prof. haim wolfson structural bionformatics 2004
TRANSCRIPT

Structural Bionformatics 2004 Prof. Haim Wolfson

Structural Bionformatics 2004 Prof. Haim Wolfson
Flexible Docking - general methodology
Major approaches :
• Rigid subpart docking (place and join):– Split the flexible molecule into rigid subparts.– Dock independently each subpart.– Pair the top hypotheses for each subpart to detect
hinge consistency.Example : Des Jarlais, Sheridan, Dixon, Kuntz,
Venkatraghavan (1986).

Structural Bionformatics 2004 Prof. Haim Wolfson
• Incremental construction method :– Position a ‘preferred’ anchor fragment. – Rotate sequentially the flexible bonds to position the
other fragments.Example: Leach & Kuntz (1992); Lengauer et al. - FLEXX.
• Hinge scoring method:– Incorporate bond information already in the initial
filtering steps by accumulating information at the hinges. – No preference for specific parts. Reminds the place and
join method yet exploits the consistency of neighboring part placement in the initial stages.
Example : Sandak, Nussinov, Wolfson (1995).

Structural Bionformatics 2004 Prof. Haim Wolfson
Search in multi-dimensional degrees of freedom (torsion angle) space :
• Evolutionary/Genetic Algorithms : – Represent degrees of freedom as strings.– Create offsprings by (genetic) combination of parents.– Re-evaluate fitness of each string and prune “weak”
hypotheses.– Jones et al. J. Mol. Bio . Vol 245 (1995), pp. 43-
….• Simulated Annealing :
AutoDock – Goodsell et al. Proteins 1990.

Structural Bionformatics 2004 Prof. Haim Wolfson
GGH based flexible docking
Applies either to flexible ligands or to flexible receptors.

Structural Bionformatics 2004 Prof. Haim Wolfson
General Algorithm outline
Can be applied either to a dataset of ligands vs a receptor or a dataset of receptors vs. a ligand.
• Calculate the molecular surface of the receptor and the ligands and their interest points (+ normals).
• Match the interest points and recover candidate multi-transformations.
• Check for inter-molecule and intra-molecule penetrations and score the amount of contact.
• Rank by energies.

Structural Bionformatics 2004 Prof. Haim Wolfson
Point Matching algorithm- prepr.• For each database molecule :
– Define r.f.’s at every hinge.– For each minimal feature (e.g. triplet) compute an r.f.
and shape signature.– For each (triplet based) reference frame compute the
transformation btwn that frame and the hinge based frame and store (molec., part, r.f., transf.) in a hash (lookup) table at an entry addressed by the r.f. shape signature.

Structural Bionformatics 2004 Prof. Haim Wolfson
Point Matching algorithm-recognition
• For the target molecule :– For each minimal feature compute an r.f. and shape
signature.– Access the table by the shape signature, and for
each transformation appearing there :• transform the r.f. to ‘hypothesized’ hinge position;• advance the counter of that hinge location for the appropriate
molecule and part.
– Check highest scoring hinges .– Verify the resulting transformations .

Structural Bionformatics 2004 Prof. Haim Wolfson
Flexible DockingCalmodulin with M13 ligand

Structural Bionformatics 2004 Prof. Haim Wolfson
Flexible Docking HIV Protease Inhibitor

Structural Bionformatics 2004 Prof. Haim Wolfson
The FlexX Algorithm
• Rarey, …, Lengauer. J. Mol. Bio., vol. 261, (1996), pp. 470-
• An incremental construction algorithm

Structural Bionformatics 2004 Prof. Haim Wolfson
The general schema
Incremental construction
Scoring function
Receptor-ligand interactions
Ligand conformational flexibility
Modeling
Algorithm
Base selection
Base placement

Structural Bionformatics 2004 Prof. Haim Wolfson
The Ligand conformational flexibility
• Approximated by a discrete set of conformations.– rotatable single bond - modeled by a
discrete set of preferred torsion angles from the MIMUMBA DB.
– Ring system - A set of ring conformations is computed with the program CORINA.

Structural Bionformatics 2004 Prof. Haim Wolfson
The model of receptor-ligand interactions
• Modeled by a few special types of interactions
• hydrogen bonds• metal acceptors bonds• hydrophobic contacts

Structural Bionformatics 2004 Prof. Haim Wolfson
The model of protein-ligand interactions – Cont.
• To each interaction group, we assign:– Interaction types – Interaction geometry ( center + surface)

Structural Bionformatics 2004 Prof. Haim Wolfson
Two groups interact if :• The centers of the groups lie approximately on the
surface of the counter group.• The interaction types are compatible
• The intermolecular interactions can be classified by the strength of their geometric constrains

Structural Bionformatics 2004 Prof. Haim Wolfson
Scoring function• Estimates the free binding energy in the complex
• The function is additive in the ligand atoms.
match score
contact score

Structural Bionformatics 2004 Prof. Haim Wolfson
Overall docking algorithm
1. Ligand fragmentation2. Select & Place a set of base fragments3. Construct the ligand by linking the
remaining fragments.

