properties of simian virus 40 transcriptional ...jvi.asm.org/content/23/1/20.full.pdf · properties...
TRANSCRIPT

JOURNAL OF VIROLOGY, JUly 1977, P. 20-28Copyright © 1977 American Society for Microbiology
Vol. 23, No. 1Printed in U.S.A.
Properties of Simian Virus 40 Transcriptional IntermediatesIsolated from Nuclei of Permissive Cells
MOSHE SHANI,* EDWARD BIRKENMEIER, EVELYNE MAY, AND NORMAN P. SALZMANLaboratory ofBiology ofViruses, National Institute ofAllergy and Infectious Diseases, National Institutes of
Health, Bethesda, Maryland 20014
Received for publication 9 February 1977
A nucleoprotein complex that is an intermediate in viral transcription hasbeen isolated from simian virus 40 (SV40)-infected BSC-1 cells after lysinginfected nuclei with Sarkosyl. It contains DNA, DNA-dependent RNA polymer-ase II, and nascent RNA chains. RNA chain elongation continues for severalhours in vitro and is dependent on exogenous ribonucleoside triphosphates. Thecomplex sediments in neutral sucrose gradients with a main peak at about 24 to26S. When the nascent RNA on the complex is treated with RNase A, a fractionof the RNA remains resistant to RNase and is hydrogen bonded to the DNAtemplate. The pulse-labeled RNase-resistant RNA can be chased into RNase-sensitive RNA, indicating that it is located at the 3' terminus of the RNA chain.The rate of RNA displacement from the DNA template is consistent with anaverage rate of RNA chain elongation of 15 to 30 nucleotides per min. At least70% of the RNA synthesized in this in vitro system is SV40 specific. Hybridiza-tion with the separated strands of SV40 DNA and with fragments of SV40 DNAgenerated with endonucleases Hindu + III indicates that this RNA is comple-mentary to all regions of the "late" SV40 DNA strand. Studies of SV40 RNAsynthesis in this partially purified preparation at early and late times afterinfection should provide a way of locating promoter sites for transcription andidentifying the form of SV40 DNA that serves as a template for late tran-scription.
RNA transcripts of the "late" strand of sim-ian virus 40 (SV40) DNA are not found in thenucleus and/or the cytoplasm at early timesafter infection, when a transcript that corre-sponds to approximately 50% of the "early"strand is detected in both cell fractions (forreview, see 1). Late in infection, after DNAsynthesis has started, both of the DNA strandsare transcribed, but there is 10 to 20 times morenuclear viral RNA complementary to the latestrand than to the early strand (2, 6, 9). Thesedata suggest that specific initiation and termi-nation sites exist on the SV40 genome and thattranscription of the late strand is more efficientthan transcription of the early strand late ininfection.Understanding the regulation of the biogene-
sis of SV40 mRNA requires purification of cellcomponents that can synthesize viral mRNA invitro. These reconstitution experiments wouldrequire the isolation and identification of theRNA polymerases and relevant regulatory pro-teins in addition to the viral DNA template.However, attempts to create an in vitro systemthat is a model for in vivo transcription havebeen unsuccessful. An alternate approach tothe problem of studying SV40 RNA synthesis in
vitro is to isolate the in vivo viral complexes ofendogenous RNA polymerase, regulatory ele-ments, and the DNA template. After purifica-tion of such complexes, one could identify theproteins associated with the template in vivoand determine the physical form of the DNAtemplate.Green and Brooks (7) have extracted a por-
tion of the intracellular SV40 transcription in-termediates (TIs). However, most of the viraltranscriptional activity remained associatedwith the cellular DNA and could not be charac-terized. Recent results of Gariglio and Mousset(5) demonstrated that the majority of SV40 TIscould be extracted from infected nuclei withSarkosyl.
In the present study, we have determined thesedimentation velocity of the SV40 TIs, mea-sured the size of the nascent RNA associatedwith the TIs before and after treatment withRNase, and hybridized the RNA synthesized invitro to restriction enzyme fragments of viralDNA.
MATERIALS AND METHODSCells and virus. A line of African green monkey
cells (BSC-1) was obtained from M. Singer, and a
20
on June 10, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

SV40 TRANSCRIPTIONAL INTERMEDIATES
plaque-purified stock of SV40 (small-plaque strain)was obtained from R. G. Martin. Cells were grownat 370C in 150-mm plastic petri dishes in Eagle me-dium supplemented with 10% fetal calf serum.
Infection and labeling of cells. Subconfluent cul-tures were infected with SV40 at an input multiplic-ity of 5 to 10 PFU/cell in Eagle medium supple-mented with 2% fetal calf serum. The infected cellswere labeled with [2-14C]thymidine (1 ACi/ml, 57mCi/mmol; Radiochemical Centre, Amersham,England) for the time specified.
Isolation ofSV40 TIs. Forty hours after infection,the cells were trypsinized and washed twice with0.15 M NaCl and 0.01 M Tris, pH 7.4. Nuclei wereprepared by a hypotonic procedure (17) and lysed at4VC with 0.25% Sarkosyl and 0.4 M NaCl (5). Thechromatin was immediately pelleted in an SW50.1rotor for 30 min at 20,000 rpm at 4VC. The superna-tant was carefully poured off and stored at 4VC.
