purification of the yeast centromere binding protein … · (32 ml), diluted with 16 ml of btp...
TRANSCRIPT

THE JOURNAL OF BIOLOGICAL CHEMISTRY 0 1989 by The American Society for Biochemistry and Molecular Biology, Inc
Vol. 264, No. 18, Issue of June 25, pp. 10843-10850,1989 Printed in U. S.A.
Purification of the Yeast Centromere Binding Protein CP1 and a Mutational Analysis of Its Binding Site*
(Received for publication, February 8, 1989)
Richard E. Baker, Molly Fitzgerald-Hayes$, and Timothy C. O’Brien From the Department of Molecular Genetics and Microbiology, University of Massachusetts Medical School, Worcester, Massachusetts 01655 and the $Department of Biochemistry, Program in Molecular and Cellular Biology, University of Massachusetts, Amherst, Massachusetts 01003
CP1 is a yeast protein which binds to the highly - conserved DNA clement I (CDEI) of yeast centromeres. We have purified CP1 to near homogeneity; it is com- prised of a single polypeptide of molecular weight 58,400. When bound to yeast CEN3 DNA, CP1 protects a 12-15-base pair region centered over CDEI. Meth- ylation interference experiments show that methyla- tions of residues located outside of the 8-base pair CDEI sequence have no detectable effect on CP1 bind- ing, suggesting that the DNA sequences important for CP1 recognition are confined to the CDEI octanucleo- tide. The equilibrium constant for CP1 binding to CEN3 DNA is relatively low, 3 X 10’ M-’. Using a novel method to determine relative DNA binding con- stants, we analyzed the effect of CDEI mutations on CP1 binding. A C to T point mutation at position 5 (CO1) reduces the equilibrium constant about 35-fold, while the insertion of an additional T at this position (CAT) reduces the equilibrium constant 1,400-fold. The effect of these mutations on mitotic centromere function in vivo was assessed using a plasmid stability assay. While the CO1 mutation had a slight effect, the CAT mutation significantly impaired function, imply- ing that CP1 binding is required for the optimal mitotic function of yeast centromeres.
The faithful segregation of chromosomes during mitosis and meiosis requires the attachment of each chromosome to the spindle apparatus. The connections between chromosome and spindle microtubules are made at the kinetochore, a distinct organelle found associated with the centromere of each chromosome (Rieder, 1982). Evidently, the centromeric DNA contains specific information, either in sequence or structure, to signal the precise location and initiate the assem- bly of the kinetochore. To investigate the structure and func- tion of centromeres and kinetochores, we have attempted to identify proteins which specifically bind to the centromeric DNA of yeast (Saccharomyces cereuisiae). Yeast is an ideal organism for such a study. Centromeres from 12 of the 16 yeast chromosomes have been cloned and sequenced, and sensitive genetic methods exist for analyzing centromere func- tion (reviewed by Fitzgerald-Hayes, 1987).
Yeast centromeres share three highly conserved DNA se- quence elements, designated CDEI, CDEII, and CDEIII (Hie- ter et al., 1985). CDEI is the octanucleotide RTCACRTG (R
* This work was supported by Grant 1554-C-1 from the American
of Health Grant GM38566. The costs of publication of this article Cancer Society, Massachusetts Chapter and by National Institutes
were defrayed in part by the payment of page charges. This article must therefore be hereby marked “aduertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
= purine), CDEII consists of 78-86 bp’ of highly (>go%) AT- rich DNA, and CDEIII is a 25-bp domain containing dyad symmetry. Together, these three elements comprise the cen- tromere core, and the presence of this 120-bp sequence is sufficient for complete centromere activity in vivo (Hegemann et al., 1988). Through genetic analysis, it has been possible to precisely identify those DNA sequences within the centromere core which are important for proper function. CDEIII is absolutely essential; point mutations within CDEIII inacti- vate the centromere (McGrew et al., 1986; Ng and Carbon, 1987; Hegemann et al., 1988). Point mutations in CDEI impair but do not abolish function (Hegemann et al., 1988). CDEII seems to play a spacing role. Small insertions or deletions are tolerated, but major alterations in the length or AT content of CDEII are detrimental to function (Gaudet and Fitzgerald- Hayes, 1987).
The highly conserved nature of the CDEI and CDEIII sequences and their functional importance strongly suggests that they are sites of interaction for sequence-specific DNA binding proteins. Proteins definitely appear to be bound there in uiuo, because nuclease sensitivity studies of yeast chromatin reveal that the centromeric core is highly resistant to digestion (Bloom and Carbon, 1982). Two groups have identified a yeast protein which binds specifically to CDEIII in vitro (Hegemann et al., 1986; Ng and Carbon, 1987). This protein appears to be present in very low amounts, at least in nuclear extracts, and no biochemical characterization or purification has been re- ported. Bram and Kornberg (1987) have identified a protein that binds in the CDEI region of yeast centromeres. Interest- ingly, this protein was originally discovered in footprinting studies with the yeast GAL2 gene. An unexpected site of protection in the GAL2 gene regulatory region was found to contain a perfect CDEI sequence, and the protein that bound to it, named CP1, was subsequently shown to bind to several yeast centromere DNAs. CP1 is heat-stable and relatively abundant; assays of eluates from SDS gels suggest a molecular weight of 57,000-64,000 (Bram and Kornberg, 1987).
We report here the complete purification of CP1. We show that CP1 recognizes CDEI, and we have measured the equi- librium constant for its binding to wild type and mutant CDEI sequences. In addition, we have analyzed the effects of the CDEI mutations on centromere function in uiuo. Our results suggest that CP1 binding, while not absolutely essen- tial, is required for the optimum mitotic function of centro- meres in yeast.
The abbreviations used are: bp, base pair(s); Hepes, 4-(2-hydrox- yethy1)-1-piperazineethanesulfonic acid BTP, 1,3-bis[tris(hydroxy- methyl)methylamino]propane; MES, 2-(N-morpho1ino)ethane- sulfonic acid SDS, sodium dodecyl sulfate.
10843

10844 Yeast Centromere Binding Protein
EXPERIMENTAL PROCEDURES Materials-Sephacryl S-200, DEAE-Sephadex A-25, Sephacryl S-
200 HR, Mono S, and Mono Q chromotography matrices were pur- chased from Pharmacia LKB Biotechnology Inc., phosphocellulose P-11 from Whatman, and restriction enzymes, Klenow polymerase, T4 DNA ligase, and poly[d(I-C)] from Boehringer Mannheim. T4 polynucleotide kinase was obtained from Bethesda Research Labo- ratories. Radiolabeled compounds were purchased from Amersham Corp. and Du Pont-New England Nuclear. All chemicals were of reagent grade or better.
