review - papyrus : université de montréal digital … · web viewimmuno-competent cells, such as...
TRANSCRIPT

07/05/2023
Title
Drug-loaded nanocarriers: passive targeting and crossing of biological barriers
Authors
Jean-Michel Rabanel, Valery Aoun, Igor Elkin, Mohamed Mokhtar & Patrice Hildgen *
Affiliation
Faculté de pharmacie
Université de Montréal
C.P. 6128, Succursale Centre-ville
Montréal, QC, H3C 3J7 Canada
* Corresponding author:
Prof. Patrice Hildgen
E-mail: [email protected]
Telephone: (514) 343-6448
Fax: 514 343-2102
1

07/05/2023
Abstract
Poor bioavailability and poor pharmacokinetic characteristics are some of the leading causes of
drug development failure. Therefore, poorly-soluble drugs, fragile proteins or nucleic acid products may
benefit from their encapsulation in nanosized vehicles, providing enhanced solubilisation, protection
against degradation, and increased access to pathological compartments. A key element for the success of
drug-loaded nanocarriers (NC) is their ability to either cross biological barriers themselves or allow loaded
drugs to traverse them to achieve optimal pharmacological action at pathological sites. Depending on the
mode of administration, NC may have to cross different physiological barriers in their journey towards
their target.
In this review, the crossing of biological barriers by passive targeting strategies will be presented
for intravenous delivery (vascular endothelial lining, particularly for tumour vasculature and blood-brain
barrier targeting), oral administration (gastrointestinal lining) and upper airway administration (pulmonary
epithelium). For each specific barrier, background information will be provided on the structure and
biology of the tissues involved as well as available pathways for nano-objects or loaded drugs (diffusion
and convection through fenestration, transcytosis, tight junction crossing, etc.). The determinants of passive
targeting − size, shape, surface chemistry, surface patterning of nanovectors − will be discussed in light of
current results. Perspectives on each mode of administration will be presented. The focus will be on
polymeric nanoparticles and dendrimers although advances in liposome technology will be also reported as
they represent the largest body in the drug delivery literature.
Keywords:
Nanocarrier; Drug delivery; Biological barriers; Passive targeting; Surface properties; Vascular
endothelium, Oral administration, Pulmonary administration
1

07/05/2023
1. INTRODUCTION
In the domain of drug delivery, 2 ways of targeting are generally differentiated − “passive” and
“active” targeting − even if the distinction could be somewhat blurred as we will see later. “Passive
targeting” is based on nanocarrier (NC) size and general surface properties, namely, surface charge, degree
of hydrophobicity and nonspecific adhesion, which direct them towards particular organs, cross biological
barriers, such as specialized epithelia, or enter the cell cytoplasm. On the other hand, “active targeting”
refers to specific ligand-receptor recognition or antibody-antigen binding, aimed to increase the selectivity
of the drug-carriers delivery. For the purpose of this review, we define “passive targeting” as including
general, nonspecific, surface-modified and internal stimuli-responsive NC; excluding any specific ligand
recognition. At the present time, most clinical trials involving NC, rely on passive targeting (see
www.clinicaltrials.gov), mainly expansion of nanoparticle-albumin-bound drugs (or “nab”) technology, for
taxane delivery in cancer and pegylated liposomes (for doxorubicin (DOX) or amphotericin B delivery).
NC can enter the body via the upper airways and the gastrointestinal tract (GIT) respectively or by
injection (intravenous (i.v.), subcutaneous, intramuscular). They have to cross different specialized
epithelia, either lung or GIT epithelia, to reach the blood compartment, tumoral vascular endothelium or the
blood-brain barrier (BBB) to access pathological tissues via the blood circulation. This is not an easy task,
even for nanometric objects (1-1,000 nm), and available pathways are limited to epithelium porosity or
transcytosis routes.
Besides size, a common feature of all administration routes is that the biological interface between
NC and the biological medium (solid/liquid interface) is a major determinant of NC fate and therapeutic
outcomes. In particular, events such as opsonisation, mononuclear phagocytosis system (MPS) uptake,
biodistribution (NC localization among organs), interactions with cell membranes and extracellular
matrices strongly depend on interface properties. These properties will be determined by surface chemistry,
shape and curvature radius, porosity, roughness, fractal dimension and hydrophobicity (as well as specific
recognition elements in case of active targeting). Moreover, additional properties depend on biological
medium composition (pH, salts, ionic strength, proteins, etc.), charge (zeta potential) and particle
aggregation. The interactive forces involved are mainly van der Waals forces, ionic and water solvatation
[1].
2

07/05/2023
Nowadays, the majority of new molecules present a delivery challenge because of solubility
issues, size or sensitivity to degradation and instability. Over the years, a lot of promising actives have seen
their development compromised for similar reasons. Other concerns hampering drug development are
adverse side-effects and narrow therapeutic indexes. These pharmacodynamic, pharmacokinetic (PK) and
solubility challenges have to be addressed to translate positive in vitro results into clinical outcomes.
Encapsulation in NC – nanosized structures carrying drug loads – is a solution to modify drug PK and
distribution profiles. Encapsulation can help to achieve one or several of these goals, such as increased
residence time (by decreasing renal and reticuloendothelial system (RES) clearance), protection from fast
degradation by inhibiting metabolic clearance in blood or inside the GIT, reduced side-effects (by
suppressing the volume of distribution or by organ targeting), and crossing specific biological barriers to
deliver actives to specific areas. Because of very unfavourable physicochemical properties (molecular
weight (MW), sensitivity to enzymatic degradation, charge, etc.), some drugs such as DNA or siRNA have
to be developed clinically along with associated NC [2]. The choice of nanometer range for drug carriers is
justified by the route of administration (injection, inhalation), increased surface-to-volume ratio for release,
mucosa-penetration properties, accessibility of pathways to cross either epithelial barriers or cellular
membranes. Moreover, minimum size is determined by renal filtration cut-off (for NC aimed at the
systemic circulation), while maximum size is limited by extensive phagocytosis of microparticles in the 1-6
µm range and the emboli properties of even bigger microparticles.
All these considerations have led to the development of several NC classes, including liposomes
[3], micelles [4], dendrimers [5] and solid polymeric particles [6, 7], among others (Fig. (1)). While first-
generation NC rely on very simple structures (single polymers or excipients) and geometry, NC to date are
becoming increasingly sophisticated, incorporating several polymers or materials to impart multiple
functions. Indeed, different properties are sought for NC: cytocompatibility, maximization of encapsulation
efficiency, elimination and, finally, the ability to cross biological barriers. Moreover, knowledge of the
biological determinants of NC fate in vivo is improving, allowing more rational development of their
physicochemical properties.
3

07/05/2023
Fig. (1). Different types of Nanocarriers
(A) Micelle: self assembly of amphiphilic molecules; (B) Liposomes vesicles primarily constituted of a
phospholipids bilayer along with another types of lipids (cholesterol or PEG-phospholipids conjugates); (C)
Dendrimers are branched symmetric polymeric structures constituted by a core and branches (the
dendrons); (D) Polymeric nanoparticles are matrix particle in which the drug is dispersed (here symbolized
with black dots); (E) Nanocapsules, are constituted by an core (generally hydrophilic containing drugs)
enclosed in a thin polymeric wall.
The present review will focus on the challenge of biological barrier crossing upon administration
via major routes (i.v., oral and pulmonary) and NC characteristics that are determinants of this goal through
passive targeting strategies. The aim is to provide a biological perspective to NC development linked with
recent experimental data. Issues regarding carrier fate and elimination will not be covered, and readers are
referred to a recent review on this aspect of NC fate in the body [8]. Intracellular delivery and trafficking
represent research fields by themselves, and are described in recent reviews [9, 10], although some
information will be provided when optimal cellular uptake properties are in conflict with upstream
targeting step requirements.
2. THE I.V. ROUTE
The i.v. route is the fastest, easiest and most reliable route of entry for all drug NC, allowing quick
and complete distribution across the body via the systemic circulation. However, even if i.v. injection
4

07/05/2023
provides fast distribution in the blood compartment, NC still have to overcome several physical and
physiological barriers (Fig. (2)), protecting the body against intruders, to reach targeted organs, such as
solid tumors or inflammation sites. The principal obstacles to NC are: 1) Clearance by the RES or MPS; 2)
The immune barrier: reaction of the immune system, activation of the complement cascade and allergic
responses to foreign materials; 3) Fast renal elimination by glomerular filtration; 4) The blood vessel wall,
particularly the endothelial cell (EC) lining and basement membranes, preventing direct access to organs
and tissues at the capillary level.
Finally, once NCs have evaded these barriers, they should be able to diffuse in the immediate
environment of targeted cells (in the interstitial space) and release their contents efficaciously. In addition,
they may have to reach targeted cell membranes for eventual internalization, if the drug needs to be
released in cytosol to exert its action. Alternatively, the vascular endothelium has been proposed as a direct
target of drug carriers mainly by active targeting to specific receptors expressed on the cell surface [11].
An exhaustive presentation of all physiological barriers (Fig. (2)) crossed by NC aimed at tumour
sites is beyond the length of this review. Our review on NC surface properties will concentrate on 2
aspects. First, the relationship between surface characteristics and opsonisation will be discussed along with
some consequences in terms of MPS uptake and biodistribution. Second, the determinants of efficacious
solid tumour passive targeting, and the extravasation (vascular wall crossing) process towards the tumour
interstitium will be presented. Readers are referred to recent reviews regarding the specific domain of
complement cascade induction by NC surfaces [12], renal filtration, NC fate after MPS uptake [8] and cell
uptake [9].
5

07/05/2023
Fig. (2). General view of biological barriers to i.v. delivery of drug-loaded NC aimed at the solid tumour
interstitium
2.1. The first barrier: NC opsonisation
Opsonisation is a process of protein adsorption occurring on the carrier surface immediately upon
injection in biological medium. In blood, different plasma proteins, such as immunoglobulin G (IgG) and
IgM, apolipoproteins (Apo), fibronectin, complement system proteins, etc., tend to absorb on NC surfaces
[13], forming a “protein corona” [14]that shapes NC surface properties in biological medium [1]. The
corona is a dynamic layer, with variable kinetics of association and dissociation for each protein or surface
type. The relative abundance of proteins in plasma or the cell interstitium and their affinity for the surface
define this dynamic [15]. Albumin, the major plasma protein (about 55%), tends to bind to NC by
6
i.v. injection
MPS UPTAKE
NC in the blood circulation
OPSONISATION
BIO-DISTRIBUTION
MARGINATION
EXTRAVASATION
DIFFUSION in the tumoral
interstitium
Urinary excretion(Renal filtration <6 nm)
Blood returnCell uptake Lymphatic clearance
Degradation
Elimination
Other organs
Specific immune reaction
Complement cascade
Celllevel
Organ
level
Systemic level

07/05/2023
hydrophobic and ionic interactions, but with lower affinity than the above-mentioned proteins, and will be
eventually displaced. Albumin is considered as a “dysopsonin”, i.e., it promotes longer circulation,
probably by binding competition with opsonins [16, 17]. As we will see later, these exchanges influence the
carrier’s fate, including NC/cell interactions, elimination, immune reactions and biodistribution, mediated
mainly by their effects on MPS uptake.
Macrophages of the MPS, mainly composed of macrophages from the liver (Kupffer cells) and
spleen, as well as peripheral macrophages are part of the immune system. Their role is to engulf and
destroy foreign particles, such as bacteria and viruses in blood, by phagocytosis [18]. Unfortunately, they
also recognize NC as foreign and clear them from blood. This clearance is dependent on opsonisation.
Generally, macrophages do not recognize NC directly. They express several opsonin receptors which
mediate recognition [13, 18]. Receptors for bacterial and fungal polysaccharides (PS) have been identified,
and “scavenger receptors” have been suggested to participate in the uptake of PS particles [19]. Ligand-
receptor recognition triggers actin rearrangement and phagosome formation.
2.1.1. Mechanisms of protein binding to NC surfaces
The physicochemical characteristics of NC, such as surface hydrophobicity, surface charge and
charge density [20], carrier size [15], and the presence of functional groups influence opsonisation and,
consequently, uptake by the MPS. Other biomaterials, such as lipids, could also bind to NC surfaces,
although the biological significance of this observation has not yet been determined [21]. Several strategies
have been explored to alter NC surface properties, including polymer coverage and charge modification to
decrease or change opsonisation patterns to curb MPS uptake, increase circulation time and transform NC
biodistribution. Interactions between proteins and NC surfaces are determined mainly by electrostatic,
hydrophobic and/or specific interactions, such as ligand-receptor recognition [1]. Generally, the degree of
surface hydrophilicity influences the amount and identity of bound proteins. On the other hand,
hydrophobic surfaces are opsonised at a higher speed than hydrophilic surfaces [13].
2.1.2. Surface properties and hydrophilic polymer surface coverage
The incorporation of neutral and hydrophilic polymers onto NC surfaces (absorbed or covalently-
linked) increases NC half-life in the systemic circulation (from minutes to hours), and this effect is related
to decreased opsonisation.
7

07/05/2023
Pegylation of NC surfaces
In the 90, linear poly(ethylene glycol) (PEG) were introduced in liposomes [22, 23] and
polymeric NP [24]. PEG decreases protein interactions with NC surfaces [25], modifies PK and
biodistribution [13], and influences NC cellular uptake [26]. PEG chains of sizes from 2 kilo Dalton (kD)
and beyond are able to greatly reduce the adsorption of opsonins and other serum proteins [27, 28].
Hydrophilic and flexible PEG chains act via a steric repulsion effect, rendering protein binding
unfavourable. This repulsion effect depends on chain length, optimal surface density and optimal chain
configuration [13]. 2 to 5 kD seems to be the minimum length for the “stealth” effect [27, 29], but longer
chains have shown improved circulation time for rigid nanocapsules [30] and changed in vivo
biodistribution and clearance [31]. PEG has been added either as an adsorbed layer on NC surfaces (NP
made of poly(styrene) (PS) or poly(D,L-lactide-co-gycolide) (PLGA)), or attached covalently to other NC
components (phospholipids, polyesters, etc.). Covalently-linked PEGs have gained prominence, as
adsorbed polymers on particles hydrophobic surfaces, such as Poloxamer® (triblock of PEG-PPG-PEG),
are subjected to shedding by competition with plasma components, particles swelling and erosion,
compromising surface repulsive properties and decreasing NC circulation time [32, 33].
Optimal protein resistance has been reported in poly(lactic) (PLA) polymeric NP with 5% w/w
PEG content. PEG chain density for optimal conformation and efficacy translates into an inter-chain
distance of around 1.5 nm on polyester NP surfaces [27]. This distance is crucial for PEG chain
organisation and the prevention of penetration of smaller opsonins between PEG chains.
In the case of liposomes, the percentage of pegylated phospholipids necessary for stealth behavior
is about 5-7% mol. with PEG 2 kD and 15-25% with smaller PEG 350 D to 1 kD [33]. Furthermore, lipids
with higher transition phase temperature or cholesterol tend to decrease nonspecific binding of opsonins by
increasing bilayer stability [34]. The addition of PEG increases the circulation time of liposomes from
minutes to hours (up to 24-48 h). It is not clear though if the effect is strictly due to the prevention of
opsonisation or mediated through steric stabilisation of the phospholipid bilayer, preventing aggregation
and their fast elimination [35].
It is generally accepted that the PEG layer should reach such a density, PEG chains are forced to
adopt an intermediate “mushroom/brush” or “brush” configuration as seen in Fig. (3), but without leaving
8

07/05/2023
exposed hydrophic or charged surfaces where opsonins can bind [12, 33]. Some conflicting results
regarding PEG coverage efficacy are likely due to misuse of the term brush and mushroom configuration
and a lack of complete physicochemical characterisation of surfaces. In particular, optimal PEG density
may vary with curvature radius and core modulus (from liposomes to hydrogel particles to rigid polymeric
NP).
Linear PEG (usually methoxy-PEG) is the standard, but other configurations have been tested.
While branched PEGs are less effective than linear PEGs of equivalent sizes [36], it had been shown that
PEG 1400 distearate in PLA NP, forming a hydrophilic loop exposed at the surface and anchored by 2
hydrophobic domains buried in the particle core (Fig. (3) panel C), decreases opsonisation and macrophage
uptake [37]. Multiblock copolymers of PLA and PEG have the same “loop” configuration. Some studies
have reported similar efficacy with loop configuration [38] whereas others have demonstrated that,
although superior to naked PLA particles in terms of opsonisation and macrophage uptake, multiblock NP
have reduced efficacy in preventing protein binding and macrophage uptake compared to NP with PEG
“mushroom” configuration [26]. Although overall PEG content was reported to be higher in multiblock NP,
PEG surface coverage was lower, as determined by XPS analysis, suggesting a hypothesis to explain the
results obtained [26].
Fig. (3). PEG NC coverage
(A) “Brush” regimen (high density), (B) “Mushroom” configuration (low density), (C) “PEG loops”:
multiblock NP (PLA-PEG-PLA) [26] or PLA particles with PEG 1400 distearate [37]. PEG chains are
presented in black, and the NC core in grey.
9

07/05/2023
A complex interplay between PEG chain length, carrier size (determining surface availability for
PEG anchorage) andweight ratio between PEG and hydrophobic components of the carrier (e.g. PLA-PEG
particles) is influencing the final surface properties of NC. For instance, for the same PEG molecular ratio,
if NC size increases, total NC surface decreases, and curvature radius declines, augmenting PEG coverage
and density. It is important to state that, even after optimisation of hydrophilic polymer coverage,
opsonisation is not completely abolished, and significant amounts of opsonins are still detected on NC
surfaces [27, 39].
PEG and immunity
The question of NC immunity is seldom addressed although some recent results have raised the
issue in relation to PEG use. For a long time, PEG was considered as non-immunogenic and to prevent
immune recognition of NC. However, antibody development against pegylated liposomes has been
observed [40]. Immune reactions to and the toxicity of pegylated liposomes seem to be linked with long-
term circulation. A diffusible PEG-lipid molecule in pegylated liposomes has been proposed so that PEG
shedding off the surface intervenes with time, rendering NC susceptible to RES clearance and thus limiting
circulation time as well as the potential side-effects linked with long-term exposure [41]. Long-circulating
pegylated liposomes can also result in complement activation and pseudo-allergic reactions [42], which can
be prevented by shielding some specific negative charges [43]. Complement activation has also been
reported for polyester/PEG nanocapsules [12], but does not show correlation with surface hydrophobicity,
as does opsonisation [44].
Surface heterogeneity
Surface heterogeneity is not easily documented but may be the cause of NC under-performance.
For instance, patchy or non homogeneous PEG coverage and the existence of a particle subpopulation with
suboptimal PEG coverage could accelerate opsonisation and elimination [33]. This surface heterogeneity
could be caused by a non-homogenous mix of pegylated ingredients during the preparation stage, leading to
different PEG contents. For liposomes, in the presence of divalent cations, the phase separation of
pegylated and acidic phospholipids could explain rapid macrophage localization owing to the increased
opsonisation of exposed, non-pegylated patches [33]. Heterogeneity can also be found in less dynamic
structures, such as PS particles [45]. For solid polymeric NP, the phase separation of polymers with
10

07/05/2023
different degrees of hydrophobicity could occur during the solvent removal step. Atomic force microscopy
(AFM) phase imaging studies in tapping mode provide mapping of some surface property variations. Non-
homogenous phase imaging of pegylated polyester NP could indicate micro-domain separation and
suboptimal coverage of PEG, even for NP, showing protein repulsion properties [26, 46]. Surface
heterogeneity is also the consequence of NC degradation, with breakdown or partitioning of PEG
conjugates [33].
Other polymers
While PEG remains the most effective polymer, other flexible hydrophilic polymers have been
tested, such as different polysaccharides (although they are more prone to immune recognition), dextrans
and heparans [25, 47, 48], poly(vinyl alcohol) (PVA), PEG-PVA combination [49], polyvinylpyrolidone
(PVP) [50], etc. PVA is also often employed as a stabilizer for the emulsion step during solid polymeric NP
preparations. Residual surfactants resulting from preparation conditions could remain bound to the particle
surface in non-negligible quantities, influencing surface properties [51].
2.1.3. Surface properties: charges and opsonisation
The nature of charge and charge density on NC can be evaluated by zeta potential, an electrostatic
potential present at a shear plane located at a distance from the particle surface. These potential values rely
on the nature of the surface material and dispersion medium. Thus, it is not a true intrinsic property of NC
as it will depend strongly on the environment: pH, ionic concentration [52], polymer coating, hydrated
layer and protein shielding. The nature of charge and charge density [53] is a determinant of the amount
and identity of proteins bound on the NC surfaces [13]. Neutral particles have a slower opsonisation rate
than either cationic or anionic surfaces. Zwitterionic or neutral surfaces have been shown to prevent protein
adsorption on particle sizes of 3-10 nm (quantum dots). With anionic or cationic surfaces, hydrodynamic
diameter increases up to 15 nm because of protein adsorption [54]. The zwitteration of NP silica surfaces
reveals comparable results with pegylation in terms of protein binding prevention in in vitro assays [55].
100 nm PS NP with different zeta potential have been observed to end up with similar zeta potential after
serum protein binding. This adsorption also causes a 15- to 25-nm diameter increase [56].
In liposomes (80-100 nm, unknown polydispersity index (PI)), heightened clearance is seen with
increased negative charges, concomitant with enhanced uptake by the liver and decreased uptake by the
11

07/05/2023
spleen [57]. On the other hand, negatively-charged PEG-PLA micelles (35 nm, PI <0.1) show lower uptake
in the liver and spleen than neutral micelles [58]. Stability, broad size distribution and opsonisation could
explain these divergent findings.
As cell membranes are negatively charged, it has been argued that cationic NC interact favourably
with them. However, electrostatic interactions between NC and macrophage membrane have been found to
differ in vitro and in vivo. This difference could be related to very different opsonisation coatings, during in
vitro uptake studies and in vivo distribution experiments [52].
Surface charge could play a role in NC toxicity. Positively- and negatively-charged NC, such as
poly(amido amine) (PAMAM) dendrimers have been linked with adverse interactions to red blood cell
(RBC) membranes, culminating in hematolysis [59]. Hematolysis assays are performed in phosphate-
buffered saline (PBS) without plasma proteins interfering in the test. In vivo, however, opsonisation could
occur and charge changed, mitigating toxicity [60] or platelet aggregation [61].
2.1.4. Opsonisation and NC size
Although this review focuses on NC surface properties, NC size is fundamental as it has a
profound effect on opsonisation and, thus, on PK, biodistribution, toxicity [60] and passive targeting (see
Section 2.2). Cyanoacrylate particles (80, 170 and 240 nm), with different PEG-derived surfaces, show that
serum protein adsorption and phagocytic uptake decrease with smaller diameter and longer PEG chains.
The half-life of pegylated carriers is extended about 20-fold and the drug concentration in tumours is
increased by about 3-fold. NC of 80 nm take up less proteins than 240-nm NC (about 6-fold less),
indicating a role for PEG density coverage and conformation [62]. Curvature radius could be involved in
binding efficacy too, as reported for PS NP [15]. NC size seems also to participate in the choice of plasma
proteins binding to PS particles [20].
Macrophages are designed to engulf particles in the range of 1-6 µm with maximum efficacy
around 2-3 µm [63]. The uptake of opsonised 1-3 µm size PS beads has been demonstrated to proceed by
phagocytosis, while smaller 200 to 750 nm beads only partially enter macrophages by phagocytosis, as an
endocytosis mechanism is also involved [64]. These results are consistent with data on PLA/PEG NP
uptake by murine macrophages, revealing a role for a non-phagocytosis mechanism [26].
12

07/05/2023
The filtering effect of RES organs (liver, spleen and kidneys), based on NC size, is a major player
in NC biodistribution [8]. Fast clearance by glomerular filtration is observed below 5.5 nm hydrodynamic
diameter [54]. Small NC (quantum dots, 5-10 nm diameter) tend to increase their diameter upon
opsonisation and, consequently, decrease their renal filtration [54]. NP with a mean diameter of 80-100 nm
show prolonged blood residence and a relatively low rate of RES uptake. For instance, the biodistribution
of non-pegylated liposomes of different sizes (30-400 nm) in blood, liver, spleen, and tumour indicates that
liposomes of 100-200 nm size are mainly located in the circulation while liposomes smaller than 50 nm and
larger than 250 are mostly cleared. Regarding organs of the RES/MPS, liposomes of 50 nm and less are
captured in the liver (liver fenestrae are about 100 nm), while those of 100-400 nm are only partially
captured (plasticity of liposomes allowing spleen escape). On the other hand, the spleen primarily captures
the biggest liposomes (400 nm) [65].
2.1.5. Particle shape
The impact of NC shape on clinical performance, such as targeting and MPS uptake, was not
studied extensively until recently [66, 67]. Particle shape certainly affects NC behaviour and motion in
blood flow, membrane adhesion strength, cell uptake pathways and efficacy [68, 69]. Particle shape
impacts phagocytosis by macrophages and, more importantly, particle shape and size impact attachment
and internalization separately [70]. Filament-like micelles or “filomicelles” have been compared to
spherical micelles in vivo: their half-time in blood is increased about 10-fold in comparison to their
spherical counterparts [71].
2.1.6. Conclusion on opsonisation and surface properties
“Naked” hydrophobic NC are opsonised in minutes, rapidly cleared from the circulation and
sequestered in macrophages. They are sequestered mainly in organs of the MPS, the liver and spleen (80%
of macrophages), and degraded. This property is advantageous to target these organs passively but
complicates the targeting of other organs. It is noteworthy that even stealth NC are opsonised and cleared
from the circulation by macrophages, albeit at a slower rate. Eventually, NC degradation by-products will
be eliminated via urine or feces. If the ingredients are not degradable, accumulation and toxicity can occur
in the liver and spleen [8].
13

07/05/2023
In a nutshell, neutral pegylated NC between 10-100 nm undergo reduced opsonisation and
decreased hepatic filtration, which results in long systemic circulation, allowing extravasation and targeting
(see below). Clearance by the MPS is not per se a bad thing. A certain level of “stealth” is necessary to
ensure that NC are not recognized prematurely by the MPS and degraded. On the other hand, very long
circulation could cause unwanted side-effects over time. It is, therefore, necessary to reach equilibrium
between stealth and opsonisation as well as between stability and degradability, therapeutic efficacy and
biocompatibility.
2.2. NC extravasation
Having discussed the determinants of NC behaviour in blood after i.v. administration, we will now
review the NC properties significant for specific extravasation toward tissues by passive mechanisms. To
achieve this goal, we have to consider the structure, organization and heterogeneity of blood vessel walls
(arteries and veins of different sizes alike) that are composed of 3 layers or “tunica” as seen in Fig. (4). The
thickness of each layer depends on vessel function.
Fig. (4). General organisation of blood vessel walls
The intima, the layer in direct contact with blood flow, is composed of an EC layer supported by a
basement layer comprised mainly of a collagen matrix. EC harbour a layer of proteoglycans, called the
14

07/05/2023
glycocalyx. The media is composed of smooth muscle cells associated with an elastic matrix of collagen
and elastin. These cells are responsible for vasodilatation and vasoconstriction. The adventitia, mainly
comprised of a collagen matrix and fibroblasts, contributes to the reinforcement of larger vessels. Unless
the target is the blood vessels EC themselves, crossing the vascular wall to reach organs is a very difficult
task for NC, even in the smaller arteries or venules
2.2.1. Anatomy of normal capillaries
Of the general organisation described above, precapillary arteries, post capillary venules and
capillaries themselves, retain only the EC lining with a basement membrane – sometimes incomplete (the
“intima”). An incomplete layer of pericytes is encountered, wrapped around EC by longitudinal and lateral
extensions. Pericytes (also called mural cells) are contractile cells that act on blood flow and play a role in
permeability (for instance, they are in larger number in the BBB where vascular permeability is tightly
regulated). The capillaries are responsible for bringing nutrients and oxygen from the blood towards the
cell interstitium and removing cell wastes. Their diameters are 4-15 µm, compared to 6-8 µm for RBC.
Exchanges with tissue interstitium are favoured in capillaries by slow blood flow, a large surface area to
volume ratio and because of their wall structure detailed below. The total surface of the EC lining is about
7,000 m2 (representing about 6. 1013 cells), underscoring the importance of regulating exchange across this
barrier [72]. Normal capillaries are classified according to their organization and sizes of pores and holes in
their structure as illustrated in Fig. (5).
15

07/05/2023
Fig. (5). Different types of normal capillaries and transport pathways
(A) Continuous capillary, (B) Fenestrated capillary, (C) Open fenestrated capillary, (D) Sinusoidal
capillary. VVO: vesicular-vacuolar organelles; TE channels: transendothelial channels (adapted and
modified from [73])
EC are very flat, asymmetric cells (about 1 µm thick or less). Their luminal surface is exposed to
blood flow, and the abluminal surface is exposed to the interstitium. EC of different tissues and organs are
morphologically and functionally distinct [72, 74]. They undergo dynamic changes upon activation (during
inflammation, for instance) or in pathological conditions (tumoural angiogenesis). Four principal structures
are illustrated in Fig. (5). More details on capillary morphology can be found elsewhere [73, 74, 75].
“Continuous walls” are formed in capillaries from skin, muscle, lung and at the BBB. They
constitute a continuous lining on the luminal part of the vessels with permeability heterogeneity in organ
function. Junctions between cells are overlapping regions (intercellular clefts) sealed by protein structures,
16

