risk regulation in the single market: the governance of pharmaceuticals and foodstuffs in the...
TRANSCRIPT

Risk Regulation in the Single Market
The Governance of Pharmaceuticals andFoodstuffs in the European Union
Sebastian Krapohl

Palgrave Studies in European Union Politics
Edited by: Michelle Egan, American University USA, Neill Nugent, ManchesterMetropolitan University, UK, William Paterson, University of Birmingham, UK
Editorial Board: Christopher Hill, Cambridge, UK, Simon Hix, London School of Economics, UK, Mark Pollack, Temple University, USA, Kalypso Nicolaïdis,Oxford UK, Morten Egeberg, University of Oslo, Norway, Amy Verdun, Universityof Victoria, Canada, Claudio M. Radaelli, University of Exeter, UK, FrankSchimmelfennig, Swiss Federal Institute of Technology, Switzerland
Following on the sustained success of the acclaimed European Union Series, whichessentially publishes research-based textbooks, Palgrave Studies in European UnionPolitics publishes cutting edge research-driven monographs.
The remit of the series is broadly defined, both in terms of subject and academicdiscipline. All topics of significance concerning the nature and operation of theEuropean Union potentially fall within the scope of the series. The series is multidisciplinary to reflect the growing importance of the EU as a political, economic and social phenomenon. We will welcome submissions from the areas ofpolitical studies, international relations, political economy, public and social policy,economics, law and sociology.
Submissions should be sent to Amy Lankester-Owen, Politics Publisher, ‘[email protected]’.
Titles include:
Ian Bache and Andrew Jordan (editors)THE EUROPEANIZATION OF BRITISH POLITICS
Richard Balme and Brian Bridges (editors)EUROPE-ASIA RELATIONSBuilding Multilateralisms
Derek Beach and Colette Mazzucelli (editors)LEADERSHIP IN THE BIG BANGS OF EUROPEAN INTEGRATION
Milena BüchsNEW GOVERNANCE IN EUROPEAN SOCIAL POLICYThe Open Method of Coordination
Dario Castiglione, Justus Schönlau, Chris Longman, Emanuela Lombardo, NievesPérez-Solórzano Borragán and Mirim AzizCONSTITUTIONAL POLITICS IN THE EUROPEAN UNIONThe Convention Moment and its Aftermath
Morten Egeberg (editor)MULTILEVEL UNION ADMINISTRATIONThe Transformation of Executive Politics in Europe
Kevin Featherstone and Dimitris PapadimitriouTHE LIMITS OF EUROPEANIZATIONReform Capacity and Policy Conflict in Greece
Stefan Gänzle and Allen G. Sens (editors)THE CHANGING POLITICS OF EUROPEAN SECURITYEurope Alone?
Isabelle GarzonREFORMING THE COMMON AGRICULTURAL POLICYHistory of a Paradigm Change
PPL-UK_RR-Krapohl_FM.qxd 9/23/2008 3:52 PM Page i

Heather GrabbeTHE EU’S TRANSFORMATIVE POWER
Katie Verlin Laatikainen and Karen E. Smith (editors)THE EUROPEAN UNION AND THE UNITED NATIONSIntersecting Multilateralisms
Esra LaGro and Knud Erik Jørgensen (editors)TURKEY AND THE EUROPEAN UNIONProspects for a Difficult Encounter
Paul G.Lewis and Zdenka Mansfeldová (editors)THE EUROPEAN UNION AND PARTY POLITICS IN CENTRAL AND EASTERN EUROPE
Ingo Linsenmann, Christoph O. Meyer and Wolfgang T. Wessels (editors)ECONOMIC GOVERNMENT OF THE EUA Balance Sheet of New Modes of Policy Coordination
Hartmut Mayer and Henri Vogt (editors)A RESPONSIBLE EUROPE?Ethical Foundations of EU External Affairs
Lauren M. McLarenIDENTITY, INTERESTS AND ATTITUDES TO EUROPEAN INTEGRATION
Christoph O. Meyer, Ingo Linsenmann and Wolfgang Wessels (editors)ECONOMIC GOVERNMENT OF THE EUA Balance Sheet of New Modes of Policy Coordination
Philomena Murray (editor)EUROPE AND ASIARegions in Flux
Daniel Naurin and Helen Wallace (editors)UNVEILING THE COUNCIL OF THE EUROPEAN UNIONGames Governments Play in Brussels
Frank Schimmelfennig, Stefan Engert and Heiko KnobelINTERNATIONAL SOCIALIZATION IN EUROPEEuropean Organizations, Political Conditionality and Democratic Change
Justus SchönlauDRAFTING THE EU CHARTER
Angelos SeposTHE EUROPEANIZATION OF CYPRUSPolity, Policies and Politics
Marc Weller, Denika Blacklock and Katherine Nobbs (editors)THE PROTECTION OF THE MINORITIES IN THE WIDER EUROPE
Palgrave Studies in European Union PoliticsSeries Standing Order ISBN 978-1-4039-9511-7 (hardback) and ISBN 978-1-4039-9512-4 (paperback)
You can receive future titles in this series as they are published by placing a standingorder. Please contact your bookseller or, in case of difficulty, write to us at theaddress below with your name and address, the title of the series and one of theISBNs quoted above.
Customer Services Department, Macmillan Distribution Ltd, Houndmills,Basingstoke, Hampshire RG21 6XS, England
PPL-UK_RR-Krapohl_FM.qxd 9/23/2008 3:52 PM Page ii

Risk Regulation in theSingle MarketThe Governance of Pharmaceuticals andFoodstuffs in the European Union
Sebastian KrapohlAssistant Professor of International Relations, Otto-Friedrich-University of Bamberg,Germany
PPL-UK_RR-Krapohl_FM.qxd 9/23/2008 3:52 PM Page iii

© Sebastian Krapohl 2008
All rights reserved. No reproduction, copy or transmission of thispublication may be made without written permission.
No portion of this publication may be reproduced, copied or transmittedsave with written permission or in accordance with the provisions of theCopyright, Designs and Patents Act 1988, or under the terms of any licencepermitting limited copying issued by the Copyright Licensing Agency,Saffron House, 6–10 Kirby Street, London EC1N 8TS.
Any person who does any unauthorized act in relation to this publicationmay be liable to criminal prosecution and civil claims for damages.
The author has asserted his right to be identified as the author of thiswork in accordance with the Copyright, Designs and Patents Act 1988.
First published 2008 byPALGRAVE MACMILLAN
Palgrave Macmillan in the UK is an imprint of Macmillan Publishers Limited,registered in England, company number 785998, of Houndmills, Basingstoke,Hampshire RG21 6XS.
Palgrave Macmillan in the US is a division of St Martin’s Press LLC,175 Fifth Avenue, New York, NY 10010.
Palgrave Macmillan is the global academic imprint of the above companiesand has companies and representatives throughout the world.
Palgrave® and Macmillan® are registered trademarks in the United States,the United Kingdom, Europe and other countries.
ISBN-13: 978-0-230-53765-1 hardbackISBN-10: 0-230-53765-0 hardback
This book is printed on paper suitable for recycling and made from fullymanaged and sustained forest sources. Logging, pulping and manufacturingprocesses are expected to conform to the environmental regulations of thecountry of origin.
A catalogue record for this book is available from the British Library.
A catalog record for this book is available from the Library of Congress.
10 9 8 7 6 5 4 3 2 117 16 15 14 13 12 11 10 09 08
Printed and bound in Great Britain byCPI Antony Rowe, Chippenham and Eastbourne
PPL-UK_RR-Krapohl_FM.qxd 9/23/2008 3:52 PM Page iv

For Sarah and Lauramay they always have safe food and medicine
PPL-UK_RR-Krapohl_FM.qxd 9/23/2008 3:52 PM Page v

This page intentionally left blank

Contents
List of Figures and Tables ix
Preface x
List of Abbreviations xiii
1 Introduction: The Need for a Systematic Analysis of `Supranational Risk Regulation 1
Part I An Institutionalist Approach to Supranational Risk Regulation
2 Functional Pressure and Path-Dependencies: The Emergence and Development of Supranational Regulatory Regimes 17
3 Efficiency and Legitimacy: The Evaluation of SupranationalRegulatory Regimes 33
Part II The Authorisation of Pharmaceuticals in the EU
4 From National Crises to a Strong Supranational Regime: The Development of Pharmaceutical Authorisation in Europe 61
5 A Strong Regulatory Network: The Evaluation of the European Regulatory Regime for Pharmaceuticals 86
Part III The Regulation of Foodstuffs in the EU
6 From an Early Single Market to a Crisis of Consumer Confidence: The Development of Foodstuff Regulation in Europe 121
7 A Weak Supranational Agency: The Evaluation of the European Regulatory Regime for Foodstuffs 151
vii
PPL-UK_RR-Krapohl_FM.qxd 9/23/2008 3:52 PM Page vii

Part IV Conclusion
8 A Comparison of Pharmaceutical and Foodstuff Regulation in Europe 183
Notes 195
Bibliography 208
Index 221
viii Contents
PPL-UK_RR-Krapohl_FM.qxd 9/23/2008 3:52 PM Page viii

Figures and Tables
Figure 2.1 Product regulation in the Single Market 19Figure 2.2 Path-dependencies of supranational regulatory
regimes 28Figure 5.1 Multi-state and concertation procedures 88Figure 5.2 Centralised authorisation procedure 92Figure 5.3 Mutual recognition and decentralised authorisation
procedures 103Figure 7.1 The committee system in the foodstuff sector 153Figure 7.2 Regulatory policy-making in the foodstuff sector
after 2002 159Figure 7.3 Authorisation of genetically modified food 165
Table 5.1 Evaluation of the two authorisation procedures in 2000 101
Table 5.2 Activities within the comitology procedure from 1995 to 2001 102
Table 5.3 Activities within the mutual recognition procedure from 1995 to 2000 106
ix
PPL-UK_RR-Krapohl_FM.qxd 9/23/2008 3:52 PM Page ix

x
Preface
The decision to conduct this research project on Risk Regulation in theSingle Market was certainly not the result of any single event, but theconsequence of a range of favourable circumstances. Nevertheless, therewas one starting point, when I began to think about health and con-sumer protection in the European Union. In the Fall of 2000 – when Ihad just started my studies at the London School of Economics with theaim of earning a Master’s degree – the BSE disease was spreading all overEurope. Before, BSE had mainly been regarded as a British problem, butsuddenly BSE cases were also found in other EU member states, includingGermany. The result was a huge public scandal: the German Ministers forHealth and Agriculture had to leave offices, and although Beef disap-peared from the Christmas menu, it was nevertheless present in people’sminds and the daily news. As a student of EU politics, I began to askmyself why the EU had not been able to protect its citizens from the riskof contaminated beef, when it was at the same time responsible forensuring the free trade of such goods across the Single Market. Mythinking about the topic has always had an analytic and a normativedimension: Why did this scandal occur and how could such problemsbe avoided in future? Besides its academic character, this book cannotand shall not deny its normative connotation. It aims not only tounderstand EU risk regulation as it is, but also to search for mechanismswhich could improve the functioning thereof.
This book is developed from my PhD thesis, which was written at the University of Bamberg, Germany. I thank the German ResearchFoundation for funding the research project ‘Rationality by Procedures’,wherein I was employed for two years. The case study on the efficiencyof pharmaceutical regulation in the EU is one result of this project.Additionally, I am indebted to the Friedrich-Ebert-Foundation which sup-ported my postgraduate studies with a generous grant. This grant allowedme to visit the European University Institute in Florence in 2004/05 andto finish the work on my PhD thesis in the years following.
Of course, this book could not have been written without the supportof many colleagues and friends, as well as my family. Foremost, I wouldlike to thank my supervisor, Prof. Dr. Thomas Gehring. In Germany, weuse the somewhat old-fashioned term ‘Doktorvater’, which can onlyimperfectly be translated into the English language. A ‘Doktorvater’ is
PPL-UK_RR-Krapohl_FM.qxd 9/23/2008 3:52 PM Page x

much more than a supervisor. He is a paternal friend, who is alwaysapproachable, and who helps with his experience whenever problemsappear. To me, Thomas Gehring has always been such a ‘Doktorvater’ inthe truest sense of the word. Without his help and encouragement, thisbook would not exist.
I had the chance to develop and discuss my own work within theResearch Programme ‘Markets and Social Systems’ at the University ofBamberg. This book profited a great deal from this interdisciplinaryenvironment. I am especially thankful to Prof. Dr. Hans W. Micklitz,who is Professor for Private Law, and to Prof. Dr. Richard Münch, whoholds a Chair for Sociological Theory. Both have supervised my work inrecent years, and their comments from perspectives outside of PoliticalScience were challenging – but also extremely helpful.
Large parts of this book are inspired by a debate between twofamous scholars whom I was lucky enough to meet personally. Prof. Dr. Giandomenico Majone strongly influenced my view on regulatorypolicy-making. I am thankful that he commented on two of my papersat a workshop in Bamberg and during my half-year visit at the EuropeanUniversity Institute in Florence. Prof. Dr. Christian Joerges providedconvincing arguments that the member states cannot be left aside insupranational risk regulation and that regulatory independence is there-fore not an option. I am very indebted to him for making my visit inFlorence possible and for becoming my supervisor during this half-year.
I thank numerous colleagues from the Chair for International Relationsat the University of Bamberg and from the Law Department at theEuropean University Institute in Florence. Prof. Dr. Sebastian Oberthürhas been an extremely helpful friend, who always encouraged mywork. I am especially thankful that he proofread the final manuscriptbefore publication. It has always been a pleasure to work with MichaelKerler, and I thank him for proofreading some chapters of this book.The same is true for Simon Fink, whose comments I always appreci-ate. Besides, I owe many thanks to Karolina Zurek, who had enoughpatience to write an article with me, and who also proofread somechapters.
Lastly, I thank my family who helped and encouraged me during thework on this book. Most important for me was – and is – the love of mywife Laura Brander. I apologise for all my bad moods when I was trappedin the underworld of EU committees, and I thank her most of all for stillloving me after that. Finally, I thank Sarah Brander for reminding herfather that there are things in life that are more important than a PhD thesis and a book publication, and that he has to hurry up a bit
Preface xi
PPL-UK_RR-Krapohl_FM.qxd 9/23/2008 3:52 PM Page xi

after five years of research. This book is dedicated to her and hermother, and I hope that they will always have safe food and medicinethroughout a long, happy and healthy life.
Bamberg, January 2008
xii Preface
PPL-UK_RR-Krapohl_FM.qxd 9/23/2008 3:52 PM Page xii
Abbreviations
AFSSAPS Agence Française de Sécurité Sanitaire des Produits deSanté (Agency for the Safety of Sanitary Products; France)
BfArM Bundesinstitut für Arzneimittel und Medizinprodukte(Federal Institute for Pharmaceuticals and MedicinalProducts; Germany)
BGA Bundesgesundheitsamt (Federal Health Office; Germany)BSE Bovine Spongiform EncephalopathyCHMP Committee for Human Medicinal Products (after 2004)COMP Committee for Orphan Medicinal ProductsCPMP Committee for Proprietary Medicinal Products (before
2004)DG Directorate-GeneralECB European Central BankECJ European Court of JusticeEFSA European Food Safety AuthorityEMEA before 2004: European Agency for the Evaluation of
Medicinal Products; after 2004: European MedicinesAgency
EP European ParliamentEU European UnionFDA Food and Drug Administration (United States of America)FVO Food and Veterinary OfficeGM food Genetically Modified FoodGMO(s) Genetically Modified Organism(s)HMPC Committee for Herbal Medicinal ProductsICH International Conference on Harmonisation of Technical
Requirements for Registration of Pharmaceuticals forHuman Use
MCA Medicines Control Agency (United Kingdom)PEI Paul-Ehrlich-Institute (Germany)TSE Transmissible Spongiform EncephalopathyUK United Kingdom of Great Britain and Northern IrelandUSA United States of AmericavCJD Variant Creutzfeld-Jakob DiseaseWTO World Trade Organization
xiii
PPL-UK_RR-Krapohl_FM.qxd 9/23/2008 3:52 PM Page xiii

This page intentionally left blank

1Introduction: The Need for aSystematic Analysis ofSupranational Risk Regulation
During the past ten years, health and safety regulation of productswithin the European Single Market1 has developed from a technocraticissue far beyond public attention to a high-priority topic on the politi-cal agenda. This increasing importance is due, among other things, tothe regulatory problems that shattered the European Union (EU)2 dur-ing the 1990s. Firstly, the handling of bovine spongiform encephalopa-thy (BSE) by EU institutions damaged people’s trust in the regulatorycapacities at the EU level (Ansell and Vogel 2006; Majone 2000; Vogel2001a; Vos 2004). Until 1996, the EU regulatory bodies seriously under-estimated the risk that BSE constituted for the health of consumers.Consequently, their regulatory measures were not strict enough to pro-tect the health of European citizens or to prevent the spread of the dis-ease all over Europe. Secondly, the increasing importation of geneticallymodified organisms (GMOs) and food (GM food) from the USA metwith strong resistance from the already alienated European consumers(Vogel 2001b). In reaction to these concerns, the EU member statesimposed a de facto moratorium on the authorisation of these products,and hazarded a trade war with the USA. At first view, the EU institutionshad no answers to the BSE and GMO/GM food problems. The spread ofBSE could not be prevented, nor could a sustainable policy on GMOsand GM food be adopted.
However, at second view, the picture is much more complex. Not onlydo the BSE and biotechnology problems affect the area of foodstuff reg-ulation, but also that of pharmaceutical authorisation. Not only beef,but also vaccines, which are produced with bovine sera, can transmitBSE to human beings. Moreover, not only food, but also pharmaceuti-cals, might be genetically modified or derived from GMOs. However,whereas both problems led to a crisis of consumer confidence in the
1
PPL-UK_RR-Krapohl_Ch01.qxd 8/29/2008 11:53 AM Page 1

foodstuff sector, pharmaceutical regulation was much less under attack.Besides, the EU regulatory institutions differ widely between these twoproduct sectors. Since 1993, an EU agency (the European MedicinesAgency; EMEA) has been responsible for the authorisation of pharma-ceuticals in the Single Market. In contrast, food safety measures wereadopted within the EU committee system3 with the participation of sev-eral expert and member state committees. Only after the BSE scandalwas the EU committee system in the foodstuff sector abolished and thenew European Food Safety Authority (EFSA) established. But its successhas yet to be proven. Overall, the regulatory institutions at the EU levelare not uniform, but differ between regulatory areas and across time.
Several questions arise from the variety of regulatory institutions inthe EU for a political-science analysis. Why did different supranationalregulatory institutions for products traded on the Single Market evolve?Are some regulatory institutions more efficient than others, and, if so,why? What are the factors that determine their democratic legitimacyand their acceptance by EU citizens? To answer these questions, thisbook first develops a historical-institutionalist approach to suprana-tional risk regulation. In doing so, it identifies the most important vari-ables for the development, efficiency and legitimacy of regulatoryinstitutions in the EU. Next, this newly developed theory is tested usingtwo important cases of EU product regulation: pharmaceutical authori-sation and foodstuff regulation. The analysis demonstrates that thevarying performance of the EU in pharmaceutical and foodstuff regula-tion can largely be traced back to differences in institutions and theirhistorical development.
1.1 Theoretical framework: An institutionalist approach to supranational risk regulation
The growing public attention to the regulatory capacities of the EU wentalong with a discussion in political science about the specific features ofEU policy-making. In contrast to traditional welfare states, the EU lacksa strong political centre that might adopt and implement regulatorypolicies. Instead, ‘softer’ forms of governance seem to emerge, wherein avariety of transnational and supranational expert networks replace – orat least complement – old state authorities (Eberlein and Grande 2005;Eberlein and Kerwer 2004). The oldest and probably most well-knowntheory for such network governance is the concept of deliberative supra-nationalism, conceived by Joerges and Neyer (1997a, 1997b, 2006).These authors analysed the EU committee system in detail (e.g. Joerges
2 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch01.qxd 8/29/2008 11:53 AM Page 2

and Falke 2000; Joerges and Vos 1999), and came to the conclusion thatit is the most appropriate answer to the problems of regulatory policy-making within the EU. On the one hand, EU policy-making shouldalways take the legitimate interests of member states into account.However, on the other, intergovernmental bargaining should not pre-vent the search for appropriate regulatory solutions. According to theseauthors, the committee system should be able to solve this tension.Within different committees at the EU level, experts and representativesof member states meet repeatedly to adopt regulatory decisions. Joergesand Neyer claim that these repeated interactions lead to the emergenceof trust, common norms and belief systems. As a result, power-based bar-gaining can be left behind and ‘deliberative problem-solving’ emerges.Member states’ interests are represented within decision-making, butthey are simultaneously ‘civilised’.
Probably the most prominent opponent of Joerges and Neyer in the academic discussion about regulatory policy-making in the EU isMajone. He states that regulatory policy-making is always in danger ofbecoming influenced by political interests (Majone 1996: 28–46). This is due to the short time horizon of politicians, who face pressures tobecome re-elected. The capture of regulatory policy-making by politicalinterests would result in the adoption of regulatory decisions, whichwould only reflect distributional conflicts and compromises, but whichwould not properly address the regulatory problem. To prevent capture,regulatory policy-making should be conducted by independent bodiesat arm’s length from direct political influence (Majone 1996: 61–79,2001a). Consequently, much of Majone’s work focuses not on the EUcommittee system, but on regulatory agencies (Majone 1997, 2002a,2004, 2005: 83–106). However, the major problem for Majone’s theoryis that the agencies at EU level are not at all independent of politicalinfluence (e.g. Chiti 2000; Gardner 1996; Geradin and Petit 2004;Kreher 1997). At least formally, the EU agencies are only advisory bod-ies which are strictly controlled by the European Commission and themember states. Thus, they resemble inflated scientific committees ratherthan real independent regulators like the American Food and DrugAdministration (FDA). It still seems difficult to imagine that the memberstates delegate so much competence to establish truly independent EUagencies (Majone 2002a; Vos 2000a).
Neither of the two existing approaches to supranational risk regulationallows for comparative analyses of different regulatory institutions withindifferent policy areas at different points in time. Whereas Joerges’ subjectis the EU committee system, Majone concentrates on supranational
Introduction 3
PPL-UK_RR-Krapohl_Ch01.qxd 8/29/2008 11:53 AM Page 3

regulatory agencies. Both approaches are not designed to compare thecommittee system with regulatory agencies. Consequently, a broadertheoretical framework is needed for a comparative analysis. Therefore,the following analysis applies the concept of ‘regulatory regimes’ (e.g.Francis 1993; Hood et al. 2001; Randall 2006). At such an abstract level,the EU committee system and supranational regulatory agencies arecomparable. It is not important whether the committee system is appliedin certain regulatory areas or whether supranational agencies are estab-lished, because both can be functional equivalents within supranationalregulatory regimes.
1.1.1 Path-dependent development of supranational regulatory regimes
The rationale for the emergence of supranational regulatory regimes inmany product sectors is the Single Market.4 Different national productstandards, which act as non-tariff trade barriers, have to be abolished toestablish the Single Market. One possibility for this is the mutual recog-nition principle, according to which every member state has to accepton its market products that fulfil the regulatory standards of at least oneother member state. However, this could lead to a lowering of productstandards in many member states if the standards of the member statewith the lowest level of health and consumer protection prevail (Scharpf1999: 84–129). Consequently, the member states do not fully committhemselves to this principle, but allow each other to deviate from it for reasons of health and consumer protection. A second possibility toachieve a single market also for risky products is the harmonisation ofproduct standards at the EU level within the usual legislative proce-dures. However, here the problem is that the member states face coor-dination problems and a negotiators’ dilemma within the Council ofthe EU (Gehring 1999; Scharpf 1997a: 116–50). They have to find appro-priate regulatory measures, and have to bargain about the distributiveconsequences of these measures at the same time. The result may be ablockade of decision-making or an inefficient political compromise. Inthe end, functional pressure emerges for the member states to establishsupranational regulatory regimes in which they delegate some aspectsof regulatory policy-making to the Commission and to scientific bodieslike expert committees or regulatory agencies. Now, the member statesno longer decide about every single standard, but only about the moregeneral substantive and procedural rules of decision-making (Gehring2002). The final decisions are then adopted within supranational regula-tory regimes on the basis of these rules. However, functional pressure
4 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch01.qxd 8/29/2008 11:53 AM Page 4

towards the establishment of supranational regulatory regimes cannotexplain the differences between different regimes in different regulatoryareas at different points in time. Functional pressure is a more or lessconstant factor, and thus cannot explain differences.
A historical-institutionalist analysis is able to provide hypotheses aboutthe differences in the institutional design of supranational regulatoryregimes (e.g. Bulmer 1998; Lindner and Rittberger 2003; Stacey andRittberger 2003). The development of supranational regulatory regimesis characterised by critical junctures and path-dependencies. The two rel-evant critical junctures are regulatory scandals, which may result in theset up of regulatory agencies, and the establishment of the Single Market,which required abolishing non-tariff barriers of trade. Both critical junc-tures lead to the establishment of institutions that develop some persist-ence, and which change only within certain paths. If path-dependenciesmatter, sequencing becomes important (Mahoney 2000), and the ques-tion is which critical juncture was first and which second. Two differentpaths towards supranational regulatory regimes can be distinguished,and the hypothesis is that these paths lead to different designs of supra-national regulatory regimes (Krapohl 2007). If regulatory scandals withincertain policy areas emerge before a single market is set up, this will mostlikely lead to the establishment of national regulatory agencies in therespective areas. These national agencies are new actors with their owninterests, and they have resources to push them. When a single marketis created later, the national regulatory agencies are stakeholders of insti-tutional change, and can influence further developments. The creationof a single market probably proves more difficult and takes more time inpolicy areas where strong national agencies exist, because these mayestablish non-tariff trade barriers. However, once a single market is setup, the national agencies are likely to influence the institutional designof the supranational regulatory regimes, because they push their ownparticipation within the regimes. The results are networks of nationalregulatory agencies. In contrast, if no national regulatory agencies exist,the regimes built up at the supranational level cannot fall back on thesupport of national regulatory agencies. Member states are probably morehesitant to delegate far-reaching competencies in such circumstances,because the newly established EU bodies cannot be controlled by theirown agencies.
1.1.2 Efficiency and legitimacy of supranational risk regulation
To evaluate the different regulatory regimes that follow from such devel-opmental paths, it is first necessary to find a benchmark for appropriate
Introduction 5
PPL-UK_RR-Krapohl_Ch01.qxd 8/29/2008 11:53 AM Page 5

risk regulation. If one cannot identify objectively – or at least inter-subjectively shared – best regulatory solutions, it is not possible to eval-uate the efficiency of regulatory regimes, because the regulatory policyoutcome would then only be the result of distributive struggles wheresome stakeholders win what other stakeholders lose. Indeed, in the shortterm, distributive interests of consumers and producers in risk regulationare directly opposed to each other (Wilson 1980). Consumers ask for precautionary regulatory standards, whereas producers want regulatorystandards that are as low as possible, in order not to endanger their com-petitiveness on the market. If these short-term interests prevailed, therewould be no shared understanding of better or worse risk regulation.However, there are good reasons to assume that the interests of con-sumers and producers converge in the long term. On the one hand,overly strict, precautionary regulatory standards waste financial resources,which have to be paid in the form of higher prices by consumers, andwhich could have been spent somewhere else to reduce greater risks(Majone 2002b, 2005: 124–37; Wildavsky 1988: 189–204). Thus, con-sumers should have a natural interest in efficient regulatory standards,which lead to the lowest level of risk at given costs. On the other hand,regulatory standards give producers a good reputation for their products(Akerlof 1970; Anania and Nisticò 2004). Thus, producers might chooseto accept efficient regulatory standards to prevent or fight crises of con-sumer confidence, and to keep markets stable. To sum up, these con-verging interests of consumers and producers towards the most efficientregulatory standards can be used as a benchmark for appropriate risk regulation.
To establish reliable supranational regulatory regimes, member statesmust not follow the particularistic short-term interests of some stake-holders, but need to commit themselves to their common long-terminterests in an efficient regulation of the Single Market. The hypothesisis that regimes are more successful the more the member states restrainthemselves from representing particularistic short-term interests. Themember states have two different possibilities for binding themselves.Firstly, they may delegate regulatory policy-making to agents that actindependently of direct political influence in the long-term interests oftheir principals (Majone 1994, 1996: 61–79, 2001a). The independenceand strengths of these agents depend on their recruitment and on thepolitical control that is exercised by the member states. Secondly, themember states can bind the regulatory bodies and themselves to sub-stantive criteria of decision-making (Gehring 2002: 155–96). Here, mem-ber states’ commitment depends on the degree of legalisation (i.e. the
6 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch01.qxd 8/29/2008 11:53 AM Page 6

precision and obligation of legal rules, as well as the review of these rulesby independent courts; Abbott et al. 2000).
The need for credible commitment in supranational risk regulationhas consequences for the legitimacy of supranational regulatory regimes.The importance of input legitimacy (i.e. ‘government by the people’;Scharpf 1970, 1999: 6–42, 2004) is necessarily reduced. If democraticallyelected bodies delegate competencies for regulatory decision-making toindependent agents, or if they commit themselves to precise and oblig-atory rules of regulatory decision-making, they limit their ability to reactto the demands of their constituencies. Thus, input legitimacy is neces-sarily restricted to the establishment of supranational regulatory regimes,but it cannot legitimise their day-to-day decision-making. As a result,output legitimacy (i.e. ‘government for the people’; Scharpf 1970, 1999:6–42, 2004) becomes much more important in legitimising the regimes’daily operations. Supranational regulatory regimes can legitimise them-selves with their policy output to the extent that they are successful inestablishing a single market for risky products and providing effectiveregulation of these products. However, to avoid agency drifts, and thuslosses of efficiency and output legitimacy, supranational regulatoryregimes must be accountable to a range of actors. The member states andthe European Parliament (EP) can hold the regime politically account-able, experts from national regulatory authorities may scrutinise theregimes’ decisions scientifically, and the European courts can legallychallenge regulatory decisions on behalf of stakeholders. To avoid thecapture of regimes by one particular group, a range of accountabilitymechanisms can create a system of checks-and-balances. As a result, nosingle group is able to control the regime, but the regime is neverthelessunder control (Majone and Everson 2001; Moe 1987a).
1.2 Case studies: Pharmaceutical and foodstuff regulation in Europe
The institutionalist approach to supranational risk regulation is used tocompare systematically the two policy areas of pharmaceutical andfoodstuff regulation in the EU. The two groups of products are wellsuited for such a comparison, because they share important characteris-tics. Pharmaceuticals and foodstuffs are incorporated by consumers.Consequently, they could both pose enormous risks to consumer health.For both products, information asymmetries between consumers andproducers are large. Usually, consumers cannot infer ingredients or pro-duction methods from the appearance of such products; and if they
Introduction 7
PPL-UK_RR-Krapohl_Ch01.qxd 8/29/2008 11:53 AM Page 7

could, they would not usually be able to evaluate this information ade-quately. Furthermore, both groups of products have to be regulatedagainst similar threats. BSE cannot only be transmitted to humans by theconsumption of contaminated beef, but also by the injection of vaccinesthat are produced with bovine sera. And not only foodstuffs, but alsopharmaceuticals could be genetically modified or produced from GMOs.
Nevertheless, structural differences also exist between the two areas.The market for pharmaceuticals is relatively homogeneous, and onlyfew suppliers compete against each other within an oligopoly (Feick2000a). In contrast, the foodstuff market is extremely heterogeneous,and many suppliers compete against each other (Bernauer and Caduff2006). For these reasons, the possibilities for regulating the safety of thetwo groups of products differ. Because of the homogeneous market,pharmaceuticals can be subject to pre-marketing control. Accordingly,every medicinal product is evaluated according to its safety, efficacy andquality before it receives market access. In contrast, the heterogeneousfoodstuff market makes pre-marketing authorisation extremely difficult.Consequently, most foodstuffs are only subject to post-marketing con-trol in Europe, i.e. regulatory authorities only react, if problems withfoodstuffs are detected.
However, not all foodstuffs share this characteristic. GM food belongsto the area of foodstuff regulation, but it shares important characteris-tics of pharmaceuticals. This makes it an interesting case for compari-son. The market for GM food is also very homogeneous, and only a fewsuppliers compete with each other. Consequently, GM food is – likepharmaceuticals – subject to pre-marketing control. The crucial ques-tion is whether the regulatory institutions for GM food and their devel-opment are more similar to those of pharmaceuticals or to those ofother foodstuffs. If the former proved to be the case, a functionalistexplanation would be supported, and the characteristics of the regu-lated product would determine the regulatory institutions. However, ifthe latter holds true – as is demonstrated in the case studies – the caseof GM food further supports the hypotheses derived from the historical-institutionalist analysis.
1.2.1 The authorisation of pharmaceuticals in the EU
In the case of pharmaceuticals, a regulatory crisis – namely the thalido-mide scandal (Kirk 1999) – marked the starting point for the develop-ment of regulatory institutions in Europe in the 1960s, long before asingle market for these products was created.5 In the next 15 years,compulsory authorisation for pharmaceuticals was introduced, and
8 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch01.qxd 8/29/2008 11:53 AM Page 8

independent regulatory authorities were set up in most EU member states.At the same time, the EU tried to establish a single market for pharma-ceuticals by partial harmonisation of authorisation criteria and mutualrecognition of national authorisations. Later, this partial harmonisationand mutual recognition was further supported by the establishment of dif-ferent expert and comitology committees. Nevertheless, the mutual recog-nition principle failed to produce a single market for medicinal products,because national regulatory authorities did not accept authorisations ofother member states (Feick 2000b; Vos 1999a: 210; Hart and Reich 1990).Market integration became only possible in the mid-1990s owing to a centralisation of the authorisation regime under the responsibility of the newly established European Agency for the Evaluation of MedicinalProducts (later renamed the European Medicines Agency; EMEA). Withinthe agency, a regulatory network of member states’ regulatory bodiesdecides about the authorisation of pharmaceuticals. Even though theagency is formally not independent – its opinions are subject to politicalscrutiny within a comitology procedure – it soon gained an extremelystrong position in its relationships with the Commission and the gov-ernments of the member states (Gehring and Krapohl 2007; Krapohl2004, 2005: 105–31). So far, not a single opinion of the agency has beenchanged or rejected by the Commission or the member states in thecomitology procedure.
Currently, two different authorisation procedures are applied in theEU for highly innovative medicinal products and less innovative prod-ucts. At least in the case of highly innovative medicinal products –which include all biotechnologically produced pharmaceuticals – theestablishment of EMEA and the centralisation of the authorisation pro-cedure can be seen as a success. The procedural and substantive rules forthe authorisation of pharmaceuticals express a rather deep commitmentof the member states to achieving the two goals of market integrationand effective risk regulation simultaneously (Gehring and Krapohl 2007;Krapohl 2004, 2005). As a result, the centralised authorisation proce-dure managed to create a single market for highly innovative medicinalproducts, and, so far, it has not experienced regulatory crises and scan-dals. In a survey conducted for the Commission, consumers and pro-ducers proved to be satisfied with the current regulatory regime, even ifproducers’ interests are somewhat favoured because of their betteraccess to the regime (Feick 2002a; Lewis and Abraham 2001).
The situation is slightly different for less innovative pharmaceuticals –including the large group of generics. These are subject to a mutualrecognition procedure which is – as the name suggests – still built upon
Introduction 9
PPL-UK_RR-Krapohl_Ch01.qxd 8/29/2008 11:53 AM Page 9

mutual recognition of member states’ authorisation. Until 2004, themutual recognition procedure seemed to be somewhat less efficient thanthe centralised one, because pharmaceutical companies often withdrewapplications from opposing member states to prevent centralised arbi-trations. In this way, centralised evaluations of safety concerns and uni-form market access were prevented. After an evaluation conducted onbehalf of the Commission, the regulatory regime for pharmaceuticalswas recently subject to a revision process, and centralised arbitrationshave become compulsory whenever at least one member state raisesobjections against authorisations. Nevertheless, the scope of the decen-tralised authorisation procedure became reduced and that of the cen-tralised procedure extended (Broscheid and Feick 2005; Feick 2005).
At first view, the legitimacy of EU pharmaceutical authorisation seemsto be very low. The creation of the supranational regulatory regime wasmainly in the interest of producers, whereas the consumer voice was notheard (Feick 2002a; Lewis and Abraham 2001). Besides, EMEA was estab-lished within a consultation procedure, wherein the EP was not able togain significant influence (Kelemen 2002). Despite this general lack ofinput legitimacy, the EU regulatory regime seems to achieve high effi-ciency and thus output legitimacy. Consumers and producers both seemto be satisfied with its function. This output legitimacy is ensured by sev-eral accountability mechanisms that allow stakeholders to hold theregime responsible in the long term. Applicants may challenge authori-sation decisions before the European courts, member states’ experts mayreview each other’s work within the agency’s expert committee, and themember states may control the regime politically. All three mechanismsbalance each other in such a way that no single group is able to capturethe regime with its particularistic interests (Majone and Everson 2001;Moe 1987a).
1.2.2 The regulation of foodstuffs in the EU
The EU regulatory regime for foodstuffs followed the opposite develop-mental path (Ugland and Veggeland 2006). Here, a single market wasalready established in the 1980s. At that time, the member states couldalready look back on a long history of foodstuff regulation. However,with the exception of the Scandinavian countries, they had not estab-lished independent regulatory agencies for foodstuffs, and regulatorypolicy-making remained in the hands of agriculture and/or health min-istries. Consequently, there were no independent actors, which couldhave influenced the institutional development towards a single marketand a supranational regulatory regime. According to the so-called ‘New
10 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch01.qxd 8/29/2008 11:53 AM Page 10

Approach’ laid down in the Single European Act, non-tariff barriers totrade in foodstuffs were basically abolished by mutual recognition ofproduct standards. Because there were no national regulatory agencies,which could have blocked mutual recognition by setting regulatory stan-dards independently from political agreement, the ‘New Approach’ wasable to create a single market for foodstuffs. However, in the 1990s, theEU regulatory regime for foodstuffs was shattered by the BSE crisis. EUregulations, which were adopted in the EU committee system, came toolate to prevent a spread of the disease across Europe. The result was a fun-damental crisis of consumer confidence in both the safety of their foodand the capacity of EU regulatory institutions (Ansell and Vogel 2006;Majone 2000; Vogel 2001a; Vos 2004). In the aftermath of the crisis (Vos2000b), the supranational regulatory regime for foodstuffs was reformed,and EFSA was established to replace the scientific advisory committees.However, the competencies of the new agency are strictly limited to riskassessment and risk communication (Chalmers 2003; Szawlowska 2004).Risk management decisions are still left to the Commission and a comi-tology committee.
Because GMOs and GM food came on the political agenda in parallelto the set up of the Single Market, they were from the beginning regu-lated at the EU level. Strong national regulatory agencies were not estab-lished for these products, which means that the respective EU regimeresembles more the regime for traditional foodstuffs than that for phar-maceuticals. In the 1990s, a relatively weak supranational regulatoryregime for GMOs and GM food, based on the mutual recognition ofassessments by the member states, was established. When this mutualrecognition failed – which happened in almost all cases – arbitrations,which led to centralised authorisation decisions, were started within the EU committee system. Until 1996, this regime proved sufficient increating a single market for the few products authorised up to then.However, when the first GMOs and GM food, which were imported inlarger quantities from the USA, were authorised in the EU in the mid-1990s, consumer resistance to these products was immense (Vogel2001b). As a reaction, member states imposed a de facto moratorium onGMOs and GM food (Shaffer and Pollack 2004). The result was a tradewar with the USA (Bernauer 2003: 118–67). The regulatory regime forGMOs and GM food had to be reformed, and the risk assessment of these products was delegated to the newly established EFSA. As aresult, the first GMOs and GM food were authorised in 2004, but againagainst the resistance of a significant minority of the member states.Overall, the case of GM food closely followed the general development
Introduction 11
PPL-UK_RR-Krapohl_Ch01.qxd 8/29/2008 11:53 AM Page 11

of foodstuff regulation. A new supranational regulatory regime had to beset up, but the problem was that it could not fall back on strong regula-tory agencies in the member states.
The mutual recognition principle and the EU committee system wereinsufficient to achieve the twin goals of market integration and effectiverisk regulation in the foodstuff sector. Although a single market was orig-inally established by the committee system, a regulatory deficit led tothe BSE crisis, which in turn endangered the functioning of the market.As a result, the regime was reformed fundamentally. Because EFSA wasonly established in 2002, its efficiency cannot yet be fully assessed.However, the institutionalist approach developed in this book allowssome reasoned hypotheses about the agency’s success to be posed.Overall, member states’ commitment to risk regulation is much weakerfor foodstuff regulation than it is for pharmaceutical authorisation,because risk management – that is, the competence to adopt regulatorymeasures – was left in the hands of the Commission, supervised by themember states within a comitology committee. The agenda-setting roleof EFSA is relatively weak, and if the credible commitment argumentholds true, this will lead to less efficient risk regulation. The situation forGM food regulation, where the food agency is also responsible for riskassessment, is very similar. Though the commitment of member stateshas become stronger since the latest reform, they still have strong vetopowers. Whether the improvements are strong enough to prevent amoratorium on GM food in the future is an open question, but the ongo-ing resistance of certain member states raises some doubts about this.
The committee system in the foodstuff sector did not only lack effi-ciency, but also legitimacy. The legislative actors did not establish thecommittee system intentionally: rather, it evolved out of increasing reg-ulatory activities at the EU level (Vos 1997, 1999b). Thus, the committeesystem lacked the input legitimacy of a founding act, and the participa-tion of the EP therein. As a consequence, the committee system was, fora long time, heavily disputed between the EP and the member states(Bradley 1992; Steunenberg et al. 1997). On the output dimension, thecommittee system lacked legitimacy because of its failure in handling the BSE crisis. Since the latest reform, the supranational regulatory regimefor foodstuffs has mainly been legitimised by input factors. EFSA wasestablished by a co-decision procedure. Consequently, the EP was moresuccessful in gaining influence within the legislative process, and in rep-resenting the diffuse interests of consumers (Kelemen 2002). However,one may expect that both efficiency and output legitimacy of the regimeare weaker than those of the regulatory regime for pharmaceuticals.
12 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch01.qxd 8/29/2008 11:53 AM Page 12

The accountability mechanisms are not as well balanced between the dif-ferent groups of actors as in the case of pharmaceuticals. In fact, the food-stuff regime can only be controlled by the governments of the memberstates, whereas scientific peer review and judicial scrutiny is rather weak.As a result, the whole policy area is more easily politicised than in thecase of pharmaceuticals, and this is likely to influence the regimes’ out-put legitimacy negatively.
1.3 Outline of the book
The book is divided into three parts. The first part develops the insti-tutionalist approach to supranational risk regulation. The second chap-ter analyses the functional need for, and the historical developmentof, EU risk regulation. When making decisions about regulatory stan-dards, member states functional pressure to establish strong regulatoryregimes at the supranational level. However, such regimes are not onlya result of functional pressure, but also of concrete historical circum-stances. Accordingly, their development is always distinguished bypath-dependencies. Next, chapter three develops analytic tools forevaluating the performance of the resulting supranational regulatoryregimes. It discusses, on an abstract level, short- and long-term interestsof consumers and producers, and concludes that an appropriate risk reg-ulation would be a policy that meets the long-term interests of both. Toachieve this, member states need to commit themselves credibly withinsupranational regulatory regimes. Such commitment is expressed in thedelegation of competencies to independent agents, and in the legalisa-tion of the respective policy areas. Additionally, criteria are establishedfor the evaluation of the legitimacy of supranational regulatory regimes.Because of the need for credible commitment in supranational risk reg-ulation, output factors and accountability mechanisms become moreimportant for legitimising the regimes’ day-to-day operation than inputfactors.
The second part of the book contains the case study of the EU regula-tory regime for pharmaceuticals. Therein, chapter four analyses the his-tory of European pharmaceutical authorisation from the 1950s until thepresent day. The developmental path is mainly influenced by the earlythalidomide catastrophe and the subsequent establishment of nationalregulatory agencies. The aim of the fifth chapter is to evaluate the EUregulatory regime for pharmaceuticals. It analyses the credible commit-ment of the member states within the regime, and assesses its efficiency.The EU agency for pharmaceuticals developed to be rather independent.
Introduction 13
PPL-UK_RR-Krapohl_Ch01.qxd 8/29/2008 11:53 AM Page 13

Consequently, the regime works rather efficiently in the interest of moststakeholders. As a result of its relative success, the regime derives its legit-imacy mainly from its policy output, whereas input factors played a lessimportant role during its establishment.
The third part contains a case study of the EU regulatory regime for foodstuffs, and is structured in parallel to the second part aboutpharmaceutical regulation. Chapter six analyses the development ofEuropean foodstuffs regulation from the 1950s onwards. In the case offoodstuffs, national regulatory agencies were missing for a long time;thus, they were not able to influence the design of the regulatory insti-tutions at the EU level. In chapter seven, efficiency and legitimacy of theregulatory regime for foodstuffs are analysed. Because both delegationand legalisation are not as strong as for pharmaceuticals, the foodstuffregime is unlikely to be as successful. Finally, during the establishmentof the foodstuff regime, input legitimacy played a much larger role thanfor pharmaceuticals, whereas output legitimacy of the foodstuff regimeis probably much lower.
The last chapter summarises the findings of the book and comparesthe results of the two case studies. It identifies further research that maybe used for testing the hypotheses of the institutionalist approach tosupranational risk regulation. It concludes with a summary of the con-sequences of this approach for current discussions in political science, asthey are the institutionalist debate in general and the approaches ofJoerges and Neyer (1997a, 1997b, 2006) and Majone (1994, 1996: 61–79,2001) in particular.
14 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch01.qxd 8/29/2008 11:53 AM Page 14

Part I An InstitutionalistApproach to SupranationalRisk Regulation
PPL-UK_RR-Krapohl_Ch02.qxd 9/15/2008 10:25 AM Page 15

This page intentionally left blank

2Functional Pressure and Path-Dependencies: TheEmergence and Development ofSupranational Regulatory Regimes
Despite the fact that health and consumer protection was not originallyamong the competencies of the EU (it was first introduced as an inde-pendent policy objective by the Amsterdam Treaty in 1997), the EU hasbecome very active in the area of risk regulation, at least since the SingleEuropean Act in 1986. The EU sets standards for technical goods, adoptsfood safety measures and authorises medicinal products (Vos 1999a).The question is why different regulatory regimes emerged at the EUlevel, even though risk regulation was originally not mentioned in thetreaties. To answer the question, this chapter proceeds in two steps.Firstly, it analyses the functional pressure that leads to the emergence ofsupranational risk regulation (see section 2.1). The emergence of supra-national regulation of risky products is a spillover process from theSingle Market. National product standards constitute non-tariff tradebarriers that hinder free trade across national borders. Thus, functionalpressure emerges to centralise risk regulation at the supranational levelin order to realise the Single Market.1
Secondly, the effects of different developmental paths of supranationalregulatory regimes are examined to explain the differences between dif-ferent regimes (section 2.2). Not only has the EU become very active inthe field of risk regulation, but several supranational bodies for it havealso emerged. Private standardisation bodies develop technical standardson behalf of the Commission (Egan 1998; Kerler 2005; Pelkmans 1987),several expert committees advise the Commission on technical aspectsof regulatory policy-making (Gehring 1999; Krapohl 2003) and regula-tory agencies have replaced scientific committees in some policy areas(Chiti 2000, 2004; Gardner 1996; Kreher 1997). If the emergence ofsupranational risk regulation in different product sectors is due to thesame functional pressure, one could expect that the resulting regulatory
17
PPL-UK_RR-Krapohl_Ch02.qxd 9/15/2008 10:25 AM Page 17

institutions would look rather similar. However, functional pressuredoes not fully determine the design of regulatory institutions (Thatcher2002). Instead, functional pressure is always mediated by historical cir-cumstances at a particular time. In the case of supranational regulatoryregimes, it is important whether national regulatory agencies, whichmight have influenced the paths towards supranational regulatoryregimes, already existed.
2.1 Risk regulation between negative and positive integration
To abolish non-tariff trade barriers in the Single Market, the memberstates have generally three opportunities. Firstly, they may recogniseeach other’s product standards (see section 2.1.1). However, this carriesthe danger of downward regulatory competition, in which the lowestregulatory standards prevail. Secondly, member states may establish har-monised product regulations at the EU level (section 2.1.2). However,here, regulatory policies cannot be passed and implemented by a politi-cal centre, but are subject to horizontal coordination between 27 mem-ber states. Thirdly, member states may delegate some competencies tosupranational bodies to solve this coordination problem (see section2.1.3). As a result, supranational regulatory regimes emerge in which dif-ferent actors – scientific or expert bodies, the Commission, comitologycommittees and the Council – fulfil different functions.
2.1.1 The single market problem and mutual recognition
Member states face the preference constellation of a prisoner’s dilemmawhen they try to create the Single Market (see the lower part of Figure 2.1;Garrett and Weingast 1993). Accordingly, each member state has todecide whether it will protect its own consumers or industries withtrade barriers, or whether it will allow free trade of foreign products. TheSingle Market emerges if all member states give up the possibility of pro-tectionism and allow free trade of goods throughout the whole com-munity. However, even if they all profit from the establishment of theSingle Market, every member state faces incentives to protect its con-sumers or industries. When other member states allow free trade ofgoods on their markets, another member state can try to be a free-rider.That is, it can profit from the export of its own goods to foreign marketswithout carrying the costs of less consumer protection or more compe-tition for its own industry (e.g. member state A tries to achieve point 0/4in Figure 2.1). When other member states choose to protect their own
18 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch02.qxd 9/15/2008 10:25 AM Page 18

consumers or industries, a member state should follow this example inorder to avoid becoming exploited by other free-riders (e.g. member stateA tries to avoid point 4/0 in Figure 2.1). This situation is the same for allmember states. Consequently, they would all choose to protect theirmarkets, at least in the short run. The result is that no Single Marketemerges (point 1/1 in the lower part of Figure 2.1), and that all memberstates face welfare losses compared with the situation of mutual cooper-ation, where free international trade would lead to profits for everyone.
If such prisoner’s dilemmas are played only once, the result is alwaysmutual defection, i.e. protectionism of domestic markets. However,within the EU, decisions between protectionism on the one hand andfree trade on the other occur repeatedly. The cooperation problem does
Functional Pressure and Path-Dependencies 19
Figure 2.1 Product regulation in the Single Market
Member State A
Single Market
Protect- ionism
Sin
gle
Mar
ket
0/4
Mem
ber
Sta
te B
Pro
tect
- io
nism
4/0 1/1
Member State A
Standard a
Standard b
Sta
ndar
da 2/3 1/1
Mem
ber
Sta
te B
Sta
ndar
db 1/1 3/2
PPL-UK_RR-Krapohl_Ch02.qxd 9/15/2008 10:25 AM Page 19

not resemble a one-shot game, but an iterated prisoner’s dilemma. Insuch situations, actors can learn to trust each other to achieve higherpay-offs by mutual cooperation (Axelrod 1984). However, such cooper-ation is potentially unstable. One or more of the 27 member statescould decide to defect if short-term gains of individual defection arehigh, or if individual defections are unlikely to be noticed. Thus, evenif the member states in principle agreed on cooperation, the implemen-tation of this agreement would still be endangered, because every singlemember state faces incentives to be a free-rider. To ensure cooperation,Moravcsik (1998: 73–7) stresses the role of institutions. Accordingly,institutions indicate credible commitments (North 1993; Shepsle 1991)of member states not to deviate from agreements. They can thus help toshift member states’ behaviour from mutual defection (point 1/1 in thelower part of Figure 2.1) to mutual cooperation, and can thereby helpto increase the pay-off for all parties in the agreement.
One institution, which should ensure cooperation and, in consequence,the Single Market, is the mutual recognition principle. Accordingly, everymember state would have to accept products on its own market, whichfulfil the regulatory standards of at least one other member state.Member states loose regulatory sovereignty if they follow this mutualrecognition principle. They are forced to accept goods, which meetsafety standards of other member states, on their own markets, but theyare not able to influence these standards. This became evident in thewell-known Cassis-de-Dijon case,2 where Germany was forced by theEuropean Court of Justice (ECJ) to allow the import of a French spirit,which did not meet its own standards for either wine or liqueurs (Alterand Meunier-Aitsahalia 1994). Scharpf (1996a, 1997b, 1999: 43–83) usesthe term ‘negative integration’ for market integration by mutual recogni-tion, because it is based on the breakup of national regulatory standards.If levels of health and consumer protection differ between member states,a strict application of the mutual recognition principle would havederegulatory effects (Scharpf 1999: 84–129), at least for high-standardcountries. Products would only have to meet standards of memberstates with the lowest level of protection, and would nevertheless beallowed to be traded on the entire Single Market.
The EU averts the danger of deregulation in high-standard countries byallowing exceptions from the mutual recognition principle for reasons ofhealth and consumer protection. Article 30 (formerly 36) of the Treatyestablishing the European Community states that trade restrictions basedon health and consumer protection measures are legitimate, but that theyshould not lead to discrimination or disguised protectionism between
20 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch02.qxd 9/15/2008 10:25 AM Page 20

member states. The result of this exception is that member states mayimplement their own product standards, as long as they can be justifiedfor reasons of health and consumer protection. In the Cassis-de-Dijoncase, Germany was not able to prove that the French liquor could endan-ger German consumers more than other liquors. Consequently, a prohi-bition of the French spirit in Germany was judged an excessive andillegitimate measure. However, aside from this concrete decision, the ECJmade clear that it would pass other decisions if such justifications seemedto be valid. As a result, the Single Market can be disturbed by non-tarifftrade barriers, even if these are set up for reasons of health of consumerprotection and not for protection of domestic industries.
In the end, these exceptions for reasons of health and consumer pro-tection mean that the mutual recognition principle is not properlyimplemented for risky products, and that the member states fall back tothe cooperation problem of the prisoner’s dilemma. Thus, the memberstates face difficulties in achieving simultaneously the goals of healthand consumer protection, as well as the establishment of the SingleMarket. If they mutually accept each other’s regulatory standards, theyface the danger of deregulation; however, if they deviate from this prin-ciple, they endanger the Single Market itself.
2.1.2 Harmonisation of product standards and the coordination problem
The second instrument of market integration is the harmonisation ofproduct standards at the EU level. New harmonised EU standards replacepreviously existing regulatory standards of the individual member states.Consequently, the standards lead to the Single Market’s re-regulation, andbelong to the so-called ‘positive integration’ (Scharpf 1996a, 1997b, 1999:43–83). However, economic deregulation is much easier to achieve thanpolitical re-regulation in the EU’s day-to-day policy-making. Whereas themutual recognition principle is grounded in constitutional principleswithin the treaties, new EU legislation needs a high degree of politicalagreement. In contrast to the enforcement of mutual recognition by theECJ, political re-regulation needs to be explicitly accepted by the memberstates (and often by the EP).3
Member states face coordination problems with distributive conse-quences (Battle-of-the-Sexes) when they try to establish common stan-dards (see the upper part of Figure 2.1; Garrett and Weingast 1993; Scharpf1996a). Originally, one can expect that member states have differenthealth and consumer protection standards on their markets. The regula-tions may differ either in their safety standards – poorer member states
Functional Pressure and Path-Dependencies 21
PPL-UK_RR-Krapohl_Ch02.qxd 9/15/2008 10:25 AM Page 21

might tend to have lower standards than wealthier ones – or in the regu-latory instruments they choose – some states may directly regulate respec-tive products whereas others may aim to inform and teach consumers.Even if all member states are interested in adopting common regulatorystandards, they have to agree on one standard from the variety ofnational ones. Of course, every member state would prefer that its ownnational standards become compulsory for all other member states (point2/3 for member state A and point 3/2 for member state B in Figure 2.1).This would mean that industries and consumers of all other memberstates would have to adapt to its own standards, whereas its own indus-try and consumers would not face any new adaptation costs. The mem-ber states must find solutions in these coordination games. If they are notable to adopt common regulatory standards, they fall back on the mutualrecognition principle, and this could result in mutual defection. Findingsolutions for such coordination problems with distributive consequencesis by no means a trivial task (Zürn 1992: 184–97). The game does not havea natural solution, because it does not have a single equilibrium, but atleast two different ones. In contrast to the preference constellation of a prisoner’s dilemma, the problem is not the implementation of agree-ments, but finding such agreements in the first place.
Coordination between the member states becomes even more prob-lematic if one keeps in mind that the different regulatory standards underconsideration usually do not only differ in their distributive conse-quences, but also in their aggregated utility for the member states. Mostlikely, some standards are more adequate to solve the respective regula-tory problems than others. As a result, struggles between the memberstates about which standards to choose as harmonised ones do not onlyhave distributive consequences, but also an efficiency dimension. Toachieve higher aggregated pay-offs, the member states have to engage inproblem-solving, i.e. they have to search for the most efficient of allavailable options (Gehring 2000; Scharpf 1993, 1997a: 134).
If harmonised product standards have to pass negotiations in theCouncil, member states face a so-called negotiator’s dilemma (Gehring1999; Lax and Sebenius 1986; Scharpf 1997a: 124). On the one hand,they have to take care of their own interests. This is best done if one holdsfixed positions in negotiations, and if one is only willing to compromisewhen the other side also moves away from its respective position. On theother, the search for more efficient solutions requires that both partiesquestion their own positions, and are willing to listen to arguments fromthe other side. Consequently, both goals are difficult to achieve simulta-neously. Negotiators cannot bargain and argue effectively at the same
22 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch02.qxd 9/15/2008 10:25 AM Page 22

time. There is always danger that more efficient, common solutions can-not be reached, because one of the negotiators tries to achieve individualgains or to avoid individual losses.
In a bargaining system, the danger of such blockades could only bereduced if the distributive consequences of regulatory standards werenot discrete, but continual and dividable. In this case, the search for anefficient solution and bargaining about its distributive consequencescould be separated. However, in reality this is more the exception thanthe rule. Costs of regulatory standards are diffuse, and are imposed onmember states’ industries. They affect industries’ competitiveness, andare only indirectly reflected in member states’ budgets (Majone andEverson 2001). Thus, side-payments for losing member states are diffi-cult to quantify, and are unusual within the EU. Thus, negotiationsabout the costs of regulatory standards cannot be separated from thechoice of the standards themselves. As a result, losing member stateswill probably try to block the adoption of new standards.
Because of the coordination problem and the negotiator’s dilemma, itis uncertain whether member states are able to adopt common regula-tory standards, and whether they are able to achieve the most efficientsolutions. Decision-making is always in danger of being blocked,because member states may insist on their own individual advantage atthe costs of common standards. Significant welfare gains may be lost, aslong as no decision-making rules exist to solve these problems withoutexploiting each other’s goodwill. Thus, functional pressure emerges forthe member states to establish institutions that solve coordinationproblems and avoid the negotiator’s dilemma.
2.1.3 Delegation to supranational regulatory regimes
To solve these problems of decision-making within an intergovernmentalbargaining system, member states may delegate some competencies forregulatory policy-making to third actors like the Commission or expertbodies. The institutional arrangements that result from such delegationalways include two types of rule (Gehring 2002: 155–96, 2005). Theyestablish procedural rules, which organise the way different expert andpolitical bodies work together in the decision-making process. Secondly,they lay down substantive criteria on which final decisions shall bebased. In the following, such sets of institutions will be called suprana-tional regulatory regimes.
Such supranational regulatory regimes can increase the efficiency ofpolicy-making, because they reduce transaction costs in several ways.Firstly, regulatory regimes allow for incomplete contracting at the
Functional Pressure and Path-Dependencies 23
PPL-UK_RR-Krapohl_Ch02.qxd 9/15/2008 10:25 AM Page 23

legislative level (Pollack 1997a), which may facilitate agreement in theCouncil. When member states decide on the establishment of regulatoryregimes, they do not have to decide on single regulatory policies, but oninstitutional rules, according to which future policies will be decided(Gehring 2003, 2005). Such a horizontal differentiation between legisla-tion and implementation could have a moderating effect on decision-making within the Council. Even if member states have particularisticinterests in single regulatory policies, they cannot fully represent theseinterests when they bargain about institutional rules (Brennan andBuchanan 1985: 28–31). They need to find consistent rules for all futurepolicies, and they cannot adapt them to every single decision. Memberstates need not find a consensus for every single policy, but only for thesum of the expected policies (Gehring 2000).
Secondly, supranational regulatory regimes also help to avoid thenegotiator’s dilemma at the implementation level. They do this by dif-ferentiating decision-making vertically according to different functions,thus separating problem-solving from bargaining about distributiveconsequences (Gehring 2002: 155–96). On the one hand, expert bodies –such as different committees, standardisation bodies or agencies – try tofind the most efficient solutions to regulatory problems. On the other,the member states within comitology committees and the Council con-trol these bodies. Therefore, it is not only important that advisory bod-ies provide technical or scientific expertise, but also that the negotiator’sdilemma is not simply copied within the expert bodies. This would bethe case if the respective experts were to search for the most efficientsolutions and to bargain about their distributive consequences at thesame time. To avoid this, expert bodies should be free of any economicor political interests in the discussed policies (Majone 1996: 28–46,2001a, 2001b). The absence of particularistic interests allows them toleave bargaining behind and to concentrate on problem-solving (Scharpf1997a: 130–2). The result is that appropriate solutions can be more eas-ily identified, because scientific or technical arguments are not rejectedfor political reasons.
And thirdly, even if the final decisions about single regulatory policiesare still passed in intergovernmental bodies like comitology committeesand the Council, supranational regulatory regimes help to solve thecoordination problem between member states, because the task ofagenda setting is delegated. Expert bodies develop scientific opinions,upon which the Commission bases policy proposals, which are thensubject to a vote of the member states. So, one solution to a particularregulatory problem is deemed to be more efficient than the others.
24 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch02.qxd 9/15/2008 10:25 AM Page 24

