ref degenerasi makula
DESCRIPTION
DEGENERASI MAKULATRANSCRIPT

http://en.wikipedia.org/wiki/Retinal_degeneration_(rhodopsin_mutation)
Retinal degeneration (rhodopsin mutation)From Wikipedia, the free encyclopedia
Fundus images of retinitis pigmentosa
Retinal degeneration is the deterioration of the retina(1) caused by the progressive and eventual death of
the cells of the retina.(2) There are several reasons for retinal degeneration, including artery or vein
occlusion, diabetic retinopathy, R.L.F./R.O.P. (retrolental fibroplasia/ retinopathy of prematurity), or disease
(usually hereditary).(3) These may present in many different ways such as impaired vision, night
blindness, retinal detachment, light sensitivity, tunnel vision, and loss of peripheral vision to total loss of
vision(4). Of the retinal degenerative diseases retinitis pigmentosa (RP) is a very important example.
Inherited retinal degenerative disorders in humans exhibit genetic and phenotypic heterogeneity in their
underlying causes and clinical outcomes* (5) (6) (7). These retinopathies affect approximately one in 2000
individuals worldwide (7). A wide variety of causes have been attributed to retinal degeneration, such as
disruption of genes that are involved in phototransduction, biosynthesis and folding of the rhodopsin
molecule, and the structural support of the retina.(6) Mutations in the rhodopsin gene account for 25% to
30% (30% to 40% according to(9)) of all cases of autosomal dominant retinitis pigmentosa (adRP) (5) (20–
22) in North America (10–12) There are many mechanisms of retinal degeneration attributed to rhodopsin
mutations or mutations that involve or affect the function of rhodopsin. One mechanism of retinal
degeneration is rhodopsin overexpression. Another mechanism, whereby a mutation caused a truncated
rhodopsin, was found to affect rod function and increased the rate of photoreceptor degeneration.(9)
*For example, a single peripherin/RDS splice site mutation was identified as the cause
of retinopathy in eight families; the phenotype in these families ranged from retinitis pigmentosa
tomacular degeneration.(7)

Photoreceptor cell death[edit]
Illustration of the structure of the mammalian retina, including photoreceptor cells ; rod cells and cone cells
Photoreceptor cell death is the eventual outcome of retinal degeneration. Without proper function of
the photoreceptor cells, vision is not possible. Irreversible loss of these cells has been attributed as a
cause of blindness in many retinal degenerative disorders, including RP. The exact mechanism of
photoreceptor cell death is not clearly understood. (5)(14) Among potential causes is
the endocytosis of stable complexes formed between rhodopsin and its regulatory protein arrestin in
certain mutants. (5) Various studies have also documented that over-expression of rhodopsin itself
(mutations in genes involved in the termination of rhodopsin signaling activity have been shown to
cause degeneration by persistent activation of the phototransduction cascade (14)) causes
photoreceptor cell death and may induce photoreceptor cell loss in transgenic animals expressing
truncated rhodopsin. Yet another mechanism may be prolonged photoreceptor responses and also
abnormal rhodopsin deactivation may induce outer segment shortening and eventual photoreceptor
death (9)
In RP photoreceptor cell death is believed to occur by programmed cell death or apoptosis. (10) (11)
(15)
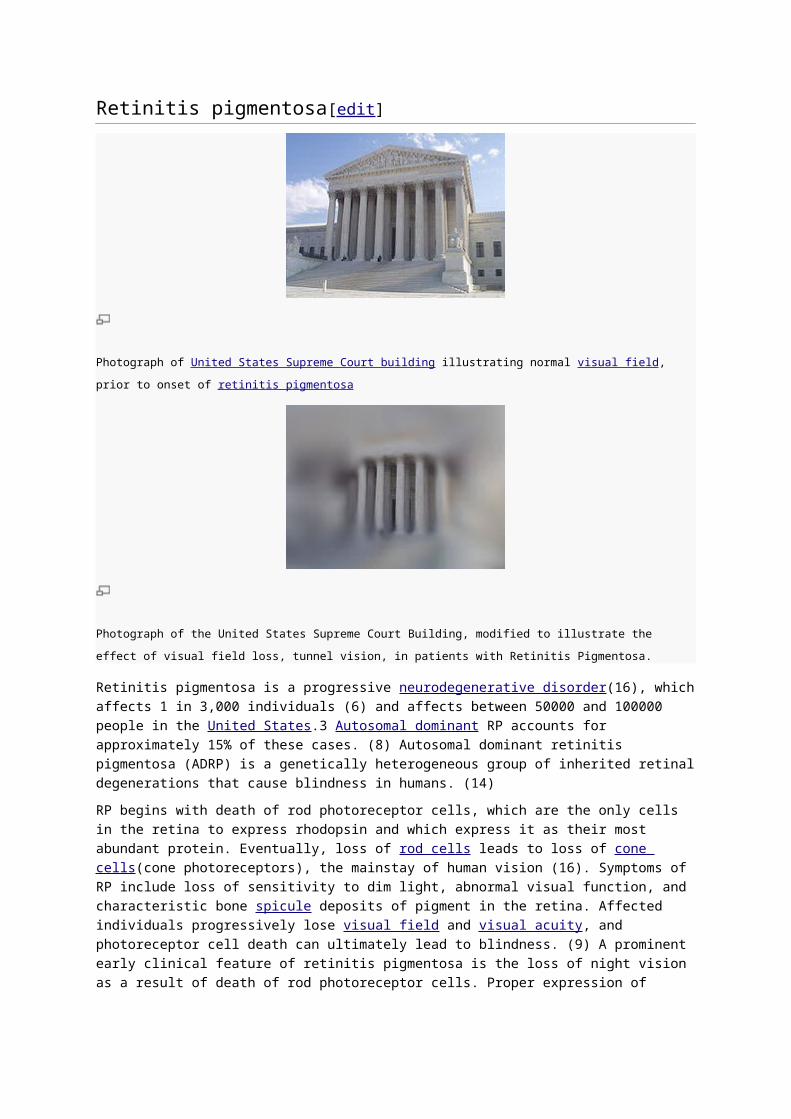
Retinitis pigmentosa[edit]
Photograph of United States Supreme Court building illustrating normal visual field, prior to onset of retinitis pigmentosa
Photograph of the United States Supreme Court Building, modified to illustrate the effect of visual field loss, tunnel
vision, in patients with Retinitis Pigmentosa.
Retinitis pigmentosa is a progressive neurodegenerative disorder(16), which affects 1 in 3,000
individuals (6) and affects between 50000 and 100000 people in the United States.3 Autosomal
dominant RP accounts for approximately 15% of these cases. (8) Autosomal dominant retinitis
pigmentosa (ADRP) is a genetically heterogeneous group of inherited retinal degenerations that
cause blindness in humans. (14)
RP begins with death of rod photoreceptor cells, which are the only cells in the retina to express
rhodopsin and which express it as their most abundant protein. Eventually, loss of rod cells leads to
loss of cone cells(cone photoreceptors), the mainstay of human vision (16). Symptoms of RP include
loss of sensitivity to dim light, abnormal visual function, and characteristic bone spicule deposits of
pigment in the retina. Affected individuals progressively lose visual field and visual acuity, and
photoreceptor cell death can ultimately lead to blindness. (9) A prominent early clinical feature of
retinitis pigmentosa is the loss of night vision as a result of death of rod photoreceptor cells. Proper
expression of the wild-type rhodopsin gene is essential for the development and sustained function of
photoreceptor cells.(10)
Mutations in the human rhodopsin that affect its folding, trafficking and activity are the most commonly
encountered causes of retinal degeneration in afflicted patients. A single base-substitution at
the codon position 23 in the human opsin gene (P23H) is the most common cause of ADRP in
American patients. (6)(17) ADRP due to rhodopsin mutations has a wide range of clinical presentation

and severity. Before 1991, phenotypic evidence pointed to different subsets of ADRP with
varying prognoses.(30–34) Molecular classification of ADRP and further sub-classification based on
the region of the mutation in the rhodopsin gene allowed better prediction of a particular disease
course. But even within these specific subsets, the prognosis is influenced by the specific mutation
itself.(8)
Rhodopsin and its function in vision[edit]
Three dimensional structure of rhodopsin
Rhodopsin is a transmembrane protein (Rh1) that is the primary visual pigment (photopigment) of rod
photoreceptors (which are the only cells in the retina to express rhodopsin and which express it as
their most abundant protein(16)) and forms an integral part of the visual cascade.(8)(13)(10) It is a G-
protein-coupled receptor activated by light that initiates the phototransduction cascade (visual
transduction cascade taking place in photoreceptor rod outer segments (13).) converting light signals
to electrophysiologicalsignals in retinal neurons. This photo-activated signal transduction process is
essential for vision. (10)
The structure and function of rhodopsin and the gene encoding it have been the subjects of intense
scrutiny for many years because rhodopsin serves as a useful model for understanding the largest
receptor family in the human genome, the G protein-coupled receptors, and because defects in the
rhodopsin gene are the most common cause of the most common inherited blinding disease, retinitis
pigmentosa. (16) (18)(19)
Rhodopsin Mutation[edit]
The human rhodopsin gene is the locus for numerous alleles linked to the neurodegenerative disease
retinitis pigmentosa. (16) Mutations in the rhodopsin gene account for 25% to 30% (30% to 40%
according to (9)) of all cases of autosomal dominant retinitis pigmentosa (ADRP).(8)(10)(20–22) Over
100 distinct mutations in the light-sensing molecule rhodopsin are known to cause (adRP). (6)(9)(10)
(13)(14) Most of these mutations aremissense mutations affecting single amino acid residues in the
rhodopsin protein. (10)(24) These mutations affect rhodopsin transport to the outer segments of rod
photoreceptor cells, rhodopsin folding, and rhodopsin endocytosis. Mutations in the human rhodopsin
that affect its folding, trafficking and activity are the most commonly encountered causes of retinal
degeneration in patients afflicted with RP. (6) A single base-substitution mutation of codon 23 of the
rhodopsin gene in which proline is changed to histidine (Pro23His) in the human opsin gene accounts

