rights / license: research collection in copyright - …33730/... · cyclischen triterpene '...
TRANSCRIPT

Research Collection
Doctoral Thesis
Ueber die Konfiguration der tetracyclischen Triterpene vomTypus des Tirucalladienols
Author(s): Ménard, Enrico
Publication Date: 1958
Permanent Link: https://doi.org/10.3929/ethz-a-000098895
Rights / License: In Copyright - Non-Commercial Use Permitted
This page was generated automatically upon download from the ETH Zurich Research Collection. For moreinformation please consult the Terms of use.
ETH Library

Prom. Nr. 2602
Über die Konfiguration
der tetracyclischen Triterpene
vom Typus des Tirucalladienols
Von der
Eidgenössischen Technischen
Hochschule in Zürich
zur Erlangung
der Würde eines Doktors der Technischen Wissenschaften
genehmigte
PROMOTIONSARBEIT
vorgelegt von
ENRICO MÉNARD
dipl. Ing.-Chem. E. T. H.
Italienischer Staatsangehöriger
Referent: Herr Prof. Dr. L. Ruzicka
Korreferent : Herr Prof. Dr. O. Jeger
Juris-Verlag Zürich
1958

Leer - Vide - Empty

Meinen lieben Eltern
und
meiner lieben Tante Klara

Leer - Vide - Empty

Meinem sehr verehrten Lehrer,
Herrn Prof. Dr. L. Ruzicka,
möchte ich für seine grosszügige Unterstützung und das stete Interesse an die¬
ser Arbeit herzlich danken.
Herrn Privat-Dozentur. O. J e g e r,unter dessen Leitung die vorliegende
Arbeit ausgeführt wurde, danke ich recht herzlich für die wertvollen und zahlrei¬
chen Ratschläge und Anregungen und das mir immer entgegengebrachte Wohlwol¬
len.
Herrn Dr. D. Ar igoni danke ich an dieser Stelle für die stets bereit¬
willigst erteilten Ratschläge und die unermüdliche Hilfe.
Der CIBA Aktiengesellschaft in Basel danke ich für die finanzielle Unter¬
stützung dieser Arbeit.

Leer - Vide - Empty

Inhaltsverzeichnis
Theoretischer Teil
Seite
Einleitung 9
Bisherige Arbeiten 13
Die Konfiguration des Euphadienols (X) 13
Die Konfiguration der oC-Elemadienolsäure (XII) 18
Die Konfiguration des Tirucalladienols (II) 20
Die Konfiguration des Euphorbadienols (XIV) 21
Die Konfiguration des Butyrospermols (XVa) oder (XVb) 21
Die Dammarendiole (IX) 22
Eigene Arbeiten 23
Herstellung der enantiomeren Acetoxy-phenol-lactone (LUI)und (LXXXI) 23
Die Konfigurationsverhältnisse am C7, Cq, Cg, Cjj bei gesättigten7,11-Diolender Tirucallan-Reihe 36
Zur Gliederzahl des Ringes C der pentacyclischen Triterpene 43
Experimenteller Teil 47
I. Herstellung des Acetoxy-phenol-lactons (LIII) aus Lanosterin (I) 47
II. Herstellung des Acetoxy-phenol-lactons (LXV) aus Euphadienol (X) 51
Iü. Herstellung des Acetoxy-phenol-lactons (LXXXI) aus ©<-Elemadienol-
säure (XII) 56
IV. Reduktion von 7,11-Diketo-euphan (XC) 62
V. Herstellung von 6-Nor-7-keto-cholestan (CXIV) 67
Zusammenfassung 69

Leer - Vide - Empty

- 9 -
THEORETISCHER TEIL
Einleitung
Auf Grund der Untersuchungen, die in den letzten Jahren in verschiedenen
Laboratorien durchgeführt wurden, lassen sich heute fast alle bekannten tetra-
cyclischen Triterpene'in zwei Gruppen einteilen, deren Grundkörper das Lano-
sterin (I) und das Tirucalladienol (II) sind.
1) Eine Ausnahme bildet das tetracyclische Triterpen«-Onocerin (VI) , lb>welches zusammen mit dem tricyclischen Triterpen Ambrein (Vu) '
strukturell und konfigurativ der squalenoiden Gruppe angehört.
..-OH
la)D.H.R. Barton &K.H. Overton, J.chem.Soc. 1955, 2639.
Vergl. K. Schaffner, R. Viterbo, D.Arigoni & ÖTJeger, Helv. 39, 174(1956).lb) L. Ruzicka & F. Lardon, Helv. 29, 912 (1946).
E. Lederer, F. Marx, D. Mercier & Pérot, Helv. 29, 1354 (1946).O. Jeger, O. Durst & L. Ruzicka, Helv. 30, 1859 (1947).E. Lederer & D. Mercier, Experientia 37188 (1947).

- 10 -
Die Formel des Tirucalladienols (II), welche sich lediglich durch die Kon
figurationen der C-Atome 13, 14, 17, 20 von derjenigen des Lanosterins (I) un¬
terscheidet, lässt sich nicht restlos in Isoprenreste aufteilen, gehorcht jedoch
der von L. Ruzicka & Mitarb. vorgeschlagenen biogenetischen Isoprenregel'
welche für alle Triterpene einen gemeinsamen biogenetischen Vorläufer, das
Squalen (V), voraussetzt.
2)
Es wurde bereits mehrmals in der Literatur hervorgehoben, dass, im Ge¬
gensatz zum Lanosterin (I), Verbindungen vom Typus des Tirucalladienols (II)
sich durch säurekatalysierte Isomerisierung in sogenannte Isoprodukte überfüh¬
ren lassen, denen das modifizierte Gerüst (VIII) zugrunde liegt. Interessanter¬
weise sind in letzter Zeit in der Natur tetracyclische Triterpene (IX) aufgefun-3)
den worden, deren Strukturen sich von diesem Isogerüst ableiten lassen ,und
die somit als Vertreter einer neuen Untergruppe aufgefasst werden können.
H W°v /«,
2) L. Ruzicka, A. Eschenmoser & H. Heusser, Experientia 9, 362 (1953).A. Eschenmoser, L. Ruzicka, O. Jeger & D. Arigoni, HeTv. 38, 1890 (1955).
3)J.S. Mills &A.E.A. Werner, J.chem.Soc. 1955, 3132.
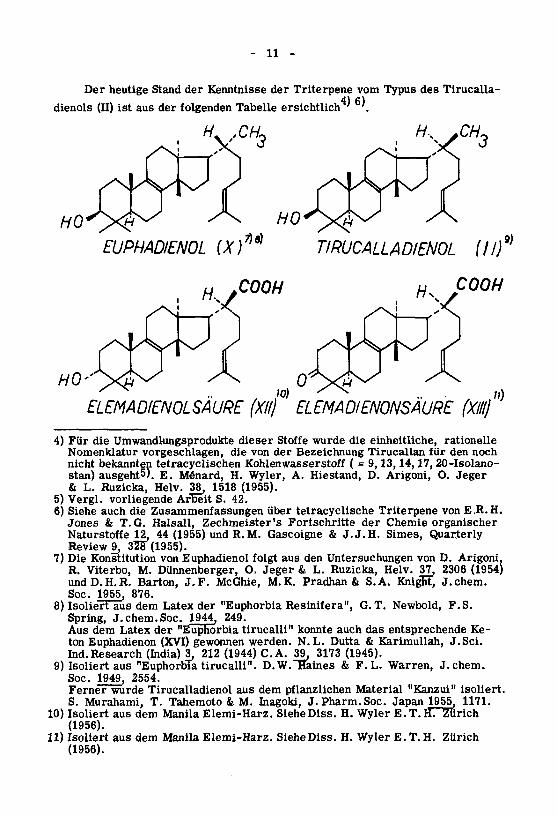
11
Der heutige Stand der Kenntnisse der Triterpene vom Typus des Tirucalla-
dienols (II) ist aus der folgenden Tabelle ersichtlich4)6)
#, AH.
EUPHADIENOL (X)
COOH
TIRUCALLADIENOL ill)
H^COOH
9)
10) ' ^ ;;)ELEMADIENOLSAURE (Xllj ELEMADIENONSAURE (XIII)
4) Für die Umwandlungsprodukte dieser Stoffe wurde die einheitliche, rationelle
Nomenklatur vorgeschlagen, die von der Bezeichnung Tirucallan für den noch
nicht bekannten tetracyclischen Kohlenwasserstoff ( = 9,13,14,17, 20-Isolano-
stan) ausgeht5). E. Ménard, H. Wyler, A. Hiestand, D. Arigoni, O. Jeger& L. Ruzicka, Helv. 38, 1518 (1955).
5) Vergl. vorliegende Arbeit S. 42.
6) Siehe auch die Zusammenfassungen über tetracyclische Triterpene von E.R.H.
Jones & T. G. Halsall, Zechmeister's Fortschritte der Chemie organischerNaturstoffe 12, 44 (1955) und R. M. Gascoigne & J.J.H. Simes, QuarterlyReview 9, 325(1955).
7) Die Konstitution von Euphadienol folgt aus den Untersuchungen von D. Arigoni.R. Viterbo, M. DUnnenberger, O. Jeger & L. Ruzicka, Helv. 37, 2306 (1954)undD.H.R. Barton, J.F. McGhie, M.K. Pradhan & S.A. KnigRT, J.chem.
Soc. 1955, 876.
8) Isoliert aus dem Latex der "Euphorbia Resinifera", G. T. Newbold, F.S.
Spring, J.chem.Soc. 1944, 249.
Aus dem Latex der "Euphorbia tirucalli" konnte auch das entsprechende Ke-
ton Euphadienon (XVI) gewonnen werden. N. L. Dutta & Karimullah, J.Sci.
Ind.Research (India) 3, 212 (1944) CA. 39, 3173 (1945).9) Isoliert aus "Euphorbia tirucalli". D.W."Haines & F.L. Warren, J.chem.
Soc. 1949, 2554.
Ferner wurde Tirucalladienol aus dem pflanzlichen Material "Kanzui" isoliert.
S. Murahami, T. Tahemoto & M. Inagoki, J. Pharm.Soc. Japan 1955, 1171.
10) Isoliert aus dem Manila Elemi-Harz. Siehe Diss. H. Wyler E.T.H. Zürich
(1956).11) Isoliert aus dem Manila Elemi-Harz. Siehe Diss. H. Wyler E. T. H. Zürich
(1956).

12
R >CM, HK sChL
H0'?<^ '^ *
HO'
EUPHORBADIENOL (Xivf BUTYROSPERMOL (XVa)
H^ ,CH~
BUTYROSPERMOL (XVb)13)H)1S)
12) Erhalten aus den Mutterlaugen der Umkristallisation von Euphadienol (X).Siehe Diss. Ch. Vogel E.T.H. Zürich (1954).
13) Isoliert aus dem "shea-nut" Oel. Siehe K. Seitz & O. Jeger, Helv. 32, 1627
(1949).14) Ausser den angeführten sind noch Dehydro-elemadienol-säure und Dehydro-
elemadienon-säure in der Natur vertreten.
T.G. Halsall, G.D. Meakins & R.E.H. Swayne, J.chem.Soc. 1953, 4139.
15) Aus "Eucalyptus microcorys" und aus "Erithrophloeum guineense" wurde
kürzlich ein tetracyclisches Triterpen isoliert, dessen Grundgerüst noch
nicht vollständig bekannt ist. I.S.G. Cox, F.E. King & T.J. King, Chem.
u. Ind. 1955, 1669.

- 13 -
Bisherige Arbeiten
Im Laufe der Arbeiten über die Konstitutionsaufklärung der tetracyclischen
Triterpene vom Typus des Tirucalladienols spielte das Euphadienol (X) bald eine
sehr wichtige Rolle, da es als erstes in seiner Konstitution voll aufgeklärt wur-
7)de
. Die gewonnenen Vorstellungen über Euphadienol (X) erlaubten die Planung
und Ausführung von Arbeiten, die bald zur gegenseitigen Verknüpfung zahlrei¬
cher Triterpene vom Typus des Tirucalladienols (II) führten. Aus diesem Grun¬
de wird hier zunächst die Stereochemie des Euphadienols (X) eingehender behan¬
delt.
Die Konfiguration des Euphadienols (X)
Die Strukturformel des Euphadienols (X) weist 7 asymmetrisch substitu-
ierte Kohlenstoffatome auf,deren Konfigurationen hier im einzelnen disku¬
tiert werden sollen.
a) Das durch milde Oxydation von Euphadienol (X) erhältliche Euphadienon (XVI)
setzt sich mit Ketonreagentien leicht um und lässt sich durch Hydrierung mit
Lithiumaluminiumhydrid wieder ins ursprüngliche Euphadienol (X) ' über¬
führen. Es ist bekannt, dass dieses Reagens bei seiner Einwirkungauf cycli-
sche Ketone mit kleiner, sterischer Hinderung zur Bildung äquatorialer Hy-18)
droxylgruppen führt '. Somit weist auch Euphadienol (X) eine äquatoriale
Oxygruppe in Stellung 3 auf.19)
Anderseits fanden Dauben & Mitarb. ' mit Hilfe der von V .P r e 1 o g
entwickelten Methode der asymmetrischen Synthese ,dass im Lanosterin
(I) und Euphadienol (X) die Hydroxylgruppe in Stellung 3 ß -Konfigura¬
tion aufweist.
Diese Konfiguration der Hydroxylgruppe in 3 wird ferner durch den Verlauf
der Wasserabspaltung mit Phosphorpentachlorid im Euphenol (XVH) bestätigt .
Die Retropinakolinumlagerung ist durch die Koplanarität der an der Reaktion
16) Die Numerierung der Kohlenstoffatome in den Produkten mit Tirucallan-Ge-
rüst entspricht derjenigen der Steroide.
17) Diss. B.C. Roth, E.T.H. Zürich, S. 47(1950).18)D.H.R. Barton & N.J. Holness, J.chem.Soc. 1952, 78.
19) W.G. Dauben, D.F. Dickel, O. Jeger & V. PreT5g7 Helv. 36, 325 (1953).20) V. Prelog, Helv. 36, 308 (1953).21) K. Christen, M. Dünnenberger, C.B. Roth, H. Heusser & O. Jeger, Helv.
35, 1756 (1952).
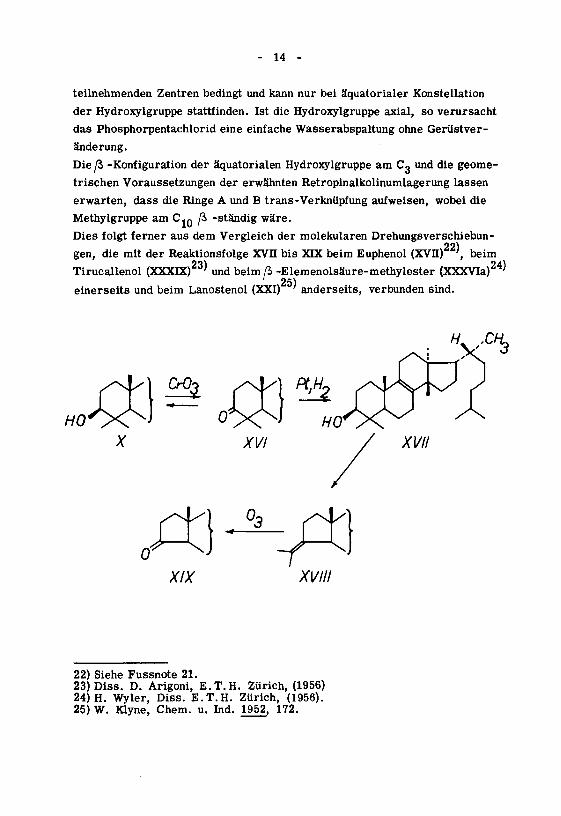
- 14 -
teilnehmenden Zentren bedingt und kann nur bei äquatorialer Konstellation
der Hydroxylgruppe stattfinden. Ist die Hydroxylgruppe axial, so verursacht
das Phosphorpentachlorid eine einfache Wasserabspaltung ohne Gerüstver¬
änderung.
Die/3 -Konfiguration der äquatorialen Hydroxylgruppe am C« und die geome¬
trischen Voraussetzungen der erwähnten Retropinalkolinumlagerung lassen
erwarten, dass die Ringe A und B trans-Verknüpfung aufweisen, wobei die
Methylgruppe am C-0 ß -ständig wäre.
Dies folgt ferner aus dem Vergleich der molekularen Drehungsverschiebun¬
gen, die mit der Reaktionsfolge XVII bis XIX beim Euphenol (XVII) ', beim
Tirucallenol (XXXIX)23' und beimß -Elemenolsäure-methylester (XXXVIa)24^25)
einerseits und beim Lanostenol (XXI) ' anderseits, verbunden sind.
XIX XVIII
22) Siehe Fussnote 21.
23) Diss. D. Arigoni, E.T.H. Zürich, (1956)24) H. Wyler, Diss. E.T.H. Zürich, (1956).25) W. Klyne, Chem. u. Ind. 1952, 172.
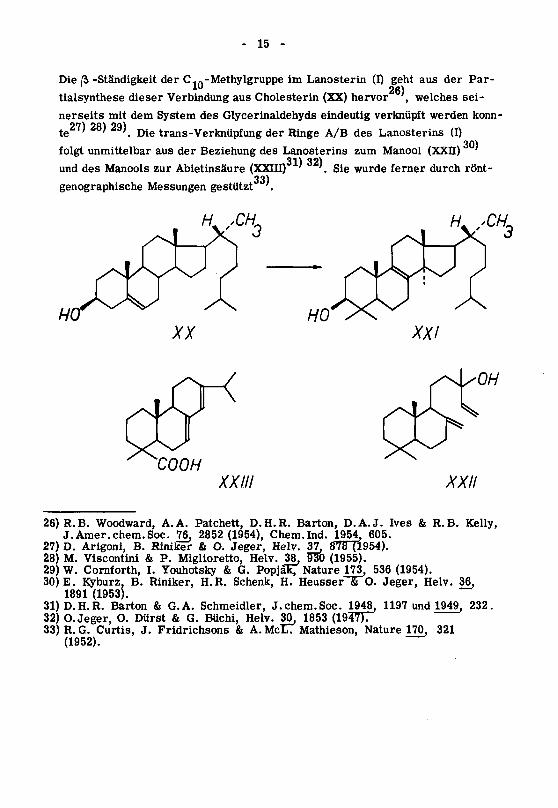
- 15 -
Die ß -Ständigkeit der C,n-Methylgruppe im Lanosterin (I) geht aus der Par-
26)tialsynthese dieser Verbindung aus Cholesterin (XX) hervor
,welches sei¬
nerseits mit dem System des Glycerinaldehyds eindeutig verknüpft werden konn¬
te' ' '. Die trans-Verknüpfung der Ringe A/B des Lanosterins (I)
folgt unmittelbar aus der Beziehung des Lanosterins zum Manool (XXII)
und des Manools zur Abietinsäure (XXIII) ' '.Sie wurde ferner durch rönt-
33)genographische Messungen gestützt '.
A/_ sCH
COOH
XXIII XXII
26) R.B. Woodward, A.A. Patchett, D.H.R. Barton, D.A.J. Ives & R.B. Kelly,J.Amer. ehem. Soc. 76, 2852(1954), Chem. Ind. 1954, 605.
27) D. Arigoni, B. RiniÊèr & O. Jeger, Helv. 37, 87811954).28) M. Viscontini & P. Miglioretto, Helv. 38, 330 (1955).29) W. Cornforth, I. Youhotsky & G. PopjäR; Nature 173, 536 (1954).30)E. Kyburz. B. Riniker, H.R. Schenk, H. HeusserTTO. Jeger, Helv. 36,
1891 (1953).31) D.H.R. Barton & G.A. Schmeidler, J.chem.Soc. 1948, 1197 und 1949, 232.
32) O. Jeger, O. Durst & G. BUchi, Helv. 30, 1853 (19377733) R. G. Curtis, J. Fridrichsons & A. McLT Mathieson, Nature 170, 321
(1952).

