renal amyloidosis
TRANSCRIPT

Renal Amyloidosis - A Review
Dr Kiran Kumar M,DM Senior Resident,Dept of Nephrology

Introduction• A generic term –
– group of diseases that are caused by the misfolding and extracellular accumulation of various proteins
• Misfolded proteins form fibrillar deposits
– Produce pathognomonic green birefringence when stained with Congo red dye and viewed under cross-polarized light

• Amyloidosis - remarkably diverse• – Hereditary or acquired, – Localized or systemic– Lethal or an incidental finding
• 27 human proteins with amyloidogenic potential in vivo have been identified
– ~15 of these proteins cause systemic amyloidosis.

Pathogenesis of amyloidosis
• For each of these amyloidogenic “precursor proteins,”
– initial step in amyloid fibril formation is a misfolding event
• Misfolding can result from
– Proteolytic cleavage (e.g., amyloid β protein)
– An amino acid substitution (e.g., transthyretin [TTR])
– Intrinsic properties that become significant only at high serum concentration or in the presence of specific local factors (e.g., β2-microglobulin)

• Combination of these factors often determines the amyloidogenic potential of a particular protein
– they are not sufficient to account for the occurrence, timing, distribution or effects of amyloid deposition in vivo
• Amyloid fibrils– avidly bind a normal circulating protein of uncertain
physiologic significance called serum amyloid P component (SAP)
• This observation may be of diagnostic utility
– SAP scintigraphy -IV injection of radiolabeled SAP

• Regardless of the protein or the trigger for misfolding, the misfolded variants are highly prone to self-aggregation
• Self-aggregation generates protofilaments that interact to form fibrils
• Amyloid fibrils have a characteristic - β pleated sheet configuration– Produces birefringence under polarized light when
stained with Congo red dye


Determinants of Renal Deposition ofAmyloid
• Factors that determine the organ distribution of amyloid deposits - not well understood
– Kidney - frequent site of amyloid deposition in AL, AA, fibrinogen, lysozyme, apoAII, and, to a lesser extent, apoAI disease
– In contrast, TTR amyloidosis typically does not involve the kidney

Variety of light chain sequences that are amyloidogenicIn AL amyloidosis, disease can be restricted to a single
tissue type in one individual and involve as many as five to six organ systems in another individual
Differences in amino acid sequence of an amyloidogenic protein– Familial TTR amyloidosis
• amino acid substitution of methionine for valine at position 30 (Val30Met) - predominant neuropathic involvement
• Val122Ile TTR variant - cardiomyopathy is the major manifestation

Position of the mutation in the apoAI protein a/w the distribution of organ involvement
– Mutations in the amino terminal portion are - kidney, liver, heart involvement
– Mutations in the carboxy terminal portion -heart, skin, laryngeal involvement

• Kidney tropism in AL disease
– Specific uptake by mesangial cells– Does not seem to be a uniform requirement for amyloid
deposition in the kidney
• Other factors that might promote or retard amyloid formation or deposition in the kidney
• – Negative charge– High glycosaminoglycan content of GBM – Presence of certain proteases that could either render a
protein amyloidogenic or affect stability of amyloid deposits– Local PH

How Does Amyloidosis Cause RenalDisease?
• Disruption of tissue architecture by amyloid deposits
• Amyloidogenic precursor proteins, folding intermediates, and protofilaments have toxicities that are independent of the amyloid deposits
– Lack of correlation between quantity of amyloid in tissue and organ dysfunction
– in vitro demonstrations of direct toxicity of amyloidogenic precursor proteins on cultured cells
– Detection of amyloidogenic precursor proteins in tissue in the absence of amyloid
– Rapid improvement in markers of organ dysfunction after treatment induced reductions in precursor protein production