for the largest fraction of rhodopsin mutations observed in the United States and is the most common
cause of ADRP in American patients.(6)(8)(17)
In 1990, the Pro23His mutation of the rhodopsin gene was reported as the first mutation associated
with RP.(8)(20)(21)(24–26) This mutation has been described only in the United States, where it
continues to be the most commonly described gene defect in RP. The phenotype of the RP
associated with the P23H mutation is characteristically relatively mild but variable.(8)(20–22)(24–28)
Multiple studies have demonstrated that the degree of severity of a given mutation in the rhodopsin
gene is based in large part on its position in the rhodopsin molecule—intradiscal,transmembrane,
or cytoplasmic.(29–36) Intradiscal mutations tend to be less severe, whereas mutations affecting
cytoplasmic domains and retinol binding sites tend to be very severe.(30)(36) The severity of
cytoplasmic mutations affecting maintenance of photoreceptor cell polarity, such as those at the C-
terminus (sorting signal of rhodopsin) may result from inappropriate intracellular transport of the
molecule. (8)(13)(37)
Some research has described transgenic mouse mutants that cause degeneration by prolonged
activation of the phototransduction cascade.(38) In this research null mutations in the
rhodopsinkinase(39) and arrestin(40) genes, each of which plays a role in terminating rhodopsin
activity, caused light-dependent retinal degeneration.(14)
Rhodopsin C-terminal mutations[edit]
A fraction of rhodopsin mutations alter the C-terminal tail of the protein, such as the point
mutations P347L, P347S, P347R, and V345M.3–5 In addition, two frameshift mutations (fs 341del
and fs 341-343del) are predicted to add additional residues to the C terminus,(43) whereas Q344ter
results in a C-terminal truncation, and an intron splice mutation (N88) is thought to remove the entire
C-terminal tail of rhodopsin.(9)(41)(42)
Data indicate that expression of truncated rhodopsin negatively affects both photoreceptor function
and health, compromising rod cell survival.The presence of truncated opsin may impair synaptic
transmission or other cellular processes and eventually cause cell death. Large quantities of
mislocalized opsin may decrease the availability of functional proteins in regions where truncated
opsin is concentrated. The presence of truncated rhodopsin in the outer segments causes functional
abnormalities and localization of rhodopsin in the outer and/or inner segments induces increased
photoreceptor cell death (9)
Rhodopsin endocytosis[edit]
Like many other G protein-coupled receptors, the rhodopsin protein undergoes endocytosis following
activation (6)(44)(45). Perturbation of endocytic regulation of rhodopsin has deleterious effects on
photoreceptor cell physiology. In certain mutants, rhodopsin and its regulatory protein arrestin form
stable complexes. As mentioned these complexes have a fatal effect; when taken up during
endocytosis these complexes causes photoreceptor cell death, because the internalized rhodopsin is
not degraded in the lysosome but instead accumulates in the late endosomes. The formation of toxic
Rhodopsin- Arrestin complexes is also reported for mutants of human rhodopsin associated with
severe forms of ADRP (6)(46)(47). For example, mutations at Arg135 are associated with severe
forms of retinitis pigmentosa and exhibit a high affinity for arrestin, undergo endocytosis, and display
endosomal abnormalities.(6)
Missense mutations in the opsin gene affecting the R135 and K296 residues of the protein product
cause ADRP and result in accumulation of Rhodopsin-Arrestin complexes in the photoreceptor cell (6)
(46)(47). The R135 mutant rhodopsin is noted to form stable complex with arrestin and undergo
endocytosis resulting in aberrant endocytic vesicles in HEK cell culture system (47). Similarly, the

K296E rhodopsin is observed to bind the visual arrestin with high affinity. This abnormal interaction is
demonstrated to have pathological consequences in the retina. Besides this, the stable rhodopsin and
arrestin complexes are shown to mislocalize and accumulate in the inner segments of rod
photoreceptors of the mouse model of ADRP.(6)
Using mutants that are defective in late endosome to lysosome trafficking, it was shown that
rhodopsin accumulates in endosomal compartments in these mutants and leads to light-dependent
retinal degeneration. It was also shown that the internalized rhodopsin in dying photoreceptor cells
were not degraded but instead showed characteristics of insoluble proteins. This data led to the
implication of rhodopsin buildup in the late endosomal system as a novel trigger of death of
photoreceptor neurons. Thus failure to degrade internalized rhodopsin in a timely manner triggers cell
death of photoreceptor neurons, suggesting that lysosomal turnover of rhodopsin is vital in
maintaining photoreceptor viability. (6)
The precise mechanisms regulating the pro-cell death signaling pathways and their interconnection
with endocytosis is not well understood. It is speculated that a component innate to the
endolysosomal system plays a crucial role in regulating the cell death signals emanating from the
endosomes. This component senses the accumulation of rhodopsin and then engages the proper
machinery to execute cell death in the retina. Failure of proper protein degradation and resultant
subsequent accumulation of proteins, as in the case of rhodopsin-arrestin complexes, is a well-
recognized cause of cell death in many neurodegenerative disorders, such as retinitis pigmentosa (6)
(48).
Non-rhodopsin mutations[edit]
Mutations non-retina-specific ADRP genes that encode for proteins essential for pre-mRNA splicing
may be a major cause of ADRP. Proteins required for the formation of stable U4/U6 snRNPs and for
assembly of the U4/U6.U5 tri-snRNP have been identified, including HPRP3, PRPC8, and PRPF31,
proper function of these proteins is necessary for spliceosome performance. (10)
The rhodopsin transcript is a pre-mRNA splicing substrate affected by PRPF31 protein, meaning that
rhodopsin (RHO) is among the target splicing substrate genes for PRPF31. Thus it can be understood
that mutations in PRPF31 can cause alternative, potentially non-functional, forms of the rhodopsin
protein. It was shown that expression of these mutant PRPF31 proteins significantly reduced
rhodopsin expression in cultured retinal cells and induced apoptosis of retinal cells, establishing a link
between mutations in proteins involved in pre-mRNA splicing and the expression of a critical retina-
specific gene, rhodopsin. (10)
This shows that non-rhodopsin mutations may also be critical in presentation of retinal degenerative
disorders. It suggests a mechanism for retinal degeneration caused by non-retinal-specific genes,
such as PRPF31 mutations. (10)
References[edit]
1. http://bioweb.uwlax.edu/bio203/s2007/berends_bets/glossary_of_terms.htm
2. http://www.optigen.com/opt9_glossary.html
3. http://www.tsbvi.edu/Education/anomalies/Retinal_degeneration.htm
4. http://www.wrongdiagnosis.com/r/retinal_degeneration/symptoms.htm
5. Sullivan LS, Daiger SP (1996) Inherited retinal degeneration: exceptional genetic and clinical
heterogeneity. Mol Med Today 2: 380–386

6. http://www.plosgenetics.org/article/info:doi/10.1371/journal.pgen.1000377
7. http://www.pubmedcentral.nih.gov/picrender.fcgi?artid=2585107&blobtype=pdf
8. http://archopht.ama-assn.org/cgi/reprint/118/9/1269.pdf
9. http://www.pubmedcentral.nih.gov/picrender.fcgi?artid=2570206&blobtype=pdf
10. http://www.pubmedcentral.nih.gov/picrender.fcgi?artid=2149905&blobtype=pdf
11. Dejneka NS, Bennett J. Gene therapy and retinitis pigmentosa: advances and future challenges.
BioEssays 2001;23:662–668. [PubMed: 11462220]
12. Dryja TP, McEvoy JA, McGee TL, Berson EL. Novel rhodopsin mutations Gly114Val and
Gln184Pro in dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci 2000;41:3124–3127. [PubMed:
10967073]
13. http://www.pnas.org/content/102/9/3301.full.pdf+html
14. http://www.pubmedcentral.nih.gov/picrender.fcgi?artid=2248236&blobtype=pdf
15. Chang GQ, Hao Y, Wong F. Apoptosis: final common pathway of photoreceptor death in rd, rds,
and rhodopsin mutant mice. Neuron 1993;11:595–605. [PubMed: 8398150]
16. http://ncmi.bcm.tmc.edu/homes/wensellab/wenselpubs/chanpnas04.pdf
17. Dryja TP, McGee TL, Reichel E, Hahn LB, Cowley GS, et al. (1990) A point mutation of the
rhodopsin gene in one form of retinitis pigmentosa. Nature 343: 364–366.
18. Rivolta, C., Sharon, D., DeAngelis, M. M. & Dryja, T. P. (2002) Hum. Mol. Genet. 11, 1219–1227.
19. Menon, S. T., Han, M. & Sakmar, T. P. (2001) Physiol. Rev. 81, 1659–1688.
20. Dryja TP, McGee TL, Hahn LB, et al. Mutations within the rhodopsin gene in patients with
autosomal dominant retinitis pigmentosa. N Engl J Med. 1990;323: 1302–1307.
21. Dryja TP, Hahn LB, Cowley GS, McGee TL, Berson EL. Mutation spectrum of the rhodopsin gene
among patients with autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci U S A.
1991;88:9370–9374.
22. Dryja TP. Doyne Lecture: rhodopsin and autosomal dominant retinitis pigmentosa. Eye. 1992;6:1–
10.
23. Vaithinathan R, Berson EL, Dryja TP. Further screening of the rhodopsin gene in patients with
autosomal dominant retinitis pigmentosa. Genomics 1994;21:461–463. [PubMed: 8088850]
24. Dryja TP, McGee TL, Reichel E, et al. A point mutation of the rhodopsin gene in one form of
retinitis pigmentosa. Nature. 1990;343:364–366.
25. Berson EL. Ocular findings in a form of retinitis pigmentosa with rhodopsin gene defect. Trans Am
Ophthalmol Soc. 1990;88:355–388.
26. Berson EL, Rosner B, Sandberg MA, Dryja TP. Ocular findings in patients with autosomal
dominant retinitis pigmentosa and a rhodopsin gene defect (Pro-23-His). Arch Ophthalmol.
1991;109:92–101.
27. Heckenlively JR, Rodriguez JA, Daiger SP. Autosomal dominant sectoral retinitis pigmentosa: two
families with transversion mutation in codon 23 of rhodopsin. Arch Ophthalmol. 1991;109:84–91.