07/05/2023
the “tight junctions”. EC rest on basement membranes composed of collagen fibrils, laminin, fibronectin,
and glycosaminoglycans responsible for additional restrictions to molecule movement. However,
intercellular clefts allow some exchange of water and small solutes with limited size of a few nanometers.
EC are covered by a proteoglycan layer, the glycocalyx, on the luminal side of the membrane. Moreover,
very active vesicle trafficking is observed in EC with a large population of caveolae (60-70 nm) and some
transcytosis activity. EC flatness limits the distance for caveolae to travel to transfer their contents from the
vessel lumen to the sub-endothelial space (tissue interstitium).
“Fenestrated walls” are constituted of EC with large intracellular pores (50 to 60 nm), with (in
exocrine glands and the GIT) or without diaphragm (kidney glomerulus). The presence of the diaphragm
seems to reduce permeability to about 5-6 nm sized macromolecules and objects. The basement membrane
limits overall permeability. A certain level of transcytosis is also seen but with fewer caveolae and more
frequent transendothelial channels (TEC) than in continuous capillaries. Diaphragms of fenestrae and TEC
are constituted of proteins arranged in octagonal geometry, including radially-organized fibrils connected to
the center of the pores. Their precise role is still unknown.
“Discontinuous, sinusoidal or leaky walls” have large holes (100 to 150 nm) between EC
(intercellular gaps), less tight junctions, and discontinuous (spleen, bone marrow) or absent (liver) basal
membranes, allowing the passage of large proteins and even cells into the interstitium.
2.2.2. Importance of the glycocalyx
As mentioned earlier, this proteoglycan layer, sitting on the luminal/apical surface of EC, is
present ubiquitously from capillaries to large arteries, albeit with varying thickness. It has a crucial
function in renal glomerular filtration, as it completely covers the fenestrae, decreasing the molecular cut-
off [76]. It likely participates in interactions between EC and NC surfaces. The importance of this structure
has been underestimated for a long time, mainly because it is difficult to image in microscopy [77]. It is
composed of 2 layers (Fig. (6)). First, the inner layer, an ordered structure extending 40 to 60 nm from the
surface, is composed of core proteins and membrane-attached molecules with side-chains up to 100-200
nm. Glycoproteins (such as cellular receptors with short and branched sugar side-chains) and proteoglycans
(proteins with long carbohydrate polymers) are molecules belonging to this glycocalyx inner layer.
Individual fibres are spaced at a maximum distance of 20-nm apart [75]. This cell-attached layer accounts
17

07/05/2023
for hindered diffusion and is an osmotic barrier to plasma proteins (and thus eventually to NC). By
regulating protein exchange with the cell interstitium, it controls fluid exchange locally [78].
Second, an outer layer (Fig. (6)), appearing as a less ordered and more dynamic structure, extends
up to 500 nm from the cell membrane in capillaries (several µm in arteries) with different elements
absorbed transiently from blood (polymers, plasma proteins, etc.) or cell secretions (soluble proteoglycans,
etc.). Secreted soluble proteoglycans are in equilibrium with blood [79]. This dynamic structure is sensitive
to shear stress from blood flow and the enzymatic cleavage of covalently-bound components, and is
continuously exposed to shedding and de novo synthesis. It seems to have a role in blood cell flow in
microvessels, acting as a lubricant and repulsing erythrocytes from EC membrane interactions [80].
Glycocalyx composition may vary from tissue to tissue and pathological conditions, including tumours.
Fig. (6). Glycocalyx structure
Protein backbones appear in black, and glycans in grey. The cell-bound layer is about 60 nm,
while the absorbed layer is about 500 nm in capillaries. Glycipans and syndecans are the 2 main families of
core protein proteoglycans anchored on the plasma membrane. Proteoglycans are associated with
glycoaminoglycans (GAG), primarily heparan sulphate (HS), representing 50-90% of GAG along with
chondroitin/dermatan sulphate (CS) and hyaluronic acid chains (HA). More details can be found in [81]
18

07/05/2023
The involvements of this dynamic structure in different vascular functions are increasingly well-understood
[81, 82]. However, its implication in trans-vascular NC delivery is still unclear. The glycocalyx could
represent a supplementary barrier that NC have to cross or elude, to either bind to endothelial membranes
(by active targeting of receptors − 20-80 nm high − “buried” in the glycocalyx) or cross the cell monolayer
via available pores and holes (passive targeting). It could be a mechanical physical barrier to carrier
diffusion, a trap acting via ionic interactions with proteoglycans (HS or CS harbour negative charges),
leading to the accumulation or repulsion of cationic or zwitterionic surface NC.
When migrating from main flow towards the EC, NC have to diffuse through this layer. Indeed, it
had been demonstrated in vivo that glycocalyx removal by enzymatic degradation increases functionalized
carrier binding (with antibodies directed to intercellular adhesion protein 1 (ICAM-1) to the rat femoral
vein endothelium [83]. Alternatively, the mesh network could capture NC and accumulate them at specific
locations, particularly by ionic interaction. Indeed, it had been reported that gene transfer by cationic
liposomes is increased in cells expressing proteoglycans [84]. It has also been postulated that the higher
tumoural vascular permeability of pegylated liposomes is due to the repulsive effect of PEG on the
glycocalyx barrier, preventing their capture, an effect not seen with “normal” liposomes [85].
2.2.3. Hemodymanic considerations
NC extravasation is conditional on the possibility of lateral drift from main blood flow towards the
vessel walls (“margination”), to be either taken up by EC endocytosis or leave capillaries via fenestrations
or intercellular gaps. This movement depends on several hemodynamic considerations, including vessel
diameter and flow rate. Minimal margination has been described for NP, while microparticles accumulate
in higher amounts on the walls of capillary models, seemingly showing preferential localization of NP in
the center of flow [61]. If NC stay in the center of the bloodstream along with RBC, there could be 2
consequences. First, NC diffusion towards the wall is in competition with blood convection, and, second,
NC have less chance of being directed to smaller vessels at the next bifurcation. Indeed, the outcome of
blood “phase separation” is preferential collection of the marginal fluid layer by smaller side branches. This
“skimming effect”, well-known for RBC, could in fact limit NC access to the microvascular bed, keeping a
substantial part of the NC dose in larger vessels where it is ineffective and eventually eliminated. It has
been speculated that NC should be designed to accumulate in the cell-free layer to more easily leave the
19

07/05/2023
large vessels. Size, density and shape have been reported to affect these properties [68, 86]. Once in the
smaller capillaries (with diameters about 5-30 µm), the situation is different as RBC (7 µm) are squeezed
in, with blood flow being slower and less structured. NC size is closer to vessel size, and the probability of
collision with the EC lining and glycocalyx is increased. Moreover, in the tumour vasculature, irregular
vessel organization and less ordered/turbulent blood flow may increase NC interactions with EC.
2.2.4. Pathways of NC transport across normal capillaries
In brief, assuming that NC are able to remain in close contact with the EC lining, the pathways
available for transportation across normal capillary walls are the following:
- Transport through the intercellular junction of intercellular clefts. Macromolecular,
paracellular transport is limited to sizes below 1-5 nm (e.g. dendrimers), depending on the tight
junction type. Zonula occludens inter-endothelial tight junctions in the BBB are an absolute barrier
for macromolecules with openings less than 1 nm (see section on BBB) [75].
- Transport through intracellular fenestrations. Fenestrations without diaphragms are found only
in kidney glomeruli with a cut-off between 40 and 60 nm, but the physiological cut-off is about 6
nm because of glycocalyx presence and podocyte contribution to permeability. Diaphragmed
fenestrations in other types of fenestrated walls (GIT, endocrine glands) have a physiological cut-
off of about 6-12 nm [75].
- Transport in discontinuous sinusoidal capillaries. Sinusoidal capillaries occur in some organs:
liver, spleen and bone marrow. The pore range in human liver is between 50 and 180 nm, average
diameter is 105 nm, and neither the glycocalyx nor the basement membrane limits this opening
[75].
- Transcytosis transport. All EC are involved in transcytosis while intracellular vesicular
trafficking is more intense in continuous capillaries than in fenestrae and sinusoids. The exact
mechanisms of transcytosis and their extent are still a controversial subject. In normal, continuous
capillary beds, macromolecular entities (>5 nm) can only cross the wall via the transcellular route,
either by vesicular transport (caveolae are dominant in EC) or special organelles. Their respective
contributions vary with capillary type and, currently, their roles are not fully reconciled with
20

07/05/2023
permeability study results. Different organelles seemingly related to transcellular transport
functions have been identified by imaging of vascular EC (Fig. (7)):
o Caveolae are cytoplasmic membrane invaginations of about 60-80 nm present at a high
rate in EC of continuous capillaries (up to 50-70% of the cell surface in vivo [73]. This
endocytosis pathway depends on protein caveolin-1 and specific membrane domains
(lipid raft). Caveolae, on their luminal front, take up the bulk of plasma and molecules
attached to lipid microdomains. After endocytosis, intracellular caveolar trafficking leads
to endosome and exocytosis on the abluminal side (transcytosis). The caveolae pathway
can bypass lysosomes. Caveolae can transcytose antibodies [87] and other blood
components across EC into the organ interstitium. Uptake can be mediated by receptor
(as for albumin or LDL) or non-receptor-mediated endocytosis (nonspecific, in which
ionic interactions at the membrane could play a role. Still, the basement membrane could
limit the diffusion of macromolecules and NC after transcytosis. Caveolae are able to
transport several types of NC across EC. Although this transport has been shown to be
highly dependant on NC size [88], its machinery is still not completely understood [89].
o Vesicular-vacuolar organelles (VVO) are present in normal post-capillary vessels and in
tumoural capillaries as caveolae-like clusters that can span the entire thickness of EC. It
is not clear whether VVO are clusters of caveolae (mean diameter around 110 nm),
separated by open stromata and diaphragm structures, or are genuine new organelles.
Although VVO have been demonstrated to contribute to the transport of macromolecular
tracers with sizes up to 11 nm [90], their contribution to vascular permeability has not
been completely elucidated [73].
o Transendothelial channels (TEC) are true, permanent pores (diameter similar to
caveolae) in fenestrae endothelium with diaphragm [74].
Other endocytic pathways, albeit present as clathrin-mediated endocytosis, seem to be marginal for
transcytosis in EC [73]. The glycocalyx contributes to the provision of negative charges to EC membranes.
These negative charges are associated with clathrin-coated membranes. Cationic molecules are
21

07/05/2023
preferentially transported by this pathway leading to lysosomes. Anionic molecules (including plasma
proteins) are excluded from it, being transported mainly via the fluid phase caveolae pathway and shuttled
to the interstitium (via a non-degradative pathway) [91]. Despite a growing body of information, the
involvement of transcytosis in passive and active targeting of drug-loaded NC is still under scrutiny. It is a
possible pathway, as demonstrated by several studies, but what is the dominant mechanism of transport
across the vascular bed? Quantitatively, which mechanism could meet the challenge of delivering a
therapeutic level of drug to the tumour interstitium? These questions remain to be investigated.
Although transcytosis occurs predominantly via the fluid phase, albumin, for instance, can bind to
albumin receptor gp60 present in lipid rafts and is transported to the interstitium by the caveolae pathway.
This property is thought to be a key element in the efficacy of Abraxane®, a paclitaxel albumin-bound
anticancer agent, by increasing its concentration in the tumour interstitium [92]. However, Abraxane®
competes with a high concentration of natural albumin (35-55 g/l), and a leaky tumoural vasculature (and
the enhanced permeability and retention (EPR) effect) could exert a role too in vascular permeability.
Blood vessels in the vicinity of capillaries could also be involved in vascular permeability to NC.
Pre-capillary arterioles are completely surrounded by a layer of smooth muscle cells, with its matrix layer
(“media”) decreasing the vascular permeability. Post-capillary venules, on the other hand, are less
organized, and NC extravasation typically occurs there, as fenestration density and pinocytosis increase
from arteries to post-capillary venules [93].
Fig. (7). Vascular permeability at the normal continuous capillary level
22

07/05/2023
MP: Macropinosis; CLME: Clathrin-mediated endocytosis; CVME: Caveolae-mediated endocytosis;
NCNC: Non-clathrin, non-caveolae pathway; TE: Transendothelial channel; VVO: Vesicular-vacuolar
organelle; Lys.: Lysosome; End.: Endosome; Ex. : Exocytosis
2.3. Tumour microvasculature structure: targeting and crossing
Healthy, normal, continuous capillaries do not allow important extravasation of NC, with sizes
above 6 nm. However, in pathological conditions, such as tumours or inflammation, capillary anatomy and
permeability change. In regard to normal vasculature structure, tumour blood vessels have distinctive
characteristics which could be of interest when designing NC. Solid tumours above the size of 2 mm3
initiate angiogenesis to respond to the increased metabolic needs of rapidly-dividing cells [94]. One of the
main angiogenesis promoters, vascular endothelial growth factor (VEGF), has been associated with
increased vascular permeability as a result of intercellular gap openings and fenestrae induction [95]. The
tumoural neovasculature is often not fully mature, resulting in a tortuous network, irregular vessel
diameters, and abnormally-branched architecture. Tumour vascular growth patterns also culminate in an
usually highly vascularised border, while the inside is deficient in vascularisation or is avascular [94, 96].
In particular, deficiency can be observed in pericytes, smooth muscle cells and abnormal basement
membranes (thinner or thicker than usual), all elements participating to the stabilization of newly-formed
vessels. The resulting tumoural capillaries are characterized by leaky and enhanced permeability with
intercellular gaps (mean size of about 1.7 µm) [97]and transcellular porosity up to 100-780 nm [98],
compared to a few nm for normal, continuous capillaries. Moreover, increases in fenestrae [98], VVO [90]
and transendothelial channels are observed [97], but variations are seen among different tumour types [99].
The filtration cut-off of tumour vascular permeability is reported to be around 400 to 600 nm in most
models [100] but may vary slightly, depending on tumour type [101]. This high cut-off could be attributed
to intercellular gaps rather than transcellular pathways limited to 60 to 120 nm organelles, even if
physiological permeability of fenestration is increased in the absence of continuous basement membranes.
It is noteworthy that fenestration seems to be linked with negative charges imparted by HS [98].
2.3.1. Passive targeting of tumours by the EPR effect
These leaky characteristics allow “passive” tumour targeting based on the enhanced EPR effect,
which was formally conceptualized in 1986 by Matsumura and Maeda [102] and schematized in Fig. (8). It
23

07/05/2023
refers to preferential accumulation of macromolecules in tumoural compared to normal tissues. The
accumulation takes place in the tumour interstitium, the extracellular compartment between the basement
membrane of capillaries and cells (tumour cells, stroma cells, etc.). The effect is recorded for
macromolecules above 40 kD or objects with an hydrodynamic radius from a few nm to around 1 µm. At
the basis of this preferential accumulation are high tumoural vascular permeability, slow venous return
from tumour tissues and diminished lymphatic drainage [103]. The effect has been recognized for many
drug carriers with sizes above renal filtration (6 nm): polymeric NP [104], liposomes [105] and micelles
[4]. Smaller molecules accumulate faster in tumour sites but larger molecules stay for a longer period of
time. NC extravasation and accumulation in the interstitium seem optimal for sizes 20-200 nm. The
magnitude of the EPR effect on NC or drug accumulation is variable, but rarely over 10% of the initial
dose. Most of the dose is still found in the liver and spleen [106, 107]. Tumour tissues show a 4-fold
increase in capture for non-pegylated liposomes of sizes between 100 and 200 nm compared to liposomes
smaller than 50 nm or larger than 300 nm [65].
24

07/05/2023
Fig. (8). Simplified view of the EPR effect
On the left: situation of normal tissue with continuous capillaries, normal lymphatic drainage, normal ECM
and cell organization. NC are excluded from interstitium. On the right: solid tumour with leaky capillaries,
increased ECM, defective lymphatic drainage and increased IFP. NC are transported by convection and/or
diffusing in the tumour interstitium, accumulating mainly in the perivascular region.
Preferential accumulation in tumours is also the result of a combination of increased circulation
time [106] and enhanced vascular permeability. The increment in circulation time gives more time to the
slow extravasation process as the maximum is reached after several hours (usually >24 h). That is why
“long-circulating NC” are crucial for the EPR effect. To increase the extent of the EPR effect, to overcome
tumour vasculature heterogeneity that limits efficacy, different strategies have been tested, including
administration of the pro-inflammatory bradykinin, generating systemic hypertension with angiotensin II
25

07/05/2023
(increased blood flow in tumours) or vasodilatation with nitric oxide generators, such as nitroglycerine [96,
108].
2.3.2. The EPR effect and inflammation
Similar observations on increased vascular permeability and the EPR effect have been reported in
inflammation. Indeed, the hallmark of inflammation is increased vascular permeability leading to the
escape of protein-rich fluid (exudate) into extravascular tissue. Rheumatoid arthritis (RA), for instance,
similarly to tumour development, is characterized by a leaky vasculature and angiogenesis. In an attempt to
improve RA management, a glucocorticoid has been encapsulated in small liposomes. This drug, which
undergoes rapid clearance, a large volume of distribution and induces side-effects, is a good candidate for
encapsulation. The liposomal formulation shows a decrease in distribution volume and diminution of the
drug clearance rate. Clinical improvements are attributed to the 7-fold increase of drug concentration in
injured joints [109], although it represents only a small percentage of the initial dose. Similar data have
been obtained with betamethasone encapsulated in PLGA/PLA-PEG stealth NP in a rat arthritis model
[110]. Non-pegylated liposomes have been found to accumulate in the chronically ischemic myocardium
and intestine [111].
2.3.3. Limits of the EPR effect on passive NC targeting
As pointed out by R.K. Jain in a recent review, approved NC (liposomes, albumin NP), to date,
show modest clinical improvement [93]. Although the EPR effect is claimed to be at the basis of increased
therapeutic efficacy of DOX pegylated lipsome formulations (marketed as Doxil® or Caelyx®) against
several cancers [105], some nuances are warranted as improved PK and biodistribution could explain, at
least in part, the observed improvements and cannot be ruled out. The 3- to 10-fold increase in tumour
accumulation, observed in most studies of this type [33], represent about 1 to 7% of the initial dose. Several
hypotheses have been proposed to explain the modest improvements in clinical outcomes, relying on the
biology of the tumour environment, and should guide future improvements in NC properties.
Uneven distribution of blood vessels, permeability and blood flow
Because of abnormal angiogenesis in tumours, vascularization and blood flow are not homogenous
across tumours, resulting in a non-uniform EPR effect. Some regions of the tumour are inaccessible to NC,
causing uneven drug distribution and accumulation of limited quantities of the initial dose (an increase in
26

07/05/2023
drug quantity is seen but the accumulated percentage of the initial dose stays low). Window chamber
(intravital microscopy) studies have shown an heterogeneous extravasation pattern of 90 nm pegylated
liposomes in a tumour model, demonstrating variations in permeability of the tumour vasculature.
However, tumour permeability to pegylated liposomes increases 3- to 4-fold in comparison to normal
liposomes [85, 112]. Vessel heterogeneity could arise, for instance, from variable pericyte coverage, from
10-20% in glioblastomas to 60% in colon and mammary gland tumours, influencing vessel permeability.
Moreover, mature (non-proliferating) vessels with different permeability status, are a non-negligible part of
tumoural vessels [101]. Heterogeneity in dose delivery could also derive from tumoural vessels with high
or low blood flow [94].
Pressure differences between blood and interstitium
Exchanges are partially driven by balanced tissue perfusion, with fluids coming from blood and
returning to capillaries (85%) and lymph (15%). Tissue perfusion is the consequence of hydrostatic and
oncotic pressures. Hydrostatic and osmotic pressures are major determinants of exchange across capillaries.
They are important considerations for exchange with the interstitium through capillary walls, particularly
for NC displaced more efficaciously by liquid convection rather than diffusion though the leaky vasculature
of fenestrae.
In tumours, the situation is different from normal capillaries as tumour capillaries are leaky,
allowing the movement of plasma proteins and macromolecules into the interstitium [113]. Oncotic
pressures increase in interstitial fluid, decreasing the convective transport of macromolecules by liquid
movement (the oncotic pressure difference between the vascular and extravascular compartments tends to
0). Moreover, an increase is observed in interstitial fluid hydrostatic pressure (IFP) due to ECM alterations
[101] and because lymphatic drainage is defective. IFP can go from 0-1 up to 50 mm Hg in some tumours
[99]. With the oncotic pressure becoming null and the difference between capillary hydrostatic pressure
(17-25 mm Hg) and IFP becoming smaller in tumours, convection flux across intercellular gaps is limited
[114]. The consequence is a decrease in extravasation as diffusive transport becomes predominant, relying
solely on a concentration gradient between blood and the tumour interstitium. Limited NC diffusion in the
interstitium determines predominant NC accumulation in the perivascular region, decreasing extravasation
even more by diffusion.
27

07/05/2023
IFP not only decreases NC uptake but also affects their homogenous distribution inside tumours.
Indeed, IFP varies from the tumour center to the periphery, generating outward flow from the tumour
center to the periphery [93, 101], opposing convective NC transport and potentially washing out NC into
peripheral tissues.
Finally, as pointed out by several authors, the results of tumour targeting by the EPR effect in
animal models should be interpreted with prudence. Indeed, it had been reported that in human tumour
xenografts, vascular permeability depends on the tumour implantation site, varying with time and treatment
course, making NC performance extrapolation. from animal to clinical studies uneasy [93, 94].
The ECM limits NC movement inside the tumour interstitium
After crossing the vascular endothelial barrier, the NC journey towards their target is not yet over.
Immediate release of the drug load too close from the blood vessels’ leaky walls may not exert maximal
efficacy. The tumour interstitium, albeit an aqueous compartment, is filled with a relatively stiff and
partially cross-linked extracellular matrix (ECM) of high collagen content along with proteoglycans and
hyalurans and similar to hydrogel, to which cells are attached [113]. The tumoural interstitium has usually
higher content in ECM that normal tissues [115]. The ECM presence in the tumour interstitium results in
slow diffusion, partly attributed to the sieving effect and interactions primarily with collagen fibers [116].
NC diffusion is not homogenous and depends on the orientation of fibres in collagenous tissues [117].
Moreover, neutral particles may diffuse faster in the ECM than charged particles, even if cationic surfaces
are advantageous for initial tumour vasculature targeting [118].
Although relatively large NC, e.g., Doxil®/Caelyx® (100 nm liposomes), are extravasated
efficaciously by the EPR effect, their further diffusive transport is limited by their size and by the
characteristics of the interstitial medium. Diffusion speed depends strongly on MW and size. The
dependence of tumour penetration on MW has been studied with dextrans linked to a fluorescent marker. It
revealed an inverse relationship between the size of linear macromolecules, vascular permeability and
tumour penetration [119]. The results disclosed tumour penetration of 35 µm for 3-10 kD dextrans, 15 µm
for 40-70 kD dextrans, and only 5 µm for 2,000 kD dextrans. 40-70 kD dextrans had the highest
accumulation compared to smaller dextrans which were able to diffuse in and out easily but with faster
clearance. The diffusion of pegylated, spherical gold NP within tumours beyond the perivascular region
28

07/05/2023
was also highly dependent on their size, with NP around 100 nm appearing to stay near the vasculature,
while smaller NP (10 nm) were rapidly diffused throughout the tumour matrix [29]. Similar results were
obtained with polymeric micelles of 25 and 60 nm, respectively [120].
NC size in biological medium
If sizes of 50 to 100 nm seem optimal for extravasation, once embedded in biological medium, NC
could experience some changes, with a size increase above this threshold. For instance, cation surface
adsorption on polyester particles (with negative zeta potential) decreases repulsive forces, resulting in
aggregation. Besides opsonisation, naked liposomes are also more prone to aggregation than pegylated
liposomes. The size increase in biological medium could explain, in part, their reduced access to the tumour
interstitium [85]. NC size and size distribution in biological medium are seldom reported but are
fundamental properties. Mayer et al. observed important size variations of PS NP measured in PBS or in
complete culture medium, resulting from opsonisation/aggregation phenomena [60]. To prevent this from
happening, the role of PEG and that of surface charge in maintaining the dispersion state of colloids should
be considered [121]. Opsonisation as well as swelling (by water uptake) could also affect NC size and size
distribution. NC size distribution is not always considered as it should, so that conflicting data and
ambiguity arise from a lack of information. In case of broad or polydisperse preparations, it is not always
possible to identify the NC size fraction responsible for positive or negative outcomes [122].
2.3.4. Conclusion on the EPR effect and NC properties
Although some nuances on the efficacy of the EPR effect are in order, it is so far the best targeting
strategy available, but, as discussed above, its extent is limited by several factors. Regarding drug
concentration, the effect is limited to some percent of the dose increase; more than 5-10% of the initial dose
is seldom found in the tumour site, the rest (90-95%) being still accumulated significantly in other organs
(primarily the liver and spleen) or excreted [107]. There are some indications that clinical improvements
are at least partially due to accumulation in targeted tissues, but the effect of slow release from long-
circulating NC cannot be ruled out [33]. Some authors are looking for ways to generate a more extensive
EPR effect, by increasing nonspecific tumour tropism (such as charge), modifying vascular permeability,
interstitial and blood pressures. To fully exploit the EPR effect, a better understanding of diverse tumour
environments and tumour capillary functions is needed.
29

07/05/2023
2.3.5. Passive targeting by surface charge modification
Aside from the addition of hydrophilic polymers (discussed earlier) and ligands ("active
targeting", which is beyond the scope of this review), surface charge seems to be involved in “nonspecific”
targeting although conflicting results have been reported. Positive charges are thought to generally
influence NC adhesion to negatively-charged cell membranes. Cationic liposomes (150±40 nm) have
higher uptake in tumoural areas compared to neutral or negatively-charged liposomes, with selectivity
towards the tumour vasculature [123]. It has been proposed that anionic phospholipids are markers of
tumoural cell membranes, resulting in increased charge density relative to normal tissues [124]. However,
the effect is quantitative rather than qualitative as negative charges are also found in the normal vasculature
[125]. Moreover, heightened uptake in the liver and augmented in vivo clearance, that could be attributed in
part to opsonisation, drastically reduce blood residence time (half-life as low as 5 min) and increase side-
effects [126]. To prevent these effects, the addition of PEG coverage to shield cationic charges has been
proposed [127, 128]. The length and density of PEG anchored on the surface seem to be determinants that
retain the dual properties of tumour vascular targeting and stealth behaviour. PEG coating adds a hydrated
layer on the surface, leading to an apparent decrease in charge (i.e. measured zeta potential). In contrast to
these results, a study of NP made of chitosan derivatives, grafting polymerization of methyl methacrylate
with different zeta potentials and sizes, showed that negatively-charged NP around 150 nm accumulated
preferentially in tumours and less in the liver and spleen than cationic NP [129]. PLGA NP (100±39 nm),
surface modified with a cationic compound, displayed a 10-fold increment in binding to arterial walls in in
vivo studies compared to unmodified anionic PLGA particles [130]. In blood, cationic carrier surfaces are
opsonised (whether PEG is present or not). Possible direct interactions of cationic charges with the anionic
EC glycocalyx, possibly explaining tumour vasculature tropism, are thus unlikely. Interaction of the tumour
vasculature with cationic NC may be mediated by one of the opsonins with specificity for cationic surfaces
[128]. This phenomenon has also been documented with ApoE exchange between circulating VLDL,
chylomicrons and NC, conferring hepatocyte tropism to otherwise untargeted NC [61, 131]. Serda et al.
reported that cationic surface-modified silicone microparticles favoured their uptake by EC, while anionic
alteration favoured macrophage uptake. Whatever the surface charge was before injection, all NP were
negatively charged after opsonisation. The uptake results could only be explained by different proteins
30

07/05/2023
binding to different charged NP surfaces, changing their cell tropism [132]. In contrast, modifying the
profile of adsorbed proteins on NC does not affect the level of EC association in vitro; thus, it seems that
cellular association does not depend on the identity of adsorbed proteins and they are consequently not
mediated by binding to specific receptors [56]. Roser et al. observed no difference in the in vivo
biodistribution of differently-charged NP while in vitro macrophage uptake changed with charge [52].
Charges are also important to control NC aggregation, which could change NC biodistribution.
DOX silica NC coated with PEG and poly(ethyleneimine) (PEI) showed increased efficacy in an animal
model. The authors attributed the improvement in EPR accumulation to controlled size of the silica core
(50 nm), the presence of a copolymer layer with PEG (5 kD) and low MW, low-toxicity PEI (1.2 kD)
conferring positive charges to the final NP, preventing their aggregation and thus exclusion from the
tumour interstitium [121]. Similarly, thiolated pegylated gelatine NP also showed slightly increased
accumulation in tumours compared to control pegylated gelatine NP [133].
The effect of charge on targeting should be interpreted cautiously as it is strongly dependent on
dispersion media, pH ionic strength as well as proteins or biological surfactant adsorption (see Section
2.1.2). The latter elements could impart their own charges to NC to the surface after adsorption. If
confirmed, these results move towards a limited role of surface charge in biological media, being limited to
affect preferential binding to NC surfaces of different sub-sets of opsonins.
2.3.6. Passive targeting with stimuli-sensitive NC
A last strategy to maximize the passive targeting effect is the addition of internal stimuli-
responsive properties to NC. Adding stimuli-responsive properties to NC can have different objectives,
including triggering drug release in specific pathological environments (and not in normal tissues);
changing surface properties for sequestration in a particular site [134]. All these approaches rely on a strong
EPR effect to accumulate NC in the tumour interstitium in the first place, before triggering specific release
of the encapsulated active, the release of a second targeting device, or a change in surface properties.
Thermal targeting of tumours
Thermal targeting relies on thermally-responsive polymers coupled with localized heating of
tumours (internal or external stimuli) to achieve targeted drug delivery [135, 136]. Several strategies have
been proposed. Among them, pegylated liposomes, prepared with phospholipids with phase transition (gel
31

