be deemedsafe if it is found to induce cancer when ingested by man or ani-mal or, if it is found, after tests which are appropriate for the eval-uation of the safely of food additives, to induce cancer in man oranimals” (National Academy of Sciences, 1987). It should be notedthat the Delaney clause does not require proof of carcinogenicityin humans. Pesticides fall under this “additive” legislation.
The Federal Insecticide, Fungicide, and Rodenticide Act(FIFRA) was originally passed by Congress in 1947 as a labelingstatute that would group all pest control products—initially onlyinsecticides, fungicides, rodenticides, and herbicides—under onelaw to be administered by the U.S. Department of Agriculture(USDA). Amendments in 1959 and 1961 added nematicides, plantgrowth regulators, defoliants, and desiccants to FIFRA jurisdiction,plus the authorizations to deny, suspend, or cancel registrations ofproducts, although it assured the registrant’s right to appeal. In1972, FIFRA was reorganized and administrative authority wasturned over to the newly formed Environmental Protection Agency(EPA). The new law, along with subsequent amendments in 1975,1978, 1980, and 1984, defines the registration requirements andappropriate chemical, toxicologic, and environmental impact stud-ies, label specifications, use restrictions, tolerances for pesticideresidues on raw agricultural products, and the responsibility tomonitor pesticide residue levels in foods. FIFRA is not all-encompassing because the Food and Drug Administration (FDA)retains the basic responsibility for both monitoring residue levelsand for seizure of foods not in compliance with the regulations;what is more, the USDA continues to be the authority responsiblefor the monitoring of meat and poultry for pesticides as well as forother chemicals. The Food Quality Protection Act (FQPA), passedby the U.S. Congress in 1996, amended federal laws regarding pes-ticides to give special consideration for children by (1) providingadditional protection when allowable levels of pesticides for foodsare set (data providing children’s food consumption patterns, recog-nition of all possible routes of exposure in risk assessment); and(2) considering whether infants and children are disproportionallysusceptible to pesticides. Where data on the pesticides are not ad-equate, pesticide tolerances for children must incorporate an addi-tional 10-fold safety factor.
FIFRA regulations set out the requirements essential beforeEPA-Office of Programs review of any pesticide and/or formulatedproduct can occur for registration purposes. This information baseincludes product and residue chemistry, environmental fate, toxi-cology, biotransformation/degradation, occupational exposure andreentry protection, spray drift, environmental impact on nontargetspecies, and product performance and efficacy. Depending on theproposed use pattern of the pesticide, results from different“groups” or toxicologic studies are required to support the regis-tration. The typical spectrum of basic pesticide toxicity data re-quired under FIFRA regulations is summarized in Table 22-2. Extensive ancillary studies of environmental impact on birds,mammals, aquatic organisms, plants, soils, environmental persist-ence, and bioaccumulation are also required. A schematic diagramshowing the “information package” required in support of a regis-tration and the appropriate time span required to develop this data-base—from the point of patenting the newly synthesized chemicaluntil its registration, production, marketing, and user acceptabil-ity—is shown in Fig. 22-3. Although the ultimate uses of the par-ticular chemical will govern the extent of the information base re-quired prior to registration, estimates of average development costson the order of $30 to $80 million are not unrealistic.
Other nations including Canada, the United Kingdom, Japan,and, more recently, the European Economic Community (EEC)have promulgated harmonizing legislation similar to that of theUnited States as safeguards in human exposure to pesticides in foodcommodities. Some developing nations, with a shortage of trainedtechnical, scientific, and legal professionals to develop their ownlegislation, have adopted the regulatory framework of one or an-other of the industrialized nations, permitting the sale and use of
Table 22-2Basic Requirements Regarding Toxicity Data for NewPesticide Registrations
AcuteOral (rat)Dermal (rabbit)Inhalation (usually rat)Irritation studiesEye (rabbit)Skin (rabbit, guinea pig)Dermal sensitization (guinea pig)Delayed neurotoxicity (hen)
Subchronic90-Day feeding studyRodent (rat, mouse)Nonrodent (dog)Dermal Dependent upon use pattern and po-Inhalation tential for occupational exposureNeurotoxicity
ChronicOne- or two-year oral studyRodent (usually rat)Nonrodent (dog)Oncogenicity study (rat or mouse)
ReproductiveIn vitro mutagenicity (microorganisms, etc.)Fertility/reproduction (rat, mouse, rabbit)Teratogenicity (rat, mouse, rabbit)
�
2996R_ch22_761-810 4/16/01 4:37 PM Page 766
Copy
right
ed M
ater
ial
Copyright © 2001 by The McGraw-Hill Companies Retrieved from: www.knovel.com

CHAPTER 22 TOXIC EFFECTS OF PESTICIDES 767
pesticides registered under the legislation of that country but pro-hibiting the use of agents unable to meet the stringent requirements.In still other countries, almost any pesticide ever manufactured isavailable, no legislation having been introduced to curb adverse ef-fects to the environment and human health.
Exposure
The evaluation of the hazards of pesticides to human health beginswith the development of a dose-effect relationship based on docu-mented and anecdotal information on human exposure (Fig. 22-4).Several populations of individuals may be identified as having ex-posure to a range of concentrations of a particular agent, includ-ing: (1) accidental and/or suicidal poisonings that no amount oflegislation or study can prevent; (2) occupational (manufacturing,mixing/loading, application, harvesting, and handling of crops) ex-posure (Albertson and Cross, 1993; Edmiston and Maddy, 1987);(3) bystander exposure to off-target drift from spraying operationswith, in some cases, the development of hypersensitivity (Bartle,1991); (4) the general public who consume food items containingpesticide residues as a consequence of the illegal use of an agent(e.g., aldicarb on melons and cucumbers) or its misuse, in termsof an incorrect application rate or picking and shipping a crop toosoon after pesticide application, resulting in residue concentrationsabove established tolerance levels. The media are replete with doc-umented incidents of environmental contamination by pesticides:(1) of surface and/or groundwater essential as sources of potabledrinking water; (2) of commercial fish stock as well as sportingfish; (3) of wildlife upon which native peoples depend as a majorsource of dietary protein; and (4) of long-distance aerial transportof undeposited and/or revolatilized pesticide.
The shape of the dose-effect curve is dependent on a detailedknowledge of the amount of exposure received by each of thesegroups. Within each group, variability will be considerable. Fre-quently, exposure evaluations begin at the top of the relationshipwhere exposure is greatest, more easily estimated, and, in most
cases, the acute biological effects are clearly observed and may beassociated with a specific agent or a class of chemicals over a rel-atively narrow dosage range. If no discernible adverse health ef-fects are seen at high levels of exposure, it is unlikely that any-thing will be observed at lower levels of exposure. Although thishypothesis may be true for acute systemic effects, it is not appli-cable to chronic effects (changes in organ function, mutagenicity,teratogenicity, carcinogenicity) that may develop after some latentperiod following either a single high-level exposure, repeated mod-erate or high-level exposure, or annual exposure to low levels ofthe agents for decades.
There is sufficiently detailed documentation on many pestici-dal poisonings to permit an estimation of exposure (Hayes, 1982).In some 48 suicide attempts by ingestion of the herbicideglyphosate, the average volume of product (concentrate containingactive ingredient and a surfactant) ingested was 120 mL [range of104 mL (nonfatal) to 206 mL (fatal)] (Swada et al., 1988). In othercases, such as one involving the insecticide fenitrothion, where theindividual experienced dermal exposure to a 7.5% solution of theagent in corn oil wiped up with facial tissues by a bare hand, ex-posure was more difficult to assess (Ecobichon et al., 1977). It isimperative that forensic and clinical toxicologists and emergencyservice personnel attempt to ascertain how much of the materialwas involved in the poisoning.
Worker exposure can be estimated within reason by consid-ering the various job functions performed (e.g., diluting concen-trated formulations, loading diluted end-use formulations intotanks, spray application, harvesting sprayed crops, postharvest han-dling of sprayed crops, etc.). The potential level(s) of pesticide en-countered in each job category and the route(s) of exposure can beestimated. The majority of occupational illnesses arising from pes-ticides involve dermal exposure enhanced, in certain job categories,by acquisition of a portion of the dosage by the inhalation of theaerosolized spray. Many exposures appear to be entirely dermal incharacter (Vercruysse et al., 1999). The surface areas of the un-clothed parts of the body of an unprotected worker are shown in
Figure 22-3. A schematic diagram depicting the generation of an appropriate toxicity database, the timeframe for data acquisition and the significant milestones in the life cycle of a pesticide in the United States.Stages I to III represent the sales of the pesticide once the commercial product enters the marketplace. (GI-FAP Bulletin, Sept. 1983, with permission.)
2996R_ch22_761-810 4/16/01 4:37 PM Page 767
Copy
right
ed M
ater
ial
Copyright © 2001 by The McGraw-Hill Companies Retrieved from: www.knovel.com

768 UNIT 5 TOXIC AGENTS
Table 22-3, being derived from the U.S. EPA (1987). Data for theentire surface area of a “50 percentile man,” as determined by Spearand coworkers (1977), are shown in Table 22-4. With surface patch(gauze, fabric) testing on various parts of the body, accurate esti-mates of dermal exposure can be obtained. The reader is referredto the following studies for details: Wolfe and colleagues (1967,1972), Wojeck and coworkers (1981), and Franklin and coworkers(1981). Where inhalation can be considered to contribute signifi-cantly to the total exposure, as in greenhouse and other structuralspraying operations in enclosed environments, drivers in tractorcabs, operators of rotary fan mist sprayers, and other operations,measurements of aerial concentrations in the working environmentcan be made and related to respiratory rates and length of timespent in that environment. Assessment of the inhalation componentof an exposure can be obtained with personal air sampling moni-tors worn during the day (Trumbull et al., 1985; Grover et al.,1986).
In the past, direct exposure was estimated by measuring dep-osition (on skin, clothing, or surrogate patch) or by ambient air
concentrations of the potentially toxic agent (Durham and Wolfe,1962). In reality, direct exposure should be attributed only to pes-ticide residues gaining entry into the body by systemic absorptionfollowing ingestion, inhalation, and/or transdermal penetration. To-tal exposure can be estimated by measuring excretory products (theparent chemical, degradation products) in urine and feces over asuitable postexposure time interval (Durham et al., 1972;Kolmodin-Hedmann et al., 1983; Frank et al., 1985; Grover et al.,1986; Takamiya, 1994; Azaroff, 1999). Potential biological effectsof exposure can be monitored, including plasma and erythrocy-tic cholinesterase measurements, �-aminolevulinic dehydratase(ALAD), superoxide dismutase (SOD) activities, cytogenic analy-
Figure 22-4. A theoretical dose–effect relationship for acute toxicity, comparing the potential for exposurein terms of occupation, level of exposure and possible biological effects.
Table 22-3Estimated Surface Area of Exposed Portions of a Body of a Casually Dressed Individual
SURFACE AREA PERCENT OF
UNCLOTHED SURFACE (SQ FT) TOTAL
Face 0.70 22.0Hands 0.87 27.6Forearms 1.30 41.3Back of neck 0.12 3.8Front of neck and “v” of chest 0.16 5.1
SOURCE: Batchelor and Walker, 1954.
Table 22-4Percent of Total Body Surface Area Represented by BodyRegions*
SURFACE AREA
BODY REGION (% OF TOTAL)
Head 5.60Neck 1.20Upper arms 9.70Forearms 6.70Hands 6.90Chest, back, shoulders 22.80Hips 9.10Thighs 18.00Calves 13.50Feet 6.40
*Estimated proportions from the “50 percentile man” having a surface area of 1.92 m2,height of 175 cm, and body weight of 78 kg.
SOURCE: Spear et al., 1977.
2996R_ch22_761-810 4/16/01 4:37 PM Page 768
Copy
right
ed M
ater
ial
Copyright © 2001 by The McGraw-Hill Companies Retrieved from: www.knovel.com

CHAPTER 22 TOXIC EFFECTS OF PESTICIDES 769
sis of lymphocytic micronuclei, semen quality, fertility, etc.(Lopez-Carillo and Lopez-Cervantes, 1993; Ciesielski et al., 1994;Davies et al., 1998; Panemangalore et al., 1998; Venegas et al.,1998; Larsen et al., 1999). A more complete discussion of the topicof occupational exposure can be found in Ecobichon (1998a).
Minimal protection of certain parts of the body can markedlyreduce exposure to an agent. Protection of the hands (5.6 percentof the body surface) by appropriate chemical-resistant gloves mayreduce contamination by 33 percent (in forest spraying with a knap-sack sprayer having a single-nozzle lance), by 66 percent (in weedcontrol using tractor-mounted booms equipped with hydraulic noz-zles), or by 86 percent (involving filling tanks on tractor-poweredsprayers) (Bdonsall, 1985). Studies monitoring the absorption ofpesticides applied to the skin of different areas of the human bodyhave revealed marked regional variations in per cutaneous absorp-tion, with the greatest uptake being in the scrotal region, followedby the axilla, forehead, face, scalp, the dorsal aspect of the hand,the palm of the hand, and the forearm in decreasing order (Feldmanand Maiback, 1974).
The exposure of a bystander, where an individual is acciden-tally sprayed directly or exposed to aerial off-target drifting aerosol,is considerably more difficult to assess. The levels encountered maybe severalfold lower than those in the occupational setting, mak-ing the analysis of residue and the detection of meaningful bio-logical changes more difficult. Greater variation in exposure esti-mates and biological effects can be anticipated. The adverse healtheffects may be subtle in appearance and nonspecific, reflecting aslow deterioration of physiologic function clouded by the individ-ual’s adjustment or adaptation to the changes, taking many yearsto develop to the point of detection. The identification of pesticide-related adverse health effects in the general population, who inad-vertently acquire low levels of pesticides daily via food and water,is extremely difficult. The residue levels in these media are oftenorders of magnitude lower than those encountered in occupationalor bystander exposure and are at or near the limits of analyticaldetection by sophisticated techniques. Any biological effects re-sulting from such low-level exposure are unlikely to be distinctiveand any causal association with a particular chemical or class ofagents is likely to be tenuous and confounded by many other fac-tors of a given lifestyle.
Many of the public concerns about pesticides are related to“older” chemicals, these having entered the market in the 1950sand 1960s without the benefit of the extensive toxicity and envi-ronmental impact studies demanded prior to the registration ofchemicals today. It must also be pointed out that many of theseolder pesticides have received little reassessment using the moredefinitive techniques and protocols required today. Although gov-ernment agencies and industry have been slow in their reevalua-tion of a vast array of pesticides in use, reassessment often comesin the wake of or concomitant with some recently disclosed ad-verse environmental or health effect. Given the above-mentionedcosts of conducting a full range of studies (introductory section,this chapter), the time frame required, and the limited market forsome of these chemicals in North America or even worldwide, theregistration of many of these pesticides will be withdrawn volun-tarily by industry, and the answers to some of the public’s con-cerns will never be obtained. Hazardous chemicals will be removedfrom use but, unfortunately, it is possible that some very benefi-cial and essential pesticides will be lost. The problems of today’ssituation, created by the last generation and inherited by the pres-ent one, still must be dealt with.
INSECTICIDES
The literature pertaining to the chemistry and development of thevarious classes of insecticides over the past 45 years is extensiveand the reader is referred to the monographs of O’Brien (1960,1967), Melnikov (1971), Fest and Schmidt (1973), Brooks (1974),Eto (1974), Hayes (1982), Kuhr and Dorough (1976), Buchel(1983), Leahey (1985), Chambers and Levi (1992), and Ecobichonand Joy (1994) for detailed discussions of the chemistry, nomen-clature (chemical, common, and trade names), biotransformationand degradation, and environmental effects as well as target andnontarget species toxicity. Compilations of LD50 values in the lab-oratory rat may be found in Gaines (1969), Frear (1969), andWorthing (1987). Acute toxicity data for laboratory animals, fish,and wildlife are recorded in a number of reports (Pickering et al.,1962; Tucker and Crabtree, 1970; Worthing, 1987). Several com-pilations of pesticide monographs exist, giving brief but succinctprofiles of the physical and chemical properties of variouspesticides as well as their environmental persistence and toxicityto wildlife, domestic animals, and humans (Kamrin, 1997;Montgomery, 1997; Tomlin, 1997). Only selected examples of theclasses of insecticides are discussed in this chapter, with emphasison their toxicity to humans.
All of the chemical insecticides in use today are neurotoxi-cants and act by poisoning the nervous systems of the targetorganisms. The central nervous system (CNS) of insects is highlydeveloped and not unlike that of the mammal (O’Brien, 1960).While the peripheral nervous system (PNS) of insects is not ascomplex as that of mammals, there are striking similarities(O’Brien, 1960). The development of insecticides has been basedon specific structure-activity relationships requiring the manipula-tion of a basic chemical structure to obtain an optimal shape andconfiguration for specificity toward a unique biochemical or phys-iologic feature of the nervous system. Given the fact that insecti-cides are not selective and affect nontarget species as readily astarget organisms, it is not surprising that a chemical that acts onthe insect’s nervous system will elicit similar effects in higher formsof life. The target sites and/or mechanism(s) of action may be sim-ilar in all species; only the dosage (level of exposure and duration)will dictate the intensity of biological effects. It is sufficient at thisstage to indicate the potential sites of action of the insecticideclasses (Fig. 22-5) and their interference with the membrane trans-port of sodium, potassium, calcium, or chloride ions; inhibition ofselective enzymatic activities; or contribution to the release and/orthe persistence of chemical transmitters at nerve endings.
Organochlorine Compounds
No longer considered an important class of insecticides in NorthAmerica and Europe, the organochlorine insecticides see contin-ued use in developing, tropical countries because they are effec-tive, inexpensive, essential chemicals in agriculture, forestry, struc-tural protection, and public health. The risk-benefit ratio is highlyweighted in favor of their continued use for the control of insectscausing devastation to crops and human health. For example,technical-grade hexachlorocyclohexane (HCH), banned in Canada,the United States, China, the Soviet Union, and Australia in 1971,1976, 1983, 1990 and 1994, respectively, still sees extensive usein a number of African nations, Brazil, India, and others (Li, 1999).While banned in the early 1970s, DDT was still being manufac-tured in the United States and exported at the rate of 1 ton per day
2996R_ch22_761-810 4/16/01 4:37 PM Page 769
Copy
right
ed M
ater
ial
Copyright © 2001 by The McGraw-Hill Companies Retrieved from: www.knovel.com

770 UNIT 5 TOXIC AGENTS
in 1994 (Smith, 1995). Global transport of such chemicals fromequatorial regions to Arctic and Antarctic regions, with contami-nation of wildlife food sources, suggests that these agents are stilltoxicologically important. Currently, major concerns are centeredon the indigenous people living in Arctic regions, where the sourcesof dietary protein (fish, seals, walruses, whales, etc.) have provento be major depositories of organochlorine insecticides and otherchlorinated hydrocarbons (PCBs, PBBs, PCDDs, PSDFs) (Berti etal., 1998; Chan, 1998).
The properties (low volatility, chemical stability, lipid solu-bility, slow rate of biotransformation and degradation) that madethese chemicals such effective insecticides also brought about their
demise because of their persistence in the environment, biocon-centration and biomagnification in food chains, and the acquisitionof biologically active body burdens at higher trophic levels (Carson,1962). Definitive studies in wild, domestic, and laboratory speciesdemonstrated potent enzyme-inducing and/or estrogenic proper-ties, with interference indirectly or directly with fertility and re-production (Stickel, 1968; McFarland and Lacy, 1969; Peakall,1970; Longcore et al., 1971; McBlain et al., 1977; Crum et al.,1993).
The organochlorine (chlorinated hydrocarbon) insecticides area diverse group of agents belonging to three distinct chemicalclasses including the dichlorodiphenylethane-, chlorinated cyclo- diene- and chlorinated benzene- and cyclohexane-related structures(Table 22-5). The historical development of these chemicals fromthe mid-1940s and their impact on agriculture and human healthcan be found in O’Brien (1967), Metcalf (1973), and Brooks(1974).
Signs and Symptoms of Poisoning Given the diversity of chem-ical structures, it is not surprising that the signs and symptoms oftoxicity and the mechanisms of action are somewhat different(Table 22-6).
Exposure of humans and animals to high oral doses of DDTresults in paresthesia of the tongue, lips, and face; apprehension;hypersusceptibilty to external (light, touch, sound) stimuli; irri-tability, dizziness, and vertigo; tremor and tonic and clonic con-vulsions. Motor unrest and fine tremors associated with voluntarymovements progress to coarse tremors without interruption in mod-erate to severe poisonings. Symptoms generally appear severalhours (6 to 24 h) after exposure to large doses. Little toxicity isseen following the dermal exposure to DDT, presumably becausethe agent is poorly absorbed through the skin, a physiologic phe-nomenon that has contributed to the fairly good safety record ofDDT despite careless handling by applicators and formulators(Hayes, 1971). It has been estimated that a dose of 10 mg/kg willcause signs of poisoning in humans. Chronic exposure to moder-ate concentrations of DDT causes somewhat milder signs to toxi-city, as listed in Table 22-6.
Figure 22-5. Potential sites of action of classes of insecticides on theaxon and terminal portions of the nerve.
Table 22-5Structural Classification of Organochlorine Insecticides
Dichlorodiphenylethanes DDT, DDDDicofolPerthaneMethoxychlorMethlochlor
Cyclodienes Aldrin, DieldrinHeptachlorChlordaneEndosulfan
Chlorinated benzenes HCB, HCHCyclohexanes Lindane (�-BHC)
C
Cl ClCH
Cl Cl
C(CCl)2
ClCl
Cl
Cl
Cl
Cl
Cl(Cl)6
Cl
Cl
2996R_ch22_761-810 4/16/01 4:37 PM Page 770
Copy
right
ed M
ater
ial
Copyright © 2001 by The McGraw-Hill Companies Retrieved from: www.knovel.com

CHAPTER 22 TOXIC EFFECTS OF PESTICIDES 771
Although the functional injury of DDT poisoning can be as-sociated with effects on the CNS in humans, few pathologicchanges can be demonstrated in CNS tissue in animals. However,following exposure to moderate or high nonfatal doses or subse-quent to subacute or chronic feeding, major pathologic changes areobserved in the liver and reproductive organs. Morphologicchanges in mammalian liver include hypertrophy of hepatocytesand subcellular organelles such as mitochondria, proliferation ofsmooth endoplasmic reticulum and the formation of inclusion bod-ies, centrolobular necrosis following exposure to high concentra-tions, and an increase in the incidence of hepatic tumors (Hayes,1959; Hansell and Ecobichon, 1974; IARC, 1974). However, therehas yet to be conclusive epidemiologic evidence linking DDT tocarcinogenicity in humans (Hayes, 1982; Baris et al., 1998;Takayama et al., 1999). When technical-grade DDT (20 percento,p�-DDT plus 80 percent p,p�-DDT) was administered to malecockerels or rats, reduced testicular size was observed and, in fe-male rats, the estrogenic effects of the o,p�-isomer were observedin the edematous, blood-engorged uteri (Hayes, 1959; Ecobichon
and MacKenzie, 1974). The o,p�-isomer has been shown to com-pete with estradiol for binding the estrogen receptors in rat uterinecytosol (Kupfer and Bulger, 1976). The DDT analog methoxychlor[1,1,1-trichloro-2,2-bis(4-methoxyphenyl) ethane] is estrogenic inthe mouse; problems in initiating and/or maintaining a pregnancyare seen, due possibly to alterations in preimplantation embryonicdevelopment and estrogenic effects on the oviduct and uterus (Hallet al., 1997; Swartz and Eroschenko, 1998).
Dicofol (p-p�-dichlorodiphenyl-2,2,2-trichloroethanol) — ananalog of DDT still registered as an agricultural miticide for cot-ton, beans, citrus, and grapes—has been associated with acute tox-icity (nausea, dizziness, double vision, ataxia, confusion, disorien-tation) in a 12-year-old male whose clothing because saturated inan accident (Lessenger and Riley, 1991). These acute effects pro-gressed to chronic signs (headaches, blurred vision, horizontal nys-tagmus, numbness/tingling in the legs with shooting pains, clum-siness, memory loss and decreasing academic performance,impulsive behavior, restlessness and fatigue), which persisted insome fashion for up to 4 months. Continuing emotional and aca-
Table 22-6Signs and Symptoms of Acute and Chronic Toxicity Following Exposure to Organochlorine Insecticides
INSECTICIDE CLASS ACUTE SIGNS CHRONIC SIGNS
DichlorodiphenylethanesDDT Parethesia (oral ingestion) Loss of weight, anorexiaDDD (Rothane) Ataxia, abnormal stepping Mild anemiaDMC (Dimite) Dizziness, confusion, headache TremorsDicofol (Kelthane) Nausea, vomiting Muscular weaknessMethoxychlor Fatigue, lethargy EEG pattern changesMethiochlor Tremor (peripheral) Hyperexcitability, anxietyChlorbenzylate Nervous tension
HexachlorocyclohexanesLindane (�-isomer)Benzene hexachloride (mixed isomers)
CyclodienesEndrin Dizziness, headache Headache, dizziness,Telodrin Nausea, vomiting hyperexcitabilityIsodrin Motor hyperexcitability Intermittent muscle twitchingEndosulfan Hyperreflexia and myoclonic jerkingHeptachlor Myoclonic jerking Psychological disordersAldrin General malaise including insomnia,Dieldrin Convulsive seizures anxiety, irritabilityChlordane Generalized convulsions EEG pattern changesToxaphene Loss of consciousness
Epileptiform convulsionsChlordecone (Kepone) Chest pains, arthralgiaMirex Skin rashes
Ataxia, incoordination, slurredspeech, opsoclonus
Visual difficulty, inability tofocus and fixate
Nervousness, irritability, depressionLoss of recent memoryMuscle weakness, tremors of
handsSevere impairment of spermatogenesis
2996R_ch22_761-810 4/16/01 4:37 PM Page 771
Copy
right
ed M
ater
ial
Copyright © 2001 by The McGraw-Hill Companies Retrieved from: www.knovel.com

772 UNIT 5 TOXIC AGENTS
demic difficulties, impairment of certain cognitive skills, poor self-esteem and depression, all of which were subtle cognitive and psy-chological changes, persisted for over 18 months. Dicofol is knownto be contaminated by a small percentage of p-p�-DDT, showingestrogenicity in birds (Peakall, 1970).
Unlike the situation with DDT, in which there have been fewrecorded fatalities following poisoning, there have been a numberof fatalities following poisoning by the cyclodiene- and hexa-chlorocyclohexane-type insecticides. The chlorinated cyclodieneinsecticides are among the most toxic and environmentally per-sistent pesticides known (Hayes, 1982). One recent study of twopatients, one with a history of chronic exposure to aldrin and theother with a chronic exposure to lindane/heptachlor, reported deathwithin 2 years of developing clinical and electromyographic signsand symptoms of chronic motor disease with aggravation of dys-phagia and weight loss resulting in the mobilization of adipose tis-sue and stored insecticide to enhance the neuronal toxicity (Fon-seca et al., 1993). Even at low doses, these chemicals tend to induceconvulsions before less serious signs of illness occur. Although thesequence of signs generally follows the appearance of headaches,nausea, vertigo, and mild clonic jerking, motor hyperexcitability,and hyperreflexia, some patients have convulsions without warn-ing symptoms (Hayes, 1982). An important difference betweenDDT and the chlorinated cyclodienes is that the latter are efficientlyabsorbed through the skin and therefore pose an appreciable haz-ard to occupationally exposed individuals. Chronic exposure to lowor moderate concentrations of these agents elicits a spectrum ofsigns and symptoms, involving both sensory and motor compo-nents of the CNS (Table 22-6). In addition to the recognized neu-rotoxicity, aldrin and dieldrin interfere with reproduction, with in-creased pup losses (vitality, viability) being reported in studies inrats and dogs (Kitselman, 1953; Treon and Cleveland 1955). Treat-ment with dieldrin during pregnancy caused a reduction in fertil-ity and increased pup mortality (Treon and Cleveland, 1955). Thetreatment of pregnant mice with dieldrin resulted in teratologic ef-fects (delayed ossification, increases in supernumerary ribs) (Cher-noff et al., 1975).
Exposure to lindane (the �-isomer of hexachlorocyclohexane,HCH) produces signs of poisoning that resemble those caused byDDT (e.g., tremors, ataxia, convulsions, stimulated respiration,and prostration). In severe cases of acute poisoning, violent tonic andclinic convulsions occur and degenerative changes in the liver andrenal tubules have been noted. Technical-grade HCH used in in-secticidal preparations contains a mixture of isomers: the �- and�-isomer are convulsant poisons; the �- and �-isomers are CNSdepressants. The mechanisms of action remain unknown. Lifetimefeeding studies in mice have revealed that technical-grade HCHand some of the isomers caused an increase in hepatocellular tu-mors (IARC, 1974). Only the �-isomer (lindane) sees any medic-inal use today, as a component of a pediculicide shampoo for headlice. One undocumented case of lindane toxicity, known to the au-thor, resulted in mild tremors in a child on whose head the sham-poo was used vigorously and repeatedly for more than a week. Thesymptoms disappeared rapidly when the treatment was terminated.
Industrial carelessness during the manufacture of anorganochlorine compound chlordecone (Kepone) brought thisagent and mirex, the closely related insecticide, to the attention oftoxicologists in 1975, when 76 of 148 workers in a factory inHopewell, Virginia, developed a severe neurologic syndrome(Cannon et al., 1978; Taylor et al., 1978; Guzelian, 1982). Thiscondition, known as the “Kepone shakes,” was characterized by
tremors, altered gait, behavioral changes, ocular flutter (opso-clonus), arthralgia, headache, chest pains, weight loss, he-patomegaly, splenomegaly, and impotence, the onset of symptomsgenerally occurring with a latency of approximately 30 days fromthe initiation of exposure and persisting for many months after thetermination of exposure (Joy, 1994a). Laboratory tests showed areduced sperm count and reduced sperm motility. Routine neuro-logic studies showed nothing untoward, but microscopic examina-tion of biopsies of the sural nerve revealed relative decreases in thepopulations of small myelinated and unmyelinated axons. Withelectron microscopy, a number of abnormalities were visible; thesignificant findings included damage to Schwann cells (membra-nous inclusions, cytoplasmic folds), prominent endoneural collagenpockets, vacuolization of unmyelinated fibers, focal degenerationof axons with condensations of neurofilaments and neurotubules,focal interlamellar splitting of myelin sheaths, and the formationof myelin bodies and a complex infolding of inner mesaxonal mem-branes into axoplasm (Martinez et al., 1977). The involvement ofunmyelinated fibers and small myelinated fibers may partially ex-plain the clinical picture. It has been suggested that chlordeconemay interfere with metabolic processes in Schwann cells. How-ever, it should be noted that all of these degenerative changes arenonspecific in nature and are commonly seen in other toxicpolyneuropathies. Many of the toxic manifestations of chlordeconepoisoning in these workers have been confirmed in animal studies,the major target organs being the CNS, liver, adrenals, and testes,as summarized by Joy (1994a). As with other organochlorine in-secticides, chlordecone is an excellent inducer of hepatic micro-somal monooxygenase enzymes and, in rats and mice, has beenassociated with the formation of hepatomas and malignant tumorsin organs other than the liver, female animals being more suscep-tible than male (Guzelian, 1982). In many ways, mirex behaveslike chlordecone, and there is evidence for the oxidative biotrans-formation of mirex to chlordecone in vivo. Mirex causes he-patomegaly and a dose-dependent increase in neoplastic nodulesand hepatocellular carcinomas, particularly in male animals (Inneset al., 1969; Waters et al., 1977).
Site and Mechanism of Toxic Actions Essential to the action oforganochlorine insecticides is an intact reflex are consisting of af-ferent (sensory) peripheral neurons impinging on interneurons inthe spinal cord, with accompanying ramifications and intercon-nections up and down the CNS and interactions with efferent mo-tor neurons, as shown schematically in Fig. 22-6. In terms of themechanism of action of the DDT-type insecticides, the most strik-ing observation in a poisoned insect or mammal is the display ofperiodic sequences of persistent tremoring and/or convulsiveseizures suggestive of repetitive discharges in neurons. These char-acteristic episodes of hyperactivity interspersed with normal func-tion were recognized as early as 1946. The second most strikingobservation is that these repetitive tremors, seizures, and electricalactivity can be initiated by tactile and auditory stimuli, suggestingthat the sensory nervous system appears to be much more respon-sive to stimuli. An examination of the sequence of electrical eventsin normal and DDT-poisoned nerves reveals that, in the latter, acharacteristic prolongation of the falling phase of the action po-tential (the negative afterpotential) occurs (Fig. 22-7). The nervemembrane remains in a partially depolarized state and is extremelysensitive to complete depolarization again by very small stimuli(Joy, 1994a). Thus, following exposure to DDT, the repetitive stim-ulation of the peripheral sensory nerves by touch or sound is mag-
2996R_ch22_761-810 4/16/01 4:37 PM Page 772
Copy
right
ed M
ater
ial
Copyright © 2001 by The McGraw-Hill Companies Retrieved from: www.knovel.com