28. Weleber RG, Murphey WH, Rodriguez JA, Lovrien EW, Litt M, Daiger SP. Phenotypic expression
of Pro-23-His mutation of rhodopsin in a large family with autosomal dominant retinitis pigmentosa
[abstract]. Invest Ophthalmol Vis Sci. 1991;32:913. ARVO abstract 1204.
29. Sung C-H, Schneider BG, Agarwal N, Papermaster DS, Nathans J. Functional heterogeneity of
mutant rhodopsins responsible for autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci U S
A. 1991;88:8840–8844.
30. Pannarale MR, Grammatico B, Iannaccone A, et al. Autosomal dominant retinitis pigmentosa
associated with an Arg-135-Trp point mutation of the rhodopsin gene: clinical features and longitudinal
observations. Ophthalmology. 1996;103: 1443–1452.
31. Sung C-H, Davenport CM, Hennessey JC, et al. Rhodopsin mutations in autosomal dominant
retinitis pigmentosa. Proc Natl Acad Sci U S A. 1991;88:6481- 6485.
32. Fishman GA, Stone EM, Gilbert LD, Kenna P, Sheffield VC. Ocular findings associated with a
rhodopsin gene codon 58 transversion mutation in autosomal dominant retinitis pigmentosa. Arch
Ophthalmol. 1991;109:1387–1393.
33. Fishman GA, Stone EM, Gilbert LD, Sheffield VC. Ocular findings associated with a rhodopsin
gene codon 106 mutation: glycine-to-arginine change in autosomal dominant retinitis pigmentosa.
Arch Ophthalmol. 1992;110:646–653.
34. Fishman GA, Stone EM, Sheffield VC, Gilbert LD, Kimura AE. Ocular findings associated with
rhodopsin gene codon 17 and codon 182 transition mutations in dominant retinitis pigmentosa. Arch
Ophthalmol. 1992;110:54–62.
35. Fishman GA, Vandenburgh K, Stone EM, Gilbert LD, Alexander KR, Sheffield VC. Ocular findings
associated with rhodopsin gene codon 267 and 190 mutations in autosomal dominant retinitis
pigmentosa. Arch Ophthalmol. 1992;110:1582- 1588.
36. Sandberg MA, Weigel-DiFranco C, Dryja TP, Berson EL. Clinical expression correlates with
location of rhodopsin mutation in dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci.
1995;36:1934–1942.
37. Berson EL, Rosner B, Sandberg MA, Weigel-DiFranco C, Dryja TP. Ocular findings in patients
with autosomal dominant retinitis pigmentosa and rhodopsin, proline-347-leucine. Am J Ophthalmol.
1991;111:614–623.
38. Lem J, Fain GL. Constitutive opsin signaling: night blindness or retinal degeneration? Trends Mol
Med 2004;10:150–157. [PubMed: 15059605]
39. Chen CK, Burns ME, Spencer M, et al. Abnormal photoresponses and light-induced apoptosis in
rods lacking rhodopsin kinase. Proc Natl Acad Sci USA 1999;96:3718–3722. [PubMed: 10097103]
40. Xu J, Dodd RL, Makino CL, Simon MI, Baylor DA, Chen J. Prolonged photoresponses in
transgenic mouse rods lacking arrestin. Nature 1997;389:505–509. [PubMed: 9333241]
41. Jacobson SG, Kemp CM, Cideciyan AV, et al. Phenotypes of stop codon and splice site rhodopsin
mutations causing retinitis pigmentosa. Invest Ophthalmol Vis Sci. 1994;35:2521–2534. [PubMed]
42. Sung CH, Schneider BG, Agarwal N, et al. Functional heterogeneity of mutant rhodopsins
responsible for autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci USA. 1991;88:8840–
8844. [PubMed]

43. Horn M, Humphries P, Kunisch M, et al. Deletions in exon 5 of the human rhodopsin gene causing
a shift in the reading frame and autosomal dominant retinitis pigmentosa. Hum Genet. 1992;90:255–
257. [PubMed]
44. Orem NR, Dolph PJ (2002) Epitope masking of rhabdomeric rhodopsin during endocytosis-
induced retinal degeneration. Mol Vis 8: 455–461.
45. Satoh AK, Ready DF (2005) Arrestin1 mediates light-dependent rhodopsin endocytosis and cell
survival. Curr Biol 15: 1722–1733.
46. Chen J, Shi G, Concepcion FA, Xie G, Oprian D, et al. (2006) Stable rhodopsin/arrestin complex
leads to retinal degeneration in a transgenic mouse model of autosomal dominant retinitis
pigmentosa. J Neurosci 26: 11929–11937.
47. Chuang JZ, Vega C, Jun W, Sung CH (2004) Structural and functional impairment of endocytic
pathways by retinitis pigmentosa mutant rhodopsinarrestin complexes. J Clin Invest 114: 131–140.
48. Taylor JP, Hardy J, Fischbeck KH (2002) Toxic proteins in neurodegenerative disease. Science
296: 1991–1995.

http://www.ffb.ca/eye_conditions/RD_diseases.html
Retinal Degenerative Diseases
Millions of people in North America live with varying degrees of irreversible vision loss because they have an untreatable, degenerative eye disorder like retinitis pigmentosa (RP), which affects an estimated 1.5 million people worldwide, or age-related macular degeneration, which is the leading cause of vision loss in Canada and North America. Retinal degenerative diseases affect the delicate layer of tissue that lines the inside back of the eye. This part of the eye -- the retina - is essential for vision.
Imagine the eye is like a camera. The shutter, like the iris of the eye, opens and closes to let in the right amount of light. The lens helps to focus light on the film. The retina is like film. Regardless of the perfection or quality of the rest of the camera, if the film is faulty, the developed pictures may be distorted, blurred or impossible to see.
This very large and diverse group of vision disorders affects young and old and people from many cultures, races and ethnicities. Age-related macular degeneration is distinguished by its prevalence in the senior population, and age is considered to be the major risk factor for developing this eye disease. Retinitis pigmentosa and other related conditions are inherited genetic conditions, even if a person who develops it has no previous family history of vision loss. The list of inherited retinal dystrophies (degenerations) is very long, but here are a few of the more common:
Retinitis pigmentosa
Choroideremia (affects males)
Retinoschisis, Juvenile
Stargardt disease
Usher disease

http://www.retina-international.org/eye-conditions/retinal-degenerative-conditions/amd/
What is Macular Degeneration?
Macular Degeneration is the name for several similar conditions that are characterised by a
breakdown of the macula.
The word "macula" comes from the Latin for "spot"; it is the centre portion of the retina that
makes central vision, the vision directly in front of you, possible. The macula is very small: only
about three by five millimetres (about the size of a ladybug) covering about 10 percent of the
retina.
What causes Macular Degeneration?
There are two basic types of Macular degeneration - Age-related and early onset or Juvenile -
and they will be explained in detail in separate sections.
Age-related Macular Degeneration (referred to as AMD) usually does not develop until the sixth
or seventh decade of life, although early onset cases are becoming more common in patients as
young as 40. Because of the later onset of this disease, it is difficult to determine if it is inherited,
but studies are showing familial patterns of the condition, indicating that there may be genetic
causes. There are also other aspects of your health that are risk factors for Age-related Macular
Degeneration; these will be discussed later.
Early onset Macular Degeneration appears to be largely genetic; that is, it is a condition that is
programmed into your cells and not caused by injury or infection or any other external agent.
Certain genes that are necessary for normal vision give faulty messages to the cells in the
macula, leading to their progressive degeneration and eventually to vision loss.
To understand why and how Macular Degenerations occur, scientists must look to a variety of
disciplines. Increasingly sophisticated research at the cellular level is providing insight into the
processes that cells undergo whey they die, and how one step leads to another.
The study of molecular genetics, cell biology, biochemistry, and how these and other fields
interact with each other has provided an abundance of avenues for researchers to pursue. The
symptoms of Macular Degeneration, like those of other retinal diseases, vary greatly and range
in severity from one person to another.
What is Dry AMD?
Dry AMD is the more common form of Macular Degeneration. It is also referred to as atrophic,
nonexcudative, or drusenoid form. This form accounts for 90 percent of Age-related Macular
Degenerations.

Dry AMD is characterised by the build-up of drusen, small yellowish deposits, beneath the
macula. The layer of photo receptor cells in the macula begin to atrophy, or die, as some of the
cells break down. These changes, may, in turn, result in a distortion of vision that is most
apparent when reading. Often if one eye has dry AMD, the other eye will also show some signs
of the condition. However, dry AMD does not usually cause total loss of reading vision.
What is Wet AMD?
Wet AMD accounts for 10 percent of patients with AMID. It is also called choroidal
neovascularisation, subretinal neovascularisation, exudative form, or disciform degeneration.
In wet AMD, new abnormal blood vessels begin to grow beneath the macula, in a thin layer of
tissue called the choroid. The choroid is the main source of oxygen and nutrients for the retinal
photo receptors, and it is the only blood supply for the macula. New fragile blood vessels develop
which may leak fluid and blood, and then cause the choroid and retina to deteriorate. This
causes the retinal layer to blister under the macula, and the photo receptor cells to degenerate.
At this stage, there is marked disturbance of vision in the affected eye.
What are the symptoms?
The most common symptoms are blurring of vision with particular difficulty discerning details,
both up close and from a distance. People with Macular Degeneration may have blind spots,
resulting in a dark or empty area in the centre of their field of vision. They may also notice
distortions of lines and shapes, either in everyday objects (e.g. crooked door frames) or in tests
given by the eye doctor.
Colour vision may also be diminished, although peripheral vision and night vision usually remain
unaffected. Because Age-related Macular Degeneration could begin in one eye, the remaining
good eye will take over on its own to compensate for vision loss. It may be some time before the
second eye is seriously affected enough for an individual to notice vision problems. Others do
notice a sudden loss of vision. If you experience a sudden vision loss or distortion, it is important
that you see your eye care professional immediately.
How are macular degenerations diagnosed?
The early signs of Macular Degeneration are usually detectable in a thorough eye exam even
before the disease begins affecting vision. The doctor will examine the eye with special lenses,
which help to show the interior of the eyeball through the pupil, the opening in the centre of the
iris through which light rays enter the eye. Tests for Macular Degeneration include:
Acuity tests which measure the accuracy of your reading and perception vision at specific
distances in specific lighting situations. This is the test that people are most familiar with and
typically involves a standard eye chart.

