primary and secondary metabolism, and post-translational...
TRANSCRIPT

Molecular Microbiology (2002)
46
(4) 917ndash932
copy 2002 Blackwell Publishing Ltd
Blackwell Science LtdOxford UKMMIMolecular Microbiology0950-382XBlackwell Science 200246Original Article
Proteomic analysis of S coelicolorA R Hesketh et al
Accepted 15 August 2002 For correspondence E-mailkeithchaterbbsrcacuk Tel (
+
44) 1603 450 297 Fax (
+
44) 1603450 778
Primary and secondary metabolism and post-translational protein modifications as portrayed by proteomic analysis of
Streptomyces coelicolor
A R Hesketh
1
G Chandra
1
A D Shaw
23
J J Rowland
3
D B Kell
2
M J Bibb
1
and K F Chater
1
1
Department of Molecular Microbiology John Innes Centre Norwich Research Park Colney Norwich NR4 7UH UK
2
Institute of Biological Sciences and
3
Department of Computer Science University of Wales Aberystwyth SY23 3DD UK
Summary
The newly sequenced genome of
Streptomycescoelicolor
is estimated to encode 7825 theoreticalproteins We have mapped approximately 10 ofthe theoretical proteome experimentally using two-dimensional gel electrophoresis and matrix-assistedlaser desorption ionization time-of-flight (MALDI-TOF)mass spectrometry Products from 770 differentgenes were identified and the types of proteinsrepresented are discussed in terms of their anno-tated functional classes An average of 12 proteinsper gene was observed indicating extensive post-translational regulation Examples of modification byN-acetylation adenylylation and proteolytic process-ing were characterized using mass spectrometryProteins from both primary and certain secondarymetabolic pathways are strongly represented on themap and a number of these enzymes were identifiedat more than one two-dimensional gel location Post-translational modification mechanisms may thereforeplay a significant role in the regulation of these path-ways Unexpectedly one of the enzymes for synthesisof the actinorhodin polyketide antibiotic appears tobe located outside the cytoplasmic compartmentwithin the cell wall matrix Of 20 gene clusters encod-ing enzymes characteristic of secondary metabolismeight are represented on the proteome map includingthree that specify the production of novel metabolites
This information will be valuable in the characteriza-tion of the new metabolites
Introduction
The genome of a plasmid-free derivative strain M145of the Gram-positive bacterium
Streptomyces coelicolor
A3(2) has recently been completely sequenced and anno-tated (Bentley
et al
2002) At 867 Mb and 7825 anno-tated genes it is nearly twice the size of the
Escherichiacoli
(Blattner
et al
1997) and
Bacillus subtilis
(Kunst
et al
1997) genomes presumably reflecting the lifestyleof the organism Streptomycetes are mycelial saprophyticsoil bacteria that undergo complex morphological differ-entiation and produce a range of diverse secondarymetabolites with important applications in human medi-cine and agriculture (for a review see Champness 2000)Spore germination is followed by the development ofbranched hyphae that grow on and into appropriate sub-strates In response to complex but still poorly definedsignals the substrate mycelium produces aerial hyphaethat eventually undergo septation to yield chains of unige-nomic spores As the aerial branches grow the substratemycelium typically begins to produce the various anti-biotics Before the genome sequencing project began
Scoelicolor
was known to produce four antibiotics (one ofthem plasmid determined) two of which actinorhodin(Act) and undecylprodigiosin (Red) are pigmented and apolyketide spore pigment It is now apparent that there areabout 20 gene clusters that are likely to direct the biosyn-thesis of what may broadly be considered as secondarymetabolites (Bentley
et al
2002) In addition Bentley
et al
(2002) predicted more than 800 secreted proteins65 sigma factors 37 SerThr kinase homologues 85 two-component sensor histidine kinases and a host of tran-scriptional regulators
The complete sequencing of an organismrsquos genomeimmediately allows study of overall gene expression at thelevels of mRNA abundance typically using DNA micro-arrays (the transcriptome) (Schena 2000 Lucchini
et al
2001) and protein profiling by two-dimensional (2D)PAGE coupled with high-throughput matrix-assisted laserdesorption ionization time-of-flight (MALDI-TOF) mass
918
A R Hesketh
et al
copy 2002 Blackwell Publishing Ltd
Molecular Microbiology
46
917ndash932
spectrometry (the proteome) (Blackstock and Weir 1999Mann
et al
2001) Analysis of the proteome is morecomplicated than transcriptome analysis because of thediverse physical and chemical properties of proteins andbecause of the need for any given protein to be extractedin sufficient quantity not only to be detected but also tobe identified Thompson and co-workers have used 2D-PAGE extensively to analyse changes in the pattern ofradiolabelled protein profiles in pulsendashchase experimentsin
S coelicolor
during growth and in response to stressbut did not have the benefit of the complete genomesequence to use mass spectrometry to identify proteinsof interest (Puglia
et al
1995 Vohradsky
et al
19972000) In this study we have mapped a substantial partof the
S coelicolor
proteome using 2D-PAGE and MALDI-TOF peptide mass fingerprint analysis Particular atten-tion is paid to the representation of proteins involved insecondary metabolic pathways in comparison with thosefrom primary metabolism and to the extent of regulationby post-translational modification mechanisms
Results
Analysis of the proteome using 2D-PAGE
Streptomyces coelicolor
M145 produces the pigmentedantibiotics actinorhodin (Act) and the prodigiosin complex(Red) in a growth phase-dependent manner (Takano
et al
1992 Gramajo
et al
1993) Thus in liquid minimalmedium supplemented with casamino acids (SMM) pig-ment production begins at the transition between expo-nential growth and stationary phase In order to observeproteins from both primary and secondary metabolic path-ways pigmented mycelium from transition phase cultureswas harvested and disrupted directly into the stronglydenaturing isoelectric focusing buffer UTCHAPS (see
Experimental procedures
) Protein extracts were sepa-
rated by 2D-PAGE using several different immobilized pHgradient (IPG) strips for the first-dimension separation(pH 4ndash7 pH 6ndash11 pH 45ndash55 and pH 55ndash67) 125SDSndashPAGE for the second and silver nitrate staining forprotein detection (Fig 1) The proteins visible on each gelwere counted after spot detection of scanned gel imagesusing
PHORETIX
2D image analysis software and com-pared with the total number expected assuming that everypotential gene was expressed (Table 1) To calculate thenumber of spots expected proteins in the theoretical pro-teome with predicted isoelectric point values within the pHrange of the IPG strip used and with molecular weightsbetween 8 kDa and 140 kDa (the detectable range using125 PAGE) were counted and multiplied by a factor of12 representing the observed average number of proteinspots per gene (see Table 1) Separation using the iso-electric point (pI) range pH 4ndash7 produced 1051 detectablespots representing 19 of the total number of proteinspredicted for this region Excluding secreted and mem-brane proteins which would not generally be expected inthe protein extract the proportion was 25 In the pIrange pH 6ndash11 these numbers were 8 for the theoreti-cal total proteome and 12 for the theoretical extractableproteome Using the narrow-range IPG strips pH 45ndash55and pH 55ndash67 1555 and 913 spots were detectedrespectively corresponding to 82 and 41 of the totalnumber of predicted extractable proteins Thus the higherresolution and higher loading possible on these lsquozoomgelsrsquo approximately doubled the number of proteinsdetectable
Identification of separated proteins towards a proteome map and database
Protein spot identification was performed exclusively ongels stained with colloidal Coomassie G250 and mapped
Table 1
Estimate of the proportion of the
S coelicolor
proteome that can be detected by silver staining of 2D-PAGE separations of transitionphase protein extracts and MALDI-TOF identification of protein spots illustrating the extent of post-translational modifications
Isoelectric point range (pH)
4ndash7 45ndash55 55ndash67 6ndash11
Protein spots detected
a
1051 1555 913 379Percentage of expected 19 63 37 8protein spots detected
b
of 5418 of 2462 of 2470 of 4792Percentage of expected 25 82 41 12extractable protein spots detected
c
of 4221 of 1886 of 2218 of 3112Protein spots identified
d
443 459 193 210Different gene products represented 374 360 170 179Ratio of protein spots per gene product 118 127 113 117
a
On silver-stained gels
b
The total number of gene products with theoretical isoelectric point values and molecular weights within the range of the gel separation(8ndash140 kDa) was multiplied by 12 the observed average number of protein spots per gene as a result of post-translational modifications
c
Same as a but excluding proteins annotated as being membrane or secreted
d
From colloidal Coomassie-stained gels
Proteomic analysis of
S coelicolor 919
copy 2002 Blackwell Publishing Ltd
Molecular Microbiology
46
917ndash932
Fig 1
A proteome map of
S coelicolor
showing 2D-PAGE separations using IPG strips pH 4ndash7 pH 6ndash11 pH 45ndash55 and pH 55-67 The number of spots identified is given at the foot of each reference gel Protein spots from metabolic pathways that were identified at positions inconsistent with their predicted amino acid sequences and suggestive of post-translational modification are marked in yellow (primary metabolism) or red (secondary metabolism) Blue spots correspond to other examples of post-translational modifications that are mentioned in the text and in Table 3 Separations have been performed on numerous occasions and generally show high spatial reproducibility
920
A R Hesketh
et al
copy 2002 Blackwell Publishing Ltd
Molecular Microbiology
46
917ndash932
back onto the analytical silver-stained gels in Fig 1Although silver staining is 5ndash10 times more sensitive thancolloidal Coomassie staining (and about 100 times moresensitive than conventional Coomassie staining) furthermass spectrometry analysis of protein spots is much moreefficient after staining with colloidal Coomassie than withsilver A total of 1305 protein spots were unambiguouslyassigned to sequenced genes using MALDI-TOF peptidemass fingerprint analysis at a success rate of
ordf
90(Table 1) There is some redundancy in these figures withseveral spots being represented in more than one pIrange On each gel some proteins were identified in morethan one position indicative of post-translational modifi-cations The average number of protein spots per geneproduct was 12 The products from 770 different geneswere assigned to the proteome map representing
ordf
10of the genome and corresponding to many differentclasses of proteins (Table 2) The master gel referencemaps are the basis for the production of a publicly acces-sible proteome database (httpqbababeracukS_coeliReferenceGelrefgelhtml see last Results section)
Analysis of co- and post-translational modifications
The results of MALDI-TOF identification of protein spotssummarized in Table 1 indicated a significant amount ofregulation at the post-transcriptional level in
S coelicolor
as in most other bacteria Table 3 lists those gene prod-ucts that were identified at pI andor molecular weight co-ordinates significantly different from predicted values Ingeneral these fell into two categories one or more spotswith lower than expected molecular weights and oftendifferent pI values indicating modification by proteolyticprocessing (Table 3A) and multiple spots with the sameapparent molecular weights but different pI values indi-
cating modification by covalent addition of a small adduct(Table 3B) In the former case the full-length protein wasoften but not always detected A similar fraction ofproteins of unknown function and annotated as lsquohypo-theticalrsquo or lsquoconserved hypotheticalrsquo was subject to post-translational modification but these will be described ina later publication and are not considered in Table 3Evidence in the peptide mass fingerprint data for the spe-cific nature of the modification was obtained in 10 out ofthe 88 proteins listed (Table 4) Figure 2 illustrates thecharacterization of an example of each type of modifica-tion by interpretation of the MALDI-TOF data
N-acetylation
The putative cellulose-binding proteinSCO5396 was identified in two positions on the proteomemap (spots labelled 10 in Fig 1) with the same apparentmolecular weight but significantly different pI values Themore basic spot gave a tryptic peptide at 137076 Dacorresponding to the N-terminal peptide 2ndash13 indicatingthat the initiator Met residue had been co-translationallyremoved (Fig 2A top) In the more acidic spot (ie on theleft in Fig 1) this peptide had increased in mass by420 Da diagnostic of the addition of an acetyl grouppresumably on the N-terminal Ser residue (Fig 2A bot-tom) A similar modification was identified for the 20Sproteasome alpha subunit (spots 12 in Fig 1) and for theputative ABC transport protein BldK-ORFD (spots 17 inFig 1) although the presumptive N-terminal acetylatedresidue is Thr in the latter (data not shown)
Adenylylation
By analogy with the nitrogen regulatorysystem in
E coli
glutamine synthetase I (GSI) wasalready believed to be modified by adenylylation on aconserved Tyr-397 residue and biochemical evidence forthis has been reported previously (Fink
et al
1999) TheMALDI-TOF data showed the tryptic peptide 395ndash420 atthe expected mass of 28074 Da for the larger more basicspot 6 in Fig 1 but this increased by 3290 Da corre-sponding to the addition of an adenylyl group in thesmaller spot on the left (Fig 2B) Adenylylation of thenitrogen regulatory protein GlnK (spot 15 in Fig 1) wasidentified similarly but this is described in detail elsewhere(Hesketh
et al
2002)
Proteolytic processing
The 50S ribosomal protein L9was identified as two spots that differed in both pI andmolecular weight (spots 30 in Fig 1) suggesting modifi-cation by proteolytic processing The spot with the highermolecular weight showed the tryptic peptide correspond-ing to amino acids 3ndash22 at the expected mass of199310 Da in agreement with this being the unproc-essed form of the protein (Fig 2C top) In the lower ofthe two spots the peptide decreased in mass by11312 Da (Fig 2C bottom) corresponding to the loss of
Table 2
Types of proteins identified on the proteome map
Protein class
a
Number ingenome
a
Number identified
b
Hypothetical 1049 68 (6)Conserved hypothetical 1322 114 (8)Secreted 530 30 (5)ABC transport ATP binding 109 16 (14)Lipoproteins 124 10 (8)Sigma factors 65 0 (0)Antisigma factors 7 2 (28)Antisigma factor antagonists 15 1 (6)Two-component system regulators 80 11 (13)Other regulators 592 36 (6)Two-component system kinases 85 0 (0)SerThr kinases 37 1 (2)Other 3923 482 (12)All 7816 770 (10)
a
Based on annotation of the
S coelicolor
genome sequence(httpwwwsangeracukProjectsS_coelicolor)
b
Percentage identified is included in parentheses
Proteomic analysis of
S coelicolor 921
copy 2002 Blackwell Publishing Ltd
Molecular Microbiology
46
917ndash932
Table 3
Proteins identified at pI and molecular weight co-ordinates on the 2D-PAGE maps that are inconsistent with their predicted amino acidsequences indicating possible post-translational modification either by proteolytic processing (A) or by covalent addition of a low-molecular-weight adduct (B)
ORF no Gene product Spot no in Fig 1
ASCO0498 Probable peptide monooxygenase 33SCO1230 Putative tripeptidylaminopeptidaseSCO1644 20S proteasome beta subunit precursor 35SCO1815 3-Oxoacyl-[acyl carrier protein] reductaseSCO1860 Putative secreted proteinSCO1947 Glyceraldehyde-3-phosphate dehydrogenase 9SCO1998 30S ribosomal protein S1SCO2619 ATP-dependent Clp protease subunit 13SCO2920 Probable secreted proteinaseSCO3236 Probable oxygenase 34SCO3671 Heat shock protein DNAKSCO3909 50S ribosomal protein L9 30SCO4296 Chaperonin GroEL2SCO4439
D
-Alanyl-
D
-alanine carboxypeptidaseSCO4643 UDP-N-acetylanoylpyruvoylglucosamine reductaseSCO4662 Elongation factor Tu1SCO4762 Chaperonin GroEL1SCO4808 Succinyl-CoA synthetase beta chain 8SCO4824 Bifunctional protein methylene tetra-hydrofolate dehydrogenasecyclohydrolaseSCO5073 Putative oxidoreductase ActVI-ORF2 29SCO5074 Putative dehydratase ActVI-ORF3 31SCO5088 Ketoacyl synthase beta subunit ActI-ORF2 24SCO5113 Oligopeptide ABC transport protein BldK-ORFBSCO5254 Superoxide dismutase precursor 32SCO5371 ATP synthase alpha chainSCO5477 Probable oligopeptide-binding lipoproteinSCO5624 30S ribosomal protein S2SCO5737 Guanosine pentaphosphate synthetaseSCO5769 Recombinase RecASCO5892 Polyketide synthase RedL 18SCO6282 3-Oxoacyl-[acyl carrier protein] reductase 11
BSCO1074 Putative peptidaseSCO1476 S-adenosylmethionine synthetaseSCO1594 Phenyl-tRNA synthetase beta chainSCO1643 20S proteasome alpha subunit 12SCO1814 Enoyl-[acyl carrier protein] reductaseSCO1935 Transketolase 3SCO1947 Glyceraldehyde-3-phosphate dehydrogenase 9SCO2180 Dihydrolipoamide dehydrogenase 21SCO2181 Probable dihydrolipoamide S-succinyltransferase 14SCO2198 Glutamine synthetase I 6SCO2390 3-Oxoacyl-[acyl carrier protein] synthase IISCO2478 Putative reductaseSCO2618 ATP-dependent Clp protease subunit 2SCO2822 Putative decarboxylaseSCO3096 Enolase 23SCO3229 4-Hydroxyphenylpyruvic acid dioxygenaseSCO3337 Probable pyrroline-5-carboxylate reductase 28SCO3409 Inorganic pyrophosphataseSCO3549 Antisigma factor antagonist BldGSCO3563 Acetyl-CoA synthaseSCO3671 Heat shock protein DNAKSCO3878 DNA polymerase III beta chainSCO3890 Thioredoxin reductaseSCO3906 30S ribosomal protein S6SCO3970 XAA-Pro aminopeptidase ISCO4164 Putative thiosulphate sulphurtransferaseSCO4209 Phosphoglycerate mutase 19SCO4240 ABC transport ATP binding protein MsiKSCO4296 Chaperonin GroEL2SCO4645 Aspartate aminotransferase 7SCO4652 50S ribosomal protein L10
Some proteins appear to be subject to both types of modification and are present in both parts Only proteins from primary or secondary metabolicpathways or with identified modifications are assigned to spots in Fig 1
922
A R Hesketh
et al
copy 2002 Blackwell Publishing Ltd
Molecular Microbiology
46
917ndash932
the isoleucine residue at its N-terminus (expected massloss 11308 Da) This indicates removal of the N-terminalMKI residues from ribosomal protein L9 The theoreticalisoelectric point (997) and molecular weight (156 kDa) ofL9 protein N-terminally truncated in this way are in goodagreement with the position of the lower spot in Fig 1(predicted values for the unprocessed protein are pH 960and 159 kDa) MALDI-TOF data indicating proteolytic
processing of the superoxide dismutase precursor protein(SCO5254 spot 32 in Fig 1) and the 20S proteasomebeta subunit precursor (SCO1644 spot 35 in Fig 1)agreed with previous reports (Kim
et al
1998 Nagy
et al
1998) Evidence for the proteolysis of
S coelicolor
ClpP1(SCO2619 spot 13 in Fig 1) has also been reportedalthough the processing site was not defined (de Crecy-Lagard
et al
1999)
SCO4661 Elongation factor GSCO4662 Elongation factor Tu1SCO4677 Putative regulatory proteinSCO4729 DNA-directed RNA polymerase alpha chainSCO4762 Chaperonin GroEL1SCO4770 Inosine-5
cent
-monophosphate dehydrogenase 25SCO4792 LuxR family two-component regulatorSCO4808 Succinyl-CoA synthetase beta chain 8SCO4809 Succinyl-CoA synthetase alpha chain 22SCO4921 Probable acyl-CoA carboxylase complex A chainSCO5071 Hydroxylacyl-CoA dehydrogenase ActVI-ORFA 16SCO5079 Hypothetical protein ActVA-ORF4 20SCO5080 Possible hydrolase ActVA-ORF5 26SCO5115 Probable ABC transporter intracellular ATPase chain BldK-ORFD 17SCO5371 ATP synthase alpha chainSCO5396 Putative cellulose-binding protein 10SCO5419 Putative thioredoxinSCO5515 Probable
D
-3-phosphoglycerate dehydrogenase 4SCO5535 Putative carboxyl transferaseSCO5584 Nitrogen regulatory protein GlnK 15SCO5625 Elongation factor TsSCO5699 Prolyl-tRNA synthetaseSCO5737 Guanosine pentaphosphate synthetaseSCO5777 Glutamate uptake system ATP-binding proteinSCO5803 SOS regulatory protein LexASCO5999 Aconitase 1SCO6198 Probable secreted proteinSCO6282 3-Oxoacyl-[acyl carrier protein] reductase 11SCO6636 Bacteriophage
f
-C31 resistance protein PglZSCO6663 Transketolase 27SCO7000 Isocitrate dehydrogenase 2SCO7036 Argininosuccinate synthase 5SCO7510 Peptidyl-prolyl
cis-trans
isomerase
ORF no Gene product Spot no in Fig 1
Some proteins appear to be subject to both types of modification and are present in both parts Only proteins from primary or secondary metabolicpathways or with identified modifications are assigned to spots in Fig 1
Table 3
cont
Table 4
Co- and post-translational modification of proteins suggested by MALDI-TOF peptide mass fingerprint analysis
Protein Modification
SCO1643 20S proteasome alpha subunit Removal of initiator Met residue and acetylation of N-terminal SerSCO5115 ABC transporter intracellular Removal of initiator Met residue and acetylation of N-terminal Thr
ATPase BldK-ORFDSCO5396 Putative cellulose-binding protein Removal of initiator Met residue and acetylation of N-terminal SerSCO2198 Glutamine synthetase I Adenylylation of peptide 395ndash420SCO5584 Nitrogen regulatory protein GlnK Adenylylation of peptide 48ndash58SCO1644 20S proteasome
b
-subunit precursor Proteolytic processing between amino acid residues 5354SCO2619 ATP-dependent Clp protease Proteolytic processing between amino acid residues 2223
proteolytic subunit ISCO3909 50S ribosomal protein L9 Proteolytic processing between amino acid residues 34SCO5074 Putative dehydratase ActVI-ORF3 Proteolytic processing between amino acid residues 3132 and 3435SCO5254 Superoxide dismutase precursor Proteolytic processing between amino acid residues 1415
Proteomic analysis of
S coelicolor 923
copy 2002 Blackwell Publishing Ltd
Molecular Microbiology
46
917ndash932
Co-translational modification by removal of the N-terminal initiator Met occurred in 149 out of 190 (78)cases in which the N-terminal peptide was detected Pro-cessing by the N-methionylaminopeptidase was observedif the second amino acid was Ser (45 out of 149 exam-ples) Thr (40) Ala (32) Pro (27) Gly (4) or Val (1) Ononly one occasion was any of these six amino acids foundas the second residue if the N-terminal methionine wasretained This is in broad agreement with observations in
B subtilis
in which two-thirds of proteins were reported tobe similarly processed (Buttner
et al
2001)
Representation of primary and secondary metabolic pathways
Streptomycetes produce
ordf
60 of all commercially usefulantibiotics (Berdy 1984 Miyodah 1993) Most antibioticsare the product of complex biosynthetic pathways oftenencoded by 20ndash30 clustered genes Production of thepigmented antibiotics actinorhodin (Act) and undecyl-
prodigiosin (Red) has been studied extensively in
Scoelicolor
(for reviews see Chater and Bibb 1997Champness 2000) and involves clusters of approximately22 genes each A third cluster producing the calcium-dependent antibiotic (CDA) and consisting of 40 geneshas also been studied (Chong
et al
1998 Huang
et al
2001) In the extensive spot identification studies for thepreparation of a proteome map of
S coelicolor
(seeabove) more than one-third of the proteins from each ofthese pathways was identified (Table 5) Proteins fromthree clusters that specify metabolic products that haveyet to be identified experimentally (Bentley
et al
2002)were also extensively represented a type I polyketidesynthase cluster (SCO6273ndash6288) a deoxysugarglycosyltransferase cluster (SCO0381ndash0401) and anon-ribosomal peptide synthetase cluster predicted toencode a novel siderophore coelichelin (SCO0489ndash0499) Several clusters were unrepresented
The primary metabolic pathways were somewhat morehighly represented with
ordf
60 or more of proteins anno-
Fig 2
Examples of MALDI-TOF peptide mass fingerprint data identifying post-translational modification of proteins by (A) acetylation (predicted mass increase of 420 Da) (B) adenylylation (predicted mass increase of 3290 Da) and (C) proteolytic processing (predicted mass loss for Ile of 11308 Da) In each fingerprint the
x
-axis is masscharge ratio (mz) and the
y
-axis is absolute intensity The observed mass accuracies in the MALDI-TOF fingerprints are (A) 60 ppm (B) 20 ppm and (C) 20 ppm
924
A R Hesketh
et al
copy 2002 Blackwell Publishing Ltd
Molecular Microbiology
46
917ndash932
tated as enzymes of glycolysis the TCA cycle thepentose phosphate pathway purine ribonucleotide bio-synthesis and pyrimidine nucleotide biosynthesis beingmapped (Table 6) About 50 of amino acid biosyntheticproteins were identified Lists of individual members of allpathways considered here including those that were andwere not identified during proteome mapping can befound in the
Supplementary material
Several proteins from both primary and secondary met-
abolic pathways were identified at more than one positionon the proteome map and may therefore be subject toregulation by post-translational modification (see Table 3and Fig 1) Primary metabolic proteins of this type (yellowspots on Fig 1) were usually detected as chains of two ormore spots with the same apparent molecular weights butdifferent pI values suggesting covalent modification bylow-molecular-weight adducts Apart from adenylylation ofGSI (see above) the nature of the modification of theseproteins was not revealed by MALDI-TOF peptide massfingerprint data Both covalent modification and proteolyticprocessing were also found in secondary metabolic pro-teins (red spots in Fig 1) For example the Act pathwaymembers SCO5071 (spot 16) and SCO5075 (spot 20)
and the putative 3-oxoacyl[acyl carrier protein] reductasefrom the type I polyketide cluster (SCO6282 spot 11)appear to be covalently modified whereas proteolytic pro-cessing was detected for RedL (SCO5892 spot 18) fromthe undecylprodigiosin cluster the putative dehydrataseActVI-ORF3 (SCO5074 spot 31) from the Act cluster theputative peptide monooxygenase from coelichelin biosyn-thesis (SCO0498 spot 34) and the putative oxygenasefrom the CDA pathway (SCO3236 spot 33) Two Act bio-synthetic proteins SCO5088 (spot 24) and SCO5073(spot 29) were detected both at their predicted pI andmolecular weight co-ordinates and at positions with simi-lar pI values but significantly higher molecular weightsThis may indicate that these proteins form stable mul-timers that are not completely denatured under theconditions used Only the modification of ActVI-ORF3(SCO5074 spot 31) could be characterized further fromthe mass spectrometry data which showed that it involvesproteolytic processing at two sites between amino acidresidues 3132 and 3435 (data not shown) Analysis of asample obtained by externally extracting proteins fromintact washed mycelium with a solution containing 2SDS and 50 mM dithiothreitol (DTT) revealed that sur-
Table 5
Summary of proteins identified on the proteome map from secondary metabolic pathways during growth in SMM
Biosynthetic pathwaygene cluster
Pathwaygenesannotated
Pathwaygene productsidentified
Percentagepathwaydetected
Calcium-dependent antibiotic CDA SCO3210ndash3249 40 16 40Actinorhodin SCO5071ndash5092 22 15 68Undecylprodigiosin SCO5877ndash5898 22 8 36Deoxysugar synthaseglycosyltransferase cluster SCO0381ndash0401 21 7 33Type I polyketide synthase SCO6273ndash6288 16 6 38Hopanoids SCO6759ndash6771 13 1 7Siderophore (coelichelin) SCO0489ndash0499 11 5 45Siderophore (desferrioxamine) SCO2783ndash2785 4 4 100
Chromosomal locations and extent of gene clusters are taken from Bentley
et al
(2002) No proteins were detected from the remaining predictedsecondary metabolic clusters ie siderophore (coelibactin) SCO7681ndash7691 (11 ORFs) siderophore SCO5799ndash5801 (three ORFs) non-ribosomal peptide synthases SCO6429ndash6438 (10 ORFs) type II fatty acid synthases SCO1265ndash1273 (nine ORFs) WhiE spore pigment clusterSCO5314ndash5320 (seven ORFs) isorenieratine SCO0185ndash0191 (seven ORFs) eicosapentaenoic acid SCO0124ndash0129 (six ORFs) chalconesynthases SCO1206ndash1208 (three ORFs) chalcone synthases SCO7669ndash7671 and SCO7222 (three ORFs) sesquiterpene cyclase SCO5222ndash5223 (two ORFs) type I polyketide synthases SCO6826ndash6827 (two ORFs) geosmin SCO6703 (one ORF)
Table 6 Summary of proteins identified on the proteome map from primary metabolic pathways during growth in SMM
Biosynthetic pathwayPathway genesannotateda
Pathway geneproducts identified
Percentage pathway detected
Glycolysisb 24 13 55Pentose phosphate pathwayb 14 10 71TCA cycleb 21 12 57Amino acid biosynthesisc 115 55 48Purine ribonucleotide biosynthesisb 18 12 67Pyrimidine ribonucleotide biosynthesisb 8 5 62
a Derived from interpretation of the annotated S coelicolor genome sequence at httpwwwsangeracukProjectsS_coelicolorb See Supplementary material for details of pathway membersc See Supplementary material for details of individual pathways
Proteomic analysis of S coelicolor 925
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
prisingly the truncated ActVI-ORF3 is located extracellu-larly presumably non-covalently associated with the cellwall matrix (Fig 4)
Expression analysis of certain primary and secondary metabolic proteins
Production of the pigmented antibiotics Act and Red inliquid minimal medium supplemented with casamino acids(SMM) begins in the transition phase between exponentialgrowth and stationary phase Figure 3 shows a compari-son of the expression of certain primary and secondarymetabolic proteins between exponentially growing andstationary phase cultures Two proteins from the Actcluster (ActVA-ORF5 and ActI-ORF2) and two from theRed (RedI and RedR) were present in the stationaryphase cultures but were not detected during exponentialgrowth Similarly the putative 3-oxoacyl-[acyl carrierprotein] reductase (SCO6282) and epoxide hydrolase(SCO6277) enzymes from a type I polyketide secondarymetabolic cluster showed a large increase in abundance
between the exponential and stationary phase samplesTriose phosphate isomerase (SCO1945) fructose bis-phosphate aldolase (SCO3649) and succinyl-CoA syn-thetase alpha chain (SCO4809) all enzymes from centralcarbon metabolism were present in approximatelyconstant amounts in both samples Interestingly two iso-forms (SCO7511 and SCO1947) of glyceraldehyde-3-phosphate dehydrogenase from the glycolytic pathwayshowed different patterns of expression SCO1947 levelswere the same in both exponential and stationary phasebut SCO7511 barely detectable in exponentially growingcultures increased markedly in stationary phase
The Streptomyces coelicolor online proteome map
The 2D gel images in Fig 1 together with the spot iden-tification results are being made available over the WorldWide Web to provide a reference map of the S coelicolorproteome At the time of writing the reference gel forthe pH 4ndash7 isoelectric point range was fully accessiblewhereas those for pH 45ndash55 pH 55ndash67 and pH 6ndash11
Fig 3 A comparison of the expression of certain primary and secondary metabolic proteins between exponentially growing and stationary phase cultures of S coelicolor M145 Larger arrows represent higher protein level in one gel than in the other The experiment was performed on duplicate biological samples and both showed similar protein expression profiles
926 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
were under construction (httpqbababeracukS_coeliReferenceGelrefgelhtml) A tool for comparing two ormore gel images with each other is also accessible fromthe same website at httpqbababeracukS_coeliflickerhtml
Discussion
Silver staining is one of the most sensitive methods fordetecting proteins on polyacrylamide gels allowing aslittle as 05ndash2 ng of an individual protein to be detected(Berggren et al 2000 Gorg et al 2000) The proportionof the S coelicolor proteome that can be analysed usingsilver staining of 2D-PAGE-separated proteins dependsat least in part on the pH range of the IPG strip used forthe first-dimension separation (see Table 1) Using theintermediate-range IPG strip covering pH 4ndash7 about 20ndash25 of the proteins in the theoretical proteome predictedto be observable in the resulting 2D-PAGE gel were actu-ally detected This figure was reduced to 10ndash12 whenanalysing the more basic pI range using the pH 6ndash11 IPGstrip Attempts to increase the percentage of the proteomedetectable using the pH 4ndash7 and pH 6ndash11 IPG strips byloading more protein sample resulted in poorer separa-tions (data not shown) This was especially true of thepH 6ndash11 strips which were generally the most difficult touse successfully However good separation of higher pro-tein loads was achieved using the narrow-range IPG stripspH 45ndash55 and 55ndash67 significantly increasing the pro-portion of theoretical proteins detected to over 40Clearly and more pertinently the limited conditions usedfor culturing S coelicolor in this study will also haverestricted the number of proteins detected as not all theproteome will be expressed In particular S coelicolordoes not sporulate in shaken liquid-grown cultures soproteins involved in this differentiation process are unlikelyto be present Nevertheless it is striking that the com-bined data for the pH 45ndash55 and 55ndash67 isoelectric pointranges indicate that 2468 out of a total of predicted 4932protein spots can be detected (see Table 1) suggestingthat transition phase mycelium in this supplementedminimal medium is expressing 50 of its genes (ie byextrapolation some 3912 of the 7825 in the genome)
Analysis of post-translational regulation
Although the regulation of cellular processes and adaptiveresponses in bacteria has been studied extensively at thetranscriptional level post-translational regulatory mecha-nisms are much less understood Proteomic analysisusing 2D-PAGE coupled with mass spectrometry offersthe opportunity to observe and identify modification ofproteins at a general cellular level The functions of pro-teins can be altered by specific covalent attachment of
adducts (eg phosphorylation glycosylation nucleotidy-lation acetylation) or by proteolytic processing Bothevents alter the pI andor mass of the protein and so canusually be observed on 2D-PAGE In the S coelicolorproteome map ordf110 out of a total of 770 gene productswere identified in more than one position or at pI andapparent mass co-ordinates significantly different frompredicted values indicating extensive regulation at thepost-translational level (88 are listed in Table 3) a propor-tion similar to that reported for other prokaryotes (Buttneret al 2001 Tonella et al 2001) The initial peptide massfingerprint analysis identified the modification of only 10of these proteins probably partly because fingerprint datatypically cover ordf30ndash60 of a proteinrsquos sequence (data notshown) so modification at positions not represented willbe missed Protein digestion with endopeptidases otherthan trypsin before mass spectrometry may help in thissituation but some covalently attached groups are labileand cannot be detected routinely Phosphorylation of Hisoccurs on nitrogen producing a high-energy phosphora-midate bond that is easily hydrolysed in acidic conditionswhereas phospho-Asp is the most labile phosphorylatedresidue with a half-life of a few hours under physiologicalconditions (reviewed by Robinson et al 2000) Thesemodifications the basis of bacterial two-component regu-latory systems are therefore unlikely to survive stainingof the 2D gels which involved the use of a fixing solutioncontaining 10 acetic acid and mass spectrometry Iden-tification of the more stable phosphoester phosphorylationthat occurs on Ser Thr or Tyr residues using PAGEfollowed by mass spectrometry has been reported(Godovac-Zimmermann et al 1999 Buttner et al 2001)but no examples were detected in this work Of the mod-ifications identified in this study three were by acetylationat the N-terminus after removal of the initiator Met residuetwo involved adenylylation and five proteins had been N-terminally truncated Modification of the 20S proteasomeby N-acetylation in yeast involves removal of the Metresidue by N-methionylaminopeptidase followed byacetylation with an Na-acetyltransferase (Kimura et al2000) Homologues of these enzymes are predicted fromthe S coelicolor genome and are presumably responsiblefor the observed modification of the 20S proteasomealpha subunit and of BldK-ORFD and the putative cellu-lose-binding protein The functional significance of Na-acetylation of these proteins is unclear
Analysis of the biosynthesis of antibiotics and other secondary metabolites
Post-translational modification The proteome mappingresults indicate that at least six enzymes in the Act clusterare subject to post-translational modifications and one
Proteomic analysis of S coelicolor 927
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
each from the undecylprodigiosin (SCO5877ndash5898)lsquocoelichelinrsquo (SCO0489ndash0499) and type I polyketidesynthase (SCO6273ndash6288) clusters (see Table 3) Thesemetabolic pathways may therefore be significantly regu-lated at the post-translational level The actVI-ORF3 gene(SCO5074) encodes a putative dehydratase enzyme thatis part of the biosynthetic pathway for Act a blue memberof the benzoisochromanequinone (BIQ) family of pig-mented antibiotics Mutation in actVI-ORF3 causes a 50decrease in the accumulation of Act (and a reddish brownphenotype) and it is believed to be responsible for pyranring closure an intermediate step in Act biosynthesis (thatmay also occur spontaneously) leading to the formationof the BIQ chromophore (Fernandez-Moreno et al 1994Ichinose et al 1998a Taguchi et al 2000) In this studyActVI-ORF3 protein was detected in two forms a majorspot corresponding to ActVI-ORF3 truncated by removalof the N-terminal 31 amino acid residues and a minor onein which the first 34 amino acids were missing (spots 31in Fig 1) The full-length protein was not detected Anal-ysis of the protein sequence using the SIGNALP server athttpwwwcbsdtudkservicesSignalP predicted a signalpeptide cleavage site between amino acids 31 and 32 inagreement with the observed results
The protein extract analysed was prepared from myce-lium that had been collected by centrifugation and sub-jected to two washes indicating that if ActVI-ORF3 isexported it is effectively retained within the cell wallmatrix This was confirmed by analysis of a preparationobtained by externally extracting proteins from intactwashed mycelium (Fig 4) Export of an enzyme requiredfor the efficient biosynthesis of an antibiotic that is thoughtto be produced intracellularly appears to be anomalousparticularly as it is involved in an intermediate biosynthetic
event Database analysis of the other members of the Actcluster revealed only one other protein with a predictedsignal peptide sequence the putative integral membranetransport-related protein ActII-ORF3 and it seemsunlikely that all the Act biosynthetic steps following BIQchromophore formation are extracellular It is possible thatfull-length ActVI-ORF3 catalyses the intermediate biosyn-thetic event intracellularly and the exported processedprotein is responsible for a different extracellular activityperhaps later in the pathway However no full-length pro-tein was detected so the exported enzyme may performthe pyran ring closure on a secreted metabolite that isthen reabsorbed by the cell for completion of the sub-sequent biosynthetic steps Only two homologues ofactVI-ORF3 are known and both are also in BIQ antibioticbiosynthetic clusters gra-ORF18 from the granaticincluster (Ichinose et al 1998b) and med-ORF5 frommedermycin biosynthesis (K Ichinose personal com-munication) Interestingly the encoded proteins of bothsequences are also predicted to contain N-terminal signalpeptides The observation that ActVI-ORF3 is exported tothe cell wall matrix may therefore be of general signifi-cance to the BIQ family of antibiotics and provide a newinsight into their biosynthetic pathways
Pathway representation Of the 20 putative secondarymetabolite gene clusters identified in the S coelicolorgenome sequence (Bentley et al 2002) eight had atleast one protein each identified on the proteome map(see Table 5) The biosynthetic clusters for the antibioticsAct Red and CDA had 68 36 and 40 of their pro-teins represented There are several possible explana-tions for failure to detect all the proteins for each pathwaySome are putative integral membrane proteins so cannotreadily be detected using conventional 2D-PAGE SomeRed and CDA proteins have molecular weights gt200 kDaoutside the range of the 125 gels used (for clustermembers see Supplementary material) The pathway-specific regulatory proteins for Act and Red biosynthesiswere not detected despite a detailed search of their pre-dicted gel locations even though the relevant genes areknown to be transcribed in equivalent cultures of the samestrain (Hesketh et al 2001) The proteins are presumablypresent but at levels below the limit of detection Howeverthe transcriptional regulator for CDA production CdaRwas identified as well as the two-component systemresponse regulator AbsA2 which is also encoded by agene in the CDA cluster (Anderson et al 2001) Interest-ingly proteins from three putative secondary meta-bolite clusters producing chemicals of as yet unknownstructure were also identified on the map correspondingto the deoxysugar synthaseglycosyltransferase clusterSCO0381ndash0401 the lsquocoelichelinrsquo cluster for a predicted(but not yet confirmed) oligopeptide siderophore
Fig 4 Processed ActVI-ORF3 is present in the cell wall matrix 2D-gel separations of protein extracts prepared (A) from whole cells or (B) by externally washing cells in a 2 SDS extraction buffer The arrow indicates ActVI-ORF3 protein The experiment was performed on duplicate biological samples and both showed similar protein expression profiles
928 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
(SCO0489ndash0499) and the type I polyketide clusterSCO6273ndash6288 The 3-oxoacyl-[acyl carrier protein]reductase (SCO6282) from the latter pathway is ex-pressed in a growth phase-dependent manner (Fig 3)and in early stationary phase cultures is perhaps themost abundant protein spot in the entire detectable pro-teome Presumably the metabolites produced by thesethree pathways are being synthesized under the condi-tions used In addition to coelichelin three other sidero-phores are postulated for S coelicolor All the proteinsfrom one of these desferrioxamine (SCO2782ndash2785)were identified but none was seen from either of the othertwo pathways The WhiE polyketide spore pigment isassociated with sporulation of surface-grown cultures(Davis and Chater 1990) and the expression of the whiEgenes depends on a series of sporulation-specific regula-tory genes (Kelemen et al 1998) no corresponding pro-teins of which were detected here It is therefore notsurprising that WhiE proteins were not detected in sam-ples prepared from liquid-grown vegetative mycelium
Analysis of primary metabolism
Proteins annotated as functioning in primary metabolicpathways were highly represented on the proteome mapwith ordf60 or more of the enzymes assigned to glycolysisthe TCA cycle the pentose phosphate pathway andpurine and pyrimidine ribonucleotide biosynthesis beingidentified (see Table 6) This figure may be deceptively lowbecause these pathways in S coelicolor often appear tohave more than one enzyme isoform for some of themetabolic steps potentially offering additional levels ofcontrol or developmental responsiveness at these pointsFor example only two steps in glycolysis have just a singlegene product assigned to them the remainder typicallyhave two or three each (Fig 5) At least one isoform waspresent for each step so the pathway is in theory 100functionally represented Understanding the regulation ofthese pathways will require analysis of the expression ofthese isoenzymes under different nutritional conditionsor different developmental stages Figure 5 shows that ofthe three predicted glyceraldehyde-3-phosphate dehydro-genase isoforms from glycolysis only two were detectedin this study (see also Supplementary material) Of theseSCO1947 appears to be constitutive whereas SCO7511is growth phase dependent (see Fig 3) Thus even thelimited conditions in this study yielded useful informationabout isoenzyme expression patterns (see Supplemen-tary material) In Streptomyces aureofaciens two genesencoding glyceraldehyde-3-phosphate dehydrogenaseactivity also appear to be differently regulated transcrip-tion of one highly homologous to SCO7511 is activatedby the regulator GapR whereas the other is GapR inde-pendent (Spusansky et al 2001)
Hood et al (1992) reported that most amino acid bio-synthetic enzymes are expressed at very low constitutivelevels in S coelicolor so it is reassuring to note that ordf50could be detected and identified using proteomics Theseinclude TrpB and TrpC1 from the tryptophan biosyntheticpathway the corresponding transcripts of which couldbarely be detected in previously reported S1 nucleaseprotection experiments (Hu et al 1999)
Perspectives
No attempt was made in the work reported here to simplifythe protein separations by subdividing the sample intosoluble and membrane fractions and this is reflected inthe 10 lipoproteins and 16 ABC transport system ATP-binding subunits identified on the proteome map (seeTable 2) No integral membrane proteins were detectedand even when a membrane protein sample was pre-pared and analysed no proteins identified possessedmore than three predicted transmembrane helices (datanot shown) This is consistent with previous studiesin other bacteria (Molloy et al 1999 Nilsson andDavidsson 2000) Another group of proteins also strik-ingly absent from the list of those identified is the sigmafactors which determine promoter selection by RNA poly-merase There are an extraordinarily high number (65) ofsigma factors encoded in the S coelicolor genome aswell as at least seven antisigma factors and 15 antisigmafactor antagonists (some of each of which were detected)Sigma factor heterogeneity in bacteria is an important wayof regulating expression of specific sets of genes under
Fig 5 The glycolytic pathway in S coelicolor illustrating the assignment of multiple gene products to individual metabolic steps based on the annotation of the genome sequence at httpwwwsangeracukProjectsS_coelicolor Gene products identified in the proteome map are underlined
Proteomic analysis of S coelicolor 929
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
different conditions and in different cell types (for a reviewsee Gross et al 1998) Although no sigma factors wereidentified about half (31 out of 65) have predicted pIvalues gtpH 7 and so can only be detected using thepH 6ndash11 IPG strips that give the poorest results (seeTable 1) However the principal vegetative sigma factorHrdB which is expected to be moderately abundanthas a predicted pI of 613 and its absence from thepH 55ndash67 proteome map may indicate that the samplepreparation method which does not remove DNA is inap-propriate Among other regulatory proteins 11 out of apossible 80 two-component system response regulatorsplus a further 36 proteins annotated as lsquoregulatorsrsquo weredetected Only one SerThr kinase and no two-componentsystem sensor kinases were detected possibly reflectingthe difficulty in analysing proteins with several transmem-brane domains Interestingly 30 secreted proteins wereidentified on the proteome map at least 10 of whichappear from comparison of their observed pI and molec-ular weight positions with predicted values to have beenprocessed already and therefore exported Presumablythese proteins do not end up exclusively in the culturemedium
Clearly this approach to proteomic analysis of S coeli-color has some important deficiencies not least theabsence of certain groups of proteins and the relativelypoor initial success rate for specifically characterizingpost-translational modifications Nevertheless it offersunique insights into the molecular biology of the organismthe expression of many proteins from both primary andsecondary metabolic pathways can be analysed post-translational regulation of proteins can be investigated ona general cellular level and the physical locations forsome proteins can be determined Approximately 35 ofthe S coelicolor genome is annotated as encoding lsquohypo-theticalrsquo proteins of unknown function and 182 proteins ofthis type were identified on the proteome map No exam-ples were found of overt disagreements between pre-dicted and observed proteins providing experimentalsupport for the general accuracy of the S coelicolorgenome sequence and its annotation Information aboutthe expression post-translational modification and physi-cal location of these (no longer) hypothetical proteinsfrom proteomic analyses is going to be important forassigning future functions As many of the hypotheticalproteins have apparent orthologues in pathogenic myco-bacteria some of the information may have an immediateapplication in the context of pathogenesis
Experimental procedures
Materials
Immobilized pH gradient (IPG) strips and IPG bufferswere purchased from Amersham Biosciences Acrylamide
duracryl and chemicals for silver staining were obtained fromGenomic Solutions All other chemicals were from Sigmaunless stated in the text
Strain and growth conditions
Streptomyces coelicolor M145 was cultivated with vigorousagitation at 30infinC in minimal medium supplemented with 02casamino acids (SMM) as described previously (Kieser et al2000) Briefly a high-density spore preparation [about 1010
colony-forming units (cfu) ml-1] was pregerminated in 2yen YTmedia for 7 h at 30infinC Germlings were harvested by centrifu-gation (5 min at 4000 g) resuspended in SMM and sonicatedbriefly to disperse any aggregates before inoculation into50 ml of SMM in 250 ml siliconized flasks containing coiledstainless steel springs Each flask received the equivalent of5 yen 107 spore cfu
Protein extraction and 2D-gel electrophoresis
The preparation of the protein sample to be separated waskept as simple as possible disrupting harvested myceliadirectly into denaturing isoelectric focusing (IEF) buffer con-taining Pefabloc SC (Roche) protease inhibitor This methodminimized unwanted protein degradation that was observedin initial studies using a Tris buffer containing Pefabloc SC forcell lysis (data not shown) Removal of DNA RNA salts andpolysaccharides from the sample was therefore not per-formed (other than the shearing of viscous DNA and RNA bysonication) and the protein extracts were sometimes difficultto focus when the sample was loaded by in-gel rehydrationLoading the sample soaked in paper strips at the anodicend of IPG strips that had already been rehydrated improvedthe reproducibility of separations significantly
Mycelium for protein extraction was harvested from cul-tures by brief centrifugation (30 s at 4000 g) at room tem-perature and immediately frozen in liquid nitrogen Typicallymycelium from 10 ml culture aliquots was collected and thetransfer time from culture flask to frozen sample was 15 minFrozen cells were thawed on ice in 5 ml of washing buffer(40 mM Tris pH 90 1 mM EGTA 1 mM EDTA) then pelletedby centrifugation (5 min at 3000 g) at 4infinC Washed cells wereresuspended in 400 ml of denaturing IEF buffer UTCHAPS[7 M urea 2 M thiourea 4 (wv) CHAPS 40 mM TrispH 90 1 mM EDTA 50 mM DTT 4 mM Pefabloc SC pro-tease inhibitor (Roche)] and disrupted by sonication (SanyoSoniprep 150 10 yen 2 s bursts at amplitude 75 mm) whilecooling in an ethanolndashice bath Cell debris was removed bycentrifugation (15 min 10 000 g 4infinC) and the protein extractwas stored frozen in aliquots at -80infinC until use For thepreparation of extracellular protein extracts washed cellswere resuspended in extraction buffer consisting of 40 mMTris pH 90 1 mM EDTA 50 mM DTT 2 SDS and 4 mMPefabloc SC protease inhibitor Cells were vortexed for 2 minbefore being pelleted by centrifugation (5 min 10 000 g4infinC) The supernatant was precipitated using the Amersham2D Clean-Up kit according to the manufacturerrsquos instructionsand the precipitated proteins were redissolved in UTCHAPSand stored at -80infinC until use
930 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
For first-dimension isoelectric focusing 18 cm IPG stripspH 4ndash7 pH 6ndash11 pH 45ndash55 or pH 55ndash67 (Amersham Bio-sciences) were rehydrated overnight in IEF buffer containing1 ampholytes according to the manufacturerrsquos instructionsusing a Phaser isoelectric focusing unit (Genomic Solutions)set at 20 V Protein samples to be separated were applied tothe rehydrated strips at the anodic end soaked in a 5ndash10 mmsection of IEF electrode strip (Amersham Biosciences)Separation was performed for 120 000 V-h with a maximumvoltage of 5000 V After isoelectric focusing IPG strips wereequilibrated for the second dimension for 15 min in IPG equil-ibration buffer (50 mM Tris pH 68 6 M urea 30 glycerol1 SDS and 001 bromophenol blue) plus 80 mM DTTthen for 10 min in IPG equilibration buffer plus 135 mMiodoacetamide Approximately 1 cm was removed from theanodic end of each equilibrated strip before applying the stripto the top of a vertical 125 SDS-PAGE gel for second-dimension separation using the Investigator 5000 systemfrom Genomic Solutions
Large-format gels (24 cm wide yen 22 cm high yen 1 mm thick)for the second-dimension separation were cast in-houseusing the system supplied by Genomic Solutions 125SDS-PAGE gels prepared using 065 NN-methylenebi-sacrylamide as cross-linking agent were used for pH 4ndash7and pH 45ndash55 IPG strips whereas pH 55ndash67 and pH 6ndash11 strips were applied to similar gels made with 08 cross-linker Electrophoresis was performed while cooling using themaximum setting (ordf 4ndash10infinC) at 20 000 mW per gel constantpower and a maximum voltage of 500 V Gels were thenstained with either colloidal Coomassie G-250 (Neuhoff et al1988) or silver nitrate (Rabilloud 1992) and scanned in aProXPRESS proteomic imaging system (Perkin-Elmer)Image analysis was performed using PHORETIX 2D version51 (NonLinear Dynamics) spot detection was optimizedautomatically using the lsquoSpot Detection Wizardrsquo and thenedited manually background subtraction was performedautomatically using the lsquoMode of Non-Spotrsquo setting imageswere then normalized to the total spot volume for quantifica-tion Spot filtering was not used
Protein identification using mass spectrometry
Protein spots of interest were excised from colloidalCoomassie-stained gels in-gel digested with trypsin andidentified by MALDI-TOF peptide mass fingerprint analysisExcised gel pieces (ordf 1-mm-diameter circles cut from 1-mm-thick gels) were washed twice with 100 ml of 50 mMammonium bicarbonate for 15 min once with 100 ml of 20acetonitrile-40 mM ammonium bicarbonate for 15 min andonce with 100 acetonitrile Washed gel pieces were allowedto air-dry for 10 min before being rehydrated in 5 ml of10 mg ml-1 trypsin (sequencing grade modified Promega) in10 mM ammonium bicarbonate Digestion (37infinC for 4 h) wasstopped by the addition of 5 ml of 5 formic acid Extractionof peptides into this solution was encouraged by a sonicwater bath treatment for 20 min Peptide extract (05 ml) wasspotted on to a thin layer of a-cyano-4-hydroxycinnamic acidapplied to a MALDI-TOF sample template and analysed byMALDI-TOF mass spectrometry (Bruker Reflex III) using anaccelerating voltage of 25 kV Samples were externally cali-brated using a standard mixture of six peptides ranging in
mass from 10465423 Da to 34946500 Da and the massaccuracy obtained was 60 ppm or better Identification ofproteins from MALDI-TOF peptide mass fingerprint data wasperformed using the lsquoMascotrsquo search engine at httpwwwmatrixsciencecom and was based on a positive resultusing their lsquoProbability Based MOWSE Scorersquo algorithm AMOWSE score of 60 or higher is significant at the 5 levelor better and proteins in this work typically gave scores gt80(frequently considerably so) In addition no identification wasaccepted unless at least five peptides representing at least20 of the protein sequence were detected in the MALDI-TOF peptide mass fingerprint
Assignment of proteins to metabolic pathways
Proteins were assigned to primary metabolic pathwaysusing the lsquoProtein Classification Schemersquo web page ofthe annotated Streptomyces coelicolor genome sequenceat httpwwwsangeracukProjectsS_coelicolor as it ap-peared in May 2002 The annotation of the genome andtherefore the Protein Classification Scheme is likely to bemodified as more experimental data are obtained about thefunction of genes that have currently been assigned on thebasis of sequence information alone It is therefore possiblethat proteins assigned to metabolic pathways in this studymay in future be shown to perform different functions in thecell
Assignments to gene clusters encoding secondary metab-olites are based on an analysis of the genome sequencereported by Bentley et al (2002) In some cases theseassignments are based on extensive experimental data (actred reviewed by Chater and Bibb 1997 Champness 2000cda Chong et al 1998 Huang et al 2001 whiE Davis andChater 1990)
Acknowledgements
This work was funded by grants (2FGT11406 208FGT11408 and 208IGF12432) under the BBSRCrsquos Tech-nologies for Functional Genomics and Investigating GeneFunction initiatives and by a competitive strategic grant fromthe BBSRC to the John Innes Centre We thank Mike Naldrettfor his extensive contribution in establishing technology forproteome analysis at the JIC
Supplementary material
The following material is available from httpwwwblackwell-sciencecomproductsjournalssuppmatmolemole3219mmi3219smhtmSection 1 Detection of gene products assigned to individualmetabolic steps in primary metabolic pathways in ScoelicolorSection 2 Identification of proteins from individual amino acidbiosynthetic pathwaysSection 3 Detection of gene products assigned to individualmetabolic steps in secondary metabolic pathways in Scoelicolor
Proteomic analysis of S coelicolor 931
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
References
Anderson TB Brian P and Champness WC (2001)Genetic and transcriptional analysis of absA an antibioticgene cluster linked two-component system that regulatesmultiple antibiotics in Streptomyces coelicolor Mol Micro-biol 39 553ndash566
Bentley SD Chater KF Cerdeno-Tarraga A-M ChallisGL Thomson NR James KD et al (2002) Completegenome sequence of the model actinomycete Streptomy-ces coelicolor A3(2) Nature 417 141ndash147
Berdy J (1984) New ways to obtain antibiotics Chin J Anti-biot 7 272ndash290
Berggren K Chernokalskaya E Steinberg TH KemperC Lopez MF Diwu Z et al (2000) Background-freehigh sensitivity staining of proteins in one- and two-dimensional sodium dodecyl sulfate-polyacrylamide gelsusing a luminescent ruthenium complex Electrophoresis21 2509ndash2521
Blackstock WP and Weir MP (1999) Proteomics quan-titative and physical mapping of cellular proteins TrendsBiotechnol 17 121ndash127
Blattner FR Plunkett G Bloch CA Perna NT BurlandV Riley M et al (1997) The complete genome sequenceof Escherichia coli K-12 Science 277 1453ndash1462
Buttner K Bernhardt J Scharf C Schmid R MaderU Eymann C et al (2001) A comprehensive two-dimensional map of cytosolic proteins of Bacillus subtilisElectrophoresis 22 2908ndash2935
Champness W (2000) Actinomycete development antibioticproduction and phylogeny questions and challenges InProkaryotic Development Brun YV and Skimketts LJ(eds) Washington DC American Society for MicrobiologyPress pp 11ndash31
Chater KF and Bibb MJ (1997) Regulation of bacterialantibiotic production In Biotechnology Vol 7 Products ofSecondary Metabolism Kleinkauf H and von Doumlhren H(eds) Weinheim Germany VCH pp 57ndash105
Chong PP Podmore SM Kieser HM Redenbach MTurgay K Marahiel M et al (1998) Physical identificationof a chromosomal locus encoding biosynthetic genesfor the lipopeptide calcium-dependent antibiotic (CDA) ofStreptomyces coelicolor A3(2) Microbiology 144 193ndash199
de Crecy-Lagard V Servant-Moisson JV Grandvalet Cand Mazodier P (1999) Alteration of the synthesis of theClp ATP-dependent protease affects morphological andphysiological differentiation in Streptomyces Mol Microbiol32 505ndash517
Davis NK and Chater KF (1990) Spore colour inStreptomyces coelicolor A3(2) involves the developmen-tally regulated synthesis of a compound biosyntheticallyrelated to polyketide antibiotics Mol Microbiol 4 1679ndash1691
Fernandez-Moreno FA Martinez E Caballero JLIchinose K Hopwood DA and Malpartida F (1994)DNA sequence and functions of the actVI region of theactinorhodin biosynthetic gene cluster of Streptomycescoelicolor A3(2) J Biol Chem 269 24854ndash24863
Fink D Falke D Wohllheben W and Engels A (1999)Nitrogen metabolism in Streptomyces coelicolor A3(2)
modification of glutamine synthase I by an adenylyltrans-ferase Microbiology 145 2313ndash2322
Godovac-Zimmermann J Soskic V Posnanovic S andBrianza F (1999) Functional proteomics of signal trans-duction by membrane receptors Electrophoresis 20 952ndash961
Gorg A Obermaier C Boguth G Harder A Scheibe BWildgruber R and Weiss W (2000) The current state oftwo-dimensional electrophoresis with immobilized pH gra-dients Electrophoresis 21 1037ndash1053
Gramajo HC Takano E and Bibb MJ (1993) Stationary-phase production of the antibiotic actinorhodin in Strepto-myces coelicolor A3(2) is transcriptionally regulated MolMicrobiol 7 837ndash845
Gross CA Chan C Dombroski A Gruber T Sharp MTupy J and Young B (1998) The functional and regula-tory roles of sigma factors in transcription Cold SpringHarb Symp Quant Biol 63 141ndash155
Hesketh A Sun J and Bibb MJ (2001) Induction ofppGpp synthesis in Streptomyces coelicolor A3(2) grownunder conditions of nutritional sufficiency elicits actII-ORF4transcription and actinorhodin biosynthesis Mol Microbiol39 136ndash144
Hesketh A Fink D Gust B Rexer H-U Scheel BChater K et al (2002) The PII nitrogen regulatory proteinGlnK in Streptomyces coelicolor A3(2) is modified by ade-nylylation rather than uridylylation and the GlnD and GlnKhomologues are functionally dissimilar to their nitrogenregulatory system counterparts Mol Microbiol (inpress)
Hood DW Heidstra R Swodoba UK and Hodgson DA(1992) Molecular genetic analysis of proline and tryp-tophan biosynthesis in Streptomyces coelicolor A3(2)interaction between primary and secondary metabolism- areview Gene 115 5ndash12
Hu DS-J Hood DW Heidstra R and Hodgson DA(1999) The expression of the trpD trpC and trpBA genesof Streptomyces coelicolor A3(2) is regulated by growthrate and growth phase but not by feedback repression MolMicrobiol 32 869ndash880
Huang J Lih CJ Pan KH and Cohen SN (2001)Global analysis of growth phase responsive gene expres-sion and regulation of antibiotic biosynthetic pathways inStreptomyces coelicolor using DNA microarrays GenesDev 15 3183ndash3192
Ichinose K Taguchi T Ebizuka Y and Hopwood DA(1998a) Biosynthetic gene clusters of benzoisochromane-quinone antibiotics in Streptomyces spp Identification ofgenes involved in post-PKS tailoring steps Actinomyceto-logica 12 99ndash109
Ichinose K Bedford DJ Tornus D Bechthold A BibbMJ Revill PW et al (1998b) The granaticin biosyntheticgene cluster of Streptomyces violaceoruber Tu22sequence analysis and expression in a heterologous hostChem Biol 5 647ndash659
Kelemen GH Brian P Flardh K Chamberlin L ChaterK and Buttner M (1998) Developmental regulation oftranscription of whiE a locus specifying the polyketidespore pigment in Streptomyces coelicolor A3(2) J Bacte-riol 180 2515ndash2521
Kieser T Bibb MJ Buttner MJ Chater KF and
932 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
Hopwood DA (2000) Practical Streptomyces GeneticsNorwich John Innes Foundation
Kim EJ Chung HJ Suh B Hah YC and Roe JH(1998) Expression and regulation of the sodF gene encod-ing iron- and zinc-containing superoxide dismutase inStreptomyces coelicolor Muumlller J Bacteriol 180 2014ndash2020
Kimura Y Takaoka M Tanaka S Sassa H Tanaka KPolevoda B et al (2000) Na-acetylation and proteolyticactivity of the yeast 20S proteasome J Biol Chem 2754635ndash4639
Kunst F Ogasawara N Moszer I Albertini AM AlloniG Azevedo V et al (1997) The complete genomesequence of the Gram-positive bacterium Bacillus subtilisNature 390 249ndash256
Lucchini S Thompson A and Hinton JCD (2001)Microarrays for microbiologists Microbiology 147 1403ndash1414
Mann M Hendrickson RC and Pandey A (2001) Analy-sis of proteins and proteomes by mass spectrometry AnnuRev Biochem 70 437ndash473
Miyodah S (1993) Research on antibiotic screening inJapan over the last decade a producing microorganismsapproach Actinomycetologica 7 100ndash106
Molloy MP Herbert BR Williams KL and Gooley AA(1999) Extraction of Escherichia coli proteins with organicsolvents prior to two-dimensional electrophoresis Electro-phoresis 20 701ndash704
Nagy I Tamura T Vanderleyden J Baumeister W andDe Mot R (1998) The 20S proteasome of Streptomycescoelicolor J Bacteriol 180 5448ndash5453
Neuhoff V Arold N Taube D and Ehrhardt W (1988)Improved staining of proteins in polyacrylamide gels includ-ing isoelectric focusing gels with clear background at nan-ogram sensitivity using Coomassie Brilliant Blue G-250and R-250 Electrophoresis 9 255ndash262
Nilsson CL and Davidsson P (2000) New separation toolsfor comprehensive studies of protein expression by massspectrometry Mass Spectrom Rev 19 390ndash397
Puglia AM Vohradsky J and Thompson CJ (1995)
Developmental control of the heat-shock stress regulon inStreptomyces coelicolor Mol Microbiol 17 737ndash746
Rabilloud T (1992) A comparison between low backgroundsilver diamine and silver nitrate protein stains Electro-phoresis 13 429ndash439
Robinson VL Buckler DR and Stock AM (2000) A taleof two components a novel kinase and a regulatory switchNature Struct Biol 7 626ndash633
Schena M (2000) Microarray Biochip Technology NatickMA Eaton Publishing
Spusansky O Rezuchova B Homerova D andKormanec J (2001) Expression of the gap gene encodingglyceraldehyde-3-phosphate dehydrogenase of Strepto-myces aureofaciens requires GapR a member of theAraCXylS family of transcriptional activators Microbiology147 1291ndash1301
Taguchi T Itou K Ebizuka Y Malpartida F HopwoodDA Surti CM et al (2000) Chemical characterisationof disruptants of the Streptomyces coelicolor A3(2) actVIgenes involved in actinorhodin biosynthesis J Antibiot 53144ndash152
Takano E Gramajo HC Strauch E Andres N WhiteJ and Bibb MJ (1992) Transcriptional regulation of theredD transcriptional activator gene accounts for growth-phase-dependent production of the antibotic undecylpro-digiosin in Streptomyces coelicolor A3(2) Mol Microbiol6 2797ndash2804
Tonella L Hoogland C Binz P-A Appel RDHochstrasser DF and Sanchez J-C (2001) New per-spectives in the Escherichia coli proteome investigationProteomics 1 409ndash423
Vohradsky J Li XM and Thompson CJ (1997) Identifi-cation of prokaryotic developmental stages by statisticalanalysis of two-dimensional gel patterns Electrophoresis18 1418ndash1428
Vohradsky J Li XM Dale G Folcher M Nguyen LViollier PH and Thompson CJ (2000) Developmentalcontrol of stress stimulons in Streptomyces coelicolorrevealed by statistical analyses of global gene expressionpatterns J Bacteriol 182 4979ndash4986

918
A R Hesketh
et al
copy 2002 Blackwell Publishing Ltd
Molecular Microbiology
46
917ndash932
spectrometry (the proteome) (Blackstock and Weir 1999Mann
et al
2001) Analysis of the proteome is morecomplicated than transcriptome analysis because of thediverse physical and chemical properties of proteins andbecause of the need for any given protein to be extractedin sufficient quantity not only to be detected but also tobe identified Thompson and co-workers have used 2D-PAGE extensively to analyse changes in the pattern ofradiolabelled protein profiles in pulsendashchase experimentsin
S coelicolor
during growth and in response to stressbut did not have the benefit of the complete genomesequence to use mass spectrometry to identify proteinsof interest (Puglia
et al
1995 Vohradsky
et al
19972000) In this study we have mapped a substantial partof the
S coelicolor
proteome using 2D-PAGE and MALDI-TOF peptide mass fingerprint analysis Particular atten-tion is paid to the representation of proteins involved insecondary metabolic pathways in comparison with thosefrom primary metabolism and to the extent of regulationby post-translational modification mechanisms
Results
Analysis of the proteome using 2D-PAGE
Streptomyces coelicolor
M145 produces the pigmentedantibiotics actinorhodin (Act) and the prodigiosin complex(Red) in a growth phase-dependent manner (Takano
et al
1992 Gramajo
et al
1993) Thus in liquid minimalmedium supplemented with casamino acids (SMM) pig-ment production begins at the transition between expo-nential growth and stationary phase In order to observeproteins from both primary and secondary metabolic path-ways pigmented mycelium from transition phase cultureswas harvested and disrupted directly into the stronglydenaturing isoelectric focusing buffer UTCHAPS (see
Experimental procedures
) Protein extracts were sepa-
rated by 2D-PAGE using several different immobilized pHgradient (IPG) strips for the first-dimension separation(pH 4ndash7 pH 6ndash11 pH 45ndash55 and pH 55ndash67) 125SDSndashPAGE for the second and silver nitrate staining forprotein detection (Fig 1) The proteins visible on each gelwere counted after spot detection of scanned gel imagesusing
PHORETIX
2D image analysis software and com-pared with the total number expected assuming that everypotential gene was expressed (Table 1) To calculate thenumber of spots expected proteins in the theoretical pro-teome with predicted isoelectric point values within the pHrange of the IPG strip used and with molecular weightsbetween 8 kDa and 140 kDa (the detectable range using125 PAGE) were counted and multiplied by a factor of12 representing the observed average number of proteinspots per gene (see Table 1) Separation using the iso-electric point (pI) range pH 4ndash7 produced 1051 detectablespots representing 19 of the total number of proteinspredicted for this region Excluding secreted and mem-brane proteins which would not generally be expected inthe protein extract the proportion was 25 In the pIrange pH 6ndash11 these numbers were 8 for the theoreti-cal total proteome and 12 for the theoretical extractableproteome Using the narrow-range IPG strips pH 45ndash55and pH 55ndash67 1555 and 913 spots were detectedrespectively corresponding to 82 and 41 of the totalnumber of predicted extractable proteins Thus the higherresolution and higher loading possible on these lsquozoomgelsrsquo approximately doubled the number of proteinsdetectable
Identification of separated proteins towards a proteome map and database
Protein spot identification was performed exclusively ongels stained with colloidal Coomassie G250 and mapped
Table 1
Estimate of the proportion of the
S coelicolor
proteome that can be detected by silver staining of 2D-PAGE separations of transitionphase protein extracts and MALDI-TOF identification of protein spots illustrating the extent of post-translational modifications
Isoelectric point range (pH)