CHAPTER 22 TOXIC EFFECTS OF PESTICIDES 773
nified in the CNS, causing generalized tremoring throughout thebody.
How does DDT elicit this effect? There are at least four mech-anisms, possibly all functioning simultaneously (Matsumura,1985), as seen in Fig. 22-8. At the level of the neuronal membrane,DDT affects the permeability to potassium ions, reducing potas-sium transport across the membrane. DDT alters the porous chan-nels through which sodium ions pass. These channels activate(open) normally, but, once open, are inactivated (closed) slowly,thereby interfering with the active transport of sodium out of thenerve axon during repolarization. DDT inhibits neuronal adenosinetriphosphatase (ATPase), particularly the Na�,K�-ATPase andCa2�-ATPase, which play vital roles in neuronal repolarization.DDT also inhibits the ability of calmodulin, a calcium mediator innerves, to transport calcium ions essential for the intraneuronal re-lease of neurotransmitters. All of these inhibited functions reducethe rate at which depolarization occurs and increase the sensitiv-ity of the neurons to small stimuli that would not elicit a responsein a fully depolarized neuron.
The chlorinated cyclodiene-, benzene-, and cyclohexane-typeinsecticides are different from DDT in many respects, both in theappearance of the intoxicated individual and possibly also in themechanism(s), which appear to be localized more in the CNS than
Figure 22-6. A simple, intact reflex arc involving a peripheral, afferent(sensory) neuron, interneurons, and a peripheral, efferent (motor) neu-ron that innervates a muscle.
Figure 22-7. A schematic diagram of an oscilloscope recording of thedepolarization and repolarization of a normal neuron ( ——— ) and onefrom a DDT-treated (---) animal, showing the prolonged, negative after-potential (AP).
Figure 22-8. Proposed sites of action of DDT on (1) reducing potassiumtransport through pores, (2) inactivating sodium channel closure, (3) in-hibiting sodium-potassium and calcium-magnesium ATPases, and (4)calmodulin-calcium binding with the release of neurotransmitter.
in the sensory division of the PNS (Table 22-6). The overall ap-pearance of the intoxicated individual is one of CNS stimulation.As shown in Fig. 22-9, the cyclodiene compounds antagonize theaction of the neurotransmitter gamma-aminobutyric acid (GABA)acting at the GABAA receptors in rat dorsal root ganglia and ef-fectively blocking the GABA-induced uptake of chloride ions bydesensitizing the current in the receptor-channel complex (Nagataand Narahashi, 1994). A more recent paper explains the fact thatdieldrin has a dual effect on the GABAA receptor-channel: an ini-tial enhancement of the GABA-induced chloride ion current (withan EC50 of 754 nM) followed by a suppression (Narahashi et al.,1995). Two types of chloride currents exist, one having a high sen-sitivity to dieldrin (IC50 � 5 nM) and the other having a lowersensitivity to dieldrin (IC50 � 92 nM). The dieldrin-suppressiveaction is responsible for the hyperactivity observed in poisoned an-imals. The nature of the GABAA receptor-channel is still beingexplored. The cyclodienes are also potent inhibitors of Na�,K�-ATPase and, more importantly, of the enzyme Ca2�, Mg2�-ATPaseessential for the transport (uptake and release) of calcium acrossmembranes (Matsumura, 1985; Wafford et al., 1989). Evidencesuggests that gamma-hexachlorocyclohexane (�-HCH, lindane)neurotoxicity is primarily related to the blockade of chloride ionflux through the inotropic GABAA receptors, resulting in depolar-ization and hyperexcitation of post-synaptic membranes (Mat-sumura and Tanaka, 1984). There is also evidence that �-HCH canalter calcium homeostasis, elevating free calcium ion levels intra-cellularly with the release of neurotransmitters (Joy, 1994a). Theinhibition of Ca2�, Mg2�-ATPase, located in the terminal ends ofneurons in synaptic membranes, results in an accumulation of in-tracellular free calcium ions with the promotion of calcium-induced
2996R_ch22_761-810 4/18/01 7:58 AM Page 773
Copy
right
ed M
ater
ial
Copyright © 2001 by The McGraw-Hill Companies Retrieved from: www.knovel.com

774 UNIT 5 TOXIC AGENTS
release of neurotransmitters from storage vesicles and the subse-quent depolarization of adjacent neurons and the propagation ofstimuli throughout the CNS.
Biotransformation, Distribution, and Storage The phenome-non of bioconcentration and biomagnification of the organochlo-rine insecticides in food chains has been mentioned. Once acquired,biotransformation/degradation proceeds at an exceptionally slowpace, in part due to the complex aromatic ring structures and thenumber of the chlorine substituents, the latter being exceedinglydifficult to remove by the enzymatic processes available in tissues.The biotransformation of the various organochlorines has been ex-tensively studied—for example, DDT (Ecobichon and Saschen-brecker, 1968), aldrin and heptachlor conversion to dieldrin andheptachlor epoxide, respectively (Keane and Zavon, 1969;Matthews and Matsumura, 1969); chlordane (Menzie, 1969); hexa-chlorocyclohexane (Egan et al., 1965; O’Brien, 1967; Abbott et al.,1969); and toxaphene (Saleh et al., 1977; Turner et al., 1977). Withthe exception of aldrin, chlordane, and heptachlor, most biotrans-formation reactions reduce the neurotoxicant activities but mar-ginally affect the weak estrogenicity of the agents.
Slow biotransformation, in addition to the highly lipophilicnature of the organochlorine compounds, guarantees that theseagents will be sequestered in body tissue having a high lipid con-tent, where some biological action may result. In the case of adi-pose tissue, the agents will remain stored and undisturbed, onlysmall amounts equilibrating with the blood and being degradedand/or excreted (Dale and Quinby, 1963; Davies et al., 1972). Thedepuration of these chemicals from systems occurs slowly, in amatter of a few weeks for chlordane, and months to years foraldrin/dieldrin, DDT, and others (Laben et al., 1965; Ecobichon
and Saschenbrecker, 1969; Craan and Haines, 1998; Delorme etal., 1999).
It is not surprising that humans acquired body burdens of thesechemicals during the 1950s and 1960s, when they were used onalmost all food crops. Depending on the region of the world, theintensity of use, the extent of occupational and accidental expo-sure, and dietary habits, the bioconcentration/bioaccumulation ofDDT in human adipose tissue resulted in levels on the order of5 ppm DDT and approximately 15 ppm of total DDT-derivedmaterial (Quinby et al., 1965; Fiserova-Bergerova et al., 1967;Abbott et al., 1968; Morgan and Roan, 1970). The levels of otherorganochlorine insecticides sequestered in body fat were never ashigh as those of DDT. With declining use and the eventual ban ofthis class on insecticides from the North American market, bodyburdens of these insecticides declined slowly. By the late 1960s,adipose levels of 2 ppm DDT(0 ppm of total DDT-derived mate-rial) were detectable. Whereas the daily intake of DDT in theUnited States was approximately 0.2 mg/day in 1958, this had de-creased to about 0.04 mg/day by 1970 (Hayes, 1971). Today, onlytrace levels of DDT, less than 2.0 ppm of total DDT-derivedmaterial, are detectable in human adipose tissue (Mes et al., 1982;Redetzke and Applegate, 1993; Stevens et al., 1993).
Treatment of Poisoning The life-threatening situation inorganochlorine insecticide poisoning is associated with tremors,motor seizures, and interference with respiratory function (hypox-emia and resulting acidosis) arising from repetitve stimulation ofthe CNS. In addition to general decontamination and supportivetreatment, diazepam (0.3 mg/kg IV; maximum dose of 10 mg) orphenobarbital (15 mg/kg IV; maximum dose of 1.0 g) may be ad-ministered by slow injection to control the convulsions. It may benecessary to repeat the treatment.
Anticholinesterase Agents
The agents comprising this type of insecticide have a commonmechanism of action but arise from two distinctly different chem-ical classes, the esters of phosphoric or phosphorothioic acid andthose of carbamic acid (Fig. 22-10). The anticholinesterase insec-ticides are represented by a vast array of structures that havedemonstrated the ultimate in structure-activity relationships in at-tempts to produce potent and selective insect toxicity while mini-mizing the toxicity toward nontarget species. Today, there are some200 different organophosphorus ester insecticides and approxi-mately 25 carbamic acid ester insecticides in the marketplace, for-mulated into literally thousands of products. For detailed discus-sions on the nomenclature, chemistry, and development of theseinsecticides, the reader is referred to the books of O’Brien (1960),Heath (1961), Melnikov (1971), Fest and Schmidt (1973), Eto(1974), Kuhr and Dorough (1976), Ecobichon and Joy (1994), andMatsumura (1985) and a review by Holmstedt (1959).
The organophosphorus ester insecticides were first synthe-sized in 1937 by a group of German chemists led by GerhardSchrader at Farbenfabriken Bayer AG (Schrader and Kukenthal,1937). Many of their trial compounds proved to be exceedinglytoxic and unfortunately, under the management of the Nazis inWorld War II, some were developed as potential chemical warfareagents. It is unwise to dismiss the chemical warfare nerve gasescompletely as the weapons of a past, more barbaric era, because itis known that at least one country has stocks of a number of these
Figure 22-9. Proposed sites of action of cyclodiene-type organochlorineinsecticides on chloride ion transport through inhibition of the GABAA
receptor channel as well as inhibition of calcium-magnesium ATPase.
2996R_ch22_761-810 4/16/01 4:37 PM Page 774
Copy
right
ed M
ater
ial
Copyright © 2001 by The McGraw-Hill Companies Retrieved from: www.knovel.com

CHAPTER 22 TOXIC EFFECTS OF PESTICIDES 775
agents (Fig. 22-11) (Clement, 1994; Gee, 1992). The physico-chemical and toxicologic properties of these agents have been re-viewed (Sidell, 1992; Somani et al., 1992). Sarin (O-isopropylmethylphosphonofluoridate) was used in Iraq against Kurdish vil-lages in northern Iraq in 1988 and residues of isopropylmethylphosphonic acid were found in soil samples along withminute traces of sarin (Webb, 1993). These potent agents shouldnot be ignored, since they are relatively easy to manufacture insmall amounts and have been the toxicants of choice of terroristsin at least two documented attacks in Japan, in Matsumoto on June27, 1994 (Morita et al., 1995), and in the Tokyo subway on March20, 1995 (Masuda et al., 1995; Nazaki and Aikowa, 1995; Nazakiet al., 1995; Suzuki et al., 1995). The acute and chronic toxicologiceffects of these incidents are discussed below.
Although it is true that all of the organoposphorus esters werederived from “nerve gases” (chemicals such as soman, sarin, andtabun), a fact that the media continually emphasizes, the insecti-cides used today are at least four generations of development awayfrom those highly toxic chemicals. The first organophosphorus es-ter insecticide to be used commercially was tetraethylpyrophos-phate (TEPP); although effective, it was extremely, toxic to allforms of life and chemical stability was a major problem in thatTEPP hydrolyzed readily in the presenced of moisture. Further de-velopment was directed toward the synthesis of more stable chem-icals having moderate environmental persistence, giving rise toparathion (O,O-diethyl-O-p-nitrophenyl phosphorothioate, E-605)in 1944 and the oxygen analog paraoxon (O,O-diethyl-O-p-nitro-phenyl phosphate) at a later date. Although these two chemicalshad the properties desired in an insecticide (low volatility, chemi-cal stability in sunlight and in the presence of water, environmen-tal persistence for efficacy), they both exhibited a marked mam-malian toxicy and were unselective with respect to target andnontarget species. The replacement of DDT with parathion in the1950s resulted in a series of fatal poisonings and bizarre accidentsarising from the fact that workers did not appreciate that this agentwas far different from the relatively innocuous organochlorine in-secticides with which they were familiar (Ecobichon, 1994a). Thenumber of severe poisonings attributed to parathion provided the
stimulus for a search for analogs more selective in their toxicity totarget species and less toxic to nontarget organisms, includingwildlife, domestic stock, and humans.
The first pesticidal carbamic acid esters were synthesized inthe 1930s and were marketed as fungicides. Since these aliphaticesters possessed poor insecticidal activity, interest lay dormant un-til the mid-1950s, when renewed interest in insecticides having an-ticholinesterase activity but reduced mammalian toxicity led to thesynthesis of several potent aryl esters of methylcarbamic acid. Theinsecticidal carbamates were synthesized on purely chemicalgrounds as analogs of the drug physostigmine, a toxic anti-cholinesterase alkaloid extracted from the seeds of the plantPhysostigma venenosum, the Calabar bean.
Signs and Symptoms of Poisoning Although their structures arediverse in nature, the mechanism by which the organophosphorusand carbamate ester insecticides elicit their toxicity is idential andis associated with the inhibition of the nervous tissue acetyl-cholinesterase (AChE), the enzyme responsible the the destructionand termination of the biological activity of the neurotransmitteracetylcholine (ACh). With the accumulation of free, unbound AChat the nerve endings of all cholinergic nerves, there is continualstimulation of electrical activity. The signs of toxicity include thoseresulting from stimulation of the muscarinic receptors of theparasympathetic autonomic nervous system (increased secretions,bronchoconstriction, miosis, gastrointestinal cramps, diarrhea, uri-nation, bradycardia); those resulting from the parasympathetic di-visions of the autonomic nervous system as well as the junctionsbetween nerves and muscles (causing tachycardia, hypertension,muscle fasciculation, tremors, muscle weakness, and/or flaccidparalysis); and those resulting from effects on the CNS (restless-ness, emotional liability, ataxia, lethargy, mental confusion, loss ofmemory, generalized weakness, convulsion, cyanosis, coma)(Table 22-7).
The classic picture of anticholinesterase insecticide intoxica-tion, first described by DuBois (DuBois, 1948; DuBois et al., 1949),has become more complicated in recent years by the recognitionof additional and persistent signs of neurotoxicity not previouslyassociated with these chemicals. First, and frequently associatedwith exposure to high concentrations of the insecticides (resultingfrom suicide attempts or drenching with dilute or concentrated
Figure 22-10. The basic backbone structures of the two types of anti-cholinesterase-class insecticides, the organophosphorus and carbamateesters.
Figure 22-11. Structures of the organophosphorus ester chemical war-fare nerve gases, the forerunners of the organophosphorus ester insecti-cides.
2996R_ch22_761-810 4/16/01 4:37 PM Page 775
Copy
right
ed M
ater
ial
Copyright © 2001 by The McGraw-Hill Companies Retrieved from: www.knovel.com

776 UNIT 5 TOXIC AGENTS
chemicals), are effects that may persist for several months follow-ing exposure and may involve neurobehavioral, cognitive, andneuromuscular functions (Marrs, 1993; Ecobichon, 1994a; Jamal,1997; Ecobichon, 1998b). One author describes this as a chronicorganophosphate-induced neuropsychiatric disorder (COPIND)(Jamal, 1997). The first evidence of this type of syndrome, delayedpsychopathologic-neurologic lesions, was reported by Spiegelberg(1963), who had been studying workers involved in the productionand handling of the highly toxic nerve gases in Germany duringWorld War II. The characteristic symptomatology subdivided thesepatients into two distinct groups. The first and largest group wascharacterized by persistently lowered vitality and ambition; defec-tive autonomic regulation leading to cephalalgia and gastrointesti-nal and cardiovascular symptoms; premature decline in potencyand libido; intolerance to alcohol, nicotine, and various medicines;and an impression of premature aging. The second group, in addi-tion to the above symptoms, showed one or more of the following:depressive or subdepressive disorders of vital function; cerebralvegetative (syncopal) attacks; slight or moderate amnestic or de-mential effects; and slight organoneurologic defects. These symp-toms developed and persisted for some 5 to 10 years followingexposure to these most toxic organophosphorus esters during thewar years. The controversial paper of Gershon and Shaw (1961),a study of 16 cases of pesticide applicators exposed primarily toorganophosphorus ester insecticides for 10 to 15 years, reported awide range of persistent signs of toxicity, including tinnitus, nys-
tagmus, pyrexia, ataxia, tremor, paresthesia, polyneuritits, paraly-sis, speech difficulty (slurring), loss of memory, insomnia, som-nambulism, excessive dreaming, drowsiness, lassitude, generalizedweakness, emotional liability, mental confusion, difficulty in con-centration, restlessness, anxiety, depression, dissociation, andschizophrenic reactions. Although the results of other studies havebeen equivocal in their support of such an array of long-term signsand symptoms, there is a persistent recurrence of the symptoma-tology in a number of anecdotal and documented reports(Ecobichon, 1994a; Marrs, 1993; Jamal, 1997). The literature onpotential, suspected, and established sequelae of organophospho-rus ester insecticide intoxications does not confirm the frequentlyseen statement that clinical recovery from nonfatal poisoning is al-ways complete in a few days. Continuous and close observation ofacutely intoxicated patients for some weeks following their recov-ery from the initial toxicity and treatment thereof would be neces-sary to identify the subtle changes indicated above. The emergencyservice physician rarely sees the patient following stabilization andinitial “recovery.” Definitive examples in which such observationhas been possible are few, but one such fortuitous case illustrateswhat can be achieved if there is close follow-up (Ecobichon et al.,1977).
While most clinical manifestations of acute poisoning are re-solved within days to weeks, some symptoms, particularly thoseof a neuropsychological nature, appear to persist for months orlonger. Complete reviews of this aspect have been published re-
Table 22-7Signs and Symptoms of Anticholinesterase Insecticide Poisoning
NERVOUS TISSUE AND RECEPTORS
AFFECTED SITE AFFECTED MANIFESTATIONS
Parasympatheic autonomic Exocrine glands Increased salivation, lacrimation, perspiration(muscarinic receptors) Eyes Miosis (pinpoint and nonreactive), ptosis, blurring ofpostganglionic nerve fibers vision, conjunctival injection, “bloody tears”
Gastrointestinal tract Nausea, vomiting, abdominal tightness, swelling andcramps, diarrhea, tenesmus, fecal incontinence
Respiratory tract Excessive bronchial secretions, rhinorrhea, wheezing,edema, tightness in chest, bronchospasms, broncho-constriction, cough, bradypnea, dyspnea
Cardiovascular system Bradycardia, decrease in blood pressureBladder Urinary frequency and incontinence
Parasympathetic and sympathetic Cardiovascular system Tachycardia, pallor, increase in blood pressureautonomic fibers(nicotinic receptors)
Somatic motor nerve fibers Skeletal muscles Muscle fasciculations (eyelids, fine facial muscles),(nicotine receptors) cramps, diminished tendon reflexes, generalized muscle
weakness in peripheral and respiratory muscles,paralysis, flaccid or rigid tone
Restlessness, generalized motor activity, reaction toacoustic stimuli, tremulousness, emotional lability,ataxia
Brain (acetylcholine receptors) Central nervous system Drowsiness, lethargy, fatigue, mental confusion, inabilityto concentrate, headache, pressure in head, generalizedweakness
Coma with absence of reflexes, tremors, Cheyne-Stokesrespiration, dyspnea, convulsions, depression ofrespiratory centers, cyanosis
SOURCE: Ecobichon and Joy, 1982.
2996R_ch22_761-810 4/16/01 4:37 PM Page 776
Copy
right
ed M
ater
ial
Copyright © 2001 by The McGraw-Hill Companies Retrieved from: www.knovel.com

CHAPTER 22 TOXIC EFFECTS OF PESTICIDES 777
cently (Ecobichon, 1994a; Jamal, 1997). A 4-month surveillanceof 19 acutely poisoned farm workers revealed many subjectivesigns and symptoms (blurred vision, muscle weakness, nausea,headaches, night sweats) to persist through the study period, ac-companied by a slow recovery of plasma and erythrocyticcholinesterases (Whorton and Obrinsky, 1983). Rosenstock andcoworkers (1991) described the neuropsychological testing of 36poisoned Nicaraguan agricultural workers some 2 years postexpo-sure, reporting that the poisoned group did much worse than con-trols on all subtests, with significantly worse performance on fiveof six subtests in the World Health Organization (WHO) neu-ropsychological test battery and on three of six additional tests thatassessed verbal and visual attention, visual memory, visuomotorspeed, sequencing and problem solving, motor steadiness, and dex-terity (Ecobichon, 1998b). More recent examples of acute intoxi-cations—e.g. “dippers’ flu” from repeated exposure to organophos-phorus esters (diazinon, propetamphos, chlorfenvinphos) used insheep dip—have revealed persistent adverse neurophysiologicaland psychological/behavioral effects that can be evaluated by suit-able test batteries (Cook, 1992; Murray et al., 1992; Sims, 1992;Stephens et al., 1995; Beach et al., 1996; Stephens et al., 1996).As has been seen with signs and symptoms in acute intoxications,those afflicted did not show adverse responses to all test batteryparameters; only some responses were significantly different fromnormal-range values. However, such poisoning incidents have pro-gressed from an anecdotal stage to a testable basis, with refinedtest parameters revealing subtle but distinct changes in memory,academic and motor skills, abstraction, and flexibility in thinking(Ecobichon, 1998b).
A second distinct manifestation of exposure to organophos-phorus ester insecticides has been described by clinicians in SriLanka involved in the treatment of suicide attempts (Senanayakeand Karalliedde, 1987). This paralytic condition, called the inter-mediate syndrome, consisted of a sequence of neurologic signs thatappeared some 24 to 96 h after the acute cholinergic crisis but be-fore the expected onset of delayed neuropathy, the major effect be-ing muscle weakness, primarily affecting muscles innervated bythe cranial nerves (neck flexors, muscles of respiration) as well asthose of the limbs. Cranial nerve palsies were common. There wasa distinct risk of death during this time interval because of respi-ratory depression and distress that required urgent ventilatory sup-port and was not responsive to atropine or oximes. The chemicalsinvolved in these distinctive intoxications included fenthion,dimethoate, monocrotophos, and methamidophos. There were noobvious clinical differences during the acute intoxication phase inthose patients who developed the intermediate syndrome and oth-ers who did not, and all patients were treated in the same manner.
A third syndrome, that of organophosphate-induced delayedneurotoxicity (OPIDN), is caused by some phosphate, phospho-nate, and phosphoramidate esters, very few of which have everbeen used as insecticides (Fig. 22-12). Historically, this syndromehas been known for almost 100 years and has been associated withthe chemical tri-o-tolyl phosphate (TOTP) (Ecobichon, 1994a). Thefirst major epidemic of OPIDN occurred during the prohibitionyears in the United States, resulting from the consumption of a par-ticular brand of alcoholic extract of Jamaican ginger contaminatedor adulterated with mixed tolyl phosphate esters. The syndrome,affecting some 20,000 individuals to varying degrees, was knownas ginger jake paralysis or jake leg and was studied in detail byMaurice Smith of the U.S. Public Health Service. He not only con-firmed that the condition could be reproduced in animals (e.g., rab-
bits, dogs, monkeys, calves) but also demonstrated that only oneof the three isomers found in commercial tri-tolyl phosphate, theortho-isomer, was responsible for the toxicity (Smith and Lillie,1931). The initial flaccidity, characterized by muscle weakness inthe arms and legs—giving rise to a clumsy shuffling gait—was re-placed by spasticity, hypertonicity, hyperreflexia, clonus, and ab-normal reflexes, indicative of damage to the pyramidal tracts anda permanent upper motor neuron syndrome (Ecobichon, 1994a). Inmany patients, recovery was limited to the arms and hands anddamage to the lower extremities (foot drop, spasticity, and hyper-active reflexes) was permanent, suggesting damage to the spinalcord (Moregan and Penovich 1978), Recent studies have demon-strated that other commercial triarylphosphates (flame retardantsin lubricants and plastics) did not elicit significant OPIDN-typeneurotoxicity at the maximum dose of 2000 mg/kg (Weiner andJortner, 1999). An OPIDN-type neuropathy occurred with an ex-perimental organophosphorus ester insecticide, mipafox, followingan accident in a manufacturing pilot plant. Details of the effectson two of the workers were described by Bidstrup and coworkers(1953) and Ecobichon (1994a). The poisoning of water buffalo inthe early 1970s in Egypt by a phosphonate insecticide, leptophos,revealed a neurologic syndrome similar to that observed followingexposure to TOTP (Abou-Donia, 1981). There was also evidenceof leptophos-induced neuropathies among workers in a manufac-turing plant in the United States, but the controversial observationswere obscured by concomitant exposure of the workers to n-hexane,another neurotoxic chemical (Xintaris et al., 1978).
A number of organophosphorus insecticides—includingomethoate, trichloronate, trichlorfon, parathion, methamidophos,fenthion, and chlorpyrifos—have been implicated in causingOPIDN in humans (Abou-Donia and Lapadula, 1990). However,it should be emphasized that these incidents all involved accidentalor suicidal exposure to excessively high levels. Concern that manyof the over 200 organophosphorus ester insecticides in use mightcause this unique neuropathy has resulted in an intensive study ofthe syndrome, the identification of the most susceptible species (thehen and the cat), the development of standard protocols to test allinsecticides, and at least a partial elucidation of the mechanismsby which agents elicit this condition. Histologic examination of the
Figure 22-12. The basic structures and nomenclature of organophos-phorus esters, with examples, capable of causing organophosphate-induced delayed neurotoxicity (OPIDN).
2996R_ch22_761-810 4/16/01 4:37 PM Page 777
Copy
right
ed M
ater
ial
Copyright © 2001 by The McGraw-Hill Companies Retrieved from: www.knovel.com

778 UNIT 5 TOXIC AGENTS
nervous systems of hens treated with a suitable agent [e.g., TOTP,O,O-diisopropyl phosphorofluoridate (DFP), mipafox, leptophos]has revealed a wallerian “dying-back” degeneration of large-diameter axons and their myelin sheaths in distal parts of the pe-ripheral nerves and of long spinal cord tracts—the rostral ends ofascending tracts and the distal ends of descending tracts (Cavanagh,1954; Sprague and Bickford, 1981). At autopsy, examination of anunfortunate victim who died 15 months after the sarin attack in theTokyo subway revealed moderate-to-severe fiber loss in the suraland sciatic nerves with little effect in the CNS, results consistentwith the dying-back degeneration of the PNS described above(Himuro et al., 1998). Surviving victims also showed a higher fre-quency of sister chromatid exchanges (SCEs) in peripheral bloodlymphocytes (Li et al., 1998). Biochemical studies have demon-strated that the above-mentioned agents inhibit a neuronal, non-specific carboxylesterase, neuropathic target esterase (NTE), whichappears to have some, as yet unknown, role in lipid metabolism inneurons (Johnson, 1982). If acute exposure to an appropriateorganophosphorus ester results in 70 percent inhibition of NTE,the characteristic OPIDN usually follows, with ataxia being ob-served some 7 to 14 days following treatment and progression tomoderate to severe muscular weakness and paralysis with con-comitant changes in neuronal morphology (Johnson, 1982; Slottand Ecobichon, 1984). It is the considered opinion of many inves-tigators that many of the commonly used phosphate and phospho-rothioate ester insecticides might be capable of causing thissyndrome if only sufficient concentrations of the agents could beattained in vivo. However, taking paraoxon as an example of sucha phosphate ester, the animal(s) would either die as a consequenceof other acute toxic effects or would rapidly detoxify the agent,thereby preventing the acquisition of sufficient paraoxon to inhibitNTE. There also appear to be subtle structure-activity relationshipsbetween organophosphorus esters and the active site on the NTEprotein, because many phosphate esters are not good inhibitors ofNTE (Ohkawa et al., 1980; Abou-Donia, 1981). Conversely, whilethe nerve gases cause a marked inhibition of NTE, the exposed an-imals do not develop OPIDN, suggesting that NTE inhibition maynot be obligatory (Johnson et al., 1985, Lotti, 1992; Marrs, 1993;Jamal, 1997). It should be emphasized that although NTE inhibi-tion remains a useful function for monitoring the potential oforganophosphorus esters to induce OPIDN, the role of this enzymein the initiation of the syndrome remains unknown and histopatho-logic evidence is a requirement of the U.S. EPA protocol. How-ever, a recently reported poisoning by methamidophos proved thatlymphocyte NTE inhibition was predictive of the subsequentOPIDN, the level of activity increasing from 3.1 to 13.3nmol/min/mg protein between day 3 after poisoning and day 52(McConnell et al., 1999). Interestingly, serum autoantibodies (IgM,IgG) to glial fibrillary acidic protein (GFAP), myelin basic protein(MBP), and cytoskeletal elements increased immediately after poi-soning in this case and persisted to day 52, suggesting that theseparameters might be useful markers. Two noninsecticidalorganophosphorus esters, the tri-S-alkyl defoliant S,S,S-tributylphosphorotrithioite (Merphos) and its oxidation product, S,S,S-trib-utyl phosphorotrithioate (DEF), have been implicated in produc-ing OPIDN in at least one agricultural worker and cause a char-acteristic delayed neurotoxicity in hens (Abou-Donia andLapadula, 1990).
The signs and symptoms of acute intoxication by carbamateinsecticides are similar to those described for organophosphoruscompounds, differing only in the duration and intensity of the tox-
icity. The most apparent reasons are that (1) carbamate insecticidesare reversible inhibitors of nervous tissue AChE, unlike most ofthe organophosphorus esters (see below, “Mechanism of Toxic Ac-tion”) and (2) they are rapidly biotransformed in vivo. Despite theextensive toxicologic short-term toxicity following acute adminis-tration, carbamate insecticide toxicity has been reported in humansand fatalities have occurred (Ecobichon, 1994b; Hayes, 1982, Feld-man, 1999). Invariably, these serious poisonings have involved car-baryl and have occurred as a consequence of accidental or pur-poseful (suicidal) exposure to high concentrations (Hayes, 1982;Cranmer, 1986). Information on the incidences of human intoxi-cation by carbaryl can be found in the Carbaryl Decision Docu-ment (EPA, 1980). For the period 1966–1980, a total of 195 hu-man intoxication cases were reported (3 fatalities, 16 hos-pitalizations, and 176 cases receiving medical attention). A singleoral dose of 250 mg of carbaryl (2.8 mg/kg body weight) is suffi-cient to elicit moderately severe poisoning in an adult man (Cran-mer, 1986). Moderate but transient toxicity has also been observedfollowing exposure to a few of the more potent carbamate ester in-secticides methomyl (Lannate) and propoxur (Baygon) (Fandekaret al., 1968, 1971; Liddle et al., 1979). More recently the illegaluse of aldicarb (Temik), a very acutely toxic carbamate ester, onwatermelons in California and on English cucumbers in British Co-lumbia, Canada, resulted in moderate to severe toxicity in con-sumers of these products, with signs and symptoms including nau-sea, vomiting, gastrointestinal cramps, and diarrhea (Goldman etal., 1990a,b).
While there is little evidence of prolonged neurotoxicity afterexposure to carbamate ester insecticides, this statement should bemade cautiously because the signal danger appears to involve acutesingle exposures to massive doses or at least repeated exposuresto large doses. Examining a number of carbamate (aldicarb,methomyl) intoxications in children and adults, Lifshitz et al.(1997) discovered that the predominant symptoms in adults weremiosis and muscle fasciculations, while the children were morelikely to reveal CNS effects (stupor and/or coma) in addition to di-arrhea and hypotension. The authors suggested that the blood-brainbarrier was more permeable in the children.
Bizarre anecdotal cases exist that are contrary to everythingthat we know about carbamates. One case, that of a farmer whohand-sprayed a vegetable garden with a water-wettable formula-tion of carbaryl, drenching himself in the process, developed achronic polyneuropathy, persistent photophobia and paresthesia,recent memory loss, muscular weakness, fatigue, and lassitude(Ecobichon, 1994b). The case study presented by Branch and Jacqz(1986) developed into a persistent, stocking-and-glove peripheralneuropathy accompanied by mental confusion and weakness in ma-jor skeletal muscle groups, with fasciculations and cerebral atro-phy. Grendon et al. (1994) presented long-term observations of agroup of men who had been exposed acutely to aldicarb, severalexperiencing symptoms 3 years after the initial severe intoxication.Feldman (1999) conducted extensive neurologic and psychologi-cal investigations on three individuals exposed accidentally to aeri-ally applied carbaryl some three to five years before. Their per-sisting subjective symptoms were confirmed by peripheral nerveconduction studies, electromyography, and neuropsychologicalassessment, both peripheral and central impairment beingdocumented.
There is evidence in animal studies, albeit at near toxic doses,of a wallerian-type degeneration of spinal cord tracts in rabbits andhens following treatment with sodium diethyldithiocarbamate
2996R_ch22_761-810 4/16/01 4:37 PM Page 778
Copy
right
ed M
ater
ial
Copyright © 2001 by The McGraw-Hill Companies Retrieved from: www.knovel.com