Colour testing which can help to determine the status of your cone cells. Since cone cells are
the retinal cells that interpret colour, your doctor will be better able to determine the health of
these cells through your performance on these tests. There are several types of colour tests
which measure various aspects of vision.
A dark adaptation test which will measure how well your eyes adjust to changes in lighting.
Information from this test can help the doctor to better understand the current function of your
rod cells, which are the retinal cells responsible for night vision.
A fluorescein angiogram which allows inner eye structures to be visualised. A non-toxic dye is
injected into the patient's arm and it moves through the bloodstream, including the blood
vessels in the eyes. Photos are then taken of the retina and macula, which will identify new
blood vessel growth and leakage from blood vessels. New blood vessel growth is a feature of
wet age-related Macular Degeneration, which is explained in detail in the section of this
booklet describing the different forms of Macular Degeneration.
Check your vision with an Amsler grid
The Amsler grid test may be helpful in revealing signs of wet Age-related Macular Degeneration
(AMD). It is not a substitute for regularly scheduled eye exams.
Directions:
Wear your reading glasses (if you use them).
Sit at a comfortable reading distance (12” or 30cm) from the grid in a well lit room.
Cover one eye with your hand. Look at the centre dot at all times. If you notice any area on the
grid that is blurred, distorted, discoloured or missing, you may be exhibiting signs of AMD and
should contact your ophthalmologist promptly.
Are there any specific risk factors related to AMD?
As we have seen, AMD is related to ageing, but it certainly does not affect all older people. There
is a growing body of evidence that suggests genetic factors can determine whether or not
someone will get AMD.
Other studies have looked for additional conditions that may be associated with AMD and have
found a number of factors besides age that are associated with an increased risk of developing
AMD.
These include a history of hypertension (high blood pressure) and/or cardiovascular disease,
smoking, a family history of AMD, hyperopia (farsightedness), light skin and eye colour, and lens
opacities (cataracts). Both men and women are at risk for this disease.
The presence of these risk factors in people with AMD has been noted, but the relationship of the
various factors to the disease itself has not been systematically studied and it is not clear just
why the links are present.

How common is AMD?
Researchers estimate that one in 2000 people in the developed world are affected by AMD.
Among non-diabetics, it is the most common problem affecting the retina, and it is the major
cause of legal blindness in individuals over the age of 65. As the population enjoys a longer life,
the number of those affected by AMD will increase as well.
How quickly does AMD progress?
Most cases of AMD progress very slowly over a period of years. Vision may remain stable
between annual eye examinations, and many patients retain a reasonable amount of peripheral
vision. However, wet AMD typically progresses much more rapidly than dry AMD. The majority of
people who experience severe vision loss from AMD have the wet form.
Do Macular Degenerations lead to total blindness?
Most people with Macular Degeneration do retain some peripheral vision, and they learn to
optimize the use of their remaining vision. Each case differs. However, many will be classified as
legally blind". Legally blind individuals are those whose best visual sharpness or acuity (with
glasses or contact lenses, if needed) is 20/200 or worse in their better eye; or whose visual field,
regardless of acuity, is restricted to a 20 degree diameter (10 degree radius).
Can people with Macular Degeneration drive?
This is a difficult question. Many people with mild forms of Macular Degeneration do drive legally,
and do not have problems. It would be best to discuss your visual limitations and their effect on
driving with your eye care professional. Driving is a symbol of independence for many people,
and individuals with progressive vision loss may be unwilling to face the fact that their vision
impairment may impede save driving. Often people with Macular and other retinal degenerations
speak of "near miss" accidents that force them to confront their vision loss and acknowledge that
it is affecting their driving. It is important to remember that your driving affects not only you but
other drivers and pedestrians, and that the results of an error can ultimately be life-threatening.
If I have drusen, will I develop AMD?
Not necessarily, although the presence of drusen may indicate that your eyes are at some risk
for developing AMD. Drusen are deposits that contain complex lipids (fats) and calcium and can
accumulate as a person ages. It is not known how they form but it has been suggested that they
are undigested waste products from RPE cells. There are two basic types of drusen: "hard" and
"soft".
Hard drusen are small, round, solid deposits that sit under the RPE without causing structural
damage. Most people start accumulating some drusen after the age of 40, and hard drusen

alone do not impair vision. However, hard drusen may advance to soft drusen, although this does
not always happen.
Soft drusen are more likely to be associated with vision loss. They are less uniform, involve a
larger area under the RPE, and contain other cellular substances in addition to lipids.
Development of soft drusen may cause the RPE to separate from other eye tissue layers.
It is important to understand the significance of drusen, particularly if you have a family history of
Macular Degeneration. There are many people with drusen in both eyes and no impairment to
their vision at all. However, it is not known who will go on to develop a vision problem and who
will not, so if your eye doctor tells you there are drusen in your eyes, you should continue to seek
eye care regularly, as well as use an Amsler grid to monitor your vision yourself.
What are retinal detachments and tears?
Retinal detachment occurs when the photo receptors (the rods and cones) separate from their
underlying tissue layers. This causes a loss of vision in the area of the separation. Retinal
detachments can sometimes be repaired or partially repaired with surgery. Retinal tears are
splits in the retina that may cause vision disturbances, such as flashes of light or floaters. Retinal
detachments and tears may be caused by physical injury or may be inherited. Although they may
occur in people with Macular Degenerations, they are not necessarily a consequence of the
disease.
Are cataracts associated with Macular Degeneration?
It is not unusual for an older individual with a Macular Degeneration to develop a cataract, which
is a clouding of the lens of the eye. Cataracts that significantly interfere with vision can be
surgically removed. While cataract surgery cannot improve the vision loss due to retinal
degeneration, it can alleviate the added loss caused by the cataract, Whether or not surgery
improves the vision often depends on the extent of retinal changes. Because surgery is not
advised for everyone, it is important to discuss the details of your individual case with an eye
care professional.
Is AMD related to other retinal diseases?
Age-related Macular Degeneration is just one type of a group of retinal degenerations that are
characterised by a breakdown of the photo receptor cells, leading to impaired vision. While AMD
is characterised by a loss of central vision, Retinitis Pigmentosa is associated with night
blindness and a progressive loss of peripheral vision leading to "tunnel" vision. These symptoms
are not the same, but as researchers get closer to uncovering the causes of these conditions,
there is increasing evidence that they are related. For example, it has been found that different
defects of the same gene are responsible for certain forms of RP and Macular Degeneration.

Will others in my family be affected?
The late onset forms of Macular Degeneration are not known to be inherited in a straightforward
pattern such as autosomal dominant or recessive. However, familial patters have been observed,
indicating that inheritance is involved to some extent. Some surveys have estimated that 15 to 20
percent of AMD patients have one or more first-degree relatives who are also affected.
AMD presents a special challenge for genetic researchers because it is difficult to determine the
exact role of genetics when a disease occurs late in life. A number of research projects are now
underway to look specifically at the role of genetics in the development of AMD.
In one study, occurrence of AMD is being compared in groups of identical and non-identical
twins. Genes from blood samples of people with Macular Degeneration are being searched for
disease-causing changes. From these and other studies, scientists hope to learn more about the
relationship between genes and Macular Degeneration.
Because it is not the same gene that causes Macular Degeneration in everyone with the
condition, but an assortment, each with its own particular variations, the hereditary pattern of
these diseases differs from family to family, although the gene mutation will be consistent within
one family. You can get the best information about the likely pattern of the disease in your own
family by consulting with a genetic counsellor or an eye care professional who specializes in
hereditary retinal degenerations. They can help you learn how the disease is inherited in your
family and the chance of passing it on to your children.
Is there any way to prevent AMD?
While research supported by the member organisations of Retina International and other private
and government agencies worldwide is adding to new understandings of AMD, at present there
is no known method of preventing its occurrence. However, there are studies that suggest that
adjustments in lifestyle may help reduce the risk of developing AMD.
Regular eye exams may allow for early diagnosis of AMD. Individuals at risk - for example, those
with degenerative vision loss in one eye, soft drusen, or positive family history - should have
regular eye examinations by an eye care professional after the age of 50 and self-monitor their
vision daily with the use of an Amsler grid. Without self-monitoring, a person may not realize his
or her vision is impaired until the disease has reached advanced stages.
How can nutrition influence the occurrence of AMD?
Researchers found that people who consumed the highest quantity of spinach, collard greens
and other dark green leafy vegetables foods that are rich in carotenoids, were less likely to have
the advanced form of AMD, compared with people in the study who ate the least amounts of
these foods. The findings also suggest that people should not rely on vitamin supplements as

their main source for vitamins, minerals and nutrients, but instead should eat a balanced diet that
includes a wide range of vegetables.
Does sunlight influence the onset of AMD?
Avoiding intense, bright sunlight may help to reduce the retinal degeneration due to AMD. Good
quality sunglasses, hats and visors can help people to protect their eyes from the sun.
Does smoking influence the onset of AMD?
Cigarette smoking has been linked to increased risk of developing AMD. It is recommended that
persons stop smoking to decrease their chance of developing AMD.
What treatment options are available for AMD?
Retina International and its members support extensive multi-disciplinary research programmes
in an effort to find the causes prevention, and possible treatment for Macular Degeneration and
other forms of retinal degenerations. Researches studying retinal degenerative diseases may
contribute to the understanding of others so that, for example, research related to RP may also
benefit those with Macular Degeneration. Information from research projects is shared with
others in the field in order to advance the goal of understanding all forms of retinal degeneration.
Listed below are details of some of the latest advances in research and treatments for AMD.
Dry AMD
Although several new drugs are being investigated and approved by regulatory agencies around
the world for the treatment of the exudative (wet) type of AMD, aside from cessation of smoking
and a healthy diet of dark green leafy vegetables and fruits supplemented by zinc and anti-
oxidant vitamins (Vitamins E, C, and beta carotene), very little is available to help patients with
atrophic or "dry" AMD to prevent progression to more serious stages of debilitating disease. A
blood filtration process, called Rheopheresis, is being marketed in Canada as a treatment for dry
AMD by OccuLogix Inc. Little is known about what causes the conversion from dry to wet AMD,
and this is the subject of ongoing research studies. Check back regularly for updates to this
website as research studies are published.
Wet AMD
At present people with macular degeneration have three possible treatment options: thermal
(heat laser); Photodynamic Therapy; or anti-VEFG drugs.