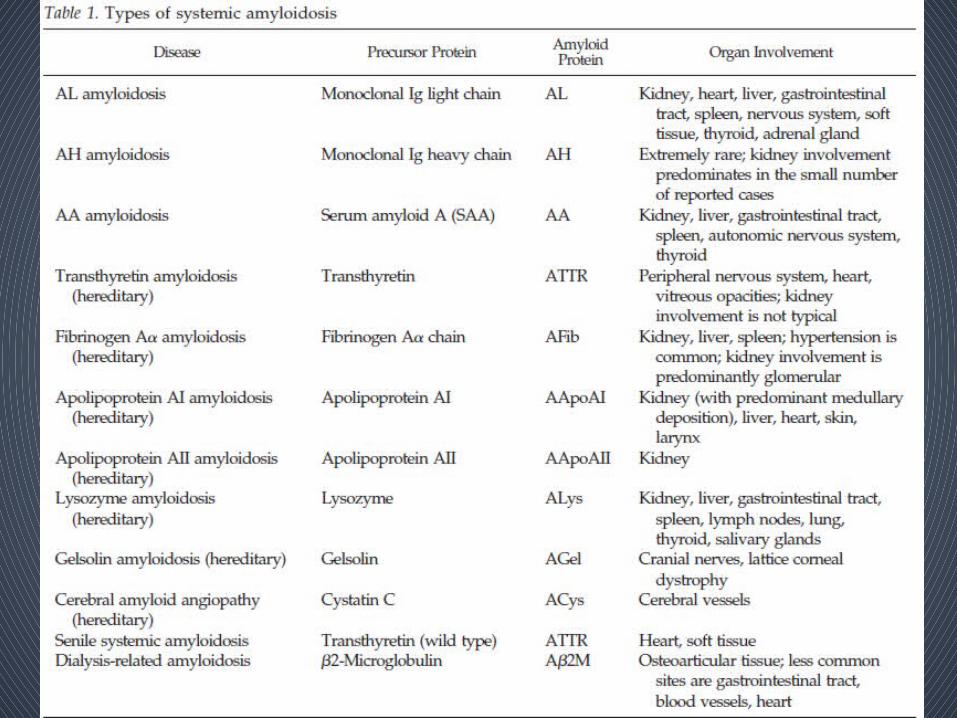
Classification • Based on – the precursor protein that forms the amyloid
fibrils– the distribution of amyloid deposition as either
systemic or localized


Epidemeology • Predominantly a disease of mid-to-late life
• Renal amyloidosis is identified in ~4% of adult renal biopsy samples – accounts for ~1.6% of pts starting dialysis
• Age-adjusted incidence of AL amyloidosis in the US and in the UK is estimated to be 5.1–12.8 cases per million persons per yr
• Disease develops in about 2% of individuals with monoclonal B cell dyscrasias‑

• AA amyloidosis - reported prevalence in patients with chronic arthritides is 3–6%
• Duration of latency b/w onset of inflammation and diagnosis of AA amyloidosis can vary from less than a year to many decades– median of approximately 17 yrs

Indian scenario
• Chugh et al – 1981
– 233 pts with renal amyloidosis– Incidence of amyloidosis - 1.01% of 6431 postmortems and
8-4% of 1980 renal biopsies– Secondary amyloidosis - 87.1 %– Primary amyloid - 9.4 % – Amyloidosis associated with multiple myeloma - 3.5 % – Tuberculosis - commonest predisposing disease accounting
for secondary amyloidosis - 59%– Chronic suppurative lung disease – 24.1 %. – RA, chronic osteomyelitis and lepromatous leprosy - small
percentage of pts (2 to 8 %)

1992-2002 2003-2010
No of biopsies 974 1524
Incidence of renal amyloidosis
1.74% 1.9%
Mean age 38 ± 17.9 39.2 ± 19
Gender predominance Male female
Renal insufficiency in patients with renal amyloidosis
n = 2; 12.8% n = 14; 48.2%
Subnephrotic proteinuria 12.8% 48.82%
MC cause of secondary amyloidosis
Infection (n = 10; 58.8%) Inflammatory disorders (n = 14; 48.2%)
Prakash J1, Brojen T, Rathore SS, Choudhury TA, Gupta T. Ren Fail. 2012;34(10):1212-6. doi: 10.3109/0886022X.2012.723514. Epub 2012 Sep 25.