4ndash7 45ndash55 55ndash67 6ndash11
Protein spots detected
a
1051 1555 913 379Percentage of expected 19 63 37 8protein spots detected
b
of 5418 of 2462 of 2470 of 4792Percentage of expected 25 82 41 12extractable protein spots detected
c
of 4221 of 1886 of 2218 of 3112Protein spots identified
d
443 459 193 210Different gene products represented 374 360 170 179Ratio of protein spots per gene product 118 127 113 117
a
On silver-stained gels
b
The total number of gene products with theoretical isoelectric point values and molecular weights within the range of the gel separation(8ndash140 kDa) was multiplied by 12 the observed average number of protein spots per gene as a result of post-translational modifications
c
Same as a but excluding proteins annotated as being membrane or secreted
d
From colloidal Coomassie-stained gels
Proteomic analysis of
S coelicolor 919
copy 2002 Blackwell Publishing Ltd
Molecular Microbiology
46
917ndash932
Fig 1
A proteome map of
S coelicolor
showing 2D-PAGE separations using IPG strips pH 4ndash7 pH 6ndash11 pH 45ndash55 and pH 55-67 The number of spots identified is given at the foot of each reference gel Protein spots from metabolic pathways that were identified at positions inconsistent with their predicted amino acid sequences and suggestive of post-translational modification are marked in yellow (primary metabolism) or red (secondary metabolism) Blue spots correspond to other examples of post-translational modifications that are mentioned in the text and in Table 3 Separations have been performed on numerous occasions and generally show high spatial reproducibility
920
A R Hesketh
et al
copy 2002 Blackwell Publishing Ltd
Molecular Microbiology
46
917ndash932
back onto the analytical silver-stained gels in Fig 1Although silver staining is 5ndash10 times more sensitive thancolloidal Coomassie staining (and about 100 times moresensitive than conventional Coomassie staining) furthermass spectrometry analysis of protein spots is much moreefficient after staining with colloidal Coomassie than withsilver A total of 1305 protein spots were unambiguouslyassigned to sequenced genes using MALDI-TOF peptidemass fingerprint analysis at a success rate of
ordf
90(Table 1) There is some redundancy in these figures withseveral spots being represented in more than one pIrange On each gel some proteins were identified in morethan one position indicative of post-translational modifi-cations The average number of protein spots per geneproduct was 12 The products from 770 different geneswere assigned to the proteome map representing
ordf
10of the genome and corresponding to many differentclasses of proteins (Table 2) The master gel referencemaps are the basis for the production of a publicly acces-sible proteome database (httpqbababeracukS_coeliReferenceGelrefgelhtml see last Results section)
Analysis of co- and post-translational modifications
The results of MALDI-TOF identification of protein spotssummarized in Table 1 indicated a significant amount ofregulation at the post-transcriptional level in
S coelicolor
as in most other bacteria Table 3 lists those gene prod-ucts that were identified at pI andor molecular weight co-ordinates significantly different from predicted values Ingeneral these fell into two categories one or more spotswith lower than expected molecular weights and oftendifferent pI values indicating modification by proteolyticprocessing (Table 3A) and multiple spots with the sameapparent molecular weights but different pI values indi-
cating modification by covalent addition of a small adduct(Table 3B) In the former case the full-length protein wasoften but not always detected A similar fraction ofproteins of unknown function and annotated as lsquohypo-theticalrsquo or lsquoconserved hypotheticalrsquo was subject to post-translational modification but these will be described ina later publication and are not considered in Table 3Evidence in the peptide mass fingerprint data for the spe-cific nature of the modification was obtained in 10 out ofthe 88 proteins listed (Table 4) Figure 2 illustrates thecharacterization of an example of each type of modifica-tion by interpretation of the MALDI-TOF data
N-acetylation
The putative cellulose-binding proteinSCO5396 was identified in two positions on the proteomemap (spots labelled 10 in Fig 1) with the same apparentmolecular weight but significantly different pI values Themore basic spot gave a tryptic peptide at 137076 Dacorresponding to the N-terminal peptide 2ndash13 indicatingthat the initiator Met residue had been co-translationallyremoved (Fig 2A top) In the more acidic spot (ie on theleft in Fig 1) this peptide had increased in mass by420 Da diagnostic of the addition of an acetyl grouppresumably on the N-terminal Ser residue (Fig 2A bot-tom) A similar modification was identified for the 20Sproteasome alpha subunit (spots 12 in Fig 1) and for theputative ABC transport protein BldK-ORFD (spots 17 inFig 1) although the presumptive N-terminal acetylatedresidue is Thr in the latter (data not shown)
Adenylylation
By analogy with the nitrogen regulatorysystem in
E coli
glutamine synthetase I (GSI) wasalready believed to be modified by adenylylation on aconserved Tyr-397 residue and biochemical evidence forthis has been reported previously (Fink
et al
1999) TheMALDI-TOF data showed the tryptic peptide 395ndash420 atthe expected mass of 28074 Da for the larger more basicspot 6 in Fig 1 but this increased by 3290 Da corre-sponding to the addition of an adenylyl group in thesmaller spot on the left (Fig 2B) Adenylylation of thenitrogen regulatory protein GlnK (spot 15 in Fig 1) wasidentified similarly but this is described in detail elsewhere(Hesketh
et al
2002)
Proteolytic processing
The 50S ribosomal protein L9was identified as two spots that differed in both pI andmolecular weight (spots 30 in Fig 1) suggesting modifi-cation by proteolytic processing The spot with the highermolecular weight showed the tryptic peptide correspond-ing to amino acids 3ndash22 at the expected mass of199310 Da in agreement with this being the unproc-essed form of the protein (Fig 2C top) In the lower ofthe two spots the peptide decreased in mass by11312 Da (Fig 2C bottom) corresponding to the loss of
Table 2
Types of proteins identified on the proteome map
Protein class
a
Number ingenome
a
Number identified
b
Hypothetical 1049 68 (6)Conserved hypothetical 1322 114 (8)Secreted 530 30 (5)ABC transport ATP binding 109 16 (14)Lipoproteins 124 10 (8)Sigma factors 65 0 (0)Antisigma factors 7 2 (28)Antisigma factor antagonists 15 1 (6)Two-component system regulators 80 11 (13)Other regulators 592 36 (6)Two-component system kinases 85 0 (0)SerThr kinases 37 1 (2)Other 3923 482 (12)All 7816 770 (10)
a
Based on annotation of the
S coelicolor
genome sequence(httpwwwsangeracukProjectsS_coelicolor)
b
Percentage identified is included in parentheses
Proteomic analysis of
S coelicolor 921
copy 2002 Blackwell Publishing Ltd
Molecular Microbiology
46
917ndash932
Table 3
Proteins identified at pI and molecular weight co-ordinates on the 2D-PAGE maps that are inconsistent with their predicted amino acidsequences indicating possible post-translational modification either by proteolytic processing (A) or by covalent addition of a low-molecular-weight adduct (B)
ORF no Gene product Spot no in Fig 1
ASCO0498 Probable peptide monooxygenase 33SCO1230 Putative tripeptidylaminopeptidaseSCO1644 20S proteasome beta subunit precursor 35SCO1815 3-Oxoacyl-[acyl carrier protein] reductaseSCO1860 Putative secreted proteinSCO1947 Glyceraldehyde-3-phosphate dehydrogenase 9SCO1998 30S ribosomal protein S1SCO2619 ATP-dependent Clp protease subunit 13SCO2920 Probable secreted proteinaseSCO3236 Probable oxygenase 34SCO3671 Heat shock protein DNAKSCO3909 50S ribosomal protein L9 30SCO4296 Chaperonin GroEL2SCO4439
D
-Alanyl-
D
-alanine carboxypeptidaseSCO4643 UDP-N-acetylanoylpyruvoylglucosamine reductaseSCO4662 Elongation factor Tu1SCO4762 Chaperonin GroEL1SCO4808 Succinyl-CoA synthetase beta chain 8SCO4824 Bifunctional protein methylene tetra-hydrofolate dehydrogenasecyclohydrolaseSCO5073 Putative oxidoreductase ActVI-ORF2 29SCO5074 Putative dehydratase ActVI-ORF3 31SCO5088 Ketoacyl synthase beta subunit ActI-ORF2 24SCO5113 Oligopeptide ABC transport protein BldK-ORFBSCO5254 Superoxide dismutase precursor 32SCO5371 ATP synthase alpha chainSCO5477 Probable oligopeptide-binding lipoproteinSCO5624 30S ribosomal protein S2SCO5737 Guanosine pentaphosphate synthetaseSCO5769 Recombinase RecASCO5892 Polyketide synthase RedL 18SCO6282 3-Oxoacyl-[acyl carrier protein] reductase 11
BSCO1074 Putative peptidaseSCO1476 S-adenosylmethionine synthetaseSCO1594 Phenyl-tRNA synthetase beta chainSCO1643 20S proteasome alpha subunit 12SCO1814 Enoyl-[acyl carrier protein] reductaseSCO1935 Transketolase 3SCO1947 Glyceraldehyde-3-phosphate dehydrogenase 9SCO2180 Dihydrolipoamide dehydrogenase 21SCO2181 Probable dihydrolipoamide S-succinyltransferase 14SCO2198 Glutamine synthetase I 6SCO2390 3-Oxoacyl-[acyl carrier protein] synthase IISCO2478 Putative reductaseSCO2618 ATP-dependent Clp protease subunit 2SCO2822 Putative decarboxylaseSCO3096 Enolase 23SCO3229 4-Hydroxyphenylpyruvic acid dioxygenaseSCO3337 Probable pyrroline-5-carboxylate reductase 28SCO3409 Inorganic pyrophosphataseSCO3549 Antisigma factor antagonist BldGSCO3563 Acetyl-CoA synthaseSCO3671 Heat shock protein DNAKSCO3878 DNA polymerase III beta chainSCO3890 Thioredoxin reductaseSCO3906 30S ribosomal protein S6SCO3970 XAA-Pro aminopeptidase ISCO4164 Putative thiosulphate sulphurtransferaseSCO4209 Phosphoglycerate mutase 19SCO4240 ABC transport ATP binding protein MsiKSCO4296 Chaperonin GroEL2SCO4645 Aspartate aminotransferase 7SCO4652 50S ribosomal protein L10
Some proteins appear to be subject to both types of modification and are present in both parts Only proteins from primary or secondary metabolicpathways or with identified modifications are assigned to spots in Fig 1
922
A R Hesketh
et al
copy 2002 Blackwell Publishing Ltd
Molecular Microbiology
46
917ndash932
the isoleucine residue at its N-terminus (expected massloss 11308 Da) This indicates removal of the N-terminalMKI residues from ribosomal protein L9 The theoreticalisoelectric point (997) and molecular weight (156 kDa) ofL9 protein N-terminally truncated in this way are in goodagreement with the position of the lower spot in Fig 1(predicted values for the unprocessed protein are pH 960and 159 kDa) MALDI-TOF data indicating proteolytic
processing of the superoxide dismutase precursor protein(SCO5254 spot 32 in Fig 1) and the 20S proteasomebeta subunit precursor (SCO1644 spot 35 in Fig 1)agreed with previous reports (Kim
et al
1998 Nagy
et al
1998) Evidence for the proteolysis of
S coelicolor
ClpP1(SCO2619 spot 13 in Fig 1) has also been reportedalthough the processing site was not defined (de Crecy-Lagard
et al
1999)
SCO4661 Elongation factor GSCO4662 Elongation factor Tu1SCO4677 Putative regulatory proteinSCO4729 DNA-directed RNA polymerase alpha chainSCO4762 Chaperonin GroEL1SCO4770 Inosine-5
cent
-monophosphate dehydrogenase 25SCO4792 LuxR family two-component regulatorSCO4808 Succinyl-CoA synthetase beta chain 8SCO4809 Succinyl-CoA synthetase alpha chain 22SCO4921 Probable acyl-CoA carboxylase complex A chainSCO5071 Hydroxylacyl-CoA dehydrogenase ActVI-ORFA 16SCO5079 Hypothetical protein ActVA-ORF4 20SCO5080 Possible hydrolase ActVA-ORF5 26SCO5115 Probable ABC transporter intracellular ATPase chain BldK-ORFD 17SCO5371 ATP synthase alpha chainSCO5396 Putative cellulose-binding protein 10SCO5419 Putative thioredoxinSCO5515 Probable
D
-3-phosphoglycerate dehydrogenase 4SCO5535 Putative carboxyl transferaseSCO5584 Nitrogen regulatory protein GlnK 15SCO5625 Elongation factor TsSCO5699 Prolyl-tRNA synthetaseSCO5737 Guanosine pentaphosphate synthetaseSCO5777 Glutamate uptake system ATP-binding proteinSCO5803 SOS regulatory protein LexASCO5999 Aconitase 1SCO6198 Probable secreted proteinSCO6282 3-Oxoacyl-[acyl carrier protein] reductase 11SCO6636 Bacteriophage
f
-C31 resistance protein PglZSCO6663 Transketolase 27SCO7000 Isocitrate dehydrogenase 2SCO7036 Argininosuccinate synthase 5SCO7510 Peptidyl-prolyl
cis-trans
isomerase
ORF no Gene product Spot no in Fig 1
Some proteins appear to be subject to both types of modification and are present in both parts Only proteins from primary or secondary metabolicpathways or with identified modifications are assigned to spots in Fig 1
Table 3
cont
Table 4
Co- and post-translational modification of proteins suggested by MALDI-TOF peptide mass fingerprint analysis
Protein Modification
SCO1643 20S proteasome alpha subunit Removal of initiator Met residue and acetylation of N-terminal SerSCO5115 ABC transporter intracellular Removal of initiator Met residue and acetylation of N-terminal Thr
ATPase BldK-ORFDSCO5396 Putative cellulose-binding protein Removal of initiator Met residue and acetylation of N-terminal SerSCO2198 Glutamine synthetase I Adenylylation of peptide 395ndash420SCO5584 Nitrogen regulatory protein GlnK Adenylylation of peptide 48ndash58SCO1644 20S proteasome
b
-subunit precursor Proteolytic processing between amino acid residues 5354SCO2619 ATP-dependent Clp protease Proteolytic processing between amino acid residues 2223
proteolytic subunit ISCO3909 50S ribosomal protein L9 Proteolytic processing between amino acid residues 34SCO5074 Putative dehydratase ActVI-ORF3 Proteolytic processing between amino acid residues 3132 and 3435SCO5254 Superoxide dismutase precursor Proteolytic processing between amino acid residues 1415
Proteomic analysis of
S coelicolor 923
copy 2002 Blackwell Publishing Ltd
Molecular Microbiology
46
917ndash932
Co-translational modification by removal of the N-terminal initiator Met occurred in 149 out of 190 (78)cases in which the N-terminal peptide was detected Pro-cessing by the N-methionylaminopeptidase was observedif the second amino acid was Ser (45 out of 149 exam-ples) Thr (40) Ala (32) Pro (27) Gly (4) or Val (1) Ononly one occasion was any of these six amino acids foundas the second residue if the N-terminal methionine wasretained This is in broad agreement with observations in
B subtilis
in which two-thirds of proteins were reported tobe similarly processed (Buttner
et al
2001)
Representation of primary and secondary metabolic pathways
Streptomycetes produce
ordf
60 of all commercially usefulantibiotics (Berdy 1984 Miyodah 1993) Most antibioticsare the product of complex biosynthetic pathways oftenencoded by 20ndash30 clustered genes Production of thepigmented antibiotics actinorhodin (Act) and undecyl-
prodigiosin (Red) has been studied extensively in
Scoelicolor
(for reviews see Chater and Bibb 1997Champness 2000) and involves clusters of approximately22 genes each A third cluster producing the calcium-dependent antibiotic (CDA) and consisting of 40 geneshas also been studied (Chong
et al
1998 Huang
et al
2001) In the extensive spot identification studies for thepreparation of a proteome map of
S coelicolor
(seeabove) more than one-third of the proteins from each ofthese pathways was identified (Table 5) Proteins fromthree clusters that specify metabolic products that haveyet to be identified experimentally (Bentley
et al
2002)were also extensively represented a type I polyketidesynthase cluster (SCO6273ndash6288) a deoxysugarglycosyltransferase cluster (SCO0381ndash0401) and anon-ribosomal peptide synthetase cluster predicted toencode a novel siderophore coelichelin (SCO0489ndash0499) Several clusters were unrepresented
The primary metabolic pathways were somewhat morehighly represented with
ordf
60 or more of proteins anno-
Fig 2
Examples of MALDI-TOF peptide mass fingerprint data identifying post-translational modification of proteins by (A) acetylation (predicted mass increase of 420 Da) (B) adenylylation (predicted mass increase of 3290 Da) and (C) proteolytic processing (predicted mass loss for Ile of 11308 Da) In each fingerprint the
x
-axis is masscharge ratio (mz) and the
y
-axis is absolute intensity The observed mass accuracies in the MALDI-TOF fingerprints are (A) 60 ppm (B) 20 ppm and (C) 20 ppm
924
A R Hesketh
et al
copy 2002 Blackwell Publishing Ltd
Molecular Microbiology
46
917ndash932
tated as enzymes of glycolysis the TCA cycle thepentose phosphate pathway purine ribonucleotide bio-synthesis and pyrimidine nucleotide biosynthesis beingmapped (Table 6) About 50 of amino acid biosyntheticproteins were identified Lists of individual members of allpathways considered here including those that were andwere not identified during proteome mapping can befound in the
Supplementary material
Several proteins from both primary and secondary met-
abolic pathways were identified at more than one positionon the proteome map and may therefore be subject toregulation by post-translational modification (see Table 3and Fig 1) Primary metabolic proteins of this type (yellowspots on Fig 1) were usually detected as chains of two ormore spots with the same apparent molecular weights butdifferent pI values suggesting covalent modification bylow-molecular-weight adducts Apart from adenylylation ofGSI (see above) the nature of the modification of theseproteins was not revealed by MALDI-TOF peptide massfingerprint data Both covalent modification and proteolyticprocessing were also found in secondary metabolic pro-teins (red spots in Fig 1) For example the Act pathwaymembers SCO5071 (spot 16) and SCO5075 (spot 20)
and the putative 3-oxoacyl[acyl carrier protein] reductasefrom the type I polyketide cluster (SCO6282 spot 11)appear to be covalently modified whereas proteolytic pro-cessing was detected for RedL (SCO5892 spot 18) fromthe undecylprodigiosin cluster the putative dehydrataseActVI-ORF3 (SCO5074 spot 31) from the Act cluster theputative peptide monooxygenase from coelichelin biosyn-thesis (SCO0498 spot 34) and the putative oxygenasefrom the CDA pathway (SCO3236 spot 33) Two Act bio-synthetic proteins SCO5088 (spot 24) and SCO5073(spot 29) were detected both at their predicted pI andmolecular weight co-ordinates and at positions with simi-lar pI values but significantly higher molecular weightsThis may indicate that these proteins form stable mul-timers that are not completely denatured under theconditions used Only the modification of ActVI-ORF3(SCO5074 spot 31) could be characterized further fromthe mass spectrometry data which showed that it involvesproteolytic processing at two sites between amino acidresidues 3132 and 3435 (data not shown) Analysis of asample obtained by externally extracting proteins fromintact washed mycelium with a solution containing 2SDS and 50 mM dithiothreitol (DTT) revealed that sur-
Table 5
Summary of proteins identified on the proteome map from secondary metabolic pathways during growth in SMM
Biosynthetic pathwaygene cluster
Pathwaygenesannotated
Pathwaygene productsidentified
Percentagepathwaydetected
Calcium-dependent antibiotic CDA SCO3210ndash3249 40 16 40Actinorhodin SCO5071ndash5092 22 15 68Undecylprodigiosin SCO5877ndash5898 22 8 36Deoxysugar synthaseglycosyltransferase cluster SCO0381ndash0401 21 7 33Type I polyketide synthase SCO6273ndash6288 16 6 38Hopanoids SCO6759ndash6771 13 1 7Siderophore (coelichelin) SCO0489ndash0499 11 5 45Siderophore (desferrioxamine) SCO2783ndash2785 4 4 100
Chromosomal locations and extent of gene clusters are taken from Bentley
et al
(2002) No proteins were detected from the remaining predictedsecondary metabolic clusters ie siderophore (coelibactin) SCO7681ndash7691 (11 ORFs) siderophore SCO5799ndash5801 (three ORFs) non-ribosomal peptide synthases SCO6429ndash6438 (10 ORFs) type II fatty acid synthases SCO1265ndash1273 (nine ORFs) WhiE spore pigment clusterSCO5314ndash5320 (seven ORFs) isorenieratine SCO0185ndash0191 (seven ORFs) eicosapentaenoic acid SCO0124ndash0129 (six ORFs) chalconesynthases SCO1206ndash1208 (three ORFs) chalcone synthases SCO7669ndash7671 and SCO7222 (three ORFs) sesquiterpene cyclase SCO5222ndash5223 (two ORFs) type I polyketide synthases SCO6826ndash6827 (two ORFs) geosmin SCO6703 (one ORF)
Table 6 Summary of proteins identified on the proteome map from primary metabolic pathways during growth in SMM
Biosynthetic pathwayPathway genesannotateda
Pathway geneproducts identified
Percentage pathway detected
Glycolysisb 24 13 55Pentose phosphate pathwayb 14 10 71TCA cycleb 21 12 57Amino acid biosynthesisc 115 55 48Purine ribonucleotide biosynthesisb 18 12 67Pyrimidine ribonucleotide biosynthesisb 8 5 62
a Derived from interpretation of the annotated S coelicolor genome sequence at httpwwwsangeracukProjectsS_coelicolorb See Supplementary material for details of pathway membersc See Supplementary material for details of individual pathways
Proteomic analysis of S coelicolor 925
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
prisingly the truncated ActVI-ORF3 is located extracellu-larly presumably non-covalently associated with the cellwall matrix (Fig 4)
Expression analysis of certain primary and secondary metabolic proteins
Production of the pigmented antibiotics Act and Red inliquid minimal medium supplemented with casamino acids(SMM) begins in the transition phase between exponentialgrowth and stationary phase Figure 3 shows a compari-son of the expression of certain primary and secondarymetabolic proteins between exponentially growing andstationary phase cultures Two proteins from the Actcluster (ActVA-ORF5 and ActI-ORF2) and two from theRed (RedI and RedR) were present in the stationaryphase cultures but were not detected during exponentialgrowth Similarly the putative 3-oxoacyl-[acyl carrierprotein] reductase (SCO6282) and epoxide hydrolase(SCO6277) enzymes from a type I polyketide secondarymetabolic cluster showed a large increase in abundance
between the exponential and stationary phase samplesTriose phosphate isomerase (SCO1945) fructose bis-phosphate aldolase (SCO3649) and succinyl-CoA syn-thetase alpha chain (SCO4809) all enzymes from centralcarbon metabolism were present in approximatelyconstant amounts in both samples Interestingly two iso-forms (SCO7511 and SCO1947) of glyceraldehyde-3-phosphate dehydrogenase from the glycolytic pathwayshowed different patterns of expression SCO1947 levelswere the same in both exponential and stationary phasebut SCO7511 barely detectable in exponentially growingcultures increased markedly in stationary phase
The Streptomyces coelicolor online proteome map
The 2D gel images in Fig 1 together with the spot iden-tification results are being made available over the WorldWide Web to provide a reference map of the S coelicolorproteome At the time of writing the reference gel forthe pH 4ndash7 isoelectric point range was fully accessiblewhereas those for pH 45ndash55 pH 55ndash67 and pH 6ndash11
Fig 3 A comparison of the expression of certain primary and secondary metabolic proteins between exponentially growing and stationary phase cultures of S coelicolor M145 Larger arrows represent higher protein level in one gel than in the other The experiment was performed on duplicate biological samples and both showed similar protein expression profiles
926 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
were under construction (httpqbababeracukS_coeliReferenceGelrefgelhtml) A tool for comparing two ormore gel images with each other is also accessible fromthe same website at httpqbababeracukS_coeliflickerhtml
Discussion
Silver staining is one of the most sensitive methods fordetecting proteins on polyacrylamide gels allowing aslittle as 05ndash2 ng of an individual protein to be detected(Berggren et al 2000 Gorg et al 2000) The proportionof the S coelicolor proteome that can be analysed usingsilver staining of 2D-PAGE-separated proteins dependsat least in part on the pH range of the IPG strip used forthe first-dimension separation (see Table 1) Using theintermediate-range IPG strip covering pH 4ndash7 about 20ndash25 of the proteins in the theoretical proteome predictedto be observable in the resulting 2D-PAGE gel were actu-ally detected This figure was reduced to 10ndash12 whenanalysing the more basic pI range using the pH 6ndash11 IPGstrip Attempts to increase the percentage of the proteomedetectable using the pH 4ndash7 and pH 6ndash11 IPG strips byloading more protein sample resulted in poorer separa-tions (data not shown) This was especially true of thepH 6ndash11 strips which were generally the most difficult touse successfully However good separation of higher pro-tein loads was achieved using the narrow-range IPG stripspH 45ndash55 and 55ndash67 significantly increasing the pro-portion of theoretical proteins detected to over 40Clearly and more pertinently the limited conditions usedfor culturing S coelicolor in this study will also haverestricted the number of proteins detected as not all theproteome will be expressed In particular S coelicolordoes not sporulate in shaken liquid-grown cultures soproteins involved in this differentiation process are unlikelyto be present Nevertheless it is striking that the com-bined data for the pH 45ndash55 and 55ndash67 isoelectric pointranges indicate that 2468 out of a total of predicted 4932protein spots can be detected (see Table 1) suggestingthat transition phase mycelium in this supplementedminimal medium is expressing 50 of its genes (ie byextrapolation some 3912 of the 7825 in the genome)
Analysis of post-translational regulation
Although the regulation of cellular processes and adaptiveresponses in bacteria has been studied extensively at thetranscriptional level post-translational regulatory mecha-nisms are much less understood Proteomic analysisusing 2D-PAGE coupled with mass spectrometry offersthe opportunity to observe and identify modification ofproteins at a general cellular level The functions of pro-teins can be altered by specific covalent attachment of
adducts (eg phosphorylation glycosylation nucleotidy-lation acetylation) or by proteolytic processing Bothevents alter the pI andor mass of the protein and so canusually be observed on 2D-PAGE In the S coelicolorproteome map ordf110 out of a total of 770 gene productswere identified in more than one position or at pI andapparent mass co-ordinates significantly different frompredicted values indicating extensive regulation at thepost-translational level (88 are listed in Table 3) a propor-tion similar to that reported for other prokaryotes (Buttneret al 2001 Tonella et al 2001) The initial peptide massfingerprint analysis identified the modification of only 10of these proteins probably partly because fingerprint datatypically cover ordf30ndash60 of a proteinrsquos sequence (data notshown) so modification at positions not represented willbe missed Protein digestion with endopeptidases otherthan trypsin before mass spectrometry may help in thissituation but some covalently attached groups are labileand cannot be detected routinely Phosphorylation of Hisoccurs on nitrogen producing a high-energy phosphora-midate bond that is easily hydrolysed in acidic conditionswhereas phospho-Asp is the most labile phosphorylatedresidue with a half-life of a few hours under physiologicalconditions (reviewed by Robinson et al 2000) Thesemodifications the basis of bacterial two-component regu-latory systems are therefore unlikely to survive stainingof the 2D gels which involved the use of a fixing solutioncontaining 10 acetic acid and mass spectrometry Iden-tification of the more stable phosphoester phosphorylationthat occurs on Ser Thr or Tyr residues using PAGEfollowed by mass spectrometry has been reported(Godovac-Zimmermann et al 1999 Buttner et al 2001)but no examples were detected in this work Of the mod-ifications identified in this study three were by acetylationat the N-terminus after removal of the initiator Met residuetwo involved adenylylation and five proteins had been N-terminally truncated Modification of the 20S proteasomeby N-acetylation in yeast involves removal of the Metresidue by N-methionylaminopeptidase followed byacetylation with an Na-acetyltransferase (Kimura et al2000) Homologues of these enzymes are predicted fromthe S coelicolor genome and are presumably responsiblefor the observed modification of the 20S proteasomealpha subunit and of BldK-ORFD and the putative cellu-lose-binding protein The functional significance of Na-acetylation of these proteins is unclear
Analysis of the biosynthesis of antibiotics and other secondary metabolites
Post-translational modification The proteome mappingresults indicate that at least six enzymes in the Act clusterare subject to post-translational modifications and one
Proteomic analysis of S coelicolor 927
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
each from the undecylprodigiosin (SCO5877ndash5898)lsquocoelichelinrsquo (SCO0489ndash0499) and type I polyketidesynthase (SCO6273ndash6288) clusters (see Table 3) Thesemetabolic pathways may therefore be significantly regu-lated at the post-translational level The actVI-ORF3 gene(SCO5074) encodes a putative dehydratase enzyme thatis part of the biosynthetic pathway for Act a blue memberof the benzoisochromanequinone (BIQ) family of pig-mented antibiotics Mutation in actVI-ORF3 causes a 50decrease in the accumulation of Act (and a reddish brownphenotype) and it is believed to be responsible for pyranring closure an intermediate step in Act biosynthesis (thatmay also occur spontaneously) leading to the formationof the BIQ chromophore (Fernandez-Moreno et al 1994Ichinose et al 1998a Taguchi et al 2000) In this studyActVI-ORF3 protein was detected in two forms a majorspot corresponding to ActVI-ORF3 truncated by removalof the N-terminal 31 amino acid residues and a minor onein which the first 34 amino acids were missing (spots 31in Fig 1) The full-length protein was not detected Anal-ysis of the protein sequence using the SIGNALP server athttpwwwcbsdtudkservicesSignalP predicted a signalpeptide cleavage site between amino acids 31 and 32 inagreement with the observed results
The protein extract analysed was prepared from myce-lium that had been collected by centrifugation and sub-jected to two washes indicating that if ActVI-ORF3 isexported it is effectively retained within the cell wallmatrix This was confirmed by analysis of a preparationobtained by externally extracting proteins from intactwashed mycelium (Fig 4) Export of an enzyme requiredfor the efficient biosynthesis of an antibiotic that is thoughtto be produced intracellularly appears to be anomalousparticularly as it is involved in an intermediate biosynthetic
event Database analysis of the other members of the Actcluster revealed only one other protein with a predictedsignal peptide sequence the putative integral membranetransport-related protein ActII-ORF3 and it seemsunlikely that all the Act biosynthetic steps following BIQchromophore formation are extracellular It is possible thatfull-length ActVI-ORF3 catalyses the intermediate biosyn-thetic event intracellularly and the exported processedprotein is responsible for a different extracellular activityperhaps later in the pathway However no full-length pro-tein was detected so the exported enzyme may performthe pyran ring closure on a secreted metabolite that isthen reabsorbed by the cell for completion of the sub-sequent biosynthetic steps Only two homologues ofactVI-ORF3 are known and both are also in BIQ antibioticbiosynthetic clusters gra-ORF18 from the granaticincluster (Ichinose et al 1998b) and med-ORF5 frommedermycin biosynthesis (K Ichinose personal com-munication) Interestingly the encoded proteins of bothsequences are also predicted to contain N-terminal signalpeptides The observation that ActVI-ORF3 is exported tothe cell wall matrix may therefore be of general signifi-cance to the BIQ family of antibiotics and provide a newinsight into their biosynthetic pathways
Pathway representation Of the 20 putative secondarymetabolite gene clusters identified in the S coelicolorgenome sequence (Bentley et al 2002) eight had atleast one protein each identified on the proteome map(see Table 5) The biosynthetic clusters for the antibioticsAct Red and CDA had 68 36 and 40 of their pro-teins represented There are several possible explana-tions for failure to detect all the proteins for each pathwaySome are putative integral membrane proteins so cannotreadily be detected using conventional 2D-PAGE SomeRed and CDA proteins have molecular weights gt200 kDaoutside the range of the 125 gels used (for clustermembers see Supplementary material) The pathway-specific regulatory proteins for Act and Red biosynthesiswere not detected despite a detailed search of their pre-dicted gel locations even though the relevant genes areknown to be transcribed in equivalent cultures of the samestrain (Hesketh et al 2001) The proteins are presumablypresent but at levels below the limit of detection Howeverthe transcriptional regulator for CDA production CdaRwas identified as well as the two-component systemresponse regulator AbsA2 which is also encoded by agene in the CDA cluster (Anderson et al 2001) Interest-ingly proteins from three putative secondary meta-bolite clusters producing chemicals of as yet unknownstructure were also identified on the map correspondingto the deoxysugar synthaseglycosyltransferase clusterSCO0381ndash0401 the lsquocoelichelinrsquo cluster for a predicted(but not yet confirmed) oligopeptide siderophore
Fig 4 Processed ActVI-ORF3 is present in the cell wall matrix 2D-gel separations of protein extracts prepared (A) from whole cells or (B) by externally washing cells in a 2 SDS extraction buffer The arrow indicates ActVI-ORF3 protein The experiment was performed on duplicate biological samples and both showed similar protein expression profiles
928 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
(SCO0489ndash0499) and the type I polyketide clusterSCO6273ndash6288 The 3-oxoacyl-[acyl carrier protein]reductase (SCO6282) from the latter pathway is ex-pressed in a growth phase-dependent manner (Fig 3)and in early stationary phase cultures is perhaps themost abundant protein spot in the entire detectable pro-teome Presumably the metabolites produced by thesethree pathways are being synthesized under the condi-tions used In addition to coelichelin three other sidero-phores are postulated for S coelicolor All the proteinsfrom one of these desferrioxamine (SCO2782ndash2785)were identified but none was seen from either of the othertwo pathways The WhiE polyketide spore pigment isassociated with sporulation of surface-grown cultures(Davis and Chater 1990) and the expression of the whiEgenes depends on a series of sporulation-specific regula-tory genes (Kelemen et al 1998) no corresponding pro-teins of which were detected here It is therefore notsurprising that WhiE proteins were not detected in sam-ples prepared from liquid-grown vegetative mycelium
Analysis of primary metabolism
Proteins annotated as functioning in primary metabolicpathways were highly represented on the proteome mapwith ordf60 or more of the enzymes assigned to glycolysisthe TCA cycle the pentose phosphate pathway andpurine and pyrimidine ribonucleotide biosynthesis beingidentified (see Table 6) This figure may be deceptively lowbecause these pathways in S coelicolor often appear tohave more than one enzyme isoform for some of themetabolic steps potentially offering additional levels ofcontrol or developmental responsiveness at these pointsFor example only two steps in glycolysis have just a singlegene product assigned to them the remainder typicallyhave two or three each (Fig 5) At least one isoform waspresent for each step so the pathway is in theory 100functionally represented Understanding the regulation ofthese pathways will require analysis of the expression ofthese isoenzymes under different nutritional conditionsor different developmental stages Figure 5 shows that ofthe three predicted glyceraldehyde-3-phosphate dehydro-genase isoforms from glycolysis only two were detectedin this study (see also Supplementary material) Of theseSCO1947 appears to be constitutive whereas SCO7511is growth phase dependent (see Fig 3) Thus even thelimited conditions in this study yielded useful informationabout isoenzyme expression patterns (see Supplemen-tary material) In Streptomyces aureofaciens two genesencoding glyceraldehyde-3-phosphate dehydrogenaseactivity also appear to be differently regulated transcrip-tion of one highly homologous to SCO7511 is activatedby the regulator GapR whereas the other is GapR inde-pendent (Spusansky et al 2001)
Hood et al (1992) reported that most amino acid bio-synthetic enzymes are expressed at very low constitutivelevels in S coelicolor so it is reassuring to note that ordf50could be detected and identified using proteomics Theseinclude TrpB and TrpC1 from the tryptophan biosyntheticpathway the corresponding transcripts of which couldbarely be detected in previously reported S1 nucleaseprotection experiments (Hu et al 1999)
Perspectives
No attempt was made in the work reported here to simplifythe protein separations by subdividing the sample intosoluble and membrane fractions and this is reflected inthe 10 lipoproteins and 16 ABC transport system ATP-binding subunits identified on the proteome map (seeTable 2) No integral membrane proteins were detectedand even when a membrane protein sample was pre-pared and analysed no proteins identified possessedmore than three predicted transmembrane helices (datanot shown) This is consistent with previous studiesin other bacteria (Molloy et al 1999 Nilsson andDavidsson 2000) Another group of proteins also strik-ingly absent from the list of those identified is the sigmafactors which determine promoter selection by RNA poly-merase There are an extraordinarily high number (65) ofsigma factors encoded in the S coelicolor genome aswell as at least seven antisigma factors and 15 antisigmafactor antagonists (some of each of which were detected)Sigma factor heterogeneity in bacteria is an important wayof regulating expression of specific sets of genes under
Fig 5 The glycolytic pathway in S coelicolor illustrating the assignment of multiple gene products to individual metabolic steps based on the annotation of the genome sequence at httpwwwsangeracukProjectsS_coelicolor Gene products identified in the proteome map are underlined
Proteomic analysis of S coelicolor 929
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
different conditions and in different cell types (for a reviewsee Gross et al 1998) Although no sigma factors wereidentified about half (31 out of 65) have predicted pIvalues gtpH 7 and so can only be detected using thepH 6ndash11 IPG strips that give the poorest results (seeTable 1) However the principal vegetative sigma factorHrdB which is expected to be moderately abundanthas a predicted pI of 613 and its absence from thepH 55ndash67 proteome map may indicate that the samplepreparation method which does not remove DNA is inap-propriate Among other regulatory proteins 11 out of apossible 80 two-component system response regulatorsplus a further 36 proteins annotated as lsquoregulatorsrsquo weredetected Only one SerThr kinase and no two-componentsystem sensor kinases were detected possibly reflectingthe difficulty in analysing proteins with several transmem-brane domains Interestingly 30 secreted proteins wereidentified on the proteome map at least 10 of whichappear from comparison of their observed pI and molec-ular weight positions with predicted values to have beenprocessed already and therefore exported Presumablythese proteins do not end up exclusively in the culturemedium
Clearly this approach to proteomic analysis of S coeli-color has some important deficiencies not least theabsence of certain groups of proteins and the relativelypoor initial success rate for specifically characterizingpost-translational modifications Nevertheless it offersunique insights into the molecular biology of the organismthe expression of many proteins from both primary andsecondary metabolic pathways can be analysed post-translational regulation of proteins can be investigated ona general cellular level and the physical locations forsome proteins can be determined Approximately 35 ofthe S coelicolor genome is annotated as encoding lsquohypo-theticalrsquo proteins of unknown function and 182 proteins ofthis type were identified on the proteome map No exam-ples were found of overt disagreements between pre-dicted and observed proteins providing experimentalsupport for the general accuracy of the S coelicolorgenome sequence and its annotation Information aboutthe expression post-translational modification and physi-cal location of these (no longer) hypothetical proteinsfrom proteomic analyses is going to be important forassigning future functions As many of the hypotheticalproteins have apparent orthologues in pathogenic myco-bacteria some of the information may have an immediateapplication in the context of pathogenesis
Experimental procedures
Materials
Immobilized pH gradient (IPG) strips and IPG bufferswere purchased from Amersham Biosciences Acrylamide
duracryl and chemicals for silver staining were obtained fromGenomic Solutions All other chemicals were from Sigmaunless stated in the text
Strain and growth conditions
Streptomyces coelicolor M145 was cultivated with vigorousagitation at 30infinC in minimal medium supplemented with 02casamino acids (SMM) as described previously (Kieser et al2000) Briefly a high-density spore preparation [about 1010
colony-forming units (cfu) ml-1] was pregerminated in 2yen YTmedia for 7 h at 30infinC Germlings were harvested by centrifu-gation (5 min at 4000 g) resuspended in SMM and sonicatedbriefly to disperse any aggregates before inoculation into50 ml of SMM in 250 ml siliconized flasks containing coiledstainless steel springs Each flask received the equivalent of5 yen 107 spore cfu
Protein extraction and 2D-gel electrophoresis
The preparation of the protein sample to be separated waskept as simple as possible disrupting harvested myceliadirectly into denaturing isoelectric focusing (IEF) buffer con-taining Pefabloc SC (Roche) protease inhibitor This methodminimized unwanted protein degradation that was observedin initial studies using a Tris buffer containing Pefabloc SC forcell lysis (data not shown) Removal of DNA RNA salts andpolysaccharides from the sample was therefore not per-formed (other than the shearing of viscous DNA and RNA bysonication) and the protein extracts were sometimes difficultto focus when the sample was loaded by in-gel rehydrationLoading the sample soaked in paper strips at the anodicend of IPG strips that had already been rehydrated improvedthe reproducibility of separations significantly
Mycelium for protein extraction was harvested from cul-tures by brief centrifugation (30 s at 4000 g) at room tem-perature and immediately frozen in liquid nitrogen Typicallymycelium from 10 ml culture aliquots was collected and thetransfer time from culture flask to frozen sample was 15 minFrozen cells were thawed on ice in 5 ml of washing buffer(40 mM Tris pH 90 1 mM EGTA 1 mM EDTA) then pelletedby centrifugation (5 min at 3000 g) at 4infinC Washed cells wereresuspended in 400 ml of denaturing IEF buffer UTCHAPS[7 M urea 2 M thiourea 4 (wv) CHAPS 40 mM TrispH 90 1 mM EDTA 50 mM DTT 4 mM Pefabloc SC pro-tease inhibitor (Roche)] and disrupted by sonication (SanyoSoniprep 150 10 yen 2 s bursts at amplitude 75 mm) whilecooling in an ethanolndashice bath Cell debris was removed bycentrifugation (15 min 10 000 g 4infinC) and the protein extractwas stored frozen in aliquots at -80infinC until use For thepreparation of extracellular protein extracts washed cellswere resuspended in extraction buffer consisting of 40 mMTris pH 90 1 mM EDTA 50 mM DTT 2 SDS and 4 mMPefabloc SC protease inhibitor Cells were vortexed for 2 minbefore being pelleted by centrifugation (5 min 10 000 g4infinC) The supernatant was precipitated using the Amersham2D Clean-Up kit according to the manufacturerrsquos instructionsand the precipitated proteins were redissolved in UTCHAPSand stored at -80infinC until use
930 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
For first-dimension isoelectric focusing 18 cm IPG stripspH 4ndash7 pH 6ndash11 pH 45ndash55 or pH 55ndash67 (Amersham Bio-sciences) were rehydrated overnight in IEF buffer containing1 ampholytes according to the manufacturerrsquos instructionsusing a Phaser isoelectric focusing unit (Genomic Solutions)set at 20 V Protein samples to be separated were applied tothe rehydrated strips at the anodic end soaked in a 5ndash10 mmsection of IEF electrode strip (Amersham Biosciences)Separation was performed for 120 000 V-h with a maximumvoltage of 5000 V After isoelectric focusing IPG strips wereequilibrated for the second dimension for 15 min in IPG equil-ibration buffer (50 mM Tris pH 68 6 M urea 30 glycerol1 SDS and 001 bromophenol blue) plus 80 mM DTTthen for 10 min in IPG equilibration buffer plus 135 mMiodoacetamide Approximately 1 cm was removed from theanodic end of each equilibrated strip before applying the stripto the top of a vertical 125 SDS-PAGE gel for second-dimension separation using the Investigator 5000 systemfrom Genomic Solutions
Large-format gels (24 cm wide yen 22 cm high yen 1 mm thick)for the second-dimension separation were cast in-houseusing the system supplied by Genomic Solutions 125SDS-PAGE gels prepared using 065 NN-methylenebi-sacrylamide as cross-linking agent were used for pH 4ndash7and pH 45ndash55 IPG strips whereas pH 55ndash67 and pH 6ndash11 strips were applied to similar gels made with 08 cross-linker Electrophoresis was performed while cooling using themaximum setting (ordf 4ndash10infinC) at 20 000 mW per gel constantpower and a maximum voltage of 500 V Gels were thenstained with either colloidal Coomassie G-250 (Neuhoff et al1988) or silver nitrate (Rabilloud 1992) and scanned in aProXPRESS proteomic imaging system (Perkin-Elmer)Image analysis was performed using PHORETIX 2D version51 (NonLinear Dynamics) spot detection was optimizedautomatically using the lsquoSpot Detection Wizardrsquo and thenedited manually background subtraction was performedautomatically using the lsquoMode of Non-Spotrsquo setting imageswere then normalized to the total spot volume for quantifica-tion Spot filtering was not used
Protein identification using mass spectrometry
Protein spots of interest were excised from colloidalCoomassie-stained gels in-gel digested with trypsin andidentified by MALDI-TOF peptide mass fingerprint analysisExcised gel pieces (ordf 1-mm-diameter circles cut from 1-mm-thick gels) were washed twice with 100 ml of 50 mMammonium bicarbonate for 15 min once with 100 ml of 20acetonitrile-40 mM ammonium bicarbonate for 15 min andonce with 100 acetonitrile Washed gel pieces were allowedto air-dry for 10 min before being rehydrated in 5 ml of10 mg ml-1 trypsin (sequencing grade modified Promega) in10 mM ammonium bicarbonate Digestion (37infinC for 4 h) wasstopped by the addition of 5 ml of 5 formic acid Extractionof peptides into this solution was encouraged by a sonicwater bath treatment for 20 min Peptide extract (05 ml) wasspotted on to a thin layer of a-cyano-4-hydroxycinnamic acidapplied to a MALDI-TOF sample template and analysed byMALDI-TOF mass spectrometry (Bruker Reflex III) using anaccelerating voltage of 25 kV Samples were externally cali-brated using a standard mixture of six peptides ranging in
mass from 10465423 Da to 34946500 Da and the massaccuracy obtained was 60 ppm or better Identification ofproteins from MALDI-TOF peptide mass fingerprint data wasperformed using the lsquoMascotrsquo search engine at httpwwwmatrixsciencecom and was based on a positive resultusing their lsquoProbability Based MOWSE Scorersquo algorithm AMOWSE score of 60 or higher is significant at the 5 levelor better and proteins in this work typically gave scores gt80(frequently considerably so) In addition no identification wasaccepted unless at least five peptides representing at least20 of the protein sequence were detected in the MALDI-TOF peptide mass fingerprint
Assignment of proteins to metabolic pathways
Proteins were assigned to primary metabolic pathwaysusing the lsquoProtein Classification Schemersquo web page ofthe annotated Streptomyces coelicolor genome sequenceat httpwwwsangeracukProjectsS_coelicolor as it ap-peared in May 2002 The annotation of the genome andtherefore the Protein Classification Scheme is likely to bemodified as more experimental data are obtained about thefunction of genes that have currently been assigned on thebasis of sequence information alone It is therefore possiblethat proteins assigned to metabolic pathways in this studymay in future be shown to perform different functions in thecell
Assignments to gene clusters encoding secondary metab-olites are based on an analysis of the genome sequencereported by Bentley et al (2002) In some cases theseassignments are based on extensive experimental data (actred reviewed by Chater and Bibb 1997 Champness 2000cda Chong et al 1998 Huang et al 2001 whiE Davis andChater 1990)
Acknowledgements
This work was funded by grants (2FGT11406 208FGT11408 and 208IGF12432) under the BBSRCrsquos Tech-nologies for Functional Genomics and Investigating GeneFunction initiatives and by a competitive strategic grant fromthe BBSRC to the John Innes Centre We thank Mike Naldrettfor his extensive contribution in establishing technology forproteome analysis at the JIC
Supplementary material
The following material is available from httpwwwblackwell-sciencecomproductsjournalssuppmatmolemole3219mmi3219smhtmSection 1 Detection of gene products assigned to individualmetabolic steps in primary metabolic pathways in ScoelicolorSection 2 Identification of proteins from individual amino acidbiosynthetic pathwaysSection 3 Detection of gene products assigned to individualmetabolic steps in secondary metabolic pathways in Scoelicolor
Proteomic analysis of S coelicolor 931
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
References
Anderson TB Brian P and Champness WC (2001)Genetic and transcriptional analysis of absA an antibioticgene cluster linked two-component system that regulatesmultiple antibiotics in Streptomyces coelicolor Mol Micro-biol 39 553ndash566
Bentley SD Chater KF Cerdeno-Tarraga A-M ChallisGL Thomson NR James KD et al (2002) Completegenome sequence of the model actinomycete Streptomy-ces coelicolor A3(2) Nature 417 141ndash147
Berdy J (1984) New ways to obtain antibiotics Chin J Anti-biot 7 272ndash290
Berggren K Chernokalskaya E Steinberg TH KemperC Lopez MF Diwu Z et al (2000) Background-freehigh sensitivity staining of proteins in one- and two-dimensional sodium dodecyl sulfate-polyacrylamide gelsusing a luminescent ruthenium complex Electrophoresis21 2509ndash2521
Blackstock WP and Weir MP (1999) Proteomics quan-titative and physical mapping of cellular proteins TrendsBiotechnol 17 121ndash127
Blattner FR Plunkett G Bloch CA Perna NT BurlandV Riley M et al (1997) The complete genome sequenceof Escherichia coli K-12 Science 277 1453ndash1462
Buttner K Bernhardt J Scharf C Schmid R MaderU Eymann C et al (2001) A comprehensive two-dimensional map of cytosolic proteins of Bacillus subtilisElectrophoresis 22 2908ndash2935
Champness W (2000) Actinomycete development antibioticproduction and phylogeny questions and challenges InProkaryotic Development Brun YV and Skimketts LJ(eds) Washington DC American Society for MicrobiologyPress pp 11ndash31
Chater KF and Bibb MJ (1997) Regulation of bacterialantibiotic production In Biotechnology Vol 7 Products ofSecondary Metabolism Kleinkauf H and von Doumlhren H(eds) Weinheim Germany VCH pp 57ndash105
Chong PP Podmore SM Kieser HM Redenbach MTurgay K Marahiel M et al (1998) Physical identificationof a chromosomal locus encoding biosynthetic genesfor the lipopeptide calcium-dependent antibiotic (CDA) ofStreptomyces coelicolor A3(2) Microbiology 144 193ndash199
de Crecy-Lagard V Servant-Moisson JV Grandvalet Cand Mazodier P (1999) Alteration of the synthesis of theClp ATP-dependent protease affects morphological andphysiological differentiation in Streptomyces Mol Microbiol32 505ndash517
Davis NK and Chater KF (1990) Spore colour inStreptomyces coelicolor A3(2) involves the developmen-tally regulated synthesis of a compound biosyntheticallyrelated to polyketide antibiotics Mol Microbiol 4 1679ndash1691
Fernandez-Moreno FA Martinez E Caballero JLIchinose K Hopwood DA and Malpartida F (1994)DNA sequence and functions of the actVI region of theactinorhodin biosynthetic gene cluster of Streptomycescoelicolor A3(2) J Biol Chem 269 24854ndash24863
Fink D Falke D Wohllheben W and Engels A (1999)Nitrogen metabolism in Streptomyces coelicolor A3(2)
modification of glutamine synthase I by an adenylyltrans-ferase Microbiology 145 2313ndash2322
Godovac-Zimmermann J Soskic V Posnanovic S andBrianza F (1999) Functional proteomics of signal trans-duction by membrane receptors Electrophoresis 20 952ndash961
Gorg A Obermaier C Boguth G Harder A Scheibe BWildgruber R and Weiss W (2000) The current state oftwo-dimensional electrophoresis with immobilized pH gra-dients Electrophoresis 21 1037ndash1053
Gramajo HC Takano E and Bibb MJ (1993) Stationary-phase production of the antibiotic actinorhodin in Strepto-myces coelicolor A3(2) is transcriptionally regulated MolMicrobiol 7 837ndash845
Gross CA Chan C Dombroski A Gruber T Sharp MTupy J and Young B (1998) The functional and regula-tory roles of sigma factors in transcription Cold SpringHarb Symp Quant Biol 63 141ndash155
Hesketh A Sun J and Bibb MJ (2001) Induction ofppGpp synthesis in Streptomyces coelicolor A3(2) grownunder conditions of nutritional sufficiency elicits actII-ORF4transcription and actinorhodin biosynthesis Mol Microbiol39 136ndash144
Hesketh A Fink D Gust B Rexer H-U Scheel BChater K et al (2002) The PII nitrogen regulatory proteinGlnK in Streptomyces coelicolor A3(2) is modified by ade-nylylation rather than uridylylation and the GlnD and GlnKhomologues are functionally dissimilar to their nitrogenregulatory system counterparts Mol Microbiol (inpress)
Hood DW Heidstra R Swodoba UK and Hodgson DA(1992) Molecular genetic analysis of proline and tryp-tophan biosynthesis in Streptomyces coelicolor A3(2)interaction between primary and secondary metabolism- areview Gene 115 5ndash12
Hu DS-J Hood DW Heidstra R and Hodgson DA(1999) The expression of the trpD trpC and trpBA genesof Streptomyces coelicolor A3(2) is regulated by growthrate and growth phase but not by feedback repression MolMicrobiol 32 869ndash880
Huang J Lih CJ Pan KH and Cohen SN (2001)Global analysis of growth phase responsive gene expres-sion and regulation of antibiotic biosynthetic pathways inStreptomyces coelicolor using DNA microarrays GenesDev 15 3183ndash3192
Ichinose K Taguchi T Ebizuka Y and Hopwood DA(1998a) Biosynthetic gene clusters of benzoisochromane-quinone antibiotics in Streptomyces spp Identification ofgenes involved in post-PKS tailoring steps Actinomyceto-logica 12 99ndash109
Ichinose K Bedford DJ Tornus D Bechthold A BibbMJ Revill PW et al (1998b) The granaticin biosyntheticgene cluster of Streptomyces violaceoruber Tu22sequence analysis and expression in a heterologous hostChem Biol 5 647ndash659
Kelemen GH Brian P Flardh K Chamberlin L ChaterK and Buttner M (1998) Developmental regulation oftranscription of whiE a locus specifying the polyketidespore pigment in Streptomyces coelicolor A3(2) J Bacte-riol 180 2515ndash2521
Kieser T Bibb MJ Buttner MJ Chater KF and
932 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
Hopwood DA (2000) Practical Streptomyces GeneticsNorwich John Innes Foundation
Kim EJ Chung HJ Suh B Hah YC and Roe JH(1998) Expression and regulation of the sodF gene encod-ing iron- and zinc-containing superoxide dismutase inStreptomyces coelicolor Muumlller J Bacteriol 180 2014ndash2020
Kimura Y Takaoka M Tanaka S Sassa H Tanaka KPolevoda B et al (2000) Na-acetylation and proteolyticactivity of the yeast 20S proteasome J Biol Chem 2754635ndash4639
Kunst F Ogasawara N Moszer I Albertini AM AlloniG Azevedo V et al (1997) The complete genomesequence of the Gram-positive bacterium Bacillus subtilisNature 390 249ndash256
Lucchini S Thompson A and Hinton JCD (2001)Microarrays for microbiologists Microbiology 147 1403ndash1414
Mann M Hendrickson RC and Pandey A (2001) Analy-sis of proteins and proteomes by mass spectrometry AnnuRev Biochem 70 437ndash473
Miyodah S (1993) Research on antibiotic screening inJapan over the last decade a producing microorganismsapproach Actinomycetologica 7 100ndash106
Molloy MP Herbert BR Williams KL and Gooley AA(1999) Extraction of Escherichia coli proteins with organicsolvents prior to two-dimensional electrophoresis Electro-phoresis 20 701ndash704
Nagy I Tamura T Vanderleyden J Baumeister W andDe Mot R (1998) The 20S proteasome of Streptomycescoelicolor J Bacteriol 180 5448ndash5453
Neuhoff V Arold N Taube D and Ehrhardt W (1988)Improved staining of proteins in polyacrylamide gels includ-ing isoelectric focusing gels with clear background at nan-ogram sensitivity using Coomassie Brilliant Blue G-250and R-250 Electrophoresis 9 255ndash262
Nilsson CL and Davidsson P (2000) New separation toolsfor comprehensive studies of protein expression by massspectrometry Mass Spectrom Rev 19 390ndash397
Puglia AM Vohradsky J and Thompson CJ (1995)
Developmental control of the heat-shock stress regulon inStreptomyces coelicolor Mol Microbiol 17 737ndash746
Rabilloud T (1992) A comparison between low backgroundsilver diamine and silver nitrate protein stains Electro-phoresis 13 429ndash439
Robinson VL Buckler DR and Stock AM (2000) A taleof two components a novel kinase and a regulatory switchNature Struct Biol 7 626ndash633
Schena M (2000) Microarray Biochip Technology NatickMA Eaton Publishing
Spusansky O Rezuchova B Homerova D andKormanec J (2001) Expression of the gap gene encodingglyceraldehyde-3-phosphate dehydrogenase of Strepto-myces aureofaciens requires GapR a member of theAraCXylS family of transcriptional activators Microbiology147 1291ndash1301
Taguchi T Itou K Ebizuka Y Malpartida F HopwoodDA Surti CM et al (2000) Chemical characterisationof disruptants of the Streptomyces coelicolor A3(2) actVIgenes involved in actinorhodin biosynthesis J Antibiot 53144ndash152
Takano E Gramajo HC Strauch E Andres N WhiteJ and Bibb MJ (1992) Transcriptional regulation of theredD transcriptional activator gene accounts for growth-phase-dependent production of the antibotic undecylpro-digiosin in Streptomyces coelicolor A3(2) Mol Microbiol6 2797ndash2804
Tonella L Hoogland C Binz P-A Appel RDHochstrasser DF and Sanchez J-C (2001) New per-spectives in the Escherichia coli proteome investigationProteomics 1 409ndash423
Vohradsky J Li XM and Thompson CJ (1997) Identifi-cation of prokaryotic developmental stages by statisticalanalysis of two-dimensional gel patterns Electrophoresis18 1418ndash1428
Vohradsky J Li XM Dale G Folcher M Nguyen LViollier PH and Thompson CJ (2000) Developmentalcontrol of stress stimulons in Streptomyces coelicolorrevealed by statistical analyses of global gene expressionpatterns J Bacteriol 182 4979ndash4986

Proteomic analysis of
S coelicolor 919
copy 2002 Blackwell Publishing Ltd
Molecular Microbiology
46
917ndash932
Fig 1
A proteome map of
S coelicolor
showing 2D-PAGE separations using IPG strips pH 4ndash7 pH 6ndash11 pH 45ndash55 and pH 55-67 The number of spots identified is given at the foot of each reference gel Protein spots from metabolic pathways that were identified at positions inconsistent with their predicted amino acid sequences and suggestive of post-translational modification are marked in yellow (primary metabolism) or red (secondary metabolism) Blue spots correspond to other examples of post-translational modifications that are mentioned in the text and in Table 3 Separations have been performed on numerous occasions and generally show high spatial reproducibility
920
A R Hesketh
et al
copy 2002 Blackwell Publishing Ltd
Molecular Microbiology
46
917ndash932
back onto the analytical silver-stained gels in Fig 1Although silver staining is 5ndash10 times more sensitive thancolloidal Coomassie staining (and about 100 times moresensitive than conventional Coomassie staining) furthermass spectrometry analysis of protein spots is much moreefficient after staining with colloidal Coomassie than withsilver A total of 1305 protein spots were unambiguouslyassigned to sequenced genes using MALDI-TOF peptidemass fingerprint analysis at a success rate of
ordf
90(Table 1) There is some redundancy in these figures withseveral spots being represented in more than one pIrange On each gel some proteins were identified in morethan one position indicative of post-translational modifi-cations The average number of protein spots per geneproduct was 12 The products from 770 different geneswere assigned to the proteome map representing
ordf
10of the genome and corresponding to many differentclasses of proteins (Table 2) The master gel referencemaps are the basis for the production of a publicly acces-sible proteome database (httpqbababeracukS_coeliReferenceGelrefgelhtml see last Results section)
Analysis of co- and post-translational modifications
The results of MALDI-TOF identification of protein spotssummarized in Table 1 indicated a significant amount ofregulation at the post-transcriptional level in
S coelicolor
as in most other bacteria Table 3 lists those gene prod-ucts that were identified at pI andor molecular weight co-ordinates significantly different from predicted values Ingeneral these fell into two categories one or more spotswith lower than expected molecular weights and oftendifferent pI values indicating modification by proteolyticprocessing (Table 3A) and multiple spots with the sameapparent molecular weights but different pI values indi-
cating modification by covalent addition of a small adduct(Table 3B) In the former case the full-length protein wasoften but not always detected A similar fraction ofproteins of unknown function and annotated as lsquohypo-theticalrsquo or lsquoconserved hypotheticalrsquo was subject to post-translational modification but these will be described ina later publication and are not considered in Table 3Evidence in the peptide mass fingerprint data for the spe-cific nature of the modification was obtained in 10 out ofthe 88 proteins listed (Table 4) Figure 2 illustrates thecharacterization of an example of each type of modifica-tion by interpretation of the MALDI-TOF data
N-acetylation
The putative cellulose-binding proteinSCO5396 was identified in two positions on the proteomemap (spots labelled 10 in Fig 1) with the same apparentmolecular weight but significantly different pI values Themore basic spot gave a tryptic peptide at 137076 Dacorresponding to the N-terminal peptide 2ndash13 indicatingthat the initiator Met residue had been co-translationallyremoved (Fig 2A top) In the more acidic spot (ie on theleft in Fig 1) this peptide had increased in mass by420 Da diagnostic of the addition of an acetyl grouppresumably on the N-terminal Ser residue (Fig 2A bot-tom) A similar modification was identified for the 20Sproteasome alpha subunit (spots 12 in Fig 1) and for theputative ABC transport protein BldK-ORFD (spots 17 inFig 1) although the presumptive N-terminal acetylatedresidue is Thr in the latter (data not shown)
Adenylylation
By analogy with the nitrogen regulatorysystem in
E coli
glutamine synthetase I (GSI) wasalready believed to be modified by adenylylation on aconserved Tyr-397 residue and biochemical evidence forthis has been reported previously (Fink
et al
1999) TheMALDI-TOF data showed the tryptic peptide 395ndash420 atthe expected mass of 28074 Da for the larger more basicspot 6 in Fig 1 but this increased by 3290 Da corre-sponding to the addition of an adenylyl group in thesmaller spot on the left (Fig 2B) Adenylylation of thenitrogen regulatory protein GlnK (spot 15 in Fig 1) wasidentified similarly but this is described in detail elsewhere(Hesketh
et al
2002)
Proteolytic processing
The 50S ribosomal protein L9was identified as two spots that differed in both pI andmolecular weight (spots 30 in Fig 1) suggesting modifi-cation by proteolytic processing The spot with the highermolecular weight showed the tryptic peptide correspond-ing to amino acids 3ndash22 at the expected mass of199310 Da in agreement with this being the unproc-essed form of the protein (Fig 2C top) In the lower ofthe two spots the peptide decreased in mass by11312 Da (Fig 2C bottom) corresponding to the loss of
Table 2
Types of proteins identified on the proteome map
Protein class
a
Number ingenome
a
Number identified
b
Hypothetical 1049 68 (6)Conserved hypothetical 1322 114 (8)Secreted 530 30 (5)ABC transport ATP binding 109 16 (14)Lipoproteins 124 10 (8)Sigma factors 65 0 (0)Antisigma factors 7 2 (28)Antisigma factor antagonists 15 1 (6)Two-component system regulators 80 11 (13)Other regulators 592 36 (6)Two-component system kinases 85 0 (0)SerThr kinases 37 1 (2)Other 3923 482 (12)All 7816 770 (10)
a
Based on annotation of the
S coelicolor
genome sequence(httpwwwsangeracukProjectsS_coelicolor)
b
Percentage identified is included in parentheses
Proteomic analysis of
S coelicolor 921
copy 2002 Blackwell Publishing Ltd
Molecular Microbiology
46
917ndash932
Table 3
Proteins identified at pI and molecular weight co-ordinates on the 2D-PAGE maps that are inconsistent with their predicted amino acidsequences indicating possible post-translational modification either by proteolytic processing (A) or by covalent addition of a low-molecular-weight adduct (B)
ORF no Gene product Spot no in Fig 1
ASCO0498 Probable peptide monooxygenase 33SCO1230 Putative tripeptidylaminopeptidaseSCO1644 20S proteasome beta subunit precursor 35SCO1815 3-Oxoacyl-[acyl carrier protein] reductaseSCO1860 Putative secreted proteinSCO1947 Glyceraldehyde-3-phosphate dehydrogenase 9SCO1998 30S ribosomal protein S1SCO2619 ATP-dependent Clp protease subunit 13SCO2920 Probable secreted proteinaseSCO3236 Probable oxygenase 34SCO3671 Heat shock protein DNAKSCO3909 50S ribosomal protein L9 30SCO4296 Chaperonin GroEL2SCO4439
D
-Alanyl-
D
-alanine carboxypeptidaseSCO4643 UDP-N-acetylanoylpyruvoylglucosamine reductaseSCO4662 Elongation factor Tu1SCO4762 Chaperonin GroEL1SCO4808 Succinyl-CoA synthetase beta chain 8SCO4824 Bifunctional protein methylene tetra-hydrofolate dehydrogenasecyclohydrolaseSCO5073 Putative oxidoreductase ActVI-ORF2 29SCO5074 Putative dehydratase ActVI-ORF3 31SCO5088 Ketoacyl synthase beta subunit ActI-ORF2 24SCO5113 Oligopeptide ABC transport protein BldK-ORFBSCO5254 Superoxide dismutase precursor 32SCO5371 ATP synthase alpha chainSCO5477 Probable oligopeptide-binding lipoproteinSCO5624 30S ribosomal protein S2SCO5737 Guanosine pentaphosphate synthetaseSCO5769 Recombinase RecASCO5892 Polyketide synthase RedL 18SCO6282 3-Oxoacyl-[acyl carrier protein] reductase 11
BSCO1074 Putative peptidaseSCO1476 S-adenosylmethionine synthetaseSCO1594 Phenyl-tRNA synthetase beta chainSCO1643 20S proteasome alpha subunit 12SCO1814 Enoyl-[acyl carrier protein] reductaseSCO1935 Transketolase 3SCO1947 Glyceraldehyde-3-phosphate dehydrogenase 9SCO2180 Dihydrolipoamide dehydrogenase 21SCO2181 Probable dihydrolipoamide S-succinyltransferase 14SCO2198 Glutamine synthetase I 6SCO2390 3-Oxoacyl-[acyl carrier protein] synthase IISCO2478 Putative reductaseSCO2618 ATP-dependent Clp protease subunit 2SCO2822 Putative decarboxylaseSCO3096 Enolase 23SCO3229 4-Hydroxyphenylpyruvic acid dioxygenaseSCO3337 Probable pyrroline-5-carboxylate reductase 28SCO3409 Inorganic pyrophosphataseSCO3549 Antisigma factor antagonist BldGSCO3563 Acetyl-CoA synthaseSCO3671 Heat shock protein DNAKSCO3878 DNA polymerase III beta chainSCO3890 Thioredoxin reductaseSCO3906 30S ribosomal protein S6SCO3970 XAA-Pro aminopeptidase ISCO4164 Putative thiosulphate sulphurtransferaseSCO4209 Phosphoglycerate mutase 19SCO4240 ABC transport ATP binding protein MsiKSCO4296 Chaperonin GroEL2SCO4645 Aspartate aminotransferase 7SCO4652 50S ribosomal protein L10
Some proteins appear to be subject to both types of modification and are present in both parts Only proteins from primary or secondary metabolicpathways or with identified modifications are assigned to spots in Fig 1
922
A R Hesketh
et al
copy 2002 Blackwell Publishing Ltd
Molecular Microbiology
46
917ndash932
the isoleucine residue at its N-terminus (expected massloss 11308 Da) This indicates removal of the N-terminalMKI residues from ribosomal protein L9 The theoreticalisoelectric point (997) and molecular weight (156 kDa) ofL9 protein N-terminally truncated in this way are in goodagreement with the position of the lower spot in Fig 1(predicted values for the unprocessed protein are pH 960and 159 kDa) MALDI-TOF data indicating proteolytic
processing of the superoxide dismutase precursor protein(SCO5254 spot 32 in Fig 1) and the 20S proteasomebeta subunit precursor (SCO1644 spot 35 in Fig 1)agreed with previous reports (Kim
et al
1998 Nagy
et al
1998) Evidence for the proteolysis of
S coelicolor
ClpP1(SCO2619 spot 13 in Fig 1) has also been reportedalthough the processing site was not defined (de Crecy-Lagard
et al
1999)
SCO4661 Elongation factor GSCO4662 Elongation factor Tu1SCO4677 Putative regulatory proteinSCO4729 DNA-directed RNA polymerase alpha chainSCO4762 Chaperonin GroEL1SCO4770 Inosine-5
cent
-monophosphate dehydrogenase 25SCO4792 LuxR family two-component regulatorSCO4808 Succinyl-CoA synthetase beta chain 8SCO4809 Succinyl-CoA synthetase alpha chain 22SCO4921 Probable acyl-CoA carboxylase complex A chainSCO5071 Hydroxylacyl-CoA dehydrogenase ActVI-ORFA 16SCO5079 Hypothetical protein ActVA-ORF4 20SCO5080 Possible hydrolase ActVA-ORF5 26SCO5115 Probable ABC transporter intracellular ATPase chain BldK-ORFD 17SCO5371 ATP synthase alpha chainSCO5396 Putative cellulose-binding protein 10SCO5419 Putative thioredoxinSCO5515 Probable
D
-3-phosphoglycerate dehydrogenase 4SCO5535 Putative carboxyl transferaseSCO5584 Nitrogen regulatory protein GlnK 15SCO5625 Elongation factor TsSCO5699 Prolyl-tRNA synthetaseSCO5737 Guanosine pentaphosphate synthetaseSCO5777 Glutamate uptake system ATP-binding proteinSCO5803 SOS regulatory protein LexASCO5999 Aconitase 1SCO6198 Probable secreted proteinSCO6282 3-Oxoacyl-[acyl carrier protein] reductase 11SCO6636 Bacteriophage
f
-C31 resistance protein PglZSCO6663 Transketolase 27SCO7000 Isocitrate dehydrogenase 2SCO7036 Argininosuccinate synthase 5SCO7510 Peptidyl-prolyl
cis-trans
isomerase
ORF no Gene product Spot no in Fig 1
Some proteins appear to be subject to both types of modification and are present in both parts Only proteins from primary or secondary metabolicpathways or with identified modifications are assigned to spots in Fig 1
Table 3
cont
Table 4
Co- and post-translational modification of proteins suggested by MALDI-TOF peptide mass fingerprint analysis
Protein Modification
SCO1643 20S proteasome alpha subunit Removal of initiator Met residue and acetylation of N-terminal SerSCO5115 ABC transporter intracellular Removal of initiator Met residue and acetylation of N-terminal Thr
ATPase BldK-ORFDSCO5396 Putative cellulose-binding protein Removal of initiator Met residue and acetylation of N-terminal SerSCO2198 Glutamine synthetase I Adenylylation of peptide 395ndash420SCO5584 Nitrogen regulatory protein GlnK Adenylylation of peptide 48ndash58SCO1644 20S proteasome
