role of disrupted gap junctional intercellular communication in detection and characterization of...
TRANSCRIPT

E L S E V I E R Mutation Research 365 (1996) 91-105 Reviews in Genetic Toxicology
Role of disrupted gap junctional intercellular communication in detection and characterization of carcinogens
H i r o s h i Y a m a s a k i *
Unit of Multistage Carcinogenesis, International Agency for Research on Cancer, 150, cours Albert Thomas, 69372 Lyon cedex 08, France
Abstract
Results from short-term tests for carcinogens and our advanced knowledge on cellular and molecular mechanisms of carcinogenesis strongly suggest that carcinogens do not induce genetic changes necessarily by directly interacting with DNA. Therefore, it is not surprising to see that many carcinogens are not detectable by available genetic toxicology tests. Thus, it has become necessary to study nongenotoxic mechanisms of carcinogenesis and to provide methods to predict those carcinogens which escape from conventional mutation tests. One possible nongenotoxic mechanism of carcinogenesis which is supported by abundant experimental evidence is inhibition of gap junctional intercellular communication. Many, but not all, tumor-promoting agents have been shown to inhibit the communication of cultured cells as well as in vivo. Molecular mechanisms of gap junctional intercellular communication control revealed that connexin (gap junction) genes form a family of tumor suppressor genes. Control mechanisms of expression as well as function of connexins are vulnerable to various carcinogenic insults, notably to nongenetoxic carcinogens. Thus, studies on the role of connexins in cell growth and carcinogenesis may prove to be useful for establishing a mechanism-based test to detect certain types of nongenotoxic carcinogens.
1. Introduct ion
Any reliable short-term test to detect carcinogens must be based on solid scientific information. During the past two decades, numerous short-term test sys- tems have been developed, almost all are based on the assumption that carcinogenesis requires muta- tional events. Recent molecular studies of human cancers have supported the idea that multiple genetic changes must be cumulated before a normal cell develops into a malignant tumor (Fearon and Vogel- stein, 1990), so, therefore, short-term tests based on mutational events may well be justified. On the other
* Corresponding author. Tel.: (33) 72 73 84 85; Fax: (33) 72 73 84 42.
hand, it has been revealed that standard short-term test results identify no more than 65% of rodent carcinogens (Ashby and Tennant, 1991).
Does such a low predictivity suggest that the assumption that mutational events are critical in car- cinogenesis is wrong? The answer is clearly no. What is wrong, however, is to assume that genetic changes required for carcinogenesis are always di- rectly induced by exogenous carcinogens and thus detectable by short-term tests. The hypothesis that cancers arise from mutagenic events is based on the observations that malignant phenotypes are heredi- tary in terms of their cellular lineage and that molec- ular analysis of cancer cells indicates the presence of specific changes in oncogenes and tumor-suppressor genes. Because many environmental carcinogens
0165-1110/96/$15.00 Copyright © 1996 Published by Elsevier Science B.V. PII SO 165- 11 10(96)0001 8-8

92 H. Yamasaki / Mutation Research 365 (1996) 91-105
(e.g., tobacco smoke, X-rays, aflatoxin, sunlight etc.) have been implicated in human cancer causation and because the genetic basis of cancer is solid, it has been assumed that environmental carcinogens must have induced these genetic changes. That assumption is probably still correct, but it cannot be easily translated into simple mutation tests. For example,
exposure to agents that induce cell proliferation, block intercellular communication, or produce oxy- gen radicals may eventually lead to genetic damage, but these indirect mutational processes are not usu- ally detected by existing short-term tests.
It is therefore important to find new short-term tests that will detect carcinogenic agents that do not
CELL N ° I
CELL N°2
N H 2 (ph / voltage gating)
CYTOP ASM Plasma [iil]',~!!!![l~]~r~[ r ~ = t ~ t ~ l ~ mem brane 1[1[
EXTRACEL'U'AR b
Plasma - membrine ~
CYTOPLASM i(
CONNEXON ~ C H A N N E L
[ ~ - -~ CONNEXlN
PROPOSED TOPOLOGY OF CONNEXIN
COOH
GAP JUNCTION
emO ane
Involved in connexon / eonnexon interaction ( E1 and E2 ) Voltage gating ( E1 ) Recognition of connexon specificity ( E2 )
Conserved among different connexin molecules
Hydrophobic surface
Polar residues proposed to be involved in forming the wall of the channel
Fig. h Schematic view of gap junctions, connexons, and connexins (modified from Yamasaki, 1990).

H. Yamasaki / Mutation Research 365 (1996) 91-105 93
directly induce DNA damage but participate indi- rectly in the process of accumulating genetic changes. These carcinogens are usually called nongenotoxic carcinogens, since their primary target of interactions with cells is not the DNA. One possible nongeno- toxic mechanism of carcinogenesis which is sup- ported by abundant experimental evidence is inhibi- tion of gap junctional intercellular communication (GJIC) (Loewenstein, 1979; Trosko and Chang, 1988; Yamasaki, 1990; Yamasaki et al., 1995b). In this article, current knowledge on the role of GJIC in carcinogenesis and its possible application to car- cinogen detection is discussed.
2. Gap junctional intercellular communication
Among various forms of intercellular communica- tion systems in multicellular organisms, GJIC is the only one by which cells exchange signals directly from the inside of one cell to that of neighboring cells (Pitts and Finbow, 1986; Bennett et al., 1991). GJIC is considered to play a crucial role in maintain- ing homeostasis by keeping growth control signals at equilibrium among GJIC-connected cells. GJIC-dif- fusible molecules include those involved in signal transduction; for example, calcium, cAMP and inosi- tol triphosphate (Lawrence et al., 1978; Saez et al., 1989). Because GJIC is considered to play an essen- tial role in homeostasis maintenance, it is reasonable to assume that its disruption will result in a variety of diseases. In fact, besides its significant role in carcinogenesis, aberrant GJIC is implicated in the etiology of heart malformations and malfunctions of the neurosystem in humans (Berghoffen et al., 1993; Britz-Cunningham et al., 1995).
GJIC is mediated by gap junction channels, which are composed of connexin molecules. Six connexin molecules form a connexon in one cell, and a gap junction channel is formed when a connexon in one cell links with one in a neighboring cell (Fig. 1). So far, 12 cDNAs coding for different connexin species have been cloned (Beyer, 1993). Connexins have four transmembrane domains and their C- and N- terminals in the cytoplasm. The amino acid se- quences of these terminal portions and of the cyto- plasmic loop vary significantly between different connexins. Protein kinase motifs are present at the C-terminal end of some of the connexins, phosphory-
lation is considered to play an important regulatory role in GJIC (Moreno et al., 1992). Connexin gene expression has a certain tissue specificity, and one cell type usually expresses several connexin gene species. It is known that gap junctions composed of one connexin species differ in their permeability from those made of another connexin species, and it is likely that the combination of different connexins may subtly differentiate the function of GJIC in specific tissues (Elfgang et al., 1995). Cis and trans
regulatory elements that control connexin gene tran- scription are not yet fully understood, although a few studies have indicated the presence of various ele- ments in the upstream region of the connexin genes (Bai et al., 1995; Yu et al., 1994; Chen et al., 1995). It also has been suggested that promoter sequences of the Cx32 gene play a crucial role in its tissue- specific expression (Neuhaus et al., 1995).
GJIC can be modulated by various mechanisms. In addition to the usual regulatory mechanisms that apply for most proteins, such as transcription, mRNA stabilization and translational control, and posttrans- lational phosphorylation, the function of connexin proteins can also be modulated at other control lev- els. For example, connexins are assembled into con- nexons in a trans-golgi apparatus, and their transloca- tion to the cytoplasmic membrane may also be regu- lated (Musil and Goodenough, 1995). Moreover, even when gap junctions are formed between two cells, it is known that gap junctional channels can be closed or opened under certain circumstances (Zampighi and Unwin, 1979). More indirectly, GJIC can be influenced by cell adhesion molecules (Jongen et al., 1991; Meyer et al., 1992) as well as by extracellular matrix (Spray et al., 1987) and growth factors (Madhuker et al., 1989; Van Zoelen and Tertoolen, 1991); mutations of connexin genes or other genes involved in the control of connexin function also may affect the regulation of GJIC (Britz-Cun- ningham et al., 1995). As described below, these various regulatory points appear to be vulnerable to carcinogenic insults.