Laser Photocoagulation
Laser photocoagulation is a surgical procedure involving the application of a hot laser to seal and
halt or slow the progression of abnormal blood vessels. In the 1990's laser treatment was the
only therapy available for AMD.
Through the use of a high-energy light that turns to heat when it hits the parts of the retina to be
treated, laser photocoagulation seals the choroidal neovascularization (CNV) and inhibits the
leaky blood vessels growth, preventing further vision deterioration. A scar forms as a result of the
treatment, and this scar creates a permanent blind spot in the field of vision. Vision does not
usually improve after laser treatment and may even be somewhat worse. However, loss of vision
following laser treatment, though immediate, is generally less severe than the eventual loss of
vision that usually occurs if laser treatment is not done. In many cases, some visual distortion will
disappear after laser treatment.
Photo Dynamic Therapy (PDT)
Photodynamic Therapy (PDT) (trade name Visudyne) uses a non-thermal (or cold) laser with an
intravenous light-sensitive drug to seal and halt or slow the progression of abnormal retina blood
vessels. This treatment does not produce a blind spot on the retina. The light is shone directly at
the targeted tissue and the drug accumulates in these cells. It therefore reduces damage to
normal surrounding tissue and allows the treatment to be given again as needed. . However,
early diagnosis of AMD is key, because once vision is lost due to of the growth of abnormal blood
vessels, it cannot be reclaimed by either treatment.
Anti-angiogenesis Therapies
As of February 2006, pegaptanib sodium (trade name Macugen) is approved for use in Canada,
the United States and Europe. The United States Food and Drug Administration (FDA) approved
Macugen for treatment of neovascular (wet) age-related macular degeneration. FDA approval
came following successful clinical trials demonstrating that the drug reduced vision loss in 70 per
cent of clinical trial patients. It is also very encouraging that the drug is effective for all kinds of
wet AMD, whether in the early or late stages.
Pegaptanib sodium (trade name Macugen) is what researchers call an anti-VEGF drug, or in
other words, a drug which works by targeting the proteins which act to trigger abnormal blood
vessel growth and leakage. Anti-VEGF drugs are delivered directly to the eye by an injection,
which is repeated every four to six weeks.
Other anti-VEFG drugs on the horizon include ranubizimab (trade name Lucentis), from
Genetech and Novartis. On June 30, 2006, the US Food and Drug Administration (FDA)
announced approval of Lucentis (Ranibizumab). AMD Alliance International (AMDAI) loudly
applauded the decision, which effectively makes available in the USA a ground breaking
treatment for wet age related macular degeneration. This approval is based on the evidence

presented from several years of rigorous clinical trials, in which Lucentis was shown to maintain
vision in 95% of trial participants, and improve vision in approximately 30 to 40% of trial
participants. This decision means that treatment with Lucentis will now be widely available in the
USA through retinal specialists. The FDA approval of course only covers the USA. Introduction of
Lucentis in Europe is expected to follow in the coming year. Fighting Blindness and the AMD
Alliance International of which it is a member, applauds the introduction of new treatments, which
bring hope and help to those with macular degeneration.
Angiostatic Therapies
In other research developments, a completely different class of AMD drugs, called angiostatic
therapies, is showing promise. This class of drugs propose yet another approach to treatment of
AMD, in this case by administering a type of steroid to stop the abnormal growth of blood vessels
in the eye. Unlike the anti-VEGF treatments, angiostatic drugs are delivered through a canula, to
the back of the eye.
One possible angiostatic treatment is anecortave acetate (Retaane), from Alcon Laboratories.
Although early clinical results were not as stellar as hoped, scientists working on the treatment
believe this may be a result of drug delivery problems, not the drug itself and are making
adjustments. On May 24, 2005, the USA Food and Drug Administration released what is called
an "approvable" letter, basically meaning that the drug is approvable but some further study is
required. Alcon recently reported that their researchers and officials will "meet with the FDA to
discuss the approvable letter, the clinical studies submitted with the NDA and other ongoing
clinical studies for RETAANE® suspension to determine the steps necessary to gain final
approval for the wet AMD indication." Retaane has received market approval for use in Australia.
In early March 2006, a request for market approval in Europe was withdrawn, by Alcon, from
regulatory consideration.
Combination Therapies
Other investigations are also showing promise, including combination therapies, which combine
traditional PDT therapy with new drugs to increase the effectiveness of PDT. Combination
treatments pair one or more existing or new AMD treatments to see if the end result might be
greater than what could be achieved individually. More and more medical practitioners believe
that combination methods are the way of the future for wet AMD treatment. Usually the idea is
that one kind of treatment will take care of existing AMD in the patient, and the other will help to
prevent any future developments.
Position Statement on Avastin (bevacizumab)
The role, efficacy, and safety of anti-vascular endothelial growth factor (VEGF) therapies for use
in the treatment of age-related macular degeneration (AMD) were first established by clinical

trials of pegaptanib sodium, (Macugen, [OSI] Eyetech/Pfizer) and later by clinical trials for
ranibizumab (Lucentis, Genentech, Inc.)
Pegaptanib Sodium
Phase 3 clinical trials for pegaptanib sodium demonstrated that after 1 year of treatment,
individuals who were treated with 0.3 mg and 1 mg pegaptanib sodium experienced less vision
loss than those who were treated with a placebo. Individuals who were treated with pegaptanib
sodium experienced lasting results for 2 years. The most common side effect (occurring in
approximately 1.3% of cases) was endophthalmitis, which was caused by the injection.
Ranibizumab
Phase 3 clinical trials for ranibizumab demonstrated superior results after 1 year of treatment,
and showed that the majority of individuals who were treated with ranibizumab improved or
maintained vision 2 years later. The improvement in visual acuity endpoints in the ranibizumab-
treated groups (0.3 mg and 0.5 mg) was maintained at year 2, while individuals in the control
group continued to experience vision deterioration. At 2 years, at least 90% of individuals who
were treated with ranibizumab maintained or improved vision compared to approximately 53% of
individuals who were treated with sham injections. Treatment side effects were mild to moderate,
affected less than 3% of individuals, and included conjunctival hemorrhage, increased IOP,
vitreous floaters, and endophthalmitis.
Broadening the Anti-VEGF Theory
Ranibizumab was developed by Genentech, Inc. The company had previously developed
bevacizumab (Avastin, Genentech, Inc.) an anti-VEGF drug that is currently approved by the
Food and Drug Administration (FDA) as an intravenous therapy for metastatic colorectal cancer
patients. Bevacizumab for use in cancer therapy is currently being investigated. Ranibizumab is
a molecular fragment of an antibody, and bevacizumab is a full-length antibody. They are both
thought to work by a similar principle - the drug blocks the production of VEGF. VEGF, which is
also produced by cancer cells, prompts the abnormal growth of blood vessels, also known as
angiogenesis. Bevacizumab binds with VEGF and interferes with its ability to stimulate blood
vessel growth.
In early 2004, Philip Rosenfeld, MD, PhD, and colleagues at the Bascom Palmer Eye Institute in
Miami, Fla, initiated the use of bevacizumab in the treatment of AMD. Their first study was called
Systemic Avastin for Neovascular AMD (SANA). In this and subsequent studies, which consisted
of intravitreal injections of bevacizumab, individuals who were clinically followed reported
improvements in visual acuity comparable to ranibizumab with no serious adverse events. It is
important to note that these clinical studies were not conducted as randomised clinical trials.4,5
Based on these results, the use of bevacizumab for the treatment of AMD appears to have been
broadly accepted by retinal specialists around the world.

The use of bevacizumab in the eyes, an indication for which it is not approved, is called off-label
use. It is reasoned conjecture on the part of the AMD Alliance International that the off-label use
of bevacizumab was first suggested for reasons of economy and availability in the face of a
significant unmet need. Until the June 30, 2006 FDA approval in the USA, treatment with
ranibizumab was not available unless an individual was registered in a clinical trial, or, as is
possible in some European countries, receives the treatment on what is called a 'named-patient'
basis.
Antioxidants and Vitamin Therapies
One working hypothesis is that a cause or contributing factor to Macular Degeneration involves
the formation of chemicals in the body called free radicals. Free radicals are thought to result, in
part, from exposure to sunlight and other forms of ultraviolet light. They cause cellular damage by
taking electrons from molecules in healthy cells. This process, called oxidation, has been linked
to a variety of health problems including heart disease and cancer. Substances called
antioxidants may counteract the oxidation process; the body produces its own antioxidants, and
these are helped by antioxidants that we ingest through food or vitamin supplements. Vitamins C,
E nd carotenoids, including beta-carotene, are examples of potent antioxidants. However, which
of these is helpful to AMD is not yet known.
The work investigating a link between vitamins and Macular Degeneration is still in preliminary
stages. Recommendations regarding nutritional supplementation and light avoidance for patients
with Macular Degeneration are expected to emerge from studies now in progress.
Can an eye transplant cure Macular Degeneration?
No. Medical technology is not yet advanced enough to transplant the entire eye. It is simply
impossible to reconnect the nerves leading from the eye to the brain. What you may have heard
referred to as an "eye transplant" is probably the process of corneal transplantation, which is a
valuable vision saving procedure for some people, but unfortunately has no relationship to the
problems in the eye caused by Macular Degeneration. However, retinal cell transplantation is a
procedure that may have promise for people with Macular Degeneration in the future, although it
is still in its early experimental stages Retinal cell transplantation is described below.
Retinal Cell Transplantation
Transplantation of retinal cells has shown some encouraging results in animals, although it is
important to emphasize that this is not yet a treatment available for use in humans. Retinal cell
transplantation is still in preliminary stages of investigation in the laboratory. Before a procedure
can be tested in humans, long-term beneficial effects must be proven and possible side effects
must be determined. Such research might take several more years.