b
-subunit precursor Proteolytic processing between amino acid residues 5354SCO2619 ATP-dependent Clp protease Proteolytic processing between amino acid residues 2223
proteolytic subunit ISCO3909 50S ribosomal protein L9 Proteolytic processing between amino acid residues 34SCO5074 Putative dehydratase ActVI-ORF3 Proteolytic processing between amino acid residues 3132 and 3435SCO5254 Superoxide dismutase precursor Proteolytic processing between amino acid residues 1415
Proteomic analysis of
S coelicolor 923
copy 2002 Blackwell Publishing Ltd
Molecular Microbiology
46
917ndash932
Co-translational modification by removal of the N-terminal initiator Met occurred in 149 out of 190 (78)cases in which the N-terminal peptide was detected Pro-cessing by the N-methionylaminopeptidase was observedif the second amino acid was Ser (45 out of 149 exam-ples) Thr (40) Ala (32) Pro (27) Gly (4) or Val (1) Ononly one occasion was any of these six amino acids foundas the second residue if the N-terminal methionine wasretained This is in broad agreement with observations in
B subtilis
in which two-thirds of proteins were reported tobe similarly processed (Buttner
et al
2001)
Representation of primary and secondary metabolic pathways
Streptomycetes produce
ordf
60 of all commercially usefulantibiotics (Berdy 1984 Miyodah 1993) Most antibioticsare the product of complex biosynthetic pathways oftenencoded by 20ndash30 clustered genes Production of thepigmented antibiotics actinorhodin (Act) and undecyl-
prodigiosin (Red) has been studied extensively in
Scoelicolor
(for reviews see Chater and Bibb 1997Champness 2000) and involves clusters of approximately22 genes each A third cluster producing the calcium-dependent antibiotic (CDA) and consisting of 40 geneshas also been studied (Chong
et al
1998 Huang
et al
2001) In the extensive spot identification studies for thepreparation of a proteome map of
S coelicolor
(seeabove) more than one-third of the proteins from each ofthese pathways was identified (Table 5) Proteins fromthree clusters that specify metabolic products that haveyet to be identified experimentally (Bentley
et al
2002)were also extensively represented a type I polyketidesynthase cluster (SCO6273ndash6288) a deoxysugarglycosyltransferase cluster (SCO0381ndash0401) and anon-ribosomal peptide synthetase cluster predicted toencode a novel siderophore coelichelin (SCO0489ndash0499) Several clusters were unrepresented
The primary metabolic pathways were somewhat morehighly represented with
ordf
60 or more of proteins anno-
Fig 2
Examples of MALDI-TOF peptide mass fingerprint data identifying post-translational modification of proteins by (A) acetylation (predicted mass increase of 420 Da) (B) adenylylation (predicted mass increase of 3290 Da) and (C) proteolytic processing (predicted mass loss for Ile of 11308 Da) In each fingerprint the
x
-axis is masscharge ratio (mz) and the
y
-axis is absolute intensity The observed mass accuracies in the MALDI-TOF fingerprints are (A) 60 ppm (B) 20 ppm and (C) 20 ppm
924
A R Hesketh
et al
copy 2002 Blackwell Publishing Ltd
Molecular Microbiology
46
917ndash932
tated as enzymes of glycolysis the TCA cycle thepentose phosphate pathway purine ribonucleotide bio-synthesis and pyrimidine nucleotide biosynthesis beingmapped (Table 6) About 50 of amino acid biosyntheticproteins were identified Lists of individual members of allpathways considered here including those that were andwere not identified during proteome mapping can befound in the
Supplementary material
Several proteins from both primary and secondary met-
abolic pathways were identified at more than one positionon the proteome map and may therefore be subject toregulation by post-translational modification (see Table 3and Fig 1) Primary metabolic proteins of this type (yellowspots on Fig 1) were usually detected as chains of two ormore spots with the same apparent molecular weights butdifferent pI values suggesting covalent modification bylow-molecular-weight adducts Apart from adenylylation ofGSI (see above) the nature of the modification of theseproteins was not revealed by MALDI-TOF peptide massfingerprint data Both covalent modification and proteolyticprocessing were also found in secondary metabolic pro-teins (red spots in Fig 1) For example the Act pathwaymembers SCO5071 (spot 16) and SCO5075 (spot 20)
and the putative 3-oxoacyl[acyl carrier protein] reductasefrom the type I polyketide cluster (SCO6282 spot 11)appear to be covalently modified whereas proteolytic pro-cessing was detected for RedL (SCO5892 spot 18) fromthe undecylprodigiosin cluster the putative dehydrataseActVI-ORF3 (SCO5074 spot 31) from the Act cluster theputative peptide monooxygenase from coelichelin biosyn-thesis (SCO0498 spot 34) and the putative oxygenasefrom the CDA pathway (SCO3236 spot 33) Two Act bio-synthetic proteins SCO5088 (spot 24) and SCO5073(spot 29) were detected both at their predicted pI andmolecular weight co-ordinates and at positions with simi-lar pI values but significantly higher molecular weightsThis may indicate that these proteins form stable mul-timers that are not completely denatured under theconditions used Only the modification of ActVI-ORF3(SCO5074 spot 31) could be characterized further fromthe mass spectrometry data which showed that it involvesproteolytic processing at two sites between amino acidresidues 3132 and 3435 (data not shown) Analysis of asample obtained by externally extracting proteins fromintact washed mycelium with a solution containing 2SDS and 50 mM dithiothreitol (DTT) revealed that sur-
Table 5
Summary of proteins identified on the proteome map from secondary metabolic pathways during growth in SMM
Biosynthetic pathwaygene cluster
Pathwaygenesannotated
Pathwaygene productsidentified
Percentagepathwaydetected
Calcium-dependent antibiotic CDA SCO3210ndash3249 40 16 40Actinorhodin SCO5071ndash5092 22 15 68Undecylprodigiosin SCO5877ndash5898 22 8 36Deoxysugar synthaseglycosyltransferase cluster SCO0381ndash0401 21 7 33Type I polyketide synthase SCO6273ndash6288 16 6 38Hopanoids SCO6759ndash6771 13 1 7Siderophore (coelichelin) SCO0489ndash0499 11 5 45Siderophore (desferrioxamine) SCO2783ndash2785 4 4 100
Chromosomal locations and extent of gene clusters are taken from Bentley
et al
(2002) No proteins were detected from the remaining predictedsecondary metabolic clusters ie siderophore (coelibactin) SCO7681ndash7691 (11 ORFs) siderophore SCO5799ndash5801 (three ORFs) non-ribosomal peptide synthases SCO6429ndash6438 (10 ORFs) type II fatty acid synthases SCO1265ndash1273 (nine ORFs) WhiE spore pigment clusterSCO5314ndash5320 (seven ORFs) isorenieratine SCO0185ndash0191 (seven ORFs) eicosapentaenoic acid SCO0124ndash0129 (six ORFs) chalconesynthases SCO1206ndash1208 (three ORFs) chalcone synthases SCO7669ndash7671 and SCO7222 (three ORFs) sesquiterpene cyclase SCO5222ndash5223 (two ORFs) type I polyketide synthases SCO6826ndash6827 (two ORFs) geosmin SCO6703 (one ORF)
Table 6 Summary of proteins identified on the proteome map from primary metabolic pathways during growth in SMM
Biosynthetic pathwayPathway genesannotateda
Pathway geneproducts identified
Percentage pathway detected
Glycolysisb 24 13 55Pentose phosphate pathwayb 14 10 71TCA cycleb 21 12 57Amino acid biosynthesisc 115 55 48Purine ribonucleotide biosynthesisb 18 12 67Pyrimidine ribonucleotide biosynthesisb 8 5 62
a Derived from interpretation of the annotated S coelicolor genome sequence at httpwwwsangeracukProjectsS_coelicolorb See Supplementary material for details of pathway membersc See Supplementary material for details of individual pathways
Proteomic analysis of S coelicolor 925
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
prisingly the truncated ActVI-ORF3 is located extracellu-larly presumably non-covalently associated with the cellwall matrix (Fig 4)
Expression analysis of certain primary and secondary metabolic proteins
Production of the pigmented antibiotics Act and Red inliquid minimal medium supplemented with casamino acids(SMM) begins in the transition phase between exponentialgrowth and stationary phase Figure 3 shows a compari-son of the expression of certain primary and secondarymetabolic proteins between exponentially growing andstationary phase cultures Two proteins from the Actcluster (ActVA-ORF5 and ActI-ORF2) and two from theRed (RedI and RedR) were present in the stationaryphase cultures but were not detected during exponentialgrowth Similarly the putative 3-oxoacyl-[acyl carrierprotein] reductase (SCO6282) and epoxide hydrolase(SCO6277) enzymes from a type I polyketide secondarymetabolic cluster showed a large increase in abundance
between the exponential and stationary phase samplesTriose phosphate isomerase (SCO1945) fructose bis-phosphate aldolase (SCO3649) and succinyl-CoA syn-thetase alpha chain (SCO4809) all enzymes from centralcarbon metabolism were present in approximatelyconstant amounts in both samples Interestingly two iso-forms (SCO7511 and SCO1947) of glyceraldehyde-3-phosphate dehydrogenase from the glycolytic pathwayshowed different patterns of expression SCO1947 levelswere the same in both exponential and stationary phasebut SCO7511 barely detectable in exponentially growingcultures increased markedly in stationary phase
The Streptomyces coelicolor online proteome map
The 2D gel images in Fig 1 together with the spot iden-tification results are being made available over the WorldWide Web to provide a reference map of the S coelicolorproteome At the time of writing the reference gel forthe pH 4ndash7 isoelectric point range was fully accessiblewhereas those for pH 45ndash55 pH 55ndash67 and pH 6ndash11
Fig 3 A comparison of the expression of certain primary and secondary metabolic proteins between exponentially growing and stationary phase cultures of S coelicolor M145 Larger arrows represent higher protein level in one gel than in the other The experiment was performed on duplicate biological samples and both showed similar protein expression profiles
926 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
were under construction (httpqbababeracukS_coeliReferenceGelrefgelhtml) A tool for comparing two ormore gel images with each other is also accessible fromthe same website at httpqbababeracukS_coeliflickerhtml
Discussion
Silver staining is one of the most sensitive methods fordetecting proteins on polyacrylamide gels allowing aslittle as 05ndash2 ng of an individual protein to be detected(Berggren et al 2000 Gorg et al 2000) The proportionof the S coelicolor proteome that can be analysed usingsilver staining of 2D-PAGE-separated proteins dependsat least in part on the pH range of the IPG strip used forthe first-dimension separation (see Table 1) Using theintermediate-range IPG strip covering pH 4ndash7 about 20ndash25 of the proteins in the theoretical proteome predictedto be observable in the resulting 2D-PAGE gel were actu-ally detected This figure was reduced to 10ndash12 whenanalysing the more basic pI range using the pH 6ndash11 IPGstrip Attempts to increase the percentage of the proteomedetectable using the pH 4ndash7 and pH 6ndash11 IPG strips byloading more protein sample resulted in poorer separa-tions (data not shown) This was especially true of thepH 6ndash11 strips which were generally the most difficult touse successfully However good separation of higher pro-tein loads was achieved using the narrow-range IPG stripspH 45ndash55 and 55ndash67 significantly increasing the pro-portion of theoretical proteins detected to over 40Clearly and more pertinently the limited conditions usedfor culturing S coelicolor in this study will also haverestricted the number of proteins detected as not all theproteome will be expressed In particular S coelicolordoes not sporulate in shaken liquid-grown cultures soproteins involved in this differentiation process are unlikelyto be present Nevertheless it is striking that the com-bined data for the pH 45ndash55 and 55ndash67 isoelectric pointranges indicate that 2468 out of a total of predicted 4932protein spots can be detected (see Table 1) suggestingthat transition phase mycelium in this supplementedminimal medium is expressing 50 of its genes (ie byextrapolation some 3912 of the 7825 in the genome)
Analysis of post-translational regulation
Although the regulation of cellular processes and adaptiveresponses in bacteria has been studied extensively at thetranscriptional level post-translational regulatory mecha-nisms are much less understood Proteomic analysisusing 2D-PAGE coupled with mass spectrometry offersthe opportunity to observe and identify modification ofproteins at a general cellular level The functions of pro-teins can be altered by specific covalent attachment of
adducts (eg phosphorylation glycosylation nucleotidy-lation acetylation) or by proteolytic processing Bothevents alter the pI andor mass of the protein and so canusually be observed on 2D-PAGE In the S coelicolorproteome map ordf110 out of a total of 770 gene productswere identified in more than one position or at pI andapparent mass co-ordinates significantly different frompredicted values indicating extensive regulation at thepost-translational level (88 are listed in Table 3) a propor-tion similar to that reported for other prokaryotes (Buttneret al 2001 Tonella et al 2001) The initial peptide massfingerprint analysis identified the modification of only 10of these proteins probably partly because fingerprint datatypically cover ordf30ndash60 of a proteinrsquos sequence (data notshown) so modification at positions not represented willbe missed Protein digestion with endopeptidases otherthan trypsin before mass spectrometry may help in thissituation but some covalently attached groups are labileand cannot be detected routinely Phosphorylation of Hisoccurs on nitrogen producing a high-energy phosphora-midate bond that is easily hydrolysed in acidic conditionswhereas phospho-Asp is the most labile phosphorylatedresidue with a half-life of a few hours under physiologicalconditions (reviewed by Robinson et al 2000) Thesemodifications the basis of bacterial two-component regu-latory systems are therefore unlikely to survive stainingof the 2D gels which involved the use of a fixing solutioncontaining 10 acetic acid and mass spectrometry Iden-tification of the more stable phosphoester phosphorylationthat occurs on Ser Thr or Tyr residues using PAGEfollowed by mass spectrometry has been reported(Godovac-Zimmermann et al 1999 Buttner et al 2001)but no examples were detected in this work Of the mod-ifications identified in this study three were by acetylationat the N-terminus after removal of the initiator Met residuetwo involved adenylylation and five proteins had been N-terminally truncated Modification of the 20S proteasomeby N-acetylation in yeast involves removal of the Metresidue by N-methionylaminopeptidase followed byacetylation with an Na-acetyltransferase (Kimura et al2000) Homologues of these enzymes are predicted fromthe S coelicolor genome and are presumably responsiblefor the observed modification of the 20S proteasomealpha subunit and of BldK-ORFD and the putative cellu-lose-binding protein The functional significance of Na-acetylation of these proteins is unclear
Analysis of the biosynthesis of antibiotics and other secondary metabolites
Post-translational modification The proteome mappingresults indicate that at least six enzymes in the Act clusterare subject to post-translational modifications and one
Proteomic analysis of S coelicolor 927
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
each from the undecylprodigiosin (SCO5877ndash5898)lsquocoelichelinrsquo (SCO0489ndash0499) and type I polyketidesynthase (SCO6273ndash6288) clusters (see Table 3) Thesemetabolic pathways may therefore be significantly regu-lated at the post-translational level The actVI-ORF3 gene(SCO5074) encodes a putative dehydratase enzyme thatis part of the biosynthetic pathway for Act a blue memberof the benzoisochromanequinone (BIQ) family of pig-mented antibiotics Mutation in actVI-ORF3 causes a 50decrease in the accumulation of Act (and a reddish brownphenotype) and it is believed to be responsible for pyranring closure an intermediate step in Act biosynthesis (thatmay also occur spontaneously) leading to the formationof the BIQ chromophore (Fernandez-Moreno et al 1994Ichinose et al 1998a Taguchi et al 2000) In this studyActVI-ORF3 protein was detected in two forms a majorspot corresponding to ActVI-ORF3 truncated by removalof the N-terminal 31 amino acid residues and a minor onein which the first 34 amino acids were missing (spots 31in Fig 1) The full-length protein was not detected Anal-ysis of the protein sequence using the SIGNALP server athttpwwwcbsdtudkservicesSignalP predicted a signalpeptide cleavage site between amino acids 31 and 32 inagreement with the observed results
The protein extract analysed was prepared from myce-lium that had been collected by centrifugation and sub-jected to two washes indicating that if ActVI-ORF3 isexported it is effectively retained within the cell wallmatrix This was confirmed by analysis of a preparationobtained by externally extracting proteins from intactwashed mycelium (Fig 4) Export of an enzyme requiredfor the efficient biosynthesis of an antibiotic that is thoughtto be produced intracellularly appears to be anomalousparticularly as it is involved in an intermediate biosynthetic
event Database analysis of the other members of the Actcluster revealed only one other protein with a predictedsignal peptide sequence the putative integral membranetransport-related protein ActII-ORF3 and it seemsunlikely that all the Act biosynthetic steps following BIQchromophore formation are extracellular It is possible thatfull-length ActVI-ORF3 catalyses the intermediate biosyn-thetic event intracellularly and the exported processedprotein is responsible for a different extracellular activityperhaps later in the pathway However no full-length pro-tein was detected so the exported enzyme may performthe pyran ring closure on a secreted metabolite that isthen reabsorbed by the cell for completion of the sub-sequent biosynthetic steps Only two homologues ofactVI-ORF3 are known and both are also in BIQ antibioticbiosynthetic clusters gra-ORF18 from the granaticincluster (Ichinose et al 1998b) and med-ORF5 frommedermycin biosynthesis (K Ichinose personal com-munication) Interestingly the encoded proteins of bothsequences are also predicted to contain N-terminal signalpeptides The observation that ActVI-ORF3 is exported tothe cell wall matrix may therefore be of general signifi-cance to the BIQ family of antibiotics and provide a newinsight into their biosynthetic pathways
Pathway representation Of the 20 putative secondarymetabolite gene clusters identified in the S coelicolorgenome sequence (Bentley et al 2002) eight had atleast one protein each identified on the proteome map(see Table 5) The biosynthetic clusters for the antibioticsAct Red and CDA had 68 36 and 40 of their pro-teins represented There are several possible explana-tions for failure to detect all the proteins for each pathwaySome are putative integral membrane proteins so cannotreadily be detected using conventional 2D-PAGE SomeRed and CDA proteins have molecular weights gt200 kDaoutside the range of the 125 gels used (for clustermembers see Supplementary material) The pathway-specific regulatory proteins for Act and Red biosynthesiswere not detected despite a detailed search of their pre-dicted gel locations even though the relevant genes areknown to be transcribed in equivalent cultures of the samestrain (Hesketh et al 2001) The proteins are presumablypresent but at levels below the limit of detection Howeverthe transcriptional regulator for CDA production CdaRwas identified as well as the two-component systemresponse regulator AbsA2 which is also encoded by agene in the CDA cluster (Anderson et al 2001) Interest-ingly proteins from three putative secondary meta-bolite clusters producing chemicals of as yet unknownstructure were also identified on the map correspondingto the deoxysugar synthaseglycosyltransferase clusterSCO0381ndash0401 the lsquocoelichelinrsquo cluster for a predicted(but not yet confirmed) oligopeptide siderophore
Fig 4 Processed ActVI-ORF3 is present in the cell wall matrix 2D-gel separations of protein extracts prepared (A) from whole cells or (B) by externally washing cells in a 2 SDS extraction buffer The arrow indicates ActVI-ORF3 protein The experiment was performed on duplicate biological samples and both showed similar protein expression profiles
928 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
(SCO0489ndash0499) and the type I polyketide clusterSCO6273ndash6288 The 3-oxoacyl-[acyl carrier protein]reductase (SCO6282) from the latter pathway is ex-pressed in a growth phase-dependent manner (Fig 3)and in early stationary phase cultures is perhaps themost abundant protein spot in the entire detectable pro-teome Presumably the metabolites produced by thesethree pathways are being synthesized under the condi-tions used In addition to coelichelin three other sidero-phores are postulated for S coelicolor All the proteinsfrom one of these desferrioxamine (SCO2782ndash2785)were identified but none was seen from either of the othertwo pathways The WhiE polyketide spore pigment isassociated with sporulation of surface-grown cultures(Davis and Chater 1990) and the expression of the whiEgenes depends on a series of sporulation-specific regula-tory genes (Kelemen et al 1998) no corresponding pro-teins of which were detected here It is therefore notsurprising that WhiE proteins were not detected in sam-ples prepared from liquid-grown vegetative mycelium
Analysis of primary metabolism
Proteins annotated as functioning in primary metabolicpathways were highly represented on the proteome mapwith ordf60 or more of the enzymes assigned to glycolysisthe TCA cycle the pentose phosphate pathway andpurine and pyrimidine ribonucleotide biosynthesis beingidentified (see Table 6) This figure may be deceptively lowbecause these pathways in S coelicolor often appear tohave more than one enzyme isoform for some of themetabolic steps potentially offering additional levels ofcontrol or developmental responsiveness at these pointsFor example only two steps in glycolysis have just a singlegene product assigned to them the remainder typicallyhave two or three each (Fig 5) At least one isoform waspresent for each step so the pathway is in theory 100functionally represented Understanding the regulation ofthese pathways will require analysis of the expression ofthese isoenzymes under different nutritional conditionsor different developmental stages Figure 5 shows that ofthe three predicted glyceraldehyde-3-phosphate dehydro-genase isoforms from glycolysis only two were detectedin this study (see also Supplementary material) Of theseSCO1947 appears to be constitutive whereas SCO7511is growth phase dependent (see Fig 3) Thus even thelimited conditions in this study yielded useful informationabout isoenzyme expression patterns (see Supplemen-tary material) In Streptomyces aureofaciens two genesencoding glyceraldehyde-3-phosphate dehydrogenaseactivity also appear to be differently regulated transcrip-tion of one highly homologous to SCO7511 is activatedby the regulator GapR whereas the other is GapR inde-pendent (Spusansky et al 2001)
Hood et al (1992) reported that most amino acid bio-synthetic enzymes are expressed at very low constitutivelevels in S coelicolor so it is reassuring to note that ordf50could be detected and identified using proteomics Theseinclude TrpB and TrpC1 from the tryptophan biosyntheticpathway the corresponding transcripts of which couldbarely be detected in previously reported S1 nucleaseprotection experiments (Hu et al 1999)
Perspectives
No attempt was made in the work reported here to simplifythe protein separations by subdividing the sample intosoluble and membrane fractions and this is reflected inthe 10 lipoproteins and 16 ABC transport system ATP-binding subunits identified on the proteome map (seeTable 2) No integral membrane proteins were detectedand even when a membrane protein sample was pre-pared and analysed no proteins identified possessedmore than three predicted transmembrane helices (datanot shown) This is consistent with previous studiesin other bacteria (Molloy et al 1999 Nilsson andDavidsson 2000) Another group of proteins also strik-ingly absent from the list of those identified is the sigmafactors which determine promoter selection by RNA poly-merase There are an extraordinarily high number (65) ofsigma factors encoded in the S coelicolor genome aswell as at least seven antisigma factors and 15 antisigmafactor antagonists (some of each of which were detected)Sigma factor heterogeneity in bacteria is an important wayof regulating expression of specific sets of genes under
Fig 5 The glycolytic pathway in S coelicolor illustrating the assignment of multiple gene products to individual metabolic steps based on the annotation of the genome sequence at httpwwwsangeracukProjectsS_coelicolor Gene products identified in the proteome map are underlined
Proteomic analysis of S coelicolor 929
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
different conditions and in different cell types (for a reviewsee Gross et al 1998) Although no sigma factors wereidentified about half (31 out of 65) have predicted pIvalues gtpH 7 and so can only be detected using thepH 6ndash11 IPG strips that give the poorest results (seeTable 1) However the principal vegetative sigma factorHrdB which is expected to be moderately abundanthas a predicted pI of 613 and its absence from thepH 55ndash67 proteome map may indicate that the samplepreparation method which does not remove DNA is inap-propriate Among other regulatory proteins 11 out of apossible 80 two-component system response regulatorsplus a further 36 proteins annotated as lsquoregulatorsrsquo weredetected Only one SerThr kinase and no two-componentsystem sensor kinases were detected possibly reflectingthe difficulty in analysing proteins with several transmem-brane domains Interestingly 30 secreted proteins wereidentified on the proteome map at least 10 of whichappear from comparison of their observed pI and molec-ular weight positions with predicted values to have beenprocessed already and therefore exported Presumablythese proteins do not end up exclusively in the culturemedium
Clearly this approach to proteomic analysis of S coeli-color has some important deficiencies not least theabsence of certain groups of proteins and the relativelypoor initial success rate for specifically characterizingpost-translational modifications Nevertheless it offersunique insights into the molecular biology of the organismthe expression of many proteins from both primary andsecondary metabolic pathways can be analysed post-translational regulation of proteins can be investigated ona general cellular level and the physical locations forsome proteins can be determined Approximately 35 ofthe S coelicolor genome is annotated as encoding lsquohypo-theticalrsquo proteins of unknown function and 182 proteins ofthis type were identified on the proteome map No exam-ples were found of overt disagreements between pre-dicted and observed proteins providing experimentalsupport for the general accuracy of the S coelicolorgenome sequence and its annotation Information aboutthe expression post-translational modification and physi-cal location of these (no longer) hypothetical proteinsfrom proteomic analyses is going to be important forassigning future functions As many of the hypotheticalproteins have apparent orthologues in pathogenic myco-bacteria some of the information may have an immediateapplication in the context of pathogenesis
Experimental procedures
Materials
Immobilized pH gradient (IPG) strips and IPG bufferswere purchased from Amersham Biosciences Acrylamide
duracryl and chemicals for silver staining were obtained fromGenomic Solutions All other chemicals were from Sigmaunless stated in the text
Strain and growth conditions
Streptomyces coelicolor M145 was cultivated with vigorousagitation at 30infinC in minimal medium supplemented with 02casamino acids (SMM) as described previously (Kieser et al2000) Briefly a high-density spore preparation [about 1010
colony-forming units (cfu) ml-1] was pregerminated in 2yen YTmedia for 7 h at 30infinC Germlings were harvested by centrifu-gation (5 min at 4000 g) resuspended in SMM and sonicatedbriefly to disperse any aggregates before inoculation into50 ml of SMM in 250 ml siliconized flasks containing coiledstainless steel springs Each flask received the equivalent of5 yen 107 spore cfu
Protein extraction and 2D-gel electrophoresis
The preparation of the protein sample to be separated waskept as simple as possible disrupting harvested myceliadirectly into denaturing isoelectric focusing (IEF) buffer con-taining Pefabloc SC (Roche) protease inhibitor This methodminimized unwanted protein degradation that was observedin initial studies using a Tris buffer containing Pefabloc SC forcell lysis (data not shown) Removal of DNA RNA salts andpolysaccharides from the sample was therefore not per-formed (other than the shearing of viscous DNA and RNA bysonication) and the protein extracts were sometimes difficultto focus when the sample was loaded by in-gel rehydrationLoading the sample soaked in paper strips at the anodicend of IPG strips that had already been rehydrated improvedthe reproducibility of separations significantly
Mycelium for protein extraction was harvested from cul-tures by brief centrifugation (30 s at 4000 g) at room tem-perature and immediately frozen in liquid nitrogen Typicallymycelium from 10 ml culture aliquots was collected and thetransfer time from culture flask to frozen sample was 15 minFrozen cells were thawed on ice in 5 ml of washing buffer(40 mM Tris pH 90 1 mM EGTA 1 mM EDTA) then pelletedby centrifugation (5 min at 3000 g) at 4infinC Washed cells wereresuspended in 400 ml of denaturing IEF buffer UTCHAPS[7 M urea 2 M thiourea 4 (wv) CHAPS 40 mM TrispH 90 1 mM EDTA 50 mM DTT 4 mM Pefabloc SC pro-tease inhibitor (Roche)] and disrupted by sonication (SanyoSoniprep 150 10 yen 2 s bursts at amplitude 75 mm) whilecooling in an ethanolndashice bath Cell debris was removed bycentrifugation (15 min 10 000 g 4infinC) and the protein extractwas stored frozen in aliquots at -80infinC until use For thepreparation of extracellular protein extracts washed cellswere resuspended in extraction buffer consisting of 40 mMTris pH 90 1 mM EDTA 50 mM DTT 2 SDS and 4 mMPefabloc SC protease inhibitor Cells were vortexed for 2 minbefore being pelleted by centrifugation (5 min 10 000 g4infinC) The supernatant was precipitated using the Amersham2D Clean-Up kit according to the manufacturerrsquos instructionsand the precipitated proteins were redissolved in UTCHAPSand stored at -80infinC until use
930 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
For first-dimension isoelectric focusing 18 cm IPG stripspH 4ndash7 pH 6ndash11 pH 45ndash55 or pH 55ndash67 (Amersham Bio-sciences) were rehydrated overnight in IEF buffer containing1 ampholytes according to the manufacturerrsquos instructionsusing a Phaser isoelectric focusing unit (Genomic Solutions)set at 20 V Protein samples to be separated were applied tothe rehydrated strips at the anodic end soaked in a 5ndash10 mmsection of IEF electrode strip (Amersham Biosciences)Separation was performed for 120 000 V-h with a maximumvoltage of 5000 V After isoelectric focusing IPG strips wereequilibrated for the second dimension for 15 min in IPG equil-ibration buffer (50 mM Tris pH 68 6 M urea 30 glycerol1 SDS and 001 bromophenol blue) plus 80 mM DTTthen for 10 min in IPG equilibration buffer plus 135 mMiodoacetamide Approximately 1 cm was removed from theanodic end of each equilibrated strip before applying the stripto the top of a vertical 125 SDS-PAGE gel for second-dimension separation using the Investigator 5000 systemfrom Genomic Solutions
Large-format gels (24 cm wide yen 22 cm high yen 1 mm thick)for the second-dimension separation were cast in-houseusing the system supplied by Genomic Solutions 125SDS-PAGE gels prepared using 065 NN-methylenebi-sacrylamide as cross-linking agent were used for pH 4ndash7and pH 45ndash55 IPG strips whereas pH 55ndash67 and pH 6ndash11 strips were applied to similar gels made with 08 cross-linker Electrophoresis was performed while cooling using themaximum setting (ordf 4ndash10infinC) at 20 000 mW per gel constantpower and a maximum voltage of 500 V Gels were thenstained with either colloidal Coomassie G-250 (Neuhoff et al1988) or silver nitrate (Rabilloud 1992) and scanned in aProXPRESS proteomic imaging system (Perkin-Elmer)Image analysis was performed using PHORETIX 2D version51 (NonLinear Dynamics) spot detection was optimizedautomatically using the lsquoSpot Detection Wizardrsquo and thenedited manually background subtraction was performedautomatically using the lsquoMode of Non-Spotrsquo setting imageswere then normalized to the total spot volume for quantifica-tion Spot filtering was not used
Protein identification using mass spectrometry
Protein spots of interest were excised from colloidalCoomassie-stained gels in-gel digested with trypsin andidentified by MALDI-TOF peptide mass fingerprint analysisExcised gel pieces (ordf 1-mm-diameter circles cut from 1-mm-thick gels) were washed twice with 100 ml of 50 mMammonium bicarbonate for 15 min once with 100 ml of 20acetonitrile-40 mM ammonium bicarbonate for 15 min andonce with 100 acetonitrile Washed gel pieces were allowedto air-dry for 10 min before being rehydrated in 5 ml of10 mg ml-1 trypsin (sequencing grade modified Promega) in10 mM ammonium bicarbonate Digestion (37infinC for 4 h) wasstopped by the addition of 5 ml of 5 formic acid Extractionof peptides into this solution was encouraged by a sonicwater bath treatment for 20 min Peptide extract (05 ml) wasspotted on to a thin layer of a-cyano-4-hydroxycinnamic acidapplied to a MALDI-TOF sample template and analysed byMALDI-TOF mass spectrometry (Bruker Reflex III) using anaccelerating voltage of 25 kV Samples were externally cali-brated using a standard mixture of six peptides ranging in
mass from 10465423 Da to 34946500 Da and the massaccuracy obtained was 60 ppm or better Identification ofproteins from MALDI-TOF peptide mass fingerprint data wasperformed using the lsquoMascotrsquo search engine at httpwwwmatrixsciencecom and was based on a positive resultusing their lsquoProbability Based MOWSE Scorersquo algorithm AMOWSE score of 60 or higher is significant at the 5 levelor better and proteins in this work typically gave scores gt80(frequently considerably so) In addition no identification wasaccepted unless at least five peptides representing at least20 of the protein sequence were detected in the MALDI-TOF peptide mass fingerprint
Assignment of proteins to metabolic pathways
Proteins were assigned to primary metabolic pathwaysusing the lsquoProtein Classification Schemersquo web page ofthe annotated Streptomyces coelicolor genome sequenceat httpwwwsangeracukProjectsS_coelicolor as it ap-peared in May 2002 The annotation of the genome andtherefore the Protein Classification Scheme is likely to bemodified as more experimental data are obtained about thefunction of genes that have currently been assigned on thebasis of sequence information alone It is therefore possiblethat proteins assigned to metabolic pathways in this studymay in future be shown to perform different functions in thecell
Assignments to gene clusters encoding secondary metab-olites are based on an analysis of the genome sequencereported by Bentley et al (2002) In some cases theseassignments are based on extensive experimental data (actred reviewed by Chater and Bibb 1997 Champness 2000cda Chong et al 1998 Huang et al 2001 whiE Davis andChater 1990)
Acknowledgements
This work was funded by grants (2FGT11406 208FGT11408 and 208IGF12432) under the BBSRCrsquos Tech-nologies for Functional Genomics and Investigating GeneFunction initiatives and by a competitive strategic grant fromthe BBSRC to the John Innes Centre We thank Mike Naldrettfor his extensive contribution in establishing technology forproteome analysis at the JIC
Supplementary material
The following material is available from httpwwwblackwell-sciencecomproductsjournalssuppmatmolemole3219mmi3219smhtmSection 1 Detection of gene products assigned to individualmetabolic steps in primary metabolic pathways in ScoelicolorSection 2 Identification of proteins from individual amino acidbiosynthetic pathwaysSection 3 Detection of gene products assigned to individualmetabolic steps in secondary metabolic pathways in Scoelicolor
Proteomic analysis of S coelicolor 931
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
References
Anderson TB Brian P and Champness WC (2001)Genetic and transcriptional analysis of absA an antibioticgene cluster linked two-component system that regulatesmultiple antibiotics in Streptomyces coelicolor Mol Micro-biol 39 553ndash566
Bentley SD Chater KF Cerdeno-Tarraga A-M ChallisGL Thomson NR James KD et al (2002) Completegenome sequence of the model actinomycete Streptomy-ces coelicolor A3(2) Nature 417 141ndash147
Berdy J (1984) New ways to obtain antibiotics Chin J Anti-biot 7 272ndash290
Berggren K Chernokalskaya E Steinberg TH KemperC Lopez MF Diwu Z et al (2000) Background-freehigh sensitivity staining of proteins in one- and two-dimensional sodium dodecyl sulfate-polyacrylamide gelsusing a luminescent ruthenium complex Electrophoresis21 2509ndash2521
Blackstock WP and Weir MP (1999) Proteomics quan-titative and physical mapping of cellular proteins TrendsBiotechnol 17 121ndash127
Blattner FR Plunkett G Bloch CA Perna NT BurlandV Riley M et al (1997) The complete genome sequenceof Escherichia coli K-12 Science 277 1453ndash1462
Buttner K Bernhardt J Scharf C Schmid R MaderU Eymann C et al (2001) A comprehensive two-dimensional map of cytosolic proteins of Bacillus subtilisElectrophoresis 22 2908ndash2935
Champness W (2000) Actinomycete development antibioticproduction and phylogeny questions and challenges InProkaryotic Development Brun YV and Skimketts LJ(eds) Washington DC American Society for MicrobiologyPress pp 11ndash31
Chater KF and Bibb MJ (1997) Regulation of bacterialantibiotic production In Biotechnology Vol 7 Products ofSecondary Metabolism Kleinkauf H and von Doumlhren H(eds) Weinheim Germany VCH pp 57ndash105
Chong PP Podmore SM Kieser HM Redenbach MTurgay K Marahiel M et al (1998) Physical identificationof a chromosomal locus encoding biosynthetic genesfor the lipopeptide calcium-dependent antibiotic (CDA) ofStreptomyces coelicolor A3(2) Microbiology 144 193ndash199
de Crecy-Lagard V Servant-Moisson JV Grandvalet Cand Mazodier P (1999) Alteration of the synthesis of theClp ATP-dependent protease affects morphological andphysiological differentiation in Streptomyces Mol Microbiol32 505ndash517
Davis NK and Chater KF (1990) Spore colour inStreptomyces coelicolor A3(2) involves the developmen-tally regulated synthesis of a compound biosyntheticallyrelated to polyketide antibiotics Mol Microbiol 4 1679ndash1691
Fernandez-Moreno FA Martinez E Caballero JLIchinose K Hopwood DA and Malpartida F (1994)DNA sequence and functions of the actVI region of theactinorhodin biosynthetic gene cluster of Streptomycescoelicolor A3(2) J Biol Chem 269 24854ndash24863
Fink D Falke D Wohllheben W and Engels A (1999)Nitrogen metabolism in Streptomyces coelicolor A3(2)
modification of glutamine synthase I by an adenylyltrans-ferase Microbiology 145 2313ndash2322
Godovac-Zimmermann J Soskic V Posnanovic S andBrianza F (1999) Functional proteomics of signal trans-duction by membrane receptors Electrophoresis 20 952ndash961
Gorg A Obermaier C Boguth G Harder A Scheibe BWildgruber R and Weiss W (2000) The current state oftwo-dimensional electrophoresis with immobilized pH gra-dients Electrophoresis 21 1037ndash1053
Gramajo HC Takano E and Bibb MJ (1993) Stationary-phase production of the antibiotic actinorhodin in Strepto-myces coelicolor A3(2) is transcriptionally regulated MolMicrobiol 7 837ndash845
Gross CA Chan C Dombroski A Gruber T Sharp MTupy J and Young B (1998) The functional and regula-tory roles of sigma factors in transcription Cold SpringHarb Symp Quant Biol 63 141ndash155
Hesketh A Sun J and Bibb MJ (2001) Induction ofppGpp synthesis in Streptomyces coelicolor A3(2) grownunder conditions of nutritional sufficiency elicits actII-ORF4transcription and actinorhodin biosynthesis Mol Microbiol39 136ndash144
Hesketh A Fink D Gust B Rexer H-U Scheel BChater K et al (2002) The PII nitrogen regulatory proteinGlnK in Streptomyces coelicolor A3(2) is modified by ade-nylylation rather than uridylylation and the GlnD and GlnKhomologues are functionally dissimilar to their nitrogenregulatory system counterparts Mol Microbiol (inpress)
Hood DW Heidstra R Swodoba UK and Hodgson DA(1992) Molecular genetic analysis of proline and tryp-tophan biosynthesis in Streptomyces coelicolor A3(2)interaction between primary and secondary metabolism- areview Gene 115 5ndash12
Hu DS-J Hood DW Heidstra R and Hodgson DA(1999) The expression of the trpD trpC and trpBA genesof Streptomyces coelicolor A3(2) is regulated by growthrate and growth phase but not by feedback repression MolMicrobiol 32 869ndash880
Huang J Lih CJ Pan KH and Cohen SN (2001)Global analysis of growth phase responsive gene expres-sion and regulation of antibiotic biosynthetic pathways inStreptomyces coelicolor using DNA microarrays GenesDev 15 3183ndash3192
Ichinose K Taguchi T Ebizuka Y and Hopwood DA(1998a) Biosynthetic gene clusters of benzoisochromane-quinone antibiotics in Streptomyces spp Identification ofgenes involved in post-PKS tailoring steps Actinomyceto-logica 12 99ndash109
Ichinose K Bedford DJ Tornus D Bechthold A BibbMJ Revill PW et al (1998b) The granaticin biosyntheticgene cluster of Streptomyces violaceoruber Tu22sequence analysis and expression in a heterologous hostChem Biol 5 647ndash659
Kelemen GH Brian P Flardh K Chamberlin L ChaterK and Buttner M (1998) Developmental regulation oftranscription of whiE a locus specifying the polyketidespore pigment in Streptomyces coelicolor A3(2) J Bacte-riol 180 2515ndash2521
Kieser T Bibb MJ Buttner MJ Chater KF and
932 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
Hopwood DA (2000) Practical Streptomyces GeneticsNorwich John Innes Foundation
Kim EJ Chung HJ Suh B Hah YC and Roe JH(1998) Expression and regulation of the sodF gene encod-ing iron- and zinc-containing superoxide dismutase inStreptomyces coelicolor Muumlller J Bacteriol 180 2014ndash2020
Kimura Y Takaoka M Tanaka S Sassa H Tanaka KPolevoda B et al (2000) Na-acetylation and proteolyticactivity of the yeast 20S proteasome J Biol Chem 2754635ndash4639
Kunst F Ogasawara N Moszer I Albertini AM AlloniG Azevedo V et al (1997) The complete genomesequence of the Gram-positive bacterium Bacillus subtilisNature 390 249ndash256
Lucchini S Thompson A and Hinton JCD (2001)Microarrays for microbiologists Microbiology 147 1403ndash1414
Mann M Hendrickson RC and Pandey A (2001) Analy-sis of proteins and proteomes by mass spectrometry AnnuRev Biochem 70 437ndash473
Miyodah S (1993) Research on antibiotic screening inJapan over the last decade a producing microorganismsapproach Actinomycetologica 7 100ndash106
Molloy MP Herbert BR Williams KL and Gooley AA(1999) Extraction of Escherichia coli proteins with organicsolvents prior to two-dimensional electrophoresis Electro-phoresis 20 701ndash704
Nagy I Tamura T Vanderleyden J Baumeister W andDe Mot R (1998) The 20S proteasome of Streptomycescoelicolor J Bacteriol 180 5448ndash5453
Neuhoff V Arold N Taube D and Ehrhardt W (1988)Improved staining of proteins in polyacrylamide gels includ-ing isoelectric focusing gels with clear background at nan-ogram sensitivity using Coomassie Brilliant Blue G-250and R-250 Electrophoresis 9 255ndash262
Nilsson CL and Davidsson P (2000) New separation toolsfor comprehensive studies of protein expression by massspectrometry Mass Spectrom Rev 19 390ndash397
Puglia AM Vohradsky J and Thompson CJ (1995)
Developmental control of the heat-shock stress regulon inStreptomyces coelicolor Mol Microbiol 17 737ndash746
Rabilloud T (1992) A comparison between low backgroundsilver diamine and silver nitrate protein stains Electro-phoresis 13 429ndash439
Robinson VL Buckler DR and Stock AM (2000) A taleof two components a novel kinase and a regulatory switchNature Struct Biol 7 626ndash633
Schena M (2000) Microarray Biochip Technology NatickMA Eaton Publishing
Spusansky O Rezuchova B Homerova D andKormanec J (2001) Expression of the gap gene encodingglyceraldehyde-3-phosphate dehydrogenase of Strepto-myces aureofaciens requires GapR a member of theAraCXylS family of transcriptional activators Microbiology147 1291ndash1301
Taguchi T Itou K Ebizuka Y Malpartida F HopwoodDA Surti CM et al (2000) Chemical characterisationof disruptants of the Streptomyces coelicolor A3(2) actVIgenes involved in actinorhodin biosynthesis J Antibiot 53144ndash152
Takano E Gramajo HC Strauch E Andres N WhiteJ and Bibb MJ (1992) Transcriptional regulation of theredD transcriptional activator gene accounts for growth-phase-dependent production of the antibotic undecylpro-digiosin in Streptomyces coelicolor A3(2) Mol Microbiol6 2797ndash2804
Tonella L Hoogland C Binz P-A Appel RDHochstrasser DF and Sanchez J-C (2001) New per-spectives in the Escherichia coli proteome investigationProteomics 1 409ndash423
Vohradsky J Li XM and Thompson CJ (1997) Identifi-cation of prokaryotic developmental stages by statisticalanalysis of two-dimensional gel patterns Electrophoresis18 1418ndash1428
Vohradsky J Li XM Dale G Folcher M Nguyen LViollier PH and Thompson CJ (2000) Developmentalcontrol of stress stimulons in Streptomyces coelicolorrevealed by statistical analyses of global gene expressionpatterns J Bacteriol 182 4979ndash4986

920
A R Hesketh
et al
copy 2002 Blackwell Publishing Ltd
Molecular Microbiology
46
917ndash932
back onto the analytical silver-stained gels in Fig 1Although silver staining is 5ndash10 times more sensitive thancolloidal Coomassie staining (and about 100 times moresensitive than conventional Coomassie staining) furthermass spectrometry analysis of protein spots is much moreefficient after staining with colloidal Coomassie than withsilver A total of 1305 protein spots were unambiguouslyassigned to sequenced genes using MALDI-TOF peptidemass fingerprint analysis at a success rate of
ordf
90(Table 1) There is some redundancy in these figures withseveral spots being represented in more than one pIrange On each gel some proteins were identified in morethan one position indicative of post-translational modifi-cations The average number of protein spots per geneproduct was 12 The products from 770 different geneswere assigned to the proteome map representing
ordf
10of the genome and corresponding to many differentclasses of proteins (Table 2) The master gel referencemaps are the basis for the production of a publicly acces-sible proteome database (httpqbababeracukS_coeliReferenceGelrefgelhtml see last Results section)
Analysis of co- and post-translational modifications
The results of MALDI-TOF identification of protein spotssummarized in Table 1 indicated a significant amount ofregulation at the post-transcriptional level in
S coelicolor
as in most other bacteria Table 3 lists those gene prod-ucts that were identified at pI andor molecular weight co-ordinates significantly different from predicted values Ingeneral these fell into two categories one or more spotswith lower than expected molecular weights and oftendifferent pI values indicating modification by proteolyticprocessing (Table 3A) and multiple spots with the sameapparent molecular weights but different pI values indi-
cating modification by covalent addition of a small adduct(Table 3B) In the former case the full-length protein wasoften but not always detected A similar fraction ofproteins of unknown function and annotated as lsquohypo-theticalrsquo or lsquoconserved hypotheticalrsquo was subject to post-translational modification but these will be described ina later publication and are not considered in Table 3Evidence in the peptide mass fingerprint data for the spe-cific nature of the modification was obtained in 10 out ofthe 88 proteins listed (Table 4) Figure 2 illustrates thecharacterization of an example of each type of modifica-tion by interpretation of the MALDI-TOF data
N-acetylation
The putative cellulose-binding proteinSCO5396 was identified in two positions on the proteomemap (spots labelled 10 in Fig 1) with the same apparentmolecular weight but significantly different pI values Themore basic spot gave a tryptic peptide at 137076 Dacorresponding to the N-terminal peptide 2ndash13 indicatingthat the initiator Met residue had been co-translationallyremoved (Fig 2A top) In the more acidic spot (ie on theleft in Fig 1) this peptide had increased in mass by420 Da diagnostic of the addition of an acetyl grouppresumably on the N-terminal Ser residue (Fig 2A bot-tom) A similar modification was identified for the 20Sproteasome alpha subunit (spots 12 in Fig 1) and for theputative ABC transport protein BldK-ORFD (spots 17 inFig 1) although the presumptive N-terminal acetylatedresidue is Thr in the latter (data not shown)
Adenylylation
By analogy with the nitrogen regulatorysystem in
E coli
glutamine synthetase I (GSI) wasalready believed to be modified by adenylylation on aconserved Tyr-397 residue and biochemical evidence forthis has been reported previously (Fink
et al
1999) TheMALDI-TOF data showed the tryptic peptide 395ndash420 atthe expected mass of 28074 Da for the larger more basicspot 6 in Fig 1 but this increased by 3290 Da corre-sponding to the addition of an adenylyl group in thesmaller spot on the left (Fig 2B) Adenylylation of thenitrogen regulatory protein GlnK (spot 15 in Fig 1) wasidentified similarly but this is described in detail elsewhere(Hesketh
et al
2002)
Proteolytic processing
The 50S ribosomal protein L9was identified as two spots that differed in both pI andmolecular weight (spots 30 in Fig 1) suggesting modifi-cation by proteolytic processing The spot with the highermolecular weight showed the tryptic peptide correspond-ing to amino acids 3ndash22 at the expected mass of199310 Da in agreement with this being the unproc-essed form of the protein (Fig 2C top) In the lower ofthe two spots the peptide decreased in mass by11312 Da (Fig 2C bottom) corresponding to the loss of
Table 2
Types of proteins identified on the proteome map
Protein class
a
Number ingenome
a
Number identified
b
Hypothetical 1049 68 (6)Conserved hypothetical 1322 114 (8)Secreted 530 30 (5)ABC transport ATP binding 109 16 (14)Lipoproteins 124 10 (8)Sigma factors 65 0 (0)Antisigma factors 7 2 (28)Antisigma factor antagonists 15 1 (6)Two-component system regulators 80 11 (13)Other regulators 592 36 (6)Two-component system kinases 85 0 (0)SerThr kinases 37 1 (2)Other 3923 482 (12)All 7816 770 (10)
a
Based on annotation of the
S coelicolor
genome sequence(httpwwwsangeracukProjectsS_coelicolor)
b
Percentage identified is included in parentheses
Proteomic analysis of
S coelicolor 921
copy 2002 Blackwell Publishing Ltd
Molecular Microbiology
46
917ndash932
Table 3
Proteins identified at pI and molecular weight co-ordinates on the 2D-PAGE maps that are inconsistent with their predicted amino acidsequences indicating possible post-translational modification either by proteolytic processing (A) or by covalent addition of a low-molecular-weight adduct (B)
ORF no Gene product Spot no in Fig 1
ASCO0498 Probable peptide monooxygenase 33SCO1230 Putative tripeptidylaminopeptidaseSCO1644 20S proteasome beta subunit precursor 35SCO1815 3-Oxoacyl-[acyl carrier protein] reductaseSCO1860 Putative secreted proteinSCO1947 Glyceraldehyde-3-phosphate dehydrogenase 9SCO1998 30S ribosomal protein S1SCO2619 ATP-dependent Clp protease subunit 13SCO2920 Probable secreted proteinaseSCO3236 Probable oxygenase 34SCO3671 Heat shock protein DNAKSCO3909 50S ribosomal protein L9 30SCO4296 Chaperonin GroEL2SCO4439
D
-Alanyl-
D
-alanine carboxypeptidaseSCO4643 UDP-N-acetylanoylpyruvoylglucosamine reductaseSCO4662 Elongation factor Tu1SCO4762 Chaperonin GroEL1SCO4808 Succinyl-CoA synthetase beta chain 8SCO4824 Bifunctional protein methylene tetra-hydrofolate dehydrogenasecyclohydrolaseSCO5073 Putative oxidoreductase ActVI-ORF2 29SCO5074 Putative dehydratase ActVI-ORF3 31SCO5088 Ketoacyl synthase beta subunit ActI-ORF2 24SCO5113 Oligopeptide ABC transport protein BldK-ORFBSCO5254 Superoxide dismutase precursor 32SCO5371 ATP synthase alpha chainSCO5477 Probable oligopeptide-binding lipoproteinSCO5624 30S ribosomal protein S2SCO5737 Guanosine pentaphosphate synthetaseSCO5769 Recombinase RecASCO5892 Polyketide synthase RedL 18SCO6282 3-Oxoacyl-[acyl carrier protein] reductase 11
BSCO1074 Putative peptidaseSCO1476 S-adenosylmethionine synthetaseSCO1594 Phenyl-tRNA synthetase beta chainSCO1643 20S proteasome alpha subunit 12SCO1814 Enoyl-[acyl carrier protein] reductaseSCO1935 Transketolase 3SCO1947 Glyceraldehyde-3-phosphate dehydrogenase 9SCO2180 Dihydrolipoamide dehydrogenase 21SCO2181 Probable dihydrolipoamide S-succinyltransferase 14SCO2198 Glutamine synthetase I 6SCO2390 3-Oxoacyl-[acyl carrier protein] synthase IISCO2478 Putative reductaseSCO2618 ATP-dependent Clp protease subunit 2SCO2822 Putative decarboxylaseSCO3096 Enolase 23SCO3229 4-Hydroxyphenylpyruvic acid dioxygenaseSCO3337 Probable pyrroline-5-carboxylate reductase 28SCO3409 Inorganic pyrophosphataseSCO3549 Antisigma factor antagonist BldGSCO3563 Acetyl-CoA synthaseSCO3671 Heat shock protein DNAKSCO3878 DNA polymerase III beta chainSCO3890 Thioredoxin reductaseSCO3906 30S ribosomal protein S6SCO3970 XAA-Pro aminopeptidase ISCO4164 Putative thiosulphate sulphurtransferaseSCO4209 Phosphoglycerate mutase 19SCO4240 ABC transport ATP binding protein MsiKSCO4296 Chaperonin GroEL2SCO4645 Aspartate aminotransferase 7SCO4652 50S ribosomal protein L10
Some proteins appear to be subject to both types of modification and are present in both parts Only proteins from primary or secondary metabolicpathways or with identified modifications are assigned to spots in Fig 1
922
A R Hesketh
et al
copy 2002 Blackwell Publishing Ltd
Molecular Microbiology
46
917ndash932
the isoleucine residue at its N-terminus (expected massloss 11308 Da) This indicates removal of the N-terminalMKI residues from ribosomal protein L9 The theoreticalisoelectric point (997) and molecular weight (156 kDa) ofL9 protein N-terminally truncated in this way are in goodagreement with the position of the lower spot in Fig 1(predicted values for the unprocessed protein are pH 960and 159 kDa) MALDI-TOF data indicating proteolytic
processing of the superoxide dismutase precursor protein(SCO5254 spot 32 in Fig 1) and the 20S proteasomebeta subunit precursor (SCO1644 spot 35 in Fig 1)agreed with previous reports (Kim
et al
1998 Nagy
et al
1998) Evidence for the proteolysis of
S coelicolor
ClpP1(SCO2619 spot 13 in Fig 1) has also been reportedalthough the processing site was not defined (de Crecy-Lagard
et al
1999)
SCO4661 Elongation factor GSCO4662 Elongation factor Tu1SCO4677 Putative regulatory proteinSCO4729 DNA-directed RNA polymerase alpha chainSCO4762 Chaperonin GroEL1SCO4770 Inosine-5
cent
-monophosphate dehydrogenase 25SCO4792 LuxR family two-component regulatorSCO4808 Succinyl-CoA synthetase beta chain 8SCO4809 Succinyl-CoA synthetase alpha chain 22SCO4921 Probable acyl-CoA carboxylase complex A chainSCO5071 Hydroxylacyl-CoA dehydrogenase ActVI-ORFA 16SCO5079 Hypothetical protein ActVA-ORF4 20SCO5080 Possible hydrolase ActVA-ORF5 26SCO5115 Probable ABC transporter intracellular ATPase chain BldK-ORFD 17SCO5371 ATP synthase alpha chainSCO5396 Putative cellulose-binding protein 10SCO5419 Putative thioredoxinSCO5515 Probable
D
-3-phosphoglycerate dehydrogenase 4SCO5535 Putative carboxyl transferaseSCO5584 Nitrogen regulatory protein GlnK 15SCO5625 Elongation factor TsSCO5699 Prolyl-tRNA synthetaseSCO5737 Guanosine pentaphosphate synthetaseSCO5777 Glutamate uptake system ATP-binding proteinSCO5803 SOS regulatory protein LexASCO5999 Aconitase 1SCO6198 Probable secreted proteinSCO6282 3-Oxoacyl-[acyl carrier protein] reductase 11SCO6636 Bacteriophage
f
-C31 resistance protein PglZSCO6663 Transketolase 27SCO7000 Isocitrate dehydrogenase 2SCO7036 Argininosuccinate synthase 5SCO7510 Peptidyl-prolyl
cis-trans
isomerase
ORF no Gene product Spot no in Fig 1
Some proteins appear to be subject to both types of modification and are present in both parts Only proteins from primary or secondary metabolicpathways or with identified modifications are assigned to spots in Fig 1
Table 3
cont
Table 4
Co- and post-translational modification of proteins suggested by MALDI-TOF peptide mass fingerprint analysis
Protein Modification
SCO1643 20S proteasome alpha subunit Removal of initiator Met residue and acetylation of N-terminal SerSCO5115 ABC transporter intracellular Removal of initiator Met residue and acetylation of N-terminal Thr
ATPase BldK-ORFDSCO5396 Putative cellulose-binding protein Removal of initiator Met residue and acetylation of N-terminal SerSCO2198 Glutamine synthetase I Adenylylation of peptide 395ndash420SCO5584 Nitrogen regulatory protein GlnK Adenylylation of peptide 48ndash58SCO1644 20S proteasome
b
-subunit precursor Proteolytic processing between amino acid residues 5354SCO2619 ATP-dependent Clp protease Proteolytic processing between amino acid residues 2223
proteolytic subunit ISCO3909 50S ribosomal protein L9 Proteolytic processing between amino acid residues 34SCO5074 Putative dehydratase ActVI-ORF3 Proteolytic processing between amino acid residues 3132 and 3435SCO5254 Superoxide dismutase precursor Proteolytic processing between amino acid residues 1415
Proteomic analysis of
S coelicolor 923
copy 2002 Blackwell Publishing Ltd
Molecular Microbiology
46
917ndash932
Co-translational modification by removal of the N-terminal initiator Met occurred in 149 out of 190 (78)cases in which the N-terminal peptide was detected Pro-cessing by the N-methionylaminopeptidase was observedif the second amino acid was Ser (45 out of 149 exam-ples) Thr (40) Ala (32) Pro (27) Gly (4) or Val (1) Ononly one occasion was any of these six amino acids foundas the second residue if the N-terminal methionine wasretained This is in broad agreement with observations in
B subtilis
in which two-thirds of proteins were reported tobe similarly processed (Buttner
et al
2001)
Representation of primary and secondary metabolic pathways
Streptomycetes produce
ordf
60 of all commercially usefulantibiotics (Berdy 1984 Miyodah 1993) Most antibioticsare the product of complex biosynthetic pathways oftenencoded by 20ndash30 clustered genes Production of thepigmented antibiotics actinorhodin (Act) and undecyl-
prodigiosin (Red) has been studied extensively in
Scoelicolor
(for reviews see Chater and Bibb 1997Champness 2000) and involves clusters of approximately22 genes each A third cluster producing the calcium-dependent antibiotic (CDA) and consisting of 40 geneshas also been studied (Chong
et al
1998 Huang
et al
2001) In the extensive spot identification studies for thepreparation of a proteome map of
S coelicolor
(seeabove) more than one-third of the proteins from each ofthese pathways was identified (Table 5) Proteins fromthree clusters that specify metabolic products that haveyet to be identified experimentally (Bentley
et al
2002)were also extensively represented a type I polyketidesynthase cluster (SCO6273ndash6288) a deoxysugarglycosyltransferase cluster (SCO0381ndash0401) and anon-ribosomal peptide synthetase cluster predicted toencode a novel siderophore coelichelin (SCO0489ndash0499) Several clusters were unrepresented
The primary metabolic pathways were somewhat morehighly represented with
ordf
60 or more of proteins anno-
Fig 2
Examples of MALDI-TOF peptide mass fingerprint data identifying post-translational modification of proteins by (A) acetylation (predicted mass increase of 420 Da) (B) adenylylation (predicted mass increase of 3290 Da) and (C) proteolytic processing (predicted mass loss for Ile of 11308 Da) In each fingerprint the
x
-axis is masscharge ratio (mz) and the
y
-axis is absolute intensity The observed mass accuracies in the MALDI-TOF fingerprints are (A) 60 ppm (B) 20 ppm and (C) 20 ppm
924
A R Hesketh
et al
copy 2002 Blackwell Publishing Ltd
Molecular Microbiology
46
917ndash932
tated as enzymes of glycolysis the TCA cycle thepentose phosphate pathway purine ribonucleotide bio-synthesis and pyrimidine nucleotide biosynthesis beingmapped (Table 6) About 50 of amino acid biosyntheticproteins were identified Lists of individual members of allpathways considered here including those that were andwere not identified during proteome mapping can befound in the
Supplementary material
Several proteins from both primary and secondary met-
abolic pathways were identified at more than one positionon the proteome map and may therefore be subject toregulation by post-translational modification (see Table 3and Fig 1) Primary metabolic proteins of this type (yellowspots on Fig 1) were usually detected as chains of two ormore spots with the same apparent molecular weights butdifferent pI values suggesting covalent modification bylow-molecular-weight adducts Apart from adenylylation ofGSI (see above) the nature of the modification of theseproteins was not revealed by MALDI-TOF peptide massfingerprint data Both covalent modification and proteolyticprocessing were also found in secondary metabolic pro-teins (red spots in Fig 1) For example the Act pathwaymembers SCO5071 (spot 16) and SCO5075 (spot 20)
and the putative 3-oxoacyl[acyl carrier protein] reductasefrom the type I polyketide cluster (SCO6282 spot 11)appear to be covalently modified whereas proteolytic pro-cessing was detected for RedL (SCO5892 spot 18) fromthe undecylprodigiosin cluster the putative dehydrataseActVI-ORF3 (SCO5074 spot 31) from the Act cluster theputative peptide monooxygenase from coelichelin biosyn-thesis (SCO0498 spot 34) and the putative oxygenasefrom the CDA pathway (SCO3236 spot 33) Two Act bio-synthetic proteins SCO5088 (spot 24) and SCO5073(spot 29) were detected both at their predicted pI andmolecular weight co-ordinates and at positions with simi-lar pI values but significantly higher molecular weightsThis may indicate that these proteins form stable mul-timers that are not completely denatured under theconditions used Only the modification of ActVI-ORF3(SCO5074 spot 31) could be characterized further fromthe mass spectrometry data which showed that it involvesproteolytic processing at two sites between amino acidresidues 3132 and 3435 (data not shown) Analysis of asample obtained by externally extracting proteins fromintact washed mycelium with a solution containing 2SDS and 50 mM dithiothreitol (DTT) revealed that sur-
Table 5
Summary of proteins identified on the proteome map from secondary metabolic pathways during growth in SMM
Biosynthetic pathwaygene cluster
Pathwaygenesannotated
Pathwaygene productsidentified
Percentagepathwaydetected
Calcium-dependent antibiotic CDA SCO3210ndash3249 40 16 40Actinorhodin SCO5071ndash5092 22 15 68Undecylprodigiosin SCO5877ndash5898 22 8 36Deoxysugar synthaseglycosyltransferase cluster SCO0381ndash0401 21 7 33Type I polyketide synthase SCO6273ndash6288 16 6 38Hopanoids SCO6759ndash6771 13 1 7Siderophore (coelichelin) SCO0489ndash0499 11 5 45Siderophore (desferrioxamine) SCO2783ndash2785 4 4 100
Chromosomal locations and extent of gene clusters are taken from Bentley
et al
(2002) No proteins were detected from the remaining predictedsecondary metabolic clusters ie siderophore (coelibactin) SCO7681ndash7691 (11 ORFs) siderophore SCO5799ndash5801 (three ORFs) non-ribosomal peptide synthases SCO6429ndash6438 (10 ORFs) type II fatty acid synthases SCO1265ndash1273 (nine ORFs) WhiE spore pigment clusterSCO5314ndash5320 (seven ORFs) isorenieratine SCO0185ndash0191 (seven ORFs) eicosapentaenoic acid SCO0124ndash0129 (six ORFs) chalconesynthases SCO1206ndash1208 (three ORFs) chalcone synthases SCO7669ndash7671 and SCO7222 (three ORFs) sesquiterpene cyclase SCO5222ndash5223 (two ORFs) type I polyketide synthases SCO6826ndash6827 (two ORFs) geosmin SCO6703 (one ORF)
Table 6 Summary of proteins identified on the proteome map from primary metabolic pathways during growth in SMM
Biosynthetic pathwayPathway genesannotateda
Pathway geneproducts identified
Percentage pathway detected
Glycolysisb 24 13 55Pentose phosphate pathwayb 14 10 71TCA cycleb 21 12 57Amino acid biosynthesisc 115 55 48Purine ribonucleotide biosynthesisb 18 12 67Pyrimidine ribonucleotide biosynthesisb 8 5 62
a Derived from interpretation of the annotated S coelicolor genome sequence at httpwwwsangeracukProjectsS_coelicolorb See Supplementary material for details of pathway membersc See Supplementary material for details of individual pathways
Proteomic analysis of S coelicolor 925
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
prisingly the truncated ActVI-ORF3 is located extracellu-larly presumably non-covalently associated with the cellwall matrix (Fig 4)
Expression analysis of certain primary and secondary metabolic proteins
Production of the pigmented antibiotics Act and Red inliquid minimal medium supplemented with casamino acids(SMM) begins in the transition phase between exponentialgrowth and stationary phase Figure 3 shows a compari-son of the expression of certain primary and secondarymetabolic proteins between exponentially growing andstationary phase cultures Two proteins from the Actcluster (ActVA-ORF5 and ActI-ORF2) and two from theRed (RedI and RedR) were present in the stationaryphase cultures but were not detected during exponentialgrowth Similarly the putative 3-oxoacyl-[acyl carrierprotein] reductase (SCO6282) and epoxide hydrolase(SCO6277) enzymes from a type I polyketide secondarymetabolic cluster showed a large increase in abundance
between the exponential and stationary phase samplesTriose phosphate isomerase (SCO1945) fructose bis-phosphate aldolase (SCO3649) and succinyl-CoA syn-thetase alpha chain (SCO4809) all enzymes from centralcarbon metabolism were present in approximatelyconstant amounts in both samples Interestingly two iso-forms (SCO7511 and SCO1947) of glyceraldehyde-3-phosphate dehydrogenase from the glycolytic pathwayshowed different patterns of expression SCO1947 levelswere the same in both exponential and stationary phasebut SCO7511 barely detectable in exponentially growingcultures increased markedly in stationary phase
The Streptomyces coelicolor online proteome map
The 2D gel images in Fig 1 together with the spot iden-tification results are being made available over the WorldWide Web to provide a reference map of the S coelicolorproteome At the time of writing the reference gel forthe pH 4ndash7 isoelectric point range was fully accessiblewhereas those for pH 45ndash55 pH 55ndash67 and pH 6ndash11
Fig 3 A comparison of the expression of certain primary and secondary metabolic proteins between exponentially growing and stationary phase cultures of S coelicolor M145 Larger arrows represent higher protein level in one gel than in the other The experiment was performed on duplicate biological samples and both showed similar protein expression profiles
926 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
were under construction (httpqbababeracukS_coeliReferenceGelrefgelhtml) A tool for comparing two ormore gel images with each other is also accessible fromthe same website at httpqbababeracukS_coeliflickerhtml
Discussion
Silver staining is one of the most sensitive methods fordetecting proteins on polyacrylamide gels allowing aslittle as 05ndash2 ng of an individual protein to be detected(Berggren et al 2000 Gorg et al 2000) The proportionof the S coelicolor proteome that can be analysed usingsilver staining of 2D-PAGE-separated proteins dependsat least in part on the pH range of the IPG strip used forthe first-dimension separation (see Table 1) Using theintermediate-range IPG strip covering pH 4ndash7 about 20ndash25 of the proteins in the theoretical proteome predictedto be observable in the resulting 2D-PAGE gel were actu-ally detected This figure was reduced to 10ndash12 whenanalysing the more basic pI range using the pH 6ndash11 IPGstrip Attempts to increase the percentage of the proteomedetectable using the pH 4ndash7 and pH 6ndash11 IPG strips byloading more protein sample resulted in poorer separa-tions (data not shown) This was especially true of thepH 6ndash11 strips which were generally the most difficult touse successfully However good separation of higher pro-tein loads was achieved using the narrow-range IPG stripspH 45ndash55 and 55ndash67 significantly increasing the pro-portion of theoretical proteins detected to over 40Clearly and more pertinently the limited conditions usedfor culturing S coelicolor in this study will also haverestricted the number of proteins detected as not all theproteome will be expressed In particular S coelicolordoes not sporulate in shaken liquid-grown cultures soproteins involved in this differentiation process are unlikelyto be present Nevertheless it is striking that the com-bined data for the pH 45ndash55 and 55ndash67 isoelectric pointranges indicate that 2468 out of a total of predicted 4932protein spots can be detected (see Table 1) suggestingthat transition phase mycelium in this supplementedminimal medium is expressing 50 of its genes (ie byextrapolation some 3912 of the 7825 in the genome)
Analysis of post-translational regulation
Although the regulation of cellular processes and adaptiveresponses in bacteria has been studied extensively at thetranscriptional level post-translational regulatory mecha-nisms are much less understood Proteomic analysisusing 2D-PAGE coupled with mass spectrometry offersthe opportunity to observe and identify modification ofproteins at a general cellular level The functions of pro-teins can be altered by specific covalent attachment of