- 16 -
b) Die konsequente Anwendung der Konstellationsanalyse bei der Isomerisierungs-
reaktion, die von Acetoxy-euphen (XXIV) zu Iso-acetoxy-euphen (XXV) führt,
zwang zur Annahme der trans-Verknüpfung der Ringe C/D, wobei die Methyl¬
gruppe in C,„ hinter die Zeichnungsebene und diejenige in C14 vor die Zeich-
34)nungsebene zu stehen kommen .
Insbesondere liess sich dieß-Ständigkeit der Methylgruppe in C-4 durch die
Beobachtung stützen, dass im Acetoxy-lanosten (XXIa) die Doppelbindung un¬
ter analogen Bedingungen der Isomerisierung lediglich in Stellung 7,8 verscho¬
ben wird und dieser Vorgang einem Gleichgewicht entspricht.
H^ sCH
H
R0'/<^^' ^
AcO
R = H XVII; R=Ac XXIV XXV
c) Während die Seitenkette des Lanosterins (I) gleich wie bei den Steioiden
ß -ständig ist, was heutzutage unmittelbar aus der Partialsynthese des Lano-26)
stenols aus Cholesterin hervorgeht ,wurde für das Euphadienol (X) die
c* -Konfiguration der langen Seitenkette postuliert. Dies setzt voraus eine völ¬
lig synchrone Reaktion für den Uebergang von Acetoxy-euphen (XXIV) zu Iso-
acetoxy-euphen (XXV), die dann mit der Abspaltung eines Protons von Cr,34)
enden müsste '. Gegen dieses Argument liess sich jedoch einwenden, dass
man bei (X-Lage des Wasserstoffatoms am C._ zum gleichen Produkt auch
dann gelangen kann, wenn man nach Wanderung der Methylgruppe von C.-
nach C14 eine Protonabspaltung mit dem /3-ständigen Wasserstoffatom am
G-, vornimmt und erst nachträglich die entstandene Doppelbindung in die
34) D. Arigoni, R. Viterbo, M. Dünnenberger, O. Jeger & L. Ruzicka, Helv.37. 2306 (1954).
876.1955,
J.chem.Soc.Knight,S.A.&PradhanM.K.McGhie,J.F.Barton,37)DTTT.R.1325.1954,
Ind.&Chem.Knight,S.A.&PradhanM.K.McGhie,J.F.Barton,D.H.R.36)
XXXXXIX
COOHCOOH
78.1952,Soc.
J.chem.Holness,N.J.&BartonD.H.R.(XXX),-OleanolsäureSin(XXDC)OleanolsäurederIsomerisierungdieZusammenhangdieseminVergleiche35)
XXVII
XXVIII
AcO
HOOC03
,JCH^H^'3
)
H00C°3
/^W°f__
|C
XXV
.-CKW.
charakterisiert.methylester
2,6-Dimethyl-önanthsäure-
D-(-)-alsundverestertwurde(XXVm)ProduktflüchtigeleichtDas(XXVm).
und(XXVII)SpaltstückezweidiesichergabenZwischenproduktesdieses
Ozonisationnachfolgendedieund(XXVI)Ketoneinemzu(XXV)acetoxy-euphens
Iso-desOxydationDurch'.bewiesenSinneklassischenimangegebene(X)34r
elFormderindiealswurdeon
CamMethylgruppederKonfigurationabsoluteDied)
'.hinArgumentesdiesesZweideutigkeitdieaufselbstspäterdoch37)
je-wiesenAutorenenglischenDie'.angenommenDrehungsverschiebungen
molekularerVergleichesunzulässigeneinesGrundaufMitarb.&Barton
vonauchanfänglichwurdeC*„inWasserstoffatomsdes-StändigkeitDie/3
'.isomerisiert13-17-Lage35)
-17-

- 18 -
Die Konfiguration der (X-Elemadienolsäure (XII)
Die im Manila Elemi-Harz vorkommende <*-Elemadienolsäure(XII) weist im
Gegensatz zu den meisten bekannten Triterpenen eine axiale «-Lage der Hydro¬
xylgruppe in Stellung 3 auf '. Dies geht zunächst aus den Ergebnissen der De-
40)hydratisierungsexperimente mit Phosphorpentachlorid hervor ', welche unter
Abspaltung der C„-ständigen Hydroxylgruppe und des antiparallel ß> -orientier¬ten Wasserstoffatoms in Stellung 2 zur A -ungesättigten Verbindung (XXXIV)
führen. u C00&
HO'
R-H XXXIII; R=Me XXXIIIa XXXIV
In der Folge ist es gelungen, in der 3/J-Elemadienolsäure (XXXV) die
räumliche Lage der Hydroxylgruppe sowie die konfigurative Identität der Koh¬
lenstoffatome 3, 5, 10, 13, 14, 17 dieser Verbindung mit den entsprechenden
asymmetrischen Kohlenstoffatomen des Euphadienols (X) durch gegenseitige Um-
41)Wandlungen zu beweisen. Aus 3ß-Elemadienolsäure (XXXV) wurde zunächst
über die Zwischenstufen der Dihydroverbindung (XXXVI) und des Säurechlorids
(XXXVn) der Aldehyd (XXXVin) bereitet. Die Reduktion dieses letzteren nach
Wolff-Kishner ergab als Nebenprodukt das Euphenol (XVII), welches in Form
38) Eine weitere Ausnahme stellt in dieser Hinsicht die Polyporensäure A(XXXI)39' dar. Beim pentacyclischen Triterpen Bre'in (XXXII) benötigen die
konfigurativen Verhältnisse am C3 immer noch eine genauere Ueberprüfung(vergl. Diss. G. Büchi, E.T.H. Zürich).
H+ ,-COOH
XXXI39) Siehe Note 6.
40) Diss. A. Hiestand, E.T.H. Zürich, S. 20 (1949).41) Erhältlich aus Elemadienonsäure (XIII) durch Reduktion mit Natrium und
n-Propanol.

19 -
RO'
R=H,R^H XXXV,R=ActR^H XXXVa
R=Ac, R^Me XXXVb
H.yCOOR4 KsCOOR, HyCOCl
R<=H XXXVIa
R,=Me XXXVIb
H CH3
'À 7W.K.
XVII
R = H XXXIX; R=Ac XXXIXa
t
XL/II
XUI
t
XU
t
XL
XXXVIII
HR,
2"
XXXVIb
XXXIXa
RO4
R = H, R^OH
R=Ac, R^OACR=Ac, R^OHR = Ac, R^CL
XLV
XXVII
XLVI

- 20 -
42)
des Acetates (XXIV) charakterisiert wurde '. D iese Versuche sind beweisend für die
konfigurative Identität der Kohlenstoffatome 3, 5, 10, 13, 14, 17. Die Konfigu¬
ration am C20 bleibt hingegen nach wie vor unbestimmt, da hier nicht entschie¬
den werden kann, ob die alkalische Reduktion des Aldehyds (XXXVm) mit oder43)
ohne Inversion aniC,. zum Euphenol (XVII) führt '.
Die Konfiguration des Tirucalladienols (II)
Die konfigurativen Verhältnisse des Tirucalladienols (II) lassen sich auf
Grund der direkten Beziehung mit der 3/3-Elemadienolsäure (XXXV) ableiten.
Der aus (XXXVn) erhältliche Aldehyd (XXXVIÜ)44^ ergibt bei der Reduktion
nach Wolff-Kishner als Hauptprodukt Tirucallenol (XXXIX). Dass bei dieser
Reaktion keine Inversion stattgefunden hat, geht aus einer zweiten Versuchs¬
reihe hervor, bei welcher die Bedingungen zur Bewahrung der ursprünglichen
Konfiguration am C2Q streng eingehalten worden sind. Die Reduktion des 3ß-
Acetoxy-elemen-säuremethylesters (XXXVIb) mit Lithiumaluminiumhydrid lie¬
ferte das 3/3,21-Diol (XL)45*, dessen 3^-Monoacetat (XLII) mittels Tosyl-
chlorid ins entsprechende Acetoxy-chlorid(XLIII)übergeführt wurde. In diesem
liess sich das Halogenatom mit Natrium und Propanol reduktiv entfernen, wo¬
bei wiederum in guter Ausbeute Tirucallenol (XXXIX) isoliert werden konnte.
Die Ueberführung der 3/i-Oxy-elemensäure (XXXVIa) einerseits in Tiru¬
callenol (XXXIX) und anderseits, unter Inversion am C„0, in Euphenol (XVII),,
ist zunächst für die Tatsache beweisend, dass die beiden letzteren Produkte
46)die gleiche Konfiguration amC.. besitzen '.
Dass Euphadienol (X) und Tirucalladienol (II) diastereomere am C„q sind,
beweist im übrigen der Abbau von Tirucallenol (XXXIX) über die Zwischenstufe
des Iso-acetoxy-tirucallens (XLIV) zu den zwei Spaltstücken (XXVII) und (XLVI).
Während das tricyclische Acetoxy-keton (XXVII) identisch ist mit dem Abbaupro¬
dukt des Euphadienols (X), liegt hier nun als leichtflüchtiges Produkt nicht mehr,
42) E. Menard, H. Wyler, A. Hiestand, D. Arigoni, O. Jeger & L. Ruzicka,Helv. 38, 1517 (1955).
43) Vergleiche jedoch die Konfiguration des Tirucalladienols (n).44) Vergleiche vorheriger Abschnitt.
45) Für die Stereospezifität dieser Reaktion vergleiche D.S. Noyce & D.B. Den-
ney, J.Amer.ehem.Soc. 72, 5743 (1950).46) D. Arigoni, O. Jeger & L. Ruzicka, Helv. 38, 222 (1955).

- 21 -
wie beim Euphadienol (X), die D-(-)-2,6-Dimethyl-önanthsäure (XXVm) vor,47)
sondern deren enantiomere L-(+)-Form (XLVI) '.
Die Konfiguration des Euphorbadienols (XIV)
Euphorbadienol (XIV) weist, ähnlich wie die Polyporensäuren mit dem La-
nostan-Gerüst (HI), 31 Kohlenstoffatome auf. Das zusätzliche, einunddreissigste
C-Atom haftet in Form einer Methylengruppe in Stellung 24 des Tirucallan-Ge¬
rüstes (IV) .Die Beziehung von XIV zum Tirucalladienol (H) ist auch in diesem
Falle durch direkte Verknüpfung aufgeklärt worden: Die Hydroxylierung der
endständigen Doppelbindung des Euphorbadienols (XTV) mit Osmium-(VHI)-oxyd
liefert das Triol (XLVII); die nachfolgende Einwirkung von Blei-(IV)-acetat
führt zum Nor-3-oxy-24-keto-euphorben (XLVm), dessen 24-Desoxo-Verbindung
mit dem Tirucallenol (XXXIX) identisch ist '.
XLVII
Die Konfiguration des Butyrospermols (XVa) oder (XVb)
51)
Nach Versuchen von H a 1 s a 11 und Jones ' stellt das Butyrospermol ein
Doppelbindungsisomere des Euphadienols (X) dar. Die ringständige Doppelbin¬
dung des Butyrospermols, deren Lage auf Stellung 7-8 oder auf Stellung 9-11
des 20-Iso-tirucallan-Gerüstes beschränkt ist (XVa) und (XVb), wandert bei mil¬
der Behandlung mit Mineralsäuren in die ditertiäre 8-9-Lage, wobei das bekann-
47) F. L. Warren und K. H. Watling, Chem. u. Ind. 1956, 24, haben diese Re¬sultate durch neue Experimente bestätigt. Sie haben m der Euphadienol- und
in der Tirucalladienol-Reihe das Asymmetriezentrumam C20 eliminiert und
sind so erwartungsgemäss auf zwei identische Produkte gestossen.48) Diese auf Grund biogenetischer Ueberlegungen abgeleitete Lage der Methy¬
lengruppe49) konnte unlängst durch W a r r e n & Mitarb. bewiesen werden^0).49) D. Arigoni, H. Wyler & O. Jeger, Helv. 37_, 1553 (1954).50) J.B. Barbour, W.A. Laurens, F. L. Warren & K.H. Wathing, J.chem.Soc.
1955, 2194.
51)m7Ö". Dawson, T.G. Halsall, E.R.H. Jones, G.D. Meakins & P.C. Phil¬
lips, Chem. u. Ind. 1955, 918.