Plasmids-Plasmid pRB45-1 contains a 624-bp Sau3A fragment carrying yeast CENB (Fitzgerald-Hayes et al., 1982) inserted at the BarnHI site of pUC8. Plasmid pRB45-2 is analogous except that it contains two copies of the Sau3A fragment as a direct tandem repeat; the insert is oriented with the upstream side of CEN3 next to the HindIII site of the polylinker. Plasmid pRB59 contains the 564-bp HindIIIISalI fragment of plasmid dl314 (McGrew et al., 1986) inserted between the HindIII and Sal1 sites of pUC8. This insert contains 211 bp of CEN3 along with pBR322 sequences. Plasmids pRB60, pRB61, and pRB73 are identical to pRB59 except they contain the CDEI mutations CO1, CAT, and ACDEI, respectively. These mutations are described by Gaudet and Fitzgerald-Hayes (1989), and their sequences are given in Fig. 4. Plasmids pUC(TRP/CEN3), pUC(TRP/COl), pUC(TRP/CAT), pUC(TRP/ACDEI), and pUC(TRP) were derived from pRB59, pRB60, pRB61, pRB73, and pUC8, respectively, by inserting the 1.45-kilobase pair yeast TRPlIARSl fragment (Tschumper and Carbon, 1980) into their EcoRI sites. In each case, the TRPl gene is oriented such that transcription proceeds away from the CEN segment.
Purification of CPl -All procedures were conducted at 4 "C except fast protein liquid chromatography which was carried out at room temperature. CP1 activity was detected using gel retardation assays (below). Material for the binding experiments of Figs. 1, 5, 7, and 8 and Table I was prepared exactly as described by Baker et al. (1986). Under these conditions, CP1 elutes from the phosphocellulose column at 300 mM KC1 and from the DEAE-Sephadex column at 190 mM KCl.
The material used for footprinting (Fig. 6) was obtained as follows: Phosphocellulose-purified CP1 (50 ml) was diluted with an equal volume of buffer F (20 mM Hepes (pH 7.9), 0.1 mM EDTA, 0.02% Nonidet P-40, 0.5 mM dithiothreitol, 0.5 mM phenylmethylsulfonyl fluoride, 20% glycerol) and applied at 1.0 ml/min to a Mono Q 5/5 column previously equilibrated with buffer F containing 0.15 M KC1. CP1 was eluted at 0.5 ml/min using a 20-ml linear gradient of buffer F increasing in KC1 concentration from 0.15 to 0.50 M. The CP1- containing fractions (eluting at 0.31 mM KCI) were pooled and applied to a 1.5 X 35-cm column of Sephacryl S-200 HR, equilibrated, and run with buffer G containing 0.10 M KC]. Buffer G is the same as buffer F except it contains only 10% glycerol. Fractions eluting near the void volume contained the CPl and were pooled and passed over a 2-ml column of Affi-Gel-heparin (Bio-Rad) equilibrated with buffer F/0.10 M KCI. The flow through fractions, which contained the CP1, were pooled and rechromatographed on Mono Q exactly as before. The final CP1 preparation was greater than 75% pure as judged by SDS-gel electrophoresis.
Ultimately, CP1 was purified as follows: The yeast strain employed was S. cerevisiae strain ZA521 (pep4) , kindly provided by Dr. Vivian MacKay of Zymogenetics, Inc. (Seattle, WA). 175 g of cells, harvested at late log phase, were broken in three batches in a Bead Beater (Biospec Products) as described by Klekamp and Weil (1982) except that the lysis buffer contained ammonium sulfate at 10% saturation. The pooled cell lysates were centrifuged at 27,000 X g for 10 min, and the resulting supernatant (466 ml) brought to 35% ammonium sulfate saturation by addition of the solid salt. After stirring for 30 min, the suspension was centrifuged 30 min at 27,000 X g and the supernatant brought to 50% ammonium sulfate saturation. After stirring and centrifugation as before, the precipitate was resuspended in 150 ml of lysis buffer lacking ammonium sulfate and dialyzed against buffer F/0.10 M KCI. The dialyzed material was applied to a 40 X 2.5-cm column of phosphocellulose, and the column was eluted with a 1.6- liter linear gradient of buffer G increasing in KC1 concentration from 0.10 to 0.70 M. CP1-containing fractions were pooled and the proteins precipitated with ammonium sulfate (55% saturation). The very fine precipitate was collected by filtration, dissolved in 18 ml of buffer G, and dialyzed against buffer F/0.10 M KC]. This sample was applied to a 25 X 1.6-cm column of DEAE-Sephadex and eluted with a 500- ml linear gradient of buffer F increasing in salt concentration from
0.10 to 0.50 M KC1. The fractions containing CP1 activity were pooled (32 ml), diluted with 16 ml of BTP buffer (20 mM BTP (pH 6.81, 0.1 mM EDTA, 0.5 mM dithiothreitol, 10% glycerol), and applied to a mono Q 5/5 column equilibrated with BTP/O.15 mM NaCl. CP1 was eluted with a 20-ml linear NaCl gradient (0.15-0.50 M) in BTP buffer at 0.5 ml/min flow rate, collecting 1-min fractions. Activity was detected in fractions eluting at approximately 0.23 M NaCl. These fractions were pooled (2.0 ml), diluted with 6.0 ml of buffer M (50 mM MES (pH 6.1), 0.5 mM dithiothreitol, 10% glycerol), and applied to a Mono S 5/5 column. The Mono S column was run as described for the Mono Q column except buffer M was used and the gradient was 0.075-0.50 M NaCI. CP1 activity eluted at 0.30 M NaC1.