Structural Bionformatics 2004 Prof. Haim Wolfson

Structural Bionformatics 2004 Prof. Haim Wolfson
Ligand fragmentation
• The ligand is decomposed into components by cutting at each acyclic bond.
• Fragmentation is a partition of the components of the molecule, such that every part, called fragment, is connected in the component tree.

Structural Bionformatics 2004 Prof. Haim Wolfson
Ligand fragmentation
• Good results are produced if the added fragments are small
• Every fragment, except for the base fragment, consists of only one component.

Structural Bionformatics 2004 Prof. Haim Wolfson
Selecting a base fragment
• The problem: Find a fragment which leads to low energy docking solution.
• Good base fragment properties:– Placeability– Specificity

Structural Bionformatics 2004 Prof. Haim Wolfson
Selecting a base fragment –Cont.• We look for fragments maximizing the
function:

Structural Bionformatics 2004 Prof. Haim Wolfson
Rules for selecting a set of fragments
• No base fragment is fully contained in another base fragment
• Each component occurs in at most two base fragments
• Each component in a base fragment must be either necessary for the connectivity of the fragment or it must have interaction centers.

Structural Bionformatics 2004 Prof. Haim Wolfson
The base placement algorithm
• Goal: find positions of the base fragment in the active site such that sufficient number of favorable interactions between the fragment and the protein can occur simultaneously.
• Solution: pose clustering.

Structural Bionformatics 2004 Prof. Haim Wolfson
The base placement algorithm –Cont.
• Preparation: Store all triangles of interaction points (IP) of the protein in a hash table.
• Find all the compatible fragment IP’s triangles.
• Clustering of the legal transformations

Structural Bionformatics 2004 Prof. Haim Wolfson
The incremental construction algorithm
• Input: solution set - set of partial placements with the ligands constructed up to and including fragment i-1
• Output: set of partial placements with the ligands constructed up to and including fragment i

Structural Bionformatics 2004 Prof. Haim Wolfson

Structural Bionformatics 2004 Prof. Haim Wolfson
The complex construction algorithm – cont.
• Adding the next fragment in all the possible conformations
• Reject extended placements that have strong overlap with the receptor or internal overlap with the ligand.
• Searching for new interactions• Optimizing the positions of the partial ligand• Selecting a new solution set• Clustering the solution set

Structural Bionformatics 2004 Prof. Haim Wolfson
Optimizing the positions of the partial ligand
• The placement is optimized when:– New interactions are found.– The placement contains slightly overlapping
atoms between the receptor and the ligand.
( )2
rlw iii −∑

Structural Bionformatics 2004 Prof. Haim Wolfson
Selecting a new solution set
• Select k best-scoring solutions• Problem: the scoring values cannot be
compared directly when different fragments are involved.
• Solution: estimate the score of the whole ligand, given a partial placement.

Structural Bionformatics 2004 Prof. Haim Wolfson
Clustering partial solutions
• If no placement contains the other, the distance is infinity
• Otherwise, the distance is defined to be the RMSD of the intersecting atoms.
• A cluster is reduced to a single placement.

Structural Bionformatics 2004 Prof. Haim Wolfson
Exploring receptor Flexibility

Structural Bionformatics 2004 Prof. Haim Wolfson
Protein flexibility - motivation• Induced fit – side chain or even backbone
adjustments upon docking of different ligands to the same protein.
• Even small conformational changes are critical for docking applications e.g. if a rotatable bond prevents a ligand from binding in the correct position.

Structural Bionformatics 2004 Prof. Haim Wolfson
Protein flexibelity• Main idea: describe the protein structure
variations with a set of protein structures representing the flexibility, mutation or alternative models of a protein.
• The variability considered by FlexE is defined by the differences within the given input structures.