Assay for RNA polymerase activity. Assays werecarried out in a total volume of 125 A.l that con-tained: 46 mM ammonium sulfate; 8 mM KCl; 1.8mM MnCl2; 1.6 mM dithiothreitol; 0.6 mM (each)GTP, CTP, and ATP; 56 iM Tris, pH 7.9; 5 ACi of[5,6-3H]UTP (47 Ci/mmol; New England NuclearCorp., Boston, Mass.); and 6 ,uM UTP at 22°C. Thistemperature was used since at higher temperaturesthere was a rapid loss of enzymatic activity. RNAsynthesis was determined by measuring the incor-poration of labeled UMP into acid-precipitable ma-terial.
Purification of RNA synthesized in vitro. At theend of the incubation period, sodium dodecyl sulfatewas added to a final concentration of 0.5%, andEDTA was added to 10 mM. The RNA was extractedwith phenol-chloroform-isoamyl alcohol (15) and col-lected by ethanol precipitation. The pellet was dis-solved in TKM (0.05 M Tris, pH 7.4; 0.025 M KCl;0.0025 M MgCl2) and digested with 25 to 50 ,g ofDNase per ml (Worthington Biochemicals Corp.,Freehold, N.J.; RNase-free and electrophoreticallypurified) at 2°C for 60 min. The digest was extractedwith sodium dodecyl sulfate-phenol and collected byethanol precipitation.DNA-RNA hybridization. For hybridization ex-
periments with whole viral DNA, SV40 DNA I wasprepared as previously described (11). Preparationand purification of SV40 DNA fragments generatedby Hind restriction endonuclease was carried out aspreviously described (13). DNA was immobilized on25-mm filters (BA85, Schleicher & Schuell Co.,Keene, N.H.) that were dried and heated at 80°C for3 h, and square minifilters (3 by 3 mm) were cutfrom them. Blank minifilters without DNA wereprepared in the same way.The minifilters containing the DNA fragments
were preincubated in the hybridization mixture(50% [vol/vol] formamide, 0.3 M NaCl, 0.03 M so-dium citrate, 0.1% sodium dodecyl sulfate, and 0.01M Tris, pH 7.4) for 24 h at 37°C. After this treatmentthe fragments were irreversibly immobilized on thefilters, as judged by the retention of [14Clthymidine-labeled fragments on the filters (13). Hybridizationof the in vitro synthesized RNA with either wholeSV40 DNA or its Hind fragments was performed, in
a final volume of 100 ,ul of the hybridization buffer,for 72 h at 37°C. At the end of the incubation, thefilters were washed with 2x SSC (SSC = 0.15 MNaCl, 0.015 M sodium citrate), treated with pan-creatic RNase (20 ,ug/ml) at 22°C for 1 h, washedwith 2x SSC, dried, and counted.
Sedimentation analysis. Samples were layeredonto 11.5-ml linear 5 to 20% (wt/wt) sucrose gra-dients in 0.3 M NaCl, 0.25% Sarkosyl, 0.001 M dithi-othreitol, and 0.01 M Tris, pH 7.4, and centrifugedfor 16 h at 24,000 rpm at 4°C in a Spinco SW41 rotor(Beckman Instruments, Inc., Fullerton, Calif.).Fractions were collected, and the amount of acid-precipitable radioactivity in each was determined.
RESULTS
Characterization of the transcriptional ac-tivity of the Sarkosyl supernatant. BSC-1 cellswere infected with plaque-purified SV40, andthe Sarkosyl supernatant was prepared 40 hlater as described in Materials and Methods. Ininfected cells that were labeled in vivo for 16 hwith [14C]thymidine before the preparation ofthe Sarkosyl supernatant, more than 85% ofthelabeled SV40 DNA I was recovered in this su-pernatant fluid. In the standard assay for en-dogenous RNA polymerase activity in the Sar-kosyl supernatant, incorporation of [3H]UMPinto acid-precipitable material continued for atleast 2 h at 22°C. Omission of ATP, CTP, andGTP completely inhibited the incorporation of[3H]UMP.Under these conditions, the UTP concentra-