DNA Binding Assays-Gel retardation assays were performed as described (Baker et al., 1986) except that the reaction volume was 15 pl, the buffer consisted of 20 mM Hepes (pH 7.9), 5 mM MgCL, 0.15 M KCl, 0.5 mg/ml bovine serum albumin, 0.05% Nonidet P-40, 5% glycerol, and the gels were run at room temperature using 0.025 M Tris, 0.19 M glycine, 0.1 mM EDTA as the buffer. Both sonicated pBR322 DNA and poly[d(I-C)] were used as nonspecific competitor DNAs; no significant difference was observed. Specific competitor DNAs containing CDEI and CDEIII' sequences were prepared by synthesizing complementary single-stranded oligonucleotides:
5'-GATCCAAATAAGTCACATGATGATA-3' CDEI 3'-GTTTATTCAGTGTACTACTATCTAG-5'
5"GATCCGTTTTCCGAAGATGT-3' CDEIII' 3'-GCAAAAGGCTTCTACACTAG-5'
(CDEIII' is the central, most conserved region of CDEIII. It corre- sponds to CDEIII positions 8-22 (Hegemann et al., 1988).) Oligonu- cleotides were phosphorylated, annealed, and oligomerized as de- scribed by Vinson et al. (1988).
Methylation interference experiments were carried out as described above except that the probe DNA, singly end-labeled, was methylated by dimethyl sulfate (Maxam and Gilbert, 1980) prior to its addition to the binding reactions. The CENB probe employed was a HindIIIl Ssp1 fragment cleaved from pRB45-2 and labeled at the HindIII site (upstream of CEN3). Labeling with [ ( U - ~ ~ P I ~ A T P and Klenow polym- erase placed the label on the bottom strand, and with [-y-32P]ATP and polynucleotide kinase, on the top strand (oriented as in Fig. 4). After resolving CP1-DNA complexes by electrophoresis, the undried
to excise the bound and free DNA bands. The gel slices were extruded gels were used to expose x-ray films which were then used as templates
through 18-gauge needles and incubated overnight at 50 "C in 10 mM Tris (pH 7.5), 1 mM EDTA, 0.1 M NaC1. Gel pieces were removed by centrifugation, and the DNA purified from the supernatants on NACS columns (Bethesda Research Laboratories). The isolated DNA was cleaved with piperidine and run on 8% sequencing gels (Maxam and Gilbert, 1980'.
FootDrintine reactions (20 ul) contained 20,000 cpm of probe, 1 pg of soniiated pBR322 DNA, 60 mM KC], and 0, 1,2, or 4 plof purified CP1; otherwise, the binding conditions were standard. After incubat- ing 15 min at room temperature, 2 p1 of 50 pg/ml DNase I (Sigma) was added, and after a further 30 s the reactions were stopped by adding 2 p1 of a solution containing 5% SDS and 0.5 mg/ml yeast tRNA. Proteins were removed by adding 1 ~l of 2 mg/ml proteinase K (EM Science) and incubating at 65 "C for 10 min. The DNA was precipitated with ethanol and analyzed on 6% sequencing gels (Maxam and Gilbert, 1980).
SDS-Gel Electrophoresis and Protein Renaturation-Aliquots (50- 200 pl) of chromatographic fractions were precipitated in microcen- trifuge tubes by adding 4 volumes of acetone and freezing on dry ice. After centrifugation for 15 min at 4 "C, pellets were rinsed with 80% acetone, dried under vacuum, and dissolved in SDS sample buffer. Electrophoresis was carried out using the buffer system of Laemmli (1970). Gels were 60 X 85 X 0.75 mm in size. Before loading, samples were incubated at 65 "C for 15 min. For renaturation experiments, thioglycolate was added to the upper reservoir buffer to a final concentration of 0.1 mM (Hunkapiller et al., 1983).
Elution and renaturation of proteins from SDS gel slices was carried out as described by Hager and Burgess (1980) with the following modifications: Protein bands were visualized by soaking the gels briefly in ice-cold 0.25 M KCl. After elution, the final protein pellets were dissolved in 50 pl of buffer F containing 0.10 M KC1 and 6 M guanidine HC1. The guanidine was removed by dialysis for 24 h at 4 "C against two changes of buffer F/0.10 M KC1 (MEGA micro- dialysis chamber, Health Products, Inc.).

Yeast Centromere Binding Protein 10845
RESULTS
Purification of CPI-CP1 activity was routinely monitored using gel-shift DNA binding assays. Specific binding was easily detected even in crude whole cell extracts (Fig. 1). Two CEN-specific complexes were observed in some crude ex- tracts, but only the major, higher mobility complex was ob- served after chromatography on phosphocellulose. Subse- quent chromatography on DEAE-Sephadex resulted in CP1 preparations which were approximately 1% pure and suffi- ciently free of nonspecific DNA binding contaminants that they could be used for quantitative binding experiments. Due to the large amount of nonspecific DNA binding activity present in crude extracts, we were unable to reliably quanti- tate CP1 activity in our starting material; therefore, we were unable to determine the actual percent recovery of CP1. However, in one preparation, the active CP1 obtained at the DEAE-Sephadex step was carefully quantitated by DNA ti- tration (see Fig. 7). Starting with 56 g of yeast (approximately 3 X 10” cells), we obtained 0.3 nmol of active CP1. This corresponds to over 600 molecules/cell.
The final purification of CP1 was achieved using Mono Q and Mono S ion exchange resins. Fig. 2 shows the analysis of Mono S column fractions. Fractions with CP1 binding activity contain predominantly a single polypeptide having a molec- ular weight of 58,400. Densitometry of the Coomassie-stained gel indicates that the peak fractions (22 and 23) are 80-90% pure. To prove that the 58,400-Da polypeptide is indeed CP1, a renaturation experiment was performed (Fig. 3). Mono S fractions 21 and 24 were pooled and electrophoresed on an SDS gel. Material eluted from slices of the gel was renatured and assayed for CP1 activity. The only fraction which con- tained significant activity derived from the slice containing the 58,400-Da polypeptide (slice E) . Binding was specific for CDEI, as shown by the fact that it was almost totally com- peted by excess unlabeled CDEI DNA, but not CDEIII DNA. We conclude that the centromere binding protein CP1 is comprised of a single 58,400-Da polypeptide.
CP l Binds to CDEZof Yeast CEN3-At the start, the probes we used to assay for binding activity contained the entire CEN3 core sequence. Nonetheless, only one CEN-specific binding activity was ever reproducibly detected in cell extracts or during the initial fractionations. Two lines of evidence (data not shown) indicated to us that the binding protein we had detected was CP1, i.e. that it bound in the region of CDEI. First, in competition experiments, only plasmids containing CDEI were able to compete for binding, and second, binding
+ + + + - extract
1 2 4 8 8 pBR322
DNA-protein complexes
4 CEN3 DNA
FIG. 1. Detection of C P l activity in crude yeast extract. Gel-shift DNA binding assays were carried out as described under “Experimental Procedures” using 0.4 pl of crude yeast extract, ”P- labeled CENS DNA (650-bp EcoRIIHindIII fragment from pRB45- l) , and the indicated amounts of unlabeled, sheared pBR322 DNA.