This advice from expert bodies can act as a focal point, on which it iseasier to find consensus than on other regulatory standards (Garrett andWeingast 1993). The member states no longer bargain about a range ofequivalent regulatory standards, but they must decide on one policyproposal from the Commission. Thus, the coordination problem disap-pears, because the range of possible alternatives declines.
2.2 Development of supranational regulatory regimes
The existence of functional pressure is not sufficient to explain theappearance of different regulatory institutions. Functional pressure is aconstant variable. Consequently, it is not able to explain variance. Toconclude that the existing institutions were erected to answer theobservable functional needs, and that they would be the best possibleanswer to these needs, would be a functionalist fallacy. It is necessary tolook at the development of supranational regulatory regimes in moredetail to explain their variety. According to historical institutionalism(e.g. Pierson 1996, 2000a, 2000b; Thelen 1999, 2003), institutionaldevelopment is generally distinguished by critical junctures (i.e. situa-tions where open policy windows allow significant institutional change)and path-dependency (i.e. institutions develop some persistence oncethey are established; see section 2.2.1). In the case of risk regulation, onecan distinguish two critical junctures that distinguish the developmen-tal paths towards supranational regulatory regimes (see section 2.2.2).Crises of consumer confidence may lead to the establishment of regula-tory authorities, and the establishment of the Single Market may lead tothe delegation of more competencies to the EU level. Because institu-tional development is path-dependent, it is important for the form ofsupranational regulatory regimes which critical juncture occurs first andwhich later.
2.2.1 Critical junctures and path-dependencies
Critical junctures occur if existing institutions are no longer ‘equilib-rium institutions’ (Shepsle 1986). Within such disequilibria, institu-tional change can, but need not necessarily, occur. ‘Policy windows’(Kingdon 1995: 165–95) open up, but it is uncertain whether the actorsconcerned will exploit these. Actors face transaction costs when they tryto reform institutions. They have to invest in the search for new andmore efficient institutions, they have to find new agreements betweenthem and they have to ensure compliance with new regimes. However,even during critical junctures, actors do not only follow functional needs
Functional Pressure and Path-Dependencies 25
PPL-UK_RR-Krapohl_Ch02.qxd 9/15/2008 10:25 AM Page 25

when they reform institutions. They are also influenced by previouslyexisting institutions. Institutions have some power of persistence, whichmakes it unlikely that they could be totally abolished and replaced.Persistence results from the fact that institutions indicate credible com-mitments of different actors not to deviate in the future from agreementsof the past (North 1993). If institutions were not stable and persistent,they could not guarantee such commitments. Such commitments can becredible either in a motivational or in an imperative sense (Shepsle1991). The former means that institutions set incentives in such a waythat actors no longer have any interests in deviating from agreements,whereas the latter means that actors cannot breach institutional rules,independently of their own will.
Once institutions are established, it may become more advantageousfor the involved actors to stick to these institutions instead of changingto completely new ones. Institutions can influence actors, because theymay lead to so-called ‘increasing returns’ (Arthur 1994: 1–12; Pierson2000a). Four factors lead to such positive feedbacks of institutions.Firstly, if the establishment of institutions involves large set-up costs, itbecomes less likely that these institutions will be abolished and newones created. Even if new institutions were more efficient, additionalgains would first have to make up for the new investment costs to makethe whole change worthwhile. Secondly, institutions lead to learningeffects of concerned actors. Routine action emerges, which makes actioncheaper. Change towards new institutions would require new invest-ments in learning. Thirdly, coordination effects occur if actors’ compli-ance with institutions has positive externalities. If institutions areresponsible for the provision of club goods, profits for the involvedactors increase when further actors join the group, because then thecosts can be shared by more actors. The larger the groups that stick tosuch institutions, the less likely it is that institutions will be replaced bynew ones. Fourthly, if institutions are expensive when they do not pre-vail in the long term, actors may feel the need to ‘pick the right horse’from the beginning. Thus, adaptive expectation of actors could reinforceearly choices for certain institutions.
Actors cannot abolish or reform institutions if they are locked in‘joint-decision traps’ (Scharpf 1985, 1989). This can happen if actorscooperate in lasting decision-making systems where they decide withunanimity (or near unanimity) and where exit options are either closedor at least very expensive (e.g. in German federalism or in the EU;Scharpf 1985). In such cases, institutions that were once set up by allactors are difficult to change, because they establish a new status quo
26 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch02.qxd 9/15/2008 10:25 AM Page 26

that can only be changed by unanimity or near unanimity. Therefore,the abolishment of such institutions is difficult as long as some actorsstill have an interest in the old institutions. Here, actors are committedto the old institutions in an imperative sense, because institutionalchange is blocked by the decision-making rules of broader institutions.The different institutions are interlocked like Russian dolls. Changes inminor institutions would also require changes of broader institutions.Consequently, institutional change becomes more unlikely.
2.2.2 Different paths towards supranational regulatory regimes
The major problem of historical institutionalism is that it is itself not atheory that leads to testable propositions (e.g. Bulmer 1998, Lindner andRittberger 2003). The finding that institutional development is distin-guished by critical junctures and path-dependencies is too broad andimprecise to be falsifiable. One cannot really imagine historical develop-ments where no critical junctures and path-dependencies can be identi-fied. However, at least in its rationalist version (Stacey and Rittberger2003), historical institutionalism may be an adequate basis on which to build theoretical models that would then arrive at hypotheses.Therefore, one must recognise the relevant critical junctures and exam-ine how they influence further paths of development.
In supranational risk regulation, two critical junctures may influencepaths of institutional development. Firstly, a crisis of consumer confidencein a certain regulatory area may lead to the establishment of regulatoryagencies.4 It can be expected that regulatory policy-making in highly sci-entific matters – such as pharmaceutical or foodstuff regulation – usuallydoes not receive much public attention. Normal consumers neither havethe education to deal with these matters nor any concrete interests indoing so as long as no scandals provoke their attention. Consequently,in a pluralist polity the policy field will mainly be dominated by interest-group politics (Wilson 1980: 357–94). However, if regulatory scandalsoccur, consumers receive new and unfavourable information, and theensuing scandal can damage consumer confidence in the safety of agroup of products and in the respective regulatory institutions. As aresult, consumers push towards stronger regulations and regulatory insti-tutions, and politicians are likely to pay more attention to the diffuseinterests of consumers. Policy windows thus open up, in which institu-tional change towards new regulatory agencies is possible.
Secondly, market integration may lead to the centralisation of regula-tory competencies at the EU level. It became clear in the aftermath ofthe Luxembourg compromise that the resistance of member states to
Functional Pressure and Path-Dependencies 27
PPL-UK_RR-Krapohl_Ch02.qxd 9/15/2008 10:25 AM Page 27

give up competencies hindered the establishment of the Single Market.Whereas negative integration was pushed forward by supranationalinstitutions – most importantly the ECJ (Stone Sweet and Caporaso1998) – positive integration was hindered by unanimous voting in theCouncil (Scharpf 1999: 43–83). Because of this ‘Euro-sclerosis’, memberstates re-considered their resistance to further integration (particularlyFrance and Britain; see Moravcsik 1991). Consequently, in the early1980s it was widely agreed that a policy project was needed that wouldrevive European integration. The Commission, as a policy-entrepreneur,was able to use this open policy window. It introduced its well-knownWhite Book on the Single Market,5 and set the agenda for the negotiationsof the Single European Act (Fligstein and Mara-Drita 1996). As a result,qualified majority voting was re-introduced for all measures belonging tothe Single Market programme, and the so-called new approach was imple-mented, which led to the establishment of supranational regimes in manyregulatory areas.
The major consequence of the persistence of institutions and theresulting path-dependencies is that the ‘specific patterns of timing andsequence matter’ (Pierson 2000a: 251). It is not only important that crit-ical junctures occur, but also at which stage of the development thishappens. If previously existing institutions influence the interests ofand restrictions on actors, it is important to know which institutions areset up first and which thereafter. There are two possible paths of insti-tutional development towards supranational regulatory regimes. Theseare illustrated in Figure 2.2. Both developments start with low-regulatednational markets (at point t0). The first possible path is that a publicscandal, which leads to the establishment of national regulatory author-ities, occurs first (at point t1), and is later followed by the establishmentof a single market (at point t2). The second possible path is that a singlemarket is established first (at point t1), and a public scandal occurs later
28 Risk Regulation in the Single Market
Figure 2.2 Path-dependencies of supranational regulatory regimes
?
t0 t1 t2 t3
Lowly Regulated National Markets
Supranational Regulatory Regime
Public Scandal
Market Integration
Market Integration
Public Scandal
PPL-UK_RR-Krapohl_Ch02.qxd 9/15/2008 10:25 AM Page 28

(at point t2). The question is whether both paths end at the same desti-nation, namely at the same form of a supranational regulatory regime(at point t3). If path-dependencies matter, this is unlikely.
If crises of consumer confidence occur for certain groups of productsbefore a single market is established, national regulatory agencies maybe set up. As a consequence of a public scandal, consumers are likely tobuild up pressure to establish strong regulatory policies and agencies. Aslong as the European market is not integrated, the addressees of thesedemands are the national governments. Once national regulatory agen-cies are established, they develop some persistence. Because consumersand producers have to adapt their behaviour, regulatory agencies bearhuge set-up costs. However, once these agencies are established, they arestrengthened by learning and coordination effects of consumers andproducers. Regulatory agencies may be difficult to change or abolish ifdomestic groups are able to veto their reform, thus making the regula-tory agencies more stable (Tsebelis 2001).
If national regulatory agencies are set up, new actors with their owninterests and resources enter the playing field. These new actors becomestakeholders in institutional developments once a related single market iscreated. In this respect, they represent two fundamental interests. Firstly,they want to ensure their continued existence. Secondly, they try toensure that their own regulatory goals and standards are not diminished,because these goals legitimise their own policy-making. Because of thepotentially deregulatory impact of the mutual recognition principle, a sin-gle market endangers both interests. Once a single market is established,national regulatory agencies are obsolete, because they could establishnon-tariff barriers of trade. Moreover, the danger of regulatory competi-tion under the mutual recognition principle threatens the regulatory goalsand standards of national regulatory agencies. Consequently, one canexpect that national regulatory agencies oppose market integration bymutual recognition, because this endangers their existence and legitimacy.
The existence of national regulatory agencies makes it more difficultto establish a single market. National regulatory agencies can set up reg-ulations that build up non-tariff barriers of trade. It is surely more diffi-cult to integrate a highly regulated market than a non-regulated one. Inaddition, the difficulties increase the more national regulatory agenciesexpress commitment. If regulatory policies cannot simply be overruledby political bodies – because regulatory agencies may enjoy independ-ence from political influence – the national regulatory agencies them-selves are able to act as veto players against institutional change, whichcan prevent the creation of a single market.
Functional Pressure and Path-Dependencies 29
PPL-UK_RR-Krapohl_Ch02.qxd 9/15/2008 10:25 AM Page 29

This leads to a second consequence: the interests of national regula-tory agencies have to be taken into account once a single market is cre-ated. Only in this way can their resistance be overcome and their vetoprevented. To allow national regulatory agencies to agree to market inte-gration, their existence and regulatory goals must not be threatened. Itis likely that this is reflected in the institutional design of supranationalregulatory regimes. Even if regulatory policy-making were centralised at supranational level, national agencies could be involved in the newsupranational regulatory regime. Therefore, networks of member states’regulatory agencies, instead of completely new supranational bodies,would be established (Dehousse 1997; Majone 1997).
Such a network design has some advantages compared with new, cen-tral bodies. Knowledge and expertise of the national agencies would notbe lost, because they could be fed into the regulatory network. Moreimportantly, member states’ governments would probably delegate morefar-reaching competencies to such regulatory networks than to com-pletely new European bodies, because the former are not such big threatsto their sovereignty as the latter. Member states’ governments would nothave to give their own powers away, because the respective competencieswould already be in the hands of their national agencies. Additionally,regulatory networks can still be controlled by the national agencies, sothat member states still have some influence over their decisions, even ifthis is not political. Consequently, under the same political circum-stances, regulatory networks could probably become more independentof direct political influence than completely new European agencies.
The question is what happens if crises of consumer confidence takeplace in an already integrated single market where no national regula-tory agencies exist. In such circumstances, national agencies cannot actas stakeholders, and cannot influence market integration. Obviously,regulatory networks cannot be established, because their basis withinthe member states is missing. Consequently, completely new Europeanregulatory agencies have to be set up. This threatens member states’ sov-ereignty more than a pooling of existing national regulatory agencies.For completely new supranational agencies, member states’ govern-ments would have to give up their own influence and delegate compe-tencies that had been given to national agencies long before. Besides,the resulting supranational regulatory regimes could not be controlledby national agencies, which may be answered with more control bymember states’ governments themselves. Overall, member states’ resist-ance to delegating far-reaching competencies to new, independent reg-ulatory agencies at the EU level is probably much stronger than in the
30 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch02.qxd 9/15/2008 10:25 AM Page 30

case of regulatory networks. As a result, faced with the same functionalpressure to establish supranational regulatory regimes, new centralisedagencies are probably much weaker than networks of national agencies,because the former are a greater threat to member states’ governmentsthan the latter.
2.3 Conclusion
If a single market for risky products is established, functional pressureemerges to establish supranational regulatory regimes. This is because asingle market conflicts with the regulatory sovereignty of the memberstates in matters of health and consumer protection. National productstandards may constitute non-tariff barriers to trade. Such barriers haveto be abolished to integrate markets. One way of doing this is themutual recognition of member states’ national standards. However, iflevels of health and consumer protection differ between the memberstates, the mutual recognition principle is probably too weak to ensuremember states’ commitment to a single market. If the principle werestrictly applied, it would have deregulatory effects (at least in high-standard countries), because these countries would have to accept ontheir markets products that fulfil the standards of low-standard coun-tries. To answer this threat of deregulation, member states are allowedto deviate from the mutual recognition principle for health and con-sumer protection. However, in these cases the Single Market is again dis-turbed. A solution to this problem is the harmonisation of regulatorystandards at the EU level. However, if member states decide about har-monised product standards within the Council, they face coordinationproblems and the negotiator’s dilemma. They have to find the best possible regulatory standards, and they have to decide about the distrib-utive consequences. Because this probably leads to blockages of decision-making in the Council, functional pressure emerges to delegate at leastsome competencies to supranational regulatory regimes. Therein, differ-ent actors like scientific advisory bodies, the Commission and the mem-ber states themselves fulfil different functions, and the differentiation ofdecision-making helps to solve coordination problems and to avoid thenegotiator’s dilemma.
The hypothesis of this chapter is that the concrete form of suprana-tional regulatory regimes does not only follow from functionalist pres-sure, but is also a result of path-dependencies. If functional pressurewere the only decisive variable, all regimes for risk regulation in the EUshould look similar, because the underlying functional pressures are
Functional Pressure and Path-Dependencies 31
PPL-UK_RR-Krapohl_Ch02.qxd 9/15/2008 10:25 AM Page 31

likely to be similar. Instead, regulatory regimes in the EU differ betweendifferent policy areas and different points in time. The hypothesisderived from a historical-institutionalist analysis is that this is due tothe sequencing of two different critical junctures. If crises of consumerconfidence occur before the establishment of a single market, they arelikely to be answered with the set up of national regulatory agencies.These national agencies could be obstacles against market integration,because they could implement non-tariff trade-barriers. To overcometheir resistance and to establish a single market, national regulatoryagencies are very likely to become included in supranational regulatoryregimes. Thus, new EU regimes can fall back on support of powerful reg-ulatory networks. However, if these national agencies are missing, thenewly established supranational regulatory regimes cannot fall back onsuch powerful assistance within the member states. Completely newregulatory agencies have to be established at the EU level, which islikely to meet more resistance from member states’ governments thanthe pooling of national regulatory agencies. Member states’ govern-ments would have to delegate their own competencies instead of thoseof their national agencies, and completely new supranational bodiescould not be controlled by regulatory networks. Because of memberstates’ resistance, the resulting regimes are likely to remain significantlyweaker and less independent of direct political influence than compa-rable regulatory networks.
32 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch02.qxd 9/15/2008 10:25 AM Page 32

3Efficiency and Legitimacy: The Evaluation of SupranationalRegulatory Regimes
Although most regulatory regimes in the EU share important characteristics – like participation of expert bodies, the Commissionand the member states – they are different if one takes a closer look.Privately organised standardisation bodies develop standards for techni-cal goods on behalf of the Commission, scientific committees advise theCommission in several policy areas, and regulatory agencies replacethese scientific advisory committees in other sectors (e.g. Vos 1999a).These regimes not only differ in their formal organisation, but also intheir procedural and substantive rules of decision-making. The questionis whether this variance in the institutional designs of supranationalregulatory regimes also leads to differences in performance. Accordingto the general assumption of institutionalism that ‘institutions matter’,it is very likely that different regulatory institutions will lead to differ-ences in decision-making and to different policy outcomes. As a result,the performance of supranational regulatory regimes depends, at leastin part, on their institutional design. Thus, it has to be possible to iden-tify institutional settings that are more appropriate for supranationalrisk regulation than others.
To derive hypotheses about the relationship between the institutionaldesign of supranational regulatory regimes and their performance, thischapter proceeds in three steps. Firstly, a benchmark is needed to evalu-ate the performance of regulatory regimes (see section 3.1). Supranationalregulatory regimes face competing demands when they decide about reg-ulatory standards. At least in the short term, consumers may ask for highregulatory standards to protect them from risks, whereas producersdemand low standards to remain competitive (Wilson 1980: 357–94).However, efficient risk regulation is possible if there is a level of regula-tory standards where consumers’ and producers’ demands converge in
33
PPL-UK_RR-Krapohl_Ch03.qxd 9/15/2008 10:26 AM Page 33

the long term. Secondly, because efficient risk regulation is alwaysendangered by particularistic, short-term interests, it is important thatthe member states credibly commit themselves to their common long-term interests (section 3.2). On the one hand, member states may dele-gate regulatory policy-making to independent agents. On the other, theymay legalise regulatory policy-making, which reduces the agents’ andtheir own discretion to influence regulatory policy-making with particu-laristic interests. Thirdly, the decision-making of supranational regulatoryregimes should be legitimate, because the regimes adopt authoritativedecisions with short-term distributive effects for concerned stakeholders(section 3.3). Even if regulatory standards are efficient, they imply costs and benefits for consumers and producers. Therefore the respec-tive regimes need to be able to justify themselves to these stakeholders(Majone 1996: 284–302).
3.1 Efficiency and distributive consequences of risk regulation
In economic textbooks, market failures are usually seen as the only jus-tification for state regulation in free-market economies (e.g. Heertjeand Wenzel 1997: 385–91). The basic rationale for such regulation isthat markets alone are not able to produce efficient outcomes underunfavourable circumstances. Regulatory policies should aim to increasethe efficiency of markets in the case of such failures. A prominentexample for market failures is information asymmetry, which leads toproblems of health and consumer protection. Information problemsoccur (Majone 1996: 28–31) if consumers cannot evaluate the qualityof products on their own, either because sellers do not provide suffi-cient information or because consumers themselves are not able toprocess this information. In both cases, the results are risks for con-sumers who are unable to make choices under these circumstances.Public regulators have two possibilities for dealing with informationproblems (e.g. Francis 1993: 149–58). They may stipulate that producersand sellers of certain products must provide information for consumersin order to reduce information asymmetries. However, the effects of theprovision of further information are rather limited if consumers are not able to process this information. Thus regulatory can also set upsafety standards that have to be fulfilled by products in order to gainaccess to the market.
Such regulations are widely applied in the EU, but they create hugedifficulties for public regulators. When setting up product standards,
34 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch03.qxd 9/15/2008 10:26 AM Page 34

regulators limit the available choices for consumers. They thereby replaceconsumers’ judgements about certain products with their own evalua-tions. This can be justified by an informational advantage of regulatorsover consumers. However, regulators usually also act in a scientificallyuncertain environment and do not have complete information (section3.1.1). Moreover, despite the fact that risk regulation is supposed toreduce market failures and thereby to increase the efficiency of marketallocations, it always leads to distributive consequences for market par-ticipants, at least in the short term (section 3.1.2). High safety standardsincrease the production costs of the respective products. These costs canbe shifted partly towards consumers who profit from the reduction ofrisks, but they also have partly to be carried by producers.
3.1.1 Objective and subjective perceptions of risk
Within the interdisciplinary literature on risk and its regulation, twoconcepts of risk face each other (Francis 1993: 132–9). On the one hand,scientists and economists follow an ‘objective’ concept of risks. On theother, psychologists and sociologists stress the ‘subjective’ perception ofrisks by individuals and society as a whole. When they make decisionsabout risk management measures, regulators are often stuck betweenthe two logics of decision-making (e.g. Krücken 1997). They need to beadvised by scientists about the ‘objective’ risks of respective products.Without such scientific risk assessments, political actors would not beable to make reasonable choices in such complex matters as pharmaceu-tical or foodstuff regulation. In addition, they have to take ‘subjective’risk perceptions of consumers into account (the interests of producersare analysed in section 3.1.2).
From an ‘objective’ point of view, risks are nothing more than productsof a potential damage and the probability of its occurrence. Scientistsshould try to quantify both the probabilities and the extent of dam-ages in order to give decision-makers clear guidance for their choice ofmeasurements (Majone 2002b). However, it is often quite difficult, if notimpossible, for scientists to detect reliable and consensually acceptedmeasurements for certain risks, which could act as clear guidance fordecision-makers. For example, it is a widespread problem for risk regula-tion that humans are usually exposed only to small doses of potentiallyharmful chemical substances – such as additives in foodstuffs or activesubstances in pharmaceuticals – but over a long period of time (e.g. Breyer1993: 3–32; Majone 2002b). This long-term toxicity of small doses can-not, for obvious ethical and practical reasons, be tested in human trials.Consequently, scientists usually refer back to high-dose trials with these
Efficiency and Legitimacy 35
PPL-UK_RR-Krapohl_Ch03.qxd 9/15/2008 10:26 AM Page 35

substances in animals. A crucial question is how findings of these animaltrials can be extrapolated to estimate risks for human health. Animalshave different constitutions to human beings, and within the trials theyare exposed to higher doses of the substance, but usually for a shorterperiod of time. Scientists need mathematical models to calculate risks forhumans based on such trials, but these models are built upon assump-tions that are far from being undisputed scientific knowledge. As a result,different scientists might arrive at different conclusions about the samerisks, which leaves decision-makers uncertain about whose guidance theyshould follow.
Another problem for risk regulation is that risks are perceived subjec-tively and differently by different consumers. Compared with scientists,consumers have less information about and less understanding of scien-tific problems. Consequently, it might be rational for them to refer backto other information from outside the world of scientific reasoning inorder to build up individual risk perceptions. This informational disad-vantage of consumers leaves room for social factors to influence risk per-ceptions, as is stressed by constructivist risk theories (e.g. Douglas andWildavsky 1982: 186–98). For example, consumers might draw attentionto their own experiences or to those of their social environment, andthey are unlikely to evaluate these experiences with the laws of proba-bility theory, as scientists would. Alternatively, if personal experiencesare missing, consumers might follow the opinion of the published media.However, to draw attention, the media often exaggerates certain riskscompared with others (e.g. Breyer 1993: 33–54; Wildavsky 1995: 375–94).Whether consumers refer back to personal experiences or to publishedopinion in the media, they might come to a different perception of riskthan scientists.
A possible reaction to simultaneous uncertainty and public attention isrisk regulation according to the ‘precautionary principle’ (e.g. O’Riordanand Cameron 1994). This principle originates from German law, where ithas been applied – under the name Vorsorgeprinzip – since the early 1970s(Rehbinder 1991). But in the meantime, it has found its way into the regulatory policy-making of other West European states and of the EU(Eckley and Selin 2004), whereas it has been applied to a more limitedextent in the UK and USA (Majone 2002b). An undisputed and coherentdefinition of the precautionary principle is still missing in both interna-tional and EU law (Gollier and Treich 2003; Majone 2002b). However,according to the general understanding, it basically proposes to adoptregulatory measures against risks, even if these cannot yet be scientificallyproven. In other words, if regulators make mistakes, they should err on
36 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch03.qxd 9/15/2008 10:26 AM Page 36

the safe side: it is better to be safe than sorrow. Of course, this principlemeets the interests of consumers, at least in the short run. If consumersare frightened by an uncertain danger – perhaps because it receives a lotof public attention in the media – they will favour strong regulatorymeasures, even if there is no clear scientific proof of risks.
Whereas the precautionary principle is very prominent on the politi-cal agenda in Europe, and is becoming the guiding regulatory principleof the Commission,1 it is greatly criticised by academics who support an‘objective’ concept of risk (e.g. Breyer 1993; Majone 2002b; Wildavsky1995: 427–48). Regulators might make two possible mistakes when theyallocate resources for risk regulation (Wildavsky 1988: 199–201). Firstly,they might not regulate risk where necessary. This can be called anunder-regulation. Secondly, they might regulate a risk where none existsor where the risk is so small that it would not be worth the costs of itsregulation. This can be called an over-regulation. The problem is that anasymmetry exists between the two mistakes. An under-regulation of risksis noticeable, because regulations fail to achieve their aim of health andconsumer protection, and, consequently, they can be adapted. On thecontrary, over-regulation leads to high costs, but is not noticed, becausethe regulations fulfil their aim of health and consumer protection. It isless likely that over-regulation is adapted, but the costs remain high. Theregulation remains effective but inefficient. If one decides to err on thesafe side, as the precautionary principle proposes, one is likely to producesystematic over-regulation, which is, in the long term, difficult to correct.
Safety is a scarce good, and other scarce resources have to be spent to get it. If the allocation of resources is inefficient, one achieves lessoverall safety than one could get with an efficient allocation of the sameresources. Consumers might sometimes prefer to accept the costs ofover-regulation rather than the risks of certain products, but theythereby ignore the fact that the costs of over-regulation could be spentto avoid other risks that might be much bigger than the regulated ones.Consequently, at least when strictly applied, the precautionary principleleads to an effective but inefficient allocation of resources, because itignores the opportunity costs of regulations, and does not pay any atten-tion to the asymmetry between under- and over-regulation (Wildavsky1988: 189–204). As a result, it should be in the interest of consumers – atleast in the long term – that the precautionary principle does not applygenerally, and that risk regulation should instead be as consistent andefficient as possible.
The contradictory consequences that follow from ‘objective’ versus‘subjective’ concepts of risks have to be brought together to develop
Efficiency and Legitimacy 37
PPL-UK_RR-Krapohl_Ch03.qxd 9/15/2008 10:26 AM Page 37

coherent assumptions about consumers’ interests in risk regulation. Todecide for only one of the two sides of the struggle is neither empiricallynor theoretically satisfactory. On the one hand, a concept of ‘objective’risk is not able to explain the different risk perceptions of consumers. Itsadvocates state that such ‘subjective’ risk perceptions are simply wrong,and that consumers make mistakes when they demand strongly pre-cautious risk regulations (Breyer 1993; Burnett 2000). However, suchstatements disapprove of consumers’ needs for additional safety, a con-clusion that is normatively questionable. On the other hand, a ‘subjec-tive’ concept of risk leads to inconsistent regulatory policies. It favoursover-regulation of risks, which receive a great deal of public attention,while other risks are neglected. In the end, this results in an inefficientoutcome, which does not achieve the greatest possible safety (Majone2002b, 2005: 124–42; Wildavsky 1988: 199–201).
The solution to this contradiction is that consumer interests areinconsistent over time. Consumers might have interests in the shortterm, which can be different from those of the long term. When con-sumers have incomplete information and restricted capacities withwhich to process it, their interests might change over time if more infor-mation becomes more easily available. When regulatory problems arenew, the gap between an ‘objective’ risk assessment and consumers’‘subjective’ risk perceptions might be large. Consequently, consumersmight wish to establish very precautious regulations in the short termto deal with new and unknown risks, whereas their long-term interestmight be to achieve the most efficient allocation of resources. There issome hope that the gap between consumers’ ‘subjective’ risk perceptionand scientists ‘objective’ risk assessment declines over time, when sci-entists, consumers and public regulators have more and more experi-ence with the respective matters (Rekaiti and van den Bergh 2000). Thisdoes not only mean that consumers change their ‘subjective’ fear of riskstowards more ‘objective’ scientific risk assessments. It can also be theother way round: that scientists, who also act under uncertainty, cometo new conclusions about risks, which might be closer to consumers‘subjective’ perceptions than to their previous ‘objective’ assessments.However, the informational advantage of scientists suggests that this isless likely to happen.
3.1.2 Costs and benefits of regulatory policies
Regulators do not only have to find efficient risk regulations in theinterests of consumers, they also have to be aware that safety standardsinflict costs on producers. The basic problem of health and consumer
38 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch03.qxd 9/15/2008 10:26 AM Page 38

protection in its distributive consequences is that they belong to the so-called ‘entrepreneurial politics’ (Wilson 1980: 357–94). In the shortterm, costs of higher product standards have to be carried by producers,whereby consumers benefit from higher protection. The costs of thiskind of regulation are concentrated on the relatively small societal groupof producers who have a concrete interest in not carrying higher bur-dens. However, the benefits are distributed to the large societal group ofconsumers who have relatively diffuse interests in health and consumerprotection. According to Olson’s (1968: 52–64) group theory, the formerinterests can be organised much more easily and effectively than the lat-ter. On the one hand, producers in a certain sector are relatively easy tocoordinate, and they have similar and, for themselves, very importantinterests, as well as the necessary resources to organise themselves or toput pressure on public regulators. On the other, consumers are a vastgroup, their interests may differ significantly, and they probably havefewer resources to mobilise. Consequently, producers’ interests can bemuch better and more powerfully articulated in a pluralist democracythan consumers’ interests. Under these circumstances, one may expectthat it is difficult to establish reliable regulatory regimes for health andconsumer protection in pluralist democracies. Opposition from power-ful producer interest groups would have to be overcome, although con-sumer support would be low.
However, this picture of directly opposite interests of consumers ver-sus producers falls short in two respects. Firstly, producers usually do notcarry the full costs for regulations in the long term. Producers may try toshift additional costs of regulatory standards to consumers by increasingprices of regulated goods. The consequences of these increased pricesdepend on the reactions of consumers (e.g. Heertje and Wenzel 1997:157–60). On the one hand, consumers might, to some degree, replacemore expensive goods with other goods. Thus, producers would not beable to sell as many goods as before, and prices would be under pressureto go down again. On the other hand, as long as consumers’ demandsare not totally price-elastic, the new equilibrium price is higher than theold one, even if fewer goods are sold for that price than before. Thus, inthe long term, higher costs for regulatory standards usually result in bothprice and quantity effects: that is, costs for higher regulatory standardswould be carried by both consumers who pay higher prices, and pro-ducers who cannot sell as many products as before. The exact distribu-tion of burdens depends on the price-elasticities of demands.2 As a result,even if new regulatory standards might have some distributive conse-quences in the short term, these are probably less dramatic in the long
Efficiency and Legitimacy 39
PPL-UK_RR-Krapohl_Ch03.qxd 9/15/2008 10:26 AM Page 39

term, because both consumers and producers have to carry some parts ofthe burden.
Secondly, producers cannot only shift parts of the burden of higherstandards towards consumers. They can also profit from public regulationsof their products, because these provide reputation. When informationasymmetries between producers and consumers exist, it is important forproducers to build up a good reputation about the quality of their prod-ucts. Information asymmetries implicate that consumers cannot fullyevaluate the quality of respective products on their own. Consequently,they might be guided by the reputation of producers, i.e. the experiencesof other consumers with these producers’ goods. A good reputation hasan advantage for producers, because it is easier to sell their products athigher prices.
As various economists demonstrate (e.g. Kreps and Wilson 1982;Shapiro 1983), such a reputation can be achieved without public regu-lation. The ‘reputation mechanism’ of the market’s ‘invisible hand’ (Hill1990) is built upon three conditions. Firstly, producers and consumersmeet repeatedly on the market to sell and buy products, respectively.Secondly, consumers need to be able to judge the quality of productsafter they have bought them. Thirdly, consumers exchange their expe-riences to gain common knowledge. Now, it is possible that producersestablish a good reputation by repeatedly selling products of high qual-ity. This is noticed and communicated among consumers. As a result,the respective producers are able to achieve higher prices on the market,because of their good reputation. Producers have to invest in a reputa-tion at the beginning, because they sell high-quality products to normalprices. Their goal though is to earn back their investments in the longterm, when they can sell their products at higher prices.
However, reputation-building is often not only an individual task, buta collective action problem. Only large firms, with trademarks that arevisible for consumers, or local suppliers who are personally known byconsumers, can build up a reputation on their own. For many small-and medium-sized enterprises this is not possible, because they are notidentifiable or distinguishable for consumers. Consequently, reputationis often a common good for an entire sector (Akerlof 1970; Anania and Nistìco 2004). Thus, every producer in the respective sector has tocooperate to build up common credibility. Of course, in the short-run,every producer faces incentives to exploit the common reputation, and toproduce comparatively cheap products. These could be sold at the sameprices as the products of other producers, as long as the common reputa-tion stands. However, ‘black sheep’, which try to milk the reputation of
40 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch03.qxd 9/15/2008 10:26 AM Page 40

the group by selling products of low quality, can endanger the reputa-tion of the whole group. If cheating is detected by consumers, but can-not be assigned to the responsible individuals, consumers will assign itto the whole branch. In the long term, it is unlikely that a common rep-utation can emerge or remain stable. If all producers face incentives toexploit the common reputation and to destroy it in this way, no rationalactor will invest in good reputation, because this goodwill would bemisused by others.
In the long term, it might be rational for producers not to carry theburden of reputation-building alone, but to shift it towards regulatoryinstitutions. Reputation is a valuable good for producers, but it is unsta-ble, because producers face incentives to exploit it in the short term. Andonce a good reputation is destroyed, it is difficult and costly to rebuild(Herbig et al. 1994). Public regulations have some advantages that couldpay off for producers (Akerlof 1970; Sinn 2003). A regulation in itselfmight carry some reputation effects. Consumers might be more willingto trust products that are controlled by public regulators. Regulationsmight make individual investment in reputation unnecessary. Now, pro-ducers can demand higher prices from the beginning, and do not haveto wait until consumers are willing to pay for the reputation. Finally,public regulations solve collective action problems. It is now compulsoryto behave according to the new regulations, and cheating can be pun-ished by regulators.
Taken together, both consumers and producers have short- and long-term interests in risk regulation. In the short term, consumers couldexaggerate certain risk, and could push for strong, precautionary regu-lations. In contrast, producers fear the costs of higher regulatory stan-dards. However, in the long term, the interests of consumers andproducers should converge. On the demand side, a wide application ofthe precautionary principle leads to inconsistencies, and consequentlyto inefficient risk regulation. It should be in the long-term interests ofconsumers to avoid such over-regulation, and to push for regulationsthat are oriented toward the most ‘objective’ risk assessment possible.On the supply side, producers could profit from public regulation ofrisky products, because regulation can secure the necessary reputationto sell these products. Additionally, producers are not necessarily theonly ones who pay the price for new regulations, because it is possible toshift parts of the costs toward consumers. Therefore, the conflict of con-sumer and producer interest is not as clear-cut as Wilson (1980: 357–94)states. In consequence, one may hypothesise that in the long term bothparties have common interests in the most efficient regulatory policies.
Efficiency and Legitimacy 41
PPL-UK_RR-Krapohl_Ch03.qxd 9/15/2008 10:26 AM Page 41

This does not mean that struggles about regulatory policies cannotoccur. In the short term, political disputes can be very hard and very fun-damental. However, many conflicts between consumers and producersabout appropriate levels of health and consumer protection exist only inthe short term, and will diminish as more and more information aboutregulatory problems becomes available in the long term.
3.2 Credible commitment and efficiency
Whenever actors face competing long- and short-term interests, theyneed to commit themselves to their long-term interests so as not toendanger these with their short-term interests. This argument is widelyapplied in the political economy literature about independent centralbanks. Here, it is argued that politicians face incentives to lower interestrates in the short-run to achieve short-lasting economic growth, whichmight help them to win the next election. However, in the long term, itis more rational to keep interest rates stable to avoid inflation, and toachieve long-lasting economic growth. As a result, politicians shouldcommit themselves to their long-term interests by delegating competen-cies for monetary policy to independent central banks, which then adoptinterest rates on their behalf and according to their long-term interests(e.g. De Grauwe 1997; Kydland and Prescott 1977). This argument alsoholds true for the case of risk regulation. In the long term, politiciansshould aim for efficient risk regulation that is in the interests of bothconsumers and producers of respective products. However, in the shortterm, politicians may be under considerable pressure from stakeholdersto adopt inefficient regulatory policies. Consumers may push towardsover-regulation in order to be protected from new threats, whereas pro-ducers may push towards under-regulation so as not to lose market share.If politicians followed the short-term interests of one particular group, thelong-term goal of efficient risk regulation would be endangered (Majone1996: 28–46, 2001a). Like Ulysses expecting the Sirens (Elster 1979),politicians have to bind themselves to the mast of institutions that pre-vent them from succumbing to short-term political pressure.
Credible commitment is all the more important for supranational riskregulation if one remembers that it interferes with the establishment ofthe Single Market (section 2.1). Within the EU, at least a qualified major-ity of 27 member states has to agree in order to adopt harmonised regu-latory standards for potentially risky products. If a blocking minority ofmember states represents the short-term interests of a particular group ofstakeholders – e.g. because they try to protect their own industry against
42 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch03.qxd 9/15/2008 10:26 AM Page 42

competition, or their consumers push for very strict regulatory standards –they may easily block the adoption of harmonised standards. In suchcases, mutual defection (i.e. the set up of non-tariff trade barriers forhealth and consumer protection) could be the result. To prevent this, themember states should bind themselves within supranational regulatoryregimes. The more they commit themselves, the better they will be ableto solve coordination problems and to avoid the negotiator’s dilemma.Thereby, the member states may either commit themselves by delegatingcompetencies to independent agents (section 3.2.1) or by legalising therespective policy area (section 3.2.2).
3.2.1 Credible commitment by delegation and the control problem
One possibility for actors to commit themselves credibly to certain pol-icy objectives is to delegate decision-making to agents (e.g. Franchino2002; Gilardi 2002; Majone 2001a). This act of delegation should bedesigned in a way that problems of time-inconsistent preferences aresolved. Agents should act in the long-term interests of their principals,but should not be influenced by particularistic short-term interests. Ifagents were influenced by such interests, they themselves would face thenegotiator’s dilemma and coordination problems, because they wouldhave to find the most efficient regulatory solutions and bargain for theirdistributive consequences simultaneously. Problems of decision-makingin the Council would thus only be passed down to agents, but theywould not be solved. Thus, for regulatory policy-making, agents shouldbe independent from any political interests, and act at arm’s length fromthe political sphere to build up ‘regulatory commitment’ (Majone 1996:28–46, 2001a; Majone and Everson 2001). The long-term interests ofprincipals should be laid down in the statutory rules for their agents, but in the following, principals should abstain from influencing day-to-day decision-making with their short-term interests. Such independentagents are sometimes also called ‘trustees’ of their principals (Majone2001a).
Because agents should not be influenced by particularistic short-terminterests, it is important to know who they are and how they arerecruited. In this respect, a great variety of different regulatory bodiescan be found at EU level. The central actor within all supranational reg-ulatory regimes is the Commission (Majone 1994). The Commissionalways owns the formal right of initiative, i.e. it develops decision pro-posals, which are then forwarded to member-state committees or theCouncil. The Commission sets the agenda of regulatory policy-making,
Efficiency and Legitimacy 43
PPL-UK_RR-Krapohl_Ch03.qxd 9/15/2008 10:26 AM Page 43

and it can easily veto regulations. However, the Commission is not theonly agent to which regulatory policy-making is delegated. Additionally,several expert or scientific committees are established to advise regula-tory policy-making. The composition of these committees may differwidely. Committees may consist of experts from member states’ regula-tory agencies, or they may comprise independent scientists. In additionto these committees, regulatory agencies are created in some policy areasand have the same task of scientific advice as expert and scientific com-mittees, but are more independent from the Commission (Shapiro 1997).Thus, the recruitment of supranational regulatory bodies differs widely,as does the independence of these bodies. Consequently, the composi-tion of regulatory bodies has to be analysed from case to case in order toevaluate member states’ commitment.
A problem inherent to any principal–agent relationship is the poten-tial drift of agents away from the interests of their principals. Within anuncertain and complex world, agents can always use some discretionarypowers and informational advantages to follow discrete interests(Calvert et al. 1989). Such agency-drifts may occur for two reasons, andboth have negative effects on the efficiency of regulatory policy-making.Firstly, agents may develop and purse discrete interests. They may try toincrease their budget (e.g. Niskanen 1973), or to enhance their owninfluence on policy-making (e.g. Dunleavy 1991; Moe 1990). For exam-ple, if agencies are created for regulating certain policy-areas, they maydevelop institutional interests in regulating these areas far beyond thepoints envisaged by their principals in order to legitimise their ownexistence. The result of such drifts would be that respective areas becomeover-regulated, and regulations would no longer be efficient (Breyer1993). Secondly, the more independent agents are, the more easily theycan be captured by particularistic interests. For example, regulatory agen-cies are in day-to-day contact with regulated producers. Agencies maybecome subject to intensive lobbying pressure from these producers, andmay take on industries’ interests in regulatory policies. The result of suchcaptures would be that respective areas would become under-regulated,and regulations would no longer protect consumers.
To counter agency-drifts, principals might choose to control their agentsmore strictly. Therefore, one possibility is to make agents’ decisions subjectto principals’ approval. Principals may establish oversight mechanismsthat scrutinise agents’ day-to-day policy-making, and which intervene ifregulations are not in the principals’ interests. Within the principal–agentliterature, such oversight mechanisms are called ‘police-patrol control’,because they imply regular checks of agents’ behaviour – just like police
44 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch03.qxd 9/15/2008 10:26 AM Page 44

patrols routinely control a certain district (e.g. Franchino 2000a;McCubbins and Schwartz 1987; Pollack 1997a). Whereas such police-patrol control surely reduces agents’ possibilities to deviate from princi-pals’ interests, it also bears one fundamental disadvantage: particularisticshort-term interests of principals might come back into day-to-day policy-making. Agents no longer act within an arm’s length of the politicalsphere, but their decisions are directly overseen by political bodies.Consequently, the stricter such oversight procedures are, the less credi-ble principals’ commitment to follow certain policy objectives becomes,and the more delegation gains are reduced (Tallberg 2002). Thus, if cred-ible commitment is the rationale behind delegation, strict oversightmechanisms are counter-productive.
In the EU, oversight over the Commission and the various expert bod-ies is performed within the so-called comitology system (e.g. Franchino2000a, 2000b; Pollack 1997a, 2003). To supervise the implementation of framework legislation, member states set up committees, which arechaired by representatives of the Commission, but which are made up of representatives from national ministerial bureaucracies. Usually, theCommission is obliged to pass its policy proposals to the relevant com-mittees before it makes regulatory decisions. Generally, committees issuetheir opinions to these proposals by qualified majority, whereby mem-ber states’ votes are weighted according to the same rules that apply tothe Council. Except in the case of an advisory procedure, proposals arepassed to the Council if committees’ opinions are negative. There are dif-ferent comitology procedures, which vary in the strictness of controlapplied to the Commission and relevant expert bodies.3 Advisory proce-dures do not include any formal competence of the member states toblock Commission proposals. Management procedures allow a qualifiedmajority of the member states to pass proposals on to the Council, andregulatory procedures even allow a qualified minority of the memberstates to block proposals and to forward them to the Council. Whereasthe strictness of control increases from advisory to management to reg-ulatory procedures, member states’ commitment declines along thissame line (Franchino 2000a, 2000b; Steunenberg et al. 1996).
Agents of the member states and the applied oversight mechanismstogether constitute supranational regulatory regimes. Therein, expertbodies – e.g. committees or agencies – give scientific opinions, theCommission develops policy proposals based on these opinions, theresulting proposals are forwarded to comitology committees, and if theseproposals are rejected by the committees they are finally decided on bythe Council. Obviously, the obligation of member states in such regimes
Efficiency and Legitimacy 45
PPL-UK_RR-Krapohl_Ch03.qxd 9/15/2008 10:26 AM Page 45

may differ widely, according to the more detailed rules of decision-making.Expert bodies may be recruited by the Commission or by the memberstates themselves. They may be relatively strong agencies or just weakercommittees. The Commission may be obliged to consider scientificadvice or may do this only voluntarily. The oversight of member statesalso differs according to the strictness of the applied comitology proce-dures. The hypothesis is that the more independent the recruitment ofexpert bodies is, and the stronger these bodies are within the followingdecision-making procedures, the better member states will be able toachieve the two policy goals of establishing a single market for poten-tially risky products and of effectively protecting consumers from therisks of these products.
3.2.2 Credible commitment by legalisation
Besides the establishment of strong oversight mechanisms, principalsmay also control their agents’ decision-making by setting up substan-tive criteria on which decisions have to be based, and by making theimplementation of these rules subject to judicial review. In the terms ofprincipal–agent theory, this strategy would be a combination of ex antecontrol by administrative statutes and ex post fire-alarm control by judicial review (e.g. Franchino 2000a; McCubbins and Schwartz 1987;Pollack 1997a). In other words, one could also call this a legalisation ofrespective policy areas (e.g. Abbott et al. 2000). Compared with oversightprocedures, this kind of control has a fundamental advantage: legalisa-tion does not only bind agents to the long-term interests of their princi-pals, but it also binds the principals and their control bodies themselves.Consequently, legalisation is not only a mean to control agents, but itmay also express principals’ commitment in certain policy objectives. Ifthis control method is applied, the whole supranational regulatoryregime – including expert bodies, the Commission and member states –operates in the shadow of law and judicial review. Here, credible com-mitment is not necessarily achieved by independent regulators, but byjudicial review of independent courts.
According to the widely accepted definition of Abbott et al. (2000),the degree of legalisation can be measured relative to three interdepen-dent dimensions. Firstly, legal rules can impose more or less obligationupon actors (i.e. they may simply be recommendations to behave in cer-tain ways, or they may impose duties on actors). Secondly, legal rulescan differ widely in their precision (i.e. they may leave much discretionfor actors, or they may describe in detail how these actors shouldbehave). Thirdly, the authority for settling disputes over the application
46 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch03.qxd 9/15/2008 10:26 AM Page 46

of legal rules can be more or less delegated (i.e. authority can be left toactors, or can be delegated to court-like bodies). One may speak of ‘hardlaw’ if legal rules are obligatory, precise and subject to judicial review.Such hard law obviously implies the most credible commitment of con-cerned actors. In contrast, one may speak of ‘soft law’ if one or more ofthe three dimensions are weakened (i.e. if legal rules are less obligatory,less precise or not subject to judicial review; Abbott and Snidal 2000).
Legalisation includes two mechanisms that are interdependent, butindividually distinct. Whereas the precision and obligation of legal rulesrestrict the discretion of actors, delegation refers to the supervision ofactors’ discretion. Firstly, the precision and obligation of legal rules influ-ence decision-making within supranational regulatory regimes, becausethey give actors criteria on which decisions will be based. When actorsplay iterated games, they have to accept relative losses from time totime to avoid gridlocks. In such circumstances, substantive criteria areimportant focal points that guide actors’ behaviour and back up coop-eration (Schelling 1995). Here, rules are upheld as long as all actorsprofit from ongoing cooperation (Stone Sweet 1999). The effect of sub-stantive criteria depends strongly on precision and obligation. If legalrules are vague, much discretion is left to pass arbitrary decisions.Consequently, cooperation is barely reinforced. However, if criteria areprecise and obligatory there is less discretionary room left, and cooper-ation becomes more stable.
Secondly, the supervision of legal rules by courts influences decision-making within supranational regulatory regimes, because it impliessome control not only over expert bodies and the Commission, but alsoover member states themselves. Actors may face incentives to break fromongoing cooperation, when short-term gains of defection are higherthan the discounted, long-term gains of cooperation. However, if rulesare reviewed by third parties (Stone Sweet 1999), actors within theregimes have to be aware that courts may later scrutinise their decisions.Thus, all actors – including the member states – must be able to justifytheir decisions based on substantive decision-making criteria (the ‘givingreasons requirement’, Shapiro 1988: 1–35, 1992). Consequently, sub-stantive criteria may significantly reduce actors’ possibilities to pushtheir own interests if these cannot be justified by valid reasons. This con-trol mechanism may ensure the obligation of member states to followtheir own long-term interests, which are laid down in the substantiverules of the regime.
The EU is probably the most legalised international organisation inthe world – even to the extent that it is not an ordinary international
Efficiency and Legitimacy 47
PPL-UK_RR-Krapohl_Ch03.qxd 9/15/2008 10:26 AM Page 47

organisation, but a unique political system that is one of a kind (Shapiroand Stone Sweet 2002; Stone Sweet and Brunell 1998; Stone Sweet andCaporaso 1998). This is mainly due to the strong position of the ECJ,which finds no equivalent in dispute settlement bodies of other inter-national regimes. With two important judgements in the 1960s, andwith the help of the Commission, as well as of national Courts, the ECJestablished the direct effect and supremacy of EU law and jurisdiction(Alter 2001: 182–208). From that point on, EU law has been superior tocontradicting national law, and it has legal effects for EU citizens, evenif it should not yet implemented by member states.
For the commitment of member states within supranational regula-tory regimes, nullity claims4 are the most important legal instruments,because they may address legal acts of EU bodies. Within such judicialdisputes, the ECJ examines whether the defendant EU bodies have beenincompetent to decide, have misused their powers, or whether theclaimed decisions infringe on treaties or any secondary legislation. Thestrength of judicial review depends on the scope of potential plaintiffs,who might have an interest in challenging the regimes’ regulatory deci-sions. Within the EU, this scope is very broad for an internationalorganisation. Not only member states, the EP, the Council and theCommission may bring nullity claims before the ECJ, but citizens mayalso institute such proceedings under certain conditions. To bring nul-lity claims before the ECJ or the Court of First Instance, citizens have todemonstrate that the legal acts in question are of ‘direct and individualconcern’ to them. The difficulty of this proof may differ between differ-ent regulatory policy areas, and so is the resulting strength of judicialreview. If regulatory decisions are directly addressed to natural or legalpersons, these addresses have no difficulty to demonstrate their con-cern. Consequently, they may always institute proceedings (Collatz1996: 135–59). However, if regulatory decisions have broader scopes, itmay be more difficult for potential plaintiffs to demonstrate that theyare individually and directly affected.
An open question is whether member states should better delegatecompetencies to independent agents, legalise the respective policy area, orboth, to commit themselves to certain policy objectives. The maximisa-tion of agents’ independence would obviously be a problem within the‘mixed polity’ of the EU (Majone 2002a, 2004, 2005: 83–106). Such inde-pendent agents would not only carry the danger of agency drifts, but evenmore importantly they would endanger the EU’s institutional balancebetween supranational bodies and member states in favour of the former.5
Because of their central position within the EU, member states are unlikely
48 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch03.qxd 9/15/2008 10:26 AM Page 48

to accept such far-reaching delegation and to give up procedural oversightin near future. Thus, it is more appropriate for the multi-level system ofthe EU if member states bind themselves to their long-term interests by legalisation of regulatory policy-making. However, legalisation alonewould probably not result in strong commitment from member states. Aslong as no delegation to agents takes place, member states themselveswould have to implement broader framework legislation by adopting reg-ulatory decisions in the Council. This means that a real differentiationbetween legislation and implementation would not take place, becausemember states could change broader framework legislations if strong par-ticularistic interests were at stake. The ECJ would not be able to imposejudicial review, because it would not be able to scrutinise misuses of com-petencies if the Council alone owned all competencies.
The solution for member states’ commitment problems has to befound between the two extremes. Commitment of member states can beachieved best if delegation and legalisation go hand in hand withinsupranational regulatory regimes. The ECJ can only scrutinise regulatorypolicy-making if the decision-making load is shared by member statesand their agents, because this is a situation in which both sides mightmisuse their competencies. If decision-making is differentiated accord-ing to legislation and implementation (Gehring 2002: 155–96), a systemof checks-and-balances between different actors (expert bodies and theCommission, member states and their control bodies, as well as the ECJ)emerges. It is of less importance whether member states establish policepatrol mechanisms to control their agents, as long as this oversight issubject to the same substantive rules as the decision-making of agentsthemselves. In such circumstances, increasing legalisation compensatesthe loss of delegation gains, and the ECJ becomes the main guarantor ofcredible commitment. Such a system of checks-and-balances, in whichdifferent bodies control each other, is much more appropriate for riskregulation in the EU than delegation of far-reaching competencies toindependent agents. The former may express the same obligation thanthe latter, but it does not require member states to give up their stakeswithin supranational regulatory regimes. The price to be paid by mem-ber states is that the greatest amount of control is delegated to the ECJ,even if member states themselves are still represented in the regimes.
3.3 Credible commitment and legitimacy
Supranational risk regulation is problematic in its democratic legiti-macy. Like all supranational policy-making, it could suffer from the EU’s
Efficiency and Legitimacy 49
PPL-UK_RR-Krapohl_Ch03.qxd 9/15/2008 10:26 AM Page 49

‘democratic deficit’. It is often argued that the EU lacks democratic legit-imacy, because it is not well-enough controlled by a strong parliament(e.g. Follesdal and Hix 2006), or because it has a neo-liberal bias (Scharpf1996b, 1997b, 1999). If these arguments hold true, this would of coursealso affect the regulation of the Single Market by the Commission andvarious expert bodies. Besides, supranational risk regulation is ofteneven more detached from public scrutiny than other areas of EU policy-making. It usually takes place in technocratic bodies like expert andmember-state committees or regulatory agencies. These bodies are notdemocratically elected and are thus not directly responsible to EU citi-zens. Moreover, their decision-making often lacks transparency or eventakes place behind closed doors. Consequently, a great deal of criticismthat is addressed to the EU as a whole may apply even more to one ofits core competencies – the regulation of the Single Market.
Consequently, it is important to ask which institutional mechanismsmay strengthen the legitimacy of supranational risk regulation. One candistinguish between input and output legitimacy, as has been repeat-edly suggested by Scharpf (e.g. 1970, 1999: 6–42, 2004). Accordingly,input legitimacy derives from ‘government by the people’, i.e. whenevercitizens are able to articulate their will within policy-making (section3.3.1). They may do so in national parliamentary elections or in theelections for the EP. In contrast, output legitimacy results from ‘govern-ment for the people’, i.e. whenever policies meet the interests of con-cerned stakeholders (section 3.3.2). Thus, this kind of legitimacy doesnot depend on the input of decision-making, but on the quality of itsoutcome.
3.3.1 The problem of input legitimacy
The most fundamental problem for the legitimacy of supranational riskregulation is that credible commitment is at odds with input legitimacy.Whereas self-binding of member states leads to efficiency gains in thelong-term policy goals of health and consumer protection, as well as theestablishment of a single market, it simultaneously leads to legitimacyproblems. If regulatory competencies are delegated to independentagents, the Weberian (Weber 1985: 551–79) ‘transmission-belt’ model ofadministration cannot be upheld (Majone and Everson 2001). Thismodel is built on the idea that non-elected bureaucracies are legitimisedby the fact that they are responsive to the will of elected politicians (inthe case of the EU, these would be member states’ governments ormembers of the EP). Consequently, the will of the people would be com-municated through elections of politicians, and from there by orders to
50 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch03.qxd 9/15/2008 10:26 AM Page 50

bureaucrats. However, this ‘delegation chain’ is interrupted if inde-pendent agencies do not respond to political orders. In this case, gov-ernment by the people – or in the words of Scharpf (1999: 6–42) ‘inputlegitimacy’ – no longer exists.
Besides, democratically elected politicians can also no longer answerthe demands of the people if they are bound by legalisation of respec-tive regulatory areas. In such cases, representatives of the member statesor the EP may participate in supranational regulatory regimes, but theyare bound by the substantive rules of decision-making and by judicialreview of the ECJ and the Court of First Instance. Thus, they can nolonger react if their constituencies ask for certain regulatory policies.The transmission-belt, which conveys people’s will into policies, is againdisturbed, and people cannot influence regulatory policies with theirdemands. Consequently, input legitimacy also does not exist in caseswhere elected politicians participate in regulatory policy-making, butare bound by detailed substantive rules.
As a result, input legitimacy has to be restricted to the adoption ofthe more general procedural and substantive rules of regulatory policy-making, at least if one does not want to endanger the efficiency ofsupranational regulatory regimes. This does not mean that democrati-cally legitimised actors should not play any role in supranational riskregulation. It is the task of these actors to establish supranational regu-latory regimes (Gehring 2003; Gehring et al. 2007). At this upper level,input legitimacy can play a decisive role. However, the implementationof the broader procedural and substantive rules should be delegated tothe respective regimes, where political actors are either excluded orbound to the rules they previously adopted. Thus, democratically electedactors could provide input legitimacy at the upper level, but not at thelower level of regulatory policy-making.
At the upper level (i.e. during the adoption of procedural and sub-stantive rules of supranational regulatory regimes), input legitimacymay differ according to different institutional settings. Firstly, the EP –the only directly elected body at the EU level – may be more or lessinvolved in the set up of supranational regulatory regimes. If the respec-tive regimes consist only of scientific and comitology committeeswhich are recruited by the Commission and the Council, the EP hasnearly no influence at the upper level, and thus input legitimacy resultsonly from the involvement of the member states. However, the delega-tion chain from national parliamentary elections to national govern-ments and finally to the Council is rather long ( Jachtenfuchs 1999),causing input legitimacy to be rather weak in such cases. However, if the
Efficiency and Legitimacy 51
PPL-UK_RR-Krapohl_Ch03.qxd 9/15/2008 10:26 AM Page 51

respective regimes include supranational agencies that have to be set upby legislative acts, the EP may be involved. Here, the EP’s influencedepends on the applied legislative procedures. Whereas the consulta-tion and cooperation procedures imply only an advisory role for the EP,the EP stands on equal footing with the Council in the co-decision pro-cedure (at least after its reform in the Amsterdam Treaty; Crombez 2003;Tsebelis and Garrrett 2000, 2001). Thus, input legitimacy is strongestwhen the establishment of supranational regulatory regimes resultsfrom constituting acts that are adopted in such a co-decision procedure.
Secondly, input legitimacy depends on the influence of various inter-est groups during the establishment of supranational regulatory regimes.Here, it is important that influences of different interests are well bal-anced. Concrete economic interests are usually favoured vis-à-vis diffuseinterest, like those of health and consumer protection, because the for-mer ones are easier to organise and to represent than the latter ones(Olson 1968: 52–64). Concrete economic interests are usually shared by a relatively small group of producers that have enough resources torepresent their interests effectively. On the contrary, diffuse interests inhealth and consumer protection are usually shared by large groups,which are not very well able to mobilise necessary resources. To increaseinput legitimacy in EU policy-making, these advantages for concreteeconomic interests should be balanced out by providing opportunitiesfor diffuse interests to raise their voices. Here again, the involvement ofthe EP becomes important. Usually, the EP is the ‘champion of diffuseinterests’ within legislative processes (Pollack 1997b). Because it is theonly directly elected body at the EU level, it is mainly dependent onbroad public support. Thus, it tries to hold positions that are favouredby large majorities. Therefore, involvement of the EP increases inputlegitimacy not only because the EP is directly elected, but also becauseit often represents the diffuse and disfavoured interests of consumers.
3.3.2 Output legitimacy and accountability
Because the day-to-day decision-making of supranational regulatoryregimes cannot be legitimised by input from democratically elected bod-ies, it must legitimise itself by the efficiency of its policy output (Majone1996: 284–301). Indeed, supranational regulatory regimes may be legiti-mate, because they provide policy outputs that cannot be achieved byindividual action of the member states (e.g. Menon and Weatherill 2002).Member states face difficulties in establishing a single market for riskyproducts when they act on their own (section 2.1). The mutual recognition
52 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch03.qxd 9/15/2008 10:26 AM Page 52

principle cannot be strictly applied, because member states would facethe danger of downward regulatory competition (Scharpf 1999: 84–120).Moreover, if member states tried to adopt harmonised standards withinthe Council, decision-making may easily be blocked and the adoption ofharmonised standards prevented. Supranational regulatory regimes maylegitimise their day-to-day policy-making, because they contribute posi-tively to the establishment of a single market and to the simultaneousprotection of consumers.
To avoid agency drifts and losses of output legitimacy, supranationalregulatory regimes have to be subject to after-the-ex post political scrutinyin the long term (e.g. Dehousse 1999; Everson 1995; Joerges 2000; Majone1996: 284–301; Majone and Everson 2001). Therefore, three differentmechanisms of accountability can be distinguished. Firstly, supranationalregulatory regimes may be embedded within networks of member states’regulatory agencies, and these national agencies may scrutinise supra-national regimes from a more scientific point of view (Dehousse 1997;Majone 1997). Of course, this possibility only exists if member stateshave established strong national regulatory agencies (section 3.2). Insuch cases, EU expert bodies could consist of representatives from mem-ber states’ regulatory agencies, instead of scientific experts from outside.Such representation of national regulatory agencies would again carrythe danger of influencing regimes with member states’ interests. However,there are two reasons that suggest regulatory networks are softer con-trol mechanisms than political oversight procedures. Representatives ofnational regulatory agencies act at some distance from their own gov-ernments. Their domestic regulatory agencies themselves are deemed to be, at least partly, independent from political influence, and withinexpert committees they should not be subject to any orders from nationalgovernments. Besides, representatives of regulatory agencies are usuallyscientists and not politicians or bureaucrats. As scientists, it is importantfor them to build up a reputation of independence and neutrality inorder to give their arguments credibility within scientific reasoning. Ifthey exploited their reputation by passing politically motivated deci-sions, their scientific arguments would lose credibility in the long term.Here, networks of regulatory agencies may work as mechanisms thatensure that a reputation can be built, and which sanction the exploita-tion of such reputation (Majone 1997).
Secondly, supranational regulatory regimes can be held politicallyaccountable by the Council and the EP. Here, the Council may controlregulatory regimes through the usual comitology procedures (Franchino2000a, 2000b; Pollack 1997a). Whenever expert bodies and the
Efficiency and Legitimacy 53
PPL-UK_RR-Krapohl_Ch03.qxd 9/15/2008 10:26 AM Page 53