adducts (eg phosphorylation glycosylation nucleotidy-lation acetylation) or by proteolytic processing Bothevents alter the pI andor mass of the protein and so canusually be observed on 2D-PAGE In the S coelicolorproteome map ordf110 out of a total of 770 gene productswere identified in more than one position or at pI andapparent mass co-ordinates significantly different frompredicted values indicating extensive regulation at thepost-translational level (88 are listed in Table 3) a propor-tion similar to that reported for other prokaryotes (Buttneret al 2001 Tonella et al 2001) The initial peptide massfingerprint analysis identified the modification of only 10of these proteins probably partly because fingerprint datatypically cover ordf30ndash60 of a proteinrsquos sequence (data notshown) so modification at positions not represented willbe missed Protein digestion with endopeptidases otherthan trypsin before mass spectrometry may help in thissituation but some covalently attached groups are labileand cannot be detected routinely Phosphorylation of Hisoccurs on nitrogen producing a high-energy phosphora-midate bond that is easily hydrolysed in acidic conditionswhereas phospho-Asp is the most labile phosphorylatedresidue with a half-life of a few hours under physiologicalconditions (reviewed by Robinson et al 2000) Thesemodifications the basis of bacterial two-component regu-latory systems are therefore unlikely to survive stainingof the 2D gels which involved the use of a fixing solutioncontaining 10 acetic acid and mass spectrometry Iden-tification of the more stable phosphoester phosphorylationthat occurs on Ser Thr or Tyr residues using PAGEfollowed by mass spectrometry has been reported(Godovac-Zimmermann et al 1999 Buttner et al 2001)but no examples were detected in this work Of the mod-ifications identified in this study three were by acetylationat the N-terminus after removal of the initiator Met residuetwo involved adenylylation and five proteins had been N-terminally truncated Modification of the 20S proteasomeby N-acetylation in yeast involves removal of the Metresidue by N-methionylaminopeptidase followed byacetylation with an Na-acetyltransferase (Kimura et al2000) Homologues of these enzymes are predicted fromthe S coelicolor genome and are presumably responsiblefor the observed modification of the 20S proteasomealpha subunit and of BldK-ORFD and the putative cellu-lose-binding protein The functional significance of Na-acetylation of these proteins is unclear
Analysis of the biosynthesis of antibiotics and other secondary metabolites
Post-translational modification The proteome mappingresults indicate that at least six enzymes in the Act clusterare subject to post-translational modifications and one
Proteomic analysis of S coelicolor 927
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
each from the undecylprodigiosin (SCO5877ndash5898)lsquocoelichelinrsquo (SCO0489ndash0499) and type I polyketidesynthase (SCO6273ndash6288) clusters (see Table 3) Thesemetabolic pathways may therefore be significantly regu-lated at the post-translational level The actVI-ORF3 gene(SCO5074) encodes a putative dehydratase enzyme thatis part of the biosynthetic pathway for Act a blue memberof the benzoisochromanequinone (BIQ) family of pig-mented antibiotics Mutation in actVI-ORF3 causes a 50decrease in the accumulation of Act (and a reddish brownphenotype) and it is believed to be responsible for pyranring closure an intermediate step in Act biosynthesis (thatmay also occur spontaneously) leading to the formationof the BIQ chromophore (Fernandez-Moreno et al 1994Ichinose et al 1998a Taguchi et al 2000) In this studyActVI-ORF3 protein was detected in two forms a majorspot corresponding to ActVI-ORF3 truncated by removalof the N-terminal 31 amino acid residues and a minor onein which the first 34 amino acids were missing (spots 31in Fig 1) The full-length protein was not detected Anal-ysis of the protein sequence using the SIGNALP server athttpwwwcbsdtudkservicesSignalP predicted a signalpeptide cleavage site between amino acids 31 and 32 inagreement with the observed results
The protein extract analysed was prepared from myce-lium that had been collected by centrifugation and sub-jected to two washes indicating that if ActVI-ORF3 isexported it is effectively retained within the cell wallmatrix This was confirmed by analysis of a preparationobtained by externally extracting proteins from intactwashed mycelium (Fig 4) Export of an enzyme requiredfor the efficient biosynthesis of an antibiotic that is thoughtto be produced intracellularly appears to be anomalousparticularly as it is involved in an intermediate biosynthetic
event Database analysis of the other members of the Actcluster revealed only one other protein with a predictedsignal peptide sequence the putative integral membranetransport-related protein ActII-ORF3 and it seemsunlikely that all the Act biosynthetic steps following BIQchromophore formation are extracellular It is possible thatfull-length ActVI-ORF3 catalyses the intermediate biosyn-thetic event intracellularly and the exported processedprotein is responsible for a different extracellular activityperhaps later in the pathway However no full-length pro-tein was detected so the exported enzyme may performthe pyran ring closure on a secreted metabolite that isthen reabsorbed by the cell for completion of the sub-sequent biosynthetic steps Only two homologues ofactVI-ORF3 are known and both are also in BIQ antibioticbiosynthetic clusters gra-ORF18 from the granaticincluster (Ichinose et al 1998b) and med-ORF5 frommedermycin biosynthesis (K Ichinose personal com-munication) Interestingly the encoded proteins of bothsequences are also predicted to contain N-terminal signalpeptides The observation that ActVI-ORF3 is exported tothe cell wall matrix may therefore be of general signifi-cance to the BIQ family of antibiotics and provide a newinsight into their biosynthetic pathways
Pathway representation Of the 20 putative secondarymetabolite gene clusters identified in the S coelicolorgenome sequence (Bentley et al 2002) eight had atleast one protein each identified on the proteome map(see Table 5) The biosynthetic clusters for the antibioticsAct Red and CDA had 68 36 and 40 of their pro-teins represented There are several possible explana-tions for failure to detect all the proteins for each pathwaySome are putative integral membrane proteins so cannotreadily be detected using conventional 2D-PAGE SomeRed and CDA proteins have molecular weights gt200 kDaoutside the range of the 125 gels used (for clustermembers see Supplementary material) The pathway-specific regulatory proteins for Act and Red biosynthesiswere not detected despite a detailed search of their pre-dicted gel locations even though the relevant genes areknown to be transcribed in equivalent cultures of the samestrain (Hesketh et al 2001) The proteins are presumablypresent but at levels below the limit of detection Howeverthe transcriptional regulator for CDA production CdaRwas identified as well as the two-component systemresponse regulator AbsA2 which is also encoded by agene in the CDA cluster (Anderson et al 2001) Interest-ingly proteins from three putative secondary meta-bolite clusters producing chemicals of as yet unknownstructure were also identified on the map correspondingto the deoxysugar synthaseglycosyltransferase clusterSCO0381ndash0401 the lsquocoelichelinrsquo cluster for a predicted(but not yet confirmed) oligopeptide siderophore
Fig 4 Processed ActVI-ORF3 is present in the cell wall matrix 2D-gel separations of protein extracts prepared (A) from whole cells or (B) by externally washing cells in a 2 SDS extraction buffer The arrow indicates ActVI-ORF3 protein The experiment was performed on duplicate biological samples and both showed similar protein expression profiles
928 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
(SCO0489ndash0499) and the type I polyketide clusterSCO6273ndash6288 The 3-oxoacyl-[acyl carrier protein]reductase (SCO6282) from the latter pathway is ex-pressed in a growth phase-dependent manner (Fig 3)and in early stationary phase cultures is perhaps themost abundant protein spot in the entire detectable pro-teome Presumably the metabolites produced by thesethree pathways are being synthesized under the condi-tions used In addition to coelichelin three other sidero-phores are postulated for S coelicolor All the proteinsfrom one of these desferrioxamine (SCO2782ndash2785)were identified but none was seen from either of the othertwo pathways The WhiE polyketide spore pigment isassociated with sporulation of surface-grown cultures(Davis and Chater 1990) and the expression of the whiEgenes depends on a series of sporulation-specific regula-tory genes (Kelemen et al 1998) no corresponding pro-teins of which were detected here It is therefore notsurprising that WhiE proteins were not detected in sam-ples prepared from liquid-grown vegetative mycelium
Analysis of primary metabolism
Proteins annotated as functioning in primary metabolicpathways were highly represented on the proteome mapwith ordf60 or more of the enzymes assigned to glycolysisthe TCA cycle the pentose phosphate pathway andpurine and pyrimidine ribonucleotide biosynthesis beingidentified (see Table 6) This figure may be deceptively lowbecause these pathways in S coelicolor often appear tohave more than one enzyme isoform for some of themetabolic steps potentially offering additional levels ofcontrol or developmental responsiveness at these pointsFor example only two steps in glycolysis have just a singlegene product assigned to them the remainder typicallyhave two or three each (Fig 5) At least one isoform waspresent for each step so the pathway is in theory 100functionally represented Understanding the regulation ofthese pathways will require analysis of the expression ofthese isoenzymes under different nutritional conditionsor different developmental stages Figure 5 shows that ofthe three predicted glyceraldehyde-3-phosphate dehydro-genase isoforms from glycolysis only two were detectedin this study (see also Supplementary material) Of theseSCO1947 appears to be constitutive whereas SCO7511is growth phase dependent (see Fig 3) Thus even thelimited conditions in this study yielded useful informationabout isoenzyme expression patterns (see Supplemen-tary material) In Streptomyces aureofaciens two genesencoding glyceraldehyde-3-phosphate dehydrogenaseactivity also appear to be differently regulated transcrip-tion of one highly homologous to SCO7511 is activatedby the regulator GapR whereas the other is GapR inde-pendent (Spusansky et al 2001)
Hood et al (1992) reported that most amino acid bio-synthetic enzymes are expressed at very low constitutivelevels in S coelicolor so it is reassuring to note that ordf50could be detected and identified using proteomics Theseinclude TrpB and TrpC1 from the tryptophan biosyntheticpathway the corresponding transcripts of which couldbarely be detected in previously reported S1 nucleaseprotection experiments (Hu et al 1999)
Perspectives
No attempt was made in the work reported here to simplifythe protein separations by subdividing the sample intosoluble and membrane fractions and this is reflected inthe 10 lipoproteins and 16 ABC transport system ATP-binding subunits identified on the proteome map (seeTable 2) No integral membrane proteins were detectedand even when a membrane protein sample was pre-pared and analysed no proteins identified possessedmore than three predicted transmembrane helices (datanot shown) This is consistent with previous studiesin other bacteria (Molloy et al 1999 Nilsson andDavidsson 2000) Another group of proteins also strik-ingly absent from the list of those identified is the sigmafactors which determine promoter selection by RNA poly-merase There are an extraordinarily high number (65) ofsigma factors encoded in the S coelicolor genome aswell as at least seven antisigma factors and 15 antisigmafactor antagonists (some of each of which were detected)Sigma factor heterogeneity in bacteria is an important wayof regulating expression of specific sets of genes under
Fig 5 The glycolytic pathway in S coelicolor illustrating the assignment of multiple gene products to individual metabolic steps based on the annotation of the genome sequence at httpwwwsangeracukProjectsS_coelicolor Gene products identified in the proteome map are underlined
Proteomic analysis of S coelicolor 929
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
different conditions and in different cell types (for a reviewsee Gross et al 1998) Although no sigma factors wereidentified about half (31 out of 65) have predicted pIvalues gtpH 7 and so can only be detected using thepH 6ndash11 IPG strips that give the poorest results (seeTable 1) However the principal vegetative sigma factorHrdB which is expected to be moderately abundanthas a predicted pI of 613 and its absence from thepH 55ndash67 proteome map may indicate that the samplepreparation method which does not remove DNA is inap-propriate Among other regulatory proteins 11 out of apossible 80 two-component system response regulatorsplus a further 36 proteins annotated as lsquoregulatorsrsquo weredetected Only one SerThr kinase and no two-componentsystem sensor kinases were detected possibly reflectingthe difficulty in analysing proteins with several transmem-brane domains Interestingly 30 secreted proteins wereidentified on the proteome map at least 10 of whichappear from comparison of their observed pI and molec-ular weight positions with predicted values to have beenprocessed already and therefore exported Presumablythese proteins do not end up exclusively in the culturemedium
Clearly this approach to proteomic analysis of S coeli-color has some important deficiencies not least theabsence of certain groups of proteins and the relativelypoor initial success rate for specifically characterizingpost-translational modifications Nevertheless it offersunique insights into the molecular biology of the organismthe expression of many proteins from both primary andsecondary metabolic pathways can be analysed post-translational regulation of proteins can be investigated ona general cellular level and the physical locations forsome proteins can be determined Approximately 35 ofthe S coelicolor genome is annotated as encoding lsquohypo-theticalrsquo proteins of unknown function and 182 proteins ofthis type were identified on the proteome map No exam-ples were found of overt disagreements between pre-dicted and observed proteins providing experimentalsupport for the general accuracy of the S coelicolorgenome sequence and its annotation Information aboutthe expression post-translational modification and physi-cal location of these (no longer) hypothetical proteinsfrom proteomic analyses is going to be important forassigning future functions As many of the hypotheticalproteins have apparent orthologues in pathogenic myco-bacteria some of the information may have an immediateapplication in the context of pathogenesis
Experimental procedures
Materials
Immobilized pH gradient (IPG) strips and IPG bufferswere purchased from Amersham Biosciences Acrylamide
duracryl and chemicals for silver staining were obtained fromGenomic Solutions All other chemicals were from Sigmaunless stated in the text
Strain and growth conditions
Streptomyces coelicolor M145 was cultivated with vigorousagitation at 30infinC in minimal medium supplemented with 02casamino acids (SMM) as described previously (Kieser et al2000) Briefly a high-density spore preparation [about 1010
colony-forming units (cfu) ml-1] was pregerminated in 2yen YTmedia for 7 h at 30infinC Germlings were harvested by centrifu-gation (5 min at 4000 g) resuspended in SMM and sonicatedbriefly to disperse any aggregates before inoculation into50 ml of SMM in 250 ml siliconized flasks containing coiledstainless steel springs Each flask received the equivalent of5 yen 107 spore cfu
Protein extraction and 2D-gel electrophoresis
The preparation of the protein sample to be separated waskept as simple as possible disrupting harvested myceliadirectly into denaturing isoelectric focusing (IEF) buffer con-taining Pefabloc SC (Roche) protease inhibitor This methodminimized unwanted protein degradation that was observedin initial studies using a Tris buffer containing Pefabloc SC forcell lysis (data not shown) Removal of DNA RNA salts andpolysaccharides from the sample was therefore not per-formed (other than the shearing of viscous DNA and RNA bysonication) and the protein extracts were sometimes difficultto focus when the sample was loaded by in-gel rehydrationLoading the sample soaked in paper strips at the anodicend of IPG strips that had already been rehydrated improvedthe reproducibility of separations significantly
Mycelium for protein extraction was harvested from cul-tures by brief centrifugation (30 s at 4000 g) at room tem-perature and immediately frozen in liquid nitrogen Typicallymycelium from 10 ml culture aliquots was collected and thetransfer time from culture flask to frozen sample was 15 minFrozen cells were thawed on ice in 5 ml of washing buffer(40 mM Tris pH 90 1 mM EGTA 1 mM EDTA) then pelletedby centrifugation (5 min at 3000 g) at 4infinC Washed cells wereresuspended in 400 ml of denaturing IEF buffer UTCHAPS[7 M urea 2 M thiourea 4 (wv) CHAPS 40 mM TrispH 90 1 mM EDTA 50 mM DTT 4 mM Pefabloc SC pro-tease inhibitor (Roche)] and disrupted by sonication (SanyoSoniprep 150 10 yen 2 s bursts at amplitude 75 mm) whilecooling in an ethanolndashice bath Cell debris was removed bycentrifugation (15 min 10 000 g 4infinC) and the protein extractwas stored frozen in aliquots at -80infinC until use For thepreparation of extracellular protein extracts washed cellswere resuspended in extraction buffer consisting of 40 mMTris pH 90 1 mM EDTA 50 mM DTT 2 SDS and 4 mMPefabloc SC protease inhibitor Cells were vortexed for 2 minbefore being pelleted by centrifugation (5 min 10 000 g4infinC) The supernatant was precipitated using the Amersham2D Clean-Up kit according to the manufacturerrsquos instructionsand the precipitated proteins were redissolved in UTCHAPSand stored at -80infinC until use
930 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
For first-dimension isoelectric focusing 18 cm IPG stripspH 4ndash7 pH 6ndash11 pH 45ndash55 or pH 55ndash67 (Amersham Bio-sciences) were rehydrated overnight in IEF buffer containing1 ampholytes according to the manufacturerrsquos instructionsusing a Phaser isoelectric focusing unit (Genomic Solutions)set at 20 V Protein samples to be separated were applied tothe rehydrated strips at the anodic end soaked in a 5ndash10 mmsection of IEF electrode strip (Amersham Biosciences)Separation was performed for 120 000 V-h with a maximumvoltage of 5000 V After isoelectric focusing IPG strips wereequilibrated for the second dimension for 15 min in IPG equil-ibration buffer (50 mM Tris pH 68 6 M urea 30 glycerol1 SDS and 001 bromophenol blue) plus 80 mM DTTthen for 10 min in IPG equilibration buffer plus 135 mMiodoacetamide Approximately 1 cm was removed from theanodic end of each equilibrated strip before applying the stripto the top of a vertical 125 SDS-PAGE gel for second-dimension separation using the Investigator 5000 systemfrom Genomic Solutions
Large-format gels (24 cm wide yen 22 cm high yen 1 mm thick)for the second-dimension separation were cast in-houseusing the system supplied by Genomic Solutions 125SDS-PAGE gels prepared using 065 NN-methylenebi-sacrylamide as cross-linking agent were used for pH 4ndash7and pH 45ndash55 IPG strips whereas pH 55ndash67 and pH 6ndash11 strips were applied to similar gels made with 08 cross-linker Electrophoresis was performed while cooling using themaximum setting (ordf 4ndash10infinC) at 20 000 mW per gel constantpower and a maximum voltage of 500 V Gels were thenstained with either colloidal Coomassie G-250 (Neuhoff et al1988) or silver nitrate (Rabilloud 1992) and scanned in aProXPRESS proteomic imaging system (Perkin-Elmer)Image analysis was performed using PHORETIX 2D version51 (NonLinear Dynamics) spot detection was optimizedautomatically using the lsquoSpot Detection Wizardrsquo and thenedited manually background subtraction was performedautomatically using the lsquoMode of Non-Spotrsquo setting imageswere then normalized to the total spot volume for quantifica-tion Spot filtering was not used
Protein identification using mass spectrometry
Protein spots of interest were excised from colloidalCoomassie-stained gels in-gel digested with trypsin andidentified by MALDI-TOF peptide mass fingerprint analysisExcised gel pieces (ordf 1-mm-diameter circles cut from 1-mm-thick gels) were washed twice with 100 ml of 50 mMammonium bicarbonate for 15 min once with 100 ml of 20acetonitrile-40 mM ammonium bicarbonate for 15 min andonce with 100 acetonitrile Washed gel pieces were allowedto air-dry for 10 min before being rehydrated in 5 ml of10 mg ml-1 trypsin (sequencing grade modified Promega) in10 mM ammonium bicarbonate Digestion (37infinC for 4 h) wasstopped by the addition of 5 ml of 5 formic acid Extractionof peptides into this solution was encouraged by a sonicwater bath treatment for 20 min Peptide extract (05 ml) wasspotted on to a thin layer of a-cyano-4-hydroxycinnamic acidapplied to a MALDI-TOF sample template and analysed byMALDI-TOF mass spectrometry (Bruker Reflex III) using anaccelerating voltage of 25 kV Samples were externally cali-brated using a standard mixture of six peptides ranging in
mass from 10465423 Da to 34946500 Da and the massaccuracy obtained was 60 ppm or better Identification ofproteins from MALDI-TOF peptide mass fingerprint data wasperformed using the lsquoMascotrsquo search engine at httpwwwmatrixsciencecom and was based on a positive resultusing their lsquoProbability Based MOWSE Scorersquo algorithm AMOWSE score of 60 or higher is significant at the 5 levelor better and proteins in this work typically gave scores gt80(frequently considerably so) In addition no identification wasaccepted unless at least five peptides representing at least20 of the protein sequence were detected in the MALDI-TOF peptide mass fingerprint
Assignment of proteins to metabolic pathways
Proteins were assigned to primary metabolic pathwaysusing the lsquoProtein Classification Schemersquo web page ofthe annotated Streptomyces coelicolor genome sequenceat httpwwwsangeracukProjectsS_coelicolor as it ap-peared in May 2002 The annotation of the genome andtherefore the Protein Classification Scheme is likely to bemodified as more experimental data are obtained about thefunction of genes that have currently been assigned on thebasis of sequence information alone It is therefore possiblethat proteins assigned to metabolic pathways in this studymay in future be shown to perform different functions in thecell
Assignments to gene clusters encoding secondary metab-olites are based on an analysis of the genome sequencereported by Bentley et al (2002) In some cases theseassignments are based on extensive experimental data (actred reviewed by Chater and Bibb 1997 Champness 2000cda Chong et al 1998 Huang et al 2001 whiE Davis andChater 1990)
Acknowledgements
This work was funded by grants (2FGT11406 208FGT11408 and 208IGF12432) under the BBSRCrsquos Tech-nologies for Functional Genomics and Investigating GeneFunction initiatives and by a competitive strategic grant fromthe BBSRC to the John Innes Centre We thank Mike Naldrettfor his extensive contribution in establishing technology forproteome analysis at the JIC
Supplementary material
The following material is available from httpwwwblackwell-sciencecomproductsjournalssuppmatmolemole3219mmi3219smhtmSection 1 Detection of gene products assigned to individualmetabolic steps in primary metabolic pathways in ScoelicolorSection 2 Identification of proteins from individual amino acidbiosynthetic pathwaysSection 3 Detection of gene products assigned to individualmetabolic steps in secondary metabolic pathways in Scoelicolor
Proteomic analysis of S coelicolor 931
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
References
Anderson TB Brian P and Champness WC (2001)Genetic and transcriptional analysis of absA an antibioticgene cluster linked two-component system that regulatesmultiple antibiotics in Streptomyces coelicolor Mol Micro-biol 39 553ndash566
Bentley SD Chater KF Cerdeno-Tarraga A-M ChallisGL Thomson NR James KD et al (2002) Completegenome sequence of the model actinomycete Streptomy-ces coelicolor A3(2) Nature 417 141ndash147
Berdy J (1984) New ways to obtain antibiotics Chin J Anti-biot 7 272ndash290
Berggren K Chernokalskaya E Steinberg TH KemperC Lopez MF Diwu Z et al (2000) Background-freehigh sensitivity staining of proteins in one- and two-dimensional sodium dodecyl sulfate-polyacrylamide gelsusing a luminescent ruthenium complex Electrophoresis21 2509ndash2521
Blackstock WP and Weir MP (1999) Proteomics quan-titative and physical mapping of cellular proteins TrendsBiotechnol 17 121ndash127
Blattner FR Plunkett G Bloch CA Perna NT BurlandV Riley M et al (1997) The complete genome sequenceof Escherichia coli K-12 Science 277 1453ndash1462
Buttner K Bernhardt J Scharf C Schmid R MaderU Eymann C et al (2001) A comprehensive two-dimensional map of cytosolic proteins of Bacillus subtilisElectrophoresis 22 2908ndash2935
Champness W (2000) Actinomycete development antibioticproduction and phylogeny questions and challenges InProkaryotic Development Brun YV and Skimketts LJ(eds) Washington DC American Society for MicrobiologyPress pp 11ndash31
Chater KF and Bibb MJ (1997) Regulation of bacterialantibiotic production In Biotechnology Vol 7 Products ofSecondary Metabolism Kleinkauf H and von Doumlhren H(eds) Weinheim Germany VCH pp 57ndash105
Chong PP Podmore SM Kieser HM Redenbach MTurgay K Marahiel M et al (1998) Physical identificationof a chromosomal locus encoding biosynthetic genesfor the lipopeptide calcium-dependent antibiotic (CDA) ofStreptomyces coelicolor A3(2) Microbiology 144 193ndash199
de Crecy-Lagard V Servant-Moisson JV Grandvalet Cand Mazodier P (1999) Alteration of the synthesis of theClp ATP-dependent protease affects morphological andphysiological differentiation in Streptomyces Mol Microbiol32 505ndash517
Davis NK and Chater KF (1990) Spore colour inStreptomyces coelicolor A3(2) involves the developmen-tally regulated synthesis of a compound biosyntheticallyrelated to polyketide antibiotics Mol Microbiol 4 1679ndash1691
Fernandez-Moreno FA Martinez E Caballero JLIchinose K Hopwood DA and Malpartida F (1994)DNA sequence and functions of the actVI region of theactinorhodin biosynthetic gene cluster of Streptomycescoelicolor A3(2) J Biol Chem 269 24854ndash24863
Fink D Falke D Wohllheben W and Engels A (1999)Nitrogen metabolism in Streptomyces coelicolor A3(2)
modification of glutamine synthase I by an adenylyltrans-ferase Microbiology 145 2313ndash2322
Godovac-Zimmermann J Soskic V Posnanovic S andBrianza F (1999) Functional proteomics of signal trans-duction by membrane receptors Electrophoresis 20 952ndash961
Gorg A Obermaier C Boguth G Harder A Scheibe BWildgruber R and Weiss W (2000) The current state oftwo-dimensional electrophoresis with immobilized pH gra-dients Electrophoresis 21 1037ndash1053
Gramajo HC Takano E and Bibb MJ (1993) Stationary-phase production of the antibiotic actinorhodin in Strepto-myces coelicolor A3(2) is transcriptionally regulated MolMicrobiol 7 837ndash845
Gross CA Chan C Dombroski A Gruber T Sharp MTupy J and Young B (1998) The functional and regula-tory roles of sigma factors in transcription Cold SpringHarb Symp Quant Biol 63 141ndash155
Hesketh A Sun J and Bibb MJ (2001) Induction ofppGpp synthesis in Streptomyces coelicolor A3(2) grownunder conditions of nutritional sufficiency elicits actII-ORF4transcription and actinorhodin biosynthesis Mol Microbiol39 136ndash144
Hesketh A Fink D Gust B Rexer H-U Scheel BChater K et al (2002) The PII nitrogen regulatory proteinGlnK in Streptomyces coelicolor A3(2) is modified by ade-nylylation rather than uridylylation and the GlnD and GlnKhomologues are functionally dissimilar to their nitrogenregulatory system counterparts Mol Microbiol (inpress)
Hood DW Heidstra R Swodoba UK and Hodgson DA(1992) Molecular genetic analysis of proline and tryp-tophan biosynthesis in Streptomyces coelicolor A3(2)interaction between primary and secondary metabolism- areview Gene 115 5ndash12
Hu DS-J Hood DW Heidstra R and Hodgson DA(1999) The expression of the trpD trpC and trpBA genesof Streptomyces coelicolor A3(2) is regulated by growthrate and growth phase but not by feedback repression MolMicrobiol 32 869ndash880
Huang J Lih CJ Pan KH and Cohen SN (2001)Global analysis of growth phase responsive gene expres-sion and regulation of antibiotic biosynthetic pathways inStreptomyces coelicolor using DNA microarrays GenesDev 15 3183ndash3192
Ichinose K Taguchi T Ebizuka Y and Hopwood DA(1998a) Biosynthetic gene clusters of benzoisochromane-quinone antibiotics in Streptomyces spp Identification ofgenes involved in post-PKS tailoring steps Actinomyceto-logica 12 99ndash109
Ichinose K Bedford DJ Tornus D Bechthold A BibbMJ Revill PW et al (1998b) The granaticin biosyntheticgene cluster of Streptomyces violaceoruber Tu22sequence analysis and expression in a heterologous hostChem Biol 5 647ndash659
Kelemen GH Brian P Flardh K Chamberlin L ChaterK and Buttner M (1998) Developmental regulation oftranscription of whiE a locus specifying the polyketidespore pigment in Streptomyces coelicolor A3(2) J Bacte-riol 180 2515ndash2521
Kieser T Bibb MJ Buttner MJ Chater KF and
932 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
Hopwood DA (2000) Practical Streptomyces GeneticsNorwich John Innes Foundation
Kim EJ Chung HJ Suh B Hah YC and Roe JH(1998) Expression and regulation of the sodF gene encod-ing iron- and zinc-containing superoxide dismutase inStreptomyces coelicolor Muumlller J Bacteriol 180 2014ndash2020
Kimura Y Takaoka M Tanaka S Sassa H Tanaka KPolevoda B et al (2000) Na-acetylation and proteolyticactivity of the yeast 20S proteasome J Biol Chem 2754635ndash4639
Kunst F Ogasawara N Moszer I Albertini AM AlloniG Azevedo V et al (1997) The complete genomesequence of the Gram-positive bacterium Bacillus subtilisNature 390 249ndash256
Lucchini S Thompson A and Hinton JCD (2001)Microarrays for microbiologists Microbiology 147 1403ndash1414
Mann M Hendrickson RC and Pandey A (2001) Analy-sis of proteins and proteomes by mass spectrometry AnnuRev Biochem 70 437ndash473
Miyodah S (1993) Research on antibiotic screening inJapan over the last decade a producing microorganismsapproach Actinomycetologica 7 100ndash106
Molloy MP Herbert BR Williams KL and Gooley AA(1999) Extraction of Escherichia coli proteins with organicsolvents prior to two-dimensional electrophoresis Electro-phoresis 20 701ndash704
Nagy I Tamura T Vanderleyden J Baumeister W andDe Mot R (1998) The 20S proteasome of Streptomycescoelicolor J Bacteriol 180 5448ndash5453
Neuhoff V Arold N Taube D and Ehrhardt W (1988)Improved staining of proteins in polyacrylamide gels includ-ing isoelectric focusing gels with clear background at nan-ogram sensitivity using Coomassie Brilliant Blue G-250and R-250 Electrophoresis 9 255ndash262
Nilsson CL and Davidsson P (2000) New separation toolsfor comprehensive studies of protein expression by massspectrometry Mass Spectrom Rev 19 390ndash397
Puglia AM Vohradsky J and Thompson CJ (1995)
Developmental control of the heat-shock stress regulon inStreptomyces coelicolor Mol Microbiol 17 737ndash746
Rabilloud T (1992) A comparison between low backgroundsilver diamine and silver nitrate protein stains Electro-phoresis 13 429ndash439
Robinson VL Buckler DR and Stock AM (2000) A taleof two components a novel kinase and a regulatory switchNature Struct Biol 7 626ndash633
Schena M (2000) Microarray Biochip Technology NatickMA Eaton Publishing
Spusansky O Rezuchova B Homerova D andKormanec J (2001) Expression of the gap gene encodingglyceraldehyde-3-phosphate dehydrogenase of Strepto-myces aureofaciens requires GapR a member of theAraCXylS family of transcriptional activators Microbiology147 1291ndash1301
Taguchi T Itou K Ebizuka Y Malpartida F HopwoodDA Surti CM et al (2000) Chemical characterisationof disruptants of the Streptomyces coelicolor A3(2) actVIgenes involved in actinorhodin biosynthesis J Antibiot 53144ndash152
Takano E Gramajo HC Strauch E Andres N WhiteJ and Bibb MJ (1992) Transcriptional regulation of theredD transcriptional activator gene accounts for growth-phase-dependent production of the antibotic undecylpro-digiosin in Streptomyces coelicolor A3(2) Mol Microbiol6 2797ndash2804
Tonella L Hoogland C Binz P-A Appel RDHochstrasser DF and Sanchez J-C (2001) New per-spectives in the Escherichia coli proteome investigationProteomics 1 409ndash423
Vohradsky J Li XM and Thompson CJ (1997) Identifi-cation of prokaryotic developmental stages by statisticalanalysis of two-dimensional gel patterns Electrophoresis18 1418ndash1428
Vohradsky J Li XM Dale G Folcher M Nguyen LViollier PH and Thompson CJ (2000) Developmentalcontrol of stress stimulons in Streptomyces coelicolorrevealed by statistical analyses of global gene expressionpatterns J Bacteriol 182 4979ndash4986

Proteomic analysis of
S coelicolor 921
copy 2002 Blackwell Publishing Ltd
Molecular Microbiology
46
917ndash932
Table 3
Proteins identified at pI and molecular weight co-ordinates on the 2D-PAGE maps that are inconsistent with their predicted amino acidsequences indicating possible post-translational modification either by proteolytic processing (A) or by covalent addition of a low-molecular-weight adduct (B)
ORF no Gene product Spot no in Fig 1
ASCO0498 Probable peptide monooxygenase 33SCO1230 Putative tripeptidylaminopeptidaseSCO1644 20S proteasome beta subunit precursor 35SCO1815 3-Oxoacyl-[acyl carrier protein] reductaseSCO1860 Putative secreted proteinSCO1947 Glyceraldehyde-3-phosphate dehydrogenase 9SCO1998 30S ribosomal protein S1SCO2619 ATP-dependent Clp protease subunit 13SCO2920 Probable secreted proteinaseSCO3236 Probable oxygenase 34SCO3671 Heat shock protein DNAKSCO3909 50S ribosomal protein L9 30SCO4296 Chaperonin GroEL2SCO4439
D
-Alanyl-
D
-alanine carboxypeptidaseSCO4643 UDP-N-acetylanoylpyruvoylglucosamine reductaseSCO4662 Elongation factor Tu1SCO4762 Chaperonin GroEL1SCO4808 Succinyl-CoA synthetase beta chain 8SCO4824 Bifunctional protein methylene tetra-hydrofolate dehydrogenasecyclohydrolaseSCO5073 Putative oxidoreductase ActVI-ORF2 29SCO5074 Putative dehydratase ActVI-ORF3 31SCO5088 Ketoacyl synthase beta subunit ActI-ORF2 24SCO5113 Oligopeptide ABC transport protein BldK-ORFBSCO5254 Superoxide dismutase precursor 32SCO5371 ATP synthase alpha chainSCO5477 Probable oligopeptide-binding lipoproteinSCO5624 30S ribosomal protein S2SCO5737 Guanosine pentaphosphate synthetaseSCO5769 Recombinase RecASCO5892 Polyketide synthase RedL 18SCO6282 3-Oxoacyl-[acyl carrier protein] reductase 11
BSCO1074 Putative peptidaseSCO1476 S-adenosylmethionine synthetaseSCO1594 Phenyl-tRNA synthetase beta chainSCO1643 20S proteasome alpha subunit 12SCO1814 Enoyl-[acyl carrier protein] reductaseSCO1935 Transketolase 3SCO1947 Glyceraldehyde-3-phosphate dehydrogenase 9SCO2180 Dihydrolipoamide dehydrogenase 21SCO2181 Probable dihydrolipoamide S-succinyltransferase 14SCO2198 Glutamine synthetase I 6SCO2390 3-Oxoacyl-[acyl carrier protein] synthase IISCO2478 Putative reductaseSCO2618 ATP-dependent Clp protease subunit 2SCO2822 Putative decarboxylaseSCO3096 Enolase 23SCO3229 4-Hydroxyphenylpyruvic acid dioxygenaseSCO3337 Probable pyrroline-5-carboxylate reductase 28SCO3409 Inorganic pyrophosphataseSCO3549 Antisigma factor antagonist BldGSCO3563 Acetyl-CoA synthaseSCO3671 Heat shock protein DNAKSCO3878 DNA polymerase III beta chainSCO3890 Thioredoxin reductaseSCO3906 30S ribosomal protein S6SCO3970 XAA-Pro aminopeptidase ISCO4164 Putative thiosulphate sulphurtransferaseSCO4209 Phosphoglycerate mutase 19SCO4240 ABC transport ATP binding protein MsiKSCO4296 Chaperonin GroEL2SCO4645 Aspartate aminotransferase 7SCO4652 50S ribosomal protein L10
Some proteins appear to be subject to both types of modification and are present in both parts Only proteins from primary or secondary metabolicpathways or with identified modifications are assigned to spots in Fig 1
922
A R Hesketh
et al
copy 2002 Blackwell Publishing Ltd
Molecular Microbiology
46
917ndash932
the isoleucine residue at its N-terminus (expected massloss 11308 Da) This indicates removal of the N-terminalMKI residues from ribosomal protein L9 The theoreticalisoelectric point (997) and molecular weight (156 kDa) ofL9 protein N-terminally truncated in this way are in goodagreement with the position of the lower spot in Fig 1(predicted values for the unprocessed protein are pH 960and 159 kDa) MALDI-TOF data indicating proteolytic
processing of the superoxide dismutase precursor protein(SCO5254 spot 32 in Fig 1) and the 20S proteasomebeta subunit precursor (SCO1644 spot 35 in Fig 1)agreed with previous reports (Kim
et al
1998 Nagy
et al
1998) Evidence for the proteolysis of
S coelicolor
ClpP1(SCO2619 spot 13 in Fig 1) has also been reportedalthough the processing site was not defined (de Crecy-Lagard
et al
1999)
SCO4661 Elongation factor GSCO4662 Elongation factor Tu1SCO4677 Putative regulatory proteinSCO4729 DNA-directed RNA polymerase alpha chainSCO4762 Chaperonin GroEL1SCO4770 Inosine-5
cent
-monophosphate dehydrogenase 25SCO4792 LuxR family two-component regulatorSCO4808 Succinyl-CoA synthetase beta chain 8SCO4809 Succinyl-CoA synthetase alpha chain 22SCO4921 Probable acyl-CoA carboxylase complex A chainSCO5071 Hydroxylacyl-CoA dehydrogenase ActVI-ORFA 16SCO5079 Hypothetical protein ActVA-ORF4 20SCO5080 Possible hydrolase ActVA-ORF5 26SCO5115 Probable ABC transporter intracellular ATPase chain BldK-ORFD 17SCO5371 ATP synthase alpha chainSCO5396 Putative cellulose-binding protein 10SCO5419 Putative thioredoxinSCO5515 Probable
D
-3-phosphoglycerate dehydrogenase 4SCO5535 Putative carboxyl transferaseSCO5584 Nitrogen regulatory protein GlnK 15SCO5625 Elongation factor TsSCO5699 Prolyl-tRNA synthetaseSCO5737 Guanosine pentaphosphate synthetaseSCO5777 Glutamate uptake system ATP-binding proteinSCO5803 SOS regulatory protein LexASCO5999 Aconitase 1SCO6198 Probable secreted proteinSCO6282 3-Oxoacyl-[acyl carrier protein] reductase 11SCO6636 Bacteriophage
f
-C31 resistance protein PglZSCO6663 Transketolase 27SCO7000 Isocitrate dehydrogenase 2SCO7036 Argininosuccinate synthase 5SCO7510 Peptidyl-prolyl
cis-trans
isomerase
ORF no Gene product Spot no in Fig 1
Some proteins appear to be subject to both types of modification and are present in both parts Only proteins from primary or secondary metabolicpathways or with identified modifications are assigned to spots in Fig 1
Table 3
cont
Table 4
Co- and post-translational modification of proteins suggested by MALDI-TOF peptide mass fingerprint analysis
Protein Modification
SCO1643 20S proteasome alpha subunit Removal of initiator Met residue and acetylation of N-terminal SerSCO5115 ABC transporter intracellular Removal of initiator Met residue and acetylation of N-terminal Thr
ATPase BldK-ORFDSCO5396 Putative cellulose-binding protein Removal of initiator Met residue and acetylation of N-terminal SerSCO2198 Glutamine synthetase I Adenylylation of peptide 395ndash420SCO5584 Nitrogen regulatory protein GlnK Adenylylation of peptide 48ndash58SCO1644 20S proteasome
b
-subunit precursor Proteolytic processing between amino acid residues 5354SCO2619 ATP-dependent Clp protease Proteolytic processing between amino acid residues 2223
proteolytic subunit ISCO3909 50S ribosomal protein L9 Proteolytic processing between amino acid residues 34SCO5074 Putative dehydratase ActVI-ORF3 Proteolytic processing between amino acid residues 3132 and 3435SCO5254 Superoxide dismutase precursor Proteolytic processing between amino acid residues 1415
Proteomic analysis of
S coelicolor 923
copy 2002 Blackwell Publishing Ltd
Molecular Microbiology
46
917ndash932
Co-translational modification by removal of the N-terminal initiator Met occurred in 149 out of 190 (78)cases in which the N-terminal peptide was detected Pro-cessing by the N-methionylaminopeptidase was observedif the second amino acid was Ser (45 out of 149 exam-ples) Thr (40) Ala (32) Pro (27) Gly (4) or Val (1) Ononly one occasion was any of these six amino acids foundas the second residue if the N-terminal methionine wasretained This is in broad agreement with observations in
B subtilis
in which two-thirds of proteins were reported tobe similarly processed (Buttner
et al
2001)
Representation of primary and secondary metabolic pathways
Streptomycetes produce
ordf
60 of all commercially usefulantibiotics (Berdy 1984 Miyodah 1993) Most antibioticsare the product of complex biosynthetic pathways oftenencoded by 20ndash30 clustered genes Production of thepigmented antibiotics actinorhodin (Act) and undecyl-
prodigiosin (Red) has been studied extensively in
Scoelicolor
(for reviews see Chater and Bibb 1997Champness 2000) and involves clusters of approximately22 genes each A third cluster producing the calcium-dependent antibiotic (CDA) and consisting of 40 geneshas also been studied (Chong
et al
1998 Huang
et al
2001) In the extensive spot identification studies for thepreparation of a proteome map of
S coelicolor
(seeabove) more than one-third of the proteins from each ofthese pathways was identified (Table 5) Proteins fromthree clusters that specify metabolic products that haveyet to be identified experimentally (Bentley
et al
2002)were also extensively represented a type I polyketidesynthase cluster (SCO6273ndash6288) a deoxysugarglycosyltransferase cluster (SCO0381ndash0401) and anon-ribosomal peptide synthetase cluster predicted toencode a novel siderophore coelichelin (SCO0489ndash0499) Several clusters were unrepresented
The primary metabolic pathways were somewhat morehighly represented with
ordf
60 or more of proteins anno-
Fig 2
Examples of MALDI-TOF peptide mass fingerprint data identifying post-translational modification of proteins by (A) acetylation (predicted mass increase of 420 Da) (B) adenylylation (predicted mass increase of 3290 Da) and (C) proteolytic processing (predicted mass loss for Ile of 11308 Da) In each fingerprint the
x
-axis is masscharge ratio (mz) and the
y
-axis is absolute intensity The observed mass accuracies in the MALDI-TOF fingerprints are (A) 60 ppm (B) 20 ppm and (C) 20 ppm
924
A R Hesketh
et al
copy 2002 Blackwell Publishing Ltd
Molecular Microbiology
46
917ndash932
tated as enzymes of glycolysis the TCA cycle thepentose phosphate pathway purine ribonucleotide bio-synthesis and pyrimidine nucleotide biosynthesis beingmapped (Table 6) About 50 of amino acid biosyntheticproteins were identified Lists of individual members of allpathways considered here including those that were andwere not identified during proteome mapping can befound in the
Supplementary material
Several proteins from both primary and secondary met-
abolic pathways were identified at more than one positionon the proteome map and may therefore be subject toregulation by post-translational modification (see Table 3and Fig 1) Primary metabolic proteins of this type (yellowspots on Fig 1) were usually detected as chains of two ormore spots with the same apparent molecular weights butdifferent pI values suggesting covalent modification bylow-molecular-weight adducts Apart from adenylylation ofGSI (see above) the nature of the modification of theseproteins was not revealed by MALDI-TOF peptide massfingerprint data Both covalent modification and proteolyticprocessing were also found in secondary metabolic pro-teins (red spots in Fig 1) For example the Act pathwaymembers SCO5071 (spot 16) and SCO5075 (spot 20)
and the putative 3-oxoacyl[acyl carrier protein] reductasefrom the type I polyketide cluster (SCO6282 spot 11)appear to be covalently modified whereas proteolytic pro-cessing was detected for RedL (SCO5892 spot 18) fromthe undecylprodigiosin cluster the putative dehydrataseActVI-ORF3 (SCO5074 spot 31) from the Act cluster theputative peptide monooxygenase from coelichelin biosyn-thesis (SCO0498 spot 34) and the putative oxygenasefrom the CDA pathway (SCO3236 spot 33) Two Act bio-synthetic proteins SCO5088 (spot 24) and SCO5073(spot 29) were detected both at their predicted pI andmolecular weight co-ordinates and at positions with simi-lar pI values but significantly higher molecular weightsThis may indicate that these proteins form stable mul-timers that are not completely denatured under theconditions used Only the modification of ActVI-ORF3(SCO5074 spot 31) could be characterized further fromthe mass spectrometry data which showed that it involvesproteolytic processing at two sites between amino acidresidues 3132 and 3435 (data not shown) Analysis of asample obtained by externally extracting proteins fromintact washed mycelium with a solution containing 2SDS and 50 mM dithiothreitol (DTT) revealed that sur-
Table 5
Summary of proteins identified on the proteome map from secondary metabolic pathways during growth in SMM
Biosynthetic pathwaygene cluster
Pathwaygenesannotated
Pathwaygene productsidentified
Percentagepathwaydetected
Calcium-dependent antibiotic CDA SCO3210ndash3249 40 16 40Actinorhodin SCO5071ndash5092 22 15 68Undecylprodigiosin SCO5877ndash5898 22 8 36Deoxysugar synthaseglycosyltransferase cluster SCO0381ndash0401 21 7 33Type I polyketide synthase SCO6273ndash6288 16 6 38Hopanoids SCO6759ndash6771 13 1 7Siderophore (coelichelin) SCO0489ndash0499 11 5 45Siderophore (desferrioxamine) SCO2783ndash2785 4 4 100
Chromosomal locations and extent of gene clusters are taken from Bentley
et al
(2002) No proteins were detected from the remaining predictedsecondary metabolic clusters ie siderophore (coelibactin) SCO7681ndash7691 (11 ORFs) siderophore SCO5799ndash5801 (three ORFs) non-ribosomal peptide synthases SCO6429ndash6438 (10 ORFs) type II fatty acid synthases SCO1265ndash1273 (nine ORFs) WhiE spore pigment clusterSCO5314ndash5320 (seven ORFs) isorenieratine SCO0185ndash0191 (seven ORFs) eicosapentaenoic acid SCO0124ndash0129 (six ORFs) chalconesynthases SCO1206ndash1208 (three ORFs) chalcone synthases SCO7669ndash7671 and SCO7222 (three ORFs) sesquiterpene cyclase SCO5222ndash5223 (two ORFs) type I polyketide synthases SCO6826ndash6827 (two ORFs) geosmin SCO6703 (one ORF)
Table 6 Summary of proteins identified on the proteome map from primary metabolic pathways during growth in SMM
Biosynthetic pathwayPathway genesannotateda
Pathway geneproducts identified
Percentage pathway detected
Glycolysisb 24 13 55Pentose phosphate pathwayb 14 10 71TCA cycleb 21 12 57Amino acid biosynthesisc 115 55 48Purine ribonucleotide biosynthesisb 18 12 67Pyrimidine ribonucleotide biosynthesisb 8 5 62
a Derived from interpretation of the annotated S coelicolor genome sequence at httpwwwsangeracukProjectsS_coelicolorb See Supplementary material for details of pathway membersc See Supplementary material for details of individual pathways
Proteomic analysis of S coelicolor 925
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
prisingly the truncated ActVI-ORF3 is located extracellu-larly presumably non-covalently associated with the cellwall matrix (Fig 4)
Expression analysis of certain primary and secondary metabolic proteins
Production of the pigmented antibiotics Act and Red inliquid minimal medium supplemented with casamino acids(SMM) begins in the transition phase between exponentialgrowth and stationary phase Figure 3 shows a compari-son of the expression of certain primary and secondarymetabolic proteins between exponentially growing andstationary phase cultures Two proteins from the Actcluster (ActVA-ORF5 and ActI-ORF2) and two from theRed (RedI and RedR) were present in the stationaryphase cultures but were not detected during exponentialgrowth Similarly the putative 3-oxoacyl-[acyl carrierprotein] reductase (SCO6282) and epoxide hydrolase(SCO6277) enzymes from a type I polyketide secondarymetabolic cluster showed a large increase in abundance
between the exponential and stationary phase samplesTriose phosphate isomerase (SCO1945) fructose bis-phosphate aldolase (SCO3649) and succinyl-CoA syn-thetase alpha chain (SCO4809) all enzymes from centralcarbon metabolism were present in approximatelyconstant amounts in both samples Interestingly two iso-forms (SCO7511 and SCO1947) of glyceraldehyde-3-phosphate dehydrogenase from the glycolytic pathwayshowed different patterns of expression SCO1947 levelswere the same in both exponential and stationary phasebut SCO7511 barely detectable in exponentially growingcultures increased markedly in stationary phase
The Streptomyces coelicolor online proteome map
The 2D gel images in Fig 1 together with the spot iden-tification results are being made available over the WorldWide Web to provide a reference map of the S coelicolorproteome At the time of writing the reference gel forthe pH 4ndash7 isoelectric point range was fully accessiblewhereas those for pH 45ndash55 pH 55ndash67 and pH 6ndash11
Fig 3 A comparison of the expression of certain primary and secondary metabolic proteins between exponentially growing and stationary phase cultures of S coelicolor M145 Larger arrows represent higher protein level in one gel than in the other The experiment was performed on duplicate biological samples and both showed similar protein expression profiles
926 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
were under construction (httpqbababeracukS_coeliReferenceGelrefgelhtml) A tool for comparing two ormore gel images with each other is also accessible fromthe same website at httpqbababeracukS_coeliflickerhtml
Discussion
Silver staining is one of the most sensitive methods fordetecting proteins on polyacrylamide gels allowing aslittle as 05ndash2 ng of an individual protein to be detected(Berggren et al 2000 Gorg et al 2000) The proportionof the S coelicolor proteome that can be analysed usingsilver staining of 2D-PAGE-separated proteins dependsat least in part on the pH range of the IPG strip used forthe first-dimension separation (see Table 1) Using theintermediate-range IPG strip covering pH 4ndash7 about 20ndash25 of the proteins in the theoretical proteome predictedto be observable in the resulting 2D-PAGE gel were actu-ally detected This figure was reduced to 10ndash12 whenanalysing the more basic pI range using the pH 6ndash11 IPGstrip Attempts to increase the percentage of the proteomedetectable using the pH 4ndash7 and pH 6ndash11 IPG strips byloading more protein sample resulted in poorer separa-tions (data not shown) This was especially true of thepH 6ndash11 strips which were generally the most difficult touse successfully However good separation of higher pro-tein loads was achieved using the narrow-range IPG stripspH 45ndash55 and 55ndash67 significantly increasing the pro-portion of theoretical proteins detected to over 40Clearly and more pertinently the limited conditions usedfor culturing S coelicolor in this study will also haverestricted the number of proteins detected as not all theproteome will be expressed In particular S coelicolordoes not sporulate in shaken liquid-grown cultures soproteins involved in this differentiation process are unlikelyto be present Nevertheless it is striking that the com-bined data for the pH 45ndash55 and 55ndash67 isoelectric pointranges indicate that 2468 out of a total of predicted 4932protein spots can be detected (see Table 1) suggestingthat transition phase mycelium in this supplementedminimal medium is expressing 50 of its genes (ie byextrapolation some 3912 of the 7825 in the genome)
Analysis of post-translational regulation
Although the regulation of cellular processes and adaptiveresponses in bacteria has been studied extensively at thetranscriptional level post-translational regulatory mecha-nisms are much less understood Proteomic analysisusing 2D-PAGE coupled with mass spectrometry offersthe opportunity to observe and identify modification ofproteins at a general cellular level The functions of pro-teins can be altered by specific covalent attachment of
adducts (eg phosphorylation glycosylation nucleotidy-lation acetylation) or by proteolytic processing Bothevents alter the pI andor mass of the protein and so canusually be observed on 2D-PAGE In the S coelicolorproteome map ordf110 out of a total of 770 gene productswere identified in more than one position or at pI andapparent mass co-ordinates significantly different frompredicted values indicating extensive regulation at thepost-translational level (88 are listed in Table 3) a propor-tion similar to that reported for other prokaryotes (Buttneret al 2001 Tonella et al 2001) The initial peptide massfingerprint analysis identified the modification of only 10of these proteins probably partly because fingerprint datatypically cover ordf30ndash60 of a proteinrsquos sequence (data notshown) so modification at positions not represented willbe missed Protein digestion with endopeptidases otherthan trypsin before mass spectrometry may help in thissituation but some covalently attached groups are labileand cannot be detected routinely Phosphorylation of Hisoccurs on nitrogen producing a high-energy phosphora-midate bond that is easily hydrolysed in acidic conditionswhereas phospho-Asp is the most labile phosphorylatedresidue with a half-life of a few hours under physiologicalconditions (reviewed by Robinson et al 2000) Thesemodifications the basis of bacterial two-component regu-latory systems are therefore unlikely to survive stainingof the 2D gels which involved the use of a fixing solutioncontaining 10 acetic acid and mass spectrometry Iden-tification of the more stable phosphoester phosphorylationthat occurs on Ser Thr or Tyr residues using PAGEfollowed by mass spectrometry has been reported(Godovac-Zimmermann et al 1999 Buttner et al 2001)but no examples were detected in this work Of the mod-ifications identified in this study three were by acetylationat the N-terminus after removal of the initiator Met residuetwo involved adenylylation and five proteins had been N-terminally truncated Modification of the 20S proteasomeby N-acetylation in yeast involves removal of the Metresidue by N-methionylaminopeptidase followed byacetylation with an Na-acetyltransferase (Kimura et al2000) Homologues of these enzymes are predicted fromthe S coelicolor genome and are presumably responsiblefor the observed modification of the 20S proteasomealpha subunit and of BldK-ORFD and the putative cellu-lose-binding protein The functional significance of Na-acetylation of these proteins is unclear
Analysis of the biosynthesis of antibiotics and other secondary metabolites
Post-translational modification The proteome mappingresults indicate that at least six enzymes in the Act clusterare subject to post-translational modifications and one
Proteomic analysis of S coelicolor 927
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
each from the undecylprodigiosin (SCO5877ndash5898)lsquocoelichelinrsquo (SCO0489ndash0499) and type I polyketidesynthase (SCO6273ndash6288) clusters (see Table 3) Thesemetabolic pathways may therefore be significantly regu-lated at the post-translational level The actVI-ORF3 gene(SCO5074) encodes a putative dehydratase enzyme thatis part of the biosynthetic pathway for Act a blue memberof the benzoisochromanequinone (BIQ) family of pig-mented antibiotics Mutation in actVI-ORF3 causes a 50decrease in the accumulation of Act (and a reddish brownphenotype) and it is believed to be responsible for pyranring closure an intermediate step in Act biosynthesis (thatmay also occur spontaneously) leading to the formationof the BIQ chromophore (Fernandez-Moreno et al 1994Ichinose et al 1998a Taguchi et al 2000) In this studyActVI-ORF3 protein was detected in two forms a majorspot corresponding to ActVI-ORF3 truncated by removalof the N-terminal 31 amino acid residues and a minor onein which the first 34 amino acids were missing (spots 31in Fig 1) The full-length protein was not detected Anal-ysis of the protein sequence using the SIGNALP server athttpwwwcbsdtudkservicesSignalP predicted a signalpeptide cleavage site between amino acids 31 and 32 inagreement with the observed results
The protein extract analysed was prepared from myce-lium that had been collected by centrifugation and sub-jected to two washes indicating that if ActVI-ORF3 isexported it is effectively retained within the cell wallmatrix This was confirmed by analysis of a preparationobtained by externally extracting proteins from intactwashed mycelium (Fig 4) Export of an enzyme requiredfor the efficient biosynthesis of an antibiotic that is thoughtto be produced intracellularly appears to be anomalousparticularly as it is involved in an intermediate biosynthetic
event Database analysis of the other members of the Actcluster revealed only one other protein with a predictedsignal peptide sequence the putative integral membranetransport-related protein ActII-ORF3 and it seemsunlikely that all the Act biosynthetic steps following BIQchromophore formation are extracellular It is possible thatfull-length ActVI-ORF3 catalyses the intermediate biosyn-thetic event intracellularly and the exported processedprotein is responsible for a different extracellular activityperhaps later in the pathway However no full-length pro-tein was detected so the exported enzyme may performthe pyran ring closure on a secreted metabolite that isthen reabsorbed by the cell for completion of the sub-sequent biosynthetic steps Only two homologues ofactVI-ORF3 are known and both are also in BIQ antibioticbiosynthetic clusters gra-ORF18 from the granaticincluster (Ichinose et al 1998b) and med-ORF5 frommedermycin biosynthesis (K Ichinose personal com-munication) Interestingly the encoded proteins of bothsequences are also predicted to contain N-terminal signalpeptides The observation that ActVI-ORF3 is exported tothe cell wall matrix may therefore be of general signifi-cance to the BIQ family of antibiotics and provide a newinsight into their biosynthetic pathways
Pathway representation Of the 20 putative secondarymetabolite gene clusters identified in the S coelicolorgenome sequence (Bentley et al 2002) eight had atleast one protein each identified on the proteome map(see Table 5) The biosynthetic clusters for the antibioticsAct Red and CDA had 68 36 and 40 of their pro-teins represented There are several possible explana-tions for failure to detect all the proteins for each pathwaySome are putative integral membrane proteins so cannotreadily be detected using conventional 2D-PAGE SomeRed and CDA proteins have molecular weights gt200 kDaoutside the range of the 125 gels used (for clustermembers see Supplementary material) The pathway-specific regulatory proteins for Act and Red biosynthesiswere not detected despite a detailed search of their pre-dicted gel locations even though the relevant genes areknown to be transcribed in equivalent cultures of the samestrain (Hesketh et al 2001) The proteins are presumablypresent but at levels below the limit of detection Howeverthe transcriptional regulator for CDA production CdaRwas identified as well as the two-component systemresponse regulator AbsA2 which is also encoded by agene in the CDA cluster (Anderson et al 2001) Interest-ingly proteins from three putative secondary meta-bolite clusters producing chemicals of as yet unknownstructure were also identified on the map correspondingto the deoxysugar synthaseglycosyltransferase clusterSCO0381ndash0401 the lsquocoelichelinrsquo cluster for a predicted(but not yet confirmed) oligopeptide siderophore
Fig 4 Processed ActVI-ORF3 is present in the cell wall matrix 2D-gel separations of protein extracts prepared (A) from whole cells or (B) by externally washing cells in a 2 SDS extraction buffer The arrow indicates ActVI-ORF3 protein The experiment was performed on duplicate biological samples and both showed similar protein expression profiles
928 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
(SCO0489ndash0499) and the type I polyketide clusterSCO6273ndash6288 The 3-oxoacyl-[acyl carrier protein]reductase (SCO6282) from the latter pathway is ex-pressed in a growth phase-dependent manner (Fig 3)and in early stationary phase cultures is perhaps themost abundant protein spot in the entire detectable pro-teome Presumably the metabolites produced by thesethree pathways are being synthesized under the condi-tions used In addition to coelichelin three other sidero-phores are postulated for S coelicolor All the proteinsfrom one of these desferrioxamine (SCO2782ndash2785)were identified but none was seen from either of the othertwo pathways The WhiE polyketide spore pigment isassociated with sporulation of surface-grown cultures(Davis and Chater 1990) and the expression of the whiEgenes depends on a series of sporulation-specific regula-tory genes (Kelemen et al 1998) no corresponding pro-teins of which were detected here It is therefore notsurprising that WhiE proteins were not detected in sam-ples prepared from liquid-grown vegetative mycelium
Analysis of primary metabolism
Proteins annotated as functioning in primary metabolicpathways were highly represented on the proteome mapwith ordf60 or more of the enzymes assigned to glycolysisthe TCA cycle the pentose phosphate pathway andpurine and pyrimidine ribonucleotide biosynthesis beingidentified (see Table 6) This figure may be deceptively lowbecause these pathways in S coelicolor often appear tohave more than one enzyme isoform for some of themetabolic steps potentially offering additional levels ofcontrol or developmental responsiveness at these pointsFor example only two steps in glycolysis have just a singlegene product assigned to them the remainder typicallyhave two or three each (Fig 5) At least one isoform waspresent for each step so the pathway is in theory 100functionally represented Understanding the regulation ofthese pathways will require analysis of the expression ofthese isoenzymes under different nutritional conditionsor different developmental stages Figure 5 shows that ofthe three predicted glyceraldehyde-3-phosphate dehydro-genase isoforms from glycolysis only two were detectedin this study (see also Supplementary material) Of theseSCO1947 appears to be constitutive whereas SCO7511is growth phase dependent (see Fig 3) Thus even thelimited conditions in this study yielded useful informationabout isoenzyme expression patterns (see Supplemen-tary material) In Streptomyces aureofaciens two genesencoding glyceraldehyde-3-phosphate dehydrogenaseactivity also appear to be differently regulated transcrip-tion of one highly homologous to SCO7511 is activatedby the regulator GapR whereas the other is GapR inde-pendent (Spusansky et al 2001)
Hood et al (1992) reported that most amino acid bio-synthetic enzymes are expressed at very low constitutivelevels in S coelicolor so it is reassuring to note that ordf50could be detected and identified using proteomics Theseinclude TrpB and TrpC1 from the tryptophan biosyntheticpathway the corresponding transcripts of which couldbarely be detected in previously reported S1 nucleaseprotection experiments (Hu et al 1999)
Perspectives
No attempt was made in the work reported here to simplifythe protein separations by subdividing the sample intosoluble and membrane fractions and this is reflected inthe 10 lipoproteins and 16 ABC transport system ATP-binding subunits identified on the proteome map (seeTable 2) No integral membrane proteins were detectedand even when a membrane protein sample was pre-pared and analysed no proteins identified possessedmore than three predicted transmembrane helices (datanot shown) This is consistent with previous studiesin other bacteria (Molloy et al 1999 Nilsson andDavidsson 2000) Another group of proteins also strik-ingly absent from the list of those identified is the sigmafactors which determine promoter selection by RNA poly-merase There are an extraordinarily high number (65) ofsigma factors encoded in the S coelicolor genome aswell as at least seven antisigma factors and 15 antisigmafactor antagonists (some of each of which were detected)Sigma factor heterogeneity in bacteria is an important wayof regulating expression of specific sets of genes under
Fig 5 The glycolytic pathway in S coelicolor illustrating the assignment of multiple gene products to individual metabolic steps based on the annotation of the genome sequence at httpwwwsangeracukProjectsS_coelicolor Gene products identified in the proteome map are underlined
Proteomic analysis of S coelicolor 929
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
different conditions and in different cell types (for a reviewsee Gross et al 1998) Although no sigma factors wereidentified about half (31 out of 65) have predicted pIvalues gtpH 7 and so can only be detected using thepH 6ndash11 IPG strips that give the poorest results (seeTable 1) However the principal vegetative sigma factorHrdB which is expected to be moderately abundanthas a predicted pI of 613 and its absence from thepH 55ndash67 proteome map may indicate that the samplepreparation method which does not remove DNA is inap-propriate Among other regulatory proteins 11 out of apossible 80 two-component system response regulatorsplus a further 36 proteins annotated as lsquoregulatorsrsquo weredetected Only one SerThr kinase and no two-componentsystem sensor kinases were detected possibly reflectingthe difficulty in analysing proteins with several transmem-brane domains Interestingly 30 secreted proteins wereidentified on the proteome map at least 10 of whichappear from comparison of their observed pI and molec-ular weight positions with predicted values to have beenprocessed already and therefore exported Presumablythese proteins do not end up exclusively in the culturemedium
Clearly this approach to proteomic analysis of S coeli-color has some important deficiencies not least theabsence of certain groups of proteins and the relativelypoor initial success rate for specifically characterizingpost-translational modifications Nevertheless it offersunique insights into the molecular biology of the organismthe expression of many proteins from both primary andsecondary metabolic pathways can be analysed post-translational regulation of proteins can be investigated ona general cellular level and the physical locations forsome proteins can be determined Approximately 35 ofthe S coelicolor genome is annotated as encoding lsquohypo-theticalrsquo proteins of unknown function and 182 proteins ofthis type were identified on the proteome map No exam-ples were found of overt disagreements between pre-dicted and observed proteins providing experimentalsupport for the general accuracy of the S coelicolorgenome sequence and its annotation Information aboutthe expression post-translational modification and physi-cal location of these (no longer) hypothetical proteinsfrom proteomic analyses is going to be important forassigning future functions As many of the hypotheticalproteins have apparent orthologues in pathogenic myco-bacteria some of the information may have an immediateapplication in the context of pathogenesis
Experimental procedures
Materials
Immobilized pH gradient (IPG) strips and IPG bufferswere purchased from Amersham Biosciences Acrylamide
duracryl and chemicals for silver staining were obtained fromGenomic Solutions All other chemicals were from Sigmaunless stated in the text
Strain and growth conditions
Streptomyces coelicolor M145 was cultivated with vigorousagitation at 30infinC in minimal medium supplemented with 02casamino acids (SMM) as described previously (Kieser et al2000) Briefly a high-density spore preparation [about 1010
colony-forming units (cfu) ml-1] was pregerminated in 2yen YTmedia for 7 h at 30infinC Germlings were harvested by centrifu-gation (5 min at 4000 g) resuspended in SMM and sonicatedbriefly to disperse any aggregates before inoculation into50 ml of SMM in 250 ml siliconized flasks containing coiledstainless steel springs Each flask received the equivalent of5 yen 107 spore cfu
Protein extraction and 2D-gel electrophoresis
The preparation of the protein sample to be separated waskept as simple as possible disrupting harvested myceliadirectly into denaturing isoelectric focusing (IEF) buffer con-taining Pefabloc SC (Roche) protease inhibitor This methodminimized unwanted protein degradation that was observedin initial studies using a Tris buffer containing Pefabloc SC forcell lysis (data not shown) Removal of DNA RNA salts andpolysaccharides from the sample was therefore not per-formed (other than the shearing of viscous DNA and RNA bysonication) and the protein extracts were sometimes difficultto focus when the sample was loaded by in-gel rehydrationLoading the sample soaked in paper strips at the anodicend of IPG strips that had already been rehydrated improvedthe reproducibility of separations significantly
Mycelium for protein extraction was harvested from cul-tures by brief centrifugation (30 s at 4000 g) at room tem-perature and immediately frozen in liquid nitrogen Typicallymycelium from 10 ml culture aliquots was collected and thetransfer time from culture flask to frozen sample was 15 minFrozen cells were thawed on ice in 5 ml of washing buffer(40 mM Tris pH 90 1 mM EGTA 1 mM EDTA) then pelletedby centrifugation (5 min at 3000 g) at 4infinC Washed cells wereresuspended in 400 ml of denaturing IEF buffer UTCHAPS[7 M urea 2 M thiourea 4 (wv) CHAPS 40 mM TrispH 90 1 mM EDTA 50 mM DTT 4 mM Pefabloc SC pro-tease inhibitor (Roche)] and disrupted by sonication (SanyoSoniprep 150 10 yen 2 s bursts at amplitude 75 mm) whilecooling in an ethanolndashice bath Cell debris was removed bycentrifugation (15 min 10 000 g 4infinC) and the protein extractwas stored frozen in aliquots at -80infinC until use For thepreparation of extracellular protein extracts washed cellswere resuspended in extraction buffer consisting of 40 mMTris pH 90 1 mM EDTA 50 mM DTT 2 SDS and 4 mMPefabloc SC protease inhibitor Cells were vortexed for 2 minbefore being pelleted by centrifugation (5 min 10 000 g4infinC) The supernatant was precipitated using the Amersham2D Clean-Up kit according to the manufacturerrsquos instructionsand the precipitated proteins were redissolved in UTCHAPSand stored at -80infinC until use
930 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
For first-dimension isoelectric focusing 18 cm IPG stripspH 4ndash7 pH 6ndash11 pH 45ndash55 or pH 55ndash67 (Amersham Bio-sciences) were rehydrated overnight in IEF buffer containing1 ampholytes according to the manufacturerrsquos instructionsusing a Phaser isoelectric focusing unit (Genomic Solutions)set at 20 V Protein samples to be separated were applied tothe rehydrated strips at the anodic end soaked in a 5ndash10 mmsection of IEF electrode strip (Amersham Biosciences)Separation was performed for 120 000 V-h with a maximumvoltage of 5000 V After isoelectric focusing IPG strips wereequilibrated for the second dimension for 15 min in IPG equil-ibration buffer (50 mM Tris pH 68 6 M urea 30 glycerol1 SDS and 001 bromophenol blue) plus 80 mM DTTthen for 10 min in IPG equilibration buffer plus 135 mMiodoacetamide Approximately 1 cm was removed from theanodic end of each equilibrated strip before applying the stripto the top of a vertical 125 SDS-PAGE gel for second-dimension separation using the Investigator 5000 systemfrom Genomic Solutions
Large-format gels (24 cm wide yen 22 cm high yen 1 mm thick)for the second-dimension separation were cast in-houseusing the system supplied by Genomic Solutions 125SDS-PAGE gels prepared using 065 NN-methylenebi-sacrylamide as cross-linking agent were used for pH 4ndash7and pH 45ndash55 IPG strips whereas pH 55ndash67 and pH 6ndash11 strips were applied to similar gels made with 08 cross-linker Electrophoresis was performed while cooling using themaximum setting (ordf 4ndash10infinC) at 20 000 mW per gel constantpower and a maximum voltage of 500 V Gels were thenstained with either colloidal Coomassie G-250 (Neuhoff et al1988) or silver nitrate (Rabilloud 1992) and scanned in aProXPRESS proteomic imaging system (Perkin-Elmer)Image analysis was performed using PHORETIX 2D version51 (NonLinear Dynamics) spot detection was optimizedautomatically using the lsquoSpot Detection Wizardrsquo and thenedited manually background subtraction was performedautomatically using the lsquoMode of Non-Spotrsquo setting imageswere then normalized to the total spot volume for quantifica-tion Spot filtering was not used