Structural Bionformatics 2004 Prof. Haim Wolfson
United protein description
• Data structure that handles the protein structures variations.
• Contains an ensemble of up to 30 possible conformation of the protein.
• Most of them are low energy conformations of the same protein.
Structural Bionformatics 2004 Prof. Haim Wolfson
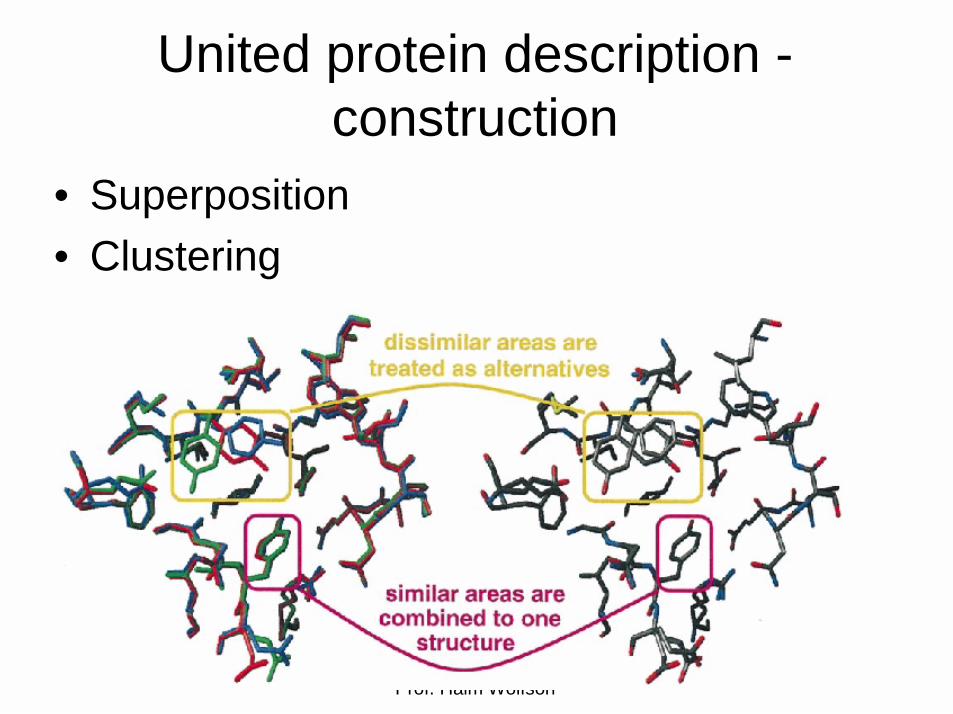
United protein description -construction
• Superposition• Clustering
Add picture - 8

Structural Bionformatics 2004 Prof. Haim Wolfson
Notation
• Component : all the atoms which belong to the same amino acid or mutation of the amino acid. Contains a backbone part and a side chain part
• Part : set of instances• Instance : one of the
alternative conformations.

Structural Bionformatics 2004 Prof. Haim Wolfson
United protein description -clustering
• The superimposed structures are combined by clustering each part separately
• Complete linkage hierarchical cluster• The clustered instances can be
recombined to form new valid protein structures.

Structural Bionformatics 2004 Prof. Haim Wolfson
Incompatibility
• Two instances of the united protein description are incompatible if they cannot be realized simultaneously. – Logical: two instances are
alternative to each other– Geometric: two logically
compatible instances overlap– Structural: two instances of
the same chain are unconnected

Structural Bionformatics 2004 Prof. Haim Wolfson
Incompatibility graph
{ }}bleincompatiaandE
cesinsV
vve jiij=
= tan

Structural Bionformatics 2004 Prof. Haim Wolfson
Incompatibility graph• The incompatibility is
internally represented as a graph by using the instances as nodes and the connecting pairs of incompatible nodes by an edge.
• Valid protein structures correspond to independent sets in the graph.

Structural Bionformatics 2004 Prof. Haim Wolfson
Selection of instances
• The ligand is placed fragment by fragment into the active site by the incremental construction algorithm.
• After each construction step, all possible interactions are determined.
• Apply the scoring function for each instance.
• We chose the IS with the highest score.

Structural Bionformatics 2004 Prof. Haim Wolfson
• The IS can be assembled from IS of the connected components.
• Apply a modified version of the Bron-Kerbosch algorithm.
Select the optimal IS

Structural Bionformatics 2004 Prof. Haim Wolfson
Evaluation
• FlexE was evaluated with ten protein structures ensembles containing 105 crystal structure from the PDB.
• The structures within the ensemble – highly similar backbone trace– Different conformations for several side
chains.