- 22 -
te Euphenol (XVII) entsteht. Dadurch bleibt die Konfiguration des Butyrosper-
mols bis auf die räumliche Lage eines Wasserstoffatoms eindeutig bestimmt.51)
Unter Zugrundelegung der Formel (XVa) schliessen Jones und Mitarb. 'aus
der Betrachtung von molaren Drehungsverschiebungen im Bezirke der Hydroxyl¬
gruppe auf die/J -Ständigkeit des Wasserstoffatoms am C„.
Die Dammarendiole (IX)
In letzter Zeit wurden aus dem Neutralteil des Dammar-Harzes sechs neue,3)
tetracyklische Triterpene isoliert '. Eine dieser Verbindungen, das Dammar-
endiol (£K) wurde durch Hydrierung und Wasserabspaltung in das Iso-tirucallenol
(XUV) übergeführt52*.
Die zusammenfassende Betrachtung der einzelnen besprochenen Abschnitte
lässt erblicken, dass wohl die erwähnten Verknüpfungen innerhalb der Tirucal-
lan-Gruppe keine Zweideutigkeit zulassen, dass hingegen das gesamte konfigura-53)
tive Bild nicht als vollkommen betrachtet werden kann '. In der Tat sind bis¬
her lediglich für die Konfiguration der Kohlenstoffatome 3 und 20 eindeutige che¬
mische Beweise erbracht worden, während sich sowohl die relative als auch die
absolute Konfiguration aller übrigen asymmetrischen Kohlenstoffatome auf Ar¬
gumente verschiedenen Gewichtes stützen. Die Resultate der Interpretation der
molekularen Drehungsverschiebungen bei C, und C.Q und die Schlüsse der Kon¬
stellationsanalyse für C|3, C14 und erst recht für C.„ sind, obwohl aufschluss¬
reich, nicht ohne weiteres im klassischen Sinne als eindeutig zu betrachten. Es
war deshalb wünschenswert, die gewonnenen Vorstellungen durch einen direkten
chemischen Beweis sicherzustellen.
52) Privatmitteilung von J. S. Mills.
53) Dies umso mehr, als in struktureller Hinsicht die meisten triftigen Experi¬mente in der sogenannten Isoreihe ausgeführt worden sind. Ueber die Unsi¬
cherheit, die damit verbunden ist, vergleiche die Diskussion von D. Arigoni,R. Viterbo, M. Dünnenberger, O. Jeger & L. Ruzicka, Helv. 37, 2306
(1954).
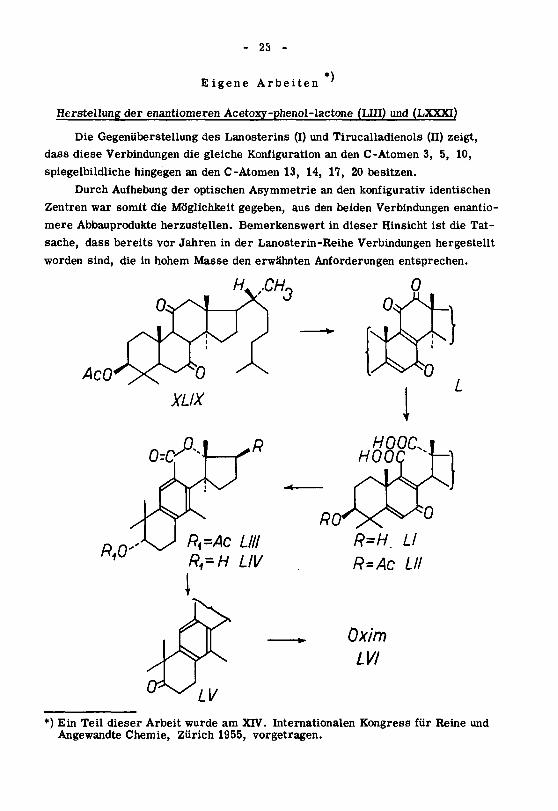
- 23 -
Eigene Arbeiten*)
Herstellung der enantiomeren Acetoxy-phenol-lactone (LIII) und (LXXXI)
Die Gegenüberstellung des Lanosterins (I) und Tirucalladienols (II) zeigt,
dass diese Verbindungen die gleiche Konfiguration an den C-Atomen 3, 5, 10,
spiegelbildliche hingegen an den C-Atomen 13, 14, 17, 20 besitzen.
Durch Aufhebung der optischen Asymmetrie an den konfigurativ identischen
Zentren war somit die Möglichkeit gegeben, aus den beiden Verbindungen enantio-
mere Abbauprodukte herzustellen. Bemerkenswert in dieser Hinsicht ist die Tat¬
sache, dass bereits vor Jahren in der Lanosterin-Reihe Verbindungen hergestellt
worden sind, die in hohem Masse den erwähnten Anforderungen entsprechen.
AcO
R<=Ac Uli
R^H UV
R=H. LI
R=Ac LH
Ox/rn
LVI
*) Ein Teil dieser Arbeit wurde am XIV. Internationalen Kongress für Reine und
Angewandte Chemie, Zürich 1955, vorgetragen.
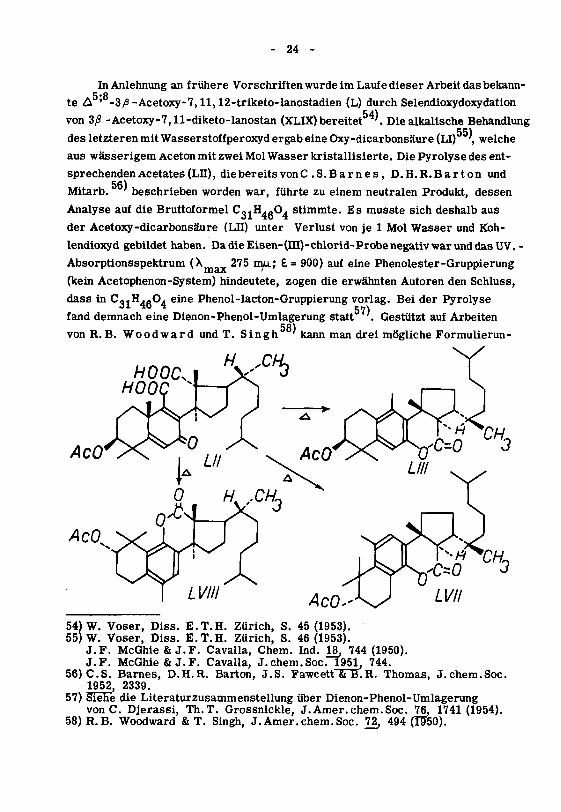
- 24 -
In Anlehnung an frühere Vorschriftenwurde im Laufe dieser Arbeit das bekann-
te ùr' -3^-Acetoxy-7,ll,12-triketo-lanostadien (L) durch Selendioxydoxydation
von 3/3 -Acetoxy-7,ll-diketo-lanostan (XLIX) bereitet '. Die alkalische Behandlung
des letzteren mit Wasserstoffperoxyd ergab eine Oxy-dicarbonsäure (LI) ,welche
aus wässerigem Aceton mit zwei Mol Wasser kristallisierte. Die Pyrolyse des ent¬
sprechenden Acetates (Lu), die bereits von C.S.Barnes, D.H.R.Barton und
Mitarb. ' beschrieben worden war, führte zu einem neutralen Produkt, dessen
Analyse auf die Bruttoformel CgjH.-O. stimmte. Es musste sich deshalb aus
der Acetoxy-dicarbonsäure (LH) unter Verlust von je 1 Mol Wasser und Koh¬
lendioxyd gebildet haben. DadieEisen-(ni)-chlorid-ProbenegativwarunddasUV.-
Absorptionsspektrum (^max 275 mti; £= 900) auf eine Phenolester-Gruppierung
(kein Acetophenon-System) hindeutete, zogen die erwähnten Autoren den Schluss,
dass in C3,H .ß04 eine Phenol-lacton-Gruppierung vorlag. Bei der Pyrolyse
fand demnach eine Dienon-Phenol-Umlagerung statt '. Gestützt auf Arbeiten58)
von R.B. Woodward und T. Singh ' kann man drei mögliche Formulierun-
AVOOCsH00
54) W. Voser, Diss. E.T.H. Zürich, S. 45 (1953).55) W. Voser, Diss. E.T.H. Zürich, S. 46 (1953).
J.F. McGhie & J.F. Cavalla, Chem. Ind. 18, 744 (1950).J.F. McGhie 6 J.F. Cavalla, J.chem.Soc."1951, 744.
56) CS. Barnes, D.H.R. Barton, J.S. FawcettlTB".R. Thomas, J.chem.Soc.
1952, 2339.
57) Siehe die Literaturzusammenstellung über Dienon-Phenol-Umlagerungvon C. Djerassi, Th.T. Grossnickle, J. Amer.chem.Soc. 76, 1741(1954).
58) R. B. Woodward & T. Singh, J.Amer.chem.Soc. 72_, 494 (T950).

- 25 -
gen für das Aromatisierungsprodukt (Lin), (LVH) und (LVm) diskutieren. Von
diesen gaben CS. Barnes, D.H.R. Barton und Mitarb. der Variante (LUI)
den Vorzug, da sie in besserem Einklang mit dem wahrscheinlichen Mechanismus
der Pyrolyse und insbesondere mit der glatten Decarboxylierung ist.
HOOC HOOC HOOC
Uli
59)A.S. Dreiding undA.I. Tomasewski 'vermuten, dass es sich hier
um eine durch das Carboxyl C10 intramolekular katalysierte Reaktion handelt,60)
was aber nur mit der Konstellation (Lila) zu erklären wäre '.
ft £ft r oHOOC.
o:
Uli
AcO-'
Einezufällige Beobachtung gestattete uns, das gleiche Produkt (LIII) auf ei¬
ne mildere Art inbesserer Ausbeute zu erhalten. Die Oxy-dicarbonsäure (LI) wurde
in Pyridin-Acetanhydrid-Lösung zwecks Acetylierung über Nacht stehen gelassen,
die Lösung dann mit Wasser bis zur leichten Trübung versetzt und weitere vier-
61)undzwanzig Stunden stehen gelassen .
Nach dieser Zeit hatten sich Kristalle
59) A.S. Dreiding &A.I. Tomasewski, J.org.Chemistry 19, 241(1954).60) Dieser Mechanismus der thermischen Aromatisierung erklärt, warum man
aus dem Anhydrid (LXXX) durch Pyrolyse nicht zum Aromatisierungsprodukt(LXXXI) gelangen konnte. Es handelt sich hier um einen eigenen Versuch,der im experimentellen Teil nicht erwähnt wurde.
61) Nach sofortiger Aufarbeitung lässt sich lediglich das Acetat (LII) isolieren.

- 26 -
ausgeschieden, die in allen Eigenschaften mit dem bereits beschriebenen Pyro¬
lyseprodukt (LUI) identisch waren '.
Aus dem Acetoxy-phenol-lacton (LIII) wurde ferner durch milde Verseifung
und Oxydation das entsprechende Keton (LV) bereitet, das als Oxim (LVI) kristallin
und analysenrein erhalten werden konnte.
Um Erfahrungen über die Aromatisierung des Ringes B bei Verbindungen
der Tirucallan-Reihe (IV) zu sammeln, wählte man zuerst, der leichteren An¬
schaffung wegen, Euphadienol (X). Euphadienon (XVI) wurde mit Platinkatalysa-Q
torinEisessig-Essigester-Gemischzu Û -3-Keto-euphen(LX) hydriert; dieses wur¬
de mit Lithiumaluminiumhydrid zu 3ß -Euphenol (XVII) reduziert. Letzteres ergab63)
nach Acetylierung'und Oxydation mit Chromsäure das 3ß -Acetoxy-7,11-dike-
to-euphen (LXI) '. Eine Selendioxydoxydation in Dioxan führte zum A ' -3j&-
Acetoxy-7,11,12-triketo-euphadien (LXII) ', das bei der weiteren Behandlung
mit alkalischem Wasserstoffperoxyd die Dicarbonsäure (LXIII) ' lieferte.
Nach der Pyrolyse der Acetoxy-dicarbonsäure (LXIIIa) ' bei 270° konnte
jedoch kein Produkt mit den gewünschten Eigenschaften gefasst werden. Erfolg¬
reich erwies sich hingegen ein Versuch nach der neuen, in dieser Untersuchung
ausgearbeiteten Vorschrift für Aromatisierung. Dabei wurde im Neutralteil ne¬
ben einem nicht in analysenreiner Form erhaltenen, unbekannten Produkt (LXIV) '
das erwartete Acetoxy-phenol-lacton (LXV) erhalten '. Dieses Produkt wurde
ebenfalls über die nicht gefassten Zwischenstufen des Alkohols (LXVI) und des
57)
62) In der Literatur ' wurde bis heute noch keine Dienon-Phenol-Umlagerungbeschrieben, die unter so milden Bedingungen abläuft. Mit dem beschränk¬
ten Tatsachenmaterial ist es noch verfrüht, einen bestimmten Reaktionsme¬
chanismus für diese Säure-Base-katalysierte Aromatisierung vorzuschlagen.63) Siehe Fussnote 17.
64) K. Christen, Diss. E.T.H. Zürich (1952).65) Siehe experimenteller Teil (S. 54).66) Anhand der physikalischen Daten (UV. X 242 myu; log £ = 3,7) (IR. 1695 cm"
,
1735 cm-1, 1780 cm-1) durfte wohl dasmax
Anhydrid-Acetat der Dicarbon¬
säure (LXIV) vorliegen. Die UV. -Kurve ist derjenigen der Oxy-dicarbonsäu-re (LXIII) ähnlich.
67) Die Behandlung der Oxy-dicarbonsäure (LXIII) mit Wasser-Pyridin-Essig¬säure-Gemisch ergibt nur das Ausgangsmaterial.

27 -
Ketons (LXVII) ins entsprechende Oxim (LXVin) übergeführt, welches erwar-
tungsgemäss vom Abbauprodukt (LVI) des Lanosterins (I) verschieden war .
H. CHn
XVII XXIV
HOOC ;HOOC
^
R=H LX//I; R=AC LXIHa LXtl
LXIV (Smp. 160°)
R0-R=AC LXV
R = H LXVI
OximLXVIII
LXVII
68) Es ist an dieser Stelle interessant zu bemerken, dass der Uebergang vomOxim (LXVIII) aus Euphadienol (X) zum antipodischen Oxim (LVI) aus Lano-
sterin (I) mit einer ähnlichen molaren Drehungsverschiebung(MD = + 191, 5°)verbunden ist, wie der Uebergang vom Euphenol (XVII) zum Tirucalle-
nol (XXXK) (MD = + 1740).

- 28 -
Besonders geeignet für eine direkte Verknüpfung mit dem Lanosterin (I)
schien das 3o<-Tirucallenol (LXXV) zu sein, da es aus der gut zugänglichen
o(-Elemadienolsäure (XII) leicht erhältlich ist, und weil bei diesem Produkt, in¬
folge der bereits vorhandenen CX-Konfiguration der Hydroxylgruppe am C-, die
Eliminierung der optischen Asymmetrie am Kohlenstoff 3 sich erübrigt. Die
<X-Elemadienolsäure (XII) wurde zunächst durch Reduktion mit Platinkatalysa¬
tor in die c<-Elemenolsäure (XXXIII) übergeführt, und aus der letzteren durch
sukzessive Behandlung mit Acetanhydrid-Pyridin und Diazomethan das Methyl-
ester-acetat (LXX) bereitet. Weiter gelangte man mittels einer Lithiumalumini--Q
umhydrid-ReduktionzumZi -3 oc, 21-Dioxy-tirucallen(LXXI), das bei der Acety-
lierung ein amorphes Diacetat (LXXII) lieferte. Durch partielle Verseifungmit einemo
Aequivalent äthanolischer Kalilauge bei Zimmertemperatur erhielt man à -3 <x - Ace-
toxy-21-oxy-tirucallen (LXXHI). Die Behandlung dieses letzteren Zwischenpro¬
duktes mit p-Toluolsulfosäurechlorid in Pyridin führte in guter Ausbeute zum Chlo¬
rid (LXXIV) ,das schliesslich mit Natrium und Butanol zum 3 ex -Tirucallenol
(LXXV) glatt reduziert werden konnte. Die Acetylierung des Alkohols (LXXV)
ergab quantitativ das 3c<-Acetoxy-tirucallen(LXXVa). Die Konstitution LXXVa
dieser Verbindung konnte bestätigt werden, indem es gelang, Tirucallenol
(XXXIX) in Stellung 3 milde zu Tirucallenon (LXXVI) 'zu oxydieren und durch
Reduktion des letzteren mit Platin in Essigester-Eisessig-(1:1)-GemischTiru¬
callenol (XXXIX) und 3<X-Tirucallenol (LXXV)71* zu isolieren.
XXXIX
LXXV
LXXVaXXXIX LXXVI
Das auf diese Weise durch Reduktion erhaltene 3<X-Tirucallenol (LXXV)
wurde als Acetat identifiziert; im Gemisch mit dem aus der CK-Elemadienol-
säure (XII) gewonnenen Präparat ergab es keine Schmelzpunktdepression.
69) Siehe Fussnote 40.
70) D.W. Haines & F.L. Warren, J.chem.Soc. 1950, 1562.
71) Siehe Fussnote 17.