3. Basis for using GJIC disruption as an endpoint for short-term tests for carcinogens
The first evidence for the involvement of aberrant GJIC in carcinogenesis was the observation that

94 1-1. Yamasaki / Mutation Research 365 (1996) 91-105
almost all tumors have aberrant GJIC (reviewed by Yamasaki, 1990). Cell lines established from tumors as well as in vitro transformed cells usually show lower GJIC ability. These include not only rodent but also human cell lines. With B A L B / c 3T3 cells, GJIC between transformed cells was unchanged, but between transformed cells and neighboring normal counterparts, it was clearly defective (Enomoto and Yamasaki, 1984). A similar lack of heterologous GJIC between transformed and normal cells was also observed with rat liver epithelial cell lines (Mesnil and Yamasaki, 1988). To extend these in vitro re- suits, we developed a simple method by which GJIC can be examined in slices of liver freshly removed from the rat (Krutovskikh and Yamasaki, 1995). Using this method, we have shown a reduced level of GJIC in rat liver tumors (Krutovskikh et al., 1991). We also applied such a method for measuring GJIC in human liver freshly removed surgically in a hospital. Again, we found that the GJIC level was much reduced in hepatocellular carcinoma cells in comparison with normal or surrounding lesions (Krutovskikh et al., 1994). It therefore appears that reduced GJIC is a common feature of many tumor cells. This is compatible with the idea that GJIC is essential for the maintenance of homeostasis and, because tumor formation inevitably requires the loss
of homeostasis, GJIC must be altered during carcino- genesis.
Further studies on changes of GJIC using multi- stage models of rat liver and mouse skin carcino- genesis have revealed that there is in general a progressive decrease in the level of GJIC during tumor progression. For example, in rat liver carcino- genesis, GJIC is already reduced in many preneo- plastic foci and further reduction was evident in tumors (Krutovskikh et al., 1991). In some of these foci, we also noted a lack of heterologous communi- cation between preneoplastic (GST-P-positive) and surrounding cells. In mouse skin carcinogenesis, cell lines established from papillomas usually communi- cate less than normal cells, but more than cells from carcinomas (Klann et al., 1989). When the levels of GJIC and tumorigenicity of a series of rat liver epithelial cell lines were examined, there was an inverse relationship (Mesnil et al., 1986), although no such relationship was observed in human esophageal cancer cell lines (Oyamada et al., 1994).
The above information indicates possible roles of GJIC blockage in carcinogenesis, but it is not clear whether the disruption of GJIC is a consequence or a cause of carcinogenesis. The most direct way to test whether GJIC is causally involved in carcinogenesis is to transfect connexin genes into GJIC-deficient
Table 1 Growth suppression by connexin genes
Gene Host cell line Growth suppression Ref.
In vitro In vivo
Cx32
Cx43
Cx43
Human hepatoma Rat g l ioma (C6) + Transformed 10T 1 / 2 cells +
+
Rat g l ioma (C6) +
HeLa cells + Rat WB-F344 cells a + Human rhabdomyosarcoma + Rat-1 cells b (antisense oligo transfection) +
+ Eghbali et al., 1991 + Bond et al., 1994
NT Mehta et al., 1991 + Rosen et al., 1993
+ Zhu et al., 1991 Naus et al., 1992
+ Mesnil et al., 1995 NT Esinduy et al., 1995 NT Lin et al., 1995 NT Goldberg et al., 1994
Cx40 HeLa cells - - Mesnil et al., 1995 Cx26 HeLa cells + + Mesnil et al., 1995
a Restoration of GJIC by Cx43 cDNA transfection was important to suppress growth of cocultured transformed cells. b Inhibition of GJIC by Cx43 antisense oligos reduced the ability of rat-I cells to suppress foci formation of cocultured transformed cells.
NT, not tested.

H. Yamasaki / Mutation Research 365 (1996) 91-105 95
cancer cells; a number of such studies have been performed. As summarized in Table 1, such transfec- tants showed an increase in communication capacity and a decrease in tumorigenicity in vivo and /or in
growth in vitro. Rat glioma cells and chemically transformed mouse fibroblasts transfected with the connexin 43 gene (Cx43) showed reduced growth both in vivo and in vitro (Naus et al., 1992; Zhu et
Table 2 Examples of GJIC inhibition by carcinogenic stimuli and stimulation by chemopreventive agents
Tissues/cells studied Methods of GJIC assay a Selected refs.
INHIBITION Tumor-promoting agents
Phorbol esters Various cultured cells EC, MC, DT, BMA DDT Various cultured cells, rat liver MC, DT~ Many others Cultured cells MC, DT, BMA
Tumor-promoting stimuli Partial hepatectomy Rat liver DT, BMA Skin wounding Mouse skin EC DNA-reactive carcinogens Cultured cells MC, DT
ONCOGENES v-src NRK cells DT c-src NIH3T3 cells DT v-Ha-ras Rat liver cell line (WB) DT v-raf Cultured rat liver epithelial DT cells v-fps Rat-2 cells DT, BMA neu Rat liver cell line (WB) DT, BMA Polyoma-middle T B A L B / c 3T3 cells DT SV-40-T Human fibroblasts MC HPV 16-E5 Human keratinocyte cell line DT
(HaCaT)
GROWTH FACTORS/HORMONES EGF Ethinyl estradiol FGF b
PDGF Testosterone
Stimulation Retinoids cAMP elevating agents Glucocorticoids
Green tea extracts Flavones Fermented soy bean (Natto) TGF-~
Cholesterol Vitamin D
Swierenga and Yamasaki, 1992 c Budunova and Williams, 1994 c
Yamasaki et al., 1993 c Loewenstein and Penn, 1967 Budunova and Williams, 1994
Atkinson et al., 1981 Azarnia et al., 1988 De Feijter et al., 1990 Kalimi et al., 1992 Kurata and Lau, 1994 Jou et al., 1995 Katoh et al., 1993 Rosen et al., 1988 Oelze et al., 1995
v79 cells MC Trosko et al., 1982 v79 cells MC Trosko et al., 1982 Rat liver cell line (T51B) DT Kanematsu and Lau, 1993 C3H10T 1 /2 cells DT Pelletier and Boynton, 1994 Human transitional cell DT Kihara et al., 1990 Carcinoma cell lines
Various cells MC, DT, BMA Goldberg and Bertram, 1994 c Various cells MC, DT, BMA Mehta et al., 1992 B A L B / c 3T3 cells DT Yamasaki and Katoh, 1988 Primary hepatocyte culture DT Kwiatkowski et al., 1994 Rat liver cell line (WB) DT Sigler and Ruch, 1993 Rat liver cell lines (REL) DT Chaumontet et al., 1994 B A L B / c 3T3 cells DT Takahashi et al., 1995 NRK cells DT Van Zoelen and Tertoolen, 1991 C3H10T 1 /2 cells DT Ubeda et al., 1995 Novikoff hepatoma cells DT Meyer et al., 1990 C3HI0 1 / 2 cells DT Stahl et al., 1994
a DT, dye-transfer (see Table 3); EC, electrical coupling; MC, metabolic cooperation; BMA, biochemical and molecular assays (e.g., immunohistological staining, expression of connexin mRNA and proteins). b Synergistic with 12-0-tetradecanoylphorbol 13-acetate. c Representative reviews.