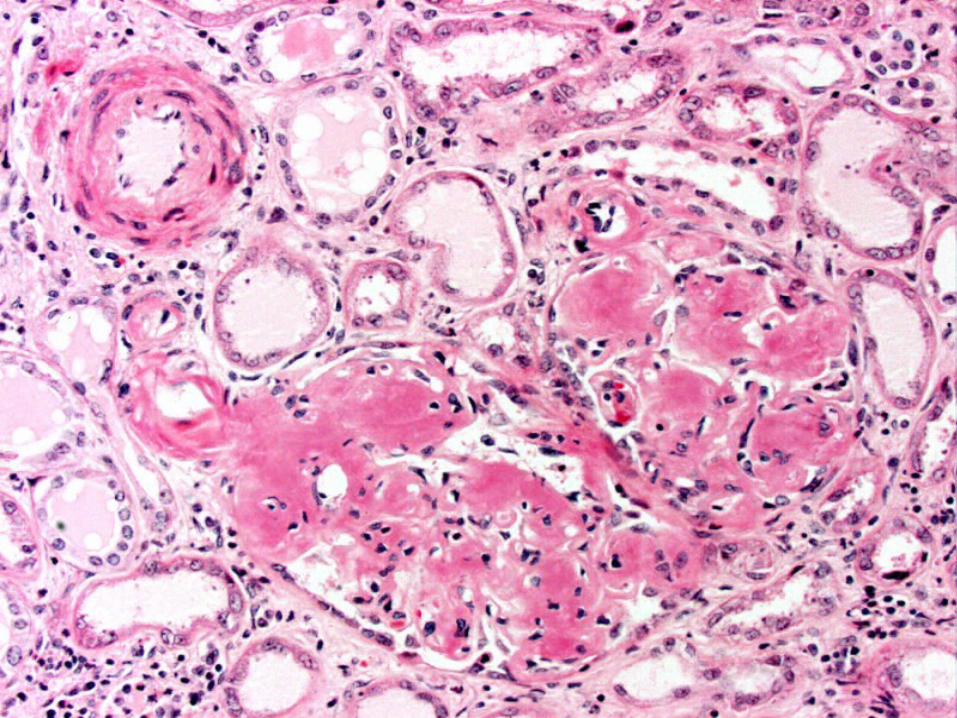
Pathology
• Histologic Demonstration of Amyloid– Usually by Congo red dye
– Congo red–stained amyloid • Orange-red appearance under light microscopy • Produces apple-green birefringence under polarized
light– birefringence results from the ordered intercalation of Congo
red dye into the amyloid fibrils– this optical property must be present to consider the staining
Congo red positive

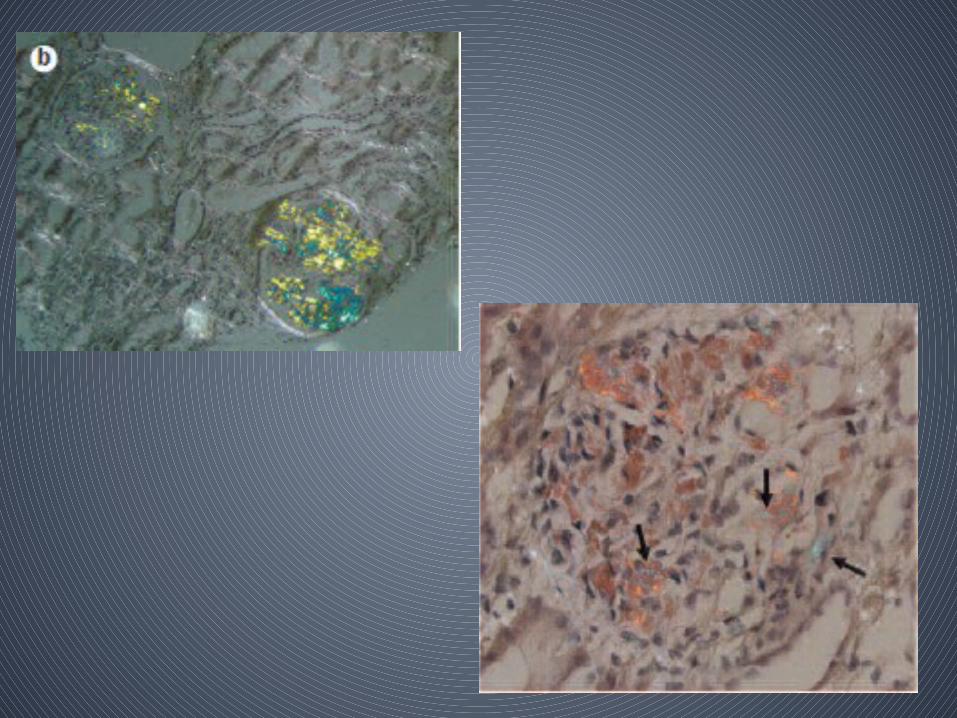
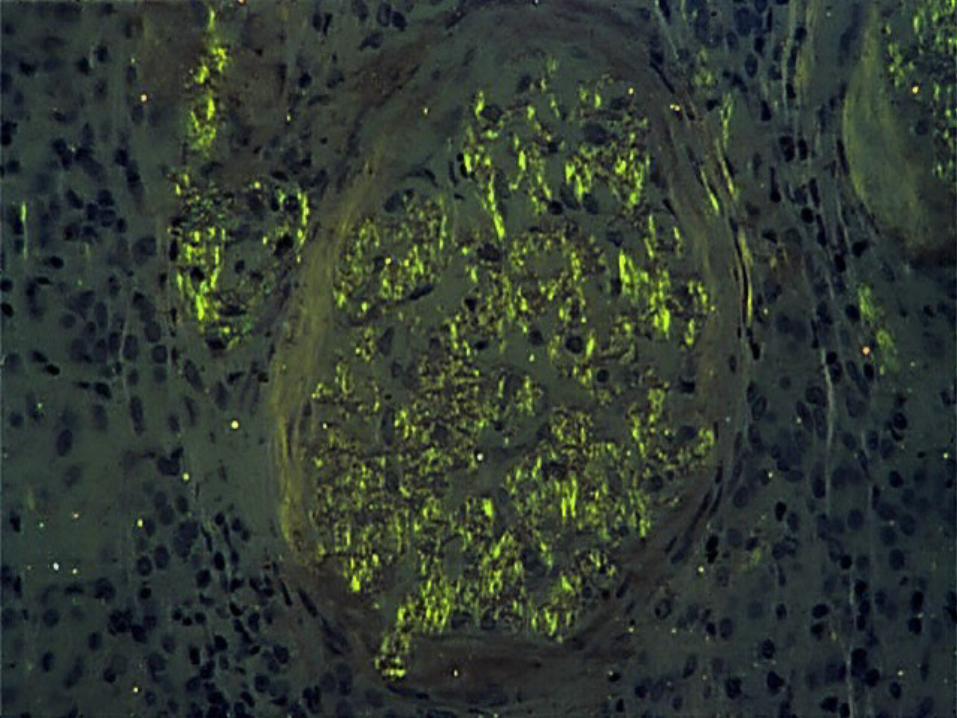