- 29 -
HO-
H XOOH
K *CHr>
RO'
RO''
R=H R4=0H LXXI
R=Ac P,= OAc LXXII
R=>AcR<=OH LXXIII
R=Ac R<=CL LXXIV
I
R=H LXXV; R=Ac LXXVa
tS?"t£p~.HOOC>. H^H3
HOOC Ni-
LXXVII LXXVHIRO'' ^
^ °
R=H LXXIX
R=AC LXXIXa
oA- v*>
AcO''
AcOLXXXI

- 30 -
Die weitere Oxydation des 3 W-Acetoxy-tirucallens (LXXVa) mit Chrom-
(VI)-oxyd lieferte das A -3*-Acetoxy-7,ll-diketo-tirucallen (LXXVII), das,c. o
mit Selendioxyd im Dioxan erhitzt, das ölige A ' -3 tf-Àcetoxy-7,11,12-trike-
to-tirucalladien (LXXVHI) ergab. Das Vorliegen der Dientrion-Gruppierung in
LXXVin wurde durch die Lage des UV. -Absorptionsmaximums (288 m/u,
72)log £ = 3,99) bestätigt. Gleiche UV.-Maxima wurden von Y.Mazur 'für
das Produkt (LXXXII) aus <X-Elemadienolsäure (Xu) und von W.Voser für73)
die Verbindung (L) aus Lanosterin (I) 'angegeben.
H. XO0H
AcO' 0
LXXXIIAcO
,5;8
Durch Oxydation des à ' -3 ix-Acetoxy-7,11,12-triketo-tirucalladiens
(LXXVin) mit Wasserstoffperoxyd erhielt man die Dicarbonsäure (LXXIX).
Wie bereits in der Euphadienol-Reihe, versagte auch hier der Versuch einer ther¬
mischen Aromatisierung. Zum gewünschten Ziel führte wiederum die bereits
besprochene Variante der Aromatisierung. Je nach der Dauer der Hydrolyse
erhielt man nach der Acetylierung verschiedene Produkte. Sechsstündiges Ste¬
hen der acetylierten Verbindung mit Wasser lieferte als Hauptprodukt des Neu¬
tralteils das Anhydrid (LXXX), nach dreitägiger Dauer der Hydrolyse konnte
aber das gesuchte Aromatisierungsprodukt (LXXXI) in massiger Ausbeute iso¬
liert werden.
Das erhaltene Produkt schmilzt bei 116 und weist eine spez. Drehung von
-143° auf, während das entsprechende Produkt aus Lanosterin (LUI) einen
Schmelzpunkt von 116 und eine[°CD von +146 hat. Die zwei Verbindungen wei¬
sen identische Absorptionskurven sowohl im UV. wie auch im IR. auf (siehe Fig. 1
und Fig. 2).
72) Y. Mazur, Diss. E.T.H. Zürich (1952) \ma% 288 m/d., log £ = 3,84.
73) W. Voser, Diss. E.T.H. Zürich (1951) \max 285 m/±, log e = 4,0.

31
/-v -~"S. /
/
2
1
logZ
280 260 240 220 mjj
Fig. l
UV. -Spektren 1. Acetoxy-phenol-lacton (LIII) aus Lanosterin (I)
2. Acetoxy-phenol-lacton (LXV) aus Euphadienol (X)
3. Acetoxy-phenol-lacton (LXXXI) aus Elemadienolsäure (XII)
Die Spektren der einzelnen Acetoxy-phenol-lactone sind gegeneinander um
je eine Einheit von log £ verschoben.
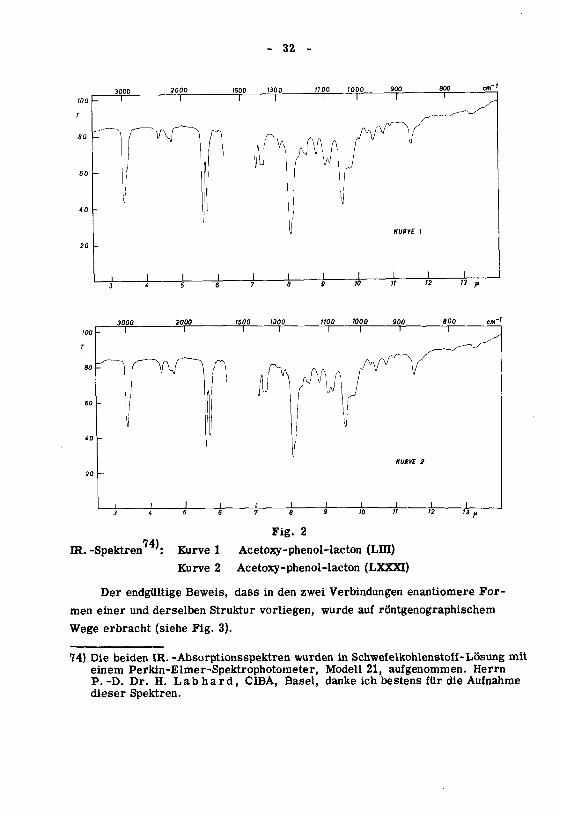
- 32 -
IR. -Spektren74).
Fig. 2
Kurve 1 Acetoxy-phenol-lacton (LID)
Kurve 2 Acetoxy-phenol-lacton (LXXXI)
Der endgültige Beweis, dass in den zwei Verbindungen enantiomere For¬
men einer und derselben Struktur vorliegen, wurde auf röntgenographischem
Wege erbracht (siehe Fig. 3).
74) Die beiden IR. -Absorptionsspektren wurden in Schwefelkohlenstoff-Lösung mit
einem Perkin-Elmer-Spektrophotometer, Modell 21, aufgenommen. Herrn
P.-D. Dr. H. Lab hard, CIBA, Basel, danke ich bestens für die Aufnahme
dieser Spektren.

- 33
•. 1•. *
t .• •
.• »
•
.. î^fr £
Verbindung (Lin) aus
Lanosterin (I)Verbindung (LXXXI) aus
oc-Elemadienolsäure (Xu)
75)
J76)Fig. 3
Die Diagramme wurden nach der Buerger'sehen Präzessionsmethode'
aufgenommen und stellen analoge Ebenen des reziproken Gitters dar. Aus der
völligen Uebereinstimmung der Reflexe, sowohl nach Lage wie nach relativer
Intensität, kann auf Gleichheit der Elementarzellen bei beiden Substanzen ge¬
schlossen werden.
Diese Versuche bestätigen zunächst die Ableitungen über die Struktur der
TriterpenederTirucalladienol-Gruppe, insbesondere sind sie für die Haftstelle
der langen Seitenkette beweisend. Zudem liefern sie eine konfigurative Ver¬
knüpfung der Kohlenstoffatome 3, 13, 14, 17, 20 des Tirucalladienols (II) mit
den entsprechenden Zentren des Lanosterins (I), somit aber auch mit dem
System des Glyzerinaldehyds.
75) Prof.Dr. F. Laves & Dr. A. Niggli, Mineralogisches Institut der Eidgenös¬sischen Technischen Hochschule, Zürich, dankeich bestens für die Aufnahmeder Röntgendiagramme.
76) M. J. Buerger, The Photography of the Reciprocal Lattice, ASXRED-Mono-
graph Nr. 1, 1944.

- 34 -
Obwohl in den zwei Abbauprodukten (Lin) und (LXXXI) die optische Asym¬
metrie des Kohlenstoffatoms 10 aufgehoben worden ist, ermöglicht die oben be¬
schriebene Abbaureihe, auch für dieses asymmetrische C-Atom des Tirucalla-
dienols (II) eine eindeutige Konfigurationsbestimmung vorzunehmen.
In der Oxy-dicarbonsäure (LXXIX), die sich aus dem 3cK-Tirucallenol (LXXV)
ableitet, muss das Kohlenstoffatom 10 eine anguläre Gruppe tragen, da sonst
das Dienon-System des Ringes B sich sofort in die tautomere phenolische Form
umwandeln würde.
» m » a a h is art
f
J5ÖÖ 30Ö5 25ÖÖ 20Ö5 JSÖÖ WÖÖ iTOÖ Ï6ÔO tSÖÖ MOD Ï5Ô5 ÖÖÖ JlÖÖ tOÖÖ 900 90Ö 7o5~ji
J5ÖÖ JOrö SSOO JoÖÖ «WO tBÖÖ I7ÖÖ Ï6ÔÔ Ï50Ô MÖÖ Ï3ÛÔ Î2Ô5 t(j00 ttOÖ 90Ö BOÖ 700 ji
Fig. 4 77)
IR. -Spektren: Kurve 1 Oxy-dienon-dicarbonsäure (LI)
Kurve 2 Oxy-dienon-dicarbonsäure (LXXDC)
77) Die beiden IR. -Absorptionsspektren wurden in Kaliumbromid-Phase mit ei¬
nem Baird-Spektrophotometer aufgenommen.

- 35 -
Da beim Lanosterin (I) die Anwesenheit einer angularen Methylgruppe am
C10 bewiesen ist,und da ferner die Dicarbonsäuren (LI) und (LXXDX), in
welchen die Kohlenstoffatome 3, 13, 14, 17, 20 eine antipodische Konfiguration79)
aufweisen, nicht enantiomere,wohl aber diastereomere Verbindungen dar¬
stellen, muss daraus gefolgert werden, dass in beiden Dicarbonsäuren (LI) und
(LXXDX) die C-g-ständige Methylgruppe die identische ß-Lage besitzt. Dies
gilt dann selbstverständlich auch für die Stammverbindung Tirucalladienol (ü)
unti für alle Vertreter der Tirucalladienol-Gruppe.
78) Für diese Methylgruppe des Lanosterins (I) steht dieß -Lage fest. Vergl.Fussnote 26.
79) Dies geht aus dem Vergleich der physikalischen Eigenschaften der zwei Ver¬
bindungen hervor.
(LI) Smp. 243° wD. -79°
(LXXDX) Smp. 208° WD- -89,5°
Vergl. auch die IR. -Spektren auf S. 34.

- 36 -
Die Konfigurationsverhältnisse am C„, Cg, Cg, C«. bei gesättigten
7,11-Diolen der Tirucallan-Reihe '
Die konfigurativen Verhältnisse an den Kohlenstoffatomen 13, 14, 17 bei
im Ringsystem gesättigten Verbindungen der Tirucallan-Reihe (IV) bedin¬
gen eine bestimmte Stereochemie am Cß und Cg, und diese gibt ihrerseits An-
lass zu Reaktionen, die einen verschiedenen Verlauf haben als die analogen
Versuche in der Lanostan-Reihe (IQ). Solche Unterschiede sollen an dieser
Stelle näher erörtert werden.o
Bereits vor Jahren wurde gezeigt, dass die Reduktion von A -7,11-Di-
keto-lanosten (LXXXin) mit Zink und Eisessig in nahezu quantitativer Ausbeute
i Fig,82)
das gesättigte 7,11-Diketo-lanostan (LXXXIV) liefert ', welches die in Fig. 5
räumlich dargestellte trans-anti-trans-anti-trans Konfiguration besitzt
Fig. 5
In Uebereinstimmung mit dieser Konfiguration haben D.H. R. Bar ton
und N.J. Ho Ine s s 'beobachtet, dass unter milden Bedingungen keine Dehy¬
drierung von 7,11-Diketo-lanostan (LXXXTV) mit Selendioxyd stattfindet, was
für die trans-Anordnung der H-Atome am Cg und Cg spricht. Anders gestalten
sich hingegen die Verhältnisse bei Verbindungen der Tirucallan-Reihe. So. z.B.Q
führt wohl die Reduktion von A -7, ll-Diketo-euphen(LXXXIX) mit Zink und Eis¬
essig zu einem gesättigten Diketon (XC), dieses besitzt aber keinesfalls trans¬
Verknüpfung der Ringe B und C (Fig. 6).
80) Siehe Fussnote 4.
81) E. Kyburz, Diss. E.T.H. Zürich, S. 28 (1954).82) Analoge Verhältnisse wurden auch bei Steroiden festgestellt: vergl. D.H.R.
Barton & N.J. Holness, J.chem.Soc. 1952, 78.

37
XCib
rwpS -ffS^LXXXIX
XCIV

- 38 -
Fig. 6
Gegen die Annahme der trans-Verknüpfung spricht zunächst die Tatsache,
dass bereits die Behandlung des gesättigten Dions (XC) mit Alkalien genügt, um
diese Verbindung zum Ausgangsprodukt (LXXXIX) zu dehydrieren '.
Die cis-Verknüpfung der Ringe B/C bei XC lässt sich ferner leicht deuten,
wenn man diese Verbindung im Lichte der Konstellationsanalyse näher betrachtet.
Würde man nämlich für das gesättigte Dion (XC) eine normale anti-trans-Verknüp-
fung der Ringe B und C voraussetzen, so hätten, sofern Ring C in Sesselform vor¬
liegen würde, sowohl diey3 -ständige Bindung am C. „ als auch die o< -ständige
Bindung am C.. axiale Lage. Da nun über solche diaxiale Bindungen der fünf-
gliedrige Ring D nicht geschlossen werden kann, wäre man gezwungen, in Ver¬
bindung (XC) den Ring C in Wannenform anzunehmen (Fig. 7) .
83) D.H.R. Barton, J.F. McGhie, M.K. Pradhan & S.A. Knight, J.ehem.Soc.
1955, 876.
KTChristen, Diss. E.T.H. Zürich (1952).Analoge Verhältnisse wurden von Y. Mazur in der Elemadienolsäurereihe
festgestellt.Y. Mazur, Diss. E.T.H. Zürich (1953).
84) Die konfigurativ symmetrische Anordnung der Zentren 5, 10 einerseits und
13, 14 anderseits zeigt, dass die zweite, formell mögliche trans-Verknüpfungder Ringe B und C (vergleiche Formel (XCb) ) Wannen-Konstellation des Rin¬
ges B zur Folge hat.

- 39 -
Fig. 7
Die beiden möglichen Raumformeln (XCc) und (XCd) erlauben hingegen die
Annahme der Sesselform für sämtliche drei Sechserringe und sind somit a p r i85)
ori gegenüber Verbindungen (XCa) und (XCb) als stabiler zu betrachten '.
XCa XCb XCc XCd
Anderseits ist die Konfiguration des gesättigten 7,11-Diketo-euphans (XC), infolge
der Anwesenheit zweier KetogruppenamC^undamC.j, thermodynamisch kontrol¬
liert und darf daher nicht auf eine sterisch einseitig verlaufende Absättigung der
86)Doppelbindung durch den naszierenden Wasserstoff zurückgeführt werden '.
Der Entscheid zwischen den beiden möglichen cis-Formen (XCc) und (XCd) (siehe
85) Es sei daran erinnert, dass, für den Fall des Cyclohexans, der UebergangSessel-Wanne mit einem Energie-Unterschied von 5,4 kcal/Mol verbunden ist.
C.W. Beckett, K.H. Pitzer & R. Spitzer, J.Amer.Chem.Soc. 69, 2488
(1947).Die höhere Stabilität des trans-anti-cis-Perhydrophenanthrens gegenüber dem
trans-syn-trans-Perhydrophenanthren wurde schon von W.S. Johnson, J. Amer.
Chem.Soc. 75_, 1498 (1953) hervorgehoben.86) Dies geht bereits aus der Tatsache hervor, dass unter analogen Versuchsbe¬
dingungen in der Lanostan-Reihe (III) lediglich diejenige Verbindung (LXXXTV)isoliert wird, deren Bildung nur durch Inversion des primären Zwischenpro¬duktes am C„ zu verstehen ist.

- 40 -
Fig. 8 und 9) kann dennoch gefällt werden. Die Modellbetrachtung zeigt nämlich,
dass bei 0<-Lage der zwei Wasserstoffatome am CQ und Cn die Methylgruppen
am C-Q und C^ sehr stark gegeneinander gedrückt werden '.
H £",
H V
Fig. 8 Fig. 9
Im Einklang mit der bevorzugten Konfiguration (XCd) des 7,11-Diketo-euphans
(XC) scheinen übrigens die in der Folge beschriebenen Versuche zu sein. Die Reduk¬
tion von 7,11-Diketo-euphan (XC) mit Lithiumaluminiumhydrid liefert in guter Aus¬
beute ein gesättigtes Diol (XCI), welches hier als (XCIa) formuliert wird. Die Zutei¬
lung der axialen «X-Lage beider Hydroxylgruppen wurde zunächst in Analogie zu
beschriebenen Experimenten getroffen, da, wie bekannt, die Reduktion von ge¬
hinderten Ketogruppen mit Lithiumaluminiumhydrid zu axialen Hydroxylen
führt ' '. In Uebereinstimmung damit lassen sich die zwei Hydroxylgruppen
unter normalen Bedingungen (Acetanhydrid- Pyridin über Nacht) nicht acetylieren.
Die Tatsache, dass die entsprechende C7-ständige Hydroxylgruppe des 7,11-Dioxy-90)
lanostans (LXXXV) ,erhalten durch Reduktion mit Lithiumaluminiumhydrid des
7,11-Diketo-lanostans (LXXXIV), äquatoriale Lage besitzt und demzufolge leicht
acetylierbar ist, spricht bereits für eine grössere Hinderung der Substituenten
87) Da bis heute unseres Wissens kein analoger Fall in der Literatur beschrie¬
ben wurde, muss hier von einer quantitativen Abschätzung der energetischenVerhältnisse abgesehen werden.
88) Siehe Fussnote 18.
89) 11-Ketone der Steroid-Reihe liefern z.B. bei Reduktion mit Lithiumaluminium -
hydrid ausschliesslich IIa -Oxyverbindungen.90) Siehe Fussnote 81.