Tab
le 3
C
ompa
riso
n of
var
ious
ass
ay s
yste
ms
to m
easu
re G
JIC
Spec
ial
equi
pmen
t re
quir
emen
t T
echn
ical
sim
plic
ity
(+ +
+,
very
sim
ple,
-,
di
ffic
ult)
App
lica
tion
A
ppli
cati
on
Qua
ntif
icat
ion
Ava
ilab
le d
ata
to v
ario
us
to i
n vi
vo
of G
JIC
lev
el
base
for
car
cino
g.
cell
typ
es
( +
+,
good
, de
tect
ion
( + +
+
, (~
_+
,poo
r)
rich
,(~
,s
carc
e)
Oth
er c
hara
cter
isti
cs
Met
abol
ic c
oope
rati
on a
H
PR
T+
/HP
RT
C
onve
ntio
nal
cell
cul
ture
+
+ D
iffi
cult
N
ot p
ossi
ble
+ +
+ +
AS
S -
/AS
L-
+ +
Dif
ficu
lt
+ _+
A
K+
/AK
+
+ D
iffi
cult
+
- D
ye-t
rans
fer
Mic
roin
ject
ion
Mic
roin
ject
ion
+ E
asy
Suc
cess
ful
+ +
+ +
Scr
ape
load
ing
Con
vent
iona
l ce
ll c
ultu
re
+ +
+ E
asy
Not
don
e, b
ut p
ossi
ble
+ +
Pho
tobl
each
ing
Pho
tobl
each
ing
syst
em
+ E
asy
Not
don
e +
+ +
Prel
oadi
ng
Con
vent
iona
l ce
ll c
ultu
re
+ E
asy
Not
don
e +
+ -
Ele
ctri
cal
coup
ling
E
lect
roph
ysio
logi
cal
appa
ratu
s -
Eas
y S
ucce
ssfu
l +
+ _+
Nee
ds t
o ac
quir
e ap
prop
riat
e m
utan
t ce
ll l
ines
Stu
dies
on
dye-
tr
ansf
er k
inet
ics
are
poss
ible
Mea
sure
s io
n tr
ansf
er
I
~' F
or p
rinc
iple
s of
met
hods
, se
e Fi
g.
2.
Abb
revi
atio
ns o
f m
utan
ts a
re:
HP
RT
, hy
poxa
nthi
ne p
hosp
hori
bosy
ltra
nsfe
rase
; A
ss
, ar
gini
nosu
ccin
ate
synt
heta
se-d
efic
ient
; A
SL
,
argi
nino
succ
inat
e ly
ased
efic
ient
; A
K,
aden
osin
e ki
nase
.

H. Yamasaki / Mutation Research 365 (1996) 91-105 97
al., 1991; Rose et al., 1993), but human liver tumor cells transfected with the Cx32 gene showed reduced tumorigenicity but no effect on growth in vitro (Eghbali et al., 1991). The transformed mouse cells also lost the ability to form foci on a normal cell monolayer after transfection of the Cx43 gene (Mehta et al., 1991). Our previous studies showed a more complicated picture when HeLa cells were trans- fected with Cx26, 40 or 43; only those transfected with the Cx26 gene became nontumorigenic and showed a reduced growth rate in vitro (Mesnil et al., 1995). Interestingly, the Cx26 gene is the major connexin gene expressed in the cervix, from which the HeLa cell line was originally isolated. These results suggest that connexin genes exert differential regulatory effects on cell growth, depending on the cell type in which they are expressed. This would be in agreement with the notion that connexin genes are in fact a family of tumor-suppressor genes, which is further supported by the emerging evidence that connexin genes are mutated in some tumors (Yamasaki et al., 1995a).
Another line of evidence that suggests a causal role of blockage of intercellular communication in carcinogenesis has come from studies in which agents or genes involved in carcinogenesis were shown to modulate GJIC. First, the mouse skin tumor-promo- ting agent 12-O-tetradecanoylphorbol 13-acetate (TPA) has been shown to inhibit GJIC in many types of cultured cells by various methods including metabolic cooperation, electrical coupling, and dye- transfer assays (Murray and Fitzgerald, 1979; Yotti et al., 1979; Enomoto et al., 1981 Enomoto et al., 1984). Many tumor-promoting agents were subse- quently reported to inhibit GJIC, and this led to the proposal that GJIC inhibition could be used as an endpoint for detecting tumor-promoting agents (Swierenga and Yamasaki, 1992). In addition to these tumor-promoting agents, other types of tumor- promoting stimuli such as partial hepatectomy and skin wounding also inhibit GJIC. Furthermore, it has been shown that various oncogenes also inhibit GJIC; such oncogenes include src, SV-40-T antigen, neu,
raf , f p s and ras oncogenes. On the other hand, certain growth factors such as PDGF, EGF, and TGF-[3 also modulate GJIC in cultured cells.
It has also been observed that chemopreventive agents enhance GJIC. Retinoids, glucocorticoids and
cAMP are known to antagonize the tumor-promoting ability of TPA in mouse skin, and they have been shown to upregulate GJIC. Other agents that upregu- late GJIC include green tea extracts, flavones, vita- min D, and components of Japanese soybean fer- mented food (Natto).
Examples of inhibition and stimulation of GJIC by agents or factors and stimuli involved in carcino- genesis are summarized in Table 2. It is important to emphasize that only those studies that gave positive results are listed. There also are many studies that provided negative results. Budunova and Williams (1994) have reviewed this subject in detail. Taken together, there are sufficient lines of support for the use of GJIC disruption as a marker to identify poten- tial carcinogens.
4. Issues associated with assays of GJIC
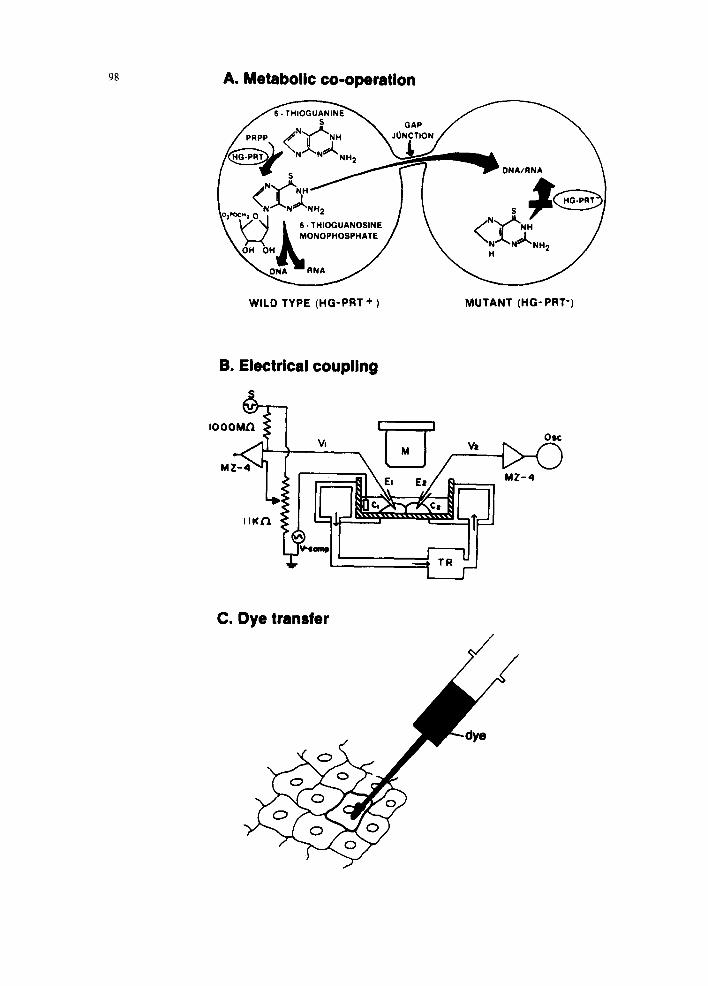
In cultured cells, GJIC capacity can be measured by three main types of methods (1) metabolic coop- eration, (2) dye-transfer, and (3) electrical coupling, as shown schematically in Fig. 2. Table 3 presents the different assays with their advantages and disad- vantages. Many laboratories are also using the micro- injection dye-transfer assay to quantitate GJIC. The biggest advantage of this procedure is that one can use any cell type that adheres to culture dishes. Because many tumor-promoting agents or so-called nongenotoxic carcinogens are likely to show a tis- sue-specific activity, it is important to study chemi- cals with some hypothesis regarding target tissues in which they may work. In this case, it is not practical to use a metabolic cooperation assay because one must prepare mutants of target cells before such an assay can be performed.