CHAPTER 22 TOXIC EFFECTS OF PESTICIDES 779
(Edingon and Howell, 1969). Carbaryl, when fed to hogs (150mg/kg per day for 72 or 83 days), caused a rear leg paralysis, min-imal at rest but, when the animals were forced to move, resultingin marked incoordination, ataxia, tremors, clonic muscle contrac-tions, and prostration, with histologic evidence of lesions in theCNS and in skeletal muscle (Smalley et al., 1969). Carbamate es-ter insecticides do no inhibit NTE or elicit OPIDN-type neurotox-icity. Behavioral changes have been noted in a number of animalstudies following the subchronic or chronic administration of dif-ferent carbamate insecticides (Santalucito and Morrison, 1971;Desi et al., 1974).
Mechanism of Toxic Action Although the anticholinesterase-type insecticides have a common mode of action, there are signif-icant difference between organophosphorus and carbamate esters.The reaction between and organophosphorus ester and the activesite in the AChE protein (a serine hydroxyl group) results in theformation of a transient intermediate complex that partially hy-drolyzes with the loss of the “Z” substituent group, leaving a sta-ble, phosphorylated, and largely unreactive inhibited enzyme that,under normal circumstances, can be reactivated only at a very slowrate (Fig. 22-13). With many organophosphorus ester insecticides,an irreversibly inhibited enzyme is formed, and the signs and symp-toms of intoxication are prolonged and persistent, requiring vigor-ous medical intervention including the reactivation of the enzymewith specific chemical antidotes (see “Treatment of Poisoning,” be-low, this section). Without intervention, the toxicity will persistuntil sufficient quantities of “new” AChE are synthesized in 20 to30 days to destroy efficiently the excess neurotransmitter. The na-ture of the substituent groups at “X,” “Y,” and “Z” plays an im-portant role in the specificity for the enzyme, the tenacity of bind-ing to the active site, and the rate at which the phosphorylatedenzyme dissociates to produce free enzyme. More recently intro-duced organophosphorus esters (acephate, temephos, dichlorvos,trichlorfon) are less tenacious inhibitors of nervous tissue AChE,the phosphorylated enzyme being more readily and spontaneouslydissociated.
In contrast, carbamic acid esters, which attach to the reactivesite of the AChE, undergo hydrolysis in two stages: the first stageis the removal of the “X” substituent (an aryl or alkyl group) withthe formation of a carbamylated enzyme; the second stage is the
decarbamylation of the inhibited enzyme with the generation offree, active enzyme (Fig. 22-13). Carbamic acid esters are nothingmore than poor substrates for the cholinesterase-type enzymes.
When the concept of the interaction between organophos-phorus and carbamic esters with AChE is presented in another man-ner (Table 22-8), one can see that the only distinctive differencebetween the two anticholinesterase-type insecticides lies in the rateat which the dephosphorylation or decarbamylation takes place.The rate is exceedingly slow for organophosphorus esters, so muchso that the enzyme is frequently considered to be irreversibly in-hibited. The rate of decarbamylation is sufficiently rapid that theseesters are often considered to be reversible inhibitors with lowturnover rates. The characteristics of the various rate constants forthe natural substrate (ACh), organophosphorus, and carbamylationesters are shown in Table 22-8. It is important to appreciate thatthe rate at which step 3 proceeds is thousands of times slower withcarbamate esters than with ACh, whereas with organophosphorusesters it is several orders of manitude slower (Ecobichon, 1979).This subject has been extensively reviewed by Aldridge and Reiner(1972). A number of organophosphorus (phosphate, phosphonate,and phosphoramidate) esters (Fig. 22-12)—the chemical warfareagents sarin, soman, and tabun, and a few other compounds suchas DFP, mipafox, and leptophos—have the ability to bind tena-ciously to the active site of AChE and NTE to produce an irre-versibly inhibited enzyme by a mechanism known as aging. Theaging process is dependent on the size and configuration of thealkyl (R) substituent, with the potency of the ester increasing inthe order of diethyl, dipropyl, and dibutyl for such analogs as DFPand mipafox (Aldridge and Johnson, 1971). The aging process isgenerally accepted as being caused by the dealkylation of the in-termediate dialkylphosphorylated enzymes by one of two possiblemechanisms (Fig. 22-14). The first involves the hydrolysis of a P–O bond following a nucleophile (base) attack on the phospho-rus atom. The second mechanism involves the hydrolysis of anO–C bond by an acid catalysis, resulting in the formation of a car-bonium ion as the leaving group (O’Brien, 1960; Eto, 1974;Johnson, 1982). The aging process is believed to fix an extra chargeto the protein, causing some perturbation to the active site andthereby preventing dephosphorylation. While the exact nature ofthis reaction has not been demonstrated for AChE and NTE, evi-dence from experiments with saligenin cyclic phosphorus esters(derivatives of TOTP) and �-chymotrypsin points to the possibil-ity of two stabilized forms of “aged” enzyme (Toia and Casida,1979). Both of the reactions utilize the imidazole group of a neigh-boring histidine. In one reaction, the hydroxylated substituent isreleased and the phosphorylated enzyme is stabilized by a hydro-gen on the imidazole group. In the other reaction, the leaving sub-stituent becomes attached to the imidazole, yielding a N–C-hy-droxylated derivative of the phospyhorylated enzyme. Johnson(1982) proposed that, in the case of NTE, if one or two of the P–Rbonds were P–O–C (as in phosphates and phosphonates), agingwould occur rapidly, whereas if the P–R bonds were P–C (as inphosphinates), aging would not be possible.
A number of acute, high-level exposures and intoxications byorganophosphorus and carbamate esters have resulted in persistentCNS effects as well as debilitating muscle weakness, particularlyin the legs, that cannot be explained on the basis of nervous tissueAChE inhibition alone. One hypothesis has been put forward thatthe excessive amount of undestroyed acetylcholine (ACh) may beinvolved by an action at nicotinic (nAChR) and muscarinic(mAChR) acetylcholine receptors (Ecobichon, 1994a). As shown
Figure 22-13. The interaction between an organophosphorus or carba-mate ester with the serine hydroxyl groups in the active site of the en-zyme acetylcholinesterase (E-OH). The intermediate, unstable complexesformed before the release of the “leaving” groups (ZH and XOH) are notshown. The dephosphorylation or decarbamoylation of the inhibited en-zyme is the rate-limiting step to forming free enzymes.
2996R_ch22_761-810 4/16/01 4:37 PM Page 779
Copy
right
ed M
ater
ial
Copyright © 2001 by The McGraw-Hill Companies Retrieved from: www.knovel.com

780 UNIT 5 TOXIC AGENTS
in Fig. 22-15, the ACh accumulating at nAChR would elicit stim-ulation of the neuromuscular junctions, causing fasciculations(repetitive stimulation) followed by a depolarizing blockade if theACh levels remained elevated, leading subsequently to a desensi-tization process with a more or less permanent reduction in nAChRnumbers and causing the persistent muscle weakness observedthrough a lack of response to stimuli. Support for this hypothesishas been seen in studies revealing chronic abnormal electromyo-graphic (EMG) activity in human intoxications by anti-cholinesterase insecticides, the details of which can be found inEcobichon (1994a) and Feldman (1999). An alternative theorybears examination.
In an acute tabun study in rats, necrotic diaphragmatic mus-cle was observed, the effect being prevented by sectioning thephrenic nerve to a portion of the diaphragm (Ariens et al., 1969).Similar diaphragmatic damage has been observed in human poi-sonings (DeRueck and Willens, 1975; Wecker et al., 1986). Earlywork by Dettbarn and colleagues demonstrated that close intra-muscular injections of parathion or paraoxon resulted in a skeletalmyopathy accompanied by necrosis of the nerve terminal mem-brane and of the underlying muscle (summarized in Ecobichon,1994a). There is evidence that organophosphate-induced AChE in-
hibition causes muscle hyperactivity as an initial step that triggersfree radical–induced lipid peroxidation before muscle injury (Yangand Dettbarn, 1996; Yang et al., 1996). More recent studies, con-ducted at the level of brain mAChR and nAChR, have demonstratedthat organophosphorus esters (soman, VX, DFP, parathion,paraoxon, chlorpyrifos) bind to both receptor types, particularly ifthe circulatory concentrations are in the micromolar range as wouldbe encountered in poisoning cases, resulting in desensitization(Bakry et al., 1988; Eldefrawi et al., 1988; Huff et al., 1994; Katzet al., 1997). There is also evidence of differential down-regulationof nAChR subtypes through reduction of messenger RNA expres-sion in rat brain following repeated injections of parathion (Jett etal., 1993, 1994; Ward and Mundy, 1996). Adult mice, exposed toDFP on postnatal day 3 or 10, showed decreases in brain nAChR
Table 22-8Kinectics of Ester Hydrolysis
EH � AB �� EHAB � BH � EA � EH � AOH
COMPLEX FORMATION ACYLATION DEACYLATION
ESTERS (KA � k1�k�1) (k2) (k3)
Substrates Small Extremely Extremelyfast fast
Organophosphorus Small Moderately Slow oresters fast extremely
slowCarbamate esters Small Slow Slow
SOURCE: Ecobichon, 1979.
Figure 22-14. A schematic diagram illustrating two mechanisms bywhich the “aging” of organophosphorus ester–inhibited acetyl-cholinesterase may occur. See text for details.
Excessive
Stimulation
Desensitization
Down-regulationof Receptor Numbers
Neuromuscular JunctionDamage or Necrosis
DepolarizingBlockade
Persistent Muscle Weakness
Paralysis
Fasciculations
ACh
Figure 22-15. A schematic diagram illustrating the impact of excessiveconcentrations of acetylcholine (ACh) on muscarinic and nicotinic acetyl-choline receptors in order to explain neuromuscular weakness and dam-age.
2996R_ch22_761-810 4/16/01 4:37 PM Page 780
Copy
right
ed M
ater
ial
Copyright © 2001 by The McGraw-Hill Companies Retrieved from: www.knovel.com

CHAPTER 22 TOXIC EFFECTS OF PESTICIDES 781
density as well as significant alterations in spontaneous motor be-havior (Ahlbom et al., 1995).
Can the carbamate esters induce muscle damage and receptoralterations through mechanisms involving ACh, direct binding toreceptors, or through receptor expression? The repeated injectionof such reversible anticholinesterase carbamates as physostigmineor neostigmine has produced myopathies qualitatively similar tothose produced by paraoxon, possibly supporting the ACh-relatedtheory (Wecker et al., 1978). Skeletal muscle lesions (myodegen-eration, vacuolization) were observed in swine fed carbaryl over aperiod of 80� days (Smalley et al., 1969). The peripheral neu-ropathy of a carbaryl-exposed elderly male has been described(Branch and Jacqz, 1986). Some interesting experiments could beperformed with some of the more potent carbamate insecticides.
Biotransformation, Distribution, and Storage Both theorganophosphorus and carbamate ester insecticides undergo ex-tensive biotransformation in all forms of life. Both the route(s) andthe rate(s) of metabolism are highly species-specific and depend-ent on the substituent chemical groups attached to the basic “back-bone” structure of these esters (Fig. 22-10). Tissue enzymes of bothphase I (oxidative, reductive, hydrolytic) and phase II (transfer orconjugative reactions with glutathione, glucuronic acid, glycine,and so forth) types are founds in a widespread pattern in plant, in-vertebrate, and vertebrate species and, indeed, are responsible forsome aspects of the species sensitivity and/or both natural andacquired restance to many of these insecticides. The phase I detox-ification processes usually form reactive metabolites, whereas
phase II processes conjugate the polar phase I metabolites withsome natural body substituent to form a product with enhancedwater solubility and excretability. The biotransformation of anti-cholinesterase-type insecticides has been extensively reviewed inthe literature and the reader is referred to such sources as O’Brien(1967), Menzie (1969), Eto (1974), Kulkarni and Hodgson (1984),and Matsumura (1985) for details on the various mechanismsinvolved.
Organophosphorus esters undergo simultaneous oxidative bio-transformation at a number of points in the molecule (Fig. 22-16),the enzymes utilizing the ubiquitous cytochrome P450 isoenzymesystem. One reaction, that of oxidative desulfuration of phospho-rothioate (parathion, methyl parathion, fenitrothion, etc.) and phos-phorodithioate (azinophos methyl, malathion, etc.) esters, resultsin a significant increase in the toxicity of the biotransformationproducts, oxygen analogs (mechanism 1). This reaction is a majorobligatory pathway in ester detoxification in mammals equippedwith tissue aryl and aliphatic hydrolases (mechanism 8), whereasinsects are deficient in these enzymes, making them more suscep-tible. Dealkylation with the formation of an aldehyde occurs read-ily (mechanism 2), but this pathway does not function efficientlywhen the alkyl group becomes longer. Dearylation occurs with theformation of a phenol and either a dialkylphosphoric or di-alkylphosphorothioic acid (mechanism 3). The cytochrome systemcan also catalyze (1) aromatic ring hydroxylation (mechanism 4);(2) thioether oxidation (mechanism 5); (3) deamination; (4) alkyland N-hydroxylation; (5) N-oxide formation; and (6) N-dealkyla-tion. A number of transferases use glutathione (gamma-glutamyl-
Figure 22-16. A schematic diagram depicting the various phase I and II biotransformation pathways of anorganophosphorus ester and the nature of the products formed as a consequence of oxidative, hydrolytic,GSH-mediated transfer and conjugation of intermediates in mammals. See text for details.
2996R_ch22_761-810 4/16/01 4:37 PM Page 781
Copy
right
ed M
ater
ial
Copyright © 2001 by The McGraw-Hill Companies Retrieved from: www.knovel.com

782 UNIT 5 TOXIC AGENTS
L-cysteinyl glycine, GSH) as a cofactor and acceptor for O-alkyl(methyl) and O-aryl groups (mechanisms 6 and 7) to yield monodesmethyl products plus S-methylglutathione or aryl-glu-tathione derivatives plus the respective phosphoric or phospho-rothioic acids.
Hydrolysis of phosphoric and phosphorothioic acid ester oc-curs via a number of different tissue hydrolases (nonspecific car-boxylesterases, arylesterases, phosphorylphosphatases, phospho-triesteraes, carboxyamidases) scattered ubiquitously throughout theplant and animal kingdoms, with activity being highly dependenton the nature of the substituents (Ecobichon, 1979). Slight struc-tural modifications to substituents on the insecticide molecule candramatically alter the specificity of these enzymes toward anagent and affect species selectivity. The arylesterases [aromatic orA-esterases (ArE), EC 3.1.1.2] preferentially hydrolyze aryl(phenol, naphtol, indole) esters of short-chain aliphatic or phos-phorus acids, particulary if there is a double bond present in thealcohol moiety in position � with respect to the ester bond (mech-anism 8). Carboxylesterases [carboxylic acid ester hydrolases (CE),EC 3.1.1.] are capable of hydrolyzing a variety of aliphatic andaryl esters of short-chain fatty acids. The most important exampleof this reaction involving organophosphorus ester insecticides iswith malathion [O,O-dimethyl-S-(1,2-dicarbethoxyethyl) phospho-rodithioate], in which one of the two available ethylated carboxylicester groups is hydrolyzed to yield malathion (or malaoxon) �-monoacids that are biologically inactive (Dauterman and Main,1966). This CE-catalyzed reaction is an important feature of re-sistance to this insecticide in insects and to tolerance in mammals.Potentiation of anticholinergic effects can be produced by the com-bined administration of certain pairs of organophosphate ester in-secticides, such as EPN (O-ethyl-O-p-nitrophenylbenzenethio-phosphonate) and malathion (Frawley et al., 1957). The mechanismfor this effect involves the inhibition of carboxyesterases by EPN(Murphy, 1969, 1972, 1980). Carboxyamidases (acylamide am-diohydrolase, EC 3.5.1.4), found extensively in plant, insect, andvertebrate tissues, are of limited current interest in the degradationof insecticides; dimethoate (O,O-dimethyl-S-(N-methylcarbamoyl-methyl) phosphorodithioate] is the only organophosphorus ester in-secticide shown to be hydrolyzed by mammalian tissue amidases(mechanism 9). Phosphorylphosphatases and phosphotriesteraseshave limited involvement in the biotransformation of organophos-phorus ester insecticides but play a role in the detoxification ofsome of the chemical warfare agents.
Phase II conjugative reactions are of limited use in the bio-transformation of organophosphorus ester insecticides, and they areusually relegated to the task of glucuronidating or sulfating the aro-matic phenols, cresols, and other substances hydrolyzed from theester (Yang, 1976). However, one must be wary of these enzymesystems because metabolism studies of chlorfenvinphos (2-chloro-1-(29,49-dichlorophenyl)vinyl diethylphosphate) revealed the pres-ence of glucuronide and glycine conjugates of several products,whereas studies with trichlorfon (O,O-dimethyl-1-hydroxy-2,2,2-trichloroethyl phosphonate) revealed direct glucuronidation of theinsecticide without prior biotransformation (Hutson et al., 1967;Bull, 1972).
Carbamate ester insecticides can undergo simultaneous at-tack at several points in the molecule, depending on the nature ofthe substituents attached to the basic structure. In addition to thehydrolysis of the carbamate ester group by tissue CE and the re-lease of a substituted phenol, carbon dioxide, and methylamine(Fig. 22-10), (mechanism 1), several oxidative and reductive reac-
tions involving cytochrome P450– related monooxygenases canproceed, the ultimate products being considerably more polar thanthe parent insecticide. The extent of hydrolysis of carbamate esterinsecticides varies greatly between species, ranging from 30 to95 percent hydrolysis. The type of oxidative reactions observedwith carbamate esters can be simplified into two main groups: (1)direct ring hydroxylation (mechanism 2) and (2) oxidation ofappropriate side chains as is shown for this “mythical” methylcar-bamate, resulting in the hydroxylation of N-methyl groups ormethyl groups to form hydroxymethyl groups (mechanism 3),N-demethylation of secondary and tertiary amines (mechanism 4),O-dealkylation of alkoxy side chains (mechanism 5), thioetheroxidation (mechanism 6), and so forth. Phase II conjugative reac-tions can occur at any free reactive grouping with glucuronide andsulfate derivatives (mechanism 7), and GSH conjugates (mercap-turates) may be formed (mechanism 8). For a comprehensive ex-position of the various mechanisms involved, the reader is referredto those reviews mentioned above, this section, as well as topertinent articles by Ryan (1971), Fukuto (1972), and Kuhr andDorough (1976).
Treatment of Poisoning Despite the qualitative and quantita-tive differences between organophosphorus and carbamate insec-ticide intoxications, all cases of anticholinesterase poisoning shouldbe treated as serious medical emergencies and the patient shouldbe hospitalized as quickly as possible. The status of the patientshould be monitored by repeated analysis of the plasma (serum)cholinesterase and the erythrocyte AChE; the inhibition of the ac-tivities of these two enzymes is a good indicator of the severity oforganophosphorus ester poisoning, because only the erythrocyticAChE is inhibited by carbamate esters (except at excessively highlevels of exposure). As a consequence of the extensive involvementof the entire nervous system, the life-threatening signs (respiratorydepression, bronchospasm, bronchial secretions, pulmonaryedema, muscular weakness) resulting in hypoxemia will requireimmediate artificial respiration and suctioning via an endotrachealtube to maintain a patent airway. Arterial blood gases and cardiacfunction should be monitored.
The regimen for the treatment of organophosphorus ester in-secticide intoxication, based on the analysis of serum pseudo-cholinesterase, is described in Table 22-9 (Namba et al., 1971;Ecobichon et al., 1977; Marrs, 1993). Atropine is used to counter-act the initial muscarinic effects of the accumulating neurotrans-mitter. However, atropine is a highly toxic antidote and great caremust be taken. Frequent small doses of atropine (subcutaneouslyor intravenously) are indicated for mild signs and symptoms fol-lowing a brief, intense exposure. Large cumulative doses of at-ropine, up to 50 mg daily, may be essential to control severe mus-carinic symptoms. The status of the patient must be monitoredcontinuously by examining for the disappearance of secretions (drymouth and nose) and sweating, facial flushing, and mydriasis (di-latation of pupils). Supplementary treatment to offset moderate tosevere nicotinic and CNS signs and symptoms usually takes theform of one of the specific antidotal chemicals, the oximes (prali-doxime chloride or 2-PAM, pralidoxime methanesulfonate or P2S),administered intravenously to reactivate the inhibited nervous tis-sue AChE. The use of oximes may not be necessary for cases ofmild intoxication and should be reserved for moderate to severepoisonings. Treatment by slow intravenous infusion of doses of 1.0 g should be initiated as soon as possible, because the longerthe interval between exposure and treatment, the less effective the
2996R_ch22_761-810 4/16/01 4:37 PM Page 782
Copy
right
ed M
ater
ial
Copyright © 2001 by The McGraw-Hill Companies Retrieved from: www.knovel.com

CHAPTER 22 TOXIC EFFECTS OF PESTICIDES 783
oxime will be. In many poisonings, a single treatment with prali-doxime will be sufficient to elicit a reversal of the signs and symp-toms and to reduce the amount of atropine needed. In severe poi-soning cases, a prodigious amount of pralidoxime may be needed.If absorption, distribution, and/or metabolism of the organophos-phorus ester is delayed in the body, pralidoxime may be adminis-tered repeatedly over several days after the initial treatment. Careshould be taken with repeated dosing because pralidoxime effec-tively binds calcium ions and causes muscle spasms not unlikethose elicited by the organophosphorus esters. Severe musclecramping, particularly in the extremities, may be alleviated by oralor intravenous calcium solutions (Ecobichon et al., 1977).
The therapeutic action of the oxime compounds resides intheir capacity to reactivate AChE without contributing markedlytoxic actions of their own. Those organophosphorus esters that pos-sess good “leaving groups” (i.e., the “X” moiety) phosphorylatethe nervous tissue AChE by a mechanism similar to that of acety-lation by the substrate ACh. These esters are frequently calledirreversible inhibitors because the hydrolysis of the phosphorylatedenzyme by water is exceedingly slow (Table 22-8). However, var-ious nucleophilic agents containing a substituted ammonium groupwill dephosphorylate the phosphorylated enzyme at a much morerapid rate than water. The basic requirements for a reactivating mol-ecule consist of a rigid structure containing a quaternary ammo-nium group and an acidic nucleophile, which would be comple-mentary with the phosphorylated enzyme, in such a way that the
nucleophilic oxygen would be positioned close to the electrophilicphosphorus atom. These structure-activity requirements led to thedevelopment of the pralidoxime compounds, with the syn-isomerof 2-PAM (2-formyl-N-methylpyridinium chloride oxime) beingparticularly active (Childs et al., 1955; Askew, 1956; Kewitz andWilson, 1956; Namba and Hiraki, 1958). The reaction of 2-PAMwith the phosphorylated enzyme proceeds as shown in Fig. 22-17.
The reactivation is an equilibrium reaction, the oxime react-ing either with the phosphorylated enzyme or with free, unboundorganophosphorus ester, and the product is a phosphorylated oximewhich in itself can be a potent cholinesterase inhibitor if it is sta-ble in an aqueous medium (Schoene, 1972). In general, the phos-phorylated oxime degrades quickly in water.
A practical limitation on the usefulness of oxime reactivatorslies in the inability of these agents to reactivate “aged” AChE, thatenzyme in which the phosphorylated enzyme has been furtherdealkylated and the phosphoryl group becomes tightly bound tothe reactive site (see Fig. 22-14). Success with the pyridiniumanalogs led to an intensive search for more effective oximes andthe discovery of the bispyridinium compounds toxogonin orobidoxime [bis(4-formyl-N-methylpyridinium oxime) ether dichlo-ride], TMB-4 [N,N-trimethylene bis(pyridine-4-aldoxime) bro-mide], and, more recently, the H-series compounds. However, theseagents are not without toxicity and only pralidoxime and toxogo-nin have seen extensive antidotal use (Engelhard and Erdmann,1964; Steinberg et al., 1977).
Table 22-9Classification and Treatment of Organophosphorus Insecticide Poisoning Based on Plasma Pseudocholinesterase ActivityMeasurement
TREATMENTCLASSIFICATION ENZYME ACTIVITY
OF POISONING (% OF NORMAL) ATROPINE PRALIDOXIME
Mild 20–50 1.0 mg SC 1.0 g IV over 20 to 30 minModerate 10–20 1.0 mg IV every 20 to 30 min 1.0 g IV over 20 to 30 min
until sweating and salivationdisappear and slight flush andmydriasis appear
Severe 10 5.0 mg IV every 20 to 30 min 1.0 g IV as above. If no improvement,until sweating and salivation administer another 1.0 g IV. If nodisappear and slight flush improvement, start IV infusion atand mydriasis appear 0.5 g/h
SOURCE: Ecobichon et al., 1977.
Figure 22-17. The pralidoxime-catalyzed reactivation of an organo-phosphate-inhibited molecule of acetyl-cholinesterase, showing the release of active enzyme and the formation of an oxime-phosphate complex.
2996R_ch22_761-810 4/16/01 4:37 PM Page 783
Copy
right
ed M
ater
ial
Copyright © 2001 by The McGraw-Hill Companies Retrieved from: www.knovel.com

784 UNIT 5 TOXIC AGENTS
With the apparent availability of organophosphorus nervegases and their known ability to form rapidly aging complexes withAChE, the relative ineffectiveness of atropine and pralidoxime insuch poisonings must be taken into account when the treatment ofindividuals exposed to these agents is confronted (Koplovitz et al.,1992; Webb, 1993; Clement, 1994). Toxogonin (obidoximechloride) appears to be effective in tabun poisoning while, of theH-series of bis-pyridinium monooximes, HI-6 appears to be effi-cacious in soman and cyclohexylmethyl phosphorofluoridate(CMPF, or CF) poisonings. However, despite striking therapeuticeffects, the issue is clouded by the fact that little reactivation oferythrocytic or brain AChE occurs (Kusic et al., 1991; Shih, 1993).
The clinical treatment of carbamate toxicity is similar to thatfor organophosphorus ester insecticide intoxication with the ex-ception that the use of oximes is contraindicated. Early reports, inwhich pralidoxime or toxogonin was used in treating carbaryl in-toxications, revealed that the oxime enhanced the carbaryl-inducedtoxicity (Sterri et al., 1979; Ecobichon and Joy, 1994). However,the oximes appear to offer some antidotal properties in patientsexposed to aliphatic oxime carbamates (aldicarb, methomyl)(Feldman, 1999).
Diazepam (10 mg SC or IV) may be included in the treatmentregimen of all but the mildest cases of organophosphorus and/orcarbamate intoxications. In addition to relieving any mental anxi-ety associated with the exposure, diazepam counteracts some as-pects of the CNS and neuromuscular signs that are not affected byatropine. Doses of 10 mg (SC or IV) are appropriate and may berepeated. Other centrally acting drugs that may depress respirationare not recommended in the absence of artificial respiration.
It is important to appreciate that vigorous treatment of anti-cholinesterase-type insecticide intoxications does not offer protec-tion against the possibility of delayed-onset neurotoxicity or thepersistent sensory, cognitive, and motor defects discussed earlier.These deficits, albeit reversible over a long time interval, appearconsistently in intoxications and are caused by mechanisms as yetunknown. Certain evidence points to severe damage to the neuro-muscular junctions in skeletal muscle, resulting in a persistent pe-ripheral muscular weakness (Wecker et al., 1978).
Pyrethroid Esters
A major class of insecticides comprises the synthetic pyrethroids,a group of chemicals that entered the marketplace in 1980 but, by1982, accounted for more than 30 percent of worldwide insecti-cide usage (Anon., 1977; Vijverberg and van den Bercken, 1982).However, these synthetics arise from a much older class of botan-ical insecticides, pyrethrum, a mixture of six insecticidal esters(pyrethrins, cinerins, and jasmolins) extracted from driedpyrethrum or chrysanthemum flowers (Chrysanthemum cinerari-aefolium, Chrysanthemum coccineum) (Hartley and West, 1969).Although it is believed that the natural pyrethroids were discov-ered by the Chinese in the first century A.D., the first written ac-counts of these agents are found in the seventeenth-century litera-ture and commercial preparations made their appearance in themid-1800s (Neumann and Peter, 1987). Japanese woodblock printsfrom the early 1800s exist in which one can see smoldering in-secticide coils of pressed pyrethrum powder not unlike those man-ufactured and used today.
In 1965, the world output of pyrethrum was approximately20,000 tons, with Kenya alone producing some 10,000 tons(Cremlyn, 1978). The ever-increasing demand for this product has
far exceeded the limited world production, leading chemists to fo-cus attention on the synthesis of new analogs, hopefully with bet-ter stability in light and air, better persistence, more selectivity intarget species, and low mammalian toxicity. In addition to exten-sive agricultural use, the synthetic pyrethroids are components ofhousehold sprays, flea preparations for pets, plant sprays for homeand greenhouse use, and other applications. For an in-depth dis-cussion of the development of the pyrethroid ester insecticides,their chemistry, and their biological activity, the reader is referredto Elliott (1976), Cremlyn (1978), Casida et al. (1983), Leahey(1985), Matsumura (1985), Narahashi (1985), Narahashi et al.(1985), and Joy (1994b).
Natural pyrethrum consists of a mixture of six esters derivedfrom two acids (chrysanthemic, pyrethric) and three alcohols(pyrethrolol, cinerolol, jasmolol), producing an effective contactand stomach poison mixture having both knockdown and lethality.When the complex structure was identified, the synthetic esterswere marketed (Fig. 22-18). Distinctive molecular structures con-vey selectivity toward certain insect species and in some cases totoxicity in mammals. Several of the pyrethroid esters exist in iso-meric forms around the cyclopropane nucleus, resulting in dis-tinctly different toxicities and potencies (Casida et al., 1983).
Signs and Symptoms of Poisoning Based on the symptoms pro-duced in animals receiving acute toxic doses, the pyrethroids fallinto two distinct classes of chemicals (Table 22-10). The type Ipoisoning syndrome or T syndrome is produced by esters lackingthe �-cyano substituent and is characterized by restlessness, inco-ordination, prostration, and paralysis in the cockroach, as comparedwith the rat, which exhibits such signs as sparring and aggressivebehavior, enhanced startle response, whole-body tremor, and pros-tration. The type II syndrome, also known as the CS syndrome, isproduced by those esters containing the �-cyano substituent andelicits intense hyperactivity, incoordination, and convulsions incockroaches, in contrast to rats, which display burrowing behav-ior, coarse tremors, clonic seizures, sinuous writhing (choreo-athetosis), and profuse salivation without lacrimation; hence the
A
B
R1
R2
C C
O R3
O R4
CCH3CH3
CH CHCH CH
CH OR5
R4
C CH
O C
N
CH3 CH3
CH
Figure 22-18. The basic structures of the synthetic pyrethroid ester in-secticides showing (A) the intact cyclopropane ring in type I esters, withR1 and R2 (methyl, bromine, chlorine, etc.), R3 (hydrogens or cyano) andR4 (3-phenoxybenzoate, other) substituents; and (B) the “open” struc-ture of type II esters with R4 (3-phenoxybenzoate, other) and R5 (substi-tuted phenyl substituents).
2996R_ch22_761-810 4/16/01 4:37 PM Page 784
Copy
right
ed M
ater
ial
Copyright © 2001 by The McGraw-Hill Companies Retrieved from: www.knovel.com

CHAPTER 22 TOXIC EFFECTS OF PESTICIDES 785
term CS (choreoathetosis/salivation) syndrome. A few of theseagents, fenpropanthrin, for example, cause a mixture of type I andII effects, depending on the species (rat or mouse) treated and pos-sibly on the route of administration (Verschoyle and Aldridge,1980; Gammon et al., 1981; Lawrence and Casida, 1982). The bulkof evidence points to the fact that the type II syndrome involvesprimarily an action in the mammalian CNS, whereas with the type Isyndrome, peripheral nerves are also involved. This hypothesis wasbased initially on the observed symptomatology, but more recentevidence has revealed a correlation between the severity of type IIresponses and brain concentrations of deltamethrin in mice, re-gardless of the route of administration (Barnes and Verschoyle,1974; Ruzo et al., 1979). Agents eliciting the type II syndromehave greater potency when injected intracerebrally, relative to in-traperitoneal injection, than those causing type I syndrome effects(Lawrence and Casida, 1982). There is no indication of a funda-mental difference between the mode of action of pyrethroids onneurons of target and nontarget species, and the neurotoxicologicresponses depend on a combination of physicochemical propertiesof the particular pyrethroid ester, the dose applied, the time inter-val after treatment, and the physiologic properties of the particu-lar model used (Leaks et al., 1985).
Although these insecticides cannot be considered to be highlytoxic to mammals, their use indoors in enclosed and poorly venti-lated spaces has resulted in some interesting signs and symptomsof toxicity to humans. Exposure to the natural pyrethrum mixtureis known to cause contact dermatitis; descriptions of the effectsrange from localized erythema to a severe vesicular eruption(McCord et al., 1921). The allergenic nature of this natural prod-uct is not surprising, with asthma-like attacks and anaphylactic re-actions and peripheral vascular collapse among the responses ob-served. Human toxicity associated with the natural pyrethrins stemsfrom their allergenic properties rather than from direct neurotoxi-
city. There has been little evidence of the allergic-type reactions inhumans exposed to synthetic pyrethroid esters.
One notable form of toxicity associated with syntheticpyrethroids has been a cutaneous paresthesia observed in workersspraying esters containing an �-cyano substituent (deltamethrin,cypermethrin, fenvalerate). The paresthesia developed several hoursfollowing exposure and was described as a stinging or burningsensation on the skin which, in some cases, progressed to a tin-gling and numbness, the effects lasting some 12 to 24 h (LeQuesneet al., 1980; Tucker and Flannigan, 1983; Zhang et al., 1991).
Reports have appeared in the literature from the People’sRepublic of China, where synthetic pyrethroids have been usedon a large scale on cotton crops since 1982 (Stuart-Harte, 1988;He et al., 1988, 1989). Associated with the sloppy handling ofdeltamethrin and fenvalerate, both of which are type II compounds,some 573 cases of acute poisoning have occurred with some 229cases of occupational exposure. Some 45 cases of intoxication in-volved cypermethrin. Occupational exposure resulted in somedizziness plus a burning, itching, or tingling sensation of the ex-posed skin, which was exacerbated by sweating and washing withwarm water. The signs and symptoms disappeared by 24 h afterexposure. Spilling these agents on the head, face, and eyes resultedin pain, lacrimation, photophobia, congestion, and edema of theconjunctiva and eyelids. Ingestion of pyrethroid esters caused epi-gastric pain, nausea and vomiting, headache, dizziness, anorexia,fatigue, tightness in the chest, blurred vision, paresthesia, palpita-tions, coarse muscular fasciculations in the large muscles of theextremities, and disturbances of consciousness. In severe poison-ings, convulsive attacks persisting from 30 to 120 s were accom-panied by flexion of the upper limbs and extension of the lowerlimbs, with opisthotonos and loss of consciousness. The frequencyof these seizures was on the order of 10 to 30 times a day in thefirst week after exposure, gradually decreasing in incidence, with
Table 22-10Classification of Pyrethroid Ester Insecticides on the Basis of Chemical Structure and Observed Biological Activity
Signs and Symptoms
SYNDROME COCKROACH RAT CHEMICALS
Type I Restlessness Hyperexcitation Allethrin(“T” syndrome) Incoordination Sparring Cismethrin
Prostration Aggressiveness PhenothrinParalysis Enhanced startle Pyrethrin I
response ResmethrinWhole-body tremor TetramethrinProstration
Type II Hyperactivity Burrowing Acrinathrin(“CS” syndrome) Incoordination Dermal tingling Cycloprothrin
Convulsions Clonic seizures CyfluthrinWrithing CyhalothrinProfuse salivation Cyphenothrin
CypermethrinDeltamethrinEsfenvalerateFenvalerateFlucynthrateFluvalinate
2996R_ch22_761-810 4/16/01 4:37 PM Page 785
Copy
right
ed M
ater
ial
Copyright © 2001 by The McGraw-Hill Companies Retrieved from: www.knovel.com