07/05/2023
to liquid crystal) temperatures around 39-40oC, are destabilized by local, mild hyperthermia induced in a
matter of seconds and release their contents [137]. Moreover, thermal treatment by itself has been shown to
increase tumour vasculature permeability to liposomes. Another approach relies on the adhesion properties
of NC. Micronized aggregates of an elastin-like polypeptide (ELP) were targeted to solid tumours. The
aggregates adhered to the tumour vasculature when the tumours were heated to 41.5oC. They dissolved at
normal body temperature, increasing the vascular concentration locally, driving ELP across tumour
capillaries and augmenting its extravascular accumulation [138].Similar results were obtained with
p(NIPAAm)-based material [139], but ELP has the advantage of being a biocompatible macromolecule.
Tumor interstitium targeting with pH-responsive devices
pH-responsive NP and liposomes have been reviewed recently [135, 140]. Only some
characteristic examples will be mentioned here and some limitations discussed. Pathological tissues tend to
have a more acidic environment (pH 6.5 to 7.2) than normal tissues (pH 7.4), particularly tumours, because
of lactate production and hypoxia. The concept of pH-responsive NC is based on the insertion of a
molecule or polymer chain having an acid group with adequate pKa. Protonation in an acidic environment
changes ionization status; if the acidic group is strategically located, the molecule undergoes pH-dependent
conformation transition, and destabilization of the internal NC structure occurs eventually, either releasing
the encapsulated drug or changing surface properties.
Drug release at the site of action
Liposomes and micelles have been studied extensively to implement different pH-responsive
strategies. The first pH-sensitive liposome was composed of dioleoylphosphatidylethanolamine associated
with an acidic amphiphile for stabilization. Increasing pH neutralized the amphiphile charge, inducing
collapse of the bilayer [141]. Another approach is the addition of a pH-responsive polymer in the
phospholipid bilayer. A copolymer of N-isopropylacrylamide, methacrylic acid and N-vinyl-2-pyrrolidone
has been proposed for specific anticancer drug delivery [142]. However, adding PEG to the construct to
increase its circulation time decreased sensitivity of the system to pH change [143].
PEG can be linked to phospholipids by an acid-sensitive cleavable bond. When the PEG chain,
stabilizing liposomes, is cleaved, the liposomes are destabilized and their contents are released [144].
Specific drug release in response to pH has been proposed with NP swelling [145], pH-dependent
32

07/05/2023
conformation transition of dendrimers or the release of covalently-linked drug moieties [5, 146],
destabilisation of micelles or drug conjugates with sensitive linkage to pH [140]. Other carriers have been
designed to be sensitive to endosome pH to escape lysosomal degradation and release their intact contents
in the cytosol [147]. We will not, however, discuss this aspect in detail, and readers are referred to the
reviews mentioned above.
Surface modification
Another strategy proposed was to enhance NP retention in tumours by changing their surface
charge in response to acidic pH [148]. Stealth NP were prepared by the layer-by-layer technique with
alternate layers of oppositely- charged polyelectrolytes, the outer layer being a PEG-conjugated polymer.
The outer layer of PEG was shed by pH change, when NC entered the tumoural interstitium, exposing a
layer of cationic poly(lysine), with the aim of improving tumour cell uptake [149]. There is another reason
for discarding the PEG layer as lower cellular uptake is reported for pegylated devices [150].
Limitations
p(NIPAM) and several other chemicals deployed in several of these studies, although they permit
elegant approaches to the problem, are not degradable and/or produce degradation by-products not accurate
for long-term administration. It is one of the major limitations for human use. Moreover, the efficacy of the
system relies on sharp changes in pH when in vivo, changes are more gradual. Several hypothesis could
explain the suboptimal results, one of them being the fact that most of the NC load stays at the periphery of
the tumour, where pH is closer to blood pH (less acidic) [141, 151], the lower pH being recorded at the
tumour center [152].
Passive targeting with redox-responsive NC
Tumor interstitium are also highly reducing environments with extracellular glutathione
concentration about 4-fold higher than in the normal interstitium. The intracelluar glutathione level is even
higher: 100 to 1,000-fold higher than the extracellular normal level [141]. Thiol ester and disulfide-
mediated redox-responsive NC have been reviewed recently [134, 141]. A good example of this potential
approach is the exploitation of disulfide links between the hydrophobic and hydrophilic moieties. For
instance, detachable PEG in the presence of glutathione elicits link reduction, breakage, PEG release from
the NC surface, liposome destabilization and content release [141]. Alternatively, PEG removal taking
33

07/05/2023
place in the tumour interstitium could lead to exposure of specific ligands (the cell-penetrating peptide
TAT), improving liposome uptake by tumour cells [153].
Enzyme-responsive NC
The enzyme-responsive NC described so far are primarily based on protease-cleavable polymers
as substrates for matrix metalloprotease (MMP) present in higher concentrations in the tumour interstitium.
This approach has been taken to remove protease-cleavable PEG to destabilize liposomes [150, 154],
remove cleavable poly-anionic peptides, neutralize cationic domains on quantum dots to improve their
cellular uptake [155], and unveil specific ligands after NC extravasation. In this last example, MMP-2 up-
regulated in angiogenesis cleaves PEG connected to NC by a short peptide-specific substrate. The MMP-2
action removes PEG, uncovering specific targeting ligands [156, 157]. 100-nm pegylated gelatin NP can
accumulate significantly in the tumour interstitium. Under enzymatic MMP-2 and MMP-9 activity, gelatine
(a collagen derivative) hydrolysis releases 10-nm model NP (quantum dots) in the interstitium with better
diffusion capabilities [158]. This approach could potentially address concerns about limited diffusion of
carriers into the tumour interstitium.
3. THE BBB
3.1. Concept and main functions
The BBB, or hematoencephalic barrier, is a sub-type of continuous capillaries (Fig. (5)), with a
very specific physiological structure (Fig. (9)) that separates the bloodstream and the central nervous
system (CNS). The main function of the BBB is to support brain homeostasis because of its high
sensitivity, vulnerability and great need for oxygen and nutrients. Thus, the BBB is a very selective
biological filter that facilitates the supply process and waste elimination. At the same time, it protects the
CNS from different agents circulating in blood: pathogenic microorganisms, toxins, hormones and other
substances capable of changing the internal environment of the brain, including fluctuations of pH or
potassium concentration [159, 160]. It should be noted that the CNS is also protected from the immune
system (antibodies and leukocytes), which considers the brain tissue as foreign [161]. Consequently,
changes in BBB functioning can cause functional disorders or diseases of the CNS [160]. On the other
hand, its protective function, including the enzymatic activity of BBB cells [162], complicates the treatment
34

07/05/2023
of many neurological diseases, because many active molecules cannot cross this obstacle [163, 164]. Thus,
research on how to overcome the BBB to enhance drug delivery is quite current.
3.2. Structure
In general, the BBB includes the basement lamina and 3 cell types: EC, pericytes and astrocytes
(Fig. (9)) which are connected differently to each other according to their type (see below).
Fig. (9). General structure of the blood brain barrier.
(1) Basement membrane, (2) Endothelial cell (EC), (3) pericyte, (4) astrocytic projection, (5) tight junction,
(6) cell nucleus.
The basement membrane is a protein film, the thickness of which varies from 40 to 50 nm. It
completely surrounds EC and pericytes on the side of the brain and so separates them partially from each
other and completely from astrocytic projections (also called astrocytic end-feet) and brain extracellular
fluid. Many studies have shown that the basement lamina contributes to the restricted passage of proteins
and is thus protective [165, 166]. Nevertheless, excellent BBB selectivity is mainly explained by the
exceptional properties of the capillary EC layer, particularly by their organization and internal structure.
The distinctive feature of blood vessels of the CNS is the absence of fenestrations and intercellular gaps
between EC, which are interconnected by tight junctions that restrict extracellular diffusion of any
relatively large objects through the capillaries. The tight junctions provide not only cell adhesion, but also
35

07/05/2023
the possibility of regulating junction permeability, depending on the biochemical environment [167]. In
addition, tightness of the capillaries can be characterized by their electrical resistance. In rats, the resistance
value of muscle capillaries is only about 30 Ω⋅cm² while that of the brain rises to about 2,000 Ω⋅cm² [168].
It should be noted that the barrier functions exercised by EC of the BBB are also explained by their specific
histological and biochemical characteristics. Endotheliocytes of the BBB have an internal thickness of
370±170 nm [169] (less than intestinal EC), and so may facilitate transcellular transport of nutrients to the
brain (see below). The number of BBB endothelial mitochondria is higher than in peripheral capillaries
[170], due to the energy needed for passage by the active transport (see below) of different compounds
through the cell. The cerebral endothelium has a low content of pinocytic vesicles [171], probably to
restrict the non-selective penetration of compounds into endotheliocytes by pinocytosis. At the same time,
the EC wall has a number of specialized channels that are responsible for the regulation of necessary
substance flows (see below). Another BBB endothelium feature of importance is high enzymatic and P-
glycoprotein (P-gp) expression [172] which is believed to prevent the entry of potentially harmful
compounds into the brain by their transformation or removal, respectively.
Pericytes, small oval cells which cover about 1/5th of the outer surface of capillaries, are the third
main BBB component [173]. Most pericytes are located at points of EC contact. They play 3 important
roles: the modulation of vessel sections because of their high actin content , the regulation of division and
differentiation of endotheliocytes that are particularly important for the formation of new blood vessels
(angiogenesis) , and, finally, macrophage activity [174] and antigen expression , which allow them to exert
their protective function [175]. Pericytes are very tightly connected to endotheliocytes. Often, their
membranes are invaginated into each other. Gap junctions, that allow various molecules and ions to pass
directly into cells, are another type of such connections [174].
Bulky, star-shaped astrocytes are a different kind of BBB cell. Their branched end-feet cover 99%
of the surface of brain capillaries, but these cells do not perform a direct barrier function, and the free
diffusion of different compounds from EC to the brain is possible [176, 177]. Their main role is the
induction of BBB formation, particularly the endothelium phenotype and its dense arrangement [178].
36

07/05/2023
3.3. Types of molecular transport
To ensure efficient oxygen and nutrient supply to the brain as well as waste evacuation, the BBB
employs different paracellular and transcellular mechanisms of molecular transport. In particular, the entry
of compounds into the brain can occur by paracellular diffusion, passive diffusion, diffusion facilitated by
active transport by membrane transporters and, finally, by several types of endocytosis. Owing to the
presence of tight junctions between EC, paracellular diffusion is suitable only for polar and very small
molecules, i.e. water, glycerine, and urea [179]. All other compounds must pass by the transcellular
pathway.
Passive or free diffusion is the simplest form of transcellular transport. It tends to establish a
concentration or chemical potential equilibrium of substances, is nonsaturable, and requires no energy
[180]. On the other hand, this type of diffusion is usually restricted to substances with higher lipophilicity
and small size. Nevertheless, some works disclosed the possibility of passive diffusion, even for relatively
large molecules. For example, Abbruscato et al. demonstrated that the passage of an 3H-labeled
penicillamine-containing octapeptide (CTAP) into the CNS was not inhibited by the addition of unlabeled
CTAP [181]. These findings concur with those of Banks et al. [182], showing that some octapeptide
analogs of somatostatin also can cross the murine BBB by diffusion.
Facilitated diffusion or transport by selective membrane channels at the BBB involves the use of
specialized membrane proteins which allow some compounds to cross the cells in both directions without
energy expenditure, according to their concentration gradient. These transport membrane proteins can act
as uniports (1 molecule in 1 direction), as symports (2 or more molecules in the same direction) or as
antiports (2 or more molecules in opposite directions) [183]. However, flux is saturated by increasing
concentrations and can be inhibited by competitive substrates. For example, to ensure the passage of
significant quantities of water through endotheliocytes of the BBB that simple paracellular diffusion is
unable to provide, hydrophilic channels are formed by aquaporin-4 (AQP4) [184]and aquaporin-9 (AQP9)
[185]. The role of AQP9 is also to form aquaglyceroporins necessary for glycerine, urea and methanoate
ion transport [186]. The other important selective channels designed for glucose and vitamin C in their
37

07/05/2023
oxidated form are glucose transporters 1 (GLUT-1) [187]. Membrane peptides of the MCT
(monocarboxylate transporter) and SLC (solute carrier) families ensure the passage of lactic, pyruvic,
mevalonic, butiyric and acetic acids (MCT-1 and MCT-2), thyroid hormones (SLC16a2 and SLCO1c1),
sulphate ions (SLC13a4), L-ascorbic acid and vitamin C (SLC23a2), amino acids (SLC 38a3), folate or
vitamin B9 (SLC19a1), the cationic amino acids arginine, lysine, ornithine (SLC7) [188], respectively, and
others [189]. Facilitated diffusion is also used during biphalin absorption [190].
The mechanism of BBB transcellular penetration described above does not require any energy
contribution on the part of the cells, but there are substances that must be transported against the
electrochemical gradient. This requires not only special channels but also some ATP energy to drive these
molecular "pumps" (active transport). For example, in the BBB, such transport is provided by influx
transporters of enkephalin [191], vasopressin [192], [D-penicillamine2,5] enkephalin [193], and many
efflux pumps produced respectively by the following gene families, ABC (MRP1-5 and BCRP pumps
[194]), SLC (OAT3, OATP-A, OATP3A1, EAAT-1 [160]), and TAUT [189]. The role of these
transporters is the elimination of foreign compounds, including many drug molecules [195]. The EC and
astrocytes of the BBB contain a very high quantity of efflux P-gp, especially to increase their protective
potential [196]. Some active transporters may be stereoselective, i.e. ASCT2 removing L-aspartic acid
[197], while others do not present any substrate specificity [180].
In addition to previously-mentioned mechanisms, endocytosis and transcytosis are an important
way to pass through the BBB, especially for comparatively large particles, macromolecules or even their
aggregates. In this case, the plasma membrane is invaginated around the object to incorporate it and form
an internalized vesicle, which crosses the cell to be opened on the opposite side by a reverse mechanism,
and release its contents. The BBB allows 3 types of endocytosis/transcytosis: pinocytosis, receptor- and
absorptive-mediated endocytosis/transcytosis.
Pinocytosis or bulk-phase endocytosis, the non-saturable, nonspecific uptake of extracellular
fluids which occurs readily and to a large extent in other cells of the body, takes place to a very limited
degree in endothelial tissue of the brain microvasculature [198]. Thus, the other 2 endocytosis mechanisms
are more frequent. In particular, receptor-mediated endocytosis/transcytosis, a highly-specific, energy-
38

07/05/2023
dependent transport pathway, needs a specific ligand at the particle surface to trigger the formation of
vesicles [199]. For example, the TFR1 membrane receptor (or transferring receptor 1) is selective for
transferrin [200], the INSR receptor and some other peptide hormones (cytokines) for insulin [159, 201],
LEPR for leptin [202], IGF1R for the insulin-like growth factors IGF-I and IGF-II [203], etc.
Absorptive-mediated endocytosis/transcytosis or cationic transport is the uptake of positively-
charged particles launched by electrostatic interaction with the negatively-charged plasma membrane
surface [204]. This type of transport across the BBB is faster than receptor-mediated
endocytosis/transcytosis and allows more flow. Moreover, owing to its nonspecific and nonsaturable
nature, cationic transport has been in focus for the development of many novel drug delivery systems [205].
3.4. Passive anticancer drug targeting across the BBB
As indicated previously, the BBB should be considered not only as a mechanical filter but also as
a very specific biochemical barrier which engages particular mechanisms of protection and transportation.
For example, the fact that passage across the BBB is normally easier for small molecules than for large
ones [206] cannot explain the penetration of very large particles, such as antibodies, proteins, RNA [207],
and even some viruses [208] and microorganisms [209], in the CNS. Thus, all factors should be taken into
account to define BBB selectivity. However, it is still very problematic, and up to now 98% of drug
molecules are not able to pass from blood to the brain [210]. Actually, one of the major challenges in
pharmaceutical sciences is efficient drug delivery for the treatment of brain tumours. The prognosis and
survival of most patients with these diseases are quite poor. For example, median survival for a patient with
glioblastoma multiform is approximately 12-14 months, and has not improved substantially over the past
30 years [211]. It has been determined that the clinical failure of many potentially effective anticancer
therapeutics is usually not due to the lack of drug potency, but rather to the inability to cross the BBB
[212]. Thereby, it is obvious that new treatment modalities must be developed for the efficient delivery of
anticancer drugs to the brain. In addition, therapeutic strategies without alteration of BBB properties, by
administration in a more patient-compliant and safe manner (i.e. oral or i.v. forms), seem to be preferable.
In this context, drug nano-encapsulation techniques (without chemical coupling between the vector and
biologically-active unit), allowing passive BBB crossing (without any highly-specific interactions between
39

07/05/2023
NC and the capillary cell wall), represent interesting ways of improving CNS anticancer drug
bioavailability. In particular, the main desirable functions of such NC are the capacity to pass the brain
capillary wall, hiding encapsulated drug molecules from metabolic enzymes and efflux transporters, and,
finally, to increase specificity towards tumour regions of the brain. Nowadays, there are different kinds of
known drug NC for passive BBB penetration, namely, micelles, liposomes, polymeric NP, nanogels, and
dendrimers.
3.4.1. Micelles
Polymeric micelles, formed by self-assembly of large amphiphilic molecules in aqueous media,
have not been tested for the delivery of chemotherapeutics to the brain in clinical practice. However, some
investigations have demonstrated great potential of these nanostructures. For example, PluronicTM unimers
(block copolymers based on ethylene oxide and propylene oxide) allowed the penetration of bovine brain
microvessel EC monolayers by such active compounds as digoxin [213] and DOX [214], particularly by
inhibition of the P-gp efflux system [215]. In vitro and in vivo toxicity studies demonstrated no apparent
toxicological issues with carriers at the human dose equivalent.
3.4.2. Liposomes
Liposomes, small vesicles consisting of uni- or multilamellar phospholipid bilayers surrounding
aqueous compartments, are some of the most widespread drug NC proposed for brain targeting. Generally,
liposomes contain antineoplastic agents, such as daunorubicin, carboplatin, etoposide [216, 217, 218], etc.
For example, the commercial liposomal formulations Doxil®/Caelyx® (Sequus Pharmaceutical Co.) with
DOX [219] and DaunoXome® (Nexstar Pharmaceutical Co.) incorporating daunorubicin [220] are actually
in clinical use, including pediatric populations, showing some effectiveness in glioblastoma and metastatic
(high-grade glioma and teratoid/rhabdoid tumour) treatments [216]. Liposomes of both these formulations
are based on modified phospholipids, i.e. PEG derivatives in the case of Doxil®/Caelyx®. For example,
Depocyt® (ScyePharma Inc.), liposomal cytarabine, is actually serving in the clinical treatment of
malignant lymphomatous meningitis. These liposomes are presented by non-concentric vesicles, each with
an internal, aqueous chamber containing encapsulated cytarabine solution surrounded by a bilayer lipid
membrane containing cholesterol, triolein, dioleoylphosphatidylcholine, and
40

07/05/2023
dipalmitoylphosphatidylglycerol [221]. Many other similar anticancer liposomal formulations are in the
preclinical or clinical phase [222]. For example, modified lipid nanoliposomes containing irinotecan (CPT-
11) were recently demonstrated to prolong tissue retention of the drug and enhance its anti-tumour effects
in an intracranial U87 glioma xenograft model [223]. However, this type of nanovector is generally
unstable in plasma due to its interaction with high- and low-density lipoproteins that results in too rapid
release of the encapsulated drug [222].
Polymeric NP
NP, prepared from polymers and having a matrix structure that releases drugs by diffusion and
degradation, are generally very promising materials to encapsulate anticancer drugs [224]. Some of these
polymeric nanosystems have also demonstrated improvement in terms of drug amounts delivered to the
brain. For instance, DOX-loaded NP, obtained by anionic polymerisation from butylcyanoacrylate (Fig.
(10) (a)) and dextran, after coating with polysorbate(PSB)-80, significantly increased survival times in rats
with glioblastoma [225]. However, this family of materials (Fig. (10) (a)(b)), coated with PSB-80,
displayed some toxic effects toward the BBB [226, 227] as their PEG-coated analogues accumulated in
healthy tissue [228]. More interesting results were obtained with PEGylated-poly(hexadecylcyanoacrylate)
NP (PEG-PHDCA NPs) (Fig. (10) (c)). In particular, after i.v. administration in rats bearing well-
established intracerebral gliosarcoma, these carriers accumulated preferentially in tumoural tissues rather
than in peritumoural brain tissues or in the healthy contralateral hemisphere. In addition, PEG-PHDCA NP
concentrated much more in the gliosarcoma than their non-PEGylated counterparts, and did not display any
toxicity towards the BBB based on the sucrose permeability test [229]. Despite very encouraging
preliminary testing, PEG-PHDCA NP loaded with DOX have presented negative preclinical results in the
rat 9L gliosarcoma model, attributed to aggregation of the encapsulated, positively-charged drug (DOX) to
negatively-charged plasma proteins [230]. Thus, the physicochemical nature of the drug also should be
taken into account in the formulation process. Moreover, the cell internalization and intracellular
distribution of PEG-coated PHDCA NP in rat brain endothelial cells (RBEC) was investigated recently.
These NP displayed different patterns of intracellular capture. Depending on their specific surface
composition, PEG-PHDCA NP were 48% in the plasma membrane, 24% in the cytoplasm, 20% in
vesicular compartments and 8% associated with fractions of the nucleus, cytoskeleton and caveolae,
41

07/05/2023
indicating that PEG-PHDCA NP uptake by RBEC is specific and presumably derived from endocytosis
[231].
Fig. (10). Structures of polymers used in polymeric NP.
(a) poly(butylcyanoacrylate), (b) PHDCA polymers, and (c) PEG-PHDCA copolymer.
Other interesting materials for NP confection were synthesized by covalent binding between the
biodegradable copolymer PLGA and 5 short peptides similar to some synthetic opioid peptides. In
particular, the authors used the peptides H2N–Gly-L-Phe-D-Thr-Gly-L-Phe-L-Leu–X–CONH2, where X is
L-Ser–OH or L-Ser–O–β-D-glucose, L-Ser–O–β-D-galactose, L-Ser–O–β-D-xylose and L-Ser–O–β-D-
lactose. These peptides bear some resemblance to the matrix metalloproteinase (MMP-2200) but the Tyr
present in that proteinase was substituted with Phe in order to avoid a potential opioid effect. The ability of
these NP to cross the BBB was assessed in vivo by the rat brain perfusion technique after i.v.
administration. Fluorescence and confocal microscopy disclosed that the vectors crossed the BBB, whereas
NP made from pure PLGA were unable to do so [232]. Similar results were obtained with PLGA
conjugated with heptapeptide H2N-Gly-L-Phe-D-Thr-Gly-L-Phe-L-Leu-L-Ser(O-β-D-Glucose)-CONH2
loaded with loperamide [233].
3.4.3. Solid lipid NP (SLN)
42

07/05/2023
SLN, obtained by mixing solid lipids and some surfactants, are also a potential antitumoural drug
delivery system for brain targeting [234]. In vivo (mice) accumulation of camptothecin and DOX was
observed in the brain after both oral and i.v. administration [235, 236, 237]. Significant paclitaxel uptake by
the CNS was also noted in a short-term in situ rat brain perfusion experiment with SLN formulated with the
biocompatible emulsifying wax Brij® [238].
3.4.4. Nanogels
Recently, a new, promising family of carrier systems was proposed for drug delivery to the brain.
These so-called “nanogel” systems are made from a network of cross-linked ionic poly(ethyleneimine (PEI)
and non-ionic PEG chains. When a biologically-active macromolecule is coupled with the nanogel by
electrostatic interaction, the PEI fragments have a tendency to collapse, which results in decreased particle
volume and size. Because of steric stabilisation of the PEG chains, the collapsed nanogel forms stable
dispersions with a mean particle size of 80 nm. The nanogel has been tested as a potential carrier for
oligonucleotide (ODN) delivery to the brain employing polarized monolayers of bovine endothelial cells.
After i.v. injection of ODN-loaded nanogel in mice, no adverse toxic effects were observed and increased
brain and decreased liver/spleen accumulations were noted, compared to free ODN [239, 240].
3.4.5. Dendrimers
Dendrimers, highly-branched, symmetrical macromolecules, represent novel, very interesting drug
encapsulation systems. These compounds possess properties considerably different from linear polymers,
such as monodispersity, globular shape, high level of surface functionality, the presence of internal cavities,
and so on. The open nature of the dendritic architecture has led several groups to investigate the possibility
of encapsulating drug molecules within the branches of dendrimers (dendrons). In particular, very
encouraging data on methotrexate (MTX) delivery in an in vitro model of the BBB for the treatment of
gliomas were reported, with polyester-polyether (PEPE) dendrimers having the same butanetetracorboxylic
core, PEG spacers, and different branching agents: 3,5-dihydroxybenzoic acid, gallic acid, and 2,2-
bis(hydroxymethyl)butyric acid [241, 242, 243]. The efficient penetration of dendrimers across bEnd.3
culture (a BBB model) and internalization into U87 MG and U343 MG-A tumour cells and their spheroids
as paradigms of solid tumour tissue have been observed. The antitumoural effect of MTX-loaded
43

07/05/2023
dendrimers was better than the result obtained with free MTX [242]. Moreover, these dendrimers did not
manifest significant cytotoxic effects evaluated by MTT cell proliferation assay, erythrocyte lysis and
bEnd.3 viability experiments, even at high concentrations [243, 244]. Study of BBB- crossing mechanisms
showed relatively rapid transcytosis from 5 to 22 µg in the first hour. The increased number of PEG
terminal chains and glucosamine grafting to the surface of dendrimers were found to augment permeability
in this BBB model [242]. Transepithelial electric resistance measurements revealed that PEPE dendrimers
did not cause significant tight junction disruption during permeation. Basal to apical efflux of dendrimers
from the endothelium was very low (1-12%). A permeation investigation in the presence of the endocytosis
inhibitor sodium fluoride (NaF) revealed inhibition of transport. In addition, the combination of NaF and
low temperature (4oC) at the same time indicated that energy-dependent endocytosis was involved in the
process [243]. Thus, the proposed PEG-containing PEPE dendrimers present a very interesting object for
further study.
In conclusion, it should be noted that all approaches to reach passive nanoscale targeting across
the BBB are able to more or less enhance the uptake of small and/or large active molecules into various
tissues, but they usually lack selective/specific homing devices needed to target the brain and to
selectively/specifically increase drug delivery to the CNS [245].
4. THE GIT
4.1. Structure and physiology of the GIT
The majority of drugs on the market are formulated for the oral route, most of them aimed at the
blood circulation for systemic action. It is the most convenient and safe administration route, particularly
for chronic delivery, but it poses a number of challenges for the formulator in terms of bioavailability
(fraction of drug actually reaching the circulation) due to degradation by enzymes and harsh pH conditions,
low solubility of some drugs or limited absorption by the GIT epithelium. The GIT is approximately 6
meters long with varying diameters. It consists of the esophagus, stomach, small intestine (the major
digestive organ) and large intestine or colon. The luminal surface is not smooth, with deep, circular folds
about 1 cm high, villi, mucosal projections about 1 mm long, and microvilli (plasma membrane
microprojections), increasing the surface area of absorption to about 200 m2. Orally-administered drug
44

07/05/2023
molecules must stay for enough time in the intestinal lumen to be efficiently absorbed by intestinal cells via
different mechanisms detailed below [246, 247, 248].
The GIT wall (at the small intestine level) is comprised of 4 main histological layers . The first
layer, the “serosa”, is the outer layer of epithelial and supporting connective tissues. The second layer, the
“muscularis externa”, contains 2 layers of smooth muscle, a thinner outer layer, with longitudinally-
oriented muscle fibres and a thicker inner layer, with fibres oriented in a circular pattern. The “submucosa”
is a connective tissue layer that consists of some secretory tissues richly supplied with blood and lymphatic
vessels, including the lacteal, a wide lymph capillary at the center of the villi . The fourth layer, the
mucosa, is composed of 3 layers: the “muscularis mucosa”, the “mucosa”, connective tissue, and
epithelium. The intestinal epithelium acts as a physical and physiological barrier to drug absorption. It
mainly consists of absorptive enterocytes and mucus-producing Goblet cells, endocrine and Paneth cells
spread along the epithelium. The GIT epithelium is covered by a layer of mucus. Immuno-competent cells,
such as B and T lymphocytes and dendritic cells are located beneath the epithelium. The small intestine
wall possesses a rich blood network, and the GIT blood circulation is nearly a third of cardiac output flow,
underlining the importance of exchanges between the GIT lumen and the blood circulation. The lymphatic
system plays an important part in fat absorption from the GIT. The areas of lymphoid tissue close to the
epithelial surface are called Peyer’s patches or the follicle-associated epithelium (FAE) and serve as
immune sampling ports in the intestine. The FAE, separating organized, mucosa-associated tissues from the
lumen, is composed of enterocytes and M-cells. Microfold “M-cells” are important in local immune
responses to pathogens [249] and could be harnessed to deliver macromolecules and oral vaccines [250].
M-cells have a disorganized brush border at the apical membrane with reduced microvilli and thinner
surface mucus. Moreover, M-cells have specific surface-adhesion molecules that might be important
features for active targeting [249].
45