The dye-transfer assay also can be applied to measure GJIC in organ slices. This allows changes in GJIC to be followed in such slices after test chemi- cals are administered in vivo. Such a method has been developed in our laboratory and has already been successfully used in at least two other laborato- ries to test various tumor-promoting agents (Tateno et al., 1994; Krutovskikh et al., 1995). Alternatively, fresh tissue slices can be exposed to test chemicals in vitro before GJIC is measured. Primary cultures of human and rodent hepatocytes can be maintained
9g A. Metabolic co-operation
GAP
\" V'-'~ L ~.,.,oQu.~os,NE / ,~ o~, /
WILD TYPE (HG-PFIT + )
N
MUTANT (HG- PRT')
B. Electrical coupling
,ooo IIKfl
! I
El Ee
I
0$¢
M Z - 4
C. Dye transfer

H. Yamasaki / Mutation Research 365 (1996) 91-105
Table 4 Examples of mechanisms involved in GJIC modulation observed in tumors or induced by tumor-promoting agents
99
Examples in tumors or by carcinogenic factors Ref.
Mutation of connexin genes
Aberrant connexin gene expression
Reduced connexin protein expression
Aberrant phosphorylation of connexins
Aberrant localization (cytoplasmic) of connexins
Lack of cell adhesion molecules
Primary rat liver tumor Lewis lung carcinoma cell line
Primary human liver tumors (appearance of Cx43) Primary rat liver tumors (reduced Cx32) Human bladder cancer cells (reduced Cx26) Rat bladder tumors (increased Cx26 and Cx43) Reduction by the tumor-promoting agent, phenobarbital
Various human tumors Mouse skin Rat liver
By tumor-promoting agents (e.g., TPA, DDT, endosulfan, dieldrin, heptachlor epoxide)
By oncogenes
Primary human liver tumors Primary rat liver tumors By tumor-promoting agents in rat liver By tumor-promoting agents in cultured cells
Mouse skin tumor cells
Yamasaki, 1995 Mandelboim et al., 1994
Oyamada et al., 1990 Fitzgerald et al., 1989, Neveu et al., 1994 Grossman et al., 1994 Asamoto et al., 1994 Mesnil et al., 1988
Wilgenbus et al., 1992 Kamibayashi et al., 1995 Sakamoto et al., 1992
Berthoud et al., 1993, Mesnil et al., 1994, Rivedal et al., 1994, Kenne et al., 1994, Matesic et al., 1994 Loo et al., 1995, Kurata and Lau, 1994
Krutovskikh et al., 1994 Yamasaki, 1995 Krutovskikh et al., 1995 Asamoto et al., 1991, Rivedal et al., 1994, Matesic et al., 1994
Jongen et al., 1991
wi th c o n t i n u e d e x p r e s s i o n o f d i f f e ren t i a t ed func t i ons
for a long t ime on ly w h e n they are cocu l tu red wi th
b i l ia ry ep i the l ia l cells, m i m i c k i n g the in v ivo si tua-
t ion ( G u g u e n - G u i l l o u z o et al., 1983). W e h a v e re-
cen t ly i m p r o v e d the m e t h o d so that p r i m a r y hepa to -
cytes can be m a i n t a i n e d in cocu l tu re wi th e s t ab l i s hed
cel l l ines and h a v e s h o w n tha t G J I C is m a i n t a i n e d in
the hepa tocy tes . G J I C was i nh ib i t ed b y p h e n o b a r b i t a l
bu t no t by T P A , s h o w i n g t i s sue-spec i f i c i nh ib i t i on
by t u m o r - p r o m o t i n g agen ts ( Mes n i l et al., 1993).
A n o t h e r in vi t ro s y s t e m is p r ima ry cu l ture of ha i r
fol l ic les . P lucked h u m a n ha i r fo l l ic les can be p l aced
in cu l ture and the g r o w i n g cel ls are su i t ab le for GJ IC
s tudy ( S w i e r e n g a et al., 1991). T h e s e m e t h o d s shou ld
ref lect in v ivo G J I C states m o r e appropr ia t e ly than
c o n v e n t i o n a l l y used cel l l ines and p r ima ry cu l ture o f
s ingle types of cells.
W h i l e it appea r s tha t the dye - t r ans fe r assay is
su i tab le for m a n y labora tor ies , it is adv i sab le no t to
fix any assay s y s t e m or pro tocol at th is s tage for
Fig. 2. (A) Metabolic cooperation. In the coculture of HPRT + and HPRT cells, only HPRT + cells metabolize 6-thioguanine to 6-thioguanine monophosphates, which kill cells upon incorporation into nucleic acids. Although HPRT- cells do not convert 6-thioguanine and could therefore survive in the presence of 6-thioguanine when they are cultured alone, in contact with HPRT + cells, 6-thioguanine monophosphates can be transferred from HPRT ÷ cells through gap junctions. Thus, in the presence of 6-thioguanine monophosphates can be transferred from HPRT + cells through gap junctions. Thus, in the presence of 6-thioguanine, both HPRT + and HPRT- cells are killed. (B) Electrical coupling. A microelectrode is inserted into each coupled cell (C1 and C2). Electrical current is then run from C1, and whether the electron transfer occurs from C1 to C2 through GJIC is measured. (C) Dye transfer. Tracer dye molecules (usually fluorescent) that are not membrane-diffusible, but gap junction-diffusible, are microinjected into a cell. Lucifer Yellow CH molecules are mostly used for this purpose. The spread of dye into neighboring cells through gap junctions is determined.

100 H. Yamasaki / Mutation Research 365 (1996) 91-105
carcinogen screening. Because each chemical differs in its properties and modes of action, it is wiser to vary the methods and protocols accordingly.
5. Mechanisms of GJIC inhibition by carcinogens
Advances in understanding the molecular biology of GJIC have enabled us to characterize the molecu- lar events associated with the inhibition of GJIC by carcinogens. Although relatively few chemicals have been studied for their mechanisms of action on GJIC, it appears that posttranslational modulation is impor- tant. For example, TPA-treated cultured rat liver epithelial cells do not show any changes in their level of mRNA for the Cx43 gene (Asamoto et al., 1991), and Cx43 protein was present in similar amounts before and after TPA treatment. However, its phosphorylated forms were changed, and the con- nexin proteins were localized in the cytoplasm rather than in cytoplasmic membranes. Therefore, it seems that TPA caused phosphorylation of connexin pro- teins, probably through protein kinase C activation, leading to their translocation from the cytoplasmic membrane to cytoplasm (Mesnil et al., 1994). Be- cause connexin proteins can function only when they are in the cytoplasmic membranes, this internaliza- tion of connexins may be responsible for GJIC inhi- bition. Changes in the phosphorylation pattern of connexins are also associated with GJIC inhibition by other tumor-promoting agents and oncogenes (Ta- ble 4).
In vivo studies also suggest that posttranslational modulation of connexins and /or aberrant localiza- tion is a common mechanism for carcinogen-induced downregulation of GJIC. We have submitted groups of Fischer-344 rats were subjected to repeated treat- ment with phenobarbital (PB), polychlorinated biphenyls (PCB), dichloro-diphenyltrichloroethane (DDT), and clofibrate (CF) for 5 weeks. All four tumor-promoting agents decreased dye coupling in rat liver. This decrease was associated with a re- duced number of gap junctions and aberrant localiza- tion of some of the Cx32 proteins in hepatocytes; Cx32 was often observed in the cytoplasm of hepato- cytes instead of at gap junctions in the plasma membrane. Western blot analysis indicated only slight changes in the level of Cx32 proteins. A1-
though levels of Cx26 proteins at gap junctions were usually decreased by tumor promoters in rat liver, local induction of Cx26 protein expression in cen- trilobular groups of hepatocytes after PCB and DDT treatment was observed. Expression of Cx43 was induced in hepatocytes after PCB, DDT, and CF exposure, but this protein was also localized intracy- toplasmically, suggesting that it had no functional role (Krutovskikh et al., 1995).