Commission develop scientific opinions and policy proposals, they aredependent on the agreement of comitology committees or the Councilto make them law. This is of course a very strong accountability mech-anism, because it not only works in the long term, but also covers day-to-day policy-making. In contrast, it is striking that the EP is usually notactively involved in supranational regulatory regimes. It is regularlyinformed about decision-making within the comitology system (Bradley1992, 1999), but it has no competencies to veto regulatory policies. Theaccountability mechanism that may be applied by the EP is compara-tively weak. The EP may try to gain influence on regimes’ recruitment,but its success is likely to depend on two factors. It is necessary thatexpert bodies within regulatory regimes are agencies that are, to somedegree, independent from the Commission. Scientific advisory commit-tees belong organisationally to the Commission, so that the EP cannotinfluence their recruitment. Besides, Parliament’s success in influencingthe recruitment of agencies depends of course on its influence withinthe legislative procedures that establish the respective regulatory regimes(Kelemen 2002).
Thirdly, and finally, supranational regulatory regimes can be held judi-cially accountable by the ECJ and the Court of First Instance on behalfof EU citizens. The first precondition, which is necessary to allow stake-holders to bring claims in front of these courts, is that decision-makingwithin regulatory regimes is open and transparent (Dehousse 1999;Héritier 2003). It is only possible to hold actors within respective regimesaccountable if the decision-making process can be followed from theoutside. To increase the transparency of regulatory policy-making and tosimplify access to information, it is often claimed that the EU needs an‘administrative procedure act’ – similar to that in the USA – which wouldprescribe the rules of openness, transparency and public participation inregulatory decision-making (e.g. Dehousse 1999; Everson 1995; Majone1996: 284–301). Besides, the efficiency of judicial accountability dependsnot only on transparency, but also on the legalisation of the respectivepolicy area. If substantive decision-making criteria are neither precisenor obligatory, the discretion of actors within regulatory regimes is wide.Thus, it is difficult for courts to scrutinise regulatory policy-making,because actors will rarely exceed their discretion. And many actorsshould have rights to take legal action against EU decisions for Europeancourts to receive enough complaints to hold the respective regimesaccountable. Here, it is important that access to European courts is sym-metrical. So far, it has been necessary for potential plaintiffs to prove thatregulatory decisions are of their direct concern, in order to challenge
54 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch03.qxd 9/15/2008 10:26 AM Page 54

them in front of the European courts. Such proof is much easier for pro-ducers to deliver than for consumers, because regulatory decisions usu-ally interfere more directly with the rights of producers, than with thoseof consumers (Collatz 1996: 135–59).
There is a whole range of instruments available for making suprana-tional regulatory regimes accountable. However, none of these mecha-nisms should be allowed to dominate the supranational regulatoryregime, because this would allow a group of actors to influence the regimewith their particularistic interests. If experts dominated regulatory policy-making, the regime might be an unresponsive technocracy. Over-regulation would occur when experts identified themselves too muchwith the tasks of risk regulation, and under-regulation would take placewhen experts are captured by producers’ interests. If political bodies dom-inated regulatory policy-making, credible commitment would be reducedand the regime would again become politicised. Depending on which par-ticular interests prevailed in the political process, under- or over-regulationcould follow. In addition, if judicial accountability was the dominantmechanism, the result would depend on the access to European courts. Aslong as it is easier for producers than for consumers to demonstrate theirdirect and individual concerns, producers would have more influence, andunder-regulation could be the outcome. To avoid regimes being capturedby the interests of one particular group, multiple mechanisms should beapplied at the same time, and should work in close association. In suchcases, the influence of one group would be balanced by that of another. A situation would emerge where no single body is able to directly controlthe regime, but where the regime is nevertheless subject to control(Majone and Everson 2001; Moe 1987a).
3.4 Conclusion
The hypothesis in this chapter is that the efficiency of supranationalregulatory regimes, in establishing a single market for risky productsand in effectively protecting consumers from the risks of these products,depends on the credible commitment of the member states, which is expressed in the delegation and legalisation of regulatory policy-making. This need for credible commitment is due to the special char-acter of risk regulation. Consumers and producers of potentially riskyproducts both face time-inconsistent preferences with such regulation.In the short term, consumers may strive for rather high regulatory stan-dards in order to be protected from new threats, whereas producers maypush for low regulatory standards so as not to endanger their market
Efficiency and Legitimacy 55
PPL-UK_RR-Krapohl_Ch03.qxd 9/15/2008 10:26 AM Page 55

shares. However, in the long-term, consumers’ and producers’ interestsmay converge towards the most efficient risk regulation, the costs andbenefits of which balance each other. Because member states may faceconsiderable pressure to represent the short-term interests of particulargroups of stakeholders at the EU level, they need to commit themselvestowards their common long-term interests in order not to endanger anefficient regulation of the Single Market.
To commit themselves, member states may choose between two insti-tutional mechanisms. Firstly, they may delegate competencies for regu-latory policy-making to independent agents. These agents could solvethe problem of time-inconsistent preferences, because they would notbe subject to the same short-term interests and could thus act on behalfof their principals’ long-term interests. However, the establishment ofindependent agents always leads to control problems if agents developdiscrete interests or are captured by third-party interests. To avoid suchagency drifts, member states might choose to establish oversight proce-dures for their agents’ day-to-day policy-making, which could, of course,reintroduce particularistic short-term interests into regulatory decision-making. However, secondly, member states may also bind their agentsand themselves to criteria defined beforehand, subject to judicial reviewby European courts. Such legalisation seems to be an appropriate answerto the commitment problem in the multi-level system EU, becausemember states would not have to be excluded from regulatory policy-making. Although they would have to give up discretion for the over-sight of supranational regulatory regimes, they could nevertheless stillbe involved in their day-to-day operation. Credible commitmenttowards long-term policy objectives would therefore not be ensured byagents’ independence, but by independent judicial review provided bythe ECJ and the Court of First Instance.
The need for credible commitment to efficient risk regulation has twofundamental consequences for the legitimacy of supranational regula-tory regimes. Firstly, the possibilities for legitimising day-to-day policy-making with input from democratically elected actors are limited. Inputlegitimacy is necessarily restricted to the adoption of general proceduraland substantive rules for regulatory policy-making. During everydayregulatory policy-making, elected actors are either absent because therespective competencies have been delegated to independent regulators,or they still participate, but are bound to detailed substantive criteriaand can no longer react to their constituencies’ demands. As a result,supranational regulatory regimes are even more dependent on their out-put legitimacy. This output legitimacy may derive from the fact that
56 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch03.qxd 9/15/2008 10:26 AM Page 56

supranational regulatory regimes help to establish a single market forrisky products and effectively to protect consumers from the risks ofthese products. Both policy objectives could not be achieved at the sametime by the member states if these acted on their own. However, toensure output legitimacy, supranational regulatory regimes need to beaccountable to different actors. It is important that this accountability isnot only addressed to one particular group of actors, because this wouldallow them to influence the regime with their particularistic interests.Instead, a system of checks-and-balances – wherein no individual bodyalone is able to control the regime, but the regime is nevertheless undercontrol – should be established.
Efficiency and Legitimacy 57
PPL-UK_RR-Krapohl_Ch03.qxd 9/15/2008 10:26 AM Page 57

This page intentionally left blank

Part II The Authorisation ofPharmaceuticals in the EU
PPL-UK_RR-Krapohl_Ch04.qxd 9/12/2008 10:55 AM Page 59

This page intentionally left blank

4From National Crises to a StrongSupranational Regime: TheDevelopment of PharmaceuticalAuthorisation in Europe
In modern states, pharmaceuticals belong to the most regulated productson markets: Their safety, efficacy and quality are controlled by regulatoryagencies before they can gain access to markets. And when they entermarkets, pharmaceuticals are strictly supervised for unknown adverseeffects (the so-called pharmacovigiliance). Finally, most states also regu-late prices and advertising for medicinal products.1 However, it is impor-tant to keep in mind that this tight regulation of pharmaceuticals is arelatively recent phenomenon, and not a natural characteristic of thisgroup of products. The starting point for extensive pharmaceutical regu-lation was, in most West European states, the thalidomide scandal in thelate 1950s and early 1960s, when the supposedly harmless sleeping pillsContergan and Distaval caused thousands of birth deformities all overEurope (Feick 2000a, 2000b, 2002b; Krücken 1997: 93–110; Permanand2006: 1–18).
This chapter aims to test the hypothesis developed from the historical-institutionalist argument (see section 2.2). It argues that European phar-maceutical regulation clearly followed the first developmental path:accordingly, a regulatory scandal occurred before the establishment ofthe Single Market, and was followed by the establishment of strongnational regulatory agencies (section 4.1). The hypothesis is that thesenational regulatory agencies, which had the interests not to endangertheir own existence and competencies, influenced the further develop-ment towards a supranational regulatory regime for pharmaceuticals.This had two consequences for the establishment of a single market forpharmaceuticals. Firstly, because of its potentially deregulatory effects,the Single Market project contravened the interests of the national reg-ulatory agencies. Consequently, these agencies tried to veto the estab-lishment of a single market for pharmaceuticals (section 4.2). Secondly,
61
PPL-UK_RR-Krapohl_Ch04.qxd 9/12/2008 10:55 AM Page 61

to overcome this blockade, the newly established supranational regula-tory regime has to take care of the interests of national regulatory agen-cies. The national agencies are included in the supranational regulatoryregime, and the resulting regulatory network is comparatively inde-pendent from political interests, because it is not as big a threat to mem-ber states’ sovereignty as a completely new supranational body would be(section 4.3).
4.1 The thalidomide scandal
The thalidomide scandal had its origin in West Germany, where thesleeping pill Contergan – which contained thalidomide – was inventedand widely distributed by the company Grünenthal. However, licenseesof Grünenthal also sold thalidomide under different brand names in 45other states, among them the UK, Sweden, Italy, Ireland, the Netherlands,Belgium, Finland, Denmark and Austria. Only a few states – most notablythe USA, France and East Germany – were not affected, because they didnot authorise these medicinal products. Altogether, ten European states(not all of them were EU members at that time) were involved in thescandal, and the problem clearly had a European dimension. However,because the European markets for pharmaceuticals were not yet inte-grated in the 1960s, the addressees of consumer demands for stricter reg-ulation were still the European nation states. Because of the importanceof the German case, the following analysis of the thalidomide scandalconcentrates on the events in West Germany (section 4.1.1), but the casesof the UK and France are also discussed (section 4.1.2).
4.1.1 Thalidomide in Germany
In the pharmaceutical sector, the likelihood of large-scale scandalsincreased throughout the 1950s. At this time, industrially producedpharmaceuticals were not regulated in West Germany, and a generalpharmaceutical law did not exist (Batz 1986: 3–14; Kirk 1999: 20–2;Scheu 2003: 730). Only the mixing of medicines by pharmacists – whichhad been the standard method of pharmaceutical production for a longtime – was regulated by a general pharmacopoeia (a compendium withreceipts for medicines). However, the post-war era in Western Europe saw a ‘pharmaceutical revolution’ (Abraham 1995: 36–86; Sjöström andNilsson 1975: 15–38; Stephens and Brynner 2001: 1–18): Increasinglyeffective medicinal products were being developed by pharmaceuticalcompanies, their production was becoming industrialised, and sales weresteadily increasing. Because of this industrialisation, manufacturing of
62 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch04.qxd 9/12/2008 10:55 AM Page 62

medicinal products within pharmacies became less important. As aresult, an increasing part of the pharmaceutical market was basicallyuncontrolled, even though industrialised production of medicinal prod-ucts and their wide distribution on large markets increased the problemsof pharmaceutical safety. Thus, large groups of consumers would beaffected by unknown adverse effects of respective products.
In reaction to this industrialisation of the pharmaceutical sector, the‘Bundestag’ adopted its first pharmaceutical law in May 1961.2 This lawbrought some general improvements to the regulation of pharmaceuti-cals (Kirk 1999: 33). General substantive criteria for pharmaceuticalswere set up, a licensing requirement for the production of pharmaceu-tical products was introduced, and the sale of pharmaceuticals becamea monopoly of pharmacies. Nevertheless, the pharmaceutical law from1961 fell short of achieving an adequate level of health and consumerprotection (Scheu 2003: 739–49). The instruments for pre- and post-marketing control of pharmaceuticals by public authorities remainedrather limited, and the responsibility to ensure pharmaceutical safetywas left to producers. Although pharmaceuticals had to be registered atthe German Federal Health Office (Bundesgesundheitsamt, BGA) inBonn, the agency did not evaluate medicinal products, and could notdeny registration. Consequently, effective pre-marketing control wasnot possible. Moreover, although pharmaceuticals and their productionwere supervised by public authorities, the threshold for public inter-ventions and withdrawals from the market was very high. Withdrawalswere only possible if scientific proof existed that certain pharmaceuti-cals were dangerous, and if this danger resulted from the respectiveproducts alone and not from coincidences with other medical factors.Thus, suspicion of adverse effects or interactive effects with othermedicinal factors did not justify public intervention. As a result, effec-tive post-marketing control was also not possible.
The story of German thalidomide began shortly before the adoptionof the first pharmaceutical law, when Grünenthal began selling thesleeping pill Contergan (which contained thalidomide) in 1957. In the two previous years, thalidomide was tested in animal experimentsand clinical trials (Kirk 1999: 52–4). The evaluation of these tests waspositive, because thalidomide was found to have no toxic effects.Consequently, it was not possible for patients to commit suicide usingthalidomide. The non-toxicity of Contergan was extensively advertisedby Grünenthal to physicians and pharmacists (Knightley et al. 1979:25–41; Monser 1993: 12–15). In the following, Contergan became ahuge commercial success for Grünenthal. It became the most frequently
From National Crises to a Strong Supranational Regime 63
PPL-UK_RR-Krapohl_Ch04.qxd 9/12/2008 10:55 AM Page 63

used sleeping pill in West Germany, and accounted for 50 per cent ofGrünenthal’s sales in 1960 and 1961. During this time the companysold around 300 million daily allowances of the supposedly ‘harmless’sleeping pill (Kirk 1999: 35, 56).
Two years after Contergan had been placed on the market, the firstadverse effects of the sleeping pill were observed. In autumn 1959, aGerman neurologist informed Grünenthal that some of his patients suf-fered from previously unknown neural damage (the so-called thalido-mide polyneuritis), which he traced back to the intake of Contergan(Kirk 1999: 60–4). And in autumn 1960, the first concerns about neuralreactions to thalidomide were made public by a British physician(Stephens and Brynner 2001: 19–38). However, Grünenthal’s first reac-tions came rather late, and were only a partial answer to steadily increas-ing concerns (Kirk 1999: 70–83; Sjöström and Nilsson 1975: 52–71). Oneyear after the first neural reactions had been communicated to the com-pany, Grünenthal added a warning to the package leaflet of Contergan.Nevertheless, observations of adverse effects and the criticism fromphysicians continued to grow. Consequently, the Länder (states) NorthRhine-Westphalia, Hesse and Badem-Wuertemberg introduced a pre-scription requirement for Contergan in August 1961 (Kirk 1999: 76–8).However, the sleeping pill was still freely available in all other German‘Länder’, even though sales declined as a result of public concerns (Kirk1999: 78–83).
The most devastating effects of Contergan were not the neural reac-tions of patients, but the birth deformities of unborn children whosemothers took Contergan during pregnancy. German physicians hadalready noticed an increase in birth deformities since 1958, approxi-mately one year after Contergan was placed on the German market. Thetask of detecting a connection between birth deformities and the intakeof Contergan by pregnant women, as well as provoking the withdrawalof Contergan from the German market, was taken up by the Germanpaediatrician Widukind Lenz (Daemmrich 2002; Kirk 1999: 83–6,136–55; Knightley et al. 1979: 96–111). In summer 1961, he began sus-pecting Contergan of causing the birth deformities. Lenz based his sus-picion on interviews with mothers who gave birth to deformedchildren, and he saw an ethical need to take action against Contergan(Daemmrich 2002; Kirk 1999: 151–5). However, it became evident thatGrünenthal was not willing to withdraw Contergan from the Germanmarket. Public authorities from the German Länder were also not will-ing to prohibit Contergan, because Lenz’s data were still insufficient sci-entific proof of the side-effects of Contergan. Consequently, public
64 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch04.qxd 9/12/2008 10:55 AM Page 64

authorities feared being taken into regress by Grünenthal if theydecided to prohibit the sleeping pill. It took the publication of a news-paper article3 about the adverse effects of Contergan for Grünenthal tochange its course. The company finally withdrew the sleeping pill fromthe German market on 27 November 1961, after it had caused between3,000 and 4,000 birth deformities and an uncounted number of still-births in Germany.
The thalidomide scandal immediately demonstrated the fundamentaldeficits of the German pharmaceutical law of May 1961. The instru-ments for post-marketing control were insufficient, and did not allowfor an intervention of public authorities to prevent the catastrophe. Thedistribution of pharmaceuticals could only have been prohibited if ithad been scientifically proven that they were harmful to consumers.Thus, suspicions alone were not sufficient to take action against theproduct, and public authorities were frightened to be taken into regressby Grünenthal if they prohibited the sleeping pill (Kirk 1999: 111–13;Scheu 2003: 739–49). And the instruments for pre-marketing controlwould also not have been able to prevent the marketing of Contergan,even if the product had been subject to them (Contergan was placed onthe market four years before the adoption of the first pharmaceuticallaw). Although pre-marketing registration of pharmaceuticals at theGerman BGA was a formal requirement, it did not include any evalua-tions of respective products by the BGA. Thus Contergan would nothave been evaluated by independent scientists.
The thalidomide scandal provoked political reactions in Germany. InJune 1964, the Bundestag amended the German pharmaceutical law,4
which included two general reforms (Daemmrich 2004: 34–46; Kirk1999: 179–82). Firstly, a general prescription requirement was intro-duced for all new active agents. This aimed at better identification of adverse effects by the prescribing physicians. Thus, possibilities forpost-marketing control were improved by this measure. Secondly, theamendment required that pharmaceutical companies demonstrate tothe BGA that newly registered products had previously been tested.However, the BGA could still not deny registration of products if theywere insufficiently tested, but could only delay registration by askingfor more information. Further, there existed no guidelines for the test-ing of new pharmaceuticals. This changed when interest groups ofpharmacists and physicians developed legally non-binding guidelinesfor the testing of new medicinal products in the early 1960s, Later, inJune 1971, such guidelines were published as a ministerial regulation(Kirk 1999: 182–5). Nevertheless, even if the amendment of the
From National Crises to a Strong Supranational Regime 65
PPL-UK_RR-Krapohl_Ch04.qxd 9/12/2008 10:55 AM Page 65

pharmaceuticals law and the new guidelines on the testing of medici-nal products improved pre- and post-marketing control, they fell shortof establishing a full authorisation requirement and a pharmacovig-iliance system for pharmaceuticals.
In Germany, a modern regulatory regime for pharmaceuticals wasonly established by the second pharmaceutical law of 1976.5 The les-sons from the Contergan catastrophe heavily influenced the discussionsabout this law (Beyer 1989: 290; Kirk 1999: 190). The law brought fun-damental improvements for both pre- and post-marketing control ofpharmaceuticals by the BGA, as well as for liability of the pharmaceuti-cal industry (Kirk 1999: 188–90). Firstly, an authorisation procedure forall new medicinal products was introduced (Hart et al. 1988: 29–104).Within that procedure, the BGA examined whether the respectiveproducts were safe, had positive therapeutic effects and were of highquality. The examination was conducted based on the records of pre-clinical and clinical trials. The trials had to be conducted by the com-pany itself, and had to comply with the up-to-date guidelines of thenewly established Ministry of Youth, Family and Health. Secondly, post-marketing control of authorised pharmaceuticals was improved (Hart et al. 1988: 105–44). Therefore, the Ministry of Youth, Family and Healthwas empowered to set up a general administrative plan for the coordi-nation and cooperation of different federal and Länder authorities.Besides, the threshold for an intervention by public authorities was sig-nificantly lowered. Whereas scientific facts were necessary under thefirst pharmaceutical law to withdraw pharmaceuticals from the market,the second pharmaceutical law allowed such action also in cases of rea-sonable suspicion. Consequently, the BGA got more discretion for post-marketing control of pharmaceuticals.
In reaction to the thalidomide scandal, these reforms established amodern regulatory regime for pharmaceuticals at the national level,long before the Single European Act and long before a single market for pharmaceuticals was established. The BGA was at the centre of thisregime.6 Even though this regulatory agency was formally not fullyindependent from political influence by the Ministry (Hart et al. 1988:31–2), it had far-reaching competencies to authorise and withdrawmedicinal products, and it became an important actor in the regulationof the German pharmaceutical market. According to the historical-institutionalist argument, the BGA could use these competencies toinfluence the further path of development towards a single market anda supranational regulatory regime. For example, it could deny theacceptance of other member states’ authorisation for pharmaceuticals
66 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch04.qxd 9/12/2008 10:55 AM Page 66

and could thereby prevent the establishment of a single market bymutual recognition.
4.1.2 Other European countries
The early development of pharmaceutical regulation in the UK was verysimilar to that in Germany (Abraham and Lewis 2000: 49–56). Until the1960s, only the quality of pharmaceutical production within pharmacieswas regulated by a British pharmacopoeia. As in Germany, safety andefficiency regulation of industrially produced medicinal products didnot exist. Consequently, pharmaceuticals did not need any authorisa-tion to gain access to the British market. From April 1958, thalidomidewas distributed in the UK by the company Distillers under the brandname Distaval (Kirk 1999: 119–24; Knightley et al. 1979; Monser 1993:269–71). Distillers was a licensee of the German company Grünenthal,and placed Distaval on the market without conducting any further pre-clinical or clinical tests of the sleeping pill. Distaval was similarly aggres-sively advertised like Contergan in Germany. Even its prescription topregnant women was actively recommended. Nevertheless, the pharma-ceutical was less widespread in the UK and the number of victims wasconsiderably lower than in Germany. Whereas 3,000 to 4,000 childrensuffered from birth deformities in Germany, there were only 400 in theUK. When suspicions about the connection between intake of thalido-mide and birth deformities were corroborated, Distaval was taken off themarket on the same day as Contergan (27 November 1961).
Like in Germany, the UK regulatory regime for pharmaceuticals wasreformed after the thalidomide scandal (Abraham 1995: 36–86; Abrahamand Lewis 2000: 49–56; Hancher 1990). As early as 1964, a voluntaryauthorisation system was set up, wherein a Committee on the Safety of Drugs – comprising scientific experts – made assessments of newlyintroduced medicinal products. Despite the fact that the Committeelacked the power to prevent unsafe products from entering the market,only two pharmaceuticals that lacked the Committee’s approval weresold before 1968. This was because the British National Health Service,as the most important customer of pharmaceuticals, was able to applypressure to industry. Four years later, the Medicines Act of 1968 replacedthe voluntary system with compulsory authorisation for pharmaceuti-cals. Subsequently, pharmaceutical producers had to apply to theMedicines Division of the Department of Health for marketing authori-sation. During the assessment of pharmaceuticals, the licensing author-ity was supported by several committees. Most important among themwere the Committee on the Safety of Medicines and the Medicines
From National Crises to a Strong Supranational Regime 67
PPL-UK_RR-Krapohl_Ch04.qxd 9/12/2008 10:55 AM Page 67

Commission. The former evaluated all pharmaceuticals containing newactive substances, whereas the latter acted as an appellate body forindustry if products did not get approval of the former committee.Additionally, the committee established a pharmacovigiliance systemfor post-marketing control of pharmaceuticals, which became known asthe ‘Yellow Card System’, because it encouraged physicians to fill outyellow cards if they observed adverse drug effects (Abraham and Lewis2000: 53–4). In summary, the Medicines Act of 1968 established pre-and post-marketing control of pharmaceuticals, like those established in1976 in Germany. The Medicines Division at the Department of Health,and later the Medicines Control Agency (MCA),7 became the mainactors in the regulation of the British pharmaceutical market, becausethey had far-reaching competencies to authorise and withdraw medici-nal products from the market.
In contrast to Germany and the UK, France already had an authorisa-tion system for pharmaceuticals before the 1960s (Hancher 1989: 73–102;Reich 1988: 15–59). In 1941 the Vichy government introduced the so-called visa system. All pharmaceuticals needed permission from theMinistry to be allowed to enter the French market. To receive such a ‘VisaMinistériel’, pharmaceuticals had to be produced in France, they had tobe novelties (generics were only allowed after the original products hadbeen on the market for at least six years) and they had to be safe.However, the aim of the visa system was more toward the protection ofthe French pharmaceutical industry against competition from abroadthan toward the promotion of the safety and efficacy of medicines.Safety was usually assessed by experts from the industry itself, and anadditional committee to advise the Minister in this respect was only setup in 1953. Despite its weakness in safety regulation, the visa systemsaved France the experience of the thalidomide catastrophe, because thesleeping pill was not authorised on the French market. However, in the1950s, France suffered from a pharmaceutical scandal which had similareffects on its regulatory system as the thalidomide catastrophe had inGermany and the UK. In 1953, the pharmaceutical Stalinon – a medicinefor the treatment of staphylococcal infections – received a Visa Ministeriel,and was distributed on the French market. In the end, the product causedthe deaths of 100 persons and serious health damage in 117 others,because it contained toxic metal compounds.
After the Stalinon scandal, a twenty-year reform process of the Frenchregulatory regime began. It ended with pre- and post-marketing con-trols similar to those in Germany and the UK (Hancher 1990: 73–102;Reich 1988: 15–59). As early as 1959, the protection of pharmaceutical
68 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch04.qxd 9/12/2008 10:55 AM Page 68

innovations was taken out of the pharmaceutical law and became regu-lated by patent law. From then on, French pharmaceutical law concen-trated on health protection. Accordingly, all applications for visas had tobe accompanied by dossiers about the testing of respective products. Thedossiers became subject to the scrutiny of the visa committee within theDirectorate for Pharmaceuticals and Medicinal Products. Later, in 1967,the old visa system was replaced by a modern authorisation system forpharmaceuticals. And in subsequent years, standards for pre-clinical andclinical tests were adopted. A further breakthrough was the establish-ment of the Commission d’Authorisation de Mise sur les Marché desMédicaments in 1978. This commission was an independent expertcommittee that advised the licensing authority – i.e. the Directorate forPharmaceuticals and Medicinal Products – on the authorisation of med-icines (Reich 1988: 36–9). Lastly, post-marketing controls were intro-duced in 1982, when the National Commission of Pharmacovigiliancewas set up to advise the Directorate for Pharmaceuticals and MedicinalProducts in this respect (Reich 1988: 47–59). After that, the French reg-ulatory regime for pharmaceuticals was quite similar to the respectiveregimes in Germany and the UK, even if some minor particularities(like the reliance on registered experts from outside administration)persisted (Hancher 1990: 82–5). The Directorate for Pharmaceuticalsand Medicinal Products, and later the French Agency for the Safety ofSanitary Products,8 as the licensing authorities for pharmaceuticals,stood at the centre of this regime.
Like Germany, the UK and France established national regulatoryregimes for pharmaceuticals during the 1960s and 1970s, long before asingle market for these products was established. The reaction to criseswas made at the national level, because no single market for pharma-ceuticals existed and European integration was not advanced enough inthe 1960s and 1970s to allow for centralised, supranational answers tothese crises. The result of the reforms at national level was that newactors entered the arena. National regulatory agencies (or agency-likebodies) were established with far-reaching competencies and resourcesto authorise and supervise pharmaceuticals on national markets.According to the historical-institutionalist argument, these agencies arelikely to influence the further development.
4.2 Mutual recognition and the committee system
Not only member states, but also the EU itself, became active in the fieldof pharmaceutical regulation after the thalidomide scandal. Whereas
From National Crises to a Strong Supranational Regime 69
PPL-UK_RR-Krapohl_Ch04.qxd 9/12/2008 10:55 AM Page 69

member states built up national regulatory agencies to answer the crisis,the EU tried to lay the foundations for the establishment of a singlemarket for pharmaceuticals. However, at this time – it was the period ofthe Luxembourg compromise and the so-called Eurosclerosis – the EUwas not yet strong enough to provide a supranational answer to the crisis. As in other product sectors, the first attempt to create a singlemarket was to establish the mutual recognition principle and to bolsterthis with framework legislation. However, in line with the historical-institutionalist argument, this mutual recognition of member states’authorisations met with the resistance of national regulatory agencies,because it endangered their regulatory sovereignty. As a result, theseagencies used their competencies to deny mutual recognition and toblock the early establishment of a single market for pharmaceuticals.
As a response to the thalidomide scandal, the EU first took action in1965, when Directive 65/65/EEC introduced a general authorisationrequirement for new pharmaceuticals (Collatz 1996: 34–9; Thompson1994: 49–62).9 According to this Directive, every member state had toestablish pre-marketing controls for pharmaceuticals on its nationalmarket. The Directive prescribed that all authorisations, as well as with-drawals or suspensions of authorisations, had to be based only on theevaluation of safety, therapeutic efficacy and quality of respective medic-inal products. Economic or political reasons were not valid in justifyingauthorisation decisions, and only ethical considerations were allowed forthe prohibition of medicinal products with a positive cost–benefit ratioin exceptional circumstances (e.g. contraceptives and abortion pills inIreland). It is important to note that this Directive had already beenadopted before most EU member states introduced such authorisationrequirements for pharmaceuticals on their domestic markets. Thus, theEU did not only react to developments within its member states, but itreinforced and canalised national reactions to the thalidomide scandal.However, it took quite some time until all member states implementedthis early EU Directive (Collatz 1996: 36). For example, general pre-marketing controls for pharmaceuticals were introduced in 1967 inFrance and in 1968 in the UK, but only in 1976 in Germany.
The next step towards a European harmonisation of pharmaceuticalregulation followed ten years later with two Directives about testingrequirements and mutual recognition of pharmaceutical authorisation.Firstly, the annex of Directive 75/318/EEC10 (Collatz 1996: 39–42;Thompson 1994: 63–6) prescribed in detail the trials that had to be conducted to prove the safety, efficacy and quality of medicinal prod-ucts. Pre-clinical examinations included physico-chemical, biological or
70 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch04.qxd 9/12/2008 10:55 AM Page 70

microbiological tests, as well as toxicological and pharmacological trialson animals. The following clinical trials on patients were controlled by the ‘double blind’ method, according to which neither physicians nor patients were to know whether active substances or placebos wereapplied. Secondly, Directive 75/319/EEC11 set up the so-called ‘CommunityProcedure’ (later renamed to ‘Multi-State Procedure’) for mutual recog-nition of member states’ authorisations (Collatz 1996: 41–6; Thompson1994: 67–80; Vos 1999a: 207–9). Within this new procedure, pharma-ceutical companies were allowed to apply for recognition of one mem-ber state’s market authorisation by at least five (later two) other memberstates. This meant that the community procedure could only be startedif the respective product had already received positive authorisationfrom the so-called reference member state. Within the community pro-cedure, the other concerned member states had to decide whether theyaccepted the authorisation of the reference member state. Together, thetwo Directives established the foundations of the regulatory regime thatin principle governed the authorisation of pharmaceuticals in the EUover the next 20 years. Directive 75/318/EEC systematically harmonisedthe substantive criteria for the authorisation of pharmaceuticals. Thiswas deemed sufficient to allow mutual recognition of member states’authorisations in the ‘Community Procedure’.
The existing EU legislation about pharmaceutical regulation was onlyslightly reformed in the early 1980s. To facilitate mutual recognition of member states’ marketing authorisations, an expert committee, theso-called Committee for Proprietary Medicinal Products (CPMP), was setup (Collatz 1996: 42). This committee comprised representatives fromthe member states’ regulatory agencies, and it gave scientific advice formutual recognition of pharmaceutical authorisation. If disagreementsbetween member states occurred within the multi-state procedure, thenewly introduced expert committee issued opinions about the safety,efficacy and quality of the respective products. However, these scientificopinions were only recommendations and had no compulsory charac-ter for the concerned member states. Consultation of the expert com-mittee was deemed an attempt at arbitration in cases where mutualrecognition was blocked by objections from one or more concernedmember states.
A further change to the EU regulatory regime for pharmaceutical wasintroduced in 1987, when two Directives set up new decision-makingprocedures for the review of legislation and the authorisation of medic-inal products. The first Directive established a classic comitology proce-dure in the area of pharmaceutical regulation (Collatz 1996: 40).12
From National Crises to a Strong Supranational Regime 71
PPL-UK_RR-Krapohl_Ch04.qxd 9/12/2008 10:55 AM Page 71

Accordingly, the Commission received competencies to amend the annexof the Directive 75/318/EEC, which contained the substantive criteriafor the authorisation of pharmaceuticals. However, the Commissionwas not independent in fulfilling this task, but it became subject tomember states’ control within a relatively strict comitology procedure(regulatory procedure IIIa). The newly established ‘Standing Committeeon Medicinal Products for Human Use’ was composed of representativesfrom member states’ governments. Whenever the Commission aimedto amend the substantive authorisation criteria, it had to pass its pro-posals to this member state committee. The committee decided by qual-ified majority vote, whether it accepted the proposals or not. If thepolicies did not gain the support of the committee, the proposals werepassed to the Council, which finally decided on them by qualifiedmajority. If the Council failed to adopt decisions, the Commission wasfree to act.
The second Directive from 1987 set up the first centralised authori-sation procedure for pharmaceuticals in the EU, which was, however,not binding for the regulatory agencies of the member states.13 The so-called ‘Concertation Procedure’ (Collatz 1996: 47–8; Thompson 1994:81–8; Vos 1999a: 209–10) did apply to all biotechnologically producedpharmaceuticals and other innovative products were voluntarilyauthorised by this procedure. However, less innovative products orgenerics were still subject to the multi-state and national authorisationprocedures. Within the concertation procedure, the first member statethat received applications for pharmaceuticals asked the expert com-mittee (CPMP) for its scientific opinions before it adopted nationalauthorisation decisions. However, the committee’s recommendationsdid not oblige the member states to take respective action, and thenational regulatory agencies were still free in their authorisation deci-sions for their own domestic markets. Positive opinions of the expertcommittee facilitated authorisation but were no guarantee for positivedecisions by all national agencies.
As a consequence of all these attempts to facilitate mutual recognitionof member states’ authorisations, the policy area of pharmaceutical regu-lation became highly legalised in the EU (Hart and Reich 1990: 14–35).The substantive criteria for the evaluation of pharmaceuticals were har-monised at the EU level by Directives 65/65/EEC, 75/318/EEC and theiramendments. Consequently, in all EU member states, pharmaceuticalshad to be authorised on the basis of their safety, efficacy and quality, andthese characteristics had to be proven by the same pre-clinical and clini-cal trials. Additionally, the expert committee harmonised the substantive
72 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch04.qxd 9/12/2008 10:55 AM Page 72

authorisation criteria even further. In its competence to inform appli-cants about EU rules on pharmaceutical authorisations, the committeepublished many legally non-binding guidelines. By the end of the 1980s,the committee had published a ‘Notice to Applicants’ with detailed rulesfor application dossiers, and principles for Good Manufacturing Practise,as well as several guidelines about the safety, efficacy and quality of phar-maceuticals (Collatz 1996: 41).14 With these guidelines, the expert com-mittee further reduced the discretion of applying companies, whichincreased their chances for positive authorisation decisions when theyfollowed the committee’s advice.
The systematic harmonisation of substantive authorisation criteriacontrasted with the weak procedural rules of the EU regulatory regimefor pharmaceuticals. Until the late 1980s, the EU had established a com-plex institutional architecture to set up a single market for pharmaceu-ticals. A comitology procedure existed to amend the substantive criteriafor the authorisation of medicinal products, a multi-state procedureexisted to facilitate the mutual recognition of member states’ authorisa-tions, and a concertation procedure aimed to achieve unified evalua-tions of highly innovative medicinal products. Two committees playeda role within these different procedures. A member state committeecontrolled the Commission within the comitology procedure for theamendment of pharmaceutical legislation, and an expert committeegave scientific opinions about the safety, efficacy and quality of medic-inal products. Despite this differentiation at the supranational level, theEU was still not able to adopt binding authorisation decisions for phar-maceuticals on the European market. All decisions of the expert com-mittee needed the approval of member states’ regulatory agencies.
The strategy of partial harmonisation and mutual recognition failedto establish a single market for pharmaceuticals in the EU. Generally,the member states’ agencies did not recognise each other’s authorisa-tions or the scientific opinions of the expert committee, but preferredto evaluate the respective products on their own (Collatz 1996: 49–50;Feick 2000a; Vos 1999a: 210). Not a single authorisation from the 122applications under the multi-state procedure between 1986 and 1990was simply recognised by the concerned member states,15 and referenceto the expert committee turned out to be the rule rather than the excep-tion. Even though the substantive criteria for evaluation of pharmaceu-ticals were widely harmonised, they still left enough discretion for theadoption of different authorisation decisions by national regulatoryagencies. In particular, the criteria of safety and efficacy often constitutea trade-off, which does not necessarily lead to a single optimal decision.
From National Crises to a Strong Supranational Regime 73
PPL-UK_RR-Krapohl_Ch04.qxd 9/12/2008 10:55 AM Page 73

The more therapeutic effects pharmaceuticals have, the more they carrythe risk of adverse side-effects (Heilmann 2002). Consequently, both cri-teria cannot be fulfilled at the same time. Their balancing was left to thenational authorisation agencies and differed widely between the mem-ber states (Glaeske et al. 1988), so that a single market for pharmaceuti-cals did not emerge.
The weakness of the EU within the two authorisation procedures wascomplemented by an even weaker influence on post-marketing controlof pharmaceuticals. Pharmacovigiliance was left to the member states,and national systems of market surveillance were not harmonised by EUlaw (Hart and Reich 1990: 26–9). Member states only had to inform theexpert committee if they wanted to take action against authorised phar-maceuticals (i.e. if they suspended or withdrew authorisations). Theexpert committee tried to replace the lack of pharmacovigiliance atsupranational level with legally non-binding action. It set up a workinggroup for pharmacovigiliance, established a rapid-alert system for adverseeffects of pharmaceuticals, and met regularly to discuss routine mattersof pharmacovigiliance (Hart and Reich 1990: 116–19). However, alldecisions adopted by the expert committee needed the approval of thenational regulatory agencies to become effective in the member states.Consequently, the weak pharmacovigiliance reinforced the problems inestablishing a single market for pharmaceuticals (Collatz 1996: 49–50;Glaeskeet al. 1988).
In line with the historical-institutionalist hypothesis, the regulatoryagencies of the member states used their competencies to prevent theearly establishment of a single market for pharmaceuticals by mutualrecognition. Until the 1990s, the EU was not able to establish such asingle market. Even though the weak supranational authorisationregime was not the only reason for this – different health systems andprice regulations in the member states had additional effects(Mossialos et al. 2004; Permanand and Altenstetter 2004; Permanand2006: 151–79; Hancher 2004) – it nevertheless contributed significantlyto the national fragmentation of the European pharmaceutical market.The national regulatory agencies were still able to make decisionsindependently from each other and from scientific advice from theexpert committee. They had no interest in establishing a single mar-ket for pharmaceuticals by mutual recognition, because this wouldhave required giving up regulatory competencies to other memberstates’ agencies or to the EU expert committee. Thus, despite the sys-tematic and detailed harmonisation of the substantive authorisationcriteria, the national regulatory agencies widely refused to accept other
74 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch04.qxd 9/12/2008 10:55 AM Page 74

member states’ authorisations (Collatz 1996: 49–50; Feick 2000a; Vos1999a: 210).
4.3 The European Medicines Agency
The failure of mutual recognition of national pharmaceutical authorisa-tion led to a failure of market integration in this sector. At the end of1992, when the Single Market programme had already been imple-mented in most other product sectors, a single market for pharmaceuti-cals still did not exist, and new strategies were needed to achieveuniform access of pharmaceuticals to the markets of all member states.A policy window opened up, which allowed for a fundamental reformof the EU regulatory regime for pharmaceuticals (section 4.3.1). Duringthe following years, the regime worked rather well without raising pub-lic concerns. It was evaluated positively by stakeholders within a survey,which was conducted on behalf of the Commission at the beginning ofthe new millennium. As a result, the latest reform led only to modestchanges, and reinforced rather than abolished the main characteristicsof the regime (section 4.3.2).
4.3.1 The centralisation of the regime in the 1990s
The European pharmaceutical industry – at least the producers of inno-vative medicinal products – had a strong interest in the establishment ofa single market (Feick 2002a). A bigger market would allow for highereconomies of scale, and uniform access to the markets of all memberstates would reduce the costs and risks of authorisation. Consequently,when it became obvious that mutual recognition failed to create a singlemarket, industry became more receptive for more centralised solutions.In 1988, the Association of the British Pharmaceutical Industry pub-lished its ‘Blueprint for Europe’ (Abraham and Lewis 2000: 80–114).Therein, industry stressed the importance of single authorisation for thewhole Single Market. It suggested making biotechnologically producedpharmaceuticals subject to a centralised authorisation procedure underthe lead of an EU agency, whereas other innovative products should be voluntarily subject to such a procedure. All other, less innovativepharmaceuticals – including the large group of generics – should still besubject to a mutual recognition procedure; but if mutual recognitionwere to fail, the new EU agency should be able to adopt binding arbitra-tion decisions. The supposed distinction between different kinds ofproduct corresponded to the scope of the old concertation procedure.The Commission took up these suggestions within a memorandum,
From National Crises to a Strong Supranational Regime 75
PPL-UK_RR-Krapohl_Ch04.qxd 9/12/2008 10:55 AM Page 75

which described the ‘Future System for the Authorization of MedicinalProducts in the European Community’ (Hart and Reich 1990: 36–48).Therein, the establishment of a European Medicines Agency (EMA), aswell as the introduction of a centralised and a mutual recognition pro-cedure, was proposed.
The Commission published its first proposal for a new EU regulatoryregime for pharmaceuticals at the end of 1990. The goals of the newregime were threefold: to protect public health within the EU, toenhance the competitiveness of the European pharmaceutical industry,and to follow the general policy goals of the community, i.e. to establisha single market for medicinal products.16 The Commission therefore pro-posed two legislative acts. A regulation set up a new European Agency forthe Evaluation of Medicinal Products (EMEA; later renamed the EuropeanMedicines Agency), which incorporated the existing expert committee(CPMP) recruited from member states’ regulatory agencies. The same reg-ulation also established a centralised authorisation procedure,17 which iscompulsory for biotechnologically produced pharmaceuticals and volun-tary for other innovative medicinal products for both human and veteri-nary use.18 Authorisations resulting from the new centralised procedurewere binding for all member states, and granted uniform access to thecomplete single market. The new agency and its expert committee advisedthe Commission and the member states within this centralised proce-dure, but all authorisation decisions were still subject to member states’approval within a comitology procedure. Furthermore, a Directive wasproposed for the reform of the multi-state procedures.19 The mutual recog-nition procedure differed from the old multi-state procedure in compul-sory arbitration. If mutual recognition failed, an arbitration proceduresimilar to the centralised authorisation procedure applied, which led tobinding decisions for all member states that were addressees of the initial applications. Both the centralised and the mutual recognitionprocedure still used the substantive authorisation criteria laid down in Directives 65/65/EEC and 75/318/EEC. Thus, the reform concernedonly procedural, but not substantive, rules of the regulatory regime.
Within the following legislative process, the EP suggested some amend-ments to centralise the new regime even further (Krapohl 2005:81–104).20 It wanted to expand the scope of application of the cen-tralised authorisation procedure, it suggested recruiting the agency’sexpert committee from independent scientists and not from delegates ofthe national regulatory agencies, and it aimed to reduce member states’control in the comitology procedure. However, the legislative proposalsof the Commission were subject to a consultation procedure, wherein
76 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch04.qxd 9/12/2008 10:55 AM Page 76

the EP had only advisory competencies (Tsebelis and Garrett 2000;Tsebelis and Kreppel 1998). As a result, the legislative process mainlyreflected a compromise between the Commission and the Council,whereas the influence of the EP was, because of its weak standing withinthe consultation procedure, rather limited (Kelemen 2002). With onlyone exception (the Council accepted two delegates of the EP within theagency’s management board), the far-reaching EP amendments wereunsuccessful. The changes remained rather modest compared with thefirst Commission proposal, and the new regime became less centralisedthan envisaged by the EP. The legislative package was finally adopted bythe Council in 1993 and came into force on 1 January 1995.21
The EMEA constitutes the core of the new supranational regime(Collatz 1996: 56–64; Permanand 2006: 117–50; Thompson 1994:89–122; Vos 1999a: 216–22). The management board is the administra-tive centre of the agency. It is composed of two representatives fromeach member state, the Commission and the EP. Thus, member staterepresentatives constitute a large majority compared with representa-tives of the two supranational organs. The management board’s respon-sibilities are the adoption of general reports, working programmes andyearly budgets for the agency. Further, the board appoints the executivedirector on a proposal of the Commission. The director is the head andlegal representative of the agency. He is responsible for day-to-dayadministration and execution of the budget. Whereas the managementboard and the executive director perform administrative tasks, theexpert committee is the scientific core of the agency. The committeewas (up to the reforms of 2004) composed of two experts from eachmember state. Committee members usually belong to the nationalauthorisation agencies, but member states may also appoint expertsfrom universities or research institutes (which they do very rarely). Theembedding of committee members within their national regulatoryauthorities leads to the network character of the European pharmaceu-tical agency (Dehousse 1997; Majone 1997). The groundwork of scien-tific evaluations of pharmaceuticals is not conducted within the EMEAitself, but by committee members within their own national agencies.The EMEA itself lacks scientific resources, and is therefore dependent onthe cooperation of the national authorisation bodies.
The EMEA plays a prominent role within the newly centralised autho-risation procedure (Collatz 1996: 65–89; Thompson 1994: 89–122; Vos1999a: 212–14). Every biotechnologically produced pharmaceuticalmust, and other innovative medicinal products may voluntarily, beauthorised within this procedure. Pharmaceutical companies have to
From National Crises to a Strong Supranational Regime 77
PPL-UK_RR-Krapohl_Ch04.qxd 9/12/2008 10:55 AM Page 77

address applications for marketing authorisations directly to the agency.Within the agency, the expert committee is responsible for scientific eval-uations of applications. As soon as scientific opinions are adopted, the sci-entific phase of the authorisation procedure is complete, and the politicalphase begins. In this phase, the Commission develops decision proposalsbased on the committee’s opinions. These proposals are then forwardedto a member state committee (the Standing Committee on HumanMedicinal Products), which makes its decisions within a written proce-dure by qualified majority. If the proposals do not receive support of a qualified majority within the committee, they are forwarded to theCouncil, which may veto them by simple majority, adopt them by quali-fied majority or amend them by unanimity (regulatory procedure IIIb).
The mutual recognition procedure is – as the name suggests – stillbased on the mutual recognition principle (Collatz 1996: 90–103;Thompson 1994: 123–40; Vos 1999a: 214–15). Accordingly, if pharma-ceutical companies apply for marketing authorisations for less innova-tive medicinal products in more than one member state, the memberstates should recognise decisions of the first member state (the so-calledreference member state). The concerned member states may deny recog-nition of authorisation only if the respective medicinal products pres-ent a risk to public health. If different positions prevail, a centralisedarbitration procedure begins, which can be avoided if applying compa-nies withdrawal their applications from reluctant member states. Thearbitration procedure is similar to the centralised authorisation proce-dure. However, in contrast to the latter, authorisation resulting from anarbitration procedure is not valid throughout the whole Single Market,but only within the member states concerned.
Finally, the pharmacovigiliance system has become more centralisedand coordinated by the new EU agency (Collatz 1996: 84–7 and 99–101;Thompson 1994: 153–92; Vos 1999a: 215–16). Accordingly, authorisa-tion holders have to collect all information about suspected adverseeffects of respective medicinal products, and have to report them regu-larly to the agency and the member states. Further, member states them-selves are obliged to establish national pharmacovigiliance systems, andto report all serious side-effects to the EU agency immediately. If adverseeffects require the adoption of further regulatory measures (i.e. suspen-sions or withdrawals of authorisations) member states are allowed totake intermediary measures to protect human health. However, in anycase, a centralised procedure should be started to re-evaluate respectiveproducts, regardless of whether these were authorised by the centralisedauthorisation or the mutual recognition procedure.
78 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch04.qxd 9/12/2008 10:55 AM Page 78

The new supranational regulatory regime for pharmaceuticals becamemuch more centralised than its predecessor: a European agency wasset up, a binding centralised authorisation procedure was introduced,the mutual recognition procedure was complemented by a centralisedarbitration procedure, and the pharmacovigiliance system became cen-tralised under the guidance of the new EU agency. Despite this funda-mental reorganisation of the regime, path dependencies of institutionaldevelopment can clearly be observed: The substantive criteria for theauthorisation of medicinal products remained the same as in the exist-ing regime, the distinction between innovative and less innovativepharmaceuticals which are subject to different authorisation procedureswas maintained, and – most importantly – the old expert committee(CPMP) composed of representatives of member states’ regulatory agen-cies became the central body of the new EU agency. Consequently, thenational agencies are represented at a prominent place within the newregime, and they constitute a strong regulatory network. The regimecould not function without their support, and their existence andimportance are therefore ensured.
4.3.2 The reform process at the beginning of the new millennium
The EU regulatory regime for pharmaceuticals was again reformed at thebeginning of the new millennium (Broscheid and Feick 2005). This wasdue to two facts. Firstly, the Regulation that set up the new regime in1993 obliged the Commission to conduct an evaluation of both the cen-tralised authorisation and mutual recognition procedures within six yearsof their operation. After an extensive survey among concerned stake-holders, an evaluation report was published in October 2000.22 Althoughthe report demonstrated overall satisfaction with the regime, it alsopointed to some issues that justified legislative review. Secondly, the EUwas already awaiting enlargement of twelve additional member statesfrom Middle and Eastern Europe. The extension of the number of mem-ber states from 15 to 27 made some reforms of the supranational regula-tory regime necessary. However, whereas the establishment of the EMEAin the 1990s can be seen as a critical juncture, the latest reform was onlya routine revision of the regime, and central features of the regime werereinforced. Overall, the reform process was more a confirmation of theregime’s success than a response to any serious deficiencies.
For the most part, the evaluation report of two consulting companieson behalf of the Commission came to very positive conclusions aboutthe EU regulatory regime for pharmaceuticals.23 Overall, the variousstakeholders in the system – e.g. supranational and national regulators,
From National Crises to a Strong Supranational Regime 79
PPL-UK_RR-Krapohl_Ch04.qxd 9/12/2008 10:55 AM Page 79

the pharmaceutical industry as well as physicians’ and patients’ organi-sations – seemed to be satisfied with its operation. Thus, there was noneed to reform the overall structure of the regime. However, on a closerlook, some differences between the performance of the two authorisa-tion procedures became visible. On the whole, the centralised procedureseemed to be more successful than the mutual recognition procedure,because the latter still failed to achieve mutual recognition of memberstates’ authorisations, and was thus not able to establish a single marketfor less innovative medicinal products. Arbitrations were seldom startedafter mutual recognition failed, because applying companies withdrewtheir applications from member states that raised serious objectionsagainst their products (Feick 2002b).
The centralised procedure was also addressed by some minor cri-tique. The pharmaceutical industry in particular regarded the politicalphase of the procedure as superfluous. Whereas the Commission andthe member states had not changed any authorisation decisions up to 2000 (Krapohl 2005: 105–32), the comitology procedure delayedmarketing of new medicinal products for another two to five months.Nevertheless, an overwhelming majority of applicants and nearly allregulatory agencies preferred an extension of the scope of the cen-tralised procedure. Moreover, the consumers of pharmaceuticals – i.e.physicians’ and patients’ associations – also preferred the centralisedauthorisation to the mutual recognition procedure. Overall, the core ofthe new EU authorisation regime for pharmaceuticals – namely thecentralised authorisation procedure – received support from the evalu-ation report. No group of stakeholders expressed any interest in takingback the centralisation of the authorisation regime. In contrast, furthercentralisation was clearly favoured.
The subsequent legislative review started in November 2001, whenthe Commission proposed a reform package to the Council and the EP.24 The most important proposals of the Commission were to extendboth the compulsory and voluntary application of the centralisedauthorisation procedure, to reduce the number of member states’ repre-sentatives within the management board and the expert committee(renamed the Committee for Human Medicinal Products (CHMP)) and toreduce member states’ control within the political phase of the authori-sation procedures. The name of the European agency was also changedto European Medicines Agency (in place of the former, rather cumber-some European Agency for the Evaluation of Medicinal Products).
The legislative process and its outcome differed in two respectsfrom that of the 1990s. On the one hand, backed up by the positive
80 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch04.qxd 9/12/2008 10:55 AM Page 80

evaluation of the centralised authorisation procedure, all three legisla-tive actors preferred more centralised solutions than they had ten yearsbefore. On the other, because of the newly applied co-decision proce-dure, the EP had much more influence on the legislative process (Tsebelisand Garrett 2000, 2001), and used its competencies to strengthen itsown position and the representation of diffuse consumer interests(Kelemen 2002). As in the 1990s, the EP opted for an even more cen-tralised regulatory regime for pharmaceuticals than that suggested bythe Commission.25 It demanded an even wider extension of the scopeof the centralised authorisation procedure, a more independent recruit-ment of the agency’s management board and expert committee, and fur-ther reduction of member states’ control in the comitology procedure.Because of the co-decision procedure, the Council had to give significantconcessions to the EP.26 The scope of the centralised authorisation pro-cedure was expanded, the recruitment of the expert committee and themanagement board were opened to independent experts and stake-holder representatives, and member states’ control in the political phaseof the authorisation procedures was reduced to a management proce-dure, wherein a qualified majority was needed to veto proposals and topass them to the Council (see also section 5.1.2).27 Overall, the EU regu-latory regime for pharmaceuticals became more centralised and moreindependent from influence of the member states.
In response to criticism contained in the evaluation report, the mutualrecognition procedure was also reformed (Broscheid and Feick 2005).28
Whereas the old mutual recognition procedure started only after the so-called reference member state issued authorisation, this order may nowbe reversed. Within a newly established decentralised procedure, the ref-erence member state may inform the other concerned member statesabout its evaluations before it issues authorisation. This leaves moreroom for discussion between the member states, because no memberstate has taken a binding decision. Another problem was that before thelatest reform, conflicts between the member states did not necessarilyneed to be solved, because applying companies were allowed to with-draw their applications selectively in member states that opposed autho-risation of respective products. However, this strategy prevented theexamination of potentially reasonable objections to medicinal products.Disputes were not solved, but only avoided. The legislative review cor-rected this shortcoming. Although it is still possible for pharmaceuticalcompanies to withdraw their applications selectively, this no longer pre-vents centralised arbitrations. Thus, the mutual recognition procedurehas also become further centralised within the legislative review.
From National Crises to a Strong Supranational Regime 81
PPL-UK_RR-Krapohl_Ch04.qxd 9/12/2008 10:55 AM Page 81

The EU pharmacovigiliance system was also subject to the evaluationreport – and was one of the very few subjects of critique.29 Most patients’associations considered the old system to be insufficient in providing anadequate level of safety, even if this judgement was not shared by indus-try and regulatory authorities. As a consequence of patients’ criticism,the pharmacovigiliance system was further strengthened and centralised.To reduce the necessity for intermediate measures of member states, theCommission itself received competencies to take emergency measureswithout the support of the member state committee or the Council. In such circumstances, a comitology procedure follows only after themeasures are already in place.
Although the evaluation report dealt extensively with the proceduralrules of the EU regulatory regime for pharmaceuticals (i.e. with the cen-tralised authorisation and mutual recognition procedures or the EUagency for pharmaceuticals), little can be learned about stakeholders’ sat-isfaction with the substantive authorisation requirements for pharma-ceuticals (i.e. the rules laid down in Directives 65/65/EEC and75/318/EEC). Nevertheless, these substantive rules were also repeatedlyamended and revised after the establishment of the new EU regime in1995. Thus, the path towards increasing legalisation of the policy area –which had already started in the 1960s to facilitate mutual recognition –was continued.
The substantive authorisation criteria were adapted to special groupsof products. As early as December 1999, special rules were adopted fororphan medicinal products, i.e. pharmaceuticals for very rare illnesses.30
Because investments in such products are often not profitable for phar-maceutical companies, the EU tries to support such investments by pro-viding additional incentives. If the Committee for Orphan MedicinalProducts (COMP; a newly established expert committee within theEMA) confirms that authorised pharmaceuticals meet the criteria oforphan medicinal products, they get additional protection against com-petitors. No other pharmaceuticals with similar indications will beauthorised for the next ten years, unless they are able to prove superiorsafety, efficacy or quality. Some years later, in 2004, specific rules werealso set up for traditional herbal pharmaceuticals.31 Even though theseproducts usually look back on a long record of therapeutic use, it isoften impossible to prove their efficacy according to scientific stan-dards. In order not to suppress these products from the Single Market, asimplified registration procedure was established. Member states shallmutually recognise registrations of herbal medicinal products, and theCommittee for Herbal Medicinal Products (HMPC; also a newly established
82 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch04.qxd 9/12/2008 10:55 AM Page 82

expert committee within the agency) supports mutual recognition byproviding expert opinions.
Besides, the substantive authorisation criteria that had so far been laiddown in various Directives were consolidated within a single legislativeact. In 2001, Directive 2001/83/EC32 established the Community coderelating to medicinal products for human use. This code included theprovisions of the old Directives 65/65/EEC and 75/318/EEC, as well asthe rules of the mutual recognition procedure. The substance of the oldDirectives was not changed by this act. However, two years later in2003, the annex of the Community code, which contained the testingrequirements for pharmaceuticals, was reformed within a comitologyprocedure.33 Together with the Standing Committee on MedicinalProducts for Human Use, the Commission adapted the testing require-ments to new international rules, which were laid down in the ‘CommonTechnical Document’ of the International Conference on Harmonisationof Technical Requirements for Registration of Pharmaceuticals for HumanUse (ICH). This conference is a regular meeting of regulatory agencies of the USA, Japan and the EU (represented by the EU pharmaceuticalagency). It tries to harmonise substantive criteria for the authorisation of medicinal products to facilitate international trade (Vogel 1998). Withthe adaptation to the ICH guideline, the annex of the Community code was fundamentally changed without participation of the legislativeactors.
Although the expert committee was set up as an advisory body for theevaluation of medicinal products, it became a rule-setting actor on itsown during the 1990s. Guidelines of various working groups of the expertcommittee became more and more important for the specification of sub-stantive authorisation criteria (Krapohl 2005: 133–46). Although most ofthe guidelines are not legally enforceable, they develop binding characterin practice, because compliance with these rules reduces the risk ofunfavourable authorisation decisions. Guidelines already existed beforethe establishment of the new regulatory regime (section 4.2), but theirimportance increased during the 1990s. By summer 2003, 146 guidelineshad been adopted by the expert committee and its working parties, andanother 47 were in preparation (Krapohl 2005: 136). Moreover, many ofthe guidelines became legally binding, because they were included in leg-islative acts by the Commission. The substantive criteria for both orphanand herbal medicinal products were developed by working groups of theexpert committee before they became legislative acts. In addition, thenew ‘Common Technical Document’, which became part of the annex ofthe Community code for human medicinal products, was developed as a
From National Crises to a Strong Supranational Regime 83
PPL-UK_RR-Krapohl_Ch04.qxd 9/12/2008 10:55 AM Page 83

guideline by the ICH with participation of representatives from theagency’s expert committee. Finally, a BSE guideline34 of the expert com-mittee, which aims to reduce the risk of BSE transmission to humans by contaminated vaccines derived from bovine sera, became included inthe Community code for human medicinal products (Krapohl 2005:158–71).35
4.4 Conclusion
The development of EU pharmaceutical regulation clearly supports the historical-institutionalist hypothesis (section 2.2). The theoreticalanalysis concluded that the development of supranational regulatoryregimes is distinguished by two different critical junctures and by theirsequencing. A crisis of consumer confidence could either precede theestablishment of a single market in one product sector, or it could hap-pen the other way round. The development of a supranational regula-tory regime clearly followed the first developmental path: thethalidomide scandal occurred 25 years before the Single European Actand 35 years before a single market for pharmaceuticals was set up. As aresult, the thalidomide scandal (and in France the Stalinon scandal) ledto the establishment of national regulatory agencies for pharmaceuti-cals. These agencies were to some degree independent of direct politicalinfluence and had the competencies to grant or deny market access forpharmaceuticals. When a single market was deemed to be establishedby mutual recognition of member states’ authorisations, the agenciesused these competencies and refused to accept authorisations mutually,despite the fact that national authorisations were all based on the sameharmonised authorisation criteria. Thereby, they protected their owncompetencies and their respective levels of health and consumer pro-tection, which were endangered by the establishment of a single marketand by the mutual recognition principle.
However, once a single market for pharmaceuticals was adopted inthe 1990s, the national regulatory agencies strengthened, rather thanweakened, the newly established supranational regulatory regime. Toovercome their resistance to market integration, the national agencieswere included in the new regime. Thus, the regime could fall back ona regulatory network, and completely new supranational bodies didnot have to be set up. As a result, the supranational regulatory regimefor pharmaceuticals became quite strong, because it did not constituteas big a threat to member states’ sovereignty as a completely new body.Although a completely new supranational agency would have been
84 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch04.qxd 9/12/2008 10:55 AM Page 84

more independent from the national regulatory agencies, it probablywould have been less independent from political control of memberstates’ governments. The reform process at the beginning of the newmillennium did not lead to fundamental changes of the regime, but it reinforced some of its most important characteristics. The regimebecame even more centralised, more independent and more legalised,but it remained a regulatory network. Thus, the regime developed fur-ther along the developmental path that was adopted in 1990 andwhich has its roots in the thalidomide scandal of the late 1950s andearly 1960s.
From National Crises to a Strong Supranational Regime 85
PPL-UK_RR-Krapohl_Ch04.qxd 9/12/2008 10:55 AM Page 85

86
5A Strong Regulatory Network: The Evaluation of the EuropeanRegulatory Regime forPharmaceuticals
The question of this chapter is how the institutional features of the EU regulatory regime for pharmaceuticals influence its function. Mostmember states established national regulatory agencies for pharmaceu-ticals, and these now constitute a regulatory network within the supra-national regime. After 40 years of EU legislation on pharmaceuticals,the regime is now highly legalised and bases its decisions on a broadrange of substantive authorisation requirements. If the hypothesis ofthe theoretical argument (see section 3.2) holds true, the strong regula-tory network and the tight legalisation should be decisive factors for theregime’s success. The regulatory network of national authorisation bod-ies allows member states to delegate far-reaching competencies toexpert bodies without giving up national stakes. Strong legalisationreinforces the regime’s independence, because all actors – includingpolitical actors who control the regime – have to justify their decisionsbased on these substantive criteria.
In this chapter, two dimensions for the evaluation of supranationalregulatory regimes are distinguished. Firstly, the EU regime for pharma-ceuticals is analysed for its efficiency (section 5.1). Here, it is importantwhether it is able to fulfil its two policy goals of creating a single mar-ket for pharmaceuticals and of establishing effective health and con-sumer protection therein. Secondly, the legitimacy of the regime isevaluated (section 5.2). If stakeholders’ commitment to the proceduraland substantive rules of the regime is high, input legitimacy is basicallyrestricted to the set up of these more general rules. It is then crucial forthe regime’s day-to-day operation to what extent it provides outputlegitimacy. Therefore, it is important that the regime can be heldaccountable by a range of different stakeholders to prevent agencydrifts, and thus a loss of efficiency and output legitimacy.
PPL-UK_RR-Krapohl_Ch05.qxd 8/29/2008 11:54 AM Page 86