Protein identification using mass spectrometry
Protein spots of interest were excised from colloidalCoomassie-stained gels in-gel digested with trypsin andidentified by MALDI-TOF peptide mass fingerprint analysisExcised gel pieces (ordf 1-mm-diameter circles cut from 1-mm-thick gels) were washed twice with 100 ml of 50 mMammonium bicarbonate for 15 min once with 100 ml of 20acetonitrile-40 mM ammonium bicarbonate for 15 min andonce with 100 acetonitrile Washed gel pieces were allowedto air-dry for 10 min before being rehydrated in 5 ml of10 mg ml-1 trypsin (sequencing grade modified Promega) in10 mM ammonium bicarbonate Digestion (37infinC for 4 h) wasstopped by the addition of 5 ml of 5 formic acid Extractionof peptides into this solution was encouraged by a sonicwater bath treatment for 20 min Peptide extract (05 ml) wasspotted on to a thin layer of a-cyano-4-hydroxycinnamic acidapplied to a MALDI-TOF sample template and analysed byMALDI-TOF mass spectrometry (Bruker Reflex III) using anaccelerating voltage of 25 kV Samples were externally cali-brated using a standard mixture of six peptides ranging in
mass from 10465423 Da to 34946500 Da and the massaccuracy obtained was 60 ppm or better Identification ofproteins from MALDI-TOF peptide mass fingerprint data wasperformed using the lsquoMascotrsquo search engine at httpwwwmatrixsciencecom and was based on a positive resultusing their lsquoProbability Based MOWSE Scorersquo algorithm AMOWSE score of 60 or higher is significant at the 5 levelor better and proteins in this work typically gave scores gt80(frequently considerably so) In addition no identification wasaccepted unless at least five peptides representing at least20 of the protein sequence were detected in the MALDI-TOF peptide mass fingerprint
Assignment of proteins to metabolic pathways
Proteins were assigned to primary metabolic pathwaysusing the lsquoProtein Classification Schemersquo web page ofthe annotated Streptomyces coelicolor genome sequenceat httpwwwsangeracukProjectsS_coelicolor as it ap-peared in May 2002 The annotation of the genome andtherefore the Protein Classification Scheme is likely to bemodified as more experimental data are obtained about thefunction of genes that have currently been assigned on thebasis of sequence information alone It is therefore possiblethat proteins assigned to metabolic pathways in this studymay in future be shown to perform different functions in thecell
Assignments to gene clusters encoding secondary metab-olites are based on an analysis of the genome sequencereported by Bentley et al (2002) In some cases theseassignments are based on extensive experimental data (actred reviewed by Chater and Bibb 1997 Champness 2000cda Chong et al 1998 Huang et al 2001 whiE Davis andChater 1990)
Acknowledgements
This work was funded by grants (2FGT11406 208FGT11408 and 208IGF12432) under the BBSRCrsquos Tech-nologies for Functional Genomics and Investigating GeneFunction initiatives and by a competitive strategic grant fromthe BBSRC to the John Innes Centre We thank Mike Naldrettfor his extensive contribution in establishing technology forproteome analysis at the JIC
Supplementary material
The following material is available from httpwwwblackwell-sciencecomproductsjournalssuppmatmolemole3219mmi3219smhtmSection 1 Detection of gene products assigned to individualmetabolic steps in primary metabolic pathways in ScoelicolorSection 2 Identification of proteins from individual amino acidbiosynthetic pathwaysSection 3 Detection of gene products assigned to individualmetabolic steps in secondary metabolic pathways in Scoelicolor
Proteomic analysis of S coelicolor 931
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
References
Anderson TB Brian P and Champness WC (2001)Genetic and transcriptional analysis of absA an antibioticgene cluster linked two-component system that regulatesmultiple antibiotics in Streptomyces coelicolor Mol Micro-biol 39 553ndash566
Bentley SD Chater KF Cerdeno-Tarraga A-M ChallisGL Thomson NR James KD et al (2002) Completegenome sequence of the model actinomycete Streptomy-ces coelicolor A3(2) Nature 417 141ndash147
Berdy J (1984) New ways to obtain antibiotics Chin J Anti-biot 7 272ndash290
Berggren K Chernokalskaya E Steinberg TH KemperC Lopez MF Diwu Z et al (2000) Background-freehigh sensitivity staining of proteins in one- and two-dimensional sodium dodecyl sulfate-polyacrylamide gelsusing a luminescent ruthenium complex Electrophoresis21 2509ndash2521
Blackstock WP and Weir MP (1999) Proteomics quan-titative and physical mapping of cellular proteins TrendsBiotechnol 17 121ndash127
Blattner FR Plunkett G Bloch CA Perna NT BurlandV Riley M et al (1997) The complete genome sequenceof Escherichia coli K-12 Science 277 1453ndash1462
Buttner K Bernhardt J Scharf C Schmid R MaderU Eymann C et al (2001) A comprehensive two-dimensional map of cytosolic proteins of Bacillus subtilisElectrophoresis 22 2908ndash2935
Champness W (2000) Actinomycete development antibioticproduction and phylogeny questions and challenges InProkaryotic Development Brun YV and Skimketts LJ(eds) Washington DC American Society for MicrobiologyPress pp 11ndash31
Chater KF and Bibb MJ (1997) Regulation of bacterialantibiotic production In Biotechnology Vol 7 Products ofSecondary Metabolism Kleinkauf H and von Doumlhren H(eds) Weinheim Germany VCH pp 57ndash105
Chong PP Podmore SM Kieser HM Redenbach MTurgay K Marahiel M et al (1998) Physical identificationof a chromosomal locus encoding biosynthetic genesfor the lipopeptide calcium-dependent antibiotic (CDA) ofStreptomyces coelicolor A3(2) Microbiology 144 193ndash199
de Crecy-Lagard V Servant-Moisson JV Grandvalet Cand Mazodier P (1999) Alteration of the synthesis of theClp ATP-dependent protease affects morphological andphysiological differentiation in Streptomyces Mol Microbiol32 505ndash517
Davis NK and Chater KF (1990) Spore colour inStreptomyces coelicolor A3(2) involves the developmen-tally regulated synthesis of a compound biosyntheticallyrelated to polyketide antibiotics Mol Microbiol 4 1679ndash1691
Fernandez-Moreno FA Martinez E Caballero JLIchinose K Hopwood DA and Malpartida F (1994)DNA sequence and functions of the actVI region of theactinorhodin biosynthetic gene cluster of Streptomycescoelicolor A3(2) J Biol Chem 269 24854ndash24863
Fink D Falke D Wohllheben W and Engels A (1999)Nitrogen metabolism in Streptomyces coelicolor A3(2)
modification of glutamine synthase I by an adenylyltrans-ferase Microbiology 145 2313ndash2322
Godovac-Zimmermann J Soskic V Posnanovic S andBrianza F (1999) Functional proteomics of signal trans-duction by membrane receptors Electrophoresis 20 952ndash961
Gorg A Obermaier C Boguth G Harder A Scheibe BWildgruber R and Weiss W (2000) The current state oftwo-dimensional electrophoresis with immobilized pH gra-dients Electrophoresis 21 1037ndash1053
Gramajo HC Takano E and Bibb MJ (1993) Stationary-phase production of the antibiotic actinorhodin in Strepto-myces coelicolor A3(2) is transcriptionally regulated MolMicrobiol 7 837ndash845
Gross CA Chan C Dombroski A Gruber T Sharp MTupy J and Young B (1998) The functional and regula-tory roles of sigma factors in transcription Cold SpringHarb Symp Quant Biol 63 141ndash155
Hesketh A Sun J and Bibb MJ (2001) Induction ofppGpp synthesis in Streptomyces coelicolor A3(2) grownunder conditions of nutritional sufficiency elicits actII-ORF4transcription and actinorhodin biosynthesis Mol Microbiol39 136ndash144
Hesketh A Fink D Gust B Rexer H-U Scheel BChater K et al (2002) The PII nitrogen regulatory proteinGlnK in Streptomyces coelicolor A3(2) is modified by ade-nylylation rather than uridylylation and the GlnD and GlnKhomologues are functionally dissimilar to their nitrogenregulatory system counterparts Mol Microbiol (inpress)
Hood DW Heidstra R Swodoba UK and Hodgson DA(1992) Molecular genetic analysis of proline and tryp-tophan biosynthesis in Streptomyces coelicolor A3(2)interaction between primary and secondary metabolism- areview Gene 115 5ndash12
Hu DS-J Hood DW Heidstra R and Hodgson DA(1999) The expression of the trpD trpC and trpBA genesof Streptomyces coelicolor A3(2) is regulated by growthrate and growth phase but not by feedback repression MolMicrobiol 32 869ndash880
Huang J Lih CJ Pan KH and Cohen SN (2001)Global analysis of growth phase responsive gene expres-sion and regulation of antibiotic biosynthetic pathways inStreptomyces coelicolor using DNA microarrays GenesDev 15 3183ndash3192
Ichinose K Taguchi T Ebizuka Y and Hopwood DA(1998a) Biosynthetic gene clusters of benzoisochromane-quinone antibiotics in Streptomyces spp Identification ofgenes involved in post-PKS tailoring steps Actinomyceto-logica 12 99ndash109
Ichinose K Bedford DJ Tornus D Bechthold A BibbMJ Revill PW et al (1998b) The granaticin biosyntheticgene cluster of Streptomyces violaceoruber Tu22sequence analysis and expression in a heterologous hostChem Biol 5 647ndash659
Kelemen GH Brian P Flardh K Chamberlin L ChaterK and Buttner M (1998) Developmental regulation oftranscription of whiE a locus specifying the polyketidespore pigment in Streptomyces coelicolor A3(2) J Bacte-riol 180 2515ndash2521
Kieser T Bibb MJ Buttner MJ Chater KF and
932 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
Hopwood DA (2000) Practical Streptomyces GeneticsNorwich John Innes Foundation
Kim EJ Chung HJ Suh B Hah YC and Roe JH(1998) Expression and regulation of the sodF gene encod-ing iron- and zinc-containing superoxide dismutase inStreptomyces coelicolor Muumlller J Bacteriol 180 2014ndash2020
Kimura Y Takaoka M Tanaka S Sassa H Tanaka KPolevoda B et al (2000) Na-acetylation and proteolyticactivity of the yeast 20S proteasome J Biol Chem 2754635ndash4639
Kunst F Ogasawara N Moszer I Albertini AM AlloniG Azevedo V et al (1997) The complete genomesequence of the Gram-positive bacterium Bacillus subtilisNature 390 249ndash256
Lucchini S Thompson A and Hinton JCD (2001)Microarrays for microbiologists Microbiology 147 1403ndash1414
Mann M Hendrickson RC and Pandey A (2001) Analy-sis of proteins and proteomes by mass spectrometry AnnuRev Biochem 70 437ndash473
Miyodah S (1993) Research on antibiotic screening inJapan over the last decade a producing microorganismsapproach Actinomycetologica 7 100ndash106
Molloy MP Herbert BR Williams KL and Gooley AA(1999) Extraction of Escherichia coli proteins with organicsolvents prior to two-dimensional electrophoresis Electro-phoresis 20 701ndash704
Nagy I Tamura T Vanderleyden J Baumeister W andDe Mot R (1998) The 20S proteasome of Streptomycescoelicolor J Bacteriol 180 5448ndash5453
Neuhoff V Arold N Taube D and Ehrhardt W (1988)Improved staining of proteins in polyacrylamide gels includ-ing isoelectric focusing gels with clear background at nan-ogram sensitivity using Coomassie Brilliant Blue G-250and R-250 Electrophoresis 9 255ndash262
Nilsson CL and Davidsson P (2000) New separation toolsfor comprehensive studies of protein expression by massspectrometry Mass Spectrom Rev 19 390ndash397
Puglia AM Vohradsky J and Thompson CJ (1995)
Developmental control of the heat-shock stress regulon inStreptomyces coelicolor Mol Microbiol 17 737ndash746
Rabilloud T (1992) A comparison between low backgroundsilver diamine and silver nitrate protein stains Electro-phoresis 13 429ndash439
Robinson VL Buckler DR and Stock AM (2000) A taleof two components a novel kinase and a regulatory switchNature Struct Biol 7 626ndash633
Schena M (2000) Microarray Biochip Technology NatickMA Eaton Publishing
Spusansky O Rezuchova B Homerova D andKormanec J (2001) Expression of the gap gene encodingglyceraldehyde-3-phosphate dehydrogenase of Strepto-myces aureofaciens requires GapR a member of theAraCXylS family of transcriptional activators Microbiology147 1291ndash1301
Taguchi T Itou K Ebizuka Y Malpartida F HopwoodDA Surti CM et al (2000) Chemical characterisationof disruptants of the Streptomyces coelicolor A3(2) actVIgenes involved in actinorhodin biosynthesis J Antibiot 53144ndash152
Takano E Gramajo HC Strauch E Andres N WhiteJ and Bibb MJ (1992) Transcriptional regulation of theredD transcriptional activator gene accounts for growth-phase-dependent production of the antibotic undecylpro-digiosin in Streptomyces coelicolor A3(2) Mol Microbiol6 2797ndash2804
Tonella L Hoogland C Binz P-A Appel RDHochstrasser DF and Sanchez J-C (2001) New per-spectives in the Escherichia coli proteome investigationProteomics 1 409ndash423
Vohradsky J Li XM and Thompson CJ (1997) Identifi-cation of prokaryotic developmental stages by statisticalanalysis of two-dimensional gel patterns Electrophoresis18 1418ndash1428
Vohradsky J Li XM Dale G Folcher M Nguyen LViollier PH and Thompson CJ (2000) Developmentalcontrol of stress stimulons in Streptomyces coelicolorrevealed by statistical analyses of global gene expressionpatterns J Bacteriol 182 4979ndash4986

922
A R Hesketh
et al
copy 2002 Blackwell Publishing Ltd
Molecular Microbiology
46
917ndash932
the isoleucine residue at its N-terminus (expected massloss 11308 Da) This indicates removal of the N-terminalMKI residues from ribosomal protein L9 The theoreticalisoelectric point (997) and molecular weight (156 kDa) ofL9 protein N-terminally truncated in this way are in goodagreement with the position of the lower spot in Fig 1(predicted values for the unprocessed protein are pH 960and 159 kDa) MALDI-TOF data indicating proteolytic
processing of the superoxide dismutase precursor protein(SCO5254 spot 32 in Fig 1) and the 20S proteasomebeta subunit precursor (SCO1644 spot 35 in Fig 1)agreed with previous reports (Kim
et al
1998 Nagy
et al
1998) Evidence for the proteolysis of
S coelicolor
ClpP1(SCO2619 spot 13 in Fig 1) has also been reportedalthough the processing site was not defined (de Crecy-Lagard
et al
1999)
SCO4661 Elongation factor GSCO4662 Elongation factor Tu1SCO4677 Putative regulatory proteinSCO4729 DNA-directed RNA polymerase alpha chainSCO4762 Chaperonin GroEL1SCO4770 Inosine-5
cent
-monophosphate dehydrogenase 25SCO4792 LuxR family two-component regulatorSCO4808 Succinyl-CoA synthetase beta chain 8SCO4809 Succinyl-CoA synthetase alpha chain 22SCO4921 Probable acyl-CoA carboxylase complex A chainSCO5071 Hydroxylacyl-CoA dehydrogenase ActVI-ORFA 16SCO5079 Hypothetical protein ActVA-ORF4 20SCO5080 Possible hydrolase ActVA-ORF5 26SCO5115 Probable ABC transporter intracellular ATPase chain BldK-ORFD 17SCO5371 ATP synthase alpha chainSCO5396 Putative cellulose-binding protein 10SCO5419 Putative thioredoxinSCO5515 Probable
D
-3-phosphoglycerate dehydrogenase 4SCO5535 Putative carboxyl transferaseSCO5584 Nitrogen regulatory protein GlnK 15SCO5625 Elongation factor TsSCO5699 Prolyl-tRNA synthetaseSCO5737 Guanosine pentaphosphate synthetaseSCO5777 Glutamate uptake system ATP-binding proteinSCO5803 SOS regulatory protein LexASCO5999 Aconitase 1SCO6198 Probable secreted proteinSCO6282 3-Oxoacyl-[acyl carrier protein] reductase 11SCO6636 Bacteriophage
f
-C31 resistance protein PglZSCO6663 Transketolase 27SCO7000 Isocitrate dehydrogenase 2SCO7036 Argininosuccinate synthase 5SCO7510 Peptidyl-prolyl
cis-trans
isomerase
ORF no Gene product Spot no in Fig 1
Some proteins appear to be subject to both types of modification and are present in both parts Only proteins from primary or secondary metabolicpathways or with identified modifications are assigned to spots in Fig 1
Table 3
cont
Table 4
Co- and post-translational modification of proteins suggested by MALDI-TOF peptide mass fingerprint analysis
Protein Modification
SCO1643 20S proteasome alpha subunit Removal of initiator Met residue and acetylation of N-terminal SerSCO5115 ABC transporter intracellular Removal of initiator Met residue and acetylation of N-terminal Thr
ATPase BldK-ORFDSCO5396 Putative cellulose-binding protein Removal of initiator Met residue and acetylation of N-terminal SerSCO2198 Glutamine synthetase I Adenylylation of peptide 395ndash420SCO5584 Nitrogen regulatory protein GlnK Adenylylation of peptide 48ndash58SCO1644 20S proteasome
b
-subunit precursor Proteolytic processing between amino acid residues 5354SCO2619 ATP-dependent Clp protease Proteolytic processing between amino acid residues 2223
proteolytic subunit ISCO3909 50S ribosomal protein L9 Proteolytic processing between amino acid residues 34SCO5074 Putative dehydratase ActVI-ORF3 Proteolytic processing between amino acid residues 3132 and 3435SCO5254 Superoxide dismutase precursor Proteolytic processing between amino acid residues 1415
Proteomic analysis of
S coelicolor 923
copy 2002 Blackwell Publishing Ltd
Molecular Microbiology
46
917ndash932
Co-translational modification by removal of the N-terminal initiator Met occurred in 149 out of 190 (78)cases in which the N-terminal peptide was detected Pro-cessing by the N-methionylaminopeptidase was observedif the second amino acid was Ser (45 out of 149 exam-ples) Thr (40) Ala (32) Pro (27) Gly (4) or Val (1) Ononly one occasion was any of these six amino acids foundas the second residue if the N-terminal methionine wasretained This is in broad agreement with observations in
B subtilis
in which two-thirds of proteins were reported tobe similarly processed (Buttner
et al
2001)
Representation of primary and secondary metabolic pathways
Streptomycetes produce
ordf
60 of all commercially usefulantibiotics (Berdy 1984 Miyodah 1993) Most antibioticsare the product of complex biosynthetic pathways oftenencoded by 20ndash30 clustered genes Production of thepigmented antibiotics actinorhodin (Act) and undecyl-
prodigiosin (Red) has been studied extensively in
Scoelicolor
(for reviews see Chater and Bibb 1997Champness 2000) and involves clusters of approximately22 genes each A third cluster producing the calcium-dependent antibiotic (CDA) and consisting of 40 geneshas also been studied (Chong
et al
1998 Huang
et al
2001) In the extensive spot identification studies for thepreparation of a proteome map of
S coelicolor
(seeabove) more than one-third of the proteins from each ofthese pathways was identified (Table 5) Proteins fromthree clusters that specify metabolic products that haveyet to be identified experimentally (Bentley
et al
2002)were also extensively represented a type I polyketidesynthase cluster (SCO6273ndash6288) a deoxysugarglycosyltransferase cluster (SCO0381ndash0401) and anon-ribosomal peptide synthetase cluster predicted toencode a novel siderophore coelichelin (SCO0489ndash0499) Several clusters were unrepresented
The primary metabolic pathways were somewhat morehighly represented with
ordf
60 or more of proteins anno-
Fig 2
Examples of MALDI-TOF peptide mass fingerprint data identifying post-translational modification of proteins by (A) acetylation (predicted mass increase of 420 Da) (B) adenylylation (predicted mass increase of 3290 Da) and (C) proteolytic processing (predicted mass loss for Ile of 11308 Da) In each fingerprint the
x
-axis is masscharge ratio (mz) and the
y
-axis is absolute intensity The observed mass accuracies in the MALDI-TOF fingerprints are (A) 60 ppm (B) 20 ppm and (C) 20 ppm
924
A R Hesketh
et al
copy 2002 Blackwell Publishing Ltd
Molecular Microbiology
46
917ndash932
tated as enzymes of glycolysis the TCA cycle thepentose phosphate pathway purine ribonucleotide bio-synthesis and pyrimidine nucleotide biosynthesis beingmapped (Table 6) About 50 of amino acid biosyntheticproteins were identified Lists of individual members of allpathways considered here including those that were andwere not identified during proteome mapping can befound in the
Supplementary material
Several proteins from both primary and secondary met-
abolic pathways were identified at more than one positionon the proteome map and may therefore be subject toregulation by post-translational modification (see Table 3and Fig 1) Primary metabolic proteins of this type (yellowspots on Fig 1) were usually detected as chains of two ormore spots with the same apparent molecular weights butdifferent pI values suggesting covalent modification bylow-molecular-weight adducts Apart from adenylylation ofGSI (see above) the nature of the modification of theseproteins was not revealed by MALDI-TOF peptide massfingerprint data Both covalent modification and proteolyticprocessing were also found in secondary metabolic pro-teins (red spots in Fig 1) For example the Act pathwaymembers SCO5071 (spot 16) and SCO5075 (spot 20)
and the putative 3-oxoacyl[acyl carrier protein] reductasefrom the type I polyketide cluster (SCO6282 spot 11)appear to be covalently modified whereas proteolytic pro-cessing was detected for RedL (SCO5892 spot 18) fromthe undecylprodigiosin cluster the putative dehydrataseActVI-ORF3 (SCO5074 spot 31) from the Act cluster theputative peptide monooxygenase from coelichelin biosyn-thesis (SCO0498 spot 34) and the putative oxygenasefrom the CDA pathway (SCO3236 spot 33) Two Act bio-synthetic proteins SCO5088 (spot 24) and SCO5073(spot 29) were detected both at their predicted pI andmolecular weight co-ordinates and at positions with simi-lar pI values but significantly higher molecular weightsThis may indicate that these proteins form stable mul-timers that are not completely denatured under theconditions used Only the modification of ActVI-ORF3(SCO5074 spot 31) could be characterized further fromthe mass spectrometry data which showed that it involvesproteolytic processing at two sites between amino acidresidues 3132 and 3435 (data not shown) Analysis of asample obtained by externally extracting proteins fromintact washed mycelium with a solution containing 2SDS and 50 mM dithiothreitol (DTT) revealed that sur-
Table 5
Summary of proteins identified on the proteome map from secondary metabolic pathways during growth in SMM
Biosynthetic pathwaygene cluster
Pathwaygenesannotated
Pathwaygene productsidentified
Percentagepathwaydetected
Calcium-dependent antibiotic CDA SCO3210ndash3249 40 16 40Actinorhodin SCO5071ndash5092 22 15 68Undecylprodigiosin SCO5877ndash5898 22 8 36Deoxysugar synthaseglycosyltransferase cluster SCO0381ndash0401 21 7 33Type I polyketide synthase SCO6273ndash6288 16 6 38Hopanoids SCO6759ndash6771 13 1 7Siderophore (coelichelin) SCO0489ndash0499 11 5 45Siderophore (desferrioxamine) SCO2783ndash2785 4 4 100
Chromosomal locations and extent of gene clusters are taken from Bentley
et al
(2002) No proteins were detected from the remaining predictedsecondary metabolic clusters ie siderophore (coelibactin) SCO7681ndash7691 (11 ORFs) siderophore SCO5799ndash5801 (three ORFs) non-ribosomal peptide synthases SCO6429ndash6438 (10 ORFs) type II fatty acid synthases SCO1265ndash1273 (nine ORFs) WhiE spore pigment clusterSCO5314ndash5320 (seven ORFs) isorenieratine SCO0185ndash0191 (seven ORFs) eicosapentaenoic acid SCO0124ndash0129 (six ORFs) chalconesynthases SCO1206ndash1208 (three ORFs) chalcone synthases SCO7669ndash7671 and SCO7222 (three ORFs) sesquiterpene cyclase SCO5222ndash5223 (two ORFs) type I polyketide synthases SCO6826ndash6827 (two ORFs) geosmin SCO6703 (one ORF)
Table 6 Summary of proteins identified on the proteome map from primary metabolic pathways during growth in SMM
Biosynthetic pathwayPathway genesannotateda
Pathway geneproducts identified
Percentage pathway detected
Glycolysisb 24 13 55Pentose phosphate pathwayb 14 10 71TCA cycleb 21 12 57Amino acid biosynthesisc 115 55 48Purine ribonucleotide biosynthesisb 18 12 67Pyrimidine ribonucleotide biosynthesisb 8 5 62
a Derived from interpretation of the annotated S coelicolor genome sequence at httpwwwsangeracukProjectsS_coelicolorb See Supplementary material for details of pathway membersc See Supplementary material for details of individual pathways
Proteomic analysis of S coelicolor 925
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
prisingly the truncated ActVI-ORF3 is located extracellu-larly presumably non-covalently associated with the cellwall matrix (Fig 4)
Expression analysis of certain primary and secondary metabolic proteins
Production of the pigmented antibiotics Act and Red inliquid minimal medium supplemented with casamino acids(SMM) begins in the transition phase between exponentialgrowth and stationary phase Figure 3 shows a compari-son of the expression of certain primary and secondarymetabolic proteins between exponentially growing andstationary phase cultures Two proteins from the Actcluster (ActVA-ORF5 and ActI-ORF2) and two from theRed (RedI and RedR) were present in the stationaryphase cultures but were not detected during exponentialgrowth Similarly the putative 3-oxoacyl-[acyl carrierprotein] reductase (SCO6282) and epoxide hydrolase(SCO6277) enzymes from a type I polyketide secondarymetabolic cluster showed a large increase in abundance
between the exponential and stationary phase samplesTriose phosphate isomerase (SCO1945) fructose bis-phosphate aldolase (SCO3649) and succinyl-CoA syn-thetase alpha chain (SCO4809) all enzymes from centralcarbon metabolism were present in approximatelyconstant amounts in both samples Interestingly two iso-forms (SCO7511 and SCO1947) of glyceraldehyde-3-phosphate dehydrogenase from the glycolytic pathwayshowed different patterns of expression SCO1947 levelswere the same in both exponential and stationary phasebut SCO7511 barely detectable in exponentially growingcultures increased markedly in stationary phase
The Streptomyces coelicolor online proteome map
The 2D gel images in Fig 1 together with the spot iden-tification results are being made available over the WorldWide Web to provide a reference map of the S coelicolorproteome At the time of writing the reference gel forthe pH 4ndash7 isoelectric point range was fully accessiblewhereas those for pH 45ndash55 pH 55ndash67 and pH 6ndash11
Fig 3 A comparison of the expression of certain primary and secondary metabolic proteins between exponentially growing and stationary phase cultures of S coelicolor M145 Larger arrows represent higher protein level in one gel than in the other The experiment was performed on duplicate biological samples and both showed similar protein expression profiles
926 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
were under construction (httpqbababeracukS_coeliReferenceGelrefgelhtml) A tool for comparing two ormore gel images with each other is also accessible fromthe same website at httpqbababeracukS_coeliflickerhtml
Discussion
Silver staining is one of the most sensitive methods fordetecting proteins on polyacrylamide gels allowing aslittle as 05ndash2 ng of an individual protein to be detected(Berggren et al 2000 Gorg et al 2000) The proportionof the S coelicolor proteome that can be analysed usingsilver staining of 2D-PAGE-separated proteins dependsat least in part on the pH range of the IPG strip used forthe first-dimension separation (see Table 1) Using theintermediate-range IPG strip covering pH 4ndash7 about 20ndash25 of the proteins in the theoretical proteome predictedto be observable in the resulting 2D-PAGE gel were actu-ally detected This figure was reduced to 10ndash12 whenanalysing the more basic pI range using the pH 6ndash11 IPGstrip Attempts to increase the percentage of the proteomedetectable using the pH 4ndash7 and pH 6ndash11 IPG strips byloading more protein sample resulted in poorer separa-tions (data not shown) This was especially true of thepH 6ndash11 strips which were generally the most difficult touse successfully However good separation of higher pro-tein loads was achieved using the narrow-range IPG stripspH 45ndash55 and 55ndash67 significantly increasing the pro-portion of theoretical proteins detected to over 40Clearly and more pertinently the limited conditions usedfor culturing S coelicolor in this study will also haverestricted the number of proteins detected as not all theproteome will be expressed In particular S coelicolordoes not sporulate in shaken liquid-grown cultures soproteins involved in this differentiation process are unlikelyto be present Nevertheless it is striking that the com-bined data for the pH 45ndash55 and 55ndash67 isoelectric pointranges indicate that 2468 out of a total of predicted 4932protein spots can be detected (see Table 1) suggestingthat transition phase mycelium in this supplementedminimal medium is expressing 50 of its genes (ie byextrapolation some 3912 of the 7825 in the genome)
Analysis of post-translational regulation
Although the regulation of cellular processes and adaptiveresponses in bacteria has been studied extensively at thetranscriptional level post-translational regulatory mecha-nisms are much less understood Proteomic analysisusing 2D-PAGE coupled with mass spectrometry offersthe opportunity to observe and identify modification ofproteins at a general cellular level The functions of pro-teins can be altered by specific covalent attachment of
adducts (eg phosphorylation glycosylation nucleotidy-lation acetylation) or by proteolytic processing Bothevents alter the pI andor mass of the protein and so canusually be observed on 2D-PAGE In the S coelicolorproteome map ordf110 out of a total of 770 gene productswere identified in more than one position or at pI andapparent mass co-ordinates significantly different frompredicted values indicating extensive regulation at thepost-translational level (88 are listed in Table 3) a propor-tion similar to that reported for other prokaryotes (Buttneret al 2001 Tonella et al 2001) The initial peptide massfingerprint analysis identified the modification of only 10of these proteins probably partly because fingerprint datatypically cover ordf30ndash60 of a proteinrsquos sequence (data notshown) so modification at positions not represented willbe missed Protein digestion with endopeptidases otherthan trypsin before mass spectrometry may help in thissituation but some covalently attached groups are labileand cannot be detected routinely Phosphorylation of Hisoccurs on nitrogen producing a high-energy phosphora-midate bond that is easily hydrolysed in acidic conditionswhereas phospho-Asp is the most labile phosphorylatedresidue with a half-life of a few hours under physiologicalconditions (reviewed by Robinson et al 2000) Thesemodifications the basis of bacterial two-component regu-latory systems are therefore unlikely to survive stainingof the 2D gels which involved the use of a fixing solutioncontaining 10 acetic acid and mass spectrometry Iden-tification of the more stable phosphoester phosphorylationthat occurs on Ser Thr or Tyr residues using PAGEfollowed by mass spectrometry has been reported(Godovac-Zimmermann et al 1999 Buttner et al 2001)but no examples were detected in this work Of the mod-ifications identified in this study three were by acetylationat the N-terminus after removal of the initiator Met residuetwo involved adenylylation and five proteins had been N-terminally truncated Modification of the 20S proteasomeby N-acetylation in yeast involves removal of the Metresidue by N-methionylaminopeptidase followed byacetylation with an Na-acetyltransferase (Kimura et al2000) Homologues of these enzymes are predicted fromthe S coelicolor genome and are presumably responsiblefor the observed modification of the 20S proteasomealpha subunit and of BldK-ORFD and the putative cellu-lose-binding protein The functional significance of Na-acetylation of these proteins is unclear
Analysis of the biosynthesis of antibiotics and other secondary metabolites
Post-translational modification The proteome mappingresults indicate that at least six enzymes in the Act clusterare subject to post-translational modifications and one
Proteomic analysis of S coelicolor 927
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
each from the undecylprodigiosin (SCO5877ndash5898)lsquocoelichelinrsquo (SCO0489ndash0499) and type I polyketidesynthase (SCO6273ndash6288) clusters (see Table 3) Thesemetabolic pathways may therefore be significantly regu-lated at the post-translational level The actVI-ORF3 gene(SCO5074) encodes a putative dehydratase enzyme thatis part of the biosynthetic pathway for Act a blue memberof the benzoisochromanequinone (BIQ) family of pig-mented antibiotics Mutation in actVI-ORF3 causes a 50decrease in the accumulation of Act (and a reddish brownphenotype) and it is believed to be responsible for pyranring closure an intermediate step in Act biosynthesis (thatmay also occur spontaneously) leading to the formationof the BIQ chromophore (Fernandez-Moreno et al 1994Ichinose et al 1998a Taguchi et al 2000) In this studyActVI-ORF3 protein was detected in two forms a majorspot corresponding to ActVI-ORF3 truncated by removalof the N-terminal 31 amino acid residues and a minor onein which the first 34 amino acids were missing (spots 31in Fig 1) The full-length protein was not detected Anal-ysis of the protein sequence using the SIGNALP server athttpwwwcbsdtudkservicesSignalP predicted a signalpeptide cleavage site between amino acids 31 and 32 inagreement with the observed results
The protein extract analysed was prepared from myce-lium that had been collected by centrifugation and sub-jected to two washes indicating that if ActVI-ORF3 isexported it is effectively retained within the cell wallmatrix This was confirmed by analysis of a preparationobtained by externally extracting proteins from intactwashed mycelium (Fig 4) Export of an enzyme requiredfor the efficient biosynthesis of an antibiotic that is thoughtto be produced intracellularly appears to be anomalousparticularly as it is involved in an intermediate biosynthetic
event Database analysis of the other members of the Actcluster revealed only one other protein with a predictedsignal peptide sequence the putative integral membranetransport-related protein ActII-ORF3 and it seemsunlikely that all the Act biosynthetic steps following BIQchromophore formation are extracellular It is possible thatfull-length ActVI-ORF3 catalyses the intermediate biosyn-thetic event intracellularly and the exported processedprotein is responsible for a different extracellular activityperhaps later in the pathway However no full-length pro-tein was detected so the exported enzyme may performthe pyran ring closure on a secreted metabolite that isthen reabsorbed by the cell for completion of the sub-sequent biosynthetic steps Only two homologues ofactVI-ORF3 are known and both are also in BIQ antibioticbiosynthetic clusters gra-ORF18 from the granaticincluster (Ichinose et al 1998b) and med-ORF5 frommedermycin biosynthesis (K Ichinose personal com-munication) Interestingly the encoded proteins of bothsequences are also predicted to contain N-terminal signalpeptides The observation that ActVI-ORF3 is exported tothe cell wall matrix may therefore be of general signifi-cance to the BIQ family of antibiotics and provide a newinsight into their biosynthetic pathways
Pathway representation Of the 20 putative secondarymetabolite gene clusters identified in the S coelicolorgenome sequence (Bentley et al 2002) eight had atleast one protein each identified on the proteome map(see Table 5) The biosynthetic clusters for the antibioticsAct Red and CDA had 68 36 and 40 of their pro-teins represented There are several possible explana-tions for failure to detect all the proteins for each pathwaySome are putative integral membrane proteins so cannotreadily be detected using conventional 2D-PAGE SomeRed and CDA proteins have molecular weights gt200 kDaoutside the range of the 125 gels used (for clustermembers see Supplementary material) The pathway-specific regulatory proteins for Act and Red biosynthesiswere not detected despite a detailed search of their pre-dicted gel locations even though the relevant genes areknown to be transcribed in equivalent cultures of the samestrain (Hesketh et al 2001) The proteins are presumablypresent but at levels below the limit of detection Howeverthe transcriptional regulator for CDA production CdaRwas identified as well as the two-component systemresponse regulator AbsA2 which is also encoded by agene in the CDA cluster (Anderson et al 2001) Interest-ingly proteins from three putative secondary meta-bolite clusters producing chemicals of as yet unknownstructure were also identified on the map correspondingto the deoxysugar synthaseglycosyltransferase clusterSCO0381ndash0401 the lsquocoelichelinrsquo cluster for a predicted(but not yet confirmed) oligopeptide siderophore
Fig 4 Processed ActVI-ORF3 is present in the cell wall matrix 2D-gel separations of protein extracts prepared (A) from whole cells or (B) by externally washing cells in a 2 SDS extraction buffer The arrow indicates ActVI-ORF3 protein The experiment was performed on duplicate biological samples and both showed similar protein expression profiles
928 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
(SCO0489ndash0499) and the type I polyketide clusterSCO6273ndash6288 The 3-oxoacyl-[acyl carrier protein]reductase (SCO6282) from the latter pathway is ex-pressed in a growth phase-dependent manner (Fig 3)and in early stationary phase cultures is perhaps themost abundant protein spot in the entire detectable pro-teome Presumably the metabolites produced by thesethree pathways are being synthesized under the condi-tions used In addition to coelichelin three other sidero-phores are postulated for S coelicolor All the proteinsfrom one of these desferrioxamine (SCO2782ndash2785)were identified but none was seen from either of the othertwo pathways The WhiE polyketide spore pigment isassociated with sporulation of surface-grown cultures(Davis and Chater 1990) and the expression of the whiEgenes depends on a series of sporulation-specific regula-tory genes (Kelemen et al 1998) no corresponding pro-teins of which were detected here It is therefore notsurprising that WhiE proteins were not detected in sam-ples prepared from liquid-grown vegetative mycelium
Analysis of primary metabolism
Proteins annotated as functioning in primary metabolicpathways were highly represented on the proteome mapwith ordf60 or more of the enzymes assigned to glycolysisthe TCA cycle the pentose phosphate pathway andpurine and pyrimidine ribonucleotide biosynthesis beingidentified (see Table 6) This figure may be deceptively lowbecause these pathways in S coelicolor often appear tohave more than one enzyme isoform for some of themetabolic steps potentially offering additional levels ofcontrol or developmental responsiveness at these pointsFor example only two steps in glycolysis have just a singlegene product assigned to them the remainder typicallyhave two or three each (Fig 5) At least one isoform waspresent for each step so the pathway is in theory 100functionally represented Understanding the regulation ofthese pathways will require analysis of the expression ofthese isoenzymes under different nutritional conditionsor different developmental stages Figure 5 shows that ofthe three predicted glyceraldehyde-3-phosphate dehydro-genase isoforms from glycolysis only two were detectedin this study (see also Supplementary material) Of theseSCO1947 appears to be constitutive whereas SCO7511is growth phase dependent (see Fig 3) Thus even thelimited conditions in this study yielded useful informationabout isoenzyme expression patterns (see Supplemen-tary material) In Streptomyces aureofaciens two genesencoding glyceraldehyde-3-phosphate dehydrogenaseactivity also appear to be differently regulated transcrip-tion of one highly homologous to SCO7511 is activatedby the regulator GapR whereas the other is GapR inde-pendent (Spusansky et al 2001)
Hood et al (1992) reported that most amino acid bio-synthetic enzymes are expressed at very low constitutivelevels in S coelicolor so it is reassuring to note that ordf50could be detected and identified using proteomics Theseinclude TrpB and TrpC1 from the tryptophan biosyntheticpathway the corresponding transcripts of which couldbarely be detected in previously reported S1 nucleaseprotection experiments (Hu et al 1999)
Perspectives
No attempt was made in the work reported here to simplifythe protein separations by subdividing the sample intosoluble and membrane fractions and this is reflected inthe 10 lipoproteins and 16 ABC transport system ATP-binding subunits identified on the proteome map (seeTable 2) No integral membrane proteins were detectedand even when a membrane protein sample was pre-pared and analysed no proteins identified possessedmore than three predicted transmembrane helices (datanot shown) This is consistent with previous studiesin other bacteria (Molloy et al 1999 Nilsson andDavidsson 2000) Another group of proteins also strik-ingly absent from the list of those identified is the sigmafactors which determine promoter selection by RNA poly-merase There are an extraordinarily high number (65) ofsigma factors encoded in the S coelicolor genome aswell as at least seven antisigma factors and 15 antisigmafactor antagonists (some of each of which were detected)Sigma factor heterogeneity in bacteria is an important wayof regulating expression of specific sets of genes under
Fig 5 The glycolytic pathway in S coelicolor illustrating the assignment of multiple gene products to individual metabolic steps based on the annotation of the genome sequence at httpwwwsangeracukProjectsS_coelicolor Gene products identified in the proteome map are underlined
Proteomic analysis of S coelicolor 929
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
different conditions and in different cell types (for a reviewsee Gross et al 1998) Although no sigma factors wereidentified about half (31 out of 65) have predicted pIvalues gtpH 7 and so can only be detected using thepH 6ndash11 IPG strips that give the poorest results (seeTable 1) However the principal vegetative sigma factorHrdB which is expected to be moderately abundanthas a predicted pI of 613 and its absence from thepH 55ndash67 proteome map may indicate that the samplepreparation method which does not remove DNA is inap-propriate Among other regulatory proteins 11 out of apossible 80 two-component system response regulatorsplus a further 36 proteins annotated as lsquoregulatorsrsquo weredetected Only one SerThr kinase and no two-componentsystem sensor kinases were detected possibly reflectingthe difficulty in analysing proteins with several transmem-brane domains Interestingly 30 secreted proteins wereidentified on the proteome map at least 10 of whichappear from comparison of their observed pI and molec-ular weight positions with predicted values to have beenprocessed already and therefore exported Presumablythese proteins do not end up exclusively in the culturemedium
Clearly this approach to proteomic analysis of S coeli-color has some important deficiencies not least theabsence of certain groups of proteins and the relativelypoor initial success rate for specifically characterizingpost-translational modifications Nevertheless it offersunique insights into the molecular biology of the organismthe expression of many proteins from both primary andsecondary metabolic pathways can be analysed post-translational regulation of proteins can be investigated ona general cellular level and the physical locations forsome proteins can be determined Approximately 35 ofthe S coelicolor genome is annotated as encoding lsquohypo-theticalrsquo proteins of unknown function and 182 proteins ofthis type were identified on the proteome map No exam-ples were found of overt disagreements between pre-dicted and observed proteins providing experimentalsupport for the general accuracy of the S coelicolorgenome sequence and its annotation Information aboutthe expression post-translational modification and physi-cal location of these (no longer) hypothetical proteinsfrom proteomic analyses is going to be important forassigning future functions As many of the hypotheticalproteins have apparent orthologues in pathogenic myco-bacteria some of the information may have an immediateapplication in the context of pathogenesis
Experimental procedures
Materials
Immobilized pH gradient (IPG) strips and IPG bufferswere purchased from Amersham Biosciences Acrylamide
duracryl and chemicals for silver staining were obtained fromGenomic Solutions All other chemicals were from Sigmaunless stated in the text
Strain and growth conditions
Streptomyces coelicolor M145 was cultivated with vigorousagitation at 30infinC in minimal medium supplemented with 02casamino acids (SMM) as described previously (Kieser et al2000) Briefly a high-density spore preparation [about 1010
colony-forming units (cfu) ml-1] was pregerminated in 2yen YTmedia for 7 h at 30infinC Germlings were harvested by centrifu-gation (5 min at 4000 g) resuspended in SMM and sonicatedbriefly to disperse any aggregates before inoculation into50 ml of SMM in 250 ml siliconized flasks containing coiledstainless steel springs Each flask received the equivalent of5 yen 107 spore cfu
Protein extraction and 2D-gel electrophoresis
The preparation of the protein sample to be separated waskept as simple as possible disrupting harvested myceliadirectly into denaturing isoelectric focusing (IEF) buffer con-taining Pefabloc SC (Roche) protease inhibitor This methodminimized unwanted protein degradation that was observedin initial studies using a Tris buffer containing Pefabloc SC forcell lysis (data not shown) Removal of DNA RNA salts andpolysaccharides from the sample was therefore not per-formed (other than the shearing of viscous DNA and RNA bysonication) and the protein extracts were sometimes difficultto focus when the sample was loaded by in-gel rehydrationLoading the sample soaked in paper strips at the anodicend of IPG strips that had already been rehydrated improvedthe reproducibility of separations significantly
Mycelium for protein extraction was harvested from cul-tures by brief centrifugation (30 s at 4000 g) at room tem-perature and immediately frozen in liquid nitrogen Typicallymycelium from 10 ml culture aliquots was collected and thetransfer time from culture flask to frozen sample was 15 minFrozen cells were thawed on ice in 5 ml of washing buffer(40 mM Tris pH 90 1 mM EGTA 1 mM EDTA) then pelletedby centrifugation (5 min at 3000 g) at 4infinC Washed cells wereresuspended in 400 ml of denaturing IEF buffer UTCHAPS[7 M urea 2 M thiourea 4 (wv) CHAPS 40 mM TrispH 90 1 mM EDTA 50 mM DTT 4 mM Pefabloc SC pro-tease inhibitor (Roche)] and disrupted by sonication (SanyoSoniprep 150 10 yen 2 s bursts at amplitude 75 mm) whilecooling in an ethanolndashice bath Cell debris was removed bycentrifugation (15 min 10 000 g 4infinC) and the protein extractwas stored frozen in aliquots at -80infinC until use For thepreparation of extracellular protein extracts washed cellswere resuspended in extraction buffer consisting of 40 mMTris pH 90 1 mM EDTA 50 mM DTT 2 SDS and 4 mMPefabloc SC protease inhibitor Cells were vortexed for 2 minbefore being pelleted by centrifugation (5 min 10 000 g4infinC) The supernatant was precipitated using the Amersham2D Clean-Up kit according to the manufacturerrsquos instructionsand the precipitated proteins were redissolved in UTCHAPSand stored at -80infinC until use
930 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
For first-dimension isoelectric focusing 18 cm IPG stripspH 4ndash7 pH 6ndash11 pH 45ndash55 or pH 55ndash67 (Amersham Bio-sciences) were rehydrated overnight in IEF buffer containing1 ampholytes according to the manufacturerrsquos instructionsusing a Phaser isoelectric focusing unit (Genomic Solutions)set at 20 V Protein samples to be separated were applied tothe rehydrated strips at the anodic end soaked in a 5ndash10 mmsection of IEF electrode strip (Amersham Biosciences)Separation was performed for 120 000 V-h with a maximumvoltage of 5000 V After isoelectric focusing IPG strips wereequilibrated for the second dimension for 15 min in IPG equil-ibration buffer (50 mM Tris pH 68 6 M urea 30 glycerol1 SDS and 001 bromophenol blue) plus 80 mM DTTthen for 10 min in IPG equilibration buffer plus 135 mMiodoacetamide Approximately 1 cm was removed from theanodic end of each equilibrated strip before applying the stripto the top of a vertical 125 SDS-PAGE gel for second-dimension separation using the Investigator 5000 systemfrom Genomic Solutions
Large-format gels (24 cm wide yen 22 cm high yen 1 mm thick)for the second-dimension separation were cast in-houseusing the system supplied by Genomic Solutions 125SDS-PAGE gels prepared using 065 NN-methylenebi-sacrylamide as cross-linking agent were used for pH 4ndash7and pH 45ndash55 IPG strips whereas pH 55ndash67 and pH 6ndash11 strips were applied to similar gels made with 08 cross-linker Electrophoresis was performed while cooling using themaximum setting (ordf 4ndash10infinC) at 20 000 mW per gel constantpower and a maximum voltage of 500 V Gels were thenstained with either colloidal Coomassie G-250 (Neuhoff et al1988) or silver nitrate (Rabilloud 1992) and scanned in aProXPRESS proteomic imaging system (Perkin-Elmer)Image analysis was performed using PHORETIX 2D version51 (NonLinear Dynamics) spot detection was optimizedautomatically using the lsquoSpot Detection Wizardrsquo and thenedited manually background subtraction was performedautomatically using the lsquoMode of Non-Spotrsquo setting imageswere then normalized to the total spot volume for quantifica-tion Spot filtering was not used
Protein identification using mass spectrometry
Protein spots of interest were excised from colloidalCoomassie-stained gels in-gel digested with trypsin andidentified by MALDI-TOF peptide mass fingerprint analysisExcised gel pieces (ordf 1-mm-diameter circles cut from 1-mm-thick gels) were washed twice with 100 ml of 50 mMammonium bicarbonate for 15 min once with 100 ml of 20acetonitrile-40 mM ammonium bicarbonate for 15 min andonce with 100 acetonitrile Washed gel pieces were allowedto air-dry for 10 min before being rehydrated in 5 ml of10 mg ml-1 trypsin (sequencing grade modified Promega) in10 mM ammonium bicarbonate Digestion (37infinC for 4 h) wasstopped by the addition of 5 ml of 5 formic acid Extractionof peptides into this solution was encouraged by a sonicwater bath treatment for 20 min Peptide extract (05 ml) wasspotted on to a thin layer of a-cyano-4-hydroxycinnamic acidapplied to a MALDI-TOF sample template and analysed byMALDI-TOF mass spectrometry (Bruker Reflex III) using anaccelerating voltage of 25 kV Samples were externally cali-brated using a standard mixture of six peptides ranging in
mass from 10465423 Da to 34946500 Da and the massaccuracy obtained was 60 ppm or better Identification ofproteins from MALDI-TOF peptide mass fingerprint data wasperformed using the lsquoMascotrsquo search engine at httpwwwmatrixsciencecom and was based on a positive resultusing their lsquoProbability Based MOWSE Scorersquo algorithm AMOWSE score of 60 or higher is significant at the 5 levelor better and proteins in this work typically gave scores gt80(frequently considerably so) In addition no identification wasaccepted unless at least five peptides representing at least20 of the protein sequence were detected in the MALDI-TOF peptide mass fingerprint
Assignment of proteins to metabolic pathways
Proteins were assigned to primary metabolic pathwaysusing the lsquoProtein Classification Schemersquo web page ofthe annotated Streptomyces coelicolor genome sequenceat httpwwwsangeracukProjectsS_coelicolor as it ap-peared in May 2002 The annotation of the genome andtherefore the Protein Classification Scheme is likely to bemodified as more experimental data are obtained about thefunction of genes that have currently been assigned on thebasis of sequence information alone It is therefore possiblethat proteins assigned to metabolic pathways in this studymay in future be shown to perform different functions in thecell
Assignments to gene clusters encoding secondary metab-olites are based on an analysis of the genome sequencereported by Bentley et al (2002) In some cases theseassignments are based on extensive experimental data (actred reviewed by Chater and Bibb 1997 Champness 2000cda Chong et al 1998 Huang et al 2001 whiE Davis andChater 1990)
Acknowledgements
This work was funded by grants (2FGT11406 208FGT11408 and 208IGF12432) under the BBSRCrsquos Tech-nologies for Functional Genomics and Investigating GeneFunction initiatives and by a competitive strategic grant fromthe BBSRC to the John Innes Centre We thank Mike Naldrettfor his extensive contribution in establishing technology forproteome analysis at the JIC
Supplementary material
The following material is available from httpwwwblackwell-sciencecomproductsjournalssuppmatmolemole3219mmi3219smhtmSection 1 Detection of gene products assigned to individualmetabolic steps in primary metabolic pathways in ScoelicolorSection 2 Identification of proteins from individual amino acidbiosynthetic pathwaysSection 3 Detection of gene products assigned to individualmetabolic steps in secondary metabolic pathways in Scoelicolor
Proteomic analysis of S coelicolor 931
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
References
Anderson TB Brian P and Champness WC (2001)Genetic and transcriptional analysis of absA an antibioticgene cluster linked two-component system that regulatesmultiple antibiotics in Streptomyces coelicolor Mol Micro-biol 39 553ndash566
Bentley SD Chater KF Cerdeno-Tarraga A-M ChallisGL Thomson NR James KD et al (2002) Completegenome sequence of the model actinomycete Streptomy-ces coelicolor A3(2) Nature 417 141ndash147
Berdy J (1984) New ways to obtain antibiotics Chin J Anti-biot 7 272ndash290
Berggren K Chernokalskaya E Steinberg TH KemperC Lopez MF Diwu Z et al (2000) Background-freehigh sensitivity staining of proteins in one- and two-dimensional sodium dodecyl sulfate-polyacrylamide gelsusing a luminescent ruthenium complex Electrophoresis21 2509ndash2521
Blackstock WP and Weir MP (1999) Proteomics quan-titative and physical mapping of cellular proteins TrendsBiotechnol 17 121ndash127
Blattner FR Plunkett G Bloch CA Perna NT BurlandV Riley M et al (1997) The complete genome sequenceof Escherichia coli K-12 Science 277 1453ndash1462
Buttner K Bernhardt J Scharf C Schmid R MaderU Eymann C et al (2001) A comprehensive two-dimensional map of cytosolic proteins of Bacillus subtilisElectrophoresis 22 2908ndash2935
Champness W (2000) Actinomycete development antibioticproduction and phylogeny questions and challenges InProkaryotic Development Brun YV and Skimketts LJ(eds) Washington DC American Society for MicrobiologyPress pp 11ndash31
Chater KF and Bibb MJ (1997) Regulation of bacterialantibiotic production In Biotechnology Vol 7 Products ofSecondary Metabolism Kleinkauf H and von Doumlhren H(eds) Weinheim Germany VCH pp 57ndash105
Chong PP Podmore SM Kieser HM Redenbach MTurgay K Marahiel M et al (1998) Physical identificationof a chromosomal locus encoding biosynthetic genesfor the lipopeptide calcium-dependent antibiotic (CDA) ofStreptomyces coelicolor A3(2) Microbiology 144 193ndash199
de Crecy-Lagard V Servant-Moisson JV Grandvalet Cand Mazodier P (1999) Alteration of the synthesis of theClp ATP-dependent protease affects morphological andphysiological differentiation in Streptomyces Mol Microbiol32 505ndash517
Davis NK and Chater KF (1990) Spore colour inStreptomyces coelicolor A3(2) involves the developmen-tally regulated synthesis of a compound biosyntheticallyrelated to polyketide antibiotics Mol Microbiol 4 1679ndash1691
Fernandez-Moreno FA Martinez E Caballero JLIchinose K Hopwood DA and Malpartida F (1994)DNA sequence and functions of the actVI region of theactinorhodin biosynthetic gene cluster of Streptomycescoelicolor A3(2) J Biol Chem 269 24854ndash24863
Fink D Falke D Wohllheben W and Engels A (1999)Nitrogen metabolism in Streptomyces coelicolor A3(2)
modification of glutamine synthase I by an adenylyltrans-ferase Microbiology 145 2313ndash2322
Godovac-Zimmermann J Soskic V Posnanovic S andBrianza F (1999) Functional proteomics of signal trans-duction by membrane receptors Electrophoresis 20 952ndash961
Gorg A Obermaier C Boguth G Harder A Scheibe BWildgruber R and Weiss W (2000) The current state oftwo-dimensional electrophoresis with immobilized pH gra-dients Electrophoresis 21 1037ndash1053
Gramajo HC Takano E and Bibb MJ (1993) Stationary-phase production of the antibiotic actinorhodin in Strepto-myces coelicolor A3(2) is transcriptionally regulated MolMicrobiol 7 837ndash845
Gross CA Chan C Dombroski A Gruber T Sharp MTupy J and Young B (1998) The functional and regula-tory roles of sigma factors in transcription Cold SpringHarb Symp Quant Biol 63 141ndash155
Hesketh A Sun J and Bibb MJ (2001) Induction ofppGpp synthesis in Streptomyces coelicolor A3(2) grownunder conditions of nutritional sufficiency elicits actII-ORF4transcription and actinorhodin biosynthesis Mol Microbiol39 136ndash144
Hesketh A Fink D Gust B Rexer H-U Scheel BChater K et al (2002) The PII nitrogen regulatory proteinGlnK in Streptomyces coelicolor A3(2) is modified by ade-nylylation rather than uridylylation and the GlnD and GlnKhomologues are functionally dissimilar to their nitrogenregulatory system counterparts Mol Microbiol (inpress)
Hood DW Heidstra R Swodoba UK and Hodgson DA(1992) Molecular genetic analysis of proline and tryp-tophan biosynthesis in Streptomyces coelicolor A3(2)interaction between primary and secondary metabolism- areview Gene 115 5ndash12
Hu DS-J Hood DW Heidstra R and Hodgson DA(1999) The expression of the trpD trpC and trpBA genesof Streptomyces coelicolor A3(2) is regulated by growthrate and growth phase but not by feedback repression MolMicrobiol 32 869ndash880
Huang J Lih CJ Pan KH and Cohen SN (2001)Global analysis of growth phase responsive gene expres-sion and regulation of antibiotic biosynthetic pathways inStreptomyces coelicolor using DNA microarrays GenesDev 15 3183ndash3192
Ichinose K Taguchi T Ebizuka Y and Hopwood DA(1998a) Biosynthetic gene clusters of benzoisochromane-quinone antibiotics in Streptomyces spp Identification ofgenes involved in post-PKS tailoring steps Actinomyceto-logica 12 99ndash109
Ichinose K Bedford DJ Tornus D Bechthold A BibbMJ Revill PW et al (1998b) The granaticin biosyntheticgene cluster of Streptomyces violaceoruber Tu22sequence analysis and expression in a heterologous hostChem Biol 5 647ndash659
Kelemen GH Brian P Flardh K Chamberlin L ChaterK and Buttner M (1998) Developmental regulation oftranscription of whiE a locus specifying the polyketidespore pigment in Streptomyces coelicolor A3(2) J Bacte-riol 180 2515ndash2521
Kieser T Bibb MJ Buttner MJ Chater KF and
932 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
Hopwood DA (2000) Practical Streptomyces GeneticsNorwich John Innes Foundation
Kim EJ Chung HJ Suh B Hah YC and Roe JH(1998) Expression and regulation of the sodF gene encod-ing iron- and zinc-containing superoxide dismutase inStreptomyces coelicolor Muumlller J Bacteriol 180 2014ndash2020
Kimura Y Takaoka M Tanaka S Sassa H Tanaka KPolevoda B et al (2000) Na-acetylation and proteolyticactivity of the yeast 20S proteasome J Biol Chem 2754635ndash4639
Kunst F Ogasawara N Moszer I Albertini AM AlloniG Azevedo V et al (1997) The complete genomesequence of the Gram-positive bacterium Bacillus subtilisNature 390 249ndash256
Lucchini S Thompson A and Hinton JCD (2001)Microarrays for microbiologists Microbiology 147 1403ndash1414
Mann M Hendrickson RC and Pandey A (2001) Analy-sis of proteins and proteomes by mass spectrometry AnnuRev Biochem 70 437ndash473
Miyodah S (1993) Research on antibiotic screening inJapan over the last decade a producing microorganismsapproach Actinomycetologica 7 100ndash106
Molloy MP Herbert BR Williams KL and Gooley AA(1999) Extraction of Escherichia coli proteins with organicsolvents prior to two-dimensional electrophoresis Electro-phoresis 20 701ndash704
Nagy I Tamura T Vanderleyden J Baumeister W andDe Mot R (1998) The 20S proteasome of Streptomycescoelicolor J Bacteriol 180 5448ndash5453
Neuhoff V Arold N Taube D and Ehrhardt W (1988)Improved staining of proteins in polyacrylamide gels includ-ing isoelectric focusing gels with clear background at nan-ogram sensitivity using Coomassie Brilliant Blue G-250and R-250 Electrophoresis 9 255ndash262
Nilsson CL and Davidsson P (2000) New separation toolsfor comprehensive studies of protein expression by massspectrometry Mass Spectrom Rev 19 390ndash397
Puglia AM Vohradsky J and Thompson CJ (1995)
Developmental control of the heat-shock stress regulon inStreptomyces coelicolor Mol Microbiol 17 737ndash746
Rabilloud T (1992) A comparison between low backgroundsilver diamine and silver nitrate protein stains Electro-phoresis 13 429ndash439
Robinson VL Buckler DR and Stock AM (2000) A taleof two components a novel kinase and a regulatory switchNature Struct Biol 7 626ndash633
Schena M (2000) Microarray Biochip Technology NatickMA Eaton Publishing
Spusansky O Rezuchova B Homerova D andKormanec J (2001) Expression of the gap gene encodingglyceraldehyde-3-phosphate dehydrogenase of Strepto-myces aureofaciens requires GapR a member of theAraCXylS family of transcriptional activators Microbiology147 1291ndash1301
Taguchi T Itou K Ebizuka Y Malpartida F HopwoodDA Surti CM et al (2000) Chemical characterisationof disruptants of the Streptomyces coelicolor A3(2) actVIgenes involved in actinorhodin biosynthesis J Antibiot 53144ndash152
Takano E Gramajo HC Strauch E Andres N WhiteJ and Bibb MJ (1992) Transcriptional regulation of theredD transcriptional activator gene accounts for growth-phase-dependent production of the antibotic undecylpro-digiosin in Streptomyces coelicolor A3(2) Mol Microbiol6 2797ndash2804
Tonella L Hoogland C Binz P-A Appel RDHochstrasser DF and Sanchez J-C (2001) New per-spectives in the Escherichia coli proteome investigationProteomics 1 409ndash423
Vohradsky J Li XM and Thompson CJ (1997) Identifi-cation of prokaryotic developmental stages by statisticalanalysis of two-dimensional gel patterns Electrophoresis18 1418ndash1428
Vohradsky J Li XM Dale G Folcher M Nguyen LViollier PH and Thompson CJ (2000) Developmentalcontrol of stress stimulons in Streptomyces coelicolorrevealed by statistical analyses of global gene expressionpatterns J Bacteriol 182 4979ndash4986

Proteomic analysis of
S coelicolor 923
copy 2002 Blackwell Publishing Ltd
Molecular Microbiology
46
917ndash932
Co-translational modification by removal of the N-terminal initiator Met occurred in 149 out of 190 (78)cases in which the N-terminal peptide was detected Pro-cessing by the N-methionylaminopeptidase was observedif the second amino acid was Ser (45 out of 149 exam-ples) Thr (40) Ala (32) Pro (27) Gly (4) or Val (1) Ononly one occasion was any of these six amino acids foundas the second residue if the N-terminal methionine wasretained This is in broad agreement with observations in
B subtilis
in which two-thirds of proteins were reported tobe similarly processed (Buttner
et al
2001)
Representation of primary and secondary metabolic pathways
Streptomycetes produce
ordf
60 of all commercially usefulantibiotics (Berdy 1984 Miyodah 1993) Most antibioticsare the product of complex biosynthetic pathways oftenencoded by 20ndash30 clustered genes Production of thepigmented antibiotics actinorhodin (Act) and undecyl-
prodigiosin (Red) has been studied extensively in
Scoelicolor
(for reviews see Chater and Bibb 1997Champness 2000) and involves clusters of approximately22 genes each A third cluster producing the calcium-dependent antibiotic (CDA) and consisting of 40 geneshas also been studied (Chong
et al
1998 Huang
et al
2001) In the extensive spot identification studies for thepreparation of a proteome map of
S coelicolor
(seeabove) more than one-third of the proteins from each ofthese pathways was identified (Table 5) Proteins fromthree clusters that specify metabolic products that haveyet to be identified experimentally (Bentley
et al
2002)were also extensively represented a type I polyketidesynthase cluster (SCO6273ndash6288) a deoxysugarglycosyltransferase cluster (SCO0381ndash0401) and anon-ribosomal peptide synthetase cluster predicted toencode a novel siderophore coelichelin (SCO0489ndash0499) Several clusters were unrepresented
The primary metabolic pathways were somewhat morehighly represented with
ordf
60 or more of proteins anno-
Fig 2
Examples of MALDI-TOF peptide mass fingerprint data identifying post-translational modification of proteins by (A) acetylation (predicted mass increase of 420 Da) (B) adenylylation (predicted mass increase of 3290 Da) and (C) proteolytic processing (predicted mass loss for Ile of 11308 Da) In each fingerprint the
x
-axis is masscharge ratio (mz) and the
y
-axis is absolute intensity The observed mass accuracies in the MALDI-TOF fingerprints are (A) 60 ppm (B) 20 ppm and (C) 20 ppm
924
A R Hesketh
et al
copy 2002 Blackwell Publishing Ltd
Molecular Microbiology
46
917ndash932
tated as enzymes of glycolysis the TCA cycle thepentose phosphate pathway purine ribonucleotide bio-synthesis and pyrimidine nucleotide biosynthesis beingmapped (Table 6) About 50 of amino acid biosyntheticproteins were identified Lists of individual members of allpathways considered here including those that were andwere not identified during proteome mapping can befound in the
Supplementary material
Several proteins from both primary and secondary met-
abolic pathways were identified at more than one positionon the proteome map and may therefore be subject toregulation by post-translational modification (see Table 3and Fig 1) Primary metabolic proteins of this type (yellowspots on Fig 1) were usually detected as chains of two ormore spots with the same apparent molecular weights butdifferent pI values suggesting covalent modification bylow-molecular-weight adducts Apart from adenylylation ofGSI (see above) the nature of the modification of theseproteins was not revealed by MALDI-TOF peptide massfingerprint data Both covalent modification and proteolyticprocessing were also found in secondary metabolic pro-teins (red spots in Fig 1) For example the Act pathwaymembers SCO5071 (spot 16) and SCO5075 (spot 20)
and the putative 3-oxoacyl[acyl carrier protein] reductasefrom the type I polyketide cluster (SCO6282 spot 11)appear to be covalently modified whereas proteolytic pro-cessing was detected for RedL (SCO5892 spot 18) fromthe undecylprodigiosin cluster the putative dehydrataseActVI-ORF3 (SCO5074 spot 31) from the Act cluster theputative peptide monooxygenase from coelichelin biosyn-thesis (SCO0498 spot 34) and the putative oxygenasefrom the CDA pathway (SCO3236 spot 33) Two Act bio-synthetic proteins SCO5088 (spot 24) and SCO5073(spot 29) were detected both at their predicted pI andmolecular weight co-ordinates and at positions with simi-lar pI values but significantly higher molecular weightsThis may indicate that these proteins form stable mul-timers that are not completely denatured under theconditions used Only the modification of ActVI-ORF3(SCO5074 spot 31) could be characterized further fromthe mass spectrometry data which showed that it involvesproteolytic processing at two sites between amino acidresidues 3132 and 3435 (data not shown) Analysis of asample obtained by externally extracting proteins fromintact washed mycelium with a solution containing 2SDS and 50 mM dithiothreitol (DTT) revealed that sur-
Table 5
Summary of proteins identified on the proteome map from secondary metabolic pathways during growth in SMM
Biosynthetic pathwaygene cluster
Pathwaygenesannotated
Pathwaygene productsidentified
Percentagepathwaydetected
Calcium-dependent antibiotic CDA SCO3210ndash3249 40 16 40Actinorhodin SCO5071ndash5092 22 15 68Undecylprodigiosin SCO5877ndash5898 22 8 36Deoxysugar synthaseglycosyltransferase cluster SCO0381ndash0401 21 7 33Type I polyketide synthase SCO6273ndash6288 16 6 38Hopanoids SCO6759ndash6771 13 1 7Siderophore (coelichelin) SCO0489ndash0499 11 5 45Siderophore (desferrioxamine) SCO2783ndash2785 4 4 100
Chromosomal locations and extent of gene clusters are taken from Bentley
et al
(2002) No proteins were detected from the remaining predictedsecondary metabolic clusters ie siderophore (coelibactin) SCO7681ndash7691 (11 ORFs) siderophore SCO5799ndash5801 (three ORFs) non-ribosomal peptide synthases SCO6429ndash6438 (10 ORFs) type II fatty acid synthases SCO1265ndash1273 (nine ORFs) WhiE spore pigment clusterSCO5314ndash5320 (seven ORFs) isorenieratine SCO0185ndash0191 (seven ORFs) eicosapentaenoic acid SCO0124ndash0129 (six ORFs) chalconesynthases SCO1206ndash1208 (three ORFs) chalcone synthases SCO7669ndash7671 and SCO7222 (three ORFs) sesquiterpene cyclase SCO5222ndash5223 (two ORFs) type I polyketide synthases SCO6826ndash6827 (two ORFs) geosmin SCO6703 (one ORF)
Table 6 Summary of proteins identified on the proteome map from primary metabolic pathways during growth in SMM
Biosynthetic pathwayPathway genesannotateda
Pathway geneproducts identified
Percentage pathway detected
Glycolysisb 24 13 55Pentose phosphate pathwayb 14 10 71TCA cycleb 21 12 57Amino acid biosynthesisc 115 55 48Purine ribonucleotide biosynthesisb 18 12 67Pyrimidine ribonucleotide biosynthesisb 8 5 62
a Derived from interpretation of the annotated S coelicolor genome sequence at httpwwwsangeracukProjectsS_coelicolorb See Supplementary material for details of pathway membersc See Supplementary material for details of individual pathways
Proteomic analysis of S coelicolor 925
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
prisingly the truncated ActVI-ORF3 is located extracellu-larly presumably non-covalently associated with the cellwall matrix (Fig 4)
Expression analysis of certain primary and secondary metabolic proteins
Production of the pigmented antibiotics Act and Red inliquid minimal medium supplemented with casamino acids(SMM) begins in the transition phase between exponentialgrowth and stationary phase Figure 3 shows a compari-son of the expression of certain primary and secondarymetabolic proteins between exponentially growing andstationary phase cultures Two proteins from the Actcluster (ActVA-ORF5 and ActI-ORF2) and two from theRed (RedI and RedR) were present in the stationaryphase cultures but were not detected during exponentialgrowth Similarly the putative 3-oxoacyl-[acyl carrierprotein] reductase (SCO6282) and epoxide hydrolase(SCO6277) enzymes from a type I polyketide secondarymetabolic cluster showed a large increase in abundance
between the exponential and stationary phase samplesTriose phosphate isomerase (SCO1945) fructose bis-phosphate aldolase (SCO3649) and succinyl-CoA syn-thetase alpha chain (SCO4809) all enzymes from centralcarbon metabolism were present in approximatelyconstant amounts in both samples Interestingly two iso-forms (SCO7511 and SCO1947) of glyceraldehyde-3-phosphate dehydrogenase from the glycolytic pathwayshowed different patterns of expression SCO1947 levelswere the same in both exponential and stationary phasebut SCO7511 barely detectable in exponentially growingcultures increased markedly in stationary phase
The Streptomyces coelicolor online proteome map