It has previously been shown that treatment of rats with phenobarbital lowers the level of Cx32 mRNA (Mesnil et al., 1988). It has also been shown that in many rat liver tumors, levels of Cx32 mRNA are decreased (Beer et al., 1988). However, in human hepatocellular carcinomas, the level of Cx32 mRNA was not changed, although that of Cx43 mRNA was significantly reduced (Oyamada et al., 1990). In contrast, carotenoids can upregulate the transcription of Cx43 mRNA (Zhang et al., 1992). These results suggest that GJIC can also be modulated at the transcriptional level.
Although the inhibition of GJIC in toxicology has always been approached as a nongenotoxic process, it is possible that genotoxic mechanisms such as mutations of connexin genes lead to GJIC impair- ment. However, among 20 human liver tumors and 22 human gastric tumors, we found no Cx32 gene mutation (Mironov et al., 1994; Krutovskikh et al., 1994). On the other hand, when several rat liver tumors induced by a nitrosamine were analyzed, we found one sample with a mutation at the 220th codon of the Cx32 gene (Yamasaki et al., 1995a). Our preliminary results also suggest that some human malignant meningiomas harbor a mutation of the Cx43 gene.
Interestingly, recent studies have shown that two human diseases are caused by germ cell mutations of connexin genes. In certain types of visceroatrial het- erotaxia syndrome, a Cx43 gene mutation was found (Britz-Cunningham et al., 1995). In X-linked Char- cot-Marie Tooth (CMTX) disease, Cx32 gene muta- tions have been found (Berghoffen et al., 1993; Fairweather et al., 1994). Thus, it seems that muta- tional events in connexin genes can lead to impair- ment of GJIC. This would imply that mutagenic carcinogens may also induce connexin gene muta- tions and inhibition of GJIC. We and others have shown that some Cx32 mutants found in CMTX

H. Yamasaki / Mutation Research 365 (1996) 91-105 101
patients are indeed defective in forming functional GJIC (Bruzzone et al., 1994; Omori et al., unpub- lished data). Moreover, these mutants showed a strong dominant-negative effect on wild-type Cx32 proteins when both of them were transfected into HeLa cells (unpublished data). It is likely that such a dominant-negative effect is due to the formation of chimeric connexons containing both mutant and wild-type connexins. Thus, mutations in one allele of connexin genes may result in complete loss of the connexin function, suggesting that a mutational event may be a powerful mechanism for GJIC inhibition.
GJIC can thus be modulated by various mecha- nisms induced by various carcinogenic stimuli. Some examples are summarized in Table 4.
6. Implications for cancer risk assessment
Mutation assays were introduced with great hopes that they could predict most human carcinogens. This expectation was not fulfilled, and it has there- fore become important to attempt to develop assays in which the endpoints are not mutations. In develop- ing new nongenotoxic mechanism-based test systems for carcinogens, we should learn from the experience of genetic toxicologists. In retrospect, it appears that validation of a protocol by assaying large numbers of numerous chemicals may not have been a sound approach. It is essential to examine the concept behind a proposed test system. For example, it is important to have solid scientific evidence that dis- rupted GJIC is causally related to carcinogenesis, rather than simply testing many chemicals in one of the GJIC assays using cultured cells. It is also un- wise to suppose that all nongenotoxic carcinogens will be detected by a single test system; it is more likely that different carcinogens will affect different cellular constituents and in different manners.
Approaches to hazard identification and risk as- sessment in cancer are changing. It is being increas- ingly recognized that mechanistic information should be considered in addition to conventional epidemio- logical data or short-term and long-term test results from model systems (IARC, 1991). Thus, in the context of this article, it would be important to know whether chemicals inhibit GJIC by modulation of gene expression or by post translational modifica- tion. It is also possible that mutation of connexin
genes will lead to inhibition of GJIC. If agents inhibit GJIC by mutations, the effect may be irre- versible, whereas posttranslational modification could be reversible. On the other hand, a mutagenic effect can occur in a very few cells and thus is localized, whereas a nongenotoxic carcinogen may affect the communication of the whole exposed cell popula- tion. Such information is important for accurate risk estimation of a test agent.
The fact that a given endpoint of a test, such as inhibition of GJIC, can be attained by either muta- genic or nonmutagenic events implies a certain diffi- culty in classifying carcinogens into genotoxic and nongenotoxic categories on the basis of short-term results. As with the example of GJIC inhibition, most of the apparently nongenotoxic endpoints can also be induced by genotoxic mechanisms (Yamasaki et al., 1996). Therefore, classification of carcinogens into genotoxic or nongenotoxic, or into initiating or promoting agents, may not only be unhelpful but even an impediment to risk assessment. Once enough relevant mechanistic information about individual chemicals is accumulated, it will be more reasonable to use this information directly for risk estimation, without expending effort in classifying them. A pos- sible negative consequence of carcinogen classifica- tion for risk assessment is that a classification can be based on only one of various activities of an agent; for example, ultraviolet light may be considered to be genotoxic (induction of gene mutations) or nongenotoxic (immunomodulation). If carcinogens are classified, the level of risk tends to be associated with such classification, so that the classification of a given agent is based on only one type of activity, an overall assessment of its risk cannot be made. Thus, a more complete risk assessment of carcinogens may be carried out in the absence of a classification.
Most of the data on inhibition of GJIC by chemi- cals have not been produced with a single protocol. This is probably not a defect of the approach, but in fact a more appropriate way to address the problem. If the protocol is fixed with one method and one cell type, it is likely that many carcinogens will not be detected. Instead, if assay systems with various cell types are used, selected on the basis of a working hypothesis, more useful information may be ob- tained. At a crossroads of changes in carcinogen screening, it is probably better to study each chemi-

102 H. Yamasaki / Mutation Research 365 (1996) 91-105
cal agent with hypotheses rather than simply test
them with a fixed protocol.
Acknowledgements
I wish to thank many of my colleagues for contin- uous helpful discussion; my special thanks go to Drs. V. Krutovskikh, M. Mesnil, and C. Naus. I am grateful to Mme. Chantal D6chaux for her efficient secretarial aid and to Dr. John Cheney for editing the manuscript.
References
Asamoto, M., M. Oyamada, A. El Aoumari, D. Gros and H. Yamasaki (1991) Molecular mechanisms of TPA-mediated inhibition of gap junctional intercellular communication; evi- dence for action on the assembly or function but not the expression of connexin 43 in rat liver epithelial cells, Mol. Carcinogen., 4, 322-327.
Asamoto, M., S. Takahashi, K. Imaida, T. Shirai and S. Fukushima (1994) Increased gap junctional intercellular communication capacity and connexin 43 and 26 expression in rat bladder carcinogenesis, Carcinogenesis, 15, 2163-2166.
Ashby, J. and R. W. Tennant (1991) Definitive relationships among chemical structure, carcinogenicity and mutagenicity for 301 chemicals tested by the US NTP, Mutation Res., 257, 229-306.
Atkinson, M.M., A.S. Menko, R.G. Johnson, J.R. Sheppard and J.D. Sheridan (1981) Rapid and reversible reduction of junc- tional permeability in cells infected with a temperature-sensi- tive mutant of avian sarcoma virus, J. Cell Biol., 91,573-578.
Azarnia, R., S. Reddy, T.E. Kmiecik, D. Shalloway and W.R. Loewenstein (1988) The cellular src gene product regulates junctional cell-to-cell communication, Science, 239, 398-401.
Bai, S., A. Schoenfeld, A. Pietrangelo and R.F. Burk (1995) Basal promoter of the rat connexin 32 gene: identification and characterization of an essential element and its DNA-binding protein, Mol. Cell Biol., 15, 1439-1445.