786 UNIT 5 TOXIC AGENTS
recovery within 2 to 3 weeks (He et al., 1989). The signs and symp-toms of acute intoxication appear to be reversible and no chronictoxicity has been reported to date.
Site and Mechanism of Toxicity Several mechanisms of actionof pyrethroid esters exist in the sensory, motor, and central nerv-ous systems of insects and vertebrates, many of these actions be-ing studied in vitro using cockroach, crayfish, and squid giant axonpreparations or various cultured cell systems (Narahashi, 1971,1976, 1985; Casida et al., 1983; Forshaw et al., 1993; Narahashi,1998; Ray, 2000).
Both type I and type II esters modify the gating kinetics ofsodium channels involved in the inward flow of sodium ions, pro-ducing the action potential in cells that are normally closed at theresting potential (Marban et al., 1998). The pyrethroids affect boththe activation (opening) and inactivation (closing) of the channel,resulting in a hyperexcitable state as a consequence of a prolongednegative afterpotential that is raised to the threshold membrane po-tential and producing abnormal repetitive discharges (Ginsburg andNarahashi, 1993; Narahashi et al., 1998). The observed differencesbetween type I and type II esters lie in the fact that the former holdsodium channels open for a relatively short time period (millisec-onds), whereas type II esters keep the channel open for a prolongedtime period (up to seconds) (Joy, 1994b). Although the repetitivedischarges could occur in any region of the nervous system, thoseat presynaptic nerve terminals would have the most dramatic ef-fect on synaptic transmission (i.e., on the CNS and peripheral sen-sory and motor ganglia), giving rise to the signs documented inTable 22-10. The depolarizing action would have a dramatic effecton the sensory nervous system because such neurons tend to dis-charge when depolarized even slightly, resulting in an increasednumber of discharges and accounting for the tingling and/or burn-ing sensation felt on the skin and observed in particular with typeII esters (van den Bercken and Vijverberg, 1983). The biologicalactions of different pyrethroid esters at sodium channels is highlyvariable in that (1) cis and trans stereospecificity exists, the cis iso-mers being as much as 10-fold more toxic than trans isomers(Soderlund, 1985); (2) an additional chiral center is produced if acyano-substituent is added to the alcohol (Fig. 22-18), giving riseto eight possible isomers; (3) the binding of cis and trans isomersdiffers, being competitive at one site and noncompetitive at another(Narahashi, 1986); (4) tetrodotoxin-resistant sodium channels are10-fold more sensitive to pyrethroids than are tetrodotoxin-sensitive channels (Ginsburg and Narahashi, 1993; Song andNarahashi, 1996); (5) affinity to sodium channels is dependent onthe variable �-subunit composition of the 10 or more differentchannels identified to date (Marban et al., 1998); (6) insect sodiumchannels are 100-fold more sensitive than mammalian channels,thereby in part explaining species susceptibility (Warmke et al.,1997); (7) low temperature (25°C) exerts a greater inhibitory ef-fect of pyrethroid esters (on sodium channels) than a higher (37°C)temperature due to increased current flow at low temperature(Narahashi et al., 1998). According to Narahashi et al. (1998), themost important factor that causes differential toxicity is the sodiumchannel of the nervous system.
Other sites of action have been noted for pyrethroid esters, asshown in Fig. 22-19. Calcium channels have been proposed tar-gets, particularly in insects at levels of 107 M (Duce et al., 1999).Mammalian calcium channels appear to be less sensitive, with ef-fects being seen only with type I esters. However, many of these
experiments were conducted on cultured neuroblastoma cells,sinoatrial node cells, and so on, with results difficult to reconcilewith neuronal actions. Several agents (permethrin, cypermethrin,deltamethrin) inhibit Ca2�, Mg2�-ATPase, the effect of whichwould result in increased intracellular calcium levels accompaniedby increased neurotransmitter release and postsynaptic depolariza-tion (Clark and Matsumura, 1982).
Type II esters at relatively high concentrations act on theGABA-gated chloride channels in mammalian brain, this actionperhaps being related to the seizures seen with type II ester intox-ication (Abalis et al., 1986; Bloomquist et al., 1986). However,other studies have shown pyrethroids to have no GABA-antago-nistic activity (Joy and Albertson, 1991; Ray, 2000).
Voltage-sensitive calcium-independent chloride channels innervous tissue control cell excitability and exist in functionally dif-ferent kinds, those sensitive to pyrethroid esters belonging to themaxi-chloride channel type (Franciolini and Petris, 1990; Forshawand Ray, 1993). The pyrethroid-induced decrease in chloride ioncurrent would result in cell excitability, synergizing with thepyrethroid effect on sodium channels. Only type II esters affectmaxi-chloride channels and may be responsible for the generationof salivation and myotonia along with those effects elicited onsodium channels (Joy, 1994; Ray et al., 1997; Ray, 2000).
Biotransformation, Distribution, and Storage Evidence to datesuggests that pyrethroid esters elicit little chronic toxicity in eitherin animals or humans. Chronic animal feeding studies yield high“no effect” levels, suggesting that there is little storage or accu-mulation of a body burden of these agents and, perhaps, an effi-cient detoxification of the chemicals.
Two ester linkages exist in pyrethroid esters, a terminal methylester (pyrethrin II) and one more centrally located ester adjacent
Figure 22-19. Other proposed cellular mechanisms by which pyrethroidesters interfere with neuronal function.
2996R_ch22_761-810 4/16/01 4:37 PM Page 786
Copy
right
ed M
ater
ial
Copyright © 2001 by The McGraw-Hill Companies Retrieved from: www.knovel.com

CHAPTER 22 TOXIC EFFECTS OF PESTICIDES 787
to the cyclopropane moiety (allethrin, tetramethrin, phenothrin,deltamethrin) and/or the �-cyano substituent (deltamethrin, cyper-methrin, fenvalerate, cyphenothrin). Pyrethroid esters are suscep-tible to degradation by hydrolytic enzymes, possibly by nonspe-cific carboxylesterases found associated with the microsomalfraction of tissue homogenates in various species (Ecobichon,1979; Casida et al., 1983). Hydrolysis of the methoxycarbonylgroup in pyrethrin II by an esterase in rat liver has been reported,but the major site of hydrolytic activity would appear to be at thecentral ester linkage (Elliott et al., 1972; Shono et al., 1979; Glick-man and Casida, 1982). The importance of ester hydrolysis as aroute of detoxification is verified by the fact that many organophos-phorus esters that are capable of inhibiting tissue esterases poten-tiate pyrethroid ester toxicity in a variety of species (Casida et al.,1983). Species susceptibility to pyrethroid ester toxicity would ap-pear to be highly dependent on the nature of the tissue esterase,the level of activity detected, the substrate specificity, and rate ofhydrolysis encountered in target and nontarget species. The mi-crosomal monooxygenase system found in the tissues of almost allspecies is extensively involved in the detoxification of everypyrethroid ester in mammals and of some of these agents in insectand fish species. Much of the research in this field has been sum-marized by Shono et al. (1979), Kulkami and Hodgson (1984), andCasida et al., (1983). The importance of the oxidative mechanismsin detoxification is demonstrated by the inclusion of the synergistpiperonyl butoxide, a classic monooxygenase inhibitor, in prepa-rations toxic to houseflies and other insects, in order to enhancethe potency of pyrethroid esters in the 10- to 300-fold range (Casidaet al., 1983; Matsumura, 1985).
Treatment of Poisoning Limited experience with pyrethroid in-toxications has restricted the development of acceptable protocolsfor treatment. Removal from exposure and lavage with vegetableand/or vitamin E cream will alleviate dermal paresthesia, presum-ably by binding the lipophilic pyrethroids (Flannigan et al., 1985).Most other therapies have been symptom-related topical steroidsfor contact dermatitis, antihistaminics, decongestants, and steroidnasal spray for rhinitis and inhaled steroids for asthma (O’Malley,1997). Systemic poisoning is more difficult to treat, symptomaticand supportive measures being appropriate (He et al., 1989). Ex-perimental therapies in animals have included local anesthetics,phenytoin, phenobarbitone, valproate, diazepam, mephenesin, andurethane, all at exceptionally high doses with interfering side ef-fects (Ray, 2000). Only the chloride channel agonists ivermectinand pentobarbitone appeared to be of any benefit, particularly intype II ester intoxications (Forshaw, 2000).
Avermectins
The avermectins were discovered in 1975, isolated from a cultureof the actinomycete Streptomyces avermitilis in a soil sample madeavailable to Merck and Co. by the Kitisato Institute in Japan(Campbell et al., 1983; Campbell, 1989; Fisher and Mrozik, 1992).Avermectins are derived from 16-member macrocyclic lactones,three of which—avermectin B1a, the homolog B1b (abamectin),and the semisynthetic ivermectin—have come into wide use in vet-erinary medicine as potent insecticidal, acaricidal, and anthelminticagents (Fisher and Mrozik, 1992) (Fig. 22-20). Ivermectin is usedfor a wide range of ecto- and endoparasites of domestic and wildanimals. Potent anthelmintic activity against most gastrointestinal
and systemic nematodes has been identified at exceptionally low(0.001 to 0.05 mg/kg body weight) dose levels, but this efficacydoes not extend to tapeworms or flukes (Campbell et al., 1983).Administered topically or subcutaneously (systemically), iver-mectin controls grubs, lice, mites, certain ticks, and biting flies oncattle, horses, sheep, swine, and dogs (except the collie breed,which appears to be uniquely susceptible) (Fisher and Mrozik,1992). Ivermectin treatment does not result in the rapid death ofbiting insects but appears to disrupt engorgement, molting, and re-production (Campbell et al., 1983). A mixture of avermectins B1aand B1b is also used currently as a foliar spray on crops againstvarious mites, Colorado potato beetle, tomato hornworm, Mexicanbean beetle, pea aphid, cabbage looper, corn earworm, and south-ern army worm (Fisher and Mrozik, 1992). Emamectin (MK-244),a derivative of avermectin B1, is more effective against Lepidopteralarvae.
The discovery that ivermectin was efficacious against the skin-dwelling microfilaria of Onchocerca species in cattle and horsessuggested that it might be of use in treating onchocerciasis (riverblindness) in humans, a debilitating disease caused by the organ-ism Onchocerca volvulus (Taylor and Green, 1989). It has becomethe drug of choice in treating lightly infected individuals, markedlyreducing the number of microfilaria in the skin though without ef-fect on the adult worms (Aziz et al., 1982; Taylor and Greene,1989). It is a safe, long-acting microfilaricide, an applied singledose (150 ug/kg body weight) killing the immature microfilariaand keeping the numbers of reappearing microfilaria well belowpretreatment levels for up to 1 year (Fisher and Mrozik, 1992).Ivermectin treatment can significantly reduce the incidence of on-chocerciasis by limiting the source of infection.
While the avermectins are highly lipid soluble, dermal ab-sorption is less than 1.0 percent of the applied dose (Fisherand Mrozik, 1992). Minimal oxidative biotransformation occurs,2,4-hydroxymethyl ivermectin and 3�-O-desmethyl ivermectin be-ing formed. Regardless of the route of administration, urinary ex-cretion accounts for only 0.5 to 2.0 percent of the dose, the re-mainder being excreted slowly in the feces (Campbell et al., 1983).While the liver and adipose tissue took up the agent(s), very littleresidue was found in muscle, kidney or brain. In the case of thebrain, presumably the agent could not penetrate the blood-brainbarrier. In 28 days, almost complete elimination of ivermectin oc-curred from the tissues of treated cattle with the exception of theliver and body fat (Fisher and Mrozik, 1992).
Toxicologically, ivermectin is acutely toxic to rodents andother species, the oral and intraperitoneal LD50 values being of theorder of 25 to 80 mg/kg body weight (Fisher and Mrozik, 1992).Dermal toxicity is low, the LD50 for rats and rabbits being 660and 406 mg/kg body weight, respectively. In subchronic studies,no treatment-related effects were observed in rats, dogs, and mon-keys at the usual therapeutic dosage range (0.1 to 1.2 mg/kg bodyweight per day). Ivermectin showed no positive genotoxic effectsin microbial assays, in a forward mutation assay (mouse lym-phoma), or in an unscheduled DNA synthesis assay. In develop-mental/reproductive studies, offspring abnormalities were observedonly at maternotoxic doses in mice, rats, and rabbits. Oncogeniceffects were not found in studies with abamectin.
Mechanism of Action Early studies, with a variety of inverte-brate species and model systems, resulted in a number of possiblemechanisms of action on neuronal transmission, chloride channels,
2996R_ch22_761-810 4/16/01 4:37 PM Page 787
Copy
right
ed M
ater
ial
Copyright © 2001 by The McGraw-Hill Companies Retrieved from: www.knovel.com

788 UNIT 5 TOXIC AGENTS
and GABA-like effects, summarized in Fisher and Mrozik (1992).However, care must be taken in interpreting such results, since in-terneuronal and neuromuscular junctions are known to differ con-siderably between species (Gerschenfeld, 1973). The low water sol-ubility and extensive (nonspecific) binding of avermectins precludeaccurate estimations of target-site concentrations, overestimatingthe actual target-site concentrations (Fisher and Mrozik, 1992).More recent binding affinity studies in rat brain and in insect andnematode nerve tissue preparations have revealed that, in inverte-brates, biological activity occurred at nanomolar and picomolarconcentrations (Schaeffer et al., 1989; Fisher and Mrozik, 1992).Higher concentrations appear to cause secondary events and/or ar-tifacts. While there may be different species-specific avermectin“receptors,” this insecticide class has a high binding affinity tochloride-channel proteins, opening the channels, reducing mem-brane resistance, and increasing conductance inward. Such bind-ing sites appear to be distinct from those of other effector mole-cules and involve GABA-insensitive chloride channels (Arena etal., 1991; Payne and Soderlund, 1991). Explaining the selectivespecificity of avermectins (wide range of nematodes, arthropods,insects but not tapeworms, flukes, and adult filaria) must await themolecular study of the receptor proteins in these species. Inmammals, in the nematode Caenorhabditis elegans, and the housefly, avermectins interact with the GABA-receptor complex, as hasbeen shown by avermectin-induced changes in GABA receptor–directed ligands (Deng and Casida, 1992; Arena et al., 1995; Lankaset al., 1997).
Newer Chemical Insecticides
With the development of insect resistance to various classes ofchemical insecticides, new approaches to insect control are needed.Utilizing a better understanding of insect neurobiology, some newand highly specific agents have been developed, capitalizing on therecognition that various receptors in insect nervous systems are sig-nificantly different from those in mammals. Shown in Fig. 22-21are the structures of three types of new agents entering or avail-able in the marketplace: (1) the nitromethylene heterocycles, de-veloped from the cyclodienes and cyclohexanes; (2) the nitroiminoderivatives (chloronicotinyl or neonicotinoids), similar to nicotine;and (3) the phenylpyrazoles. These chemicals are effective at lowapplication rates (8 to 10 g/ha), are not environmentally persistent,and show negligible toxicity toward vertebrates. They possessunique and selective mechanisms of action and perhaps point theway to future insecticide development. To date, there have been noreports of toxicity in humans.
Nitromethylenes A search for new classes of insecticides led toa study of various aromatic heterocycles containing a nitrometh-ylene substituent, only those containing a pyridine system beinginsecticidal (Fig. 22-21A and B) (Soloway et al., 1979). The ni-tromethylene heterocycle (NMH) insecticides are fast-acting neu-rotoxicants, effective by both contact or oral ingestion; they are rel-atively safe to vertebrates and degrade rapidly in the environment(Schroeder and Flattum, 1984).
HO
OCH3
CH3 O
O
O
O
H
O
OH
OCH3
O
OH
H
CH3O
CH3 H
CH3
CH3
H
H
O OH
HCH3
C2H5
CH3
22–23
25
CommonName
22–23 25
Avermectin Bla As above As above
CH(CH3)2
80% As above20% CH(CH3)2
As aboveAvermectin BlbAbamectin
Ivermectin CH2 CH2
Structural Positions
Figure 22-20. A structural representation of the macrocyclic lactone avermectin (B1a), abamectin (B1b), andthe semisynthetic insecticide ivermectin showing the structural differences at positions 22 to 23 and 25 of thering.
2996R_ch22_761-810 4/16/01 4:38 PM Page 788
Copy
right
ed M
ater
ial
Copyright © 2001 by The McGraw-Hill Companies Retrieved from: www.knovel.com

CHAPTER 22 TOXIC EFFECTS OF PESTICIDES 789
Using the cockroach ventral nerve cord (the sixth abdominalganglion nerve) preparation, various NMHs at micromolar con-centrations produced a biphasic effect characterized by an initialincrease in the frequency of spontaneous discharges followed by ablockade of nerve impulse conduction (Schroeder and Flattum,1984). Elegant, classic neuropharmacologic experiments con-ducted by these authors demonstrated that the NMHs acted as neu-rotransmitter mimics and had both excitatory depressant effects,eventually blocking postsynaptic nicotinic receptors (nAChR).More recent experiments with 2(nitromethylene)tetrahydro-1,3-thiazine (NMTHT) (Fig. 22-21B) confirmed the biphasic action atnAChRs in several insect species and suggested that a binding siteresided on the �-subunits of insect nAChRs (Leech et al., 1991).
Chloronicotinyl An outgrowth of studies of the nitromethylene-type insecticides was the discovery of the nitroimino heterocyclesbest known by the compound imidacloprid (Fig. 22-21C), devel-oped in Japan in the mid-1980s. This agent combines high potencyto insects with exceptionally low mammalian toxicity and a favor-able persistence (Liu et al., 1993; Bayer Corp., 1994). The potencyof imidacloprid and several analogs can be correlated with the bind-ing affinity to house fly head membranes and mouse brain mem-branes, these agents having a low affinity to the latter (Liu et al.,1993, 1995; Chao and Casida, 1997). Extensive studies havedemonstrated that imidacloprid binds specifically to nAChRs invarious insects’ nervous systems (Liu and Casida, 1993; Nishimuraet al., 1994; Liu et al., 1995). Imidacloprid acts as a partial ago-nist at the nAChR channel, generating subconductance-state cur-rents (Nagata et al., 1997). Characterization of cloned nAChRsfrom insects (Myzus persicae, peach aphid) revealed that imida-cloprid affinity was influenced strongly by the �-subunit content
of the receptor (Huang et al., 1999). Investigations of a nAChRstructure and function will prove to be a boon to further insecti-cide development.
Phenylpyrazoles The phenylpyrazole derivatives show extensivebiological activity, including insecticidal, herbicidal, and miticidalproperties. The compound fipronil (Fig. 22-21D) is insecticidal andhas been shown to be highly effective against a broad range of in-sect pests at low-level foliar or soil application. It was first stud-ied by Rhone-Poulenc Agro in 1987 and is now available com-mercially (Gant et al., 1998). It is considered to be a secondgeneration removed from the earlier organochlorine insecticides(lindane, �-endosulfan) acting at the GABA receptor to block thechloride channel (Damgaard et al., 1999).
Fipronil has a unique mode of action in that it blocks thepassage of chloride ions through the GABA-regulated chloridechannel, disrupting CNS activity, since GABA is an inhibitory neu-rotransmitter. Fipronil is a noncompetitive inhibitor of GABA-induced effects, the effects in neurons being an increase in rapidbursts of electrical activity (Gant et al., 1998). Fipronil, a sulfox-ide, is biotransformed to the corresponding sulfone in biologicalsystems, the latter compound still being a potent insecticide. Thisconversion can be blocked by piperonyl butoxide, the cytochrome-P450 inhibitor. There is also a photoproduct, desulfinyl fipronil.Fipronyl and the sulfone selctively bind with high affinity to in-sect GABA receptors and with much lower affinity to vertebratebrain receptors (Cole et al., 1993; Hainzl et al., 1998). Comparingfipronil and its derivatives to earlier chloride-channel blockers, theIC50 vertebrate/IC50 insect ratios are 158 for fipronil, 140 forlindane, 31 for the desulfinyl product, 19 for fipronil sulfone, and4 for �-endosulfan (Hainzl et al., 1998). The selectivity ratios rel-ative to human GABA receptor are 135 for fipronil, 17 for the sul-fone, and 16 for the desulfinyl photoproduct (Hainzl et al., 1998).
All of the above receptor-specific insecticides have providednew ligand probes for neurotransmitter receptors as well as a newapproach to insecticide development. The only worrisome concernis that, with increasing use and reliance on such agents, target in-sects may develop resistance to these new compounds throughmechanisms similar to those seen for other neurotransmitters. Inthe meantime, the high insect selectivity, low mammalian toxicity,and low rates of application may provide a breathing space in theongoing search for efficacious, nontoxic chemical insecticides.
BOTANICAL INSECTICIDES
Naturally occurring agents of plant origin have been used to con-trol insect pests. These chemicals ranged from highly toxic agents(to both target and nontarget species), such as nicotine, to relativelyinnocuous substances, such as derris root. Interestingly, despite theoverwhelming number of synthetic insecticide formulations on themarket, the two above-mentioned agents can still be purchased andare still considered effective insecticides.
Nicotine
Nicotine, first used as an insecticide in 1763, has been used as acontact insecticide, stomach poison, and fumigant in the form ofnicotine alkaloid, the sulfate salt, or in the form of other deriva-tives. Commercially, nicotine is extracted from the leaves of Nico-tiana tabacum and Nicotiana rustica by alkali treatment and steamdistillation or by extraction with benzene, trichloroethylene, or di-
N
NH2
NCF3
Cl
Cl
CN
SO
CF3
D
FIPRONIL
Cl
N
CH2
CH
N NHA
NMINO2
N
S
H
B
NMTHT
CH NO2
Cl
N
CH2 N NHC
IMIDACLOPRID(IMI)N NO2
Figure 22-21. Representative structures of the nitromethylene, chlor-onicotinyl, and phenylpyrazole insecticides.
2996R_ch22_761-810 4/16/01 4:38 PM Page 789
Copy
right
ed M
ater
ial
Copyright © 2001 by The McGraw-Hill Companies Retrieved from: www.knovel.com

790 UNIT 5 TOXIC AGENTS
ethyl ether. Nicotine makes up some 97 percent of the alkaloid con-tent of commercial tobacco. It is marketed under the trade nameof Black Leaf 40, an aqueous solution of the sulfate salt of nico-tine, containing 40 percent nicotine.
Nicotine is extremely toxic, the acute oral LD50 in rats beingon the order of 50 to 60 mg/kg. It is readily absorbed through theskin, and any contact with nicotine solutions should be washed offimmediately. Anecdotal accounts of experiences by people whosprayed this chemical as an agricultural insecticide make an inter-esting collection of stories, all pointing to the fact that nicotinemimics the action of acetylcholine at all ganglionic synapses andat neuromuscular junctions, causing muscular fasciculations, con-vulsions, and death from paralysis of the respiratory muscles viablockade of the neuromuscular junctions (see Table 22-7). It func-tions as an insecticide in much the same manner, causing a block-ade of synapses associated with motor nerves in insects.
Rotenoids
Six rotenoid esters occur naturally and are isolated from the plantDerris eliptica found in Southeast Asia or from the plant Lon-chocarpus utilis or Lonchocarpus urucu, native to South America.Rotenone, one of the alkaloids, is the most potent and can be pu-rified by solvent extraction and recrystallization. It can be used ei-ther as a contact or a stomach poison. However, it is unstable inlight and heat and almost all toxicity can be lost after 2 to 3 daysduring the summer. Rotenone is very toxic to fish, and one of itsmain uses by native people over the centuries was to paralyze fishfor capture and consumption. The mammalian toxicity variesgreatly with the species exposed, the method of administration, andthe type of formulation. Crystalline rotenone has an acute oral LD50
of 60, 132, and 3000 mg/kg for guinea pigs, rats, and rabbits, re-spectively (Matsumura, 1985). Because the toxicity of derris pow-ders exceeds that of the equivalent content of rotenone, it is obvi-ous that the other esters in crude preparations have significantbiological activity. Acute poisoning in animals is characterized byan initial respiratory stimulation followed by respiratory depres-sion, ataxia, convulsions, and death by respiratory arrest (Shimkinand Anderson, 1936). The anesthetic-like action on nerves appearsto be related to the ability of rotenone to block electron transportin mitochondria by inhibiting oxidation linked to NADH2, this re-sulting in nerve conduction blockade (O’Brien, 1967; Corbett,1974). Although toxicity in laboratory and domestic animals hasbeen reported with acute LD50 values of 10 to 30 mg/kg, humanintoxications are rare. The estimated fatal oral dose for a 70-kgman is of the order of 10 to 100 g. Rotenone has been used topicallyfor treatment of head lice, scabies, and other ectoparasites, but thedust is highly irritating to the eyes (potentially causing conjunc-tivitis), the skin (causing contact dermatitis), and the upper respi-ratory tract (causing rhinitis) and throat (linked with pharyngitis).
HERBICIDES
A herbicide, in the broadest definition, is any compound that is ca-pable of either killing or severely injuring plants; it may be usedfor the elimination of plant growth or the killing off of plant parts(Jager, 1983). Many of the early chemicals—such as sulfuric acid,sodium chlorate, arsenic trioxide, sodium arsenite, petroleum oils,iron and copper sulfate, or sodium borate—were frequently hardto handle and/or were very toxic, relatively nonspecific, or phyto-toxic to the crop as well as the unwanted plant life if not applied
at exactly the proper time. In the late 1930s, many studies wereinitiated to find agents that would selectively destroy certain plantspecies. Many of these early chemicals were more effective butstill possessed considerable mammalian toxicity. However, a fewcompounds served as prototype chemicals for further development.Summaries of the early days of herbicide development are pre-sented by Cremlyn (l978), McEwen and Stephenson (1979), Kirby(1980), and Jager (1983).
In the past two decades, the herbicides have represented themost rapidly growing section of the agrochemical pesticide busi-ness due in part to (1) movement into monocultural practices, wherethe risk of weed infestation has increased because fallowing andcrop rotation, which would change weed species, are no longer invogue; and (2) mechanization of agricultural practices (planting,tending, harvesting) because of increased labor costs. The annualrate of growth of herbicide production on a worldwide basis be-tween 1980 and 1985 was 1.9 percent per year, more than doublethe rate of growth for insecticides during the same period (Marquis,1986). The result has been a plethora of chemically diverse struc-tures rivaling the innovative chemistry of the insecticides, the aimbeing to protect desirable crops and obtain high yields by selec-tively eliminating unwanted plant species, thereby reducing thecompetition for nutrients.
Herbicides may be classified by chemical structure, althoughthis is not very enlightening because of overlapping biological ef-fects for a variety of chemical structures. The second method ofclassification pertains to how and when the agents are applied. Pre-planting herbicides are applied to the soil before a crop is seeded.Preemergent herbicides are applied to the soil before the usual timeof appearance of the unwanted vegetation. Postemergent herbicidesare applied to the soil or foliage after the germination of the cropand/or weeds. Plant biochemists classify herbicides according totheir mechanism of toxicity in plants; their action is referred to asselective (toxic to some species), contact (act when impinging onthe plant foliage), or translocated (being absorbed via the soil orthrough the foliage into the plant xylem and phloem).
ln this chapter, herbicides are classified by their ability to in-terfere with specific biochemical processes essential for normalgrowth and development—interactions that result in severe injuryto the plant and its eventual death. In Table 22-11, the various mech-anisms by which herbicides exert their biological effects are shown,along with the generic and chemical names of the classes of her-bicides and some examples of each class. The claim has been madethat, because the modes of action involve biochemical phyto-processes having no counterparts in mammalian systems, no riskof mammalian toxicity is associated with these chemicals. With theexception of a few chemicals, the herbicides have demonstratedlow toxicity in mammals.
However, the current controversy around these chemicals cen-ters on demonstrated or suspected mutagenicity, teratogenicity,and/or carcinogenicity associated either with the agent(s) or withcontaminants and by-products of manufacture found in traceamounts in technical-grade material. The presence of some of thesecontaminants has been largely ignored without any recognition thatthe toxicities associated with them are different from those ob-served with the herbicidal chemical and frequently occur at farlower dosages.
In terms of general toxicity, because the major route of ex-posure to herbicides is dermal and these agents tend to be strongacids, amines, esters, and phenols, they are dermal irritants, caus-ing skin rashes and contact dermatitis even when exposure involves
2996R_ch22_761-810 4/16/01 4:38 PM Page 790
Copy
right
ed M
ater
ial
Copyright © 2001 by The McGraw-Hill Companies Retrieved from: www.knovel.com

CHAPTER 22 TOXIC EFFECTS OF PESTICIDES 791
diluted formulations. There appear to be subpopulations of indi-viduals who are hypersensitive to dermal contact with solutions oraerosolized mists of certain types of herbicides, and moderate tosevere urticaria has been observed to persist for 5 to 10 days fol-lowing exposure. Certain individuals, particularly those prone toallergic reactions, may experience severe contact dermatitis,asthma-like attacks, and even anaphylactic reactions following der-mal or inhalation contact with formulated herbicides. Whetherthese effects are chemical-specific for the herbicide or for emulsi-fiers, cosolvents, and so-called inerts found in formulations has notbeen established. One such example is discussed below. Althoughskin patch testing of herbicidal chemicals has usually proven to benegative, it is possible that patients’ responses may be associatedwith a generalized, nonspecific irritant effect of the formulation.Many of these dermal and pulmonary reactions respond satisfac-torily to treatment with antihistaminic agents.
In contrast, there are other herbicides that can elicit a rangeof acute and chronic effects following exposure, and it is on thesechemicals that attention is focused here.
Chlorophenoxy Compounds
During World War II, considerable effort was directed toward thedevelopment of effective, broad-spectrum herbicides in both theUnited States and the United Kingdom with a view to bothincreasing food production and finding potential chemical war-fare agents (Kirby, 1980). The chlorophenoxy compounds (Fig. 22-22)—including the acids, salts, amines, and esters—werethe first commercially available products evolving from this re-search in 1946. This class of herbicides has seen continuous, ex-
tensive, and uninterrupted use since 1947 in agriculture for broad-leafed weeds and in the control of woody plants along roadside,railway, and utilities’ rights of way and in reforestation programs.In plants, these chemicals mimic the action of auxins, hormoneschemically related to indoleacetic acid, that stimulate growth. Nohormonal activity is observed in mammals and other species, andbeyond target organ toxicity that can be associated with the phar-macokinetics, biotransformation, and/or elimination of these chem-icals, their mechanisms of toxic action are poorly understood. Thechlorophenoxy herbicides are no longer the agents of choice be-cause of concerns over the formation of chlorinated dibenzofuransand dibenzodioxins, particularly 2,3,7,8-tetrachlorodibenzo-p-dioxin(TCDD), as a consequence of poorly monitored manufacturingpractices or improper product storage in steel drums sitting beside
Table 22-11Mechanisms of Action of Herbicides
MECHANISM(S) CHEMICAL CLASSES
Inhibition of photosynthesis by disruption of light Ureas, 1,3,5-triazines, 1,4-triazines, uracils, pyridazones,reactions and blockade of electron transport 4-hydroxybenzonitriles, N-arylcarbamates, acylanilides
Inhibition of respiration by blockade of electron transfer Dinitrophenolsfrom NADH or blocking the coupling of electron transfer Halophenolsto ADP to form ATP
Growth stimulants, “auxins” Aryloxyalkylcarboxylic acids, benzoic acidsInhibitors of cell and nucleus division Alkyl N-arylcarbamatesInhibitors of protein synthesis DinitroanilinesInhibition of carotenoid synthesis, protective pigments in Chloracetamide, hydrazines, o-substituted diphenyl ethers
chloroplasts to prevent chlorophyll from being destroyedby oxidative reactions
Inhibition of lipid synthesis S-alkyl dialkylcarbamodithioatesAliphatic chlorocarboxylic acids
Inhibition of acetolase synthase Sulfonylureas, imidazolines, triazolopyrimidines,sulfonamides
Inhibition of protoporphyrinogen oxidase Diphenyl ethers, heterocyclic phenyl ethers (benzotriazoles,indolinones, benzisoxazoles, quinoxalindones,benzoxazines)
Inhibition of enolpyruvylshikimate-3-phosphate synthetase GlyphosateInhibition of glutamine synthetase GlufosinateUnknown mechanisms, nonselective chemicals Inorganic agents (copper sulfate, sulfuric acid, sodium
chlorate, sodium borate)Organic agents (dichlobenil, benzoylpropethyl,
chlorthiamid, bentazone)
Figure 22-22. The molecular structure of the three most commonchlorophenoxyacetic acid herbicides: 2,4-dichlorophenoxyacetic acid(2,4-D), 2,4,5-trichlorophenoxyacetic acid (2,4,5-T), and 4-chloro-o-toloxyacetic acid (MCPA). Ester and amine salt derivatives are also mar-keted.
2996R_ch22_761-810 4/16/01 4:38 PM Page 791
Copy
right
ed M
ater
ial
Copyright © 2001 by The McGraw-Hill Companies Retrieved from: www.knovel.com