The 2D gel images in Fig 1 together with the spot iden-tification results are being made available over the WorldWide Web to provide a reference map of the S coelicolorproteome At the time of writing the reference gel forthe pH 4ndash7 isoelectric point range was fully accessiblewhereas those for pH 45ndash55 pH 55ndash67 and pH 6ndash11
Fig 3 A comparison of the expression of certain primary and secondary metabolic proteins between exponentially growing and stationary phase cultures of S coelicolor M145 Larger arrows represent higher protein level in one gel than in the other The experiment was performed on duplicate biological samples and both showed similar protein expression profiles
926 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
were under construction (httpqbababeracukS_coeliReferenceGelrefgelhtml) A tool for comparing two ormore gel images with each other is also accessible fromthe same website at httpqbababeracukS_coeliflickerhtml
Discussion
Silver staining is one of the most sensitive methods fordetecting proteins on polyacrylamide gels allowing aslittle as 05ndash2 ng of an individual protein to be detected(Berggren et al 2000 Gorg et al 2000) The proportionof the S coelicolor proteome that can be analysed usingsilver staining of 2D-PAGE-separated proteins dependsat least in part on the pH range of the IPG strip used forthe first-dimension separation (see Table 1) Using theintermediate-range IPG strip covering pH 4ndash7 about 20ndash25 of the proteins in the theoretical proteome predictedto be observable in the resulting 2D-PAGE gel were actu-ally detected This figure was reduced to 10ndash12 whenanalysing the more basic pI range using the pH 6ndash11 IPGstrip Attempts to increase the percentage of the proteomedetectable using the pH 4ndash7 and pH 6ndash11 IPG strips byloading more protein sample resulted in poorer separa-tions (data not shown) This was especially true of thepH 6ndash11 strips which were generally the most difficult touse successfully However good separation of higher pro-tein loads was achieved using the narrow-range IPG stripspH 45ndash55 and 55ndash67 significantly increasing the pro-portion of theoretical proteins detected to over 40Clearly and more pertinently the limited conditions usedfor culturing S coelicolor in this study will also haverestricted the number of proteins detected as not all theproteome will be expressed In particular S coelicolordoes not sporulate in shaken liquid-grown cultures soproteins involved in this differentiation process are unlikelyto be present Nevertheless it is striking that the com-bined data for the pH 45ndash55 and 55ndash67 isoelectric pointranges indicate that 2468 out of a total of predicted 4932protein spots can be detected (see Table 1) suggestingthat transition phase mycelium in this supplementedminimal medium is expressing 50 of its genes (ie byextrapolation some 3912 of the 7825 in the genome)
Analysis of post-translational regulation
Although the regulation of cellular processes and adaptiveresponses in bacteria has been studied extensively at thetranscriptional level post-translational regulatory mecha-nisms are much less understood Proteomic analysisusing 2D-PAGE coupled with mass spectrometry offersthe opportunity to observe and identify modification ofproteins at a general cellular level The functions of pro-teins can be altered by specific covalent attachment of
adducts (eg phosphorylation glycosylation nucleotidy-lation acetylation) or by proteolytic processing Bothevents alter the pI andor mass of the protein and so canusually be observed on 2D-PAGE In the S coelicolorproteome map ordf110 out of a total of 770 gene productswere identified in more than one position or at pI andapparent mass co-ordinates significantly different frompredicted values indicating extensive regulation at thepost-translational level (88 are listed in Table 3) a propor-tion similar to that reported for other prokaryotes (Buttneret al 2001 Tonella et al 2001) The initial peptide massfingerprint analysis identified the modification of only 10of these proteins probably partly because fingerprint datatypically cover ordf30ndash60 of a proteinrsquos sequence (data notshown) so modification at positions not represented willbe missed Protein digestion with endopeptidases otherthan trypsin before mass spectrometry may help in thissituation but some covalently attached groups are labileand cannot be detected routinely Phosphorylation of Hisoccurs on nitrogen producing a high-energy phosphora-midate bond that is easily hydrolysed in acidic conditionswhereas phospho-Asp is the most labile phosphorylatedresidue with a half-life of a few hours under physiologicalconditions (reviewed by Robinson et al 2000) Thesemodifications the basis of bacterial two-component regu-latory systems are therefore unlikely to survive stainingof the 2D gels which involved the use of a fixing solutioncontaining 10 acetic acid and mass spectrometry Iden-tification of the more stable phosphoester phosphorylationthat occurs on Ser Thr or Tyr residues using PAGEfollowed by mass spectrometry has been reported(Godovac-Zimmermann et al 1999 Buttner et al 2001)but no examples were detected in this work Of the mod-ifications identified in this study three were by acetylationat the N-terminus after removal of the initiator Met residuetwo involved adenylylation and five proteins had been N-terminally truncated Modification of the 20S proteasomeby N-acetylation in yeast involves removal of the Metresidue by N-methionylaminopeptidase followed byacetylation with an Na-acetyltransferase (Kimura et al2000) Homologues of these enzymes are predicted fromthe S coelicolor genome and are presumably responsiblefor the observed modification of the 20S proteasomealpha subunit and of BldK-ORFD and the putative cellu-lose-binding protein The functional significance of Na-acetylation of these proteins is unclear
Analysis of the biosynthesis of antibiotics and other secondary metabolites
Post-translational modification The proteome mappingresults indicate that at least six enzymes in the Act clusterare subject to post-translational modifications and one
Proteomic analysis of S coelicolor 927
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
each from the undecylprodigiosin (SCO5877ndash5898)lsquocoelichelinrsquo (SCO0489ndash0499) and type I polyketidesynthase (SCO6273ndash6288) clusters (see Table 3) Thesemetabolic pathways may therefore be significantly regu-lated at the post-translational level The actVI-ORF3 gene(SCO5074) encodes a putative dehydratase enzyme thatis part of the biosynthetic pathway for Act a blue memberof the benzoisochromanequinone (BIQ) family of pig-mented antibiotics Mutation in actVI-ORF3 causes a 50decrease in the accumulation of Act (and a reddish brownphenotype) and it is believed to be responsible for pyranring closure an intermediate step in Act biosynthesis (thatmay also occur spontaneously) leading to the formationof the BIQ chromophore (Fernandez-Moreno et al 1994Ichinose et al 1998a Taguchi et al 2000) In this studyActVI-ORF3 protein was detected in two forms a majorspot corresponding to ActVI-ORF3 truncated by removalof the N-terminal 31 amino acid residues and a minor onein which the first 34 amino acids were missing (spots 31in Fig 1) The full-length protein was not detected Anal-ysis of the protein sequence using the SIGNALP server athttpwwwcbsdtudkservicesSignalP predicted a signalpeptide cleavage site between amino acids 31 and 32 inagreement with the observed results
The protein extract analysed was prepared from myce-lium that had been collected by centrifugation and sub-jected to two washes indicating that if ActVI-ORF3 isexported it is effectively retained within the cell wallmatrix This was confirmed by analysis of a preparationobtained by externally extracting proteins from intactwashed mycelium (Fig 4) Export of an enzyme requiredfor the efficient biosynthesis of an antibiotic that is thoughtto be produced intracellularly appears to be anomalousparticularly as it is involved in an intermediate biosynthetic
event Database analysis of the other members of the Actcluster revealed only one other protein with a predictedsignal peptide sequence the putative integral membranetransport-related protein ActII-ORF3 and it seemsunlikely that all the Act biosynthetic steps following BIQchromophore formation are extracellular It is possible thatfull-length ActVI-ORF3 catalyses the intermediate biosyn-thetic event intracellularly and the exported processedprotein is responsible for a different extracellular activityperhaps later in the pathway However no full-length pro-tein was detected so the exported enzyme may performthe pyran ring closure on a secreted metabolite that isthen reabsorbed by the cell for completion of the sub-sequent biosynthetic steps Only two homologues ofactVI-ORF3 are known and both are also in BIQ antibioticbiosynthetic clusters gra-ORF18 from the granaticincluster (Ichinose et al 1998b) and med-ORF5 frommedermycin biosynthesis (K Ichinose personal com-munication) Interestingly the encoded proteins of bothsequences are also predicted to contain N-terminal signalpeptides The observation that ActVI-ORF3 is exported tothe cell wall matrix may therefore be of general signifi-cance to the BIQ family of antibiotics and provide a newinsight into their biosynthetic pathways
Pathway representation Of the 20 putative secondarymetabolite gene clusters identified in the S coelicolorgenome sequence (Bentley et al 2002) eight had atleast one protein each identified on the proteome map(see Table 5) The biosynthetic clusters for the antibioticsAct Red and CDA had 68 36 and 40 of their pro-teins represented There are several possible explana-tions for failure to detect all the proteins for each pathwaySome are putative integral membrane proteins so cannotreadily be detected using conventional 2D-PAGE SomeRed and CDA proteins have molecular weights gt200 kDaoutside the range of the 125 gels used (for clustermembers see Supplementary material) The pathway-specific regulatory proteins for Act and Red biosynthesiswere not detected despite a detailed search of their pre-dicted gel locations even though the relevant genes areknown to be transcribed in equivalent cultures of the samestrain (Hesketh et al 2001) The proteins are presumablypresent but at levels below the limit of detection Howeverthe transcriptional regulator for CDA production CdaRwas identified as well as the two-component systemresponse regulator AbsA2 which is also encoded by agene in the CDA cluster (Anderson et al 2001) Interest-ingly proteins from three putative secondary meta-bolite clusters producing chemicals of as yet unknownstructure were also identified on the map correspondingto the deoxysugar synthaseglycosyltransferase clusterSCO0381ndash0401 the lsquocoelichelinrsquo cluster for a predicted(but not yet confirmed) oligopeptide siderophore
Fig 4 Processed ActVI-ORF3 is present in the cell wall matrix 2D-gel separations of protein extracts prepared (A) from whole cells or (B) by externally washing cells in a 2 SDS extraction buffer The arrow indicates ActVI-ORF3 protein The experiment was performed on duplicate biological samples and both showed similar protein expression profiles
928 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
(SCO0489ndash0499) and the type I polyketide clusterSCO6273ndash6288 The 3-oxoacyl-[acyl carrier protein]reductase (SCO6282) from the latter pathway is ex-pressed in a growth phase-dependent manner (Fig 3)and in early stationary phase cultures is perhaps themost abundant protein spot in the entire detectable pro-teome Presumably the metabolites produced by thesethree pathways are being synthesized under the condi-tions used In addition to coelichelin three other sidero-phores are postulated for S coelicolor All the proteinsfrom one of these desferrioxamine (SCO2782ndash2785)were identified but none was seen from either of the othertwo pathways The WhiE polyketide spore pigment isassociated with sporulation of surface-grown cultures(Davis and Chater 1990) and the expression of the whiEgenes depends on a series of sporulation-specific regula-tory genes (Kelemen et al 1998) no corresponding pro-teins of which were detected here It is therefore notsurprising that WhiE proteins were not detected in sam-ples prepared from liquid-grown vegetative mycelium
Analysis of primary metabolism
Proteins annotated as functioning in primary metabolicpathways were highly represented on the proteome mapwith ordf60 or more of the enzymes assigned to glycolysisthe TCA cycle the pentose phosphate pathway andpurine and pyrimidine ribonucleotide biosynthesis beingidentified (see Table 6) This figure may be deceptively lowbecause these pathways in S coelicolor often appear tohave more than one enzyme isoform for some of themetabolic steps potentially offering additional levels ofcontrol or developmental responsiveness at these pointsFor example only two steps in glycolysis have just a singlegene product assigned to them the remainder typicallyhave two or three each (Fig 5) At least one isoform waspresent for each step so the pathway is in theory 100functionally represented Understanding the regulation ofthese pathways will require analysis of the expression ofthese isoenzymes under different nutritional conditionsor different developmental stages Figure 5 shows that ofthe three predicted glyceraldehyde-3-phosphate dehydro-genase isoforms from glycolysis only two were detectedin this study (see also Supplementary material) Of theseSCO1947 appears to be constitutive whereas SCO7511is growth phase dependent (see Fig 3) Thus even thelimited conditions in this study yielded useful informationabout isoenzyme expression patterns (see Supplemen-tary material) In Streptomyces aureofaciens two genesencoding glyceraldehyde-3-phosphate dehydrogenaseactivity also appear to be differently regulated transcrip-tion of one highly homologous to SCO7511 is activatedby the regulator GapR whereas the other is GapR inde-pendent (Spusansky et al 2001)
Hood et al (1992) reported that most amino acid bio-synthetic enzymes are expressed at very low constitutivelevels in S coelicolor so it is reassuring to note that ordf50could be detected and identified using proteomics Theseinclude TrpB and TrpC1 from the tryptophan biosyntheticpathway the corresponding transcripts of which couldbarely be detected in previously reported S1 nucleaseprotection experiments (Hu et al 1999)
Perspectives
No attempt was made in the work reported here to simplifythe protein separations by subdividing the sample intosoluble and membrane fractions and this is reflected inthe 10 lipoproteins and 16 ABC transport system ATP-binding subunits identified on the proteome map (seeTable 2) No integral membrane proteins were detectedand even when a membrane protein sample was pre-pared and analysed no proteins identified possessedmore than three predicted transmembrane helices (datanot shown) This is consistent with previous studiesin other bacteria (Molloy et al 1999 Nilsson andDavidsson 2000) Another group of proteins also strik-ingly absent from the list of those identified is the sigmafactors which determine promoter selection by RNA poly-merase There are an extraordinarily high number (65) ofsigma factors encoded in the S coelicolor genome aswell as at least seven antisigma factors and 15 antisigmafactor antagonists (some of each of which were detected)Sigma factor heterogeneity in bacteria is an important wayof regulating expression of specific sets of genes under
Fig 5 The glycolytic pathway in S coelicolor illustrating the assignment of multiple gene products to individual metabolic steps based on the annotation of the genome sequence at httpwwwsangeracukProjectsS_coelicolor Gene products identified in the proteome map are underlined
Proteomic analysis of S coelicolor 929
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
different conditions and in different cell types (for a reviewsee Gross et al 1998) Although no sigma factors wereidentified about half (31 out of 65) have predicted pIvalues gtpH 7 and so can only be detected using thepH 6ndash11 IPG strips that give the poorest results (seeTable 1) However the principal vegetative sigma factorHrdB which is expected to be moderately abundanthas a predicted pI of 613 and its absence from thepH 55ndash67 proteome map may indicate that the samplepreparation method which does not remove DNA is inap-propriate Among other regulatory proteins 11 out of apossible 80 two-component system response regulatorsplus a further 36 proteins annotated as lsquoregulatorsrsquo weredetected Only one SerThr kinase and no two-componentsystem sensor kinases were detected possibly reflectingthe difficulty in analysing proteins with several transmem-brane domains Interestingly 30 secreted proteins wereidentified on the proteome map at least 10 of whichappear from comparison of their observed pI and molec-ular weight positions with predicted values to have beenprocessed already and therefore exported Presumablythese proteins do not end up exclusively in the culturemedium
Clearly this approach to proteomic analysis of S coeli-color has some important deficiencies not least theabsence of certain groups of proteins and the relativelypoor initial success rate for specifically characterizingpost-translational modifications Nevertheless it offersunique insights into the molecular biology of the organismthe expression of many proteins from both primary andsecondary metabolic pathways can be analysed post-translational regulation of proteins can be investigated ona general cellular level and the physical locations forsome proteins can be determined Approximately 35 ofthe S coelicolor genome is annotated as encoding lsquohypo-theticalrsquo proteins of unknown function and 182 proteins ofthis type were identified on the proteome map No exam-ples were found of overt disagreements between pre-dicted and observed proteins providing experimentalsupport for the general accuracy of the S coelicolorgenome sequence and its annotation Information aboutthe expression post-translational modification and physi-cal location of these (no longer) hypothetical proteinsfrom proteomic analyses is going to be important forassigning future functions As many of the hypotheticalproteins have apparent orthologues in pathogenic myco-bacteria some of the information may have an immediateapplication in the context of pathogenesis
Experimental procedures
Materials
Immobilized pH gradient (IPG) strips and IPG bufferswere purchased from Amersham Biosciences Acrylamide
duracryl and chemicals for silver staining were obtained fromGenomic Solutions All other chemicals were from Sigmaunless stated in the text
Strain and growth conditions
Streptomyces coelicolor M145 was cultivated with vigorousagitation at 30infinC in minimal medium supplemented with 02casamino acids (SMM) as described previously (Kieser et al2000) Briefly a high-density spore preparation [about 1010
colony-forming units (cfu) ml-1] was pregerminated in 2yen YTmedia for 7 h at 30infinC Germlings were harvested by centrifu-gation (5 min at 4000 g) resuspended in SMM and sonicatedbriefly to disperse any aggregates before inoculation into50 ml of SMM in 250 ml siliconized flasks containing coiledstainless steel springs Each flask received the equivalent of5 yen 107 spore cfu
Protein extraction and 2D-gel electrophoresis
The preparation of the protein sample to be separated waskept as simple as possible disrupting harvested myceliadirectly into denaturing isoelectric focusing (IEF) buffer con-taining Pefabloc SC (Roche) protease inhibitor This methodminimized unwanted protein degradation that was observedin initial studies using a Tris buffer containing Pefabloc SC forcell lysis (data not shown) Removal of DNA RNA salts andpolysaccharides from the sample was therefore not per-formed (other than the shearing of viscous DNA and RNA bysonication) and the protein extracts were sometimes difficultto focus when the sample was loaded by in-gel rehydrationLoading the sample soaked in paper strips at the anodicend of IPG strips that had already been rehydrated improvedthe reproducibility of separations significantly
Mycelium for protein extraction was harvested from cul-tures by brief centrifugation (30 s at 4000 g) at room tem-perature and immediately frozen in liquid nitrogen Typicallymycelium from 10 ml culture aliquots was collected and thetransfer time from culture flask to frozen sample was 15 minFrozen cells were thawed on ice in 5 ml of washing buffer(40 mM Tris pH 90 1 mM EGTA 1 mM EDTA) then pelletedby centrifugation (5 min at 3000 g) at 4infinC Washed cells wereresuspended in 400 ml of denaturing IEF buffer UTCHAPS[7 M urea 2 M thiourea 4 (wv) CHAPS 40 mM TrispH 90 1 mM EDTA 50 mM DTT 4 mM Pefabloc SC pro-tease inhibitor (Roche)] and disrupted by sonication (SanyoSoniprep 150 10 yen 2 s bursts at amplitude 75 mm) whilecooling in an ethanolndashice bath Cell debris was removed bycentrifugation (15 min 10 000 g 4infinC) and the protein extractwas stored frozen in aliquots at -80infinC until use For thepreparation of extracellular protein extracts washed cellswere resuspended in extraction buffer consisting of 40 mMTris pH 90 1 mM EDTA 50 mM DTT 2 SDS and 4 mMPefabloc SC protease inhibitor Cells were vortexed for 2 minbefore being pelleted by centrifugation (5 min 10 000 g4infinC) The supernatant was precipitated using the Amersham2D Clean-Up kit according to the manufacturerrsquos instructionsand the precipitated proteins were redissolved in UTCHAPSand stored at -80infinC until use
930 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
For first-dimension isoelectric focusing 18 cm IPG stripspH 4ndash7 pH 6ndash11 pH 45ndash55 or pH 55ndash67 (Amersham Bio-sciences) were rehydrated overnight in IEF buffer containing1 ampholytes according to the manufacturerrsquos instructionsusing a Phaser isoelectric focusing unit (Genomic Solutions)set at 20 V Protein samples to be separated were applied tothe rehydrated strips at the anodic end soaked in a 5ndash10 mmsection of IEF electrode strip (Amersham Biosciences)Separation was performed for 120 000 V-h with a maximumvoltage of 5000 V After isoelectric focusing IPG strips wereequilibrated for the second dimension for 15 min in IPG equil-ibration buffer (50 mM Tris pH 68 6 M urea 30 glycerol1 SDS and 001 bromophenol blue) plus 80 mM DTTthen for 10 min in IPG equilibration buffer plus 135 mMiodoacetamide Approximately 1 cm was removed from theanodic end of each equilibrated strip before applying the stripto the top of a vertical 125 SDS-PAGE gel for second-dimension separation using the Investigator 5000 systemfrom Genomic Solutions
Large-format gels (24 cm wide yen 22 cm high yen 1 mm thick)for the second-dimension separation were cast in-houseusing the system supplied by Genomic Solutions 125SDS-PAGE gels prepared using 065 NN-methylenebi-sacrylamide as cross-linking agent were used for pH 4ndash7and pH 45ndash55 IPG strips whereas pH 55ndash67 and pH 6ndash11 strips were applied to similar gels made with 08 cross-linker Electrophoresis was performed while cooling using themaximum setting (ordf 4ndash10infinC) at 20 000 mW per gel constantpower and a maximum voltage of 500 V Gels were thenstained with either colloidal Coomassie G-250 (Neuhoff et al1988) or silver nitrate (Rabilloud 1992) and scanned in aProXPRESS proteomic imaging system (Perkin-Elmer)Image analysis was performed using PHORETIX 2D version51 (NonLinear Dynamics) spot detection was optimizedautomatically using the lsquoSpot Detection Wizardrsquo and thenedited manually background subtraction was performedautomatically using the lsquoMode of Non-Spotrsquo setting imageswere then normalized to the total spot volume for quantifica-tion Spot filtering was not used
Protein identification using mass spectrometry
Protein spots of interest were excised from colloidalCoomassie-stained gels in-gel digested with trypsin andidentified by MALDI-TOF peptide mass fingerprint analysisExcised gel pieces (ordf 1-mm-diameter circles cut from 1-mm-thick gels) were washed twice with 100 ml of 50 mMammonium bicarbonate for 15 min once with 100 ml of 20acetonitrile-40 mM ammonium bicarbonate for 15 min andonce with 100 acetonitrile Washed gel pieces were allowedto air-dry for 10 min before being rehydrated in 5 ml of10 mg ml-1 trypsin (sequencing grade modified Promega) in10 mM ammonium bicarbonate Digestion (37infinC for 4 h) wasstopped by the addition of 5 ml of 5 formic acid Extractionof peptides into this solution was encouraged by a sonicwater bath treatment for 20 min Peptide extract (05 ml) wasspotted on to a thin layer of a-cyano-4-hydroxycinnamic acidapplied to a MALDI-TOF sample template and analysed byMALDI-TOF mass spectrometry (Bruker Reflex III) using anaccelerating voltage of 25 kV Samples were externally cali-brated using a standard mixture of six peptides ranging in
mass from 10465423 Da to 34946500 Da and the massaccuracy obtained was 60 ppm or better Identification ofproteins from MALDI-TOF peptide mass fingerprint data wasperformed using the lsquoMascotrsquo search engine at httpwwwmatrixsciencecom and was based on a positive resultusing their lsquoProbability Based MOWSE Scorersquo algorithm AMOWSE score of 60 or higher is significant at the 5 levelor better and proteins in this work typically gave scores gt80(frequently considerably so) In addition no identification wasaccepted unless at least five peptides representing at least20 of the protein sequence were detected in the MALDI-TOF peptide mass fingerprint
Assignment of proteins to metabolic pathways
Proteins were assigned to primary metabolic pathwaysusing the lsquoProtein Classification Schemersquo web page ofthe annotated Streptomyces coelicolor genome sequenceat httpwwwsangeracukProjectsS_coelicolor as it ap-peared in May 2002 The annotation of the genome andtherefore the Protein Classification Scheme is likely to bemodified as more experimental data are obtained about thefunction of genes that have currently been assigned on thebasis of sequence information alone It is therefore possiblethat proteins assigned to metabolic pathways in this studymay in future be shown to perform different functions in thecell
Assignments to gene clusters encoding secondary metab-olites are based on an analysis of the genome sequencereported by Bentley et al (2002) In some cases theseassignments are based on extensive experimental data (actred reviewed by Chater and Bibb 1997 Champness 2000cda Chong et al 1998 Huang et al 2001 whiE Davis andChater 1990)
Acknowledgements
This work was funded by grants (2FGT11406 208FGT11408 and 208IGF12432) under the BBSRCrsquos Tech-nologies for Functional Genomics and Investigating GeneFunction initiatives and by a competitive strategic grant fromthe BBSRC to the John Innes Centre We thank Mike Naldrettfor his extensive contribution in establishing technology forproteome analysis at the JIC
Supplementary material
The following material is available from httpwwwblackwell-sciencecomproductsjournalssuppmatmolemole3219mmi3219smhtmSection 1 Detection of gene products assigned to individualmetabolic steps in primary metabolic pathways in ScoelicolorSection 2 Identification of proteins from individual amino acidbiosynthetic pathwaysSection 3 Detection of gene products assigned to individualmetabolic steps in secondary metabolic pathways in Scoelicolor
Proteomic analysis of S coelicolor 931
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
References
Anderson TB Brian P and Champness WC (2001)Genetic and transcriptional analysis of absA an antibioticgene cluster linked two-component system that regulatesmultiple antibiotics in Streptomyces coelicolor Mol Micro-biol 39 553ndash566
Bentley SD Chater KF Cerdeno-Tarraga A-M ChallisGL Thomson NR James KD et al (2002) Completegenome sequence of the model actinomycete Streptomy-ces coelicolor A3(2) Nature 417 141ndash147
Berdy J (1984) New ways to obtain antibiotics Chin J Anti-biot 7 272ndash290
Berggren K Chernokalskaya E Steinberg TH KemperC Lopez MF Diwu Z et al (2000) Background-freehigh sensitivity staining of proteins in one- and two-dimensional sodium dodecyl sulfate-polyacrylamide gelsusing a luminescent ruthenium complex Electrophoresis21 2509ndash2521
Blackstock WP and Weir MP (1999) Proteomics quan-titative and physical mapping of cellular proteins TrendsBiotechnol 17 121ndash127
Blattner FR Plunkett G Bloch CA Perna NT BurlandV Riley M et al (1997) The complete genome sequenceof Escherichia coli K-12 Science 277 1453ndash1462
Buttner K Bernhardt J Scharf C Schmid R MaderU Eymann C et al (2001) A comprehensive two-dimensional map of cytosolic proteins of Bacillus subtilisElectrophoresis 22 2908ndash2935
Champness W (2000) Actinomycete development antibioticproduction and phylogeny questions and challenges InProkaryotic Development Brun YV and Skimketts LJ(eds) Washington DC American Society for MicrobiologyPress pp 11ndash31
Chater KF and Bibb MJ (1997) Regulation of bacterialantibiotic production In Biotechnology Vol 7 Products ofSecondary Metabolism Kleinkauf H and von Doumlhren H(eds) Weinheim Germany VCH pp 57ndash105
Chong PP Podmore SM Kieser HM Redenbach MTurgay K Marahiel M et al (1998) Physical identificationof a chromosomal locus encoding biosynthetic genesfor the lipopeptide calcium-dependent antibiotic (CDA) ofStreptomyces coelicolor A3(2) Microbiology 144 193ndash199
de Crecy-Lagard V Servant-Moisson JV Grandvalet Cand Mazodier P (1999) Alteration of the synthesis of theClp ATP-dependent protease affects morphological andphysiological differentiation in Streptomyces Mol Microbiol32 505ndash517
Davis NK and Chater KF (1990) Spore colour inStreptomyces coelicolor A3(2) involves the developmen-tally regulated synthesis of a compound biosyntheticallyrelated to polyketide antibiotics Mol Microbiol 4 1679ndash1691
Fernandez-Moreno FA Martinez E Caballero JLIchinose K Hopwood DA and Malpartida F (1994)DNA sequence and functions of the actVI region of theactinorhodin biosynthetic gene cluster of Streptomycescoelicolor A3(2) J Biol Chem 269 24854ndash24863
Fink D Falke D Wohllheben W and Engels A (1999)Nitrogen metabolism in Streptomyces coelicolor A3(2)
modification of glutamine synthase I by an adenylyltrans-ferase Microbiology 145 2313ndash2322
Godovac-Zimmermann J Soskic V Posnanovic S andBrianza F (1999) Functional proteomics of signal trans-duction by membrane receptors Electrophoresis 20 952ndash961
Gorg A Obermaier C Boguth G Harder A Scheibe BWildgruber R and Weiss W (2000) The current state oftwo-dimensional electrophoresis with immobilized pH gra-dients Electrophoresis 21 1037ndash1053
Gramajo HC Takano E and Bibb MJ (1993) Stationary-phase production of the antibiotic actinorhodin in Strepto-myces coelicolor A3(2) is transcriptionally regulated MolMicrobiol 7 837ndash845
Gross CA Chan C Dombroski A Gruber T Sharp MTupy J and Young B (1998) The functional and regula-tory roles of sigma factors in transcription Cold SpringHarb Symp Quant Biol 63 141ndash155
Hesketh A Sun J and Bibb MJ (2001) Induction ofppGpp synthesis in Streptomyces coelicolor A3(2) grownunder conditions of nutritional sufficiency elicits actII-ORF4transcription and actinorhodin biosynthesis Mol Microbiol39 136ndash144
Hesketh A Fink D Gust B Rexer H-U Scheel BChater K et al (2002) The PII nitrogen regulatory proteinGlnK in Streptomyces coelicolor A3(2) is modified by ade-nylylation rather than uridylylation and the GlnD and GlnKhomologues are functionally dissimilar to their nitrogenregulatory system counterparts Mol Microbiol (inpress)
Hood DW Heidstra R Swodoba UK and Hodgson DA(1992) Molecular genetic analysis of proline and tryp-tophan biosynthesis in Streptomyces coelicolor A3(2)interaction between primary and secondary metabolism- areview Gene 115 5ndash12
Hu DS-J Hood DW Heidstra R and Hodgson DA(1999) The expression of the trpD trpC and trpBA genesof Streptomyces coelicolor A3(2) is regulated by growthrate and growth phase but not by feedback repression MolMicrobiol 32 869ndash880
Huang J Lih CJ Pan KH and Cohen SN (2001)Global analysis of growth phase responsive gene expres-sion and regulation of antibiotic biosynthetic pathways inStreptomyces coelicolor using DNA microarrays GenesDev 15 3183ndash3192
Ichinose K Taguchi T Ebizuka Y and Hopwood DA(1998a) Biosynthetic gene clusters of benzoisochromane-quinone antibiotics in Streptomyces spp Identification ofgenes involved in post-PKS tailoring steps Actinomyceto-logica 12 99ndash109
Ichinose K Bedford DJ Tornus D Bechthold A BibbMJ Revill PW et al (1998b) The granaticin biosyntheticgene cluster of Streptomyces violaceoruber Tu22sequence analysis and expression in a heterologous hostChem Biol 5 647ndash659
Kelemen GH Brian P Flardh K Chamberlin L ChaterK and Buttner M (1998) Developmental regulation oftranscription of whiE a locus specifying the polyketidespore pigment in Streptomyces coelicolor A3(2) J Bacte-riol 180 2515ndash2521
Kieser T Bibb MJ Buttner MJ Chater KF and
932 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
Hopwood DA (2000) Practical Streptomyces GeneticsNorwich John Innes Foundation
Kim EJ Chung HJ Suh B Hah YC and Roe JH(1998) Expression and regulation of the sodF gene encod-ing iron- and zinc-containing superoxide dismutase inStreptomyces coelicolor Muumlller J Bacteriol 180 2014ndash2020
Kimura Y Takaoka M Tanaka S Sassa H Tanaka KPolevoda B et al (2000) Na-acetylation and proteolyticactivity of the yeast 20S proteasome J Biol Chem 2754635ndash4639
Kunst F Ogasawara N Moszer I Albertini AM AlloniG Azevedo V et al (1997) The complete genomesequence of the Gram-positive bacterium Bacillus subtilisNature 390 249ndash256
Lucchini S Thompson A and Hinton JCD (2001)Microarrays for microbiologists Microbiology 147 1403ndash1414
Mann M Hendrickson RC and Pandey A (2001) Analy-sis of proteins and proteomes by mass spectrometry AnnuRev Biochem 70 437ndash473
Miyodah S (1993) Research on antibiotic screening inJapan over the last decade a producing microorganismsapproach Actinomycetologica 7 100ndash106
Molloy MP Herbert BR Williams KL and Gooley AA(1999) Extraction of Escherichia coli proteins with organicsolvents prior to two-dimensional electrophoresis Electro-phoresis 20 701ndash704
Nagy I Tamura T Vanderleyden J Baumeister W andDe Mot R (1998) The 20S proteasome of Streptomycescoelicolor J Bacteriol 180 5448ndash5453
Neuhoff V Arold N Taube D and Ehrhardt W (1988)Improved staining of proteins in polyacrylamide gels includ-ing isoelectric focusing gels with clear background at nan-ogram sensitivity using Coomassie Brilliant Blue G-250and R-250 Electrophoresis 9 255ndash262
Nilsson CL and Davidsson P (2000) New separation toolsfor comprehensive studies of protein expression by massspectrometry Mass Spectrom Rev 19 390ndash397
Puglia AM Vohradsky J and Thompson CJ (1995)
Developmental control of the heat-shock stress regulon inStreptomyces coelicolor Mol Microbiol 17 737ndash746
Rabilloud T (1992) A comparison between low backgroundsilver diamine and silver nitrate protein stains Electro-phoresis 13 429ndash439
Robinson VL Buckler DR and Stock AM (2000) A taleof two components a novel kinase and a regulatory switchNature Struct Biol 7 626ndash633
Schena M (2000) Microarray Biochip Technology NatickMA Eaton Publishing
Spusansky O Rezuchova B Homerova D andKormanec J (2001) Expression of the gap gene encodingglyceraldehyde-3-phosphate dehydrogenase of Strepto-myces aureofaciens requires GapR a member of theAraCXylS family of transcriptional activators Microbiology147 1291ndash1301
Taguchi T Itou K Ebizuka Y Malpartida F HopwoodDA Surti CM et al (2000) Chemical characterisationof disruptants of the Streptomyces coelicolor A3(2) actVIgenes involved in actinorhodin biosynthesis J Antibiot 53144ndash152
Takano E Gramajo HC Strauch E Andres N WhiteJ and Bibb MJ (1992) Transcriptional regulation of theredD transcriptional activator gene accounts for growth-phase-dependent production of the antibotic undecylpro-digiosin in Streptomyces coelicolor A3(2) Mol Microbiol6 2797ndash2804
Tonella L Hoogland C Binz P-A Appel RDHochstrasser DF and Sanchez J-C (2001) New per-spectives in the Escherichia coli proteome investigationProteomics 1 409ndash423
Vohradsky J Li XM and Thompson CJ (1997) Identifi-cation of prokaryotic developmental stages by statisticalanalysis of two-dimensional gel patterns Electrophoresis18 1418ndash1428
Vohradsky J Li XM Dale G Folcher M Nguyen LViollier PH and Thompson CJ (2000) Developmentalcontrol of stress stimulons in Streptomyces coelicolorrevealed by statistical analyses of global gene expressionpatterns J Bacteriol 182 4979ndash4986

924
A R Hesketh
et al
copy 2002 Blackwell Publishing Ltd
Molecular Microbiology
46
917ndash932
tated as enzymes of glycolysis the TCA cycle thepentose phosphate pathway purine ribonucleotide bio-synthesis and pyrimidine nucleotide biosynthesis beingmapped (Table 6) About 50 of amino acid biosyntheticproteins were identified Lists of individual members of allpathways considered here including those that were andwere not identified during proteome mapping can befound in the
Supplementary material
Several proteins from both primary and secondary met-
abolic pathways were identified at more than one positionon the proteome map and may therefore be subject toregulation by post-translational modification (see Table 3and Fig 1) Primary metabolic proteins of this type (yellowspots on Fig 1) were usually detected as chains of two ormore spots with the same apparent molecular weights butdifferent pI values suggesting covalent modification bylow-molecular-weight adducts Apart from adenylylation ofGSI (see above) the nature of the modification of theseproteins was not revealed by MALDI-TOF peptide massfingerprint data Both covalent modification and proteolyticprocessing were also found in secondary metabolic pro-teins (red spots in Fig 1) For example the Act pathwaymembers SCO5071 (spot 16) and SCO5075 (spot 20)
and the putative 3-oxoacyl[acyl carrier protein] reductasefrom the type I polyketide cluster (SCO6282 spot 11)appear to be covalently modified whereas proteolytic pro-cessing was detected for RedL (SCO5892 spot 18) fromthe undecylprodigiosin cluster the putative dehydrataseActVI-ORF3 (SCO5074 spot 31) from the Act cluster theputative peptide monooxygenase from coelichelin biosyn-thesis (SCO0498 spot 34) and the putative oxygenasefrom the CDA pathway (SCO3236 spot 33) Two Act bio-synthetic proteins SCO5088 (spot 24) and SCO5073(spot 29) were detected both at their predicted pI andmolecular weight co-ordinates and at positions with simi-lar pI values but significantly higher molecular weightsThis may indicate that these proteins form stable mul-timers that are not completely denatured under theconditions used Only the modification of ActVI-ORF3(SCO5074 spot 31) could be characterized further fromthe mass spectrometry data which showed that it involvesproteolytic processing at two sites between amino acidresidues 3132 and 3435 (data not shown) Analysis of asample obtained by externally extracting proteins fromintact washed mycelium with a solution containing 2SDS and 50 mM dithiothreitol (DTT) revealed that sur-
Table 5
Summary of proteins identified on the proteome map from secondary metabolic pathways during growth in SMM
Biosynthetic pathwaygene cluster
Pathwaygenesannotated
Pathwaygene productsidentified
Percentagepathwaydetected
Calcium-dependent antibiotic CDA SCO3210ndash3249 40 16 40Actinorhodin SCO5071ndash5092 22 15 68Undecylprodigiosin SCO5877ndash5898 22 8 36Deoxysugar synthaseglycosyltransferase cluster SCO0381ndash0401 21 7 33Type I polyketide synthase SCO6273ndash6288 16 6 38Hopanoids SCO6759ndash6771 13 1 7Siderophore (coelichelin) SCO0489ndash0499 11 5 45Siderophore (desferrioxamine) SCO2783ndash2785 4 4 100
Chromosomal locations and extent of gene clusters are taken from Bentley
et al
(2002) No proteins were detected from the remaining predictedsecondary metabolic clusters ie siderophore (coelibactin) SCO7681ndash7691 (11 ORFs) siderophore SCO5799ndash5801 (three ORFs) non-ribosomal peptide synthases SCO6429ndash6438 (10 ORFs) type II fatty acid synthases SCO1265ndash1273 (nine ORFs) WhiE spore pigment clusterSCO5314ndash5320 (seven ORFs) isorenieratine SCO0185ndash0191 (seven ORFs) eicosapentaenoic acid SCO0124ndash0129 (six ORFs) chalconesynthases SCO1206ndash1208 (three ORFs) chalcone synthases SCO7669ndash7671 and SCO7222 (three ORFs) sesquiterpene cyclase SCO5222ndash5223 (two ORFs) type I polyketide synthases SCO6826ndash6827 (two ORFs) geosmin SCO6703 (one ORF)
Table 6 Summary of proteins identified on the proteome map from primary metabolic pathways during growth in SMM
Biosynthetic pathwayPathway genesannotateda
Pathway geneproducts identified
Percentage pathway detected
Glycolysisb 24 13 55Pentose phosphate pathwayb 14 10 71TCA cycleb 21 12 57Amino acid biosynthesisc 115 55 48Purine ribonucleotide biosynthesisb 18 12 67Pyrimidine ribonucleotide biosynthesisb 8 5 62
a Derived from interpretation of the annotated S coelicolor genome sequence at httpwwwsangeracukProjectsS_coelicolorb See Supplementary material for details of pathway membersc See Supplementary material for details of individual pathways
Proteomic analysis of S coelicolor 925
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
prisingly the truncated ActVI-ORF3 is located extracellu-larly presumably non-covalently associated with the cellwall matrix (Fig 4)
Expression analysis of certain primary and secondary metabolic proteins
Production of the pigmented antibiotics Act and Red inliquid minimal medium supplemented with casamino acids(SMM) begins in the transition phase between exponentialgrowth and stationary phase Figure 3 shows a compari-son of the expression of certain primary and secondarymetabolic proteins between exponentially growing andstationary phase cultures Two proteins from the Actcluster (ActVA-ORF5 and ActI-ORF2) and two from theRed (RedI and RedR) were present in the stationaryphase cultures but were not detected during exponentialgrowth Similarly the putative 3-oxoacyl-[acyl carrierprotein] reductase (SCO6282) and epoxide hydrolase(SCO6277) enzymes from a type I polyketide secondarymetabolic cluster showed a large increase in abundance
between the exponential and stationary phase samplesTriose phosphate isomerase (SCO1945) fructose bis-phosphate aldolase (SCO3649) and succinyl-CoA syn-thetase alpha chain (SCO4809) all enzymes from centralcarbon metabolism were present in approximatelyconstant amounts in both samples Interestingly two iso-forms (SCO7511 and SCO1947) of glyceraldehyde-3-phosphate dehydrogenase from the glycolytic pathwayshowed different patterns of expression SCO1947 levelswere the same in both exponential and stationary phasebut SCO7511 barely detectable in exponentially growingcultures increased markedly in stationary phase
The Streptomyces coelicolor online proteome map
The 2D gel images in Fig 1 together with the spot iden-tification results are being made available over the WorldWide Web to provide a reference map of the S coelicolorproteome At the time of writing the reference gel forthe pH 4ndash7 isoelectric point range was fully accessiblewhereas those for pH 45ndash55 pH 55ndash67 and pH 6ndash11
Fig 3 A comparison of the expression of certain primary and secondary metabolic proteins between exponentially growing and stationary phase cultures of S coelicolor M145 Larger arrows represent higher protein level in one gel than in the other The experiment was performed on duplicate biological samples and both showed similar protein expression profiles
926 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
were under construction (httpqbababeracukS_coeliReferenceGelrefgelhtml) A tool for comparing two ormore gel images with each other is also accessible fromthe same website at httpqbababeracukS_coeliflickerhtml
Discussion
Silver staining is one of the most sensitive methods fordetecting proteins on polyacrylamide gels allowing aslittle as 05ndash2 ng of an individual protein to be detected(Berggren et al 2000 Gorg et al 2000) The proportionof the S coelicolor proteome that can be analysed usingsilver staining of 2D-PAGE-separated proteins dependsat least in part on the pH range of the IPG strip used forthe first-dimension separation (see Table 1) Using theintermediate-range IPG strip covering pH 4ndash7 about 20ndash25 of the proteins in the theoretical proteome predictedto be observable in the resulting 2D-PAGE gel were actu-ally detected This figure was reduced to 10ndash12 whenanalysing the more basic pI range using the pH 6ndash11 IPGstrip Attempts to increase the percentage of the proteomedetectable using the pH 4ndash7 and pH 6ndash11 IPG strips byloading more protein sample resulted in poorer separa-tions (data not shown) This was especially true of thepH 6ndash11 strips which were generally the most difficult touse successfully However good separation of higher pro-tein loads was achieved using the narrow-range IPG stripspH 45ndash55 and 55ndash67 significantly increasing the pro-portion of theoretical proteins detected to over 40Clearly and more pertinently the limited conditions usedfor culturing S coelicolor in this study will also haverestricted the number of proteins detected as not all theproteome will be expressed In particular S coelicolordoes not sporulate in shaken liquid-grown cultures soproteins involved in this differentiation process are unlikelyto be present Nevertheless it is striking that the com-bined data for the pH 45ndash55 and 55ndash67 isoelectric pointranges indicate that 2468 out of a total of predicted 4932protein spots can be detected (see Table 1) suggestingthat transition phase mycelium in this supplementedminimal medium is expressing 50 of its genes (ie byextrapolation some 3912 of the 7825 in the genome)
Analysis of post-translational regulation
Although the regulation of cellular processes and adaptiveresponses in bacteria has been studied extensively at thetranscriptional level post-translational regulatory mecha-nisms are much less understood Proteomic analysisusing 2D-PAGE coupled with mass spectrometry offersthe opportunity to observe and identify modification ofproteins at a general cellular level The functions of pro-teins can be altered by specific covalent attachment of
adducts (eg phosphorylation glycosylation nucleotidy-lation acetylation) or by proteolytic processing Bothevents alter the pI andor mass of the protein and so canusually be observed on 2D-PAGE In the S coelicolorproteome map ordf110 out of a total of 770 gene productswere identified in more than one position or at pI andapparent mass co-ordinates significantly different frompredicted values indicating extensive regulation at thepost-translational level (88 are listed in Table 3) a propor-tion similar to that reported for other prokaryotes (Buttneret al 2001 Tonella et al 2001) The initial peptide massfingerprint analysis identified the modification of only 10of these proteins probably partly because fingerprint datatypically cover ordf30ndash60 of a proteinrsquos sequence (data notshown) so modification at positions not represented willbe missed Protein digestion with endopeptidases otherthan trypsin before mass spectrometry may help in thissituation but some covalently attached groups are labileand cannot be detected routinely Phosphorylation of Hisoccurs on nitrogen producing a high-energy phosphora-midate bond that is easily hydrolysed in acidic conditionswhereas phospho-Asp is the most labile phosphorylatedresidue with a half-life of a few hours under physiologicalconditions (reviewed by Robinson et al 2000) Thesemodifications the basis of bacterial two-component regu-latory systems are therefore unlikely to survive stainingof the 2D gels which involved the use of a fixing solutioncontaining 10 acetic acid and mass spectrometry Iden-tification of the more stable phosphoester phosphorylationthat occurs on Ser Thr or Tyr residues using PAGEfollowed by mass spectrometry has been reported(Godovac-Zimmermann et al 1999 Buttner et al 2001)but no examples were detected in this work Of the mod-ifications identified in this study three were by acetylationat the N-terminus after removal of the initiator Met residuetwo involved adenylylation and five proteins had been N-terminally truncated Modification of the 20S proteasomeby N-acetylation in yeast involves removal of the Metresidue by N-methionylaminopeptidase followed byacetylation with an Na-acetyltransferase (Kimura et al2000) Homologues of these enzymes are predicted fromthe S coelicolor genome and are presumably responsiblefor the observed modification of the 20S proteasomealpha subunit and of BldK-ORFD and the putative cellu-lose-binding protein The functional significance of Na-acetylation of these proteins is unclear
Analysis of the biosynthesis of antibiotics and other secondary metabolites
Post-translational modification The proteome mappingresults indicate that at least six enzymes in the Act clusterare subject to post-translational modifications and one
Proteomic analysis of S coelicolor 927
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
each from the undecylprodigiosin (SCO5877ndash5898)lsquocoelichelinrsquo (SCO0489ndash0499) and type I polyketidesynthase (SCO6273ndash6288) clusters (see Table 3) Thesemetabolic pathways may therefore be significantly regu-lated at the post-translational level The actVI-ORF3 gene(SCO5074) encodes a putative dehydratase enzyme thatis part of the biosynthetic pathway for Act a blue memberof the benzoisochromanequinone (BIQ) family of pig-mented antibiotics Mutation in actVI-ORF3 causes a 50decrease in the accumulation of Act (and a reddish brownphenotype) and it is believed to be responsible for pyranring closure an intermediate step in Act biosynthesis (thatmay also occur spontaneously) leading to the formationof the BIQ chromophore (Fernandez-Moreno et al 1994Ichinose et al 1998a Taguchi et al 2000) In this studyActVI-ORF3 protein was detected in two forms a majorspot corresponding to ActVI-ORF3 truncated by removalof the N-terminal 31 amino acid residues and a minor onein which the first 34 amino acids were missing (spots 31in Fig 1) The full-length protein was not detected Anal-ysis of the protein sequence using the SIGNALP server athttpwwwcbsdtudkservicesSignalP predicted a signalpeptide cleavage site between amino acids 31 and 32 inagreement with the observed results
The protein extract analysed was prepared from myce-lium that had been collected by centrifugation and sub-jected to two washes indicating that if ActVI-ORF3 isexported it is effectively retained within the cell wallmatrix This was confirmed by analysis of a preparationobtained by externally extracting proteins from intactwashed mycelium (Fig 4) Export of an enzyme requiredfor the efficient biosynthesis of an antibiotic that is thoughtto be produced intracellularly appears to be anomalousparticularly as it is involved in an intermediate biosynthetic
event Database analysis of the other members of the Actcluster revealed only one other protein with a predictedsignal peptide sequence the putative integral membranetransport-related protein ActII-ORF3 and it seemsunlikely that all the Act biosynthetic steps following BIQchromophore formation are extracellular It is possible thatfull-length ActVI-ORF3 catalyses the intermediate biosyn-thetic event intracellularly and the exported processedprotein is responsible for a different extracellular activityperhaps later in the pathway However no full-length pro-tein was detected so the exported enzyme may performthe pyran ring closure on a secreted metabolite that isthen reabsorbed by the cell for completion of the sub-sequent biosynthetic steps Only two homologues ofactVI-ORF3 are known and both are also in BIQ antibioticbiosynthetic clusters gra-ORF18 from the granaticincluster (Ichinose et al 1998b) and med-ORF5 frommedermycin biosynthesis (K Ichinose personal com-munication) Interestingly the encoded proteins of bothsequences are also predicted to contain N-terminal signalpeptides The observation that ActVI-ORF3 is exported tothe cell wall matrix may therefore be of general signifi-cance to the BIQ family of antibiotics and provide a newinsight into their biosynthetic pathways
Pathway representation Of the 20 putative secondarymetabolite gene clusters identified in the S coelicolorgenome sequence (Bentley et al 2002) eight had atleast one protein each identified on the proteome map(see Table 5) The biosynthetic clusters for the antibioticsAct Red and CDA had 68 36 and 40 of their pro-teins represented There are several possible explana-tions for failure to detect all the proteins for each pathwaySome are putative integral membrane proteins so cannotreadily be detected using conventional 2D-PAGE SomeRed and CDA proteins have molecular weights gt200 kDaoutside the range of the 125 gels used (for clustermembers see Supplementary material) The pathway-specific regulatory proteins for Act and Red biosynthesiswere not detected despite a detailed search of their pre-dicted gel locations even though the relevant genes areknown to be transcribed in equivalent cultures of the samestrain (Hesketh et al 2001) The proteins are presumablypresent but at levels below the limit of detection Howeverthe transcriptional regulator for CDA production CdaRwas identified as well as the two-component systemresponse regulator AbsA2 which is also encoded by agene in the CDA cluster (Anderson et al 2001) Interest-ingly proteins from three putative secondary meta-bolite clusters producing chemicals of as yet unknownstructure were also identified on the map correspondingto the deoxysugar synthaseglycosyltransferase clusterSCO0381ndash0401 the lsquocoelichelinrsquo cluster for a predicted(but not yet confirmed) oligopeptide siderophore
Fig 4 Processed ActVI-ORF3 is present in the cell wall matrix 2D-gel separations of protein extracts prepared (A) from whole cells or (B) by externally washing cells in a 2 SDS extraction buffer The arrow indicates ActVI-ORF3 protein The experiment was performed on duplicate biological samples and both showed similar protein expression profiles
928 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
(SCO0489ndash0499) and the type I polyketide clusterSCO6273ndash6288 The 3-oxoacyl-[acyl carrier protein]reductase (SCO6282) from the latter pathway is ex-pressed in a growth phase-dependent manner (Fig 3)and in early stationary phase cultures is perhaps themost abundant protein spot in the entire detectable pro-teome Presumably the metabolites produced by thesethree pathways are being synthesized under the condi-tions used In addition to coelichelin three other sidero-phores are postulated for S coelicolor All the proteinsfrom one of these desferrioxamine (SCO2782ndash2785)were identified but none was seen from either of the othertwo pathways The WhiE polyketide spore pigment isassociated with sporulation of surface-grown cultures(Davis and Chater 1990) and the expression of the whiEgenes depends on a series of sporulation-specific regula-tory genes (Kelemen et al 1998) no corresponding pro-teins of which were detected here It is therefore notsurprising that WhiE proteins were not detected in sam-ples prepared from liquid-grown vegetative mycelium
Analysis of primary metabolism
Proteins annotated as functioning in primary metabolicpathways were highly represented on the proteome mapwith ordf60 or more of the enzymes assigned to glycolysisthe TCA cycle the pentose phosphate pathway andpurine and pyrimidine ribonucleotide biosynthesis beingidentified (see Table 6) This figure may be deceptively lowbecause these pathways in S coelicolor often appear tohave more than one enzyme isoform for some of themetabolic steps potentially offering additional levels ofcontrol or developmental responsiveness at these pointsFor example only two steps in glycolysis have just a singlegene product assigned to them the remainder typicallyhave two or three each (Fig 5) At least one isoform waspresent for each step so the pathway is in theory 100functionally represented Understanding the regulation ofthese pathways will require analysis of the expression ofthese isoenzymes under different nutritional conditionsor different developmental stages Figure 5 shows that ofthe three predicted glyceraldehyde-3-phosphate dehydro-genase isoforms from glycolysis only two were detectedin this study (see also Supplementary material) Of theseSCO1947 appears to be constitutive whereas SCO7511is growth phase dependent (see Fig 3) Thus even thelimited conditions in this study yielded useful informationabout isoenzyme expression patterns (see Supplemen-tary material) In Streptomyces aureofaciens two genesencoding glyceraldehyde-3-phosphate dehydrogenaseactivity also appear to be differently regulated transcrip-tion of one highly homologous to SCO7511 is activatedby the regulator GapR whereas the other is GapR inde-pendent (Spusansky et al 2001)
Hood et al (1992) reported that most amino acid bio-synthetic enzymes are expressed at very low constitutivelevels in S coelicolor so it is reassuring to note that ordf50could be detected and identified using proteomics Theseinclude TrpB and TrpC1 from the tryptophan biosyntheticpathway the corresponding transcripts of which couldbarely be detected in previously reported S1 nucleaseprotection experiments (Hu et al 1999)
Perspectives
No attempt was made in the work reported here to simplifythe protein separations by subdividing the sample intosoluble and membrane fractions and this is reflected inthe 10 lipoproteins and 16 ABC transport system ATP-binding subunits identified on the proteome map (seeTable 2) No integral membrane proteins were detectedand even when a membrane protein sample was pre-pared and analysed no proteins identified possessedmore than three predicted transmembrane helices (datanot shown) This is consistent with previous studiesin other bacteria (Molloy et al 1999 Nilsson andDavidsson 2000) Another group of proteins also strik-ingly absent from the list of those identified is the sigmafactors which determine promoter selection by RNA poly-merase There are an extraordinarily high number (65) ofsigma factors encoded in the S coelicolor genome aswell as at least seven antisigma factors and 15 antisigmafactor antagonists (some of each of which were detected)Sigma factor heterogeneity in bacteria is an important wayof regulating expression of specific sets of genes under
Fig 5 The glycolytic pathway in S coelicolor illustrating the assignment of multiple gene products to individual metabolic steps based on the annotation of the genome sequence at httpwwwsangeracukProjectsS_coelicolor Gene products identified in the proteome map are underlined
Proteomic analysis of S coelicolor 929
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
different conditions and in different cell types (for a reviewsee Gross et al 1998) Although no sigma factors wereidentified about half (31 out of 65) have predicted pIvalues gtpH 7 and so can only be detected using thepH 6ndash11 IPG strips that give the poorest results (seeTable 1) However the principal vegetative sigma factorHrdB which is expected to be moderately abundanthas a predicted pI of 613 and its absence from thepH 55ndash67 proteome map may indicate that the samplepreparation method which does not remove DNA is inap-propriate Among other regulatory proteins 11 out of apossible 80 two-component system response regulatorsplus a further 36 proteins annotated as lsquoregulatorsrsquo weredetected Only one SerThr kinase and no two-componentsystem sensor kinases were detected possibly reflectingthe difficulty in analysing proteins with several transmem-brane domains Interestingly 30 secreted proteins wereidentified on the proteome map at least 10 of whichappear from comparison of their observed pI and molec-ular weight positions with predicted values to have beenprocessed already and therefore exported Presumablythese proteins do not end up exclusively in the culturemedium
Clearly this approach to proteomic analysis of S coeli-color has some important deficiencies not least theabsence of certain groups of proteins and the relativelypoor initial success rate for specifically characterizingpost-translational modifications Nevertheless it offersunique insights into the molecular biology of the organismthe expression of many proteins from both primary andsecondary metabolic pathways can be analysed post-translational regulation of proteins can be investigated ona general cellular level and the physical locations forsome proteins can be determined Approximately 35 ofthe S coelicolor genome is annotated as encoding lsquohypo-theticalrsquo proteins of unknown function and 182 proteins ofthis type were identified on the proteome map No exam-ples were found of overt disagreements between pre-dicted and observed proteins providing experimentalsupport for the general accuracy of the S coelicolorgenome sequence and its annotation Information aboutthe expression post-translational modification and physi-cal location of these (no longer) hypothetical proteinsfrom proteomic analyses is going to be important forassigning future functions As many of the hypotheticalproteins have apparent orthologues in pathogenic myco-bacteria some of the information may have an immediateapplication in the context of pathogenesis
Experimental procedures
Materials
Immobilized pH gradient (IPG) strips and IPG bufferswere purchased from Amersham Biosciences Acrylamide
duracryl and chemicals for silver staining were obtained fromGenomic Solutions All other chemicals were from Sigmaunless stated in the text
Strain and growth conditions
Streptomyces coelicolor M145 was cultivated with vigorousagitation at 30infinC in minimal medium supplemented with 02casamino acids (SMM) as described previously (Kieser et al2000) Briefly a high-density spore preparation [about 1010
colony-forming units (cfu) ml-1] was pregerminated in 2yen YTmedia for 7 h at 30infinC Germlings were harvested by centrifu-gation (5 min at 4000 g) resuspended in SMM and sonicatedbriefly to disperse any aggregates before inoculation into50 ml of SMM in 250 ml siliconized flasks containing coiledstainless steel springs Each flask received the equivalent of5 yen 107 spore cfu
Protein extraction and 2D-gel electrophoresis
The preparation of the protein sample to be separated waskept as simple as possible disrupting harvested myceliadirectly into denaturing isoelectric focusing (IEF) buffer con-taining Pefabloc SC (Roche) protease inhibitor This methodminimized unwanted protein degradation that was observedin initial studies using a Tris buffer containing Pefabloc SC forcell lysis (data not shown) Removal of DNA RNA salts andpolysaccharides from the sample was therefore not per-formed (other than the shearing of viscous DNA and RNA bysonication) and the protein extracts were sometimes difficultto focus when the sample was loaded by in-gel rehydrationLoading the sample soaked in paper strips at the anodicend of IPG strips that had already been rehydrated improvedthe reproducibility of separations significantly
Mycelium for protein extraction was harvested from cul-tures by brief centrifugation (30 s at 4000 g) at room tem-perature and immediately frozen in liquid nitrogen Typicallymycelium from 10 ml culture aliquots was collected and thetransfer time from culture flask to frozen sample was 15 minFrozen cells were thawed on ice in 5 ml of washing buffer(40 mM Tris pH 90 1 mM EGTA 1 mM EDTA) then pelletedby centrifugation (5 min at 3000 g) at 4infinC Washed cells wereresuspended in 400 ml of denaturing IEF buffer UTCHAPS[7 M urea 2 M thiourea 4 (wv) CHAPS 40 mM TrispH 90 1 mM EDTA 50 mM DTT 4 mM Pefabloc SC pro-tease inhibitor (Roche)] and disrupted by sonication (SanyoSoniprep 150 10 yen 2 s bursts at amplitude 75 mm) whilecooling in an ethanolndashice bath Cell debris was removed bycentrifugation (15 min 10 000 g 4infinC) and the protein extractwas stored frozen in aliquots at -80infinC until use For thepreparation of extracellular protein extracts washed cellswere resuspended in extraction buffer consisting of 40 mMTris pH 90 1 mM EDTA 50 mM DTT 2 SDS and 4 mMPefabloc SC protease inhibitor Cells were vortexed for 2 minbefore being pelleted by centrifugation (5 min 10 000 g4infinC) The supernatant was precipitated using the Amersham2D Clean-Up kit according to the manufacturerrsquos instructionsand the precipitated proteins were redissolved in UTCHAPSand stored at -80infinC until use
930 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
For first-dimension isoelectric focusing 18 cm IPG stripspH 4ndash7 pH 6ndash11 pH 45ndash55 or pH 55ndash67 (Amersham Bio-sciences) were rehydrated overnight in IEF buffer containing1 ampholytes according to the manufacturerrsquos instructionsusing a Phaser isoelectric focusing unit (Genomic Solutions)set at 20 V Protein samples to be separated were applied tothe rehydrated strips at the anodic end soaked in a 5ndash10 mmsection of IEF electrode strip (Amersham Biosciences)Separation was performed for 120 000 V-h with a maximumvoltage of 5000 V After isoelectric focusing IPG strips wereequilibrated for the second dimension for 15 min in IPG equil-ibration buffer (50 mM Tris pH 68 6 M urea 30 glycerol1 SDS and 001 bromophenol blue) plus 80 mM DTTthen for 10 min in IPG equilibration buffer plus 135 mMiodoacetamide Approximately 1 cm was removed from theanodic end of each equilibrated strip before applying the stripto the top of a vertical 125 SDS-PAGE gel for second-dimension separation using the Investigator 5000 systemfrom Genomic Solutions
Large-format gels (24 cm wide yen 22 cm high yen 1 mm thick)for the second-dimension separation were cast in-houseusing the system supplied by Genomic Solutions 125SDS-PAGE gels prepared using 065 NN-methylenebi-sacrylamide as cross-linking agent were used for pH 4ndash7and pH 45ndash55 IPG strips whereas pH 55ndash67 and pH 6ndash11 strips were applied to similar gels made with 08 cross-linker Electrophoresis was performed while cooling using themaximum setting (ordf 4ndash10infinC) at 20 000 mW per gel constantpower and a maximum voltage of 500 V Gels were thenstained with either colloidal Coomassie G-250 (Neuhoff et al1988) or silver nitrate (Rabilloud 1992) and scanned in aProXPRESS proteomic imaging system (Perkin-Elmer)Image analysis was performed using PHORETIX 2D version51 (NonLinear Dynamics) spot detection was optimizedautomatically using the lsquoSpot Detection Wizardrsquo and thenedited manually background subtraction was performedautomatically using the lsquoMode of Non-Spotrsquo setting imageswere then normalized to the total spot volume for quantifica-tion Spot filtering was not used
Protein identification using mass spectrometry
Protein spots of interest were excised from colloidalCoomassie-stained gels in-gel digested with trypsin andidentified by MALDI-TOF peptide mass fingerprint analysisExcised gel pieces (ordf 1-mm-diameter circles cut from 1-mm-thick gels) were washed twice with 100 ml of 50 mMammonium bicarbonate for 15 min once with 100 ml of 20acetonitrile-40 mM ammonium bicarbonate for 15 min andonce with 100 acetonitrile Washed gel pieces were allowedto air-dry for 10 min before being rehydrated in 5 ml of10 mg ml-1 trypsin (sequencing grade modified Promega) in10 mM ammonium bicarbonate Digestion (37infinC for 4 h) wasstopped by the addition of 5 ml of 5 formic acid Extractionof peptides into this solution was encouraged by a sonicwater bath treatment for 20 min Peptide extract (05 ml) wasspotted on to a thin layer of a-cyano-4-hydroxycinnamic acidapplied to a MALDI-TOF sample template and analysed byMALDI-TOF mass spectrometry (Bruker Reflex III) using anaccelerating voltage of 25 kV Samples were externally cali-brated using a standard mixture of six peptides ranging in
mass from 10465423 Da to 34946500 Da and the massaccuracy obtained was 60 ppm or better Identification ofproteins from MALDI-TOF peptide mass fingerprint data wasperformed using the lsquoMascotrsquo search engine at httpwwwmatrixsciencecom and was based on a positive resultusing their lsquoProbability Based MOWSE Scorersquo algorithm AMOWSE score of 60 or higher is significant at the 5 levelor better and proteins in this work typically gave scores gt80(frequently considerably so) In addition no identification wasaccepted unless at least five peptides representing at least20 of the protein sequence were detected in the MALDI-TOF peptide mass fingerprint
Assignment of proteins to metabolic pathways
Proteins were assigned to primary metabolic pathwaysusing the lsquoProtein Classification Schemersquo web page ofthe annotated Streptomyces coelicolor genome sequenceat httpwwwsangeracukProjectsS_coelicolor as it ap-peared in May 2002 The annotation of the genome andtherefore the Protein Classification Scheme is likely to bemodified as more experimental data are obtained about thefunction of genes that have currently been assigned on thebasis of sequence information alone It is therefore possiblethat proteins assigned to metabolic pathways in this studymay in future be shown to perform different functions in thecell
Assignments to gene clusters encoding secondary metab-olites are based on an analysis of the genome sequencereported by Bentley et al (2002) In some cases theseassignments are based on extensive experimental data (actred reviewed by Chater and Bibb 1997 Champness 2000cda Chong et al 1998 Huang et al 2001 whiE Davis andChater 1990)
Acknowledgements
This work was funded by grants (2FGT11406 208FGT11408 and 208IGF12432) under the BBSRCrsquos Tech-nologies for Functional Genomics and Investigating GeneFunction initiatives and by a competitive strategic grant fromthe BBSRC to the John Innes Centre We thank Mike Naldrettfor his extensive contribution in establishing technology forproteome analysis at the JIC
Supplementary material
The following material is available from httpwwwblackwell-sciencecomproductsjournalssuppmatmolemole3219mmi3219smhtmSection 1 Detection of gene products assigned to individualmetabolic steps in primary metabolic pathways in ScoelicolorSection 2 Identification of proteins from individual amino acidbiosynthetic pathwaysSection 3 Detection of gene products assigned to individualmetabolic steps in secondary metabolic pathways in Scoelicolor
Proteomic analysis of S coelicolor 931
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
References
Anderson TB Brian P and Champness WC (2001)Genetic and transcriptional analysis of absA an antibioticgene cluster linked two-component system that regulatesmultiple antibiotics in Streptomyces coelicolor Mol Micro-biol 39 553ndash566
Bentley SD Chater KF Cerdeno-Tarraga A-M ChallisGL Thomson NR James KD et al (2002) Completegenome sequence of the model actinomycete Streptomy-ces coelicolor A3(2) Nature 417 141ndash147
Berdy J (1984) New ways to obtain antibiotics Chin J Anti-biot 7 272ndash290
Berggren K Chernokalskaya E Steinberg TH KemperC Lopez MF Diwu Z et al (2000) Background-freehigh sensitivity staining of proteins in one- and two-dimensional sodium dodecyl sulfate-polyacrylamide gelsusing a luminescent ruthenium complex Electrophoresis21 2509ndash2521
Blackstock WP and Weir MP (1999) Proteomics quan-titative and physical mapping of cellular proteins TrendsBiotechnol 17 121ndash127