07/05/2023
Fig. (11). Structure GIT barrier: drug and NC transportation pathways. On the left, enterocytes, on the right
M cell. A mucus layer is found on the apical side of enterocytes and M cell. (1) Paracellular route, (2)
Transcellular route, (3) M-cell phagocytosis
4.2. Routes of and barriers to drug absorption
Drug molecules encounter many barriers in the GIT once released from their dosage forms after
they have dissolved in GI fluids. Drug molecules must be in solution and not bound to food or other
materials within the GIT, chemically stable to withstand the GIT pH, and resistant to enzymatic
degradation in the lumen. Finally, drug molecules need to diffuse across the mucus (“unstirred water
layer”) and across the GlT membrane, the main cellular barrier in order to reach blood circulation [248].
The main drug absorption pathways from the GIT include carrier-mediated transcellular transport, vesicular
transport, passive paracellular transport, passive transcellular transport through enterocytes and lymphatic
uptake by M-cells.
4.2.1. GIT environment
46

07/05/2023
The GIT environment is characterized by peristalsis, variable pH, the presence of surfactants (bile
salts), enzymes, bacteria, food and different types of secretions. GIT pH affects the dissolution rate and
absorption of drug molecules. The GIT has a wide range of pH values in the stomach (pH 2-3), small
intestine (pH 5-6) and colon (pH7). Moreover, pH depends of some variables, such as time, meal volume
and content, and volume of secretions. In addition, the presence of enzymes in the GIT could have an effect
on drug molecules that might be degraded, even before being absorbed. To provide effective treatment, the
delivery system should offer good protection against these enzymes [247, 251].
4.2.2. Mucus layer (“unstirred water layer”)
Mucus is a viscoelastic, translucent, aqueous, protective gel that is secreted throughout the GIT
and different mucosal surfaces (pulmonary, nasal, etc.). Its thickness varies from 50 to 150 µm in the
stomach to 15-150 µm in the intestine. It has a large water component (~95%), and its primary constituents,
which are responsible for its physical and functional properties, are largely glycoproteins called mucins.
Mucus acts as a protective layer and a mechanical barrier. It is a constantly changing mix of many
secretions, proteoglycans and exfoliated epithelial cells. It is continually replaced from beneath as it is
being removed from the GIT surface through acidic, enzymatic breakdown and abrasion (peristaltic
movement of food towards the colon). Mucus is a strong barrier that could catch and immobilize NP before
they have access to epithelial surfaces. GIT mucus is comprised of 2 layers, a firmly-adherent layer, slowly
cleared of an “unstirred layer” close to the epithelium surface, and a luminal mucus layer, a “stirred layer”,
a more rapidly-cleared layer [252]. It is secreted continuously and digested, so for drugs to be delivered to
mucosal surfaces, they need to diffuse through the unstirred mucus layers adhering to cells [252, 253].
In addition to the mucus layer, not unlike the glycocalyx in blood vessels, a glycocalyx composed
of glycoconjugate protein (that can be used for recognition), is present on cell surfaces, on microvilli in the
small intestine. It could also provide adsorption sites on the surface [247, 254].
4.2.3. Tight junctions and the paracellular route
The apical compartment of the lateral membrane consists of 3 components: tight junctions,
adherent junctions and desmosomes (Fig. (11)). For desmosomes and adherent junctions, adjacent cell
membranes are 15-20 nm apart, but are in contact at tight junctions, with the intercellular spaces being
completely absent. Tight junctions act as gatekeepers of the paracellular route, regulating drug molecule
47

07/05/2023
flux and preventing the free movement of molecules in the paracellular space. Understanding the barrier
function of tight junctions is necessary for developing absorption enhancers to deliver drugs via the
paracellular route. Tight junctions separate the cell surface into apical and basolateral membranes.
Diffusion is regulated by concentration differences and by electrical and hydrostatic pressure
gradients between 2 sides of the epithelium. Tight junctions are the main barriers to this type of absorption
[255, 256, 257].
4.2.4. Transcellular route and P-gp efflux
The transcellular route comprises passive and active transport. For carrier-mediated transcellular
transport, drugs are moved across membranes by protein transporters present on the apical membrane of
enterocytes (generally energy-dependent). The vesicular transport route consists of fluid-phase endocytosis
and receptor-mediated endocytosis (caveolae-dependent, clathrin-dependent, caveolae-clathrin-
independent, etc.) already detailed in Section 2.2. Vesicular transport could lead to the degradative
pathaway (via lysosomes) or to exocytosis, transport across the epithelium (transcytosis) to the basolateral
side of enterocytes near capillaries. For passive transcellular transport, drugs should be able to diffuse
across the apical membrane, cytoplasm and basal membrane. The surface area available for passive
transcellular transport makes up 99.9% versus 0.01% for the passive paracellular pathway.
P-gp is a membrane-bound transporter which excludes relatively lipophilic substrates from cell
cytosol by active transport. It belongs to the super-family of ATP-binding cassette transporters expressed in
cancer cells and responsible for chemotherapy resistance. P-gp is expressed in many normal tissues, such as
the intestine, and is localized in the enterocytes, limiting absorption of drugs from the intestine by pumping
them back to the lumen. NC have the ability to inhibit the effect of P-gp by limiting the presence of free
drugs in cell cytosol (drugs confined inside NC) and/or by including P-gp inhibitors, such as PEG, on their
surface. The mechanism of PEG P-gp inhibition is unknown, although direct interactions of the PEG chain
inside the channel as well as phospholipid membrane action have been proposed [258, 259, 260].
4.2.5. M-cells
Finally, transportation by Peyer’s patches, which contain M-cells found in the FAE at the base of
villi, is important in the phagocytosis and endocytosis of NP and microparticles [249]. The physiological
role of M-cells is to sample antigens present in the lumen to transport them to immune cells on their basal
48

07/05/2023
side, participating in protection against pathogens. They have high transcytosis activity, but represent less
than 1% of the intestinal mucosa surface (but more in rodents). Particles could be taken up by M-cells via a
specific (active endocytosis) or nonspecific (adsorptive endocytosis) mechanism [246].
4.2.6. Pre-systemic metabolism
Drug molecules have to be stable to cross the GIT epithelium and to resist degradation and
metabolism during passage. Drug molecules, which are absorbed from the GIT, are presented to the liver
through the hepatic portal system before reaching the systemic circulation. While some metabolizing
enzymes in the gut wall degrade drugs before they reach the systemic circulation, drug metabolism in the
liver is very extensive. Drugs may be completely absorbed but incompletely available to the systemic
circulation because of first-pass or pre-systemic metabolism by the gut wall and/or liver [261]. NC may
mitigate these deleterious effects and increase bioavailability by protecting drug molecules from enzymatic
degradation during intestinal passage and in the blood circulation [262].
4.3. NC translocation and drug bioavailability
As a consequence of GIT morphology and physiology, drugs must overcome 3 barriers to be
adsorbed efficaciously: conditions prevailing in the GIT lumen favouring degradation, the barrier mucus
layer and, finally, transport across the epithelium itself toward the blood or lymphatic circulation. The same
applies to drug-loaded NC. Depending on the encapsulation objective, NC could be either confined to the
GIT or they could participate in drug delivery beyond drug translocation across the GIT intestinal mucosa.
Their role could be limited, to protect against degradation and/or enhance drug solubilisation. In
this scenario, intact NC are confined to the GIT lumen, but contribute to increased bioavailability by
releasing drugs near the epithelium, acting as permeability enhancers, favouring adhesion and penetration
of the mucus layer, and/or contributing to the opening of tight junctions.
On the other hand, drug encapsulation could involve NC translocation [263], along with the drug
load, across the epithelium. It could even involve a role for the orally administered NC in drug distribution
in the systemic circulation. Indeed, intact NC introduced in the blood circulation could provide the
advantages of protection from metabolism, long circulation (if peggylated), targeting and improved PK, as
discussed for i.v. administrated drug-loaded NC (see Section 2-1). While most studies have focused on
blood drug levels or pharmacodynamic effects, few have followed the fate of NC [264]. However, in
49

07/05/2023
animal experiments, several types of NC, administered by the oral route, have been found in blood or
organs [265], but, in general, they are poorly uptaken, the order of magnitude being a few % of the initial
dose. There are also stability issues, as polymeric NC (polymeric NP, capsules) are more likely to be
discerned in blood compared to liposomes or micelles that are most likely to undergo dissociation and
degradation in the GIT lumen or during translocation across the epithelium. Designing this type of NC is
an engineering challenge and there is a limited set of data regarding NC translocated across epithelium to
be discharged in blood circulation. The 2 type of NC (role limited to GIT lumen or aimed at blood
circulation) may require different properties and they will be discussed below.
4.4. Optimizing NC physiochemical properties for oral delivery
4.4.1. Increased drug absorption, drug dissolution and protection from degradation
NC could address the challenges identified above, by offering protection to drug molecules, and
could also decrease the amount of drug needed for therapeutic action, thus reducing drug side-effects. This
could be particularly significant for peptide and protein delivery, with insulin, for instance. Unprotected
drugs, when introduced into the GIT, might not achieve the expected pharmacological effect because of
many factors, such as drug stability in GIT fluid, incomplete absorption and permeability. Particles in the
nano-scale range could improve bioavailability by increasing drug absorption by augmenting the
dissolution rate [266], due to favourable surface/volume ratios and favourable physical drug states
(crystallinity, amorphous or dispersed molecularly) in NC for release dissolution [246, 251, 267, 268].
Drug-loading efficacy varies with NC type and physicochemical properties of the compound to be
encapsulated. It should be considered in regard to therapeutic dose to be achieved in blood. Lastly, NC
stability is variable, and some are more sensitive than others (micelles, liposomes) to the GIT environment
(acidity, bile salts, lipases, etc.).
4.4.2. Mucus adhesion and penetrating properties
Mucoadhesion of NC
Increasing the residence time of drug-loaded NC in the small intestine might result in greater
absorption of the drug or/and of the carrier itself. Adhesion to mucus has been proposed to extend NC
residence time. For instance, mucoadhesion has been documented for NC surface modification with
chitosan, the cationic charges of the polymer interacting with negatively-charged mucus [269]. The creation
50

07/05/2023
of disulfide bonds on mucus glycoprotein, with surface NC derived from thiols, has been proposed as a
mucoadhesion strategy [270]. Broomberg et al. studied mucoadhesive anionic micelles, composed of the
copolymer Pluronic-(poly (acrylic acid)), for oral delivery [271]. The mucoadhesive properties of the
copolymer were linked to ionic interactions between mucins and carboxyl groups on the polymer, and the
entanglement of Pluronic (PPG-PEG-PPG block) with the mucin network. Mucoadhesion is size-
dependent, with maximum adhesion around 100 nm [272]. Examination of the behaviour of NC with
different surfaces, i.e. PLA-PEG NP (200 nm, zeta potential -24 mv), PS NP (200 nm, +1 mv), chitosan NP
(30 nm, +17 mv), revealed the effects of mucus adhesion (via interactions of hydrophobic surfaces with
hydrophobic domains of mucus glycoproteins) on NC uptake in vitro [273]. pH-responsive NP (250 nm) of
chitosan and poly(glutamic acid) encapsulating modified insulin were prepared. It was found that the drug
carrier was retained in the GIT, while modified insulin show an increase in absorption into the systemic
circulation, a result consistent with the mucoadhesion properties of chitosan NC and indicative of a
protective role of NC toward the active [274]
Mucoadhesion increases residence time. On the other hand, NC bound to mucin fibres are subject
to natural and continuous clearance of the mucus layer (stirred layer) with as a consequence a maximum
residence of about 4-5 h [252]. Moreover, adhesion to mucus hinders NC movement toward enterocytes or
M-cells surfaces, limiting their cellular uptake and transcytosis to subepithelial tissues, and course in
capillaries or lymph vessels. Strategies involving mucoadhesion (at least in case of strong interactions) may
be limited to NC aimed at releasing drugs in the GIT lumen, increasing drug concentration at the site of
absorption (near the mucosa epithelium).
Mucus-penetrating NC
Penetration of mucus is limited by gel porosity (NC size should be below gel mesh size to diffuse)
and interactions between NC surfaces and gel components. Conflicting data on charges required to avoid
ionic interactions with mucus resulted in an attempt to develop neutral hydrophilic surface modifications
[252].
PEG, a hydrophilic polymer described in Section 2, has been used to coat NP. The result of
coating is a decrease in zeta potential and improved NC stability. PEG prevents opsonisation by proteins
present in the GIT and charge interactions. PEG (2 kD) minimizes interactions with anionic-charged
51

07/05/2023
hydrogel, allowing faster transport [275]. Pegylated 100 nm particles show slower transport and diffusion
than 200 or 500 nm particles through the mucus layer. This could be explained by the existence of large
pores in gel, allowing large NP to be transported, while smaller ones enter the more densely-packed gel (a
process similar to size exclusion chromatography). It could also be explained by differences in the PEG
layer structure in smaller NC, with a higher radius of curvature, permitting direct interactions between the
NC surface and mucus [158]. Indeed, lowering PEG coverage to 40% and increasing PEG length to 10 kD,
decreases NC diffusion in the mucus layer [252]. PEG’s effect on further translocation across the
epithelium by different pathways remains to be clarified [265, 273, 276].
4.4.3. Translocation across tight junctions
In normal physiological conditions, translocation of NC through tight junctions is severely limited
by their relatively small surface area and tightness. In fact, NC passage is dependent on junction opening.
NC made of chitosan or with chitosan on the surface (and also co-administration of free chitosan) have
displayed the ability to transiently open tight junctions between epithelial cells, facilitating the transport of
drug molecules [255, 257]. Other permeability enhancers have been proposed [277], but all of them have
intrinsic toxicity and indiscriminately open the junctions to all kinds of GIT contents [256].
4.4.4. Translocation across enterocytes
NC translocation across enterocytes can occur by transcytosis [265], consisting of 3 steps, i.e.
uptake of the NC at the apical side, intracellular trafficking toward the basolateral side and exocytosis. The
different endocytosis routes have been described in Section 2.2 for EC and are similar in enterocytes. Size
and surface properties are the main determinants of transport efficacy and pathway chosen by NC.
Particle size, a critical characteristic of NC, has a direct effect on cellular uptake. It has been well-
accepted in research that small-size particles could be taken up by enterocytes and M-cells, enterocytes
having a maximum uptake of 50-100 nm particles, while M-cells can engulf particles up to the micrometer
range [249, 265, 278]. Peyer’s patches have up to 200-fold higher uptake of PLGA NC than non-patch
tissues. 100-nm NC diffuse in the submucosal layers while larger size particles (micrometer range) are
predominantly localized in the epithelial lining of tissues [267, 279]. Size, being a key parameter, PS NP
uptake increases with a decrease in particle diameter. Moreover, Peyer's patch uptake predominates over
enterocytes uptake, even if the former represents only about 1% of the intestinal mucosal surface [265].
52

07/05/2023
The presence of hydrophilic polymers on the surface of NP might increase transport through
mucosal surfaces [280, 281], although conflicting results have been reported. The adsorption of hydrophilic
block-co-polymers (poloxamer) onto polystyrene NC markedly reduces uptake in the small intestine [265].
For instance, 300-nm PLGA particles were found to migrate from the GIT into different tissues and organs,
especially the liver [282]. When modified on the surface with PEG and chitosan, they migrated from the
GIT into peritoneal macrophages [283]. These results are consistent with the importance of polymeric NC
stability as well as surface modification to cross the GIT epithelium. Endocytosis pathway depends on
surface properties. PLGA particles, or chitosan NC, for instances, are predominantly endocytosed via
clathrin-dependent pathways [264]. In contrast, it has been demonstrated that the bioavailability of
paclitaxel loaded in lipid nanocapsules (from 25 to 135 nm) is improved across CaCo-2 cell monolayers
because of this type of NC capacity to improve transport, predominantly via caveolae-dependent pathways
[284, 285].
Nonspecific adhesion properties might improve NC localization and transport. Lectins are
saccharide-binding membrane glycoproteins, which can bind reversibly to sugars either in free form or
supported on other membranes. They might adhere to the GIT surface to improve absorption and
permeability. Lectin binding was found to be favoured at neutral pH and reduced at acidic pH. Tomato
lectin has been reported to increase bioadhesion and conjugate with mucus gel. Surface modification with
covalent attachment of tomato lectin molecules indicated widespread uptake of PS NP by enterocytes rather
than by Peyer’s patches [265]. Wheat-germ agglutinin, a type of lectin conjugated with PLGA NP, showed
improved intestinal absorption of thymopentin as a result of lectin’s bio-adhesive effect. Lectin conjugation
with the NP surface ameliorated NP transport across the intestinal mucosa by increasing interaction with
mucus or epithelial cells [286, 287]. Gliadin, another mucoadhesive agent, was conjugated with NP
carrying amoxicillin. These NP were more effective for local treatment of H. pylori than conventional GIT
therapy [288].
4.4.5. Translocation across M-cells
The ability of M-cells to transport different materials by transcytosis (either phagocytosis or
endocytosis) has made them a good target to deliver vaccines and drugs [289]. Moreover, steric hindrance
by mucus layer is lowered as the layer over M-cells is thinner than over enterocytes. Bioadhesive materials,
53

07/05/2023
which prolong the residence time of drugs or vaccines and increase uptake by binding to intestinal mucus
or the apical surface, have served to improve NC delivery efficiency. M-cells in the intestine might be
targeted for mucosal vaccination to transport antigen to induce mucosal immunity. PEGylated PLGA NP
were also designed to target M-cells for oral vaccination [250]. Naked PLA particles (in the 200 nm range),
followed by fluorescence, showed initial accumulation in mucus and then moved to M-cells (in 15 min).
Further examination disclosed NP in immune cells in subepithelial tissue, confirming barrier crossing
[290]. In summary, optimal NC size seems to be below 1 µm with maximal uptake between 100 and 200
nm. NC with an hydrophobic surface are transported more than NC with a hydrophilic surface, and
hydrophobic surface NC are transported more than charged surface NC [277].
4.5. NC as effective oral drug carriers
The following examples have been selected to illustrate the challenge of delivery with different
types of NC and do not represent an exhaustive compilation of studies produced in this field.
Polymeric NP
Polymeric NC are among the most promising strategies to improve oral delivery, because of their
stability in the GIT, protection of encapsulated actives and the ease with which their physicochemical and
drug release characteristics can be modulated [277]. A large variety of materials could be exploited to
prepare NP (matrix-structure nanospheres or capsules) and to modify their surfaces. Synthetic (polyesters,
acrylates, etc.) and natural materials, such as chitosan, dextran, gelatin, etc., are being incorporated to
prepare these NC. Polymeric NP have several advantages for GIT delivery over other NC, especially
because they are more stable. Polymeric NP are distributed more uniformally in the GIT; therefore, the
drug is absorbed more uniformally and the risk of local irritation is reduced (compared to unique dosage
forms). Insulin has been the focus of research for formulation in effective NC for oral delivery. Increasing
the hydrophilicity of chitosan-triphosphate NP containing insulin leads to a prolonged hypoglycemic effect
attributed to the mucoadhesive and protective action of NP [291]. Similarly, poly(isobutylcyanoacrylate)
NC encapsulating insulin manifest improved effects on glycemia in a rat model over oral free insulin [292].
It is noteworthy that the percentage of dose actually crossing the GIT mucosa is increased but stays low (a
few % of bioavailability), requiring higher doses increasing potential toxic effects and underlining the
limitations of these approaches [277].
54
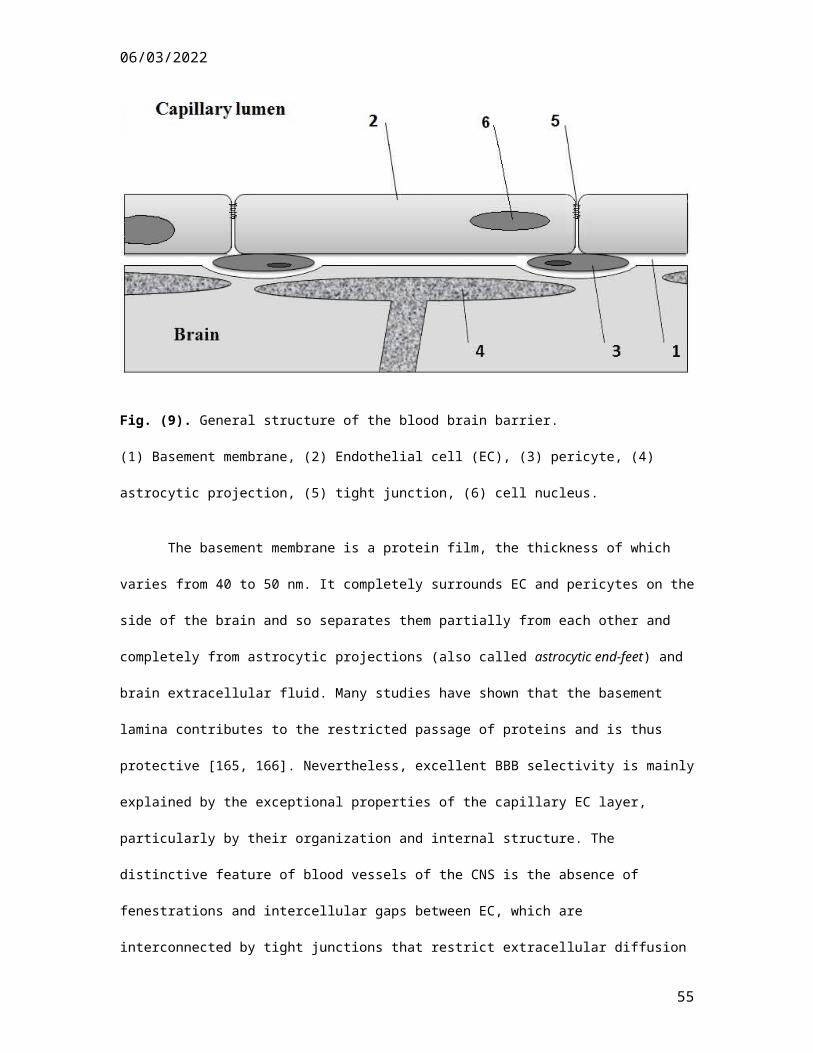
07/05/2023
Besides mucus penetration, PEG addition to the PLA NP surface has been reported to enhance
particle stability in biological fluids, preventing protein and enzyme absorption and thus protecting from
the degradation of NC and their load [293].
Liposomes and lipid-based NC
Liposomes can serve as efficient oral drug carriers to improve drug bioavailability because of their
ability to pass through lipid bilayers and the cell membrane. Because of their structure, liposomes can be
designed for water- and lipid-soluble drugs. Unfortunately, for oral drug delivery, they have limited
applications because of their instability in the GIT environment [294]. Their role seems confined to
increasing the bioavailability of low-solubility drugs. They appear to accumulate in mucus, prolonging their
residence time but limiting their diffusion. They have been proposed as vaccination adjuvants and as
antigen presentation to M-cells [295].
Lipids and phospholipids in the form of solid lipid particles (SLPs) are an interesting alternative to
polymeric matrix NC for hydrophobic drug and protein encapsulation [294]. For instance, camptothecin-
loaded SLPs coated with poloxamer 188 (200-nm particles with zeta potential around -70 mV) display
increased bioavailability compared to the oral, soluble drug form [235]. Similarly, insulin encapsulated in
lectin-modified SLPs presents increased bioavailability [296].
Micelles
Micelle polymers are composed of a hydrophilic shell (usually PEG) and a hydrophobic core.
Micelles have been studied as drug delivery systems to improve solubility, absorption and protect drug
molecules. Although polymeric micelles [297, 298] have a longer lifespan than surfactant micelles, there
are still some challenges facing their preparation, such as stability, improved drug loading, resistance to
dilution in the GIT, and narrow size distribution,. Micelles have been mainly studied to improve the drug
solubilisation of highly hydrophobic drugs, such as taxanes, for oral chemotherapy [301]. There are no
clear indications that they can cross the musocal epithelium other than in the form of isolated copolymers.
Hydrogel NC
NC delivery systems, hydrogel nanospheres, fabricated from poly(methacrylic acid) (PMAA) and
PEG, and loaded with the chemotherapeutic agent bleomycin, have disclosed an improved effect because of
their mucoadhesive properties and P-gp inhibition [303]. Negatively-charged 700-nm NC of alginate and
55

07/05/2023
chitosan encapsulating insulin were prepared. The results indicate a decrease of glycemia of about 40% in
diabetic rats. Encapsulation into mucoadhesive NC was a key factor in the improvement of oral absorption
and activity [304]. 400-nm alginate and dextran sulphate NC encapsulating insulin augmented GIT uptake
(13% bioavailability) and decreased glycemia in a rat model. This superior uptake seems to be linked to the
uptake of NC by small intestine epithelial cells [305].
Dendrimers
Dendrimers have a distinctive small particle size and could serve as solubilizing agents for both
hydrophilic and hydrophobic drugs. Poly(amido amine) (PAMAM) dendrimers have been reported to
improve the intestinal absorption of poorly soluble drugs but appear less efficient with macromolecular
drugs. Moreover, the dendrimer dose should be kept low to reduce toxicity [306]. In vitro, PAMAM
toxicity was diminished by surface modification by lauroyl chloride, while their permeation through Caco-
2 cell monolayers was increased. Both PAMAM and lauroyl dendrimers can cross epithelial cell
monolayers via paracellular and transcellular pathways [307]. Drug solubilization in dendrimers occurs
either by single drug molecule encapsulation or drug molecule attachment at the surface. PAMAM and
poly(propyleneimine) dendrimers have been studied to enhance the solubility of some drugs for oral
administration [308]. Dendrimers might be tested to improve drug permeability and bioavailability because
their small size allows them to enter cells and to cross the intestinal epithelium [309].
4.6. Conclusion
Encouraging results with some NC and actives have been reported in the literature on oral
delivery. However, low bioavailability (percentage of the initial dose) and lack of control of the absorbed
dose (crucial, for instance, in the case of insulin) still hinder the development of drug-loaded NC for oral
delivery. The physicochemical characteristics of NC need to be optimized to achieve the desired goal,
particularly in the perspective to deliver intact and functional NC into blood circulation. The challenge is to
integrate all the properties necessary to cross each barrier toward blood delivery, the properties sought for
blood circulation and targeting (see section 2.1 and 2.2) in the same NC.
5. PASSIVE PULMONARY TARGETING
56

07/05/2023
In addition to being a fast pathway for drug administration, pulmonary drug delivery is an
interesting and very promising approach to drug therapy as it avoids first-pass metabolism (in contrast with
the oral route) and is non-invasive (as i.v. could be). Also, the lungs have the advantage of large surface
contact over the alveoli, possessing a thin air-blood barrier and excellent pulmonary perfusion [261].
NC for the pulmonary route provide a viable alternative to drugs intended for parenteral
administration. Indeed, this delivery route transports drugs directly to the site of action (on the lung wall),
thus reducing side-effects. Despite their many benefits, NC must overcome several obstacles when inhaled,
such as mucociliary clearance, respiratory secretions (mucus and alveolar fluid), branching of the airways
and macrophages uptake. To develop effective NC, it is important to understand the anatomical and
physiological properties of the respiratory tract. In this section, we will first discuss lung anatomy and
physiology, then the mechanisms of particle deposition, the possible pathways of particle internalisation,
the impact of mucus on NC delivery, and finally physicochemical properties and some examples of NC
currently under study.
5.1. Lung structure and physiology
The airways are divided into 2 parts: the upper airway includes the nose, throat, pharynx and
larynx, while the lower tract contains the trachea, bronchi and bronchioles, which are connected to channels
leading to alveolar sacs and alveoli. The lungs are organs of respiration. The oxygen introduced into the
lungs goes directly to the alveolar cells and diffuse through the thin barrier to reach alveolar capillaries. As
for carbon dioxide it follows the reverse path [310]. The lungs contain about 2-6 x 108 cells, providing an
area of approximately 100 m2 in humans. The alveolar surface area exposed is normally covered by a
surface film of surfactant. Branching of the airways does not favor the inhalation of foreign particles and
microorganisms [310, 311].
At the physiological level, epithelial cells of the alveoli are called pneumocytes. They cover the
interior of the cells and contribute to their function. There are mainly 2 types of pneumocytes. Type I
pneumocytes are responsible for gas exchange (passive diffusion and active oxygen and carbon dioxide).
They are fragile and degrade rapidly on account of germs and pollutants; thus, they are difficult to repair.
They are relatively thin (about 5 µm) and cover about 90% of the total alveolar surface. A single cell covers
up to 400 m2 of the alveoli interior, but the total number of type I cells does not exceed the number of type
57

07/05/2023
II pneumocytes. Type I pneumocytes are closely coupled to capillaries from which they are separated by
the basement membrane, allowing the diffusion of respiratory gases [261]. Type II pneumocytes (or
granular pneumocytes, and large alveolar cells), in turn, have the distinction of being cube-shaped or
rounded. Their cytoplasm is rich in organelles, a sign of active metabolism, confirming the presence of
overdeveloped endoplasmic reticulum and Golgi apparatus. These cells are characterized by specific
organelles, the lamellar bodies, secreting pulmonary surfactant. There are 6-7 type II cells per alveolus.
Type II pneumocytes are believed to be essential for cellular repair after damage caused by viruses or
chemical agents [312]. They divide, by losing their lamellar bodies, and flatten themselves to replace type I
pneumocytes when they die.
Fig. (12). Structure of alveoli. Cross-section of an alveolus showing a capillary, alveolar macrophage,
surfactant layer, type I and type II cells (adapted and modified from [313])
5.2. Deposition mechanism of particles
Whether solid or liquid, nebulization and aerosol inhalation are 2 forms of pulmonary
administration. Several factors, such as respiratory rate, lung volume and health status must be taken into
account when designing a formulation intended for the pulmonary delivery. However, the size of inhaled
particles is one of the most important elements in the development of forms intended for inhalation. Indeed,
particles must have size distribution less than about 5 or 6 µm and less than 2 µm for deposition in the
alveolar region. The 4 main mechanisms affecting deposition and the pathway of particles in the lungs are
58