600
500
s 400
-
- d .- > .-
n 0 200 - 7
” 17 18 19 20 21 22 23 24 25 26
Fraction
kDa 20 21 22 23 24 25 26 - - 97.4 - 66.2 - 42.?-
31.0-
21.5-
FIG. 2. SDS-gel analysis of purified C P l . Upper, 0.15 p1 of fractions from the Mono S fast protein liquid chromatography column were assayed for binding activity using the standard gel-shift assay. CP1-CEN3 complexes were quantitated by scanning an autoradi- ogram of the gel using a Hoeffer model GS300 densitometer and activity is expressed in arbitrary density units. Lower, 50 rl of the indicated fractions were precipitated and analyzed on a 10% SDS- polyacrylamide gel. The gel was stained with Coomassie Blue. Outside hnes contain molecular weight standards (Bio-Rad).
was observed in gel-shift assays using a shorter probe con- taining only CDEI and 26 bp of CDEII (the 63-bp AluI-DraI fragment, see Fig. 4).
To precisely locate the CP1 binding site on CEN3 DNA, methylation interference and DNase I footprinting experi- ments were carried out. For methylation interference, end- labeled CEN3 probes were partially methylated at G residues and used in the gel-shift protocol. Bound and unbound DNA populations were isolated and their G ladders analyzed on sequencing gels. Comparing the G ladders of the bound and unbound DNAs reveals that four bands are absent in the bound fractions, two in each strand (Fig. 5). We infer that methylation a t these four positions has interfered with CP1 binding and, therefore, that these G residues lie within the binding site. All four affected G residues are located in CDEI. Significantly, methylation of G residues located 7 bp upstream and 3 bp downstream of CDEI does not noticeably affect binding.
Fig. 6 shows the DNase I footprint of CP1 on CEN3 DNA. The probe contained the entire CEN3 core sequence. Protec- tion of a 12-15-bp region centered over CDEI is observed. No protection of any other sites within the probe is detected. The clear conclusion from both footprinting and methylation pro- tection is that CP1 binds to CDEI.
Determination of the Equilibrium Constants for CPl Bind- ing to Wild Type and Mutant CDEZ Sequences-The equilib- rium constant for CP1 binding was determined by titrating a constant amount of CP1 with increasing amounts of CEN3 DNA and analyzing the binding reactions using the gel shift assay (Baker et al., 1986). A typical titration curve is shown in Fig. 7A. From these data, both the apparent equilibrium constant (Kapp) and the concentration of active CP1 in the
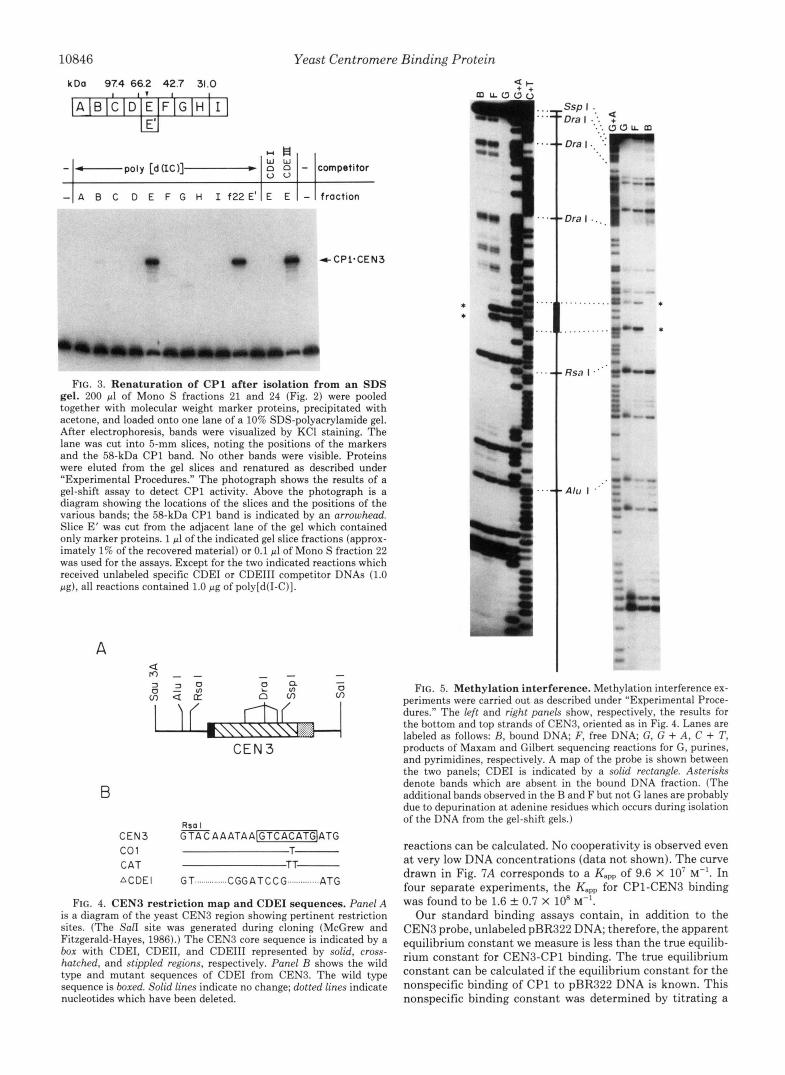
10846 Yeast Centromere Binding Protein
koa 97.4 66.2 42.7 31.0
tCPl.CEN3
FIG. 3. Renaturation of CP1 after isolation from an SDS gel. 200 pl of Mono S fractions 21 and 24 (Fig. 2) were pooled together with molecular weight marker proteins, precipitated with acetone, and loaded onto one lane of a 10% SDS-polyacrylamide gel. After electrophoresis, bands were visualized by KC1 staining. The lane was cut into 5-mm slices, noting the positions of the markers and the 58-kDa CP1 band. No other bands were visible. Proteins were eluted from the gel slices and renatured as described under “Experimental Procedures.” The photograph shows the results of a gel-shift assay to detect CP1 activity. Above the photograph is a diagram showing the locations of the slices and the positions of the various bands; the 58-kDa CP1 band is indicated by an arrowhead. Slice E’ was cut from the adjacent lane of the gel which contained only marker proteins. 1 pl of the indicated gel slice fractions (approx- imately 1% of the recovered material) or 0.1 pl of Mono S fraction 22 was used for the assays. Except for the two indicated reactions which received unlabeled specific CDEI or CDEIII competitor DNAs (1.0 pg), all reactions contained 1.0 pg of poly[d(I-C)].