Blattner FR Plunkett G Bloch CA Perna NT BurlandV Riley M et al (1997) The complete genome sequenceof Escherichia coli K-12 Science 277 1453ndash1462
Buttner K Bernhardt J Scharf C Schmid R MaderU Eymann C et al (2001) A comprehensive two-dimensional map of cytosolic proteins of Bacillus subtilisElectrophoresis 22 2908ndash2935
Champness W (2000) Actinomycete development antibioticproduction and phylogeny questions and challenges InProkaryotic Development Brun YV and Skimketts LJ(eds) Washington DC American Society for MicrobiologyPress pp 11ndash31
Chater KF and Bibb MJ (1997) Regulation of bacterialantibiotic production In Biotechnology Vol 7 Products ofSecondary Metabolism Kleinkauf H and von Doumlhren H(eds) Weinheim Germany VCH pp 57ndash105
Chong PP Podmore SM Kieser HM Redenbach MTurgay K Marahiel M et al (1998) Physical identificationof a chromosomal locus encoding biosynthetic genesfor the lipopeptide calcium-dependent antibiotic (CDA) ofStreptomyces coelicolor A3(2) Microbiology 144 193ndash199
de Crecy-Lagard V Servant-Moisson JV Grandvalet Cand Mazodier P (1999) Alteration of the synthesis of theClp ATP-dependent protease affects morphological andphysiological differentiation in Streptomyces Mol Microbiol32 505ndash517
Davis NK and Chater KF (1990) Spore colour inStreptomyces coelicolor A3(2) involves the developmen-tally regulated synthesis of a compound biosyntheticallyrelated to polyketide antibiotics Mol Microbiol 4 1679ndash1691
Fernandez-Moreno FA Martinez E Caballero JLIchinose K Hopwood DA and Malpartida F (1994)DNA sequence and functions of the actVI region of theactinorhodin biosynthetic gene cluster of Streptomycescoelicolor A3(2) J Biol Chem 269 24854ndash24863
Fink D Falke D Wohllheben W and Engels A (1999)Nitrogen metabolism in Streptomyces coelicolor A3(2)
modification of glutamine synthase I by an adenylyltrans-ferase Microbiology 145 2313ndash2322
Godovac-Zimmermann J Soskic V Posnanovic S andBrianza F (1999) Functional proteomics of signal trans-duction by membrane receptors Electrophoresis 20 952ndash961
Gorg A Obermaier C Boguth G Harder A Scheibe BWildgruber R and Weiss W (2000) The current state oftwo-dimensional electrophoresis with immobilized pH gra-dients Electrophoresis 21 1037ndash1053
Gramajo HC Takano E and Bibb MJ (1993) Stationary-phase production of the antibiotic actinorhodin in Strepto-myces coelicolor A3(2) is transcriptionally regulated MolMicrobiol 7 837ndash845
Gross CA Chan C Dombroski A Gruber T Sharp MTupy J and Young B (1998) The functional and regula-tory roles of sigma factors in transcription Cold SpringHarb Symp Quant Biol 63 141ndash155
Hesketh A Sun J and Bibb MJ (2001) Induction ofppGpp synthesis in Streptomyces coelicolor A3(2) grownunder conditions of nutritional sufficiency elicits actII-ORF4transcription and actinorhodin biosynthesis Mol Microbiol39 136ndash144
Hesketh A Fink D Gust B Rexer H-U Scheel BChater K et al (2002) The PII nitrogen regulatory proteinGlnK in Streptomyces coelicolor A3(2) is modified by ade-nylylation rather than uridylylation and the GlnD and GlnKhomologues are functionally dissimilar to their nitrogenregulatory system counterparts Mol Microbiol (inpress)
Hood DW Heidstra R Swodoba UK and Hodgson DA(1992) Molecular genetic analysis of proline and tryp-tophan biosynthesis in Streptomyces coelicolor A3(2)interaction between primary and secondary metabolism- areview Gene 115 5ndash12
Hu DS-J Hood DW Heidstra R and Hodgson DA(1999) The expression of the trpD trpC and trpBA genesof Streptomyces coelicolor A3(2) is regulated by growthrate and growth phase but not by feedback repression MolMicrobiol 32 869ndash880
Huang J Lih CJ Pan KH and Cohen SN (2001)Global analysis of growth phase responsive gene expres-sion and regulation of antibiotic biosynthetic pathways inStreptomyces coelicolor using DNA microarrays GenesDev 15 3183ndash3192
Ichinose K Taguchi T Ebizuka Y and Hopwood DA(1998a) Biosynthetic gene clusters of benzoisochromane-quinone antibiotics in Streptomyces spp Identification ofgenes involved in post-PKS tailoring steps Actinomyceto-logica 12 99ndash109
Ichinose K Bedford DJ Tornus D Bechthold A BibbMJ Revill PW et al (1998b) The granaticin biosyntheticgene cluster of Streptomyces violaceoruber Tu22sequence analysis and expression in a heterologous hostChem Biol 5 647ndash659
Kelemen GH Brian P Flardh K Chamberlin L ChaterK and Buttner M (1998) Developmental regulation oftranscription of whiE a locus specifying the polyketidespore pigment in Streptomyces coelicolor A3(2) J Bacte-riol 180 2515ndash2521
Kieser T Bibb MJ Buttner MJ Chater KF and
932 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
Hopwood DA (2000) Practical Streptomyces GeneticsNorwich John Innes Foundation
Kim EJ Chung HJ Suh B Hah YC and Roe JH(1998) Expression and regulation of the sodF gene encod-ing iron- and zinc-containing superoxide dismutase inStreptomyces coelicolor Muumlller J Bacteriol 180 2014ndash2020
Kimura Y Takaoka M Tanaka S Sassa H Tanaka KPolevoda B et al (2000) Na-acetylation and proteolyticactivity of the yeast 20S proteasome J Biol Chem 2754635ndash4639
Kunst F Ogasawara N Moszer I Albertini AM AlloniG Azevedo V et al (1997) The complete genomesequence of the Gram-positive bacterium Bacillus subtilisNature 390 249ndash256
Lucchini S Thompson A and Hinton JCD (2001)Microarrays for microbiologists Microbiology 147 1403ndash1414
Mann M Hendrickson RC and Pandey A (2001) Analy-sis of proteins and proteomes by mass spectrometry AnnuRev Biochem 70 437ndash473
Miyodah S (1993) Research on antibiotic screening inJapan over the last decade a producing microorganismsapproach Actinomycetologica 7 100ndash106
Molloy MP Herbert BR Williams KL and Gooley AA(1999) Extraction of Escherichia coli proteins with organicsolvents prior to two-dimensional electrophoresis Electro-phoresis 20 701ndash704
Nagy I Tamura T Vanderleyden J Baumeister W andDe Mot R (1998) The 20S proteasome of Streptomycescoelicolor J Bacteriol 180 5448ndash5453
Neuhoff V Arold N Taube D and Ehrhardt W (1988)Improved staining of proteins in polyacrylamide gels includ-ing isoelectric focusing gels with clear background at nan-ogram sensitivity using Coomassie Brilliant Blue G-250and R-250 Electrophoresis 9 255ndash262
Nilsson CL and Davidsson P (2000) New separation toolsfor comprehensive studies of protein expression by massspectrometry Mass Spectrom Rev 19 390ndash397
Puglia AM Vohradsky J and Thompson CJ (1995)
Developmental control of the heat-shock stress regulon inStreptomyces coelicolor Mol Microbiol 17 737ndash746
Rabilloud T (1992) A comparison between low backgroundsilver diamine and silver nitrate protein stains Electro-phoresis 13 429ndash439
Robinson VL Buckler DR and Stock AM (2000) A taleof two components a novel kinase and a regulatory switchNature Struct Biol 7 626ndash633
Schena M (2000) Microarray Biochip Technology NatickMA Eaton Publishing
Spusansky O Rezuchova B Homerova D andKormanec J (2001) Expression of the gap gene encodingglyceraldehyde-3-phosphate dehydrogenase of Strepto-myces aureofaciens requires GapR a member of theAraCXylS family of transcriptional activators Microbiology147 1291ndash1301
Taguchi T Itou K Ebizuka Y Malpartida F HopwoodDA Surti CM et al (2000) Chemical characterisationof disruptants of the Streptomyces coelicolor A3(2) actVIgenes involved in actinorhodin biosynthesis J Antibiot 53144ndash152
Takano E Gramajo HC Strauch E Andres N WhiteJ and Bibb MJ (1992) Transcriptional regulation of theredD transcriptional activator gene accounts for growth-phase-dependent production of the antibotic undecylpro-digiosin in Streptomyces coelicolor A3(2) Mol Microbiol6 2797ndash2804
Tonella L Hoogland C Binz P-A Appel RDHochstrasser DF and Sanchez J-C (2001) New per-spectives in the Escherichia coli proteome investigationProteomics 1 409ndash423
Vohradsky J Li XM and Thompson CJ (1997) Identifi-cation of prokaryotic developmental stages by statisticalanalysis of two-dimensional gel patterns Electrophoresis18 1418ndash1428
Vohradsky J Li XM Dale G Folcher M Nguyen LViollier PH and Thompson CJ (2000) Developmentalcontrol of stress stimulons in Streptomyces coelicolorrevealed by statistical analyses of global gene expressionpatterns J Bacteriol 182 4979ndash4986
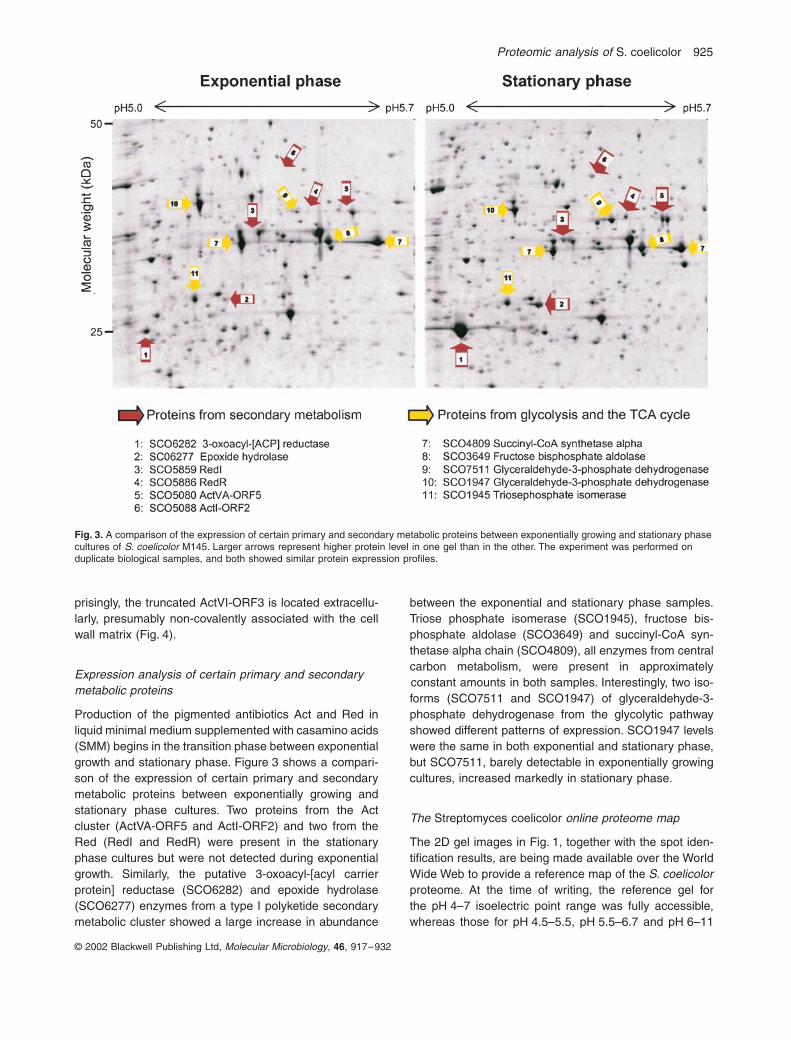
Proteomic analysis of S coelicolor 925
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
prisingly the truncated ActVI-ORF3 is located extracellu-larly presumably non-covalently associated with the cellwall matrix (Fig 4)
Expression analysis of certain primary and secondary metabolic proteins
Production of the pigmented antibiotics Act and Red inliquid minimal medium supplemented with casamino acids(SMM) begins in the transition phase between exponentialgrowth and stationary phase Figure 3 shows a compari-son of the expression of certain primary and secondarymetabolic proteins between exponentially growing andstationary phase cultures Two proteins from the Actcluster (ActVA-ORF5 and ActI-ORF2) and two from theRed (RedI and RedR) were present in the stationaryphase cultures but were not detected during exponentialgrowth Similarly the putative 3-oxoacyl-[acyl carrierprotein] reductase (SCO6282) and epoxide hydrolase(SCO6277) enzymes from a type I polyketide secondarymetabolic cluster showed a large increase in abundance
between the exponential and stationary phase samplesTriose phosphate isomerase (SCO1945) fructose bis-phosphate aldolase (SCO3649) and succinyl-CoA syn-thetase alpha chain (SCO4809) all enzymes from centralcarbon metabolism were present in approximatelyconstant amounts in both samples Interestingly two iso-forms (SCO7511 and SCO1947) of glyceraldehyde-3-phosphate dehydrogenase from the glycolytic pathwayshowed different patterns of expression SCO1947 levelswere the same in both exponential and stationary phasebut SCO7511 barely detectable in exponentially growingcultures increased markedly in stationary phase
The Streptomyces coelicolor online proteome map
The 2D gel images in Fig 1 together with the spot iden-tification results are being made available over the WorldWide Web to provide a reference map of the S coelicolorproteome At the time of writing the reference gel forthe pH 4ndash7 isoelectric point range was fully accessiblewhereas those for pH 45ndash55 pH 55ndash67 and pH 6ndash11
Fig 3 A comparison of the expression of certain primary and secondary metabolic proteins between exponentially growing and stationary phase cultures of S coelicolor M145 Larger arrows represent higher protein level in one gel than in the other The experiment was performed on duplicate biological samples and both showed similar protein expression profiles
926 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
were under construction (httpqbababeracukS_coeliReferenceGelrefgelhtml) A tool for comparing two ormore gel images with each other is also accessible fromthe same website at httpqbababeracukS_coeliflickerhtml
Discussion
Silver staining is one of the most sensitive methods fordetecting proteins on polyacrylamide gels allowing aslittle as 05ndash2 ng of an individual protein to be detected(Berggren et al 2000 Gorg et al 2000) The proportionof the S coelicolor proteome that can be analysed usingsilver staining of 2D-PAGE-separated proteins dependsat least in part on the pH range of the IPG strip used forthe first-dimension separation (see Table 1) Using theintermediate-range IPG strip covering pH 4ndash7 about 20ndash25 of the proteins in the theoretical proteome predictedto be observable in the resulting 2D-PAGE gel were actu-ally detected This figure was reduced to 10ndash12 whenanalysing the more basic pI range using the pH 6ndash11 IPGstrip Attempts to increase the percentage of the proteomedetectable using the pH 4ndash7 and pH 6ndash11 IPG strips byloading more protein sample resulted in poorer separa-tions (data not shown) This was especially true of thepH 6ndash11 strips which were generally the most difficult touse successfully However good separation of higher pro-tein loads was achieved using the narrow-range IPG stripspH 45ndash55 and 55ndash67 significantly increasing the pro-portion of theoretical proteins detected to over 40Clearly and more pertinently the limited conditions usedfor culturing S coelicolor in this study will also haverestricted the number of proteins detected as not all theproteome will be expressed In particular S coelicolordoes not sporulate in shaken liquid-grown cultures soproteins involved in this differentiation process are unlikelyto be present Nevertheless it is striking that the com-bined data for the pH 45ndash55 and 55ndash67 isoelectric pointranges indicate that 2468 out of a total of predicted 4932protein spots can be detected (see Table 1) suggestingthat transition phase mycelium in this supplementedminimal medium is expressing 50 of its genes (ie byextrapolation some 3912 of the 7825 in the genome)
Analysis of post-translational regulation
Although the regulation of cellular processes and adaptiveresponses in bacteria has been studied extensively at thetranscriptional level post-translational regulatory mecha-nisms are much less understood Proteomic analysisusing 2D-PAGE coupled with mass spectrometry offersthe opportunity to observe and identify modification ofproteins at a general cellular level The functions of pro-teins can be altered by specific covalent attachment of
adducts (eg phosphorylation glycosylation nucleotidy-lation acetylation) or by proteolytic processing Bothevents alter the pI andor mass of the protein and so canusually be observed on 2D-PAGE In the S coelicolorproteome map ordf110 out of a total of 770 gene productswere identified in more than one position or at pI andapparent mass co-ordinates significantly different frompredicted values indicating extensive regulation at thepost-translational level (88 are listed in Table 3) a propor-tion similar to that reported for other prokaryotes (Buttneret al 2001 Tonella et al 2001) The initial peptide massfingerprint analysis identified the modification of only 10of these proteins probably partly because fingerprint datatypically cover ordf30ndash60 of a proteinrsquos sequence (data notshown) so modification at positions not represented willbe missed Protein digestion with endopeptidases otherthan trypsin before mass spectrometry may help in thissituation but some covalently attached groups are labileand cannot be detected routinely Phosphorylation of Hisoccurs on nitrogen producing a high-energy phosphora-midate bond that is easily hydrolysed in acidic conditionswhereas phospho-Asp is the most labile phosphorylatedresidue with a half-life of a few hours under physiologicalconditions (reviewed by Robinson et al 2000) Thesemodifications the basis of bacterial two-component regu-latory systems are therefore unlikely to survive stainingof the 2D gels which involved the use of a fixing solutioncontaining 10 acetic acid and mass spectrometry Iden-tification of the more stable phosphoester phosphorylationthat occurs on Ser Thr or Tyr residues using PAGEfollowed by mass spectrometry has been reported(Godovac-Zimmermann et al 1999 Buttner et al 2001)but no examples were detected in this work Of the mod-ifications identified in this study three were by acetylationat the N-terminus after removal of the initiator Met residuetwo involved adenylylation and five proteins had been N-terminally truncated Modification of the 20S proteasomeby N-acetylation in yeast involves removal of the Metresidue by N-methionylaminopeptidase followed byacetylation with an Na-acetyltransferase (Kimura et al2000) Homologues of these enzymes are predicted fromthe S coelicolor genome and are presumably responsiblefor the observed modification of the 20S proteasomealpha subunit and of BldK-ORFD and the putative cellu-lose-binding protein The functional significance of Na-acetylation of these proteins is unclear
Analysis of the biosynthesis of antibiotics and other secondary metabolites
Post-translational modification The proteome mappingresults indicate that at least six enzymes in the Act clusterare subject to post-translational modifications and one
Proteomic analysis of S coelicolor 927
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
each from the undecylprodigiosin (SCO5877ndash5898)lsquocoelichelinrsquo (SCO0489ndash0499) and type I polyketidesynthase (SCO6273ndash6288) clusters (see Table 3) Thesemetabolic pathways may therefore be significantly regu-lated at the post-translational level The actVI-ORF3 gene(SCO5074) encodes a putative dehydratase enzyme thatis part of the biosynthetic pathway for Act a blue memberof the benzoisochromanequinone (BIQ) family of pig-mented antibiotics Mutation in actVI-ORF3 causes a 50decrease in the accumulation of Act (and a reddish brownphenotype) and it is believed to be responsible for pyranring closure an intermediate step in Act biosynthesis (thatmay also occur spontaneously) leading to the formationof the BIQ chromophore (Fernandez-Moreno et al 1994Ichinose et al 1998a Taguchi et al 2000) In this studyActVI-ORF3 protein was detected in two forms a majorspot corresponding to ActVI-ORF3 truncated by removalof the N-terminal 31 amino acid residues and a minor onein which the first 34 amino acids were missing (spots 31in Fig 1) The full-length protein was not detected Anal-ysis of the protein sequence using the SIGNALP server athttpwwwcbsdtudkservicesSignalP predicted a signalpeptide cleavage site between amino acids 31 and 32 inagreement with the observed results
The protein extract analysed was prepared from myce-lium that had been collected by centrifugation and sub-jected to two washes indicating that if ActVI-ORF3 isexported it is effectively retained within the cell wallmatrix This was confirmed by analysis of a preparationobtained by externally extracting proteins from intactwashed mycelium (Fig 4) Export of an enzyme requiredfor the efficient biosynthesis of an antibiotic that is thoughtto be produced intracellularly appears to be anomalousparticularly as it is involved in an intermediate biosynthetic
event Database analysis of the other members of the Actcluster revealed only one other protein with a predictedsignal peptide sequence the putative integral membranetransport-related protein ActII-ORF3 and it seemsunlikely that all the Act biosynthetic steps following BIQchromophore formation are extracellular It is possible thatfull-length ActVI-ORF3 catalyses the intermediate biosyn-thetic event intracellularly and the exported processedprotein is responsible for a different extracellular activityperhaps later in the pathway However no full-length pro-tein was detected so the exported enzyme may performthe pyran ring closure on a secreted metabolite that isthen reabsorbed by the cell for completion of the sub-sequent biosynthetic steps Only two homologues ofactVI-ORF3 are known and both are also in BIQ antibioticbiosynthetic clusters gra-ORF18 from the granaticincluster (Ichinose et al 1998b) and med-ORF5 frommedermycin biosynthesis (K Ichinose personal com-munication) Interestingly the encoded proteins of bothsequences are also predicted to contain N-terminal signalpeptides The observation that ActVI-ORF3 is exported tothe cell wall matrix may therefore be of general signifi-cance to the BIQ family of antibiotics and provide a newinsight into their biosynthetic pathways
Pathway representation Of the 20 putative secondarymetabolite gene clusters identified in the S coelicolorgenome sequence (Bentley et al 2002) eight had atleast one protein each identified on the proteome map(see Table 5) The biosynthetic clusters for the antibioticsAct Red and CDA had 68 36 and 40 of their pro-teins represented There are several possible explana-tions for failure to detect all the proteins for each pathwaySome are putative integral membrane proteins so cannotreadily be detected using conventional 2D-PAGE SomeRed and CDA proteins have molecular weights gt200 kDaoutside the range of the 125 gels used (for clustermembers see Supplementary material) The pathway-specific regulatory proteins for Act and Red biosynthesiswere not detected despite a detailed search of their pre-dicted gel locations even though the relevant genes areknown to be transcribed in equivalent cultures of the samestrain (Hesketh et al 2001) The proteins are presumablypresent but at levels below the limit of detection Howeverthe transcriptional regulator for CDA production CdaRwas identified as well as the two-component systemresponse regulator AbsA2 which is also encoded by agene in the CDA cluster (Anderson et al 2001) Interest-ingly proteins from three putative secondary meta-bolite clusters producing chemicals of as yet unknownstructure were also identified on the map correspondingto the deoxysugar synthaseglycosyltransferase clusterSCO0381ndash0401 the lsquocoelichelinrsquo cluster for a predicted(but not yet confirmed) oligopeptide siderophore
Fig 4 Processed ActVI-ORF3 is present in the cell wall matrix 2D-gel separations of protein extracts prepared (A) from whole cells or (B) by externally washing cells in a 2 SDS extraction buffer The arrow indicates ActVI-ORF3 protein The experiment was performed on duplicate biological samples and both showed similar protein expression profiles
928 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
(SCO0489ndash0499) and the type I polyketide clusterSCO6273ndash6288 The 3-oxoacyl-[acyl carrier protein]reductase (SCO6282) from the latter pathway is ex-pressed in a growth phase-dependent manner (Fig 3)and in early stationary phase cultures is perhaps themost abundant protein spot in the entire detectable pro-teome Presumably the metabolites produced by thesethree pathways are being synthesized under the condi-tions used In addition to coelichelin three other sidero-phores are postulated for S coelicolor All the proteinsfrom one of these desferrioxamine (SCO2782ndash2785)were identified but none was seen from either of the othertwo pathways The WhiE polyketide spore pigment isassociated with sporulation of surface-grown cultures(Davis and Chater 1990) and the expression of the whiEgenes depends on a series of sporulation-specific regula-tory genes (Kelemen et al 1998) no corresponding pro-teins of which were detected here It is therefore notsurprising that WhiE proteins were not detected in sam-ples prepared from liquid-grown vegetative mycelium
Analysis of primary metabolism
Proteins annotated as functioning in primary metabolicpathways were highly represented on the proteome mapwith ordf60 or more of the enzymes assigned to glycolysisthe TCA cycle the pentose phosphate pathway andpurine and pyrimidine ribonucleotide biosynthesis beingidentified (see Table 6) This figure may be deceptively lowbecause these pathways in S coelicolor often appear tohave more than one enzyme isoform for some of themetabolic steps potentially offering additional levels ofcontrol or developmental responsiveness at these pointsFor example only two steps in glycolysis have just a singlegene product assigned to them the remainder typicallyhave two or three each (Fig 5) At least one isoform waspresent for each step so the pathway is in theory 100functionally represented Understanding the regulation ofthese pathways will require analysis of the expression ofthese isoenzymes under different nutritional conditionsor different developmental stages Figure 5 shows that ofthe three predicted glyceraldehyde-3-phosphate dehydro-genase isoforms from glycolysis only two were detectedin this study (see also Supplementary material) Of theseSCO1947 appears to be constitutive whereas SCO7511is growth phase dependent (see Fig 3) Thus even thelimited conditions in this study yielded useful informationabout isoenzyme expression patterns (see Supplemen-tary material) In Streptomyces aureofaciens two genesencoding glyceraldehyde-3-phosphate dehydrogenaseactivity also appear to be differently regulated transcrip-tion of one highly homologous to SCO7511 is activatedby the regulator GapR whereas the other is GapR inde-pendent (Spusansky et al 2001)
Hood et al (1992) reported that most amino acid bio-synthetic enzymes are expressed at very low constitutivelevels in S coelicolor so it is reassuring to note that ordf50could be detected and identified using proteomics Theseinclude TrpB and TrpC1 from the tryptophan biosyntheticpathway the corresponding transcripts of which couldbarely be detected in previously reported S1 nucleaseprotection experiments (Hu et al 1999)
Perspectives
No attempt was made in the work reported here to simplifythe protein separations by subdividing the sample intosoluble and membrane fractions and this is reflected inthe 10 lipoproteins and 16 ABC transport system ATP-binding subunits identified on the proteome map (seeTable 2) No integral membrane proteins were detectedand even when a membrane protein sample was pre-pared and analysed no proteins identified possessedmore than three predicted transmembrane helices (datanot shown) This is consistent with previous studiesin other bacteria (Molloy et al 1999 Nilsson andDavidsson 2000) Another group of proteins also strik-ingly absent from the list of those identified is the sigmafactors which determine promoter selection by RNA poly-merase There are an extraordinarily high number (65) ofsigma factors encoded in the S coelicolor genome aswell as at least seven antisigma factors and 15 antisigmafactor antagonists (some of each of which were detected)Sigma factor heterogeneity in bacteria is an important wayof regulating expression of specific sets of genes under
Fig 5 The glycolytic pathway in S coelicolor illustrating the assignment of multiple gene products to individual metabolic steps based on the annotation of the genome sequence at httpwwwsangeracukProjectsS_coelicolor Gene products identified in the proteome map are underlined
Proteomic analysis of S coelicolor 929
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
different conditions and in different cell types (for a reviewsee Gross et al 1998) Although no sigma factors wereidentified about half (31 out of 65) have predicted pIvalues gtpH 7 and so can only be detected using thepH 6ndash11 IPG strips that give the poorest results (seeTable 1) However the principal vegetative sigma factorHrdB which is expected to be moderately abundanthas a predicted pI of 613 and its absence from thepH 55ndash67 proteome map may indicate that the samplepreparation method which does not remove DNA is inap-propriate Among other regulatory proteins 11 out of apossible 80 two-component system response regulatorsplus a further 36 proteins annotated as lsquoregulatorsrsquo weredetected Only one SerThr kinase and no two-componentsystem sensor kinases were detected possibly reflectingthe difficulty in analysing proteins with several transmem-brane domains Interestingly 30 secreted proteins wereidentified on the proteome map at least 10 of whichappear from comparison of their observed pI and molec-ular weight positions with predicted values to have beenprocessed already and therefore exported Presumablythese proteins do not end up exclusively in the culturemedium
Clearly this approach to proteomic analysis of S coeli-color has some important deficiencies not least theabsence of certain groups of proteins and the relativelypoor initial success rate for specifically characterizingpost-translational modifications Nevertheless it offersunique insights into the molecular biology of the organismthe expression of many proteins from both primary andsecondary metabolic pathways can be analysed post-translational regulation of proteins can be investigated ona general cellular level and the physical locations forsome proteins can be determined Approximately 35 ofthe S coelicolor genome is annotated as encoding lsquohypo-theticalrsquo proteins of unknown function and 182 proteins ofthis type were identified on the proteome map No exam-ples were found of overt disagreements between pre-dicted and observed proteins providing experimentalsupport for the general accuracy of the S coelicolorgenome sequence and its annotation Information aboutthe expression post-translational modification and physi-cal location of these (no longer) hypothetical proteinsfrom proteomic analyses is going to be important forassigning future functions As many of the hypotheticalproteins have apparent orthologues in pathogenic myco-bacteria some of the information may have an immediateapplication in the context of pathogenesis
Experimental procedures
Materials
Immobilized pH gradient (IPG) strips and IPG bufferswere purchased from Amersham Biosciences Acrylamide
duracryl and chemicals for silver staining were obtained fromGenomic Solutions All other chemicals were from Sigmaunless stated in the text
Strain and growth conditions
Streptomyces coelicolor M145 was cultivated with vigorousagitation at 30infinC in minimal medium supplemented with 02casamino acids (SMM) as described previously (Kieser et al2000) Briefly a high-density spore preparation [about 1010
colony-forming units (cfu) ml-1] was pregerminated in 2yen YTmedia for 7 h at 30infinC Germlings were harvested by centrifu-gation (5 min at 4000 g) resuspended in SMM and sonicatedbriefly to disperse any aggregates before inoculation into50 ml of SMM in 250 ml siliconized flasks containing coiledstainless steel springs Each flask received the equivalent of5 yen 107 spore cfu
Protein extraction and 2D-gel electrophoresis
The preparation of the protein sample to be separated waskept as simple as possible disrupting harvested myceliadirectly into denaturing isoelectric focusing (IEF) buffer con-taining Pefabloc SC (Roche) protease inhibitor This methodminimized unwanted protein degradation that was observedin initial studies using a Tris buffer containing Pefabloc SC forcell lysis (data not shown) Removal of DNA RNA salts andpolysaccharides from the sample was therefore not per-formed (other than the shearing of viscous DNA and RNA bysonication) and the protein extracts were sometimes difficultto focus when the sample was loaded by in-gel rehydrationLoading the sample soaked in paper strips at the anodicend of IPG strips that had already been rehydrated improvedthe reproducibility of separations significantly
Mycelium for protein extraction was harvested from cul-tures by brief centrifugation (30 s at 4000 g) at room tem-perature and immediately frozen in liquid nitrogen Typicallymycelium from 10 ml culture aliquots was collected and thetransfer time from culture flask to frozen sample was 15 minFrozen cells were thawed on ice in 5 ml of washing buffer(40 mM Tris pH 90 1 mM EGTA 1 mM EDTA) then pelletedby centrifugation (5 min at 3000 g) at 4infinC Washed cells wereresuspended in 400 ml of denaturing IEF buffer UTCHAPS[7 M urea 2 M thiourea 4 (wv) CHAPS 40 mM TrispH 90 1 mM EDTA 50 mM DTT 4 mM Pefabloc SC pro-tease inhibitor (Roche)] and disrupted by sonication (SanyoSoniprep 150 10 yen 2 s bursts at amplitude 75 mm) whilecooling in an ethanolndashice bath Cell debris was removed bycentrifugation (15 min 10 000 g 4infinC) and the protein extractwas stored frozen in aliquots at -80infinC until use For thepreparation of extracellular protein extracts washed cellswere resuspended in extraction buffer consisting of 40 mMTris pH 90 1 mM EDTA 50 mM DTT 2 SDS and 4 mMPefabloc SC protease inhibitor Cells were vortexed for 2 minbefore being pelleted by centrifugation (5 min 10 000 g4infinC) The supernatant was precipitated using the Amersham2D Clean-Up kit according to the manufacturerrsquos instructionsand the precipitated proteins were redissolved in UTCHAPSand stored at -80infinC until use
930 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
For first-dimension isoelectric focusing 18 cm IPG stripspH 4ndash7 pH 6ndash11 pH 45ndash55 or pH 55ndash67 (Amersham Bio-sciences) were rehydrated overnight in IEF buffer containing1 ampholytes according to the manufacturerrsquos instructionsusing a Phaser isoelectric focusing unit (Genomic Solutions)set at 20 V Protein samples to be separated were applied tothe rehydrated strips at the anodic end soaked in a 5ndash10 mmsection of IEF electrode strip (Amersham Biosciences)Separation was performed for 120 000 V-h with a maximumvoltage of 5000 V After isoelectric focusing IPG strips wereequilibrated for the second dimension for 15 min in IPG equil-ibration buffer (50 mM Tris pH 68 6 M urea 30 glycerol1 SDS and 001 bromophenol blue) plus 80 mM DTTthen for 10 min in IPG equilibration buffer plus 135 mMiodoacetamide Approximately 1 cm was removed from theanodic end of each equilibrated strip before applying the stripto the top of a vertical 125 SDS-PAGE gel for second-dimension separation using the Investigator 5000 systemfrom Genomic Solutions
Large-format gels (24 cm wide yen 22 cm high yen 1 mm thick)for the second-dimension separation were cast in-houseusing the system supplied by Genomic Solutions 125SDS-PAGE gels prepared using 065 NN-methylenebi-sacrylamide as cross-linking agent were used for pH 4ndash7and pH 45ndash55 IPG strips whereas pH 55ndash67 and pH 6ndash11 strips were applied to similar gels made with 08 cross-linker Electrophoresis was performed while cooling using themaximum setting (ordf 4ndash10infinC) at 20 000 mW per gel constantpower and a maximum voltage of 500 V Gels were thenstained with either colloidal Coomassie G-250 (Neuhoff et al1988) or silver nitrate (Rabilloud 1992) and scanned in aProXPRESS proteomic imaging system (Perkin-Elmer)Image analysis was performed using PHORETIX 2D version51 (NonLinear Dynamics) spot detection was optimizedautomatically using the lsquoSpot Detection Wizardrsquo and thenedited manually background subtraction was performedautomatically using the lsquoMode of Non-Spotrsquo setting imageswere then normalized to the total spot volume for quantifica-tion Spot filtering was not used
Protein identification using mass spectrometry
Protein spots of interest were excised from colloidalCoomassie-stained gels in-gel digested with trypsin andidentified by MALDI-TOF peptide mass fingerprint analysisExcised gel pieces (ordf 1-mm-diameter circles cut from 1-mm-thick gels) were washed twice with 100 ml of 50 mMammonium bicarbonate for 15 min once with 100 ml of 20acetonitrile-40 mM ammonium bicarbonate for 15 min andonce with 100 acetonitrile Washed gel pieces were allowedto air-dry for 10 min before being rehydrated in 5 ml of10 mg ml-1 trypsin (sequencing grade modified Promega) in10 mM ammonium bicarbonate Digestion (37infinC for 4 h) wasstopped by the addition of 5 ml of 5 formic acid Extractionof peptides into this solution was encouraged by a sonicwater bath treatment for 20 min Peptide extract (05 ml) wasspotted on to a thin layer of a-cyano-4-hydroxycinnamic acidapplied to a MALDI-TOF sample template and analysed byMALDI-TOF mass spectrometry (Bruker Reflex III) using anaccelerating voltage of 25 kV Samples were externally cali-brated using a standard mixture of six peptides ranging in
mass from 10465423 Da to 34946500 Da and the massaccuracy obtained was 60 ppm or better Identification ofproteins from MALDI-TOF peptide mass fingerprint data wasperformed using the lsquoMascotrsquo search engine at httpwwwmatrixsciencecom and was based on a positive resultusing their lsquoProbability Based MOWSE Scorersquo algorithm AMOWSE score of 60 or higher is significant at the 5 levelor better and proteins in this work typically gave scores gt80(frequently considerably so) In addition no identification wasaccepted unless at least five peptides representing at least20 of the protein sequence were detected in the MALDI-TOF peptide mass fingerprint
Assignment of proteins to metabolic pathways
Proteins were assigned to primary metabolic pathwaysusing the lsquoProtein Classification Schemersquo web page ofthe annotated Streptomyces coelicolor genome sequenceat httpwwwsangeracukProjectsS_coelicolor as it ap-peared in May 2002 The annotation of the genome andtherefore the Protein Classification Scheme is likely to bemodified as more experimental data are obtained about thefunction of genes that have currently been assigned on thebasis of sequence information alone It is therefore possiblethat proteins assigned to metabolic pathways in this studymay in future be shown to perform different functions in thecell
Assignments to gene clusters encoding secondary metab-olites are based on an analysis of the genome sequencereported by Bentley et al (2002) In some cases theseassignments are based on extensive experimental data (actred reviewed by Chater and Bibb 1997 Champness 2000cda Chong et al 1998 Huang et al 2001 whiE Davis andChater 1990)
Acknowledgements
This work was funded by grants (2FGT11406 208FGT11408 and 208IGF12432) under the BBSRCrsquos Tech-nologies for Functional Genomics and Investigating GeneFunction initiatives and by a competitive strategic grant fromthe BBSRC to the John Innes Centre We thank Mike Naldrettfor his extensive contribution in establishing technology forproteome analysis at the JIC
Supplementary material
The following material is available from httpwwwblackwell-sciencecomproductsjournalssuppmatmolemole3219mmi3219smhtmSection 1 Detection of gene products assigned to individualmetabolic steps in primary metabolic pathways in ScoelicolorSection 2 Identification of proteins from individual amino acidbiosynthetic pathwaysSection 3 Detection of gene products assigned to individualmetabolic steps in secondary metabolic pathways in Scoelicolor
Proteomic analysis of S coelicolor 931
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
References
Anderson TB Brian P and Champness WC (2001)Genetic and transcriptional analysis of absA an antibioticgene cluster linked two-component system that regulatesmultiple antibiotics in Streptomyces coelicolor Mol Micro-biol 39 553ndash566
Bentley SD Chater KF Cerdeno-Tarraga A-M ChallisGL Thomson NR James KD et al (2002) Completegenome sequence of the model actinomycete Streptomy-ces coelicolor A3(2) Nature 417 141ndash147
Berdy J (1984) New ways to obtain antibiotics Chin J Anti-biot 7 272ndash290
Berggren K Chernokalskaya E Steinberg TH KemperC Lopez MF Diwu Z et al (2000) Background-freehigh sensitivity staining of proteins in one- and two-dimensional sodium dodecyl sulfate-polyacrylamide gelsusing a luminescent ruthenium complex Electrophoresis21 2509ndash2521
Blackstock WP and Weir MP (1999) Proteomics quan-titative and physical mapping of cellular proteins TrendsBiotechnol 17 121ndash127
Blattner FR Plunkett G Bloch CA Perna NT BurlandV Riley M et al (1997) The complete genome sequenceof Escherichia coli K-12 Science 277 1453ndash1462
Buttner K Bernhardt J Scharf C Schmid R MaderU Eymann C et al (2001) A comprehensive two-dimensional map of cytosolic proteins of Bacillus subtilisElectrophoresis 22 2908ndash2935
Champness W (2000) Actinomycete development antibioticproduction and phylogeny questions and challenges InProkaryotic Development Brun YV and Skimketts LJ(eds) Washington DC American Society for MicrobiologyPress pp 11ndash31
Chater KF and Bibb MJ (1997) Regulation of bacterialantibiotic production In Biotechnology Vol 7 Products ofSecondary Metabolism Kleinkauf H and von Doumlhren H(eds) Weinheim Germany VCH pp 57ndash105
Chong PP Podmore SM Kieser HM Redenbach MTurgay K Marahiel M et al (1998) Physical identificationof a chromosomal locus encoding biosynthetic genesfor the lipopeptide calcium-dependent antibiotic (CDA) ofStreptomyces coelicolor A3(2) Microbiology 144 193ndash199
de Crecy-Lagard V Servant-Moisson JV Grandvalet Cand Mazodier P (1999) Alteration of the synthesis of theClp ATP-dependent protease affects morphological andphysiological differentiation in Streptomyces Mol Microbiol32 505ndash517
Davis NK and Chater KF (1990) Spore colour inStreptomyces coelicolor A3(2) involves the developmen-tally regulated synthesis of a compound biosyntheticallyrelated to polyketide antibiotics Mol Microbiol 4 1679ndash1691
Fernandez-Moreno FA Martinez E Caballero JLIchinose K Hopwood DA and Malpartida F (1994)DNA sequence and functions of the actVI region of theactinorhodin biosynthetic gene cluster of Streptomycescoelicolor A3(2) J Biol Chem 269 24854ndash24863
Fink D Falke D Wohllheben W and Engels A (1999)Nitrogen metabolism in Streptomyces coelicolor A3(2)
modification of glutamine synthase I by an adenylyltrans-ferase Microbiology 145 2313ndash2322
Godovac-Zimmermann J Soskic V Posnanovic S andBrianza F (1999) Functional proteomics of signal trans-duction by membrane receptors Electrophoresis 20 952ndash961
Gorg A Obermaier C Boguth G Harder A Scheibe BWildgruber R and Weiss W (2000) The current state oftwo-dimensional electrophoresis with immobilized pH gra-dients Electrophoresis 21 1037ndash1053
Gramajo HC Takano E and Bibb MJ (1993) Stationary-phase production of the antibiotic actinorhodin in Strepto-myces coelicolor A3(2) is transcriptionally regulated MolMicrobiol 7 837ndash845
Gross CA Chan C Dombroski A Gruber T Sharp MTupy J and Young B (1998) The functional and regula-tory roles of sigma factors in transcription Cold SpringHarb Symp Quant Biol 63 141ndash155
Hesketh A Sun J and Bibb MJ (2001) Induction ofppGpp synthesis in Streptomyces coelicolor A3(2) grownunder conditions of nutritional sufficiency elicits actII-ORF4transcription and actinorhodin biosynthesis Mol Microbiol39 136ndash144
Hesketh A Fink D Gust B Rexer H-U Scheel BChater K et al (2002) The PII nitrogen regulatory proteinGlnK in Streptomyces coelicolor A3(2) is modified by ade-nylylation rather than uridylylation and the GlnD and GlnKhomologues are functionally dissimilar to their nitrogenregulatory system counterparts Mol Microbiol (inpress)
Hood DW Heidstra R Swodoba UK and Hodgson DA(1992) Molecular genetic analysis of proline and tryp-tophan biosynthesis in Streptomyces coelicolor A3(2)interaction between primary and secondary metabolism- areview Gene 115 5ndash12
Hu DS-J Hood DW Heidstra R and Hodgson DA(1999) The expression of the trpD trpC and trpBA genesof Streptomyces coelicolor A3(2) is regulated by growthrate and growth phase but not by feedback repression MolMicrobiol 32 869ndash880
Huang J Lih CJ Pan KH and Cohen SN (2001)Global analysis of growth phase responsive gene expres-sion and regulation of antibiotic biosynthetic pathways inStreptomyces coelicolor using DNA microarrays GenesDev 15 3183ndash3192
Ichinose K Taguchi T Ebizuka Y and Hopwood DA(1998a) Biosynthetic gene clusters of benzoisochromane-quinone antibiotics in Streptomyces spp Identification ofgenes involved in post-PKS tailoring steps Actinomyceto-logica 12 99ndash109
Ichinose K Bedford DJ Tornus D Bechthold A BibbMJ Revill PW et al (1998b) The granaticin biosyntheticgene cluster of Streptomyces violaceoruber Tu22sequence analysis and expression in a heterologous hostChem Biol 5 647ndash659
Kelemen GH Brian P Flardh K Chamberlin L ChaterK and Buttner M (1998) Developmental regulation oftranscription of whiE a locus specifying the polyketidespore pigment in Streptomyces coelicolor A3(2) J Bacte-riol 180 2515ndash2521
Kieser T Bibb MJ Buttner MJ Chater KF and
932 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
Hopwood DA (2000) Practical Streptomyces GeneticsNorwich John Innes Foundation
Kim EJ Chung HJ Suh B Hah YC and Roe JH(1998) Expression and regulation of the sodF gene encod-ing iron- and zinc-containing superoxide dismutase inStreptomyces coelicolor Muumlller J Bacteriol 180 2014ndash2020
Kimura Y Takaoka M Tanaka S Sassa H Tanaka KPolevoda B et al (2000) Na-acetylation and proteolyticactivity of the yeast 20S proteasome J Biol Chem 2754635ndash4639
Kunst F Ogasawara N Moszer I Albertini AM AlloniG Azevedo V et al (1997) The complete genomesequence of the Gram-positive bacterium Bacillus subtilisNature 390 249ndash256
Lucchini S Thompson A and Hinton JCD (2001)Microarrays for microbiologists Microbiology 147 1403ndash1414
Mann M Hendrickson RC and Pandey A (2001) Analy-sis of proteins and proteomes by mass spectrometry AnnuRev Biochem 70 437ndash473
Miyodah S (1993) Research on antibiotic screening inJapan over the last decade a producing microorganismsapproach Actinomycetologica 7 100ndash106
Molloy MP Herbert BR Williams KL and Gooley AA(1999) Extraction of Escherichia coli proteins with organicsolvents prior to two-dimensional electrophoresis Electro-phoresis 20 701ndash704
Nagy I Tamura T Vanderleyden J Baumeister W andDe Mot R (1998) The 20S proteasome of Streptomycescoelicolor J Bacteriol 180 5448ndash5453
Neuhoff V Arold N Taube D and Ehrhardt W (1988)Improved staining of proteins in polyacrylamide gels includ-ing isoelectric focusing gels with clear background at nan-ogram sensitivity using Coomassie Brilliant Blue G-250and R-250 Electrophoresis 9 255ndash262
Nilsson CL and Davidsson P (2000) New separation toolsfor comprehensive studies of protein expression by massspectrometry Mass Spectrom Rev 19 390ndash397
Puglia AM Vohradsky J and Thompson CJ (1995)
Developmental control of the heat-shock stress regulon inStreptomyces coelicolor Mol Microbiol 17 737ndash746
Rabilloud T (1992) A comparison between low backgroundsilver diamine and silver nitrate protein stains Electro-phoresis 13 429ndash439
Robinson VL Buckler DR and Stock AM (2000) A taleof two components a novel kinase and a regulatory switchNature Struct Biol 7 626ndash633
Schena M (2000) Microarray Biochip Technology NatickMA Eaton Publishing
Spusansky O Rezuchova B Homerova D andKormanec J (2001) Expression of the gap gene encodingglyceraldehyde-3-phosphate dehydrogenase of Strepto-myces aureofaciens requires GapR a member of theAraCXylS family of transcriptional activators Microbiology147 1291ndash1301
Taguchi T Itou K Ebizuka Y Malpartida F HopwoodDA Surti CM et al (2000) Chemical characterisationof disruptants of the Streptomyces coelicolor A3(2) actVIgenes involved in actinorhodin biosynthesis J Antibiot 53144ndash152
Takano E Gramajo HC Strauch E Andres N WhiteJ and Bibb MJ (1992) Transcriptional regulation of theredD transcriptional activator gene accounts for growth-phase-dependent production of the antibotic undecylpro-digiosin in Streptomyces coelicolor A3(2) Mol Microbiol6 2797ndash2804
Tonella L Hoogland C Binz P-A Appel RDHochstrasser DF and Sanchez J-C (2001) New per-spectives in the Escherichia coli proteome investigationProteomics 1 409ndash423
Vohradsky J Li XM and Thompson CJ (1997) Identifi-cation of prokaryotic developmental stages by statisticalanalysis of two-dimensional gel patterns Electrophoresis18 1418ndash1428
Vohradsky J Li XM Dale G Folcher M Nguyen LViollier PH and Thompson CJ (2000) Developmentalcontrol of stress stimulons in Streptomyces coelicolorrevealed by statistical analyses of global gene expressionpatterns J Bacteriol 182 4979ndash4986

926 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
were under construction (httpqbababeracukS_coeliReferenceGelrefgelhtml) A tool for comparing two ormore gel images with each other is also accessible fromthe same website at httpqbababeracukS_coeliflickerhtml
Discussion
Silver staining is one of the most sensitive methods fordetecting proteins on polyacrylamide gels allowing aslittle as 05ndash2 ng of an individual protein to be detected(Berggren et al 2000 Gorg et al 2000) The proportionof the S coelicolor proteome that can be analysed usingsilver staining of 2D-PAGE-separated proteins dependsat least in part on the pH range of the IPG strip used forthe first-dimension separation (see Table 1) Using theintermediate-range IPG strip covering pH 4ndash7 about 20ndash25 of the proteins in the theoretical proteome predictedto be observable in the resulting 2D-PAGE gel were actu-ally detected This figure was reduced to 10ndash12 whenanalysing the more basic pI range using the pH 6ndash11 IPGstrip Attempts to increase the percentage of the proteomedetectable using the pH 4ndash7 and pH 6ndash11 IPG strips byloading more protein sample resulted in poorer separa-tions (data not shown) This was especially true of thepH 6ndash11 strips which were generally the most difficult touse successfully However good separation of higher pro-tein loads was achieved using the narrow-range IPG stripspH 45ndash55 and 55ndash67 significantly increasing the pro-portion of theoretical proteins detected to over 40Clearly and more pertinently the limited conditions usedfor culturing S coelicolor in this study will also haverestricted the number of proteins detected as not all theproteome will be expressed In particular S coelicolordoes not sporulate in shaken liquid-grown cultures soproteins involved in this differentiation process are unlikelyto be present Nevertheless it is striking that the com-bined data for the pH 45ndash55 and 55ndash67 isoelectric pointranges indicate that 2468 out of a total of predicted 4932protein spots can be detected (see Table 1) suggestingthat transition phase mycelium in this supplementedminimal medium is expressing 50 of its genes (ie byextrapolation some 3912 of the 7825 in the genome)
Analysis of post-translational regulation
Although the regulation of cellular processes and adaptiveresponses in bacteria has been studied extensively at thetranscriptional level post-translational regulatory mecha-nisms are much less understood Proteomic analysisusing 2D-PAGE coupled with mass spectrometry offersthe opportunity to observe and identify modification ofproteins at a general cellular level The functions of pro-teins can be altered by specific covalent attachment of
adducts (eg phosphorylation glycosylation nucleotidy-lation acetylation) or by proteolytic processing Bothevents alter the pI andor mass of the protein and so canusually be observed on 2D-PAGE In the S coelicolorproteome map ordf110 out of a total of 770 gene productswere identified in more than one position or at pI andapparent mass co-ordinates significantly different frompredicted values indicating extensive regulation at thepost-translational level (88 are listed in Table 3) a propor-tion similar to that reported for other prokaryotes (Buttneret al 2001 Tonella et al 2001) The initial peptide massfingerprint analysis identified the modification of only 10of these proteins probably partly because fingerprint datatypically cover ordf30ndash60 of a proteinrsquos sequence (data notshown) so modification at positions not represented willbe missed Protein digestion with endopeptidases otherthan trypsin before mass spectrometry may help in thissituation but some covalently attached groups are labileand cannot be detected routinely Phosphorylation of Hisoccurs on nitrogen producing a high-energy phosphora-midate bond that is easily hydrolysed in acidic conditionswhereas phospho-Asp is the most labile phosphorylatedresidue with a half-life of a few hours under physiologicalconditions (reviewed by Robinson et al 2000) Thesemodifications the basis of bacterial two-component regu-latory systems are therefore unlikely to survive stainingof the 2D gels which involved the use of a fixing solutioncontaining 10 acetic acid and mass spectrometry Iden-tification of the more stable phosphoester phosphorylationthat occurs on Ser Thr or Tyr residues using PAGEfollowed by mass spectrometry has been reported(Godovac-Zimmermann et al 1999 Buttner et al 2001)but no examples were detected in this work Of the mod-ifications identified in this study three were by acetylationat the N-terminus after removal of the initiator Met residuetwo involved adenylylation and five proteins had been N-terminally truncated Modification of the 20S proteasomeby N-acetylation in yeast involves removal of the Metresidue by N-methionylaminopeptidase followed byacetylation with an Na-acetyltransferase (Kimura et al2000) Homologues of these enzymes are predicted fromthe S coelicolor genome and are presumably responsiblefor the observed modification of the 20S proteasomealpha subunit and of BldK-ORFD and the putative cellu-lose-binding protein The functional significance of Na-acetylation of these proteins is unclear
Analysis of the biosynthesis of antibiotics and other secondary metabolites
Post-translational modification The proteome mappingresults indicate that at least six enzymes in the Act clusterare subject to post-translational modifications and one
Proteomic analysis of S coelicolor 927
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
each from the undecylprodigiosin (SCO5877ndash5898)lsquocoelichelinrsquo (SCO0489ndash0499) and type I polyketidesynthase (SCO6273ndash6288) clusters (see Table 3) Thesemetabolic pathways may therefore be significantly regu-lated at the post-translational level The actVI-ORF3 gene(SCO5074) encodes a putative dehydratase enzyme thatis part of the biosynthetic pathway for Act a blue memberof the benzoisochromanequinone (BIQ) family of pig-mented antibiotics Mutation in actVI-ORF3 causes a 50decrease in the accumulation of Act (and a reddish brownphenotype) and it is believed to be responsible for pyranring closure an intermediate step in Act biosynthesis (thatmay also occur spontaneously) leading to the formationof the BIQ chromophore (Fernandez-Moreno et al 1994Ichinose et al 1998a Taguchi et al 2000) In this studyActVI-ORF3 protein was detected in two forms a majorspot corresponding to ActVI-ORF3 truncated by removalof the N-terminal 31 amino acid residues and a minor onein which the first 34 amino acids were missing (spots 31in Fig 1) The full-length protein was not detected Anal-ysis of the protein sequence using the SIGNALP server athttpwwwcbsdtudkservicesSignalP predicted a signalpeptide cleavage site between amino acids 31 and 32 inagreement with the observed results
The protein extract analysed was prepared from myce-lium that had been collected by centrifugation and sub-jected to two washes indicating that if ActVI-ORF3 isexported it is effectively retained within the cell wallmatrix This was confirmed by analysis of a preparationobtained by externally extracting proteins from intactwashed mycelium (Fig 4) Export of an enzyme requiredfor the efficient biosynthesis of an antibiotic that is thoughtto be produced intracellularly appears to be anomalousparticularly as it is involved in an intermediate biosynthetic
event Database analysis of the other members of the Actcluster revealed only one other protein with a predictedsignal peptide sequence the putative integral membranetransport-related protein ActII-ORF3 and it seemsunlikely that all the Act biosynthetic steps following BIQchromophore formation are extracellular It is possible thatfull-length ActVI-ORF3 catalyses the intermediate biosyn-thetic event intracellularly and the exported processedprotein is responsible for a different extracellular activityperhaps later in the pathway However no full-length pro-tein was detected so the exported enzyme may performthe pyran ring closure on a secreted metabolite that isthen reabsorbed by the cell for completion of the sub-sequent biosynthetic steps Only two homologues ofactVI-ORF3 are known and both are also in BIQ antibioticbiosynthetic clusters gra-ORF18 from the granaticincluster (Ichinose et al 1998b) and med-ORF5 frommedermycin biosynthesis (K Ichinose personal com-munication) Interestingly the encoded proteins of bothsequences are also predicted to contain N-terminal signalpeptides The observation that ActVI-ORF3 is exported tothe cell wall matrix may therefore be of general signifi-cance to the BIQ family of antibiotics and provide a newinsight into their biosynthetic pathways
Pathway representation Of the 20 putative secondarymetabolite gene clusters identified in the S coelicolorgenome sequence (Bentley et al 2002) eight had atleast one protein each identified on the proteome map(see Table 5) The biosynthetic clusters for the antibioticsAct Red and CDA had 68 36 and 40 of their pro-teins represented There are several possible explana-tions for failure to detect all the proteins for each pathwaySome are putative integral membrane proteins so cannotreadily be detected using conventional 2D-PAGE SomeRed and CDA proteins have molecular weights gt200 kDaoutside the range of the 125 gels used (for clustermembers see Supplementary material) The pathway-specific regulatory proteins for Act and Red biosynthesiswere not detected despite a detailed search of their pre-dicted gel locations even though the relevant genes areknown to be transcribed in equivalent cultures of the samestrain (Hesketh et al 2001) The proteins are presumablypresent but at levels below the limit of detection Howeverthe transcriptional regulator for CDA production CdaRwas identified as well as the two-component systemresponse regulator AbsA2 which is also encoded by agene in the CDA cluster (Anderson et al 2001) Interest-ingly proteins from three putative secondary meta-bolite clusters producing chemicals of as yet unknownstructure were also identified on the map correspondingto the deoxysugar synthaseglycosyltransferase clusterSCO0381ndash0401 the lsquocoelichelinrsquo cluster for a predicted(but not yet confirmed) oligopeptide siderophore
Fig 4 Processed ActVI-ORF3 is present in the cell wall matrix 2D-gel separations of protein extracts prepared (A) from whole cells or (B) by externally washing cells in a 2 SDS extraction buffer The arrow indicates ActVI-ORF3 protein The experiment was performed on duplicate biological samples and both showed similar protein expression profiles
928 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
(SCO0489ndash0499) and the type I polyketide clusterSCO6273ndash6288 The 3-oxoacyl-[acyl carrier protein]reductase (SCO6282) from the latter pathway is ex-pressed in a growth phase-dependent manner (Fig 3)and in early stationary phase cultures is perhaps themost abundant protein spot in the entire detectable pro-teome Presumably the metabolites produced by thesethree pathways are being synthesized under the condi-tions used In addition to coelichelin three other sidero-phores are postulated for S coelicolor All the proteinsfrom one of these desferrioxamine (SCO2782ndash2785)were identified but none was seen from either of the othertwo pathways The WhiE polyketide spore pigment isassociated with sporulation of surface-grown cultures(Davis and Chater 1990) and the expression of the whiEgenes depends on a series of sporulation-specific regula-tory genes (Kelemen et al 1998) no corresponding pro-teins of which were detected here It is therefore notsurprising that WhiE proteins were not detected in sam-ples prepared from liquid-grown vegetative mycelium
Analysis of primary metabolism
Proteins annotated as functioning in primary metabolicpathways were highly represented on the proteome mapwith ordf60 or more of the enzymes assigned to glycolysisthe TCA cycle the pentose phosphate pathway andpurine and pyrimidine ribonucleotide biosynthesis beingidentified (see Table 6) This figure may be deceptively lowbecause these pathways in S coelicolor often appear tohave more than one enzyme isoform for some of themetabolic steps potentially offering additional levels ofcontrol or developmental responsiveness at these pointsFor example only two steps in glycolysis have just a singlegene product assigned to them the remainder typicallyhave two or three each (Fig 5) At least one isoform waspresent for each step so the pathway is in theory 100functionally represented Understanding the regulation ofthese pathways will require analysis of the expression ofthese isoenzymes under different nutritional conditionsor different developmental stages Figure 5 shows that ofthe three predicted glyceraldehyde-3-phosphate dehydro-genase isoforms from glycolysis only two were detectedin this study (see also Supplementary material) Of theseSCO1947 appears to be constitutive whereas SCO7511is growth phase dependent (see Fig 3) Thus even thelimited conditions in this study yielded useful informationabout isoenzyme expression patterns (see Supplemen-tary material) In Streptomyces aureofaciens two genesencoding glyceraldehyde-3-phosphate dehydrogenaseactivity also appear to be differently regulated transcrip-tion of one highly homologous to SCO7511 is activatedby the regulator GapR whereas the other is GapR inde-pendent (Spusansky et al 2001)
Hood et al (1992) reported that most amino acid bio-synthetic enzymes are expressed at very low constitutivelevels in S coelicolor so it is reassuring to note that ordf50could be detected and identified using proteomics Theseinclude TrpB and TrpC1 from the tryptophan biosyntheticpathway the corresponding transcripts of which couldbarely be detected in previously reported S1 nucleaseprotection experiments (Hu et al 1999)
Perspectives
No attempt was made in the work reported here to simplifythe protein separations by subdividing the sample intosoluble and membrane fractions and this is reflected inthe 10 lipoproteins and 16 ABC transport system ATP-binding subunits identified on the proteome map (seeTable 2) No integral membrane proteins were detectedand even when a membrane protein sample was pre-pared and analysed no proteins identified possessedmore than three predicted transmembrane helices (datanot shown) This is consistent with previous studiesin other bacteria (Molloy et al 1999 Nilsson andDavidsson 2000) Another group of proteins also strik-ingly absent from the list of those identified is the sigmafactors which determine promoter selection by RNA poly-merase There are an extraordinarily high number (65) ofsigma factors encoded in the S coelicolor genome aswell as at least seven antisigma factors and 15 antisigmafactor antagonists (some of each of which were detected)Sigma factor heterogeneity in bacteria is an important wayof regulating expression of specific sets of genes under
Fig 5 The glycolytic pathway in S coelicolor illustrating the assignment of multiple gene products to individual metabolic steps based on the annotation of the genome sequence at httpwwwsangeracukProjectsS_coelicolor Gene products identified in the proteome map are underlined
Proteomic analysis of S coelicolor 929
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
different conditions and in different cell types (for a reviewsee Gross et al 1998) Although no sigma factors wereidentified about half (31 out of 65) have predicted pIvalues gtpH 7 and so can only be detected using thepH 6ndash11 IPG strips that give the poorest results (seeTable 1) However the principal vegetative sigma factorHrdB which is expected to be moderately abundanthas a predicted pI of 613 and its absence from thepH 55ndash67 proteome map may indicate that the samplepreparation method which does not remove DNA is inap-propriate Among other regulatory proteins 11 out of apossible 80 two-component system response regulatorsplus a further 36 proteins annotated as lsquoregulatorsrsquo weredetected Only one SerThr kinase and no two-componentsystem sensor kinases were detected possibly reflectingthe difficulty in analysing proteins with several transmem-brane domains Interestingly 30 secreted proteins wereidentified on the proteome map at least 10 of whichappear from comparison of their observed pI and molec-ular weight positions with predicted values to have beenprocessed already and therefore exported Presumablythese proteins do not end up exclusively in the culturemedium
Clearly this approach to proteomic analysis of S coeli-color has some important deficiencies not least theabsence of certain groups of proteins and the relativelypoor initial success rate for specifically characterizingpost-translational modifications Nevertheless it offersunique insights into the molecular biology of the organismthe expression of many proteins from both primary andsecondary metabolic pathways can be analysed post-translational regulation of proteins can be investigated ona general cellular level and the physical locations forsome proteins can be determined Approximately 35 ofthe S coelicolor genome is annotated as encoding lsquohypo-theticalrsquo proteins of unknown function and 182 proteins ofthis type were identified on the proteome map No exam-ples were found of overt disagreements between pre-dicted and observed proteins providing experimentalsupport for the general accuracy of the S coelicolorgenome sequence and its annotation Information aboutthe expression post-translational modification and physi-cal location of these (no longer) hypothetical proteinsfrom proteomic analyses is going to be important forassigning future functions As many of the hypotheticalproteins have apparent orthologues in pathogenic myco-bacteria some of the information may have an immediateapplication in the context of pathogenesis
Experimental procedures
Materials
Immobilized pH gradient (IPG) strips and IPG bufferswere purchased from Amersham Biosciences Acrylamide
duracryl and chemicals for silver staining were obtained fromGenomic Solutions All other chemicals were from Sigmaunless stated in the text
Strain and growth conditions
Streptomyces coelicolor M145 was cultivated with vigorousagitation at 30infinC in minimal medium supplemented with 02casamino acids (SMM) as described previously (Kieser et al2000) Briefly a high-density spore preparation [about 1010
colony-forming units (cfu) ml-1] was pregerminated in 2yen YTmedia for 7 h at 30infinC Germlings were harvested by centrifu-gation (5 min at 4000 g) resuspended in SMM and sonicatedbriefly to disperse any aggregates before inoculation into50 ml of SMM in 250 ml siliconized flasks containing coiledstainless steel springs Each flask received the equivalent of5 yen 107 spore cfu
Protein extraction and 2D-gel electrophoresis
The preparation of the protein sample to be separated waskept as simple as possible disrupting harvested myceliadirectly into denaturing isoelectric focusing (IEF) buffer con-taining Pefabloc SC (Roche) protease inhibitor This methodminimized unwanted protein degradation that was observedin initial studies using a Tris buffer containing Pefabloc SC forcell lysis (data not shown) Removal of DNA RNA salts andpolysaccharides from the sample was therefore not per-formed (other than the shearing of viscous DNA and RNA bysonication) and the protein extracts were sometimes difficultto focus when the sample was loaded by in-gel rehydrationLoading the sample soaked in paper strips at the anodicend of IPG strips that had already been rehydrated improvedthe reproducibility of separations significantly
Mycelium for protein extraction was harvested from cul-tures by brief centrifugation (30 s at 4000 g) at room tem-perature and immediately frozen in liquid nitrogen Typicallymycelium from 10 ml culture aliquots was collected and thetransfer time from culture flask to frozen sample was 15 minFrozen cells were thawed on ice in 5 ml of washing buffer(40 mM Tris pH 90 1 mM EGTA 1 mM EDTA) then pelletedby centrifugation (5 min at 3000 g) at 4infinC Washed cells wereresuspended in 400 ml of denaturing IEF buffer UTCHAPS[7 M urea 2 M thiourea 4 (wv) CHAPS 40 mM TrispH 90 1 mM EDTA 50 mM DTT 4 mM Pefabloc SC pro-tease inhibitor (Roche)] and disrupted by sonication (SanyoSoniprep 150 10 yen 2 s bursts at amplitude 75 mm) whilecooling in an ethanolndashice bath Cell debris was removed bycentrifugation (15 min 10 000 g 4infinC) and the protein extractwas stored frozen in aliquots at -80infinC until use For thepreparation of extracellular protein extracts washed cellswere resuspended in extraction buffer consisting of 40 mMTris pH 90 1 mM EDTA 50 mM DTT 2 SDS and 4 mMPefabloc SC protease inhibitor Cells were vortexed for 2 minbefore being pelleted by centrifugation (5 min 10 000 g4infinC) The supernatant was precipitated using the Amersham2D Clean-Up kit according to the manufacturerrsquos instructionsand the precipitated proteins were redissolved in UTCHAPSand stored at -80infinC until use
930 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
For first-dimension isoelectric focusing 18 cm IPG stripspH 4ndash7 pH 6ndash11 pH 45ndash55 or pH 55ndash67 (Amersham Bio-sciences) were rehydrated overnight in IEF buffer containing1 ampholytes according to the manufacturerrsquos instructionsusing a Phaser isoelectric focusing unit (Genomic Solutions)set at 20 V Protein samples to be separated were applied tothe rehydrated strips at the anodic end soaked in a 5ndash10 mmsection of IEF electrode strip (Amersham Biosciences)Separation was performed for 120 000 V-h with a maximumvoltage of 5000 V After isoelectric focusing IPG strips wereequilibrated for the second dimension for 15 min in IPG equil-ibration buffer (50 mM Tris pH 68 6 M urea 30 glycerol1 SDS and 001 bromophenol blue) plus 80 mM DTTthen for 10 min in IPG equilibration buffer plus 135 mMiodoacetamide Approximately 1 cm was removed from theanodic end of each equilibrated strip before applying the stripto the top of a vertical 125 SDS-PAGE gel for second-dimension separation using the Investigator 5000 systemfrom Genomic Solutions
Large-format gels (24 cm wide yen 22 cm high yen 1 mm thick)for the second-dimension separation were cast in-houseusing the system supplied by Genomic Solutions 125SDS-PAGE gels prepared using 065 NN-methylenebi-sacrylamide as cross-linking agent were used for pH 4ndash7and pH 45ndash55 IPG strips whereas pH 55ndash67 and pH 6ndash11 strips were applied to similar gels made with 08 cross-linker Electrophoresis was performed while cooling using themaximum setting (ordf 4ndash10infinC) at 20 000 mW per gel constantpower and a maximum voltage of 500 V Gels were thenstained with either colloidal Coomassie G-250 (Neuhoff et al1988) or silver nitrate (Rabilloud 1992) and scanned in aProXPRESS proteomic imaging system (Perkin-Elmer)Image analysis was performed using PHORETIX 2D version51 (NonLinear Dynamics) spot detection was optimizedautomatically using the lsquoSpot Detection Wizardrsquo and thenedited manually background subtraction was performedautomatically using the lsquoMode of Non-Spotrsquo setting imageswere then normalized to the total spot volume for quantifica-tion Spot filtering was not used
Protein identification using mass spectrometry
Protein spots of interest were excised from colloidalCoomassie-stained gels in-gel digested with trypsin andidentified by MALDI-TOF peptide mass fingerprint analysisExcised gel pieces (ordf 1-mm-diameter circles cut from 1-mm-thick gels) were washed twice with 100 ml of 50 mMammonium bicarbonate for 15 min once with 100 ml of 20acetonitrile-40 mM ammonium bicarbonate for 15 min andonce with 100 acetonitrile Washed gel pieces were allowedto air-dry for 10 min before being rehydrated in 5 ml of10 mg ml-1 trypsin (sequencing grade modified Promega) in10 mM ammonium bicarbonate Digestion (37infinC for 4 h) wasstopped by the addition of 5 ml of 5 formic acid Extractionof peptides into this solution was encouraged by a sonicwater bath treatment for 20 min Peptide extract (05 ml) wasspotted on to a thin layer of a-cyano-4-hydroxycinnamic acidapplied to a MALDI-TOF sample template and analysed byMALDI-TOF mass spectrometry (Bruker Reflex III) using anaccelerating voltage of 25 kV Samples were externally cali-brated using a standard mixture of six peptides ranging in