- 41 -
am C„ in den gesättigten Tirucallan-Abkömmlingen. Zur gleichenAnnahme zwin¬
gen Ketalisierungsexperimente mit Aethylendithioglykol, welche nur bei 7-Keto-
verbindungen mit Lanostan-Gerüst(III) ', nicht aber bei solchen mit Tirucallan-92)
Gerüst (IV) 'zur Bildung von Thioketalen führen.
Zugunsten der Konfiguration (XCIa) des beschriebenen 7,11-Dioxy-euphans
(XCI) kann schliesslichfolgendes Argument aufgeführt werden: Während, wie be¬
reits erwähnt, kurze Behandlung des Diols (XCI) mit Acetanhydrid und Pyridin nicht
die acetylierte Verbindung (XCIb) ergibt, führt überraschenderweise eine drei¬
tägige Einwirkung des gleichen Gemisches auf das Diol (XCI) zur Ausbildung ei¬
nes konjugierten Diens, für welches aus dem UV. -Spektrum ( X„„
240 mu;
qq\ max /
log £ = 4,2) die Konstitution (XCII) folgt '.
Zu diesem Resultat lässt sich folgendes bemerken:
a) Die glatte Bildung des Diens (XCII) ist, infolge des hier in Betracht zu ziehen¬
den jonischen Reaktionsmechanismus, dann am besten zu verstehen, wenn in
Verbindung (XCI) je eine der zwei Hydroxylgruppen antiparallel zu einem Was¬
serstoffatom der Ringverknüpfungsstelle zu liegen kommt. Dies verlangt aber
axiale Lage beider Wasserstoffatome am Cß und Cg, eine Forderung, die un¬
ter all den theoretisch möglichen Konfigurationen desAn-dions(XC)nur im Iso¬
meren (XCd) erfüllt ist94* 95*.
b) Der glatte Uebergang des Diols (XCI) in das Dien (XCII) unter so milden Be¬
dingungen scheint ferner für eine höhere sterische Stabilität der ungesättigten
Verbindung (XCII) gegenüber der gesättigten Verbindung (XCI) zu sprechen,
was wiederum auf eine gezwungene cis-Verknüpfung der Ringe B und C hinwei¬
sen könnte .
91) W. Voser, Diss. E.T.H. Zürich (1952).92) Privatmitteilung von P.-D. Dr. O. Jeger.93) Das A7; 9(H)-3ß -Acetoxy-euphadien gibt im UV. folgende Werte: (X
240 m/j; log £ =4,2). M. Vilkas, Bull.Soc.chim.France 1950 582.ax
94) Es ist in diesem Zusammenhang von Bedeutung, dass eine ähnliche trans-
Eliminierung bei nicht acetylierbaren llcx-Oxy-Verbindungen der Steroid-Rei-he mit unnatürlicher ß -Konfiguration am C9 beobachtet worden ist. A. Craw-
shaw, H.B. Henbest, E.R.H. Jones & A.A. Wagland, J.ehem.Soc. 1955,3420.
95) Sind die zwei Wasserstoffe beide in der (X-Lage, so können sie nur äquatori¬al sein (vergl. Fig. 8). 8. g
96) Diese Schlussfolgerung lässt sich aber nicht auf die A ' -ungesättigten Ver¬
bindungen mit Tirucallan-Gerüst (IV) übertragen.

- 42 -
Dass die Abspaltung der zwei Hydroxylgruppen in der Verbindung (XCI)
wohl stereospezifisch verläuft, wird ferner bestätigt durch das Verhalten des iso¬
meren Diols (XCin) mit diäquatorialer Lage der Hydroxylgruppen, welches aus
7,11-Diketo-euphan (XC) durch Reduktion mit Natrium und Propanol erhältlich ist.
Unter normalen Bedingungen der Acetylierung liefert diese Verbindung das Diacetat
(XCIV).
Die wichtigsten sterischen Einwirkungen auf die zwei Ketogruppen im 7,11-Di¬
keto-euphan (XC) sind aus Fig. 9 ersichtlich und in Tabelle 2 zusammengestellt.
Tabelle 2
Ketogruppe am C Wechselwirkung mit
a C„ Methyl am C^.b Methylen C
15
c Methyl am C<3a' C.j Methyl am C-o
b' Methylen C15
Während die sterischen Effekte a, a' und b, b' ungefähr die gleiche Grösse
aufweisen dürften, ist die Ketogruppe am C7 noch zusätzlich durch die Methy1-97)
gruppe am C.o stark beeinflusst '. Unter Zugrundelegung der hier wahrschein¬
lich gemachten Konfiguration für das 7,11-Diketo-euphan (XCd) darf daher darauf hin¬
gewiesen werden, dass in dieser Verbindung die Ketogruppe am C» sich infolge der
grösseren sterischen Hinderung reaktionsträger als die Ketogruppe am C«« er¬
weisen wird.
Auf Grund der obigen Ueberlegungen scheint es zweckmässig, für den noch
unbekannten, gesättigten Kohlenwasserstoff Tirucallan die Konfiguration (IV) vor¬
zuschlagen.
97) Die Wechselwirkung der C»-Ketogruppe mit dem Methyl am C13 im Tirucal-
lan-Gerüst (IV) ist gleich gross wie diejenige der Ci2-Ketogruppe mit dem
Methyl am Cjg im Gerüst des cx-Amyrins (XCVI). Diese letztere Keto¬
gruppe erweist sich gegenüber den üblichen Carbonylreagentien als inert.
(Vgl. T. Lyssy, Diss. E.T.H. Zürich, 1955.)
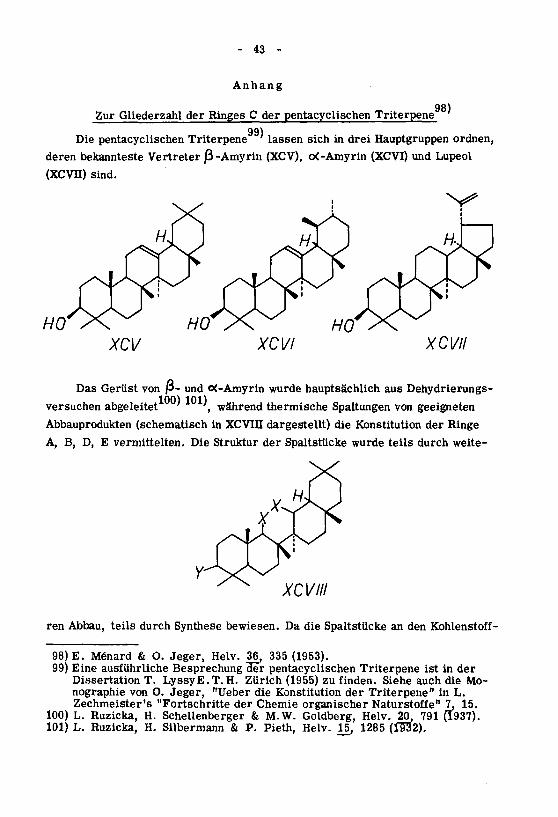
- 43 -
Anhang
Zur Gliederzahl der Ringes C der pentacyclischen Triterpene
99)
98)
Die pentacyclischen Triterpene""' lassen sich in drei Hauptgruppen ordnen,
deren bekannteste Vertreter /3-Amyrin (XCV), <X-Amyrin (XCVI) und Lupeol
(XCVn) sind.
N^
XCV XCVI XCVII
Das Gerüst von ß- und o<-Amyrin wurde hauptsächlich aus Dehydrierungs¬
versuchen abgeleitet '
,während thermische Spaltungen von geeigneten
Abbauprodukten (schematisch in XCVIII dargestellt) die Konstitution der Ringe
A, B, D, E vermittelten. Die Struktur der Spaltstücke wurde teils durch weite-
XCVIII
ren Abbau, teils durch Synthese bewiesen. Da die Spaltstücke an den Kohlenstoff -
98) E. Ménard & O. Jeger, Helv. 36, 335 (1953).99) Eine ausführliche Besprechung 3er pentacyclischen Triterpene ist in der
Dissertation T. LyssyE.T.H. Zürich (1955) zu finden. Siehe auch die Mo¬
nographie von O. Jeger, "Ueber die Konstitution der Triterpene" in L.
Zechmeister's "Fortschritte der Chemie organischer Naturstoffe" 7, 15.
100) L. Ruzicka, H, Schellenberger & M.W. Goldberg, Helv. 20, 791 (1937).101) L. Ruzicka, H. Silbermann & P. Pieth, Helv. 15, 1285 (1532).

- 44 -
atomen 8 und 14 (nach der Numerierung der Stammverbindung) Methylgruppen
tragen, war die Gliederzahl des Ringes C nicht sicher, weil die eine der
Methylgruppen aus einem Brückenmethylen hätte entstanden sein können.
Um Klarheit über die Zahl der Kohlenstoffatome im Ring C des ß- und
des of-Amyrins zu erlangen, wurden die Acetate der zwei Amyrine mit Persäu¬
ren oxydiert, wobei die Verbindungen (XCIX) und (C) entstanden '.
IR. (>CO) = 1700 cm-1 IR. (>CO) = 1694 cm"1
Die IR. -Absorptionsspektren von (XCIX) und (C) deuten auf Sechsringketo-
ne hin. Weitere Oxydation der Ketone (XCIX) und (C) mittels Salpetersäure führ
te zu den Dicarbonsäuren (CI) und (CII), deren Anhydride (CHI) und (CIV) pyro-
lysiert wurden, wobei die Norketone (CV) und (CVI) entstanden '.
\>' cv \>' CVI'
cvii
IR. (>CO) = 1735 cm'1 IR. (>CO) = 1735 cm
102) A. Meyer, O. Jeger, V. Prelog & L. Ruzicka, Helv. 34, 747 (1951).
- 45 -
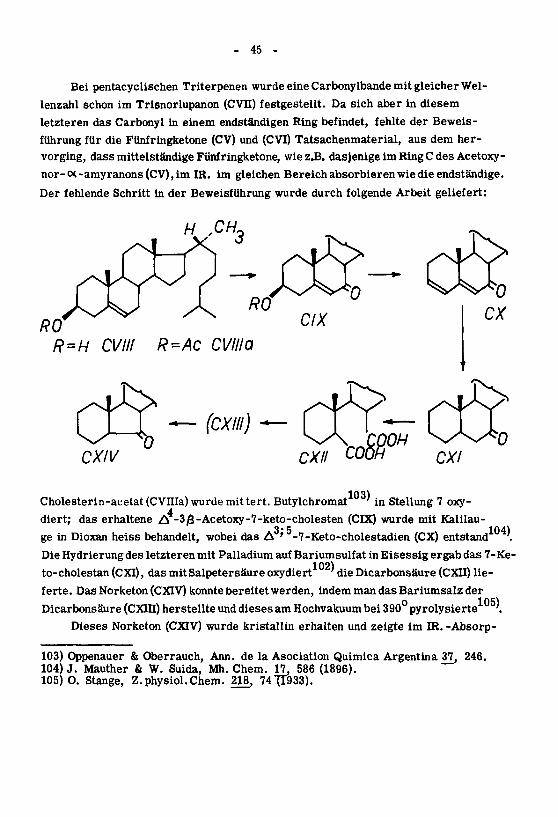
Bei pentacyclischen Triterpenen wurde eine Carbonylbande mit gleicher Wel¬
lenzahl schon im Trisnorlupanon (CVII) festgestellt. Da sich aber in diesem
letzteren das Carbonyl in einem endständigen Ring befindet, fehlte der Beweis¬
führung für die Fünfringketone (CV) und (CVI) Tatsachenmaterial, aus dem her¬
vorging, dass mittelständige Fünfringketone, wie z.B. dasjenige im RingC des Acetoxy-
nor-<X-amyranons(CV),im IR. im gleichen Bereich absorbieren wie die endständige.
Der fehlende Schritt in der Beweisführung wurde durch folgende Arbeit geliefert:
HsSCH3
R=H CI//// R=Ac Milla
(CXIIl)
CXIV CX/I COÖH
Cholesterin-acetat (CVIIIa) wurde mit tert. Butylchromat'in Stellung 7 oxy-
4diert; das erhaltene A -3/3-Acetoxy-7-keto-cholesten (CIX) wurde mit Kalilau¬
ge in Dioxan heiss behandelt, wobei das A ' -7-Keto-cholestadien (CX) entstand '.
Die Hydrierungdes letzteren mit Palladium auf Bariumsulfat in Eisessig ergab das 7-Ke-
to-cholestan (CXI), das mit Salpetersäure oxydiert'die Dicarbonsäure (CXII) lie¬
ferte. Das Norketon (CXIV) konnte bereitetwerden, indem man das Bariumsalz der
Dicarbonsäure (CXIII) herstellte und dieses am Hochvakuum bei 390° pyrolysierte '.
Dieses Norketon (CXIV) wurde kristallin erhalten und zeigte im IR. -Absorp-
103) Oppenauer & Oberrauch, Ann. de la Asociation Quimica Argentina 37, 246.
104) J. Mauther & W. Suida, Mh. Chem. 17, 586 (1896).105)0. Stange, Z.physiol.Chem. 218, 747J933).

- 46 -
tionsspektrum'luu; eine Carbonylbande bei 1736 cm ±. Somit wurde der Beweis
erbracht, dass Ketone sowohl an endständigen wie auch an mittelständigen Fünf¬
ringen im Infrarot bei gleicher Wellenlänge absorbieren. Dies beweist ferner,dass die Ringe C der Verbindungen (XCV) und (XCVI) und derjenigen, die mit
den letzteren auf chemischem Wege verknüpft worden sind, ohne Zweifel sechs-
gliedrig sind.
106) In Schwefelkohlenstoff-Lösung mit einem Perkin-Elmer-Spektrographen auf¬genommen.

- 47 -
EXPERIMENTELLER TEIL1*
I. Herstellung des Acetoxy-phenol-lactons (LUI)
aus Lanosterin (I)
A5;8-3/3-Acetoxy-7,ll, 12-triketo-lanostadien (L)2*
300 mg 3/3-Acetoxy-7,ll-diketo-lanostan (XLIX) wurden im Mörser mit 400 mg3
Selendioxyd verrieben, dann in 7 cm abs. Dioxan aufgeschlemmt und im Ein¬
schlussrohr 4 Stunden auf 230 erhitzt. Das entstandene Selen mit dem Selen¬
dioxyd wurde durch Watte abfiltriert, und die Lösung am Vakuum eingedampft,
der Rückstand in Aether aufgenommen und mit verd. Natronlauge und Wasser
gewaschen. Man erhielt 316 mg Oel, das teilweise aus Methanol kristallisierte.
Nach zweimaligem Umlösen aus Methanol erhielt man 150 mg schöne, orange
Nadeln vom Smp. 187-188,die mit einem vorhandenen Präparat von A ' -
3ß -Acetoxy-7,ll,12-triketo-lanostadien(L) keine Schmelzpunktdepression er¬
gaben.
A 5;8-3/3 -Oxy-7-keto-ll, 12-seco-lanostadien-ll. 12-disäure (LI) 2)
3 3150 mg der Verbindung (L) wurden in 7, 5 cm Feinsprit und 1, 5 cm
30%igem Wasserstoffperoxyd gelöst. In der Siedehitze wurden 15 cm 1-n.
äthanolischer Kalilauge zugetropft, und man Hess 30 Minuten am RUckfluss
kochen. Als die Sauerstoffentwicklung aufgehört hatte, wurde am Vakuum zur
Trockene eingedampft und der Rückstand mit Aether und Wasser aufgenom¬
men. Aus dem aufgearbeiteten sauren Teil erhielt man 100 mg eines weissen
1) Die Schmelzpunkte sind korrigiert und wurden in einer am Hochvakuum
evakuierten Glaskapillare bestimmt. Die optischen Drehungen wurden in
Chloroform in einem Rohr von 1 dm Länge gemessen. Die UV. -Spektrenwurden in Feinsprit auf einem Beckmann-Spektrophotometer aufgenom¬men.
2)W.Voser, Diss. E.T.H. Zürichs. 46(1953).J.F. McGhie &F.Cavalla, Chem. & Ind. 18, 744(1950).J.F. McGhie & F. Cavalla, J.chem.Soc. 1551, 744.