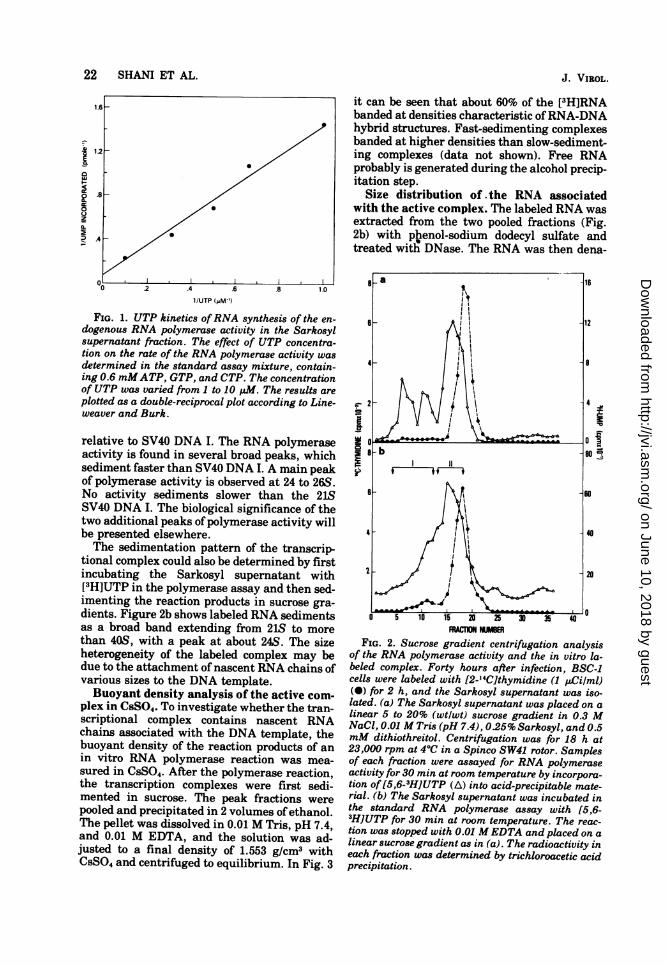
tion was rate limiting. The UTP dependencykinetics for the Sarkosyl supernatant RNA po-lymerase activity is illustrated in Fig. 1. Theeffect of the concentration of UTP displaysclassical Michaelis-Menten kinetics. From thisdata, the Km value for UTP is 19 uM.The presence of a low concentration of a-
amanitin (0.4 ,Lg/ml), an inhibitor of RNA po-lymerase II (18), inhibited 96% of the syntheticactivity. There was no significant effect causedby high concentrations ofrifamycin AF/013 (100,ug/ml), an inhibitor that blocks initiation butdoes not affect RNA chain elongation (14).These results indicate that RNA polymerase
II is involved in the transcriptional activity ofthe Sarkosyl supernatant and that RNA syn-thesis has been initiated in vivo. These ob-served data are in agreement with previousreports (5).Sedimentation pattern of the active com-
plex. Forty hours after infection, a Sarkosylsupernatant was prepared and sedimented in asucrose gradient. Each fraction of the gradientwas then assayed for its RNA polymerase activ-ity. Figure 2a shows the sedimentation patternof the endogenous RNA polymerase activity
VOL. 23, 1977 21
on June 10, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from
22 SHANI ET AL.
1.2
2 .80
(.4
1/UTP (MM I
FIG. 1. UTP kinetics ofRNA synthesis of the en-dogenous RNA polymerase activity in the Sarkosylsupernatant fraction. The effect of UTP concentra-tion on the rate of the RNA polymerase activity wasdetermined in the standard assay mixture, contain-ing 0.6 mMATP, GTP, and CTP. The concentrationof UTP was varied from 1 to 10 M. The results areplotted as a double-reciprocal plot according to Line-weaver and Burk.
relative to SV40 DNA I. The RNA polymeraseactivity is found in several broad peaks, whichsediment faster than SV40 DNA I. A main peakof polymerase activity is observed at 24 to 26S.No activity sediments slower than the 21SSV40 DNA I. The biological significance of thetwo additional peaks of polymerase activity willbe presented elsewhere.The sedimentation pattern of the transcrip-
tional complex could also be determined by firstincubating the Sarkosyl supernatant with[3H]UTP in the polymerase assay and then sed-imenting the reaction products in sucrose gra-dients. Figure 2b shows labeled RNA sedimentsas a broad band extending from 21S to morethan 40S, with a peak at about 24S. The sizeheterogeneity of the labeled complex may bedue to the attachment of nascent RNA chains ofvarious sizes to the DNA template.Buoyant density analysis of the active com-
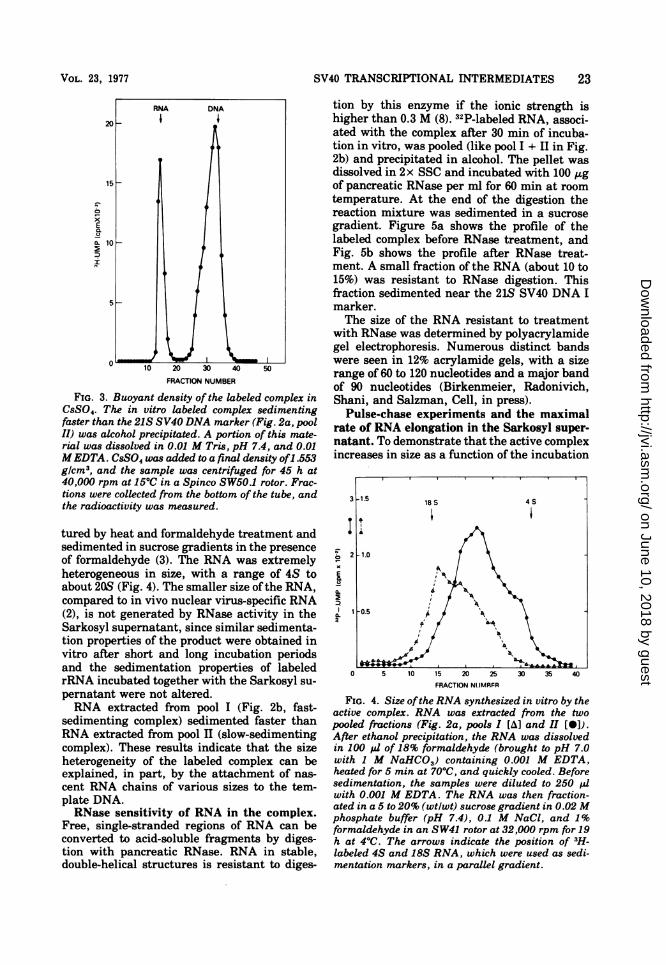
plex in CSS04. To investigate whether the tran-scriptional complex contains nascent RNAchains associated with the DNA template, thebuoyant density of the reaction products of anin vitro RNA polymerase reaction was mea-sured in CsS04. After the polymerase reaction,the transcription complexes were first sedi-mented in sucrose. The peak fractions werepooled and precipitated in 2 volumes ofethanol.The pellet was dissolved in 0.01 M Tris, pH 7.4,and 0.01 M EDTA, and the solution was ad-justed to a final density of 1.553 g/cm3 withCsS04 and centrifuged to equilibrium. In Fig. 3
it can be seen that about 60% of the [3H]RNAbanded at densities characteristic ofRNA-DNAhybrid structures. Fast-sedimenting complexesbanded at higher densities than slow-sediment-ing complexes (data not shown). Free RNAprobably is generated during the alcohol precip-itation step.