A
I I f h\,\Q,<Y ..:.:.:.:; ...... .......... ........ . . . . j- CEN 3
B
C E N 3 G % A A A T A A v ] A T G co1 C A T
T- TT-
A C D E l G T ............... CGGATCCG .............. ATG
FIG. 4. CENB restriction map and CDEI sequences. Panel A is a diagram of the yeast CENB region showing pertinent restriction sites. (The Sal1 site was generated during cloning (McGrew and Fitzgerald-Hayes, 1986).) The CEN3 core sequence is indicated by a box with CDEI, CDEII, and CDEIII represented by solid, cross- hatched, and stippled regions, respectively. Panel B shows the wild type and mutant sequences of CDEI from CEN3. The wild type sequence is boxed. Solid lines indicate no change; dotted lines indicate nucleotides which have been deleted.
..
I ..
0
”
.....
. . . . .
Rsa I
Alu I
FIG. 5. Methylation interference. Methylation interference ex- periments were carried out as described under “Experimental Proce- dures.” The left and right panels show, respectively, the results for the bottom and top strands of CEN3, oriented as in Fig. 4. Lanes are labeled as follows: R, bound DNA; F, free DNA; G, G + A , C + T, products of Maxam and Gilbert sequencing reactions for G, purines, and pyrimidines, respectively. A map of the probe is shown between the two panels; CDEI is indicated by a solid rectangle. Asterisks denote bands which are absent in the bound DNA fraction. (The additional bands observed in the B and F but not G lanes are probably due to depurination a t adenine residues which occurs during isolation of the DNA from the gel-shift gels.)
reactions can be calculated. No cooperativity is observed even a t very low DNA concentrations (data not shown). The curve drawn in Fig. 7A corresponds to a Kapp of 9.6 X 10’ M”. In four separate experiments, the Kapp for CP1-CEN3 binding was found to be 1.6 f 0.7 X 10” M”.
Our standard binding assays contain, in addition to the CEN3 probe, unlabeled pBR322 DNA; therefore, the apparent equilibrium constant we measure is less than the true equilib- rium constant for CEN3-CP1 binding. The true equilibrium constant can be calculated if the equilibrium constant for the nonspecific binding of CP1 to pBR322 DNA is known. This nonspecific binding constant was determined by titrating a
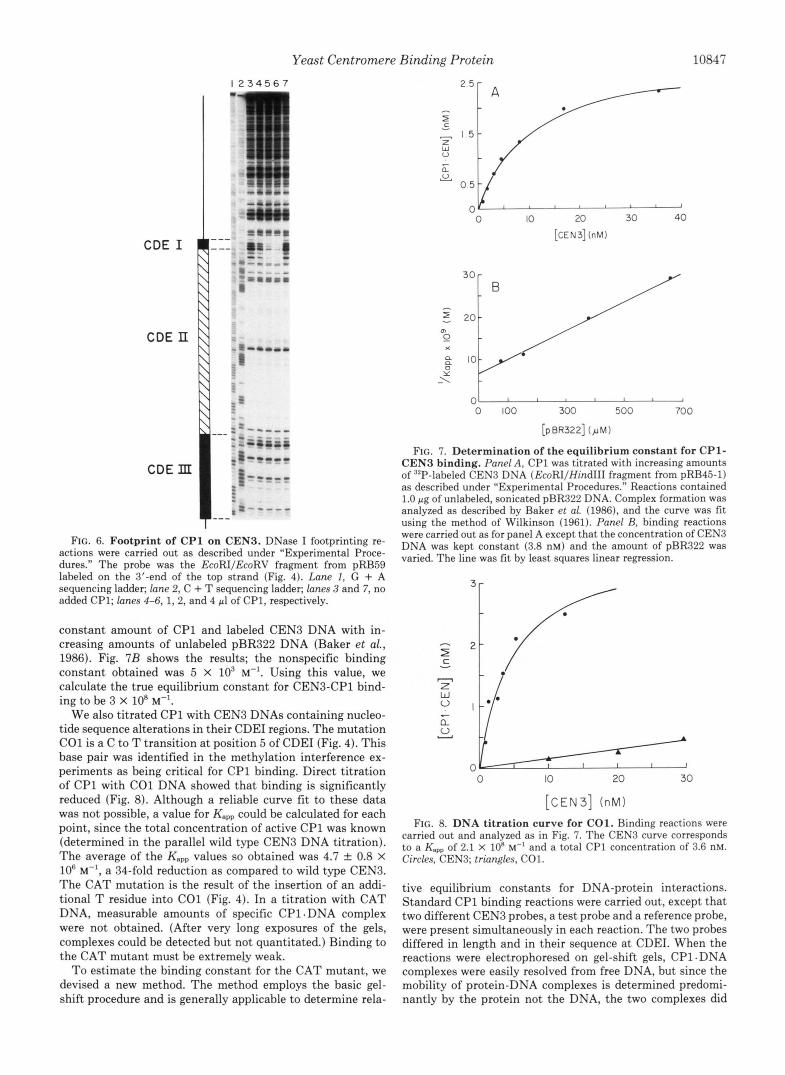
Yeast Centromere Binding Protein 10847
I 2 3 4 5 6 7
CDE II
cDEmr "_ FIG. 6. Footprint of CP1 on CEN3. DNase I footprinting re-
actions were carried out as described under "Experimental Proce- dures." The probe was the EcoRIIEcoRV fragment from pRB59 labeled on the 3'-end of the top strand (Fig. 4). Lane I , G + A sequencing ladder; lane 2, C + T sequencing ladder; lanes 3 and 7, no added CP1; lanes 4-6, 1, 2, and 4 pl of CP1, respectively.
constant amount of CP1 and labeled CEN3 DNA with in- creasing amounts of unlabeled pBR322 DNA (Baker et al., 1986). Fig. 7 B shows the results; the nonspecific binding constant obtained was 5 X lo3 M-'. Using this value, we calculate the true equilibrium constant for CEN3-CP1 bind- ing to be 3 X loR "'.