- 48 -
Schaumes, der aus Aceton-Hexan farblose Nädelchen vom Smp. 243 (Zers. )
lieferte.
MD
= -79° (c = 1,04 in Methanol)^
ÜV' Amax246T ' log£ = 4'°-
Es liegt die tricyclische Oxy-dicarbonsäure (LI) vor.
Pyrolyse der Oxy-dicarbonsäure (LI)
300 mg Substanz (Smp. 243-244°) wurden in ein Kugelrohr gebracht, das
am Wasserstrahlvakuum während 20 Minuten auf 270° erhitzt wurde, wobei
man deutlich die Kohlendioxyd-Entwicklung beobachten konnte. Man spülte mit
Methylenchlorid aus, dampfte zur Trockene ein, und das Reaktionsprodukt
wurde über Nacht acetyliert. Nach Aufarbeitung wurde das Produkt (215 mg)
an 6 g Aluminiumoxyd (Akt. II) chromatographiert. 240 cm Petroläther
eluierten 133 mg Oel, das erneut an 4 g Aluminiumoxyd (Akt. II) chromatogra¬
phiert wurde.
3120 cm Petroläther 8 mg Oel
200 cm3 Petroläther-Benzol (9 : 1) 31 mg Nadeln
80 cm3 Petroläther-Benzol (1 : 1) 46 mg Nadeln
Die kristallinen Fraktionen wurden aus Methanol umkristallisiert.
Smp. 115°.
CHjj = +144° (c = 0, 56 in Methanol)
UV. ^max276"1^ ' l0&E = 3'°
Xmax 284 mAi ' log £ = 2.98
Es liegt das Acetoxy-phenol-lacton (LIE) vor.
1) W. Voser (loc. cit.) [<=*]„ = - 67° (c = 1,05 in Methanol).

- 49 -
Acetylierung der Oxy-dicarbonsäure (LI)
3300 mg der Oxy-dicarbonsäure (LI) wurden über Nacht mit 4 cm Pyridin
3und 4 cm Acetanhydrid stehen gelassen. Das Acetanhydrid wurde mit Eis
hydrolysiert und alles mit Aether aufgenommen. Man schüttelte zweimal mit
verd. Schwefelsäure, und viermal wurde mit Wasser neutral gewaschen.
Nach Trocknung mit Natriumsulfat und Abdampfen des Aethers erhielt man
335 mg eines weissen Schaumes. Dieser wurde aus Aceton-Hexan umkristal¬
lisiert. Man erhielt feine Kristalle vom Smp. 222° (Zers.).
UV. \nax236myj. , log £ = 3,90.
Es liegt die Acetoxy-dicarbonsäure (LH) vor.
Pyrolyse der Acetoxy-dicarbonsäure (Lu)
202 mg Substanz wurden während 5 Minuten am Wasserstrahlvakuum auf
270-272° erhitzt. Das Reaktionsprodukt wurde in Methylenchlorid aufgenom¬
men, durch Watte filtriert, zur Trockene eingedampft, in Aether gelöst, mit
verd. Soda und dann mit Wasser gewaschen. Man erhielt 190 mg Neutralteil,
der gut löslich in Methanol, in Hexan und in Benzol war. In Petrolaether ge¬
löst, wurde das Produkt an 4 g Aluminiumoxyd (Akt. III) chromatographiert.3
120 cm Petroläther eluierten 89 mg eines farblosen Oels, das mit Methanol
bespritzt nach längerem Stehen kristallisierte. Smp. 115 .
IX1D = +149° (c = 1,39)
UV. Xfflax 276 m/jL , log 6 = 2,87.
Xmax284m/x , log£ = 2,86.
Es liegt das Acetoxy-phenol-lacton (Lin) vor.
Aromatisierung der Oxy-dicarbonsäure (LI)
32 g kristalline Oxy-dicarbonsäure (LI) wurden in 25 cm Acetanhydrid
3und 25 cm Pyridin 12 Stunden bei Zimmertemperatur stehen gelassen. Das
Reaktionsgemisch wurde dann bis zur leichten Trübung mit Eis versetzt und
noch weitere 24 Stunden bei Zimmertemperatur stehen gelassen. Die ent-

- 50 -
standenen Kristalle wurden abfiltriert, in Aether gelöst und gewöhnlich aufge¬
arbeitet. Man erhielt somit 478 mg eines farblosen Oels, das beim Bespritzen
mit Methanol kleine Kristalle vom Smp 115 lieferte. Zur Analyse gelangte ein
1 Tag am Hochvakuum getrocknetes Präparat vom Smp. 116.
C31H46°4 Ber* C 77'13 H 9'69 % Gef- C 77' n H 9'61
MD
= +146° (c = 1,39)
UV. ^max275m/a log £ = 2,85
^max2841^ l0«e = 2'84
IR. (CS2) 1745 cm"1 (Acetat)
1785 cm"1 ( S -Lacton)
Es liegt das Aromatisierungsprodukt (LIII) vor.
Aromatisierung der Oxy-dicarbonsäure (LI) im siedenden Acetanhydrid
3 350 mg Substanz in 5 cm Acetanhydrid und 5 cm Eisessig gelöst wurden
3 Stunden am RUckfluss gekocht. Nach der Aufarbeitung erhielt man 50 mg
Neutralteil, der aus Methanol in Nadeln kristallisierte, deren Schmelzpunkt
von 112 keine Erniedrigung im Gemisch mit dem Aromatisierungsprodukt
(Lin) erfuhr. Das UV. -Spektrum zeigte den gleichen Kurvenverlauf wie das¬
jenige von Acetoxy-phenol-lacton (LUI).
Verseifung des Acetoxy-phenol-lactons (LIII)
387 mg Substanz (LIII) wurden in 10 cm Methanol gelöst, mit einer Lö-
3sung von 150 mg Kaliumhydroxyd in 5 cm Methanol versetzt und 48 Stunden
stehen gelassen. Nach üblicher Aufarbeitung erhielt man 74 mg eines farb¬
losen Oels (LIV), das in Methanol, in Hexan und in Petroläther sehr gut lös¬
lich war, und das man auf keine Weise kristallin erhalten konnte.

- 51 -
Oxydation des Oxy-phenol-lactons (LIV) mit Chromsäure
3244 mg Substanz wurden in 106 cm Eisessig gelöst, dazu wurden 248 mg
3Chromsäure in 19 cm Wasser zugegeben, und die Reaktionslösung blieb 3 Stun¬
den bei Zimmertemperatur stehen. Nach gewöhnlicher Aufarbeitung erhielt man
230 mg eines gelben Oels, das an 5 g Aluminiumoxyd (Akt. HI) chromatographiert3
wurde. 120 cm Petroläther eluierten 181 mg eines Oels. Dieses konnte man
nicht zur Kristallisation bringen. Das Dinitro-phenyl-hydrazon dieser Verbin¬
dung schmolz bei 211-212°, das Oxim bei 163°.
C29H43°3N Ber- C 76'78 H 9> 58% Gef •C 76'80 H 9> 57%
[<*3D = +141,5° (c = 1,3)
UV. Xmax276m^ , log£ = 3,06
Xmax284mM , l°S t = 3,03
Es liegt das Oxim des Keto-phenol-lactons (LVI) vor.
II. Herstellung des Acetoxy-phenol-lactons (LXV)
aus Euphadienol (X)
A8-3/3-Oxy-euphen (XVH)
8 ^21 g A -3-Keto-euphen (LX) in 1050 cm abs. Aether wurden unter
o
Vibrieren zu 16 g Lithiumaluminiumhydrid in 500 cm Aether zugetropft. Nach
3den ersten 100 cm begann der Aether leicht zu sieden, und man regulierte die
weitere Zugabe so, dass der Rückfluss konstant blieb. Man liess das Reaktions¬
gemisch noch 4 Stunden auf dem Wasserbad stehen, und nach der Zerstörung
des überschüssigen Reagens mit Essigester wurde wie gewöhnlich aufgearbeitet.
Man erhielt 21 g kristallinen Alkohol (XVII) vom Smp. 119°.

- 52 -
A8-3/3 -Acetoxy-euphen (XXIV)^
3 321 g Euphenol (XVII) wurden in 105 cm Pyridin und 105 cm Acetanhydrid
gelöst und über Nacht stehen gelassen. Nach üblicher Aufarbeitung erhielt man
15, 5 g 3/3 -Acetoxy-euphen (XXIV) vom Smp. 127°.
A8-3/3-Acetoxy-7,11-diketo-euphen (LXI)^
o 8Bei einer Temperatur von 50 wurden zu einer Lösung von 26 g A -3/3 -
~_3 3
Acetoxy-euphen (XXIV) in 540 cm Eisessig und 130 cm Methylenchlorid 26 g
Chromsäure in 390 cm 90%igem Eisessig unter Vibrieren zugetropft. Bei der
gleichen Temperatur liess man das Reaktionsgemisch noch 4 Stunden stehen,
und nachher arbeitete man in üblicher Weise auf. Man erhielt 22 g eines gelben3
Oels, das an 350 g Aluminiumoxyd (Akt. III) chromatographiert wurde. 4250 cm
Petroläther eluierten 15,8 g gelbes Oel, das aus Methylenchlorid-Methanol
umkristallisiert 14, 2 g gelbe Kristalle vom Smp. 112° lieferte.
A,5;8-3ß-Acetoxy-7,11,12-triketo-euphadien (LXII)2*
35 g der Verbindung (LXI) wurden mit 8 g Selendioxyd und 70 cm abs.
Dioxan im Einschlussrohr 4 Stunden auf 170° erwärmt. Nach dem Erkalten
wurden das Selen und das überschüssige Selendioxyd durch Watte abfiltriert,
die Lösung am Wasserstrahlvakuum zur Trockene eingeengt, und der Rück¬
stand wurde in Aether aufgenommen, mit verd. Natronlauge und darauf mit
Wasser neutral gewaschen. Das erhaltene Oel (5,1 g ) wurde mit der gleichen
Menge aus einem früheren Ansatz an 200 g Aluminiumoxyd (Akt. III) chroma-3
tographiert. 3250 cm Petroläther eluierten 5,55 g dunkelgelbe Kristalle, di(
aus Methylenchlorid-Methanol umkristallisiert 1,9 g schöne, lange, dunkel¬
gelbe Nadeln vom Smp. 182 lieferten.
UV. Xmax28Bm^ . log£ = 4,078
IR. (CHClg)3*
1652 cm"1, 1680 cm"1, 1730 cm"1.
1)B.C. Roth, Diss. E.T.H. Zürich (1950).2) K. Christen, Diss. E.T.H. Zürichs. 41 (1952).
3) Auf einem Baird-Spektrophotometer aufgenommen.

- 53 -
A5>8-3/a-Oxy-7-keto-ll>12-seco-euphadien-ll,12-disäure (LXIII)*'
3 3300 mg der Verbindung (LXII) wurden in 15 cm Aethanol und 3 cm
30%igem Wasserstoffperoxyd gelöst und unter Rückfluss auf 90 erwärmt. Bei
dieser Temperatur wurden 30 cm 1-n. äthanolischer Kalilauge zugetropft, und man
erhitzte bis zur Beendigung der Sauerstoffentwicklung. Man dampfte die Flüssig¬
keit am Vakuum ein, versetzte den Rückstand mit verdünnter Schwefelsäure bis
zur Kongoreaktion, dann trennte man auf übliche Art in neutrale und saure
(231 mg) Teile auf. Nach einmaliger Umkristallisation der sauren Teile aus Metha¬
nol erhielt man 199 mg weisse Nadeln vom Smp. 180 (Zers. ).
C30H46°6 ' 2 H2° Ber- C 66>88 H 9.36 % Gef- c 66> 34 H 8. "%
[<=*]D
= -59, 5° (c = 0,886) (in Methanol) ^
m- ^ax246^ ' log£ = 4'°
Es liegt die 3 ji -Oxy-dicarbonsäure (LXIII) vor.
e• o
Aa'°-3/3-Acetoxy-7-keto-ll, 12-seco-euphadien-ll, 12-
disäure (LXHIa)
q
873 mg der Oxy-dicarbonsäure (LXIII) wurden in 10 cm Pyridin und3
10 cm Acetanhydrid über Nacht stehen gelassen. Nachdem das Acetanhydrid
mit Eis hydrolysiert war, wurde der Kolbeninhalt am Vakuum zur Trockene
eingedampft. In üblicher Weise wurde das Reaktionsprodukt in neutrale (101 mg)
und saure (690 mg) Teile getrennt. Aus Hexan umkristallisiert erhielt man die
Acetoxy-disäure als feines, kristallines Pulver vom Smp. 205° (Zers.).
UV- Xmax240m^ ' log£ = 3'97
Es liegt die Acetoxy-dicarbonsäure (LXIIIa) vor.
1) K. Christen, Diss. E.T.H. Zürichs. 41 (1952).K. Christen gibt eine Drehung von[o<] = + 6° (in Pyridin) an.

- 54 -
Verseifung der Acetoxy-dicarbonsäure (LXTIIa)
350 mg Substanz wurden in 20 cm verd. Sodalösung gelöst und 2 Tage bei
Zimmertemperatur stehen gelassen. Nach Ansäuern und Aufarbeiten erhielt
man feine Nadeln (aus Aceton) vom Smp. 190,die keine Mischschmelzpunkt¬
depression mit der Oxy-dicarbonsäure (LXI1I) ergaben.
UV- Xmax 246 mAL » l0& Ê = 3' 9-
Pyrolyse der Acetoxy-dicarbonsäure (LXIIIa)
117 mg Substanz wurden amWasserstrahlvakuum während 5 Minuten auf
270 erhitzt. Man beobachtete eine starke Kohlendioxyd-Entwicklung. Das
Reaktionsprodukt wurde in Methylenchlorid gelöst, durch Watte filtriert, zur
Trockene eingedampft, in Aether gelöst, mit verd. Soda und dann mit Wasser
gewaschen. Man erhielt somit 100 mg Neutralteil, der an 2 g Aluminiumoxyd3
chromatographies wurde. 60 cm Petroläther eluierten 22 mg Substanz, die
aus Methanol kristallisierte und einen Smp. von 158° aufwies.
UV- Xmax 310m^ . log£ = 3>08
^Schulter 234 mH- ' l0*£= 3'57
Es liegt ein unbekanntes Produkt vor '.
Aromatisierung der Oxy-dicarbonsäure (LXtll)
3 31, 427 g Substanz wurden in 18 cm Pyridin und 18 cm Acetanhydrid ge¬
löst, über Nacht stehen gelassen, mit Wasser bis zur leichten Trübung ver¬
setzt und weitere 24 Stunden stehen gelassen. Die entstandenen Kristalle wur¬
den abfiltriert. Nach mehreren Umkristallisationen aus Methanol war ihr
Schmelzpunkt konstant bei 160° (unter Zers. bei 260°).
UV- Xmax 244 mAL ' log £ = 3' 72
2}
Es liegt das unbekannte Produkt (LXIV) vor'
1) Siehe auf der nächsten Seite den Versuch der Aromatisierung in siedendem
Acetanhydrid.2) Siehe Fussnote 66) des theoretischen Teils.