• ThioflavinT staining - yellow-green fluorescence
– Congo red positivity greatest from sites with clinical evidence of involvement
– If amyloidosis is suspected, abd fat aspiration rather than an invasive Bx
– The sensitivity of Congo red staining of abd fat
• 80 to 90% - AL amyloidosis• 65 to 75% - AA amyloidosis• substantially lower in many of the familial amyloidoses
– Relatively noninvasive - Salivary gland and rectal biopsies

• Determination of the Type of Amyloidosis
– Types of amyloid - indistinguishable by LM or EM
– Most direct method for identifying the amyloidogenic protein - Mass spectrometry or Amino acid sequencing of proteins • Not available routinely• Usually not necessary unless other approaches are
unrevealing
– The most definitive method used in the clinical setting • Immunofluorescence • Immunohistochemical
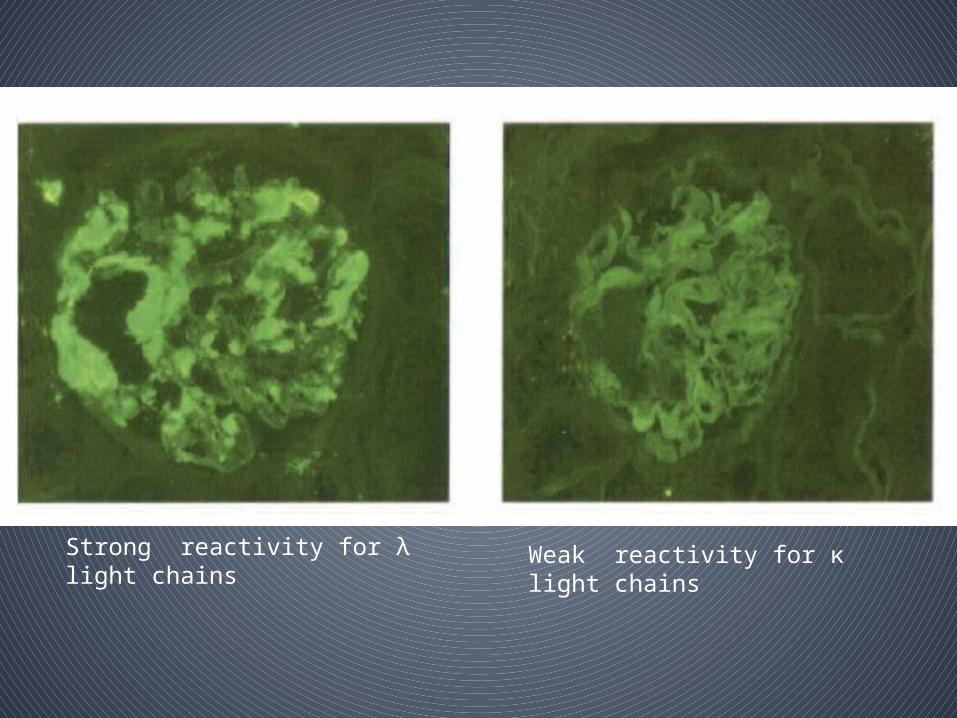
Strong reactivity for λ light chains Weak reactivity for κ light chains