We also titrated CP1 with CEN3 DNAs containing nucleo- tide sequence alterations in their CDEI regions. The mutation CO1 is a C to T transition a t position 5 of CDEI (Fig. 4). This base pair was identified in the methylation interference ex- periments as being critical for CP1 binding. Direct titration of CP1 with CO1 DNA showed that binding is significantly reduced (Fig. 8). Although a reliable curve fit to these data was not possible, a value for Kapp could be calculated for each point, since the total concentration of active CP1 was known (determined in the parallel wild type CEN3 DNA titration). The average of the Kspp values so obtained was 4.7 f 0.8 x lo6 M-', a 34-fold reduction as compared to wild type CEN3. The CAT mutation is the result of the insertion of an addi- tional T residue into CO1 (Fig. 4). In a titration with CAT DNA, measurable amounts of specific CP1.DNA complex were not obtained. (After very long exposures of the gels, complexes could be detected but not quantitated.) Binding to the CAT mutant must be extremely weak.
To estimate the binding constant for the CAT mutant, we devised a new method. The method employs the basic gel- shift procedure and is generally applicable to determine rela-
I
0 10 20 30 40
[CEN3] (nM)
30 -
m
0 \ 0 100 300 500 700
[pBR322] ( p M )
FIG. 7. Determination of the equilibrium constant for CP1- CEN3 binding. Panel A, CP1 was titrated with increasing amounts of 32P-labeled CEN3 DNA (EcoRIIHindIII fragment from pRB45-1) as described under "Experimental Procedures." Reactions contained 1.0 pg of unlabeled, sonicated pBR322 DNA. Complex formation was analyzed as described by Baker et al. (1986), and the curve was fit using the method of Wilkinson (1961). Panel R, binding reactions were carried out as for panel A except that the concentration of CEN3 DNA was kept constant (3.8 nM) and the amount of pBR322 was varied. The line was fit by least squares linear regression.
0 IO 20 30
[CEN3] (nM)
FIG. 8. DNA titration curve for CO1. Binding reactions were carried out and analyzed as in Fig. 7. The CEN3 curve corresponds to a Kapp of 2.1 X 10' M" and a total CP1 concentration of 3.6 nM. Circles, CEN3; triangles, CO1.
tive equilibrium constants for DNA-protein interactions. Standard CP1 binding reactions were carried out, except that two different CEN3 probes, a test probe and a reference probe, were present simultaneously in each reaction. The two probes differed in length and in their sequence at CDEI. When the reactions were electrophoresed on gel-shift gels, CP1. DNA complexes were easily resolved from free DNA, but since the mobility of protein-DNA complexes is determined predomi- nantly by the protein not the DNA, the two complexes did

10848 Yeast Centromere Binding Protein
TABLE I Determination of equilibrium constants relative to CO1
Binding reactions were carried out as described under "Experimen- tal Procedures." The concentrations of the two DNA probes in each reaction are given in the table. The CP1 concentration was 3.3 nM (determined by titration). Probes for the test DNAs were 560-bp EcoRI/HindIII fragments obtained from plasmids pRB59 (CEN3), pRB61 (CAT), and pRB73 (ACDEI). The CO1 probe was a 410-bp EcoRI/EcoRV fragment from pRB6O. After electrophoresis to sepa- rate the CP1.DNA complexes from unbound DNA, bands were visualized by ethidium bromide staining. The CP1-bound DNA (a closely spaced doublet) was excised from the gel, eluted into 400 wl of 10 mM Tris-HC1 (pH 81, 1 mM EDTA, 1 M NaCl by incubation at 50 "C overnight, and isolated using a NACS column (Bethesda Re- search Laboratories). The eluted DNA was then electrophoresed on a 4% nondenaturing polyacrylamide gel to resolve the two fragments. The gel was dried and its autoradiograph scanned using a Helena Quickscan R & D densitometer to quantitate the DNA. Because the specific radioactivity of the probes varied, known amounts of each were run on the same gel in order to standardize the densitometric data. Relative equilibrium constants were calculated as described in the text.
Test DNA Input Ratio of
complexesb Relative [DNA,,J [DNAco,] Ratio"
nM
CEN3 20.0 174 0.115 4.2 37 CAT 213 20.3 10.5 0.26 0.025 ACDEI 192 20.3 9.4 0.16 0.017
not resolve from each other. By excising both complexes together from the gel, purifying the DNA, and rerunning it on a polyacrylamide gel to resolve the two DNA species, it was possible to determine the relative amount of each complex formed in the original binding reaction. By carrying out the binding reactions under conditions of DNA excess, the rela- tive equilibrium constant for the test DNA could be calculated from the following equation2:
" KteBt - [DNAmst.CPl] .- [DNA21 Kref [DNAmr.CPl] [DNA;,,]
[DNA,,.CP1]/[DNAref.CPl] is the ratio of the CP1 com- plexes, and [ DNAKd/ is the ratio of the input DNAs.
Table I shows the results of an experiment in which the binding constants for CEN3 DNAs containing the wild type CDEI, the CAT mutation, and a total deletion of CDEI (ACDEI) were measured relative to CEN3 containing the CO1 mutation. The CO1 mutant was used as the reference, because
The binding reaction contains CP1 and two different DNA probes (DNA1, DNA2), each of which contains a CP1 binding site. Reaction conditions are chosen such the total amount of each DNA (DNA;, DNA;) is in large excess over the total amount of protein (CP1"). The equilibrium constants for the binding reactions are:
K1 = [DNAl. CPl]
[DNAl]. [CPl] and
The ratio of equilibrium constants is
But, because DNA is in excess, [DNA11 = [DNA;], [DNA21 [DNA;], and the above equation can be rewritten as
K1 [DNAl.CPl] [DNA;] " - .- K2 [DNA?.CPl] [DNA;]'
TABLE I1 Mitotic stability of plasmids containing CEN3 CDEI mutations
pUC(TRP/CEN3), pUC(TRP/COl), pUC(TRP/CAT), pUC(TRP/ Yeast strain YP47 (ATRPI) was transformed with the plasmids
ACDEI), and pUC(TRP) using the lithium acetate procedure (Ito et al., 1983). Individual transformant colonies were picked from the selective plates (lacking tryptophan) and grown at 30 "C in 10 ml of nonselective medium (YEPD) for 16 h (10 generations). The cultures were then diluted and plated on YEPD plates to obtain 50-150 colonies/plate. After 2 days at 30 "C, the colonies were tested for the presence of plasmid by replicating onto minimal plates with or without added tryptophan and scoring for a Trp+ phenotype 24 h later. The number of independent transformants tested is given in parentheses.