5.1 Commitment and efficiency of Europeanpharmaceutical authorisation
The following analysis explores whether the assumed correlationbetween member states’ commitment and the efficiency of EU pharma-ceutical authorisation is supported by empirical evidence. The hypoth-esis is tested on three different procedures that have been or are appliedfor EU pharmaceutical authorisation (chapter four). Firstly, from 1975to 1993, national authorisation of pharmaceuticals aimed at beingmutually recognised (section 5.1.1). Secondly, in 1993, a centralisedauthorisation procedure, which still applies today, was established forhighly innovative medicinal products (including all biotechnologicallyproduced pharmaceuticals; section 5.1.2). Finally, parallel to the cen-tralised procedure, a mutual recognition procedure was established forless innovative medicinal products (including the large group of generics;section 5.1.3).
5.1.1 Mutual recognition and the committee system
Before the centralisation of pharmaceutical authorisation in the 1990s,the EU tried to establish a single market for pharmaceuticals by facili-tating mutual recognition of national authorisations in the multi-stateprocedure (section 4.2 and Figure 5.1). Within this procedure, pharma-ceutical companies could apply for recognition of previously obtainednational marketing authorisations in at least two other EU memberstates. Because all member states had to base their decisions on the samesubstantive authorisation criteria, the concerned member states weremeant to recognise authorisations of the reference member state (thefirst member state that issued authorisations) without additional evalua-tions of respective medicinal products. However, if disagreements betweenthe member states occurred and were not settled, arbitrations were started,and scientific recommendations – concerning the safety, efficacy andquality of the respective products – were issued by the CPMP. These sci-entific opinions were forwarded to the concerned member states, whichwere expected, but not obliged, to follow this advice.
The concertation procedure was set up some years later (section 4.2and Figure 5.1). This new procedure was compulsory for biotechnolog-ically produced pharmaceuticals and voluntary for other highly inno-vative medicinal products, whereas less innovative products remainedunder the scope of the old multi-state procedure. Within the concerta-tion procedure, the expert committee presented scientific opinions aboutmedicinal products before member states issued or denied national
A Strong Regulatory Network 87
PPL-UK_RR-Krapohl_Ch05.qxd 8/29/2008 11:54 AM Page 87
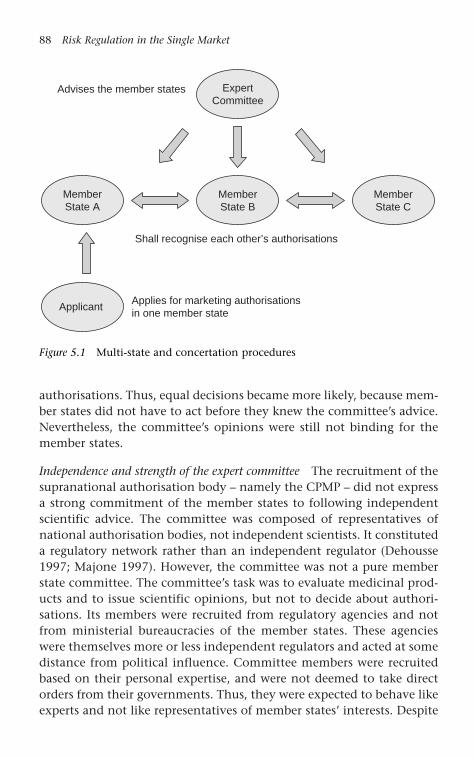
authorisations. Thus, equal decisions became more likely, because mem-ber states did not have to act before they knew the committee’s advice.Nevertheless, the committee’s opinions were still not binding for themember states.
Independence and strength of the expert committee The recruitment of thesupranational authorisation body – namely the CPMP – did not expressa strong commitment of the member states to following independentscientific advice. The committee was composed of representatives ofnational authorisation bodies, not independent scientists. It constituteda regulatory network rather than an independent regulator (Dehousse1997; Majone 1997). However, the committee was not a pure memberstate committee. The committee’s task was to evaluate medicinal prod-ucts and to issue scientific opinions, but not to decide about authori-sations. Its members were recruited from regulatory agencies and notfrom ministerial bureaucracies of the member states. These agencieswere themselves more or less independent regulators and acted at somedistance from political influence. Committee members were recruitedbased on their personal expertise, and were not deemed to take directorders from their governments. Thus, they were expected to behave likeexperts and not like representatives of member states’ interests. Despite
88 Risk Regulation in the Single Market
Figure 5.1 Multi-state and concertation procedures
Advises the member states
Shall recognise each other’s authorisations
Applies for marketing authorisationsin one member state
Expert Committee
Member State B
Member State C
MemberState A
Applicant
PPL-UK_RR-Krapohl_Ch05.qxd 8/29/2008 11:54 AM Page 88

these characteristics of the committee, it was still a body solely recruitedby the member states. Even though member states’ governments werenot deemed to give direct orders to committee members, they were stillable to influence the committee with their recruitment decisions. Thisstood in contrast to other scientific committees, and even more to EUagencies, which were and still are recruited by the Commission or bycooperation between the Commission, the Council and the EP.
The expert committee’s scientific opinions also did not have a bindingcharacter, and member states maintained full discretion about authori-sations of respective products on their own markets (Collatz 1996: 48;Vos 1999a: 210). This situation was even worse than that within a usualcomitology procedure. Within the latter, member states were only ableto deviate collectively from Commission proposals, i.e. they could rejectthem by majority decisions in the Council. However, within the multi-state or the concertation procedure, each member state was able to devi-ate individually from the committee’s scientific advice. Thus, memberstates did not commit themselves at all to the advice of the expert bodyand to common authorisation decisions.
Legalisation of the policy area
Member states’ weak commitment in the procedural rules of the multi-state and concertation procedures stood in sharp contrast to the exten-sive legalisation of pharmaceutical authorisation (Hart and Reich 1990:14–35). The first substantive authorisation requirements – namely, thatmedicinal products need to be evaluated according to their safety, effi-cacy and quality – had already been laid down in Directive 65/65/EEC,even before most EU member states had adopted national regulatoryregimes for pharmaceuticals. Ten years later, these first criteria were fur-ther substantiated by Directive 75/318/EEC, which laid down the neces-sary requirements for physico-chemical, biological or microbiological,toxicological and pharmacological, as well as clinical, tests of pharma-ceuticals. Applicants had to provide the results of these tests to allow foran evaluation of their products by the competent agencies of the mem-ber states or by the EU expert committee. During the next 20 years, thedetailed and comprehensive criteria were repeatedly amended and mademore concrete (section 4.2). Apart from these legally binding rules, theexpert committee began to adopt various guidelines aimed at advisingapplying companies and regulatory agencies of the member states (sec-tion 4.2; Collatz 1996: 41).1 Thus, a very detailed and comprehensivecatalogue of substantive authorisation criteria existed, even thoughsome of these guidelines were legally non-binding.
A Strong Regulatory Network 89
PPL-UK_RR-Krapohl_Ch05.qxd 8/29/2008 11:54 AM Page 89

Despite thight legalisation, the national character of authorisationdecisions significantly limited the possibilities of judicial review by the ECJ. If authorisation decisions of the member states were deemedto violate legally binding authorisation criteria of EU legislation, theycould only be challenged by other member states concerned or by theCommission within infringement procedures before the Europeancourts. The possibilities of concerned private persons – e.g. applicantsfor marketing authorisations – were limited, because they could onlybring claims before national courts, which could then ask the ECJ forpreliminary rulings (Collatz 1996: 172–86). Outcomes of the multi-state or concertation procedure could not be subject to nullity claims,because they were not decisions of EU bodies. Consequently, the scopeof potential plaintiffs was either limited to the Commission and othermember states within infringement procedures, or private plaintiffshad to take the detour through national courts and possible prelimi-nary proceedings.
To evaluate the judicial review of regulatory policy-making, it is notonly important to examine potential plaintiffs and their possibilities forchallenging authorisation decisions, but also to analyse the discretionof the ECJ if it were confronted with such claims. The question iswhether the ECJ is able to force member states to allow products thathad been previously authorised within other member states onto theirmarkets. Although member states are generally not allowed to establishnon-tariff barriers to trade, exceptions to this rule are allowed to protectthe health of domestic consumers. Such exceptions are usually deemedillegal if respective matters are already harmonised by EU law. However,in pharmaceutical authorisation, it was not the authorisation itself thatwas harmonised, but only the preconditions for authorisation. Withinthe jurisprudential literature, most scholars argue that the harmonisa-tion of substantive authorisation requirements did not preclude mem-ber states from denying recognitions of other member states’authorisations (Collatz 1996: 48; Hart and Reich 1990: 29–34). In par-ticular, the weighting of safety and efficacy of medicinal products leavessome discretion for regulators, which is not fully closed by substantiverules. Consequently, authorisation decisions could not a priori be deter-mined by substantive rules, and member states maintained some dis-cretion on whether to accept other member states’ authorisations.
Efficiency of the multi-state procedure and the concertation procedure
The Commission stated within an evaluation report2 that the strategy ofpartial harmonisation and mutual recognition failed to establish the
90 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch05.qxd 8/29/2008 11:54 AM Page 90

preconditions for a single market of pharmaceuticals in Europe. Generally,member states’ regulatory agencies did not recognise each other’s autho-risations or the scientific opinions of the expert committee, but preferredevaluating respective products on their own (Collatz 1996: 49–50; Feick2000a; Vos 1999a: 210). Of 122 applications under the multi-state pro-cedure between 1986 and 1990, not a single authorisation was recog-nised by the other member states. Consequently, the expert committeehad to issue scientific opinions on 92 products (the missing 30 applica-tions had been withdrawn from the rejecting member states). Thus, thesafeguard procedure (with the participation of the expert committee)had become the rule rather than the exception. By the end of 1990, onlyfor 45 of these 92 applications had all concerned member states notifiedtheir final decisions to the expert committee, and this did not evenimply that the decisions followed the advice of the committee.
Even though the concertation procedure was judged somewhat moresuccessful in establishing a single market, it nevertheless suffered fromthe same non-binding nature of experts’ recommendations (Broscheidand Feick 2005). Until 1990, 30 pharmaceuticals were subject to theconcertation procedure. Eleven positive opinions were issued by theexpert committee, whereas two applications were withdrawn because ofimminent negative decisions. The eleven positive opinions concerned121 applications within the different EU member states. For these 121national applications affected by the concertation procedure, 79 posi-tive decisions had been adopted, whereas 42 were still waiting fornational decisions at the end of 1990.
Appraisal
All indicators (independence and strength of supranational bodies, aswell as judicial review) with one exception (precision of authorisationcriteria) demonstrate weak commitment of the member states withinthe committee system. The expert committee was recruited by themember states themselves from experts of their national regulatoryagencies, and the member states were not bound to the advice of thiscommittee. The regime was far from constituting an independent reg-ulator as it would have been favoured by Majone (e.g. 1996, 2001a).An exception to this was the advanced precision of EU pharmaceuti-cal legislation. However, this legislation did not express strong com-mitment of the member states to mutually recognise authorisations,because the influence of the ECJ was limited. Possibilities of potentialplaintiffs for challenging national authorisation decisions in front ofthe ECJ were rather limited, and substantive authorisation criteria for
A Strong Regulatory Network 91
PPL-UK_RR-Krapohl_Ch05.qxd 8/29/2008 11:54 AM Page 91

pharmaceuticals still left enough discretion for member states to pro-tect their consumers individually against health risk. As a result, theregime was not successful in establishing the preconditions for a sin-gle market, and the task of health and consumer protection remainedin the hands of the member states. The weakness of the regime wasalso criticised by the Commission and various stakeholders, whichfinally led to the establishment of a more centralised regime under thelead of a new EU agency for pharmaceuticals.
5.1.2 The centralised authorisation procedure
The centralised authorisation procedure (section 4.3 and Figure 5.2) forhighly innovative medicinal products – including all biotechnologi-cally produced pharmaceuticals – starts when pharmaceutical compa-nies address official applications for marketing authorisations to the EUagency for pharmaceuticals. Since June 2003, application dossiersmust take the form of the ‘Common Technical Document’, which wasdeveloped within the International Conference on Harmonisation ofTechnical Requirements for Registration of Pharmaceuticals for HumanUse (ICH). Altogether, such dossiers cover up to 250,000 written pages.The application dossiers are evaluated by two rapporteurs from theCHMP (formerly CPMP).3 To scrutinise dossiers, rapporteurs usually relyon the expertise of their national regulatory agencies, but they may alsorefer to other experts registered at the agency within a ‘List of EuropeanExperts’.4 The rapporteurs write two assessment reports independentlyfrom each other. Subsequently, they try to settle possible disputes, andinform the other members of the expert committee, who may also givetheir comments on the preliminary assessments. Pharmaceutical com-panies may withdraw their applications during the evaluation process ifthey expect negative opinions about their products (which happens in
92 Risk Regulation in the Single Market
Figure 5.2 Centralised authorisation procedure
Expert Phase: Political Phase:
Applications Scientific opinions Decision proposals Decisions
Agency
Council
Commission
Comitology Committee
Expert Committee
Applicant
PPL-UK_RR-Krapohl_Ch05.qxd 8/29/2008 11:54 AM Page 92

approximately 19 of 20 cases, because applicants fear the negative pub-licity of unfavourable scientific opinions, Krapohl 2005: 123).
The scientific opinions of the expert committee should, wheneverpossible, be adopted by consensus (which happens in approximatelynine out of ten cases, Krapohl 2005: 128). However, if consensus cannotbe reached, the committee’s opinions may be adopted by simple major-ity of its members. Generally, the committee may issue three kinds ofscientific opinion. It may support marketing authorisation, it may rec-ommend authorisation under exceptional circumstances, or it may pro-pose to deny authorisation. Positive scientific opinions contain drafts ofthe summaries of product characteristics, manufacturing and marketingconditions, drafts of labels and package leaflets, full assessment reports,and, where relevant, divergent opinions of committee members. In thecase of authorisation under specific circumstances, the expert commit-tee additionally proposes specific obligations and follow-up measuresfor respective pharmaceutical companies. If scientific opinions are neg-ative or impose obligations, applicants may appeal against them (whichhappened in only six cases up to 31 December 2002, Krapohl 2005:128–9). In such circumstances, the expert committee appoints new rap-porteurs, and has to decide on the applications a second time.
The scientific opinions of the expert committee are formally notthe final authorisation decisions, but are subject to political scrutinywithin a comitology procedure.5 At the first stage of this procedure, theCommission must develop decision proposals out of the scientific opin-ions. If new scientific questions emerge at this stage, the Commissionmust refer matters back to the agency. When Commission proposalsdeviate from the expert committee’s opinions, the Commission has togive detailed reasons for these differences. At the second stage,Commission proposals are subject to a vote of a member state commit-tee (the Standing Committee on Medicinal Products for Human Use).This committee decides according to a management procedure (a reg-ulatory IIIb procedure before November 2005), i.e. it may rejectCommission proposals by qualified majority. Usually, the member statecommittee decides by a written procedure. Meetings of the memberstate committee may be called if this is demanded by one of the mem-ber states, or if decisions are very urgent and measures have to beapplied immediately. The third stage of the comitology procedure onlyproceeds if Commission proposals are rejected by a qualified majoritywithin the member state committee. In such cases, proposals arereferred to the Council, which has the final say. It may adopt or rejectproposals by qualified majority, or amend them by unanimity. The final
A Strong Regulatory Network 93
PPL-UK_RR-Krapohl_Ch05.qxd 8/29/2008 11:54 AM Page 93

authorisation decisions are binding for all member states; the onlyexceptions are contraceptives or abortifacients, which may be forbiddenby the member states for ethical reasons.
Independence and strength of the EU agency for pharmaceuticals
Member states’ commitment within the recruitment of the Europeanpharmaceutical agency and its expert committee increased only slightlyin compared with the committee system. The recruitment of both theagency’s management board and expert committee remained fully underthe control of the member states until November 2005, when the mostrecent reforms were implemented. According to the regulation of 1993,the management board of the agency consisted of two representatives ofeach member state, the Commission and the EP. Thus, member states’representatives outweighed representatives of the other two legislativeactors by a wide margin. Besides, the expert committee was not recruitedby the agency’s management board, but consisted of representativesfrom the member states. However, the expert committee was (and still is)not a normal member state committee, but it comprises a network ofmember states’ regulatory agencies (Dehousse 1997; Majone 1997). Theagency’s independence improved slightly as a result of the latest reform,when the management board was opened up for two representativesfrom patients’ organisations, and one representative each from physi-cians’ and veterinarians’ organisations. However, member states’ repre-sentatives still constitute a majority vis-à-vis other management boardmembers. The composition of the expert committee also changed. Now,each member state sends only one expert to the committee, and thecommittee may co-opt up to five additional members. However, despitethis reform, the recruitment of both the agency’s management boardand the expert committee can still be dominated by the member states,even though their large majority decreased somewhat.
Theoretically, the agency’s expert committee could also be composed ofscientist independent from the member states. However, the question ishow such a committee could get enough resources to fulfil its task ofevaluating applications (Gehring and Krapohl 2007). As mentioned, suchapplications may contain up to 250,000 written pages. Thus far, the eval-uation work is mainly done within the national regulatory agencies ofthe two rapporteurs. If committee members can no longer fall back on this expertise, new scientific resources need to be built up. This couldbe done in two ways. Firstly, experts within the committee could berecruited from privately financed research institutes. The disadvantage ofthis solution is that pharmaceutical companies could gain more influence
94 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch05.qxd 8/29/2008 11:54 AM Page 94

over the committee if they finance the respective research institutes.Thus, more independence from member states might in fact lead to lessindependence from industry’s interests. Secondly, the EU agency itselfcould build up the necessary scientific resources. This would mean fur-ther centralisation of the supranational regulatory regime. So far, suchcentralisation has met strong resistance from the member states and espe-cially from their national regulatory agencies. One may expect that a moreindependent and centralised regulatory agency would be more stronglycontrolled by the member states in its day-to-day decision-making. Thus,more independent recruitment could in fact lead to less independentdecision-making.
Member states’ control of the regime’s day-to-day policy-making hasdecreased significantly compared with the multi-state and the concerta-tion procedures. The results of the centralised procedure are Commissiondecisions (or Council decisions, if the member state committee refersproposals to the Council) that are binding for the member states.Consequently, the member states can no longer individually deviatefrom the agency’s advice anymore. The member states can only blockCommission proposals collectively within the comitology procedure.Before the latest reforms, the member states controlled the agency andthe Commission within the framework of a regulatory IIIb procedure.Accordingly, decision proposals were referred to the Council if they werenot supported by a qualified majority within the comitology committee,and the Council was able to reject them by simple majority. Because of the high majority requirement within the member state committeeand the low threshold for rejection within the Council, the regulatoryIIIb procedure was the most restrictive of all comitology procedures(Franchino 2000a, 2000b; Steunenberg et al. 1996). Thus, the memberstates could still control the agency comparatively strictly. However,these possibilities have declined because of the latest reforms. Now,member states control the agency and the Commission only by a man-agement procedure. Therein, the members of the comitology committeemust reject Commission proposals by qualified majority in order to refermatters to the Council. The Council can now reject or adopt proposalsby qualified majority, or amend them by unanimity. Thus, the thresholdfor blocking Commission proposals has increased, and member states’commitment has become stronger.
Unlike other comitology procedures, the control procedure in thearea of pharmaceutical authorisation is directed towards passivity ofpolitical actors. Three differences from usual comitology proceduresmake interventions of political actors in the centralised authorisation
A Strong Regulatory Network 95
PPL-UK_RR-Krapohl_Ch05.qxd 8/29/2008 11:54 AM Page 95

procedure more difficult, and thus express a higher commitment of themember states than one would normally expect. Firstly, within the cen-tralised authorisation procedure, it is the regulatory agency and not theCommission that sets the agenda. For pharmaceuticals, applications formarketing authorisations are directly addressed to the agency. Theagency always issues its scientific opinions before any other actors are ableto express their preferences. Secondly, it is difficult for the Commission todeviate from the agency’s expert advice. If the Commission does not fol-low the scientific opinions, it must give detailed reasons. And if new sci-entific questions emerge, matters are given back to the agency. Thirdly, thewritten procedure imposes a higher threshold on member states’ inter-vention than regular meetings of the comitology committee. Decisionproposals are simply sent to the member states, which may reactwithin 22 days. If they do not react, this is counted as an approval ofthe proposals.
Legalisation of the policy area
The detailed substantive rules that already governed the authorisation ofpharmaceuticals within the multi-state and the concertation procedurealso applied after the reform of the supranational regulatory regime inthe early 1990s. The regulation that set up the EU pharmaceutical agencyand the centralised authorisation procedure stated that medicinal prod-ucts may only be authorised on the basis of their safety, efficacy andquality. To prove these characteristics, applications for authorisationshad to pass the tests that were laid down in Directive 75/318/EEC(Collatz 1996: 39–41; Thompson 1994: 49–66). In the years after thisreform, the substantive authorisation requirements were repeatedlyadapted, and thus the legalisation of the policy area increased evenfurther (section 4.3). Firstly, the general substantive rules became con-solidated. The authorisation requirements were summarised within anew ‘Community Code Relating to Medicinal Products for HumanUse’. This was amended by the Commission to include requirementsof the ‘Common Technical Document’. Secondly, specific authorisa-tion requirements were adopted for orphan, as well as for herbal medic-inal, products. These groups of products usually do not fulfil theregular criteria for safety and efficacy, because they can either not betested on the same sample of patients (orphan medicinal products), orbecause their efficacy is generally low according to purely scientificstandards (herbal and traditional medicinal products). Specific authori-sation requirements for these products allow these characteristics to betaken into account.
96 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch05.qxd 8/29/2008 11:54 AM Page 96

Besides, the number and importance of legally non-binding guide-lines and recommendations of the expert committee increased greatlyafter the centralisation of pharmaceutical authorisation (Krapohl 2005:133–46). Although committee guidelines are not legally binding, theirimportance has actually increased, because the expert committeebecame the main authorisation body for highly innovative pharmaceu-ticals. The more powerful the committee is, the more important it is forapplicants to stick to its guidance to improve the chances of positiveevaluations of their products. Besides, some of the expert committee’sguidelines became legally binding EU law, as was the case with the BSEguideline (Krapohl 2005: 158–72). To reduce the risk of BSE transmis-sion through pharmaceuticals produced with bovine sera, the expertcommittee published its first guideline in 1991. This was five yearsbefore the human risk of BSE was acknowledged by the British govern-ment, and the first regulatory measures were adopted in the EU food-stuff sector (section 6.2). Basically, the BSE guideline recommendedusing only bovine materials from countries without BSE infections,abstaining from specified risk materials and using certain productionmethods to deactivate potential agents. Of course, this guideline was –at the beginning – not legally enforceable. However, in 1999 – someyears after the risk of BSE for humans became evident – a CommissionDirective included the BSE guideline within the legally enforceableannex of Directive 75/318/EEC. Following this step, all authorisedmedicinal products within the EU were checked for their accordancewith this guideline, and one vaccine was withdrawn from the Britishmarket where it held a national authorisation.
The importance of the ECJ and the Court of First Instance for theauthorisation of pharmaceuticals increased because of the centralisationof the regime. Since the early 1990s, all central authorisations of highlyinnovative medicinal products are Commission decisions. As such, theycan be subject to nullity claims before the Court of First Instance and,in second instance, before the ECJ (Collatz 1996: 135–59). Now, privatepersons need no longer take the detour of national proceedings andpossible preliminary rulings, but they can bring their claims directlybefore the European courts. In the case of authorisation decisions, it iseasy for applicants to demonstrate their direct and individual concern,which is necessary to be eligible to sue the Commission (Collatz 1996:143–6). Authorisation decisions are directly addressed to them, andintervene in their personal rights. Consequently, applicants are in needof legal protection, and always have the right to bring nullity claimsagainst negative decisions in front of the European courts.
A Strong Regulatory Network 97
PPL-UK_RR-Krapohl_Ch05.qxd 8/29/2008 11:54 AM Page 97

The increasing importance of the European courts became evident inthe case of weight control pharmaceuticals (Krapohl 2005: 148–58),when the producers of older anorectics successfully challenged theCommission decision to withdraw marketing authorisation of theirproducts.6 The producers claimed before the Court of First Instance thatthe Commission did not have the competence to withdraw these autho-risations (because they were issued by the member states in the oldmulti-state procedure), and that the respective decisions were substan-tially not justified, because new side-effects of the old anorectics werenot detected. The authorisation holders stated that the decisions werebased on comparative efficiency evaluations in relation to a newanorectic (Xenical, which was authorised two years before), but that thesubstantive authorisation requirements of EU legislation did not pro-vide for such comparative assessments.7 The Court of First Instance con-firmed this argument.8 Accordingly, the Commission decisions were notbased on valid reasons. The Commission challenged this judgementbefore the ECJ, but it also lost this case and the judgement was con-firmed.9 Two lessons can be learned from this case. Firstly, the challengeof authorisation decisions before the European courts is not only ahypothetical possibility. Instead, the pharmaceutical industry uses thisoption if its rights are endangered. Secondly, the European courts arewilling to scrutinise the substantive reasons for authorisation decisionsin detail. They not only judge whether the Commission has the com-petence to pass decisions, but also whether decisions are in accordancewith the substantive authorisation requirements.
However, the situation is completely reversed if consumers want to chal-lenge positive authorisation decisions. Unlike producers, consumers – i.e.patients and physicians – are not directly and individually concerned byauthorisation decisions, because their personal rights are not influenced.Within a recent judgement, the Court of First Instance interpreted thescope of potential plaintiffs rather restrictively (Krapohl 2005: 116–19).10
In this case, a former employer of the company Apotex brought a claimagainst the authorisation of a new medicinal product of Apotex beforethe Court of First Instance. According to the plaintiff, the clinical testsunder her own responsibility did not demonstrate sufficient safety of the product to justify its authorisation. Consequently, she demanded therevocation of the positive authorisation decision. According to thecourt’s judgement, the fact that the plaintiff was responsible for the clin-ical tests of the product did not constitute a direct and individual con-cern. Thus, the court rejected this claim as inadmissible. Accordingly, thechallenging of positive authorisation decisions by private persons is
98 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch05.qxd 8/29/2008 11:54 AM Page 98

much more difficult than the challenging of negative authorisation deci-sions. However, every member state, the Commission or the EP can alwaysbring nullity claims before the European courts, and thus they could inter-vene if they see public health to be at risk.
Efficiency of the centralised authorisation procedure
The centralised authorisation procedure undoubtedly contributes to theestablishment of a single market for pharmaceuticals (Feick 2002b; Vogel1998). Authorisations that follow from this procedure are Commissiondecisions and are valid in all EU member states. As a result, the supply ofhighly innovative pharmaceuticals has become increasingly homoge-nous throughout the Single Market. However, whereas authorisation of highly innovative pharmaceuticals became highly centralised in theEU, the competencies for health policy remained largely in the hands ofthe member states (Kotzian 2003; Permanand and Altenstetter 2004;Permanand and Mossialos 2005; Permanand 2006: 151–79). In particu-lar, the prices of pharmaceuticals and their reimbursement by nationalhealth systems are regulated by the member states. Consequently, thedemand for certain pharmaceuticals may differ considerably throughoutEurope. Even if all highly innovative pharmaceuticals theoretically haveequal access to all national markets, they may not be sold everywhere,because some health systems may deny reimbursement. Such differ-ences on the demand side may hinder the establishment of a singlemarket for pharmaceuticals, but they cannot be reduced by the supra-national regulatory regime, which already provides the preconditionsfor market integration.
It is much more difficult to assess the efficiency of the EU regulatoryregime for pharmaceuticals in its second policy goal: the establishment ofadequate health and consumer protection throughout the Single Market.Comprehensive data in this respect are lacking (Feick 2002b). However, itis possible to find some anecdotal evidence that indicates that the regu-latory standards of the supranational regime are quite high, and thatEuropean regulators and consumers are satisfied with the functioning ofthe centralised authorisation procedure. The pharmaceutical sector dealtquite well with two regulatory problems that shattered the foodstuff sec-tor during the 1990s and at the beginning of the new millennium (sections 6.2 and 6.3). Both BSE and biotechnology are also potentialproblems for medicinal products. Firstly, vaccines that are produced frombovine sera could be contaminated with the BSE agent if animals of origin suffered from ‘mad cow disease’. However, in contrast to the food-stuff sector, BSE did not lead to a crisis of consumer confidence in the
A Strong Regulatory Network 99
PPL-UK_RR-Krapohl_Ch05.qxd 8/29/2008 11:54 AM Page 99

pharmaceutical sector. The agency’s expert committee reacted swiftly,and published a regulatory guideline even before the risk to humans wasacknowledged by the British government (Krapohl 2005: 158–72). Later,all medicinal products in Europe were checked for potential BSE contam-inations. Secondly, whereas the EU regulatory regime for foodstuffs hasdifficulties regulating ‘green’ biotechnology – i.e. GM food – and findingconsumers’ trust, the regulatory regime for pharmaceuticals deals quitewell with ‘red’ biotechnology – i.e. biotechnologically produced medici-nal products – and does not meet consumer resistance. Thus, whereas EUfoodstuff regulation suffers from a crisis of consumer confidence (Anselland Vogel 2006; Majone 2000; Vogel 2001a; Vos 2004), EU pharmaceuti-cal regulation is rather unaffected by this problem. This may not only bedue to the respective regulatory regimes: other factors like different pub-lic risk perceptions may play a role as well. However, one can at leastassess that the more centralised regime in the pharmaceutical sectorallows quicker and more homogeneous replies to regulatory problemsthan the weaker regime in the foodstuff sector (chapter seven).
In general, all stakeholders – including regulatory agencies and con-sumer organisations – seem to be satisfied with the centralised authorisa-tion procedure (Table 5.1). In an extensive survey conducted on behalf ofthe Commission, nearly all authorisation holders and regulatory agencies(member states’ agencies plus the EU agency) expressed satisfaction withthe centralised authorisation procedure. An overwhelming majority werealso in favour of an extension of the scope of the centralised procedure.Although consumers were not that enthusiastic, they generally favouredthe centralised to the decentralised authorisation procedure. Because ofthis positive evaluation, core features of the centralised procedure werefurther reinforced, and its scope was expanded during the reform processat the beginning of the new millennium. The centralised procedure nowapplies voluntarily to all new active substances, and its compulsory appli-cation is also extended to a wider group of products. It is striking that theEP favoured an even wider extension of the compulsory scope of the cen-tralised procedure and even more independence for the pharmaceuticalagency within the legislative process. This behaviour of the traditionallyconsumer-friendly EP (Pollack 1997b) would not have been plausible ifthe centralised procedure had systematically failed to establish a highlevel of health and consumer protection.
Appraisal
Commitment of the member states to a common regulation of phar-maceuticals in the Single Market increased greatly after centralisation of
100 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch05.qxd 8/29/2008 11:54 AM Page 100
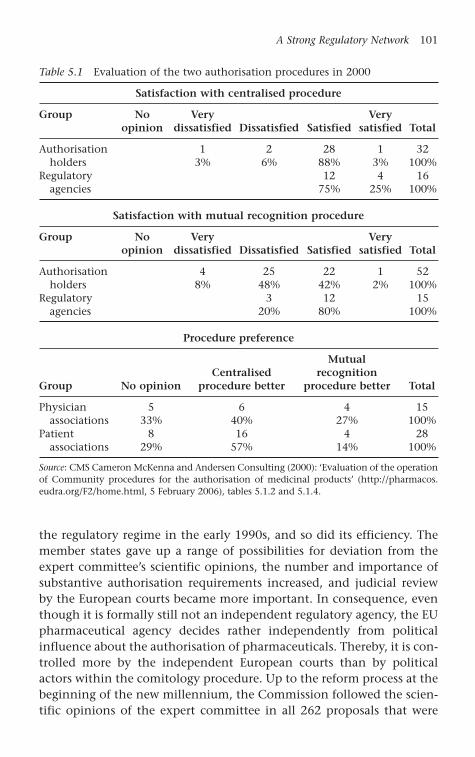
the regulatory regime in the early 1990s, and so did its efficiency. Themember states gave up a range of possibilities for deviation from theexpert committee’s scientific opinions, the number and importance ofsubstantive authorisation requirements increased, and judicial reviewby the European courts became more important. In consequence, eventhough it is formally still not an independent regulatory agency, the EUpharmaceutical agency decides rather independently from politicalinfluence about the authorisation of pharmaceuticals. Thereby, it is con-trolled more by the independent European courts than by politicalactors within the comitology procedure. Up to the reform process at thebeginning of the new millennium, the Commission followed the scien-tific opinions of the expert committee in all 262 proposals that were
A Strong Regulatory Network 101
Table 5.1 Evaluation of the two authorisation procedures in 2000
Satisfaction with centralised procedure
Group No Very Very opinion dissatisfied Dissatisfied Satisfied satisfied Total
Authorisation 1 2 28 1 32holders 3% 6% 88% 3% 100%
Regulatory 12 4 16agencies 75% 25% 100%
Satisfaction with mutual recognition procedure
Group No Very Very opinion dissatisfied Dissatisfied Satisfied satisfied Total
Authorisation 4 25 22 1 52holders 8% 48% 42% 2% 100%
Regulatory 3 12 15agencies 20% 80% 100%
Procedure preference
MutualCentralised recognition
Group No opinion procedure better procedure better Total
Physician 5 6 4 15associations 33% 40% 27% 100%
Patient 8 16 4 28associations 29% 57% 14% 100%
Source: CMS Cameron McKenna and Andersen Consulting (2000): ‘Evaluation of the operationof Community procedures for the authorisation of medicinal products’ (http://pharmacos.eudra.org/F2/home.html, 5 February 2006), tables 5.1.2 and 5.1.4.
PPL-UK_RR-Krapohl_Ch05.qxd 8/29/2008 11:54 AM Page 101

subject to a comitology procedure before 1 May 2001 (Table 5.2; Krapohl2005: 120–1). Only nine of these proposals were discussed at meetings of the standing committee, and of these, four received unanimous sup-port from the member states, whereas only five were adopted by quali-fied majority within the comitology committee. No decision proposalswere forwarded to the Council. Consequently, not a single proposal waschanged or rejected within the comitology procedure. So the final policyoutcome before 1 May 2001 reflected, one-to-one, the scientific opinionsof the expert committee (unfortunately, more current numbers are notavailable).
As a result of deepening commitment, the efficiency of the regimealso increased significantly. Authorisation resulting from the centralisedauthorisation procedure is valid in all EU member states, and providespreconditions for a single market, at least for highly innovative medic-inal products. The centralised procedure also seems to establish an ade-quate level of health and consumer protection within this market.Unlike other regulatory areas, the regulatory regime for pharmaceuticalsdoes not suffer from a crisis of consumer confidence. Overall, stakehold-ers seem to be satisfied with the centralised procedure. Consequently, itsscope was expanded and its central features were reinforced by the mostrecent reform process at the beginning of the new millennium.
5.1.3 The mutual recognition procedure
The mutual recognition procedure (section 4.3 and Figure 5.3) for lessinnovative pharmaceuticals is a mixture of the old multi-state and thenew centralised procedures. Within the first phase of the mutual recog-nition procedure, member states are expected to recognise each other’snational marketing authorisations. Only if the member states cannotfind common positions during this mutual recognition phase, bindingarbitrations take place. This arbitration procedure of the second phaseworks in the same way as the centralised authorisation procedure. Thus,to analyse the mutual recognition procedure, it is important to have a
102 Risk Regulation in the Single Market
Table 5.2 Activities within the comitology procedure from 1995 to 2001
Authorisation decisions before 1/5/2001 262Deviations between scientific opinions and Commission proposals 0Rejections by the member state committee 0Meetings of the member state committee 9 (3.44%)Majority decisions within the member state committee 5 (1.91%)
Source: Explanatory memorandum of the Commission, 26 November 2001 (COM(2001) 404final), footnotes 13 and 14.
PPL-UK_RR-Krapohl_Ch05.qxd 8/29/2008 11:54 AM Page 102

close look at the conditions under which binding arbitration occurs. Ifarbitration only applies exceptionally, the mutual recognition proce-dure is likely to come close to the example of the multi-state procedure.However, if arbitration applies generally, the mutual recognition proce-dure is more likely to bring similar results to those of the centralisedprocedure.
Generally, the mutual recognition procedure is built on assessments ofmedicinal products by regulatory agencies of only one member state (theso-called reference member state). Because such evaluations are based onthe detailed substantive authorisation requirements as prescribed byEuropean law, all concerned member states (i.e. member states wherecompanies apply for recognition of the initial authorisation of the refer-ence member state) are expected to trust the judgement of the referencemember states. Before the reforms at the beginning of the new millen-nium, the mutual recognition procedure could only begin if medicinalproducts had already been authorised by the reference member states.11
A Strong Regulatory Network 103
Figure 5.3 Mutual recognition and decentralised authorisation procedures
Applies for marketing authorisations
Shall recognise each other’sauthorisations or assessments
Scientific opinions Decision proposals Decisions
Member State B
Member State C
Member State A
Applicant
Council
Commission
Comitology Committee Agency
Expert Committee
PPL-UK_RR-Krapohl_Ch05.qxd 8/29/2008 11:54 AM Page 103

As part of the latest reform, a second option to start a procedure wasintroduced. Now, it is also possible to start the newly established decen-tralised authorisation procedure before reference member states haveissued national authorisations for respective products.12 Reference mem-ber states need not issue national authorisations, but must only preparedraft assessment reports. Thereby, they already cooperate with the con-cerned member states and clarify outstanding issues with the applicants.If spontaneous consensus between the reference and the concernedmember states is reached at this stage, national authorisation is issued.
If the reference member states have issued national authorisations orif no spontaneous consensuses could be reached between the memberstates at an early stage, the mutual recognition phase follows. Therein,the concerned member states should recognise the reference memberstates’ national authorisations or the draft assessment reports. Suchmutual recognition may only be refused if the concerned member statesexpect serious risks to public health in consequence of the referencemember states’ decisions. If divergent decisions of the member statesimpend, the reference member states may call breakout sessions of the so-called ‘Coordination Group’. This group was established informally in1995 by the so-called Heads of Agencies – a regular meeting of executivedirectors or their alternates from national regulatory agencies – under thename Mutual Recognition Facilitation Group.13 During the reforms at thebeginning of the new millennium, the group was renamed CoordinationGroup, received a formal status and became included in the mutualrecognition/decentralised authorisation procedure. If consensus isreached within breakout sessions of the Coordination Group, the refer-ence member states close the procedures, and national authorisation isissued. Binding arbitrations are only started if this Coordination Groupprocedure fails.
The circumstances under which binding arbitrations, led by the EMEA,begin were fundamentally reformed at the beginning of the new millen-nium. Before November 2005, pharmaceutical companies could preventarbitrations by withdrawing their applications from concerned memberstates that disfavoured authorisations. In such cases, consensus betweenthe member states emerged, because rejecting member states were nolonger affected by the applications. Thereby, applicants avoided the riskand negative reputation of time-consuming arbitration. However, afterNovember 2005, arbitrations must always be started if one of the con-cerned member states fears serious risks to public health.14 Withdrawalsof applications from disfavouring member states no longer preventarbitrations. This ensures that serious concerns related to public health
104 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch05.qxd 8/29/2008 11:54 AM Page 104

cannot be ignored, and that they are always be scrutinised within a centralised procedure.
If a binding arbitration procedure is started, it works like the cen-tralised authorisation procedure.15 The only major difference betweenthe centralised authorisation procedure and the arbitration procedurerests with the final authorisation decisions. The results of a centralisedprocedure are EU authorisations that automatically apply to the wholeSingle Market. In contrast, decisions of the binding arbitration proce-dure need to be implemented only by the concerned member stateswithin 30 days.
Credible commitment in the mutual recognition procedure
Commitment of member states to create a single market for pharmaceu-ticals and to protect consumers effectively differs widely between the twophases of the mutual recognition/decentralised authorisation procedure.The mutual recognition phase follows very much the example of the oldmulti-state procedure. Consequently, member states’ commitment is sup-posedly very low. No independent regulatory body exists, but the onlyadvisory body – the Mutual Recognition Facilitation or CoordinationGroup – is composed of representatives from member states’ regulatoryagencies. Further, this group cannot issue binding decisions on the autho-risation of medicinal products, but can only advise the member states.Every single member state is able to block common decisions at thisstage. Although member states decide on the basis of the substantiveauthorisation requirements, the effect of this legalisation is reduced bytwo factors: Firstly, guidelines of the expert committee do not apply toauthorisation decisions of the member states. Consequently, legalisationis weaker during the mutual recognition phase than during the arbitra-tion phase or a centralised authorisation procedure. And secondly, autho-risation decisions made in the mutual recognition phase are nationaldecisions. Thus, private plaintiffs cannot directly challenge such deci-sions before the EU courts.
In contrast, the arbitration phase comes close to the centralised pro-cedure, and member states’ commitment is rather high. Even though itis also composed of representatives from the member states’ regulatoryagencies, the expert committee is much stronger than the coordinationgroup. Arbitration leads to binding decisions, which have to be imple-mented by the member states. Because of the written comitology pro-cedure and the downgrading from a regulatory to a managementprocedure in November 2005, member states’ collective control of thearbitration procedure is rather restricted. Additionally, the arbitration
A Strong Regulatory Network 105
PPL-UK_RR-Krapohl_Ch05.qxd 8/29/2008 11:54 AM Page 105
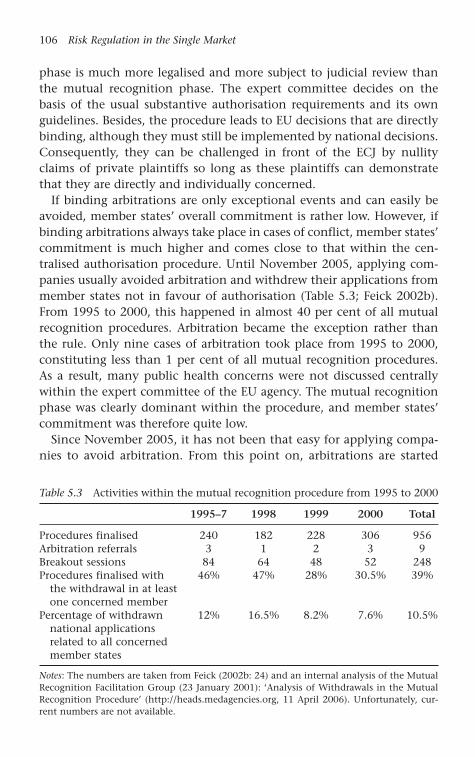
phase is much more legalised and more subject to judicial review thanthe mutual recognition phase. The expert committee decides on thebasis of the usual substantive authorisation requirements and its ownguidelines. Besides, the procedure leads to EU decisions that are directlybinding, although they must still be implemented by national decisions.Consequently, they can be challenged in front of the ECJ by nullityclaims of private plaintiffs so long as these plaintiffs can demonstratethat they are directly and individually concerned.
If binding arbitrations are only exceptional events and can easily beavoided, member states’ overall commitment is rather low. However, ifbinding arbitrations always take place in cases of conflict, member states’commitment is much higher and comes close to that within the cen-tralised authorisation procedure. Until November 2005, applying com-panies usually avoided arbitration and withdrew their applications frommember states not in favour of authorisation (Table 5.3; Feick 2002b).From 1995 to 2000, this happened in almost 40 per cent of all mutualrecognition procedures. Arbitration became the exception rather thanthe rule. Only nine cases of arbitration took place from 1995 to 2000,constituting less than 1 per cent of all mutual recognition procedures. As a result, many public health concerns were not discussed centrallywithin the expert committee of the EU agency. The mutual recognitionphase was clearly dominant within the procedure, and member states’commitment was therefore quite low.
Since November 2005, it has not been that easy for applying compa-nies to avoid arbitration. From this point on, arbitrations are started
106 Risk Regulation in the Single Market
Table 5.3 Activities within the mutual recognition procedure from 1995 to 2000
1995–7 1998 1999 2000 Total
Procedures finalised 240 182 228 306 956Arbitration referrals 3 1 2 3 9Breakout sessions 84 64 48 52 248Procedures finalised with 46% 47% 28% 30.5% 39%
the withdrawal in at leastone concerned member
Percentage of withdrawn 12% 16.5% 8.2% 7.6% 10.5%national applicationsrelated to all concernedmember states
Notes: The numbers are taken from Feick (2002b: 24) and an internal analysis of the MutualRecognition Facilitation Group (23 January 2001): ‘Analysis of Withdrawals in the MutualRecognition Procedure’ (http://heads.medagencies.org, 11 April 2006). Unfortunately, cur-rent numbers are not available.
PPL-UK_RR-Krapohl_Ch05.qxd 8/29/2008 11:54 AM Page 106

whenever one of the concerned member states fears serious risks to pub-lic health. Partial withdrawals from member states not in favour ofauthorisation are no longer sufficient for the prevention of such proce-dures. Only when applying companies withdraw their applicationsfrom all member states may the procedure be stopped. Even then it is atthe discretion of the agency’s expert committee to decide whetherassessments of respective products are nevertheless in the public inter-est. With this reform, the possibility that potentially serious risks can-not be discussed centrally at the EU agency was abolished (Broscheidand Feick 2005). After the reform was implemented, 44 arbitrations tookplace in 2006 and 2007 (27 per cent of all mutual recognition/decen-tralised procedures).16 This is a rapid increase compared with the ninearbitrations between 1995 and 2000. Thus, member states’ commitmentin the mutual recognition procedure has significantly grown with thelatest reform.
Efficiency of the mutual recognition procedure
Because the latest reform of the mutual recognition/decentralised pro-cedure was only implemented in the autumn of 2005, it is not yet pos-sible to evaluate the efficiency of the procedure as it operates today.Consequently, the following empirical analysis has to concentrate onthe procedure as it worked before the latest reform. In addition, somehypotheses can be made about the likely effects of this latest change. Aswith the centralised procedure, comprehensive data about the effi-ciency of the mutual recognition procedure are lacking (Feick 2002b).However, there is some anecdotal evidence that the mutual recognitionprocedure is less efficient than the centralised procedure.
Firstly, the mutual recognition procedure does not necessarily lead toa homogenous supply of pharmaceuticals throughout the Single Market.The more member states are affected by mutual recognition, the morehomogeneous access of pharmaceuticals to the Single Market becomes.However, as Feick (2002b: 41) calculated, the mutual recognition proce-dure usually led to authorisations in less than half of the member statesbetween 1998 and 2001. On average, between 8.69 and 6.53 of the then15 EU member states were involved in mutual recognition. Additionally,pharmaceutical companies withdrew their applications from around 10per cent of all concerned member states. Thus, the homogenising effectof the mutual recognition procedure was rather limited, and equal accessto all national markets within the EU was not achieved.
For the second policy goal – to establish efficient risk regulationthroughout the Single Market – the mutual recognition procedure, as it
A Strong Regulatory Network 107
PPL-UK_RR-Krapohl_Ch05.qxd 8/29/2008 11:54 AM Page 107

operated before 2005, suffered from an important shortcoming. The pos-sibility for pharmaceutical companies to withdraw their applicationsselectively often prevented potentially serious risks to public health frombeing centrally evaluated by the expert committee of the EU agency(Feick 2000b). Thus, medicinal products were authorised in some mem-ber states, whereas serious concerns of other member states were notaddressed and led to withdrawals. In the end, the level of health andconsumer protection was not uniform throughout the Single Market.
One example where this shortcoming of the mutual recognition pro-cedure may have been at work is the case of Vioxx. This pharmaceuticalwas the first of a new group of pain relievers – the so-called ‘COX-2inhibitors’ – which were deemed to have fewer side effects on the stom-ach than older pain relievers like aspirin and ibuprofen. Physicianshoped that the new pain relievers would allow for a new treatment forchronic pain, especially in cases of rheumatic and arthritic diseases.Consequently, and because of intensive advertising by the producingcompany Merck, Vioxx became a so-called ‘blockbuster’ and was widelydistributed in the USA and Europe. Vioxx was authorised for the SingleMarket within a mutual recognition procedure. Therein, the UK was thereference member state, but its authorisation was recognised withoutarbitration by other EU member states (Austria, Finland, France,Germany, Italy, Luxembourg and the Netherlands). The Vioxx scandalbegan when Merck had to withdraw its product worldwide, because newscientific studies found that it imposed serious health risks on con-sumers. Most notably, the risk of cardiovascular diseases – e.g. heartattacks and strokes – increased significantly after therapies with Vioxx.In the end, the US FDA estimated that Vioxx had caused 27,785 deathsalone in the USA because of such diseases.17 A scientific analysis pub-lished after the withdrawal of Vioxx from the US market accused Merckand the regulatory agencies – including, indirectly, the European ones –of neglecting the side-effects of the new pain reliever, which had beenmade evident in previous studies (Jüni et al. 2004). Following this, up to10,000 claims for compensation were brought before US courts.18
Although it is an open question whether the centralised procedurewould have denied the authorisation of Vioxx, the scandal neverthelesscasts a damning light on the mutual recognition procedure and itscapacity for effective health and consumer protection.
In general, the mutual recognition procedure was evaluated less posi-tively than the centralised procedure by various stakeholders (Table 5.1).Within the evaluation at the beginning of the new millennium, morethan half of authorisation holders and still one-fifth of the regulatory
108 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch05.qxd 8/29/2008 11:54 AM Page 108

agencies (member states’ agencies plus the EU agency) expressed dissat-isfaction with the mutual recognition procedure. These numbers are sig-nificantly worse than those for the centralised procedure. The evaluationof physicians’ and patients’ associations pointed in the same direction.Most of them preferred the centralised procedure to the mutual recogni-tion procedure.
Appraisal
The mutual recognition procedure, as it operated before November 2005,was clearly dominated by the mutual recognition phase, because apply-ing companies could easily avoid centralised arbitration. Thus, memberstates’ commitment within the procedure was rather low: no independ-ent regulatory body existed, member states were not bound by commondecisions, the expert committee’s guidelines did not bind the memberstates, and judicial review by the European courts was rather weak.Consequently, the efficiency of the procedure in achieving its two mainpolicy goals was also rather low: mutual recognition was still not able toestablish the preconditions for a single market of pharmaceuticals, it wasunable to guarantee uniform and effective health and consumer protec-tion, and, consequently, the procedure was generally less positively eval-uated than the centralised one.
However, because of the reform at the beginning of the new millen-nium, the centralised arbitration phase within the mutual recogni-tion/decentralised procedure was strengthened, and member states’commitment within the procedure has subsequently increased. As therapid increase of arbitration procedures in 2006 and 2007 indicates, it isno longer as easy for pharmaceutical companies to prevent arbitration.Consequently, the mutual recognition/decentralised procedure willprobably lead to more uniform market access, as well as to more con-sistent health and consumer protection in the future.
5.2 Legitimacy of European pharmaceutical authorisation
The following analysis examines how the EU regulatory regime forpharmaceuticals legitimises its decision-making (section 3.3). To achievehigh-input legitimacy, the EP should be involved, and stakeholders’influence should be well balanced between different interests (section5.2.1). However, because all participants in the regime’s day-to-day regu-latory policy-making are committed to the tight procedural and sub-stantive rules of decision-making, input legitimacy is restricted to theestablishment of the regime, whereas it is necessarily low during its daily
A Strong Regulatory Network 109
PPL-UK_RR-Krapohl_Ch05.qxd 8/29/2008 11:54 AM Page 109

operation. Thus, the second question is whether the policy output canlegitimise EU pharmaceutical authorisation. The regime may be legiti-mate if it provides a policy outcome that is in the interest of the stake-holders concerned. To achieve such output legitimacy, the extent towhich the regime may be held accountable by different stakeholders isimportant (section 5.2.2).
5.2.1 Input factors
For input legitimacy, two imbalances can be observed throughout theentire development of EU pharmaceutical authorisation. Beginning in the 1960s, the entire regime was established by the Commission and theCouncil with little influence from the EP. At the same time, stakeholders’influence was also unbalanced in favour of producers’ interests. Because ofthese imbalances, the overall input legitimacy of the EU regulatory regimefor pharmaceuticals can be regarded as relatively low, a fact that changedonly little during the reform at the beginning of the new millennium.
From the 1960s to the 1990s, the Council was the crucial actor for theadoption of EU legislation on pharmaceuticals, whereas parliament’sinfluence was marginal. All elements of the supranational regulatoryregime for pharmaceuticals – the substantive authorisation requirementsfor pharmaceuticals, the multi-state and the concertation procedure, thecentralised authorisation procedure, the mutual recognition and thedecentralised procedure, the expert committee, and the EU agency forpharmaceuticals – were originally adopted within consultation or coop-eration procedures. Within these legislative procedures, the Commissionand the Council were not bound by opinions of the EP (Tsebelis 1994;Tsebelis and Kreppel 1998). Thus, nearly no EP amendments were everincluded in final legislation (the only significant exception to this wasthe acceptance of EP representatives to the management board of theagency; section 4.3). As a result, intergovernmental decision-making,with its long delegation chain from member states’ citizens throughnational parliaments to member states’ governments within theCouncil, was the main source of legitimacy during the establishment ofthe EU regulatory regime for pharmaceuticals.
The situation changed slightly during the reform process at the begin-ning of the new millennium, because the reform package was adoptedwithin the co-decision procedure. Within such a procedure, legislativeproposals must have the approval of both legislative actors – an absolutemajority of EP members and a qualified majority within the Council –to come into force (Tsebelis and Garrett 2000, 2001). As a result, the EPwas more successful in influencing the final legislation of the reform
110 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch05.qxd 8/29/2008 11:54 AM Page 110

package. The Council had to accept an extension of the scope of thecentralised authorisation procedure, the management board of theagency was supplemented with representatives of stakeholders, andpolitical control by the member states during the comitology procedurebecame reduced (section 4.3). Consequently, the overall level of inputlegitimacy was, for the latest reform, somewhat higher than for the orig-inal establishment of the regime.
The interests of pharmaceutical producers can be more easily organisedand articulated than those of patients (Abraham and Lewis 2000: 44–9).Producers constitute a small group of actors (especially in the pharma-ceutical sector, which is distinguished by only a few large companies),their interests in pharmaceutical (de-)regulation are very concrete andthey have vast resources for lobbying. In contrast, consumers constitutea much larger group, their interests are much more diffuse and they havefewer resources to spend. This imbalance between the two groups ofstakeholders characterised the development of the EU regulatory regimefor pharmaceuticals. The establishment of a single market for pharma-ceuticals and the supranational regulatory regime in its current form wasat least partly a result of lobbying by the pharmaceutical industry (sec-tion 4.3). The increasingly internationalised pharmaceutical industry – atleast innovative and competitive producers – had a strong interest in asingle market for medicinal products (Feick 2002a). In 1988, theAssociation of the British Pharmaceutical Industry initiated the estab-lishment of the new authorisation regime in the EU when it publishedits ‘Blueprint for Europe’ (Abraham and Lewis 2000: 80–3). Therein, itproposed the establishment of a European pharmaceutical agency, a cen-tralised authorisation procedure for biotechnologically produced phar-maceuticals, and a decentralised procedure for less innovative products.The Commission seized the opportunity in 1991, and submitted to theCouncil and the EP a proposal that closely followed the suggestions ofindustry. Within the following consultation procedure, the basic featuresof this proposal remained unchanged, and thus the industry’s ‘Blueprintfor Europe’ became European law five years after its publication. As aresult, the establishment of the regime was dominated by input from thepharmaceutical industry, and the legitimising potential of consumergroups was missing. It is a crucial question whether this asymmetry isalso reflected within the regime’s day-to-day decision-making.
5.2.2 Output factors
Whereas the efficiency and output legitimacy of the old committee sys-tem in the pharmaceutical sector was low, it increased in consequence
A Strong Regulatory Network 111
PPL-UK_RR-Krapohl_Ch05.qxd 8/29/2008 11:54 AM Page 111

of the regime’s centralisation in the 1990s. The regime is made account-able for its policy output to a range of actors. Most important amongthem are the member states, the regulatory network of national agen-cies and the applicants for marketing authorisation through theEuropean courts. Although the EP and consumers of pharmaceuticalsare relatively weak, the other actors constitute a system of checks-and-balances which controls the regime, whereas no single group is able tocapture the regime (Majone and Everson 2001; Moe 1987a). As a result,the efficiency, and thus output legitimacy, of the EU pharmaceuticalauthorisation is quite high. Whereas policy-making of the regime clearlymeets the interests of the pharmaceutical industry, consumers also seemto be satisfied with its central features. Despite the asymmetric influenceof producers and consumers, the widespread hypothesis of a one-sidedcapture of the regime by industry’s interests (e.g. Abraham and Lewis2002; Lewis and Abraham 2001) lacks empirical support. On the contrary,the various controls inherent in the system seem to counterbalance a dis-proportionate influence of the pharmaceutical industry (Gehring andKrapohl 2007).
The political accountability of the EU regulatory regime for pharma-ceuticals is rather strong, but it is unbalanced in favour of the memberstates. A large majority of the agency’s management board and the expertcommittee are directly recruited by the member states. Thus, they areaccountable to the member states in the long term, because they need tobe reappointed after a certain time. Moreover, the Council may directlycontrol the regime during the political phase of the centralised or arbi-tration procedure (section 5.1.2). The comitology committee could rejectCommission proposals, in which case the matters would be referred tothe Council. The Council would then have the final say on the authori-sation of medicinal products. Within this political phase of the proce-dures, the supranational regime is even accountable to the member statesin the short term, because the comitology committee and the Councilcan intervene directly in the regime’s day-to-day operation. However, asthe empirical analysis of the centralised procedure demonstrates, theCouncil usually does not use this control mechanism (Table 5.2). Withinthe mutual recognition phase of the mutual recognition/decentralisedauthorisation procedure, the regime is, of course, even more accountableto the member states, because authorisation decisions remain at the dis-cretion of the national regulatory agencies.
In contrast, the regime’s accountability to the EP is rather weak,which mirrors parliament’s weak position during the establishment andreform of the regime. The EP recruits only two members of the agency’s
112 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch05.qxd 8/29/2008 11:54 AM Page 112

management board and no members of the expert committee. Moreover,it is not involved in the day-to-day decision-making of the regime. Theonly control mechanism given to the EP in the reforms at the beginningof the new millennium was budgetary monitoring. Together with theCouncil, the EP has to adopt the yearly budgets of the EU pharmaceuti-cal agency, which are prepared by the agency’s executive director. Afterimplementation of these budgets, the EP may discharge the executivedirector. However, compared with the competencies of the Council andthe Commission, the EP’s budgetary competencies are likely to beweak (Moe 1987b; Pollack 1997b). Most parts of the agency’s budget arebound to steady expenses like personnel. These cannot practically bechanged by the EP within budgetary procedures. And the refusal to discharge the executive director is the ‘nuclear option’ of budgetarycontrol. It can only be used in cases of massive mismanagement or corruption.
Expert accountability of the EU regulatory regime for pharmaceuticalsis ensured by the strong network of member states’ regulatory agencies(Dehousse 1997; Majone 1997). Within the centralised or arbitrationprocedures, this expert accountability is reached through the recruit-ment of the agency’s expert committee from members of the nationalregulatory agencies. Within mutual recognition, expert accountability isreached through the acceptance of national authorisation by the regula-tory agencies of other member states. Therefore, the member states’authorisation bodies have to trust the scientific evaluations of the refer-ence member states’ agencies. To sum up, the national authorisationbodies for pharmaceuticals establish a regulatory network either inside(centralised or arbitration procedure) or outside (mutual recognition) theEuropean agency. Within this network, all experts of the national agen-cies require a good scientific reputation. Without that, they would eithernot be chosen as rapporteurs (centralised or arbitration procedure), ortheir national authorisation decisions would not be accepted by con-cerned member states (mutual recognition). As a result of this reputationmechanism, the experts of the different national authorisation bodiesare accountable to each other, and control themselves mutually.
Because of the strong legalisation of the policy area, the legal account-ability of the regulatory regime vis-à-vis its stakeholders is rather strong,but it is unbalanced in favour of producer interests. Firstly, it is impor-tant that the regime’s decision-making is transparent so that stakehold-ers can monitor authorisation decisions to challenge them later beforethe European courts. In this regard, the regime is often criticised foroperating in apparent secrecy (Abraham and Lewis 2000: 172–201).
A Strong Regulatory Network 113
PPL-UK_RR-Krapohl_Ch05.qxd 8/29/2008 11:54 AM Page 113

Generally, this is due to the regulatory subject. Applications for market-ing authorisations contain information that is the intellectual propertyof the applying companies, and which constitutes important competi-tive advantages for them. Nevertheless, the question is where the linebetween private intellectual property and information of public interestshould be drawn. In some member states – e.g. the UK, France andGermany – no information about applications and their evaluations ispublished at all (Abraham and Lewis 2000: 172–201). Thus, decision-making within the mutual recognition procedure is, mostly, not partic-ularly transparent and cannot be scrutinised from the outside. Thesituation is somewhat better within the centralised authorisation proce-dure. Here, the EU pharmaceutical agency has made some effort to openits decision-making to public scrutiny. However, some important infor-mation is still missing: withdrawn applications are not listed individu-ally on the website, and minority views and voting behaviour ofcommittee members are usually not published. This lack of transparencyof the regulatory regime favours the interests of producers vis-à-vis thoseof consumers. As applicants for marketing authorisation, producers haveprivileged access to information, whereas consumers have to scrutiniseregulatory decision-making from outside closed doors.
Secondly, the extensive legalisation of European pharmaceutical autho-risation and the relatively wide scope of possible plaintiffs lead to poten-tially strong judicial review of the supranational regulatory regime – atleast for decisions passed within the centralised or arbitration procedures.However, one problem is that access to European courts is asymmetricallydistributed among stakeholders, because potential plaintiffs have todemonstrate their individual and direct concerns about EU decisions inorder to raise claims. Producers of pharmaceuticals may easily demon-strate such individual and direct concerns, because authorisation deci-sions are directly addressed to individual companies and directlyinfluence their legal positions (Collatz 1996: 143–6). In contrast, con-sumers have difficulties bringing claims against authorisation decisionsbefore the European courts, because they are not directly and individu-ally affected by authorisation decisions that are addressed to other legalpersons. Here again, the interests of producers are favoured vis-à-vis thoseof consumers. However, one must keep in mind that the EU organs andthe member states are always entitled to bring claims before the ECJ.
Despite some asymmetries, the accountability of the regime is, insummary, relatively strong. The regime is held responsible by three mech-anisms: firstly, it is politically controlled by the member states within the comitology procedure; secondly, it is embedded within a regulatory
114 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch05.qxd 8/29/2008 11:54 AM Page 114

network, and thus, it is accountable to the authorisation bodies of themember states; and thirdly, owing to the high legalisation of Europeanpharmaceutical regulation, the regime is subject to strong judicialreview by the European courts. Although no one of these three mecha-nisms is able to control the regime fully, they complement each other(Majone and Everson 2001; Moe 1987a). If the agency’s expert commit-tee deviates from its mandate, its decisions may be revoked by the polit-ical bodies within the comitology procedure. However, if these bodiesdecide on the basis of particularistic short-term interests, their decisionsmay be challenged before the European courts. To scrutinise regulatorydecision-making, the courts themselves are dependent on the expertiseof the European regulatory network. Altogether, the various controlmechanisms constitute a ‘magic triangle’, in which the three anglescheck and balance each other. If one of these angles does not functionproperly, the other two might step in and bring the regime back ontrack. To capture regulatory policy-making, interest groups would haveto capture all three of these angles, a task that should be very difficulteven for the powerful pharmaceutical industry.
As a result of the strong accountability mechanism, the output legiti-macy of the EU regulatory regime for pharmaceuticals is rather high.Within the evaluation report from 2000, both producers and consumersexpressed a high level of satisfaction with central features of the EU reg-ulatory regime for pharmaceuticals (Table 5.1 and section 5.1.2). Thereby,the centralised authorisation procedure – the more ‘Europeanised’ proce-dure and surely the core of the supranational regulatory regime – wasevaluated more positively than the mutual recognition procedure. Atleast for industry interests, the problem-solving capacity of the regime isgenerally high, but the available evidence of consumer satisfaction pointsin the same direction. Such a preference for the centralised procedurewould not be rational if regulatory standards were systematically loweredwithin the supranational regulatory regime. In such a case, patients andphysicians would opt for the mutual recognition procedure, wherein theywould be better protected by their national regulatory agencies. Thus,regulatory standards of the centralised procedure are likely to be at leastas high as those of the various national procedures.
It is often argued that the EU’s pharmaceutical authorisation suffersfrom a bias in favour of the pharmaceutical industry, i.e. that it servesmore the interests of producers than those of consumers (e.g. Abrahamand Lewis 2002; Feick 2003, 2005; Lewis and Abraham 2001; Permanandand Mossialos 2005; Permanand 2006: 1–19). Accordingly, because thepharmaceutical industry is much better organised in Europe than patient
A Strong Regulatory Network 115
PPL-UK_RR-Krapohl_Ch05.qxd 8/29/2008 11:54 AM Page 115

groups, it had many more possibilities to influence the establishment ofan EU authorisation regime for medicinal products. In particular, theCommission was subject to industry lobbying and has developed intoindustry’s strongest ally – not least because both of them share an inter-est in the establishment of a single market for pharmaceuticals. The tra-ditional ally of diffuse consumer interests, namely the EP, had much lessinfluence in the legislative process than the Commission, and this fur-ther disadvantaged consumers. Thus, the establishment of a single mar-ket for pharmaceuticals became a major policy-goal – which is assumedby critics to cause a decline in safety standards. Although the analysis ofthis chapter supports the claim of asymmetric influence of producers vis-à-vis consumers, it is at least questionable whether this is reflected inthe regime’s policy output.
Critics argue that the establishment of a single market by the cen-tralised and the mutual recognition/decentralised procedures leads toregulatory competition between the various regulatory agencies at bothnational and supranational level (e.g. Abraham and Lewis 2000: 147–68).Accordingly, because they are dependent on application fees, nationalregulatory agencies and – in cases where the centralised and the mutualrecognition procedure apply alternatively – the EU agency compete forapplications from the pharmaceutical industry. To attract applications,regulatory agencies could be tempted to lower their evaluation stan-dards. The result would be regulatory competition, in which regulatorystandards are reciprocally lowered. However, the effects of such regula-tory competition are countervailed by a reputation mechanism (Majone1997; Majone and Everson 2001). The regulatory agencies of the mem-ber states constitute a regulatory network in which they control eachother mutually. Within the centralised or arbitration procedure, themembers of the expert committee must be appointed by their peers tothe position of rapporteur. Moreover, within the mutual recognition pro-cedure, the reference member states’ initial authorisation must beaccepted by the concerned member states. Thus, the national regulatoryagencies need a good reputation among their neighbours. It is doubtfulthat it pays off for regulatory agencies to lower their standards systemat-ically, because they would thereby damage their reputation.
5.3 Conclusion
The analysis of the different authorisation procedures for pharmaceuti-cals clearly supports the hypothesis that member states’ commitment ispositively correlated with the efficiency of regulatory policy-making
116 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch05.qxd 8/29/2008 11:54 AM Page 116