07/05/2023
gravitational sedimentation, impaction, diffusion and interception [261]. They are developed below [312,
314].
5.2.1. Gravitational sedimentation
Sedimentation corresponds to the gravitational pull of particles on the airway wall. When particles
are moving in air, gravitational attraction and air resistance force their deposition on the lung surface,
mainly in the bronchi and bronchioles. The gravitational settling of inhaled particles depends on their size,
density and residence time in the respiratory pathway. Thus, particles with aerodynamic diameters less than
0.5 µm will not be affected by this settling. On the other hand, the probability of hygroscopic particles
settling down by sedimentation increases as their size and mass grow with moisture in the lung airways.
5.2.2. Impaction
Impaction is the projection of particles against the airway wall, usually occurring in the upper
lungs. Thus, when confronted at the junction of 2 airways, many of them continue straight ahead and hit
lung wall surfaces on their trajectory (sometimes adhering to the wall), rather than follow airflow. The
probability of impaction depends on air velocity and particle mass. It is also closely linked to particle
inertia. This mechanism of deposition is particularly important for particles with a diameter greater than 5
µm, and even more for those above 10 µm.
5.2.3. Diffusion
This delivery mechanism, also known as Brownian motion, is a major problem involving the
random motion of particles less than 0.5 µm in diameter from a region of high particle concentration to a
lower one. The smaller the particle, the more significant is its agitation energy, and it randomly deposits on
walls. Unlike the mechanism of impaction, diffusion occurs mainly in the lower lungs, i.e., in the
bronchioles and alveolar region.
5.2.4. Interception
Interception is when a particle comes into contact with a surface of the respiratory organs because
of its size or shape. It is also related to electrostatic forces and arises when the distance between the particle
and the wall is less than the size of the particle.
5.3. Respiratory secretions as a barrier to NP deliverance
59

07/05/2023
Mucus, i.e., respiratory secretion, is usually located in the upper airways. The alveolar area
contains no mucus but rather alveolar fluid, maintaining surface tension in the alveoli. Respiratory mucus,
mainly secreted by glandular cells, forms a viscoelastic and continuous layer on the respiratory epithelium
surface, thus becoming a natural barrier that protects the lungs from hazardous particles that could enter
during normal breathing through the nose. Mucus is involved in mucosal defense through its anti-infectious
and protease inhibitors as well as its mechanical and rheological properties. However, the alveolar area
does not contain such defense but can rely on macrophages to trap and eliminate foreign bodies that try to
penetrate the alveoli [252, 275, 315]. Residues from this operation are pushed to the mucociliary escalator,
then to the bronchioles for expulsion from the lungs. As GIT mucus, lung mucus is composed of 95 to 97%
water containing proteins (1% of glycoproteins), lipids (1%) and ions. Glycoproteins, or mucins, are large
molecules (0.5-40 000 KDa) with polypeptide chains that are able to connect hundreds of glycan chains.
The mucin network can form pores, with diameters ranging from 20 nm to 800 nm. In general, upper
airway lung mucus thickness is 15 m compared to 55 m in bronchi [252, 316].
Mucus characterization, whether in terms of its composition, structural organization, thickness,
flow rate or time of disposal, is crucial in the development of NC [317]. Similarly, NC surface properties
and size characterization are important factors to consider for barrier crossing. NC must be able to penetrate
mucus faster than mucus renewal and mucus clearance to overcome the barrier. For example, particles with
sizes larger than mucus pores will likely have difficulty diffusing. Particles with no affinity for mucus (i.e.
adhesive forces, electrostatic interactions with carboxyl groups or sulphate on mucin, hydrophobic forces,
interpenetration of polymer chain and hydrogen bonds) will have trouble adhering on it and will be quickly
eliminated. Most experiments on mucus focused on GIT mucus. Studies conducted on lung mucus were
distinctively limited to patients with cystic fibrosis. These patients suffer from over-excretion of pulmonary
mucus, which facilitates sampling. To our knowledge, only a few groups have been interested in
investigating the impact of mucus on pulmonary NC, and rarely did they focus on alveolar fluid. For more
details on this subject, however, readers are encouraged to consult the following reviews [252, 310, 318].
Below is a non-exhaustive list of studies that were conducted.
Wang et al. reported that NP as large as 500 nm in diameter rapidly diffuse through mucus to the
extent that they are densely covered with low MW PEG [319]. They showed that polystyrene NP are
60

07/05/2023
trapped in mucus because of bonds formed between hydrophobic polystyrene beads and the hydrophobic
domains in mucin fibers. They believe that coating particles with PEG creates a hydrophilic and neutral
shell that minimizes hydrophobic adhesive interactions with mucus. Particles coated with 5-kD PEG
retained quick mucus-penetrating properties, but particles coated with 10-kD PEG lost them, indicating that
MW between 5 and 10 kD was critical, with transition of dense PEG coating from muco-inert to
mucoadhesive. High MW PEG (≥10 kD) can be highly mucoadhesive due to the interpenetration of PEG
fibers with mucus and hydrogen bonds between oxygen atoms in PEG and sugars on glycosylated mucins.
Although this study was performed on cervico-vaginal mucus, the same conclusion can be drawn for
mucous membranes of the respiratory pathway as they have similar rheological properties.
As already mentioned for GIT mucus, it is well-known that PEGylation enhances the transport of
NP across the mucosal barrier and decreases clearance by alveolar macrophages. Lai et al. discerned that
larger PEGylated polystyrene NP (500-nm diameter) can cross fresh, undiluted human mucus more
efficiently than smaller ones (100-nm diameter) [252].
5.4. Internalization pathways of inhaled NP to the blood circulation
Translocation mechanisms of NC have been the subject of much research over several years, often
generating contradictory results. Patton et al. suggested that there are 2 different mechanisms of
translocation from the lungs to the systemic circulation [320]. In general, paracellular transport occurs fast
(between 5 and 90 min) for macromolecules with MW >40 kD and size >5-6 nm, and transport is much
slower ("receptor-mediated transcytosis") when the particles have MW <40 kD and size <5-6 nm (see Fig.
(13)). Although, studies are conflicting, researchers agree that 2 physicochemical parameters − size and
surface charge − influence the translocation of particles to the systemic route.
5.4.1. Size
By deploying 13C-labeled NP of 20-29 nm, Oberdörster et al. confirmed a rapid
clearance/translocation of particle during inhalation exposure. After 18 and 24 h, authors noticed increased
13C hepatic levels, showing that a large amount of NP reached the blood circulation. However, authors do
not explain this uptake by pulmonary epithelium crossing but rather by a significant particle absorption by
the GIT due to mucociliary clearance as well as post exposure GI uptake of NP as the animals licked their
contaminated fur [321].
61

07/05/2023
Meiring et al. (2005) stipulated that NP could follow various cellular pathways of translocation,
such as clathrin, pinocytosis and caveolae. They could have been the route for iridium NP (18 nm in size)
in a model of isolated perfused rabbit lungs [322]. Heckel et al. showed that 7% of colloidal gold NP (4-nm
size) were internalized by EC and lung epithelia in New Zealand white rabbits. Infusion of
lipopolysaccharide (LPS) caused mild pulmonary edema, and transendothelial passage was multiplied by a
factor of 5, while 14% of particles accumulated in the interstitium and 11% reached the alveoli [323].
Furuyama et al. (2009) administered gold colloid or fluorescein-labeled polystyrene NP (20 and
200 nm) [324]. Gold colloid particles were detected on the EC surface, on the alveolar surface, in
endocytotic vesicles of alveolar epithelial cells, and in the lung basement membrane after 15-min of
intratracheally instillation. A small but noteworthy amount of gold was detected in the extrapulmonary
organs. After the administration of 20- or 200-nm fluorescent particles, free particles were noted
infrequently in blood vessels, on the endocardial surface and in the kidneys and liver only in mice that
received 20-nm particles, whereas phagocytes containing 20- or 200-nm particles were found in
extrapulmonary tissues. Thus, small amounts of ultrafine particles were transported across the alveolar wall
into the blood circulation via endocytotic pathways, but particle-laden alveolar macrophages translocated
both ultrafine and fine particles from the lungs to the extrapulmonary organs [324].
In their study, Takenaka et al. exposed rats to 16-nm gold particles for 6 h [325]. After 7 days,
they observed a high percentage of gold NP in lung tissue and a low quantity in blood. NP were also
encountered in the vesicles of alveolar macrophages. They concluded that gold particles in alveolar
macrophages are internalized by endocytotic pathways and the uptake of gold particles by alveolar
macrophages is limited [325].
Chen et al. tested radiolabeled PS particles uncoated and coated with LPS with average diameters
of 56.4 and 202 nm, respectively [326]. The results indicated that the pulmonary deposition of radioactivity
was virtually similar for both sizes. Only small amounts of radioactivity (~2%) were recovered in blood
shortly after the administration of both particle types in healthy rats. However, the extent of ultrafine size
particle translocation into blood after pretreatment with LPS was significantly higher (~4%) in comparison
to larger particles (~2%). These authors concluded that only a small fraction of intratracheally-instilled
62

07/05/2023
ultrafine particles can pass rapidly into the systemic circulation, but this translocation is markedly increased
after LPS pretreatment.
Yacobi et al. investigated the translocation mechanisms of fluorescently-labeled polystyrene NP
(20 and 100 nm) through rat alveolar epithelial cell monolayers (RAECMs) [327]. These results of confocal
laser scanning microscopy revealed no intracellular co-localization of NP with early endosome antigen-1,
caveolin-1, clathrin heavy chain, cholera toxin B, or wheat germ agglutinin. They concluded that NP
translocate primarily transcellularly across RAECMs, but not via known major endocytic pathways, with
the contribution of the lipid bilayer of cell plasma membranes [327].
5.4.2. Surface charge
In addition to studying the impact of particle size, some authors analyzed the impact of surface
charge. In previously-described work, Yacobi et al. tracked NP translocation according to different surface
charges. Fluorescently-labeled polystyrene NP were either negatively charged (with a carboxylate function)
or positively charged (with an amidine function). Trafficking data revealed that the surface charge on NP
was largely affected by transport rates, with positively-charged NP being 20-40 times more rapid than
negatively-charged ones across the alveolar epithelium (RAECMs) [328].
Choi et al. (2010) studied fluorescent NP with varying chemical composition, shape, size and
surface charge. They demonstrated that NP with hydrodynamic diameter (HD) less than ≈34 nm and a non-
cationic surface charge translocate rapidly from the lungs to mediastinal lymph nodes. NP with HD <6 nm
were able to traffic rapidly from the lungs to the lymph nodes and bloodstream, and then cleared by the
kidneys [329]. Studies showed the presence of albumin and lecithin in alveolar epithelial fluid to be
essential constituents to facilitate NC epithelial cell uptake. PS 240 nm NP have been shown to be
translocated across the alveolo-capillary barrier when coated with lecithin while uncoated PS NP were not
transported [330].
63

07/05/2023
Fig. (13). Proposed NC transport pathways across alveolar and vascular endothelial cells. Possible
translocation mechanisms across the 2 cell layers: (A) Alveolar epithelial tight junctions (paracellular
transport), (B) Caveolar receptor-mediated transcytosis, (C) Caveolar trans-endothelial transport, (D)
Trans-endothelial channel, (E) Vascular endothelial cell tight junction
5.5 Physicochemical characteristics of NC
Particle size is a critical factor that determines the location of particles deposited in the lungs
[331]. It is expressed by aerodynamic diameter that takes size, density and particle shape into account.
Large particles (HD >6 µm) have strong momentum and tend to settle in the upper regions of the
respiratory path instead of following the course of breathing. The movement of small particles (HD <1 m)
is governed by Brownian motion, and most of them will be exhaled during normal breathing. For effective
deposition in the lower respiratory path, optimal particle size must have an aerodynamic diameter between
1 and 5 µm. On the other hand, for smaller size particles, the probability of deposition in the lower regions
of the lungs will be increased (due to diffuse mobility). NP smaller than 100 nm seek refuge in the alveolar
region with fractional deposition of about 50%. These fine particles enter the lungs agglomerated and can
easily break into smaller particles during deposition (Fig. (14)) [332, 333].
64

07/05/2023
Fig. (14). NC Predicted fractional deposition of inhaled particles in the human respiratory tract. (data
reformatted from [332])
In addition to the complex anatomy and physiology of the lungs, air sacs are rich in macrophages
that constitute a vital factor in immune defense. Particles between 1 and 5 µm are typical of bacteria size
and are easily engulfed by phagocytosis; of course, this limits NP translocation in the bloodstream. To
remedy the problem, Edwards et al. improved insulin absorption in the lungs by developing large porous
particles (LPP) with geometric diameter (gd) of ~10 µu and density (ρ<0.1 g cm -3), facilitating deposition
in the alveoli and decreasing the chances of phagocytosis [331].
5.6 Impact of key physicochemical properties on NC efficacy
5.6.1 Microparticles and NP
Compared to liposomes, microspheres have more stable physicochemical behaviour in vivo and in
vitro and should provide slower release and longer pharmacological activity of encapsulated drugs.
Microspheres are less hygroscopic and are thus less liable to swell in the presence of moisture located in
the lungs [334].
65

07/05/2023
There have been significant improvements in aerosol particle technology in recent years [331].
Particles with mass density (ρ<0.4 g/ml) and increasing particle geometric size (10–20 µm) were inspired
deep into the lungs and escaped alveolar macrophages until they delivered their drug. LPP, by virtue of
their porosity, display an aerodynamic diameter much lower than GD, facilitating their deep lung
deposition. Inhalation of large porous particles elevated systemic levels of both insulin and testosterone in
comparison to small non-porous particles. Edwards et al. demonstrated that inhalation of large porous
insulin particles resulted in elevated systemic levels of insulin and suppressed systemic glucose levels for
96 h, whereas small non-porous insulin particles had such an effect for only 4 h. High systemic
bioavailability of testosterone was also achieved by inhalation delivery of porous particles with a mean
diameter (20 µm) approximately 10 times that of conventional inhaled therapeutic particles. Large, porous
PLGA particles (density (ρ)=0.4 g.cm-3, diameter 5 μm) appeared to be more efficient for the pulmonary
drug delivery of inhaled particles than small porous or non-porous particles (ρ 1-0.5 g.cm-3, d=5 μm) [331,
335].
This efficacy was attributed to the fact that large particles aggregate less and disaggregate more
easily under shear forces than smaller, non-porous particles – all other considerations being equal [336,
337]. Hence, they appear to aerosolize more efficiently from a given inhaler device than conventional
therapeutic particles. Particle size is crucial in the appearance of inflammatory symptoms. Indeed, ultrafine
polystyrene particles (60 nm) induced much more pro-inflammatory activities than NP with a size range
between 200 and 500 nm. And it could be attributed to their much more pronounced specific area [338].
5.6.2 Liposomes
The utilization of liposomal drug formulations for aerosol delivery has many potential advantages,
including aqueous compatibility, sustained pulmonary release to maintain therapeutic drug levels and
facilitated intracellular delivery, particularly to alveolar macrophages [339]. Liposomal aerosols have
proven to be non-toxic in acute human and animal studies [340, 341, 342, 343, 344]. These results suggest
that drug-liposome aerosols should be more effective for the delivery, deposition and retention of water-
insoluble, hydrophobic, lipophilic compounds in contrast to water-soluble compounds [345, 346, 347]. The
goal of drug-liposomal aerosol therapy is to maximize delivery and retention while minimizing clearance.
5.4.3. Micelles
66

07/05/2023
It is well-known that polymeric micelles are often more stable than surfactant micelles [348]. In
general, they are smaller in size than polymeric microspheres and liposomes, their surface properties allow
them to escape from macrophage uptake, they are easy to prepare, and they are capable of targeting specific
tissues.
In this field, Jones and Leroux designed beclomethasone dipropionate-loaded polymeric micelles
for pulmonary delivery [4]. The concept was developed since polymeric micelles are able to evade the MPS
due to their bulky hydrophilic outer shell and sustained drug release. In addition, drug-loaded polymeric
micelles are strongly suggested to pass directly to their receptors in epithelial cells through the mucus layer
to treat bronchial inflammatory diseases. For these reasons, polymeric micelles are very beneficial in
delivering hydrophobic corticosteroids, such as beclomethasone dipropionate, for COPD theory since the
inability of such drugs to penetrate the mucus layer to reach the target site is largely the result of treatment
failure in the past.
6. PERSPECTIVES
Drug NC, able to deliver high doses of therapeutic compounds in specific areas while lowering the
exposure of healthy tissues to toxic side-effects, are still an unreached goal. The preparation of such entities
is an engineering challenge as several properties must be imparted: stability in biological medium, high
drug loading, long circulation in blood (i.v. administration), immune system evasion, and targeting
pathological tissues while avoiding healthy ones. Size and surface properties are key elements of biological
barrier crossing, at the pulmonary, GI and vascular levels.
Size, hydrodynamic radius and aggregation status are parameters that have been largely explored,
but investigations often lack adequate characterization of size and size dispersion [122]. When NC
targeting and biodistribution studies are based on inadequately-reported size dispersion or wide size
dispersion, the informative value of the studies is limited. Several NC properties are dependant on their
environments. The size is one of them as aggregation and opsonisation may change size and size
distributions in biological medium. This underlines that NC size is also a surface chemistry problem to be
resolved.
The effects of surface charge and surface hydrophobicity have also to be considered in situ.
Indeed, once a NC is immersed in biological medium, it is no longer the originally-designed artificial
67

07/05/2023
construct. At this point, it is the original construct along with adsorbed proteins (and other substances such
as lipids) that profoundly affect its surface characteristics. The properties defined in vitro, before
administration, are no longer valid, whether the entry pathway is the upper airways, the GIT or the vascular
compartment. For instance, protein coating (either discrete or complete) may not only alter carrier size, but
also alter surface charge and hydrophobicity, may turn it into specific-recognition NC (adsorbed protein-
mediated recognition), or may change its morphology and surface roughness.
The modification of generally hydrophobic NC surfaces by PEG has found its way and utility in
all type of deliveries, i.v., pulmonary or GI. Despite its widespread use, some concerns regarding its
toxicity and interactions with the complement and immune systems have renewed interest in the
development of alternatives to pegylation [55, 349]. Regarding surface properties, other challenges ahead
are better assessment of polymer architecture effects on surface properties (opsonisation, cellular uptake),
and the study and control of surface chemistry heterogeneity at the nanoscale level (as heterogeneity can
cause NC failure).
Some passive NC targeting technologies are currently approved for tumour chemotherapies, and
many are either based on liposomes or “nab” technology (both by i.v. administration). But the fraction of
dose of these NC accumulated in the tumour still stays low. A similar situation can be found in oral
delivery or pulmonary delivery, as NC transport across both epithelium is also weak. Addition of a specific
ligand on NC surfaces has been proposed to increase these values. However, no active, targeted drug NC
systems have yet been approved, as they fail to show clinically significant improvements over passive
targeted systems [93].
Clearly, certain factors have been ignored or misinterpreted, and some of them could be related to
NC surface properties. Aside from these properties explored (albeit perhaps incompletely), there are many
other parameters, ignored or less investigated, that may have their say in the limited success of current NC
approaches. For instance, shape, curvature radius, surface roughness, patterning, porosity, density and
Young’s modulus of NC may be some parameters worth exploring.
Surfaces with very high curvature radius may suppress protein adsorption, starting with larger
proteins [350]. Some authors have identified the topographic effect of surface curvature on protein binding,
demonstrating small (30-nm) particles are stabilizing globular protein conformation (albumin, lysozyme),
68

07/05/2023
while rod-like proteins are denatured (fibrinogen). It seems possible to favour the binding of some type of
proteins by surface design [351, 352]. Surface topography effect is also at work with porous cationic
polysaccharides NP showing no change of size and zeta potential or complement activation even after
extensive lipid and protein adsorption [353]. Softness and rigidity of particles (combined with size) have an
effect on localization to the lung and filtration by the spleen [354, 355]. Young’s modulus of NP has been
found to affect endocytosis efficacy and the choice of uptake pathways [356] with potential consequences
for transcytosis efficacy. Moreover, this modulus could change with temperature (above polymer glass
transition) or with the state of hydration alteration in polymeric NP. Finally, it is noteworthy that the
development of NC, as sophisticated they may be, should yield fully biocompatible and easily eliminated
materials, whether as intact NC or after degradations of their components.
7. CONCLUSION
The pursuit of well-designed NC, optimized for size with narrow size distribution, shape and
surface modification for advanced passive targeting capabilities is not trivial. Combination with active
targeting, if necessary, may further improve targeting capabilities. Nevertheless, an adequate passive
targeting effect is required to achieve efficient active targeting. The development of NC and their passage
to the clinical stage is dependent on thorough physicochemical characterization, restriction of nonspecific
uptake, improvement of stimuli-responsive systems and full toxicity assessment. Finally, there is still a
need to further knowledge on targeted diseases and tissues as well as their microenvironments (tumour
interstitium, intestinal mucosa, pulmonary alveolae) and biological barriers that hinder drug delivery and
endosomal trafficking pathways.
List of abbreviations
Apo apolipoprotein
AQP4 aquaporin-4
AQP9 aquaporin-9
BBB blood-brain barrier
CNS central nervous system
COPD chronic obstructive pulmonary disease
CS chondroitin/dermatan sulphate
69

07/05/2023
DOX doxorubicin
EC endothelial cells
ECM extracellular matrix
ELP elastin-like polypeptide
EPR enhanced permeability and retention
FAE follicle-associated epithelium
GAG Glycoaminoglycans
gd geometric diameter
GIT gastrointestinal tract
HD hydrodynamic diameter
HS heparan sulphate
IFP interstitial fluid hydrostatic pressure
IgG immunoglobulin G
i.v. intravenous
kD kilo Dalton
LPP large porous particles
LPS lipopolysaccharide
MMP matrix metalloprotease
MPS mononuclear phagocyte system (idem RES)
MTX methotrexate
MW molecular weight
NC nanocarriers
NP nanoparticles
ODN oligonucleotide
P-gp P-glycoprotein
PBS phosphate buffer saline
PEG poly(ethylene glycol)
PEI poly(ethyleneimine)
PEPE polyester-polyether
PHDCA poly(hexadecylcyanoacrylate)
PI poly-dispersity index
PK pharmacokinetics
PLA poly(D,L-lactide)
PLGA poly(D,L-lactide-co-gycolide)
PVA poly(vinyl alcohol)
PNIPAAm Poly(N-isopropylacrylamide)
PS poly(styrene)
70

07/05/2023
RA rheumatoid arthritis
RAECMs rat alveolar epithelial cell monolayers
RBC red blood cells
RBEC rat brain endothelial cells
RES reticuloendothelial system (idem MPS)
SLN solid lipid nanoparticles
SLP solid lipid particles
TEC transendothelial channels
VVO vesicular-vacuolar organelles
Conflict of interest
The authors declare that they have no conflict of interest.
Acknowledgements
IE and JMR are the recipients of doctoral support from « Fonds Québécois de Recherche sur la
Nature et les Technologies » (QC, Canada). Editing of this text by Ovid Da Silva is acknowledged.
Supportive/supplementary material (if any)
No supportive material
71

07/05/2023
Figures captions
Fig. (1). Different types of Nanocarriers
(A) Micelle: self assembly of amphiphilic molecules; (B) Liposomes vesicles primarily constituted of a
phospholipids bilayer along with another type of lipids (cholesterol or PEG-phospholipids conjugate); (C)
Dendrimers are branched symmetric polymeric structures constituted by a core and branches (the
dendrons); (E) Polymeric nanoparticles are matrix particle in which the drug is dispersed (here symbolized
with black dots); (F) Nanocapsules, are constituted by an core (generally hydrophilic) enclosed in a thin
polymeric wall.
Fig. (2). General view of biological barriers to i.v. delivery of drug-loaded NC aimed at the solid tumour
interstitium
Fig. (3). PEG NC coverage
(A) “Brush” regimen (high density), (B) “Mushroom” configuration (low density), (C) “PEG loops”:
multiblock NP (PLA-PEG-PLA) [26]or PLA particles with PEG 1400 distearate [37]. PEG chains are
presented in black, and the NC core in grey.
Fig. (4). General organisation of blood vessel walls
Fig. (5). Different types of normal capillaries and transport pathways
(A) Continuous capillary, (B) Fenestrated capillary, (C) Open fenestrated capillary, (D) Sinusoidal
capillary. VVO: vesicular-vacuolar organelles; TE channels: transendothelial channels (adapted and
modified from [73])
Fig. (6). Glycocalyx structure
Protein backbones appear in black, and glycans in grey. The cell-bound layer is about 60 nm,
while the absorbed layer is about 500 nm in capillaries. Glycipans and syndecans are the 2 main families of
core protein proteoglycans anchored on the plasma membrane. Proteoglycans are associated with
glycoaminoglycans (GAG), primarily heparan sulphate (HS), representing 50-90% of GAG along with
chondroitin/dermatan sulphate (CS) and hyaluronic acid chains (HA) [81].
Fig. (7). Vascular permeability at the normal continuous capillary level
MP: Macropinosis; CLME: Clathrin-mediated endocytosis; CVME: Caveolae-mediated endocytosis;
NCNC: Non-clathrin, non-caveolae pathway; TE: Transendothelial channel; VVO: Vesicular-vacuolar
72

07/05/2023
organelle; Lys.: Lysosome; End.: Endosome; Ex. : Exocytosis
Fig. (8). Simplified view of the EPR effect
On the left: situation of normal tissue with continuous capillaries, normal lymphatic drainage, normal ECM
and cell organization. NC are excluded from interstitium. On the right: solid tumour with leaky capillary,
increased ECM, defective lymphatic drainage and increased IFP. NC are transported by convection and/or
diffusing in the tumour interstitium, accumulating mainly in the perivascular region.
Fig. (9). General structure of the blood-brain barrier
(1) Basement membrane, (2) Endothelial cells (EC), (3) pericyte, (4) astrocytic projection, (5) tight
junction, (6) cell nucleus.
Fig. (10). Structures of polymers used in polymeric NP
(a) poly(butylcyanoacrylate), (b) PHDCA polymers, and (c) PEG-PHDCA copolymer.
Fig. (11). Structure GIT barrier: drug and NC transportation pathways. On the left, enterocytes, on the right
M cell. A mucus layer is found on the apical side of enterocytes and M cell. (1) Paracellular route, (2)
Transcellular route, (3) M-cell phagocytosis
Fig. (12). Structure of alveoli
Cross-section of an alveolus showing a capillary, alveolar macrophage, surfactant layer, type I and type II
cells (adapted and modified from [313])
Fig. (13). Proposed NC transport pathways across alveolar and vascular endothelial cells. Possible
translocation mechanisms across the 2 cell layers: (A) Alveolar epithelial tight junctions (paracellular
transport), (B) Caveolar receptor-mediated transcytosis, (C) Caveolar trans-endothelial transport, (D)
Trans-endothelial channel, (E) Vascular endothelial cell tight junction
Fig. (14). NC Predicted fractional deposition of inhaled particles in the human respiratory tract. (data
reformatted from [332])
73

07/05/2023
References
[1] Nel, A. E.; Madler, L.; Velegol, D.; Xia, T.; Hoek, E. M.; Somasundaran, P.; Klaessig, F.; Castranova, V.; Thompson, M., Understanding biophysicochemical interactions at the nano-bio interface. Nat. Mater. 2009, 8 (7), 543-57.
[2] Wang, J.; Lu, Z.; Wientjes, M. G.; Au, J. L. S., Delivery of siRNA Therapeutics: Barriers and Carriers. AAPS J. 2010, 12 (4), 492-503.
[3] Torchilin, V. P., Recent advances with liposomes as pharmaceutical carriers. Nat. Rev. Drug Discov. 2005, 4 (2), 145-60.
[4] Jones, M. C.; Leroux, J. C., Polymeric micelles - a new generation of colloidal drug carriers. Eur. J. Pharm. Biopharm. 1999, 48 (2), 101-111.
[5] Kojima, C., Design of stimuli-responsive dendrimers. Expert Opin. Drug Deliv. 2010, 7 (3), 307-19.
[6] Bala, I.; Hariharan, S.; Kumar, M. N., PLGA nanoparticles in drug delivery: the state of the art. Crit. Rev. Ther. Drug Carrier Syst. 2004, 21 (5), 387-422.
[7] Ratzinger, G.; Fillafer, C.; Kerleta, V.; Wirth, M.; Gabor, F., The Role of Surface Functionalization in the Design of PLGA Micro- and Nanoparticles. Crit. Rev. Ther. Drug Carrier Syst. 2010, 27 (1), 1-83.
[8] Bertrand, N.; Leroux, J. C., The journey of a drug carrier in the body: An anatomo-physiological perspective. J. Control. Release 2011, Available on line (october 5th, 2011).
[9] Hillaireau, H.; Couvreur, P., Nanocarriers' entry into the cell: relevance to drug delivery. Cell. Mol. Life Sci. 2009, 66 (17), 2873-96.
[10] Grdisa, M., The Delivery of Biologically Active (Therapeutic) Peptides and Proteins into Cells. Curr. Med. Chem. 2011, 18 (9), 1373-1379.
[11] Simone, E.; Ding, B. S.; Muzykantov, V., Targeted delivery of therapeutics to endothelium. Cell Tissue Res. 2009, 335 (1), 283-300.
[12] Vonarbourg, A.; Passirani, C.; Saulnier, P.; Benoit, J. P., Parameters influencing the stealthiness of colloidal drug delivery systems. Biomaterials 2006, 27 (24), 4356-73.
[13] Owens, D. E., 3rd; Peppas, N. A., Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int. J. Pharm. 2006, 307 (1), 93-102.
[14] Lynch, I.; Dawson, K. A., Protein-nanoparticle interactions. Nano Today 2008, 3 (1-2), 40-47.[15] Cedervall, T.; Lynch, I.; Lindman, S.; Berggard, T.; Thulin, E.; Nilsson, H.; Dawson, K. A.; Linse,
S., Understanding the nanoparticle-protein corona using methods to quantify exchange rates and affinities of proteins for nanoparticles. Proc. Natl. Acad. Sci. U. S. A. 2007, 104 (7), 2050-2055.
[16] Tabata, Y.; Ikada, Y., Phagocytosis of Polymer Microspheres by Macrophages. Adv. Polym. Sci. 1990, 94, 107-141.
[17] Ogawara, K.; Furumoto, K.; Nagayama, S.; Minato, K.; Higaki, K.; Kai, T.; Kimura, T., Pre-coating with serum albumin reduces receptor-mediated hepatic disposition of polystyrene nanosphere: implications for rational design of nanoparticles. J. Control. Release 2004, 100 (3), 451-455.
[18] Aderem, A.; Underhill, D. M., Mechanisms of phagocytosis in macrophages. Annu. Rev. Immunol. 1999, 17, 593-623.
[19] Kanno, S.; Furuyama, A.; Hirano, S., A murine scavenger receptor MARCO recognizes polystyrene nanoparticles. Toxicol. Sci. 2007, 97 (2), 398-406.
[20] Lundqvist, M.; Stigler, J.; Elia, G.; Lynch, I.; Cedervall, T.; Dawson, K. A., Nanoparticle size and surface properties determine the protein corona with possible implications for biological impacts. Proc. Natl. Acad. Sci. U. S. A. 2008, 105 (38), 14265-14270.
[21] Hellstrand, E.; Lynch, I.; Andersson, A.; Drakenberg, T.; Dahlback, B.; Dawson, K. A.; Linse, S.; Cedervall, T., Complete high-density lipoproteins in nanoparticle corona. FEBS J. 2009, 276 (12), 3372-3381.
[22] Papahadjopoulos, D.; Allen, T. M.; Gabizon, A.; Mayhew, E.; Matthay, K.; Huang, S. K.; Lee, K. D.; Woodle, M. C.; Lasic, D. D.; Redemann, C.; et al., Sterically stabilized liposomes:
74