% plasmid-containing cells after Plasmid 10 generations"
ExDeriment 1 ExDeriment 2 b _______
pUC(TRP/CEN3) pUC(TRP/COl)
63 f 9 (3) 73 f 3 (5) 59 f 4 (3) 58 f 5 (5)
pUC(TRP/CAT) 48 f 4 (3) pUC(TRP/ACDEI)
38 * 4 (5)
pUC(TRP) 41 f 2 (3) 42 f 4 (5) 13 f 7 (3) 15 f 3 (5)
Mean f S.E. * An F test (Lewis, 1966) was performed on the data from Experi-
ment 2 to assess statistical significance. At the 95% confidence level, the stability of pUC(TRP/CAT) was not significantly different from that of pUC(TRP/ACDEI), but was different from that of both pUC(TRP/COl) and pUC(TRP/CEN3). The stabilities of pUC(TRP/COl) and pUC(TRP/CEN3) were not statistically differ- ent at this level of confidence, while the very low stability of pUC(TRP) was significantly different from that of all other plasmids.
the very weak binding of CAT and ACDEI made it impossible to measure them relative to the wild type. By this analysis, the wild type CDEI (i.e. CEN3) was found to have an equilib- rium constant 37-fold higher than that for the CO1 mutant. This finding is in excellent agreement with the result obtained from direct titration of CO1 (Fig. 8). The equilibrium con- stants for CAT and ACDEI are further reduced 40- and 59- fold, respectively, from that of CO1. Thus, the CAT substi- tution/insertion mutation reduces CP1 binding approxi- mately 1400-fold relative to the wild type centromere. The residual CP1 binding detected with the CAT probe is only 1.5-fold better than that detected with ACDEI, a CEN frag- ment totally lacking, the CP1 recognition sequence.
Functional Analysis of CEN3 CDEI Mutants-The mitotic function of the CEN3 CDEI mutants was assessed using a plasmid stability assay. Centromere fragments containing either wild type CEN3 or CEN3 mutated a t CDEI were inserted into a pUC-derived vector containing the yeast TRPl gene and its associated replication origin. The plasmids were then introduced into a trpl yeast strain and their presence monitored by following the TRPl genetic marker. The pres- ence of a functional centromere confers mitotic stability to such plasmids (Fitzgerald-Hayes, 1987, and references therein).
The results of two separate experiments are given in Table 11. In both experiments, the stability of pUC(TRP/COl) was marginally lower than that of the wild type control plasmid pUC(TRP/CEN3), although the difference was not statisti- cally significant at the 95% confidence level. Plasmid pUC(TRP/CAT) was significantly less stable than the control and no more stable than pUC(TRP/ACDEI), indicating that the CAT mutation is as detrimental to CEN function as a total CDEI deletion. Nonetheless, pUC(TRP/CAT) and pUC(TRP/ACDEI) still retain significant centromere func- tion; both are considerably more stable than the acentric plasmid pUC(TRP). Overall, the level of centromere function inferred from these in uiuo stability assays is CEN3 2 CO1 >

Yeast Centromere Binding Protein 10849
CAT = ACDEI. This relationship correlates with the relative CP1 binding constants for these CDEI sequences.
DISCUSSION
Bram and Kornberg (1987) first identified the yeast cen- tromere binding protein CP1 and showed that it bound in the region of CDEI. Although they did not purify the protein, they estimated its molecular weight to be between 57,000 and 64,000 based on the recovery of activity from SDS gels. We have purified CP1 to near homogeneity. In agreement with Bram and Kornberg (1987), we find that CP1 is fairly abun- dant, present in excess of 600 molecules/logrithmically grow- ing cell. It is comprised of a single polypeptide of molecular weight 58,400 which when isolated from an SDS gel and renatured regains its sequence specific DNA binding activity. On a Sephacryl S-200 column, purified CP1 elutes just after the void volume (between blue dextran and apoferritin), in- dicating a native molecular weight in excess of 250,000. When a crude extract is chromatographed on the same column, the peak of CP1, again eluting near the void volume, is hetero- disperse and skewed toward the high molecular weight side, suggesting aggregation.
The nucleotide sequences critical for CP1 recognition ap- pear to be confined to the conserved CDEI octanucleotide RTCACRTG (R = purine; in CEN3, G, and A, respectively). No G residues outside of CDEI affect CP1 binding when methylated, and the region of DNase protection afforded by CP1 in footprinting experiments is limited to 12-15 bp cen- tered over this sequence. The CDEI octanucleotide would be expected to occur at random in DNA once every 8,192 bp (allowing it to be on either strand) or about 1,700 times in the 14,000 kilobase pairs of the haploid yeast genome. A computer search of 509 kilobase pairs of S. cereuisiae DNA sequences in GenBank (release 56) found 27 matches not counting the centromeres, about half the number expected. The CDEI sequence was found in coding regions on both coding and noncoding strands, in 5'-nontranscribed regions, and in 3"nontranscribed regions. The yeast 2p plasmid also contains a CDEI sequence (at position 5,271; Hartley and Donelson, 1980). While GenBank obviously contains a biased sampling of the yeast genome, it does seem clear that the CDEI octanucleotide is not limited solely to centromeres nor is its occurrence elsewhere in the genome strongly selected against. On the other hand, there is no evidence to indicate that CP1 is actually bound to any of these non-centromere sites in uiuo.
The role of CDEI in mitotic centromere function is obscure, but this element does not appear to be absolutely essential (Cumberledge and Carbon, 1987; Gaudet and Fitzgerald- Hayes, 1989). In our plasmid-based assay, the deletion of CDEI results in significantly decreased plasmid stability; how- ever, the resultant centromere still retains considerable mi- totic function. The CAT mutation reduces plasmid stability to about the same level as the CDEI deletion, while the CO1 mutation has little or no effect. The same results are obtained when these mutations are tested on the chromosome (Gaud& and Fitzgerald-Hayes, 1989). Mitotic chromosome stability is not affected by the CO1 mutation, but the frequency of chromosome loss per generation increases from 1 X (wild type CEN3) to 1 X and 6 X for the CAT and ACDEI mutations, respectively.