- 55 -
Das Filtrat wurde am Wasserstrahlvakuum eingeengt, der Rückstand
in Aether gelöst und in saure und neutrale Teile (109 mg) getrennt. Letztere
3wurden an 1,1 g Aluminiumoxyd (Akt. HI) chromatographies. 80 cm Petrol-
äther eluierten 80 mg Oel, das aus Methanol als kleine Kristalle vom Smp. 116
ausfiel. Nach vierfacher Umkristallisation stieg der Smp. auf 122.
C31H46°4 Ber-C77»13 H 9,61% Gef. C 77,10 H 9, 62%
[o<]D = -64,7° (c = 0,99)
UV' Nnax 276 mP ' log £ = 2' 90
\nax 284 m^ ' log £ = 2'88
IR. (CS2) 1745 cm"1 (Acetat)
1785 cm"1 (S-Lacton)Es liegt das Aromatisierungsprodukt (LXV) vor.
Versuch der Aromatisierung der Oxy-dicarbonsäure (LXIII) im
siedenden Acetanhydrid
3 3105 mg Substanz wurden in 2, 5 cm Eisessig und 0, 5 cm Acetanhydrid
während 1 Stunde am Rückfluss gekocht. Nach der Aufarbeitung erhielt man
60 mg Neutralteile, die beim Bespritzen mit Methanol sofort kristallisierten. Smp. 160 .
UV. Xmax 318 m/J. , log £ = 3,35
XSchulter235n^ • l°*e = 3>92
Es liegt ein unbekanntes Produkt vor.
Verseifung von Acetoxy-phenol-laeton (LXV)
310 mg Substanz (LXV) wurden in 35 cm Methanol gelöst, mit einer Lö-
3sung von 525 mg Kaliumhydroxyd in 17 cm Methanol versetzt und 48 Stunden
stehen gelassen. Nach üblicher Aufarbeitung erhielt man 260 mg eines farblosen
Oels (LXVI), das nicht kristallisierte.
Oxydation von Oxy-phenol-lacton (LXVI) mit Chromsäure
3260 mg Substanz wurden in 106 cm Eisessig gelöst. Dazu wurden 260 mg
3Chromsäure in 20 cm Wasser zugegeben, und die Reaktionslösung blieb 3 Stun-

- 56 -
157 mg eines gelben Oels, das an 4, 5 g Aluminiumoxyd (Akt. HI) chromatogra-3
phiert wurde. 120 cm Petroläther eluierten 95 mg eines Oels, dessen Oxim
bei 105 schmolz.
[e*]D = -98,5° (c = 0,96)
m- Xmax276m^ ' log£ = 2'94
^max286^ ' logÊ = 2'84
Es liegt das Oxim des Keto-phenol-lactons (LXVIII) vor.
III. Herstellung des Acetoxy-phenol-lactons (LXXXI)
aus <x-Elemadienolsäure (XII)
p
A -3o<-Oxy-elemen-21-säure (XXXIII)
8* 24 35 g A
' -3o<-Oxy-elemadiensäure (XII) wurden in 100 cm Feinsprit
mit 250 mg Platinoxyd als Katalysator hydriert. Nach Aufnahme eines Mols wur¬
de aufgearbeitet, und man erhielt in quantitativer Ausbeute die Dihydro-säure
(XXXin) vom Smp. 230°.
Q
A -3o<-Acetoxy-elemen-21-säure (LXIX)
33, 5 g der Verbindung (XXXIII) wurden über Nacht mit 36 cm Pyridin und
336 cm Acetanhydrid acetyliert. Nach Aufarbeitung und zwei Umkristallisatio-
nen aus Methanol erhielt man 3,1 g Nadeln vom Smp. 250°.
Q
A -3o<-Acetoxy-elemen-21-säure-methylester (LXX)
3 g Acetoxy-säure (LXDC), gelöst in Aether, wurden mit einem Ueber-
schuss von 40% Diazomethan unter Eiskühlung versetzt. Nach 15-minutigem
Stehenlassen wurde der Aether am Vakuum abgedampft. Das erhaltene Produkt
wurde aus Methanol umkristallisiert, und man erhielt 2, 95 g Nadeln vom Smp.
134°.
A8-3o<21-Dioxy-tirucallen (LXXI)
Q
2,97 g des kristallinen A -3o£Acetoxy-elemen-21-säure-methylesters (LXX),

- 57 -
3in 160 cm abs. Aether gelöst, wurden unter Vibrieren zu einer Aufschwem-
mung von 3 g Lithiumaluminiumhydrid in 300 cm abs. Aether langsam zuge¬
tropft. Nach 2-stündigem Vibrieren bei einer Temperatur des Wasserbades von
50 wurde das überschüssige Reagens zuerst mit Essigester, dann mit verd.
Schwefelsäure zersetzt. Auf übliche Weise aufgearbeitet, erhielt man 2,9 g ei¬
nes feinkristallinen Produktes, das nach einmaliger Umkristallisation aus Me¬
thylenchlorid-Hexan bei 254° schmolz.
[c<]D = -17, 9° (c = 0,831 in Aceton)
Es liegt das Diol (LXXI) vor.
A8-3o< 21-Diacetoxy-tirucallen (LXXII)
3 3640 mg der Verbindung (LXXI) wurden in 5 cm Pyridin und 5 cm Acetan-
hydrid gelöst und über Nacht verschlossen bei Zimmertemperatur stehen gelas¬
sen. Man hydrolysierte das überschüssige Acetanhydrid mit Eis, und die Reak¬
tionslösung wurde auf übliche Weise aufgearbeitet. Man erhielt 750 mg eines
farblosen Oels, das aus keinem Lösungsmittel kristallin erhalten werden konnte.
A8-3e<-Acetoxy-21-oxy-tirucallen (LXXIII)
8 o
750 mg öliges A -3<x, 21-Diacetoxy-tirucallen (LXXII) wurden in 30 cm
Benzol gelöst, und die Lösung wurde auf Phenolphthalein mit 0, 21-n. äthanoli-3
scher Kalilauge neutralisiert. Dann wurden weitere 7,35 cm der Kalilauge
zugegeben (5% Ueberschuss) und das Gemisch 22 Stunden bei Zimmertempe-3
ratur stehen gelassen. Verbrauch der Lauge: 6,8 cm . Die neutrale Lösung
wurde am Wasserstrahlvakuum eingeengt, und die Aufarbeitung ergab im Neu¬
tralteil 730 mg Oel, das an 21 g neutralem Aluminiumoxyd (Akt. II) chromato-3
graphiert wurde. 100 cm Petroläther eluierten 77 mg des Ausgangsmaterials3
(LXXII), und 175 cm Petroläther-Benzol (1:1) eluierten 420 mg eines in klei¬
nen, schönen Rhomboedern kristallisierenden Produktes vom Smp. 115°. Zur
Analyse gelangte ein 2 Tage am Hochvakuum getrocknetes Präparat.
C32H54°3 Ber< C 79'18 H 10,89% Gef. C 78,96 H 11,18%
£°<lD = -54,40 (C = l,12)G
Es liegt das A -3<<-Acetoxy-21-oxy-tirucallen (LXXIII) vor.

- 58 -
A8-3o<-Acetoxy-21-chlor-tirucallen (LXXIV)^
120 mg des Mono-acetates (LXXIII) wurden mit 120 mg p-Toluolsulfo-3
säurechlorid in 3 cm Pyridin während 6 Stunden am Rückfluss gekocht. Nach
dem Erkalten wurde die braune Lösung in Aether aufgenommen, mit verd.
Schwefelsäure, mit verd. Sodalösung und mit Wasser gewaschen. Die ätheri¬
sche Lösung wurde über Natriumsulfat getrocknet und abdestilliert. Man er¬
hielt ein braun-gelbes Oel, das beim Bespritzen mit Methanol sofort kristalli¬
sierte. Nach zweimaligem Umlösen aus Methanol erhielt man 95 mg schöne,
weisse Nädelchen vom Smp. 156°. Das Analysepräparat wurde 3 Tage am
Hochvakuum getrocknet.
C32H53°2C1 Ber.C 76,07 H 10,57% Gef. C 75,71 H 10,78%
[=<]D = -34,40 (c=0,66)
A8-3o<-Oxy-tirucallen (LXXV)2^
507 mg Chlorid (LXXIV) wurden mit 5 g Natrium während zwei Stunden in
3100 cm abs. Butanol am RUckfluss gekocht. Das Butanol wurde am Wasserstrahl-
vakuum abgedampft und der Rückstand in einem Gemisch von Aether und Wasser auf¬
genommen. Die ätherische Schicht wurde neutral gewaschen, mit Natriumsulfat ge¬
trocknet und eingedampft, wobei 430 mg eines Oels erhalten wurden, welches aus
Methanol in verfilzten Nadeln vom Smp. 96 kristallisierte.
Zur Analyse gelangte ein dreimal umkristallisiertes Präparat, das bei 111
schmolz.
C30H52° Ber- C84>04 H 12,23% Gef. C 83,98 H 12,18%
[©<]D = -17, 6° (c = 0, 724)
A8-3o<-Acetoxy-tirucallen (LXXVa)
3 3313 mg Alkohol (LXXV) wurden über Nacht in 6 cm Pyridin und 6 cm
Acetanhydrid stehen gelassen. Nach gewöhnlicher Aufarbeitung erhielt man
300 mg Oel, das nach dreimaligem Umlösen aus Aceton-Wasser 207 mg feine,
1)A. Hiestand, Diss. E.T.H. Zürich (1949).2) Vorschrift beschrieben von H. Wyler, Diss. E.T.H. Zürich (1956).

- 59 -
bei 131° schmelzende Nadeln ergab. Das Analysenpräparat wurde 4 Tage am
Hochvakuum getrocknet.
C32H54°2 Ber- C81>64 H 11, 56% Gef. C81,42 H 11, 44%
t^ljj = -60,5 (c = 0,77)
Oxydation von A8-3ß-Oxy-tirucallen (XXXIX) zu A8-3-Keto-tirucallen (LXXVI)
3 3272 mg Substanz wurden in einer Lösung von 108 cm Eisessig, 21 cm
Wasser und 272 mg Chromsäure drei Stunden lang bei Zimmertemperatur ge¬
schüttelt. Mit Natriumbisulfitlösung wurde die überschüssige Chromsäurelö¬
sung reduziert, dann dampfte man am Vakuum ein und löste das Produkt in
Aether-Wasser. Nach der gewöhnlichen Aufarbeitung erhielt man 271 mg Oel,
das an 8,1 g neutralem Aluminiumoxyd (Akt. II) chromatographiert wurde.
200 cm Petroläther eluierten 206 mg kristalline Substanz vom Smp. 65.Das
Analysenpräparat wurde einen Tag am Hochvakuum getrocknet.
C30H50° Ber- C84>44 H 11,81% Gef. C 84,07 H 11, 56%
[c*]D = +30° (c = 0, 53)
8 8
Hydrierung von A -3-Keto-tirucallen(LXXVI) zu A -3o<-Oxy-tirucallen (LXXV)
150 mg Substanz (LXXVI) wurden unter Rühren bei Zimmertemperatur in
einer Lösung von 10 cm Eisessig-Essigester (1:1) mit 5 mg Platinoxyd als Ka-
3talysator hydriert. Nach Aufnahme von 6 cm Wasserstoff wurde die Reaktions¬
lösung aufgearbeitet, und man erhielt 145 mg Oel, das an 4, 5 g neutralem Alu-
miniumoxyd (Akt. n) chromatographiert wurde. Mit 200 cm Petroläther wur¬
den 93 mg Substanz eluiert, die, mit Methanol bespritzt, Kristalle vom Smp.
64 ergab. Nach optischer Drehung und Mischschmelzpunkt liegt das Ausgangs¬
produkt A8-3-Keto-tirucallen (LXXVI) vor. Mit 40 cm3 Petroläther-Benzol (1:1)
erhielt man 6 mg Kristalle vom Smp. 103°, die acetyliert Nadeln vom Smp.
125 ergaben. Diese zeigten keine Mischschmelzpunktdepression mit dem
A8-3 «.-Acetoxy-tirucallen(LXXVa), während û8-3ô -Acetoxy-tirucallen (XXXK)Q _
und A -3<X-Acetoxy-tirucallengemischteineSchmelzpunktdepressionvon 14 er¬
geben.

- 60 -
A8-3cK-Acetoxy-7,11-diketo-tirucallen (LXXVII)
Q Q
143 mg A -3 <X -Acetoxy-tirucallen (LXXVI) wurden in 12,7 cmEisessiggelöst.
Bei 45 wurde eine Lösung von 95 mg Chromsäure in 9 cm 90%igem Eisessig
unter Vibrieren langsam zugetropft. Man vibrierte noch 4 Stunden bei der glei¬
chen Temperatur. Nach Stehenlassen über Nacht wurde die überschüssige Chrom¬
säure mit Methanol zerstört, und die übliche Aufarbeitung ergab 145 mg eines
gelben Oels. Dieses wurde an 4, 5 g neutralem Aluminiumoxyd (Akt. II) chroma-3
tographiert, wobei 200 cm Petroläther 105 mg Oel eluierten, das, mit Metha¬
nol bespritzt, kleine, gelbe Kristalle vom Smp. 124° ergab. Nach zweimaligem
Umlösen aus Methanol gelangte ein Präparat, das 2 Tage am Hochvakuum ge¬
trocknet wurde, zur Analyse.
C32H50°4 Ber- C77'06 H 10,11% Gef. C 77,16 H 10,17%
[<=<]D
= -48° (c = 0, 902)
UV- Xmax213m^ ' loߣ = 3'9
A5;8-3c<-Acetoxy-7,11,12-triketo-tirucalladien (LXXVIII)
3468 mg Substanz (LXXVII) wurden mit 1,17 g Selendioxyd und 23 cm abs.
Dioxan im Einschlussrohr zugeschmolzen und während 14 Stunden im Bomben¬
rohr auf 195 gelassen. Nach dem Erkalten wurde das metallische Selen abfil¬
triert, Dioxan wurde am Wasserstrahlvakuum abgedampf, man löste den Trok-
kenrückstand in Aether und wusch diesen bei 0 zuerst dreimal mit verd. Na¬
tronlauge, dann neutral mit Wasser. Der Aether wurde über Natriumsulfat ge¬
trocknet, abdestilliert, und man erhielt 350 mg eines dunklen Oels. Dieses wur-
3de an 3, 5 g neutralem Aluminiumoxyd (Akt. II) chromatographiert, wobei 60 cm
Petroläther 218 mg Oel eluierten, das im UV. ein Maximum bei A 288 m/u° '
max /
mit log £ = 3, 99 hatte.
Alle Versuche, dieses Produkt kristallin zu erhalten, gingen fehl.
Es liegt A5;8-3c<-Acetoxy-7,11,12-triketo-tirucalladien (LXXVIII) vor.
A5;8-3o<-Oxy-7-keto-ll, 12-seco-tirucalladien-ll, 12-disäure (LXXK)
3 3190 mg Substanz wurden in 20 cm Aethanol und 4 cm 30% igem Wasserstoff-
3peroxyd gelöst. Darauf versetzte man das Gemisch bei Siedehitze mit 20 cm
einer 1-n. äthanolischen Kaliumhydroxydlösung. Als die Sauerstoff-Entwicklung

- 61 -
beendet war, wurde die Reaktionsflüssigkeit am Wasserstrahlvakuum abge¬
dampft, der Rückstand nach Ansäuern in Aether-Wasser gelöst, neutral gewa¬
schen, und der Aether nach Trocknen über Natriumsulfat abdestilliert. Die
164 mg Oel kristallisierten aus Aceton-Wasser in dünnen Nadeln vom Smp. 208
(Zers.), der bei den nachherigen Umkristallisationen konstant blieb.
C30H46°6' 2 H2° Ber- C 66>88 H 9' 36% Gef •C 66>93 H 8' 96%
[°<]D = -89, 5° (c = 0, 76) (in Methanol)
UV- Xmax244m^ ' log£ = 3'94
Acetoxy-phenol-lacton (LXXXI)
3 3879 mg der Oxy-dicarbonsäure (LXXK) wurden in 9 cm Pyridin und 9 cm
Acetanhydrid gelöst und über Nacht bei Zimmertemperatur stehen gelassen. Das
Reaktionsgemisch wurde dann bis zur leichten Trübung mit Eis versetzt und noch
weitere drei Tage bei Zimmertemperatur stehen gelassen. Darauf wurde es am
Wasserstrahlvakuum zur Trockene eingedampft, der Rückstand in Aether gelöst
und auf übliche Weise in saure und neutrale Teile getrennt. Der Neutralteil (249
mg) wurde mit Petroläther versetzt, die Lösung durchWatte filtriert, und das Filtrat
an4,29 gneutralem Aluminiumoxyd (Akt. in) chromatographiert. 40 cm Petroläther
eluiertenl9mgeinesOels, das aus Methanol sofort kleine Kristalle von Smp. 116
lieferte. Zur Analyse gelangte ein 24 Stunden am Hochvakuum getrocknetes Präparat.
C31H46°4 Ber- C 77'13 H 9' 61% Gef•C 76' 96 H 9>68%
t<=<]D = -143° (c =0,87)
UV. ^max2761»^ , log t = 3,035
Xmax284m^JU , log £ = 3,045
IR. (CS2) 1745 cm"1 (Acetat)
1785 cm"1 ( S -Lacton)
Es liegt das Aromatisierungsprodukt (LXXXf) vor.

- 62 -
C a Q
A ' -3o<-Acetoxy-7-keto-ll, 12-seco-tirucalladien-
11,12-disäureanhydrid (LXXX)
1,07 g der Oxy-dicarbonsäure (LXXIX) mit 10 cm3 Acetanhydrid und 10 cm3
Pyridin versetzt wurden über Nacht bei Zimmertemperatur stehen gelassen.
Dann wurde Eis bis zur leichten Trübung zugegeben. Nach 6 Stunden wurde am
Wasserstrahlvakuum zur Trockene eingedampft, die Reaktionsprodukte in
Aether gelöst und auf übliche Weise in saure und neutrale Teile aufgetrennt. Man
erhielt somit 358 mg Neutralteile, die mit Petroläther versetzt wurden. Das Un¬
lösliche wurde abfiltriert und getrocknet (340 mg). Dieses Produkt kristallisierte
aus Aether-Benzol und wies einen Schmelzpunkt von 230 (Zers. ) auf.
UV- Xmax 244 mAL ' log £ = 3> 94
Es liegt das Acetoxy-anhydrid (LXXX) vor.
Verseifung des Acetoxy-anhydrides (LXXX)
50 mg des trockenen Acetoxy-anhydrides (LXXX) wurden in 5 cm Feinsprit
gelöst und mit 250 mg Kaliumhydroxyd zwei Stunden am Rückfluss gekocht. Nach
üblicher Aufarbeitung erhielt man aus Aceton-Wasser Nadeln vom Smp. 208°
(Zers. ), deren Mischschmelzpunkt mit der Oxy-dicarbonsäure (LXXIX) keine
Depression ergab.
UV. Xmax 244 mAL , log £ = 3,94
IV. Reduktion von 7, 11-Diketo-euphan (XC)
A8;24-Euphadien (LXXXVII)X)
8 gEuphadienon (XVI) wurden mit 8 cm Hydrazinhydrat, 24 g Natrium äthylat
und 140 cm Aethanol im Einschlussrohr 15 Stunden auf 220° erwärmt. Das Reaktions¬
produkt wurde auf Eis gegossen, mit Schwefelsäure (1:1) auf Kongorot angesäuert, in
Petroläther aufgenommen, neutral gewaschen und zur Trockene gebracht. Der
Rückstand wurde einmal aus Methylenchlorid-Methanol umkristallisiert. Man
erhielt 5, 56 g farblose, derbe Nadeln vom Smp. 102.
1)B.C. Roth, Diss. E.T.H. Zürich (1950).