07/05/2023
improvements in pharmacokinetics and antitumor therapeutic efficacy. Proc. Natl. Acad. Sci. U. S. A. 1991, 88 (24), 11460-4.
[23] Gref, R.; Minamitake, Y.; Peracchia, M. T.; Trubetskoy, V.; Torchilin, V.; Langer, R., Biodegradable long-circulating polymeric nanospheres. Science 1994, 263 (5153), 1600-3.
[24] Bazile, D.; Prud'homme, C.; Bassoullet, M. T.; Marlard, M.; Spenlehauer, G.; Veillard, M., Stealth Me.PEG-PLA nanoparticles avoid uptake by the mononuclear phagocytes system. J. Pharm. Sci. 1995, 84 (4), 493-8.
[25] Torchilin, V. P.; Omelyanenko, V. G.; Papisov, M. I.; Bogdanov, A. A., Jr.; Trubetskoy, V. S.; Herron, J. N.; Gentry, C. A., Poly(ethylene glycol) on the liposome surface: on the mechanism of polymer-coated liposome longevity. Biochim. Biophys. Acta 1994, 1195 (1), 11-20.
[26] Sant, S.; Poulin, S.; Hildgen, P., Effect of polymer architecture on surface properties, plasma protein adsorption, and cellular interactions of pegylated nanoparticles. J. Biomed. Mater. Res., Part A 2008, 87A (4), 885-895.
[27] Gref, R.; Luck, M.; Quellec, P.; Marchand, M.; Dellacherie, E.; Harnisch, S.; Blunk, T.; Muller, R. H., 'Stealth' corona-core nanoparticles surface modified by polyethylene glycol (PEG): influences of the corona (PEG chain length and surface density) and of the core composition on phagocytic uptake and plasma protein adsorption. Colloids Surf B Biointerfaces 2000, 18 (3-4), 301-313.
[28] Aggarwal, P.; Hall, J. B.; McLeland, C. B.; Dobrovolskaia, M. A.; McNeil, S. E., Nanoparticle interaction with plasma proteins as it relates to particle biodistribution, biocompatibility and therapeutic efficacy. Adv Drug Deliv Rev 2009, 61 (6), 428-37.
[29] Perrault, S. D.; Walkey, C.; Jennings, T.; Fischer, H. C.; Chan, W. C., Mediating tumor targeting efficiency of nanoparticles through design. Nano Lett. 2009, 9 (5), 1909-15.
[30] Mosqueira, V. C. F.; Legrand, P.; Morgat, J. L.; Vert, M.; Mysiakine, E.; Gref, R.; Devissaguet, J. P.; Barratt, G., Biodistribution of long-circulating PEG-grafted nanocapsules in mice: Effects of PEG chain length and density. Pharm. Res. 2001, 18 (10), 1411-1419.
[31] Choi, H. S.; Ipe, B. I.; Misra, P.; Lee, J. H.; Bawendi, M. G.; Frangioni, J. V., Tissue- and organ-selective biodistribution of NIR fluorescent quantum dots. Nano Lett. 2009, 9 (6), 2354-9.
[32] Neal, J. C.; Stolnik, S.; Schacht, E.; Kenawy, E. R.; Garnett, M. C.; Davis, S. S.; Illum, L., In vitro displacement by rat serum of adsorbed radiolabeled poloxamer and poloxamine copolymers from model and biodegradable nanospheres. J. Pharm. Sci. 1998, 87 (10), 1242-8.
[33] Moghimi, S. M.; Hunter, A. C.; Murray, J. C., Long-Circulating and Target-Specific Nanoparticles: Theory to Practice. Pharmacol. Rev. 2001, 53 (2), 283-318.
[34] Allen, T. M.; Chonn, A., Large unilamellar liposomes with low uptake into the reticuloendothelial system. FEBS Lett. 1987, 223 (1), 42-6.
[35] Dos Santos, N.; Allen, C.; Doppen, A. M.; Anantha, M.; Cox, K. A. K.; Gallagher, R. C.; Karlsson, G.; Edwards, K.; Kenner, G.; Samuels, L.; Webb, M. S.; Bally, M. B., Influence of poly(ethylene glycol) grafting density and polymer length on liposomes: Relating plasma circulation lifetimes to protein binding. Biochim. Biophys. Acta-Biomembr. 2007, 1768 (6), 1367-1377.
[36] Bergstrom, K.; Osterberg, E.; Holmberg, K.; Hoffman, A. S.; Schuman, T. P.; Kozlowski, A.; Harris, J. M., Effects of branching and molecular-weight of surface bound poly(ethylene oxide) on protein rejection. J. Biomater. Sci.-Polym. Ed. 1994, 6 (2), 123-132.
[37] Lacasse, F. X.; Filion, M. C.; Phillips, N. C.; Escher, E.; McMullen, J. N.; Hildgen, P., Influence of surface properties at biodegradable microsphere surfaces: effects on plasma protein adsorption and phagocytosis. Pharm. Res. 1998, 15 (2), 312-7.
[38] De Jaeghere, F.; Allemann, E.; Feijen, J.; Kissel, T.; Doelker, E.; Gurny, R., Cellular uptake of PEO surface-modified nanoparticles: Evaluation of nanoparticles made of PLA : PEO diblock and triblock copolymers. J. Drug Target. 2000, 8 (3), 143-153.
[39] Moghimi, S. M.; Szebeni, J., Stealth liposomes and long circulating nanoparticles: critical issues in pharmacokinetics, opsonization and protein-binding properties. Prog. Lipid Res. 2003, 42 (6), 463-78.
[40] Ishida, T.; Kiwada, H., Accelerated blood clearance (ABC) phenomenon upon repeated injection of PEGylated liposomes. Int. J. Pharm. 2008, 354 (1-2), 56-62.
[41] Li, S. D.; Huang, L., Stealth nanoparticles: high density but sheddable PEG is a key for tumor targeting. J. Control. Release 2010, 145 (3), 178-81.
75

07/05/2023
[42] Moghimi, S. M.; Andersen, A. J.; Hashemi, S. H.; Lettiero, B.; Ahmadvand, D.; Hunter, A. C.; Andresen, T. L.; Hamad, I.; Szebeni, J., Complement activation cascade triggered by PEG-PL engineered nanomedicines and carbon nanotubes: The challenges ahead. J. Control. Release 2010, 146 (2), 175-181.
[43] Moghimi, S. M.; Hamad, I.; Andresen, T. L.; Jorgensen, K.; Szebeni, J., Methylation of the phosphate oxygen moiety of phospholipid-methoxy(polyethylene glycol) conjugate prevents PEGylated liposome-mediated complement activation and anaphylatoxin production. FASEB J. 2006, 20 (14), 2591-+.
[44] Mosqueira, V. C. F.; Legrand, P.; Gulik, A.; Bourdon, O.; Gref, R.; Labarre, D.; Barratt, G., Relationship between complement activation, cellular uptake and surface physicochemical aspects of novel PEG-modified nanocapsules. Biomaterials 2001, 22 (22), 2967-2979.
[45] Gbadamosi, J. K.; Hunter, A. C.; Moghimi, S. M., PEGylation of microspheres generates a heterogeneous population of particles with differential surface characteristics and biological performance. FEBS Lett. 2002, 532 (3), 338-44.
[46] Essa, S.; Rabanel, J. M.; Hildgen, P., Effect of polyethylene glycol (PEG) chain organization on the physicochemical properties of poly(D, L-lactide) (PLA) based nanoparticles. Eur. J. Pharm. Biopharm. 2010, 75 (2), 96-106.
[47] Passirani, C.; Barratt, G.; Devissaguet, J. P.; Labarre, D., Long-circulating nanoparticles bearing heparin or dextran covalently bound to poly(methyl methacrylate). Pharm. Res. 1998, 15 (7), 1046-50.
[48] De Sousa Delgado, A.; Léonard, M.; Dellacherie, E., Surface Properties of Polystyrene Nanoparticles Coated with Dextrans and Dextran−PEO Copolymers. Effect of Polymer Architecture on Protein Adsorption. Langmuir 2001, 17 (14), 4386-4391.
[49] Shehata, T.; Ogawara, K.; Higaki, K.; Kimura, T., Prolongation of residence time of liposome by surface-modification with mixture of hydrophilic polymers. Int. J. Pharm. 2008, 359 (1-2), 272-9.
[50] Gaucher, G.; Asahina, K.; Wang, J. H.; Leroux, J. C., Effect of Poly(N-vinyl-pyrrolidone)-block-poly(D,L-lactide) as Coating Agent on the Opsonization, Phagocytosis, and Pharmacokinetics of Biodegradable Nanoparticles. Biomacromolecules 2009, 10 (2), 408-416.
[51] Sahoo, S. K.; Panyam, J.; Prabha, S.; Labhasetwar, V., Residual polyvinyl alcohol associated with poly (d,l-lactide-co-glycolide) nanoparticles affects their physical properties and cellular uptake. J. Control. Release 2002, 82 (1), 105-114.
[52] Roser, M.; Fischer, D.; Kissel, T., Surface-modified biodegradable albumin nano- and microspheres. II: effect of surface charges on in vitro phagocytosis and biodistribution in rats. Eur. J. Pharm. Biopharm. 1998, 46 (3), 255-63.
[53] Gessner, A.; Lieske, A.; Paulke, B. R.; Muller, R. H., Influence of surface charge density on protein adsorption on polymeric nanoparticles: analysis by two-dimensional electrophoresis. Eur. J. Pharm. Biopharm. 2002, 54 (2), 165-170.
[54] Choi, H. S.; Liu, W.; Misra, P.; Tanaka, E.; Zimmer, J. P.; Itty Ipe, B.; Bawendi, M. G.; Frangioni, J. V., Renal clearance of quantum dots. Nat. Biotechnol. 2007, 25 (10), 1165-70.
[55] Estephan, Z. G.; Schlenoff, P. S.; Schlenoff, J. B., Zwitteration As an Alternative to PEGylation. Langmuir 2011, 27 (11), 6794-6800.
[56] Ehrenberg, M. S.; Friedman, A. E.; Finkelstein, J. N.; Oberdorster, G.; McGrath, J. L., The influence of protein adsorption on nanoparticle association with cultured endothelial cells. Biomaterials 2009, 30 (4), 603-610.
[57] Gabizon, A.; Papahadjopoulos, D., The role of surface charge and hydrophilic groups on liposome clearance in vivo. Biochim. Biophys. Acta 1992, 1103 (1), 94-100.
[58] Yamamoto, Y.; Nagasaki, Y.; Kato, Y.; Sugiyama, Y.; Kataoka, K., Long-circulating poly(ethylene glycol)-poly(D,L-lactide) block copolymer micelles with modulated surface charge. J. Control. Release 2001, 77 (1-2), 27-38.
[59] Domanski, D. M.; Klajnert, B.; Bryszewska, M., Influence of PAMAM dendrimers on human red blood cells. Bioelectrochemistry 2004, 63 (1-2), 189-91.
[60] Mayer, A.; Vadon, M.; Rinner, B.; Novak, A.; Wintersteiger, R.; Frohlich, E., The role of nanoparticle size in hemocompatibility. Toxicology 2009, 258 (2-3), 139-47.
[61] Huang, R. B.; Mocherla, S.; Heslinga, M. J.; Charoenphol, P.; Eniola-Adefeso, O., Dynamic and cellular interactions of nanoparticles in vascular-targeted drug delivery. Mol. Membr. Biol. 2010, 27 (7), 312-27.
76

07/05/2023
[62] Fang, C.; Shi, B.; Pei, Y.-Y.; Hong, M.-H.; Wu, J.; Chen, H.-Z., In vivo tumor targeting of tumor necrosis factor-[alpha]-loaded stealth nanoparticles: Effect of MePEG molecular weight and particle size. Eur. J. Pharm. Sci. 2006, 27 (1), 27-36.
[63] Allen, L. A.; Aderem, A., Mechanisms of phagocytosis. Curr. Opin. Immunol. 1996, 8 (1), 36-40.[64] Koval, M.; Preiter, K.; Adles, C.; Stahl, P. D.; Steinberg, T. H., Size of IgG-Opsonized Particles
Determines Macrophage Response during Internalization. Exp. Cell Res. 1998, 242 (1), 265-273.[65] Liu, D.; Mori, A.; Huang, L., Role of liposome size and RES blockade in controlling
biodistribution and tumor uptake of GM1-containing liposomes. Biochim. Biophys. Acta-Biomembr. 1992, 1104 (1), 95-101.
[66] Champion, J. A.; Katare, Y. K.; Mitragotri, S., Particle shape: A new design parameter for micro- and nanoscale drug delivery carriers. J. Control. Release 2007, 121 (1-2), 3-9.
[67] Tao, L.; Hu, W.; Liu, Y.; Huang, G.; Sumer, B. D.; Gao, J., Shape-specific polymeric nanomedicine: emerging opportunities and challenges. Exp. Biol. Med. 2011, 236 (1), 20-29.
[68] Decuzzi, P.; Pasqualini, R.; Arap, W.; Ferrari, M., Intravascular Delivery of Particulate Systems: Does Geometry Really Matter? Pharm. Res. 2009, 26 (1), 235-243.
[69] Devarajan, P. V.; Jindal, A. B.; Patil, R. R.; Mulla, F.; Gaikwad, R. V.; Samad, A., Particle Shape: A New Design Parameter for Passive Targeting In Splenotropic Drug Delivery. J. Pharm. Sci. 2010, 99 (6), 2576-2581.
[70] Sharma, G.; Valenta, D. T.; Altman, Y.; Harvey, S.; Xie, H.; Mitragotri, S.; Smith, J. W., Polymer particle shape independently influences binding and internalization by macrophages. J. Control. Release 2010, 147 (3), 408-412.
[71] Geng, Y.; Dalhaimer, P.; Cai, S. S.; Tsai, R.; Tewari, M.; Minko, T.; Discher, D. E., Shape effects of filaments versus spherical particles in flow and drug delivery. Nat. Nanotechnol. 2007, 2 (4), 249-255.
[72] Simionescu, M.; Popov, D.; Sima, A., Endothelial transcytosis in health and disease. Cell Tissue Res. 2009, 335 (1), 27-40.
[73] Stan, R. V., Channels across endothelial cells. In Cell-Cell Channels, Baluška, F.; Volkmann, D.; Barlow, P. W., Eds. Landes Biosciences and Springer Science: Georgetown, TX (USA), 2006; pp 251-266.
[74] Tse, D.; Stan, R. V., Morphological Heterogeneity of Endothelium. Semin. Thromb. Hemost. 2010, 36 (3), 236-245.
[75] Sarin, H., Physiologic upper limits of pore size of different blood capillary types and another perspective on the dual pore theory of microvascular permeability. Journal of Angiogenesis Research 2010, 2 (1), 14.
[76] Singh, A.; Satchell, S. C.; Neal, C. R.; McKenzie, E. A.; Tooke, J. E.; Mathieson, P. W., Glomerular endothelial glycocalyx constitutes a barrier to protein permeability. J. Am. Soc. Nephrol. 2007, 18 (11), 2885-2893.
[77] Pries, A. R.; Secomb, T. W.; Gaehtgens, P., The endothelial surface layer. Pflugers Arch. 2000, 440 (5), 653-666.
[78] Rehm, M.; Zahler, S.; Lotsch, M.; Welsch, U.; Conzen, P.; Jacob, M.; Becker, B. F., Endothelial glycocalyx as an additional barrier determining extravasation of 6% hydroxyethyl starch or 5% albumin solutions in the coronary vascular bed. Anesthesiology 2004, 100 (5), 1211-23.
[79] Reitsma, S.; Slaaf, D. W.; Vink, H.; van Zandvoort, M. A.; oude Egbrink, M. G., The endothelial glycocalyx: composition, functions, and visualization. Pflugers Arch. 2007, 454 (3), 345-59.
[80] Curry, F. R.; Adamson, R. H., Vascular permeability modulation at the cell, microvessel, or whole organ level: towards closing gaps in our knowledge. Cardiovasc. Res. 2010, 87 (2), 218-29.
[81] Weinbaum, S.; Tarbell, J. M.; Damiano, E. R., The structure and function of the endothelial glycocalyx layer. Annu. Rev. Biomed. Eng. 2007, 9, 121-167.
[82] Raviv, U.; Giasson, S.; Kampf, N.; Gohy, J. F.; Jerome, R.; Klein, J., Lubrication by charged polymers. Nature 2003, 425 (6954), 163-5.
[83] Mulivor, A. W.; Lipowsky, H. H., Role of glycocalyx in leukocyte-endothelial cell adhesion. Am. J. Physiol. Heart Circ. Physiol. 2002, 283 (4), H1282-H1291.
[84] Mounkes, L. C.; Zhong, W.; Cipres-Palacin, G.; Heath, T. D.; Debs, R. J., Proteoglycans mediate cationic liposome-DNA complex-based gene delivery in vitro and in vivo. J. Biol. Chem. 1998, 273 (40), 26164-70.
77

07/05/2023
[85] Wu, N. Z.; Da, D.; Rudoll, T. L.; Needham, D.; Whorton, A. R.; Dewhirst, M. W., Increased Microvascular Permeability Contributes to Preferential Accumulation of Stealth Liposomes in Tumor Tissue. Cancer Res. 1993, 53 (16), 3765-3770.
[86] Toy, R.; Hayden, E.; Shoup, C.; Baskaran, H.; Karathanasis, E., The effects of particle size, density and shape on margination of nanoparticles in microcirculation. Nanotechnology 2011, 22 (11), 115101.
[87] McIntosh, D. P.; Tan, X. Y.; Oh, P.; Schnitzer, J. E., Targeting endothelium and its dynamic caveolae for tissue-specific transcytosis in vivo: a pathway to overcome cell barriers to drug and gene delivery. Proc. Natl. Acad. Sci. U. S. A. 2002, 99 (4), 1996-2001.
[88] Wang, Z. J.; Tiruppathi, C.; Minshall, R. D.; Malik, A. B., Size and Dynamics of Caveolae Studied Using Nanoparticles in Living Endothelial Cells. ACS Nano 2009, 3 (12), 4110-4116.
[89] Wang, Z.; Tiruppathi, C.; Cho, J.; Minshall, R. D.; Malik, A. B., Delivery of nanoparticle: complexed drugs across the vascular endothelial barrier via caveolae. IUBMB Life 2011, 63 (8), 659-67.
[90] Dvorak, A. M.; Feng, D., The vesiculo-vacuolar organelle (VVO). A new endothelial cell permeability organelle. J. Histochem. Cytochem. 2001, 49 (4), 419-32.
[91] Tuma, P. L.; Hubbard, A. L., Transcytosis: crossing cellular barriers. Physiol. Rev. 2003, 83 (3), 871-932.
[92] Desai, N. P.; Trieu, V.; Hwang, L. Y.; Wu, R. J.; Soon-Shiong, P.; Gradishar, W. J., Improved effectiveness of nanoparticle albumin-bound (nab) paclitaxel versus polysorbate-based docetaxel in multiple xenografts as a function of HER2 and SPARC status. Anticancer. Drugs 2008, 19 (9), 899-909.
[93] Jain, R. K.; Stylianopoulos, T., Delivering nanomedicine to solid tumors. Nat. Rev. Clin. Oncol. 2010, 7 (11), 653-664.
[94] Narang, A. S.; Varia, S., Role of tumor vascular architecture in drug delivery. Adv Drug Deliv Rev 2011, 63 (8), 640-58.
[95] Roberts, W. G.; Palade, G. E., Increased microvascular permeability and endothelial fenestration induced by vascular endothelial growth factor. J. Cell Sci. 1995, 108 ( Pt 6), 2369-79.
[96] Fang, J.; Nakamura, H.; Maeda, H., The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev 2011, 63 (3), 136-51.
[97] Hashizume, H.; Baluk, P.; Morikawa, S.; McLean, J. W.; Thurston, G.; Roberge, S.; Jain, R. K.; McDonald, D. M., Openings between Defective Endothelial Cells Explain Tumor Vessel Leakiness. Am J Pathol 2000, 156 (4), 1363-1380.
[98] Roberts, W. G.; Palade, G. E., Neovasculature induced by vascular endothelial growth factor is fenestrated. Cancer Res. 1997, 57 (4), 765-72.
[99] Hobbs, S. K.; Monsky, W. L.; Yuan, F.; Roberts, W. G.; Griffith, L.; Torchilin, V. P.; Jain, R. K., Regulation of transport pathways in tumor vessels: role of tumor type and microenvironment. Proc. Natl. Acad. Sci. U. S. A. 1998, 95 (8), 4607-12.
[100] Yuan, F.; Dellian, M.; Fukumura, D.; Leunig, M.; Berk, D. A.; Torchilin, V. P.; Jain, R. K., Vascular Permeability in a Human Tumor Xenograft: Molecular Size Dependence and Cutoff Size. Cancer Res. 1995, 55 (17), 3752-3756.
[101] Bouzin, C.; Feron, O., Targeting tumor stroma and exploiting mature tumor vasculature to improve anti-cancer drug delivery. Drug Resist. Updat. 2007, 10 (3), 109-120.
[102] Matsumura, Y.; Maeda, H., A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46 (12 Pt 1), 6387-92.
[103] Maeda, H., Tumor-Selective Delivery of Macromolecular Drugs via the EPR Effect: Background and Future Prospects. Bioconjug. Chem. 2010, 21 (5), 797-802.
[104] Acharya, S.; Sahoo, S. K., PLGA nanoparticles containing various anticancer agents and tumour delivery by EPR effect. Adv Drug Deliv Rev 2011, 63 (3), 170-83.
[105] Gabizon, A.; Catane, R.; Uziely, B.; Kaufman, B.; Safra, T.; Cohen, R.; Martin, F.; Huang, A.; Barenholz, Y., Prolonged Circulation Time and Enhanced Accumulation in Malignant Exudates of Doxorubicin Encapsulated in Polyethylene-Glycol Coated Liposomes. Cancer Res. 1994, 54 (4), 987-992.
78

07/05/2023
[106] Gabizon, A.; Goren, D.; Horowitz, A. T.; Tzemach, D.; Lossos, A.; Siegal, T., Long-circulating liposomes for drug delivery in cancer therapy: A review of biodistribution studies in tumor-bearing animals. Adv. Drug Deliv. Rev. 1997, 24 (2-3), 337-344.
[107] Bae, Y. H.; Park, K., Targeted drug delivery to tumors: Myths, reality and possibility. J. Control. Release 2011, 153 (3), 198-205.
[108] Maeda, H., Nitroglycerin enhances vascular blood flow and drug delivery in hypoxic tumor tissues: analogy between angina pectoris and solid tumors and enhancement of the EPR effect. J. Control. Release 2010, 142 (3), 296-8.
[109] Metselaar, J. M.; Wauben, M. H. M.; Wagenaar-Hilbers, J. P. A.; Boerman, O. C.; Storm, G., Complete remission of experimental arthritis by joint targeting of glucocorticoids with long-circulating liposomes. Arthritis Rheum. 2003, 48 (7), 2059-2066.
[110] Higaki, M.; Ishihara, T.; Kubota, T.; Choi, T., Treatment of Experimental Arthritis with Stealth-Type Polymeric Nanoparticles Encapsulating Betamethasone Phosphate. J. Pharmacol. Exp. Ther. 2009, 329 (2), 412-417.
[111] Palmer, T. N.; Caride, V. J.; Caldecourt, M. A.; Twickler, J.; Abdullah, V., The mechanism of liposome accumulation in infarction. Biochim. Biophys. Acta 1984, 797 (3), 363-8.
[112] Yuan, F.; Leunig, M.; Huang, S. K.; Berk, D. A.; Papahadjopoulos, D.; Jain, R. K., Mirovascular Permeability and Interstitial Penetration of Sterically Stabilized (Stealth) Liposomes in a Human Tumor Xenograft. Cancer Res. 1994, 54 (13), 3352-3356.
[113] Holback, H.; Yeo, Y., Intratumoral drug delivery with nanoparticulate carriers. Pharm. Res. 2011, 28 (8), 1819-30.
[114] Boucher, Y.; Jain, R. K., Microvascular pressure is the principal driving force for interstitial hypertension in solid tumors: implications for vascular collapse. Cancer Res. 1992, 52 (18), 5110-4.
[115] Danhier, F.; Feron, O.; Preat, V., To exploit the tumor microenvironment: Passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J. Control. Release 2010, 148 (2), 135-46.
[116] Netti, P. A.; Berk, D. A.; Swartz, M. A.; Grodzinsky, A. J.; Jain, R. K., Role of extracellular matrix assembly in interstitial transport in solid tumors. Cancer Res. 2000, 60 (9), 2497-503.
[117] Stylianopoulos, T.; Diop-Frimpong, B.; Munn, L. L.; Jain, R. K., Diffusion Anisotropy in Collagen Gels and Tumors: The Effect of Fiber Network Orientation. Biophys. J. 2010, 99 (10), 3119-3128.
[118] Stylianopoulos, T.; Poh, M. Z.; Insin, N.; Bawendi, M. G.; Fukumura, D.; Munn, L. L.; Jain, R. K., Diffusion of Particles in the Extracellular Matrix: The Effect of Repulsive Electrostatic Interactions. Biophys. J. 2010, 99 (5), 1342-1349.
[119] Dreher, M. R.; Liu, W.; Michelich, C. R.; Dewhirst, M. W.; Yuan, F.; Chilkoti, A., Tumor Vascular Permeability, Accumulation, and Penetration of Macromolecular Drug Carriers. J. Natl. Cancer Inst. 2006, 98 (5), 335-344.
[120] Lee, H.; Hoang, B.; Fonge, H.; Reilly, R. M.; Allen, C., In vivo distribution of polymeric nanoparticles at the whole-body, tumor, and cellular levels. Pharm. Res. 2010, 27 (11), 2343-55.
[121] Meng, H.; Xue, M.; Xia, T.; Ji, Z.; Tarn, D. Y.; Zink, J. I.; Nel, A. E., Use of Size and a Copolymer Design Feature To Improve the Biodistribution and the Enhanced Permeability and Retention Effect of Doxorubicin-Loaded Mesoporous Silica Nanoparticles in a Murine Xenograft Tumor Model. ACS Nano 2011, 5 (5), 4131-4144.
[122] Gaumet, M.; Vargas, A.; Gurny, R.; Delie, F., Nanoparticles for drug delivery: the need for precision in reporting particle size parameters. Eur. J. Pharm. Biopharm. 2008, 69 (1), 1-9.
[123] Campbell, R. B.; Fukumura, D.; Brown, E. B.; Mazzola, L. M.; Izumi, Y.; Jain, R. K.; Torchilin, V. P.; Munn, L. L., Cationic charge determines the distribution of liposomes between the vascular and extravascular compartments of tumors. Cancer Res. 2002, 62 (23), 6831-6836.
[124] Ran, S.; Downes, A.; Thorpe, P. E., Increased exposure of anionic phospholipids on the surface of tumor blood vessels. Cancer Res. 2002, 62 (21), 6132-40.
[125] Li, Y.; Wang, J.; Wientjes, M. G.; Au, J. L. S., Delivery of nanomedicines to extracellular and intracellular compartments of a solid tumor. Adv. Drug Deliv. Rev. In press, available online 3 may 2011.
[126] Thurston, G.; McLean, J. W.; Rizen, M.; Baluk, P.; Haskell, A.; Murphy, T. J.; Hanahan, D.; McDonald, D. M., Cationic liposomes target angiogenic endothelial cells in tumors and chronic
79