Size distribution of. the RNA associatedwith the active complex. The labeled RNA wasextracted from the two pooled fractions (Fig.2b) with phenol-sodium dodecyl sulfate andtreated with DNase. The RNA was then dena-
Fm9LUacm
I-3
FcIU NUMBERFIG. 2. Sucrose gradient centrifugation analysis
of the RNA polymerase activity and the in vitro la-beled complex. Forty hours after infection, BSC-1cells were labeled with [2-'4C]thymidine (1 uCi/ml)(@) for 2 h, and the Sarkosyl supernatant was iso-lated. (a) The Sarkosyl supernatant was placed on alinear 5 to 20% (wtlwt) sucrose gradient in 0.3 MNaCl, 0.01 M Tris (pH 7.4), 0.25% Sarkosyl, and 0.5mM dithiothreitol. Centrifugation was for 18 h at23,000 rpm at 4°C in a Spinco SW41 rotor. Samplesof each fraction were assayed for RNA polymeraseactivity for 30 min at room temperature by incorpora-tion of[5,6-3H]UTP (A) into acid-precipitable mate-rial. (b) The Sarkosyl supernatant was incubated inthe standard RNA polymerase assay with [5,6-3HlUTP for 30 min at room temperature. The reac-tion was stopped with 0.01 MEDTA and placed on alinear sucrose gradient as in (a). The radioactivity ineach fraction was determined by trichloroacetic acidprecipitation.
J. VIROL.
on June 10, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from
SV40 TRANSCRIPTIONAL INTERMEDIATES 23
E
10
5-
10 20 30 40 50
FRACTION NUMBER
FIG. 3. Buoyant density of the labeled complex inCsS04. The in vitro labeled complex sedimentingfaster than the 21S SV40 DNA marker (Fig. 2a, poolII) was alcohol precipitated. A portion of this mate-rial was dissolved in 0.01 M Tris, pH 7.4, and 0.01MEDTA. CsS04 was added to a final density of1.553g/cm3, and the sample was centrifuged for 45 h at40,000 rpm at 15°C in a Spinco SW50.1 rotor. Frac-tions were collected from the bottom of the tube, andthe radioactivity was measured.
tured by heat and formaldehyde treatment andsedimented in sucrose gradients in the presenceof formaldehyde (3). The RNA was extremelyheterogeneous in size, with a range of 4S toabout 20S (Fig. 4). The smaller size ofthe RNA,compared to in vivo nuclear virus-specific RNA(2), is not generated by RNase activity in theSarkosyl supernatant, since similar sedimenta-tion properties of the product were obtained invitro after short and long incubation periodsand the sedimentation properties of labeledrRNA incubated together with the Sarkosyl su-
pernatant were not altered.RNA extracted from pool I (Fig. 2b, fast-
sedimenting complex) sedimented faster thanRNA extracted from pool II (slow-sedimentingcomplex). These results indicate that the sizeheterogeneity of the labeled complex can beexplained, in part, by the attachment of nas-cent RNA chains of various sizes to the tem-plate DNA.RNase sensitivity of RNA in the complex.
Free, single-stranded regions of RNA can beconverted to acid-soluble fragments by diges-tion with pancreatic RNase. RNA in stable,double-helical structures is resistant to diges-
tion by this enzyme if the ionic strength ishigher than 0.3 M (8). 32P-labeled RNA, associ-ated with the complex after 30 min of incuba-tion in vitro, was pooled (like pool I + II in Fig.2b) and precipitated in alcohol. The pellet wasdissolved in 2x SSC and incubated with 100 ,ugof pancreatic RNase per ml for 60 min at roomtemperature. At the end of the digestion thereaction mixture was sedimented in a sucrosegradient. Figure 5a shows the profile of thelabeled complex before RNase treatment, andFig. 5b shows the profile after RNase treat-ment. A small fraction of the RNA (about 10 to15%) was resistant to RNase digestion. Thisfraction sedimented near the 21S SV40 DNA Imarker.The size of the RNA resistant to treatment
with RNase was determined by polyacrylamidegel electrophoresis. Numerous distinct bandswere seen in 12% acrylamide gels, with a sizerange of 60 to 120 nucleotides and a major bandof 90 nucleotides (Birkenmeier, Radonivich,Shani, and Salzman, Cell, in press).Pulse-chase experiments and the maximal
rate of RNA elongation in the Sarkosyl super-natant. To demonstrate that the active complexincreases in size as a function of the incubation
3 -1.5 18S 4S
O 2 -1.0
1 0.5
0 5 10 15 20 25 30 35 40FRACTION NLJMRFR
FIG. 4. Size ofthe RNA synthesized in vitro by theactive complex. RNA was extracted from the twopooled fractions (Fig. 2a, pools I [A] and II []).After ethanol precipitation, the RNA was dissolvedin 100 p1 of 18% formaldehyde (brought to pH 7.0with 1 M NaHC03) containing 0.001 M EDTA,heated for 5 min at 700C, and quickly cooled. Beforesedimentation, the samples were diluted to 250 p1with 0.001 M EDTA. The RNA was then fraction-ated in a 5 to 20% (wt/wt) sucrose gradient in 0.02 Mphosphate buffer (pH 7.4), 0.1 M NaCl, and 1%formaldehyde in an SW41 rotor at 32,000 rpm for 19h at 4°C. The arrows indicate the position of 3H-labeled 4S and 18S RNA, which were used as sedi-mentation markers, in a parallel gradient.