(section 3.2). The old multi-state procedure and the concertation proce-dure, which applied until the early 1990s, were the weakest of all analysedprocedures. Member states were not bound to the advice of the EU expertcommittee, and their national authorisation decisions could only be scru-tinised weakly by the ECJ. Consequently, the procedures failed to create asingle market for pharmaceuticals, and health and consumer protectionwas dependent on national regulatory agencies. The centralised authori-sation procedure, which has applied since 1995 for highly innovativemedicinal products, is the strongest of all procedures. Therein, memberstates are actually bound to the decisions of the newly established EUagency for pharmaceuticals. As a result, the procedure proves quite effi-cient. It creates the preconditions for the establishment of a single marketfor highly innovative medicinal products, and so far no evidence has beenfound that it fails to achieve a high level of health and consumer protec-tion. Finally, the mutual recognition procedure is a mixture of the oldmulti-state procedure and the new centralised procedure. Its characterdepends strongly on whether decisions are passed by mutual recognitionor centralised arbitration. Until the reforms at the beginning of the newmillennium, mutual recognition was clearly the dominant mode ofdecision-making, and centralised arbitration occurred only rarely. Thus,the procedure was rather inefficient, and it did not contribute to a homog-enous supply of pharmaceuticals in Europe. The most recent reformaimed at strengthening centralised arbitrations, and the growing numbersof arbitrations in 2006 and 2007 indicates that this was successful.
Owing to the efficiency of EU pharmaceutical authorisation – at leastwithin the centralised authorisation procedure – the regime clearly derivesits legitimacy from its policy output. Throughout the establishment andreform of the regime, influence was asymmetrically distributed amongseveral actors, and input legitimacy was therefore low. The Council andproducer interests had strong positions, whereas the EP and consumergroups remained rather weak. As a result of this asymmetry, one couldassume that the regime has been captured by producers and mainlyserves the interests of the pharmaceutical industry. However, this is notthe case. In fact, the regime’s output legitimacy is relatively high. Theregime can be held accountable by the member states, by a network ofexperts and by the European courts. Thus, it is embedded within a sys-tem of checks-and-balances wherein the regime is controlled by a vari-ety of different actors, but not by one particular group (Majone andEverson 2001; Moe 1987a). These control mechanisms hamper system-atic agency drift and regulatory capture by the pharmaceutical industry.
A Strong Regulatory Network 117
PPL-UK_RR-Krapohl_Ch05.qxd 8/29/2008 11:54 AM Page 117

This page intentionally left blank

Part III The Regulation ofFoodstuffs in the EU
PPL-UK_RR-Krapohl_Ch06.qxd 9/15/2008 10:28 AM Page 119

This page intentionally left blank

6From an Early Single Market to a Crisis of Consumer Confidence:The Development of FoodstuffRegulation in Europe
The EU and its member states can look back on a long history of food-stuff regulation and on several attempts at establishing a single marketfor these products (Alemanno 2006; Ugland and Veggeland 2006). Evenbefore the foundation of the European Economic Community in 1958,all member states regulated some aspects of the quality and safety offood on their national markets (Vos 1999a: 131–9). A single market forfoodstuffs was later achieved with the mutual recognition of thesenational standards and with the partial harmonisation of some meas-ures of health and consumer protection at the EU level. Despite this rel-atively long history of foodstuff regulation, the sector suffered from aregulatory crisis in the 1990s (Ansell and Vogel 2006; Majone 2000;Vogel 2001a; Vos 2004). The BSE scandal damaged consumer trust inthe safety of foodstuffs and in the regulatory capacities of the EU. Inaddition, struggles about the authorisation of GMOs and GM food fur-ther harmed consumer confidence (Vogel 2001b). In the aftermath ofthe crisis (Vos 2000b), the regulatory regime for foodstuffs was funda-mentally reformed.
This chapter aims to test the hypothesis derived from a historical-institutionalist approach (see section 2.2). The development of Europeanfoodstuff regulation clearly followed the second path: a major regulatoryscandal occurred only after a single market was established, and, conse-quently, a regulatory regime was only recently set up at the suprana-tional level. No independent regulatory agencies for foodstuffs, whichcould have acted as stakeholders of institutional change, existed at thenational level. The absence of national agencies had important conse-quences for market integration and for the establishment of a suprana-tional regulatory regime. Firstly, the creation of a single market did notmeet with resistance from strong national regulatory agencies, and thus
121
PPL-UK_RR-Krapohl_Ch06.qxd 9/15/2008 10:28 AM Page 121

was implemented relatively early (section 6.1). However, secondly, whena supranational regulatory regime for foodstuffs had to be set up in reac-tion to the BSE crisis (section 6.2), it could not fall back on support ofregulatory agencies within the member states, and a totally new regula-tory body was set up at the EU level (section 6.3). As a result, it is likelythat the new supranational regime is relatively weak, because such a newbody constitutes a bigger threat for member states’ sovereignty than aregulatory network of national regulatory agencies would.
6.1 Mutual recognition and the committee system
In the second half of the 20th century, almost all Western democracieshad established some kind of food law (World Health Organization1988). With the exception of Portugal, all EU member states had eithera general food law or a general trade law on which food regulationscould be based. All member states supplemented these general laws withmore specific horizontal and vertical laws and bylaws, issued by gov-ernmental bodies. Some countries also used legally non-binding guide-lines, like the Austrian and German Codices Alimentarii or the guidelinesfrom the Swedish Food Administration. Traditionally, foodstuff regula-tion was a political responsibility in most EU member states. This marksa huge difference from the USA, where the independent FDA has beenresponsible for the regulation of foodstuffs and pharmaceuticals since1906. However, within a large majority of the EU member states, gov-ernment ministries alone – mostly the ministries for agriculture orhealth – were responsible for the regulation of foodstuffs (World HealthOrganization 1988). Only Scandinavian countries – Denmark, Swedenand, from 1990 onwards, Finland – were familiar with independent reg-ulatory agencies for foodstuffs, which worked at some distance frompolitical bodies.
In a first attempt to abolish non-tariff barriers of trade in the foodstuffsector, the EU tried to completely harmonise the regulation of certainfoodstuffs at the supranational level. In a resolution of 1969,1 theCouncil published a list of nearly 50 measures of food law, which itplanned to harmonise before 1 January 1971. Most of these were stan-dards, which aimed to regulate the quality of specific foodstuffs. Thefirst measure adopted was the Council Directive for cacao and chocolateproducts in July 1973 (O’Rourke 1998: 57–60).2 It contained detaileddefinitions and compositional requirements of cacao powder, drinkingchocolate, cacao butter, chocolate, plain chocolate, chocolate flakes andmilk chocolate. The Directive for cacao and chocolate already showed
122 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch06.qxd 9/15/2008 10:28 AM Page 122

the fundamental problem with the total harmonisation of food stan-dards: planned for 1 July 1969, the Directive was delayed for four years.The reason for this was a struggle in the Council about certain additivesin chocolate. Whereas some member states allowed the use of vegetablefats other than cacao-butter in chocolate products, others disagreedstrongly with this practice. The result was a delay of the legislation andan unsatisfying outcome: the final Directive made no provision for theuse of vegetable fats in chocolate, but it contained an exception forDenmark, Ireland and the UK, which allowed the use of up to 5 per centvegetable fats other than cacao butter in chocolate products.Consequently, the use of this additive in chocolate remained a point ofconflict even after the Directive came into force (O’Rourke 1998: 59).
One result of such problems during the adoption of food standardswas that the timing of the harmonisation programme envisioned bythe Council Resolution proved not to be viable. Consequently, theCouncil set up a new, revised harmonisation programme in 1973.3
However, this new programme neither reduced the number of meas-ures that had to be adopted, nor did it establish a new approach to theharmonisation of food standards. It only delayed the deadline for theharmonisation programme to 1 January 1978. However, even this newschedule was over-optimistic. In 1985, only 14 Directives (two-fifths ofthe planned measures) were adopted. In fact, it turned out that thestandards for food additives, materials in contact with foodstuffs,labelling of foodstuffs and foodstuffs for particular nutritional use weremuch easier to adopt than standards for specific products. The memberstates were able to agree on general health requirements laid down inthese standards, whereas they faced difficulties finding consensus ondetailed provisions of standards for specific products.
The door towards a new approach to the creation of a single marketfor foodstuffs was opened by a preliminary ruling of the ECJ. With itsfamous Cassis-de-Dijon ruling,4 the ECJ invented the so-called mutualrecognition principle. The background of this case was that Germanyprohibited the import of French Cassis-de-Dijon, because it did not ful-fil German standards for liquors. In Germany, fruit liquors had to havea minimum alcohol content of 25 per cent, whereas Cassis-de-Dijoncontains only 15–20 per cent alcohol. The importer challenged this pro-hibition before the German courts, which asked for a preliminary rulingof the ECJ. The court decided that the German import restriction was an unjustifiable barrier to trade. Generally, every product that fulfils the regulatory standards in one member state should be allowed on themarkets of all member states. In other words, member states should
From an Early Single Market to a Crisis of Confidence 123
PPL-UK_RR-Krapohl_Ch06.qxd 9/15/2008 10:28 AM Page 123

mutually recognise each other’s regulatory standards. Nevertheless, thecourt laid down that it would tolerate exceptions for reasons of healthand consumer protection (Alter and Meunier-Aitsahalia 1994; Dehousse1998: 85). However, Germany was not able to rely on these exceptions,because the ECJ judged that a minimum content of alcohol would notbe justifiable for reasons of health and consumer protection.
Later, the mutual recognition principle became the fundament of theCommission’s Single Market Programme. In the late 1970s and early1980s, the government policies of the three largest member stateschanged. In Germany and the UK, socialist governments were replaced byconservative ones, and the socialist government in France fundamentallychanged its economic policy. The result was a new liberal consensusbetween the most important member states, which fitted well with themutual recognition principle supported by the Commission (Moravcsik1991, 1998: 314–78). In this situation, the Commission came forwardwith its well-known White Paper on ‘Completing the Internal Market’.5
The paper contained around 300 legislative measures, which theCommission wanted to get adopted to create the Single Market. The SingleMarket Programme was approved in June 1985 by the European Councilin Milan. Later, it found its way into the Single European Act of 1987. Thistreaty amendment incorporated the concept of the Single Market and set31 December 1992 as a deadline for its completion. Further, it introducedqualified majority voting in the Council for matters relating to the SingleMarket, in order to increase the efficiency of decision-making.
The Single Market programme had a deep impact on the integration ofthe European foodstuff market. In a communication,6 the Commissionlaid down its new approach to the elimination of barriers to trade in food-stuffs, which followed roughly the example of the new approach in thesector of technical goods (Kerler 2005: 197–222; Vos 1999a: 133–4). TheCommission left the strategy of a total harmonisation of food standardsbehind. Instead, it planned to rely mainly on the mutual recognitionprinciple to establish a single market for foodstuffs. Future EU foodstufflegislation was only aimed at regulating necessary aspects of health andconsumer protection, fair competition and foodstuff control. New legisla-tion should not address specific products, but should provide horizontalmeasures for all foodstuffs. Experiences during the 1970s and early 1980sdemonstrated that the adoption of such horizontal standards was mucheasier, because member states only had to agree on general rules of healthand consumer protection instead of detailed recipes for certain foodstuffs.Within its communication and the White Paper, the Commission listed arange of horizontal provisions that it planned to get adopted before the
124 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch06.qxd 9/15/2008 10:28 AM Page 124

end of 1992. These lists included measures such as a new regulatoryregime for food additives and a directive on the official control of food-stuffs by the member states. In fact, the Commission was able to push 25important Directives and Regulations through the legislative procedurebefore December 1992 (Nentwich 1995; O’Rourke 1998: 24–133).
With the new approach for the regulation of foodstuffs, theCommission aimed to achieve a new distribution of competenciesbetween the Council and itself. In a so-called ‘simplified procedure’, theCouncil only decided on the basic rules of foodstuff regulation, whereasthe implementation of this framework legislation was left to theCommission. However, the Commission was not left alone with thistask: several committees advised or controlled the Commission (Vos1997, 1999b). Firstly, several member state committees were establishedby the Council to control the Commission during the implementationof framework legislation. The committees consisted of representativesfrom the member states’ bureaucracies, and decided within a so-calledregulatory procedure (IIIa or IIIb).7 If Commission proposals were notsupported by at least a qualified majority in the relevant committee,they were passed on to the Council, which had the final say. Secondly,different scientific committees were set up by the Commission to givescientific advice on regulatory policy-making. The scientific committeesrecruited independent scientists and experts who were usually notmembers of their home countries’ bureaucracies. The Commission wasnot bound by scientific opinions of the committees, but was free to pro-pose any policies to the member state committees and possibly to theCouncil. Finally, the Commission established the Advisory Committeeon Foodstuffs to give representatives of the most important stakehold-ers a voice in regulatory policy-making (Vos 1999a: 148–50).8 The com-mittee consisted of representatives of industry, consumers, agriculture,commerce and labour. However, the advisory committee had no right ofinitiative and was only able to meet and to discuss topics if it was askedto by the Commission. The result was that the advisory committeeremained very passive, and that it was not able to exercise influence onregulatory policy-making in the foodstuff sector (Falke 2000: 147–8).
At the beginning of the 1990s, the EU regulatory regime for foodstuffswas shaped by the new approach (Alemanno 2006). Even though the newapproach led to harmonised standards for some aspects of foodstuff reg-ulation, it carried the danger of producing regulatory gaps in other areas.EU foodstuff regulation was still an incomplete ‘patchwork’ (Héritier1996). A general and coherent food law, which could have closed regu-latory gaps, was still missing at the EU level at the beginning of the
From an Early Single Market to a Crisis of Confidence 125
PPL-UK_RR-Krapohl_Ch06.qxd 9/15/2008 10:28 AM Page 125

1990s (Nentwich 1994: 200–6). At the time when the EU foodstuff reg-ulation was still an uncompleted patchwork, application of the mutualrecognition principle reduced member states’ competencies for the reg-ulation of food safety. In the absence of harmonised standards, memberstates generally had to accept foodstuffs that fulfilled the product stan-dards of at least one other member state, and their autonomy in estab-lishing individually high regulatory standards was reduced. Althoughthe application of the mutual recognition principle was moderated bythe fact that the Cassis-de-Dijon ruling allowed for exceptions in casesof health and consumer protection, the ECJ reserved the right to scruti-nise the reasons for such individual measures. In fact, the situation wasan example of a gap between positive and negative integration (Scharpf1996a, 1997b, 1999: 43–83). Negative integration, in the form of themutual recognition principle, reduced member states’ ability to regulatefoodstuffs, whereas delayed positive integration was not yet able to sub-stitute for this loss.
6.2 The BSE scandal
During the 1990s, the weakness of the EU regulatory regime for food-stuffs was mainly responsible for the fact that the EU had not been ableto adopt strong regulatory measures against BSE for 14 years, and thatthe disease was therefore able to spread all over Europe. From 1986 to1996, the UK was the only member state affected by BSE. To avoid a banon its beef in the Single Market, it had a strong interest in sticking to itsinitial position: that BSE was not transmissible to humans (section6.2.1). The situation changed for the first time when the UK had toadmit that BSE could cause variant Creutzfeldt-Jakob disease (vCJD) inhumans. Consequently, the EU banned the export of beef from the UK(later also from Portugal) to protect the other member states. In con-trast, common regulatory policies were difficult to adopt, because alarge majority of the member states believed these were not necessary(section 6.2.2). The situation changed for the second time in 2000,when it transpired that BSE was not only a British, and to some extenta Portuguese, problem, but that nearly the entire EU was affected. Atthat point, most member states preferred common regulatory measuresto fight the disease (section 6.2.3).
6.2.1 Britain versus the rest of Europe
The BSE story dates back to February 1985, when cow 133 died at afarm in Sussex, England, after it had developed a head tremor and
126 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch06.qxd 9/15/2008 10:28 AM Page 126

uncoordination. At this time, nobody knew the ominous disease thatcaused this death. The symptoms of the new disease were very similarto scrapie in sheep, which had been known for more than 200 years.Scrapie belongs to a group of illnesses called transmissible spongiformencephalopathy (TSE), includings CJD which affects humans. Nowadays,most scientists believe that these diseases are caused by mutated prions(Prusiner 1997), which can either be transmitted from mother to off-spring or by the consumption of meat, especially of brain and neural tis-sues. TSEs lead to brain degeneration and finally to the death of theaffected human or animal. It soon transpired that the new ‘scrapie’ incattle was not only a single case, but a new disease, which was sooncalled bovine spongiform encephalopathy, or, in short, BSE. Even today,the reasons for its outbreak in the 1980s are disputed. However, it islikely that the meat- and bone-meal fed to cattle was an important vec-tor of the disease, which led to its rapid spread; first across the UK andlater across Europe. In the early years of BSE, the hope was that ‘mad cowdisease’ would behave like scrapie, which was known as not being trans-missible to humans. However, it transpired (in 1988) that BSE is trans-missible to mice (later it was also detected in cats and pigs). It couldtherefore cross the species barrier, at least to some mammals.
From 1989 to 1996, BSE was regulated in the EU based on secondaryveterinary legislation, because the disease was thought to affect mainlythe health of farmed animals (Knipschild 2003). According to this leg-islation,9 the Commission and the member states had some safeguardcompetencies to react to the outbreak of animal diseases constituting arisk for animal or human health, and to implement trade restrictions toprevent the spread of such diseases. However, the Commission was notfree to act independently. It was advised by the Scientific VeterinaryCommittee10 and controlled by the Standing Veterinary Committee11
within a regulatory procedure (either IIIa or IIIb). Owing to the strictcomitology procedure, a majority of the member states was always ableto block regulatory measures.
Within the EU committee system, BSE led to a classic intergovern-mental cooperation problem in which the interests of different EUmember states directly opposed each other. On the one hand, theBritish interest was to protect the domestic beef industry. On the other,both consumers and producers of beef in the other member states hadan interest in protectionist measures against British beef to be shieldedfrom health risks or competition from the UK. As long as governance ofthis issue was based on the mutual recognition principle, the UK gov-ernment had every interest in insisting that BSE was not transmissible
From an Early Single Market to a Crisis of Confidence 127
PPL-UK_RR-Krapohl_Ch06.qxd 9/15/2008 10:28 AM Page 127

to humans. It had to expect that British beef would immediately bebanned by the other member states, as soon as these were able todemonstrate health risks before the ECJ. In this situation, scientificarguments were in danger of becoming politicised (Dressel 2002:65–129; Millstone and van Zwanenberg 2001; Krapohl and Zurek 2006).
The first EU policies on BSE reflected the British position that BSEcould not be transmitted to humans. Only in July 1989 – three yearsafter BSE had been detected in the UK – did the EU prohibit the exportof cattle born before July 1988 (the date of the British feeding ban ofmeat- and bone-meal to ruminants) and offspring of BSE suspects fromthe UK. Half a year later, this ban was extended to all cattle older thansix months. The next measures were adopted in spring 1990, when BSEwas made a notifiable disease in the EU, and the export from the UK ofspecified bovine offal of cattle aged over six months at slaughter wasforbidden. Later that year, the first political struggles about BSE tookplace (Scheu 2003: 591–7; Vincent 2004), because France and Germanyannounced that they intended to block imports of British beef. Thisprovoked heavy resistance from both the UK and the Commission,which tried to uphold the Single Market. They argued that this unilat-eral action was unlawful, because BSE would not be of risk to humanhealth. The Commission threatened to bring a claim before the ECJ.12
The conflict was on the agenda of an extraordinary meeting of theAgriculture Council where a compromise was reached. Accordingly,France and Germany abstained from banning imports of British beef,whereas the UK was required to establish an identification system forcattle, and to provide quality certificates for exported meat.
Surprisingly, the first political struggle on BSE was followed by a four-year period of inactivity at the EU level. With the exception of a regula-tion of the export of bovine embryos from the UK, there was totallegislative inactivity for BSE during this period. This passivity is evenmore striking if one considers that the peak of the BSE epidemic in theUK took place during that very period. The EU only became active againin 1994, when Germany raised doubts on the non-transmissibility of BSEto humans, and wanted to adopt stronger regulatory measures.13
Although Germany found itself in a minority position, the EU subse-quently adopted significant regulatory measures for BSE. In June 1994, itimplemented an EU-wide feeding ban of meat- and bone-meal to rumi-nants,14 and set up conditions for the processing of meat- and bone-meal, which was still allowed to be fed to other farmed animals. Onlyone month later, the conditions for the export of certain live cattle andbeef products were again amended, now requiring that specified bovine
128 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch06.qxd 9/15/2008 10:28 AM Page 128

offal be destroyed directly after the slaughtering of the animal. From thatdate until March 1996, the EU was only able to amend previouslyadopted policies, but did not adopt any new regulatory measures.
6.2.2 The rest of Europe versus Britain
The situation changed fundamentally in March 1996, when it tran-spired that BSE is in fact transmissible to humans. Since May 1995, sev-eral atypical cases of CJD in humans had been detected. The patientswere either farmers with BSE-infected cattle in their herds, or young per-sons aged between 20 and 30 years. In contrast, the classic form of thedisease affects mainly persons who are older than 60 years of age. Itturned out that these cases belonged to a new variant of CJD (termedvCJD). And in March 1996, the British government stated officially thatthe possibility of BSE being transmissible to humans could not be ruledout (Scheu 2003: 616). The reaction of the EU was swift. On 22 March1996, the Scientific Veterinary Committee met to discuss the new situ-ation, but it concluded that – even in the light of the new, disturbinginformation – the regulatory measures already applied were sufficient tohandle the disease (Westlake 1997). However, the political bodies wereunder more pressure to act and came to a different conclusion. On 25March, the Standing Veterinary Committee agreed (with 14 memberstates against 1) with a proposal of the Commission to ban the exportof cattle, beef, beef products and meat- and bone-meal from the UK.15
The export ban led to deep political struggles at the EU level. The UKgovernment saw the ban as an exaggerated measure which was not sup-ported by scientific evidence, and which favoured the economic interestsof the other member states. After the UK was not able to achieve at leasta partial lifting of the ban, the British Prime Minister, John Major,announced that the UK government would start legal proceedings againstthe ban before the ECJ, and that it would follow a non-cooperation pol-icy in all EU matters. Accordingly, the UK did not vote on any decisionsin the EU, and made actions by unanimity impossible (Westlake 1997).The political deadlock at the EU level was only solved at the EuropeanCouncil in Florence in June 1996, where a Commission plan for thegradual lifting of the ban was approved. In reaction, the UK governmentgave up its non-cooperation policy and got back to business (Westlake1997). Two years later, in March 1998, the Council lifted the exportprohibition for certain beef and beef products under the strict condi-tions of the Export Certified Herds Scheme. A few months later, the banwas almost completely lifted, and beef and beef products could beexported again under the newly established Date-Based Export Scheme.
From an Early Single Market to a Crisis of Confidence 129
PPL-UK_RR-Krapohl_Ch06.qxd 9/15/2008 10:28 AM Page 129

The decisions to impose and to lift bans on exports of beef and beefproducts from the UK were not fought only politically, but also judi-cially by both sides involved in the conflict. In the beginning, the UKproceeded against the export ban on British beef and beef products.Generally, the position of the UK was that the Commission misused itspower when it imposed the ban. In fact, the UK’s claim before the ECJwas later dismissed, because the ECJ regarded the safeguard clauses ofthe veterinary control Directives as sufficient and legitimate grounds forthe Commission’s decision.16 Two years later, after the ban on theexport of British beef and beef products had been lifted, France tried tooverthrow this decision. It came forward with new scientific evidencefrom its then newly founded food agency (Agence française de sécuritésanitaire des aliments, Afssa), which raised doubts about the safety ofbeef and beef products under the Date-Based Export Scheme. The Frenchgovernment proceeded against the unwillingness of the Commission tore-impose the ban, but the claim was deemed inadmissible for formalreasons.17 Despite the loss of this case, the French government went onto ban imports of British beef and beef products on its domestic market.Thus, the Commission started an infringement procedure against France.The ECJ generally confirmed the Commission’s position that the Frenchgovernment failed to fulfil its obligations by its refusal to lift the ban(Szawlowska 2004).18,19 All in all, much judicial activity with respect toBSE can be observed. However, all these cases were only related to theexport ban and not to regulatory policy-making by the Commissionand the various committees.
Whereas the previously discussed claims were all addressed againstthe imposing or lifting of the export ban, there was only one case thataddressed an alleged failure of the Commission to act during the firstphase of the BSE saga. In September 1996, a group of Italian farmersbrought a claim of compensation against the Commission before theCourt of First Instance. The farmers argued that, in reaction to theBritish statement that BSE is transmissible to humans, the Europeanmarket for beef collapsed, beef prices fell and the farmers themselvessuffered significant economic losses. The farmers claimed that this col-lapse of the European beef market could have been prevented by theCommission if it had reacted earlier to the BSE disease (e.g. if it hadimposed a ban on British beef in 1990). The claimants drew a direct linefrom the Commission’s shortcomings in regulating the disease to theirown economic losses in 1996, and asked for compensation from theCommission. However, the claim of the Italian farmers was dismissedby the Court of First Instance.20 According to the court, the claimants
130 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch06.qxd 9/15/2008 10:28 AM Page 130

were able to prove neither faults of the Commission in handling the BSEdisease, nor the direct link between the Commission’s action and theireconomic losses. The court argued that the collapse of the Europeanbeef market was a result of the British statement about the likely trans-missibility of BSE to humans, and not of the Commission’s inactivity inthe years before that statement. Accordingly, the court had seriousdoubts about whether earlier measures to fight the disease would haveprevented such a collapse of the European beef market. This was theonly judicial dispute in which the Commission was to be held respon-sible for its (in-)activity in the first phase of BSE regulation from 1989to 1996. However, the claim for compensation turned out to be a ‘bluntsword’, because it was difficult to demonstrate a direct link betweenfaults of the Commission and the claimants’ losses (Wakefield 2002).
The handling of BSE during the first phase of the saga was more suc-cessfully scrutinised by the EP (Chambers 1999). In July 1996, the EP setup – for the first time in its history – a temporary committee of inquiryto investigate mismanagement in relation to BSE (Westlake 1997; Vos1999a: 143–5).21 The EP had only recently received the respective com-petencies in the Maastricht Treaty of 1993, and the handling of the BSE scandal was a prominent case in which to use this new power. The report of the EP inquiry committee heavily criticised all bodiesinvolved in the regulation of BSE. Firstly, the Scientific VeterinaryCommittee established a working group on BSE, which was dominatedand chaired by British experts, because they were thought to have themost expertise on the disease. However, these experts did not workindependently, so the position of the Scientific Veterinary Committeereflected that of the British Ministry of Agriculture, Fisheries and Food.22
Secondly, the committee system, with its various bodies involved in reg-ulatory policy-making – namely the Scientific Veterinary Committee,the Commission, the Standing Veterinary Committee and finally theCouncil – led to a lack of transparency and diffusion of responsibili-ties.23 Consequently, one body did not always know what the otherswere doing, and everyone was able to hide behind the inaction of theothers. Thirdly, the Commission, especially the Directorate-General (DG)for Agriculture, always gave priority to the management of the SingleMarket, instead to the protection of consumers’ health.24 The report con-cluded with a detailed list of recommendations to the Commission forreforms of the EU regulatory regime for foodstuffs: it recommended anincrease in the transparency of regulatory policy-making by restructuringthe committee system; it suggested strengthening the monitoring andinspection resources of the EU by establishing a European Agency for
From an Early Single Market to a Crisis of Confidence 131
PPL-UK_RR-Krapohl_Ch06.qxd 9/15/2008 10:28 AM Page 131

Veterinary and Phytosanitary Inspections; and it proposed the adoptionof a range of measures aimed at generating a stronger legislative basis forEU foodstuff regulation.25
According to the recommendation of its inquiry committee, the EP held the Commission responsible for the mismanagement of BSE. In February 1997, the BSE inquiry report was subject to debates of the EP, which eventually threatened a motion of censure against theCommission. The EP announced that it would check whether theCommission was following the recommendations of the inquiry reportby October 1997 (Westlake 1997). Because of this political pressure, theCommission quickly answered the criticism of the EP. In April, it pub-lished its plan to reform the EU regulatory regime for foodstuffs. Thisreform was envisioned as a two-step process (Vincent 2004), accordingto which the Commission would develop its own new approach to foodsafety immediately, followed by a general revision of the EU foodstufflegislation.26 The Commission stated in a communication that it aimedto separate more clearly the different functions of scientific advice, leg-islation and control from each other in order to increase the overalltransparency of the regulatory process.27 The basic measures for achiev-ing these goals were a reorganisation of scientific advice in the foodstuffsector and a strengthening of the EU resources for veterinary and food-stuff controls. Additionally, the Commission stated that it aimed toplace all competencies for food safety regulation under the authority ofthe DG for Consumer Policy and Health Protection.
After its communication, the Commission reorganised its internaladministrative structure in the area of foodstuff regulation. Before theBSE crisis, the competencies for health and consumer protection weredivided between the DGs for Agriculture, Internal Market, Enterprise andConsumer Policy. In reaction to the EP inquiry report, the Commissionconcentrated the respective competencies within the DG for ConsumerPolicy and Health Protection (Vos 1999a: 145–7, 2000b). Besides, theCommission strengthened and reorganised veterinary and foodstuffcontrol. The Office of Veterinary and Phytosanitary Inspection andControl (established in 1991) was strengthened, renamed the Food andVeterinary Office (FVO), and moved from the DG for Agriculture to theDG for Consumer Policy and Health Protection (Chambers 1999).Finally, the Commission fundamentally restructured the scientific com-mittees in the foodstuff sector by setting up the Scientific SteeringCommittee28 under the authority of the DG for Consumer Policy andHealth Protection. The aim of the Scientific Steering Committee was todeal exclusively with BSE and other TSEs, and to coordinate the work of
132 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch06.qxd 9/15/2008 10:28 AM Page 132

the other scientific committees. The number of the other scientific com-mittees was reduced to eight,29 and they were moved to the DG forConsumer Policy and Health Protection. In the end, the measures takenby the Commission were evaluated positively by the EP, and it decidednot to table a motion of censure against the Commission (Vincent 2004).
Aside from the struggles concerning the export bans on British beefand the institutional reforms in reaction to the EP inquiry committee,surprisingly few common regulatory measures were adopted by the EUbetween 1996 and 1999. BSE was still regarded as a British (and later alsoa Portuguese), but not a European, problem. The other member statesthought that there was no reason for regulatory measures on their ownterritory as long as imports of British beef could be prevented. Regulatorymeasures were expensive, and a ban on British beef would impose thecosts of regulatory measures solely on the UK, whereas common EUmeasures would distribute the costs to all member states (Krapohl andZurek 2006). The first substantive BSE policies in 1996 were eradicationprogrammes in member states that had BSE in their domestic cattle pop-ulations. These were not only the UK and Portugal, but also France andIreland, which had already detected non-imported BSE cases. Further,the EU adopted some new measures for feedstuffs – especially on meat-and bone-meal. Specific processing systems were prescribed for theproduction of meat- and bone-meal for feeding animals other than ruminants in order to prevent an outbreak of other TSEs. Further restric-tions on the trade with meat- and bone-meal were introduced, and someanimal proteins – the so-called hydrolysed proteins – were excluded fromthe feeding ban of meat- and bone-meal. The EU also set up an epidemio-surveillance programme for all TSEs.
A prominent example of the EU’s difficulties in adopting strong com-mon measures on BSE during the second phase of the saga was the failedattempt to regulate the use of specified risk materials (Chalus and Peutz2000). These specified risk materials are tissues that are highly infectiousif the respective animals (cattle, sheep or goats) from which they werederived suffered from TSE. In the UK, the handling of these tissues hadalready been regulated since November 1989. In October 1996 – sevenyears after the UK measure – the Scientific Veterinary Committeeadopted its first opinion on this matter and recommended the destruc-tion of the skull, brain, eyes, tonsils and spinal cord of cattle, sheep and goats aged over twelve months directly after slaughtering, so thatthe materials could not enter animal or human food chains. After somestruggles in the Standing Veterinary Committee and the Council, theCommission was able to adopt a respective decision, albeit against the
From an Early Single Market to a Crisis of Confidence 133
PPL-UK_RR-Krapohl_Ch06.qxd 9/15/2008 10:28 AM Page 133

resistance of some member states. However, despite the fact that a banon specified risk materials was formally adopted in July 1997, it was notset into force until October 2000, because the implementation of thepolicy was repeatedly postponed by the member states. At the end of1999, no regulation of specified risk materials had been implemented inthe EU, ten years after such a policy was adopted in the UK.
6.2.3 The Europeanisation of the disease
During 2000, the BSE problem finally became Europeanised (Krapohl2003). Since 1998, the Scientific Steering Committee had been workingon a method to calculate the geographical risk of BSE in states otherthan the UK and Portugal. This calculation was based on two factors:the challenge of national cattle herds by imports of potentially infectedcattle and meat- and bone-meal from the UK; and the stability of thenational regulatory system (i.e. whether it would have reduced or ampli-fied BSE infectiousness). In July 2000, the Scientific Steering committeepublished a report in which the risk of BSE in each EU member state wasclassified according to this scheme.30 According to this report, nearly allEU member states faced a high risk of BSE infections in their domesticcattle population, even if these had not yet been confirmed. Only inAustria, Finland and Sweden – member states that had only joined theEU in 1995 – were BSE infections in the domestic cattle populationunlikely. The report was an affront for member states like Germany,which had stressed very much that they would not be affected by BSE and that their beef would be safe (Dressel 2002: 129–56). However,owing to reinforced epidemio-surveillance throughout the next sixmonths, the prognosis of the Scientific Steering Committee turnedout to be true. The first BSE cases were detected in Denmark, Germanyand Spain. Other member states like Belgium, France, Ireland and theNetherlands – which had recorded sporadic BSE cases before – noticedan increase in infected cattle. In Greece and Italy, the first BSE cases fol-lowed in 2001.31 The result was a new BSE scandal, which peakedaround the end of 2000, when beef disappeared from Christmas menusall over Europe.
The Europeanisation of the BSE problem led to a fundamental changein the preference constellation of the EU member states in the StandingVeterinary Committee and in the Council. From then on, a large major-ity of the member states was affected and had a clear interest in coordi-nating regulatory measures and in sharing the burden thereof. As aresult, the EU was able to impose three important regulatory measures
134 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch06.qxd 9/15/2008 10:28 AM Page 134

on BSE (Krapohl 2003). Firstly, the surveillance of the epidemic wasreinforced and backed up by financial contributions to the memberstates. The first measure in this respect was the introduction of com-pulsory BSE tests on a sample of fallen stock and emergency-slaughteredanimals in June 2000. The epidemio-surveillance programme was thenreinforced twice in November and December, and the EU providedfinancial support for the surveillance measures. All these decisions wereadopted by a qualified majority in the Standing Veterinary Committee,i.e. they were not passed forward to the Council. Conflict between themember states was modest, even though the financial support for mon-itoring programmes by the EU had some distributional impact.
Secondly, after the long struggle between 1996 and 1999 about a decision on specified risk materials, a respective policy was finallyadopted and implemented in June 2000. The list of specified risk mate-rials was amended, slaughter techniques were prescribed and officialcontrols were set up. Nevertheless, the regulation of specified riskmaterials was still a highly disputed issue between the member states.The Commission’s proposal did not receive the support of a qualifiedmajority in both the Standing Veterinary Committee and the Council,but the Council was also no longer able to block the policy by simplemajority. Consequently, the Commission was free to act, and adoptedthe decision, based on the support of only a simple majority of themember states in the Council. In the following nine months, the list ofspecified risk materials was adjusted twice. These two decisions wereadopted by the Commission after favourable opinions from the StandingVeterinary Committee.
Finally, the feeding ban of meat- and bone-meal was extended to allfarmed animals, because the previous ban – which was restricted to aprohibition of feeding ruminants with meat- and bone-meal – hadturned out to be insufficiently implemented. In November 2000, theScientific Steering Committee stated that the implementation of arestricted ban proved difficult and cross-contamination of differentfeedstuffs for different farmed animals could not be ruled out.32 Inaccordance with this statement, the Commission proposed a temporarytotal ban of meat- and bone-meal throughout the entire EU. The pro-posal did not receive a qualified majority in the Standing VeterinaryCommittee and had to be passed to the Council, which finally adoptedthe measure against the votes of Finland and Germany. The temporaryban on all meat- and bone-meal was amended twice in the next twomonths. Some animal proteins were excluded from the ban, but theywere only allowed to be produced in plants that did not produce other
From an Early Single Market to a Crisis of Confidence 135
PPL-UK_RR-Krapohl_Ch06.qxd 9/15/2008 10:28 AM Page 135

meat- and bone-meal. Both decisions set up these exceptions and con-ditions were adopted by the Commission after they received support ofa qualified majority in the Standing Veterinary Committee.
The various decisions from 1990 to 2001 on the governance of BSEwere summarised and consolidated in a Regulation of the EP and theCouncil on certain animal TSEs.33 The Regulation contains a scheme forthe classification of member states or third countries according to theirBSE and scrapie status, provisions for the monitoring and notificationof TSEs, prohibitions for animal feeding and specified risk materials,measures for the eradication of TSEs, conditions for the trading of cat-tle, sheep and goats, and rules for inspections and controls. Further, theRegulation delegates competencies to the Commission, for amendmentsto the Regulation and within a safeguard procedure. However, as before,the Commission cannot act independently based on these competen-cies, but is embedded within the committee system. Whenever ques-tions with an impact on public health arise, the Commission shouldconsult the appropriate scientific committee. The resulting policy pro-posals must still pass a regulatory committee, which may adopt them byqualified majority or pass them forward to the Council. Overall, thenew Regulation on TSE has led neither to significant changes to the sub-stantive policies on BSE, nor to procedural reforms within the EU’s reg-ulatory regime.
6.3 The reform of the European regulatory regime for foodstuffs
The EU’s handling of the BSE scandal clearly demonstrated the weak-ness of the committee system in protecting consumers from the risks ofcertain foodstuffs. The result of the scandal was a crisis of consumerconfidence (Ansell and Vogel 2006; Majone 2000; Vogel 2001a; Vos2004), wherein consumers’ trust in the safety of their food and in theregulatory capacities at the EU level were fundamentally shattered. As aresult, both consumers and producers had an interest in a stronger reg-ulatory regime for food safety. Whereas the former feared for their ownhealth, the latter saw the necessity to restore consumers’ confidence intheir products. A policy window opened up, which allowed a funda-mental reform of the EU regulatory regime for foodstuffs (section 6.3.1).In parallel to the reorganisation of the general regulatory regime forfoodstuffs, the EU also established a new regulatory regime for GMOsand GM food during the 1990s and at the beginning of the new mil-lennium (section 6.3.2). As a result of consumer scepticism against these
136 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch06.qxd 9/15/2008 10:28 AM Page 136

products (Bredahl 2001; Gaskell and Allum 2001; Toke 2004:141–89),the regulatory regime for GMOs and GM food is rather restrictive andprecautious, which led to conflicts with trading partners in the WorldTrade Organization (WTO).
6.3.1 The European Food Safety Authority
Although the BSE scandal led to a crisis of consumer confidence and thusto an open policy window in the foodstuff sector, the answer to that cri-sis was not determined only by functional pressure. According to the his-torical-institutionalist hypothesis (section 2.2), the responses to crises ofconsumer confidence are dependent on already existing regulatory insti-tutions. Because national regulatory agencies for foodstuffs did not existin most member states before the BSE scandal, they obviously could notact as stakeholders of institutional change, and could not influence thereform of the supranational regime. Although national regulatory agen-cies were set up in most member states in reaction to the BSE scandaland to the reorganisation of the supranational regulatory regime, theywere not able to influence the new regime effectively. Thus, the newregime is, to a great extent, built on a new supranational agency, but noton a strong network of national regulatory agencies. The new regime islikely to be weaker than a comparable regulatory network, because anew EU body represents a bigger threat to member states’ governmentsthan a network of national agencies would.
An important precondition for an intensive review of food safety legislation was addressed by the Intergovernmental Conference inAmsterdam in 1997. The treaty amendments from Amsterdam estab-lished that health and consumer protection is no longer a mere side-effect of the Single Market, but an independent policy objective of theEU (Krapohl and Zurek 2006; Vos 2000b). The treaty now contains anarticle that requires the Commission and other legislative actors tostrive for a high level of health, safety and environmental protectionwhen they adopt harmonised EU measures. In addition, other articlesprescribe that a high level of health and consumer protection shouldbe ensured in the definition and implementation of all Communitylegislation. Together, these amendments established a new legal basison which future EU measures of health and consumer protection couldbe based.
The first step towards the fundamental reform of the supranationalregulatory regime for foodstuffs was the Commission’s Green Paper onFood Law,34 which was published as early as April 1997 in reaction to theEP inquiry report. Only when the new Commission, under President
From an Early Single Market to a Crisis of Confidence 137
PPL-UK_RR-Krapohl_Ch06.qxd 9/15/2008 10:28 AM Page 137

Romano Prodi, entered office in autumn 1999 did the reform of the EUregulatory regime for foodstuffs receive an additional boost. Prodi waswell aware of the reputational loss that the Santer Commission experi-enced because of the BSE crisis in 1997. Consequently, food safetybecame a top priority of the new Commission (Kelemen 2002). Just threemonths after the new Commission entered office, a group of three lead-ing scientific advisers from the Scientific Steering Committee publisheda report on the future of scientific advice in the EU (Buonanno 2006).35
The three scientists opted for a very independent and powerful ‘EuropeanFood and Public Health Authority’, even more independent and power-ful than the FDA in the USA. They proposed that the agency should,among other things, include an effective monitoring system, its ownresearch resources and a policy analysis unit. Further, they explicitlystated that the agency should not only deal with risk assessment, butalso with risk management and risk communication. This would haverequired the agency to adopt legally binding decisions, a competencethath has never been given to any EU agency.
The Commission reacted to the suggestions of the scientific adviserswith its White Paper on Food Safety from January 2000 (Vincent 2004).36
Therein, the Commission proposed for the first time the establishmentof a European Food Agency. However, in contrast to the advice of thethree scientists, the Commission did not want to delegate far-reachingcompetencies to the agency. The agency was not expected to take respon-sibility for risk management, but only for risk assessment and risk com-munication. According to the White Paper, the main tasks of the agencywere the provision of scientific advice, the gathering and analysis ofinformation and the communication of risks to consumers. For thesetasks, the agency did not need to receive competencies for the adoptionof legally binding decisions. Responsibilities to adopt implementationand safeguard decisions, as well as to conduct food and veterinary con-trols, were to be left to the Commission. In addition to the establishmentof a European Food Agency, the Commission planned to revise the sub-stantive EU foodstuff legislation fundamentally. It therefore presented aprogramme of more than 80 legislative proposals in the Annex of theWhite Paper. Besides, the Commission aimed to propose a new generalfood law that would contain the basic principles of food safety regula-tions. Such general framework legislation for regulatory policy-makinghad been missing for a long time in the foodstuff sector (Nentwich 1995:200–6). Thus, a problematic regulatory gap would be closed.
In November 2000, the Commission brought forward its legislativeproposal for a general reform of the EU regulatory regime for foodstuffs.37
138 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch06.qxd 9/15/2008 10:28 AM Page 138

The Commission’s proposal followed, in large parts, the suggestions ofthe White Paper. The proposed European Food Authority was designedas a scientific advisory body that incorporated the existing scientificcommittees in its administrative structure. The authority received onlycompetencies for risk assessment and risk communication, but not forrisk management. According to this first proposal, a rapid alert systemfor food and feed would be established under the responsibility of thenew authority. Generally, risk management remained under the respon-sibility of the Commission, which was controlled by the member stateswithin a comitology procedure. The proposed food law contained arange of universal principles, which should govern food safety regula-tions in the future (among others, the precautionary principle, thetraceability of food ‘form the stable to the table’ or ‘from the farm to thefork’, and the responsibility of food producers, traders and suppliers forthe safety of their products).
The Regulation for the general EU food law and a European FoodAuthority was adopted within the co-decision procedure, wherein theCouncil and the EP are coequal legislators (Tsebelis and Garrett 2000,2001). Consequently, the EP was a relatively strong player in the leg-islative process, and it used its new power successfully (Kelemen 2002).It acted as a representative of the diffuse interests of consumers, but italso had an interest in increasing its influence on the future EU regula-tory regime for foodstuffs.38 Most importantly, the EP managed to influ-ence the future recruitment of the supranational regulatory regime, andit has now the same competencies as the Council during the recruit-ment process of the agency’s management board. Furthermore, the EPsuccessfully proposed to give the rapid alert system into the hands of theCommission and to change the name of the agency to ‘European FoodSafety Authority’ (EFSA). Generally, all legislative actors agreed on thenecessity and desirability of the regime’s reorganisation. Consequently,the final regulation was already adopted after the second reading of the Council and the EP, without having to go through conciliation. The new EFSA was established in October 2002, and it became operativeduring 2003.
The new general food law is seen as a correction to the unsystematicand ad hoc character of the previous EU food law. It is aimed at being acoherent and systematic basis of future EU action in that field. The gen-eral food law prescribes that not only the effective functioning of theSingle Market, but also a high level of health and consumer protection,should be the main goals of EU foodstuff regulations. To achieve thesegoals, future food regulations should be based on risk analysis, which
From an Early Single Market to a Crisis of Confidence 139
PPL-UK_RR-Krapohl_Ch06.qxd 9/15/2008 10:28 AM Page 139

itself contains risk assessment, risk management and risk communica-tion. In cases of scientific uncertainty, the precautionary principleapplies. Generally, foodstuffs should not be placed on the market if theyare unsafe, i.e. harmful to health or unfit for human consumption. Thisresponsibility rests with the producers, traders and suppliers of food-stuffs. To allow for a clear assignment of responsibilities, foodstuffsshould be traceable at all stages of production, processing and distri-bution. Given their vagueness, these provisions left a high degree ofdiscretion to future regulatory policy-making. Many phrases like ‘ahigh level of health and consumer protection’, ‘scientific uncertainty’or ‘unsafe food’ must still be interpreted.
The new EFSA was aimed to be a response to the failing of the old EUcommittee system during the BSE crisis (section 6.2). During the firstphase of the BSE crisis (1989–96), the committee system allowed Britisheconomic interests to dominate scientific advice of regulatory policy-making. Thus, the main mission of EFSA is to improve this scientificadvice. EFSA was detached from the Commission in order to make itsadvice more independent and its responsibilities more transparent toconsumers. However, the mission of EFSA reflects the artificial separa-tion of risk assessment, risk management and risk communication.Because risk management is left completely to the Commission, theauthority is only responsible for tasks connected to risk assessment andrisk communication. The most important one is probably the issuing of scientific opinions in matters of foodstuff regulation. EFSA mustdevelop such opinions following requests from the Commission, but itmay also act on its own initiative, or following requests from the EP orthe member states.
The most powerful organ of EFSA is the management board, becauseit recruits both the agency’s executive director and the members of thescientific committee and panels. The management board consists of 14members who are appointed for a term of four years by the Council,after consultation of the EP. The members are recruited from a list pro-posed by the Commission, which must include more candidates thanposts. In addition to these 14 members, the Commission sends one rep-resentative to the board. The management board acts with a majority ofits members and must ensure that the authority fulfils the tasks laiddown in the regulation. The management board appoints the author-ity’s executive director, for a term of five years, from a list proposed bythe Commission. The executive director is the legal representative of theauthority and is responsible for its day-to-day administration, as well asfor the implementation of the budget.
140 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch06.qxd 9/15/2008 10:28 AM Page 140

The authority also includes an advisory forum, which consist of rep-resentatives from member states’ regulatory agencies for foodstuffs.Whereas before the 1990s independent regulatory agencies for food-stuffs were common only in the Scandinavian countries, most EU mem-ber states only established such regulatory agencies in the aftermath ofthe BSE crisis. In some countries (like the UK and France; Borraz et al.2006; Rothstein 2006) this was a direct answer to previous administra-tions’ failure to handle the BSE crisis. Other countries (like Germanyand Austria) followed the example of the EU and established advisorybodies similar to EFSA (Fleischer 2005; Fuchs 2004: 117–55; Steiner2006). Of the ‘old member states’ (the countries that had joined the EUbefore the 2004 enlargement), only Italy and Luxembourg did not estab-lish independent food agencies at the beginning of the new millennium.The advisory forum within EFSA has the task of organising a networkbetween all these regulatory agencies of the member states. It advises theexecutive director and it is a forum for communication and coopera-tion. Such a network is supposed to reduce problems that occur duringthe decentralised implementation of EU regulation in the memberstates. However, the regulatory network for foodstuff is much weakerthan its counterpart in the pharmaceutical sector. Whereas the nationalregulatory agencies for pharmaceuticals participate directly in suprana-tional policy-making, the national agencies for foodstuffs have onlyadvisory competencies and are not involved in the day-to-day operationof the EU agency.
Within EFSA, the bodies responsible for the development of scientificopinions are one scientific committee and nine scientific panels.39 Thescientific committee is responsible for the tasks that were previouslyassigned to the Scientific Steering Committee: that is, it should coordi-nate the work of the eight scientific panels and should act on questionsthat require multidisciplinary expertise. The nine scientific panels(including one for GMOs) are more specialised and are responsible for thetasks that were previously assigned to the different scientific committees.The members of the authority’s scientific committee and scientific pan-els should be independent scientific experts. They are appointed by theauthority’s management board on the proposal of the executive direc-tor for three years. The scientific committee consists of the chairmen ofthe nine scientific panels plus six additional independent experts. Whenadopting scientific opinions, the scientific committee and panels may actwith a majority of their members, but minority views must be reported.
As a result of the separation between risk assessment (EFSA) and riskmanagement (Commission), the Commission must develop regulatory
From an Early Single Market to a Crisis of Confidence 141
PPL-UK_RR-Krapohl_Ch06.qxd 9/15/2008 10:28 AM Page 141

policies from the scientific opinions of the scientific committee andpanels. The Commission is free to follow EFSA’s scientific advice or not.Thus, it is the more powerful agenda-setter for regulatory policy-makingin the foodstuff sector. However, the Commission is not independent inits risk management decisions. Whenever it wants to adopt policies, itis controlled by the member states within a strong comitology proce-dure (the so-called regulatory procedure).40 The regulation established aStanding Committee on the Food Chain and Animal Health, which iscomposed of representatives from the member states and is chaired bya representative of the Commission. If Commission proposals do notreceive support of a qualified majority in the committee, matters arereferred to the Council, which may finally act by qualified majority.Even though the Commission is the agenda-setter for regulatory policy-making, the member states still have a strong veto power. The improvedscientific advice by EFSA may address the problems of risk assessmentinherent in the old committee system, but the situation for risk man-agement remains the same as before.
6.3.2 Genetically Modified organisms and food
On first view, GMOs and GM food have characteristics similar to those ofpharmaceuticals. Traditional foodstuffs are a very heterogeneous group ofproducts, their development does not require expensive research andthey are subject to post-marketing control. In contrast, GMOs and GMfood are – like pharmaceuticals – purchased by only a few companies,their development is very research-intensive, and they are subject topre-marketing authorisations, at least in the EU (Patterson 2000;Patterson and Josling 2001). If only functionalist pressure determinedthe design and function of regulatory institutions, the regulatoryregime for GMOs and GM food would look more like that for pharma-ceuticals than that for foodstuffs. However, if path dependencies mat-tered, the design of the regime would be dependent on the sequencingof critical junctures and on the existence or non-existence of nationalregulatory agencies. GMOs and GM food entered the Single Market at the beginning of the 1990s. Thus, a crisis of consumer confidenceoccurred within an already integrated market. Strong national regula-tory agencies for GMOs and GM food were never established. Althoughthe member states had to name competent authorities for GMOs andGM food authorisation, these were by far not as strong as the nationalagencies in the foodstuff sector. Overall, the developmental path of thesupranational regulatory regime for GMOs and GM food comes closerto that for traditional foodstuffs than to that for pharmaceuticals. If the
142 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch06.qxd 9/15/2008 10:28 AM Page 142

historical-institutionalist hypothesis held true, the regime would resem-ble its counterpart for traditional foodstuffs.
The EU started the regulation of GMOs as early as 1990, even beforethe first products entered the Single Market and before member stateshad any experience with the regulation of these products. The firstDirective on the deliberate release of GMOs into the environmentalready included pre-marketing control for GMOs (Schenek 1995:199–222).41 All member states had to name competent authorities forthe evaluation of GMOs, and every release of GMOs into the environ-ment had to be notified to these competent authorities. Together withthe notifications, applicants had to submit technical dossiers on whichthe national authorities could base assessments of the environmentaland consumer risks of the deliberate release of the GMOs. If the GMOswere only to be released for experimental use or research, consent fromthe concerned member states was sufficient. However, if the respectiveGMOs were to be marketed within the EU (e.g. as seeds or animal feeds),agreement of the Commission and the competent authorities of allother member states was also necessary. Usually, this should have beenachieved by mutual recognition of the first member states’ assessment.To facilitate this mutual recognition, the competent authorities of themember states informally established a committee, in which they triedto settle disputes (Töller 2002: 419–29). Nevertheless, mutual recogni-tion rarely occurred, and objections against initial assessments werearticulated in 15 out of 18 decisions between 1992 and 1998. Thus, cen-tralised arbitrations at the EU level were the rule rather than the excep-tion (Sheridan 2001: 63–4).
Within the centralised arbitration procedure, the competent authori-ties of the member states did not play any role (Töller 2002: 375–461).The Commission undertook its own risk assessments, for which it usuallyconsulted the Scientific Committee on Plants (Sheridan 2001: 62–90).The Commission proposals were then forwarded to a comitologycommittee. If the committee did not support the measures by qualifiedmajority, the proposals were referred to the Council, which finally decidedby qualified majority (regulatory IIIa procedure). Positive authorisationdecisions at the EU level implied that the competent authorities of theconcerned member states had to express their written consent about the release of the GMOs to the applying companies. However, even ifthe release of GMOs had been authorised, the member states still had considerable competencies within a safeguard procedure. Accordingly,every member state had the right to prohibit provisionally the sale ofGMOs on its territory as long as there were good reasons to assume that
From an Early Single Market to a Crisis of Confidence 143
PPL-UK_RR-Krapohl_Ch06.qxd 9/15/2008 10:28 AM Page 143

they constituted risks to human health or the environment. EU deci-sions on the validity of such safeguard measures were supposed to beadopted within three months under the same comitology procedure asthe original authorisations.
Because the focus of the Deliberate Release Directive lay on the pro-tection of the environment, it did not explicitly regulate the marketingof GMOs as foodstuffs and was thus inadequate for the protection ofconsumers (Töller 2002: 375–461). To close this gap, the Novel FoodRegulation was adopted in 1997 (Sheridan 2001: 114–69).42 The term‘Novel Food’ included foods consisting of or containing GMOs andfoods produced from, but not containing, GMOs.43 The authorisationprocedure for novel food was very similar to that of the deliberaterelease of GMOs (Christoforou 2004). Generally, every novel food whichwas to be sold on the Single Market had to be notified to the competentauthorities of the concerned member states and to the Commission.Together with the notifications, the applicants had to submit technicaldossiers of which the content was further specified by a CommissionRecommendation.44 The competent authorities conducted initial assess-ments of the products, and submitted the results to the other memberstates and the Commission. If the Commission or other member statesobjected to authorisation, the Commission conducted additional assess-ments in cooperation with the Scientific Committee for Food. TheCommission proposals were subject to the same comitology procedure asapplied within the older directive on the deliberate release of GMOs (reg-ulatory IIIa). Like the older directive, the Novel Food Regulation con-tained a safeguard clause. The member states were allowed to prohibitalready authorised products on their territory if they had good reason toassume that novel foods constituted risks for human health or the envi-ronment. The validity of such measures was examined by the Commissionwithin the aforementioned comitology procedure. The Novel FoodRegulation also included, for the first time, some provisions for thelabelling of GM food. Hence, any existence of GMOs within a novel foodhad to be labelled on the respective products.
The Novel Food Regulation also provided an exception to the gen-eral rule of pre-marketing authorisation. The marketing of novel foodsthat were ‘substantially equivalent’ to already-existing foodstuffs onlyhad to be notified to the Commission and did not require full riskassessments (Sheridan 2001: 132–50). Whereas foods produced from,but not containing, GMOs could be equivalent to traditional products,foods that consisted of or contained GMOs were always subject to themore demanding authorisation procedure. The vagueness of the term
144 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch06.qxd 9/15/2008 10:28 AM Page 144

‘substantial equivalence’ led to conflicts between member states andapplicants. In 2001, the ECJ had to decide on a preliminary rulingabout whether a novel food was ‘substantially equivalent’ to tradi-tional products or not.45 The Italian competent authority had prohib-ited foods produced from GM corn on its market, despite the fact thatthese were evaluated as substantially equivalent to traditionally pro-duced foods by the UK. The Court ruled that the Italian governmentwas nevertheless allowed to take safeguard measures, as long as no EUdecision prohibited this (Dabrowska 2004). Despite these problemswith ‘substantial equivalence’ and despite the fact that the possibilityfor a simplified notification procedure was only an exceptional clause,this developed into the more important authorisation procedure fornovel foods in the following years. Until 2004, all products authorisedunder the Novel Food Regulation were subject to the notification pro-cedure for substantially equivalent foodstuffs, and only two GM foodswere authorised regularly in 2004.46
From 1990 to 1998, 18 different GMOs were approved under theDirective on the deliberate release of GMOs, whereas only one applica-tion did not get the support of the Scientific Committee for Plants andwas subsequently rejected by the Commission (Sheridan 2001: 62–90). Inaddition, from 1997 to 1998, eight GM foods were authorised under thesimplified notification procedure of the Novel Food Regulation. However,major problems with the approval of GMOs and GM food started in themid-1990s (Toke 2004: 141–89). In 1996, the first two major commoditycrops – genetically modified soybeans from Monsanto and corn fromCiba-Geigy – were subject to an authorisation procedure for deliberaterelease into the environment (Shaffer and Pollack 2004). Whereas mostauthorised products were previously only used on a small scale in Europe,these two crops were expected to be extensively imported from the USA,where they were widely distributed and often mixed with traditionallyproduced crops. Although many member states raised objections to theauthorisation of these crops, they were not able to form a coalition thatcould have rejected the Commission proposals in the last stage of thecomitology procedure. In this situation, the Commission authorised theproducts, in one case even against an expressed will of a majority of the member states (Shaffer and Pollack 2004; Sheridan 2001: 77–81).
The Commission’s disregard for member states’ objections, lessthan a year after the BSE crisis, was seen as a provocation (Patterson2000; Patterson and Josling 2001; Toke 2004: 141–89; Vogel 2001b).In reaction to the authorisation of the GM corn from Ciba-Geigy,Greenpeace started legal proceedings against the written consent of
From an Early Single Market to a Crisis of Confidence 145
PPL-UK_RR-Krapohl_Ch06.qxd 9/15/2008 10:28 AM Page 145

the French competent authority, and argued that the precautionaryprinciple was breached by the authorisation. However, within a pre-liminary ruling, the ECJ judged that the French authorities had nodiscretion to deny written consent after a positive Commission deci-sion had been adopted.47
The member states came under increasing pressure of public resistanceand responded by imposing a de facto moratorium on GMOs and GMfood (Skogstad 2003; Shaffer and Pollack 2004; Young 2003). Austria,France, Greece, Germany, Italy, Luxembourg and the UK applied thesafeguard clauses of the EU legislation to prohibit the marketing ofGMOs and GM foods on their territory, despite the fact that the respec-tive products had been authorised regularly under EU law (Shaffer andPollack 2004; Sheridan 2001: 81–7). The Commission was not able tooverrule these safeguard measures, because it did not find the necessarysupport in the Council. And at a Council meeting in June 1999, a rangeof member states declared that they would oppose every authorisation ofGMOs and GM food as long as a reform of respective legislation was notadopted.48 Because the respective member states constituted a qualifiedmajority in the Council, they were able to veto every authorisation deci-sion of the Commission. Consequently, from April 1998 to July 2004, noGMOs or GM food were authorised in the EU, with the exception ofsome novel foods that were deemed to be substantially equivalent to tra-ditionally produced foods, and which needed only be notified to theCommission.
The moratorium on GMOs and GM food had a huge influence oninternational trade with these products. Only a few of the GMOs andGM foods that were allowed in the USA were also authorised in the EU.Because GM and traditionally produced crops are often mixed in theUSA, the entire trade of crops was heavily affected (Young 2003). Theexport of soy from the USA to the EU fell dramatically from 1998 to2003 (Shaffer and Pollack 2004). This was the beginning of a trade war,in which the USA accused the EU authorities of being captured one-sidedly by ill-justified consumer protectionism, whereas the EU was notwilling to give up regulatory sovereignty in favour of economic interestsin the USA. In August 2003, the conflict became especially virulent inthe context of the WTO, when the USA and other WTO member statesstarted legal proceedings against the EU before a WTO dispute settle-ment body. Generally, WTO rules do not allow WTO member states toestablish trade barriers that are not scientifically justifiable for reasonsof health and consumer protection (Bernauer 2003: 118–167; Shafferand Pollack 2004). The problem for the EU was that its measures against
146 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch06.qxd 9/15/2008 10:28 AM Page 146