07/05/2023
inflammation in mice. J. Clin. Invest. 1998, 101 (7), 1401-13.[127] Levchenko, T. S.; Rammohan, R.; Lukyanov, A. N.; Whiteman, K. R.; Torchilin, V. P., Liposome
clearance in mice: the effect of a separate and combined presence of surface charge and polymer coating. Int. J. Pharm. 2002, 240 (1-2), 95-102.
[128] Ho, E. A.; Ramsay, E.; Ginj, M.; Anantha, M.; Bregman, I.; Sy, J.; Woo, J.; Osooly-Talesh, M.; Yapp, D. T.; Bally, M. B., Characterization of cationic liposome formulations designed to exhibit extended plasma residence times and tumor vasculature targeting properties. J. Pharm. Sci. 2010, 99 (6), 2839-53.
[129] He, C.; Hu, Y.; Yin, L.; Tang, C.; Yin, C., Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials 2010, 31 (13), 3657-66.
[130] Labhasetwar, V.; Song, C.; Humphrey, W.; Shebuski, R.; Levy, R. J., Arterial uptake of biodegradable nanoparticles: effect of surface modifications. J. Pharm. Sci. 1998, 87 (10), 1229-34.
[131] Akinc, A.; Querbes, W.; De, S. M.; Qin, J.; Frank-Kamenetsky, M.; Jayaprakash, K. N.; Jayaraman, M.; Rajeev, K. G.; Cantley, W. L.; Dorkin, J. R.; Butler, J. S.; Qin, L. L.; Racie, T.; Sprague, A.; Fava, E.; Zeigerer, A.; Hope, M. J.; Zerial, M.; Sah, D. W. Y.; Fitzgerald, K.; Tracy, M. A.; Manoharan, M.; Koteliansky, V.; de Fougerolles, A.; Maier, M. A., Targeted Delivery of RNAi Therapeutics With Endogenous and Exogenous Ligand-Based Mechanisms. Mol. Ther. 2010, 18 (7), 1357-1364.
[132] Serda, R. E.; Gu, J.; Bhavane, R. C.; Liu, X.; Chiappini, C.; Decuzzi, P.; Ferrari, M., The association of silicon microparticles with endothelial cells in drug delivery to the vasculature. Biomaterials 2009, 30 (13), 2440-8.
[133] Kommareddy, S.; Amiji, M., Biodistribution and pharmacokinetic analysis of long-circulating thiolated gelatin nanoparticles following systemic administration in breast cancer-bearing mice. J. Pharm. Sci. 2007, 96 (2), 397-407.
[134] Ganta, S.; Devalapally, H.; Shahiwala, A.; Amiji, M., A review of stimuli-responsive nanocarriers for drug and gene delivery. J. Control. Release 2008, 126 (3), 187-204.
[135] Schmaljohann, D., Thermo- and pH-responsive polymers in drug delivery. Adv. Drug Deliv. Rev. 2006, 58 (15), 1655-1670.
[136] Hak, S.; Reitan, N. K.; Haraldseth, O.; de Lange Davies, C., Intravital microscopy in window chambers: a unique tool to study tumor angiogenesis and delivery of nanoparticles. Angiogenesis 2010, 13 (2), 113-30.
[137] Needham, D.; Dewhirst, M. W., The development and testing of a new temperature-sensitive drug delivery system for the treatment of solid tumors. Adv Drug Deliv Rev 2001, 53 (3), 285-305.
[138] Dreher, M. R.; Liu, W.; Michelich, C. R.; Dewhirst, M. W.; Chilkoti, A., Thermal cycling enhances the accumulation of a temperature-sensitive biopolymer in solid tumors. Cancer Res. 2007, 67 (9), 4418-24.
[139] Chilkoti, A.; Dreher, M. R.; Meyer, D. E.; Raucher, D., Targeted drug delivery by thermally responsive polymers. Adv Drug Deliv Rev 2002, 54 (5), 613-30.
[140] Gao, W.; Chan, J. M.; Farokhzad, O. C., pH-Responsive Nanoparticles for Drug Delivery. Mol. Pharm. 2010, 7 (6), 1913-1920.
[141] Sawant, R. R.; Torchilin, V. P., Liposomes as 'smart' pharmaceutical nanocarriers. Soft Matter 2010, 6 (17), 4026-4044.
[142] Leroux, J. C.; Roux, E.; Le Garrec, D.; Hong, K. L.; Drummond, D. C., N-isopropylacrylamide copolymers for the preparation of pH-sensitive liposomes and polymeric micelles. J. Control. Release 2001, 72 (1-3), 71-84.
[143] Roux, E.; Lafleur, M.; Lataste, E.; Moreau, P.; Leroux, J. C., On the characterization of pH-sensitive liposome/polymer complexes. Biomacromolecules 2003, 4 (2), 240-8.
[144] Shin, J.; Shum, P.; Thompson, D. H., Acid-triggered release via dePEGylation of DOPE liposomes containing acid-labile vinyl ether PEG-lipids. J. Control. Release 2003, 91 (1-2), 187-200.
[145] Griset, A. P.; Walpole, J.; Liu, R.; Gaffey, A.; Colson, Y. L.; Grinstaff, M. W., Expansile nanoparticles: synthesis, characterization, and in vivo efficacy of an acid-responsive polymeric drug delivery system. J. Am. Chem. Soc. 2009, 131 (7), 2469-71.
[146] Criscione, J. M.; Le, B. L.; Stern, E.; Brennan, M.; Rahner, C.; Papademetris, X.; Fahmy, T. M., Self-assembly of pH-responsive fluorinated dendrimer-based particulates for drug delivery and
80

07/05/2023
noninvasive imaging. Biomaterials 2009, 30 (23-24), 3946-55.[147] Bertrand, N.; Simard, P.; Leroux, J. C., Serum-stable, long-circulating, pH-sensitive PEGylated
liposomes. Methods Mol. Biol. 2010, 605, 545-58.[148] Mok, H.; Park, J. W.; Park, T. G., Enhanced Intracellular Delivery of Quantum Dot and
Adenovirus Nanoparticles Triggered by Acidic pH via Surface Charge Reversal. Bioconjug. Chem. 2008, 19 (4), 797-801.
[149] Poon, Z.; Chang, D.; Zhao, X.; Hammond, P. T., Layer-by-Layer Nanoparticles with a pH-Sheddable Layer for in Vivo Targeting of Tumor Hypoxia. ACS Nano 2011, 5 (6), 4284-4292.
[150] Hatakeyama, H.; Akita, H.; Kogure, K.; Oishi, M.; Nagasaki, Y.; Kihira, Y.; Ueno, M.; Kobayashi, H.; Kikuchi, H.; Harashima, H., Development of a novel systemic gene delivery system for cancer therapy with a tumor-specific cleavable PEG-lipid. Gene Ther. 2007, 14 (1), 68-77.
[151] Danquah, M. K.; Zhang, X. A.; Mahato, R. I., Extravasation of polymeric nanomedicines across tumor vasculature. Adv. Drug Deliv. Rev. 2011, 63 (8), 623-639.
[152] Helmlinger, G.; Yuan, F.; Dellian, M.; Jain, R. K., Interstitial pH and pO2 gradients in solid tumors in vivo: high-resolution measurements reveal a lack of correlation. Nat. Med. 1997, 3 (2), 177-82.
[153] Kuai, R.; Yuan, W.; Qin, Y.; Chen, H.; Tang, J.; Yuan, M.; Zhang, Z.; He, Q., Efficient Delivery of Payload into Tumor Cells in a Controlled Manner by TAT and Thiolytic Cleavable PEG Co-Modified Liposomes. Mol. Pharm. 2010.
[154] Zhang, J. X.; Zalipsky, S.; Mullah, N.; Pechar, M.; Allen, T. M., Pharmaco attributes of dioleoylphosphatidylethanolamine/cholesterylhemisuccinate liposomes containing different types of cleavable lipopolymers. Pharmacol. Res. 2004, 49 (2), 185-198.
[155] Zhang, Y.; So, M. K.; Rao, J., Protease-Modulated Cellular Uptake of Quantum Dots. Nano Lett. 2006, 6 (9), 1988-1992.
[156] Harris, T. J.; von Maltzahn, G.; Lord, M. E.; Park, J. H.; Agrawal, A.; Min, D. H.; Sailor, M. J.; Bhatia, S. N., Protease-triggered unveiling of bioactive nanoparticles. Small 2008, 4 (9), 1307-12.
[157] Mok, H.; Bae, K. H.; Ahn, C.-H.; Park, T. G., PEGylated and MMP-2 Specifically DePEGylated Quantum Dots: Comparative Evaluation of Cellular Uptake. Langmuir 2008, 25 (3), 1645-1650.
[158] Wong, C.; Stylianopoulos, T.; Cui, J.; Martin, J.; Chauhan, V. P.; Jiang, W.; Popovic, Z.; Jain, R. K.; Bawendi, M. G.; Fukumura, D., Multistage nanoparticle delivery system for deep penetration into tumor tissue. Proc. Natl. Acad. Sci. U. S. A. 2011, 108 (6), 2426-31.
[159] Wolf, S.; Seehaus, B.; Minol, K.; Günter Gassen, H., Die Blut-Hirn-Schranke: Eine besonderheit des cerebralen mikrozirkulationssystems. Naturwissenschaften 1996, 83 (7), 302-311.
[160] Ohtsuki, S., New aspects of the blood-brain barrier transporters; its physiological roles in the central nervous system. Biol. Pharm. Bull. 2004, 27 (10), 1489-96.
[161] Risau, W.; Engelhardt, B.; Wekerle, H., Immune function of the blood-brain barrier: incomplete presentation of protein (auto-)antigens by rat brain microvascular endothelium in vitro. J. Cell Biol. 1990, 110 (5), 1757-66.
[162] el-Bacha, R. S.; Minn, A., Drug metabolizing enzymes in cerebrovascular endothelial cells afford a metabolic protection to the brain. Cell. Mol. Biol. 1999, 45 (1), 15-23.
[163] Gilgun-Sherki, Y.; Melamed, E.; Offen, D., Oxidative stress induced-neurodegenerative diseases: the need for antioxidants that penetrate the blood brain barrier. Neuropharmacology 2001, 40 (8), 959-75.
[164] Pardridge, W. M., The blood-brain barrier and neurotherapeutics. Neurorx 2005, 2 (1), 1-2.[165] Bouchard, P.; Ghitescu, L. D.; Bendayan, M., Morpho-functional studies of the blood-brain barrier
in streptozotocin-induced diabetic rats. Diabetologia 2002, 45 (7), 1017-25.[166] Mooradian, A. D., Central nervous system complications of diabetes mellitus -- a perspective from
the blood-brain barrier. Brain Res. Rev. 1997, 23 (3), 210-218.[167] Huber, J. D.; Egleton, R. D.; Davis, T. P., Molecular physiology and pathophysiology of tight
junctions in the blood-brain barrier. Trends Neurosci. 2001, 24 (12), 719-725.[168] Butt, A. M.; Jones, H. C.; Abbott, N. J., Electrical resistance across the blood-brain barrier in
anaesthetized rats: a developmental study. J. Physiol. 1990, 429, 47-62.[169] Cornford, E. M.; Hyman, S.; Cornford, M. E.; Landaw, E. M.; Delgado-Escueta, A. V., Interictal
seizure resections show two configurations of endothelial Glut1 glucose transporter in the human blood-brain barrier. J. Cereb. Blood Flow Metab. 1998, 18 (1), 26-42.
81

07/05/2023
[170] Oldendorf, W. H.; Cornford, M. E.; Brown, W. J., The large apparent work capability of the blood-brain barrier: a study of the mitochondrial content of capillary endothelial cells in brain and other tissues of the rat. Ann. Neurol. 1977, 1 (5), 409-17.
[171] Bell, R. D.; Zlokovic, B. V., Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer's disease. Acta Neuropathol. 2009, 118 (1), 103-13.
[172] Bendayan, R.; Lee, G.; Bendayan, M., Functional expression and localization of P-glycoprotein at the blood brain barrier. Microsc. Res. Tech. 2002, 57 (5), 365-80.
[173] Dore-Duffy, P., Pericytes: pluripotent cells of the blood brain barrier. Curr. Pharm. Des. 2008, 14 (16), 1581-93.
[174] Rucker, H. K.; Wynder, H. J.; Thomas, W. E., Cellular mechanisms of CNS pericytes. Brain Res. Bull. 2000, 51 (5), 363-9.
[175] Krause, D.; Kunz, J.; Dermietzel, R., Cerebral pericytes--a second line of defense in controlling blood-brain barrier peptide metabolism. Adv. Exp. Med. Biol. 1993, 331, 149-52.
[176] Pardridge, W. M., Molecular biology of the blood-brain barrier. Mol. Biotechnol. 2005, 30 (1), 57-70.
[177] Abbott, N. J.; Ronnback, L.; Hansson, E., Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7 (1), 41-53.
[178] Tran, N. D.; Schreiber, S. S.; Fisher, M., Astrocyte regulation of endothelial tissue plasminogen activator in a blood-brain barrier model. J. Cereb. Blood Flow Metab. 1998, 18 (12), 1316-24.
[179] Sauer, I. Apolipoprotein E abgeleitete Peptide als Vektoren zur Überwindung der Blut-Hirn-Schranke. PhD thesis. Freie Universität, Berlin, 2004.
[180] Egleton, R. D.; Davis, T. P., Development of neuropeptide drugs that cross the blood-brain barrier. Neurorx 2005, 2 (1), 44-53.
[181] Abbruscato, T. J.; Thomas, S. A.; Hruby, V. J.; Davis, T. P., Blood-brain barrier permeability and bioavailability of a highly potent and mu-selective opioid receptor antagonist, CTAP: comparison with morphine. J. Pharmacol. Exp. Ther. 1997, 280 (1), 402-9.
[182] Banks, W. A.; Schally, A. V.; Barrera, C. M.; Fasold, M. B.; Durham, D. A.; Csernus, V. J.; Groot, K.; J., K. A., Permeability of the murine blood-brain barrier to some octapeptide analogs of somatostatin. Proc. Natl. Acad. Sci. USA 1990, 87 (17), 6762-6766.
[183] Cornford, E. M.; Hyman, S., Blood-brain barrier permeability to small and large molecules. Adv. Drug Deliv. Rev. 1999, 36 (2-3), 145-163.
[184] Bloch, O.; Manley, T. G., The role of aquaporin-4 in cerebral water transport and edema 1. Neurosurg. Focus 2007, 22 ((5):E3), 1-7.
[185] Badaut, J.; Brunet, J. F.; Petit, J. M.; Guerin, C. F.; Magistretti, P. J.; Regli, L., Induction of brain aquaporin 9 (AQP9) in catecholaminergic neurons in diabetic rats. Brain Res. 2008, 1188, 17-24.
[186] Badaut, J.; Brunet, J. F.; Regli, L., Aquaporins in the brain: from aqueduct to "multi-duct". Metab. Brain Dis. 2007, 22 (3-4), 251-63.
[187] Agus, D. B.; Gambhir, S. S.; Pardridge, W. M.; Spielholz, C.; Baselga, J.; Vera, J. C.; Golde, D. W., Vitamin C crosses the blood-brain barrier in the oxidized form through the glucose transporters. J Clin Invest. 1997, 100 (11), 2842-2848.
[188] Dahlin, A.; Royall, J.; Hohmann, J. G.; Wang, J., Expression profiling of the solute carrier gene family in the mouse brain. J. Pharmacol. Exp. Ther. 2009, 329 (2), 558-70.
[189] Pardridge, W. M., Blood-brain barrier delivery. Drug Discov. Today 2007, 12 (1-2), 54-61.[190] Abbruscato, T. J.; Thomas, S. A.; Hruby, V. J.; Davis, T. P., Brain and spinal cord distribution of
biphalin: correlation with opioid receptor density and mechanism of CNS entry. J. Neurochem. 1997, 69 (3), 1236-45.
[191] Zlokovic, B. V.; Mackic, J. B.; Djuricic, B.; Davson, H., Kinetic analysis of leucine-enkephalin cellular uptake at the luminal side of the blood-brain barrier of an in situ perfused guinea-pig brain. J. Neurochem. 1989, 53 (5), 1333-40.
[192] Zlokovic, B. V.; Hyman, S.; McComb, J. G.; Lipovac, M. N.; Tang, G.; Davson, H., Kinetics of arginine-vasopressin uptake at the blood-brain barrier. Biochim. Biophys. Acta 1990, 1025 (2), 191-8.
[193] Thomas, S. A.; Abbruscato, T. J.; Hruby, V. J.; Davis, T. P., The entry of [D-penicillamine2,5]enkephalin into the central nervous system: saturation kinetics and specificity. J. Pharmacol. Exp. Ther. 1997, 280 (3), 1235-40.
82

07/05/2023
[194] Begley, D. J., ABC transporters and the blood-brain barrier. Curr. Pharm. Des. 2004, 10 (12), 1295-312.
[195] Scherrmann, J.-M., Expression and function of multidrug resistance transporters at the blood brain barriers. Expert Opin. Drug Metab. Toxicol. 2005, 1 (2), 233-246.
[196] Löscher, W.; Potschka, H., Blood-Brain Barrier Active Efflux Transporters: ATP-Binding Cassette Gene Family. Neurorx 2005, 2 (1), 86-98.
[197] Hosoya, K.-i.; Sugawara, M.; Asaba, H.; Terasaki, T., Blood-Brain Barrier Produces Significant Efflux of L-Aspartic Acid but Not D-Aspartic Acid. J. Neurochem. 1999, 73 (3), 1206-1211.
[198] Pardridge, W. M., Transport of small molecules through the blood-brain barrier: biology and methodology. Adv. Drug Deliv. Rev. 1995, 15 (1-3), 5-36.
[199] Stahl, P.; Schwartz, A. L., Receptor-mediated endocytosis. J. Clin. Invest. 1986, 77 (3), 657-62.[200] Roberts, R. L.; Fine, R. E.; Sandra, A., Receptor-mediated endocytosis of transferrin at the blood-
brain barrier. J. Cell Sci. 1993, 104 ( Pt 2), 521-32.[201] Duffy, K. R.; Pardridge, W. M., Blood-brain barrier transcytosis of insulin in developing rabbits.
Brain Res. 1987, 420 (1), 32-8.[202] Banks, W. A.; Kastin, A. J.; Huang, W.; Jaspan, J. B.; Maness, L. M., Leptin enters the brain by a
saturable system independent of insulin. Peptides 1996, 17 (2), 305-11.[203] Duffy, K. R.; Pardridge, W. M.; Rosenfeld, R. G., Human blood-brain barrier insulin-like growth
factor receptor. Metabolism. 1988, 37 (2), 136-40.[204] Tamai, I.; Sai, Y.; Kobayashi, H.; Kamata, M.; Wakamiya, T.; Tsuji, A., Structure-internalization
relationship for adsorptive-mediated endocytosis of basic peptides at the blood-brain barrier. J. Pharmacol. Exp. Ther. 1997, 280 (1), 410-5.
[205] Herve, F.; Ghinea, N.; Scherrmann, J. M., CNS delivery via adsorptive transcytosis. AAPS J. 2008, 10 (3), 455-72.
[206] Fischer, H.; Gottschlich, R.; Seelig, A., Blood-brain barrier permeation: molecular parameters governing passive diffusion. J. Membr. Biol. 1998, 165 (3), 201-11.
[207] Pardridge, W. M., The blood-brain barrier: bottleneck in brain drug development. Neurorx 2005, 2 (1), 3-14.
[208] Banks, W. A.; Freed, E. O.; Wolf, K. M.; Robinson, S. M.; Franko, M.; Kumar, V. B., Transport of human immunodeficiency virus type 1 pseudoviruses across the blood-brain barrier: role of envelope proteins and adsorptive endocytosis. J. Virol. 2001, 75 (10), 4681-91.
[209] Weiss, N.; Miller, F.; Cazaubon, S.; Couraud, P. O., The blood-brain barrier in brain homeostasis and neurological diseases. Biochim. Biophys. Acta 2009, 1788 (4), 842-57.
[210] Pardridge, W. M., Blood-brain barrier drug targeting: the future of brain drug development. Mol. Interv. 2003, 3 (2), 90-105, 51.
[211] Newton, H. B., Advances in strategies to improve drug delivery to brain tumors. Expert Rev. Neurother. 2006, 6 (10), 1495-509.
[212] Doolittle, N. D.; Peereboom, D. M.; Christoforidis, G. A.; Hall, W. A.; Palmieri, D.; Brock, P. R.; Campbell, K. C.; Dickey, D. T.; Muldoon, L. L.; O'Neill, B. P.; Peterson, D. R.; Pollock, B.; Soussain, C.; Smith, Q.; Tyson, R. M.; Neuwelt, E. A., Delivery of chemotherapy and antibodies across the blood-brain barrier and the role of chemoprotection, in primary and metastatic brain tumors: report of the Eleventh Annual Blood-Brain Barrier Consortium meeting. J. Neurooncol. 2007, 81 (1), 81-91.
[213] Batrakova, E. V.; Miller, D. W.; Li, S.; Alakhov, V. Y.; Kabanov, A. V.; Elmquist, W. F., Pluronic P85 enhances the delivery of digoxin to the brain: in vitro and in vivo studies. J. Pharmacol. Exp. Ther. 2001, 296 (2), 551-7.
[214] Alakhov, V.; Klinski, E.; Li, S.; Pietrzynski, G.; Venne, A.; Batrakova, E.; Bronitch, T.; Kabanov, A., Block copolymer-based formulation of doxorubicin. From cell screen to clinical trials. Colloids Surf., B 1999, 16 (1-4), 113-134.
[215] Batrakova, E. V.; Li, S.; Vinogradov, S. V.; Alakhov, V. Y.; Miller, D. W.; Kabanov, A. V., Mechanism of pluronic effect on P-glycoprotein efflux system in blood-brain barrier: contributions of energy depletion and membrane fluidization. J. Pharmacol. Exp. Ther. 2001, 299 (2), 483-93.
[216] Fiorillo, A.; Maggi, G.; Greco, N.; Migliorati, R.; D'Amico, A.; De Caro, M. D.; Sabbatino, M. S.; Buffardi, F., Second-line chemotherapy with the association of liposomal daunorubicin,
83

07/05/2023
carboplatin and etoposide in children with recurrent malignant brain tumors. J. Neurooncol. 2004, 66 (1-2), 179-85.
[217] Albrecht, K. W.; de Witt Hamer, P. C.; Leenstra, S.; Bakker, P. J.; Beijnen, J. H.; Troost, D.; Kaaijk, P.; Bosch, A. D., High concentration of Daunorubicin and Daunorubicinol in human malignant astrocytomas after systemic administration of liposomal Daunorubicin. J. Neurooncol. 2001, 53 (3), 267-71.
[218] Fabel, K.; Dietrich, J.; Hau, P.; Wismeth, C.; Winner, B.; Przywara, S.; Steinbrecher, A.; Ullrich, W.; Bogdahn, U., Long-term stabilization in patients with malignant glioma after treatment with liposomal doxorubicin. Cancer 2001, 92 (7), 1936-42.
[219] Koukourakis, M. I.; Koukouraki, S.; Fezoulidis, I.; Kelekis, N.; Kyrias, G.; Archimandritis, S.; Karkavitsas, N., High intratumoural accumulation of stealth liposomal doxorubicin (Caelyx) in glioblastomas and in metastatic brain tumours. Br. J. Cancer 2000, 83 (10), 1281-6.
[220] Zucchetti, M.; Boiardi, A.; Silvani, A.; Parisi, I.; Piccolrovazzi, S.; D'Incalci, M., Distribution of daunorubicin and daunorubicinol in human glioma tumors after administration of liposomal daunorubicin. Cancer Chemother. Pharmacol. 1999, 44 (2), 173-6.
[221] DepoCyt(e)® (cytarabine liposome injection). Prescribing information. ScyPharma Inc. 2000.[222] Immordino, M. L.; Dosio, F.; Cattel, L., Stealth liposomes: review of the basic science, rationale,
and clinical applications, existing and potential. Int. J. Nanomedicine 2006, 1 (3), 297-315.[223] Noble, C. O.; Krauze, M. T.; Drummond, D. C.; Yamashita, Y.; Saito, R.; Berger, M. S.; Kirpotin,
D. B.; Bankiewicz, K. S.; Park, J. W., Novel nanoliposomal CPT-11 infused by convection-enhanced delivery in intracranial tumors: pharmacology and efficacy. Cancer Res. 2006, 66 (5), 2801-6.
[224] Arayne, M. S.; Sultana, N., Review: nanoparticles in drug delivery for the treatment of cancer. Pak J Pharm Sci 2006, 19 (3), 258-68.
[225] Steiniger, S. C.; Kreuter, J.; Khalansky, A. S.; Skidan, I. N.; Bobruskin, A. I.; Smirnova, Z. S.; Severin, S. E.; Uhl, R.; Kock, M.; Geiger, K. D.; Gelperina, S. E., Chemotherapy of glioblastoma in rats using doxorubicin-loaded nanoparticles. Int. J. Cancer 2004, 109 (5), 759-67.
[226] Olivier, J. C.; Fenart, L.; Chauvet, R.; Pariat, C.; Cecchelli, R.; Couet, W., Indirect evidence that drug brain targeting using polysorbate 80-coated polybutylcyanoacrylate nanoparticles is related to toxicity. Pharm. Res. 1999, 16 (12), 1836-42.
[227] Gulyaev, A. E.; Gelperina, S. E.; Skidan, I. N.; Antropov, A. S.; Kivman, G. Y.; Kreuter, J., Significant transport of doxorubicin into the brain with polysorbate 80-coated nanoparticles. Pharm. Res. 1999, 16 (10), 1564-9.
[228] Brigger, I.; Morizet, J.; Aubert, G.; Chacun, H.; Terrier-Lacombe, M. J.; Couvreur, P.; Vassal, G., Poly(ethylene glycol)-coated hexadecylcyanoacrylate nanospheres display a combined effect for brain tumor targeting. J. Pharmacol. Exp. Ther. 2002, 303 (3), 928-36.
[229] Calvo, P.; Gouritin, B.; Villarroya, H.; Eclancher, F.; Giannavola, C.; Klein, C.; Andreux, J. P.; Couvreur, P., Quantification and localization of PEGylated polycyanoacrylate nanoparticles in brain and spinal cord during experimental allergic encephalomyelitis in the rat. Eur. J. Neurosci. 2002, 15 (8), 1317-26.
[230] Brigger, I.; Morizet, J.; Laudani, L.; Aubert, G.; Appel, M.; Velasco, V.; Terrier-Lacombe, M. J.; Desmaele, D.; d'Angelo, J.; Couvreur, P.; Vassal, G., Negative preclinical results with stealth nanospheres-encapsulated Doxorubicin in an orthotopic murine brain tumor model. J. Control. Release 2004, 100 (1), 29-40.
[231] Garcia-Garcia, E.; Andrieux, K.; Gil, S.; Kim, H. R.; Le Doan, T.; Desmaele, D.; d'Angelo, J.; Taran, F.; Georgin, D.; Couvreur, P., A methodology to study intracellular distribution of nanoparticles in brain endothelial cells. Int. J. Pharm. 2005, 298 (2), 310-4.
[232] Costantino, L.; Gandolfi, F.; Tosi, G.; Rivasi, F.; Vandelli, M. A.; Forni, F., Peptide-derivatized biodegradable nanoparticles able to cross the blood-brain barrier. J. Control. Release 2005, 108 (1), 84-96.
[233] Tosi, G.; Costantino, L.; Rivasi, F.; Ruozi, B.; Leo, E.; Vergoni, A. V.; Tacchi, R.; Bertolini, A.; Vandelli, M. A.; Forni, F., Targeting the central nervous system: in vivo experiments with peptide-derivatized nanoparticles loaded with Loperamide and Rhodamine-123. J. Control. Release 2007, 122 (1), 1-9.
[234] Blasi, P.; Giovagnoli, S.; Schoubben, A.; Ricci, M.; Rossi, C., Solid lipid nanoparticles for targeted brain drug delivery. Adv. Drug Deliv. Rev. 2007, 59 (6), 454-77.
84