VOL. 23, 1977
on June 10, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

24 SHANI ET AL.
b
E
aj
b8B
6
21 S4-
2-
0 10 20 30 40 50
FRACTION NUMBER
FIG. 5. Effect of RNase treatment on the attach-ment of nascent RNA to the DNA template. (a) TheSarkosyl supernatant was incubated for 15 min atroom temperature with [a-32P]UTP in the standardpolymerase assay. The reaction was stopped by 0.01M EDTA, and the reaction mixture was placed on a36-ml linear 5 to 20% (wt/wt) sucrose gradient andsedimented for 19 h at 23,000 rpm at 40C in a SpincoSW27 rotor. (b) The main peak (fractions 10 to 19)from the gradient shown in (a) was pooled and alco-hol precipitated. The pellet was dissolved in 100 /4 of2 x SSC and treated with 100 pg of pancreaticRNase per ml for 1 h at room temperature. The digestwas placed on a 12-ml 5 to 20% (wt/wt) sucrosegradient and centrifuged for 16 h at 26,000 rpm at4°C in a Spinco SW41 rotor. The arrows indicate thesedimentation values relative to '4C-labeled SV40DNA I sedimented in a parallel gradient.
time, pulse-chase experiments were performed.If a 40-fold excess of unlabeled UTP was addedto the reaction to dilute the [32P]UTP used tostart the reaction, no further 3H label was in-corporated into RNA (data not shown). Theexperiment outlined in Fig. 6 shows that afraction ofthe radioactivity in the 15-min pulse-labeled RNA was in larger complex moleculesafter 10- and 30-min chases with 2 mM coldUTP. The peak at 24S decreased while therewas an increase in the proportion of faster-sedimenting complexes. In the previous sectionwe have shown that faster-sedimenting com-plex molecules contain larger RNA chains thanslow-sedimenting complex molecules. There-fore, the increase in the size of the complexunder chase conditions is a result ofRNA chainelongation.
The rate ofRNA chain elongation was deter-mined by following the fraction of the incorpo-rated label that was RNase resistant after var-ious chase periods. After 15 min of synthesis inthe presence of [3H]UTP, a sample was taken,and the reaction was stopped with EDTA. Tothe rest of the reaction mixture cold UTP wasadded to a final concentration of 2 mM. Sam-ples were removed at various chase periods andsedimented in neutral sucrose gradients. Thelabeled complexes in these gradients werepooled and ethanol precipitated. The pelletswere dissolved in 2 x SSC and digested with 100lig of pancreatic RNase per ml for 1 h at roomtemperature. The digestion was stopped with5% trichloroacetic acid, and the RNase-resist-ant counts were collected on GF/C filters,washed extensively with 2% trichloroaceticacid, dried, and counted. Figure 7 summarizesthe results of two such experiments. The rate ofdecrease of the RNase-resistant counts duringthe chase approximates first-order kinetics.After about 2 min, 50% of the RNase-resistantfraction became sensitive to RNase digestion.Only 60% of the RNA that was initially re-
sistant to RNase could be chased, and longerchase periods did not alter that percentage. Thereason for this is not clear, but it may reflectinactivation of enzyme molecules on the DNAor dissociation of the enzyme from DNA.
15
21 S
106
EaL
5
5 10 15 20 25
FRACTION NUMBER
30 35 400
FIG. 6. Sedimentation pattern of the in vitro la-beled complex in a pulse-chase experiment. The Sar-kosyl supernatant was incubated in the standardRNA polymerase assay with [5,6-3H]UTP for 15 minat room temperature. A sample was taken (zero time)(0), and the reaction was stopped with 0.01 MEDTA. To the remaining reaction mixture cold UTPwas added to a final concentration of 2 mM, andsamples were taken after 10 (0) and 30 min (A) ofincubation. The three samples were placed on linear5 to 20% (wt/wt) sucrose gradients and sedimentedfor 18 h at 23,000 rpm at 4°C.
J. VIROL.
on June 10, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

SV40 TRANSCRIPTIONAL INTERMEDIATES 25
60
0
°40
20
5 10 15
CHASE TIME IMINJ
FIG. 7. Kinetics of the decrease of the remaininglabeled fraction that is RNase resistant in pulse-chase experiments. The data are from sucrose gra-dient analyses of two experiments such as shown inFig. 5 and 6. The radioactive RNA that was RNaseresistant at zero time was taken as 100%.