792 UNIT 5 TOXIC AGENTS
runways under the tropical sun. However, since they are still usedin developing nations around the world, their toxicology cannot beignored.
A tremendous volume of mammalian toxicity data has beencollected over the past 42 years from both animal studies and in-cidents of human exposure (Hayes, 1982; Stevens and Sumner,1991). It is of interest to note that, in a recent toxicologic reeval-uation of 2,4-D for the purposes of providing the U.S. EPA with anew toxicity database for the chemical, an industry task force dis-covered nothing of toxicologic significance that was not alreadyknown about the chemical, with one exception (Mullison, 1986).The exceptional finding was the appearance of astrocytomas in thebrains of male Fischer 344 strain rats exposed to the highest(45 mg/kg per day) dosage. A subsequent review of the findingssuggested that this tumor incidence was not treatment-related(Koestner, 1986; Solleveld et al., 1984). The acute toxicity elicitedby chlorophenoxy herbicides has been described by Hayes (1982).The oral LD50 values ranged from 300 to 1000 mg/kg in differ-ent animal species, and only the dog appeared to be particularlysensitive, possibly on the basis that it has considerable difficultyin the renal elimination of such organic acids (Gehring et al., 1976).
Accidental and/or occupational intoxications have been re-viewed (Hayes, 1982; Stevens and Sumner, 1991; Ecobichon,1996). An industrial accident in 1949 in a 2,4,5-T manufacturingplant in Nitro, West Virginia, presented the first large occupationalexposure, with acute symptoms of exposure to the reaction prod-ucts, including skin, eye, and respiratory tract irritation; headache;dizziness; nausea; acneiform eruptions; severe muscle pain in thethorax, shoulders and extremities; fatigue; nervousness; irritabil-ity; dyspnea; complaints of decreased libido; and intolerance tocold (Ashe and Suskind, 1953). In an epidemiologic study of thesesame workers conducted in 1984, clinical evidence of chloracnepersisted in some 55.7 percent of those exposed (113 out of 204),and an association was found between the persistence of chloracneand the presence and severity of actinic elastosis of the skin(Suskind and Hertzberg, 1984). There was no evidence of increasedrisk of cardiovascular, hepatic, or renal disease or of central or pe-ripheral nervous tissue damage. One study documented neurotox-icity, decreased peripheral conduction velocities being observed inworkers employed in the manufacture of 2,4-D and 2,4,5-T (Singeret al., 1982). Earlier literature reported significant peripheral neu-ropathies in three sprayers using 2,4-D ester, signs and symptomsprogressing from muscle pain and paresthesias to severe paralysis,all of which was supported by electromyographic analysis(Goldstein et al., 1959). Chloracne, or “weed bumps,” has been themost persistent effect observed in almost all incidents of chlorophe-noxy herbicide exposure, although this is not a specific effect, beingcaused by a number of halogenated aromatic compounds includ-ing polyhalogenated biphenyls, dibenzo-p-dioxins, dibenzofurans,and naphthalenes (Schultz, 1968; Poland et al., 1971).
A wide range of human lethal dosages of 2,4-D has been re-ported, the average being in excess of 300 mg/kg, although it maybe as low as 80 mg/kg. The oral dose required to elicit symptomsis of the order of 50 to 60 mg/kg. Since these chemicals are irri-tants, oral ingestion has caused focal submucosal hemorrhage,moderate congestion, and edema as well as necrosis of the intes-tinal tract, fatty infiltration and necrosis of the liver, degenerationof the convoluted tubules of the kidney, and pneumonitis and in-flammation in the terminal bronchioles (Hayes, 1982).
In hindsight, many of the above-mentioned signs and symp-toms might be attributed to contaminants found in chlorophenoxy
herbicides from early production times, principally from a mixtureof polychlorinated dibenzo-p-dioxins, the main one being TCDD,found in samples of 2,4,5-T at levels of 30 to 50 ug/g (Hay, 1982;Gough, 1986). The teratogenicity (cleft palate, renal anomalies)produced by commercial, TCDD-contaminated (30 ug/g) 2,4,5-Twas not reproduced when highly purified 2,4,5-T was used(Courtney et al., 1970; Courtney and Moore, 1971). Recent in vitroand in vivo genotoxicity studies of 2,4-D have proven negative(Charles et al., 1999a, b; Gollapudi et al., 1999). Recent acciden-tal/occupational exposures to chlorophenoxy herbicides have notresulted in peripheral neuropathies, a fact confirmed in animal stud-ies using purified 2,4-D dimethylamine salt (Mattsson et al., 1986).While the carcinogenicity of purified 2,4-D and 2,4,5-T has notbeen established in rodent studies, that of TCDD has (Van Milleret al., 1977; Kociba et al., 1978). The toxicology of the polychlo-rinated dioxins and furans is discussed in other chapters.
Public concerns about the chlorophenoxy herbicides have fo-cused on birth defects, cancers, and the spurious illnesses reportedamong military personnel exposed to the defoliant Agent Orange(a 50:50 mixture of the n-butyl esters of 2,4-D and 2,4,5-T) sprayedextensively during the Vietnam conflict and found to be contami-nated with TCDD to a maximum of 47 ug/g (Hay, 1982; Greenwaldet al., 1984; Gough, 1986; CDC, 1988; Stellman et al., 1988;Tamburro, 1992). While exposure to this persistent contaminantcan be verified by blood analysis of veterans, the multifaceted na-ture of the adverse effects has precluded a definitive association(Ketchum et al., 1999). Epidemiologic studies of cancer in farm-ers and others occupationally exposed to chlorophenoxy herbicidesfor long periods of time have suggested an association with softtissue sarcomas, non-Hodgkin’s lymphoma (NHL), and Hodgkin’slymphoma (HL) without any definitive conclusions (Hardell andSandstrom, 1979; Ott et al., 1980; Eriksson et al., 1981; Hardell etal., 1981; Cantor, 1982; Theiss et al., 1982; Lynge, 1985;Schumacher, 1985; Hoar et al., 1986; Pearce et al., 1986; Bond etal., 1988; Bond and Rossbacher, 1993). A study of wheat farmersin western Canada, where 2,4-D has been used almost exclusivelyfrom 1947, showed an overall lower mortality and cancer rate thanexpected (Wigle et al., 1990). A review of published studies pre-sented evidence of an association between occupational exposureto chlorophenoxy herbicides and an increased risk of NHL(Morrison et al., 1992). The study of an international database setup by IARC/NIEHS revealed no clearly detectable excess for NHLand HL but a sixfold excess of soft tissue sarcomas in the cohortand a ninefold excess among sprayers (Saracci et al., 1991). An in-crease in prostate cancer mortality among Canadian farmers hasbeen reported (Morrison et al., 1993). Other studies of databaseshave suggested that the carcinogenic impact of chlorophenoxycompounds was negligible (Munro et al., 1992; Bond andRossbacher, 1993). Lacking the ideal “definitive, clean” study, thiscontroversy will continue in the literature. In the meantime, thechlorophenoxy herbicides have been phased out of use in devel-oped nations, being replaced by other agents. However, the con-cerns persist, particularly in developing countries where use con-tinues and in Vietnam, where exposure was so heavy.
Animals will tolerate repeated oral exposure to doses ofchlorophenoxy herbicides marginally below the single, toxic oraldose without showing significant signs of toxicity, an observationsuggesting that there is little cumulative effect on target organs. Atdosages causing toxicity, few specific signs other than muscularand neuromuscular involvement were observed in animals, al-though tenseness, stiffness in extremities, muscular weakness,
2996R_ch22_761-810 4/16/01 4:38 PM Page 792
Copy
right
ed M
ater
ial
Copyright © 2001 by The McGraw-Hill Companies Retrieved from: www.knovel.com

CHAPTER 22 TOXIC EFFECTS OF PESTICIDES 793
ataxia, and paralysis have been reported. Hepatic and renal injuryin addition to irritation of the gastrointestinal mucosa have beenobserved in acute lethality studies in animals.
Bipyridyl Derivatives
One chemical class of herbicides deserving of particular attentionis the bipyridyl group, specifically paraquat (1,1�-dimethyl-4,4�-bipyridylium dichloride, methyl viologen) and diquat (1,1�-ethyl-ene-2,2�-bipyridylium dibromide) (Fig. 22-23). Paraquat was firstsynthesized in 1882, but its pesticidal properties were not discov-ered until 1959 (Haley, 1979). This agent, a nonselective contactherbicide, is one of the most specific pulmonary toxicants knownand has been the subject of intensive investigation because of thestartling toxicity observed in humans. A high mortality rate is en-countered in poisoning cases. Many countries have banned or se-verely restricted the use of paraquat because of the debilitating orlife-threatening hazards from occupational exposure and the largenumber of reported accidental and suicidal fatalities (Campbell,1968; Davies et al., 1977; Haley, 1979; Tinoco et al., 1993). How-ever, paraquat is still used in some 130 countries and, in third-world nations, is available to 98 percent of the agricultural work-ers (WHO, 1984; Ramasamy et al., 1988). In Taiwan, paraquataccounted for 54 percent of the pesticide-related poisonings in theyears 1985 to 1993 (Yang et al., 1996). The toxicology of this classof herbicides should not be ignored.
In animals, paraquat shows moderate acute toxicity, the oralLD50 values for various species ranging from 22 to 262 mg/kg.Intoxication involves a combination of signs and symptoms thatinclude lethargy, hypoxia, dyspnea, tachycardia, hyperpnea, adip-sia, diarrhea, ataxia, hyperexcitability, and convulsions, dependingon the dosage and the species studied (Smith and Heath, 1976;Haley, 1979). Necropsy reveals hemorrhagic and edematous lungs,intraalveolar hemorrhage, congestion and pulmonary fibrosis,centrilobular hepatic necrosis, and renal tubular necrosis. Lungweights of intoxicated animals increase significantly despitemarked losses in body weight. From a catalog of all the signs andsymptoms, it is obvious that the lung is the most susceptible tar-get organ, and the same histopathologic picture of pulmonary le-sions is observed in mice, rats, dogs, and humans (Clark et al.,1966). In poisonings, immediate effects are usually not seen inanimals; within 10 to 14 days, however, respiration becomesimpaired, rapid, and shallow and the morphologic changes seen in-clude degeneration and vacuolization of pneumocytes, damage to
type I and type II alveolar epithelial cells, destruction of theepithelial membranes, and proliferation of fibrotic cells.
Paraquat, a highly polar compound, is poorly absorbed fromthe gastrointestinal tract (5 to 10 percent) (Haley, 1979). Experi-ments in rats showed that 52 percent of the administered oral dosewas still localized in the intestine some 32 h postadministration,although 45 percent of the dose had been excreted in the urine andfeces within 48 h, with some detected in urine for up to 21 days(Murray and Gibson, 1974). Intoxication by the ingestion of for-mulation concentrates may be enhanced by increased absorptiondue to the presence of emulsifiers and cosolvents. Metabolism bymammalian tissue is not extensive, although intestinal microfloramay account for 30 percent of the excreted metabolites in animalstudies (Daniel and Gage, 1966). Elevated levels of paraquat in re-nal tissue suggest the kidney as a primary route of excretion (Roseet al., 1976).
Pulmonary tissue, both in vivo and in vitro, acquires muchhigher concentrations of paraquat than other tissues with the ex-ception of the kidney. Over a 30-h posttreatment period, dispro-portionately high levels are found in the lung (Sharp et al., 1972;Rose et al., 1976). Acquisition is due to a unique diamine/polyamine transport system in the alveolar cells. Upon uptake,paraquat undergoes a NADPH-dependent one-electron reduction toa free radical that reacts with molecular oxygen to regenerate theparaquat cation plus a reactive superoxide anion (O2
. ), which isconverted into hydrogen peroxide (H2O2) by the enzyme superox-ide dismutase. Both O2 and H2O2 can attack polyunsaturated lipids,forming lipid hydroperoxides that, in turn, react with unsaturatedlipids to form more lipid-free radicals, perpetuating the destructivereaction (Smith, 1987). Alveolar cell membrane damage results inalveolitis, the destruction of alveolar cells, invasion of the spaceby fibrotic cells accompanied by a loss of pulmonary elasticity andrespiratory impairment, with an inefficient gas (O2, CO2) exchange.Cellular events can be modulated by the availability of oxygen, an-imals kept in air with only 10% oxygen faring better than thosekept in room air (Rhodes, 1974).
Paraquat poisonings in children and adults have been de-scribed in detail in the literature (Almog and Tal, 1967; Davies etal., 1977; Haley, 1979; Hayes, 1982; Tinoco et al., 1993). Paraquatis a favored agent in suicide attempts in many parts of the world(Wesseling et al., 1993, 1997; Yang et al., 1996). Paraquat-inducedintoxications should be divided into two categories: (1) poisoningsby accidental or intentional ingestion, usually involving formula-tion concentrates containing up to 20 percent active ingredient, and(2) functional intoxications, associated with dermal and/or inhala-tion exposure of diluted spray formulations.
The ingestion of commercial paraquat concentrates is invari-ably fatal and runs a time course of 3 to 4 weeks. The initial irri-tation and burning of the mouth and throat, the necrosis and slough-ing of the oral mucosa, severe gastroenteritis with esophageal andgastric lesions, abdominal and substernal chest pains, and bloodystools give way to the characteristic dominant pulmonary symp-toms, including dyspnea, anoxia, opacity in the lungs seen in chestx-rays, progressive fibrosis, coma, and death. While the pulmonarylesions are the most life-threatening, paraquat induces multiorgantoxicity with necrotic damage to the liver, kidneys, and myocar-dial muscle plus extensive hemorrhagic incidents throughout thebody.
Survivors of moderate-to-severe paraquat poisonings showedsignificant impairment in respiratory function tests, which im-proved dramatically or partially with time (Lin et al., 1995). How-
Figure 22-23. The chemical structures of paraquat and diquat, marketedas the dichloride and dibromide salts, respectively.
2996R_ch22_761-810 4/16/01 4:38 PM Page 793
Copy
right
ed M
ater
ial
Copyright © 2001 by The McGraw-Hill Companies Retrieved from: www.knovel.com

794 UNIT 5 TOXIC AGENTS
ever, it is as yet unknown whether long-term respiratory healtheffects occur with repeated and/or seasonal inhalation and/ordermal exposure to diluted paraquat aerosols (Wesseling et al.,1997). Swan (1969) found no detectable changes in workers ex-posed to sprays 6 days per week for 12 weeks. Senanayake et al.(1993) found no changes in standard spirometric tests in Sri Lankantea plantation workers continually exposed to paraquat aerosols.More recent studies have confirmed that no correlation existsbetween exposure and parameters such as FEV1.0, FVC, etc.(Castro-Gutierrez et al., 1997; Dalvie et al., 1999). However, inone study, severity of dyspnea and a threefold increase in episodicwheezing accompanied by shortness of breath was found amongthe most highly exposed workers (Castro-Gutierrez et al., 1997).Perhaps the standard, spirometric, functional tests are not sensitiveenough since, in the second study, there was a significant rela-tionship between long-term exposure and arterial oxygen desatu-ration measured by oximetry during an exercise test (Dalvie et al.,1999). More sensitive tests may be needed to assess long-term res-piratory disabilities.
Treatment of paraquat poisoning should be vigorous and ini-tiated as quickly as possible. Gastric lavage should be followed bythe administration of mineral adsorbents such as Fuller’s earth(kaolin), bentonite clay, or activated charcoal to bind any unab-sorbed paraquat remaining in the gastrointestinal tract. Purgativesmay be given. Absorbed paraquat may be removed from the blood-stream by aggressive, lengthy hemoperfusion through charcoal orby hemodialysis. To avoid excessive pulmonary damage, supple-mental oxygen should be reduced to a level just sufficient to main-tain acceptable arterial oxygen tension (40 to 50 mmHg) (Haley,1979; Hayes, 1982). Even though these patients may suffer fromhypoxia and respiratory insufficiency, hyperbaric oxygen is con-traindicated because it appears to promote cellular toxicity.
Diquat is a rapid-acting contact herbicide used as a desiccant,for the control of aquatic weeds, and to destroy potato halums be-fore harvesting. Diquat is slightly less toxic than paraquat; the oralLD50 values in various species are on the order of 100 to 400 mg/kg.Part of the reduced toxicity may be related to the fact that it ispoorly absorbed from the gastrointestinal tract; only 6 percent ofan ingested dose is excreted in the urine, whereas 90 to 98 percentof the dose is eliminated via the urine following subcutaneous ad-ministration (Daniel and Gage, 1966). A latency period of 24 h isseen prior to visible toxic effects.
Following acute, high-dose exposure or chronic exposure ofanimals to diquat, the major target organs were the gastrointesti-nal tract, the liver, and the kidneys (Hayes, 1982; Morgan, 1982).Chronic feeding studies resulted in an increased incidence ofcataracts in both dogs and rats (Clark and Hurst, 1970). It is con-sidered that diquat can form free radicals and that the tissue necro-sis is associated with the same mechanism(s) of superoxide-induced peroxidation as observed with paraquat. Unlike paraquat,diquat shows no special affinity for the lung and does not appearto involve the same mechanism that selectively concentratesparaquat in the lung (Rose and Smith, 1977).
Few diquat-related human intoxications have been reported todate (Schonborn et al., 1971; Narita et al., 1978; Hayes, 1982). Inthe few cases of suicidal intent described, ulceration of mucosalmembranes, gastrointestinal symptoms, acute renal failure, hepaticdamage, and respiratory difficulties were observed. CNS effectswere more severe. Interestingly, no fibrosis was evident in thelungs. One individual died of cardiac arrest.
Chloroacetanilides
Alachlor (Lasso) (Fig. 22-24) is an aniline herbicide used to con-trol annual grasses and broad-leaf weeds in a number of crops(corn, soybeans, peanuts) as a systemic herbicide absorbed by thegerminating shoots and roots by interfering with protein synthe-sis and root elongation. It is a slightly toxic (EPA class III) herbi-cide, the oral LD50 (rat) being 930 to 1350 mg/kg, and is a slight-to-moderate skin irritant (WSSA, 1994; Kamrin, 1997). Subchronicstudies in rats and dogs, with doses of 1 to 100 mg/kg/day, showedno adverse effects, but a 6-month dog study showed hepatic toxicityat all doses above 5 mg/kg/day, while a year-long study revealedhepatic, renal, and splenic effects above a dosage of 1 mg/kg/day.In reproductive studies, maternal and fetal toxicity was observedat the high oral doses used (150 and 400 mg/kg/day), but there wasno effect on reproduction. Neither were teratogenic effects seen inrats and rabbits, confirming the negative results in microbial mu-tagenicity studies. Revisiting the toxicity database and providingreplacement studies for carcinogenicity revealed thyroid tumorsand adenocarcinomas of the stomach and nasal turbinates of Long-Evans rats and in the lungs of CD-1 mice receiving relatively highdoses (126 and 260 mg/kg/day for rats and mice, respectively(Alachlor Review Board, 1987). Alachlor is considered to be acategory 2B (probable) human carcinogen by the U.S. EPA, a riskto agricultural workers during mixing and loading, with levels ofexposure ranging from 0.00038 to 2.7 mg/kg/day depending on theexposure model used. The putative carcinogen metabolite,2.6-diethylbenzoquinonimine (DEBQ1), is formed by oxidativeand nonoxidative reactions in mice, rats, and monkeys but not inhumans (Coleman et al., 1999). However, the human may formDEBQ1 via another route. Studies of thyroid tumors in rats sug-gest that alachlor-induced follicular cell tumors were related to in-creased metabolism of thyroxine via hepatic enzyme conjugation(Wilson et al., 1996). To date, there has been no evidence of anappreciable effect of alachlor exposure on worker mortality or can-cer rates (Acquavella et al., 1996).
The discovery of alachlor in well water at levels of 0.11 to2.11 �g/L, the highest being 9.1 �g/L, led to the cancellation ofits registration in Canada in 1985. Concerns about one analog ofa series has led to the examination of other chloracetanilides andclosely related agents—e.g., acetochlor, amidochlor, butachlor,
Figure 22-24. The chemical structures of alachlor (2-chloro-2�, 6�-diethyl-N-(methoxymethyl)acetanilide) and metolachlor (2-chloro-6�-ethyl-ethyl-N-(2-methoxy-1-methylethyl) acet-o-toluidide.
2996R_ch22_761-810 4/16/01 4:38 PM Page 794
Copy
right
ed M
ater
ial
Copyright © 2001 by The McGraw-Hill Companies Retrieved from: www.knovel.com

CHAPTER 22 TOXIC EFFECTS OF PESTICIDES 795
metalaxyl, metolachlor (Fig. 22-24), propachlor, etc.—all of theagents showing a consistent pattern of mutagenic activity mediatedvia metabolites, possibly even species-specific intermediates(Dearfield et al., 1999). One example acetochlor is converted intoa rat-specific metabolite that may be related to the nasal tumors,thus posing no genetic or carcinogenic hazard to humans (Ashbyet al., 1996). However, changes in leopard frog metamorphosis byacetochlor appears to be related to an interaction with thyroid hor-mone via a nonthyroid receptor mechanism (Cheek et al., 1999).No doubt, detailed mechanistic studies will reveal one or moremodes of action.
Phosphonomethyl Amino Acids
Two agents, N-phosphonomethyl glycine (glyphosate, Roundup,Vision) and N-phosphonomethyl homoalanine (glufosinate, Basta)must be considered because of their use in attempted suicides insoutheastern Asia (Fig. 22-25). Both agents are broad-spectrumnonselective systemic herbicides for postemergent control of an-nual and perennial plants (grasses, sedges, broad-leaf weeds) andwoody plants. While they exist as free acids, these agents are mar-keted as the isopropylamine or trimethylsulfonium salts ofglyphosate and the ammonium salt of glufosinate in formulationconcentrates of 41 percent and 18.5 percent, respectively. In con-sidering any toxicity data, one must make the distinction whetherthe data pertain to the acid, the salt, or the complete formulation(s)containing what appear to be biologically active surfactants(Kamrin, 1997; Watanabe and Sano, 1998).
Glyphosate Glyphosate binds to and inhibits the enzyme 5-enolpyruvyl-shikimate-3-phosphate synthetase (EPSPS), an en-zyme of the aromatic amino acid biosynthesis pathway essentialfor protein synthesis in plants (Haslam, 1993). In mammals, in-gestion, with oral LD50 values in the rat being 5600 mg/kg, al-though that of the trimethylsulfonium salt is 750 mg/kg (WSSA,1994). Formulations of glyphosate show moderate toxicity, LD50
values being between 1000 and 5000 mg/kg (Monsanto, 1985;WSSA, 1994). Glyphosate is nontoxic by the dermal route, withdermal LD50 values being in excess of 5000 mg/kg for the acidand the isopropylamine salt. While glyphosate is not a dermal ir-ritant in animals and does not induce photosensitization, formu-lations can cause severe occupational contact dermatitis, althoughpatch testing in humans resulted in no visible skin changes or sen-sitization (WSSA, 1994; Maibach, 1986). Glyphosate is an ocu-
lar irritant in the rabbit and human (Acquavella et al., 1999).Chronic studies with rats, dogs, and rabbits resulted in noglyphosate-related effects up to the highest doses tested (400 to500 mg/kg/day) (WSSA, 1994). Reproductive problems were ob-served in test animals only at very high (150 mg/kg/day) dosesthat elicited maternal toxicity. Glyphosate was not teratogenic atelevated doses in rabbits (350 mg/kg/day) or rats (175 mg/kg/day),although other signs of toxicity in dams and fetuses were observed.Glyphosate was not mutagenic in standard tests (Ames and otherbacterial assays, dominant lethal test in mouse) (Li and Long,1998; WSSA, 1994). However, one study, with glyphosate iso-propylamine salt and Roundup formulation, showed weak muta-genic activity in the Ames test and a significant increase in chro-mosomal aberrations in the Allium root cell when Roundup wastested (Rank et al., 1993). Carcinogenicity was not observed inmice, rats or dogs, glyphosate being classified by the U.S. EPAas a class E agent. Moses (1989) has reported tumors in the pitu-itary and mammary glands in glyphosate-treated rats. Glyphosatedoes not inhibit cholinesterases.
Unfortunately, glyphosate has become the agent of choice toreplace paraquat as a suicidal agent in many countries, the inci-dence of such poisonings increasing as paraquat is banned or ismore tightly controlled (Talbot et al., 1991; Tominack et al., 1991;Mendes et al., 1991; Leveridge, 1996; Yang et al., 1996). The for-mulation concentrate is used. Mild intoxications are characterizedby gastrointestinal symptoms (nausea, vomiting, diarrhea, abdom-inal pain) due to mucosal irritation and injury, with resolutionwithin 24 h (Talbot et al., 1991). In moderate intoxications, moresevere and persistent intestinal symptoms (ulceration, esophagitis,hemorrhage) are seen, along with hypotension, some pulmonarydysfunction, acid-base disturbance, and evidence of hepatic and re-nal damage. Severe poisoning is characterized by pulmonary dys-function requiring intubation, renal failure requiring dialysis, hy-potension and vascular shock, cardiac arrest, repeated seizures,coma, and death (Talbot et al., 1991; Tominack et al., 1991). A rea-sonable dose-effect relationship was established by Talbot et al.(1991), who estimated the volume(s) of formulation (concentrate)ingested causing asymptomatic, mild, moderate or severe poison-ing in humans to be 17, 58, 128 and 184 mL, respectively. Thiscould be converted to glyphosate concentrations of 87, 298, 658,and 946 mg/kg based on the formulation containing 360 g/L offree glyphosate and a 70-kg body weight.
Several reviews of intoxication cases, particularly the Japan-ese and Taiwanese series, have raised suspicions about the toxic-ity of the surfactant polyoxyethyleneamine (POEA) in the formu-lations used currently (Sawada and Nagai, 1987; Sawada et al.,1988; Talbot et al., 1991; Tominack et al. 1991). The median lethaldose of POEA is less than one-third that of the formulation orglyphosate (Sawada et al., 1988). Tai et al. (1990) found that POEAwas responsible for the hypotensive effect in dogs. This class ofsurfactants has been associated with hemolysis and with gastroin-testinal and CNS effects (Grugg et al., 1960; Sawada et al., 1988).As of 1999, efforts were being made to reformulate the Roundupproduct to replace POEA (personal communication).
Glufosinate N-phosphonomethyl homoalanine acts as a suicidesubstrate, interfering with glutamate synthesis in plants by irre-versibly inhibiting the enzyme glutamine synthetase, which playsan important role in ammonia detoxification and amino acid me-tabolism (Ebert et al., 1990). The herbicidal effect is caused by cy-
HO C PCH2 CH2NH OH
O O
OH
HO C PCH CH2 CH3CH2
O O
OHNH2
GLYPHOSATE
GLUPHOSINATE
ROUNDUP™VISION™
BASTA™TOTAL™
Figure 22-25. The chemical structures of glyphosate (N-phospho-nomethyl glycine) and glufosinate (N-phosphonomethyl homoalanine).
2996R_ch22_761-810 4/16/01 4:38 PM Page 795
Copy
right
ed M
ater
ial
Copyright © 2001 by The McGraw-Hill Companies Retrieved from: www.knovel.com

796 UNIT 5 TOXIC AGENTS
totoxicity from increased ammonia levels and impairment of pho-torespiration and photosynthesis (Hack et al., 1994).
The acute oral toxicity of glufosinate in mice, rats, and rab-bits was low, the oral LD50 lying between 1500 and 2000 mg/kg(Ebert et al., 1990; Hack et al., 1994). In subchronic feeding stud-ies in rats, no agent-related deaths occurred, although body weightgains in males were retarded in the 5000-ppm group. Food con-sumption was reduced and water consumption increased and, atthis high dose, signs of CNS excitation and hypothermia werenoted (Hack et al., 1994). Glufosinate was not considered to bemutaginic, teratogenic, or carcinogenic in animal studies, althoughin more recent studies using whole-embryo culture, teratogeniceffects in mice have been observed, the effect being induced apop-tosis in the neuroepithelium of developing embryos (Watanabeand Iwase, 1996; Watanabe, 1997). While glufosinate did not in-hibit brain cholinesterase, reductions in erythrocytic and serumcholinesterases have been seen in 7 of 16 patients affected by thisherbicide (Watanabe and Iwase, 1998). In contrast to plant metab-olism, glufosinate ammonium inhibited glutamine synthetase in an-imals, but this did not lead to a problem in ammonia metabolism,the mammal obviously compensating by using other metabolicpathways (Hack et al., 1994).
Glufosinate ammonium has been involved in a number ofpoisoning cases, particularly in Japan (Koyama et al., 1994;Watanabe and Sano, 1998; Tanaka et al., 1998). Early clinicalsymptoms included nausea, vomiting, and diarrhea associated withintestinal mucosal irritation but were followed in 24 h by neuro-logic signs, including impaired respiration, seizures, muscle weak-ness (post–status epileptic myopathy), convulsions, and deathwithin 4 days in 6 of 31 patients (Koyama, 1995). Some survivorsshowed either a brief loss of memory or amnesia for 7 to 10 daysafter intoxication (Koyama, 1995). Any significant role of gluta-mate in the neurologic events has not been confirmed. Limited in-formation is available regarding glufosinate persistence in vivo, buturinary levels higher than concomitant blood levels have been re-ported and urinary excretion persisted for 3 to 5 days after inges-tion (Watanabe and Iwase, 1998).
Once again, concerns have been raised about the role of thesurfactant. The direct cause of death seemed to be circulatory dis-turbance, especially cardiac insufficiency, possibly related to thesurfactant in the formulation involved in the intoxications (Koyamaet al., 1995; Watanabe and Sano, 1998). The absorption of glufos-inate in animals was 25 to 30 percent higher than the absorptionrate of the agent when given alone (Watanabe and Sano, 1998).This suggested that the glufosinate-related effects might be en-hanced by surfactant-induced penetration of the CNS, while thecardiovascular system effects were surfactant-related.
Many herbicides representative of several chemical classifi-cations and diverse structures have recently been introduced intoagricultural practice (Table 22-11). In general, these chemicals haverelatively low acute toxicity, the oral LD50 values in rats being ofthe order of 100 to 10,000 mg/kg, and they are toxicologically un-interesting, since—in subchronic and chronic studies—large dosescan be administered without eliciting significant biological effects.Many of these newer chemicals are applied to crops or soil atexceedingly low application rates, minimizing nontarget speciestoxicity and avoiding environmental contamination. Present con-cerns focus on groundwater contamination and closer scrutiny ofminor contaminants for mutagenic, teratogenic, and carcinogeniceffects. Poisonings in humans have usually been associated with
occupational exposure or with a few atypical but sometimes well-publicized incidents (Stevens and Sumner, 1991).
FUNGICIDES
Fungicidal chemicals are derived from a variety of structures rang-ing from simple inorganic compounds, such as sulfur and coppersulfate, through the aryl- and alkyl-mercurial compounds and chlo-rinated phenols to metal-containing derivatives of thiocarbamicacid (Fig. 22-26). The chemistry of fungicides and their propertieshave been discussed by Cremlyn (1978), Kramer (1983), and Ed-wards et al. (1991). Foliar fungicides are applied as liquids or pow-ders to the aerial green parts of plants, producing a protective bar-rier on the cuticular surface and systemic toxicity in the developing
Figure 22-26. Chemical structures of fungicides representative of vari-ous chemical classifications.
2996R_ch22_761-810 4/16/01 4:38 PM Page 796
Copy
right
ed M
ater
ial
Copyright © 2001 by The McGraw-Hill Companies Retrieved from: www.knovel.com