• Absence of immunoreactivity of tissue amyloid for λ or κ light chain, evidence for AL disease –
– demonstration of monoclonal Ig protein in the blood or urine or clonal plasma cells in the bone marrow
– Immunofixation electrophoresis vs protein electrophoresis (monoclonal protein is less in AL than in multiple myeloma)
– Nephelometric quantification of serum free light chains • presence of a monoclonal protein • disease progression• response to treatment

• Renal impairment- – Ratio of the serum conc of two light-chain
isotypes rather than the absolute serum concentrations
• Bone marrow biopsy – assessing – plasma cell clonality– plasma cell burden

• Renal Pathology• Risk for procedure-related bleeding as a result
of vascular fragility- little evidence
• Glomerular deposition typically predominates– appears as amorphous material in the mesangium
and capillary loops• Substantial mesangial deposition produce nodules – vs
diabetic nephropathy or LCDD
– periodic acid-Schiff (PAS) and methanamine silver- negative ( not extracellular tissue)

• Amyloid deposition in the tubulointerstitium produces tubular atrophy and interstitial fibrosis
• Small proportion of pts– glomerular deposition is scant or absent – amyloid is confined to the tubulointerstitium or
vasculature

• Immunofluorescence or immunohistochemical studies arenegative for intact Ig, complement, and fibrin– AL disease – will reveal Ig light chain
• Reactivity - restricted to a single light chain isotype– Some degree of background staining for light
chains, albumin
• Absence of reactivity for either or light chain does not rule out AL disease

• Commercially available reagents do not always detect amyloidogenic light chains because of– conformational change or fragmentation that
masks or eliminates the relevant epitopes
• AA amyloid - detected with available antibodies against AA protein
• Loss of Congo red staining after treatment with potassium permanganate is a property of AA amyloid

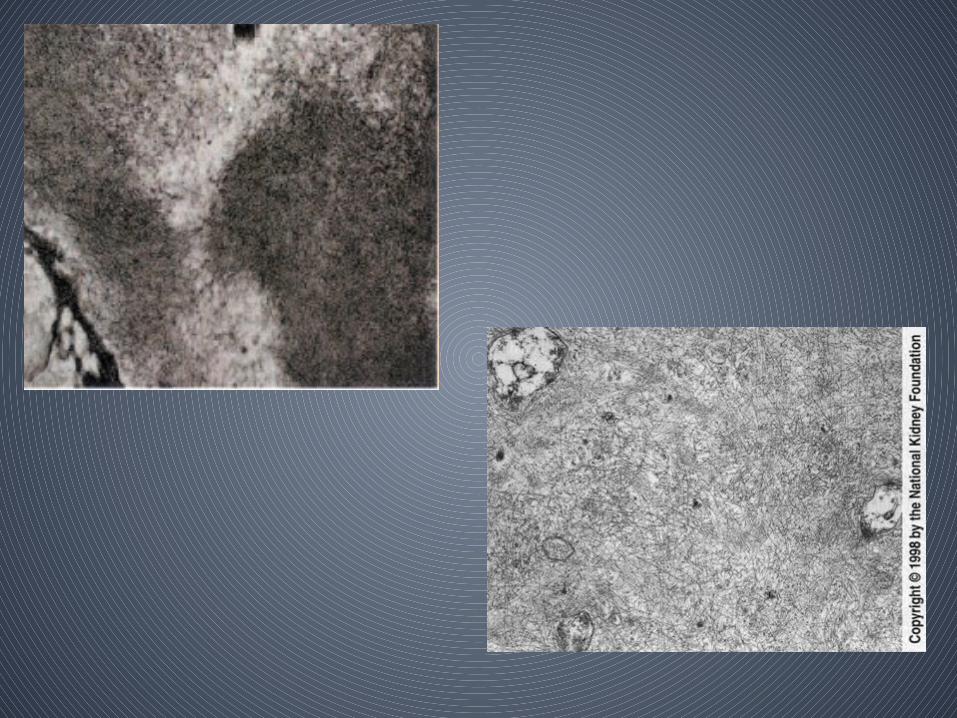
• Electron microscopy
– Amyloid appears as nonbranching fibrils with a diameter of 8 to 10 nm
– randomly arrayed without a specific orientation in the mesangium, basement membranes, interstitium, and vessels
– electron micrographic appearance of amyloid fibrils is sufficiently characteristic that, if present, thediagnosis of amyloidosis should continue to be considered even when Congo red staining is negative