GMOs and GM food were based on the precautionary principle and noton purely scientific reasoning. It is a highly disputed issue, whethersuch precautionary measures are allowed under WTO rules (Young andHolmes 2006). After repeated delays, the WTO dispute settlement bodydecided in the GMO case against the EU in November 2006.49 Withinits judgement, the body stated that the EU’s general moratorium againstGMOs and GM food was inconsistent with WTO rules.
The trade disputes with the USA placed the EU under increasing pres-sure to reform the regulatory regime for GMOs and GM food (Skogstad2001, 2003). The Commission responded to this pressure by developinga new strategy for the regulation of biotechnology, which was publishedin a consultation paper and a communication on biotechnology.50 Onthe one hand, the Commission again stressed the economic importanceof biotechnology, and declared that international regulatory standardshad to be taken into account to overcome the differences in regulatoryapproaches between the EU and some of its major trading partners. Onthe other, it stated, for the first time, that the EU’s potential in biotech-nology could be realised only if public opinion and the interests of allstakeholders were considered. To meet consumer demand for morehealth protection, the Commission announced the strengthening of thescientific basis of regulatory decisions, the application of the precau-tionary principle in case of scientific uncertainty, and the establishmentof a stricter and more coherent labelling system for GMOs and GM foodto enable consumers to make informed choices.
In March 2001, a new Directive on the deliberate release of GMOsinto the environment was finally adopted (Sheridan 2001: 29–94).51
During the legislative process, the EP and the Council had pushedtowards stricter regulatory standards than those proposed by theCommission (Shaffer and Pollack 2004). Both organs strongly repre-sented the interests of consumers, whereas the Commission tried toreduce the influence of the European GMO regulation on internationaltrade. The Directive still distinguishes between an experimental releaseof GMOs, which needs only the consent of the concerned memberstates, and a marketing of GMOs, which needs the agreement of allmember states and which can be subject to centralised arbitrationwithin a comitology procedure (still a regulatory procedure). A modifi-cation of the new Directive compared with the old one is that consul-tations of the relevant scientific committees are made compulsorywhenever member states or the Commission raise objections to authori-sation. Since the establishment of EFSA in 2002, the relevant committeeis the Scientific Panel on GMOs within the new agency. This provision
From an Early Single Market to a Crisis of Confidence 147
PPL-UK_RR-Krapohl_Ch06.qxd 9/15/2008 10:28 AM Page 147

strengthens the role of science in the regulatory process and centralisesthe risk assessment in the hands of the new agency.
One year after the adoption of the new Directive on the deliberaterelease of GMOs, the Commission started to review the authorisationregime for GM food by proposing a Regulation on GM food and feed.This new Regulation aimed to replace the provisions of the Novel FoodRegulation for products containing or derived from GMOs (Christoforou2004). Again, the Regulation was adopted by a co-decision procedurethat enabled the EP to represent the diffuse interests of consumers andto push for strict regulatory standards. The Regulation on GM food andfeed was adopted in September 2003. It differed significantly from itspredecessor, the Novel Food Regulation.52 The new Regulation no longerprovides a simplified authorisation procedure for GM food and feed thatis deemed to be substantially equivalent to traditionally produced prod-ucts. Because of its arbitrary nature, this clause of the Novel FoodRegulation was highly disputed and subject to judicial conflicts. Underthe new Regulation, all GM food and feed products are subject to the fullauthorisation procedure. In addition, the new Regulation specifies whichlevel of GMOs is allowed in foodstuffs without being labelled as such.Accordingly, food and feed products that contain GMOs need not belabelled if this presence does not exceed 0.9 per cent of the food ingre-dients (Shaffer and Pollack 2004).
Most importantly, the new Regulation on GM food and feed provideda much more centralised authorisation system and fewer safeguardcompetencies for the member states. Both the authorisation and thesafeguard procedure are the same as already provided by new EU foodsafety legislation. Even if the member states are still the addressees ofapplications, they are obliged to refer them directly to EFSA. WithinEFSA, the scientific panel on GMOs issues opinions on the safety of therespective products, which are then forwarded to the Commission. TheCommission proposals are then subject to a comitology procedure inwhich the Standing Committee on the Food Chain and Animal Healthand the Council scrutinise policy-making within a regulatory proce-dure. For emergencies, the Commission is usually responsible for takingsafeguard measures, which are subject to the same comitology proce-dure. If the Commission fails to act, the member states may adoptinterim protective measures, but only as long as the Commission doesnot adopt decisions. Taking both the centralised authorisation proce-dure and the limited safeguard competencies together, the possibilitiesof the member states to impose a de facto moratorium on GM food arereduced compared with the previously existing legislation.
148 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch06.qxd 9/15/2008 10:28 AM Page 148

On first view, the new strategy on biotechnology was a success for theCommission. In May 2004, because of the approval of GM corn, themoratorium on GMOs and GM food was effectively lifted. Later that year,another GM corn was authorised for deliberate release into the environ-ment under the reviewed Directive and as a foodstuff under the NovelFood Regulation. However, on second view, the results are much moreambivalent. No legislation of the new strategy on biotechnology receivedunified support from all member states. In particular, the reviewedDirective on the deliberate release of GMOs did not satisfy all memberstates, and after the Directive was adopted, Austria, Denmark, France,Greece, Italy and Luxembourg expressed their will to continue themoratorium on GMOs (Shaffer and Pollack 2004). Of all five GM foodsthat were authorised up to 2006, not a single one received the approvalof a qualified majority in the Council.53 In the absence of majority posi-tions in the Council, the Commission was free to act, and authorisedthe GM foods (Shaffer and Pollack 2004). Under these circumstances, itis doubtful that the Commission’s new strategy for biotechnology isreally sustainable.
6.4 Conclusion
The development of EU foodstuff regulation supports the hypothesisderived from a historical-institutionalist argument. Although memberstates had adopted a range of different food standards before the estab-lishment of the Single Market, most of them had not set up independ-ent regulatory agencies. As a result of the absence of national agencies,a single market for foodstuffs was established by mutual recognition ofmember states’ food standards. This mutual recognition was flanked bypartial harmonisation of food safety measures in the EU committee sys-tem. However, when BSE entered the agenda in Europe, it transpiredthat this committee system was too weak to handle the disease. It took14 years – from the detection of BSE in the UK in 1986 – for the EU toadopt strong regulatory measures against BSE in 2000. Before, either theUK or the other member states prevented effective regulation with theirown particularistic interests. The BSE scandal opened a policy windowthat was used by the Commission to propose a complete reorganisationof the EU regulatory regime for foodstuffs. At the same time as the EUreformed the supranational regime, many member states also estab-lished national regulatory agencies for foodstuffs. However, these agen-cies were still too weak to influence reform at the supranational level.As a result, the new supranational regime is based on a completely new
From an Early Single Market to a Crisis of Confidence 149
PPL-UK_RR-Krapohl_Ch06.qxd 9/15/2008 10:28 AM Page 149

supranational body – the EFSA – and not on a regulatory network ofstrong national agencies. Such a new body constitutes some threat forthe sovereignty of the member states, which consequently establishedstrong political control over the new supranational regime.
The case of GMOs and GM food further supports the historical-institutionalist hypothesis. This is not only because it is an additionalcase, but also because it is a rather critical one. GMOs and GM food haveto be authorised before they gain access to the Single Market. In thisrespect, they more closely resemble pharmaceuticals than traditionalfoodstuffs. If the design of regulatory institutions depended only onfunctionalist pressure, one would assume that the regulatory regime forGMOs and GM foods would also resemble that for pharmaceuticals.However, the opposite is the case, and the similarities to traditional food-stuffs are striking. As in the case of traditional foodstuffs, strong nationalregulatory agencies for GMOs and GM food do not exist. Although theEU legislation asked the member states to establish so-called ‘competentauthorities’ for the evaluation of GMOs and GM food, these were by nomeans as strong as the agencies in the pharmaceutical sector. They werenot able to prevent the authorisation of GMOs and GM foods on theirown markets, because they could always be overruled within centralisedarbitrations at the EU level, which was the rule rather than the excep-tion. When the crisis of consumer confidence shattered the Europeanfoodstuff sector, the supranational regime proved unable to uphold a sin-gle market. The member states used their competencies in the Comitologyand safeguard procedures to establish a de facto moratorium on GMOsand GM food, which led to heavy conflict with major trading partnersin the WTO. As a result of this crisis, the supranational regime was fur-ther centralised. The regime now includes the EFSA, which is responsiblefor the risk assessment of GMOs and GM food, and a strong Comitologycommittee, which controls the agency and the Commission on behalf of the member states. Consequently, the regime looks very much likethat for traditional foodstuffs, which clearly supports the historical-institutionalist hypothesis.
150 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch06.qxd 9/15/2008 10:28 AM Page 150

7A Weak Supranational Agency: The Evaluation of the EuropeanRegulatory Regime for Foodstuffs
The development of the EU regulatory regime for foodstuffs followed theopposite path to that of the regulatory regime for pharmaceuticals. As aresult, some of the institutional features of the foodstuff regime are insharp contrast to the pharmaceutical regime. National regulatory agen-cies were not established in most member states. Thus, they cannot estab-lish a strong regulatory network. The result is that supranational expertbodies in the foodstuff sector are likely to be relatively weak, because themember states are more hesitant to delegate far-reaching competencies topurely supranational bodies than to regulatory networks under control ofnational regulatory agencies. In addition, because of the market-drivenapproach to foodstuff regulation, the EU food safety legislation lacks acoherent and systematic food law, and remains an uncompleted patch-work. The question this chapter addresses is how these institutional fea-tures influence the regime’s efficiency and legitimacy in regulating the EUfoodstuff sector.
In this chapter, two dimensions of the evaluation of the EU regulatoryregime for foodstuffs are distinguished. Firstly, the regime is assessed for itsefficiency (see section 7.1). Here, the question is whether the regime is ableto achieve the two policy goals of establishing a single market for food-stuffs, and of providing effective regulation of these products. The hypoth-esis to be tested is that the regime’s efficiency strongly depends on thecommitment of the member states expressed in the procedural and sub-stantive rules of decision-making. The second question is how legitimatethe regime is, and which factors contribute to its legitimacy (section 7.2).Because of the need for credible commitment in supranational risk regu-lation, input legitimacy is necessarily restricted to the set up of the regu-latory regime for foodstuffs. Consequently, the importance of outputlegitimacy for the regime’s day-to-day operation increases significantly.
151
PPL-UK_RR-Krapohl_Ch07.qxd 9/12/2008 10:29 AM Page 151

To achieve high output legitimacy, the EU regulatory regime should beheld accountable by a range of different stakeholders whose influence isbalanced in a way that they cannot dominate the regime one-sidedlywith their particularistic interests.
7.1 Commitment and efficiency of European foodstuff regulation
The following sections evaluate the extent to which member states havecommitted themselves within the procedural and substantive rules ofthe EU regulatory regime foodstuffs, and whether this commitment isinterrelated with the regime’s efficiency. As in the pharmaceutical sector,the hypothesis is tested on three different procedures that have beenapplied for the regulation of foodstuffs in the EU (chapter six). Firstly,until 2002 foodstuff regulation was developed and adopted within theEU committee system (section 7.1.1). Secondly, after suffering the BSEscandal and the subsequent crisis of consumer confidence, the EU estab-lished the EFSA, which replaced the scientific committees, and which isnow responsible for all risk assessment and risk communication in thefoodstuff sector. However, the Commission is still responsible for riskmanagement, and it is thereby still controlled by the member stateswithin a comitology procedure (section 7.1.2). Finally, parallel to thereform of the regulatory regime for traditional foodstuffs, the EU estab-lished and reformed a regulatory regime for GM food. Therein, all GMfood has to be evaluated by the EFSA on a case-by-case basis, just as phar-maceuticals are evaluated by the EMEA (section 7.1.3).
7.1.1 The committee system
The committee system in the foodstuff sector was rather complex (section6.1 and Figure 7.1), because it was not established by a single and coher-ent legislative act, but developed from different pieces of foodstuff legis-lation, starting in the late 1960s (Vos 1997, 1999b). Even though mostcommittees in the foodstuff sector had been established before, theybecame much more important in the course of the implementation ofthe Single Market Programme. The Commission’s new approach to food-stuff regulation was distinguished by two important characteristics.Firstly, the idea of harmonising all possible food standards at the EU levelwas given up. Instead, the new strategy was to establish a single marketfor foodstuffs by mutual recognition of national food standards.Foodstuff regulations were only aimed at being partly harmonised to pre-vent individual measures of the member states in health and consumer
152 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch07.qxd 9/12/2008 10:29 AM Page 152

protection. Secondly, the new approach intended to avoid detailed tech-nical legislation for food standards. Instead, EU Directives and Regulationswere aimed at establishing only framework legislation, whereas com-pletion of this framework was delegated to the Commission. TheCommission was thereby controlled and advised by the various comi-tology and scientific committees of the foodstuff sector. As a result, thevarious committees and the Commission became the main regulatorybodies in the field (Majone 1994, 1996: 61–82; Vos 1999b).
The Commission itself (not the member states) established the scien-tific committees to get advice on scientific aspects of foodstuff regula-tion. The aim of these committees was not to control, but to support theCommission during the implementation of framework legislation. Thecommittees were composed of supposedly independent experts and the Commission provided their secretariat. The committees were usu-ally asked for their scientific opinions by the Commission, but other EUorgans, like the Council or the EP, also occasionally requested suchadvice. The committees were expected to find scientific consensus, butminority views were reported. Before the BSE crisis, four major scientificcommittees worked in the field of foodstuff regulation. The most impor-tant of these was the Scientific Committee on Food, which was respon-sible for advice on all general matters relating to food safety (Vos 1999a:141–3).1 Other scientific committees in the field were the ScientificVeterinary Committee (later the Scientific Committee on VeterinaryMeasures relating to Public Health), the Scientific Committee forAnimal Nutrition and the Scientific Committee for Pesticides (later theScientific Committee on Plant Health). In reaction to the BSE crisis, theCommission reformed the committee system in the foodstuff sector,and set up a Scientific Steering Committee, which coordinated the work
A Weak Supranational Agency 153
Figure 7.1 The committee system in the foodstuff sector
Advise the Commission
Decision proposals Decisions
CouncilCommission
StC on Foodstuffs
Scientific Committee on
Food
Scientific Veterinary Committee
Standing Veterinary Committee
PPL-UK_RR-Krapohl_Ch07.qxd 9/12/2008 10:29 AM Page 153

of the other scientific committees in the field, and which advised theCommission exclusively on all matters relating to BSE (Vos 2000b).
The agenda-setter of regulatory policy-making in the foodstuff sectorwas not the several scientific committees, but the Commission. Withinthe scope of framework legislation, the Commission was free to proposeany policies to the member states. It could, but did not have to, consultthe relevant scientific committees before drafting proposals. And it could,but again did not have to, follow the committees’ opinions. Commissionproposals were always passed to the relevant comitology committees.The fields of responsibility of the member state committees mirroredthose of the respective scientific committees (with the exception beingthe Scientific Steering Committee, which had no equivalent among the member state committees). There was also a Standing Committeeon Foodstuffs (Vos 1999a: 152–4),2 a Standing Veterinary Committee, aStanding Committee on Feedingstuff (later the Standing Committee onAnimal Nutrition) and a Standing Committee on Plant Health. All thesemember state committees decided according to a regulatory procedure(IIIa or IIIb).3 If proposals did not receive the support of a qualifiedmajority within the relevant committees, they were forwarded to theCouncil, which could either amend them by unanimity, or accept orreject them by qualified majority. Within the regulatory IIIb procedure,the Council was also able to reject proposals by simple majority. In anycase, the Commission was free to implement its proposals when theCouncil failed to reach decisions.
Independence and strength of the scientific committees
The recruitment of the several scientific committees for foodstuffs was,at least formally, rather independent from the member states. The mem-bers of the scientific committees were all appointed by the Commissionand not by the member states themselves. In contrast to the pharma-ceutical sector, the experts within the scientific committees of the food-stuff sector were supposed to be independent experts, and were notinvolved in the bureaucracies or regulatory agencies of the memberstates. Consequently, member states’ influence on the scientific basis ofregulatory policy-making should have been rather weak.
However, such a positive assessment of the scientific committees’ inde-pendence was disproved during the BSE scandal (section 6.2.1). Duringthe first phase of the crisis (from 1986 to 1996), the Scientific VeterinaryCommittee was captured by British agricultural interests (Krapohl and Zurek 2006; Millstone and van Zwanenberg 2001). To advise theCommission on BSE, the Scientific Veterinary Committee had established
154 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch07.qxd 9/12/2008 10:29 AM Page 154

a specialised working group, which became dominated and chaired byBritish experts. However, these British experts relied, for a considerabletime, on the misleading assumption that BSE would not be transmissi-ble to humans, despite the fact that transmission to other mammalslike mice, cats and pigs had already been proven. The ScientificVeterinary Committee was composed of veterinarians, who naturallyhad a better understanding of the consequences of the disease forEuropean agriculture than for consequences for consumers’ health, afact that was reinforced by the placement of the committee under theresponsibility of the DG Agriculture. Because of these two mechanisms,the Scientific Veterinary Committee reflected the position of the BritishMinistry of Agriculture, Fisheries and Food, and scientific advice wasnot independent from political influence. The European Parliamentlater heavily criticised this lack of independent scientific advice, andargued that this was due to lack of transparency during the committee’srecruitment.4
Not only was the independence of the scientific committees in thefoodstuff sector low, but the scientific committees were also weak agenda-setters within regulatory policy-making. The scientific committees onlygave scientific advice, which was not at all binding for political actors. Inaddition, even if the Commission stuck to the committees’ advice, it wasitself controlled by the member states within a strict comitology proce-dure (Franchino 2000a, 2000b; Steunenberg et al. 1996). A qualified orsingle majority of the member states was always able to veto regulatorypolicy-making.
Politicisation of regulatory policy-making in the comitology systembecame evident in the second phase of the BSE scandal, from 1996 to2000 (section 6.2.2; Krapohl and Zurek 2006). After it became clear in thespring of 1996 that BSE is transmissible to humans, the EU imposed anexport ban on British beef in the Single Market. However, in addition tothis export ban, the EU was not able to adopt strong regulatory measuresfor fighting the disease, because a majority of member states rejectedsuch policies. This way, the majority of member states imposed the costsof fighting the disease solely onto the UK, while protecting their ownagricultural industry and consumers from beef imports from the UK.With this strategy, the other member states did not follow scientificadvice, but protected the interests of their domestic industry. On thecontrary, advice of the Scientific Veterinary Committee suggested, forexample, prohibiting the sale of specified risk materials like bovinebrain, nerves and intestines (Chalus and Peutz 2000). The Commissiontook up this advice and proposed a corresponding policy to the member
A Weak Supranational Agency 155
PPL-UK_RR-Krapohl_Ch07.qxd 9/12/2008 10:29 AM Page 155

states, but these vetoed the decision repeatedly in the Standing VeterinaryCommittee and the Council.
Legalisation of the policy area
The policy area of EU foodstuff regulation was also hardly legalised. Thedevelopment of the EU food law was driven by several attempts to createa single market and became an uncompleted patchwork (Héritier 1996).Although a variety of different vertical and horizontal food standardsexisted, a coherent and systematic food law containing general goals ofregulatory policy-making was still missing (Nentwich 1995).
This lack of a general food law became evident in the BSE case (section6.2.1). To adopt policies on BSE, the Commission had to rely on twoDirectives for veterinary checks in the trade of live animals and animalproducts in the Single Market. Only in the case of outbreaks of animaldiseases had the Commission competencies to take safeguard measuresagainst the spread of these diseases. It was important that the twoDirectives did not contain any substantive criteria for when and how theCommission should use these safeguard competencies. The respectiveclauses only stated that the Commission could take protective measureswhenever it deemed this necessary. Thus, the decisions were not guidedby general criteria of regulatory policy-making, because such criteriawere widely absent.
Corresponding to the weak legalisation, the influence of the ECJ onsupranational regulatory policy-making was also rather limited. The lackof a coherent food law left a great deal of discretion to the Commissionand the member states. As a result, the ECJ was not able to scrutinise reg-ulatory policy-making effectively. The only issues where the ECJ was ableto intervene were those on free trade within the Single Market. Based onthe treaties, the ECJ was able to judge whether trade barriers of individ-ual member states were justified or not. Probably the most importantexample of such judgements was the well-known Cassis-de-Dijon casefrom 1979, which established the mutual recognition principle withinthe Single Market (Alter and Meunier-Aitsahalia 1994). However, this didnot address regulatory policy-making at the EU level. Besides weak legal-isation, the scope of potential plaintiffs was rather limited in the food-stuff sector. In contrast to the pharmaceutical sector, there were noapplicants for marketing authorisations who would have been entitledto bringing claims directly before the European courts. Neither produc-ers nor consumers of foodstuffs were individually and directly affectedby regulatory measures, so it was difficult for them to bring claims beforethe ECJ. The only exceptions were, again, issues of free trade. If the
156 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch07.qxd 9/12/2008 10:29 AM Page 156

Commission had established trade restrictions for certain products, com-panies would have been individually and directly affected by these deci-sions. Consequently, they would have been able to challenge suchdecisions in the European courts. Again, regulatory policy-making at theEU level is not affected by such claims.
The weak judicial review of regulatory policy-making in the foodstuffsector was again visible in the BSE case (section 6.2.2). Although a fullrange of BSE-related claims were brought before the European courts,nearly all of them dealt with the export ban on British beef and not withregulatory policy-making by the Commission. The implementation andlater also the lifting of the ban were repeatedly fought in front of theECJ and the Court of First Instance,5 but these disputes only referred tothe question of free trade. The only case, in which the Commission wasto be held accountable for its mismanagement of the BSE crisis, was aclaim for compensation by Italian farmers (Wakefield 2002).6 Thesefarmers argued that the collapse of the beef market in 1996 was due tothe Commission’s inactivity during the first phase of the BSE crisis from1986 to 1996. Consequently, they demanded compensation for theireconomic losses. Although such claims would be principally a way tohold the Commission accountable, they turned out to be ineffective.The farmers were not able to demonstrate the causal link between theCommission’s inactivity and their economic losses in 1996. Thus, theirclaim was rejected by the Court of First Instance.
Efficiency of the committee system
The EU committee system of the foodstuff sector was not able to regu-late the BSE disease adequately (section 6.2). During the first phase of thescandal, the Scientific Veterinary Committee was captured by Britishagricultural interests. Consequently, EU reactions to BSE came rather lateand were generally insufficient for fighting it and protecting consumers.During the second phase of the scandal, the member states blocked regu-latory measures that were proposed by the Scientific Steering Committeeand the Commission. Thereby, a majority of the member states tried toimpose the regulatory costs of fighting the disease solely on the UK –which was affected by an export ban on its beef – whereas they them-selves protected their own industry and consumers. During both phases,the failure of the EU to fight the disease was due to the prevalence ofmember states’ particularistic short-term interests, either that of the UK, or that of the other member states. Only when it became evident in2000 that a majority of the member states had already been affected bythe disease were common regulatory measures adopted, because their
A Weak Supranational Agency 157
PPL-UK_RR-Krapohl_Ch07.qxd 9/12/2008 10:29 AM Page 157

distributive consequences were no longer asymmetrically distributed(Krapohl 2003). Because the EU committee system was not able to pre-vent member states’ falling back on protectionism between 1986 and2000, the foodstuff sector suffered from a crisis of consumer confidencein the 1990s (Ansell and Vogel 2006; Majone 2000; Vogel 2001a; Vos2004).
The EU committee system not only failed to guarantee an adequatelevel of health and consumer protection in the foodstuff sector, but itwas also unable to uphold a single market for the affected products.After it became known that BSE could harm consumers, the supposedlynon-affected member states banned British beef from their markets.Thus, the Single Market for beef products was disturbed. In the follow-ing years, there were conflicts about trade restrictions within theCouncil and before the ECJ. Whereas the UK (and later Portugal) aimedtoward a swift lifting of the ban, other member states – most notablyFrance – wanted to uphold it, and they even prohibited British beef ontheir markets after the ban had been lifted by the Commission in col-laboration with the Council. These events demonstrate the close inter-relation between market integration and risk regulation. Withoutefficient risk regulation, a single market cannot be upheld, becausemember states cannot be forced to accept potentially dangerous prod-ucts onto their domestic markets. Thus, to achieve a lasting single mar-ket for foodstuffs, member states have to commit themselves to itsadequate regulation.
Appraisal
The findings of this section support the hypothesis that member states’commitment is positively correlated with the efficiency of supranationalrisk regulation. Overall, member states’ commitment in the EU commit-tee system of the foodstuff sector was extremely weak. Independentrecruitment of neutral experts for the scientific committees was not guar-anteed, the scientific committees’ influence on regulatory policy-makingwas weak, a general and comprehensive food law was missing and theEuropean courts were not able to effectively scrutinise regulatory policy-making. Thus, nothing could prevent the member states from followingtheir particularistic short-term interests during regulatory policy-makingas soon as regulatory problems with distributive consequences appearedon the agenda. As a result, member states were not bound to followbroader rules, but could always influence policy-making with their par-ticularistic interests. In the BSE case, the member states represented theirparticularistic interests in the EU committee system. The EU was therefore
158 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch07.qxd 9/12/2008 10:29 AM Page 158

neither able to achieve an adequate level of health and consumer protec-tion, nor to uphold a single market for beef. The committee system failedto achieve both of its major policy goals. Consequently, the supranationalregulatory regime for foodstuffs was fundamentally reformed in the yearsafter the BSE scandal.
7.1.2 The European Food Safety Agency
The new EU regulatory regime for foodstuffs, which was established in2002, differs from its predecessor in two important aspects. Firstly, theEFSA, which replaced the previously existing scientific committees inthe foodstuff sector, was established (section 6.3 and Figure 7.2). Toadvise the Commission on scientific aspects of foodstuff regulation, thenew agency contains a scientific committee that took over the coordi-native functions of the Scientific Steering Committee, and several sci-entific panels that replaced the more specialised scientific committees.These scientific bodies are supported by a management board and anexecutive director who is responsible for the day-to-day management ofthe agency. In addition, an advisory forum was established, which con-sists of representatives from the member states’ regulatory agencies for foodstuffs, but which has no vote in regulatory policy-making.Secondly, the same legislation that set up EFSA also contained a newand general EU food law. The general objectives of this law are the estab-lishment of a high level of health and consumer protection and theupholding of a single market for foodstuffs. To achieve these objectives,food safety regulations should rely on scientific risk assessment, butshould also be based on the precautionary principle in cases of scientificuncertainty.
A Weak Supranational Agency 159
Figure 7.2 Regulatory policy-making in the foodstuff sector after 2002
Conducts risk assessmentsand advises the Commission
Decision proposals Decisions
Agency
ScientificCommittee
ScientificPanels
Advisory Forum
Council
Comitology Committee
Commission
PPL-UK_RR-Krapohl_Ch07.qxd 9/12/2008 10:29 AM Page 159

The division of labour between EFSA and the political bodies of theEU follows the distinction between risk assessment, risk communica-tion and risk management. Risk assessment is deemed a scientifictask. Risks that could emerge from the consumption of certain food-stuffs need to be detected and quantified to allow reasonable choicesabout their regulation. The aim of risk communication is to informpolitical actors and the public about the risks of certain foodstuffs.Finally, risk management is deemed to be a political task. Here, itmust be decided which risks should be regulated in which way toincrease the overall level of food safety. EFSA has no competencies inrisk management – which is left to the political actors – only in riskassessment and risk communication. The most important task of theagency and its scientific bodies is to advise the legislative actors of theEU – most notably the Commission – on all scientific aspects relatedto food safety. Thus, in a narrow sense, EFSA cannot be called a regu-latory agency.
The real agenda-setter for food safety regulation is still the Commission.Whenever it develops proposals, it is free to ask EFSA and its scientific bod-ies for advice, or not. The agency may try to set the agenda for regulatorypolicy-making by issuing scientific opinions that were not requested bythe Commission, but it is always the Commission that must formally ini-tiate the decision-making process. Whenever the Commission suggestspolicies, it is – as in the previous committee system – subject to strongcontrol by the member states. The applied comitology procedure is theregulatory procedure, wherein the Standing Committee on the FoodChain and Animal Health must agree to Commission proposals by qual-ified majority. When proposals fail to receive such support, the Councilmay adopt or reject measures by qualified majority or amend them byunanimity. If the Council is not able to adopt decisions, the Commissionis free to act and may implement the measures.
Independence and strength of the EU agency for foodstuffs
In the recruitment of its personnel, the newly established EFSA expressesa strong commitment of the member states. The scientific bodies of the EFSA – namely the scientific committee and the different scientificpanels – are not only independent from the member states, but also fromthe Commission. This is because they are recruited by the managementboard of the agency on a proposal of the executive director, who is him-self appointed by the management board. The management board is set up by the Council and the EP on a proposal of the Commission.Consequently, no single legislative actor – neither the Council, nor the
160 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch07.qxd 9/12/2008 10:29 AM Page 160

EP, nor the Commission – is able to dominate the recruitment of themanagement board. Scientists with seats on the committee or panelsare supposed to be recruited based on their expertise; national quotasshould not play any role. Unlike the pharmaceutical agency, memberstates’ experts have seats only in the advisory forum, but not in thedecisive scientific bodies. The scientific committee and the scientificpanels are supported by the administration of the new agency. They areno longer dependent on the goodwill of the Commission, but havetheir own administrative resources, which improves the possibilities forself-tasking.
Within two recent evaluations conducted by consulting companieson behalf of the Commission, the personnel of the agency were praisedfor their scientific expertise by all stakeholders. EFSA’s staff was evalu-ated as competent, communicative and service-minded, and the qualityof scientific opinions was regarded as excellent.7 It was judged thatEFSA’s opinions are relevant, and that EFSA is succeeding in establishingitself as an independent centre of expertise.8 However, some critique ofthe agency’s scientific bodies was also raised. Both reports state that theagency and its scientific bodies are significantly understaffed and under-funded. During the starting phase, the members of the scientific bodieshad to work overtime, and the workload for each employee was judgedunsustainable. Of course, such an overload made initiatives by theagency more or less impossible. Thus, part of the agency’s capacity hasbeen undermined by a lack of resources.
Even though the recruitment of EFSA and its scientific bodies is quiteindependent from political influence, the agency is – as a result of the strict separation between risk assessment and risk management –comparatively weak within the following decision-making process(Krapohl 2004). The agency is dependent on the Commission to set theagenda for regulatory policy-making. Unlike in the pharmaceutical sec-tor, the foodstuff agency is not necessarily the first stage of the decision-making process. It is the Commission that initiates the procedure withits policy proposals.9 The Commission can reject the agency’s advice anddoes not face any restrictions in such circumstances. Hence, regulatorypolicy-making should be based on risk analysis, but EFSA’s risk assess-ment does not govern the Commission’s risk management. Besides, themember states strongly control both the agency and the Commissionwithin the strictest comitology procedure. Therein, regulatory decisionsmay be vetoed by member states’ particularistic short-term interests. Infact, the situation is not significantly different to that before the estab-lishment of EFSA.
A Weak Supranational Agency 161
PPL-UK_RR-Krapohl_Ch07.qxd 9/12/2008 10:29 AM Page 161

The strict separation between risk assessment in the hands of EFSA andrisk management in the hands of the Commission and the member stateswas evaluated ambivalently by stakeholders. Most stakeholders regardedthe separation as the best possible solution at the EU level, and a delega-tion of more competencies to the agency was seen as unrealistic or unde-sirable. However, some stakeholders criticised the relationship betweenEFSA, the Commission and the member states. It is hotly debated wherethe line between risk assessment and risk management should be drawn.Obviously, there is a grey area between the two tasks. Whereas somestakeholders favour that EFSA’s scientific opinions should already includerisk management options, others – especially the Commission – fear thatthis would be too directive for risk managers. Moreover, the coordinationbetween the different actors was seen as insufficient and lacking trans-parency. In cases of diverging opinions between risk assessors and riskmanagers, the reputation of the whole regulatory regime suffers. It isoften not clear from outside which actors have to be held responsible forfinal regulatory decisions. Risk assessors and risk managers can thereforeblame each other for regulatory failures.
Legalisation of the policy area
Compared with the old committee system, legalisation of foodstuff reg-ulation certainly increased with the adoption of the general food law.With this legislation, a fundamental shortcoming of the previous situa-tion was abolished. However, the new food law contains rather vagueand broad principles, and it does not prescribe the substance of futurefood safety regulations. According to the general food law, food safetymeasures should uphold a single market for foodstuffs and aim to estab-lish a high level of health and consumer protection throughout all stagesof the food chain (‘from the stable to the table’ or ‘from the farm to thefork’). Therefore, regulatory measures should consider scientific riskassessment, but they should also be based on the precautionary princi-ple in cases of scientific uncertainty. Thus, risk management decisionscould deviate from scientific advice, especially if they are more precau-tious. The only substantive rule given to risk assessors and risk managersis that food should not be placed on the market if it is unsafe (i.e. if it isinjurious to health or unfit for human consumption). Because of thesebroad rules, large discretion for regulatory policy-making is left. In theend, this is likely to favour political actors –the Commission and themember states – rather than scientific actors.
The judicial review of foodstuff regulation is likely to remain weakwithin the new EU regime. Firstly, if legalisation of the policy area is
162 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch07.qxd 9/12/2008 10:29 AM Page 162

weak, there is little ground on which the European courts may scrutiniseregulatory policy-making. In other words, if the discretion of regulatoryactors is large, the courts can less easily assess abuse of competencies. Forexample, in food safety regulation, political actors may easily point tothe precautionary principle – which still lacks a proper legal defini-tion (Majone 2002b, 2005: 124–42) – to justify very restrictive regula-tory measures. Secondly, the scope of potential plaintiffs is still ratherrestricted. To gain access to the European courts, potential plaintiffs mustdemonstrate their individual and direct concerns. However, food safetymeasures are usually not addressed to single individuals, but to a broaderpublic.10 Consequently, it could be difficult for affected stakeholders todemonstrate their direct and individual concerns. It is likely that thescope of potential plaintiffs will remain restricted to the member statesand EU legislative actors that can always bring their claims before theEuropean courts. In the end, the situation has not changed significantlycompared with the old regulatory regime that existed before 2002. Thegeneral food law reduced the discretion of regulatory actors only slightly,and the scope of plaintiffs was not broadened. Thus, future case lawabout foodstuff regulation will probably continue to concentrate onquestions of free trade and trade restrictions by individual member statesrather than on supranational regulatory policy-making.
Efficiency of the new regime
Because EFSA took up its work in 2002 and had some problems duringthe set up phase (it was significantly understaffed and had to move fromBrussels to Parma in 2005), it is probably too early to measure the effi-ciency of the new regulatory regime. However, it is possible to draw somereasonable hypotheses about EFSA’s future performance. Accordingly, thenew regulatory regime is likely to solve only the problems that distin-guished the first phase of the BSE scandal from 1986 to 1996 (Krapohland Zurek 2006; section 6.2.1). At that time, the Scientific VeterinaryCommittee, which advised the Commission on BSE, was captured byBritish agricultural interests. Consequently, Commission decisions werebased on very industry-friendly scientific opinions, which was laterheavily criticised by the EP. Such easy interference in science has becomeless likely within the new regime, because the new agency has moreresources and is more independent than the old scientific committees.However, the new regime is less likely to solve the problems that distin-guished the second phase of the BSE scandal from 1996 to 2000 (Krapohland Zurek 2006; section 6.2.2). During that time, British beef was bannedfrom the Single Market. However, the other member states blocked
A Weak Supranational Agency 163
PPL-UK_RR-Krapohl_Ch07.qxd 9/12/2008 10:29 AM Page 163

further harmonised regulatory measures on BSE within the StandingVeterinary Committee, against the advice of the then newly establishedScientific Steering Committee. Such behaviour from the member statesmay still prevail in the new regime for foodstuffs, because neither astrong agency, nor strong European courts can prevent it.
The new agency has already been subject to two small evaluations,which were based on stakeholder interviews conducted by consultingcompanies on behalf of the Commission. The two reports point in thesame direction as the above reasoning. Generally, stakeholders appreciatethe establishment of EFSA and are satisfied with its improved risk assess-ment and risk communication.11 However, some stakeholders do not seea great difference between the new agency and the old committee system.For example, the general level of food safety standards seems neither tohave increased nor decreased, and the separation of risk assessment awayfrom the Commission is judged as a more symbolic than substantial step.Most stakeholders share the opinion that the new regulatory regime forfoodstuffs has to demonstrate its additional value compared with the oldregime within the next food safety crisis in the EU. Thus, the crucial ques-tion yet to be answered is whether the new regime will be able to avoidlarge regulatory scandals like the BSE crisis in future.
7.1.3 The authorisation of GM food
The regulation of GMOs and GM food shares one important characteris-tic with pharmaceuticals: both groups of products have to be assessedand approved before they enter the Single Market. Whereas the USA fol-lows the approach that GMOs and GM food are generally equivalent tonon-modified products and thus need not be additionally regulated, theEU regards GMOs and GM food as fundamentally different from otherproducts and requires pre-marketing authorisation for each product(Patterson and Josling 2001; Shaffer and Pollack 2004; Skogstad 2006;Young 2003). Thus, if functionalist pressure alone were the only deter-mining variable for the design and function of regulatory institutions,the supranational regulatory regime for GMOs and GM food should lookand work similar to that for pharmaceuticals. However, as the historicalanalysis demonstrates, the regulatory regime for GMOs and GM fooddeveloped according to a path similar to that of traditional foodstuffs,and its institutional design therefore looks rather similar to that of thefoodstuff regime (section 6.3).
Several different procedures were applied in the EU for the authori-sation of GMOs and GM food. In 1990, the Deliberate Release Directiveset up the first authorisation procedure for the release of GMOs into
164 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch07.qxd 9/12/2008 10:29 AM Page 164

the environment (Schenek 1995: 199–222). In 1997, this was supple-mented by the Novel Food Regulation, which addressed all new food-stuffs that were not produced according to traditional methods(Sheridan 2001: 114–69). Both the Deliberate Release Directive and theNovel Food Regulation were reformed due to the de facto moratoriumon GMOs and GM food, and the resulting international struggles. Inthe following, the institutional analysis mainly deals with the latestRegulation on GM food and feed, which forms the centrepiece of thenew regime.12 However, because of limited experience with the new leg-islation, the empirical evaluation of the regime must mainly be basedon the old procedures.
The authorisation procedure for GM food starts when companies sub-mit applications for marketing authorisation to the competent authori-ties of the member states where the products shall be marketed for thefirst time (section 6.3 and Figure 7.3). Applications for marketing autho-risation have to be accompanied by dossiers, which must include allnecessary information demonstrating that the foods in question do notpresent a danger or mislead consumers, and that they are not disadvan-tageous compared with traditional products. The new Regulation onGM food and feed lists several points that have to be addressed in appli-cations for marketing authorisation: among others, detailed descrip-tions of production methods, scientific studies, labelling proposals and
A Weak Supranational Agency 165
Figure 7.3 Authorisation of genetically modified food
Applies for marketing authorisations
Scientific opinions Decision proposals Decisions
Member State B
Member State C
Member State A
Applicant
Council
Commission
Comitology Committee Agency
Scientific Panel on GMOs
PPL-UK_RR-Krapohl_Ch07.qxd 9/12/2008 10:29 AM Page 165

proposals for post-marketing monitoring. The legal rules are furtherspecified by legally non-binding guidelines from the newly establishedEFSA. Under the old Deliberate Release Directive and the Novel FoodRegulation, the competent authorities of the member states conductedthe initial assessments of the dossiers. However, according to the newGM food and feed Regulation, the competent authorities of the mem-ber states no longer do this, but forward the application dossiers directlyto EFSA. Within six months, EFSA’s Scientific Panel on GMOs issues sci-entific opinions on the safety of respective foods and on the labellingproposals of the applicants. The final scientific opinions are then for-warded to the Commission and the member states.
When the Scientific Panel on GMOs has submitted its scientific opin-ions about applications, the Commission develops decision proposalsbased on these opinions. These proposals are then forwarded to theStanding Committee on the Food Chain and Animal Health, whichdecides by a regulatory procedure. If Commission approvals are not sup-ported by a qualified majority within the committee, the member statesare able to adopt or to block authorisations by qualified majority in theCouncil. If the member states are unable to gain a qualified majorityeither in favour or against marketing authorisation, the Commission isfree to implement its proposals. Compared with the old DeliberateRelease Directive and the Novel Food Regulation, member states’ safe-guard competencies have been reduced and are now the same as apply to all other foodstuffs. Accordingly, it is the task of the Commission – possibly on request of the member states – to adopt measures if new risksfor consumers or the environment become evident. In such cases, theCommission is usually supported by EFSA and controlled by the memberstates’ committee. However, in cases of emergency, the Commission mayact provisionally and without member states’ approval. The memberstates are only allowed to take individual measures if the Commissionfails to act after official requests.
Independence and strength of the scientific panel on GMOs
Recruitment of the supranational body responsible for the evaluation ofGM food is as independent from member states’ interests as in otherareas of foodstuff regulation. The members of EFSA’s scientific panel onGMOs – which is responsible for the assessment of applications underthe new GM food and feed regulation – are not only recruited inde-pendently from the member states, but also from the Commission, asthey are appointed by the agency’s management board on advice of theexecutive director. The management board is in turn set up by all three
166 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch07.qxd 9/12/2008 10:29 AM Page 166

legislative actors (the Council, the Commission and the EP). It thereforecannot be dominated by the particularistic interests of any one group.
Despite this formal independence, the composition of EFSA’s scientificpanel on GMOs has been heavily criticised in recent years. In November2004, Friends of the Earth Europe – an international environmental andconsumer interest group – published a very critical review of the GMOpanel.13 It accused the panel of being heavily influenced by industryinterest. According to the review, one-third of the panel members areinvolved in national evaluations of GMOs, the panel chair and fouradditional members participate in an industry-dominated research proj-ect on GMOs, and the panel once co-opted an ad hoc expert from thebiotechnology industry. As a result, Friends of the Earth accused EFSA ofissuing scientific opinions on GM food that are not based on the pre-cautionary principle, and which are much more industry-friendly thanthose of the member states. However, this negative evaluation by aninterest group has to be qualified in light of another evaluation of EFSAas a whole, which was conducted by a consulting company on behalf ofthe Commission.14 The latter assessment is based on interviews with abroader range of stakeholders and comes to more positive conclusions.Accordingly, EFSA’s scientific opinions were regarded as relevant orhighly relevant, and most stakeholders expressed high or very high con-fidence in the availability and excellence of panel members’ expertise.
As EFSA as a whole, the Scientific Panel on GMOs is only a weakagenda-setter, because the Commission and the member states are notbound by its opinions. If the Commission chooses to deviate from sci-entific advice, it faces no restrictions, but must only explain its reasons.Decision proposals are then forwarded to a member state committeeand in some cases to the Council, which then decide according to arestrictive regulatory procedure. The power of the member states is evenmore striking if one takes their safeguard competencies into account.Even under the new Regulation, member states are still allowed to adoptindividual safeguard measures if the Commission is unable to react tonew problems with already-authorised GM foods.
The power of the member states became clearly visible during the de facto moratorium on GMOs and GM food between 1998 and 2004(Shaffer and Pollack 2004; Sheridan 2001: 81–7). As a reaction to increas-ing consumer concerns, a wide range of member states used their com-petencies under the old legislation to ban already-authorised GMOs andGM food from their national markets and to oppose every new authori-sation of GMOs and GM food within the Standing Committee onFoodstuffs. Because of this resistance, the Commission was neither able
A Weak Supranational Agency 167
PPL-UK_RR-Krapohl_Ch07.qxd 9/12/2008 10:29 AM Page 167

to condemn unilateral safeguard measures, nor to authorise any GMOsand GM food. In the end, it was only a narrow decision that ended themoratorium on GMOs and GM food under the new legislation in 2004(Shaffer and Pollack 2004). Of the five GM foods authorised between2004 and 2006, not one received the support of a qualified majority ofthe member states.15 In all five cases, both the comitology committeeand the Council were split between supporters and opponents of mar-keting authorisation. The Commission was free to adopt its proposals,and thus the respective products were finally authorised without supportof the member states. Even though these five products have been mar-keted, the example demonstrates the continuing power of the memberstates. If coalitions in the Council changed only slightly, the memberstates would again be able to impose a moratorium on GMOs and GMfoods.
Legalisation of the policy area
Not only the procedural but also the substantial rules for GM foodauthorisation leave a great deal of discretion to the member states,because legalisation of the policy area is only rudimentary. The GM foodand feed Regulation prescribes that the products in question should not present a danger for consumers, should not mislead them and shouldnot be disadvantageous compared with traditional products. Besides, the new Regulation lists several points that should be addressed by appli-cations for marketing authorisation. These points are further specified by legally non-binding guidelines of the GMO panel within EFSA.16
Although some similarities between the authorisation procedures forGM food and pharmaceuticals exist (e.g. the reliance on pre-marketingauthorisation), the amount of substantive authorisation criteria differswidely. Pharmaceutical authorisation must be based on a whole com-munity code for human medicinal products, which is totally lacking inthe case of GM food. The discretion left by substantive criteria for thepolitical actors within the authorisation process is much larger for GMfood than for pharmaceuticals. Consequently, member states can moreeasily deviate from scientific opinions, and their commitment to followscientific advice is correspondingly weak.
As a result of weak legalisation, one can expect that the judicial reviewof GM food regulation is also weak, because the ECJ and the Court ofFirst Instance can only scrutinise risk managers if they exceed their dis-cretion. If this discretion is large, the courts cannot judge risk managers.The discretion of risk managers (i.e. the Commission and the memberstates) seems even wider if one keeps in mind that the precautionary
168 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch07.qxd 9/12/2008 10:29 AM Page 168

principle has become a leading principle of foodstuff regulation withinthe EU. The precautionary principle always empowers risk managers(Majone 2002b, 2005: 124–42), because it allows non-scientific argu-ments to be comsidered. This effect was made clear by an ECJ judgementin 2000.17 In this case, Greenpeace brought a claim against the authori-sation of a GMO before a French court, which, in turn, forwarded thecase to the ECJ and asked for a preliminary ruling. Greenpeace accusedthe competent authorities of France that the authorisation of GM cornfrom the company Ciba-Geigy breached the precautionary principle.However, the authorisation of the corn by French authorities was basedon a positive Commission decision. Thus, the ECJ judged that theFrench authorities had no discretion to deny a marketing authorisation,and that it was up to the Commission to interpret the precautionaryprinciple in its decision. Though this judgement widened the discretionof risk managers in cases of positive authorisation decisions, the same isalso imaginable in opposite cases (i.e. where companies claim againstnegative authorisation decisions).
Efficiency of GM food authorisation
The Commission’s efforts to establish a single market for GMOs and GMfood suffered a clear setback during the de facto moratorium on theseproducts between 1998 and 2004 (Shaffer and Pollack 2004; Sheridan2001: 81–7). During this time, no GMO and GM food was authorised onthe European Single Market. Further, the market for already-authorisedGMOs and GM foods was disturbed by individual safeguard measures ofsome member states. Both the blockade of the authorisation process andthe safeguard measures could not be prevented by the Commission,because it was confronted with an objecting qualified majority of mem-ber states within the comitology committee and the Council. The situa-tion improved somewhat in May 2004, when the moratorium was liftedby the authorisation of GM corn from Syngenta Seeds under the newGM food and feed Regulation. Between 2004 and 2006, five GM foodswere authorised by the Commission. However, despite this success, thesituation is still precarious. So far, not a single authorisation has gainedthe support of a qualified majority of member states within the comitol-ogy committee or the Council. The Commission was only able to autho-rise these products because the Council was split between supporters andopponents, and was therefore not able to gain a qualified majority eitherin favour of or against authorisation. And the disputes about individualmeasures of some member states remain unsettled. In October 2005, theCourt of First Instance decided on an Austrian claim that strived for the
A Weak Supranational Agency 169
PPL-UK_RR-Krapohl_Ch07.qxd 9/12/2008 10:29 AM Page 169

annulment of a Commission decision.18 The Commission decision pro-hibited Austria from banning the use of GMOs in some parts of its terri-tory. Even though Austria was unsuccessful with its claim against thisdecision, the case demonstrates how disputed the release of GMOs or themarketing of GM food still is in some member states. The supranationalregulatory regime is a long way from establishing a stable single marketfor these products. A new moratorium would still be possible if onlysome member states were to change their positions.
Not only has the EU regulatory regime been unable to establish a sta-ble single market for GMOs and GM food, it also has failed to restoreconsumer confidence in the respective products. There is a strong anti-GMO movement in Europe (Ansell et al. 2006), and European con-sumers are generally very sceptical about GMOs and GM food (Bredahl2001; Gaskell and Allum 2001; Toke 2004: 141–89). So far, the regula-tory regime has been unable to dispel these doubts. On the contrary,some interest groups accuse the supranational regulatory regime of hav-ing a pro-industry bias. In fact, the member states are often regarded byconsumers as the only barrier against industry-friendly policies of atechnocratic Commission. Overall, the GMO and GM food sector seemsto be highly affected by the crisis of consumer confidence.
Appraisal
The case of GM food supports the hypothesis that member states’ com-mitment is positively correlated with the efficiency of supranational riskregulation. The procedural and substantive rules of the authorisationprocedure for GM food indicate a weak commitment of member statesto following scientific advice. Although the recruitment of the suprana-tional regulatory bodies is at least formally independent, all three otherdimensions demonstrate only weak commitment. The supranationalexpert bodies are weak agenda-setters in the authorisation procedure,the legalisation of the policy area is rather weak, and the precautionaryprinciple makes judicial scrutiny of risk managers difficult. Thus, itshould be easy for the member states to deviate from scientific adviceand to adopt political decisions that are distinguished more by com-promises than by scientific reasoning. As a result, the regime was nei-ther able to produce a stable single market, nor to restore consumerconfidence: The single market is continually disturbed by the applica-tion of safeguard measures, and consumers’ doubts are neglected. Theregime’s performance comes closer to that of its counterpart in the tra-ditional foodstuff sector than to that of its counterpart in the pharma-ceutical sector.
170 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch07.qxd 9/12/2008 10:29 AM Page 170

7.2 Legitimacy of European foodstuff regulation
This section analyses the legitimacy of the EU regulatory regime for food-stuffs. For this reason, it distinguishes between input and output factors(section 3.3). For input legitimacy, an assessment of how European citi-zens were able to influence the establishment of the regulatory regimethrough elections and interest group lobbying is presented (section7.2.1). However, the regime’s day-to-day operation should legitimiseitself more by way of its policy output. Here, the extent to which theregime’s policy output meets the long-term interests of the most impor-tant stakeholders is analysed (section 7.2.2). A crucial question for out-put legitimacy is how far the regime can, in the long term, be heldaccountable by different actors for its policy output. Efficient accounta-bility mechanisms should ensure that the regime does not drift awayfrom the converging long-term interests of stakeholders.
7.2.1 Input factors
Before the BSE scandal shattered the fundaments of EU foodstuff regula-tion, regulatory policy-making took place within the EU committee sys-tem. A range of secondary legislation had delegated some competenciesfor the adoption of implementation decisions to the Commission, whichwas advised by scientific committees and controlled by comitology com-mittees. This committee system scored relatively badly for input legiti-macy. The member state committees were all set up by the Council,without co-decision or cooperation of the EP. Worse still, the scientificcommittees were established solely by the Commission without evenpassing a consultation procedure. The EP had no influence on the recruit-ment of both types of committee. As a result, there was enduring conflictbetween the Council and the EP about the committee system (Bradley1992, 1999; Steunenberg et al. 1997). Whenever the Council wanted tocontrol the Commission by a comitology procedure, this was opposed bythe EP, which was excluded from this control mechanism. As the EPbecame stronger within the cooperation and co-decision procedures, amodus vivendi became necessary, according to which the EP was, at least,regularly informed about decision-making within the committee system(Falke 2000). This modus vivendi later became confirmed by the newcomitology Decision, which grants the EP some consultative rights forall delegation acts after 1999.19
For stakeholders’ interests, the Commission’s Single Market pro-gramme was supported by European industry (section 6.1; Alter andMeunier-Aitsahalia 1994; Fligstein and Mara-Drita 1996). Naturally, the
A Weak Supranational Agency 171
PPL-UK_RR-Krapohl_Ch07.qxd 9/12/2008 10:29 AM Page 171

Single Market was in the interest of producers, and thus it was easy forthe Commission to find support for its project among these interestgroups. However, the influence of foodstuff producers was supposed tobe counterbalanced by the Advisory Committee on Foodstuffs. Thiscommittee was established by the Commission and consisted of 30 rep-resentatives from industry, consumer groups, agriculture, commerceand labour. No single group could have been outvoted, because theadvisory committee did not vote on policy proposals, but all differentviews were reported to the Commission. The committee constituted abalanced representation of different social interests, which could havesignificantly increased the input legitimacy of EU foodstuff regulation.However, the weakness of the committee was that it did not have theright of initiative, but could only act on request of the Commission,which hardly ever asked the committee for its advice. As a result, thecommittee remained rather passive and was unable to gain significantinfluence on EU foodstuff regulation (Falke 2000: 147–8). Thus, thischance to increase the legitimacy of the committee system was lost.
The situation changed fundamentally during and after the BSE crisis,when food safety raised the attention of consumers, and when the EPgained much more influence over EU policy-making (sections 6.2.2 and6.3.1). After the British government announced, in 1996, that BSE couldendanger the health of consumers, the EP became very active and rep-resented the diffuse interests of consumers (Kelemen 2002; Vos 2000b;Westlake 1997). It set up a temporary committee of inquiry to scrutinisemismanagement in the BSE case, and threatened to adopt a motion ofcensure against the Commission if it would not follow the recommen-dations of the inquiry committee.20 On its advice, the Commissionimmediately reorganised the committee system in the foodstuff sector,especially the Scientific Steering Committee was set up to advise theCommission in all matters relating to BSE, established the Food andVeterinary Office and concentrated the relevant competencies in theDG Consumer Policy and Health Protection. Thus, input from the EPcan be seen as the starting signal for the reorganisation of the regulatoryregime. The EP also had significant influence over the more fundamen-tal reforms that followed some years later. In the inquiry report, the EPdemanded the inclusion of a legal basis for food safety regulation withinthe treaties and the general application of the co-decision procedure forsuch legislation. Indeed, the treaty amendments of Amsterdam madehealth and consumer protection an independent policy-objective of theEU, and made the application of the co-decision procedure compulsoryfor such measures (Vos 2000b). Consequently, the EP and the Council
172 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch07.qxd 9/12/2008 10:29 AM Page 172

were co-equal legislators (Tsebelis and Garrett 2000, 2001) for all legis-lation that was adopted after 1997 (i.e. the set up of EFSA, the estab-lishment of a general food law, and the reform of the GM food regime).
7.2.2 Output factors
As a result of the EU’s mismanagement of BSE, the output legitimacy ofthe EU committee system was very low. During the BSE crisis, Europeanconsumers were not made aware of the risk of British beef for ten years.Between 1986 and 1996, the British government and the EU institutionpublished the opinion that BSE was not transmissible to humans despitethe fact that some scientific studies already pointed to the opposite con-clusion (the transmission to mice, cats and swine was already proven).Moreover, after the British government had to admit that British beefcould be dangerous, the other member states imposed a ban of Britishbeef from the Single Market, but they still blocked the adoption of strongcommon regulatory measures against the disease. It took another fouryears until the other member states themselves adopted strong regula-tory measures (section 6.2; Krapohl 2003; Krapohl and Zurek 2006). Inreaction to fourteen years of insufficient protection against the new cat-tle disease, consumers lost confidence in the safety of their food and inthe regulatory capacities of the EU (Ansell and Vogel 2006; Majone 2000;Vogel 2001a; Vos 2004). This crisis of consumer confidence also affectedthe market for beef. After the risks of BSE to consumers became evidentin 1996, the following years were distinguished by hard political andjudicial battles about the export ban on British (and later Portuguese)beef. Whereas the UK (and later Portugal) pushed towards a swift liftingof the ban, the other member states – most notably France – even refusedto accept British beef on their markets after the EU had lifted the ban.Thus, a single market could not be hold up.
The crisis of consumer confidence also affected the authorisation ofGMOs and GM food in Europe (section 6.3.2). During the peak of theBSE scandal, the Commission began authorising the first GMOs and GMfoods, which were widely distributed within the Single Market (Vogel2001b), but which were met with heavy scepticism of European con-sumers. Because the Commission did not react to this scepticism, themember states adopted a de facto moratorium against GMOs and GMfood in reaction to consumer demands. Like the BSE case, the GMO andGM food case demonstrates the close relationship between suprana-tional risk regulation and the establishment of a single market. If the EUis unable to establish risk regulation that meets the long-term interestsof all stakeholders, it will also be unable to uphold a single market for
A Weak Supranational Agency 173
PPL-UK_RR-Krapohl_Ch07.qxd 9/12/2008 10:29 AM Page 173

the respective products, because the member states will then use alltheir discretion to ban such products from their markets.
The case of GMOs and GM food also demonstrates the limits of supra-national regulatory regimes in providing output legitimacy. Outputlegitimacy derives from efficient risk regulation, i.e. risk regulation thatis in the long-term interests of both consumers and producers (section3.1). For efficient risk regulation to provide output legitimacy, thereneeds to be one level of regulatory standards where the long-term inter-ests of stakeholders converge. If there is no such point of convergence,risk regulation is a distributive battle where some stakeholders win whatother stakeholders lose. Such distributive battles cannot be legitimisedby output factors, because there is no policy that is generally better thanthe others. Although it can be assumed that usual risk regulation can bemore or less efficient and can thus be legitimised by output factors, theauthorisation of GMOs and GM food may be the exception to this rule.In Europe, GMOs and GM food meet with fundamental oppositionfrom consumers (Ansell et al. 2006; Bredahl 2001; Gaskell and Allum2001; Toke 2004: 141–89). This opposition seems not only to be basedon health risks or environmental concerns, but also on cultural and eth-ical values. GMOs and GM food are seen as ‘unnatural’ or ‘artificial’, andthey are judged to contravene basic principles of nature itself. As aresult, a more market-friendly regulation of GMOs and GM food wouldnot necessarily lead to more output legitimacy, because it would notsolve the underlying cultural and ethical conflicts. In this case, a polit-ical decision about these underlying conflicts is needed. However, sucha decision should not be delegated to the respective supranational reg-ulatory regime, but it should be made at the legislative level where theCouncil and the EP provide the necessary input legitimacy. The currentproblem seems to be that the legislative actors – under pressure from theWTO – have not been able to agree on how to deal with GMOs and GMfood in general, and have therefore adopted a compromise that passesthe underlying conflicts on to the supranational regulatory regime.Here, the regime cannot provide any output legitimacy, because risk reg-ulation becomes necessarily politicised.
Given the low output legitimacy of the EU regulatory regime forfoodstuffs in the 1990s, the question is to what extent stakeholders areable to hold the regime accountable in order to bring it back on course.The political accountability of the current regime is rather strong, butit is unbalanced in favour of the member states. The Commission andEFSA are controlled within a strict comitology procedure. The involvedStanding Committee on the Food Chain and Animal Health is recruited
174 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch07.qxd 9/12/2008 10:29 AM Page 174

by the member states and acts according to their direct orders. WheneverCommission proposals do not meet the support of a qualified majoritywithin this committee, matters are forwarded to the Council. Thus,the day-to-day decision-making of the regime is strongly controlledby the Council, whereas the EP is only informed about proposals thatare adopted in the comitology procedure. Although the EP tried toimprove its own standing during the set up of the new foodstuff regime(Kelemen 2002), it gained only some influence over the recruitment ofEFSA’s management board, which appoints the executive director andthe members of the scientific committee and panels. Today, the Counciland the EP on a proposal of the Commission appoint fourteen membersof the management board, and one additional member represents theCommission itself. As a result, the EP is – like the Council – able to holdthe regime responsible in the long term, because it influences EFSA’srecruitment. However, in contrast to the Council, the EP still cannotcontrol the regime’s day-to-day operation.
The regime’s accountability to national experts from member statesregulatory agencies for foodstuffs is rather weak. Within EFSA, anAdvisory Forum was established to create a link between the suprana-tional regime and the national agencies, which had been established inmost member states in reaction to the BSE crisis and the set up of thesupranational regime. The Advisory Forum follows the example of theexpert committee of the pharmaceutical sector, and, like the expertcommittee, it consists of representatives from the national regulatoryagencies. However, in contrast to the expert committee in the pharma-ceutical sector, the Advisory Forum in the foodstuff sector has no formaldecision-making competencies. It only advises the management board,as well as the scientific committee and panels. Consequently, thisaccountability mechanism is much weaker in the foodstuff sector thanit is in the pharmaceutical sector. If member states’ experts want to holdthe EU regulatory regime for foodstuffs accountable, they cannot relyon the Advisory Forum, because it has no formal power. Instead, theymust ask their national governments to use their political power withinthe comitology committee. As a result, the regime can surely be heldaccountable by the member states, but the political mechanism is muchstronger than scientific peer-review. Thus, scientific disputes betweendifferent regulatory authorities are more easily politicised.
For the regime’s accountability to stakeholders, the picture is mixed.On the one hand, many efforts were made during and after the BSE cri-sis to open the regime to the public. Today, nearly all information aboutthe regime’s decision-making – including EFSA’s scientific opinions,
A Weak Supranational Agency 175
PPL-UK_RR-Krapohl_Ch07.qxd 9/12/2008 10:29 AM Page 175

agendas and reports of the comitology committee, as well as the finaldecisions – are published and can be easily accessed on the Internet.21
The foodstuff regime is much more transparent than that concerningpharmaceuticals, where a great deal of information is confidential inorder to protect intellectual property of the pharmaceutical industry.Besides, the EFSA has established a range of regular stakeholder consul-tations. An annual general colloquium gives all stakeholders the chanceto access EFSA, regular public consultations and technical meetingsallow stakeholders to give their opinions on specific topics (e.g. therisk assessment of GM food), further colloquia deal with specific issuesin a more scientific way, and a consultative platform, which consistsof 20 to 30 representatives from interest groups, advises the agency ona regular basis.22 Additionally, the Commission set up an AdvisoryGroup on the Food Chain and Animal and Plant Health.23 As a resultof all this, the new regulatory regime for foodstuffs can be accessed by stakeholders more easily than the old one. Information is easilyavailable and stakeholders can raise their voices during all the differ-ent consultations.
On the other hand, the regime’s legal accountability to stakeholders isnevertheless weak, because judicial scrutiny of regulatory policy-makingin the foodstuff sector is much more difficult than in the pharmaceuti-cal sector. Most regulatory decisions of the foodstuff regime are notaddressed to single applicants, with the effect that there are no naturalplaintiffs against such decisions. Stakeholders affected by broader regu-latory policies will have more difficulty in demonstrating their directand individual concerns if they wish to bring claims against these poli-cies before the ECJ or the Court of First Instance. Thus, the possibilitiesfor judicial review of regulatory policy-making are reduced, and it islikely that only the member states and the EU legislative actors have arealistic chance of bringing claims before the ECJ. The only exceptionto this is the case of GM foods, where applicants for marketing authori-sations are the addressees of authorisation decisions. Thus, they arealways directly and individually concerned by these decisions and maychallenge them before the European courts. However, judicial review isfurther weakened for both general foodstuff regulation and GM foodauthorisation by imprecise rules in the policy area. The European courtscan only intervene in regulatory policy-making if the responsible actorsbreach their discretion. The larger the discretion of the responsibleactors, the more difficult it is for the courts to scrutinise their decisions.Both the general food law and the substantive rules for GM food autho-risation are still rather broad, and thus leave a great deal of discretion to
176 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch07.qxd 9/12/2008 10:29 AM Page 176

the regulatory actors. This effect is even reinforced by the wide applica-tion of the precautionary principle, which always increases the discre-tion of risk managers. As a result, there is little ground on which thecourts could annul regulatory decisions.
Thus, although some of the regime’s accountability mechanisms arequite strong, they remain, on the whole, very unbalanced. The regimecan easily be held responsible by the member states through the comi-tology committee and the Council. In contrast, all other actors (theEP, national experts within member states’ regulatory agencies, theEuropean courts and EU citizens) are relatively weak and cannot effec-tively scrutinise regulatory policy-making. As a result, opposition againstthe regime must always take the political route, and conflicts are alwaysin danger of becoming politicised. If national experts disagree with sci-entific opinions of EFSA, they can only effectively influence regulatorypolicy-making through their government representatives in the comitol-ogy committee and the Council. And stakeholders can only effectivelychallenge regulatory decisions if they lobby their national governments,the Commission or the EP. Judicial review is less likely to solve such dis-putes, because most stakeholders do not have standing in front of theEuropean courts, and the courts would have difficulties intervening inthe face of regulators’ vast discretion. The political accountability mech-anisms are therefore the only ones left, but they are also the most dan-gerous for efficient regulatory policy-making. They always bear the riskof regulatory issues becoming politicised and being influenced by par-ticularistic short-term interests.
7.3 Conclusion
The case of the EU regulatory regime for foodstuffs again supports thehypothesis that the efficiency of supranational risk regulation dependson the credible commitment of the member states. Member states’commitment within EU foodstuff regulation was and is comparativelylow. Thus, distributive struggles between the member states continu-ally precluded efficient risk regulation. Before and during the BSE cri-sis, the Commission and several committees were in charge of EUfoodstuff regulation. Although the recruitment of the scientific com-mittees within that regime was formally independent, the committees’standing was weak during the following decision-making process. Thiswas due to strong procedural control by the member states, and to thelack of a coherent food law, which would have allowed Europeancourts to scrutinise regulatory policy-making more closely. Because of
A Weak Supranational Agency 177
PPL-UK_RR-Krapohl_Ch07.qxd 9/12/2008 10:29 AM Page 177

this weak commitment, the committee system proved unable to ade-quately deal with the BSE problem. In fact, EU decision-making in thismatter was always dominated by member states’ particularistic short-term interests. At the beginning, the UK actively downplayed the prob-lem to avoid a product ban from the Single Market. Later, the othermember states banned British beef without adopting harmonised regu-latory measures in order to support their own industry. Subsequently,EFSA and the new EU food law seem to be making (albeit small)progress towards a more efficient regime. With the establishment of theagency, the independence of scientific advice was strengthened, andwith the new food law, some general criteria for risk regulation in thefield were established. However, the new agency is neither as strong asits counterpart in the pharmaceutical sector, nor are the substantivecriteria for foodstuff regulation as tight as those for pharmaceuticalauthorisation.
The result of the GM food case is similar to that of the traditionalfoodstuff sector. Again, the formally independent scientific body is a rel-atively weak agenda-setter within the authorisation procedure, and thepolicy area is not particularly legalised. Consequently, the Commissionand the member states are still able to dominate the authorisation process.The regime is therefore neither able to establish a stable single market forGM food, nor to restore consumer confidence in these products. This sim-ilarity to traditional foodstuffs is especially important, because it invali-dates an important criticism of the historical-institutionalist argument.This criticism would argue that the success of EU pharmaceutical autho-risation and the failure of foodstuff regulation are not due to differencesin the developmental paths and the resulting institutional designs ofthe regimes, but to differences between pre- and post-marketing con-trol. Like pharmaceuticals, GM food is also subject to pre-marketingauthorisation. Nevertheless, the respective regime is far less efficientthan that for pharmaceuticals. Instead, it mirrors many weaknesses ofthe regulatory regime for traditional foodstuffs. Thus, the case of GMfoods is an important indicator of the validity of the entire historical-institutionalist analysis.
Since the BSE crisis, the input legitimacy of the EU regulatory regimefor foodstuffs has increased significantly. The EP fired the starting shotfor the reform of the regime with its inquiry report on the Commission’smismanagement in the BSE case. And because of the newly introducedco-decision procedure for all legislation relating to food safety, the EPhad a strong standing during the establishment of the EFSA, the adop-tion of the general food law and the reform of the GM food regime.
178 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch07.qxd 9/12/2008 10:29 AM Page 178