Beer, D.C., M.J. Neveu, D.L. Paul, U.R. Rapp et al. (1988) Expression of the c-raf protooncogene: (~/-glutamyltrans- peptidase and gap junction protein in rat liver neoplasms, Cancer Res., 48, 1610-1617.
Bennett, M.V.L., L.C. Barrio, T.A. Bargiello, D.C. Spray et al. (1991) Gap junctions: new tools, new answers, new questions, Neuron, 6, 305-320.
Berghoffen, J., S.S. Scherer, S. Wang, L.J. Oronzi-Scott et al. (1993) Connexin mutation in X-linked Charcot-Marie-Tooth disease, Science, 262, 2039-2041.
Berthoud, V.M., M.B. Rook, O. Traub, E.L. Hertzberg and J.C. Saez (1993) On the mechanisms of cell uncoupling induced by a tumor promoter phorbol ester in clone 9 cells, a rat liver epithelial cell line, Eur. J. Cell Biol., 62, 384-396.
Beyer, E.C. (1993) Gap junctions, Int. Rev. Cytol., 137C, 1-37. Bond, S.L., J.F. Bechberger, N.K. Khoo and C.C. Naus (1994)
Transfection of C6 glioma cells with connexin 32: the effects of expression of a nonendogenous gap junction protein, Cell Growth Differ., 5, 179-186.
Britz-Cunningham, S.H., M.M. Chan, C.W. Zuppan and W.H. Fletcher (1995) Mutations of the connexin 43 gap-junction gene in patients with heart malformations and defects of laterality, N. Engl. J. Med., 332, 1323-1329.
Bruzzone, R., T.W. White, S.S. Scherer, K.H. Fischbeck and D.L. Paul (1994) Null mutations of connexin 32 in patients with X-linked Charcot-Marie Tooth disease, Neuron, 13, 1253- 1260.
Budunova, I.V. and G.M. Williams (1994) Cell culture assays for chemicals with tumor-promoting or tumor-inhibiting activity based on the modulation of intercellular communication, Cell Biol. Toxicol., 10, 71-116.
Chaumontet, C., V. Bex, I. Gaillard-Sanchez, C. Seillan-Heberden et al. (1994) Apigenin and tangeretin enhance gap junctional intercellular communication in rat liver epithelial cells, Car- cinogenesis, 15, 2325-2330.
Chen, Z.Q., D. Lefebvre, X.H. Bai, A. Reaume et al. (1995) Identification of two regulatory elements within the promoter region of the mouse connexin 43 gene, J. Biol. Chem., 270, 3863-3868.
De Feijter, A.W., J.S. Ray, C.M. Weghorst, J.E. Klaunig et al. (1990) Infection of rat liver epithelial cells with v-Ha-ras; correlation between oncogene expression, gap junctional com- munication and tumorigenicity, Mol. Carcinogen., 3, 54-67.
Eghbali, B., J.A. Kessler, L.M. Raid, C. Roy and D.S. Spray ( 1991) Involvement of gap junction in tumorigenesis: transfec- tion of tumor cells with connexin 32 cDNA retards growth in viuo, Proc. Natl. Acad. Sci. USA, 88, 10701-10705.
Elfgang, C., R. Eckert, H. Lichtenberg-Frate, A. Butterweck et al. (1995) Specific permeability and selective formation of gap junction channels in connexin-transfected HeLa cells, J. Cell Biol., 129, 805-817.
Enomoto, T. and H. Yamasaki (1984) Lack of intercellular com- munication between chemically-transformed and surrounding non-transformed B A L B / c 3T3 cells, Cancer Res., 44, 5200- 5203.
Enomoto, T., Y. Sasaki, Y. Shiba, Y. Kanno and H. Yamasaki (1981) Tumor promoters cause a rapid and reversible inhibi- tion of the formation and maintenance of electrical cell cou- pling in culture, Proc. Natl. Acad. Sci. USA, 78, 5628-5632.
Enomoto, T., N. Martel, Y. Kanno and H. Yamasaki (1984) Inhibition of cell-cell communication between BALB/c 3T3 cells by tumor promoters and protection by cAMP, J. Cell Physiol., 121,323-333.
Esinduy, C.B., C.C. Chang, J.E. Trosko and R.J. Ruch (1995) In vitro growth inhibition of neoplastically-transformed cells by non-transformed cells: requirement for gap junctional intercel- lular communication, Carcinogenesis, 16, 915-921.

H. Yamasaki / Mutation Research 365 (1996) 91-105 103
Fairweather N., C. Bell, S. Cochrane, J. Chelly, S. Wang et al. (1994) Mutations in the connexin 32 gene in X-linked domi- nant Charcot-Marie-Tooth disease (CMTX1), Hum. Mol. Genet. 3, 29-34.
Fearon, E.R. and B.A. Vogelstein (1990) A genetic model for colorectal tumorigenesis, Cell, 61,759-767.
Fitzgerald, D.J., M. Mesnil, M. Oyamada, H. Tsuda et al. (1989) Changes in gap junction protein (connexin 32) gene expres- sion during rat liver carcinogenesis, J. Cell Biochem., 41, 97-102.
Goldberg, G.S. and J.S. Bertram (1994) Retinoids, gap junctional communication and suppression of epithelial tumors, In Vivo, 8, 745-754.
Goldberg, G.S., K.D. Martyn and A.F. Lau (1994) A connexin 43 antisense vector reduces the ability of normal ceils to inhibit the foci formation of transformed cells, Mol. Carcinogen., 11, 106-114.
Grossman, H.B., M. Liebert, I.W. Lee and S.W. Lee (1994) Decreased connexin expression and intercellular communica- tion in human bladder cancer cells, Cancer Res., 54, 3062- 3065.
Guguen-Guillouzo, C., B. Cl6ment, G. Baffet, C. Beaumont et al. (1983) Maintenance and reversibility of active albumin secre- tion by adult rat hepatocytes cocultured with another liver epithelial type, Exp. Cell Res., 143, 47.
IARC (1991) Mechanisms of carcinogenesis in risk identification, IARC Internal Technical Report 91/002, Lyon.
Jongen, W.M.F., D.J. Fitzgerald, M. Asamoto, C. Piccoli et al. (1991) Regulation of connexin43-mediated gap junctional in- tercellular communication by Ca 2+ in mouse epidermal cells is controlled by E-cadherin, J. Cell Biol., 114, 545-555.
Jou, Y.S., B. Layhe, D.F. Matesic, C.C. Chang et al. (1995) Inhibition of gap junctional intercellular communication and malignant transformation of rat liver epithelial cells by neu oncogene, Carcinogenesis, 16, 311-317.
Kalimi, G.H., L.L. Hampton, J.E. Trosko, S.S. Thorgeirsson and A.C. Huggett (1992) Homologous and heterologous gap-junc- tional intercellular communication in v-raf-, v-myc, and v- raf/v-myc-transduced rat liver epithelial cell lines, Mol. Car- cinogen., 5, 301-310.
Kamibayashi, Y., Y. Oyamada, M. Mori and M. Oyamada (1995) Aberrant expression of gap junction proteins (connexins) is associated with tumor progression during multistage mouse skin carcinogenesis in vivo. Carcinogenesis, 16, 1287-1297.
Kanematsu, M.Y. and A.F. Lau (1993) Epidermal growth factor stimulates the disruption of gap junctional communication and connexin 43 phosphorylation independent of 12-O-tetrade- canoylphorbol 13-acetate-sensitive protein kinase C: the possi- ble involvement of mitogen-activated protein kinase, Mol. Biol. Cell., 4, 837-848.
Katoh, F., J.-L. Klein, M. Bignami and H. Yamasaki (1993) Association of viral oncogene-induced changes in gap junc- tional intercellular communication and morphological transfor- mation in BALB/c 3T3 cells, Carcinogenesis, 14, 435-440.