Hybridization of RNA synthesized by theSarkosyl supernatant with SV40 DNA I. RNAsynthesized by the Sarkosyl supernatant is ameasure of SV40 transcription. This was sug-
gested by its occurrence in infected cells and notin mock-infected cells (data not shown) and wasclearly established by RNA-DNA hybridizationexperiments. [3H]UMP-labeled reaction prod-ucts synthesized by the Sarkosyl supernatantwere phenol extracted, DNase treated, and sed-imented through a sucrose gradient containing50%o formamide. Labeled RNA sedimentingfaster and slower than 18S rRNA marker was
pooled separately and hybridized to an excess ofSV40 DNA immobilized on filters. Table 1shows that 69% of the RNA sedimenting slowerthan 188 rRNA and 25% of the RNA sediment-ing faster than 188 rRNA hybridized to SV40DNA I, compared to 67% hybridization withSV40 complementary RNA. Therefore, the ma-jority of this RNA is virus specific.
Green and Brooks (7) demonstrated thatmost of the virus TIs cannot be solubilized by avariety of extraction procedures, including Sar-kosyl. Instead, they remain associated with thecellular DNA in the pellet. In order to comparethis with our method, the following experimentwas performed. A reaction mixture containingnuclei from infected cells was lysed with 0.25%Sarkosyl, and the entire lysate was incubatedin the standard RNA polymerase assay. Thesecond reaction mixture contained a Sarkosylsupernatant obtained from the same number ofinfected nuclei, which was added to a number ofuninfected nuclei equal to that contained in thefirst reaction mixture, and was assayed underthe same conditions. RNA was extracted from
these reactions and hybridized with SV40 DNAI immobilized on filters. The results (Table 2)clearly show that both RNA preparations hy-bridized to the same extent with SV40 DNA I.About 90% of the SV40-hybridizable counts ininfected nuclei were recovered in the Sarkosylsupernatant. This indicates that the majority ofthe viral TIs are solubilized by Sarkosyl extrac-tion. These results are in agreement with thoseof Gariglio and Mousset (5). The apparent dis-crepancy between our results and those ofGreen and Brooks (7) is probably due to dif-ferent cell lines and differences in extractionprocedures.
Hybridization of RNA synthesized by SV40TIs with Hind fragments of SV40 DNA. Theregions of the SV40 genome being transcribed
TABLE 1. RNA-DNA hybridization between 3H-labeled RNA from the RNA polymerase assay and
excess amounts ofSV40 DNA I
Origin of 3H-la- Input hybridizedbeled RNA Input (cpm) cpm %
>18Sa 7,020 1,814 25.8<18S 3,950 2,740 69.3SV40 cRNAb 10,230 6,915 67.5
a RNA synthesized in the standard RNA polymer-ase assay was extracted and sedimented in 5 to 20%sucrose gradients containing 50% formamide. Frac-tions sedimenting faster than 18S (>18S, represent-ing about 10% of the total RNA synthesized) orslower than 18S (<188) were pooled and hybridizedas described in the text.
b 3H-labeled complementary RNA (cRNA) wasprepared as detailed in reference 12.
TABLE 2. Distribution of SV40 TIs in whole nucleiand in Sarkosyl supernatant
% of totalOrigin of 'H-labeled cpm hybrid- SV40-hy-
RNA ized bridizablecpm
Infected nuclei 14,000 100Sarkosyl supernatantb 12,690 91+ uninfected nuclei
a Nuclei were isolated from infected cells as de-scribed in the text. Sarkosyl was added to 0.25%,and NaCl was added to 0.4 M. One half of the totallysate was incubated in the standard polymeraseassay for 30 min.
b The second halfof the lysate was used to preparea Sarkosyl supernatant. This supernatant was thenadded to uninfected nuclei. The number of nucleiwas equal to that used to prepare the Sarkosyl su-pernatant. This lysate was incubated in the stan-dard polymerase assay for 30 min. RNA was ex-tracted by sodium dodecyl sulfate-phenol-isoamylalcohol and hybridized to filters containing SV40DNA I, as described in the text.
VOL. 23, 1977
on June 10, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

26 SHANI ET AL.
in vitro were determined by hybridizing in vitrosynthesized RNA to DNA fragments made bythe Hindll + III restriction enzymes. TheSarkosyl supernatant was incubated for 15 minin the standard polymerase reaction in thepresence of [32P]UTP. The RNA was extractedand hybridized to filters containing an excess ofseparated SV40 HindII + III fragments (8).Table 3 shows that the 32P-labeled RNA hybrid-ized to DNA fragments corresponding to thelate region ofSV40 (fragments C, D, E, K, F, J,and G) as well as to the early region of SV40(fragments A, H, I, and B). These results alsoindicate that RNA polymerase molecules asso-ciated with the TI are scattered all over thegenome, ruling out the possibility that the Sar-kosyl procedure selects for specific types oftem-plate molecules. Hybridization of this RNA tothe separated strands of SV40 DNA revealedthat about 10% of the RNA synthesized wascomplementary to the E-strand, whereas 90%was complementary to the L-strand (data notshown). This proportion is in agreement withthat found in vivo (10).