The good news is that studies so far have found that when photo receptor cells are transplanted
into the retinas of animals, some features of normal photo receptors are either maintained or
develop after transplantation. However, there is not yet conclusive evidence that retinal cell
transplants or similar procedures in animals with a retinal degeneration result in long-term
improved or restored vision. Nevertheless, the research done so far has been promising enough
for the American Foundation Fighting Blindness to expand a grant award programme aimed at
scientists who are investigating several areas of basic science that could lead to new therapies
that might repair or replace damaged retinal cells.
Special challenges in AMD research
Because AMD does not develop until late in life, and all body tissues undergo changes
associated with ageing, it is difficult to determine which eye findings are normal in those over the
age of 50, and which may be predictive of AMD. The American Foundation, along with the
American National Eye Institute, is working to define normal ageing, classify AMD types, define
genetic components, define risk factors, develop new diagnostic techniques, analyse eye tissue
layers and how they interact, and develop animal models that imitate human AMD.
Gene Therapy
As researchers identify more of the mutant genes that contribute to Macular Degeneration, it
becomes possible to think about curing the defect at the most basic cellular level. Gene therapy
is what many scientists feel is the answer of the future for many forms of retinal degenerations. It
is based on a simple logic: if a gene is defective, replace it with one that is not defective. While
this may sound simple, the actual procedure of gene therapy is very complex.
Gene therapy might be described as a form of drug therapy in which the "good" gene itself is the
drug, which is introduced into the body to replace the "bad" gene. There are a number of reasons
that retinal degenerations are diseases that seem particularly suited to the use of gene therapy.
First and foremost, some of the defective genes for early onset inherited Macular Degeneration
have been identified. Also, there are a number of applicable animal models in which gene
therapy can be tested for effectiveness and safety. And the outcomes of gene therapy can be
tested by reliable and non-invasive visual examination of the retina. Finally, a treated eye can be
compared to an untreated eye in the same patient, giving researchers the ideal conditions for
conducting a controlled scientific experiment.
While all of the above factors make gene therapy a promising future approach for treating
Macular Degeneration, there are still many obstacles. One key question is how to actually
introduce the DNA of the good gene into diseased cells. Researchers have found that a
neutralized virus can act as a transporter of the gene to the degenerating photo receptor cells,
which seem to be particularly good targets for this type of gene transfer.

What assistance is available to help cope with AMD?
This information is helpful in learning how to physically cope with Macular Degeneration and
similar diseases. But what about emotional aspects? What assistance is available to help me and
my family cope with Macular Degeneration?
There are many devices and techniques that help people with Macular Degeneration maximize
the use of their remaining vision.
The white cane is probably the most visible aid, and some people with Macular Degeneration find
it a useful navigational aid if their vision loss progresses beyond a certain stage. The cane is a
form of non-optical aid; that is, it does not have an actual effect on your eyes, although it may
help you see or cope with vision loss. Other non-optical aids include guide dogs, audio tapes,
and large print books.
There are also electronic aids. These include closed-circuit televisions (CCTV), reading
machines, and talking computers, An increasing number of computer programmes are
addressing the needs of the visually impaired, making it possible to easily enlarge type on the
screen or provide an audio or Braille version to go with what is shown on the screen.
Optical aids are devices that work to improve your vision to some extent. These include Corning
and NOIR glasses, the Fresnel prism, telescopes and magnifiers. With advancing technology,
some of these devices are becoming increasingly sophisticated and offering new opportunities
for people with retinal degenerations to maximize their usable vision.
To determine which aids may be most useful for you, it is suggested that you get a thorough low
vision evaluation from a specialist, Contact the RP Foundation for more information on low vision
services in your area.
While research findings provide hope for the future, there is no actual treatment for people with
macular degeneration. Are there other ways that people with this condition can enhance the
quality of their lives? It is important that individuals and families know that there are resources
available to help them cope with the life-changing conditions Macular Degeneration may bring.
People with Macular Degenerations may rind it helpful to discuss their questions and concerns
with other people who have similar experiences. The RP Foundation Fighting Blindness can help
people get together to exchange information about these common issues in their lives and
explore possible solutions for some of their problems. You may also wish to speak with a mental
health professional to assist you and/or your family in dealing with the many changes that can be
related to Macular Degenerations.
How can I assist a person with AMD?
Age-related macular degeneration (AMD) is a difficult condition to understand, largely because
the eyes of a person with AMD look normal. The person with the conditions may still able to

manoeuvre around obstacles; can see a white piece of fluff on a dark carpet and yet we will walk
right by a neighbour or best friend without recognising them.
Those with AMD want people to understand that they are "visually impaired," not "clumsy,"
"standoffish," or "illiterate." They will sometimes fake "seeing" because it is easier than explaining
that they have no central vision. If a companion says, "Just look at that picture," many will reply,
"Oh yes!" rather than explaining, yet again, that they cannot see that kind of detail. Also it is
difficult to locate items for example, one could ask a family member 'Have you seen my umbrella'
answer 'yes, it's over there'. Now where is over there? The person may be pointing in the general
direction but the affected individual can not see where they are pointing so needs to a more
detailed explanation. Is it left, or is it right etc. So sometimes the entire family will need to be re-
educated.
Caregivers need to get as much information as they can about vision loss, and share that
information with the people they are caring for and their families. If you are a caregiver to
someone with any form of vision loss, we urge you to:
Become informed; learn as much as you can about the condition.
Be patient - adjusting to vision loss takes time.
Contact a member organization of Retina International.
Ensure the person you are caring for receives skills training and assessments for adaptive
tools.
Meet other people with the same condition.
Meet other caregivers.
When a person is diagnosed with AMD, it changes not only their own lives, but the lives of their
families and friends. Upon diagnosis painful emotions such as disbelief, panic, anger, and
frustration can be experienced by an individual and often family and caregivers are on the
receiving end of these feelings. What can really help an individual to come to terms with vision
loss is an increase in confidence and skill level - and that takes time.
As a carer it is important to be as informed as you can about the condition of the person
receiving care and educate their network of family and friends. Eventually, these steps will help
the affected individual feel confident, in control, and willing to accept help when needed without
feeling dependent on others.
By working together with caregivers and family members, those with AMD or any form of vision
loss can live with dignity, confidence, safety, and a strong feeling of self-worth.

http://idmgarut.wordpress.com/2009/02/02/referat-degenerasi-makula/
Referat : DEGENERASI MAKULA Posted on Februari 2, 2009 by idmgarut
PENDAHULUAN
Degenerasi macula adalah suatu keadaan dimana macula mengalami kemunduran sehingga terjadi penurunan
ketajaman penglihatan dan kemungkinan akan menyebabkan hilangnya fungsi penglihatan sentral. Macula
adalah pusat dari retina dan merupakan bagian yang paling vital dari retina yang memungkinkan mata melihat
detil-detil halus pada pusat lapang pandang. Tanda utama dari degenerasi pada makula adalah didapatkan
adanya bintik-bintik abu-abu atau hitam pada pusat lapangan pandang. Kondisi ini biasanya berkembang secara
perlahan-lahan, tetapi kadang berkembang secara progresif, sehingga menyebabkan kehilangan penglihatan
yang sangat berat pada satu atau kedua bola mata.(1,2)
Berdasarkan American Academy of Oftalmology penyebab utama penurunan penglihatan atau kebutaan di AS
yaitu umur yang lebih dari 50 tahun. Data di Amerika Serikat menunjukkan, 15 persen penduduk usia 75 tahun
ke atas mengalami degenerasi makula itu. Terdapat 2 jenis tipe dasar dari penyakit-penyakit tersebut yakni
Standar Macular Degeneration dan Age Related Macular Degeneration (ARMD). Bentuk yang paling sering
terjadi adalah ARMD.(3,4)
Degenerasi makula terkait usia merupakan kondisi generatif pada makula atau pusat retina. Terdapat 2 macam
degenarasi makula yaitu tipe kering (atrofik) dan tipe basah (eksudatif). Kedua jenis degenerasi tersebut
biasanya mengenai kedua mata secara bersamaan. Degenerasi makula terjadi sebagai akibat dari kerusakan
pada epitel pigmen retina.(1,2,3,4)
Degenerasi makula menyebabkan kerusakan penglihatan yang berat (misalnya kehilangan kemampuan untuk
membaca dan mengemudi) tetapi jarang menyebabkan kebutaan total. Penglihatan pada tepi luar dari lapang
pandang dan kemampuan untuk melihat biasanya tidak terpengaruh, yang terkena hanya penglihatan pada
pusat lapang pandang. Gejala klinis biasa ditandai terjadinya kehilangan fungsi penglihatan secara tiba-tiba
ataupun secara perlahan tanpa rasa nyeri. Kadang gejala awalnya berupa gangguan penglihatan pada salah
satu mata, dinilai garis yang sesungguhnya lurus terlihar bergelombang.(1,3)
Diagnosis dapat ditegakkan berdasarkan gejala klinis dan hasil pemeriksaan mata. Sejauh ini belum ada terapi
untuk degenerasi makula tipe kering. Suplemen seng hanya mampu membantu memperlambat progresivitas
gangguan. Untuk beberapa kasus basah, terapi laser bisa membersihkan pembuluh darah abnormal sehingga
kekaburan penglihatan dapat dicegah. Tetapi, tidak semua kasus bisa diatasi dengan terapi laser. Saat ini
sedang dikembangkan berbagai obat dan prosedur operasi baru antara lain terapi foto dinamik.(2,3,4,5)
Faktor resiko gangguan ini selain karena usia tua, juga riwayat keluarga (genetik), ras kaukasia serta merokok.
(1,5)
II. ANATOMI DAN FISIOLOGI RETINA
Anatomi Retina
Retina atau selaput jala merupakan bagian mata yang mengandung reseptor yang menerima rangsang cahaya.
Retina berbatas dengan koroid dengan sel epitel pigmen retina dan terdiri atas lapisan : (6,7)
1. Lapisan epitel pigmen
2. Lapisan fotoreseptor merupakan lesi terluar retina terdiri atas sel batang yang mempunyai bentuk ramping,