Kenne, K., R. Fransson-Steen, S. Honkasalo and L. Warngard (1994) Two inhibitors of gap junctional intercellular communi- cation, TPA and endosulfan: different effects on phosphoryla-
tion of connexin 43 in the rat liver epithelial cell line, IAR 20, Carcinogenesis, 15, 1161-1165.
Kihara, K., I. Fukui, Y. Higashi and H. Oshima (1990) Inhibitory effect of testosterone on gap junctional intercellular communi- cation of human transitional cell carcinoma cell lines, Cancer Res., 50, 2848-2852.
Klann, R.C., D.J. Fitzgerald, C. Piccoli, T.J. Slaga and H. Ya- masaki (1989) Characterization of gap-junctional intercellular communication in SENCAR mouse epidermal cell lines, Can- cer Res., 49, 699-705.
Krutovskikh, V. and H. Yamasaki (1995) Ex-vivo dye transfer assay as an approach to study gap junctional intercellular communication disorders in hepatocarcinogenesis, Prog. Cell Res., 4, 93-97.
Krutovskikh, V., M. Oyamada and H. Yamasaki (1991) Sequen- tial changes of gap-junctional intercellular communications during multistage rat liver carcinogenesis: direct measurement of communication in vivo, Carcinogenesis, 12, 1701-1706.
Krutovskikh, V., G. Mazzoleni, N. Mironov, Y. Omori et al. (1994) Altered homologous and heterologous gap junctional intercellular communication in primary human liver tumors associated with aberrant protein localization but not gene mutation of connexin 32, Int. J. Cancer, 56, 87-94.
Krutovskikh, V., M. Mesnil, G. Mazzoleni and H. Yamasaki (1995) Inhibition of rat liver gap junction intercellular commu- nication by tumor-promoting agents in t~iL,o; association with aberrant localization of connexin proteins, Lab. Invest., 72, 571-577.
Kurata, W.E. and A.F. Lau (1994) pl30gag-fps disrupts gap junctional communication and induces phosphorylation of con- nexin 43 in a manner similar to that of pp60v-src, Oncogene, 9, 329-335.
Kwiatkowski, A.P., T.K. Baker and J.E. Klaunig (1994) Compari- son of glucocorticoid-mediated changes in the expression and function of rat hepatocyte gap junctional proteins, Carcinogen- esis, 15, 1753-1757.
Lawrence, T.S., W.H. Beers and N.B. Gilula (1978) Hormonal stimulation and cell communication in cocultures, Nature, 272, 501-506.
Lin, Z.X., Z.Q. Zhang, K.R. Yu, D.G. Zhu and C.C.G. Naus (1995) Increased junctional communication and forced expres- sion of connexin 43 retards cell growth and enhances myo- genic differentiation in rhabdomyosarcoma cells, Prog. Cell Res., 4, 31-35.
Loewenstein, W.R. (1979) Junctional intercellular communication and the control of growth, Biochim. Biophys. Acta, 560, 1-65
Loewenstein, W.R. and R.D. Penn (1967) Intercellular communi- cation and tissue growth. II. Tissue regeneration and 33 growth. II. Tissue regeneration, J. Cell Biol., 33, 235-242.
Loo, L.W., J.M. Berestecky, M.Y. Kanemitsu and A.F. Lau (1995) pp60src-mediated phosphorylation of connexin 43, a gap junction protein, J. Biol. Chem., 270, 12751-12761
Madhuker, B.V., S.Y. Oh, C.C. Chang, M. Wade and J.E. Trosko (1989) Altered regulation of intercellular communication by epidermal growth factor, transforming growth factor 13 and peptide hormones in normal human keratinocytes, Carcinogen- esis, 10, 13-20.

104 H. Yamasaki / Mutation Research 365 (1996) 91-105
Mandelboim, O., G. Berke, M. Fridkin, M. Feldman et al. (1994) CTL induction by a tumor-associated antigen octapeptide de- rived from a murine lung carcinoma, Nature, 369, 67-71.
Matesic, D.F, H.L. Rupp, W.J. Bonney, R.J. Ruch and J.E. Trosko (1994) Changes in gap-junction permeability, phosphorylation, and number mediated by phorbol ester and non-phorbol-ester tumor promoters in rat liver epithelial cells, Mol. Carcinohen., 10, 226-36.
Mehta, P.P., A. Hotz-Wagenblatt, B. Rose, D. Shalloway and W.R. Loewenstein (1991) Incorporation of the gene for a cell-cell channel protein into transformed cells leads to nor- malization of growth, J. Membr. Biol., 124, 207-225.
Mehta, P.P., M. Yamamoto and B. Rose (1992) Transcription of the gene for the gap junctional protein connexin 43 and expression of functional cell-to-cell channels are regulated by cAMP, Mol. Biol. Cell, 3, 839-850.
Mesnil, M. and H. Yamasaki (1988) Selective gap junctional communication capacity of transformed and non-transformed rat liver epithelial cell lines, Carcinogenesis, 9, 1499-1502.
Mesnil, M., R. Montesano and H. Yamasaki (1986) Intercellular communication of transformed and non-transformed rat liver epithelial cells. Modulation by 12-O-tetradecanoylphorbol 13- acetate, Exp. Cell Res., 165, 391-402.
Mesnil, M., D.J. Fitzgerald and H. Yamasaki (1988) Phenobarbital specifically reduces gap junction protein mRNA level in rat liver, Mol. Carcinogen, 1, 79-81.
Mesnil, M., C. Piccoli and H. Yamasaki (1993) An improved long-term culture of rat hepatocytes to detect liver tumor-pro- moting agents: results with phenobarbital, Eur. J. Pharmacol., Envir. Toxicol. Pharmacol. Section, 248, 59-66.
Mesnil, M., M. Asamoto, C. Piccoli and H. Yamasaki (1994) Possible molecular mechanisms of loss of homologus and heterologous gap junctional intercellular communication in rat liver epithelial cell lines, Cell Adhes. Commun., 2, 377-384.
Mesnil, M., V. Krutovskikh, C. Piccoli, C. Elfgang et al. (1995) Negative growth control of HeLa cells by connexin genes: connexin species specificity, Cancer Res., 55, 629-639.
Meyer, R., B. Malewicz, W.J. Baumann and R.G. Johnson (1990) Increased gap junction assembly between cultured cells upon cholesterol supplementation, J. Cell. Sci., 96, 231-238.
Meyer, R.A., D.W. Laird, J.P. Revel and R.G. Johnson (1992) Inhibition of gap junction and adherens junction assembly by connexin and A-CAM antibodies, J. Cell Biol., 119, 179-189.
Mironov, N., A.-M. Aguelon. G.I. Potapova, Y. Omori et al. (1994) Alterations of (CA)n DNA repeats and tumor suppres- sor genes in human gastric cancer, Cancer Res., 54, 41-44.
Moreno, A.P., G.I. Fishman and D.C. Spray (1992) Phosphoryla- tion shifts unitary conductance and modifies voltage depen- dent kinetics of human connexin 43 gap junction channels, Biophys. J., 62, 51-53.
Murray, A.W. and D.J. Fitzgerald (1979) Tumor promoters inhibit metabolic cooperation in cocultures of epidermal and 3T3 cells, Biochem. Biophys. Res. Commun., 91, 395-401.
Musil, L.S. and D.A. Goodenough (1995) Biochemical analysis of connexon assembly. In: Y. Kanno et al. (Eds.), Intercellular Communication Through Gap Junctions, Prog. Cell Res.. 4, 327-330.
Naus, C., K. Elisevich, D. Zhu, D. Belliveau and R. Del Maestro
(1992) In vivo growth of C6 glioma cells transfected with connexin 43 cDNA, Cancer Res., 52, 4208-4213.
Neuhaus, I.M., G. Dahl and R. Werner (1995) Use of alternate promoters for tissue-specific expression of the gene coding for connexin 32, Gene, 158, 257-262.
Neveu, M.J., C.A. Sattler, G.L. Sattler, J.R. Hully et al. (1994) Differences in the expression of connexin genes in rat hep- atomas in vivo and in vitro, Mol. Carcinogen., 11, 145-154.