CHAPTER 22 TOXIC EFFECTS OF PESTICIDES 797
fungus. Soil fungicides are applied as liquids, dry powders, or gran-ules, acting either through the vapor phase or by systemic proper-ties. Dressing fungicides are applied to the postharvest crop (ce-real grains, tubers, corms, etc.) as liquids or dry powders to preventfungal infestation of the crop, particularly if it may be stored un-der less than optimum conditions of temperature and humidity. Thepostharvest loss of food crops to disease is a serious worldwideproblem (Table 22-1). Fungicides may be described as protective,curative, or eradicative, according to their mode of action. Protec-tive fungicides, applied to the plant before the appearance of anyphytopathic fungi, prevent infection by either sporicidal activity orby changing the physiologic environment on the leaf surface. Cu-rative fungicides are used when an infestation has already begunto invade the plant, and these chemicals function by penetratingthe plant cuticle and destroying the young fungal mycelium (thehyphae) growing in the epidermis of the plant, preventing furtherdevelopment. Eradicative fungicides control fungal developmentfollowing the appearance of symptoms, usually after sporulation,by killing both the new spores and the mycelia and by penetratingthe cuticle of the plant to the subdermal level (Kramer, 1983).
An effective fungicide must possess the following properties:(1) low toxicity to the plant but high toxicity to the particularfungus; (2) activity per se or ability to convert itself (by plant orfungal enzymes) into a toxic intermediate; (3) ability to penetratefungal spores or the developing mycelium to reach a site of action;and (4) formation of a protective, tenacious deposit on the plantsurface that will be resistant to weathering by sunlight, rain, andwind (Cremlyn, 1978). This list of properties is never fulfilled en-tirely by any single fungicide, and all commercially available com-pounds show some phytotoxicity, lack of persistence due to envi-ronmental degradation, and so forth. Thus, the timing of theapplication is critical in terms of the development of the plant aswell as the fungus.
The topic of fungicidal toxicity has been extensively reviewedby Hayes (1982) Edwards et al. (1991), and Kamrin (1997). Witha few exceptions, most of these chemicals have a low order of tox-icity to mammals (Table 22-12). However, all fungicides are cyto-toxic and most produce positive results in the usual in vitro mi-crobial mutagenicity test systems. Such results are not surprisingbecause the microorganisms (Salmonella, coliforms, yeasts, and
Table 22-12Acute Toxicity of Fungicides
IRRITATION* ORAL LD50, RAT
COMMON NAME CLASS (EYE/SKIN) (mg/kg)
Anilazine Triazine I 2,710Benomyl Imidazole I 10,000Captan Phthalimide I 8,400–15,000Carboxin Oxathiin NI 3,820Chinomethionate Quinomethionate NI 2,500–3,000Chlorothalonil Organochlorine I 10,000Dichloropropene Chlorinated alkene I 130–713Dinocap Dinitrophenol NI 980Dodine Aliphatic nitrogen I 1,000EPTC Thiocarbamate I 1,632Etridiazole Thiadiazole NI 1,000Fenarimol Pyrimidine I 2,500Hexachloroben- Organochlorine I 3,500Imazalil Imidazole I 227–334Iprodione Dicarboximide I 3,500Maneb Dithiocarbamate I 5,000–8,000Mancozeb Dithiocarbamate I 5,000–11,200Metalaxyl Benzenoid I 669Metiram Dithiocarbamate I 6,180–10,000Nabam Dithiocarbamate I 395Oxycarboxin Oxathiin NI 2,000Pyrazophos Phosphorothionate NI 151–632Quintozene Organochlorine I 1,710Thiabendazole Imidazole NI 3,100–3,600Thiophanate–Me Dithiocarbamate I 10,000Thiram Thiocarbamate I 800–1,900Triallate Thiocarbamate I 800–2,165Vinclozolin Dicarboximide I 10,000Zineb Dithiocarbamate I 7,600–8,900Ziram Dithiocarbamate I 1,400
*KEY: I, irritant properties; NI, nonirritation.
2996R_ch22_761-810 4/16/01 4:38 PM Page 797
Copy
right
ed M
ater
ial
Copyright © 2001 by The McGraw-Hill Companies Retrieved from: www.knovel.com

798 UNIT 5 TOXIC AGENTS
fungi) used in these test systems are not dissimilar from those cellsystems that fungicides are designed to kill, either through a directlethal effect or via lethal genetic mutations (Lukens, 1971). A safefungicide (nonmutagenic in test cell systems) would be useless forthe protection of food and health. Public concern has focused onthe positive mutagenicity tests obtained with many fungicides andthe predictive possibility of both teratogenic and carcinogenic potential. The fact that nearly 90 percent of all agricultural fungi-cides are carcinogenic in animal models has not reassured the pub-lic, especially when this is translated into the fact that some 75million pounds of the fungicides used annually fall into this cate-gory (NAS, 1987). An evaluation of 11 fungicides concluded that,although the areas treated with these chemicals represented only10 percent of the acreage treated annually with pesticides, theycould account for 60 percent of the total estimated dietary car-cinogenic risk.
While a number of different chemicals are shown in Fig. 22-26 to illustrate the diversity of structures, agents such aspentachlorophenol (PCP), hexachlorobenzene (HCB), captafol, andfolpet have been deregistered or banned in many countries but stillsee some use in other, less regulated parts of the world. Other fungi-cides are undergoing reevaluation because of suspected toxicity,particularly as teratogens or carcinogens and incomplete or out-dated toxicity databases.
Hexachlorobenzene
From the 1940s through the 1950s, HCB saw extensive use as afungidical dressing applied to seed grain as a dry powder. Between1955 and 1959, an epidemic of poisoning occurred in Turkey, ul-timately involving some 4000 individuals who consumed treatedgrain during times of crop failure. The syndrome, called black sore,was characterized by dermal blistering and epidermolysis, pig-mentation and scarring, alopecia, photosensitivity, hepatomegaly,porphyria, suppurative arthritis, osteomyelitis, and osteoporosis ofthe bones of the hands (Cam and Nigogosyan, 1963). Both adultsand children were afflicted, young children and nursing infants be-ing at particular risk (Schmid, 1960; Wray et al., 1962; Hayes,1982). While this agent has largely fallen by the wayside, it is stillbeing used in developing countries and still presents a health haz-ard.
The mammalian toxicity of HCB has been reviewed by Hayes(1982) and Hayes and Laws (1991). Like other organochlorinecompounds, HCB possesses all of the properties of chemical sta-bility, slow degradation and biotransformation, environmental per-sistence, bioaccumulation in adipose tissue and organs containinga high content of lipid membranes, and the ability to induce a rangeof tissue cytochrome-P450 as well as conjugative enzymes. Re-peated exposure of animals results in hepatomegaly and porphyriaas well as focal alopecia with itching and eruptions, followed bypigmented scars, anorexia, and neurotoxicity expressed as irri-tability, ataxia, and tremors. Immunosuppression was observed inboth mice and rats and a dose-dependent increase in hepatic andthyroid tumors was observed in hamsters (Lambrecht et al., 1982).While not mutagenic in microbial test systems and negative in dom-inant lethal studies, HCB was teratogenic in mice (renal and palatemalformations) and in rats (increased incidence of 14th rib). Hexa-chlorobenzene was particularly toxic to developing perinatal ani-mals, acquisition transplacentally and via the milk causing he-patomegaly, enlarged kidneys, hydronephrosis, and possible effectson the immune system.
Pentachlorophenol
Once used in tremendous volumes as a biocide in leather tanning,wood preservation, the paper and cellulose industry, and in paints,this chemical has been phased out of use because many commer-cial products were contaminated by polychlorinated dibenzo-dioxins and dibenzofurans, predominantly by hexachlorinated,heptachlorinated, and octachlorinated congeners. While thesecongeners are considerably less toxic than TCDD, evidence fromanimal studies has pointed to the fact that the contaminants in com-mercial- or technical-grade PCP were responsible for the toxicityobserved. Technical-grade PCP fed to rats caused altered plasmaenzymes, increased hepatic and renal weights, and caused hepato-cellular degeneration in addition to changes in blood biochemistry(decreased erythrocyte count, decreased hemoglobin and serum al-bumin). The administration of purified PCP resulted only in in-creased liver and kidney weight. Prolonged treatment of femalerats with technical PCP caused hepatic porphyria, increased mi-crosomal monooxygenase activity, and increased liver weight,whereas purified PCP caused no changes over the dosage rangestudied (Goldstein et al., 1977). Pentachlorophenol was not ter-atogenic in rats and is not considered to be carcinogenic in miceor rats (Innes et al., 1969; Johnson et al., 1973; Schwetz et al.,1977). A number of environmental problems have been associatedwith PCP (Eisler, 1989).
Human poisoning by commercial PCP has occurred, usuallyassociated with occupational exposure and instances of sloppy han-dling and neglect of hygienic principles (Jorens and Schepens,1993). The chemical is absorbed readily through the skin, the mostusual route of acquisition, with several products, including PCP,detected in the urine.
High-level exposure can result in death, preceded by an ele-vated body temperature (42°C or 108°F), profuse sweating and de-hydration, marked loss of appetite, decrease in body weight, tight-ness in the chest, dyspnea following exercise, rapid pulse, nauseaand vomiting, headache, incoordination, generalized weakness, andearly coma (Hayes, 1982). Pentachlorophenol acts cellularly to uncouple oxidative phosphorylation, the target enzyme beingNa�,K�-ATPase (Desaiah, 1977). Survivors frequently displaydermal irritation and exfoliation, irritation of the upper respiratorytract, and possible impairment of autonomic function andcirculation.
Phthalimides
Of this class of chemicals, folpet and captofol, true phthalimides,have been deregistered and only captan, being structurally differ-ent with a cyclohexene ring (Fig. 22-26), sees any use today. Allof the agents were embroiled in a prolonged controversy concern-ing mutagenic and possible teratogenic and carcinogenic proper-ties. All three chemicals were recognized as effective, persistentfoliar fungicides for rusts and smut, for Botrytis mold on soft fruit,apple and pear scab, black spot on roses, and as seed dressings(Cremlyn,1978). All three chemicals have high oral LD50 valuesof approximately 10,000 mg/kg in the rat. Mutagenicity associatedwith these agents was confirmed, but—because of the exception-ally high doses (up to 500 mg/kg) required to elicit biologicaleffects—teratogenicity was not proven or was masked by mater-nal toxicity and possible nutritional deficits.
Although the mechanism(s) by which captan and its analogsexerted their cellular toxicity has never been established, captan is
2996R_ch22_761-810 4/16/01 4:38 PM Page 798
Copy
right
ed M
ater
ial
Copyright © 2001 by The McGraw-Hill Companies Retrieved from: www.knovel.com

CHAPTER 22 TOXIC EFFECTS OF PESTICIDES 799
known to react with cellular thiols to produce thiophosgene, a po-tent and unstable chemical capable of reacting with sulfhydryl-,amino-, or hydroxyl-containing enzymes (Cremlyn, 1978). Thiolsreduce the potency of captan. A volatile breakdown product of cap-tan was responsible for mutagenic activity, the intermediate beingshort-lived and forming more quickly at higher levels at an alka-line pH. It is possible that there may be several mechanisms bywhich these chemicals can induce cellular toxicity.
Dithiocarbamates
Dimethyl- and ethylene-bisdithiocarbamate (EBDC) compoundshave been employed since the early 1950s as fungicides, and theEBDC chemicals saw widespread use on a large variety of smallfruits and vegetables. The nomenclature of these agents arises fromthe metal cations with which they are associated; for instance, di-methyldithio-carbamic acid bound to iron or zinc are ferbam andziram, respectively, whereas EBDC compounds associated withsodium, manganese, or zinc are nabam, maneb, and zineb, respec-tively. As is shown in Fig. 22-24, these chemicals are polymericstructures that possess environmental stability and yield good fo-liar protection as well as a low order of acute toxicity, with LD50
values in excess of 6000 mg/kg with the exception of nabam (395mg/kg). Mancozeb is a polymeric mixture of a zinc salt and thechemical maneb.
Although toxicity is negligible in animal feeding trials evenat high doses, acceptance of these agents has been marred by re-ported adverse health effects. Maneb, nabam, and zineb have beenreported to be teratogenic (Petrova-Vergieva and Ivanova-Chemischanka, 1973). Mancozeb has not been demonstrated to beteratogenic in the rat but has been associated with abnormallyshaped sperm (Hemavathi and Ratiman, 1993). Maneb has beenassociated with adverse reproductive outcomes (embryotoxicity:changes in usual number of offspring per litter, pregnancy rate,estrous cycle, and fetal development) (Lu and Kennedy, 1986).Maneb caused pulmonary tumors in mice, but studies in the rathave been equivocal (IARC, 1976). Environmental and mammaliandegradation of the EBDC compounds into ethylene thiourea (ETU),a known mutagen, teratogen, and carcinogen, as well as an an-tithyroid compound, has raised suspicions about these agents andfostered requests for more in-depth studies (IARC, 1976). Recentchronic studies of mancozeb administered to rats at high (500 to1500 mg/kg/day) doses demonstrated increased thyroid weight, re-duced iodine uptake, reduced protein-bound iodine and thyroxine,as well as a dose-related decrease in thyroid peroxidase activity(Kackar et al., 1997). Significant histopathologic changes were ob-served, including thyroid hyperplasia and hypertrophy of thefollicular mass with a loss of colloid mass. A study of EBDC fun-gicide–exposed backpack sprayers in Mexico demonstrated thatETU was formed in vivo and excreted in the urine and that thyroid-stimulating hormone levels were increased, although there was anonsignificant reduction in thyroxine and significant increases insister chromatic exchanges and chromosomal translocations inblood lymphocytes (Steenland et al., 1997).
Neurotoxicity has not been attributed to EBDC fungicides ineither experimental animals or humans except at excessively highdoses (Ecobichon, 1994c). A double (within 2 weeks) acute occu-pational, dermal exposure to Mandizan (mixture of maneb andzineb) resulted in initial complaints of muscle weakness, dizziness,and fatigue, with disorientation, slurred speech, muscle incoordi-nation, loss of consciousness, and tonic/clonic convulsions ap-
pearing rapidly following the second exposure (Israeli et al., 1983).A recent report from Brazil on two apparent Parkinson patients re-vealed that, as sprayers, they had experienced significant annualexposure to maneb over 4 to 5 years (Ferraz et al., 1988). Signsand symptoms included inability to walk, difficulty in talking,tremors in hands and feet, a short-stepped gait with cogwheeling,and bradykinesia. An extended study of 50 rural workers, 84 per-cent of whom admitted to using maneb improperly or carelessly,revealed milder but similar signs and symptoms. It was suggestedthat the effects might be related to the manganese content, althoughblood manganese levels were not elevated. Other evidence mightpoint to breakdown products of EBDC, such as carbon disulfide,as the neurotoxicant, although it is hard to accept such a high levelof absorption. It is also known that dithiocarbamates can bind var-ious divalent metals to form more lipophilic complexes capable ofentering the CNS (Ecobichon, 1994c).
FUMIGANTS
Such agents are used to kill insects, nematodes, weed seeds, andfungi in soil as well as in silo-stored cereal grains, fruits and veg-etables, clothes, and other consumables, generally with the treat-ment carried out in enclosed spaces because of the volatility ofmost of the products. Fumigants range from acrylonitrile and car-bon disulfide to carbon tetrachloride, ethylene dibromide, chloropi-crin, and ethylene oxide; their toxicologic properties are discussedunder other headings because many have other uses. Attention inthis section is directed only to a very few agents, although all ofthe chemicals mentioned have the potential for inhalation exposureand, for some of them, dermal and ingestion exposure.
Fumigants may be liquids (ethylene dibromide, dibro-mochloropropane, formaldehyde) that readily vaporize at ambienttemperature, solids that can release a toxic gas on reacting withwater (Zn2P3, AlP) or with acid [NACN, Ca(CN)2], or gases(methylbromide, hydrogen cyanide, ethylene oxide). These chem-icals are nonselective, highly reactive, and cytotoxic. The physico-chemical properties of these agents and hence their pattern(s) ofuse vary considerably (Cremlyn, 1978). With proper attention touse and appropriate safety precautions, there should be little effectother than occasional occupational exposure, because the volatil-ity of the agents is such that, when the enclosed space is opened,the gas or vapor escapes readily. However, reports in the literaturehave indicated the presence of low residual levels of ethylene dibro-mide, methylbromide, and other chemicals in various samples oftreated foods. More extensive descriptions of fumigant toxicity can befound in Hayes (1982), Morgan (1982), and Hayes and Laws (1991).
Phosphine
Used extensively as a grain fumigant, phosphine (PH3) is releasedfrom aluminum phosphide (AlP) by the natural moisture in thegrain over a long period of time, giving continual protection dur-ing transhipment of the grain. One serious accident with this chem-ical has been reported, in which this author played a small role inidentifying the causative agent, as the problem originated in theport of Montreal, Canada (Wilson et al., 1980). Grain leavingCanada for European destinations is fumigated by adding a certainnumber of sachets of AlP per ton of grain in the hold of the shipwhile loading. Phosphine (PH3), being heavier than air, sinksslowly through the grain. The particular ship in question ran intoa bad storm off Nova Scotia and began to leak, hastening the break-
2996R_ch22_761-810 4/16/01 4:38 PM Page 799
Copy
right
ed M
ater
ial
Copyright © 2001 by The McGraw-Hill Companies Retrieved from: www.knovel.com

800 UNIT 5 TOXIC AGENTS
down of the AlP to form PH3. The toxicant penetrated the quartersof the crew and officers, where 29 out of 31 crew members be-came acutely ill and two children, family members of one of theofficers, were seriously affected, one dying before reaching a hos-pital in Boston. Symptoms of PH3 intoxication in the adults in-cluded shortness of breath, cough and pulmonary irritation, nau-sea, headache, jaundice, and fatigue. The highest concentrations ofPH3 (20 to 30 ppm) were measured in a void space on the maindeck near the air intake for the ship’s ventilation system. In someof the living quarters, PH3 levels of 0.5 ppm were detected. Al-though this could be considered a bizarre situation, it does illus-trate an apparent problem with the use of this type of agent in anatmosphere of excess moisture.
Ethylene Dibromide/Dibromochloropropane
When inhaled at relatively high (200 ppm) concentrations, eth-ylene dibromide can cause pulmonary edema and inflammation inthe exposed animals. As one might expect, repeated exposures tolower concentrations produced hepatic and renal damage visual-ized as morphologic changes. Centrolobular hepatic necrosis andproximal tubular damage in the kidneys were observed in one fa-tal poisoning in which the individual ingested 4.5 ml of ethylenedibromide. This chemical, along with 1,2-dibromo-3-chloro-propane (DBCP), was found to elicit malignant gastric squamouscell carcinomas in mice and rats (IARC, 1977). DBCP was alsofound to cause sterility in male animals, and concentrations as lowas 5 ppm had an adverse effect on testicular morphology and sper-matogenesis. However, these results in animals came to light onlywhen a similar situation was detected in workers who manufac-tured the agent. Equivocal results have been reported for the mu-tagenicity of DBCP, the agent that causes base-pair substitution butnot a frame-shift mutation in Salmonella strains. In animal studiesof the dominant lethal assay, DBCP was positive (mutagenic) inrats but not in mice. DBCP was a reproductive toxicant in rabbitsand rats but not in mice (IARC, 1977).
RODENTICIDES
Many vertebrates—including rats, mice, squirrels, bats, rabbits,skunks, monkeys, and even elephants—on occasion can be con-sidered to be pests. Rodents, the most important of which are theblack rat (Rattus rattus), the brown or Norway rat (Rattus norvegi-cus), and the house mouse (Mus musculus), are particularly seri-ous problems because they act as vectors for several human dis-eases. They can consume large quantities of postharvest stored foodand/or foul or contaminate even greater amounts of foodstuffs withurine, feces, hair, and bacteria that cause diseases.
To be effective yet safe, a rodenticide must satisfy the fol-lowing criteria: (1) it must not be unpalatable to the target speciesand therefore must be potent; (2) it must not induce bait shyness,so that the animal will continue to eat it; (3) death should occur ina manner that does not raise the suspicions of the survivors; (4) itshould make the intoxicated animal go out into the open to die(otherwise the rotting corpses create health hazards); and (5) itshould be species-specific, with considerably lower toxicity toother animals that might inadvertently consume the bait or eat thepoisoned rodent (Cremlyn, 1978). The agents used constitute a di-verse range of chemical structures having a variety of mechanismsof action for at least partially successful attempts to attain species
selectivity (Fig. 22-27). With some chemicals, advantage has been taken of the physiology and biochemistry unique to rod-ents. With other rodenticides, the sites of action are common tomost mammals but advantage is taken of the habits of the pest animal and/or the dosage, thereby minimizing toxicity to nontar-get species.
Although most rodenticides are formulated in baits that areunpalatable to humans, thereby minimizing the potential hazard,there are surprising numbers of rodenticide intoxications each year.With only a few exceptions, the accidental or intentional ingestionof most rodenticides poses a serious, acute toxicologic problem be-cause the dosage ingested is invariably high and the signs andsymptoms of intoxication are generally well advanced and severewhen the patient is seen by a physician. As with other householdproducts, rodenticide poisoning is more frequently seen in chil-dren, whose added hazard is a small body weight in relation to thedosage ingested. The toxicology of the various classes of rodenti-cides has been extensively reviewed and the reader is referred toHayes and Laws (1991) and to Ellenhorn and Barceloux (1988) forin-depth coverage of the subject.
Figure 22-27. Representative structures of inorganic and organic ro-denticides from various chemical classifications.
2996R_ch22_761-810 4/16/01 4:38 PM Page 800
Copy
right
ed M
ater
ial
Copyright © 2001 by The McGraw-Hill Companies Retrieved from: www.knovel.com

CHAPTER 22 TOXIC EFFECTS OF PESTICIDES 801
A number of inorganic compounds—including thallium sul-fate, arsenious oxide, other arsenic salts, barium carbonate, yellowphosphorus, aluminum phosphide, and zinc phosphide—have beenused as rodenticides. A mixture of sodium cyanide with magne-sium carbonate and anhydrous magnesium sulfate has been usedin rabbit and mole burrows, causing hydrogen cyanide gas to beliberated slowly on contact with moisture. Natural or synthetic or-ganic chemicals, including strychnine, red squill (scillaren glyco-sides), and DDT have been used in the past. All of these agents arenonselective, highly toxic, and hazardous to other forms of life and,with the exception of zinc phosphide, have been abandoned in fa-vor of target-specific, selective chemicals.
Zinc Phosphide
This agent is used in developing nations because it is both cheapand effective. The toxicity of the chemical can be accounted forby the phosphine (PH3) formed following a hydrolytic reaction withwater in the stomach on ingestion. Phosphine causes widespreadcellular toxicity with necrosis of the gastrointestinal tract and in-jury to other organs, such as the liver and kidneys. Although moistzinc phosphide emits an unpleasant, rotten-fish odor, it is acceptedby rodents at concentrations of 0.5 or 1.0 percent in baits.
Accidental poisonings are rare in adults but a definite problemin children. Hayes (1982) recounts a poisoning attributed to the in-halation of zinc phosphide dust from treated grain, with signs ofintoxication that included vomiting, diarrhea, cyanosis, tachycar-dia, rhales, restlessness, fever, and albuminuria several hours fol-lowing exposure. It is a favorite chemical in suicides in Egypt (A.Amr, personal communication). The signs and symptoms includenausea, vomiting, headache, light-headedness, dyspnea, hyperten-sion, pulmonary edema, dysrrhythmias, and convulsions. Doses ofthe order of 4000 to 5000 mg have been fatal, but other individu-als have survived doses of 25,000 to 100,000 mg if early vomitinghas occurred. The usual decontamination measures and supportivetherapy are often successful if initiated early.
Fluoroacetic Acid and Derivatives
Sodium fluoroacetate (compound 1080) and fluoroacetamide(compound 1081) are white in color, odorless, and tasteless. Theextreme toxicity of these two chemicals has restricted their use toprepared baits. Both agents are well absorbed from the gastroin-testinal tract. Acute oral toxicity of fluoroacetate in the rat is of theorder of 0.2 mg/kg, whereas that of fluoroacetamide is 4 to 15mg/kg. The mechanism of action involves the incorporation of thefluoroacetate into fluoroacetyl–coenzyme A, which condenses withoxaloacetate to form fluorocitrate, which inhibits the enzymeaconitase and prevents the conversion of citrate to isocitrate in thetricarboxylic (Krebs) cycle. Inhibition of this system by fluoroci-trate results in reduced glucose metabolism and cellular respirationand affects tissue energy stores. These chemicals are uniquely ef-fective in mice and rats because of the high metabolic rate in thetissues susceptible to inhibition.
Estimates of the lethal dose of fluoroacetate in humans lie inthe range of 2 to 10 mg/kg. Gastrointestinal symptoms are seeninitially at some 30 to 100 min following ingestion. Initial nausea,vomiting, and abdominal pain are replaced by sinus tachycardia,ventricular tachycardia or fibrillation, hypotension, renal failure,muscle spasms, and such CNS symptoms as agitation, stupor,seizures, and coma. Histopathologic examination of postmortem
samples has revealed cerebellar degeneration and atrophy. Thereare no known antidotes to fluoroacetate intoxication, although glyc-erol monoacetate has proved beneficial in the treatment of poisonedmonkeys.
A-Naphthyl Thiourea
Following the discovery that phenylthiourea was lethal to rats butnot toxic to humans, �-naphthyl thiourea (ANTU) was introducedas a relatively selective rodenticide (Richter, 1946). A wide rangeof acute oral LD50 values has been reported for different species,the rat being the most sensitive at 3 mg/kg and the monkey theleast susceptible at 4 g/kg. The exact mechanism of action is notknown, but it is suspected that ANTU must be biotransformed invivo into a reactive intermediate. Young rats are resistant to thechemical, whereas older rats become tolerant to it, suggesting thatperhaps microsomal monooxygenases in young rats metabolize theagent too rapidly into nontoxic products, whereas in older rats ei-ther the lower levels of monooxygenases or the inhibition of theseenzymes results in less activation and affords protection (Boyd andNeal, 1976). ANTU causes extensive pulmonary edema and pleu-ral effusion as a consequence of action on the pulmonary capillar-ies. Studies with 35S- and 14C-labeled ANTU revealed that cova-lent binding to macromolecules in the lung and liver occurredfollowing treatment (Boyd and Neal, 1976). Following exposure toANTU, there are a number of biochemical effects, such as alter-ations in carbohydrate metabolism, adrenal stimulation, and inter-action of the chemical with sulfhydryl groups, but none of theseappear to bear any relationship to the observed signs of toxicity.
Although it would appear that the human is very resistant toANTU intoxication, probably because insufficient quantities areingested, poisonings have occurred, with tracheobronchial hyper-secretion of a white, nonmucous froth containing little protein, pul-monary edema, and respiratory difficulty (Hayes, 1982).
Anticoagulants
With the discovery that coumadin [3-(�-acetonylbenzyl)-4-hydroxycoumarin, warfarin), isolated from spoiled sweet clover,acted as an anticoagulant by antagonizing the actions of vitaminK in the synthesis of clotting factors (factors II, VII, IX, and X),it was introduced as a rodenticide. The onset of anticoagulation isdelayed 8 to 12 h after the ingestion of warfarin, with this latentperiod of onset dependent on the half-lives of the various clottingfactors (Katona and Wason, 1986). The safety of warfarin as a ro-denticide rests with the fact that multiple doses are required beforetoxicity develops and that single doses have little effect. However,the development of resistance to warfarin in rats in the 1950sprompted research into newer compounds, and the exploration ofstructure-activity relationships led to the development of the su-perwarfarins (brodifacoum, bromadiolone, coumachlor, diphen-coumarin) and the indanediones (diphacinone, chlorophacinone,pindone), a new class of anticoagulant compounds that are morewater-soluble. All of these newer agents differ from one another interms of acute toxicity, rapidity of action, and acceptance by therodent.
Human poisonings by these agents are rare because they aredispensed in grain-based baits. However, there are sufficient num-bers of suicide attempts, attempted murders, and a famous classiccase of the inadvertent consumption of warfarin-laden cornmealbait by an unsuspecting Korean family to provide adequate docu-
2996R_ch22_761-810 4/16/01 4:38 PM Page 801
Copy
right
ed M
ater
ial
Copyright © 2001 by The McGraw-Hill Companies Retrieved from: www.knovel.com

802 UNIT 5 TOXIC AGENTS
mentation of the signs and symptoms of poisoning (Lange andTerveer, 1954; Hayes, 1982; Jones, 1984; Lipton, 1984; Katonaand Wason, 1986). Following consumption over a period of days,bleeding of the gingiva and nose occurs, with bruising andhematomas developing at the knee and elbow joints and on the but-tocks, gastrointestinal bleeding with dark tarry stools, hematuriaaccompanied by abdominal or low back (flank) pain, epistaxis, andcerebrovascular accidents. The signs and symptoms will persist formany days after cessation of exposure, particularly so in the caseof the superwarfarins, which have prolonged biological half-lives(e.g., brodifacoun with 156 h compared to 37 h for warfarin)(Katona and Wason, 1986). In the Korean episode, consumption ofwarfarin was estimated to be on the order of 1 to 2 mg/kg/day fora period of 15 days, and signs and symptoms appeared 7 to 10 daysafter initial exposure; 2 out of the 14 affected individuals died asa consequence of not receiving any treatment (Lange and Terveer,1954).
CONCLUSIONS
With the advent of the chemical pesticides with their diverse na-ture, structures, and biological activity, the problem of ranking thehazard that each one poses to health has arisen. Should a classifi-cation system be based on acute toxicity alone or should some nu-merical scoring system be used to evaluate other endpoints of tox-icity? Should the classification scheme be based on the oral,dermal, or inhalation routes of exposure to the active ingredient orto a formulation concentrate? If one chooses acute toxicity and adefinitive endpoint expressed as the LD50, one must be cognizantof the fact that the LD50 is an estimate, with the range and confi-dence limits for any particular chemical possibly overlapping aclass boundary. To establish a classification system on the basis ofother toxicologic endpoints would be impossible given the vari-ability of biological effects, the dosages required to attain them,and the significance of such results in terms of human exposure.
In 1972, the WHO Expert Committee on Insecticides recom-mended the preparation of a classification of pesticides that wouldserve as a guide for developing countries (WHO, 1973). The clas-sification was to distinguish between the more and the less haz-ardous forms of each pesticide and was meant to permit formula-tions to be classified according to the percentage of the activeingredient and its physical state. Only acute hazards to health wereconsidered, meaning those resulting from single or multiple expo-sures over a relatively short period of time, from handling theproduct in accordance with the manufacturer’s directions. In 1975,the categories of the classification were established and, with onlyone modification to class 111, they are essentially the same as thosethat appear in Table 22-13. It is important to appreciate that theLD50 value quoted for any pesticide is not the median value butthe lower confidence limit value for the most sensitive sex, therebyensuring that a large safety factor has been built into the classifi-cation. A recent paper by Copplestone (1988) discusses the ad-vantages and disadvantages of the system and the placement ofproblem chemicals such as rodenticides (highly toxic to rats butnot presenting the same hazard to humans) and paraquat (havinga low dermal toxicity but causing fatal effects if ingested).
From experience, the WHO is of the opinion that this classi-fication scheme has worked well in practice, faithfully reflectingthe toxicity of these chemicals for humans. Only a few changes inclassification have been made for chemicals and/or their formula-tions since the introduction of the scheme, signifying that the sys-tem functions effectively. It would appear that acute toxicity is themost effective parameter by which to judge the hazard to humanhealth. With the move away from animal experimentation to invitro testing, this classification system can be modified to reflectother endpoints of toxicity if they can be quantitated, correlated,and validated to be equivalent to the LD50 results. As Dr. Copple-stone described it, “the classification has been a meeting point be-tween science and administration and a useful tool in the arma-mentarium of preventive medicine” (Copplestone, 1988).
Table 22-13The WHO Recommended Classification of Pesticides by Hazard
LD50 FOR THE RAT (mg/kg BODY WEIGHT)
ORAL DERMAL
CLASS SOLIDS LIQUIDS SOLIDS LIQUIDS
Ia Extremely hazardous �5 �20 �10 �40Ib Highly hazardous 5–50 20–200 10–100 40–400II Moderately hazardous 50–500 200–2000 100–1000 400–4000III Slightly hazardous 500 2000 1000 4000III� Unlikely to present 2000 3000 — —
hazard in normal use
REFERENCES
Abalis IM, Eldefrawi EM, Eldefrawi AT: Effects of insecticide on GABA-induced chloride flux into rat brain microsacs. J Toxicol EnvironHealth 18:13–23, 1986.
Abbott DC, Goulding R, Tatton JO’G: Organochlorine pesticide residuesin human fat in Great Britain. Br Med J 3:146–149, 1968.
Abou-Donia MB: Organophosphorus ester-induced delayed neurotoxicity.Annu Rev Pharmacol Toxicol 21:511–548, 1981.
Abou-Donia MB, Lapadula D: Mechanisms of organophosphorus ester-induced delayed neurotoxicity: Type 1 and type 11. Annu Rev Phar-macol Toxicol 30:405–440, 1990.
2996R_ch22_761-810 4/16/01 4:38 PM Page 802
Copy
right
ed M
ater
ial
Copyright © 2001 by The McGraw-Hill Companies Retrieved from: www.knovel.com