dan sel kerucut.
3. Membran limitan eksterna yang merupakan membrane ilusi.
4. Lapisan nucleus luar, merupakan susunan lapis nucleus sel kerucut dan batang.
5. Lapisan pleksiform luar merupakan lapis aselular dan merupakan tempat sinapsis sel fotoreseptor dengan sel
bipolar dan sel horizontal.
6. Lapis nucleus dalam, merupakan tubuh sel bipolar, sel horizontal dan sel Muller.
7. Lapisan pleksiform dalam, merupakan lapis aselular merupakan tempat sinaps sel bipolar, sel amakrin dengan
sel ganglion.
8. Lapis sel ganglion yang merupakan lapis badan sel daripada neuron kedua,
9. Lapis serabut saraf, merupakan lapis akson sel ganglion menuju kearah saraf optic.
10. Membran limitan interna, merupakan membrane hialin antara retina dan badan kecil.
Retina adalah selembar tipis jaringan saraf yang semitransparan, dan multilapis yang melapisi bagian dalam dua
per tiga posterior dinding bola mata. Retina membentang ke depan hampir sama jauhnya dengan korpus siliare,
dan akhirnya di tepi ora serrata. Pada orang dewasa, ora serrata berada sekitar 6,5 mm di belakang garis
Schwalbe pada system temporal dan 5,7 mm di belakang garis ini pada sisi nasal. Permukaan luar retina
sensorik bertumpuk dengan membran Bruch, khoroid, dan sclera. Retina menpunyai tebal 0,1 mm pada ora
serrata dan 0.23 mm pada kutub posterior. Ditengah-tengah retina posterior terdapat makula. Di tengah makula
terdapat fovea yang secara klinis merupakan cekungan yang memberikan pantulan khusus bila dilihat dengan
oftalmoskop.(3,8)
Retina menerima darah dari dua sumber : khoriokapiler yang berada tepat di luar membrana Bruch, yang
mendarahi sepertiga luar retina, termasuk lapisan pleksiformis luar dan lapisan inti luar, fotoreseptor, dan lapisan
epitel pigmen retina, serta cabang-cabang dari arteri retina sentralis yang memperdarahi dua per tiga sebelah
dalam. (6,7)
Fisiologi Retina
Untuk melihat, mata harus berfungsi sebagai suatu alat optis, sebagai suatu reseptor kompleks, dan sebagai
suatu transducer yang efektif. Sel-sel batang dan kerucut di lapisan fotoreseptor mampu mengubah rangsangan
cahaya menjadi suatu impuls saraf yang dihantarkan oleh lapisan serat saraf retina melalui saraf optikus dan
akhirnya ke korteks penglihatan. Makula bertanggung jawab untuk ketajaman penglihatan yang terbaik dan untuk
penglihatan warna, dan sebagian besar selnya adalah sel kerucut. Di fovea sentralis, terdapat hubungan hampir
1:1 antara fotoreseptor kerucut, sel ganglionnya, dan serat saraf yang keluar, dan hal ini menjamin penglihatan
yang paling tajam. Di retina perifer, banyak fotoreseptor dihubungkan ke sel ganglion yang sama, dan diperlukan
sistem pemancar yang lebih kompleks. Akibat dari susunan seperti itu adalah bahwa makula terutama digunakan
untuk penglihatan sentral dan warna ( penglihatan fototopik) sedangkan bagian retina lainnya, yang sebagian
besar terdiri dari fotoreseptor batang, digunakan terutama untuk penglihatan perifer dan malam (skotopik). (6,7)
Fotoreseptor kerucut dan batang terletak di lapisan terluar yang avaskuler pada retina sensorik dan merupakan
tempat berlangsungnya reaksi kimia yang mencetuskan proses penglihatan. Setiap sel fotoreseptor kerucut
mengandung redopsin, yang merupakan suatu pigmen penglihatan fotosensitif yang terbentuk sewaktu molekul
protein opsin bergabung dengan 11-sis-retinal. Sewaktu foton cahaya diserap oleh rodopsin, 11-sis-retinal
segera mengalami isomerisasi menjadi bentuk ali-trans. Redopsin adalah suatu glikolipid membran yang
separuh terbenam di lempeng membram lapis ganda pada segmen paling luar fotoreseptor. Penyerapan cahaya
puncak oleh terjadi pada panjang gelombang sekitar 500 nm, yang terletak di daerah biru-hijau pada spektrum
cahaya. Penelitian-penelitian sensitivitas spektrum fotopigmen kerucut memperlihatkan puncak penyerapan
panjang gelombang di 430, 540, dan 575 nm masing-masing untuk sel kerucut peka-biru, -hijau, dan –merah.
Fotopigmen sel kerucut terdiri dari 11-sis-retinal yang terikat ke berbagai protein opsin. (7)
Penglihatan skotopik seluruhnya diperantarai oleh fotoreseptor sel batang. Pada bentuk penglihatan adaptasi
gelap ini, terlihat bermacam-macam nuansa abu-abu, tetapi warna tidak dapat dibedakan. Sewaktu retina telah
beradaptasi penuh terhadap cahaya, sensitivitas spektral retina bergeser dari puncak dominasi rodopsin 500 nm
ke sekitar 560 nm, dan muncul sensasi warna. Suatu benda akan berwarna apabila benda tersebut mengandung
fotopigmen yang menyerap panjang-panjang gelombang dan secara selektif memantulkan atau menyalurkan
panjang-panjang gelombang tertentu di dalam spektrum sinar tampak (400-700 nm). Penglihatan siang hari

terutama diperantarai oleh fotoreseptor kerucut, senjakala oleh kombinasi sel kerucut dan batang, dan
penglihatan malam oleh fotoreseptor batang.(7)
III. PATOFISIOLOGI
Degenerasi makula yang terkait usia tipe kering ditandai oleh adanya atrofi dan degenerasi retina bagian luar,
epitel pigmen retina, membran Bruch, dan koriokapilaris dengan derajat yang bervariasi. Dari perubahan-
perubahan di epitel pigmen retina dan membran Bruch yang dapat dilihat secara oftalmoskopi adalah drusen
yang sangat khas. Drusen adalah endapan putih kuning, bulat, diskret, dengan ukuran bervariasi di belakang
epitel pigmen dan tersebar di seluruh makula dan kutub posterior. Seiring dengan waktu, drusen dapat
membesar, menyatu, mengalami kalsifikasi dan meningkat jumlahnya. Secara histopatologis sebagian besar
drusen terdiri dari kumpulan lokal bahan eosinifilik yang terletak di antara epitel pigmen dan membran Bruch;
drusen mencerminkan pelepasan fokal epitel pigmen.(4,7,9)
Walaupun pasien dengan degenerasi makula biasanya hanya memperlihatkan kelainan non eksudatif, sebagian
besar pasien yang menderita gangguan penglihatan berat akibat penyakit ini mengalami bentuk eksudatif akibat
terbentuknya neovaskularisasi subretina dan makulopati eksudatif terkait. Cairan serosa dari koroid di bawahnya
dapat bocor melalui defek defek kecil di membran Bruch sehingga mengakibatkan pelepasan-pelepasan lokal
epitel pigmen. Peningkatan cairan tersebut dapat semakin menarik retina sensorik di bawahnya dan penglihatan
biasanya menurun apabila fovea terkena. Pelepasan epitel pigmen retina dapat secara spontan menjadi datar
dengan bermacam-macam akibat penglihatan dan meninggalkan daerah geografik depigmentasi pada daerah
yang terkena. Dapat terjadi pertumbuhan pemubulu-pembuluh darah baru ke arah dalam yang meluas ke koroid
sampai ruang subretina dan merupakan perubahan histopatologik terpenting yang memudahkan timbulnya
pelepasan makula dan gangguan penglihatan sentral yang bersifat ireversivel pada pasien dengan drusen.
Pembuluh pembuluh darah ini akan tumbuh dalam konfigurasi roda-roda pedati datar atau sea-fan menjauhi
tempat masuk ke dalam ruang sub retina.(4,7,9)
IV. ETIOLOGI
Degenerasi macula dapat disebabkan oleh beberapa factor dan dapat diperberat oleh beberapa factor resiko,
diantaranya : (6,7,8,9)
1. Umur, faktor resiko yang paling berperan pada terjadinya degenerasi makula adalah umur. Meskipun
degenerasi makula dapat terjadi pada orang muda, penelitian menunjukkan bahwa umur di atas 60 tahun
beresiko lebih besar terjadi di banding dengan orang muda. 2% saja yang dapat menderita degenerasi makula
pada orang muda, tapi resiko ini meningkat 30% pada orang yang berusia di atas 70 tahun.
2. Genetik, penyebab kerusakan makula adalah CFH, gen yang telah bermutasi atau faktor komplemen H yang
dapat dibawa oleh para keturunan penderita penyakit ini. CFH terkait dengan bagian dari sistem kekebalan tubuh
yang meregulasi peradangan.
3. Merokok, Merokok dapat meningkatkan terjadinya degenrasi makula.
4. Ras kulit putih (kaukasia) adalah sangat rentan terjadinya degenerasi makula di banding dengan orang Afrika
atau yang berkulit hitam.
5. Riwayat keluarga, resiko seumur hidup terhadap pertumbuhan degenerasi makula adalah 50% pada orang-
orang yang mempunyai hubungan keluarga penderita dengan degenerasi makula, dan hanya 12 % pada mereka
yang tidak memiliki hubungan dengan degenerasi makula.
6. Hipertensi dan diabetes.
Degenerasi Makula menyerang para penderita penyakit diabetes, atau tekanan darah tinggi gara-gara mudah
pecahnya pembuluh-pembuluh darah kecil (trombosis) sekitar retina. Trombosis mudah terjadi akibat
penggumpalan sel-sel darah merah dan penebalan pembuluh darah halus.
7. Paparan terhadap sinar Ultraviolet
8. Obesitas dan kadar kolesterol tinggi
V. KLASIFIKASI
1. Degenerasi Makula tipe non-eksudatif (tipe kering)
Rata-rata 90% kasus degenerasi makula terkait usia adalah tipe kering. Kebanyakan kasus ini bisa memberikan
efek berupa kehilangan penglihatan yang sedang. Tipe ini bersifat multipel, kecil, bulat, bintik putih kekuningan
yang di sebut drusen dan merupakan kunci identifikasi untuk tipe kering. Bintik tersebut berlokasi di belakang
mata pada level retina bagian luar. Adapun lesi klasik yang bisa ditemukan adanya atrofi geografik. Terdapat