DISCUSSIONWe have characterized SV40 TIs isolated by
Sarkosyl extraction of infected nuclei. The viralTIs are associated with endogenous RNA
TABLE 3. Hybridization of[32P]RNA synthesized bySarkosyl supernatant to Hind fragments ofSV40
DNA ja
thyfrag- % 8V40-hybri- % Hybridiz-Fragment imedto frsag dizable cpmb able cpm/%ments ~~SV40 DNMc
CD 686 21.5 1.0E 362 11.3 1.3K 60 1.9 0.5F 187 5.9 0.8J 115 3.6 0.8G 148 4.6 0.7B 478 15.0 1.0I 57 1.8 0.4H 67 2.8 0.4A 518 16.0 0.7
a The Sarkosyl supernatant was incubated in vi-tro for 15 min in the standard polymerase reactionwith [32P]UTP. The extracted [32PJRNA was thenhybridized to fragments bound to minifilters as de-scribed in the text.bThe radioactivity hybridized to a given frag-
ment(s) was expressed as the percentage of the ra-dioactivity hybridized to whole SV40 DNA.
c To make allowance for the relative molecularweight of the fragments, we calculated the ratio ofpercent hybridizable counts per minute to percentSV40 DNA in the fragment.
polymerase molecules that continue to synthe-size SV40 RNA in vitro for at least 2 h at roomtemperature. The inhibition of the polymerasereaction by a-amanitin and its insensitivity toSarkosyl and rifamycin derivative AF/013 in-dicate that SV40 DNA is transcribed by RNApolymerase II and that RNA synthesis was ini-tiated in vivo before the isolation. Similar find-ings have been reported by Gariglio and Mous-set (5) and Green and Brooks (7).When the Sarkosyl supernatant is sedi-
mented through sucrose gradients, severalpeaks ofRNA polymerase activity are found inthe gradient sedimenting faster than 21S SV40DNA I. A more heterogeneous distribution ofthe complex is obtained if the TI molecules arelabeled by allowing RNA synthesis to occur inthe presence of [3H]UTP before sedimentation.The proportion of the in vivo '4C-prelabeledSV40 DNA that is associated with the viral TIsis small, so it has not been possible to charac-terize the DNA template that is used for tran-scription by labeling this DNA.The RNA associated with the TI molecules
has been characterized by sedimentation in su-crose gradients. The RNA has a maximum sizeof about 20S under denaturing conditions. ThisRNA is smaller than the RNA obtained in vivo(2), but this cannot be explained based on thepresence of RNase activity in the Sarkosyl ex-tracts. It may result from the dissociation oflarger RNA molecules from the TI complexesduring preparation. However, most of the RNAis attached to the DNA template, since themajority of the [3H]UTP counts band togetherwith the DNA as RNA-DNA hybrids in CsS04gradients (Fig. 3).During transcription ofDNA by Escherichia
coli RNA polymerase, nascent RNA moleculesare attached to the DNA by a limited region ofhydrogen bonding. On the average, no morethan 20 nucleotides per chain can be renderedRNase resistant (16). The formation of theRNase-resistant complex between nascentRNA and DNA is more efficient in the presenceof protein-denaturing agents if the DNA is su-percoiled (16). In the present studies, alcoholwas used as a mild protein-dissociating agent.After alcohol precipitation of the TIs labeledduring a 30-min reaction, nearly 10% of thelabeled RNA was resistant to RNase treat-ment. The size range of the majority of theRNase-resistant RNA was 60 to 120 nucleo-tides. The size of this RNA fragment, togetherwith pulse-chase experiments, provided us withan efficient method to directly measure the rateof RNA chain elongation. The calculated elon-
J. VIROL.
on June 10, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

SV40 TRANSCRIPTIONAL INTERMEDIATES 27
gation rate ranged from 15 to 30 nucleotides permin, with an average rate of about 20 nucleo-tides per min per chain. It is of interest tocompare this rate with the rate obtained inother systems. The rate of RNA chain elonga-tion in isolated adenovirus 2 TIs was estimatedto be 120 nucleotides per min per chain (19),and the rate of transcription of calfthymus chro-matin by E. coli RNA polymerase was 35 nu-cleotides per min per chain (4). The lower rateof chain elongation in our system can be ex-plained in part by the lower incubation temper-ature (22 compared to 370C), by the removal ofessential factors for chain elongation by theSarkosyl, or by direct inhibition by Sarkosylitself.The inability to chase 40% of the RNA chains
out of the DNA template cannot be due to inac-tivation by Sarkosyl only. A pulse-chase experi-ment carried out with a transcriptional com-plex of SV40 DNA I and E. coli RNA polymer-ase in the presence of Sarkosyl resulted in acomplete displacement of the RNase-resistantRNA from the DNA (unpublished data).At least 70% (corrected for efficiency of hy-
bridization) of the RNA synthesized by the Sar-kosyl supernatant is SV40 specific. Other re-ports have shown about the same level of hy-bridization to SV40 DNA (5, 7). This RNA hy-bridized to all fragments of the SV40 genomegenerated with endonucleases HindII + HI. Thelevel of hybridization to early DNA fragments(fragments A, H, I, and B) and late DNA frag-ments (fragments C, D, E, K, F, J, and G) wasabout the same. Since 90% of the synthesizedRNA originates from the late strand of SV40,these results indicate that the entire latestrand is transcribed. This is in agreement withprevious studies with in vivo RNA (10).