• D/D of monoclonal Ig light-chain disorders
• LCDD– Congo red–negative deposits - granular pattern along the glomerular
and tubular basement membranes
– Immunoreactivity with anti-λ or anti- κ light-chain antibody usually is positive • light-chain epitopes are maintained to a greater extent in LCDD than in AL
amyloidosis
– In LCDD, the κ light-chain isotype is more common than the λ isotype
– Compared with AL amyloidosis, there usually is less background staining with nonpathologic lightchain isotypes or albumin
– PAS staining is much more intense than in amyloidosis• deposition of light chains stimulates production of collagen and other
extracellular matrix components; as a result,

• Cast nephropathy or myeloma kidney –
– Light chains form intratubular casts
– light-chain casts are PAS negative
– they are highly refractile, and they often appear lamellated and fractured
– Inflammatory cell infiltration and, in some cases, granuloma formation often are present in the interstitium that surrounds the affected tubules



Clinico-pathologic Correlates• Proteinuria
– Present in the majority
– Ranges from subnephrotic to massive with urinary protein excretion rates as high as 20 to 30 g/d
– Composed mostly of albumin
– Proteinuria usually is accompanied by other components of the nephrotic syndrome
– Hypoalbuminemia can be profound
– Edema is severe and refractory to diuretics• Cardiac and autonomic nervous system involvement can cause hemodynamic
fragility that limits the effectiveness or tolerability of diuretics

• When amyloid is confined to the tubulointerstitium or vasculature
– Proteinuria is minimal and reduced GFR is the principal clinical manifestation.
– Renal impairment tends to progress less rapidly
• Normal or low BP– Vascular involvement often is accompanied by
hypertension

• Unusual manifestations
– Nephrogenic diabetes insipidus caused by amyloid deposition in the peri-collecting duct tissue
– Fanconi’s syndrome- injury to proximal tubular cells by filtered light chains
• Amyloidosis can cause enlargement of the kidneys– However, in most patients, the kidneys seem to be of
normal size by imaging studies
• Clear relationships b/w extent of amyloid deposition evident by kidney Bx and severity of clinical manifestations have not been demonstrated