07/05/2023
[235] Yang, S.; Zhu, J.; Lu, Y.; Liang, B.; Yang, C., Body distribution of camptothecin solid lipid nanoparticles after oral administration. Pharm. Res. 1999, 16 (5), 751-7.
[236] Yang, S. C.; Lu, L. F.; Cai, Y.; Zhu, J. B.; Liang, B. W.; Yang, C. Z., Body distribution in mice of intravenously injected camptothecin solid lipid nanoparticles and targeting effect on brain. J. Control. Release 1999, 59 (3), 299-307.
[237] Zara, G. P.; Cavalli, R.; Fundaro, A.; Bargoni, A.; Caputo, O.; Gasco, M. R., Pharmacokinetics of doxorubicin incorporated in solid lipid nanospheres (SLN). Pharmacol. Res. 1999, 40 (3), 281-6.
[238] Koziara, J. M.; Lockman, P. R.; Allen, D. D.; Mumper, R. J., Paclitaxel nanoparticles for the potential treatment of brain tumors. J. Control. Release 2004, 99 (2), 259-69.
[239] Vinogradov, S. V.; Batrakova, E. V.; Kabanov, A. V., Nanogels for oligonucleotide delivery to the brain. Bioconjug. Chem. 2004, 15 (1), 50-60.
[240] Kabanov, A. V.; Batrakova, E. V., New technologies for drug delivery across the blood brain barrier. Curr. Pharm. Des. 2004, 10 (12), 1355-63.
[241] Dhanikula, R. S.; Hildgen, P., Influence of molecular architecture of polyether-co-polyester dendrimers on the encapsulation and release of methotrexate. Biomaterials 2007, 28 (20), 3140-52.
[242] Dhanikula, R. S.; Argaw, A.; Bouchard, J. F.; Hildgen, P., Methotrexate loaded polyether-copolyester dendrimers for the treatment of gliomas: enhanced efficacy and intratumoral transport capability. Mol. Pharm. 2008, 5 (1), 105-16.
[243] Dhanikula, R. S.; Hammady, T.; Hildgen, P., On the mechanism and dynamics of uptake and permeation of polyether-copolyester dendrimers across an in vitro blood-brain barrier model. J. Pharm. Sci. 2009, 98 (10), 3748-60.
[244] Dhanikula, R. S.; Hildgen, P., Synthesis and evaluation of novel dendrimers with a hydrophilic interior as nanocarriers for drug delivery. Bioconjug. Chem. 2006, 17 (1), 29-41.
[245] de Boer, A. G.; Gaillard, P. J., Drug targeting to the brain. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 323-55.
[246] Lambkin, I.; Pinilla, C., Targeting approaches to oral drug delivery. Expert Opin. Biol. Ther. 2002, 2 (1), 67-73.
[247] Mudie, D. M.; Amidon, G. L.; Amidon, G. E., Physiological Parameters for Oral Delivery and in Vitro Testing. Mol. Pharm. 2010, 7 (5), 1388-1405.
[248] O'Neill, M. J.; Bourre, L.; Melgar, S.; O'Driscoll, C. M., Intestinal delivery of non-viral gene therapeutics: physiological barriers and preclinical models. Drug Discov. Today 2011, 16 (5-6), 203-218.
[249] Shakweh, M.; Ponchel, G.; Fattal, E., Particle uptake by Peyer`s patches: a pathway for drug and vaccine delivery. Expert Opin. Drug Deliv. 2004, 1 (1), 141-163.
[250] Garinot, M.; Fievez, V.; Pourcelle, V.; Stoffelbach, F.; des Rieux, A.; Plapied, L.; Theate, I.; Freichels, H.; Jerome, C.; Marchand-Brynaert, J.; Schneider, Y. J.; Preat, V., PEGylated PLGA-based nanoparticles targeting M cells for oral vaccination. J. Control. Release 2007, 120 (3), 195-204.
[251] Laroui, H.; Wilson, D. S.; Dalmasso, G.; Salaita, K.; Murthy, N.; Sitaraman, S. V.; Merlin, D., Nanomedicine in GI. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300 (3), G371-G383.
[252] Lai, S. K.; Wang, Y.-Y.; Hanes, J., Mucus-penetrating nanoparticles for drug and gene delivery to mucosal tissues. Adv. Drug Deliv. Rev. 2009, 61 (2), 158-171.
[253] Cone, R. A., Barrier properties of mucus. Adv. Drug Deliv. Rev. 2009, 61 (2), 75-85.[254] Frey, A.; Giannasca, K. T.; Weltzin, R.; Giannasca, P. J.; Reggio, H.; Lencer, W. I.; Neutra, M.
R., Role of the glycocalyx in regulating access of microparticles to apical plasma membranes of intestinal epithelial cells: implications for microbial attachment and oral vaccine targeting. J. Exp. Med. 1996 184 (3 ), 1045-1059
[255] Gumbiner, B. M., Breaking through the tigh junction barrier. J. Cell Biol. 1993, 123 (6), 1631-1633.
[256] Kondoh, M.; Yagi, K., Progress in absorption enhancers based on tight junction. Expert Opin. Drug Deliv. 2007, 4 (3), 275-286.
[257] Matsuhisa, K.; Kondoh, M.; Takahashi, A.; Yagi, K., Tight junction modulator and drug delivery. Expert Opin. Drug Deliv. 2009, 6 (5), 509-515.
[258] Hugger, E. D.; Novak, B. L.; Burton, P. S.; Audus, K. L.; Borchardt, R. T., A comparison of commonly used polyethoxylated pharmaceutical excipients on their ability to inhibit P-
85

07/05/2023
glycoprotein activity in vitro. J. Pharm. Sci. 2002, 91 (9), 1991-2002.[259] McDevitt, C. A.; Callaghan, R., How can we best use structural information on P-glycoprotein to
design inhibitors? Pharmacol. Ther. 2007, 113 (2), 429-441.[260] del Amo, E. M.; Heikkinen, A. T.; Monkkonen, J., In vitro-in vivo correlation in p-glycoprotein
mediated transport in intestinal absorption. Eur. J. Pharm. Sci. 2009, 36 (2-3), 200-211.[261] Aulton, M. E. Aulton's pharmaceutics : the design and manufacture of medicines, 3rd; Churchill
Livingstone Elsevier: Edinburgh; Toronto, 2007.[262] Devalapally, H.; Chakilam, A.; Amiji, M. M., Role of nanotechnology in pharmaceutical product
development. J. Pharm. Sci. 2007, 96 (10), 2547-2565.[263] Florence, A. T., The oral absorption of micro- and nanoparticulates: Neither exceptional nor
unusual. Pharm. Res. 1997, 14 (3), 259-266.[264] Plapied, L.; Duhem, N.; des Rieux, A.; Préat, V., Fate of polymeric nanocarriers for oral drug
delivery. Curr. Opin. Colloid Interface Sci. 2011, 16 (3), 228-237.[265] Florence, A. T.; Hillery, A. M.; Hussain, N.; Jani, P. U., Factors affecting the oral uptake and
translocation of polystyrene nanoparticles: histological and analytical evidence. J. Drug Target. 1995, 3 (1), 65-70.
[266] Kesisoglou, F.; Panmai, S.; Wu, Y. H., Nanosizing - Oral formulation development and biopharmaceutical evaluation. Adv. Drug Deliv. Rev. 2007, 59 (7), 631-644.
[267] Li, S.-D.; Huang, L., Pharmacokinetics and Biodistribution of Nanoparticles. Mol. Pharm. 2008, 5 (4), 496-504.
[268] Mozafari, M. R.; Pardakhty, A.; Azarmi, S.; Jazayeri, J. A.; Nokhodchi, A.; Omri, A., Role of nanocarrier systems in cancer nanotherapy. J. Liposome Res. 2009, 19 (4), 310-321.
[269] Bravo-Osuna, I.; Vauthier, C.; Farabollini, A.; Palmieri, G. F.; Ponchel, G., Mucoadhesion mechanism of chitosan and thiolated chitosan-poly(isobutyl cyanoacrylate) core-shell nanoparticles. Biomaterials 2007, 28 (13), 2233-2243.
[270] Bernkop-Schnürch, A.; Weithaler, A.; Albrecht, K.; Greimel, A., Thiomers: Preparation and in vitro evaluation of a mucoadhesive nanoparticulate drug delivery system. Int. J. Pharm. 2006, 317 (1), 76-81.
[271] Bromberg, L.; Temchenko, M.; Alakhov, V.; Hatton, T. A., Bioadhesive properties and rheology of polyether-modified poly(acrylic acid) hydrogels. Int. J. Pharm. 2004, 282 (1-2), 45-60.
[272] Lamprecht, A.; Schafer, U.; Lehr, C. M., Size-dependent bioadhesion of micro- and nanoparticulate carriers to the inflamed colonic mucosa. Pharm. Res. 2001, 18 (6), 788-793.
[273] Behrens, I.; Pena, A. I. V.; Alonso, M. J.; Kissel, T., Comparative uptake studies of bioadhesive and non-bioadhesive nanoparticles in human intestinal cell lines and rats: The effect of mucus on particle adsorption and transport. Pharm. Res. 2002, 19 (8), 1185-1193.
[274] Sonaje, K.; Lin, K. J.; Wey, S. P.; Lin, C. K.; Yeh, T. H.; Nguyen, H. N.; Hsua, C. W.; Yen, T. C.; Juang, J. H.; Sung, H. W., Biodistribution, pharmacodynamics and pharmacokinetics of insulin analogues in a rat model: Oral delivery using pH-Responsive nanoparticles vs. subcutaneous injection. Biomaterials 2010, 31 (26), 6849-6858.
[275] Lai, S. K.; O'Hanlon, D. E.; Harrold, S.; Man, S. T.; Wang, Y.-Y.; Cone, R.; Hanes, J., Rapid transport of large polymeric nanoparticles in fresh undiluted human mucus. Proc. Natl. Acad. Sci. U. S. A. 2007, 104 (5), 1482-1487.
[276] Jung, T.; Kamm, W.; Breitenbach, A.; Kaiserling, E.; Xiao, J. X.; Kissel, T., Biodegradable nanoparticles for oral delivery of peptides: is there a role for polymers to affect mucosal uptake? Eur. J. Pharm. Biopharm. 2000, 50 (1), 147-60.
[277] des Rieux, A.; Fievez, V.; Garinot, M.; Schneider, Y.-J.; Préat, V., Nanoparticles as potential oral delivery systems of proteins and vaccines: A mechanistic approach. J. Control. Release 2006, 116 (1), 1-27.
[278] des Rieux, A.; Ragnarsson, E. G.; Gullberg, E.; Preat, V.; Schneider, Y. J.; Artursson, P., Transport of nanoparticles across an in vitro model of the human intestinal follicle associated epithelium. Eur. J. Pharm. Sci. 2005, 25 (4-5), 455-65.
[279] Desai, M. P.; Labhasetwar, V.; Amidon, G. L.; Levy, R. J., Gastrointestinal uptake of biodegradable microparticles: effect of particle size. Pharm. Res. 1996, 13 (12), 1838-45.
[280] Vila, A.; Gill, H.; McCallion, O.; Alonso, M. J., Transport of PLA-PEG particles across the nasal mucosa: effect of particle size and PEG coating density. J. Control. Release 2004, 98 (2), 231-44.
86

07/05/2023
[281] Otsuka, H.; Nagasaki, Y.; Kataoka, K., PEGylated nanoparticles for biological and pharmaceutical applications. Adv. Drug Deliv. Rev. 2003, 55 (3), 403-419.
[282] Semete, B.; Booysen, L.; Lemmer, Y.; Kalombo, L.; Katata, L.; Verschoor, J.; Swai, H. S., In vivo evaluation of the biodistribution and safety of PLGA nanoparticles as drug delivery systems. Nanomed. 2010, 6 (5), 662-71.
[283] Semete, B.; Booysen, L. I.; Kalombo, L.; Venter, J. D.; Katata, L.; Ramalapa, B.; Verschoor, J. A.; Swai, H., In vivo uptake and acute immune response to orally administered chitosan and PEG coated PLGA nanoparticles. Toxicol. Appl. Pharmacol. 2010, 249 (2), 158-65.
[284] Peltier, S.; Oger, J.-M.; Lagarce, F.; Couet, W.; Benoit, J.-P., Enhanced Oral Paclitaxel Bioavailability After Administration of Paclitaxel-Loaded Lipid Nanocapsules. Pharm. Res. 2006, 23 (6), 1243-1250.
[285] Roger, E.; Lagarce, F.; Garcion, E.; Benoit, J. P., Lipid nanocarriers improve paclitaxel transport throughout human intestinal epithelial cells by using vesicle-mediated transcytosis. J. Control. Release 2009, 140 (2), 174-181.
[286] Irache, J. M.; Durrer, C.; Duchêne, D.; Ponchel, G., Bioadhesion of Lectin-Latex Conjugates to Rat Intestinal Mucosa. Pharm. Res. 1996, 13 (11), 1716-1719.
[287] Yin, Y.; Chen, D.; Qiao, M.; Lu, Z.; Hu, H., Preparation and evaluation of lectin-conjugated PLGA nanoparticles for oral delivery of thymopentin. J. Control. Release 2006, 116 (3), 337-345.
[288] Umamaheshwari, R. B.; Ramteke, S.; Jain, N. K., Anti-Helicobacter pylori effect of mucoadhesive nanoparticles bearing amoxicillin in experimental gerbils model. AAPS PharmSciTech 2004, 5 (2).
[289] Clark, M. A.; Jepson, M. A.; Hirst, B. H., Exploiting M cells for drug and vaccine delivery. Adv. Drug Deliv. Rev. 2001, 50 (1-2), 81-106.
[290] Primard, C.; Rochereau, N.; Luciani, E.; Genin, C.; Delair, T.; Paul, S.; Verrier, B., Traffic of poly(lactic acid) nanoparticulate vaccine vehicle from intestinal mucus to sub-epithelial immune competent cells. Biomaterials 2010, 31 (23), 6060-6068.
[291] Pan, Y.; Li, Y. J.; Zhao, H. Y.; Zheng, J. M.; Xu, H.; Wei, G.; Hao, J. S.; Cui, F. D., Bioadhesive polysaccharide in protein delivery system: chitosan nanoparticles improve the intestinal absorption of insulin in vivo. Int. J. Pharm. 2002, 249 (1-2), 139-147.
[292] Damge, C.; Michel, C.; Aprahamian, M.; Couvreur, P.; Devissaguet, J. P., Nanocapsules as Carriers for Oral Peptide Delivery. J. Control. Release 1990, 13 (2-3), 233-239.
[293] Tobio, M.; Sanchez, A.; Vila, A.; Soriano, I. I.; Evora, C.; Vila-Jato, J. L.; Alonso, M. J., The role of PEG on the stability in digestive fluids and in vivo fate of PEG-PLA nanoparticles following oral administration. Colloids Surf., B 2000, 18 (3-4), 315-323.
[294] Fricker, G.; Kromp, T.; Wendel, A.; Blume, A.; Zirkel, J.; Rebmann, H.; Setzer, C.; Quinkert, R. O.; Martin, F.; Muller-Goymann, C., Phospholipids and Lipid-Based Formulations in Oral Drug Delivery. Pharm. Res. 2010, 27 (8), 1469-1486.
[295] Jain, S.; Khomane, K.; Jain, A. K.; Dani, P., Nanocarriers for Transmucosal Vaccine Delivery. Curr. Nanosci. 2011, 7 (2), 160-177.
[296] Zhang, N.; Ping, Q.; Huang, G.; Xu, W.; Cheng, Y.; Han, X., Lectin-modified solid lipid nanoparticles as carriers for oral administration of insulin. Int. J. Pharm. 2006, 327 (1-2), 153-9.
[297] Bromberg, L., Polymeric micelles in oral chemotherapy. J. Control. Release 2008, 128 (2), 99-112.
[298] Gaucher, G.; Satturwar, P.; Jones, M. C.; Furtos, A.; Leroux, J. C., Polymeric micelles for oral drug delivery. Eur. J. Pharm. Biopharm. 2010, 76 (2), 147-58.
[299] Blanchette, J.; Kavimandan, N.; Peppas, N. A., Principles of transmucosal delivery of therapeutic agents. Biomed. Pharmacother. 2004, 58 (3), 142-151.
[300] Kim, S.; Shi, Y.; Kim, J. Y.; Park, K.; Cheng, J.-X., Overcoming the barriers in micellar drug delivery: loading efficiency, in vivo stability, and micelle-cell interaction. Expert Opin. Drug Deliv. 2010, 7 (1), 49-62.
[301] Gaucher, G.; Marchessault, R. H.; Leroux, J. C., Polyester-based micelles and nanoparticles for the parenteral delivery of taxanes. J. Control. Release 2010, 143 (1), 2-12.
[302] Mathot, F.; van Beijsterveldt, L.; Préat, V.; Brewster, M.; Ariën, A., Intestinal uptake and biodistribution of novel polymeric micelles after oral administration. J. Control. Release 2006, 111 (1-2), 47-55.
[303] Blanchette, J.; Peppas, N. A., Cellular evaluation of oral chemotherapy carriers. J. Biomed. Mater. Res., Part A 2005, 72 (4), 381-8.
87

07/05/2023
[304] Sarmento, B.; Ribeiro, A.; Veiga, F.; Sampaio, P.; Neufeld, R.; Ferreira, D., Alginate/chitosan nanoparticles are effective for oral insulin delivery. Pharm. Res. 2007, 24 (12), 2198-206.
[305] Woitiski, C. B.; Neufeld, R. J.; Veiga, F.; Carvalho, R. A.; Figueiredo, I. V., Pharmacological effect of orally delivered insulin facilitated by multilayered stable nanoparticles. Eur. J. Pharm. Sci. 2010, 41 (3-4), 556-563.
[306] Yamamoto, A., Improvement of Intestinal Absorption of Poorly Absorbable Drugs by Polyamidoamine (PAMAM) Dendrimers as Novel Absorption Enhancers. Yakugaku Zasshi 2010, 130 (9), 1123-1127.
[307] Jevprasesphant, R.; Penny, J.; Attwood, D.; McKeown, N. B.; D'Emanuele, A., Engineering of Dendrimer Surfaces to Enhance Transepithelial Transport and Reduce Cytotoxicity. Pharm. Res. 2003, 20 (10), 1543-1550.
[308] Gupta, U.; Agashe, H. B.; Jain, N. K., Polypropylene imine. dendrimer mediated solubility enhancement: Effect of pH and functional groups of hydrophobes. J. Pharm. Pharm. Sci. 2007, 10 (3), 358-367.
[309] Al-Jamal, K. T.; Ramaswamy, C.; Florence, A. T., Supramolecular structures from dendrons and dendrimers. Adv. Drug Deliv. Rev. 2005, 57 (15), 2238-2270.
[310] Todoroff, J.; Vanbever, R., Fate of nanomedicines in the lungs. Curr. Opin. Colloid Interface Sci. 2011, 16 (3), 246-254.
[311] Crapo, J. D.; Barry, B. E.; Gehr, P.; Bachofen, M.; Weibel, E. R., Cell number and cell characteristics of the normal human lung. Am. Rev. Respir. Dis. 1982, 125 (6), 740-5.
[312] Smola, M.; Vandamme, T.; Sokolowski, A., Nanocarriers as pulmonary drug delivery systems to treat and to diagnose respiratory and non respiratory diseases. Int. J. Nanomedicine 2008, 3 (1), 1-19.
[313] Ware, L. B.; Matthay, M. A., The Acute Respiratory Distress Syndrome. N. Engl. J. Med. 2000, 342 (18), 1334-1349.
[314] Newman, S. P., Aerosol deposition considerations in inhalation therapy. Chest 1985, 88 (2), S152-S160.
[315] Knowles, M. R.; Boucher, R. C., Mucus clearance as a primary innate defense mechanism for mammalian airways. J. Clin. Invest. 2002, 109 (5), 571-577.
[316] Sanders, N.; Rudolph, C.; Braeckmans, K.; De Smedt, S. C.; Demeester, J., Extracellular barriers in respiratory gene therapy. Adv. Drug Deliv. Rev. 2009, 61 (2), 115-127.
[317] Roy, I.; Vij, N., Nanodelivery in airway diseases: Challenges and therapeutic applications. NanoMed. Nanotech, biol & Med. 2010, 6 (2), 237-244.
[318] Geiser, M.; Schurch, S.; Gehr, P., Influence of surface chemistry and topography of particles on their immersion into the lung's surface-lining layer. J. Appl. Physiol. 2003, 94 (5), 1793-1801.
[319] Wang, Y.-Y.; Lai, S. K.; Suk, J. S.; Pace, A.; Cone, R.; Hanes, J., Addressing the PEG Mucoadhesivity Paradox to Engineer Nanoparticles that “Slip” through the Human Mucus Barrier. Angew. Chem., Int. Ed. Engl. 2008, 47 (50), 9726-9729.
[320] Patton, J. S., Mechanisms of macromolecule absorption by the lungs. Adv. Drug Deliv. Rev. 1996, 19 (1), 3-36.
[321] Oberdorster, G.; Sharp, Z.; Atudorei, V.; Elder, A.; Gelein, R.; Lunts, A.; Kreyling, W.; Cox, C., Extrapulmonary translocation of ultrafine carbon particles following whole-body inhalation exposure of rats. J of Tox and Env Health-Part A 2002, 65 (20), 1531-1543.
[322] Meiring, J.; Borm, P.; Bagate, K.; Semmler, M.; Seitz, J.; Takenaka, S.; Kreyling, W., The influence of hydrogen peroxide and histamine on lung permeability and translocation of iridium nanoparticles in the isolated perfused rat lung. Part. Fibre Toxicol. 2005, 2 (1), 3.
[323] Heckel, K.; Kiefmann, R.; Dorger, M.; Stoeckelhuber, M.; Goetz, A. E., Colloidal gold particles as a new in vivo marker of early acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 287 (4), L867-L878.
[324] Furuyama, A.; Kanno, S.; Kobayashi, T.; Hirano, S., Extrapulmonary translocation of intratracheally instilled fine and ultrafine particles via direct and alveolar macrophage-associated routes. Arch. Toxicol. 2009, 83 (5), 429-437.
[325] Takenaka, S.; Karg, E.; Kreyling, W. G.; Lentner, B.; Moeller, W.; Behnke-Semmler, M.; Jennen, L.; Walch, A.; Michalke, B.; Schramel, P.; Heyder, J.; Schulz, H., Distribution pattern of inhaled ultrafine gold particles in the rat lung. Inhal. Toxicol. 2006, 18 (10), 733-740.
88

07/05/2023
[326] Chen, J. M.; Tan, M. G.; Nemmar, A.; Song, W. M.; Dong, M.; Zhang, G. L.; Li, Y., Quantification of extrapulmonary translocation of intratracheal-instilled particles in vivo in rats: Effect of lipopolysaccharide. Toxicology 2006, 222 (3), 195-201.
[327] Yacobi, N. R.; Phuleria, H. C.; Dernaio, L.; Liang, C. H.; Peng, C.-A.; Sioutas, C.; Borok, Z.; Kim, K.-J.; Crandall, E. D., Nanoparticle effects on rat alveolar epithelial cell monolayer barrier properties. Toxicol. In Vitro 2007, 21 (8), 1373-1381.
[328] Yacobi, N.; Fazllolahi, F.; Kim, Y.; Sipos, A.; Borok, Z.; Kim, K.-J.; Crandall, E., Nanomaterial interactions with and trafficking across the lung alveolar epithelial barrier: implications for health effects of air-pollution particles. Air Quality, Atmosphere & Health 2011, 4 (1), 65-78.
[329] Choi, H. S.; Ashitate, Y.; Lee, J. H.; Kim, S. H.; Matsui, A.; Insin, N.; Bawendi, M. G.; Semmler-Behnke, M.; Frangioni, J. V.; Tsuda, A., Rapid translocation of nanoparticles from the lung airspaces to the body. Nat. Biotechnol. 2010, 28 (12), 1300-U113.
[330] Kato, T.; Yashiro, T.; Murata, Y.; Herbert, D.; Oshikawa, K.; Bando, M.; Ohno, S.; Sugiyama, Y., Evidence that exogenous substances can be phagocytized by alveolar epithelial cells and transported into blood capillaries. Cell Tissue Res. 2003, 311 (1), 47-51.
[331] Edwards, D. A.; Ben-Jebria, A.; Langer, R., Recent advances in pulmonary drug delivery using large, porous inhaled particles. J. Appl. Physiol. 1998, 85 (2), 379-385.
[332] Oberdorster, G.; Oberdorster, E.; Oberdorster, J., Nanotoxicology: An emerging discipline evolving from studies of ultrafine particles. Environ. Health Perspect. 2005, 113 (7), 823-839.
[333] Moller, W.; Felten, K.; Sommerer, K.; Scheuch, G.; Meyer, G.; Meyer, P.; Haussinger, K.; Kreyling, W. G., Deposition, retention, and translocation of ultrafine particles from the central airways and lung periphery. Am. J. Respir. Crit. Care Med. 2008, 177 (4), 426-432.
[334] ElBaseir, M. M.; Phipps, M. A.; Kellaway, I. W., Preparation and subsequent degradation of poly(L-lactic acid) microspheres suitable for aerosolisation: A physico-chemical study. Int. J. Pharm. 1997, 151 (2), 145-153.
[335] Edwards, D. A.; Hanes, J.; Caponetti, G.; Hrkach, J.; Ben-Jebria, A.; Eskew, M. L.; Mintzes, J.; Deaver, D.; Lotan, N.; Langer, R., Large Porous Particles for Pulmonary Drug Delivery. Science 1997, 276 (5320), 1868-1872.
[336] French, D. L.; Edwards, D. A.; Niven, R. W., The influence of formulation on emission, deaggregation and deposition of dry powders for inhalation. J. Aerosol Sci. 1996, 27 (5), 769-783.
[337] Li, W. I.; Perzl, M.; Heyder, J.; Langer, R.; Brain, J. D.; Englemeier, K. H.; Niven, R. W.; Edwards, D. A., Aerodynamics and aerosol particle deaggregation phenomena in model oral-pharyngeal cavities. J. Aerosol Sci. 1996, 27 (8), 1269-1286.
[338] Brown, D. M.; Wilson, M. R.; MacNee, W.; Stone, V.; Donaldson, K., Size-dependent proinflammatory effects of ultrafine polystyrene particles: A role for surface area and oxidative stress in the enhanced activity of ultrafines. Toxicol. Appl. Pharmacol. 2001, 175 (3), 191-199.
[339] Schreier, H.; Gonzalezrothi, R. J.; Stecenko, A. A., Pulmonary delivery of liposomes. J. Control. Release 1993, 24 (1-3), 209-223.
[340] Gilbert, B. E.; Six, H. R.; Wilson, S. Z.; Wyde, P. R.; Knight, V., Small particle aerosols of enviroxime-containing liposomes. Antiviral Res. 1988, 9 (6), 355-365.
[341] Thomas, D. A.; Myers, M. A.; Wichert, B.; Schreier, H.; Gonzalezrothi, R. J., Acute effects of liposome aerosol inhalation on pulmonary function in healthy human volunteers. Chest 1991, 99 (5), 1268-1270.
[342] Myers, M. A.; Thomas, D. A.; Straub, L.; Soucy, D. W.; Niven, R. W.; Kaltenbach, M.; Hood, C. I.; Schreier, H.; Gonzalezrothi, R. J., Pulmonary effects of chronic exposure to liposome aerosols in mice. Exp. Lung Res. 1993, 19 (1), 1-19.
[343] Waldrep, J. C.; Arppe, J.; Jansa, K. A.; Knight, V., High dose cyclosporin A and budesonide-liposome aerosols. Int. J. Pharm. 1997, 152 (1), 27-36.
[344] Waldrep, J. C.; Gilbert, B. E.; Knight, C. M.; Black, M. B.; Scherer, P. W.; Knight, V.; Eschenbacher, W., Pulmonary delivery of beclomethasone liposome aerosol in volunteers - Tolerance and safety. Chest 1997, 111 (2), 316-323.
[345] Niven, R. W.; Schreier, H., Nebulization of liposomes. I. effects of lipid composition. Pharm. Res. 1990, 7 (11), 1127-1133.
[346] Taylor, K. M. G.; Farr, S. J., Liposomes for drug delivery to the respiratory tract. Drug Dev. Ind. Pharm. 1993, 19 (1-2), 123-142.
89

07/05/2023
[347] Leung, K. K. M.; Bridges, P. A.; Taylor, K. M. G., The stability of liposomes to ultrasonic nebulisation. Int. J. Pharm. 1996, 145 (1-2), 95-102.
[348] Torchilin, V., Micellar Nanocarriers: Pharmaceutical Perspectives. Pharm. Res. 2007, 24 (1), 1-16.
[349] Knop, K.; Hoogenboom, R.; Fischer, D.; Schubert, U. S., Poly(ethylene glycol) in drug delivery: pros and cons as well as potential alternatives. Angew. Chem. Int. Ed. Engl. 2010, 49 (36), 6288-308.
[350] Klein, J., Probing the interactions of proteins and nanoparticles. Proc. Natl. Acad. Sci. U. S. A. 2007, 104 (7), 2029-2030.
[351] Roach, P.; Farrar, D.; Perry, C. C., Surface tailoring for controlled protein adsorption: Effect of topography at the nanometer scale and chemistry. J. Am. Chem. Soc. 2006, 128 (12), 3939-3945.
[352] Deng, Z. J.; Liang, M.; Monteiro, M.; Toth, I.; Minchin, R. F., Nanoparticle-induced unfolding of fibrinogen promotes Mac-1 receptor activation and inflammation. Nat Nano 2011, 6 (1), 39-44.
[353] Paillard, A.; Passirani, C.; Saulnier, P.; Kroubi, M.; Garcion, E.; Benoit, J. P.; Betbeder, D., Positively-charged, porous, polysaccharide nanoparticles loaded with anionic molecules behave as 'stealth' cationic nanocarriers. Pharm. Res. 2010, 27 (1), 126-33.
[354] Merkel, T. J.; Jones, S. W.; Herlihy, K. P.; Kersey, F. R.; Shields, A. R.; Napier, M.; Luft, J. C.; Wu, H. L.; Zamboni, W. C.; Wang, A. Z.; Bear, J. E.; DeSimone, J. M., Using mechanobiological mimicry of red blood cells to extend circulation times of hydrogel microparticles. Proc. Natl. Acad. Sci. U. S. A. 2011, 108 (2), 586-591.
[355] Kutscher, H. L.; Chao, P.; Deshmukh, M.; Sundara Rajan, S.; Singh, Y.; Hu, P.; Joseph, L. B.; Stein, S.; Laskin, D. L.; Sinko, P. J., Enhanced passive pulmonary targeting and retention of PEGylated rigid microparticles in rats. Int. J. Pharm. 2010, 402 (1-2), 64-71.
[356] Banquy, X.; Suarez, F.; Argaw, A.; Rabanel, J. M.; Grutter, P.; Bouchard, J. F.; Hildgen, P.; Giasson, S., Effect of mechanical properties of hydrogel nanoparticles on macrophage cell uptake. Soft Matter 2009, 5 (20), 3984-3991.
90