endapan pigmen di dalam retina tanpa disertai pembentukan jaringan parut , darah atau perembesan cairan.
(11,12,13,14)
Degenerasi makula terkait usia noneksudatif ditandai oleh atrofi dan degenerasi retina bagian luar, epitel pigmen
retina, membran Bruch, dan koriokapilaris dengan derajat yang bervariasi. Dari perubahan-perubahan di epitel
pigmen retina dan membran Bruch yang dapat dilihat secara oftalmoskopis, drusen adalah yang paling khas.
Drusen adalah endapan putih kuning, bulat, diskret, dengan ukuran bervariasi di belakang epitel pigmen dan
tersebar di seluruh makula dan kutub posterior. Seiring dengan waktu, drusen dapat membesar, menyatu,
mengalami kalsifikasi dan meningkat jumlahnya. Secara histopatologis sebagian besar drusen terdiri dari
kumpulan lokal bahan eosinifilik yang terletak di antara epitel pigmen dan membran Bruch; drusen
mencerminkan pelepasan fokal epitel pigmen.(7,10)
Drusen dapat di bagi berdasarkan klinik dan histopatologi yakni drusen keras ( nodular), drusen diffus
( konfluent), drusen halus ( granular ), dan drusen kalsifikasi . Selain drusen, dapat muncul secara progresif
gumpalan-gumpalan pigmen yang tersebar secara tidak merata di daerah-daerah depigmentasi atrofi di seluruh
makula.(7,10)
2. Degenerasi Makula tipe eksudatif ( tipe basah)
Degenerasi makula tipe ini adalah jarang terjadi namun lebih berbahaya di bandingkan dengan tipe kering. Kira
kira didapatkan adanya 10% dari semua degenerasi makula terkait usia dan 90% dapat menyebabkan kebutaan.
Tipe ini ditandai dengan adanya neovaskularisasi subretina dengan tanda-tanda degenerasi makula terkait usia
yang mendada atau baru mengalami gangguan penglihatan sentral termasuk penglihatan kabur, distorsi atau
suatu skotoma baru. Pada pemeriksaan fundus, terlihat darah subretina, eksudat, lesi koroid hijau abu-abu di
makula. Neovaskularisasi koroid merupakan perkembangan abnormal dari pembuluh darah pada epitel pigmen
retina pada lapisan retina. Pembuluh darah ini bisa mengalami perdarahan dan menyebabkan terjadinya scar
yang dapat menghasilkan kehilangan pusat penglihatan. Scar ini disebut dengan Scar Disciform dan biasanya
terletak di bagian sentral dan menimbulkan gangguan penglihatan sentral permanen.(4,7,11,15,16)
VI. GEJALA KLINIS
Gejala-gejala klinik yang biasa didapatkan pada penderita degenerasi makula antara lain : (1,4,8,9)
1. Distorsi penglihatan, obyek-obyek terlihat salah ukuran atau bentuk
2. Garis-garis lurus mengalami distorsi (membengkok) terutama dibagian pusat penglihatan
3. Kehilangan kemampuan membedakan warna dengan jelas
4. Ada daerah kosong atau gelap di pusat penglihatan
5. Kesulitan membaca, kata-kata terlihat kabur atau berbayang
6. Secara tiba-tiba ataupun secara perlahan akan terjadi kehilangan fungsi penglihatan tanpa rasa nyeri.
VII. DIAGNOSIS
Diagnosis dapat ditegakkan berdasarkan gejala klinik dan hasil pemeriksaan oftalmoskopi yang mencakup ruang
lingkup pemeriksaan sebagai berikut : (1,2,3,4,5)
1. Test Amsler Grid, dimana pasien diminta suatu halaman uji yang mirip dengan kertas milimeter grafis untuk
memeriksa luar titik yang terganggu fungsi penglihatannya. Kemudian retina diteropong melalui lampu senter
kecil dengan lensa khusus.
2. Test penglihatan warna, untuk melihat apakah penderita masih dapat membedakan warna, dan tes-tes lain
untuk menemukan keadaan yang dapat menyebabkan kerusakan pada makula.
3. Kadang-kadang dilakukan angiografi dengan zat warna fluoresein. Dokter spesialis mata menyuntikan zat
warna kontras ini ke lengan penderita yang kemudian akan mengalir ke mata dan dilakukan pemotretan retina
dan makula. Zat warna ini memungkinkan melihat kelainan pembuluh darah dengan lebih jelas.
e
VIII. DIAGNOSIS BANDING
Degenerasi macula khususnya tipe eksudat dapat di diagnosis banding dengan: (4)
1. Makroneurisme
2. Vaskulopati koroid polipoid
3. Khorioretinopati serous sentral
4. Kasus inflamasi
5. Tumor kecil seperti melanoma koroid

IX. PENATALAKSANAAN
Tidak ada terapi khusus untuk AMD noneksudatif Penglihatan dimaksimalkan dengan alat bantu penglihatan
termasuk alat pembesar dan teleskop. Pasien diyakinkan bahwa meski penglihatan sentral menghilang, penyakit
ini tidak menyebabkan hilangnya penglihatan perifer. Ini penting karena banyak pasien takut mereka akan
menjadi buta total. (2,4,8,9)
Pada sebagian kecil pasien dengan AMD eksudatif yang pada angiogram fluorosen memperlihatkan membrane
neovaskular subretina yang terletak eksentrik (tidak sepusat) terhadap fovea, mungkin dapat dilakukan obliterasi
membrane tersebut dengan terapi laser argon. Membrane vascular subfovea dapat diobliterasi dengan terapi
fotodinamik (PDT) karena laser argon konvensional akan merusak fotoreseptor di atasnya. PDT dilakukan
dengan menyuntikkan secara intravena bahan kimia serupa porfirin yang diaktivasi oleh sinar laser nontermal
saat sinar laser berjalan melalui pembuluh darah di membrane subfovea. Molekul yang teraktivasi
menghancurkan pembuluh darah namun tidak merusak fotoreseptor. Sayangnya kondisi ini dapat terjadi kembali
bahkan setelah terapi laser. (2,4,8,9)
Apabila tidak ada neovaskularisasi retina, tidak ada terapi medis atau bedah untuk pelepasan epitel pigmen
retina serosa yang terbukti bermanfaat. Pemakaian interferon alfa parenteral, misalnya, belum terbukti efektif
untuk penyakit ini. Namun apabila terdapat membrane neovaskular subretina ekstrafovea yang berbatas tegas (?
200 um dari bagian tengah zona avaskular fovea), diindikasikan fotokoagulasi laser. Dengan angiografi dapat
ditentukan dengan tepat lokasi dan batas-batas membrane neovaskular yang kemudian diablasi secara total oleh
luka-luka bakar yang ditimbulkan oleh laser. Fotokoagulasi juga menghancurkan retina di atasnya tetapi
bermanfaat apabila membrane subretina dapat dihentikan tanpa mengenai fovea. (4,8)
Fotokoagulasi laser krypton terhadap neovaskularisasi subretina avaskular fovea (? 200 um dari bagian tengah
zona avaskular fovea) dianjurkan untuk pasien nonhipertensif. Setelah fotokoagulasi membrane neovaskular
subretina berhasil dilakukan, neovaskularisasi rekuren di dekat atau jauh dari jaringan parut laser dapat dapat
terjadi pada separuh kasus dalam 2 tahun. Rekurensi sering disertai penurunan penglihatan berat sehingga
pemantauan yang cermat dengan Amsler grid, oftalmoskopi dan angiografi perlu dilakukan. Pasien dengan
gangguan penglihatan sentral di kedua matanya mungkin memperoleh manfaat dari pemakaian berbagai alat
bantu penglihatan kurang. (4,8)
Selain itu terapi juga dapat dilakukan di rumah berupa pembatasan kegiatan dan follow up pasien dengan
mengevaluasi daya penglihatan yang rendah. Selain itu dengan mengkomsumsi multivitamin dan antioksidan
( berupa vitamin E , vitamin C, beta caroten, asam cupric dan zinc), karena diduga dapat memperbaiki dan
mencegah terjadinya degenerasi makula. Sayuran hijau terbukti bisa mencegah terjadinya degenerasi makula
tipe kering. Selain itu kebiasaan merokok dikurangi dan dan pembatasn hipertensi.(4,15)
X. PROGNOSIS
Bentuk degenerasi makula yang progresif dapat menyebakan kebutaan total sehingga aktivitas dapat menurun.
Prognosis dari degenerasi makula dengan tipe eksudat lebih buruk di banding dengan degenerasi makula tipe
non eksudat. Prognosis dapat didasarkan pada terapi, tetapi belum ada terapi yang bernilai efektif sehingga
kemungkinan untuk sembuh total sangat kecil.(10)
DAFTAR PUSTAKA
1. Degenerasi Makula. Medicastore Online.http://www.medicastore.com/med/detail_pyk.php?
id=&iddtl=983&idktg=16&idobat=&UID=20070306192649125.162.255.115
2. Macular Degeneration. [ Online ]. [ Cited on 2007, Januari 17th ].Available from : URL: http://en.wikipedia.org/.
3. Degenerasi Makula. IDI Online-Iptek Kedokteran. http://www.idionline.org/iptek-isi.php?news_id=623
4. Liesegang TJ., Skuta GL., Cantor LB,. Retina and Vitreous. Basic and Clinical Course.Section 12 . San
Fransisco, California : American Academy of Ophthalmology. 2003-2004.
5. Degenerasi Makula. IDI Online-Iptek Kedokteran. http://www.idionline.org/iptek-isi.php?news_id=623
6. Sidarta I,. Anatomi dan Fisiologi Mata. Dalam : Ilmu Penyakit Mata Edisi kedua. Jakarta : BP-FKUI. 2002
7. Hardy RA,. Retina dan Tumor Intraokuler. Dalam : Vaughan D.G, Asbury T., Riordan E.P, Editor. Oftalmologi
Umum Edisi 14. Jakarta : Widya Medika. 2000.
8. Degenerasi Makula dan Mata Anda. Klinik Mata Nusantara
Online.http://www.klinikmatanusantara.com/degenerasi.php (diakses tanggal 6 Maret 2007)

9. James C., Chew C., Bron A. Retina dan Koroid. Dalam : Oftalmologi Edisi Kesembilan. Yakarta : Penerbit
Erlangga. 2006
10. Maturi R.K,. Aging Relation Macular Degeneration. [Online]. [ Cited on 2007, Januari 17th ]. Available from :
URL: http://www.emedicine.com/.
11. Macular Degeneration. [Online]. [ Cited on 2007, Januari 17th ]. Available from :
URL: http://www.stlukeseye.com/. In : Eye Condition.
12. Age-Related Macular Degeneration. [Online]. [ Cited on 2007, Januari 17th ]. Available from :
URL: http://www.nationaleyeinstitute.com/
13. Macular Degeneration. [Online]. [ Cited on 2007, Januari 17th ]. Available from :
URL:http://www.eyemdlink.com/.
14. Cure Macular Degeneration-Prevent Age Related Macular Degeneration. [Online].[Cited on 2007, Januari
15th]. Available from : URL:http://www.nei.nih.gov/health.
15. Macular Degeneration. [Online]. [ Cited on 2007, Januari 17th ] .Available from :
URL: http://www.emedicinehealth.com/.
16. Sarks SH, Killingsworth MC, Penfold PH, Driei DV,. Patterns in Macular Degeneration. In: Ryan SJ., Dawson
AK., Little HL,Editor. Retinal Diseases. 5th Edition. New York : Grune & Stratton, Inc. 1985.
17. Image of Eye, Retina, and Laser Theraphy of Macular Degeneration. [Online].[ Cited on 2007, Januari 17th ].
Available from:URL: http://www.google.com/.