CHAPTER 22 TOXIC EFFECTS OF PESTICIDES 803
Acquavella JF, Riordan SG, Anne M, et al: Evaluation of mortality andcancer incidence among alachlor manufacturing workers. EnvironHealth Perspect 104:728–733, 1996.
Acquavella JF, Weber JA, Cullen MR, et al: Human ocular effects fromself-reported exposures to Roundup herbicides. Hum Exp Toxicol18:479–486, 1999.
Ahlbom J, Fredriksson A, Eriksson P: Exposure to an organophosphate(DFP) during a defined period in neonatal life induces permanentchanges in brain muscarinic receptors and behavior in adult mice.Brain Res 677:13–19, 1995.
Alachlor Review Board: Report of the Alachlor Review Board. Ottawa,Canada: Agriculture Canada, Canadian Government Publishing Cen-tre, 1987.
Albert A: Xenobiosis, Food, Drugs and Poisons in the Human Body. Lon-don: Chapman and Hall, 1987, pp 113–116.
Albertson TE, Cross CE: Pesticides in the workplace: A worldwide issue.Arch Environ Health 48:364–365, 1993.
Aldridge WN, Johnson MK: Side effects of organophosphorus compounds:Delayed neurotoxicity. Bull WHO 44:259–263, 1971.
Aldridge WN, Reiner E: Enzyme Inhibitors as Substrates. Amsterdam andNew York: North-Holland/American Elsevier, 1972.
Almog C, Tal E: Death from paraquat after subcutaneous injection. Br MedJ 3:721, 1967.
Anon.: A look at world pesticide markets. Farm Chem 141:38–42, 1977.Arena J, Liu K, Paress PS, et al: Avermectin-sensitive chloride currents in-
duced by Caenorhabditis elegans RNA in Xenopus oocytes. Mol Phar-macol 40:368–374, 1991.
Arena J, Liu K, Paress P, et al: The mechanism of action of avermectins inCaenorhabditis elegans: Correlation between activation of glutamate-sensitive chloride current, membrane binding and biological activity.J Parasitol 81:286–294, 1995.
Ariens ATh, Meeter E, Wolthius OL, et al: Reversible necrosis at the end-plate region in striated muscle of the rat poisoned with cholinesteraseinhibitors. Experientia 25:57–59, 1969.
Ashby J, Kier L, Wilson AG, et al: Evaluation of the potential carcino-genicity and genetic toxicity to humans of the herbicide acetochlor.Hum Exp Toxicol 15:702–735, 1996.
Ashe W, Suskind RR: Chloracne cases of the Monsanto Chemical Com-pany, Nitro, West Virginia, in Reports of the Kettering Laboratory.Cincinnati, OH: University of Cincinnati, October 1949, April 1950,July 1953.
Askew BM: Oximes and hydroxamic acids as antidotes in anti-cholinesterase poisoning. Br J Pharmacol Chemother 11:417–423,1956.
Azaroff LS: Biomarkers of exposure to organophosphorus insecticidesamong farmers’ families in rural El Salvador: Factors associated withexposure: Environ Res A 80:138–147, 1999.
Aziz MA, Diallo S, Diopp IM, et al: Efficacy and tolerance of ivermecticin human onchocerciasis. Lancet 2:171–173, 1982.
Bakry NM, El-Rashidy AH, Eldefrawi AT, et al: Direct actions oforganophosphate anticholinesterases on nicotinic and muscarinicacetylcholine receptors. J Biochem Toxicol 3:235–259, 1988.
Baris D, Zahm SH, Cantor KP, et al: Agricultural use of DDT and the riskof non-Hodgkin’s lymphoma: Pooled analysis of three case-controlstudies in the United States. Occup Environ Med 55:522–527, 1998.
Barnes JM, Verschoyle RD: Toxicity of new pyrethroid insecticides. Na-ture 248:711, 1974.
Bartle H: Quiet sufferers of the silent spring. New Scientist 130:30–35,1991.
Batchelor GS, Walker KC: Health hazards involved inuse of parathion infruit orchards on north central Washignton. AMA Arch Ind Hyg Oc-cup Health 10:522, 529,1954.
Bayer Corp: Material Safety Data Sheet—Admire™. Bayer Corp Agri-culture Division, 1994.
Beach JR, Spurgeon A, Stephens R, et al: Abnormalities on neurologicalexamination among sheep farmers exposed to organophosphorus pes-ticides. Occup Environ Med 53:520–525, 1996.
Berti PR, Receveur O, Chan H-M, et al: Dietary exposure to chemical contaminants from traditional food among adult Dene/Metis in thewestern Northwest Territories, Canada. Environ Res A 76:131–142,1998.
Bidstrup PL, Bonner JA, Beckett AG: Paralysis following poisoning by anew organic phosphorus insecticide (Mipafox). Br Med J 1:1068–1072, 1953.
Bloomquist JR, Adams PM Soderlund DM: Inhibition of gamma-aminobutyric acid–stimulated chloride flux in mouse brain vesiclesby polychloroalkane and pyrethroid insecticides. Neurotoxicology7:11–20, 1986.
Bond GG, Rossbacher R: A review of potential human carcinogenicity ofthe chlorophenoxy herbicides MCPA, MCPP and 2,4-DP. Br J lnd Med50:340–348, 1993.
Bond GG, Wetterstroem NH, Roush GJ, et al: Cause specific mortalityamong employees engaged in the manufacture, formulation or pack-aging of 2,4-dichlorophenoxyacetic acid and related salts. Br J IndMed 45:98–105, 1988.
Bonsall JL: Measurement of occupational exposure to pesticides, inTurnbull GS (ed): Occupational Hazards of Pesticide Use. London:Taylor & Francis, 1985, pp 13–33.
Boon-Long J, Glinsukon T, Pothisiri P, et al: Toxicological problems inThailand, in Ruchirawat M, Shank RC (eds): Environmental Toxicityand Carcinogenesis. Bangkok: Text and Journal Corp, 1986, pp 283–293.
Boyd MR, Neal RA: Studies on the mechanism of toxicity and of devel-opment of tolerance to the pulmonary toxic �-naphthylthiourea(ANTU). Drug Metab Dispos 4:314–322, 1976.
Branch RA, Jacqz E: Subacute neurotoxicity following long-term exposureto carbaryl. Am J Med 80:741–746, 1986.
Brooks GT: Chlorinated Insecticides. Technology, and Application. Vol 1.Cleveland, OH: CRC Press, 1974, pp 12–13.
Büchel KH (ed): Chemistry of Pesticides. New York: Wiley, 1983.Bull DL: Metabolism of organophosphorus insecticides in animals and
plants. Residue Rev 43:1–22, 1972.Calamari D, Yameogo L, Hougard J-M, et al: Environmental assessment of
larvacide use in the onchocerciasis control programme. Parasitol To-day 14:485–489, 1998.
Cam C, Nigogosyan G: Acquired toxic porphyria cutanea tarda due to hexa-chlorobenzene. JAMA 183:88–91, 1963.
Campbell S: Paraquat poisoning. Clin Toxicol 1:245–249, 1968.Campbell WC: Ivermectin and Abamectin. New York, Springer-Verlag,
1989.Campbell WC, Fisher MH, Stapley EO, et al: Ivermectin: A potent an-
tiparasitic agent. Science 221:823–828, 1983.Cannon SB, Veasey JM Jr, Jackson RS, et al: Epidemic kepone poisoning
in chemical workers. Am J Epidemiol 107:529–537, 1978.Cantor KP: Farming and mortality from non-Hodgkin’s lymphoma: A case-
control study. lnt J Cancer 29:239–247, 1982.Carson R: Silent Spring. Boston: Houghton Mifflin, 1962.Casida JE, Gammon DW, Glockman AH, Lawrence LJ: Mechanisms of se-
lective action of pyrethroid insecticides. Annu Rev Pharmacol Toxicol23:413–438, 1983.
Castro-Gutierrez N, McConnell R, Andersson K, et al: Respiratory symp-toms, spirometry and chronic occupational paraquat exposure. ScandJ Work Environ Health 23:421–427, 1997.
Cavanagh JB: The toxic effects of tri-ortho-cresyl phosphate on the nerv-ous system, an experimental study in hens. J Neurol Neurosurg Psy-chiatry 17:163–172, 1954.
Centers for Disease Control Veterans Health Studies: Serum 2,3,7,8-tetrachlorodibenzo-p-dioxin levels in U.S. Army Vietnam-era veter-ans. JAMA 260:1249–1254, 1988.
Chambers JE, Levi PE: Organophosphates. Chemistry, Fate and Effects.New York: Academic Press, 1992.
Chan H-M: A database for environmental contaminants in traditional foodsin northern and arctic Canada. Food Add Contam 15:127–134, 1998.
Chao SL, Casida JE: Interaction of imidacloprid metabolites and analogs
2996R_ch22_761-810 4/16/01 4:38 PM Page 803
Copy
right
ed M
ater
ial
Copyright © 2001 by The McGraw-Hill Companies Retrieved from: www.knovel.com

804 UNIT 5 TOXIC AGENTS
with the nicotinic acetylcholine receptor of mouse brain in relation totoxicity. Pestic Biochem Physiol 58:77–88, 1997.
Charles JM, Cunny HC, Wilson RD, et al: Ames assays and unscheduledDNA synthesis assays on 2,4-dichloro-phenoxyacetic acid and its de-rivatives. Mutat Res 444:207–216, 1999a.
Charles JM, Cunny HC, Wilson RD, et al: In vivo micronucleus assay on2,4-dichlorophenoxyacetic acid and its derivatives. Mutat Res444:227–234, 1999b.
Cheek AO, Ide CF, Bollinger JE, et al: Alteration of leopard frog (Ranapipiens) metamorphosis by the herbicide acetochlor. Arch EnvironContam Toxicol 37:70–77, 1999.
Chernoff N, Kavlock RJ, Kathrein JR, et al: Prenatal effects of dieldrin andphotodieldrin in mice and rats. Toxicol Appl Pharmacol 31:302–308,1975.
Childs AF, Davies DR, Green AL, Rutland JP: The reactivation by oximesand hydroxamic acids of cholinesterase inhibited by organophospho-rus compounds. Br J Pharmacol Chemother 10:462–465, 1955.
Ciesielski S, Loomis DP, Mims SR, et al: Pesticide exposures, cholin-esterase depression and symptoms among North Carolina migrantfarmworkers. Am J Pub Health 84:446–451, 1994.
Clark DG, Hurst EW: The toxicity of diquat. Br J Ind Med 27:51–55, 1970.Clark DG, McElligott TF, Hurst EW: The toxicity of paraquat. Br J Ind
Med 23:126–132, 1966.Clark JM, Matsumura F: Two different types of inhibitory effects of
pyrethroids on nerve Ca and Ca-Mg-ATPase activity in the squid,Loligo pealei. Pestic Biochem Physiol 18:180–190, 1982.
Clement JG: Toxicity of the combined nerve agents GB/GF in mice: Effi-cacy of atropine and various oximes as antidotes. Arch Toxicol 68:64–66, 1994.
Cole LM, Nicholson RA and Casida JE: Action of phenylpyrazole insec-ticides in the GABA-gated chloride channel. Pestic Biochem Physiol46:47–54, 1993.
Coleman S, Liu S, Linderman R, et al: In vitro metabolism of alachlor byhuman liver microsomes and human cytochrome P450 isoforms.Chemico-Biol Int 122:27–39, 1999.
Committee on Scientific and Regulatory Issues Underlying Pesticide UsePatterns and Agricultural Innovation: Regulating Pesticides in Food.Washington, DC: National Academy Press, 1987.
Cook RR: Health effects of organophosphate sheep dips. Br Med J305:1502–1503, 1992.
Copplestone JF: The development of the WHO Recommended Classifica-tion of Pesticides by Hazard. Bull WHO 66:545–551, 1988.
Corbett JR: The Biochemical Mode of Action of Pesticides. New York: Aca-demic Press, 1974.
Courtney KD, Gaylor DW, Hogan MD, et al: Teratogenic evaluation of2,4,5-T. Science 168:864–866, 1970.
Courtney KD, Moore JA: Teratology studies with 2,4,5-trichlorophenoxy-acetic acid and 2,3,7,8-tetra-chlorodibenzo-dioxin. Toxicol Appl Phar-macol 20:396–403, 1971.
Coye MJ, Lowe IA, Maddy KT: Biological monitoring of agricultural work-ers exposed to pesticides: I. Cholinesterase activity determinators. J Occup Med 28:619–627, 1986.
Craan AG, Haines DA: Twenty-five years of surveillance for contaminantsin human breast milk. Arch Environ Cont Toxicol 35:702–710, 1998.
Cranmer MF: Carbaryl. A toxicological review and risk analysis. Neuro-toxicology 1:247–332, 1986.
Cremlyn R: Pesticides. Preparation and Mode of Action. New York: Wiley,1978.
Crum JA, Bursian SJ, Aulerich RJ, Brazelton WE: The reproductive effectsof dietary heptachlor in mink (Mustela vison). Arch Environ ContamToxicol 24:156–164, 1993.
Dale WE, Quinby GE: Chlorinated insecticides in the body fat of peoplein the United States. Science 142:593–595, 1963.
Dalvie MA, White N, Raine R, et al: Long term respiratory health effectsof the herbicide paraquat among workers in the Western Cape. OccupEnviron Med 56:391–396, 1999.
Damgaard I, Nyitrai G, Kovacs I, et al: Possible involvement of GABAA
and GABAB receptors in the inhibitory action of lindane on transmit-ter release from cerebellar granule neurons. Neurochem Res 24:1189–1193, 1999.
Daniel JW, Gage JC: Absorption and excretion of diquat and paraquat inrats, Br J Ind Med 23:133–136, 1966.
Dauterman WC, Main AR: Relationship between acute toxicity and m vitroinhibition and hydrolysis of a serves of homologs of malathion. Tox-icol Appl Pharmacol 9:408–418, 1966.
Davies DS, Hawksworth GM, Bennett PN: Paraquat poisoning. Proc EurSoc Toxicol 18:21–26, 1977.
Davies HW, Kennedy SM, Teschke K, et al: Cytogenetic analysis of SouthAsian berry pickers in British Columbia using the micronucleus as-say in peripheral lymphocytes. Mutat Res 416:101–113, 1998.
Davies JE, Edmundson WF, Schneider NJ, Cassady JC: Problems ofprevalence of pesticide residues in humans, in Davies JE, EdmondsonWF (eds): Epidemiology of DDT. Mount Kisco, NY: Futura, 1972, pp27–37.
Dearfield KL, McCarroll NE, Protzel A, et al: A survey of EPA/OPP andopen literature on selected pesticide chemicals: II Mutagenicity andcarcinogenicity of selected chloro-acetanilides and related compounds.Mutat Res 443:183–221, 1999.
Delorme PD, Lockhart WL, Mills KH, et al: Long-term effects of toxapheneand depuration in lake trout and white sucker in a natural ecosystem.Environ Toxicol Chem 18:1992–2000, 1999.
Deng Y, Casida JE: House fly GABA-gated chloride channel: Toxicologi-cally relevant binding site for avermectins coupled to site for ethynyl-bicycloorthobenzoate. Pestic Biochem Physiol 43:116–122, 1992.
DeRueck J, Willems J: Acute parathion poisoning: myopathic changes inthe diaphragm. J Neurol 208:309–314, 1975.
Desaiah D: Effects of pentachlorophenol on the ATPases in rat tissue, inRao KR (ed): Pentachlorophenol. New York: Plenum Press, 1977, pp277–283.
Desi I, Gonczi L, Simon G, et al: Neurotoxicologic studies of two carba-mate pesticides in subacute animal experiments. Toxicol Appl Phar-macol 27:465–476, 1974.
DuBois KP: New rodenticidal compounds. J Am Pharm Assoc 37:307–310,1948.
DuBois KP, Doull J, Salerno PR, Coon JM: Studies on the toxicity andmechanisms of action of p-nitrophenyl-diethyl-thionophosphate(Parathion). J Pharmacol Exp Ther 95:75–91, 1949.
Duce IR, Khan TR, Green AC, et al: Calcium channels in insects, in Bea-dle JD (ed): Progress in Neuropharmacology and Neurotoxicology ofPesticides and Drugs. London: Royal Society of Chemistry, 1999, pp55–66.
Durham WF, Wolfe HR: Measurement of the exposure of workers to pes-ticides. Bull WHO 26:75–91, 1962.
Durham WF, Wolfe HR, Elliott JW: Absorption and excretion of parathionby spraymen. Arch Environ Health 24:381–387, 1972.
Ebert E, Leist KH, Mayer D: Summary of safety evaluation of toxicity stud-ies of glufosinate ammonium. Food Chem Toxicol 28:339–349, 1990.
Ecobichon DJ: Biological monitoring: Neurophysiological and behavioralassessments, in Ecobichon DJ (ed): Occupational Hazards of Pesti-cide Exposure. Sampling, Monitoring, Measuring. Philadelphia, Taylor& Francis, 1998b, pp 209–230.
Ecobichon DJ: Carbamic acid ester insecticides, in Ecobichon DJ, Joy RM:Pesticides and Neurological Diseases, 2d ed. Boca Raton, FL: CRCPress, 1994b, pp 251–289.
Ecobichon DJ: Fungicides, in Ecobichon DJ, Joy RM: Pesticides and Neu-rological Diseases, 2d ed. Boca Raton, FL: CRC Press, 1994c, pp313–351.
Ecobichon DJ: Hydrolytic mechanisms of pesticide degradation, in Geiss-buhler H (ed): Advances in Pesticide Science. Biochemistry of Pestsand Mode of Action of Pesticides, Pesticide Degradation, PesticideResidues and Formulation Chemistry. New York: Pergamon, 1979,part 3, pp 516–524.
2996R_ch22_761-810 4/16/01 4:38 PM Page 804
Copy
right
ed M
ater
ial
Copyright © 2001 by The McGraw-Hill Companies Retrieved from: www.knovel.com

CHAPTER 22 TOXIC EFFECTS OF PESTICIDES 805
Ecobichon DJ (ed): Occupational Hazards of Pesticide Exposure. Sampling,Monitoring, Measuring. Philadelphia, Taylor & Francis, 1998a.
Ecobichon DJ: Organophosphorus ester insecticides, in Ecobichon DJ, JoyRM: Pesticides and Neurological Diseases, 2d ed. Boca Raton, FL:CRC Press, 1994a, pp 171–249.
Ecobichon DJ: Toxic effects of pesticides, in Klaassen CD (ed): Casarettand Doull’s Toxicology. The Basic Science of Poisons. 5th ed. NewYork, McGraw-Hill, 1996; pp 643–689.
Ecobichon DJ, Joy RM: Pesticides and Neurological Diseases, 2d ed. BocaRaton, FL: CRC Press, 1994.
Ecobichon DJ, MacKenzie DO: The uterotropic activity of commercial andisomerically pure chlorobiphenyls in the rat. Res Commun ChemPathol Pharmacol 9:85–95, 1974.
Ecobichon DJ, Ozere RL, Reid E, Crocker JFS: Acute fenitrothion poi-soning. Can Med Assoc J 116:377–379, 1977.
Ecobichon DJ, Saschenbrecker PW: Pharmacodynamic study of DDT incockerels. Can J Physiol Pharmacol 46:785–794, 1968.
Ecobichon DJ, Saschenbrecker PW: The redistribution of stored DDT incockerels under the influence of food deprivation. Toxicol Appl Phar-macol 5:420–432, 1969.
Edington N, Howell JM: The neurotoxicity of sodium diethyl-diethio-carbamate in the rabbit. Acta Neuropathol 12:339–346, 1969.
Edmiston S, Maddy KT: Summary of illnesses and injuries reported inCalifornia by physicians in 1986 as potentially related to pesticides.Vet Hum Toxicol 29:391–397, 1987.
Edwards R, Ferry DG, Temple WA: Fungicides and related compounds, inHayes WJ Jr, Laws ER Jr (eds): Handbook of Pesticide Toxicology.Classes of Pesticides. Vol 3. New York: Academic Press, 1991, pp1409–1470.
Egan H, Goulding R, Toburn J, Tatton JO’G: Organochlorine residues inhuman fat and human milk. Br Med J 2:66–69, 1965.
Eldefrawi ME, Schweizer C, Bakry NM, et al: Desensitization of thenicotinic acetylcholine receptor by diisopropyl-fluorophosphate. J Biochem Toxicol 3:21–32, 1988.
Ellenhorn MJ, Barceloux DG: Pesticides, in Medical Toxicology. Diagno-sis and Treatment of Human Poisoning. New York: Elsevier, 1988, pp1081–1108.
Elliott M: Future use of natural and synthetic pyrethroids, in Metcalf RL,McKelvey JJ Jr (eds): The Future for Insecticides: Needs and Pros-pects. New York: Wiley, 1976, pp 163–193.
Elliott M, Janes NF, Kimmel EC, Casida JE: Metabolic fate of pyrethrin1, pyrethrin II and allethrin administered orally to rats. J Agric FoodChem 20:300–313, 1972.
Englehard H, Erdmann WD: Beziehangen zwischen chemischer struk-tur und cholinesterase reaktivierendes wirksamkeit bei einen reiheneuer bisquartarer pyridin-4-aldoxime. Arznem Forsch 14:870–875,1964.
EPA: Carbaryl Decision Document. Washington, DC: U.S. EnvironmentalProtection Agency. Government Printing Office, 1980.
Eriksson M, Hardell L, Berg NO, et al: Soft-tissue sarcomas and exposureto chemical substances: a case-referent study. Br J Ind Med 38:27–33, 1981.
Eto M: Organophosphorus Pesticides: Organic and Biological Chemistry.Cleveland, OH: CRC Press, 1974.
Feldman RC: Carbamates. Occupational and Environmental Neurotoxi-cology. Philadelphia: Lippincott-Raven, 1999, pp 442–465.
Feldman RJ, Maiback HI: Percutaneous penetration of some pesticides andherbicides in man. Toxicol Appl Pharmacol 28:126–132, 1974.
Ferraz HB, Bertolucci PHF, Pereira JS, et al: Chronic exposure to the fun-gicide maneb may produce symptoms and signs of CNS manganeseintoxication. Neurology 38:550–553, 1988.
Fest C, Schmidt K-J: The Chemistry of Organophosphorus Pesticides. NewYork: Springer-Verlag, 1973.
Fiserova-Bergerova V, Radomski JL, Davies JE, Davis JH: Levels ofchlorinated hydrocarbon pesticides in human tissues. Ind Med Surg36:65–70, 1967.
Fisher MH, Mrozik H: The chemistry and pharmacology of avermectins.Annu Rev Pharmacol Toxicol 32:537–553, 1992.
Flannigan SA, Tucker SB, Key MM., et al: Synthetic pyrethroid insecti-cides: A dermatological evaluation. Br J Ind Med 42:363–372, 1985.
Fonseca RG, Resende LAL, Silva MD, Camargo A: Chronic motor neurondisease possibly related to intoxication with organochlorine insecti-cides. Acta Neurol Scand 88:56–58, 1993.
Forget G: Pesticides: Necessary but dangerous poisons. IDRC Rep 18:4–5,1989.
Forget G, Goodman T, deVilliers A (eds): Impact of Pesticide Use on Healthin Developing Countries. Ottawa, Canada: International DevelopmentResearch Centre, 1993.
Forshaw PJ, Lister T, Ray DE: Inhibition of a neuronal voltage-dependentchloride channel by the type II pyrethroid, deltamethrin. Neurophar-macology 32:105–111, 1993.
Forshaw PJ, Ray DE: A voltage-dependent chloride channel in NIE115 neu-roblastoma cells is activated by protein-kinase-C and also by thepyrethroid deltamethrin. J Physiol 467:252P, 1993.
Franciolini F, Petris A: Chloride channels of biological membranes.Biochim Biophys Acta 1031:247–259, 1990.
Frank R, Campbell RA, Sirons GJ: Forestry workers involved in aerial ap-plication of 2,4-dichlorophenoxyacetic acid (2,4-D): Exposure and uri-nary excretion. Arch Environ Contam Toxicol 14:427–435, 1985.
Franklin CA, Fenske RA, Greenhalgh R, et al: Correlation of urinarypesticide metabolite excretion with estimated dermal contact in thecourse of occupational exposure to guthion. J Toxicol Environ Health7:715–731, 1981.
Frawley JP, Fuyat HN, Hagan EC, et al: Marked potentiation in mammaliantoxicity from simultaneous administration of two anticholinesterasecompounds. J Pharmacol Exp Ther 121:96–106, 1957.
Frear DEH: Pesticide Index, 4th ed. State College, PA: College Science,1969.
Fukuto TR: Metabolism of carbamate insecticides. Drug Metab Rev 1:117–147, 1972.
Gaines TB: Acute toxicity of pesticides. Toxicol Appl Pharmacol 14:515–534, 1969.
Gammon DW, Brown MA, Casida JE: Two classes of pyrethroid action inthe cockroach. Pestic Biochem Physiol 15:181–191, 1981.
Gant DB, Chalmers AE, Wolff MA, et al: Fipronil: Action at the GABAreceptor. Rev Toxicol 2:147–156, 1998.
Gee J: Iraqui declarations of chemical weapons: How much did they reallyhave and what is it? Fourth International Symposium on ProtectionAgainst Chemical Warfare Agents, Stockholm, June 8–12, 1992.
Gehring PJ, Watanabe PG, Blau GE: Pharmacokinetic studies in evaluationof the toxicological and environmental hazard of chemicals, inMehlman MA, Shapiro RE, Blumenthal LL (eds): New Concepts inSafety Evaluation. New York: Wiley, 1976, pp 195–270.
Gershenfeld HM: Chemical transmission in invertebrate central nervoussystems and neuromuscular junctions. Physiol Rev 53:1–119, 1973.
Gershon S, Shaw FH: Psychiatric sequelae of chronic exposure toorganophosphorus insecticides. Lancet 1:1371–1374, 1961.
Ginsburg KS, Narahashi T: Differential sensitivity of tetrodotoxin-sensitiveand tetrodotoxin-resistant sodium channels to the insecticide allethrinin rat dorsal root ganglion neurons. Brain Res 627:239–248, 1993.
Glickman Ali, Casida JE: Species and structural variations affectingpyrethroid neurotoxicity. Neurobehav Toxicol Teratol 4:793–799,1982.
Gochfeld M: New light on the health of Vietnam veterans. Environ Res47:109–111, 1988.
Goldman LR, Better M, Jackson RJ: Aldicarb food poisonings in Califor-nia, 1985–1988: Toxicity estimates for humans. Arch Environ Health45:141–148, 1990a.
Goldman LR, Smith DF, Neutra RR, et al: Pesticide food poisoning fromcontaminated watermelons in California, 1985. Arch Environ Health45:229–236, 1990b.
Goldstein JA, Fridsen M, Linder PE, et al: Effects of pentachlorophenol on
2996R_ch22_761-810 4/16/01 4:38 PM Page 805
Copy
right
ed M
ater
ial
Copyright © 2001 by The McGraw-Hill Companies Retrieved from: www.knovel.com

806 UNIT 5 TOXIC AGENTS
hepatic drug-metabolizing enzymes and porphyria related to contam-ination with chlorinated dibenzo p-dioxins and dibenzofurans.Biochem Pharmacol 26:1549–1557, 1977.
Goldstein NP, Jones PH, Brown JR: Peripheral neuropathy after exposureto an ester of dichlorophenoxyacetic acid. JAMA 171:1306–1309,1959.
Gollapudi BB, Charles JM, Linscombe VA, et al: Evaluation of the geno-toxicity of 2,4-dichlorophenoxyacetic acid and its derivatives in mam-malian cell cultures. Mutat Res 444:217–225, 1999.
Gough M: Dioxin, Agent Orange. The Facts. New York: Plenum Press,1986.
Greenwald W, Kovasznay B, Collins DN, Therriault G: Sarcomas of softtissues after Vietnam service. J Natl Cancer Inst 73:1107–1109, 1984.
Grendon J, Frost F, Baum L: Chronic health effects among sheep and hu-mans surviving an aldicarb poisoning incident. Vet Hum Toxicol36:218–223, 1994.
Grover R, Cessna AJ, Muir NI, et al: Factors affecting the exposure ofground-rig applicators to 2,4-D dimethylamine salt. Arch Environ Con-tam Toxicol 15:677–686, 1986.
Grubb TC, Dick LC, Oser M: Studies on the toxicity of polyoxyethylenedodecanol. Toxicol Appl Pharmacol 2:133–143, 1960.
Guzelian PS: Comparative toxicology of chlordecone (kepone) in humansand experimental animals. Annu Rev Pharmacol Toxicol 22:89–113,1982.
Hack R, Ebert E, Ehling G, et al: Glufosinate ammonium—Some aspectsof its mode of action in mammals. Food Chem Toxicol 32:461–470,1994.
Hainzl D, Cole LM, Casida JE: Mechanisms for selective toxicity of fipronilinsecticide and its sulfone metabolite and desulfinyl photoproduct.Chem Res Toxicol 11:1529–1535, 1998.
Haley TJ: Review of the toxicology of paraquat (1,19-dimethyl-4,49-bipyri-dinium chloride). Clin Toxicol 14:1–46, 1979.
Hall R, Payne LA, Putnam JM, et al: Effect of methoxychlor on implan-tation and embryo development in the mouse. Reprod Toxicol 11:703–708, 1997.
Hardell L, Eriksson M, Lenner P, Lundgren E: Malignant lymphoma andexposure to chemicals, especially organic solvents, chlorophenols andphenoxy acids: A case-control study. Br J Cancer 43:169–176, 1981.
Hardell L, Sandstrom A: Case-control study: Soft-tissue sarcomas and ex-posure to phenoxyacetic acids or chlorophenols. Br J Cancer 39:711–717, 1979.
Hartley GS, West TF: Chemicals for Pest Control. Oxford, England: Perg-amon Press, 1969, p 26.
Haslam E: Shikimic Acid: Metabolism and Metabolites. Chichester, UK:Wiley, 1993.
Hay A: The Chemical Scythe. Lessons of 2,4,5,-T and Dioxin. New York:Plenum Press, 1982.
Hayes WJ Jr: Pesticides Studied in Man. Baltimore: Williams & Wilkins,1982.
Hayes WJ Jr: The pharmacology and toxicology of DDT, in Muller P (ed):The Insecticide DDT and Its Importance. Vol 2. Basel: BirkhauserVerlag, 1959, pp 9–247.
Hayes WJ Jr, Dale WE, Pirkle CI: Evidence of the safety of long-term,high, oral doses of DDT for man. Arch Environ Health 22:19–35,1971.
He F, Sun J, Han K, et al: Effects of pyrethroid insecticides on subjects en-gaged in packaging pyrethroids. Br J Ind Med 45:548–551, 1988.
He F, Wang S, Liu L, et al: Clinical manifestations and diagnosis of acutepyrethroid poisoning. Arch Toxicol 63:54–58, 1989.
Heath DF: Organophosphorus Poisons. Anticholinesterases and RelatedCompounds. London: Pergamon Press, 1961.
Hemavathi E, Rahiman MA: Toxicological effects of ziram, thiram andDithane M-45 assessed by sperm shape abnormalities in mice. J Toxicol Environ Health 38:393–398, 1993.
Himuro K, Murayame S, Nishiyama K, et al: Distal sensory axonopathyafter sarin intoxication. Neurology 51:1195–1197, 1998.
Hoar SK, Blair A, Holmes FF, et al: Agricultural herbicide use and risk oflymphoma and soft-tissue sarcoma. JAMA 256:1141–1147, 1986.
Holmstedt B: Pharmacology of organophosphorus cholinesterase inhibitors.Pharmacol Rev 11:567–688, 1959.
Huang Y, Williamson MS, Devonshire AL, et al: Molecular characteriza-tion and imidacloprid selectivity of nicotinic acetylcholine receptorsubunits from the peach-potato aphid Myzus persicae. J Neurochem73:380–389, 1999.
Huff RA, Corcoran JJ, Anderson JK, et al: Chlorpyrifos oxon binds directlyto muscarinic receptors and inhibits cAMP accumulation in rat stria-tum. J Pharmacol Exp Ther 269:329–335, 1994.
Hutson DH, Akintonwa DAA, Hathway DE: The metabolism of 2-chloro-l-(29,49-dichlorophenyl) vinyl diethylphosphate (chlorfenvinphos) inthe dog and rat. Biochem J 102:133–142, 1967.
IARC: Monograph on the Evaluation of Carcinogenic Risk of Chemicalsto Man. Some Organochlorine Pesticides. Vol 5. Lyons, France:International Agency for Research on Cancer, 1974.
IARC: Monographs on the Evaluation of Carcinogenic Risk of Chemicalsto Man. Some Carbamates, Thiocarbamates and Carbazines. Vol 12.Lyons, France: International Agency for Research on Cancer, 1976.
IARC: Monographs on the Evaluation of Carcinogenic Risk of Chemicalsto Man. Some Fumigants, the Herbicides 2,4-D and 2,4,5-T, Chlori-nated Dibenzodioxins and Miscellaneous Industrial Chemicals. Vol15. Lyons, France: International Agency for Research on Cancer, 1977.
Innes JRM, Ulland BM, Valerio MG, et al: Bioassay of pesticides and in-dustrial chemicals for tumorigenicity in mice: a preliminary note. J Natl Cancer Inst 42:1101–1114, 1969.
Israeli R, Sculsky M, Tiberin P: Acute intoxication due to exposure tomaneb and zineb. A case with behavioral and central nervous systemchanges. Scand J Work Environ Health 9:47–51, 1983.
Jager G: Herbicides, in Buchel KH (ed): Chemistry of Pesticides. New York:Wiley, 1983, pp 322–392.
Jamal GA: Neurological syndromes of organophosphorus compounds. Ad-verse Drug React Toxicol Rev 16:133–170, 1997.
Jett DA, Fernando JC, Eldefrawi ME, et al: Differential regulation of mus-carinic receptor subtypes in rat brain regions by repeated injections ofparathion. Toxicol Lett 73:33–41, 1994.
Jett DA, Hill EF, Fernando JC, et al: Down-regulation of muscarinic re-ceptors and the m3 subtype in white-footed mice by dietary exposureto parathion. J Toxicol Environ Health 39:395–415, 1993.
Jeyaratnam J: Occupational health issues in developing countries. EnvironRes 60:207–212, 1993.
Jeyaratnam J, De Alwis Senevirathe RS, Copplestone JF: Survey of pesti-cide poisoning in Sri Lanka. Bull WHO 60:615–619, 1982.
Johnson MK: The target for initiation of delayed neurotoxicity byorganophosphorus esters: Biochemical studies and toxicological ap-plications, in Hodgson E, Bend JR, Philpot RM (eds): Reviews of Bio-chemical Toxicology. Vol 4. New York: Elsevier, 1982, pp 141–212.
Johnson MK, Willems JL, DeBisschop HC, et al: Can soman cause delayedneuropathy? Fundam Appl Toxicol 5:SI80–SI81, 1985.
Johnson RL, Gehring PJ, Kociba RJ, Schwetz BA: Chlorinated dibenzodi-oxins and pentachlorophenol. Environ Health Perspect 5:171–175,1973.
Jones EC, Growe GH, Naiman SC: Prolonged anticoagulation in rat poi-soning. JAMA 252:3005–3007, 1984.
Jorens PG, Schepens PJC: Human pentachlorophenol poisoning. Hum ExpToxicol 12:479–495, 1993.
Joy RM: Chlorinated hydrocarbon insecticides, in Ecobichon DJ, Joy RM:Pesticides and Neurological Diseases, 2d ed. Boca Raton, FL: CRCPress, 1994a, pp 81–170.
Joy RM: Pyrethrins and pyrethroid insecticides, in Ecobichon DJ, Joy RM:Pesticides and Neurological Diseases, 2d ed. Boca Raton, FL: CRC,1994b, pp 291–312.
Joy RM, Albertson TE: Interactions of GABA-A antagonists withdeltamethrin, diazepam, phenobarbital and SKF 100330A in the ratdentate gyrus. Toxicol Appl Pharmacol 109:251–262, 1991.
2996R_ch22_761-810 4/16/01 4:38 PM Page 806
Copy
right
ed M
ater
ial
Copyright © 2001 by The McGraw-Hill Companies Retrieved from: www.knovel.com