The EP used this new influence to strengthen its own standing withinthe new regime and to represent the diffuse interests of consumers. Incontrast, output legitimacy of the regime’s day-to-day operation was,and is likely to remain, rather weak. The BSE crisis and the disputesabout the authorisation of GMOs and GM food damaged consumer con-fidence in the safety of their food and in the regulatory capacities of theEU. In the face of this crisis of consumer confidence, the EU was unableto uphold a single market for the affected products, because the memberstates used all their discretion to protect their consumers. The questionremains, whether the new foodstuff regime is able to fight the crisis ofconsumer confidence effectively and to improve its output legitimacy.The institutional analysis casts some doubt on this. The supranationalregulatory regime for foodstuffs is still mainly controlled by the mem-ber states, whereas other accountability mechanisms are comparativelyweak. Whenever experts or stakeholders want to challenge the regime’sdecisions, they have to take the political route and lobby their govern-ments. Thus, such conflicts are easily politicised. At least for GMOs andGM food, it is also doubtful whether efficient risk regulation wouldindeed increase the output legitimacy of the regime. If consumersrejected these products for reasons other than the risks they impose onhealth, it is unlikely that more efficient regulation would really meettheir interests.
A Weak Supranational Agency 179
PPL-UK_RR-Krapohl_Ch07.qxd 9/12/2008 10:29 AM Page 179

This page intentionally left blank

Part IV Conclusion
PPL-UK_RR-Krapohl_Ch08.qxd 9/12/2008 10:20 AM Page 181

This page intentionally left blank

8A Comparison of Pharmaceuticaland Foodstuff Regulation in Europe
The analysis of this book started from the surprising observation that theEU is confronted with similar challenges of regulatory policy-making indifferent product sectors, but that the form and function of supranationalregulatory regimes nevertheless differ. Pharmaceuticals and foodstuffs areboth incorporated by consumers and could pose enormous health risks.Information asymmetries between producers and consumers are large forboth groups of products. Finally, both pharmaceuticals and foodstuffs hadto be regulated against similar threats over the course of the past twentyyears (i.e. BSE and biotechnology). Nevertheless, the success of the EU inregulating the two product sectors is strikingly different. Whereas the reg-ulation of foodstuffs was shattered by major regulatory scandals that pro-duced a crisis of consumer confidence in the 1990s, the authorisation of pharmaceuticals has worked since 1995 to satisfy most stakeholderswithout raising public concerns. Thus, the introductory chapter posed thequestions of why different regulatory regimes in the EU have distinctinstitutional designs, why their success in fulfilling their policy goals hasdiffered, and whether they can be regarded as differently legitimate.
To answer these questions, this book developed and tested a middle-range theory to supranational risk regulation. Thereby, it followed arational–institutionalist approach, which is based on the assumption ofbounded rationality (Simon 1972). Accordingly, actors behave ration-ally, but they act under incomplete and asymmetrically distributedinformation. Besides, the rational–institutionalist approach is comple-mented by a historical dimension. Institutions never emerge from a tabula rasa – i.e. an institution-free world – but are always influenced byalready-existing institutions. Thus, institutional development is alwayspath-dependent, and inefficient institutions may prevail if they leadto increasing returns for the actors concerned (Arthur 1994; Pierson
183
PPL-UK_RR-Krapohl_Ch08.qxd 9/12/2008 10:20 AM Page 183

2000a). Only this historical-institutionalist argument is able to explaindifferences between supranational regulatory regimes in different policyareas when the underlying functional reasons are similar. Based on this‘rational choice historical institutionalism’ (Stacey and Rittberger 2003),the theoretical part set up three hypotheses, which were then tested forthe two cases of pharmaceutical and foodstuff regulation in the EU.
Hypothesis 1: Institutional Designs of Supranational Regulatory Regimes areInfluenced by Path-Dependencies
The first hypothesis, following a historical-institutionalist argument, isthat the institutional design of supranational regulatory regimes is notonly determined by functional pressures in respective policy areas, butalso by paths of institutional development (see section 2.2). Accordingto historical-institutionalism, form does not merely follow function andnew institutions are always influenced by already-existing institutions.As a result, it is important to consider which institutions emerge firstand which later, because the older ones are likely to influence the estab-lishment of the newer ones. In the case of supranational regulatoryregimes, two critical junctures are decisive. Crisis of consumer confi-dence might lead to the set up of regulatory agencies, market integra-tion might result in shifting competencies to the EU level. Two distinctpaths can be identified, which are likely to influence the institutionaldesign of supranational regulatory regimes. Firstly, a crisis of consumerconfidence may occur before market integration. In such circumstances,strong national regulatory agencies are likely to be established, and arethen able to influence the process of market integration. Because oftheir independence from political influence, they are able to veto mar-ket integration by establishing non-tariff barriers to trade until theirinterests are taken into account. Most likely, they will demand inclusionin new supranational regulatory regimes. In contrast, if national regula-tory agencies do not exist, a single market can be established more eas-ily, because it does not face opposition from such strong stakeholders.In such circumstances, supranational regulatory regimes cannot fallback on the support of national regulatory agencies once they have tobe established. Thus, the establishment of strong supranational regula-tory regimes proves much more difficult.
The development of European pharmaceutical authorisation clearlyfollowed the first path, i.e. a regulatory scandal occurred in mostEuropean countries before a single market was created. In the early 1960s,the thalidomide scandal shattered consumer confidence in the safety ofpharmaceuticals. As a result, the EU member states established regulatory
184 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch08.qxd 9/12/2008 10:20 AM Page 184

agencies that became responsible for the authorisation of medicinal prod-ucts. Later, these agencies were an obstacle for the early establishment ofa single market for pharmaceuticals. The mutual recognition principledid not function in this sector, because the national regulatory agencieswidely refused to accept products on their markets without scrutinisingthem on their own. A single market could only be established in the early1990s by at least partly centralising the authorisation regime under guid-ance of a new agency at the EU level. The national agencies were includedin the new supranational regime, and thus a strong network of memberstates’ experts emerged. Because of this strong network, the new supra-national regime became rather independent from political influence.
In contrast, European foodstuff regulation followed the second path ofdevelopment, i.e. a regulatory scandal occurred only after a single mar-ket had already been set up. Throughout the 1980s, the Commissionmanaged to create a single market with its new approach to foodstuff reg-ulation. Accordingly, member states recognised food standards mutually,and the EU only set up harmonised standards within the committee sys-tem when issues of health and consumer protection endangered mutualrecognition. However, in the 1990s, this regime was shattered by the BSEscandal and by conflicts concerning over the authorisation of GMOs andGM food. As a result, the supranational regulatory regime was reformedat the beginning of the new millennium. A rudimentary EU food law wasadopted, and a new supranational agency for foodstuffs was set up. Theagency is responsible for advising the Commission on all matters relatingto food safety regulation, including the authorisation of GMOs and GMfood. Compared with the pharmaceutical sector, the reformed regime forfoodstuff regulation still suffers from an important shortcoming. A strongnetwork of national regulators is lacking, with the effect that the regimehas become much less independent from political control.
Hypothesis 2: Efficiency of Supranational Regulatory Regimes is Dependent onCredible Commitment of the Member States
The second hypothesis is that the member states have to commit them-selves within strong supranational regulatory regimes to achieve the twopolicy goals of establishing a stable single market for potentially riskyproducts, as well as simultaneously providing effective regulation ofthese products (section 3.2). To commit themselves, member states havetwo possibilities. Firstly, they may delegate regulatory policy-making toindependent regulatory agencies. The independence of these agenciesresults from the recruitment of their personnel, as well as from thestrength of political control. Agencies’ personnel can be composed of
Conclusion 185
PPL-UK_RR-Krapohl_Ch08.qxd 9/12/2008 10:20 AM Page 185

independent scientists or of member states’ representatives. And agen-cies’ policy-making can be independent from or scrutinised by politicaloversight. Secondly, member states may also commit themselves andtheir agents by legalising the respective policy area. Here, it is importantto consider how precise the substantive rules of regulatory policy-makingare, and whether they are subject to judicial review by the Europeancourts. The substantive rules can either prescribe regulatory policy-makingin detail, or they can leave a large amount of discretion to all actorsinvolved in the decision-making process. Substantive rules can also benon-binding guidelines or ‘hard’ EU law, which can be used to challengeregulatory decisions before the European courts.
Since the reform of the early 1990s, the EU regulatory regime for phar-maceuticals has indicated a deep commitment of the member statestowards market integration and risk regulation. Although the old com-mittee system suffered from the weaknesses of supranational regulatorybodies and judicial review, the reform of the early 1990s overcame theseshortcomings, at least within the centralised authorisation procedure forhighly innovative medicinal products. Here, the supranational agencyEMEA is quite strong (although its recruitment is not independent fromthe member states), the policy area is highly legalised and the Europeancourts play an important role in scrutinising the regime. As a result, polit-ical control by the Commission and the member states remains indirect,and EMEA in fact operates like an independent regulatory agency. Thecentralised authorisation procedure has not yet produced any regulatoryscandals, and it was positively evaluated by stakeholders in a recent eval-uation conducted by two consulting companies. Consequently, centralfeatures of the centralised procedure were strengthened by the reformprocess at the beginning of the new millennium, and the scope of appli-cation was widened to additional medicinal products.
For commitment and efficiency, the new mutual recognition proce-dure for less innovative medicinal products recognition can be locatedsomewhere between the old mutual recognition and the new centralisedprocedure. The overall efficiency of this procedure depends on the fre-quency of centralised arbitration. Before the reforms at the beginning of the new millennium, centralised arbitration occurred only rarely,because applying companies were able to avoid arbitration by selectivelywithdrawing applications from member states that opposed authorisa-tion. Since the latest reform, this is no longer possible. Applicants maystill withdraw their applications, but this no longer precludes arbitration.Thus, one can expect that the overall efficiency of the mutual recogni-tion procedure will increase.
186 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch08.qxd 9/12/2008 10:20 AM Page 186

In contrast, member states’ commitment in the foodstuff sector was,and still is, quite weak, as is the efficiency of the regulatory regime.Although recruitment of the scientific committees was at least formallyindependent before the BSE crisis, these bodies had a very weak stand-ing in the following decision-making procedure. As advisory bodies, thescientific committees were dependent on the Commission for settingthe agenda of regulatory policy-making. Additionally, Commission pro-posals were further scrutinised by the member states within severalcomitology committees. The legalisation of foodstuff regulation wasalso rather weak. No general food law existed in the EU, and a highdegree of discretion was left to the Commission and the member states.Consequently, regulatory policies were not effectively scrutinised byjudicial review. The weakness of the committee system in the foodstuffsector resulted in the failure to handle the BSE crisis, which led to a fun-damental crisis of consumer confidence.
In both respects – the strength of supranational regulatory bodies andthe density of substantive decision-making criteria – the reform after theBSE scandal did not change much. Although the establishment of EFSAstrengthened independence of scientific risk assessment, risk manage-ment remained in the hands of the Commission and the member states.Furthermore, the establishment of an EU food law did not significantlyreduce the discretion of the Commission and the member states. Thesubstantive criteria for foodstuff regulation are still quite broad, and riskmanagers’ discretion is further broadened by the application of the pre-cautionary principle. Overall, the efficiency of the EU regulatory regimefor foodstuffs cannot be expected to increase much.
At least in one area of foodstuff regulation, one can still observe ongo-ing disputes. Though the reforms at the beginning of the new millen-nium led to a lifting of the de facto moratorium on GM food, the matteris still highly disputed. All five authorisations that were issued for GMfood since 2004 were adopted without the support of a qualified major-ity of the member states. The Commission was only able to push themthrough the authorisation procedure because the Council was split onthe issue. If some member states changed their position in the future, anew moratorium against GM food could be implemented.
Hypothesis 3: Output Legitimacy of Supranational Regulatory RegimesDepends on Multiple Accountability Mechanisms
As a result of the need for credible commitment, supranational regula-tory regimes may legitimise their day-to-day policy-making more byoutput than by input factors (section 3.3). Although input legitimacy
Conclusion 187
PPL-UK_RR-Krapohl_Ch08.qxd 9/12/2008 10:20 AM Page 187

may legitimise the set up and reform of supranational regulatoryregimes, its importance for the regimes’ daily operation is limited. Ifmember states credibly commit themselves to their long-term policygoals, they can no longer react to political demands of their con-stituencies. Either democratically elected bodies are absent in regulatorypolicy-making (if competencies are delegated to agents), or they arepresent but bound to detailed criteria that limit their discretion (if thepolicy area is highly legalised). As a result, the importance of outputlegitimacy increases for the regimes’ day-to-day policy-making. Therefore,the regimes’ final policy output should meet the common interest ofstakeholders, which is an efficiently regulated and stable market for therespective products. The better the regime is able to identify converginglong-term interests of stakeholders, the stronger is its output legitimacy.To avoid agency drifts and thus losses of efficiency and output legiti-macy, supranational regulatory regimes need to be accountable to arange of actors. The regimes should be politically accountable to themember states and the EP, they should be accountable to experts ofnational regulatory authorities and they should legally be accountableto affected stakeholders. Thereby, different accountability mechanismsmust be applied at the same time to balance each other and to avoiddominance of one particular group.
Whereas input legitimacy during the establishment of the EU regula-tory regime for pharmaceuticals was, for a long time, quite low, theregime provides a high degree of output legitimacy. Leading up to theregime’s reform at the beginning of the new millennium, the EP was notinvolved in the adoption of general procedural and substantive rules ofregulatory policy-making, and the regime was mainly a result of inter-governmental decision-making in the Council. The situation improvedonly slightly with the most recent reforms, where the EP and theCouncil were co-equal legislators within the co-decision procedure. Inaddition, the influence of stakeholders was rather unbalanced in favourof producers’ interests. Producers lobbied intensively during the set upof the regime, whereas the consumer voice was almost completelyabsent. Despite this weak input legitimacy, output legitimacy of theregime’s day-to-day operation is very strong. The regime is responsibleto a range of actors by different accountability mechanisms. Memberstates may scrutinise the committee within the comitology procedure,national experts may hold the regime accountable by peer reviewwithin the agency’s expert committee, and producers may challengeauthorisation decisions before the courts. These different mechanismsbalance each other and lead to the fact that the regime is under control,
188 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch08.qxd 9/12/2008 10:20 AM Page 188

although no single party is directly able to dominate it. As a result, thepolicy output of the regime seems to reflect a long-term consensusbetween both producers and consumers of pharmaceuticals. The regimehas not yet produced a crisis of consumer confidence. Instead, a rangeof stakeholders have expressed their satisfaction with the regime in arecent evaluation conducted on behalf of the Commission.
In contrast, although input legitimacy during the establishment ofthe EU regulatory regime for foodstuffs has increased since the BSE scan-dal, the regime suffered – and still suffers – from low output legitimacy.Before the BSE scandal, the committee system was set up by theCommission and the member states, and the EP did not have a say dur-ing its establishment. Additionally, mismanagement during the BSEscandal demonstrated the shortcomings of the committee system andthus destroyed its output legitimacy. However, input legitimacy wasstrengthened by the reform process at the beginning of the new mil-lennium. The EP became a co-equal legislator to the Council within theco-decision procedure, and it used this power to present itself as anadvocate of consumer interests. However, this increase in input legiti-macy is unlikely to find its equivalent in an increase of output legiti-macy. The regime is still mainly responsible to the member states,whereas accountability to other actors like national experts or stake-holders is quite weak. The political control of the member states is notbalanced by expert or legal control. Thus member states are still able todominate the regime with their interests. Whereas the success of thenew regime concerning general foodstuff regulation is an open ques-tion, the regime’s weakness in the case of GM food authorisation hasalready become visible. The authorisation of GM food still meets withresistance from consumers and from a large group of member states,and authorisations can only be issued because the Council is split in thisrespect.
8.1 Areas of further research
A broad range of cases may be used to test the institutionalist approachof this book further. One example is the case of standardisation of tech-nical goods in the EU, which comes close to that of pharmaceuticals(e.g. Egan 1998; Kerler 2005; Pelkmans 1987). Standardisation bodies fortechnical goods had already existed in many EU member states beforethe Single Market was set up although this was not due to regulatoryscandals, but to the industry’s own need for standardisation. In the1970s and 1980s, different national standards adopted by these bodies
Conclusion 189
PPL-UK_RR-Krapohl_Ch08.qxd 9/12/2008 10:20 AM Page 189

proved to be a major obstacle to the establishment of a single market fortechnical goods. The problem was solved in the mid-1980s with theCommission’s new approach to technical harmonisation, which alsoserved as a role model for other policy areas (e.g. foodstuff regulation,section 6.1). According to the new approach, the Commission restricteditself to laying down only fundamental safety requirements in EU legis-lation. The implementation of these general rules became delegated toEuropean standardisation bodies. These new European bodies organisenetworks of the old national standardisation bodies, just like theEuropean agency in the pharmaceutical sector. However, the main dif-ference from the EU agency for pharmaceuticals is that the Europeanstandardisation bodies are organised under private law, and that theirdecisions – i.e. the technical standards – are not legally binding.Nevertheless, a presumption of conformity of these standards with themore general EU legislation gives them, de facto, a binding character.
Because of this development of the standardisation of technical goodsin Europe, the institutionalist approach to supranational risk regulationwould expect that the EU regime is rather independent from direct polit-ical influence, that member states’ commitment leads to high efficiencyof regulatory policy-making, and that the regime is legitimate due to itspolicy output. Indeed, first empirical evidence points in this direction.Firstly, the European standardisation bodies are indeed very independentfrom direct political control by the member states. In contrast to thepharmaceutical sector, this is not so much due to legalisation and judi-cialisation, but to the independence of these bodies from the govern-ments of the member states. The controlling member state committeesare only advisory committees, which cannot overrule the standardisa-tion bodies and the Commission, as long as these work hand-in-hand.Secondly, the standardisation of technical goods in Europe seems tofunction rather efficiently. The new approach of technical standardisa-tion successfully managed to establish a single market for these goods,and large regulatory scandals like those of thalidomide or BSE have so farnot occurred. Finally, the regime seems to provide a high degree of out-put legitimacy, although the accountability mechanisms appear to bedifferent from those in the pharmaceutical sector. The member statescannot simply overrule the standardisation bodies and the Commission,but they may challenge standards within a safeguard procedure. In addi-tion, because standards are not legally binding, stakeholders cannotchallenge them before the courts. Nevertheless, the regime is dependenton stakeholders’ acceptance, because otherwise the standards would notbe implemented.
190 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch08.qxd 9/12/2008 10:20 AM Page 190

Besides the case of standardisation of technical goods, the approachof this book should also be applicable to other areas of EU risk regula-tion. Such areas could be, for example, chemical regulation or the regu-lation of financial services. Here, the questions to be asked would bewhether any national regulatory agencies existed before the SingleMarket was integrated, whether these agencies constitute a strong regu-latory network independent from direct political control, and whetherthe respective regimes provide efficient risk regulation and conse-quently high output legitimacy.
With some adjustments, the theory should also be applicable to othercases of supranational regulation. Economic regulation (as in the areas oftelecommunication or energy) does not aim to reduce risks for consumers,but to avoid monopolies and to ensure fair competition between differentsuppliers. In such cases, the crises that led to the establishment of respec-tive regulatory regimes are probably not crises of consumer confidence.Instead, it is more likely that new regulatory regimes emerge in reaction toeconomic crises in order to fight private or state monopolies in specificsectors. Thus, the path-dependency hypothesis must be adapted to thesespecific circumstances. However, the need for credible commitment toefficient regulatory policies is the same. As a result, the efficiency ofrespective regulatory regimes could depend on the same institutionalmechanisms as in the case of risk regulation. And the regimes may derivetheir legitimacy from the same factors as the regimes for supranationalrisk regulation.
8.2 Theoretical implications
The above analysis of pharmaceutical and foodstuff regulation in theEU clearly demonstrates that functional pressure alone is not always(and probably rarely) able to explain fully the specific design of institu-tions. Many theoretical approaches focus on functionalist reasons forthe emergence and development of institutions. The most prominentphrase in this respect is ‘form follows function’ from neo-functionalism,but rational–institutionalists also explicitly or implicitly assume thatinstitutions are primarily a consequence of functionalist pressures (e.g.Franchino 2000a, 2000b; Pollack 1997a, 2003). Accordingly, institutionsare set up, because they fulfil specific functions for actors. This mightexplain the rationale for the establishment of institutions, but it doesnot fully account for their specific form, or for their development overtime. This is because the rationalist argument often suffers from a func-tionalist fallacy. There is no market mechanism in political life that
Conclusion 191
PPL-UK_RR-Krapohl_Ch08.qxd 9/12/2008 10:20 AM Page 191

guarantees that inefficient institutions are abolished, and that only themost efficient ones prevail. Historical institutionalism (e.g. Arthur 1994;Pierson 1996, 2000a; Thelen 1999, 2003) takes up this critique. Its basicstatement is that institutions always develop out of previously exist-ing institutions. Institutions create incentives for certain actors, dis-tribute influence between them and set the rules for decision-making.Consequently, they at least partly influence the factors that determinetheir change. The consequence is a path-dependent development thatmight also reinforce inefficient institutions.
The two analysed supranational regulatory regimes function differ-ently, even though the functionalist pressures behind the regulation ofthese two product sectors are very similar. To analyse and explain thesedifferences, it is necessary to look at the history and development of therespective institutions in more detail. The EU regulatory regime forpharmaceuticals differs from that for foodstuffs, because the former isbased on a strong network of national regulatory agencies. These wereestablished in reaction to the thalidomide catastrophe long before a sin-gle market for pharmaceuticals was established. In contrast, the supra-national regulatory regime for foodstuffs could not be built upon suchnational agencies. One would not notice this important difference ifone compared only the current regimes without looking at their history.In the end, a historical-institutionalist approach not only examines theimpact of current institutions on actors’ behaviour, but it also considersthe effect of previously existing institutions on the emergence of cur-rent institutions.
The findings of this book led to serious doubts about the concept ofdeliberative supranationalism (e.g. Joerges and Neyer 1997a, 1997b,2006), which develops a rather optimistic view of the EU committee sys-tem. Joerges and Neyer state that repeated meetings of representatives,scientists and stakeholders within different committees lead to a com-mon identity of these actors. They develop common norms about theirbehaviour as experts, and common belief systems pertaining to therespective regulatory task. Besides, they act before the background ofEuropean law, and not in an institution-free bargaining situation. Allthis allows them to leave interest-based, intergovernmental bargainingbehind, and to concentrate on common ‘deliberative problem-solving’.Joerges and Neyer assume that this deliberation, in a Habermasian sense,leads to more efficient and legitimate risk regulation than one wouldexpect from intergovernmental negotiations. However, the concept ofdeliberative supranationalism has one fundamental shortcoming: itlacks a micro-analytical foundation. Committee members – especially
192 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch08.qxd 9/12/2008 10:20 AM Page 192

representatives within comitology committees – not only act at thesupranational level, but they are also embedded in a national context.The question is why the supranational community should influencetheir behaviour more than their national political system. Delegateswithin comitology committees complete national careers, are recruitedby their national governments and are constrained by the governments’direct instructions. It is unlikely that a supranational expert identitybecomes more important for these delegates than their national context.As a result, it is more likely that these delegates represent the particular-istic interests of their government instead of deliberating about commonregulatory solutions.
The empirical analysis of this book found that the committee system(in both the pharmaceutical and foodstuff sectors) was neither very effi-cient nor notably legitimate. Within the pharmaceutical sector, thecommittee system was not able to establish the preconditions for a sin-gle market, because resistance from national regulatory authorities wastoo strong. And in the foodstuff sector, the committee system was notable to handle the BSE problem adequately. At the same time that effi-ciency, and thus output legitimacy, was weak, input legitimacy of thecommittee system was only based on input from member states’ gov-ernments. Neither the EP nor stakeholder groups could significantlyinfluence regulatory policy-making. Keeping in mind the long delega-tion chain from national elections to national parliaments, nationalgovernments and finally to the comitology committees, the legitimacyof the committee system can be doubted. In the end, the preconditionsfor efficient – and thus more legitimate – policy-making within the EUcommittee system should be further specified. Legalisation of respectivepolicy areas could ‘civilise’ the interaction of representatives within thedifferent committees. However, the degree of legalisation is an openempirical question in each policy field. It must be analysed case-by-caseand cannot be taken for granted for the whole EU committee system.
In contrast to Joerges and Neyer, Majone (e.g. 1996: 61–79, 2001a,2005: 64–82) does not opt for a broad inclusion of stakeholders in EUpolicy-making. Rather, Majone stresses that regulatory policies should beadopted independently from the influence of political actors. Regulatorypolicies should only address market failures to increase the efficiency ofthe Single Market. Re-distributive concerns should not be the subject ofregulatory policies, as they would reduce the commitment to specificregulatory objectives. Consequently, influence of short-sighted politicalactors, which represent particularistic and distributive interests, shouldbe avoided by delegating the task of regulation to independent agents,
Conclusion 193
PPL-UK_RR-Krapohl_Ch08.qxd 9/12/2008 10:20 AM Page 193

for example a powerful and independent Commission (Majone 1994) orindependent regulatory agencies at the EU level (Majone 1997). Thehanding over of regulatory competencies to such independent agentsleads to delegation gains, as long as the agents act in the common long-term interests of their principals. However, as soon as principals dele-gate competencies to new actors, problems of control problems. Newagents might develop their own interests or might be captured by theinterests of some stakeholders. Agents might use their competencies tofollow these interests instead of the long-term interests of their princi-pals. Consequently, agents need to be controlled to avoid agency drifts.Thus, delegation always takes place in a tension between delegationgains, i.e. regulatory policy-making by specialised agents, and agencylosses, i.e. costs that occur when agents deviate from the member states’interests (Tallberg 2002).
The analysis presented in this book demonstrates that the legalisa-tion of a policy area can be at least as efficient in committing politicalactors as delegation to an independent regulator. Detailed substantiverules of decision-making are also suitable for committing politicalactors, even though these political actors may still be involved in day-to-day regulatory policy-making. The necessary precondition for suchan effect is the existence of independent courts, which scrutinisewhether the specific regulatory policies are in line with substantive cri-teria laid down in general legislation. In this case, not the independ-ence of the regulator, but the independence of courts guarantees thecommitment of political actors. One could well argue that the legalisa-tion of a policy area is even more important in the case of suprana-tional risk regulation. The EU can be regarded as a ‘mixed polity’(Majone 2002a) in which different interests are balanced vis-à-vis eachother. The willingness of the member states to delegate wide compe-tencies to independent agents is very low, because this could endangerthe precarious balance between member states and supranational bod-ies. However, legalisation does not affect this balance as much as dele-gation, because competencies may remain in the hands of the memberstates. Thus, legalisation of a policy field may be more easily acceptedby the member states. Consequently, it is a promising solution to thecommitment problem within the EU context.
194 Risk Regulation in the Single Market
PPL-UK_RR-Krapohl_Ch08.qxd 9/12/2008 10:20 AM Page 194

Notes
1 Introduction: The Need for a Systematic Analysis ofSupranational Risk Regulation
1. In the following, the term ‘the Single Market’ is used when the wholeEuropean Single Market is meant. The term ‘a single market’ refers to a sin-gle market for one or more groups of products, but not to the Single Marketin total.
2. All activity analysed in this book took place either within the EuropeanEconomic Community or within the European Community – the first pillarof the European Union. Nevertheless, for convenience, the term ‘EuropeanUnion’ (EU) is used throughout the book.
3. There exists some confusion in the academic literature about the terms ‘com-mittee system’ or ‘comitology’. Throughout this book, the term ‘committeesystem’ is used whenever the entity of executive committees – member statecommittees or scientific advisory committees – are meant. The term ‘comitol-ogy’ refers to all committees that are established to control the Commission,and which decide according to the comitology procedures as laid down inCouncil Decision 1999/468/EC (formerly 87/373/EEC). Thus, the comitologysystem is a smaller part of the committee system.
4. In the following, this book mainly deals with product standards, namelyhealth and safety requirements that protect the consumers of certain products.The argument does not apply to process standards, namely requirements thatprotect workers or the environment during the production process. There isless functional pressure to harmonise process standards; thus, supranationalregimes do not necessarily emerge (Scharpf 1996a).
5. Because this book deals with risk regulation, the following analysis concen-trates on the regulation of safety, efficacy and quality of pharmaceuticals inEurope. Market and price regulations are not considered. Whereas productregulation of pharmaceuticals is widely harmonised within the EU, marketregulation remains mainly with member states and national health systems(e.g. Kotzian 2003; Permanand 2006: 151–79).
2 Functional Pressure and Path-Dependencies: The Emergence and Development of SupranationalRegulatory Regimes
1. This book mainly deals with product standards, i.e. health and safetyrequirements that protect the consumers of certain products. The argumentdoes not apply to process standards, i.e. requirements that protect workersor the environment during the production process. There is less functionalpressure to harmonise process standards, and thus supranational regimes donot necessarily emerge (Scharpf 1996a).
195
PPL-UK_RR-Krapohl_Notes.qxd 9/12/2008 10:12 AM Page 195

2. Judgement of the European Court of Justice of 20 February 1979: Rewe-Zentral AG v. Bundesmonopolverwaltung für Branntwein; prelimi-nary ruling: Hessisches Finanzgericht – Germany; case 120/78 (ECR 1979: 649).
3. After the Single European Act, most legislation in respect to the SingleMarket was established within a cooperation procedure. Therein, the EPcould sometimes act as a ‘conditional agenda-setter’ (Tsebelis 1994; Tsebelisand Kreppel 1998), but its final agreement was not necessary to adoptpolicies. Later, after the Maastricht and Amsterdam treaties, the co-deci-sion procedure usually applied. Therein, the EP is able to veto all EU legislation (Tsebelis and Garrett 2000, 2001). Nevertheless, the followingargument deals mainly with decision-making in the Council, and neglectsthe role of the EP. Its aim is not to analyse policy-making within the differ-ent legislative procedures, but to understand the intergovernmental logic ofdecision-making within the Council.
4. For economic regulation, where market creation is more important than riskregulation, the crises that lead to the establishment of regulatory institutionsare probably not crises of consumer confidence. Here, monopolies (as a kindof market failure) or over-bureaucratisation (as a kind of state failure) couldbe more important.
5. European Commission (1985): ‘Completing the Internal Market: WhitePaper of the Commission of the European Community’, COM(85) 310.
3 Efficiency and Legitimacy: The Evaluation ofSupranational Regulatory Regimes
1. See, for example, the ‘Communication from the Commission on thePrecautionary Principle’ (COM(2000) 1 final), 2/2/2000.
2. The only exceptions, in which costs are concentrated on one group, are caseswhere consumers’ demands are either fully elastic or totally inflexible. In thefirst case, increased prices would only result in fewer demands, and marketprices would sink again to the old level. In the second case, consumerswould pay higher prices without reducing their demands.
3. Council Decision 1999/468/EC of 28 June 1999 laying down the proceduresfor the exercise of implementing powers conferred on the Commission, OJ L184, 17/7/1999, 23–6.
4. Art. 230 (formerly 173) of the Treaty establishing the European Community.5. This view of the institutional balance and the resulting non-delegation doc-
trine was expressed very early by the ECJ. In a decision from the 1950s, itestablished the so-called Meroni doctrine, which states that delegation ofcompetencies to institutions not mentioned in the treaties shall be subject tovery strict conditions (Everson et al. 1999: 53; Vos 1999a: 200). Accordingly,the Commission may only delegate powers of its own, delegation must belimited to the preparation and performance of executive competencies, inde-pendent bodies should not receive discretionary power, the Commissionmust retain oversight and be judicially responsible, and delegation must notdisturb the ‘balance of powers’ within the EU.
196 Notes
PPL-UK_RR-Krapohl_Notes.qxd 9/12/2008 10:12 AM Page 196

4 From National Crises to a Strong Supranational Regime:The Development of Pharmaceutical Authorisation inEurope
1. Because this book deals with risk regulation, the following analysis concen-trates on the regulation of safety, efficacy and quality of pharmaceuticals inEurope. Market and price regulations are not considered. Whereas productregulation of pharmaceuticals is widely harmonised within the EU, marketregulation remains mainly in the hands of the member states and nationalhealth systems (e.g. Kotzian 2003; Permanand 2006: 151–79).
2. Gesetz über den Verkehr mit Arzneimitteln (Arzneimittelgesetz) vom 16. Mai1961, Bundesgesetzblatt I (1961), 533–47.
3. ‘Welt am Sonntag’ from 25 November 1961: ‘Missgeburten durch Tabletten?Alarmierender Verdacht eines Arztes gegen ein weitverbreitetes Medikament’.
4. Gesetz zur Änderung des Arzneimittelgesetzes vom 29 April 1964,Bundesgesetzblatt I (1964), 275–82.
5. Gesetz zur Neuordnung des Arzneimittelrechts vom 24 August 1976,Bundesgesetzblatt I (1976), 2445–83.
6. Because of a regulatory scandal of HIV-contaminated blood products (Kirk1999: 214–28; Scheu 2003: 337–564), the BGA was dissolved in 1994. Thecompetencies for the authorisation of pharmaceuticals were delegated to themore independent Paul-Ehrlich-Institute (responsible only for sera and vac-cines) and the newly founded Federal Institute for Pharmaceuticals andMedicinal Products (Bundesinstitut für Arzneimittel und Medizinprodukte;BfArM).
7. In 1989, the Medicines Division of the Department of Health was reorgan-ised in the Medicines Control Agency (MCA; Gilardi 2004: 314).
8. In 1988, the Directorate for Pharmaceuticals and Medicinal Products wasreorganised in the Agence Française de Sécurité Sanitaire des Produits deSanté (French Agency for the Safety of Sanitary Products; AFSSAPS; Gilardi2004: 312).
9. Council Directive 65/65/EEC of 26 January 1965 on the approximation ofprovisions laid down by Law, Regulation or Administrative Action relating toproprietary medicinal products, Official Journal 22, 9/2/1965, 369–73.
10. Council Directive 75/318/EEC of 20 May 1975 on the approximation of thelaws of Member States relating to analytical, pharmaco-toxicological andclinical standards and protocols in respect of the testing of proprietarymedicinal products, Official Journal L 147, 9/6/1975, 1–12.
11. Second Council Directive 75/319/EEC of 20 May 1975 on the approximationof provisions laid down by Law, Regulation or Administrative Action relatingto proprietary medicinal products, Official Journal L 147, 9/6/1975, 13–22.
12. Council Directive 87/19/EEC of 22 December 1986 amending Directive75/318/EEC on the approximation of the laws of the Member States relatingto analytical, pharmaco-toxicological and clinical standards and protocols inrespect of the testing of proprietary medicinal products, Official Journal L15,17/1/1987, 31–3.
13. Council Directive 87/22/EEC of 22 December 1986 on the approximation ofnational measures relating to the placing on the market of high-technology
Notes 197
PPL-UK_RR-Krapohl_Notes.qxd 9/12/2008 10:12 AM Page 197

medicinal products, particularly those derived from biotechnology, OfficialJournal L 15, 17/1/1987, 38–41.
14. Report from the Commission to the Council on the Activities of theCommittee for Proprietary Medicinal Products, 15/2/1991 (COM(91)39 final).
15. Ibid.16. Erläuterndes Memorandum der Kommission: ‘Künftiges System für den
freien Arzneimittelverkehr in der Europäischen Gemeinschaft’, 14/11/1990(KOM(90)283 endg.).
17. Vorschlag für eine Verordnung des Rates (EWG) zur Festlegung vonGemeinschaftsverfahren für die Zulassung und überwachung von Human-und Tierarzneimitteln und zur Schaffung einer europäischen Agentur für dieBeurteilung von Arzneimitteln (KOM(90) 283 endg.), ABl. C 330, 31/12/1990,1–17.
18. From the early 1980s onwards, medicinal products for veterinary use wereauthorised in the EU according to procedures similar to those of pharma-ceuticals for human use. In the following, this book does not deal any fur-ther with the regulation of veterinary medicinal products, but concentratesonly on products for human use.
19. Vorschlag für eine Richtlinie (EWG) des Rates zur Änderung der Richtlinien65/65/EWG, 75/318/EWG und 75/319/EWG betreffend Arzneimittel (KOM(90)283 endg.), ABl. C 330, 31/12/1990, 18–24.
20. Stellungnahme des Europäischen Parlaments zu dem Vorschlag derKommission für eine Verordnung des Rates (EWG) zur Festlegung vonGemeinschaftsverfahren für die Zulassung und überwachung von Human-und Tierarzneimitteln und zur Schaffung einer europäischen Agentur für dieBewertung von Arzneimitteln, ABl. C 183, 15/7/1991, 145–78.
21. Council Regulation (EEC) No 2309/93 of 22 July 1993 laying downCommunity procedures for the authorization and supervision of medicinalproducts for human and veterinary use and establishing a European Agencyfor the Evaluation of Medicinal Products, Official Journal L 214, 24/8/1993,and Council Directive 93/39/EEC of 14 June 1993 amending Directives65/65/EEC, 75/318/EEC and 75/319/EEC in respect of medicinal products,Official Journal L 214, 24/8/1993, 22–30.
22. CMS Cameron McKenna and Andersen Consulting (2000): ‘Evaluation ofthe operation of Community procedures for the authorisation of medicinalproducts’ (http://pharmacos.eudra.org/F2/home.html, 5 February 2006).
23. The following assessments are derived from: Ibid., 11–21, 72, 122, 75, 76.24. Proposal for a Regulation of the European Parliament and of the Council lay-
ing down Community procedures for the authorisation and supervision ofmedicinal products for human and veterinary use and establishing a EuropeanAgency for the Evaluation of Medicinal Products, Proposal for a Directive ofthe European Parliament and of the Council amending Directive 2001/83/ECon the Community code relating to medicinal products for human use, andProposal for a Directive of the European Parliament and of the Councilamending Directive 2001/82/EC on the Community code relating to veteri-nary medicinal products, 26/11/2001 (COM(2001) 404 final).
25. European Parliament legislative resolution of 23 October 2002 on the proposal for a European Parliament and Council regulation laying down
198 Notes
PPL-UK_RR-Krapohl_Notes.qxd 9/12/2008 10:12 AM Page 198

Community procedures for the authorisation and supervision of medicinalproducts for human and veterinary use and establishing a European Agencyfor the Evaluation of Medicinal Products, Official Journal C 300, 11/12/2003,166–308.
26. Common position adopted by the Council on 29 September 2003 with aview to the adoption of a Regulation of the European Parliament and of theCouncil laying down Community procedures for the authorisation andsupervision of medicinal products for human and veterinary use and estab-lishing a European Medicines Agency, Official Journal C 297, 9/12/2003, and Regulation (EC) No 726/2004 of the European Parliament and of theCouncil of 31 March 2004 laying down Community procedures for theauthorisation and supervision of medicinal products for human and veteri-nary use and establishing a European Medicines Agency, Official Journal L 136, 30/4/2004, 1–33.
27. In 1999, the comitology system was reformed and simplified by CouncilDecision 1999/468/EC of 28 June 1999 laying down the procedures for the exercise of implementing powers conferred on the Commission, OfficialJournal L 184, 17/7/1999, 23–6. Within the reformed comitology system,which applies for all new delegation acts after 1999, the distinctions betweendifferent management procedures (IIa and IIb), different regulatory proce-dures (IIIa and IIIb) and different safeguard procedures (IVa and IVb) areabolished. Now, there exists only one advisory, one management, one regu-latory and one safeguard procedure.
28. Directive 2004/27/EC of the European Parliament and of the Council of 31March 2004 amending Directive 2001/83/EC on the Community code relat-ing to medicinal products for human use, Official Journal L 136, 30/4/2004,34–57.
29. European Commission: ‘Review of Pharmaceutical Legislation: DiscussionDocument’, 22/1/2001 (http://pharmacos.eudra.org/F2/home.html, 5/2/2006),222–33.
30. Regulation (EC) No 141/2000 of the European Parliament and of the Councilof 16 December 1999 on orphan medicinal products, Official Journal L 18,22/1/2000, 1–5.
31. Directive 2004/24/EC of the European Parliament and the Council of 31March 2004 amending, as regards traditional herbal medicinal products,Directive 2001/83/EC on the Community code relating to medicinal prod-ucts for human use, Official Journal L 136, 30/4/2004, 85–90.
32. Directive 2001/83/EC of the European Parliament and of the Council of 6November 2001 on the Community code relating to medicinal products forhuman use, Official Journal L 311, 28/11/2001, 67–128.
33. Commission Directive 2003/63/EC of 25 June 2003 amending Directive2001/83/EC of the European Parliament and of the Council on theCommunity code relating to medicinal products for human use, OfficialJournal L 159, 27/6/2003, 46–94.
34. Committee for Proprietary Medicinal Products and Committee forVeterinary Medicinal Products: Note for Guidance on Minimising the Risk ofTransmitting Animal Spongiform Encephalopathy Agents via Human andVeterinary Medicinal Products (EMEA/410/01 rev 2, Official Journal C 24,
Notes 199
PPL-UK_RR-Krapohl_Notes.qxd 9/12/2008 10:12 AM Page 199

28/1/2004, 6–19). This ‘BSE-guideline’ was later tightened by two revisions:Committee for Proprietary Medicinal Products and Committee for VeterinaryMedicinal Products (31/5/2001): Note for Guidance on Minimising the Riskof Transmitting Animal Spongiform Encephalopathy Agents via Human and Veterinary Medicinal Products (EMEA/410/01 rev 1, Official Journal C286, 12/10/2001, 4), and Committee for Proprietary Medicinal Products andCommittee for Veterinary Medicinal Products (31/10/2003): Note for Guidanceon Minimising the Risk of Transmitting Animal Spongiform EncephalopathyAgents via Human and Veterinary Medicinal Products (EMEA/410/01 rev 2,Official Journal C 24, 28/1/2004, 6).
35. Commission Directive 1999/82/EC of 8 September 1999 amending theAnnex to Council Directive 75/318/EEC on the approximation of the laws ofthe Member States relating to analytical, pharmacotoxicological and clinicalstandards and protocols in respect of the testing of medicinal products,Official Journal L 243, 15/9/1999, 7–8.
5 A Strong Regulatory Network: The Evaluation of theEuropean Regulatory Regime for Pharmaceuticals
1. Report from the Commission to the Council on the Activities of theCommittee for Proprietary Medicinal Products, 15/2/1991 (COM(91)39final).
2. Ibid.3. European Commission (2002): ‘Chapter 4: Centralised Procedure’, in:
‘EudraLex Collection: The Rules governing Medicinal Products in theEuropean Community, Volume 2A: Notice to Applicants (Medicinal Productsfor Human Use)’ (http://pharmacos.eudra.org/F2/eudralex/index.htm,29/3/2006).
4. http://www.emea.eu.int/htms/aboutus/experts.htm, 29/3/2006.5. European Commission (2005): ‘Chapter 6: Decision Making Procedure for
the Adoption of Commission Decisions’, in: ‘EudraLex Collection: The Rulesgoverning Medicinal Products in the European Community, Volume 2A:Notice to Applicants (Medicinal Products for Human Use)’ (http://pharmacos.eudra.org/F2/eudralex/index.htm, 29/3/2006).
6. Decision of the Commission from 9/3/2000 (C(2000)452), (C(2000)453) and(C(2000)608).
7. The fact that comparative efficiency evaluations are not provided by EUlegislation is often criticised by patients’ and physicians’ associations. Theyargue that the lack of comparative evaluations leads to the authorisation ofso-called ‘me-too’ products, which have no additional therapeutic value(e.g. Permanand and Mossialos 2005). In contrast, producers and most reg-ulators claim that comparative evaluations would reduce legal certainty,and would thus reduce the incentives for industry to conduct pharmaceu-tical research.
8. Judgement of the Court of First Instance of 26 November 2002: ArtegodanGmbH and Others v Commission of the European Communities, joinedcases T-74/00, T-76/00, T-83/00, T-84/00, T-85/00, T-132/00, T-137/00 and T-141/00, ECR 2002: II-4945.
200 Notes
PPL-UK_RR-Krapohl_Notes.qxd 9/12/2008 10:12 AM Page 200

9. Judgement of the Court of 24 July 2003: Commission of the EuropeanCommunities v Artegodan GmbH and Others, case C-39/03 P, ECR 2003: I-7885.
10. Judgement of the Court of First Instance of 18 December 2003: Nancy FernOlivieri v Commission of the European Communities and European Agencyfor the Evaluation of Medicinal Products, case T-326/99, ECR 2003: II-6053.
11. European Commission (1998): ‘Chapter 2: Mutual Recognition’, in: ‘EudraLexCollection: The Rules governing Medicinal Products in the EuropeanCommunity, Volume 2A: Notice to Applicants (Medicinal Products for HumanUse)’ (Luxembourg: Office for Official Publications of the EuropeanCommunities).
12. European Commission (2005): ‘Chapter 2: Mutual Recognition’, in: ‘EudraLexCollection: The Rules governing Medicinal Products in the EuropeanCommunity, Volume 2A: Notice to Applicants (Medicinal Products for HumanUse)’ (http://pharmacos.eudra.org/F2/eudralex/index.htm, 29/3/2006).
13. See the homepage of the ‘Heads of Agencies’: http://heads.medagencies.org/,29/3/2006.
14. Referrals to the binding arbitration procedure may also occur, if differentnational authorisation decisions are passed, if Community interests are atstake, or if existing authorisations need to be modified, suspended or with-drawn.
15. European Commission (2004): ‘Chapter 3: Community Referral’, in: ‘EudraLexCollection: The Rules governing Medicinal Products in the EuropeanCommunity, Volume 2A: Notice to Applicants (Medicinal Products for HumanUse)’ (http://pharmacos.eudra.org/F2/eudralex/index.htm, 29/3/2006).
16. These numbers are published by the coordination group (http://www.hma.eu,26/2/2008).
17. Graham, D. J. (2004): ‘Risk of Acute Myocardial Infarction and Sudden CardiacDeath in Patients Treated with COX-2 Selective and Non-Selective NSAIDs’(Internal Memorandum of the United States Food and Drug Administrationfrom 30 September 2004, http://www.fda.gov/cder/drug/infopage/vioxx/vioxxgraham.pdf, 6/4/2006).
18. See the homepage of the company: http://www.merck.com/newsroom/vioxx_withdrawal, 14/4/2006.
6 From an Early Single Market to a Crisis of ConsumerConfidence: The Development of Foodstuff Regulation in Europe
1. Council Resolution of 28 May 1969 drawing up a programme for the elimi-nation of technical barriers to trade in foodstuffs which result from dispari-ties between the provisions laid down by Law, Regulation or AdministrativeAction in Member States, Official Journal C 76, 17/06/1969, 5–7.
2. Council Directive 73/241/EEC of 24 July 1973 on the approximation of thelaws of the member states relating to cacao and chocolate products intendedfor human consumption, Official Journal L 228, 16/8/1973, 23–35.
3. Council Resolution of 17 December 1973 on industrial policy, OfficialJournal C 117, 16/8/1973, 1–14.
Notes 201
PPL-UK_RR-Krapohl_Notes.qxd 9/12/2008 10:12 AM Page 201

4. Judgement of the Court of 20 February 1979: Rewe-Zentral AG vBundesmonopolverwaltung für Branntwein, Reference for a preliminary rul-ing: Hessisches Finanzgericht – Germany, case 120/78, ECR 1979: 649.
5. White Paper from the Commission to the European Council (Milan, 28–29June 1985): Completing the Internal Market (Com(86) 310 final).
6. Communication from the Commission to the Council and to the EuropeanParliament from 8/11/1985: Completion of the Internal Market: Communitylegislation on foodstuffs (COM(85) 603 final).
7. 87/373/EEC: Council Decision of 13 July 1987 laying down the proceduresfor the exercise of implementing powers conferred on the Commission,Official Journal L 197, 18/7/1987, 33–5.
8. 75/420/EEC: Commission Decision of 26 June 1975 setting up an AdvisoryCommittee on Foodstuffs, Official Journal L 182, 12/7/75, 35.
9. Council Directive 89/662/EEC of 11 December 1989 concerning veterinarychecks in intra-Community trade with a view to the completion of theinternal market, Official Journal L 295, 30/12/1989, 13–22, and CouncilDirective 90/425/EEC of 26 June 1990 concerning veterinary and zootechni-cal checks applicable in intra-Community trade in certain live animals andproducts with a view to the completion of the internal market, OfficialJournal L 224, 18/8/1990, 29–41.
10. Commission Decision 81/651/EEC setting up a Scientific VeterinaryCommittee, Official Journal L 233, 19/08/1981, 32–3.
11. Council Decision 68/361/EEC setting up a Standing Veterinary Committee,Official Journal L 255, 18/10/1968, 23.
12. European Parliament (1997): ‘BSE Inquiry Report: Report on alleged contra-ventions or maladministration in the implementation of Community law inrelation to BSE, without prejudice to the jurisdiction of the Community andnational courts’ (A4–0020/97), I.1.C.
13. Ibid., I.4.14. The feeding ban of meat- and bone-meal was introduced five years later than
in the UK. One has to keep in mind that after the British ban was set up in1989, exports were still allowed for another year. In the following, exports ofpotentially infected meat- and bone-meal from the UK increased about100%. Up to 1994, these feedstuffs were still allowed to be fed to cattle in theEU, as long as the member states did not impose national feeding bans. Theexports of meat- and bone-meal from the UK, and the gap of five yearsbetween the British and the EU ban, were later seen as one major factorresponsible for the spread of the disease across Europe.
15. Later, Portugal was also affected by an export ban on its beef, even thoughthe political consequences of this measure did not reach the same scale. InPortugal, the number of detected BSE cases in domestic (not imported) cat-tle rose from 12 in 1994, to 15 in 1995, to 31 in 1996, to 30 in 1997 and to127 in 1998. In reaction to this new threat, the EU adopted a ban on exportsof cattle, beef and beef products from Portugal.
16. Judgement of the Court of 5 May 1998: United Kingdom of Great Britain and Northern Ireland v Commission of the European Communities, case C-180/96, ECR 1998: I-2265.
17. Order of the Court of 21 June 2000: French Republic v Commission of theEuropean Communities, case C-514/99, ECR 2000: I-4705.
202 Notes
PPL-UK_RR-Krapohl_Notes.qxd 9/12/2008 10:12 AM Page 202

18. Judgement of the Court of 13 December 2001: Commission of the EuropeanCommunities v French Republic, case C-1/00, ECR 2001: I-9989.
19. The story was repeated in the case of the export ban on beef and beef productsin Portugal. Like the UK, the Portuguese government proceeded against the banbefore the ECJ, but the claim was unsuccessful in the end. After the ban on theexport of Portuguese beef and beef products was lifted under a Date-Basedexport scheme, France successfully tried to overthrow this decision.
20. Judgement of the Court of First Instance (Fifth Chamber) of 30 September1998: Confederazione Nazionale Coltivatori Diretti (Coldiretti) and 110farmers v Council of the European Union and Commission of the EuropeanCommunities, case T-149/96, ECR 1998: II-3841.
21. European Parliament (1997): ‘BSE Inquiry Report: Report on alleged contra-ventions or maladministration in the implementation of Community law inrelation to BSE, without prejudice to the jurisdiction of the Community andnational courts’ (A4–0020/97).
22. European Parliament (1997): ‘BSE Inquiry Report: Report on alleged contra-ventions or maladministration in the implementation of Community law inrelation to BSE, without prejudice to the jurisdiction of the Community andnational courts’ (A4–0020/97), I.2.5.
23. Ibid., I.3.2.24. Ibid., I.5.1.25. Ibid., II.1 – II.5.26. The second step of the institutional reforms is discussed in section 6.3.1. Here,
only the reform of the committee system by the Commission is analysed.27. Communication of the European Commission: Consumer Health and Food
Safety (COM(97) 183 final).28. 97/404/EC: Commission Decision of 10 June 1997 setting up a Scientific
Steering Committee, Official Journal L 169, 27/6/1997, 85–7.29. 97/579/EC: Commission Decision of 23 July 1997 setting up Scientific
Committees in the field of consumer health and food safety, Official JournalL 237, 28/8/1997, 18–23.
30. Final Opinion of the Scientific Steering Committee on the Geographical Riskof Bovine Spongiform Encephalopathy, adopted on 6 July 2000, http://europa.eu.int/comm/food/fs/sc/index_en.html (1/8/2000).
31. The numbers are published on the homepage of the World Organization forAnimal Health (http://www.oie.int/eng/info/en_esbmonde.htm; 25/7/2005).
32. Opinion of the Scientific Steering Committee on the scientific basis for: (1) import bans proposed by three member states with regard to BSE inFrance and the Republic of Ireland; (2) several measures proposed by Francewith regards to BSE risks; and (3) banning animal protein from the feed forall farmed animals, including pig, poultry, fish and pet animals, adopted bythe Scientific Steering Committee at its meeting of 27–28 November 2000,http://europa.eu.int/comm/food/fs/sc/index_en.html (1/8/2000).
33. Regulation (EC) No. 999/2001 of the European Parliament and of theCouncil of 22 May 2001 laying down rules for the prevention, control anderadication of certain transmissible spongiform encephalopathies, OfficialJournal L 147, 31/5/2001, 1–40.
34. Commission Green Paper: The General Principles of Food Law in theEuropean Union (COM(97) 176 final).
Notes 203
PPL-UK_RR-Krapohl_Notes.qxd 9/12/2008 10:12 AM Page 203

35. James, P., F. Kemper and G. Pascal (1999). ‘A European Food and PublicHealth Authority: The future of scientific advice in the EU’ (report presentedto the European Commission).
36. Commission of the European Communities: ‘White Paper on Food Safety’(COM(1999) 719 final; 12/1/2000).
37. Commission Proposal for a Regulation of the European Parliament and ofthe Council laying down the general principles and requirements of foodlaw, establishing the European Food Authority and laying down proceduresin matters of food (COM(2000) 716 final; 8/11/2000).
38. European Parliament legislative resolution on the proposal for a EuropeanParliament and Council regulation laying down the general principles andrequirements of food law, establishing the European Food Authority and lay-ing down procedures in matter of food safety (A5–0198/2001).
39. See the homepage of EFSA: www.efsa.europa.eu/en/science.html.40. In 1999, the comitology system was reformed and simplified by Council
Decision 1999/468/EC of 28 June 1999 laying down the procedures for the exercise of implementing powers conferred on the Commission, OfficialJournal L 184, 17/7/1999, 23–6. Within the reformed comitology system,which applies for all new delegation acts after 1999, the distinctions betweendifferent management procedures (IIa and IIb), different regulatory proce-dures (IIIa and IIIb) and different safeguard procedures (IVa and IVb) areabolished. Now, there exists only one advisory, one management, one regu-latory and one safeguard procedure.
41. Council Directive 90/220/EEC of 23 April 1990 on the deliberate release intothe environment of genetically modified organisms, Official Journal L 117,8/5/1990, 15–27.
42. Regulation (EC) No. 258/97 of the European Parliament and of the Councilof 27 January 1997 concerning novel foods and food ingredients, OfficialJournal L 43, 14/2/1997, 1–6.
43. Other novel foods are foods with a modified molecular structure, food con-sisting of microorganisms, food derived from plants or animals that are notproduced by traditional farming or breeding practices, or food where a newproduction process changes its composition or structure.
44. 97/618/EC: Commission Recommendation of 29 July 1997 concerning thescientific aspects and the presentation of information necessary to supportapplications for the placing on the market of novel foods and food ingredi-ents and the preparation of initial assessment reports under Regulation (EC)No. 258/07 of the European Parliament and the Council, Official Journal L 253, 16/9/1997, 1–36.
45. Judgement of the Court of 9 September 2003: Monsanto Agricoltura ItaliaSpA and Others v Presidenza del Consiglio die Ministri and Others, case C-236/01, ECR 2003: I-8105.
46. The numbers are published on homepage of the DG Health and ConsumerProtection of the Commission (http://europa.eu.int/comm/food/food/biotechnology/index_en.htm; 14/10/2005).
47. Judgement of the Court of 21 May 2000: Association Greenpeace France andOthers v Ministère de l’Agriculture et de la Pêche and Others, case C-6/99,ECR 2000: I-1651.
204 Notes
PPL-UK_RR-Krapohl_Notes.qxd 9/12/2008 10:12 AM Page 204

48. Council of Ministers, 2194th Council Meeting – Environment – Luxembourg,24–25 June 1999, Press 203 – No. 9406/99.
49. World Trade Organization (2006): ‘Dispute Settlement: Dispute DS291’(http://www.wto.org/english/ tratop_e/dispu_e/cases_e/ds291_e.htm, 5.2.2006).
50. Communication from the Commission: ‘Towards a Strategic Vision of LifeSciences and Biotechnology: Consultation Document’ (COM(2001) 454final, 4/9/2001), and Communication from the Commission to the Council,the European Parliament, the Economic and Social Committee and theCommittee of the Regions: ‘Life Sciences and biotechnology – A Strategy forEurope’, (Com(2002) 27, 23/1/2002).
51. Directive 2001/18/EC of the European Parliament and of the Council of 12March 2001 on the deliberate release into the environment of geneticallymodified organisms and repealing Council Directive 90/220/EEC, OfficialJournal L 106, 17/4/2001, 1–29.
52. Regulation (EC) No. 1829/2003 of the European Parliament and of theCouncil of 22 September 2003 on genetically modified food and feed,Official Journal L 268, 18/10/2003, 1–23.
53. The cases are published on the homepage of the DG Health and ConsumerProtection of the Commission (http://europa.eu.int/comm/food/food/biotechnology/index_en.htm; 14/10/2005).
7 A Weak Supranational Agency: The Evaluation of theEuropean Regulatory Regime for Foodstuffs
1. 74/234/EEC: Commission Decision of 16 April 1974 relating to the institu-tion of a Scientific Committee for Food,Official Journal L 136, 20/5/1974,1–2.
2. 69/414/EEC: Council Decision of 13 November 1969 setting up a StandingCommittee for Foodstuffs,Official Journal L 291, 19/11/1969, 9–10.
3. 87/373/EEC: Council Decision of 13 July 1987 laying down the procedures forthe exercise of implementing powers conferred on the Commission,OfficialJournal L 197, 18/7/1987, 33–5.
4. European Parliament (1997): ‘BSE Inquiry Report: Report on alleged contra-ventions or maladministration in the implementation of Community law inrelation to BSE, without prejudice to the jurisdiction of the Community andnational courts’ (A4–0020/97), I.C.5.
5. Judgement of the Court of 5 May 1998: United Kingdom of Great Britain and Northern Ireland v Commission of the European Communities, case C-180/96, ECR 1998: I-2265, Order of the Court of 21 June 2000: FrenchRepublic v Commission of the European Communities, case C-514/99, ECR2000: I-4705, and Judgement of the Court of 22 October 2002: NationalFarmers’ Union v Secrétariat general du government, Reference for a prelim-inary ruling: Conseil d’Etat – France, case C-241/00, ECR 2002: I-9079.
6. Judgement of the Court of First Instance (Fifth Chamber) of 30 September1998: Confederazione Nazionale Coltivatori Diretti (Coldiretti) and 110farmers v Council of the European Union and Commission of the EuropeanCommunities, case T-149/96, ECR 1998: II-3841.
Notes 205
PPL-UK_RR-Krapohl_Notes.qxd 9/12/2008 10:12 AM Page 205