In our extraction procedure, 85% of 14C-la-beled viral DNA I could be recovered in theSarkosyl supernatant. This agrees with the re-sults of Gariglio and Mousset (5) and Green andBrooks (7). However, our data strongly suggestthat at least 90% of the viral TIs can be solubi-lized by Sarkosyl. Comparison of the total viraltranscriptional activity in the Sarkosyl-treatedinfected nuclei and in a Sarkosyl supernatantmixed with control uninfected nuclei gave thesame value. The reasons for the discrepancybetween our results and those of Green andBrooks (7) are probably due to the higher saltconcentration in the Sarkosyl extraction usedin the present study. It is impossible to comparethe reaction of the supernatant fraction thatcontains only SV40 genomes to the reaction ofthe pellet, where the conditions and the concen-
tration of the transcribing DNA are different.The mixing experiment allowed a better analy-sis of the viral RNA in these fractions.
Purification of transcriptionally active SV40chromosomes has several significant conse-quences. It provides a means of purifying RNApolymerase II. Isolated TI molecules will makepossible the study of SV40 RNA synthesis in apartially purified preparation at early and latetimes after infection. Since initiation of thetranscriptional complex occurs in vivo, itshould also provide a way of locating promotersites for transcription. The characterization ofTIs has made possible a characterization of theform of SV40 DNA that serves as a template forlate transcription (Birkenmeier et al., in press).
LITERATURE CITED
1. Acheson, N. H. 1976. Transcription during productiveinfection with polyoma virus and simian virus 40.Cell 8:1-12.
2. Aloni, Y., M. Shani, and Y. Reuveni. 1975. RNAs ofsimian virus 40 in productively infected monkey cells;kinetics of formation and decay in enucleate cells.Proc. Natl. Acad. Sci. U.S.A. 72:2587-2591.
3. Attardi, B., and G. Attardi. 1971. Expression of themitochondrial genome in HeLa cells. 1. Properties ofthe discrete RNA components from the mitochondriafraction. J. Mol. Biol. 55:231-249.
4. Cedar, H., and G. Felsenfeld. 1973. Transcription ofchromatin in-vitro. J. Mol. Biol. 77:237-254.
5. Gariglio, P., and S. Mousset. 1975. Isolation and partialcharacterization of a nuclear RNA polymerase-SV40DNA complex. FEBS Lett. 56:149-155.
6. Gilboa, E., and H. Aviv. 1976. Preferential synthesis ofviral late RNA by nuclei isolated from SV40 lyricallyinfected cells. Cell 7:567-573.
7. Green, M. H., and T. L. Brooks. 1976. Isolation of twoforms of SV40 nucleoprotein containing RNA po-lymerase from infected monkey cells. Virology72:110-120.
8. Hayashi, M., and S. Spiegelman. 1961. The selectivesynthesis of informational RNA in bacteria. Proc.Natl. Acad. Sci. U.S.A. 47:1564-1580.
9. Khoury, G., P. Howley, D. Nathans, and M. Martin.1975. Posttranscriptional selection of simian virus 40-specific RNA. J. Virol. 15:433-437.
10. Laub, O., and Y. Aloni. 1975. Transcription of simianvirus 40. V. Regulation of simian virus 40 geneexpression. J. Virol. 16:1171-1183.
11. Lebowitz, J., C. G. Garon, M. C. Y. Chen, and N. P.Salzman. 1976. Chemical modification of simian vi-rus 40 DNA by reaction with a water-soluble carbodi-imide. J. Virol. 18:205-210.
12. Lebowitz, P., and R. Bloodgood. 1975. Transcription ofsimian virus, 40 DNA by E. coli RNA polymerase:synthesis of a DNA-RNA hybrid and discrete RNAsunder restrictive transcription conditions. J. Mol.Biol. 94:183-101.
13. May, E., H. Kopecka, and P. May. 1975. Mapping thetranscription sites of the SV40-specific late mRNA.Nucleic Acid Res. 2:1955-2005.
14. Meilhac, M., Z. Tysper, and P. Chambon. 1972. AnimalDNA-dependent RNA polymerases. 4. Studies on in-hibition by rifamycin derivatives. Eur. J. Biochem.28:291-300.
VOL. 23, 1977
on June 10, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

28 SHANI ET AL.
15. Pennman, S. 1966. RNA metabolism in HeLa cell nu-
cleus. J. Mol. Biol. 17:117-130.16. Richardson, J. P. 1975. Attachment of nascent RNA
molecules to supercoiled DNA. J. Mol. Biol. 98:656-579.
17. Shmookler, R. J., J. Buss, and M. H. Green. 1974.Properties ofthe polyoma virus transcription complexobtained from mouse nuclei. Virology 57:122-127.
18. Weinmann, R., and R. Roeder. 1974. Role of DNAdependent RNA polymerase III in the transcription ofthe t-RNA and 5S RNA genes. Proc. Natl. Acad. Sci.U.S.A. 71:1790-1794.
19. Wilhelm, J., 0. Brison, C. Kedinger, and P. Chambon.1976. Characterization of adenovirus type 2 transcrip-tional complexes isolated from infected HeLa cell nu-clei. J. Virol. 19:61-81.
J. VIROL.
on June 10, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from