CHAPTER 22 TOXIC EFFECTS OF PESTICIDES 807
Kackar R, Srivastava MK, Raizada RB: Studies on rat thyroid after oraladministration of mancozeb: Morphological and biochemical evalua-tions. J Appl Toxicol 17:369–375, 1997.
Kamrin MA (ed): Pesticide Profiles. Toxicity, Environmental Impact andFate. Boca Raton, FL: Lewis Publishers, 1997.
Katona B, Wason S: Anticoagulant rodenticides. Clin Toxicol Rev 8:1–2,1986.
Katz EJ, Cortes VI, Eldefrawi ME: Chlorpyrifos, parathion and their ox-ons bind to and desensitize a nicotinic acetylcholine receptor: Rele-vance to their toxicities. Toxicol Appl Pharmacol 146:227–236, 1997.
Keane WT, Zavon MR: The total body burden of dieldrin. Bull EnvironContam Toxicol 4:1–16, 1969.
Ketchum NS, Michalek JE, Burton JE: Serum dioxin and cancer in veter-ans of Operation Ranch Hand. Am J Epidemiol 149:630–639, 1999.
Kewitz H, Wilson IB: A specific antidote against lethal alkylphosphate in-toxication. Arch Biochem Biophys 60:261–263, 1956.
Kilgore W: Human exposure to pesticides, in Newberne PM, Shank RC,Ruchirawat M (eds): International Toxicology Seminar: Environmen-tal Toxicology. Bangkok: Chulabhorn Research Institute and MahidolUniversity, 1988.
Kilgore WW, Akesson NB: Minimizing occupational exposure to pesti-cides; populations at exposure risk. Residue Rev 75:21–31, 1980.
Kirby C: The Hormone Weedkillers. Croydon, UK: BCPC Publications,1980.
Kitselman CH: Long-term studies on dogs fed aldrin and dieldrin in sub-lethal dosages with reference to the histopathological findings and re-production. J Am Vet Med Assoc 123:28–36, 1953.
Kociba RI, Keyes DG, Beyer JE: Results of a two year chronic toxicity andoncogenicity study of 2,3,7,8-tetrechlorodibenzo-p-dioxin in rats. Tox-icol Appl Pharmacol 46:279–303, 1978.
Koestner A: The brain-tumor issue in long-term toxicity studies in rats.Food Chem Toxicol 24:139–143, 1986.
Kolmodin-Hedman B, Hoglund S, Ak-erblom M: Studies on phenoxy acidherbicides. 1. Field Study. Occupational exposure to phenoxy acid her-bicides (MCPA, dichlorprop, mecoprop and 2,4-D) in agriculture. ArchToxicol 54:257–275, 1983.
Koos BJ, Longo LD: Mercury toxicity in the pregnant woman, fetus andnewborn infant. Am J Obstet Gynecol 126:390–409, 1976.
Koplovitz I, Gresham VC, Dochterman LW, et al: Evaluation of the toxic-ity, pathology and treatment of cyclohexylmethylphosphonofluoridate(CMPF) poisoning in rhesus monkeys. Arch Toxicol 66:622–628,1992.
Koyama K: Acute oral poisoning caused by a herbicide containing glufos-inate. Jpn J Toxicol 8:391–398, 1995.
Koyama K, Andou Y, Saruki K, et al: Delayed and severe toxicities of aherbicide containing glufosinate and a surfactant. Vet Hum Toxicol36:17–18, 1994.
Kramer W: Fungicides and bacteriocides, in Buchel KH (ed): Chemistry ofPesticides. New York: Wiley, 1983, pp 227–321.
Kuhr RJ, Dorough HW: Carbamate Insecticides: Chemistry, Biochemistryand Toxicology. Boca Raton, FL: CRC Press, 1976.
Kulkami AP, Hodgson E: The metabolism of insecticides: The role ofmonooxygenase enzymes. Annu Rev Pharmacol 24:19–42, 1984.
Kupfer D, Bulger WH: Studies on the mechanism of estrogenic actions ofo,p-DDT: Interactions with the estrogen receptor. Pestic BiochemPhysiol 6:461–470, 1976.
Kusic R, Jovanovic D, Randjelovic S, et al: HI-6 in man: Efficacy of theoxime in poisoning by organophosphorus insecticides. Human ExpToxicol 10:113–118, 1991.
Laben RC, Archer TE, Crosby DG, Peoples SA: Lactational output of DDTfed postpartum to dairy cattle. J Dairy Sci 48:701–708, 1965.
Lambrecht RW, Erturk E, Grunden E, et al: Hepatotoxicity and tumori-genicity of hexachlorobenzene (HCB) in Syrian golden hamsters af-ter subchronic administration. Fed Proc 41:329, 1982.
Lange PF, Terveer J: Warfarin poisoning. US Armed Forces J 5:872–877,1954.
Lankas GR, Cartwright ME, Umbenhauer D: P-glycoprotein deficiency ina subpopulation of CF-1 mice enhances avermectin-induced neuro-toxicity. Toxicol Appl Pharmacol 143:357–365, 1997.
Larsen SB, Spano M, Giwercman A, et al: Semen quality and sex hormonesamong organic and traditional Danish farmers. Occup Environ Med56:139–144, 1999.
Lawrence LJ, Casida JE: Pyrethroid toxicology: Mouse intracerebral structure-toxicity relationships. Pestic Biochem Physiol 18:9–14,1982.
Leahey JP: The Pyrethroid Insecticides. London: Taylor & Francis, 1985.Leake LD, Buckley DS, Ford MG, Salt DW: Comparative effects of
pyrethroids on neurones of target and non-target organisms. Neuro-toxicology 6:99–116, 1985.
Leech CA, Jewess P, Marshall J, et al: Nitromethylene actions on in situand expressed insect nicotinic acetylcholine receptors. FEBS Lett290:90–94, 1991.
LeQuesne PM, Maxwell IC, Butterworth ST: Transient facial sensory symp-toms following exposure to synthetic pyrethroids: A clinical and elec-trophysiological assessment. Neurotoxicology 2:1–11, 1980.
Lessenger JE, Riley N: Neurotoxicities and behavioral changes in a12-year-old male exposed to dicofol, an organochlorine pesticide. J Toxicol Environ Health 33:255–261, 1991.
Leveridge YR: Pesticide poisoning in Costa Rica during 1996. Vet HumToxicol 40:42–44, 1998.
Li AP, Long TJ: An evaluation of the genotoxic potential of glyphosate.Fundam Appl Toxicol 10:537–546, 1988.
Li Q, Minami M, Clement JG, et al: Elevated frequency of sister chromatidexchanges in lymphocytes of victims of the Tokyo sarin disaster andin experiments exposing lymphocytes to by-products of sarin synthe-sis. Toxicol Lett 98:95–103, 1998.
Li Y-F: Global technical hexachlorocyclohexane usage and its contamina-tion consequences in the environment from 1948 to 1997. Sci TotalEnviron 232:121–158, 1999.
Liddle JA, Kimbrough RD, Needham LL, et al: A fatal episode of acci-dental methomyl poisoning. Clin Toxicol 15:159–167, 1979.
Lifshitz M, Shahak E, Bolotin A, et al: Carbamate poisoning in early child-hood and in adults. Clin Toxicol 53:25–27, 1997.
Lin J-L, Liu L, Leu M-L: Recovery of respiratory function in survivorswith paraquat intoxication. Arch Environ Health 50:432–439, 1995.
Lipton RA, Klass EM: Human ingestion of a “superwarfarin” rodenticideresulting in prolonged anticoagulant effect. JAMA 252:3004–3005,1984.
Liu M-Y, Casida JE: High affinity binding of [3H] imidacloprid in the in-sect acetylcholine receptor. Pestic Biochem Physiol 46:40–46, 1993.
Liu M-Y, Lanford J, Casida JE: Relevance of [3H] imidacloprid bindingsite in house fly head acetylcholine receptor to insecticidal activity of2-nitromethylene and 2-nitroimino-imidazolines. Pestic BiochemPhysiol 46:200–206, 1993.
Lui M-Y, Latli B, Casida JE: Imidacloprid binding site in Musca nicotinicacetylcholine receptor: Interactions with physostigmine and a varietyof nicotinic agonists with cloropyridyl and chlorothiazolyl sub-stituents. Pestic Biochem Physiol 52:170–181, 1995.
Longcore JR, Samson FB, Whittendale TW Jr: DDE thins eggshells andlowers reproductive success of captive black ducks. Bull Environ Con-tamin Toxicol 6:485–490, 1971.
Lopez-Carillo L, Lopez-Cervantes M: Effect of exposure to organophos-phate pesticides on serum cholinesterase levels. Arch Environ Health48:359–363, 1993.
Lotti M: The pathogenesis of organophosphate polyneuropathy. Crit RevToxicol 21:465–487, 1992.
Lu M-H, Kennedy GL Jr: Teratogenic evaluation of mancozeb in the ratfollowing inhalation exposure. Toxicol Appl Pharmacol 84:355–368,1986.
Lukens RJ: Chemistry of Fungicidal Action. New York: Springer-Verlag,1971.
Lynge E: A follow-up study of cancer incidence among workers in manu-
2996R_ch22_761-810 4/16/01 4:38 PM Page 807
Copy
right
ed M
ater
ial
Copyright © 2001 by The McGraw-Hill Companies Retrieved from: www.knovel.com

808 UNIT 5 TOXIC AGENTS
facture of phenoxy herbicides in Denmark. Br J Cancer 52:259–270,1985.
Maibach H: Irritation, sensitization, photoirritation and photosensitizationassays with a glyphosate herbicide. Contact Dermatitis 15:152–156,1986.
Marban E, Yamagishi T, Tomaselli GF: Structure and function of voltage-gated sodium channels. J Physiol 508:647–657, 1998.
Marquis JK: Contemporary Issues in Pesticide Toxicology and Pharma-cology. Basel: Karger, 1982, pp 87–95.
Marrs TC: Organophosphate poisoning. Pharmacol Ther 58:51–66, 1993. Martinez AJ, Taylor JR, Houff SA, Isaacs ER: Kepone poisoning: Clini-
coneuropathological study, in Roizin L, Shiraki H, Greevic N (eds):Neurotoxicology. New York: Raven Press, 1977, pp 443–156.
Masuda N, Takatsu M, Morinari H, et al: Sarin poisoning in Tokyo sub-way. Lancet 345:1446, 1995.
Matsumura F: Toxicology of Insecticides. New York: Plenum Press, 1985,pp 122–128.
Matthews HB, Matsumura F: Metabolic fate of dieldrin in the rat. J AgricFood Chem 17:845–852, 1969.
Mattsson JL, Johnson KA, Albee RR: Lack of neuropathologic conse-quences of repeated dermal exposure to 2,4-dichloro-phenoxy acid inrats. Fundam Appl Toxicol 6:175–181, 1986.
Mbiapo F, Youovop G: Regulation of pesticides in Cameroon. J Toxicol En-viron Health 39:1–10, 1993.
McBlain WA, Lewin V, Wolfe FH: Estrogenic effects of the enantiomersof o,p� -DDT in Japanese quail. Can J Zool 55:562–568, 1977.
McConnell R, Delgado-Tellez E, Cuadra R, et al: Organophosphate neu-ropathy due to methamidophos: Biochemical and neurophysiologicalmarkers. Arch Toxicol 73:296–300, 1999.
McCord CP, Kilker CH, Minster DK: Pyrethrum dermatitis: A record ofthe occurrence of occupational dermatoses among workers in thepyrethrum industry. JAMA 77:448–449, 1921.
McEwen FL, Stephenson GR: The Use and Significance of Pesticides inthe Environment. New York: Wiley, 1979, pp 91–154.
McFarland LZ, Lacy PB: Physiologic and endocrinologic effects of the in-secticide kepone in the Japanese quail. Toxicol Appl Pharmacol15:441–450, 1969.
Melnikov NN: Chemistry of pesticides. Residue Rev 36:1–480, 1971. Menkes DB, Temple WA, Edwards IR: Intentional self-poisoning with
glyphosate-containing herbicides. Hum Exp Toxicol 10:103–107,1991.
Menzie CM: Metabolism of Pesticides. Special Scientific Report. WildlifeNo. 127. Washington, DC: Bureau of Sport Fisheries and Wildlife,1969.
Mes J, Davies DJ, Turton D: Polychlorinated biphenyl and other chlori-nated hydrocarbon residues in adipose tissue of Canadians. Bull Env-iron Contam Toxicol 28:97–104, 1982.
Metcalfe RL: A century of DDT. J Agric Food Chem 21:511–519, 1973.Metcalfe RL: Development of selective and biodegradable pesticides, in
Pest Control Strategies for the Future. Washington, DC: AgricultureBoard, Division of Biology and Agriculture, National Research Coun-cil, National Academy of Science, 1972, pp 137–156.
Monsanto Company: Toxicology of Glyphosate and Roundup Herbicide. StLouis, 1985.
Montgomery JH: Agrochemicals Desk Reference, 2d ed. Boca Raton, FL:CRC Press, 1997.
Morgan DP: Recognition and Management of Pesticide Poisonings, 3d ed.Publication EPA-540/9-80-005. Washington, DC: U.S. EnvironmentalProtection Agency, 1982.
Morgan DP, Roan CC: Chlorinated hydrocarbon pesticide residue in hu-man tissues. Arch Environ Health 20:452–457, 1970.
Morgan JP, Penovich P: Jamaica ginger paralysis. Forty-seven year follow-up. Arch Neurol 35:530–532, 1978.
Morita H, Yanagisawa N, Nakajima T, et al: Sarin poisoning in Matsumoto,Japan. Lancet 346:290–293, 1995.
Morrison HI, Wilkins K, Semenciw R, et al: Herbicides and cancer. J NatlCancer Inst 84:1866–1874, 1992.
Moses M: Glyphosate herbicide toxicity. JAMA 261:2549, 1989.Mullison WR: An Interim Report Summarizing 2,4-D Toxicological Re-
search Sponsored by the Industry Task Force on 2,4-D Research Dataand a Brief Review of 2,4-D Environmental Effects. Technical and Tox-icology Committees of the Industry Task Force on 2,4-D ResearchData, 1986.
Munro IC, Carlo GL, Orr JC, et al: A comprehensive, integrated reviewand evaluation of the scientific evidence relating to the safety of theherbicide 2,4-D. J Am Coll Toxicol 11:559–664, 1992.
Murphy SD: Mechanisms of pesticide interactions in vertebrates. ResidueRev 25:201–221, 1969.
Murphy SD: The toxicity of pesticides and their metabolites, in Degrada-tion of Synthetic Organic Molecules in the Biosphere. Proceedings ofa Conference. Washington, DC: National Academy of Sciences, 1972,pp 313–335.
Murray V, Wiseman HM, Dawling S, et al: Health effects of organophos-phate sheep dips. Br Med J 305:1090, 1992.
Nagata K, Iwanaga Y, Shono T, et al: Modulation of the neuronal nicotinicacetylcholine receptor channel by imidacloprid and cartap. PesticBiochem Physiol 59:119–128, 1997.
Nagata K, Narahashi T: Dual action of the cyclodiene insecticide dieldrinon the aminobutyric acid receptor chloride ion channel complex of ratdorsal root ganglion neurons. J Pharmacol Exp Ther 269:164–171,1994.
Narahashi T: Mechanisms of action of pyrethroids on sodium and calciumchannel gating, in Ford MG, Lunt GG, Reay RC, Usherwood PN (eds):Neuropharmacology of Pesticide Action. Chichester, UK: EllisHorwood, 1986, pp 36–40.
Narahashi T, Ginsburg KS, Nagata K, et al: Ion channels as targets for in-secticides. Neurotoxicology 19:581–590, 1998.
Nishimura K, Kanda Y, Okazawa A, et al: Relationship between insectici-dal and neurophysiological activities of imidacloprid and related com-pounds. Pestic Biochem Physiol 50:51–59, 1994.
Nozaki H, Aikawa N: Sarin poisoning in Tokyo subway. Lancet 345:1446–1447, 1995.
Nozaki H, Aikawa N, Shinozawa Y, et al: Sarin poisoning in Tokyo sub-way. Lancet 345:980–981, 1995.
O’Malley M: Clinical evaluation of pesticide exposure and poisonings.Lancet 349:1161–1166, 1997.
Panemangalore M, Dowla HA, Byers ME: Occupational exposure to agri-cultural chemicals: Effect on the activities of some enzymes in theblood of farm workers. Int Arch Occup Environ Health 72:84–88,1999.
Payne GT, Soderlund DM: Activation of �-aminobutyric acid insensitivechloride channels in mouse brain synaptic vesicles by avermectin B1a.J Biochem Toxicol 6:283–292, 1991.
Ramasamy S, Tajol Akos NM: A survey of pesticide use and associated in-cidences of poisoning in Peninsular Malaysia. J Plant Protect Trop5:1–9, 1988.
Rank J, Jensen A-G, Skov B, et al: Genotoxicity testing of the herbicideRoundup and its active ingredient glyphosate isopropylamine usingthe mouse bone marrow micronucleus test, Salmonella mutagenicitytest and Allium anaphase-telophase test. Mutat Res 300:29–36,1993.
Ray DE: Pyrethroid insecticides: Mechanisms of toxicity, systemic poi-soning syndromes, paresthesia and therapy, in Krieger R (ed.): Hayes’and Laws’ Handbook of Pesticide Toxicology, 3d ed. San Diego, CA:Academic Press, 2000.
Ray DE, Sutharsan S, Forshaw PJ: Actions of pyrethroid insecticides onvoltage-gated chloride channels in neuroblastoma cells. Neurotoxicol-ogy 18:755–760, 1997.
Sawada Y, Nagai Y: Roundup poisoning—its clinical observation: Possibleinvolvement of surfactant. J Clin Exp Med 143:25–27, 1987.
Schaeffer JM, Haines HW: Avermectin binding in Caenorhabditis elegans.A two-state model for the avermectin-binding site. Biochem Pharma-col 38:2329–2338, 1989.
Shroeder ME, Flattum RF: The mode of action and neurotoxic properties
2996R_ch22_761-810 4/16/01 4:38 PM Page 808
Copy
right
ed M
ater
ial
Copyright © 2001 by The McGraw-Hill Companies Retrieved from: www.knovel.com

CHAPTER 22 TOXIC EFFECTS OF PESTICIDES 809
of the nitromethylene heterocycle insecticides. Pestic Biochem Phys-iol 22:148–160, 1984.
Sims P: Letter to the Editor. Br Med J 305:1503, 1992.Smith K: Pesticides exported from the U.S., 1992–1994. Los Angeles: Foun-
dation for Advancement in Sciences and Education, 1995.Soderlund DM: Pyrethroid-receptor interactions: Stereo-specific binding
and effects on sodium channels in mouse brain preparation. Neuro-toxicology 6:35–46, 1985.
Soloway SB, Henry AC, Kollmeyer WD, et al: Nitromethylene insecticides,in Geissbuhler H, Brooks GT, Kearney PC (eds): Advances in Pesti-cide Science, Part 2. New York: Pergamon Press, 1978, pp 206–217.
Song J-H, Narahashi T: Differential effects of the pyrethroid tetramethrinon tetrodotoxin-sensitive and tetrodotoxin-resistant single sodiumchannels. Brain Res 712:258–264, 1996.
Spiegelberg U: Psychopathologisch-neurologische spat und dauerschadennach gewerblicher Intoxikation durch Phosporsaureester (alkylphos-phate). Proc l4th Int Congr Occup Health Exerpta Med Found IntCongr Ser 62, 1963, pp 1778–1780.
Steenland K, Cedillo L, Tucker J, et al: Thyroid hormones and cytogeneticoutcomes in backpack sprayers using ethylenebis(dithiocarbamate)(EBDC) fungicides in Mexico. Environ Health Perspect 105:1126–1130, 1997.
Stephens R, Spurgeon A, Berry H: Organophosphates. The relationship be-tween chronic and acute exposure effects. Neurotoxicol Teratol18:449–453, 1996.
Stephens R, Spurgeon IA, Calvert J, et al: Neuro-psychological effects oflong-term exposure to organophosphates in sheep dip. Lancet345:1135–1139, 1995.
Stevens MF, Ebell GF, Psaila-Savona P: Organochlorine pesticides in West-ern Australia nursing mothers. Med J Aust 158:238–241, 1993.
Stickel LF: Organochlorine Pesticides in the Environment. Special Scien-tific Report-Wildlife No. 119. Washington, DC: United States De-partment of the Interior, Fish and, Wildlife Service. 1968.
Suskind RR, Hertzberg VS: Human health effects of 2,4,5-T and its toxiccontaminants. JAMA 251:2372–2380, 1984.
Suzuki T, Morita H, Ono K, et al: Sarin poisoning in Tokyo subway. Lancet345:980, 1995.
Swan AAB: Exposure of spray operators to paraquat. Br J Ind Med 26:322–329, 1969.
Swartz WJ, Eroschenko VP: Neonatal exposure to technical methoxychloralters pregnancy outcome in female mice. Reprod Toxicol 12:565–573,1998.
Tai T, Yamashita M, Wakimori H: Hemodynamic effects of Roundup,glyphosate and surfactant in dogs. Jpn J Toxicol 3:63–68, 1990.
Takamiya K: Monitoring of urinary alkyl phosphates in pest control oper-ators exposed to various organophosphorus insecticides. Bull EnvironContam Toxicol 52:190–195, 1994.
Takayama S, Sieber SM, Dalgard DW, et al: Effects of long-term oral ad-ministration of DDT on nonhuman primates. J Cancer Res Clin On-col 125:219–225, 1999.
Talbot AR, Shiaw M-H, Huang J-S, et al: Acute poisoning with aglyphosate-surfactant herbicide (Roundup): A review of 93 cases. HumExp Toxicol 10:1–8, 1991.
Tamburro CH: Chronic liver injury in phenoxy herbicide-exposed Vietnamveterans. Environ Res 59:175–188, 1992.
Tanaka J, Yamashita M, Yamashita M, et al: Two cases of glufosinate poi-soning with late onset convulsions. Vet Hum Toxicol 40:219–222,1998.
Taylor HR, Greene BM: The status of ivermectin in the treatment of on-chocerciasis. Am J Trop Med 41:460–466, 1989.
Thiess AM, Frentzel-Beyme R, Link R: Mortality study of persons exposedto dioxin in a trichlorophenol process accident that occurred in theBASF AG on November 17, 1953. Am J lnd Med 3:179–189, 1982.
Tinoco R, Halperin D, Tinoco R, Parsonhet J: Paraquat poisoning in south-ern Mexico: A report of 25 cases. Arch Environ Health 48:78–80,1993.
Toia RF, Casida JE: Phosphorylation, “aging” and possible alkylation re-actions of saligenin cyclic phosphorus esters with �-chymotrypsin.Biochem Pharmacol 28:211–216, 1979.
Tominack RL, Yang G-Y, Tsai W-J, et al: Taiwan National Poison Centersurvey of glyphosate-surfactant herbicide ingestions. Clin Toxicol29:91–109, 1991.
Tomlin CD (ed), The Pesticide Manual, 11th ed. Farnham, UK, British CropProtection Council, 1997.
Treon JF, Cleveland FP: Toxicity of certain chlorinated hydrocarbon in-secticides for laboratory animals with special reference to aldrin anddieldrin. J Agric Food Chem 3:402–408, 1955.
Tucker RK, Crabtree DG: Handbook of Toxicity of Pesticides to Wildlife.Resource Publication No. 84. Washington, DC: US Department of In-terior, Fish and Wildlife Service, U.S. Government Printing Office,1970.
Tucker SB, Flannigan SA: Cutaneous effects from occupational exposureto fenvalerate. Arch Toxicol 54:195–202, 1983.
Turnbull GJ, Sanderson DM, Crome SJ: Exposure to pesticides during ap-plication, in Turnbull GJ (ed): Occupational Hazards of Pesticide Use.London: Taylor & Francis, 1985, pp 35–49.
Turner WA, Engel JL, Casida JE: Toxaphene components and relatedcompounds: Preparation and toxicity of some hepta-, octa- andnonachlorobomanes, hexa- and heptachlorobomenes and a hexa-chlorobomadiene. J Agric Food Chem 25:1394–1401, 1977.
Vandekar M, Heyadat S, Plestina R, Ahmady G: A study of the safety ofo-isopropoxyphenylmethylcarbamate in an operational field-trial inIran. Bull WHO 38:609–623, 1968.
Vandekar M, Plestina R, Wilhelm K: Toxicity of carbamates for mammals.Bull WHO 44:241–248, 1971.
Van den Bercken J, Vijverberg HPM: Interaction of pyrethroids and DDT-like compounds with the sodium channels in the nerve membrane, inMiyamoto J, Kearney PC (eds): Pesticide Chemistry. Human Welfareand the Environment. Mode of Action, Metabolism and Toxicology.Vol 3. Oxford, England: Pergamon Press, 1983, pp 115–121.
Van Miller JP, Lalich JJ, Allen JR: Increased incidence of neoplasm in ratsexposed to low levels of 2,3,7,8-tetrachlorodibenzo-p-dioxin. Chemo-sphere 6:537–544, 1977.
Venegas W, Zapata I, Carbonell E, et al: Micronuclei analysis in lympho-cytes of pesticide sprayers from Concepcion, Chile. TeratogenesisCarcinog Mutagen 18:123–129, 1998.
Vercruysse F, Driegne S, Steurbaut W, et al: Exposure assessment of pro-fessional pesticide users during treatment of potato fields. Pestic Sci55:467–473, 1999.
Verschoyle RD, Aldridge WN: Structure-activity relationships of somepyrethroids in rats. Arch Toxicol 45:325–329, 1980.
Wafford KA, Sattelle DB, Gant DB, et al: Non competitive inhibition ofGABA receptors in insect and vertebrate CNS by endrin and lindane.Pestic Biochem Physiol 33:213–219, 1989.
Ward TR, Mundy WR: Organophosphorus compounds preferentially affectsecond messenger systems coupled to M2/M4 receptors in rat frontalcortex. Brain Res Bull 39:49–55, 1996.
Warmke JW, Reenan RA, Wang P, et al: Functional expression ofDrosophila para sodium channels. Modulation by the membrane pro-tein TipE and toxin pharmacology. J Gen Physiol 110:119–138, 1997.
Watanabe T: Apoptosis induced by glufosinate ammonium in the neuro-epithelium of developing mouse embryos in culture. Neurosci Lett222:17–20, 1997.
Watanabe T, Iwase T: Developmental effects of glufosinate ammonium onmouse embryos in culture. Teratogenesis Carcinog Mutagen 16:287–299, 1996.
Watanabe T, Sano T: Neurological effects of glufosinate poisoning with abrief review. Hum Exp Toxicol 17:35–39, 1998.
Waters EM, Huff JE, Gerstner HB: Mirex. An overview. Environ Res14:212–222, 1977.
Webb J: Iraq caught out over nerve gas attack. New Scientist 138: May 1,p 4, 1993.
Wecker L, Kiauta T, Dettbarn W-D: Relationship between acetyl-
2996R_ch22_761-810 4/16/01 4:38 PM Page 809
Copy
right
ed M
ater
ial
Copyright © 2001 by The McGraw-Hill Companies Retrieved from: www.knovel.com

810 UNIT 5 TOXIC AGENTS
cholinesterase inhibition and the development of a myopathy. J Phar-macol Exp Ther 206:97–104, 1978.
Wecker L, Mrak RE, Dettbarn WD: Evidence of necrosis in human inter-costal muscle following inhalation of an organophosphate insecticide.Fundam Appl Toxicol 6:172–174, 1986.
Weed Science Society of America (WSSA): Herbicide Handbook, 7th ed.Champaign, IL: WSSA, 1994.
Weiner ML, Jortner BS: Organophosphate-induced delayed neurotoxicityof triarylphosphates. Neurotoxicology 20:653–674, 1999.
Weir S, Minton N, Murray V: Organophosphate poisoning: The UK Na-tional Poisons Unit experience during 1984–1987, in Ballantyne B,Barrs TC (eds): Clinical and Experimental Toxicology of Organophos-phates and Carbamates. Oxford, England: Butterworth-Heinemann,1992, pp 463–470.
Wesseling C, Castillo L, Elinder CC: Pesticide poisonings in Costa Rica.Scand J Work Environ Health 19:227–235, 1993.
Wesseling C, Hogstedt C, Picado, et al: Unintentional fatal paraquat poi-sonings among agricultural workers in Costa Rica: Report of 15 cases.Am J Ind Med 32:433–441, 1997.
Whorton MD, Obrinsky DL: Persistence of symptoms after mild to mod-erate acute organophosphate poisoning among 19 farm field workers.J Toxicol Environ Health 11:347–354, 1983.
Wigle DT, Semenciw RM, Wilkins K, et al: Mortality study of Canadianmale farm operators: Non-Hodgkin’s lymphoma mortality and agri-cultural practices in Saskatchewan. J Natl Cancer Inst 82:575–582,1990.
Wilson AG, Thake DC, Heydens WE, et al: Mode of action of thyroid tumorformation in the male Long-Evans rat administered high doses ofalachlor. Fundam Appl Toxicol 33:16–23, 1996.
Wilson R, Lovejoy FH, Jaeger RJ, Landrigan PL: Acute phosphine poi-soning aboard a grain freighter. JAMA 244:148–150, 1980.
Wojeck GA, Nigg FfN, Stamper JH, Bradway DE: Worker exposure toethion in Florida citrus. Arch Environ Contam in Toxicol 10:725–735,1981.
Wolfe HR, Armstrong JF, Staiff DC, Comer SW: Exposure of spraymen topesticides. Arch Environ Health 25:29–31, 1972.
Wolfe HR, Durham WF, Armstrong JF: Exposure of workers to pesticides.Arch Environ Health 14:622–633, 1967.
World Health Organization (WHO): International Programme on Chemi-cal Safety: Environmental Health Criteria 39. Paraquat and Diquat.Geneva: WHO, 1984.
World Health Organization (WHO): Public Health Impact of PesticidesUsed in Agriculture. Geneva: WHO, 1990.
World Health Organization (WHO): Twenty Years of OnchocerciasisControl in West Africa. Review of the Work of the OnchocerciasisControl Programme in West Africa 1974 – 1994. Geneva: WHO,1997.
World Health Organization (WHO): WHO Technical Report Series 513(Safe Use of Pesticides: Twentieth Report of the WHO Expert Com-mittee on Insecticides). Geneva: WHO, 1973, pp 43–44.
Worthing CR (ed): The Pesticide Manual. A World Compendium, 8th ed.British Crop Protection Council. Lavenham, UK: Lavenham Press,1987.
Wray JE, Mufti Y, Dogramaci I: Hexachlorobenzene as a cause of por-phyria turcica. Turk J Pediatr 4:132–137, 1962.
Xintaris C, Burg JR, Tanaka S, et al: Occupational Exposure to Lep-tophos and Other Chemicals. DHEW (NIOSH) Publication No. 78-136. Washington, DC: DHEW, U.S. Government Printing Office,1978.
Yang C-C, Wu J-F, Ong H-C, et al: Taiwan national poison control center:Epidemiologic data. 1985–1993. Clin Toxicol 34:651–663, 1996.
Yang RSH: Enzymatic conjugation and insecticide metabolism, in Wilkin-son CF (ed): Insecticide Biochemistry and Physiology. New York:Plenum Press, 1976, pp 177–225.
Yang ZP, Dettbarn WD: Diisopropylphosphorofluoridate-induced choliner-gic hyperactivity and lipid peroxidation. Toxicol Appl Pharmacol138:48–53, 1996.
Yang ZP, Morrow J, Wu A, et al: Diisopropylphosphoro-fluoridate inducedmuscle hyperactivity associated with enhanced lipid peroxidation invivo. Biochem Pharmacol 52:357–361, 1996.
Zhang Z, Sun J, Chen S, et al: Levels of exposure and biological monitor-ing of pyrethroids in spraymen. Br J Indus Med 48:82–86, 1991.
2996R_ch22_761-810 4/16/01 4:38 PM Page 810
Copy
right
ed M
ater
ial
Copyright © 2001 by The McGraw-Hill Companies Retrieved from: www.knovel.com