Treatment
• The goal of current treatment approaches for AL amyloidosis is to eradicate the clonal plasma cells that produce the amyloidogenic light chain
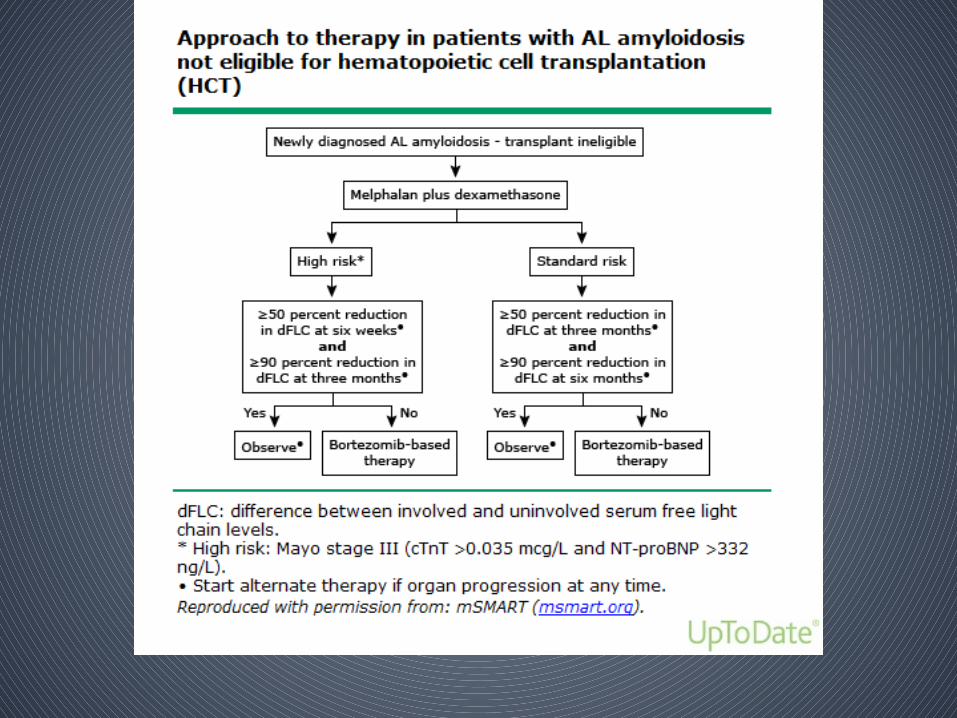
• Depends on whether pts are eligible to pursue high dose melphalan followed by autologous hematopoietic cell transplantation (HCT)

• Criteria for eligibility for HCT
– Physiologic age ≤70 years– Troponin T <0.06 ng/mL– NT-proBNP <5000 ng/L– Creatinine clearance ≥30 mL/min (unless on chronic stable
dialysis)– Eastern Cooperative Oncology Group (ECOG) performance
status ≤2 – New York Heart Association functional status Class I or II – No more than two organs significantly involved (liver, heart,
kidney, or autonomic nerve)– No large pleural effusions– No dependency on oxygen therapy


• Achieving a haematologic response in AL amyloidosis translates into improved overall survival
• Complete haematologic responses are a/w best clinical outcomes

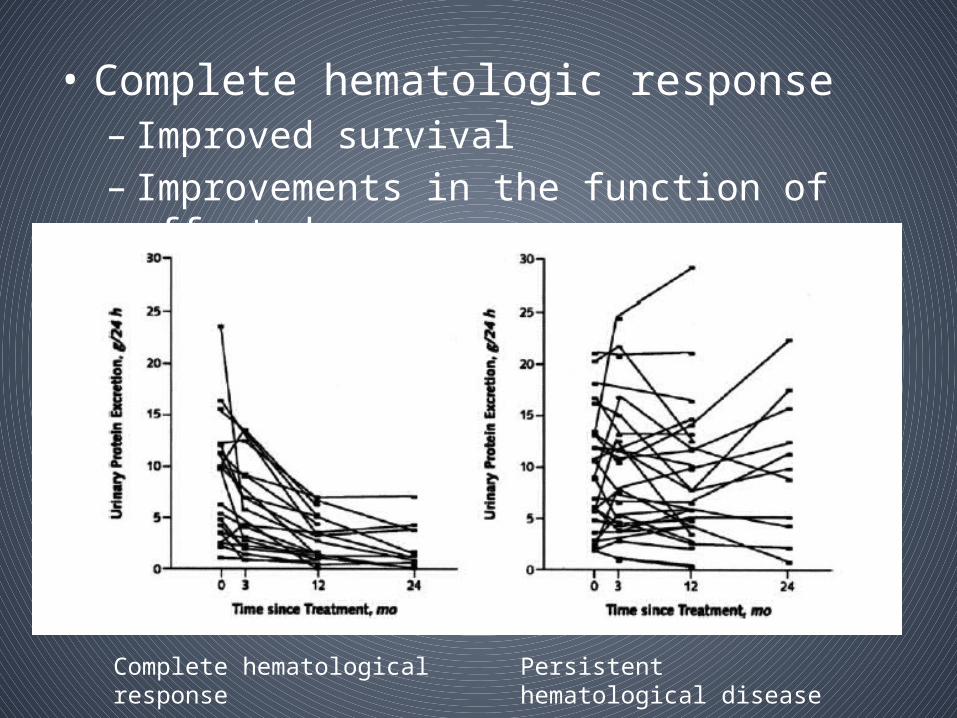
• Complete hematologic response– Improved survival– Improvements in the function of affected organs
Complete hematological response Persistent hematological disease