Structural Bionformatics 2004 Prof. Haim Wolfson

Structural Bionformatics 2004 Prof. Haim Wolfson
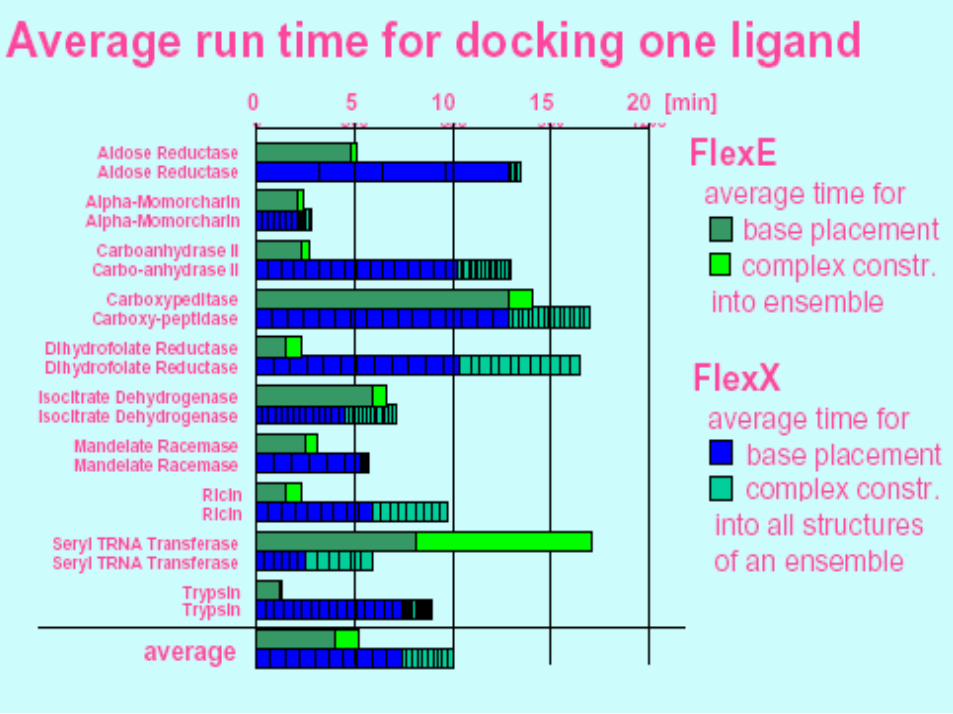
Evaluation – Cont.
• FlexE finds a ligand position with RMSD below 2 A in 67% of the cases.
• Average CPU time for the incremental construction algorithm is 5.5 minutes.
Structural Bionformatics 2004 Prof. Haim Wolfson

Structural Bionformatics 2004 Prof. Haim Wolfson
Discussion
• The ensemble approach is able to cope with several side-chains conformations and even movements of loops.
• Motions of larger backbone segments or even domains movements are not covered by this approach.

Structural Bionformatics 2004 Prof. Haim Wolfson
FlexDockFlexDock: Algorithm Stages: Algorithm Stages
Rigid Parts Docking via Geometric Hashing
BB
Assembly of partial dockings into a flexible result
AA
AA
AAAA
BBAA
AA

Structural Bionformatics 2004 Prof. Haim Wolfson
Flexible Assembly StageFlexible Assembly Stage
NODE: NODE: transformation, scoretransformation, score
Part 1 resultsPart 1 results Part 2 resultsPart 2 results Part 3 resultsPart 3 results

Structural Bionformatics 2004 Prof. Haim Wolfson
Results CompatibilityResults Compatibility
BB22
BB11
AA AA
Two docking results are compatible if and only if:
(1) Their transformations superimpose the hinge point into the same location (approximately).
(2) The parts are not penetrating.
AA
BB11BB22
Note: compatible results may have some shape complementarity

Structural Bionformatics 2004 Prof. Haim Wolfson
Flexible AssemblyFlexible Assembly
ss tt
NODE: NODE: transformation, scoretransformation, score
Part 1 resultsPart 1 results Part 2 resultsPart 2 results Part 3 resultsPart 3 results
EDGE: EDGE: parts docking scoreparts docking score

Structural Bionformatics 2004 Prof. Haim Wolfson
Flexible Assembly GraphFlexible Assembly Graph
DAG:DAG: Directed Acyclic Graph.Directed Acyclic Graph.
NODE:NODE: part transformation, score.part transformation, score.
EDGE:EDGE: connects compatible parts, score of connects compatible parts, score of docking between the parts.docking between the parts.
DOCKING PATH:DOCKING PATH: a path between s and t.a path between s and t.
PATH SCORE:PATH SCORE: sum of nodes and edges scores.sum of nodes and edges scores.
Goal:Goal: find find KK best paths in the assembly graph.best paths in the assembly graph.
Solution:Solution: dynamic programming.dynamic programming.