The observed phenotypic effects of the CO1 and CAT mutations correlate with the apparent equilibrium constants for CP1 binding to these mutant CDEI sequences. While the CO1 mutation reduces the Kapp for CP1 binding by about 35- fold, this effect is small in comparison to the extreme effect
of the CAT mutation, which reduces Kapp by 1400-fold. CP1 binding to the CAT centromere is little better than to ACDEI, and both CAT and ACDEI impair function to about the same extent. The observation that binding seems to correlate with function leads us to conclude that optimal centromere func- tion requires the binding of CP1 at CDEI. It should be recognized that since chromosome loss is lethal for a haploid organism, any mechanism which improves the accuracy of mitotic chromosome segregation will be strongly selected. The fact that CDEI is 100% conserved in the 12 yeast centromeres sequenced to date further supports our conclusion that CP1 is important for optimal centromere function.
It is surprising that the CO1 mutation has little or no detectable phenotypic effect. Possibly, the nuclear concentra- tion of CP1 is so high that, despite its lowered equilibrium constant, the mutant centromere still has CP1 bound a large percentage of the time. Alternatively, it may be that the centromere only requires CP1 to be bound transiently, and given a sufficiently long window for a productive interaction to occur, a 35-fold reduction in binding constant can be tolerated. The fact that the cellular concentration of CP1 appears to be relatively high is consistent with the former possibility.
We feel that it is unlikely that the cellular role of CP1 is confined solely to centromere structure and/or function. CP1 is an abundant protein, and the equilibrium constant for binding to its cognate site in DNA, 3 X los M-', is relatively low. (For comparison, transcription factor 7 of yeast, the 5 S rRNA transcription factor TFIIIA of Xenopus, and the ade- novirus major late transcription factor, have equilibrium con- stants of 5 X 10" "' (Baker et al., 1986), 1 X lo9 "' (Hanas et al., 1983), and 1 X 10" M-' (Chodosh et al., 1986), respec- tively.) Furthermore, there appear to be many CP1 binding sites in the genome not directly associated with centromeres. Bram and Kornberg (1987) suggested that CP1 binding facil- itates the binding of other proteins near it. For example, when bound near a gene promoter, CP1 might "attract" transcrip- tion factors, in the case of centromeres, other centromere binding proteins. At least one other protein is known to bind in the core region; it binds to CDEIII (Hegemann et al., 1986; Ng and Carbon, 1987). But how or if CP1 interacts with this or other centromere DNA binding proteins in assembling a functional centromere is not known. It is conceivable that CP1 is a general chromatin protein, important for organizing the overall structure of chromatin in the yeast nucleus. It could nucleate the assembly of nucleosomes or possibly anchor DNA to the nuclear matrix.
We recently have obtained partial amino acid sequences from the purified CP1 polypeptide. DNA probes derived from these sequences should enable us to obtain clones of the CP1 gene. We hope that by analyzing the gene and studying the phenotypic effects of CP1 mutations, we can begin to elucidate the function of CP1 in the yeast cell.
Acknowledgment-R. B. is indebted to Benjamin Hall for his support and encouragement during the initial stages of the project.
REFERENCES
Baker, R. E., Gabrielsen, O., and Hall, B. D. (1986) J. Biol. Chem.
Bloom, K. S., and Carbon, J. (1982) Cell 29, 305-317 Bram, R. J., and Kornberg, R. D. (1987) Mol. Cell. Biol. 7,403-409 Chodosh, L. A., Carthew, R. W., and Sharp, P. A. (1986) Mol. Cell.
Cumberledge, S., and Carbon, J. (1987) Genetics 117 , 203-212 Fitzgerald-Hayes, M. (1987) Yeast 3, 187-200 Fitzgerald-Hayes, M., Buhler, J.-M., Cooper, T. G., and Carbon, J.
261,5275-5282
Biol. 6,4723-4733
(1982) Mol. Cell. Biol. 2, 82-87

10850 Yeast Centromere Binding Protein Gaudet, A., and Fitzgerald-Hayes, M. (1987) Mol. Cell. Biol. 7,68-75 Gaudet, A., and Fitzgerald-Hayes, M. (1989) Genetics 121, 477-489 Hager, D. A., and Burgess, R. R. (1980) Anal. Biochem. 109 , 76-86 Hanas, J. S., Bogenhagen, D. F., and Wu, C.-W. (1983) Proc. Nutl.
Hartley, J. L., and Donelson, J. E. (1980) Nature 286,860-865 Hegemann, J. H., Pridmore, R. D., Schneider, R., and Philippsen, P.
Hegemann, J. H., Shero, J. H., Cottarel, G., Philippsen, P., and
Hieter, P., Pridmore, D., Hegemann, J. H., Thomas, M., Davis, R.
Hunkapiller, M. W., Lujan, E., Ostrander, F., and Hood, L. E. (1983)
Ito, H., Jukuda, Y., Murata, K., and Kimura, A. (1983) J. Bacteriol.
Acad. Sci. U. S. A. 80, 2142-2145
(1986) Mol. Gen. Genet. 206 , 305-311
Hieter, P. (1988) Mol. Cell. Biol. 8, 2523-2535
W., and Philippsen, P. (1985) Cell 42,913-921
Methods Enzymol. 91,227-236
163,163-168
Klekamp, M. S., and Weil, P. A. (1982) J. Biol. Chem. 2 5 7 , 8432-
Laemmli, U. K. (1970) Nature 227,680-685 Lewis, A. E. (1966) Biostatistics, pp. 125-149 Reinhold, New York Maxam, A. M., and Gilbert, W. (1980) Methods Enzymol. 6 6 , 499-
McGrew, J., Diehl, B., and Fitzgerald-Hayes, M. (1986) Mol. Cell.
Ng, R., and Carbon, J . (1987) Mol. Cell. Biol. 7,4522-4534 Rieder, C. L. (1982) Znt. Reu. Cytol. 7 9 , 1-58 Tschumper, G., and Carbon, J. (1980) Gene (Amst.) 10, 157-166 Vinson, C. R., LaMarco, K. L., Johnson, P. F., Landschultz, W. H.,
Wilkinson, G. N. (1961) Biochem. J. 80,324-332
8441
560
BWl. 6,530-538
and McKnight, S. L. (1988) Genes & Deu. 2,801-806