Bortezomib-based regimens
• Newly diagnosed or relapsed/refractory AL amyloidosis– Proteosome inhibitors (eg bortezomib) in combination
with other agents
• Demonstrated promise with rapid responses seen in a majority of pts– 43 pts - cyclophosphamide, bortezomib,
and dexamethasone (CyBorD) - hematologic response rate of 81 %• Median follow-up of 14 mths, the estimated 2-year
progression-free survival was 67 percent and 41 percent for newly diagnosed and relapsed patients, respectively

– 17 patients with AL amyloidosis treated with another dosing variant of CyBorD, 16 patients demonstrated a hematologic response (12 complete) with a median time to response of two months
• Relapsed disease – – the use of bortezomib-based or
immunomodulatory-based regimens is a reasonable approach
– There are no good data to determine which of these regimens will be of most benefit


Treatment of AA amyloidosis• Current treatment approach for AA
amyloidosis is to treat the underlying inflammatory disease– Reduce the levels of SAA
• Suppression of inflammation can result in reduction in the clinical manifestations of amyloidosis and improved survival

• AA amyloidosis–associated kidney disease– cytotoxic agents or TNF antagonists
• MOA: – Suppression of SAA production and resultant
reduction in AA amyloid formation– additional anti-amyloid effects through • suppression of cytokine production or • by altering the expression of specific mediatorsof
amyloid fibril–induced cellular toxicity

• Cytotoxic and immunosuppressive agents– Azathioprine, chlorambucil, methotrexate,
and cyclophosphamide– Only few reports– 6 monthly cycles of cyclophosphamide
• Anticytokine therapy
• Much of experience – in RA pts and on first-generation TNF-alpha antagonists (ie, etanercept and infliximab)
• Less information is available regarding the newer TNF-alpha antagonists -adalimumab, certolizumab pegol, and golimumab

• Effectiveness enhanced by pulse therapy with glucocorticoids for induction
• Regression of tissue amyloid occurring as early as three months
• Humanized anti-IL-6 receptor antibody – tocilizumab– IL-6 blockade may be more efficient than TNF
blockade in normalizing SAA levels in pts with rheumatic disease

• Eprodisate - a negatively charged sulfonated molecule with in vivo activity against experimentally induced AA amyloidosis– Interfere with interactions between GAGs and amyloid
proteins
• Tafamidis and Diflunisal
– stabilizes circulating transthyretin in its normal conformation
– Used in ATTR amyloidosis (familial amyloid polyneuropathy, hereditary amyloid cardiomyopathy and senile systemic amyloidosis)

• Amyloidosis-Associated ESRD• Large cohort of pts with amyloidosis-asstd
dialysis dependence– Median survival after initiation of dialysis was 8.5
months – Mortality – cardiac amyloidosis and malnutrition–
• Dialysis dependence, in and of itself, should not preclude aggressive treatment that aims to reduce ongoing amyloid production

• Appropriateness of offering HDM/SCT to dialysis-dependent patients with AL amyloidosis
– Hematologic response rate and treatment-asstd mortality are similar in dialysis-dependent pts compared with the overall population of pts who undergo this treatment
• Kidney transplantation in amyloidosis-associated ESRD in pts with AA amyloidosis
– Outcomes that were worse as well as outcomes that were similar compared with the general renal transplant population
– Recurrence of 71%

• AL amyloidosis– kidney transplantation is a good option for
patients who attain a complete hematologic response and do not have significant extrarenal disease

Dialysis-related amyloidosis
• Levels of β2-microglobulin are increased in patients on dialysis
• β2-microglobulin is preferentially deposited in articular and periarticular structures
• Clinical manifestations are largely confined to the locomotor system– shoulders, knees, wrists and the small joints of the
hand, and results in swelling, chronic tenosynovitis and occasionally haemarthroses

– Spondyloarthropathies and cervical cord compression
– deposition of β2 microglobulin within the ‑periarticular bone results in the appearance of subchondral erosions and cysts, which can contribute to pathological fractures,
• Manifestations outside the musculoskeletal systemic are rare – congestive cardiac failure, gastrointestinal
bleeding, perforation and pseudo-obstruction,

• Treatment – • Renal tx -only effective treatment
• Greater removal of β2-microglobulin is attained in patients undergoing high-flux haemodiafiltration