- 63 -
A8-Euphen (LXXXVm)^
316,2 g Euphadien (LXXXVH) wurden in 600 cm Essigester-Eisessig (1:1)
gelöst, zu 0, 4 g vorhydriertem Platinoxyd gegeben und an der Schüttelmaschine
hydriert. Nach der Aufnahme eines Mols Wasserstoff wurde aufgearbeitet, und
man erhielt 16,1 g Oel. Dieses wurde in Methylenchlorid gelöst, und dann gab
man warmes Methanol hinzu bis zur leichten Trübung. Das ausgefallene Oel wur-o
de abdekantiert, und die klare Lösung geimpft. Man erhielt so 13, 5 g A -Euphen
(LXXXVHI) vom Smp. 57°.
C30H52 Ber- C87>30 H 12, 70% Gef. C 87, 24 H 12,82%
[ex] = +32,5° (c = 1)
A8-7,11-Diketo-euphen (LXXXLX)1*
8 1
13,6 g A -Euphen (LXXXVIII) wurden in 1000 cm Eisessig gelöst, dannwurde
0 3bei 40 eine Lösung von 13,5 gChromsäure in 190 cm 90%igem Eisessig unter
Rühren hineingetropft. Man rührte noch 4 Stunden bei 40°, und nach der übli¬
chen Aufarbeitung chromatographierte man die 14, 2 g Rohprodukt an 420 g Alu¬
miniumoxyd (Akt. HI). 2, 4 1 Petroläther eluierten 9,8 g gelbes Oel, das aus
Methanol 5,3 g gelbe Nadeln vom Smp. 104° gab.
C30H48°2 Ber" C 81> 76 H 10' 98^> Gef *C 81» 56 H 10' 91%
MD = +73,6° (c = 0,528)
"V- \nax274m/a ' l0S £ =3>93
1)B.C. Roth, Diss. E.T.H. Zürich (1950).

- 64 -
7,11-Diketo-euphan (XC)
31 g Euphen-dion (LXXXIX) wurde in 40 cm siedendem Eisessig gelöst.
Dazu gab man noch 1 g Zinkpulver und liess eine Stunde am Rückfluss kochen.
Man dampfte den Eisessig am Vakuum ab, löste den Rückstand in Aether-Was¬
ser auf und wusch mit verd. Natronlauge und mit Wasser. Man erhielt somit
ein in langen, weissen Nadeln kristallisierendes Produkt vom Smp. 114.
C30H50°2 Ber' C 81,39 H 11,39% Gef. C 81, 26 H 11,14%
Cc<]D = -104,3° (c = 1,0)
LR. (Nujol)^
1706 cm"1 (Carbonyl)
Es liegt das 7,11-Diketo-euphan (XC) vor.
Hydrierung von 7,11-Diketo-euphan (XC) mit Lithiumaluminiumhydrid
3500 mg Substanz in 30 cm Aether wurden unter Vibrieren zu 500 mg
3Lithiumaluminiumhydrid in 50 cm Aether zugetropft. Man kochte am Rückfluss
noch während zwei Stunden, und nach der Aufarbeitung erhielt man weisse
Kristalle, die nach 5 Umkristallisationen aus Methylenchlorid-Hexan den kon¬
stanten Schmelzpunkt von 212 hatten.
C30H54°2 Ber. C 80,65 H 12,18% Gef. C 80,27 H 12,03%
£°<ID
= -28, 6° (c = 0, 95)
IR. (Nujol)1) 3200 cm"1 (Hydroxyl) 1700-2500 - cm"1 - Region leer.
Es liegt das 7,11-Dioxy-euphan (XCI) vor.
Erster Versuch der Acetylierung von 7,11-Dioxy-euphan (XCI)
3 3520mg 7,11-Dioxy-euphan (XCI) wurdenüber Nacht in 5 cm Pyridinund 5 cm
Acetanhydrid stehen gelassen. Nach der Aufarbeitung (das Anhydrid wurde zuerst
mit Eis zerstört) erhielt man weisse Nadeln, die, dreimal aus Methanol umkristal¬
lisiert, den Schmelzpunkt von 220 hatten, und die mit dem Ausgangsmaterial gemischt
keine Schmelzpunktdepression zeigten. Die Tetranitromethanprobe war negativ.
C30H54°2 Ber> C 80' 65 H 12'18% Gef • C 80' 28 H 12>16%
[<*]D = -33,4° (c = 1,02)
Es liegt das Ausgangsmaterial (XCI) vor.
1) Auf einem Perkin-Elmer-Apparat aufgenommen.

- 65 -
Zweiter Versuch der Acetylierung von 7,11-Dioxy-euphan (XCI)
3 3200mgSubstanz(XCI)wurdenln2cm Pyridin und 2 cm Acetanhydrid 5 Tage
bei Zimmertemperatur im Dunkeln gehalten. Nach der Hydrolyse des Acetan-
hydrids mit Eis und üblicher Aufarbeitung erhielt man 188 mg Oel, die an 6, 5 g3
saurem Aluminiumoxyd schnell chromatographiert wurden. 60 cm Petroläther
eluierten 98 mg Nadeln, die, mehrmals aus Methanol umkristallisiert, den
Schmelzpunkt von 65 nicht überschritten. Die Tetranitromethanprobe war
positiv.
C30H50 Ber- C 87> 73 H 12' 27% Gef•C 87' 64 H 12' 39%
[CX]D = -133° (c = 1,08)
UV. Amax 240 myu , log £ = 4,244
IR.(CHC13)^ 3000-4000 cm_1-Region leer; 1500-2700 cm_1-Region leer.
Es liegt das A?' 9-Euphadien (XCU) vor.
Reduktion von 7,11-Diketo-euphan (XC) mit Natrium und Propanol
3700 mgSubstanz (XC) wurden in 70 cm n-Propanol gelöst, und am Rückfluss
wurden portionsweise 3, 5 g Natrium zugegeben. Nach totalem Verbrauch des
Natriums wurde das Propanol am Vakuum abgedampft, man zersetzte das
Natriumpropanolat mit Wasser und dampfte am Vakuum zur Trockene ein. Der
Rückstand wurde in Aether gelöst und die ätherische Schicht zuerst mit verd. Schwefel¬
säure, dann mitWasser gewaschen. Man erhielt ein Produkt, das nach 5 Umkristal-
lisationen aus Methylenchlorid-Hexan einen konstanten Schmelzpunkt von 159 zeigte.
C30H54°2 Ber' C 80'65 H 12'18% Gef• C 80' 53 H 12'15%
[CX]D = -101° (c = 1,12)
IR. (CHClg)*'
3600 cm-1 (Hydroxyl)
Es liegt das 7,11-Dioxy-euphan (XCIII) vor.
1) Auf einem Baird-Spektrophotometer aufgenommen.

- 66 -
Acetylierung des 7,11-Dioxy-euphans (XCM)
3 3280 mgSubstanz (XCm) wurden in 2,8 cm Pyridinund 2,8 cm Acetanhydrid
über Nacht stehen gelassen. Nach der üblichen Aufarbeitung erhielt man 310mg Oel,
die an 9 g Aluminiumoxyd chromatographiert wurden. Nach 13 öligen Fraktionen
eluierten 30 cm Benzol-Aether (1:1) 105 mg Kristalle vom Smp. 123°, die mit
dem Ausgangsmaterial eine Mischschmelzpunktdepression von 13 lieferten.
C34H58°4 Ber" C76>93 H 11,01% Gef. C 76,78 H 10,66%
0]D = -135,2° (c = 1,06)
IR. (Nujol)^
1727 cm"1 (Acetat)
Es liegt das 7,11-Diacetoxy-euphan (XCIV) vor.
Reduktion von 7,11-Diketo-euphan (XC) nach Wolff-Kishner
3 3200 mgSubstanz (XC) wurden mit 2 cm Hydrazinhydrat, 10 cm abs. Feinsprit
und 750 mg Natriumäthylat während 14 Stunden im Einschlussrohr auf 230° er¬
wärmt. Nach der üblichen Aufarbeitung erhielt man 230 mg eines gelben Oels,3
das an 15 g Aluminiumoxyd (Akt. I) chromatographiert wurde. 30 cm Petrol-
äther-Benzol (4:1) eluierten 79 mg eines kristallinen Produktes vom Smp. 107.
Die Tetranitromethanprobe war negativ.
C30H52° Ber. C 84,04 H 12, 23% Gef. C 83,84 H 11, 77%
[<X]D = +21,4° (c = 0,99)
m- ^max2601"/-1 ' log £ = 3'99
Es liegt das A8-7-Keto-euphen (XCIVa) vor.
1) Auf einem Perkin-Elmer-Apparat aufgenommen.

- 67 -
V. Herstellung von 6-Nor-7-keto -cholestan (CXIV)
Acetylierung des Cholesterins (CVM)
Durch einstündiges Erhitzen von Cholesterin (CVHI) mit der vierfachen
Menge Acetanhydrid unter Rückfluss wurde Cholesterin-acetat (CVHIa) vom
Smp. 113 in guter Ausbeute erhalten.
A5-3ß -Acetoxy-7-keto-cholestan (CK)
Cholesterin-acetat (CVHIa) wurde nach der Vorschrift von K. Heussler und A.5 1^
Wettstein (Beispiel von A -3^,17/3-Diacetoxy-7-keto-androsten) 'mittert. Butyl-
chromatlösung oxydiert. Man erhielt in 40%iger Ausbeute das CEC vom Smp.
151°.
A3;5-7-Keto-cholestadien (CX)
3 328 g von CIX wurden in 560 cm Dioxan und 280 cm 10%iger methanolischer
Kaliumhydroxydlösung während drei Tagen am Rückfluss gekocht. Nach Abdampfen
der Flüssigkeit am Vakuum und Aufarbeitung des Rückstandes erhielt man ein Oel,
welches nach dreimaligem Umlösen aus Methylenchlorid-Methanol 16, 5 g Kristalle
vom Smp. 113 ergab.
k 3' 5Hydrierung von A
' -7-Keto-cholestadien (CX)
315,7 g Substanz (CX) wurden in 400 cm Eisessig mit 7 g Palladium auf Barium¬
sulfat unter Wasserstoffatmosphäre geschüttelt. Nach Aufnahme von 1, 28 Mol
Wasserstoff kam die Hydrierung zum Stillstand. Der Katalysator wurde abfiltriert,
der Eisessig am Vakuum abdestilliert und der Rückstand wie gewöhnlich aufge-3
arbeitet. 900 cm Petroläther eluierten aus der Säule von 390 g Aluminiumoxyd
(Akt. II) 1, 56 g Kristalle, die nach siebenmaligem Umkristallisieren aus Metha¬
nol einen Schmelzpunkt von 115 zeigten.
C27H46° Ber- C83»87 H 11,99% Gef. C 83,86 H 11,92%
[o<]D = -48,5° (c = 1,00)
1) K. Heussler & A. Wettstein, Helv. 35, 284 (1952).

- 68 -
Es liegt das 7-Keto-cholestan (CXI) vor.
3Weitere 7.200 cm Petroläther lieferten 7, 4 g Oel, das aus Methanol um¬
kristallisiert 5,4 g Kristalle vom Smp. 127 ergab.
[<X]D = -137,4° (c = 1,02)
Es liegt das A -7-Keto-cholesten vor.
Eine erneute Hydrierung nach der gleichen Vorschrift führte quantitativ
zum 7-Keto-cholestan (CXI).
6,7-Seco-cholestan-6,7-disäure (CXII)
3200 mg 7-Keto-cholestan (CXI) wurden mit 3, 5 cm rauchender Salpeter¬
säure und 1 cm Methylenchlorid bei 0° versetzt. Innert 20 Stunden brachte man
die Temperatur auf 10°, und während einer Stunde wurde auf 65 erwärmt. NachAb-
3kühlung goss man das Reaktionsgemisch auf 200 cm Wasser und extrahierte mit
Aether, der darauf neutral gewaschen, getrocknet und destilliert wurde. Man
erhielt ein gelbes Oel, das, aus Methanol-Essigester zweimal umkristallisiert,
100 mg weisse Nadeln vom Smp. 268 ergab.
6-Nor-7-keto-cholestan (CXIV)
31 g der Dicarbonsäure (CXII) wurde in 200 cm eines Methanol-Wasser-(3:1)
Gemisches gelöst und mit 5% iger wässeriger Kaliumhydroxydlösungauf Phenol¬
phthalein neutralisiert. Darauf versetzte man die Lösung mit einem Ueberschuss an
Bariumchlorid; das ausgefallene Bariumsalz der Dicarbonsäure wurde abfiltriert,
im Exsiccator getrocknet und in einem Kragenkolben am Hochvakuum 10 Minuten
auf 390° erhitzt. Das entstandene Oel wurde in Petroläther gelöst und an 30 g3
Aluminiumoxyd (Akt.II)chromatographiert, wobei 200 cm Petroläther 85 mg
eines Oels eluierten, das aus Methanol Kristalle vom Smp. 88 ergab. Nach drei¬
maliger Umkristallisation stieg der Schmelzpunkt auf 95 .
WD = -35°
IR. (CS2) 1736 cm"1 (Carbonyl) 5-Ringketon
Es liegt das 6-Nor-7-keto-cholestan (CXIV) vor.
******

- 69 -
Sämtliche Analysen wurden im mikrochemischen Laboratorium der Eidg.
Technischen Hochschule ausgeführt. Dem Leiter des Mikrolaboratoriums Herrn
W. Manser danke ich bestens für seine wertvolle Mithilfe.
Die Aufnahme der IR. -Spektren verdanke ich Herrn A. Hübscher und Frl.
E. Aeberli.
Herrn Hs. Grossmann danke ich ebenfalls für die Mithilfe, die er bei der
Isolierung des Euphadienols und der (X-Elemadienolsäure leistete.
ZUSAMMENFASSUNG
1. Durch die Gewinnung der enantiomeren Acetoxy-phenol-lactone Cg.H.-O,(LIII und LXXXI) aus den tetracyclischen Triterpenen Lanosterin (I) und A^-3C<-
Oxy-tirucallen (LXXV) wurde ein neuer Beweis für die Konstitution des Ringes D
und die Haftstelle der langen Seitenkette bei Verbindungen der Tirucalladienol-
Gruppe erhalten.
Durch diese und frühere Untersuchungen ist die Konfiguration der zwei
stereoisomeren Grundtypen Lanosterin (I) und Tirucalladienol (II) endgültig fest¬
gelegt.
2. Es wurde der sterische Verlauf der Hydrierung der 8,9-Doppelbindung
bei Verbindungen der Tirucalladienol-Gruppe erörtert.
3. Im Hinblick auf die Beweisführung für die Grösse des Ringes C der penta-
cyclischen Triterpene, konnte gezeigt werden, dass im IR. -Spektrum die Valenz¬
schwingung der Fünfring-Ketogruppe von der Lage des Ringes im alicyclischen Ge¬
rüst weitgehend unabhängig ist.

Lebenslauf
Als Sohn von Alessandro Ménard und Marie, geb. Siegfried, wurde ich am
2. Dezember 1927 in Pola geboren. Dort besuchte ich die Primarschule und das
"Liceo Giusoé Carducci", das ich im Jahre 1945 mit der "Maturita scientifica"
abschloss. Nach halbjähriger Tätigkeit in der staatlichen Bauverwaltung meiner
Geburtsstadt kam ich im Juli 1946 nach Zürich, wo ich im Herbst 1947 die Auf¬
nahmeprüfung an der Eidgenössischen Technischen Hochschule bestand. Im Früh¬
jahr 1951 erwarb ich das Diplom als Ingenieur-Chemiker. Nach einem Praktikum
in der Textilhilfsmittelabteilung der Firma Sandoz, Basel, begann ich im Februar
1952 meine Arbeit im organisch-chemischen Institut der Eidgenössischen Techni¬
schen Hochschule unter der Leitung von Herrn Prof. Dr. L. Ruzicka und Herrn
Privat-Dozent Dr. O. Jeger.