7. FPA (Frederic Paeps Advice), Market and Management Advice (2004):‘Assessment of the Current Image of the European Food Safety Authority:Interviews with Interested Parties and Stakeholders, March–April 2004’, http://www.efsa.europa.eu/mboard/mb_meetings/479/image_mb15_doc4_annex1_en1.pdf, 3/7/2006.
8. Bureau van Dijk Ingénieurs Conseils with Arcadia International EEIG (2005):‘Evaluation of EFSA: Final Report’, http://www.efsa.europa.eu/mboard/122/final_report_evaluation1.pdf, 3/7/2006.
9. The only exception is the authorisation procedure for GM food, which isalways started by applying companies (section 7.1.3).
10. The only exception is again the authorisation of GM food, where final autho-risation decisions are directly addressed to applying companies (section 7.1.3).
11. FPA (Frederic Paeps Advice), Market and Management Advice (2004):‘Assessment of the Current Image of the European Food Safety Authority:Interviews with Interested Parties and Stakeholders, March–April 2004’, http://www.efsa.europa.eu/mboard/mb_meetings/479/image_mb15_doc4_annex1_en1.pdf, 3/7/2006; Bureau van Dijk Ingénieurs Conseils with ArcadiaInternational EEIG (2005): ‘Evaluation of EFSA: Final Report’, http://www.efsa.europa.eu/mboard/122/final_report_evaluation1.pdf, 3/7/2006.
12. Regulation (EC) No 1829/2003 of the European Parliament and of theCouncil of 22 September 2003 on genetically modified food and feed,OfficialJournal L 268, 18/10/2003, 1–23.
13. Friends of the Earth (2004): ‘Throwing Caution to the Wind: A Review of theEuropean Food Safety Authority and its Work on Genetically Modified Foodsand Crops’ (http://www.foeeurope.org/GMOs/publications/EFSAreport.pdf).
14. Bureau van Dijk Ingénieurs Conseils with Arcadia International EEIG (2005):‘Evaluation of EFSA: Final Report’, http://www.efsa.europa.eu/mboard/122/final_report_evaluation1.pdf, 3/7/2006.
15. The cases are published on the Homepage of the DG Health and ConsumerProtection of the Commission (http://europa.eu.int/comm/food/food/biotechnology/index_en.htm; 14/10/2005).
16. EFSA (2005): ‘Guidance Document of the Scientific Panel on GeneticallyModified Organisms for the Risk Assessment of Genetically Modified Plantsand Derived Food and Feed’ (http://www.efsa.europa.eu/etc/medialib/efsa/science/gmo/gmo_guidance/660.Par.0004.File.dat/guidance_docfinal1.pdf;27/9/2006).
17. Judgement of the Court of 21 May 2000: Association Greenpeace France andOthers v Ministère de l’Agriculture et de la Pêche and Others, case C-6/99,ECR 2000: I-1651.
18. Judgement of the Court of First Instance of 5 October 2005: LandOberösterreich and Republic of Austria v Commission of the EuropeanCommunities, cases T-366/03 and T-235/04, ECR 2004: 00000.
19. Council Decision 1999/468/EC of 28 June 1999 laying down the proceduresfor the exercise of implementing powers conferred on the Commission,Official Journal L 184, 17/7/1999, 23–6.
20. European Parliament (1997): ‘BSE Inquiry Report: Report on alleged contra-ventions or maladministration in the implementation of Community law inrelation to BSE, without prejudice to the jurisdiction of the Community andnational courts’ (A4–0020/97).
206 Notes
PPL-UK_RR-Krapohl_Notes.qxd 9/12/2008 10:12 AM Page 206

21. Information about the scientific phase of decision-making can be found onthe website of EFSA (http://www.efsa.europa.eu/en.html, 7/10/06), whereasinformation about the political phase are published by the Commission DGHealth and Consumer protection (http://ec.europa.eu/food/index_en.htm,7/10/06).
22. Information about stakeholder consultations of EFSA can be obtained formthe website: http://www.efsa.europa.eu/en/stakeholder_stakeholder.html,5/10/06.
23. 2004/613/EC: Commission Decision of 6 August 2004 concerning the cre-ation of an advisory group on the food chain and animal and plant health,Official Journal L 275, 25/8/2004, 17–19.
Notes 207
PPL-UK_RR-Krapohl_Notes.qxd 9/12/2008 10:12 AM Page 207

208
Bibliography
Abbott, K. W. and D. Snidal (2000): ‘Hard and Soft Law in InternationalGovernance’, in: International Organisation, 54:3, 421–56.
Abbott, K. W., R. O. Keohane, A. Moravcsik, A.-M. Slaughter and D. Snidal (2000):‘The Concept of Legalization’, in: International Organization, 54:3, 401–19.
Abraham, J. (1995): Science, Politics and the Pharmaceutical Industry (New York: St. Martin’s Press).
Abraham, J. and G. Lewis (2000): Regulating Medicines in Europe: Competition,Expertise and Public Health (London: Routledge).
Abraham, J. and G. Lewis (2002): ‘Citizenship, Medical Expertise and theCapitalist Regulatory State in Europe’, in: Sociology, 36:1, 67–88.
Akerlof, G. A. (1970): ‘The Market for “Lemons”: Quality Uncertainty and theMarket Mechanism’, in: The Quarterly Journal of Economics, 84:3, 488–500.
Alemanno, A. (2006): ‘Food Safety and the Single European Market’, in: C. Anselland D. Vogel (eds): What’s the Beef? The Contested Governance of European FoodSafety (Cambridge: MIT Press), 237–58.
Alter, K. J. (2001): Establishing the Supremacy of European Law: The Making of anInternational Rule of Law in Europe (Oxford: University Press).
Alter, K. J. and S. Meunier-Aitsahalia (1994): ‘Judicial Politics in the EuropeanCommunity: European Integration and the Pathbreaking Cassis de DijonDecision’, in: Comparative Political Studies, 26:4, 535–61.
Anania, G. and R. Nisticò (2004): ‘Public Regulation as a Substitute for Trust inQuality Food Markets: What if the Trust Substitute cannot be Fully Trusted?’,in: Journal of Institutional and Theoretical Economics, 160, 681–701.
Ansell, C. and D. Vogel (2006) (eds): What’s the Beef? The Contested Governance ofEuropean Food Safety (Cambridge: MIT Press).
Ansell, C., R. Maxwell and D. Sicurelli (2006): ‘Protesting Food: NGOs andPolitical Mobilization in Europe’, in: C. Ansell and D. Vogel (eds): What’s theBeef? The Contested Governance of European Food Safety (Cambridge: MIT Press),97–122.
Arthur, W. B. (1994): Increasing Returns and Path Dependence in the Economy(Michigan: University Press).
Axelrod, R. (1984): The Evolution of Cooperation (New York: Basic Books).Batz, K. (1986): ‘Die Zulassungsvoraussetzungen für Arzneimittel’ (Gießen:
Dissertation).Bernauer, T. (2003): Genes, Trade, and Regulation: The Seeds of Conflict in Food
Biotechnology (Princeton: University Press).Bernauer, T. and L. Caduff (2006): ‘Food Safety and the Structure of the European
Food Industry’, in: C. Ansell and D. Vogel (eds): What’s the Beef? The ContestedGovernance of European Food Safety (Cambridge: MIT Press), 81–96.
Beyer, C. (1989): Die Grenzen der Arzneimittelhaftung dargestellt am Beispiel desContergan-Falles (München: Verlag von Florentz).
Borraz, O., J. Besançon and C. Clergeau (2006): ‘Is it just about Trust? The PartialReform of French Food Safety Regulation’, in: C. Ansell and D. Vogel (eds): What’s
PPL-UK_RR-Krapohl_Biblio.qxd 9/12/2008 10:00 AM Page 208

the Beef? The Contested Governance of European Food Safety (Cambridge: MIT Press),125–52.
Bradley, K. S. C. (1992): ‘The European Parliament and Comitology: On the Roadto Nowhere?’, in: European Law Journal, 3:3, 230–54.
Bradley, K. S. C. (1999): ‘Institutional Aspects of Comitology: Scenes from theCutting Room Floor’, in: C. Joerges and E. Vos (eds): EU Committees: SocialRegulation, Law and Politics (Oxford: Hart Publishing), 71–94.
Bredahl, L. (2001): ‘Determinants of Consumer Attitudes and Purchase Intentionswith Regard to Genetically Modified Foods: Results of a Cross-National Survey’,in: Journal of Consumer Policy, 24, 23–61.
Brennan, G. and J. Buchanan (1985): The Reasons of Rules: Constitutional PoliticalEconomy (Cambridge: University Press).
Breyer, S. (1993): Breaking the Vicious Circle: Towards Effective Risk Regulation(Cambridge: University Press).
Broscheid, A. and J. Feick (2005): ‘Towards a European FDA? The Review ofEuropean Pharmaceuticals Authorization’ (paper prepared for the 2005 Meetingof the European Union Studies Association, March 21 to April 2 in Austin,Texas).
Bulmer, S. J. (1998): ‘New Institutionalism and the Governance of the SingleEuropean Market’, in: Journal of European Public Policy, 5:3, 365–86.
Buonanno, L. (2006): ‘The Creation of the European Food Safety Authority’, in:C. Ansell and D. Vogel (eds): What’s the Beef ? The Contested Governance ofEuropean Food Safety (Cambridge: MIT Press), 259–78.
Burnett, H. S. (2000): ‘Progress at Risk: Using the Precautionary Principle as aStandard for Regulatory Policy’, in: L. Jones (ed.): Safe Enough? Managing Riskand Regulation (Vancouver: The Fraser Institute), 155–64.
Calvert, R. L., M. D. McCubbins and B. R. Weingast (1989): ‘A Theory of PoliticalControl and Agency Discretion’, in: American Journal of Political Science, 33:3,588–611.
Chalmers, D. (2003): ‘Food for Thought: Reconciling European Risks andTraditional Ways of Life’, in: Modern Law Review, 66:4, 532–64.
Chalus, T. and I. Peutz (2000): ‘BSE: The European Regulatory Context’, in:Eurosurveillance Monthly, 5:10, 107–22.
Chambers, G. R. (1999): ‘The BSE Crisis and the European Parliament’, in: C. Joerges and E. Vos (eds): EU Committees: Social Regulation, Law and Politics(Oxford: Hart Publishing), 95–106.
Chiti, E. (2000): ‘The Emergence of a Community Administration: The Case ofEuropean Agencies’, in: Common Market Law Review, 37, 309–43.
Chiti, E. (2004): ‘Decentralisation and Integration into CommunityAdministrations: A New Perspective on European Agencies’, in: European LawJournal, 10:4, 402–38.
Christoforou, T. (2004): ‘The Regulation of Genetically Modified Organisms inthe EU: The Interplay of Science, Law and Politics’, in: Common Market LawReview, 41, 637–709.
Collatz, B. (1996): Die neuen europäischen Zulassungsverfahren für Arzneimittel:Insbesondere Verfahren und Rechtsschutz des Antragstellers und Zulassungsinhabersbei Zulassungsentscheidungen (Aulendorf: Editio Cantor Verlag).
Crombez, C. (2003): ‘The Democratic Deficit in the European Union: Much Adoabout Nothing?’, in: European Union Politics, 4:1, 101–20.
Bibliography 209
PPL-UK_RR-Krapohl_Biblio.qxd 9/12/2008 10:00 AM Page 209

Dabrowska, P. (2004): ‘GM Foods, Risk, Precaution and the Internal Market: DidBoth Sides Win the Day in the recent Judgement of the European Court ofJustice?’, in: German Law Journal, 5:2.
Daemmrich, A. A. (2002): ‘A Tale of Two Experts: Thalidomide and PoliticalEngagement in the United States and West Germany’, in: Social History ofMedicine, 15:1, 137–58.
Daemmrich, A. A. (2004): Pharmacopolitics: Drug Regulation in the United States andGermany (Chapel Hill: The University of North Carolina Press).
De Grauwe, P. (1997): ‘The Design of a European Central Bank’, in: The Economicsof Monetary Integration in Europe (Oxford: Hart Publishing), 3rd edition.
Dehousse, R. (1997): ‘Regulation by Networks in the European Community: TheRole of European Agencies’, in: Journal of European Public Policy, 4:2, 246–61.
Dehousse, R. (1998): The European Court of Justice (London: Macmillan).Dehousse, R. (1999): ‘Towards a Regulation of Transnational Governance?
Citizen’s Rights and the Reform of Comitology Procedures’, in: C. Joerges andE. Vos (eds): EU Committees: Social Regulation, Law and Politics (Oxford: HartPublishing), 109–28.
Douglas, M. and A. Wildavsky (1982): Risk and Culture: An Essay on the Selectionof Technical and Environmental Dangers (Berkeley: University Press).
Dressel, K. (2002): BSE – The New Dimension of Uncertainty: The Cultural Politics ofScience and Decision-Making (Berlin: Sigma).
Dunleavy, P. (1991): Democracy, Bureaucracy and Public Choice: EconomicExplanations in Political Science (London: Harvester).
Eberlein, B. and D. Kerwer (2004): ‘New Governance in the European Union: ATheoretical Perspective’, in: Journal of Common Market Studies, 42:1, 121–42.
Eberlein, B. and E. Grande (2005): ‘Beyond Delegation: Transnational RegulatoryRegimes and the EU Regulatory State’, in: Journal of European Public Policy, 12:1,89–112.
Eckley, N. and H. Selin (2004): ‘All Talk, Little Action: Precaution and EuropeanChemicals Regulation’, in: Journal of European Public Policy, 11:1, 78–105.
Egan, M. (1998): ‘Regulatory Strategies, Delegation and European MarketIntegration’, in: Journal of European Public Policy, 5:3, 485–506.
Elster, J. (1979): Ulysses and the Sirens: Studies in Rationality and Irrationality(Cambridge: University Press).
Everson, M. (1995): ‘Independent Agencies: Hierarchy Beaters?’, in: European LawJournal, 1:2, 180–204.
Everson, M., G. Majone, L. Metcalfe and A. Schout (1999): ‘The Role of SpecialisedAgencies in Decentralising EU Governance’ (Florence: Report Presented to theCommission).
Falke, J. (2000): ‘Komitologie – Entwicklung, Rechtsgrundlagen und erste empirischeAnnäherung’, in: C. Joerges and J. Falke (eds): Das Ausschusswesen derEuropäischen Union: Praxis der Risikoregulierung im Binnenmarkt und ihre rechtlicheVerfassung (Baden-Baden: Nomos), 43–160.
Feick, J. (2000a): ‘Wissen, Expertise und regulative Politik: Das Beispiel derArzneimittelkontrolle’, in: U. Schimank and R. Werle (eds): GesellschaftlicheKomplexität und kollektive Handlungsfähigkeit (Frankfurt: Campus Verlag),208–38.
Feick, J. (2000b): ‘Marktzugangsregulierung: Nationale Regulierung, interna-tionale Harmonisierung und europäische Integration’, in: R. Czada and S. Lütz
210 Bibliography
PPL-UK_RR-Krapohl_Biblio.qxd 9/12/2008 10:00 AM Page 210

(eds): Die politische Konstitution von Märkten (Opladen: Westdeutscher Verlag),228–49.
Feick, J. (2002a): ‘Der Interessensbezug der europäischen Arzneimittelzulassung’(Cologne: unpublished working paper).
Feick, J. (2002b): ‘Regulatory Europeanization, National Autonomy and RegulatoryEffectiveness: Marketing Authorization for Pharmaceuticals’ (Cologne: Max-Planck-Institute for the Study of Societies, Discussion Paper 02/6).
Feick, J. (2003): ‘Regulatory Rationalisation and Legitimation in the Face ofInterests, Influence and Institutional De-Politicisation: Market Entry Regulationfor Pharmaceuticals in the EU’ (Paper Prepared for the Workshop ‘GoodGovernance in Supranational Market Regulation: How do RegulatoryInstitutions Matter?’ from 16 to 17 January 2004 in Bamberg).
Feick, J. (2005): ‘Learning and Interest Accommodation in Policy and InstitutionalChange: EC Risk Regulation in the Pharmaceutical Sector’ (London: DiscussionPaper No. 25 of the ESCR Centre for Analysis of Risk and Regulation).
Fleischer, J. (2005): ‘European Agencies as Engines of Regulation? On DifferentArchitectural Strategies of the Regulatory State’ (Paper prepared for the 3rdECPR Annual Conference, 8–10 September 2005 in Budapest).
Fligstein, N. and I. Mara-Drita (1996): ‘How to Make a Market: Reflections on theAttempt to Create a Single Market in the European Union’, in: American Journalof Sociology, 102:1, 1–33.
Follesdal, A. and S. Hix (2006): ‘Why there is a Democratic Deficit in the EU: AResponse to Majone and Moravcsik’, in: Journal of Common Market Studies, 44:3,533–62.
Franchino, F. (2000a): ‘Control of the Commission’s Executive Functions:Uncertainty, Conflict and Decision Rules’, in: European Union Politics, 1:1, 59–88.
Franchino, F. (2000b): ‘The Commission’s Executive Discretion, Information andComitology’, in: Journal of Theoretical Politics, 12, 155–81.
Franchino, F. (2002): ‘Efficiency or Credibility? Testing the Two Logics ofDelegation to the European Commission’, in: Journal of European Public Policy,9:5, 677–94.
Francis, J. (1993): The Politics of Regulation: A Comparative Perspective (Oxford:Blackwell).
Fuchs, L. O. (2004): Lebensmittelsicherheit in der Mehrebenenverwaltung derEuropäischen Gemeinschaft: Eine Analyse am Beispiel des deutschen und europäis-chen Lebensmittelrechts unter besonderer Betrachtung der Europäischen Behörde fürLebensmittelsicherheit (Bayreuth: Verlag P.C.O).
Gardner, J. S. (1996): ‘The European Agency for the Evaluation of MedicinalProducts and European Regulation of Pharmaceuticals’, in: European LawJournal, 2:1, 48–82.
Garrett, G. and B. R. Weingast (1993): ‘Ideas, Interests and Institutions:Constructing the European Community’s Internal Market’, in: J. Goldstein andR. O. Keohane (eds): Ideas and Foreign Policy: Beliefs, Institutions, and PoliticalChange (Ithaca: Cornell University Press).
Gaskell, G. and N. Allum (2001): ‘Two Cultures of Risk’ (Working Paper of the Centre for the Analysis of Risk and Regulation at the London School ofEconomics).
Gehring, T. (1999): ‘Bargaining, Arguing and Functional Differentiation ofDecision-Making: The Role of Committees in European Environmental Process
Bibliography 211
PPL-UK_RR-Krapohl_Biblio.qxd 9/12/2008 10:00 AM Page 211

Regulation’, in: C. Joerges and E. Voss (eds): EU Committees: Social Regulation,Law and Politics (Oxford: Hart Publishing), 195–218.
Gehring, T. (2000): ‘Die Bedeutung spezialisierter Entscheidungsprozesse für dieProblemlösungsfähigkeit der Europäischen Union’, in: E. Grande and M.Jachtenfuchs (eds): Wie problemlösungsfähig ist die EU? Regieren im europäischenMehrebenensystem (Baden-Baden: Nomos), 77–112.
Gehring, T. (2002): Die Europäische Union als komplexe internationale Organisation.Wie durch Kommunikation und Entscheidung soziale Ordnung entsteht (Baden-Baden:Nomos).
Gehring, T. (2003): ‘Communicative Rationality in European Governance?Interests and Communicative Action in Functionally Differentiated SingleMarket Regulation’, in: E. O. Erikson, C. Joerges and J. Neyer (eds): EuropeanGovernance, Deliberation and the Quest for Democratisation (Oslo: Arena ReportNo 02/2003), 57–140.
Gehring, T. (2005): ‘Gesellschaftliche Rationalität durch die Differenzierung von Entscheidungsverfahren’, in: T. Gehring, S. Krapohl, M. Kerler and S.Stefanova (eds): Rationalität durch Verfahren in der Europäischen Union:Europäische Arzneimittelzulassung und Normung technischer Güter (Baden-Baden:Nomos), 27–62.
Gehring, T., M. Kerler and S. Krapohl (2007): ‘Risikoregulierung im Europäischen Binnenmarkt: Normungsinstitute, Komitologieausschüsse undRegulierungsinstitute’, in: I. Tömmel (ed.): Die Europäische Union: Governanceund Policy-Making, in: Politische Vierteljahresschrift, Special Edition 2007/2(forthcoming).
Gehring, T. and S. Krapohl (2007): ‘Supranational Regulatory Agencies betweenIndependence and Control: The EMEA and the Authorisation of Pharmaceuticalsin the European Single Market’, in: Journal of European Public Policy, 14:2, 208–26.
Gehring, T., S. Krapohl, M. Kerler and S. Stefanova (2005): Rationalität durchVerfahren in der Europäischen Union: Europäische Arzneimittelzulassung undNormung technischer Güter (Baden-Baden: Nomos).
Geradin, D. and N. Petit (2004): ‘The Development of Agencies at EU andNational Levels: Conceptual Analysis and Proposals for Reform’ ( Jean MonnetWorking Paper 01/04).
Gilardi, F. (2002): ‘Policy Credibility and Delegation to Independent RegulatoryAgencies: A Comparative Empirical Analysis’, in: Journal of European PublicPolicy, 9:6, 873–93.
Gilardi, F. (2004): ‘Delegation in the Regulatory State: Origins and Diffusion ofIndependent Regulatory Agencies in Western Europe’ (PhD Thesis at theUniversity of Lausanne, unpublished).
Glaeske, G., D. Hart and H. Merkel (1988): ‘Regulierung des europäischenArzneimittelmarktes durch nationales und europäisches Zulassungs- undNachmarktkontrollrecht’, in: N. Reich (ed.): Die Europäisierung desArzneimittelmarktes: Chancen und Risiken (Baden-Baden: Nomos), 13–23.
Gollier, C. and N. Treich (2003): ‘Decision-Making under Scientific Uncertainty:The Economics of the Precautionary Principle’, in: The Journal of Risk andUncertainty, 27:1, 77–103.
Haas, E. (1958): The Uniting of Europe (Stanford: University Press).Hancher, L. (1990): Regulating for Competition: Government, Law, and the
Pharmaceutical Industry in the United Kingdom and France (Oxford: Clarendon Press).
212 Bibliography
PPL-UK_RR-Krapohl_Biblio.qxd 9/12/2008 10:00 AM Page 212

Hancher, L. (2004): ‘The European Community Dimension: CoordinatingDivergence’, in: E. Mossialos, M. Mrazek and T. Walley (eds): RegulatingPharmaceuticals in Europe: Striving for Efficiency, Equity and Quality (Maidenhead:Open University Press), 55–79.
Hart, D., A. Hilken, H. Merkel and O. Woggan (1988): Das Recht desArzneimittelmarktes (Baden-Baden: Nomos).
Hart, D. and N. Reich (1990): Integration und Recht des Arzneimittelmarktes in derEG: Eine Untersuchung zum Produkt- und Marktrecht der Gemeinschaft und aus-gewählter Mitgliedstaaten (Baden-Baden: Nomos).
Heertje, A. and H.-D. Wenzel (1997): Grundlagen der Volkswirtschaftslehre (Berlin:Springer), 5th edition.
Heilmann, K. (2002): ‘Risiko und Sicherheit’, in: Deutsche Apotheker Zeitung,142:11, 85–93.
Herbig, P., J. Milewicz and J. Golden (1994): ‘A Model of Reputation Building andDestruction’, in: Journal of Business Research, 31, 23–31.
Héritier, A. (1996): ‘The Accommodation of Diversity in European Policy Makingand Its Outcomes: Regulatory Policy as a Patchwork’ (EUI Working Paper SPSNo. 96/2).
Héritier, A. (2003): ‘Composite Democracy in Europe: The Role of Transparencyand Access to Information’, in: Journal of European Public Policy, 10:5, 814–33.
Hill, C. W. L. (1990): ‘Cooperation, Opportunism, and the Invisible Hand:Implications for Transaction Cost Theory’, in: Academy of Management Review,15:3, 500–13.
Hood, C., H. Rothstein and R. Baldwin (2001): The Government of Risk:Understanding Risk Regulation Regimes (Oxford: University Press).
Jachtenfuchs, M. (1999): ‘Die Zukunft der Demokratie im Rahmen derEuropäischen Union’, in: M. Kaase and G. Schmid (eds): Eine lernendeDemokratie: 50 Jahre Bundesrepublik Deutschland, WZB-Jahrbuch 1999 (Berlin:edition sigma).
Joerges, C. (2000): ‘Zusammenfassung und Perspektiven: “Gutes Regieren” imBinnenmarkt’, in: C. Joerges and J. Falke (eds): Das Ausschusswesen derEuropäischen Union: Praxis der Risikoregulierung im Binnenmarkt und ihre rechtlicheVerfassung (Baden-Baden: Nomos), 349–82.
Joerges, C. (2001): ‘Law, Science and the Management of Risks to Health at theNational, European and International Level – Stories on Baby Dummies, MadCows and Hormones in Beef’, in: Columbia Journal of European Law, 7, 1–9.
Joerges, C. and E. Vos (1999) (eds): EU Committees: Social Regulation, Law andPolitics (Oxford: Hart Publishing).
Joerges, C. and J. Falke (2000) (eds): Das Ausschusswesen der Europäischen Union:Praxis der Risikoregulierung im Binnenmarkt und ihre rechtliche Verfassung (Baden-Baden: Nomos).
Joerges, C. and J. Neyer (1997a): ‘From Intergovernmental Bargaining toDeliberative Political Processes: The Constitutionalisation of Comitology’, in:European Law Journal, 3:3, 273–99.
Joerges, C. and J. Neyer (1997b): ‘Transforming Strategic Interaction intoDeliberative Problem-Solving: European Comitology in the Foodstuffs Sector’,in: Journal of European Public Policy, 4:4, 1350–763.
Joerges, C. and J. Neyer (2006): ‘“Deliberative Supranationalism” Revisited’ (EUIWorking Paper Law No. 2006/20).
Bibliography 213
PPL-UK_RR-Krapohl_Biblio.qxd 9/12/2008 10:00 AM Page 213

Jüni, P., L. Nartey, S. Reichenbach, R. Sterchi, P. A. Dieppe and M. Egger (2004):‘Risk of Cardiovascular Events and Rofecoxib: Cumulative Meta-Analysis’, in:The Lancet, 364, 2021–9.
Kelemen, D. R. (2002): ‘The Politics of “Eurocratic” Structure and the NewEuropean Agencies’, in: West European Politics, 25:4, 93–118.
Kerler, M. (2005): ‘Der neue Ansatz zur technischen Harmonisierung’, in: T. Gehring, S. Krapohl, M. Kerler and S. Stefanova (eds): Rationalität durchVerfahren in der Europäischen Union: Europäische Arzneimittelzulassung undNormung technischer Güter (Baden-Baden: Nomos), 197–222.
Kingdon, J. W. (1995): Agendas, Alternatives, and Public Policies (New York:Longman), 2nd edition.
Kirk, B. (1999): Der Contergan-Fall: Eine unvermeidbare Arzneimittelkatastrophe? Zur Geschichte des Arzneistoffs Thalidomid (Stuttgart: WissenschaftlicheVerlagsgesellschaft).
Knightley, P., H. Evans, E. Potter and M. Wallace (1979): Suffer the Children: TheStory of Thalidomide (London: Andre Deutsch).
Knipschild, K. (2003): Lebensmittelsicherheit als Aufgabe des Veterinär- undLebensmittelrechts: Risikoverwaltung im Europäischen Binnenmarkt (Baden-Baden:Nomos).
Kotzian, P. (2003): Verhandlungen im europäischen Arzneimittelsektor: Initiierung –Institutionalisierung – Ergebnisse (Baden-Baden: Nomos).
Krapohl, S. (2003): ‘Risk Regulation in the EU between Interests and Expertise:The Case of BSE’, in: Journal of European Public Policy, 10:2, 189–207.
Krapohl, S. (2004): ‘Credible Commitment in Non-Independent RegulatoryAgencies: A Comparative Analysis of the European Agencies for Foodstuffs andPharmaceuticals’, in: European Law Journal, 10:5, 518–38.
Krapohl, S. (2005): ‘Die europäische Arzneimittelzulassung’, in: T. Gehring, S.Krapohl, M. Kerler and S. Stefanova (eds): Rationalität durch Verfahren in derEuropäischen Union: Europäische Arzneimittelzulassung und Normung technischerGüter (Baden-Baden: Nomos), 81–196.
Krapohl, S. (2007): ‘Thalidomide, BSE and the Single Market: A Historical-Institutionalist Approach to Regulatory Regimes in the European Union’, in:European Journal of Political Research, 46:1, 25–46.
Krapohl, S. and K. Zurek (2006): ‘The Perils of Committee Governance:Intergovernmental Bargaining during the BSE Scandal in the European Union’,in: European Integration Online Papers, 10:2.
Kreher, A. (1997): ‘Agencies in the European Community: A Step towardsAdministrative Integration in Europe’, in: Journal of European Public Policy, 4:2,225–45.
Kreps, D. M. and R. Wilson (1982): ‘Reputation and Imperfect Information’, in:Journal of Economic Theory, 27, 253–79.
Krücken, G. (1997): Risikotransformation: Die politische Regulierung technisch-ökolo-gischer Gefahren in der Risikogesellschaft (Opladen: Westdeutscher Verlag).
Kydland, F. E. and E. C. Prescott (1977): ‘Rules Rather than Discretion: TheInconsistency of Optimal Plans’, in: Journal of Political Economy, 85:3, 137–60.
Lax, D. A. and J. K. Sebenius (1986): The Manager as Negotiator: Bargaining forCooperation and Competitive Gain (New York: Free Press).
Lewis, G. and J. Abraham (2001): ‘The Creation of Neo-Liberal Corporate Bias inTransnational Medicines Control: The Industrial Shaping and Interest Dynamics
214 Bibliography
PPL-UK_RR-Krapohl_Biblio.qxd 9/12/2008 10:00 AM Page 214

of the European Regulatory State’, in: European Journal of Political Research, 39:1,53–80.
Lindner, J. and B. Rittberger (2003): ‘The Creation, Interpretation andContestation of Institutions: Revisiting Historical Institutionalism’, in: Journalof Common Market Studies, 41:3, 445–73.
Mahoney, J. (2000): ‘Path Dependence in Historical Sociology’, in: Theory andSociety, 29, 507–48.
Majone, G. (1994): ‘The European Community: An “Independent Fourth Branchof Government”?’, in: G. Brüggemeier (ed.): Verfassungen für ein ziviles Europa(Baden-Baden: Nomos), 23–43.
Majone, G. (1996): Regulating Europe (London: Routledge).Majone, G. (1997): ‘The New European Agencies: Regulation by Information’, in:
Journal of European Public Policy, 4:2, 262–75.Majone, G. (2000): ‘The Credibility Crisis of Community Regulation’, in: Journal
of Common Market Studies, 38:2, 273–302.Majone, G. (2001a): ‘Two Logics of Delegation: Agency and Fiduciary Relations
in EU Governance’, in: European Union Politics, 2:1, 103–22.Majone, G. (2001b): ‘Nonmajoritarian Institutions and the Limits of Democratic
Governance: A Political Transaction-Cost Approach’, in: Journal of Institutionaland Theoretical Economics, 157, 57–78.
Majone, G. (2002a): ‘Delegation of Regulatory Powers in a Mixed Polity’, in:European Law Journal, 8:3, 319–39.
Majone, G. (2002b): ‘What Price Safety? The Precautionary Principle and itsPolicy Implications’, in: Journal of Common Market Studies, 40:1, 89–109.
Majone, G. (2004): ‘European Regulatory Agencies: The Dilemma of Delegation ofPowers in the European Union’ (Working paper presented at the workshop‘Good Governance in Single Market Regulation’, Bamberg, 16–17 January 2004.
Majone, G. (2005): Dilemmas of European Integration: The Ambiguities and Pitfallsof Integration by Stealth (Oxford: University Press).
Majone, G. and M. Everson (2001): ‘Institutional Reform: Independent Agencies,Oversight, Coordination and Procedural Control’, in: O. de Schutter, M. Lebessisand J. Paterson (eds): Governance in the European Union (Luxembourg: Office forOfficial Publications of the European Communities), 129–68.
McCubbins, M. D. and T. Schwartz (1987): ‘Congressional Oversight Overlooked:Police Patrol versus Fire Alarms’, in: American Journal of Political Science, 28:1,165–79.
Menon, A. and S. Weatherill (2002): ‘Legitimacy, Accountability, and Delegationin the European Union’, in: A. Arnull and D. Wincott (eds): Accountability andLegitimacy in the European Union (Oxford: University Press), 113–32.
Millstone, E. and P. van Zwanenberg (2001): ‘Politics of Expert Advice: Lessonsfrom the Early History of the BSE Saga’, in: Science and Public Policy, 28:2,99–112.
Moe, T. M. (1987a): ‘Interests, Institutions and Positive Theory: The Politics ofthe NLRN’, in: Studies in American Development, 2, 236–58.
Moe, T. M. (1987b): ‘An Assessment of the Positive Theory of CongressionalDominance’, in: Legislative Studies Quarterly, 12, 475–520.
Moe, T. M. (1990): ‘The Politics of Structural Choice: Toward a Theory of PublicBureaucarcy’, in: O. E. Williamson (ed.): Organization Theory: From ChesterBarnard to the Present and Beyond (Oxford: University Press), 116–53.
Bibliography 215
PPL-UK_RR-Krapohl_Biblio.qxd 9/12/2008 10:00 AM Page 215

Monser, C. (1993): Contergan/Thalidomid: Ein Unglück kommt selten allein(Düsseldorf: Eggcup).
Moravcsik, A. (1991): ‘Negotiating the Single European Act: National Interestsand Conventional Statecraft in the European Community’, in: InternationalOrganization, 45, 19–46.
Moravcsik, A. (1998): The Choice for Europe: Social Purpose and State Power fromMessina to Maastricht (Ithaca: UCL Press).
Mossialos, E., T. Walley and M. Mrazek (2004): ‘Regulating Pharmaceuticals inEurope: An Overview’, in: E. Mossialos, M. Mrazek and T. Walley (eds):Regulating Pharmaceuticals in Europe: Striving for Efficiency, Equity and Quality(Maidenhead: Open University Press), 1–37.
Nentwich, M. (1995): Das Lebensmittelrecht der Europäischen Union: Entstehung,Rechtsprechung, Sekundärrecht, Nationale Handlungsspielräume (Wien: ServiceFachverlag).
Niskanen, W. A. (1973): Bureaucracy: Servant or Master?: Lessons from America(London: Hobart Paperbacks).
North, D. C. (1993): ‘Institutions and Credible Commitment’, in: Journal ofInstitutional and Theoretical Economics, 149:1, 11–23.
O’Riordan, T. and J. Cameron (1994) (eds): Interpreting the Precautionary Principle(London: Earthscan).
O’Rourke, R. (1998): European Food Law (Bembridge: Palladian Law Publishing).Olson, M. (1968): Die Logik des kollektiven Handelns: Kollektivgüter und die Theorie
der Gruppen (Tübingen: Mohr Siebeck).Patterson, L. A. (2000): ‘Biotechnology Policy’, in: H. Wallace and W. Wallace
(eds): Policy-Making in the European Union (Oxford: University Press), 317–44.Patterson, L. A. and T. Josling (2001): ‘Regulating Biotechnology: Comparing
EU and US Approaches’ (Paper prepared for Presentation at the WesternEconomic Association International 76th Annual Conference, 8 July 2001 inSan Francisco).
Pelkmans, J. (1987): ‘The New Approach to Technical Harmonization andStandardization’, in: Journal of Common Market Studies, 25:3, 249–69.
Permanand, G. (2006): EU Pharmaceutical Regulation: The Politics of Policy-Making(Manchester: University Press).
Permanand, G. and C. Altenstetter (2004): ‘The Politics of Pharmaceuticals in theEuropean Union’, in: E. Mossialos, M. Mrazek and T. Walley (eds): RegulatingPharmaceuticals in Europe: Striving for Efficiency, Equity and Quality (Maidenhead:Open University Press), 38–54.
Permanand, G. and E. Mossialos (2005): ‘Constitutional Asymmetry andPharmaceutical Policy-Making in the European Union’, in: Journal of EuropeanPublic Policy, 12:4, 687–709.
Pierson, P. (1996): ‘The Path to European Integration: A Historical InstitutionalistAnalysis’, in: Comparative Political Studies, 29:2, 123–63.
Pierson, P. (2000a): ‘Increasing Returns, Path Dependence, and the Study ofPolitics’, in: American Political Science Review, 94:2, 251–67.
Pierson, P. (2000b): ‘The Limits of Design: Explaining Institutional Origins andChange’, in: Governance: An International Journal of Policy and Administration,13:4, 475–99.
Pollack, M. A. (1997a): ‘Delegation, Agency and Agenda-Setting in the EuropeanCommunity’, in: International Organisations, 51, 99–134.
216 Bibliography
PPL-UK_RR-Krapohl_Biblio.qxd 9/12/2008 10:00 AM Page 216

Pollack, M. A. (1997b): ‘Representing Diffuse Interests in EC Policy-Making’, in:Journal of European Public Policy, 4:4, 572–90.
Pollack, M. A. (2003): The Engines of European Integration: Delegation, Agency, andAgenda Setting in the EU (Oxford: University Press).
Prusiner, S. B. (1997): ‘Prion Diseases and the BSE Crisis’, in: Science, 278,245–251.
Randall, E. (2006): ‘Not that Soft or Informal: A Response to Eberlein andGrande’s Account of Regulatory Governance in the EU with Special Referenceto the European Food Safety Authority (EFSA)’, in: Journal of European PublicPolicy, 13:3, 402–19.
Rehbinder, E. (1991): Das Vorsorgeprinzip im internationalen Vergleich (Baden-Baden: Nomos).
Reich, N. (1988): Arzneimittelregulierung in Frankreich (Baden-Baden: Nomos).Rekaiti, P. and R. van den Bergh (2000): ‘Cooling-Off Periods in the Consumer
Laws of the EC Member States: A Comparative Law and Economics Approach’,in: Journal of Consumer Policy, 23, 371–401.
Rosamond, B. (2000): ‘Neofunctionalism’, in: Theories of European Integration(London: Macmillan), 50–97.
Rothstein, H. (2006): ‘From Precautionary Bans to DIY Poison Tasting: Reform ofthe UK Food Safety Regulation Regime’, in: C. Ansell and D. Vogel (eds): What’sthe Beef? The Contested Governance of European Food Safety (Cambridge: MITPress), 153–80.
Rowell, A. (2003): Don’t Worry, It’s Safe to Eat: The True Story of GM Food, BSE &Foot and Mouth (London: Earthscan Publications).
Scharpf, F. W. (1970): Demokratietheorie zwischen Utopie und Anpassung (Konstanz:Universitätsverlag).
Scharpf, F. W. (1985): ‘Die Politikverflechtungs-Falle: Europäische Integrationund deutscher Föderalismus im Vergleich’, in: Politische Vierteljahresschrift,26:4, 323–56.
Scharpf, F. W. (1989): ‘The Joint Decision Trap: Lessons from German Federalismand European Integration’, in: Public Administration, 66, 239–78.
Scharpf, F. W. (1993): ‘Positive und negative Koordination inVerhandlungssystemen’, in: Héritier, A. (ed.) ‘Policy Analyse: Kritik undNeuorientierung’, in: Politische Vierteljahresschrift, Special Edition 24, 57–83.
Scharpf, F. W. (1996a): ‘Politische Optionen im vollendeten Binnenmarkt’, in: M. Jachtenfuchs and B. Kohler-Koch (eds): Europäische Integration (Opladen:Leske + Budrich), 109–40.
Scharpf, F. W. (1996b): ‘Democratic Policy in Europe’, in: European Law Journal,2:2, 136–55.
Scharpf, F. W. (1997a): Games Real Actors Play: Actor-Centered Institutionalism inPolicy Research (Oxford: Westview).
Scharpf, F. W. (1997b): ‘Economic Integration, Democracy and the Welfare State’,in: Journal of European Public Policy, 4:1, 18–36.
Scharpf, F. W. (1999): Governing in Europe: Effective and Democratic? (Oxford:University Press).
Scharpf, F. W. (2004): ‘Legitimationskonzepte jenseits des Nationalstaats’ (MPIfGWorking Paper 04/6).
Schelling, T. (1995): The Strategy of Conflict (Cambridge: University Press), 15thedition.
Bibliography 217
PPL-UK_RR-Krapohl_Biblio.qxd 9/12/2008 10:00 AM Page 217

Schenek, M. (1995): Das Gentechnikrecht der Europäischen Gemeinschaft:Gemeinschaftliche Biotechnologiepolitik und Gentechnikregulierung (Berlin: Dunker &Humblot).
Scheu, G. (2003): In Dubio Pro Securitate: Contergan, Hepatitis-/AIDS-Blutprodukte,Spongiformer Humaner Wahn und kein Ende? (Baden-Baden: Nomos).
Schneider, G. (2001): ‘Ideas, Mad Cows and European Integration: AnInstitutionalist Analysis of the BSE Crisis’ (Paper prepared for presentation atthe biannual convention of the European Community Studies Association,Madison, Wisconsin, May 31–June 2, 2001).
Shaffer, G. C. and M. A. Pollack (2004): ‘Reconciling (or Failing to Reconcile)Regulatory Differences: The Ongoing Transatlantic Dispute over the Regulationof Biotechnology’ (Paper prepared for presentation at the ‘Workshop on TheNew Transatlantic Agenda and the Future of Transatlantic EconomicGovernance’, 18–19 June 2004 in Florence).
Shapiro, C. (1983): ‘Premiums for High Quality Products as Returns toReputation’, in: Quarterly Journal of Economics, 97, 659–79.
Shapiro, M. (1988): Who Guards the Guardians? Judicial Control of Administration(Athens: The University of Georgia Press).
Shapiro, M. (1992): ‘The Giving-Reasons Requirement’, in: M. Shapiro and A.Stone Sweet (eds): On Law, Politics and Judicialisation (Oxford: University Press),228–58.
Shapiro, M. (1997): ‘The Problems of Independence: Agencies in the United Statesand the European Union’, in: Journal of European Public Policy, 4:2, 276–91.
Shapiro, M. and A. Stone Sweet (2002): On Law, Politics and Judicialisation(Oxford: University Press).
Shepsle, K. A. (1986): ‘Institutional Equilibrium and Equilibrium Institutions’, in:H. Weisberg (ed.): Political Science: The Science of Politics (New York: Agathon),51–82.
Shepsle, K. A. (1989): ‘Studying Institutions: Some Lessons from the RationalChoice Approach’, in: Journal of Theoretical Politics, 1:2, 131–47.
Shepsle, K. A. (1991): ‘Discretion, Institutions and the Problem of GovernmentCommitment’, in: P. Bourdieu and J. S. Coleman (eds): Social Theory for aChanging Society (Boulder: Westview).
Sheridan, B. (2001): EU Biotechnology: Law & Practise (Bembridge: Palladin LawPublishing).
Simon, H. A. (1972): ‘Theories of Bounded Rationality’, in: C. B. McGuire and R.Radner (eds): Decision and Organization (Amsterdam: University Press), 161–76.
Sinn, H.-W. (2003): ‘Verbraucherschutz als Staatsaufgabe’, in: Perspektiven derWirtschaftspolitik, 4:2, 282–94.
Sjöström, H. and R. Nilsson (1975): Contergan oder die Macht derArzneimittelkonzerne (Berlin: VEB Verlag Volk und Gesundheit).
Skogstad, G. (2001): ‘The WTO and Food Safety Regulatory Policy Innovation inthe European Union’, in: Journal of Common Market Studies, 39:3, 485–505.
Skogstad, G. (2003): ‘Legitimacy and/or Policy Effectiveness? Network Governanceand GMO Regulation in the European Union’, in: Journal of European PublicPolicy, 10:3, 321–38.
Skogstad, G. (2006): ‘Regulating Food Safety Risks in the European Union: AComparative Perspective’, in: C. Ansell and D. Vogel (eds): What’s the Beef? TheContested Governance of European Food Safety (Cambridge: MIT Press), 213–36.
218 Bibliography
PPL-UK_RR-Krapohl_Biblio.qxd 9/12/2008 10:00 AM Page 218

Stacey, J. and B. Rittberger (2003): ‘Dynamics of Formal and InformalInstitutional Change in the EU’, in: Journal of European Public Policy, 10:6,858–83.
Steiner, B. (2006): ‘Governance Reform of German Food Safety Regulation:Cosmetic or Real?’, in: C. Ansell and D. Vogel (eds): What’s the Beef ? TheContested Governance of European Food Safety (Cambridge: MIT Press), 181–210.
Stephens, T. and R. Brynner (2001): Dark Remedy: The Impact of Thalidomide andIts Revival as a Vital Medicine (Cambridge: Perseus Publishing).
Steunenberg, C., C. Koboldt and D. Schmidtchen (1996): ‘Policy-Making,Comitology, and the Balance of Power in the European Union’, in: InternationalReview of Law and Economics, 16:3, 329–44.
Steunenberg, C., C. Koboldt and D. Schmidtchen (1997): ‘Beyond Comitology: A Comparative Analysis of Implementation Procedures with ParliamentaryInvolvement’, in: Aussenwirtschaft, 52:1/22, 87–112.
Stone Sweet, A. (1999): ‘Judicialisation and the Construction of Governance’, in:Comparative Political Studies, 31, 147–84.
Stone Sweet, A. and J. A. Caporaso (1998): ‘From Free Trade to SupranationalPolity: The European Court and Integration’, in: W. Sandholtz and A. StoneSweet (eds): European Integration and Supranational Governance (Oxford: UniversityPress), 92–133.
Stone Sweet, A. and T. Brunell (1998): ‘Constructing a Supranational Constitution:Dispute Resolution and Governance in the European Community’, in: AmericanPolitical Science Review, 92, 63–95.
Szawlowska, K. (2004): ‘Risk Assessment in the European Food Safety Regulation:Who is to Decide Whose Science is better? Commission v. France and Beyond . . . ’,in: German Law Journal, 5:10, 1259–74.
Tallberg, J. (2002): ‘Delegation to Supranational Institutions: Why, How andWith What Consequences’, in: West European Politics, 25:1, 23–46.
Thatcher, M. (2002): ‘Delegation to Independent Regulatory Agencies: Pressures,Functions and Contextual Mediation’, in: West European Politics, 25:1, 125–47.
Thelen, K. (1999): ‘Historical Institutionalism in Comparative Politics’, in:Annual Review of Political Science, 2, 169–404.
Thelen, K. (2003): ‘How Institutions Evolve: Insights from ComparativeHistorical Analysis’, in: J. Mahoney and D. Rueschemeyer (eds): ComparativeHistorical Analysis in the Social Sciences (Cambridge: University Press), 208–40.
Thompson, R. (1994): The Single Market for Pharmaceuticals (London: Butterworths).Toke, D. (2004): The Politics of GM Food: A Comparative Study of the UK, USA and
EU (London: Routledge).Töller, A. E. (2002): Komitologie: Theoretische Bedeutung und praktische Funktionsweise
von Durchführungsausschüssen der Europäischen Union am Beispiel der Umweltpolitik(Opladen: Leske + Budrich).
Tsebelis, G. (1994): ‘The Power of the European Parliament as a ConditionalAgenda-Setter’, in: American Political Science Review, 88:1, 128–42.
Tsebelis, G. (2001): Veto Players: How Political Institutions Work (Los Angeles:University Press).
Tsebelis, G. and A. Kreppel (1998): ‘The History of Conditional Agenda-Setting inEuropean Institutions’, in: European Journal of Political Research, 33:1, 41–71.
Tsebelis, G. and G. Garrett (2000): ‘Legislative Politics in the European Union’,in: European Union Politics, 1:1, 9–36.
Bibliography 219
PPL-UK_RR-Krapohl_Biblio.qxd 9/12/2008 10:00 AM Page 219

Tsebelis, G. and G. Garrett (2001): ‘The Institutional Foundations ofIntergovernmentalism and Supranationalism in the European Union’,International Organization, 55:2, 357–90.
Ugland, T. and F. Veggeland (2006): ‘Experiments in Food Safety PolicyIntegration in the European Union’, in: Journal of Common Market Studies, 44:3,607–24.
Vincent, K. (2004): ‘Mad Cows and Eurocrats: Community Responses to the BSECrisis’, in: European Law Journal, 10:5, 499–517.
Vogel, D. (1998): ‘The Globalization of Pharmaceutical Regulation’, in:Governance: An International Journal of Policy Administration, 11:1, 1–22.
Vogel, D. (2001a): ‘The New Politics of Risk Regulation in Europe’ (London:Working Paper for the Centre for Analysis of Risk and Regulation at theLondon School of Economics and Political Science).
Vogel, D. (2001b): ‘Ships Passing in the Night: The Changing Politics of RiskRegulation in Europe and the United States’ (Florence: EUI Working Paper RSCNo. 2001/16).
Vos, E. (1997): ‘The Rise of Committees’, in: European Law Journal, 3, 210–29.Vos, E. (1999a): Institutional Frameworks of Community Health and Safety
Legislation: Committees, Agencies and Private Bodies (Oxford: Hart Publishing).Vos, E. (1999b): ‘EU Committees: The Evolution of Unforeseen Institutional
Actors in European Product Regulation’, in: C. Joerges and E. Vos (eds): EUCommittees: Social Regulation, Law and Politics (Oxford: Hart Publishing), 19–50.
Vos, E. (2000a): ‘Reforming the European Commission: What Role to Play for EUAgencies?’, in: Common Market Law Review, 37, 1113–34.
Vos, E. (2000b): ‘EU Food Safety Regulation in the Aftermath of the BSE Crisis’,in: Journal of Consumer Policy, 23, 227–55.
Vos, E. (2004): ‘Overcoming the Crisis of Confidence: Risk Regulation in an EnlargedEuropean Union’ (Maastricht: Working Paper of the Universiteit Maastricht).
Wakefield, J. (2002): ‘BSE: A Lesson in Containment? Avoiding Responsibilityand Accountability in the Compensation Action’, in: European Law Review, 27,426–44.
Weber, M. (1985): Wirtschaft und Gesellschaft (Tübingen: Mohr Siebeck), 5th edition.Westlake, M. (1997): ‘Mad Cows and Englishmen: The Institutional Consequences
of the BSE Crisis’, in: Journal of Common Market Studies, 35:1, Annual Review,11–36.
Wildavsky, A. (1988): Searching for Safety (New Brunswick: Transaction Books).Wildavsky, A. (1995): But Is It True? A Citizen’s Guide to Environmental Health and
Safety (Cambridge: University Press).Wilson, J. Q. (1980) (ed.): The Politics of Regulation (New York: Basic Books).World Health Organisation (1988): Public Health in Europe 14: Food Safety Services
(Copenhagen: Regional Office for Europe), 2nd edition.Young, A. R. (2003): ‘Political Transfer and “Trading Up”? Transatlantic Trade in
Genetically Modified Food and U.S. Politics’, in: World Politics 55, 457–84.Young, A. R., and P. Holmes (2006): ‘Protection or Protectionism? EU Food Safety
and the WTO’, in: C. Ansell and D. Vogel (eds): What’s the Beef? The ContestedGovernance of European Food Safety (Cambridge: MIT Press), 281–306.
Zürn, M. (1992): Interessen und Institutionen in der internationalen Politik: Grundlegungund Anwendungen des situationsstrukturellen Ansatzes (Opladen: Leske + Budrich).
220 Bibliography
PPL-UK_RR-Krapohl_Biblio.qxd 9/12/2008 10:00 AM Page 220

221
accountability, 7, 52, 114–5, 171, 177,187–9
expert accountability, 53–5, 112–3,175
judicial accountability, 53–5, 113, 176
political accountability, 53–5, 112,174, 177
Advisory Committee on Foodstuffs,172, 175
advisory forum (within EFSA), 11, 141,159, 161, 175
agencies, see regulatory agenciesagency capture, 3, 44, 115, 154, 194agency drift, 7, 44, 53, 188agenda-setting, 24, 28, 96, 154–5,
160–1, 167, 187anorectics, 98authorisation criteria, 9, 72–4, 76,
82–3, 89, 96, 168
bargaining system, 23–4, 192Blueprint for Europe (of the
pharmaceutical industry), 75, 111
bounded rationality, 183BSE (Bovine Spongiform
Encephalopathy) crisis, 1–2, 97,99, 126–36, 152–9, 171–5,185–90
Cassis-de-Dijon case, 20–1, 123, 156centralised authorisation procedure,
9–10, 75–82, 92–102, 92,110–16, 186
centralised arbitration procedure,for GMOs and GM food, 143, 147in the pharmaceutical sector, 10,
78, 81, 104–9, 186checks-and-balances, 49, 112comitology, see committee system,
comitology procedures, andmember state committees
comitology procedures, 45–6, 71–3,80, 93–5, 101–2, 127, 152–5,159–61, 171, 174, 188
advisory procedure, 45management procedure, 45, 81, 95,
105regulatory procedure, 45, 72,
78, 125, 127, 148, 154, 160,166
Commission, 3, 23–4, 44–6, 75–83,92, 93–101, 103, 124–49,152–70, 153, 159, 165, 185–90
committee system, 11–12, 69–74,87–91, 122–7, 131, 152–9,171–2, 185–9, 192–3
Common Technical Document, 83,92, 96
Community code relating tomedicinal products for humanuse, see Directive 2001/83/EC
Community Procedure (in thepharmaceutical sector), see Multi-State Procedure
concerned member states (in themutual recognition procedurefor pharmaceuticals), 71–3,87–91, 103–7, 106
Concertation Procedure (in the pharmaceutical sector), 72–5,87–91, 87
Contergan, see thalidomide scandalcontrol problem, 5–7, 43–9, 111–15,
173–7, 189–90, 194Coordination Group (in the
pharmaceutical sector), 104–5coordination problem, 4, 21–5, 43co-rapporteur, see rapporteursCourt of First Instance, 48, 54, 97–8,
130, 157, 168–9, 176credible commitment, 6–7, 20,
42–50, 87–109, 152–70, 185–7,193–4
Index
PPL-UK_RR-Krapohl_Index.qxd 9/9/2008 1:35 PM Page 221

crisis of consumer confidence, 1, 11,136–7, 142, 157–9, 169–70,173, 183–9, 191
critical junctures, 5, 25–8, 184
decentralised authorisation procedure,10, 102–9, 103
delegation, 6–7, 23–5, 43–6, 194delegation chain, 51, 110, 193delegation gains, 45, 49, 194delegation problem, see control
problemdeliberative supranationalism, 2, 192democratic deficit, 50differentiation of decision-making,
24, 46, 49direct and individual concern, 48, 55,
97–8, 163, 176Directorate General for Consumer
Policy and Health Protection,132–3
Directivesin the foodstuff sector, 124–5
for cacao and chocolate products,122
on the deliberate release of GMOsinto the environment, 143–9,164–5
in the pharmaceutical sector,65/65/EEC, 70–2, 76, 80, 82–3, 8975/318/EEC, 70–2, 76, 82–3, 89,
96–775/319/EEC, 712001/83/EC, 83–4, 96, 168
Distaval, see thalidomide scandal
ECJ (European Court of Justice), 20–1,48–9, 54, 90–8, 114, 123–30,145–6, 156–8, 168–9, 176
efficient risk regulation, see riskregulation
EFSA (European Food Safety Agency),11–12, 137–42, 148, 159–70,159, 165, 187
EMEA (before 2004: European Agencyfor the Evaluation of MedicinalProducts, after 2004: EuropeanMedicines Agency), 9–10, 75–84,92–102, 92, 103, 104, 186
entrepreneurial politics, 39EP (European Parliament), 51–4,
76–81, 109–16, 131–2, 139–40,147–8, 171–5, 188–9
epidemio-surveillance programme (onBSE), 135
EP inquiry committee on BSE, 131–3,172
Europeanisation of the BSE crisis,134–6
evaluation report (of the EU regulatoryregime for pharmaceuticals),79–82, 90, 115
executive director, of the EFSA, 159–60, 166, 175of the EMEA, 77, 104, 113
expert committees in thepharmaceutical sector, 71–84,87–116, 88, 92, 103, 188
CHMP (Committee for HumanMedicinal Products), 80, 92
COMP (Committee for OrphanMedicinal Products), 82
CPMP (Committee for ProprietaryMedicinal Products), 71–2,76–9, 87–8, 92
HMPC (Committee for HerbalMedicinal Products), 82
export ban on British beef, 126–33,155–7
fire alarm control, 46free rider, 18–20functionalist fallacy, 25, 191functional pressure, 4–5, 18–25, 19,
184, 191FVO (Food and Veterinary Office),
132, 172
general food law, 138–9, 156, 162–3,176–8, 187
generics, 9, 72, 75GM food (Genetically Modified Food),
8, 11–2, 142–9, 164–76, 185–9GMOs (Genetically Modified
Organisms), 8, 11, 164–70, 174,185–9
Green Paper on Food Law, 137group theory, 39
222 Index
PPL-UK_RR-Krapohl_Index.qxd 9/9/2008 1:35 PM Page 222

guidelines,for GMOs and GM food, 166–8in the pharmaceutical sector, 73,
83, 97, 105
harmonised standards, 4, 21–3, 73,123–6, 185, 190
herbal medicinal products, 82–3
ICH (International Conference onHarmonisation of TechnicalRequirements for Registrationof Pharmaceuticals for HumanUse), 83–4, 92
incomplete information, 30, 183increasing returns, 26, 183information asymmetry, 34, 183infringement procedure, 90, 130institutionalism, 7, 12, 190
historical institutionalism, 5, 25–32,184, 192
rational institutionalism, 183, 191iterated games, 3, 20, 47interests,
concrete interests, 27, 39, 52, 111consumers’ interests, 6, 35–41, 81,
116, 139, 147–8, 189diffuse interests, 12, 39, 52, 139,
148, 172long-term interests, 13, 41–9,
171–7, 188, 194producers’ interests, 6, 39–41, 55–6,
95, 110–15, 188short-term interests, 6, 41–6, 56,
115, 157–61
joint-decision trap, 26judicial review, 46–55, 90, 97–9, 106–9,
114–5, 157, 162, 168, 177, 186
legalisation, 6, 46–9, 89–90, 96, 105,113–5, 156–7, 162–3, 168–70,187, 190–4
legislative procedures,co-decision procedure, 52, 81, 110,
139, 148, 172, 189consultation procedure, 52, 76–7,
110–11cooperation procedure, 52, 110
legitimacy, 5–7, 49–55, 109–16,171–5, 187–9, 193
input legitimacy, 7, 50–2, 109–11,171–4, 187–9, 193
output legitimacy, 7, 52–3, 111–16,173–4, 187–9, 193
management board,of the EFSA, 139–41, 159–60of the EMEA, 77–81, 94
marketing authorisation, see pre-marketing control
market failures, 34–5, 193market integration, 18–25, 27–30, 28,
75, 122–5, 184–6meat- and bone-meal, 127–9, 133–5member state committees, 24, 45,
51–4, 92, 95–6, 103, 153, 154–6,159, 160–2, 165, 166–8, 187, 193
Standing Veterinary Committee,127–37, 153, 154, 164
Standing Committee on Foodstuffs,154, 167
Standing Committee on MedicinalProducts for Human Use, 72,78, 83, 93, 102
Standing Committee on the FoodChain and Animal Health, 142,148, 160, 166, 174
mixed polity, 48, 194moratorium on the authorisation of
GMOs and GM food, 11–2,146–9, 165–73, 187
Multi-State Procedure (in thepharmaceutical sector), 71–3,87–92, 88
mutual recognition, 4, 18–22, 29,69–75, 90, 121–6, 143, 152, 185
mutual recognition procedure (inthe pharmaceutical sector),9–10, 75–83, 102–9, 186
Mutual Recognition FaciliationGroup (in the pharmaceuticalsector), see Coordination Group
negative integration, 20, 28, 126negotiators’ dilemma, 4, 21–23New Approach, 11, 28, 124–5, 152–3,
185, 190
Index 223
PPL-UK_RR-Krapohl_Index.qxd 9/9/2008 1:35 PM Page 223

non-tariff barriers of trade, 5, 11, 29,90, 127, 184
notification procedure (for novelfood), 145
nullity claims, 48, 97–99
orphan medicinal products, 82–3, 96over-regulation, 37–42
partial harmonisation, 9, 73, 90path-dependency, 25, 191police-patrol control, 44policy entrepreneur, 28policy windows, 25–8, 136–7positive feedback, 26positive integration, 18–25, 126post-marketing control, 8, 63–9, 74,
142, 166precautionary principle, 36–41, 139,
159, 162, 167–70, 187preliminary rulings, 90, 97, 123, 145,
169pre-marketing control, 8, 63–70, 78,
92–102, 111–14, 142–4, 164–9,176
principal-agent theory, 44–6prisoners’ dilemma, 18–21product standards, 11, 18–23, 19, 34
rapporteurs, 92–4, 113, 116reference member state (in the
mutual recognition procedurefor pharmaceuticals), 71,78–81, 87, 103–8, 113, 116
Regulations,on GM food and feed, 144, 165–8on Novel Food, 144, 148–9, 165–6on TSE, 136
regulatory agencies, 3–5, 18–29, 44,50, 53
EU agency for foodstuffs, see EFSAEU agency for pharmaceuticals, see
EMEA independent regulatory agencies,
10, 30, 101, 122, 141, 185, 194
national regulatory agencies, 5,27–30, 44, 53, 69–79, 89–95,104–5, 112–6, 141–2, 184–5
supranational regulatory agencies,4, 12, 116
regulatory competition, 18, 29, 53,74, 116, 147
regulatory networks, 9, 30, 53, 79, 88,111–15, 137, 141, 191
regulatory scandals, 5, 27, 164, 183–5,190–1
reputation, 6, 40–1, 53, 104, 113–16,162
reputation mechanism, 40, 113–16risk analysis, 136–49, 161
risk assessment, 11, 35–41, 138–48,159, 160–4, 187
risk communication, 11, 138–40,160, 164
risk management, 11, 35, 138–41,160–2, 168–70, 187
risk perception, 35–8, 100objective, 35–8subjective, 35–8
risk regulation, 5–10, 18–27, 34–42,49–51, 186–8, 191–2
safeguard clauses (for GMOs and GMfood), 143–8, 166–70
scientific committees in the foodstuffsector, 17, 125, 139–41, 153–6,159, 161–3, 171, 175
scientific committee of the EFSA,139–41, 152, 159
Scientific Committee on Food, 144,153, 153
Scientific Committee on Plants,143–45
Scientific Steering Committee,132–5, 153–59, 164, 172
Scientific Veterinary Committee,127–31, 153, 154–7, 163
scientific panels of the EFSA, 141,159–61, 159
Scientific Panel on GMOs, 147–8,165, 166–7
Single European Act, 11, 28, 66, 124
Single Market, 1–7, 18–25, 19, 42,124, 189–93
single market for foodstuffs, 10–11,123–4, 152–9, 185
224 Index
PPL-UK_RR-Krapohl_Index.qxd 9/9/2008 1:35 PM Page 224

single market for pharmaceuticals,9, 70–75, 87–91, 99, 105–9,116, 185, 192
Single Market programme, 28, 75,124, 152, 171
specified risk materials, 97, 133–6,155
Stalinon scandal, 68stakeholders, 6–7, 42, 50–4, 75,
79–82, 100–2, 108–14, 161–7,171–7, 184–9, 193–4
of institutional change, 5, 29–30,184–5
standardisation of technical goods,189–91
standardisation bodies, 24, 189–90substantial equivalence (of Novel
Food), 145substantive decision-making criteria,
4–6, 23, 46–55, 71–83, 89–90,96, 105–6, 156, 162, 168,186–8, 194
supranational regulatory regimes, 4–7,23–31, 43–55, 183–94
in the foodstuff sector, 11–13,122–6, 137–49, 152–77
in the pharmaceutical sector, 8–10,69–84, 87–116
thalidomide scandal, 8, 62–70, 184total harmonisation, 123–4TSE (Transmissible Spongiform
Encephalopathy), 127, 132–3,136
under-regulation 37, 42, 55
vCJD (Variant Creutzfeld-JakobDisease), 126, 129
Vioxx, 108
White Paperon Completing the Internal Market,
124on Food Safety, 138–9
written procedure (in the centralisedauthorisation or arbitrationprocedure for pharmaceuticals),78, 93–6, 105
WTO (World Trade Organisation),146–7, 174
dispute settlement body, 146–7
Index 225
PPL-UK_RR-Krapohl_Index.qxd 9/9/2008 1:35 PM Page 225