Oelze, I., J. Kartenbeck, K. Crusius and A. Alonso (1995) Human papillomavirus type 16 E5 protein affects cell-cell communi- cation in an epithelial cell line, J. Virol., 69, 4489-4494.
Oyamada, M., V. Krutovskikh, M. Mesnil, C. Partensky et al. (1990) Aberrant expression of gap junction gene in primary human hepatocellular carcinomas: increased expression of car- diac-type gap junction gene connexin 43, Mol. Carcinogen., 3, 273-278.
Oyamada, Y., M. Oyamada, A. Fusco and H. Yamasaki (1994) Aberrant expression, function and localization of connexins in human esophageal carcinoma cell lines with different degrees of tumorigenicity, J. Cancer Res. Clin. Oncol., 120, 445-453.
Pelletier, D.B. and A.L. Boynton (1994) Dissociation of PDGF receptor tyrosine kinase activity from PDGF-mediated inhib- tion of gap junctional communication, J. Cell. Physiol,, 158, 427-434.
Pitts, J.D. and M.E. Finbow (1986) The gap junction, J. Cell Sci. (Suppl.), 4, 239-266.
Rivedal, E., H. Yamasaki and T. Sanner (1994) Inhibition of gap junctional intercellular communication in Syrian hamster em- bryo cells by TPA, retinoic acid and DDT, Carcinogenesis, 15, 689-694.
Rose, B., P.P. Mehta and W.R. Loewenstein (1993) Gap-junction protein gene suppresses tumorigenicity, Carcinogenesis, 14, 1073-1075.
Rosen, A., P.A. van der Merwe and J.S. Davidson (1988) Effects of SV40 transformation on intercellular gap junctional com- munication in human fibroblasts, Cancer Res., 48, 3485-3489.
Rosen, B., P.P. Mehta and W.R. Loewenstein (1993) Gap-junction protein gene suppresses tumorigenicity, Carcinogenesis, 14, t 073-1075.
Saez, J.C., J.A. Conner, D.C. Spray and M.V.L. Bennett (1989) Hepatocyte gap junctions are permeable to a second messen- ger, inositol 1,4,5-triphosphate and to calcium ions, Proc. Natl. Acad. Sci. USA, 86, 2708-2712.
Sakamoto, H., M. Oyamada, K. Enomoto and M. Mori (1992) Differential changes in expression of gap junction proteins connexin 26 and 32 during hepatocarcinogenesis in rats, Jap. J. Cancer Res., 83, 1210-1215.
Sigler, K. and R.J. Ruch (1993) Enhancement of gap junctional intercellular communication in tumor promoter-treated cells by components of green tea, Cancer Lett., 69, 15-19.
Spray, D.C., M. Fujita, J.C. Saez, T. Watanabe et al. (1987) Proteoglycans and glycosaminoglycans induce gap junction synthesis and function in primary liver cultures, J. Cell. Biol., 105, 541-551.
Stahl, W., S. Nicolai, M. Hanusch and H. Sies (1994) Vitamin D influences gap junctional communication in C3H/10T 1/2 murine fibroblast cells, FEBS Lett., 352, 1-3.
Swierenga, S.H.H. and H. Yamasaki (1992) Performance of tests

H. Yamasaki /Mutation Research 365 (1996) 91-105 105
for cell transformation and gap-junction intercellular commu- nication for detecting nongenotoxic carcinogenic activity, In: H. Vainio, P. Magee, D.B. McGregor and A.J. McMichael (Eds.), Mechanisms of Carcinogenesis in Risk Identification, Lyon, IARC, pp. 165-193.
Swierenga, S.H.H., D.J. Fitzgerald, H. Yamasaki, C. Piccoli and M. Goldberg (1991) Use of primary keratinocyte cultures from plucked human hairs for analysis of gap junctional intercellu- lar communication. Toxicol. In Vitro, 5, 411-418.
Takahashi, C., N. Kikuchi, N. Katou, T. Miki et al. (1995) Possible anti-tumor-promoting activity of components in Japanese soybean fermented food, Natto: effect on gap junc- tional intercellular communication, Carcinogenesis, 16, 471- 473.
Tateno, C., S. Ito, M. Tanaka, M. Oyamada and A. Yoshitake (1994) Effect of DDT on hepatic gap junctional intercellular communication in rats, Carcinogenesis, 15, 517-521.
Trosko, J.E. and C.C. Chang (1988) Nongenotoxic mechanisms in carcinogenesis: role of inhibited intercellular communication, Banbury Rep., 31, 139-170.
Trosko, J.E., L.P. Yotti, S.T. Warren et al. (1982) Inhibition of cell-cell communication by tumor promoters. In: E. Hecker et al. (Eds.), Carcinogenesis: Cocarcinogenesis and Biological Effects of Tumor Promoters, Vol. 7, New York, Raven Press, pp. 565-585.
Ubeda, A., M.A. Trillo, D.E. House and C.F. Blackman (1995) Melatonin enhances junctional transfer in normal C3H/10T1/2 cells, Cancer Lett., 91, 241-245.
van Zoelen, E.J. and L.G. Tertoolen (1991) Transforming growth factor-beta enhances the extent of intercellular communication between normal rat kidney ceils, J. Biol. Chem., 266, 12705- 12781.
Wilgenbus, K.K., C.J. Kirkpatrick, R. Knuechel, K. Willecke and O. Traub (1992) Expression of Cx26, Cx32 and Cx43 gap junction proteins in normal and neoplastic human tissues, Int. J. Cancer, 51,522-529.
Yamasaki, H. (1990) Gap junctional intercellular communication and carcinogenesis, Carcinogenesis, 11, 1051-1058.
Yamasaki, H. (1995) Nongenotoxic mechanisms of carcino- genesis: studies of cell transformation and gap junctional intercellular communication, Toxicol. Lett., 77, 55-61.
Yamasaki, H. and F. Katoh (1988) Further evidence for the involvement of gap-junctional intercellular communication in induction and maintenance of transformed loci in BALB/c 3T3 cells, Cancer Res., 48, 3490-3495.
Yamasaki, H., V. Krutovskikh, M. Mesnil, A. Columbano et al. (1993) Gap junctional intercellular communication and cell proliferation during rat liver carcinogenesis, Environ. Health Perspect., 101 (Suppl. 5), 191-198.
Yamasaki, H., M. Mesnil, Y. Omori, N. Mironov and V. Kru- tovskikh (1995a) Aberrant control of connexin expression and functions in multistage rat and human hepatocarcinogenesis, Prog. Cell Res., 4, 79-82.
Yamasaki, H., M. Mesnil, Y. Omori, N. Mironov and V. Kru- tovskikh (1995b) Intercellular communication and carcino- genesis, Mutation Res., 333, 181-188.
Yamasaki, H., J. Ashby, M. Bignami, W. Jongen et al. (1996) Nongenotoxic carcinogens: development of detection methods based on mechanisms - a European project, Mutation Res. (in press).
Yotti, L.P., C.C. Chang and J.E. Trosko (1979) Elimination of metabolic cooperation in Chinese hamster cells by a tumor promoter, Science, 206, 1089-1091.
Yu, W., G. Dahl and R. Werner (1994) The connexin 43 gene is responsive to oestrogen, Proc. R. Soc. Lond. B. Biol. Sci., 255, 125-132.
Zampighi, G. and P.N.T. Unwin (1979) Two forms of isolated gap junctions, J. Mol. Biol., 135, 451-464.
Zhang, L.X., R,V. Cooney and J.S. Bertram (1992) Carotenoids up-regulate connexin 43 gene expression independent of their provitamin A or antioxidant properties, Cancer Res., 52, 5707-5712.
Zhu, D., S. Caveney, G.M. Kidder and C.C.G. Naus (1991) Transfection of C6 glioma cells with connexin 43 cDNA: analysis of expression, intercellular coupling and cell prolifera- tion, Proc. Natl. Acad. Sci. USA, 88, 1883-1887.