mass from 10465423 Da to 34946500 Da and the massaccuracy obtained was 60 ppm or better Identification ofproteins from MALDI-TOF peptide mass fingerprint data wasperformed using the lsquoMascotrsquo search engine at httpwwwmatrixsciencecom and was based on a positive resultusing their lsquoProbability Based MOWSE Scorersquo algorithm AMOWSE score of 60 or higher is significant at the 5 levelor better and proteins in this work typically gave scores gt80(frequently considerably so) In addition no identification wasaccepted unless at least five peptides representing at least20 of the protein sequence were detected in the MALDI-TOF peptide mass fingerprint
Assignment of proteins to metabolic pathways
Proteins were assigned to primary metabolic pathwaysusing the lsquoProtein Classification Schemersquo web page ofthe annotated Streptomyces coelicolor genome sequenceat httpwwwsangeracukProjectsS_coelicolor as it ap-peared in May 2002 The annotation of the genome andtherefore the Protein Classification Scheme is likely to bemodified as more experimental data are obtained about thefunction of genes that have currently been assigned on thebasis of sequence information alone It is therefore possiblethat proteins assigned to metabolic pathways in this studymay in future be shown to perform different functions in thecell
Assignments to gene clusters encoding secondary metab-olites are based on an analysis of the genome sequencereported by Bentley et al (2002) In some cases theseassignments are based on extensive experimental data (actred reviewed by Chater and Bibb 1997 Champness 2000cda Chong et al 1998 Huang et al 2001 whiE Davis andChater 1990)
Acknowledgements
This work was funded by grants (2FGT11406 208FGT11408 and 208IGF12432) under the BBSRCrsquos Tech-nologies for Functional Genomics and Investigating GeneFunction initiatives and by a competitive strategic grant fromthe BBSRC to the John Innes Centre We thank Mike Naldrettfor his extensive contribution in establishing technology forproteome analysis at the JIC
Supplementary material
The following material is available from httpwwwblackwell-sciencecomproductsjournalssuppmatmolemole3219mmi3219smhtmSection 1 Detection of gene products assigned to individualmetabolic steps in primary metabolic pathways in ScoelicolorSection 2 Identification of proteins from individual amino acidbiosynthetic pathwaysSection 3 Detection of gene products assigned to individualmetabolic steps in secondary metabolic pathways in Scoelicolor
Proteomic analysis of S coelicolor 931
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
References
Anderson TB Brian P and Champness WC (2001)Genetic and transcriptional analysis of absA an antibioticgene cluster linked two-component system that regulatesmultiple antibiotics in Streptomyces coelicolor Mol Micro-biol 39 553ndash566
Bentley SD Chater KF Cerdeno-Tarraga A-M ChallisGL Thomson NR James KD et al (2002) Completegenome sequence of the model actinomycete Streptomy-ces coelicolor A3(2) Nature 417 141ndash147
Berdy J (1984) New ways to obtain antibiotics Chin J Anti-biot 7 272ndash290
Berggren K Chernokalskaya E Steinberg TH KemperC Lopez MF Diwu Z et al (2000) Background-freehigh sensitivity staining of proteins in one- and two-dimensional sodium dodecyl sulfate-polyacrylamide gelsusing a luminescent ruthenium complex Electrophoresis21 2509ndash2521
Blackstock WP and Weir MP (1999) Proteomics quan-titative and physical mapping of cellular proteins TrendsBiotechnol 17 121ndash127
Blattner FR Plunkett G Bloch CA Perna NT BurlandV Riley M et al (1997) The complete genome sequenceof Escherichia coli K-12 Science 277 1453ndash1462
Buttner K Bernhardt J Scharf C Schmid R MaderU Eymann C et al (2001) A comprehensive two-dimensional map of cytosolic proteins of Bacillus subtilisElectrophoresis 22 2908ndash2935
Champness W (2000) Actinomycete development antibioticproduction and phylogeny questions and challenges InProkaryotic Development Brun YV and Skimketts LJ(eds) Washington DC American Society for MicrobiologyPress pp 11ndash31
Chater KF and Bibb MJ (1997) Regulation of bacterialantibiotic production In Biotechnology Vol 7 Products ofSecondary Metabolism Kleinkauf H and von Doumlhren H(eds) Weinheim Germany VCH pp 57ndash105
Chong PP Podmore SM Kieser HM Redenbach MTurgay K Marahiel M et al (1998) Physical identificationof a chromosomal locus encoding biosynthetic genesfor the lipopeptide calcium-dependent antibiotic (CDA) ofStreptomyces coelicolor A3(2) Microbiology 144 193ndash199
de Crecy-Lagard V Servant-Moisson JV Grandvalet Cand Mazodier P (1999) Alteration of the synthesis of theClp ATP-dependent protease affects morphological andphysiological differentiation in Streptomyces Mol Microbiol32 505ndash517
Davis NK and Chater KF (1990) Spore colour inStreptomyces coelicolor A3(2) involves the developmen-tally regulated synthesis of a compound biosyntheticallyrelated to polyketide antibiotics Mol Microbiol 4 1679ndash1691
Fernandez-Moreno FA Martinez E Caballero JLIchinose K Hopwood DA and Malpartida F (1994)DNA sequence and functions of the actVI region of theactinorhodin biosynthetic gene cluster of Streptomycescoelicolor A3(2) J Biol Chem 269 24854ndash24863
Fink D Falke D Wohllheben W and Engels A (1999)Nitrogen metabolism in Streptomyces coelicolor A3(2)
modification of glutamine synthase I by an adenylyltrans-ferase Microbiology 145 2313ndash2322
Godovac-Zimmermann J Soskic V Posnanovic S andBrianza F (1999) Functional proteomics of signal trans-duction by membrane receptors Electrophoresis 20 952ndash961
Gorg A Obermaier C Boguth G Harder A Scheibe BWildgruber R and Weiss W (2000) The current state oftwo-dimensional electrophoresis with immobilized pH gra-dients Electrophoresis 21 1037ndash1053
Gramajo HC Takano E and Bibb MJ (1993) Stationary-phase production of the antibiotic actinorhodin in Strepto-myces coelicolor A3(2) is transcriptionally regulated MolMicrobiol 7 837ndash845
Gross CA Chan C Dombroski A Gruber T Sharp MTupy J and Young B (1998) The functional and regula-tory roles of sigma factors in transcription Cold SpringHarb Symp Quant Biol 63 141ndash155
Hesketh A Sun J and Bibb MJ (2001) Induction ofppGpp synthesis in Streptomyces coelicolor A3(2) grownunder conditions of nutritional sufficiency elicits actII-ORF4transcription and actinorhodin biosynthesis Mol Microbiol39 136ndash144
Hesketh A Fink D Gust B Rexer H-U Scheel BChater K et al (2002) The PII nitrogen regulatory proteinGlnK in Streptomyces coelicolor A3(2) is modified by ade-nylylation rather than uridylylation and the GlnD and GlnKhomologues are functionally dissimilar to their nitrogenregulatory system counterparts Mol Microbiol (inpress)
Hood DW Heidstra R Swodoba UK and Hodgson DA(1992) Molecular genetic analysis of proline and tryp-tophan biosynthesis in Streptomyces coelicolor A3(2)interaction between primary and secondary metabolism- areview Gene 115 5ndash12
Hu DS-J Hood DW Heidstra R and Hodgson DA(1999) The expression of the trpD trpC and trpBA genesof Streptomyces coelicolor A3(2) is regulated by growthrate and growth phase but not by feedback repression MolMicrobiol 32 869ndash880
Huang J Lih CJ Pan KH and Cohen SN (2001)Global analysis of growth phase responsive gene expres-sion and regulation of antibiotic biosynthetic pathways inStreptomyces coelicolor using DNA microarrays GenesDev 15 3183ndash3192
Ichinose K Taguchi T Ebizuka Y and Hopwood DA(1998a) Biosynthetic gene clusters of benzoisochromane-quinone antibiotics in Streptomyces spp Identification ofgenes involved in post-PKS tailoring steps Actinomyceto-logica 12 99ndash109
Ichinose K Bedford DJ Tornus D Bechthold A BibbMJ Revill PW et al (1998b) The granaticin biosyntheticgene cluster of Streptomyces violaceoruber Tu22sequence analysis and expression in a heterologous hostChem Biol 5 647ndash659
Kelemen GH Brian P Flardh K Chamberlin L ChaterK and Buttner M (1998) Developmental regulation oftranscription of whiE a locus specifying the polyketidespore pigment in Streptomyces coelicolor A3(2) J Bacte-riol 180 2515ndash2521
Kieser T Bibb MJ Buttner MJ Chater KF and
932 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
Hopwood DA (2000) Practical Streptomyces GeneticsNorwich John Innes Foundation
Kim EJ Chung HJ Suh B Hah YC and Roe JH(1998) Expression and regulation of the sodF gene encod-ing iron- and zinc-containing superoxide dismutase inStreptomyces coelicolor Muumlller J Bacteriol 180 2014ndash2020
Kimura Y Takaoka M Tanaka S Sassa H Tanaka KPolevoda B et al (2000) Na-acetylation and proteolyticactivity of the yeast 20S proteasome J Biol Chem 2754635ndash4639
Kunst F Ogasawara N Moszer I Albertini AM AlloniG Azevedo V et al (1997) The complete genomesequence of the Gram-positive bacterium Bacillus subtilisNature 390 249ndash256
Lucchini S Thompson A and Hinton JCD (2001)Microarrays for microbiologists Microbiology 147 1403ndash1414
Mann M Hendrickson RC and Pandey A (2001) Analy-sis of proteins and proteomes by mass spectrometry AnnuRev Biochem 70 437ndash473
Miyodah S (1993) Research on antibiotic screening inJapan over the last decade a producing microorganismsapproach Actinomycetologica 7 100ndash106
Molloy MP Herbert BR Williams KL and Gooley AA(1999) Extraction of Escherichia coli proteins with organicsolvents prior to two-dimensional electrophoresis Electro-phoresis 20 701ndash704
Nagy I Tamura T Vanderleyden J Baumeister W andDe Mot R (1998) The 20S proteasome of Streptomycescoelicolor J Bacteriol 180 5448ndash5453
Neuhoff V Arold N Taube D and Ehrhardt W (1988)Improved staining of proteins in polyacrylamide gels includ-ing isoelectric focusing gels with clear background at nan-ogram sensitivity using Coomassie Brilliant Blue G-250and R-250 Electrophoresis 9 255ndash262
Nilsson CL and Davidsson P (2000) New separation toolsfor comprehensive studies of protein expression by massspectrometry Mass Spectrom Rev 19 390ndash397
Puglia AM Vohradsky J and Thompson CJ (1995)
Developmental control of the heat-shock stress regulon inStreptomyces coelicolor Mol Microbiol 17 737ndash746
Rabilloud T (1992) A comparison between low backgroundsilver diamine and silver nitrate protein stains Electro-phoresis 13 429ndash439
Robinson VL Buckler DR and Stock AM (2000) A taleof two components a novel kinase and a regulatory switchNature Struct Biol 7 626ndash633
Schena M (2000) Microarray Biochip Technology NatickMA Eaton Publishing
Spusansky O Rezuchova B Homerova D andKormanec J (2001) Expression of the gap gene encodingglyceraldehyde-3-phosphate dehydrogenase of Strepto-myces aureofaciens requires GapR a member of theAraCXylS family of transcriptional activators Microbiology147 1291ndash1301
Taguchi T Itou K Ebizuka Y Malpartida F HopwoodDA Surti CM et al (2000) Chemical characterisationof disruptants of the Streptomyces coelicolor A3(2) actVIgenes involved in actinorhodin biosynthesis J Antibiot 53144ndash152
Takano E Gramajo HC Strauch E Andres N WhiteJ and Bibb MJ (1992) Transcriptional regulation of theredD transcriptional activator gene accounts for growth-phase-dependent production of the antibotic undecylpro-digiosin in Streptomyces coelicolor A3(2) Mol Microbiol6 2797ndash2804
Tonella L Hoogland C Binz P-A Appel RDHochstrasser DF and Sanchez J-C (2001) New per-spectives in the Escherichia coli proteome investigationProteomics 1 409ndash423
Vohradsky J Li XM and Thompson CJ (1997) Identifi-cation of prokaryotic developmental stages by statisticalanalysis of two-dimensional gel patterns Electrophoresis18 1418ndash1428
Vohradsky J Li XM Dale G Folcher M Nguyen LViollier PH and Thompson CJ (2000) Developmentalcontrol of stress stimulons in Streptomyces coelicolorrevealed by statistical analyses of global gene expressionpatterns J Bacteriol 182 4979ndash4986

Proteomic analysis of S coelicolor 927
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
each from the undecylprodigiosin (SCO5877ndash5898)lsquocoelichelinrsquo (SCO0489ndash0499) and type I polyketidesynthase (SCO6273ndash6288) clusters (see Table 3) Thesemetabolic pathways may therefore be significantly regu-lated at the post-translational level The actVI-ORF3 gene(SCO5074) encodes a putative dehydratase enzyme thatis part of the biosynthetic pathway for Act a blue memberof the benzoisochromanequinone (BIQ) family of pig-mented antibiotics Mutation in actVI-ORF3 causes a 50decrease in the accumulation of Act (and a reddish brownphenotype) and it is believed to be responsible for pyranring closure an intermediate step in Act biosynthesis (thatmay also occur spontaneously) leading to the formationof the BIQ chromophore (Fernandez-Moreno et al 1994Ichinose et al 1998a Taguchi et al 2000) In this studyActVI-ORF3 protein was detected in two forms a majorspot corresponding to ActVI-ORF3 truncated by removalof the N-terminal 31 amino acid residues and a minor onein which the first 34 amino acids were missing (spots 31in Fig 1) The full-length protein was not detected Anal-ysis of the protein sequence using the SIGNALP server athttpwwwcbsdtudkservicesSignalP predicted a signalpeptide cleavage site between amino acids 31 and 32 inagreement with the observed results
The protein extract analysed was prepared from myce-lium that had been collected by centrifugation and sub-jected to two washes indicating that if ActVI-ORF3 isexported it is effectively retained within the cell wallmatrix This was confirmed by analysis of a preparationobtained by externally extracting proteins from intactwashed mycelium (Fig 4) Export of an enzyme requiredfor the efficient biosynthesis of an antibiotic that is thoughtto be produced intracellularly appears to be anomalousparticularly as it is involved in an intermediate biosynthetic
event Database analysis of the other members of the Actcluster revealed only one other protein with a predictedsignal peptide sequence the putative integral membranetransport-related protein ActII-ORF3 and it seemsunlikely that all the Act biosynthetic steps following BIQchromophore formation are extracellular It is possible thatfull-length ActVI-ORF3 catalyses the intermediate biosyn-thetic event intracellularly and the exported processedprotein is responsible for a different extracellular activityperhaps later in the pathway However no full-length pro-tein was detected so the exported enzyme may performthe pyran ring closure on a secreted metabolite that isthen reabsorbed by the cell for completion of the sub-sequent biosynthetic steps Only two homologues ofactVI-ORF3 are known and both are also in BIQ antibioticbiosynthetic clusters gra-ORF18 from the granaticincluster (Ichinose et al 1998b) and med-ORF5 frommedermycin biosynthesis (K Ichinose personal com-munication) Interestingly the encoded proteins of bothsequences are also predicted to contain N-terminal signalpeptides The observation that ActVI-ORF3 is exported tothe cell wall matrix may therefore be of general signifi-cance to the BIQ family of antibiotics and provide a newinsight into their biosynthetic pathways
Pathway representation Of the 20 putative secondarymetabolite gene clusters identified in the S coelicolorgenome sequence (Bentley et al 2002) eight had atleast one protein each identified on the proteome map(see Table 5) The biosynthetic clusters for the antibioticsAct Red and CDA had 68 36 and 40 of their pro-teins represented There are several possible explana-tions for failure to detect all the proteins for each pathwaySome are putative integral membrane proteins so cannotreadily be detected using conventional 2D-PAGE SomeRed and CDA proteins have molecular weights gt200 kDaoutside the range of the 125 gels used (for clustermembers see Supplementary material) The pathway-specific regulatory proteins for Act and Red biosynthesiswere not detected despite a detailed search of their pre-dicted gel locations even though the relevant genes areknown to be transcribed in equivalent cultures of the samestrain (Hesketh et al 2001) The proteins are presumablypresent but at levels below the limit of detection Howeverthe transcriptional regulator for CDA production CdaRwas identified as well as the two-component systemresponse regulator AbsA2 which is also encoded by agene in the CDA cluster (Anderson et al 2001) Interest-ingly proteins from three putative secondary meta-bolite clusters producing chemicals of as yet unknownstructure were also identified on the map correspondingto the deoxysugar synthaseglycosyltransferase clusterSCO0381ndash0401 the lsquocoelichelinrsquo cluster for a predicted(but not yet confirmed) oligopeptide siderophore
Fig 4 Processed ActVI-ORF3 is present in the cell wall matrix 2D-gel separations of protein extracts prepared (A) from whole cells or (B) by externally washing cells in a 2 SDS extraction buffer The arrow indicates ActVI-ORF3 protein The experiment was performed on duplicate biological samples and both showed similar protein expression profiles
928 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
(SCO0489ndash0499) and the type I polyketide clusterSCO6273ndash6288 The 3-oxoacyl-[acyl carrier protein]reductase (SCO6282) from the latter pathway is ex-pressed in a growth phase-dependent manner (Fig 3)and in early stationary phase cultures is perhaps themost abundant protein spot in the entire detectable pro-teome Presumably the metabolites produced by thesethree pathways are being synthesized under the condi-tions used In addition to coelichelin three other sidero-phores are postulated for S coelicolor All the proteinsfrom one of these desferrioxamine (SCO2782ndash2785)were identified but none was seen from either of the othertwo pathways The WhiE polyketide spore pigment isassociated with sporulation of surface-grown cultures(Davis and Chater 1990) and the expression of the whiEgenes depends on a series of sporulation-specific regula-tory genes (Kelemen et al 1998) no corresponding pro-teins of which were detected here It is therefore notsurprising that WhiE proteins were not detected in sam-ples prepared from liquid-grown vegetative mycelium
Analysis of primary metabolism
Proteins annotated as functioning in primary metabolicpathways were highly represented on the proteome mapwith ordf60 or more of the enzymes assigned to glycolysisthe TCA cycle the pentose phosphate pathway andpurine and pyrimidine ribonucleotide biosynthesis beingidentified (see Table 6) This figure may be deceptively lowbecause these pathways in S coelicolor often appear tohave more than one enzyme isoform for some of themetabolic steps potentially offering additional levels ofcontrol or developmental responsiveness at these pointsFor example only two steps in glycolysis have just a singlegene product assigned to them the remainder typicallyhave two or three each (Fig 5) At least one isoform waspresent for each step so the pathway is in theory 100functionally represented Understanding the regulation ofthese pathways will require analysis of the expression ofthese isoenzymes under different nutritional conditionsor different developmental stages Figure 5 shows that ofthe three predicted glyceraldehyde-3-phosphate dehydro-genase isoforms from glycolysis only two were detectedin this study (see also Supplementary material) Of theseSCO1947 appears to be constitutive whereas SCO7511is growth phase dependent (see Fig 3) Thus even thelimited conditions in this study yielded useful informationabout isoenzyme expression patterns (see Supplemen-tary material) In Streptomyces aureofaciens two genesencoding glyceraldehyde-3-phosphate dehydrogenaseactivity also appear to be differently regulated transcrip-tion of one highly homologous to SCO7511 is activatedby the regulator GapR whereas the other is GapR inde-pendent (Spusansky et al 2001)
Hood et al (1992) reported that most amino acid bio-synthetic enzymes are expressed at very low constitutivelevels in S coelicolor so it is reassuring to note that ordf50could be detected and identified using proteomics Theseinclude TrpB and TrpC1 from the tryptophan biosyntheticpathway the corresponding transcripts of which couldbarely be detected in previously reported S1 nucleaseprotection experiments (Hu et al 1999)
Perspectives
No attempt was made in the work reported here to simplifythe protein separations by subdividing the sample intosoluble and membrane fractions and this is reflected inthe 10 lipoproteins and 16 ABC transport system ATP-binding subunits identified on the proteome map (seeTable 2) No integral membrane proteins were detectedand even when a membrane protein sample was pre-pared and analysed no proteins identified possessedmore than three predicted transmembrane helices (datanot shown) This is consistent with previous studiesin other bacteria (Molloy et al 1999 Nilsson andDavidsson 2000) Another group of proteins also strik-ingly absent from the list of those identified is the sigmafactors which determine promoter selection by RNA poly-merase There are an extraordinarily high number (65) ofsigma factors encoded in the S coelicolor genome aswell as at least seven antisigma factors and 15 antisigmafactor antagonists (some of each of which were detected)Sigma factor heterogeneity in bacteria is an important wayof regulating expression of specific sets of genes under
Fig 5 The glycolytic pathway in S coelicolor illustrating the assignment of multiple gene products to individual metabolic steps based on the annotation of the genome sequence at httpwwwsangeracukProjectsS_coelicolor Gene products identified in the proteome map are underlined
Proteomic analysis of S coelicolor 929
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
different conditions and in different cell types (for a reviewsee Gross et al 1998) Although no sigma factors wereidentified about half (31 out of 65) have predicted pIvalues gtpH 7 and so can only be detected using thepH 6ndash11 IPG strips that give the poorest results (seeTable 1) However the principal vegetative sigma factorHrdB which is expected to be moderately abundanthas a predicted pI of 613 and its absence from thepH 55ndash67 proteome map may indicate that the samplepreparation method which does not remove DNA is inap-propriate Among other regulatory proteins 11 out of apossible 80 two-component system response regulatorsplus a further 36 proteins annotated as lsquoregulatorsrsquo weredetected Only one SerThr kinase and no two-componentsystem sensor kinases were detected possibly reflectingthe difficulty in analysing proteins with several transmem-brane domains Interestingly 30 secreted proteins wereidentified on the proteome map at least 10 of whichappear from comparison of their observed pI and molec-ular weight positions with predicted values to have beenprocessed already and therefore exported Presumablythese proteins do not end up exclusively in the culturemedium
Clearly this approach to proteomic analysis of S coeli-color has some important deficiencies not least theabsence of certain groups of proteins and the relativelypoor initial success rate for specifically characterizingpost-translational modifications Nevertheless it offersunique insights into the molecular biology of the organismthe expression of many proteins from both primary andsecondary metabolic pathways can be analysed post-translational regulation of proteins can be investigated ona general cellular level and the physical locations forsome proteins can be determined Approximately 35 ofthe S coelicolor genome is annotated as encoding lsquohypo-theticalrsquo proteins of unknown function and 182 proteins ofthis type were identified on the proteome map No exam-ples were found of overt disagreements between pre-dicted and observed proteins providing experimentalsupport for the general accuracy of the S coelicolorgenome sequence and its annotation Information aboutthe expression post-translational modification and physi-cal location of these (no longer) hypothetical proteinsfrom proteomic analyses is going to be important forassigning future functions As many of the hypotheticalproteins have apparent orthologues in pathogenic myco-bacteria some of the information may have an immediateapplication in the context of pathogenesis
Experimental procedures
Materials
Immobilized pH gradient (IPG) strips and IPG bufferswere purchased from Amersham Biosciences Acrylamide
duracryl and chemicals for silver staining were obtained fromGenomic Solutions All other chemicals were from Sigmaunless stated in the text
Strain and growth conditions
Streptomyces coelicolor M145 was cultivated with vigorousagitation at 30infinC in minimal medium supplemented with 02casamino acids (SMM) as described previously (Kieser et al2000) Briefly a high-density spore preparation [about 1010
colony-forming units (cfu) ml-1] was pregerminated in 2yen YTmedia for 7 h at 30infinC Germlings were harvested by centrifu-gation (5 min at 4000 g) resuspended in SMM and sonicatedbriefly to disperse any aggregates before inoculation into50 ml of SMM in 250 ml siliconized flasks containing coiledstainless steel springs Each flask received the equivalent of5 yen 107 spore cfu
Protein extraction and 2D-gel electrophoresis
The preparation of the protein sample to be separated waskept as simple as possible disrupting harvested myceliadirectly into denaturing isoelectric focusing (IEF) buffer con-taining Pefabloc SC (Roche) protease inhibitor This methodminimized unwanted protein degradation that was observedin initial studies using a Tris buffer containing Pefabloc SC forcell lysis (data not shown) Removal of DNA RNA salts andpolysaccharides from the sample was therefore not per-formed (other than the shearing of viscous DNA and RNA bysonication) and the protein extracts were sometimes difficultto focus when the sample was loaded by in-gel rehydrationLoading the sample soaked in paper strips at the anodicend of IPG strips that had already been rehydrated improvedthe reproducibility of separations significantly
Mycelium for protein extraction was harvested from cul-tures by brief centrifugation (30 s at 4000 g) at room tem-perature and immediately frozen in liquid nitrogen Typicallymycelium from 10 ml culture aliquots was collected and thetransfer time from culture flask to frozen sample was 15 minFrozen cells were thawed on ice in 5 ml of washing buffer(40 mM Tris pH 90 1 mM EGTA 1 mM EDTA) then pelletedby centrifugation (5 min at 3000 g) at 4infinC Washed cells wereresuspended in 400 ml of denaturing IEF buffer UTCHAPS[7 M urea 2 M thiourea 4 (wv) CHAPS 40 mM TrispH 90 1 mM EDTA 50 mM DTT 4 mM Pefabloc SC pro-tease inhibitor (Roche)] and disrupted by sonication (SanyoSoniprep 150 10 yen 2 s bursts at amplitude 75 mm) whilecooling in an ethanolndashice bath Cell debris was removed bycentrifugation (15 min 10 000 g 4infinC) and the protein extractwas stored frozen in aliquots at -80infinC until use For thepreparation of extracellular protein extracts washed cellswere resuspended in extraction buffer consisting of 40 mMTris pH 90 1 mM EDTA 50 mM DTT 2 SDS and 4 mMPefabloc SC protease inhibitor Cells were vortexed for 2 minbefore being pelleted by centrifugation (5 min 10 000 g4infinC) The supernatant was precipitated using the Amersham2D Clean-Up kit according to the manufacturerrsquos instructionsand the precipitated proteins were redissolved in UTCHAPSand stored at -80infinC until use
930 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
For first-dimension isoelectric focusing 18 cm IPG stripspH 4ndash7 pH 6ndash11 pH 45ndash55 or pH 55ndash67 (Amersham Bio-sciences) were rehydrated overnight in IEF buffer containing1 ampholytes according to the manufacturerrsquos instructionsusing a Phaser isoelectric focusing unit (Genomic Solutions)set at 20 V Protein samples to be separated were applied tothe rehydrated strips at the anodic end soaked in a 5ndash10 mmsection of IEF electrode strip (Amersham Biosciences)Separation was performed for 120 000 V-h with a maximumvoltage of 5000 V After isoelectric focusing IPG strips wereequilibrated for the second dimension for 15 min in IPG equil-ibration buffer (50 mM Tris pH 68 6 M urea 30 glycerol1 SDS and 001 bromophenol blue) plus 80 mM DTTthen for 10 min in IPG equilibration buffer plus 135 mMiodoacetamide Approximately 1 cm was removed from theanodic end of each equilibrated strip before applying the stripto the top of a vertical 125 SDS-PAGE gel for second-dimension separation using the Investigator 5000 systemfrom Genomic Solutions
Large-format gels (24 cm wide yen 22 cm high yen 1 mm thick)for the second-dimension separation were cast in-houseusing the system supplied by Genomic Solutions 125SDS-PAGE gels prepared using 065 NN-methylenebi-sacrylamide as cross-linking agent were used for pH 4ndash7and pH 45ndash55 IPG strips whereas pH 55ndash67 and pH 6ndash11 strips were applied to similar gels made with 08 cross-linker Electrophoresis was performed while cooling using themaximum setting (ordf 4ndash10infinC) at 20 000 mW per gel constantpower and a maximum voltage of 500 V Gels were thenstained with either colloidal Coomassie G-250 (Neuhoff et al1988) or silver nitrate (Rabilloud 1992) and scanned in aProXPRESS proteomic imaging system (Perkin-Elmer)Image analysis was performed using PHORETIX 2D version51 (NonLinear Dynamics) spot detection was optimizedautomatically using the lsquoSpot Detection Wizardrsquo and thenedited manually background subtraction was performedautomatically using the lsquoMode of Non-Spotrsquo setting imageswere then normalized to the total spot volume for quantifica-tion Spot filtering was not used
Protein identification using mass spectrometry
Protein spots of interest were excised from colloidalCoomassie-stained gels in-gel digested with trypsin andidentified by MALDI-TOF peptide mass fingerprint analysisExcised gel pieces (ordf 1-mm-diameter circles cut from 1-mm-thick gels) were washed twice with 100 ml of 50 mMammonium bicarbonate for 15 min once with 100 ml of 20acetonitrile-40 mM ammonium bicarbonate for 15 min andonce with 100 acetonitrile Washed gel pieces were allowedto air-dry for 10 min before being rehydrated in 5 ml of10 mg ml-1 trypsin (sequencing grade modified Promega) in10 mM ammonium bicarbonate Digestion (37infinC for 4 h) wasstopped by the addition of 5 ml of 5 formic acid Extractionof peptides into this solution was encouraged by a sonicwater bath treatment for 20 min Peptide extract (05 ml) wasspotted on to a thin layer of a-cyano-4-hydroxycinnamic acidapplied to a MALDI-TOF sample template and analysed byMALDI-TOF mass spectrometry (Bruker Reflex III) using anaccelerating voltage of 25 kV Samples were externally cali-brated using a standard mixture of six peptides ranging in
mass from 10465423 Da to 34946500 Da and the massaccuracy obtained was 60 ppm or better Identification ofproteins from MALDI-TOF peptide mass fingerprint data wasperformed using the lsquoMascotrsquo search engine at httpwwwmatrixsciencecom and was based on a positive resultusing their lsquoProbability Based MOWSE Scorersquo algorithm AMOWSE score of 60 or higher is significant at the 5 levelor better and proteins in this work typically gave scores gt80(frequently considerably so) In addition no identification wasaccepted unless at least five peptides representing at least20 of the protein sequence were detected in the MALDI-TOF peptide mass fingerprint
Assignment of proteins to metabolic pathways
Proteins were assigned to primary metabolic pathwaysusing the lsquoProtein Classification Schemersquo web page ofthe annotated Streptomyces coelicolor genome sequenceat httpwwwsangeracukProjectsS_coelicolor as it ap-peared in May 2002 The annotation of the genome andtherefore the Protein Classification Scheme is likely to bemodified as more experimental data are obtained about thefunction of genes that have currently been assigned on thebasis of sequence information alone It is therefore possiblethat proteins assigned to metabolic pathways in this studymay in future be shown to perform different functions in thecell
Assignments to gene clusters encoding secondary metab-olites are based on an analysis of the genome sequencereported by Bentley et al (2002) In some cases theseassignments are based on extensive experimental data (actred reviewed by Chater and Bibb 1997 Champness 2000cda Chong et al 1998 Huang et al 2001 whiE Davis andChater 1990)
Acknowledgements
This work was funded by grants (2FGT11406 208FGT11408 and 208IGF12432) under the BBSRCrsquos Tech-nologies for Functional Genomics and Investigating GeneFunction initiatives and by a competitive strategic grant fromthe BBSRC to the John Innes Centre We thank Mike Naldrettfor his extensive contribution in establishing technology forproteome analysis at the JIC
Supplementary material
The following material is available from httpwwwblackwell-sciencecomproductsjournalssuppmatmolemole3219mmi3219smhtmSection 1 Detection of gene products assigned to individualmetabolic steps in primary metabolic pathways in ScoelicolorSection 2 Identification of proteins from individual amino acidbiosynthetic pathwaysSection 3 Detection of gene products assigned to individualmetabolic steps in secondary metabolic pathways in Scoelicolor
Proteomic analysis of S coelicolor 931
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
References
Anderson TB Brian P and Champness WC (2001)Genetic and transcriptional analysis of absA an antibioticgene cluster linked two-component system that regulatesmultiple antibiotics in Streptomyces coelicolor Mol Micro-biol 39 553ndash566
Bentley SD Chater KF Cerdeno-Tarraga A-M ChallisGL Thomson NR James KD et al (2002) Completegenome sequence of the model actinomycete Streptomy-ces coelicolor A3(2) Nature 417 141ndash147
Berdy J (1984) New ways to obtain antibiotics Chin J Anti-biot 7 272ndash290
Berggren K Chernokalskaya E Steinberg TH KemperC Lopez MF Diwu Z et al (2000) Background-freehigh sensitivity staining of proteins in one- and two-dimensional sodium dodecyl sulfate-polyacrylamide gelsusing a luminescent ruthenium complex Electrophoresis21 2509ndash2521
Blackstock WP and Weir MP (1999) Proteomics quan-titative and physical mapping of cellular proteins TrendsBiotechnol 17 121ndash127
Blattner FR Plunkett G Bloch CA Perna NT BurlandV Riley M et al (1997) The complete genome sequenceof Escherichia coli K-12 Science 277 1453ndash1462
Buttner K Bernhardt J Scharf C Schmid R MaderU Eymann C et al (2001) A comprehensive two-dimensional map of cytosolic proteins of Bacillus subtilisElectrophoresis 22 2908ndash2935
Champness W (2000) Actinomycete development antibioticproduction and phylogeny questions and challenges InProkaryotic Development Brun YV and Skimketts LJ(eds) Washington DC American Society for MicrobiologyPress pp 11ndash31
Chater KF and Bibb MJ (1997) Regulation of bacterialantibiotic production In Biotechnology Vol 7 Products ofSecondary Metabolism Kleinkauf H and von Doumlhren H(eds) Weinheim Germany VCH pp 57ndash105
Chong PP Podmore SM Kieser HM Redenbach MTurgay K Marahiel M et al (1998) Physical identificationof a chromosomal locus encoding biosynthetic genesfor the lipopeptide calcium-dependent antibiotic (CDA) ofStreptomyces coelicolor A3(2) Microbiology 144 193ndash199
de Crecy-Lagard V Servant-Moisson JV Grandvalet Cand Mazodier P (1999) Alteration of the synthesis of theClp ATP-dependent protease affects morphological andphysiological differentiation in Streptomyces Mol Microbiol32 505ndash517
Davis NK and Chater KF (1990) Spore colour inStreptomyces coelicolor A3(2) involves the developmen-tally regulated synthesis of a compound biosyntheticallyrelated to polyketide antibiotics Mol Microbiol 4 1679ndash1691
Fernandez-Moreno FA Martinez E Caballero JLIchinose K Hopwood DA and Malpartida F (1994)DNA sequence and functions of the actVI region of theactinorhodin biosynthetic gene cluster of Streptomycescoelicolor A3(2) J Biol Chem 269 24854ndash24863
Fink D Falke D Wohllheben W and Engels A (1999)Nitrogen metabolism in Streptomyces coelicolor A3(2)
modification of glutamine synthase I by an adenylyltrans-ferase Microbiology 145 2313ndash2322
Godovac-Zimmermann J Soskic V Posnanovic S andBrianza F (1999) Functional proteomics of signal trans-duction by membrane receptors Electrophoresis 20 952ndash961
Gorg A Obermaier C Boguth G Harder A Scheibe BWildgruber R and Weiss W (2000) The current state oftwo-dimensional electrophoresis with immobilized pH gra-dients Electrophoresis 21 1037ndash1053
Gramajo HC Takano E and Bibb MJ (1993) Stationary-phase production of the antibiotic actinorhodin in Strepto-myces coelicolor A3(2) is transcriptionally regulated MolMicrobiol 7 837ndash845
Gross CA Chan C Dombroski A Gruber T Sharp MTupy J and Young B (1998) The functional and regula-tory roles of sigma factors in transcription Cold SpringHarb Symp Quant Biol 63 141ndash155
Hesketh A Sun J and Bibb MJ (2001) Induction ofppGpp synthesis in Streptomyces coelicolor A3(2) grownunder conditions of nutritional sufficiency elicits actII-ORF4transcription and actinorhodin biosynthesis Mol Microbiol39 136ndash144
Hesketh A Fink D Gust B Rexer H-U Scheel BChater K et al (2002) The PII nitrogen regulatory proteinGlnK in Streptomyces coelicolor A3(2) is modified by ade-nylylation rather than uridylylation and the GlnD and GlnKhomologues are functionally dissimilar to their nitrogenregulatory system counterparts Mol Microbiol (inpress)
Hood DW Heidstra R Swodoba UK and Hodgson DA(1992) Molecular genetic analysis of proline and tryp-tophan biosynthesis in Streptomyces coelicolor A3(2)interaction between primary and secondary metabolism- areview Gene 115 5ndash12
Hu DS-J Hood DW Heidstra R and Hodgson DA(1999) The expression of the trpD trpC and trpBA genesof Streptomyces coelicolor A3(2) is regulated by growthrate and growth phase but not by feedback repression MolMicrobiol 32 869ndash880
Huang J Lih CJ Pan KH and Cohen SN (2001)Global analysis of growth phase responsive gene expres-sion and regulation of antibiotic biosynthetic pathways inStreptomyces coelicolor using DNA microarrays GenesDev 15 3183ndash3192
Ichinose K Taguchi T Ebizuka Y and Hopwood DA(1998a) Biosynthetic gene clusters of benzoisochromane-quinone antibiotics in Streptomyces spp Identification ofgenes involved in post-PKS tailoring steps Actinomyceto-logica 12 99ndash109
Ichinose K Bedford DJ Tornus D Bechthold A BibbMJ Revill PW et al (1998b) The granaticin biosyntheticgene cluster of Streptomyces violaceoruber Tu22sequence analysis and expression in a heterologous hostChem Biol 5 647ndash659
Kelemen GH Brian P Flardh K Chamberlin L ChaterK and Buttner M (1998) Developmental regulation oftranscription of whiE a locus specifying the polyketidespore pigment in Streptomyces coelicolor A3(2) J Bacte-riol 180 2515ndash2521
Kieser T Bibb MJ Buttner MJ Chater KF and
932 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
Hopwood DA (2000) Practical Streptomyces GeneticsNorwich John Innes Foundation
Kim EJ Chung HJ Suh B Hah YC and Roe JH(1998) Expression and regulation of the sodF gene encod-ing iron- and zinc-containing superoxide dismutase inStreptomyces coelicolor Muumlller J Bacteriol 180 2014ndash2020
Kimura Y Takaoka M Tanaka S Sassa H Tanaka KPolevoda B et al (2000) Na-acetylation and proteolyticactivity of the yeast 20S proteasome J Biol Chem 2754635ndash4639
Kunst F Ogasawara N Moszer I Albertini AM AlloniG Azevedo V et al (1997) The complete genomesequence of the Gram-positive bacterium Bacillus subtilisNature 390 249ndash256
Lucchini S Thompson A and Hinton JCD (2001)Microarrays for microbiologists Microbiology 147 1403ndash1414
Mann M Hendrickson RC and Pandey A (2001) Analy-sis of proteins and proteomes by mass spectrometry AnnuRev Biochem 70 437ndash473
Miyodah S (1993) Research on antibiotic screening inJapan over the last decade a producing microorganismsapproach Actinomycetologica 7 100ndash106
Molloy MP Herbert BR Williams KL and Gooley AA(1999) Extraction of Escherichia coli proteins with organicsolvents prior to two-dimensional electrophoresis Electro-phoresis 20 701ndash704
Nagy I Tamura T Vanderleyden J Baumeister W andDe Mot R (1998) The 20S proteasome of Streptomycescoelicolor J Bacteriol 180 5448ndash5453
Neuhoff V Arold N Taube D and Ehrhardt W (1988)Improved staining of proteins in polyacrylamide gels includ-ing isoelectric focusing gels with clear background at nan-ogram sensitivity using Coomassie Brilliant Blue G-250and R-250 Electrophoresis 9 255ndash262
Nilsson CL and Davidsson P (2000) New separation toolsfor comprehensive studies of protein expression by massspectrometry Mass Spectrom Rev 19 390ndash397
Puglia AM Vohradsky J and Thompson CJ (1995)
Developmental control of the heat-shock stress regulon inStreptomyces coelicolor Mol Microbiol 17 737ndash746
Rabilloud T (1992) A comparison between low backgroundsilver diamine and silver nitrate protein stains Electro-phoresis 13 429ndash439
Robinson VL Buckler DR and Stock AM (2000) A taleof two components a novel kinase and a regulatory switchNature Struct Biol 7 626ndash633
Schena M (2000) Microarray Biochip Technology NatickMA Eaton Publishing
Spusansky O Rezuchova B Homerova D andKormanec J (2001) Expression of the gap gene encodingglyceraldehyde-3-phosphate dehydrogenase of Strepto-myces aureofaciens requires GapR a member of theAraCXylS family of transcriptional activators Microbiology147 1291ndash1301
Taguchi T Itou K Ebizuka Y Malpartida F HopwoodDA Surti CM et al (2000) Chemical characterisationof disruptants of the Streptomyces coelicolor A3(2) actVIgenes involved in actinorhodin biosynthesis J Antibiot 53144ndash152
Takano E Gramajo HC Strauch E Andres N WhiteJ and Bibb MJ (1992) Transcriptional regulation of theredD transcriptional activator gene accounts for growth-phase-dependent production of the antibotic undecylpro-digiosin in Streptomyces coelicolor A3(2) Mol Microbiol6 2797ndash2804
Tonella L Hoogland C Binz P-A Appel RDHochstrasser DF and Sanchez J-C (2001) New per-spectives in the Escherichia coli proteome investigationProteomics 1 409ndash423
Vohradsky J Li XM and Thompson CJ (1997) Identifi-cation of prokaryotic developmental stages by statisticalanalysis of two-dimensional gel patterns Electrophoresis18 1418ndash1428
Vohradsky J Li XM Dale G Folcher M Nguyen LViollier PH and Thompson CJ (2000) Developmentalcontrol of stress stimulons in Streptomyces coelicolorrevealed by statistical analyses of global gene expressionpatterns J Bacteriol 182 4979ndash4986

928 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
(SCO0489ndash0499) and the type I polyketide clusterSCO6273ndash6288 The 3-oxoacyl-[acyl carrier protein]reductase (SCO6282) from the latter pathway is ex-pressed in a growth phase-dependent manner (Fig 3)and in early stationary phase cultures is perhaps themost abundant protein spot in the entire detectable pro-teome Presumably the metabolites produced by thesethree pathways are being synthesized under the condi-tions used In addition to coelichelin three other sidero-phores are postulated for S coelicolor All the proteinsfrom one of these desferrioxamine (SCO2782ndash2785)were identified but none was seen from either of the othertwo pathways The WhiE polyketide spore pigment isassociated with sporulation of surface-grown cultures(Davis and Chater 1990) and the expression of the whiEgenes depends on a series of sporulation-specific regula-tory genes (Kelemen et al 1998) no corresponding pro-teins of which were detected here It is therefore notsurprising that WhiE proteins were not detected in sam-ples prepared from liquid-grown vegetative mycelium
Analysis of primary metabolism
Proteins annotated as functioning in primary metabolicpathways were highly represented on the proteome mapwith ordf60 or more of the enzymes assigned to glycolysisthe TCA cycle the pentose phosphate pathway andpurine and pyrimidine ribonucleotide biosynthesis beingidentified (see Table 6) This figure may be deceptively lowbecause these pathways in S coelicolor often appear tohave more than one enzyme isoform for some of themetabolic steps potentially offering additional levels ofcontrol or developmental responsiveness at these pointsFor example only two steps in glycolysis have just a singlegene product assigned to them the remainder typicallyhave two or three each (Fig 5) At least one isoform waspresent for each step so the pathway is in theory 100functionally represented Understanding the regulation ofthese pathways will require analysis of the expression ofthese isoenzymes under different nutritional conditionsor different developmental stages Figure 5 shows that ofthe three predicted glyceraldehyde-3-phosphate dehydro-genase isoforms from glycolysis only two were detectedin this study (see also Supplementary material) Of theseSCO1947 appears to be constitutive whereas SCO7511is growth phase dependent (see Fig 3) Thus even thelimited conditions in this study yielded useful informationabout isoenzyme expression patterns (see Supplemen-tary material) In Streptomyces aureofaciens two genesencoding glyceraldehyde-3-phosphate dehydrogenaseactivity also appear to be differently regulated transcrip-tion of one highly homologous to SCO7511 is activatedby the regulator GapR whereas the other is GapR inde-pendent (Spusansky et al 2001)
Hood et al (1992) reported that most amino acid bio-synthetic enzymes are expressed at very low constitutivelevels in S coelicolor so it is reassuring to note that ordf50could be detected and identified using proteomics Theseinclude TrpB and TrpC1 from the tryptophan biosyntheticpathway the corresponding transcripts of which couldbarely be detected in previously reported S1 nucleaseprotection experiments (Hu et al 1999)
Perspectives
No attempt was made in the work reported here to simplifythe protein separations by subdividing the sample intosoluble and membrane fractions and this is reflected inthe 10 lipoproteins and 16 ABC transport system ATP-binding subunits identified on the proteome map (seeTable 2) No integral membrane proteins were detectedand even when a membrane protein sample was pre-pared and analysed no proteins identified possessedmore than three predicted transmembrane helices (datanot shown) This is consistent with previous studiesin other bacteria (Molloy et al 1999 Nilsson andDavidsson 2000) Another group of proteins also strik-ingly absent from the list of those identified is the sigmafactors which determine promoter selection by RNA poly-merase There are an extraordinarily high number (65) ofsigma factors encoded in the S coelicolor genome aswell as at least seven antisigma factors and 15 antisigmafactor antagonists (some of each of which were detected)Sigma factor heterogeneity in bacteria is an important wayof regulating expression of specific sets of genes under
Fig 5 The glycolytic pathway in S coelicolor illustrating the assignment of multiple gene products to individual metabolic steps based on the annotation of the genome sequence at httpwwwsangeracukProjectsS_coelicolor Gene products identified in the proteome map are underlined
Proteomic analysis of S coelicolor 929
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
different conditions and in different cell types (for a reviewsee Gross et al 1998) Although no sigma factors wereidentified about half (31 out of 65) have predicted pIvalues gtpH 7 and so can only be detected using thepH 6ndash11 IPG strips that give the poorest results (seeTable 1) However the principal vegetative sigma factorHrdB which is expected to be moderately abundanthas a predicted pI of 613 and its absence from thepH 55ndash67 proteome map may indicate that the samplepreparation method which does not remove DNA is inap-propriate Among other regulatory proteins 11 out of apossible 80 two-component system response regulatorsplus a further 36 proteins annotated as lsquoregulatorsrsquo weredetected Only one SerThr kinase and no two-componentsystem sensor kinases were detected possibly reflectingthe difficulty in analysing proteins with several transmem-brane domains Interestingly 30 secreted proteins wereidentified on the proteome map at least 10 of whichappear from comparison of their observed pI and molec-ular weight positions with predicted values to have beenprocessed already and therefore exported Presumablythese proteins do not end up exclusively in the culturemedium
Clearly this approach to proteomic analysis of S coeli-color has some important deficiencies not least theabsence of certain groups of proteins and the relativelypoor initial success rate for specifically characterizingpost-translational modifications Nevertheless it offersunique insights into the molecular biology of the organismthe expression of many proteins from both primary andsecondary metabolic pathways can be analysed post-translational regulation of proteins can be investigated ona general cellular level and the physical locations forsome proteins can be determined Approximately 35 ofthe S coelicolor genome is annotated as encoding lsquohypo-theticalrsquo proteins of unknown function and 182 proteins ofthis type were identified on the proteome map No exam-ples were found of overt disagreements between pre-dicted and observed proteins providing experimentalsupport for the general accuracy of the S coelicolorgenome sequence and its annotation Information aboutthe expression post-translational modification and physi-cal location of these (no longer) hypothetical proteinsfrom proteomic analyses is going to be important forassigning future functions As many of the hypotheticalproteins have apparent orthologues in pathogenic myco-bacteria some of the information may have an immediateapplication in the context of pathogenesis
Experimental procedures
Materials
Immobilized pH gradient (IPG) strips and IPG bufferswere purchased from Amersham Biosciences Acrylamide
duracryl and chemicals for silver staining were obtained fromGenomic Solutions All other chemicals were from Sigmaunless stated in the text
Strain and growth conditions
Streptomyces coelicolor M145 was cultivated with vigorousagitation at 30infinC in minimal medium supplemented with 02casamino acids (SMM) as described previously (Kieser et al2000) Briefly a high-density spore preparation [about 1010
colony-forming units (cfu) ml-1] was pregerminated in 2yen YTmedia for 7 h at 30infinC Germlings were harvested by centrifu-gation (5 min at 4000 g) resuspended in SMM and sonicatedbriefly to disperse any aggregates before inoculation into50 ml of SMM in 250 ml siliconized flasks containing coiledstainless steel springs Each flask received the equivalent of5 yen 107 spore cfu
Protein extraction and 2D-gel electrophoresis
The preparation of the protein sample to be separated waskept as simple as possible disrupting harvested myceliadirectly into denaturing isoelectric focusing (IEF) buffer con-taining Pefabloc SC (Roche) protease inhibitor This methodminimized unwanted protein degradation that was observedin initial studies using a Tris buffer containing Pefabloc SC forcell lysis (data not shown) Removal of DNA RNA salts andpolysaccharides from the sample was therefore not per-formed (other than the shearing of viscous DNA and RNA bysonication) and the protein extracts were sometimes difficultto focus when the sample was loaded by in-gel rehydrationLoading the sample soaked in paper strips at the anodicend of IPG strips that had already been rehydrated improvedthe reproducibility of separations significantly
Mycelium for protein extraction was harvested from cul-tures by brief centrifugation (30 s at 4000 g) at room tem-perature and immediately frozen in liquid nitrogen Typicallymycelium from 10 ml culture aliquots was collected and thetransfer time from culture flask to frozen sample was 15 minFrozen cells were thawed on ice in 5 ml of washing buffer(40 mM Tris pH 90 1 mM EGTA 1 mM EDTA) then pelletedby centrifugation (5 min at 3000 g) at 4infinC Washed cells wereresuspended in 400 ml of denaturing IEF buffer UTCHAPS[7 M urea 2 M thiourea 4 (wv) CHAPS 40 mM TrispH 90 1 mM EDTA 50 mM DTT 4 mM Pefabloc SC pro-tease inhibitor (Roche)] and disrupted by sonication (SanyoSoniprep 150 10 yen 2 s bursts at amplitude 75 mm) whilecooling in an ethanolndashice bath Cell debris was removed bycentrifugation (15 min 10 000 g 4infinC) and the protein extractwas stored frozen in aliquots at -80infinC until use For thepreparation of extracellular protein extracts washed cellswere resuspended in extraction buffer consisting of 40 mMTris pH 90 1 mM EDTA 50 mM DTT 2 SDS and 4 mMPefabloc SC protease inhibitor Cells were vortexed for 2 minbefore being pelleted by centrifugation (5 min 10 000 g4infinC) The supernatant was precipitated using the Amersham2D Clean-Up kit according to the manufacturerrsquos instructionsand the precipitated proteins were redissolved in UTCHAPSand stored at -80infinC until use
930 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
For first-dimension isoelectric focusing 18 cm IPG stripspH 4ndash7 pH 6ndash11 pH 45ndash55 or pH 55ndash67 (Amersham Bio-sciences) were rehydrated overnight in IEF buffer containing1 ampholytes according to the manufacturerrsquos instructionsusing a Phaser isoelectric focusing unit (Genomic Solutions)set at 20 V Protein samples to be separated were applied tothe rehydrated strips at the anodic end soaked in a 5ndash10 mmsection of IEF electrode strip (Amersham Biosciences)Separation was performed for 120 000 V-h with a maximumvoltage of 5000 V After isoelectric focusing IPG strips wereequilibrated for the second dimension for 15 min in IPG equil-ibration buffer (50 mM Tris pH 68 6 M urea 30 glycerol1 SDS and 001 bromophenol blue) plus 80 mM DTTthen for 10 min in IPG equilibration buffer plus 135 mMiodoacetamide Approximately 1 cm was removed from theanodic end of each equilibrated strip before applying the stripto the top of a vertical 125 SDS-PAGE gel for second-dimension separation using the Investigator 5000 systemfrom Genomic Solutions
Large-format gels (24 cm wide yen 22 cm high yen 1 mm thick)for the second-dimension separation were cast in-houseusing the system supplied by Genomic Solutions 125SDS-PAGE gels prepared using 065 NN-methylenebi-sacrylamide as cross-linking agent were used for pH 4ndash7and pH 45ndash55 IPG strips whereas pH 55ndash67 and pH 6ndash11 strips were applied to similar gels made with 08 cross-linker Electrophoresis was performed while cooling using themaximum setting (ordf 4ndash10infinC) at 20 000 mW per gel constantpower and a maximum voltage of 500 V Gels were thenstained with either colloidal Coomassie G-250 (Neuhoff et al1988) or silver nitrate (Rabilloud 1992) and scanned in aProXPRESS proteomic imaging system (Perkin-Elmer)Image analysis was performed using PHORETIX 2D version51 (NonLinear Dynamics) spot detection was optimizedautomatically using the lsquoSpot Detection Wizardrsquo and thenedited manually background subtraction was performedautomatically using the lsquoMode of Non-Spotrsquo setting imageswere then normalized to the total spot volume for quantifica-tion Spot filtering was not used
Protein identification using mass spectrometry
Protein spots of interest were excised from colloidalCoomassie-stained gels in-gel digested with trypsin andidentified by MALDI-TOF peptide mass fingerprint analysisExcised gel pieces (ordf 1-mm-diameter circles cut from 1-mm-thick gels) were washed twice with 100 ml of 50 mMammonium bicarbonate for 15 min once with 100 ml of 20acetonitrile-40 mM ammonium bicarbonate for 15 min andonce with 100 acetonitrile Washed gel pieces were allowedto air-dry for 10 min before being rehydrated in 5 ml of10 mg ml-1 trypsin (sequencing grade modified Promega) in10 mM ammonium bicarbonate Digestion (37infinC for 4 h) wasstopped by the addition of 5 ml of 5 formic acid Extractionof peptides into this solution was encouraged by a sonicwater bath treatment for 20 min Peptide extract (05 ml) wasspotted on to a thin layer of a-cyano-4-hydroxycinnamic acidapplied to a MALDI-TOF sample template and analysed byMALDI-TOF mass spectrometry (Bruker Reflex III) using anaccelerating voltage of 25 kV Samples were externally cali-brated using a standard mixture of six peptides ranging in
mass from 10465423 Da to 34946500 Da and the massaccuracy obtained was 60 ppm or better Identification ofproteins from MALDI-TOF peptide mass fingerprint data wasperformed using the lsquoMascotrsquo search engine at httpwwwmatrixsciencecom and was based on a positive resultusing their lsquoProbability Based MOWSE Scorersquo algorithm AMOWSE score of 60 or higher is significant at the 5 levelor better and proteins in this work typically gave scores gt80(frequently considerably so) In addition no identification wasaccepted unless at least five peptides representing at least20 of the protein sequence were detected in the MALDI-TOF peptide mass fingerprint
Assignment of proteins to metabolic pathways
Proteins were assigned to primary metabolic pathwaysusing the lsquoProtein Classification Schemersquo web page ofthe annotated Streptomyces coelicolor genome sequenceat httpwwwsangeracukProjectsS_coelicolor as it ap-peared in May 2002 The annotation of the genome andtherefore the Protein Classification Scheme is likely to bemodified as more experimental data are obtained about thefunction of genes that have currently been assigned on thebasis of sequence information alone It is therefore possiblethat proteins assigned to metabolic pathways in this studymay in future be shown to perform different functions in thecell
Assignments to gene clusters encoding secondary metab-olites are based on an analysis of the genome sequencereported by Bentley et al (2002) In some cases theseassignments are based on extensive experimental data (actred reviewed by Chater and Bibb 1997 Champness 2000cda Chong et al 1998 Huang et al 2001 whiE Davis andChater 1990)
Acknowledgements
This work was funded by grants (2FGT11406 208FGT11408 and 208IGF12432) under the BBSRCrsquos Tech-nologies for Functional Genomics and Investigating GeneFunction initiatives and by a competitive strategic grant fromthe BBSRC to the John Innes Centre We thank Mike Naldrettfor his extensive contribution in establishing technology forproteome analysis at the JIC
Supplementary material
The following material is available from httpwwwblackwell-sciencecomproductsjournalssuppmatmolemole3219mmi3219smhtmSection 1 Detection of gene products assigned to individualmetabolic steps in primary metabolic pathways in ScoelicolorSection 2 Identification of proteins from individual amino acidbiosynthetic pathwaysSection 3 Detection of gene products assigned to individualmetabolic steps in secondary metabolic pathways in Scoelicolor
Proteomic analysis of S coelicolor 931
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
References
Anderson TB Brian P and Champness WC (2001)Genetic and transcriptional analysis of absA an antibioticgene cluster linked two-component system that regulatesmultiple antibiotics in Streptomyces coelicolor Mol Micro-biol 39 553ndash566
Bentley SD Chater KF Cerdeno-Tarraga A-M ChallisGL Thomson NR James KD et al (2002) Completegenome sequence of the model actinomycete Streptomy-ces coelicolor A3(2) Nature 417 141ndash147
Berdy J (1984) New ways to obtain antibiotics Chin J Anti-biot 7 272ndash290
Berggren K Chernokalskaya E Steinberg TH KemperC Lopez MF Diwu Z et al (2000) Background-freehigh sensitivity staining of proteins in one- and two-dimensional sodium dodecyl sulfate-polyacrylamide gelsusing a luminescent ruthenium complex Electrophoresis21 2509ndash2521
Blackstock WP and Weir MP (1999) Proteomics quan-titative and physical mapping of cellular proteins TrendsBiotechnol 17 121ndash127
Blattner FR Plunkett G Bloch CA Perna NT BurlandV Riley M et al (1997) The complete genome sequenceof Escherichia coli K-12 Science 277 1453ndash1462
Buttner K Bernhardt J Scharf C Schmid R MaderU Eymann C et al (2001) A comprehensive two-dimensional map of cytosolic proteins of Bacillus subtilisElectrophoresis 22 2908ndash2935
Champness W (2000) Actinomycete development antibioticproduction and phylogeny questions and challenges InProkaryotic Development Brun YV and Skimketts LJ(eds) Washington DC American Society for MicrobiologyPress pp 11ndash31
Chater KF and Bibb MJ (1997) Regulation of bacterialantibiotic production In Biotechnology Vol 7 Products ofSecondary Metabolism Kleinkauf H and von Doumlhren H(eds) Weinheim Germany VCH pp 57ndash105
Chong PP Podmore SM Kieser HM Redenbach MTurgay K Marahiel M et al (1998) Physical identificationof a chromosomal locus encoding biosynthetic genesfor the lipopeptide calcium-dependent antibiotic (CDA) ofStreptomyces coelicolor A3(2) Microbiology 144 193ndash199
de Crecy-Lagard V Servant-Moisson JV Grandvalet Cand Mazodier P (1999) Alteration of the synthesis of theClp ATP-dependent protease affects morphological andphysiological differentiation in Streptomyces Mol Microbiol32 505ndash517
Davis NK and Chater KF (1990) Spore colour inStreptomyces coelicolor A3(2) involves the developmen-tally regulated synthesis of a compound biosyntheticallyrelated to polyketide antibiotics Mol Microbiol 4 1679ndash1691
Fernandez-Moreno FA Martinez E Caballero JLIchinose K Hopwood DA and Malpartida F (1994)DNA sequence and functions of the actVI region of theactinorhodin biosynthetic gene cluster of Streptomycescoelicolor A3(2) J Biol Chem 269 24854ndash24863
Fink D Falke D Wohllheben W and Engels A (1999)Nitrogen metabolism in Streptomyces coelicolor A3(2)
modification of glutamine synthase I by an adenylyltrans-ferase Microbiology 145 2313ndash2322
Godovac-Zimmermann J Soskic V Posnanovic S andBrianza F (1999) Functional proteomics of signal trans-duction by membrane receptors Electrophoresis 20 952ndash961
Gorg A Obermaier C Boguth G Harder A Scheibe BWildgruber R and Weiss W (2000) The current state oftwo-dimensional electrophoresis with immobilized pH gra-dients Electrophoresis 21 1037ndash1053
Gramajo HC Takano E and Bibb MJ (1993) Stationary-phase production of the antibiotic actinorhodin in Strepto-myces coelicolor A3(2) is transcriptionally regulated MolMicrobiol 7 837ndash845
Gross CA Chan C Dombroski A Gruber T Sharp MTupy J and Young B (1998) The functional and regula-tory roles of sigma factors in transcription Cold SpringHarb Symp Quant Biol 63 141ndash155
Hesketh A Sun J and Bibb MJ (2001) Induction ofppGpp synthesis in Streptomyces coelicolor A3(2) grownunder conditions of nutritional sufficiency elicits actII-ORF4transcription and actinorhodin biosynthesis Mol Microbiol39 136ndash144
Hesketh A Fink D Gust B Rexer H-U Scheel BChater K et al (2002) The PII nitrogen regulatory proteinGlnK in Streptomyces coelicolor A3(2) is modified by ade-nylylation rather than uridylylation and the GlnD and GlnKhomologues are functionally dissimilar to their nitrogenregulatory system counterparts Mol Microbiol (inpress)
Hood DW Heidstra R Swodoba UK and Hodgson DA(1992) Molecular genetic analysis of proline and tryp-tophan biosynthesis in Streptomyces coelicolor A3(2)interaction between primary and secondary metabolism- areview Gene 115 5ndash12
Hu DS-J Hood DW Heidstra R and Hodgson DA(1999) The expression of the trpD trpC and trpBA genesof Streptomyces coelicolor A3(2) is regulated by growthrate and growth phase but not by feedback repression MolMicrobiol 32 869ndash880
Huang J Lih CJ Pan KH and Cohen SN (2001)Global analysis of growth phase responsive gene expres-sion and regulation of antibiotic biosynthetic pathways inStreptomyces coelicolor using DNA microarrays GenesDev 15 3183ndash3192
Ichinose K Taguchi T Ebizuka Y and Hopwood DA(1998a) Biosynthetic gene clusters of benzoisochromane-quinone antibiotics in Streptomyces spp Identification ofgenes involved in post-PKS tailoring steps Actinomyceto-logica 12 99ndash109
Ichinose K Bedford DJ Tornus D Bechthold A BibbMJ Revill PW et al (1998b) The granaticin biosyntheticgene cluster of Streptomyces violaceoruber Tu22sequence analysis and expression in a heterologous hostChem Biol 5 647ndash659
Kelemen GH Brian P Flardh K Chamberlin L ChaterK and Buttner M (1998) Developmental regulation oftranscription of whiE a locus specifying the polyketidespore pigment in Streptomyces coelicolor A3(2) J Bacte-riol 180 2515ndash2521
Kieser T Bibb MJ Buttner MJ Chater KF and
932 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
Hopwood DA (2000) Practical Streptomyces GeneticsNorwich John Innes Foundation
Kim EJ Chung HJ Suh B Hah YC and Roe JH(1998) Expression and regulation of the sodF gene encod-ing iron- and zinc-containing superoxide dismutase inStreptomyces coelicolor Muumlller J Bacteriol 180 2014ndash2020
Kimura Y Takaoka M Tanaka S Sassa H Tanaka KPolevoda B et al (2000) Na-acetylation and proteolyticactivity of the yeast 20S proteasome J Biol Chem 2754635ndash4639
Kunst F Ogasawara N Moszer I Albertini AM AlloniG Azevedo V et al (1997) The complete genomesequence of the Gram-positive bacterium Bacillus subtilisNature 390 249ndash256
Lucchini S Thompson A and Hinton JCD (2001)Microarrays for microbiologists Microbiology 147 1403ndash1414
Mann M Hendrickson RC and Pandey A (2001) Analy-sis of proteins and proteomes by mass spectrometry AnnuRev Biochem 70 437ndash473
Miyodah S (1993) Research on antibiotic screening inJapan over the last decade a producing microorganismsapproach Actinomycetologica 7 100ndash106
Molloy MP Herbert BR Williams KL and Gooley AA(1999) Extraction of Escherichia coli proteins with organicsolvents prior to two-dimensional electrophoresis Electro-phoresis 20 701ndash704
Nagy I Tamura T Vanderleyden J Baumeister W andDe Mot R (1998) The 20S proteasome of Streptomycescoelicolor J Bacteriol 180 5448ndash5453
Neuhoff V Arold N Taube D and Ehrhardt W (1988)Improved staining of proteins in polyacrylamide gels includ-ing isoelectric focusing gels with clear background at nan-ogram sensitivity using Coomassie Brilliant Blue G-250and R-250 Electrophoresis 9 255ndash262
Nilsson CL and Davidsson P (2000) New separation toolsfor comprehensive studies of protein expression by massspectrometry Mass Spectrom Rev 19 390ndash397
Puglia AM Vohradsky J and Thompson CJ (1995)
Developmental control of the heat-shock stress regulon inStreptomyces coelicolor Mol Microbiol 17 737ndash746
Rabilloud T (1992) A comparison between low backgroundsilver diamine and silver nitrate protein stains Electro-phoresis 13 429ndash439
Robinson VL Buckler DR and Stock AM (2000) A taleof two components a novel kinase and a regulatory switchNature Struct Biol 7 626ndash633
Schena M (2000) Microarray Biochip Technology NatickMA Eaton Publishing
Spusansky O Rezuchova B Homerova D andKormanec J (2001) Expression of the gap gene encodingglyceraldehyde-3-phosphate dehydrogenase of Strepto-myces aureofaciens requires GapR a member of theAraCXylS family of transcriptional activators Microbiology147 1291ndash1301
Taguchi T Itou K Ebizuka Y Malpartida F HopwoodDA Surti CM et al (2000) Chemical characterisationof disruptants of the Streptomyces coelicolor A3(2) actVIgenes involved in actinorhodin biosynthesis J Antibiot 53144ndash152
Takano E Gramajo HC Strauch E Andres N WhiteJ and Bibb MJ (1992) Transcriptional regulation of theredD transcriptional activator gene accounts for growth-phase-dependent production of the antibotic undecylpro-digiosin in Streptomyces coelicolor A3(2) Mol Microbiol6 2797ndash2804
Tonella L Hoogland C Binz P-A Appel RDHochstrasser DF and Sanchez J-C (2001) New per-spectives in the Escherichia coli proteome investigationProteomics 1 409ndash423
Vohradsky J Li XM and Thompson CJ (1997) Identifi-cation of prokaryotic developmental stages by statisticalanalysis of two-dimensional gel patterns Electrophoresis18 1418ndash1428
Vohradsky J Li XM Dale G Folcher M Nguyen LViollier PH and Thompson CJ (2000) Developmentalcontrol of stress stimulons in Streptomyces coelicolorrevealed by statistical analyses of global gene expressionpatterns J Bacteriol 182 4979ndash4986

Proteomic analysis of S coelicolor 929
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
different conditions and in different cell types (for a reviewsee Gross et al 1998) Although no sigma factors wereidentified about half (31 out of 65) have predicted pIvalues gtpH 7 and so can only be detected using thepH 6ndash11 IPG strips that give the poorest results (seeTable 1) However the principal vegetative sigma factorHrdB which is expected to be moderately abundanthas a predicted pI of 613 and its absence from thepH 55ndash67 proteome map may indicate that the samplepreparation method which does not remove DNA is inap-propriate Among other regulatory proteins 11 out of apossible 80 two-component system response regulatorsplus a further 36 proteins annotated as lsquoregulatorsrsquo weredetected Only one SerThr kinase and no two-componentsystem sensor kinases were detected possibly reflectingthe difficulty in analysing proteins with several transmem-brane domains Interestingly 30 secreted proteins wereidentified on the proteome map at least 10 of whichappear from comparison of their observed pI and molec-ular weight positions with predicted values to have beenprocessed already and therefore exported Presumablythese proteins do not end up exclusively in the culturemedium
Clearly this approach to proteomic analysis of S coeli-color has some important deficiencies not least theabsence of certain groups of proteins and the relativelypoor initial success rate for specifically characterizingpost-translational modifications Nevertheless it offersunique insights into the molecular biology of the organismthe expression of many proteins from both primary andsecondary metabolic pathways can be analysed post-translational regulation of proteins can be investigated ona general cellular level and the physical locations forsome proteins can be determined Approximately 35 ofthe S coelicolor genome is annotated as encoding lsquohypo-theticalrsquo proteins of unknown function and 182 proteins ofthis type were identified on the proteome map No exam-ples were found of overt disagreements between pre-dicted and observed proteins providing experimentalsupport for the general accuracy of the S coelicolorgenome sequence and its annotation Information aboutthe expression post-translational modification and physi-cal location of these (no longer) hypothetical proteinsfrom proteomic analyses is going to be important forassigning future functions As many of the hypotheticalproteins have apparent orthologues in pathogenic myco-bacteria some of the information may have an immediateapplication in the context of pathogenesis
Experimental procedures
Materials
Immobilized pH gradient (IPG) strips and IPG bufferswere purchased from Amersham Biosciences Acrylamide
duracryl and chemicals for silver staining were obtained fromGenomic Solutions All other chemicals were from Sigmaunless stated in the text
Strain and growth conditions
Streptomyces coelicolor M145 was cultivated with vigorousagitation at 30infinC in minimal medium supplemented with 02casamino acids (SMM) as described previously (Kieser et al2000) Briefly a high-density spore preparation [about 1010
colony-forming units (cfu) ml-1] was pregerminated in 2yen YTmedia for 7 h at 30infinC Germlings were harvested by centrifu-gation (5 min at 4000 g) resuspended in SMM and sonicatedbriefly to disperse any aggregates before inoculation into50 ml of SMM in 250 ml siliconized flasks containing coiledstainless steel springs Each flask received the equivalent of5 yen 107 spore cfu
Protein extraction and 2D-gel electrophoresis
The preparation of the protein sample to be separated waskept as simple as possible disrupting harvested myceliadirectly into denaturing isoelectric focusing (IEF) buffer con-taining Pefabloc SC (Roche) protease inhibitor This methodminimized unwanted protein degradation that was observedin initial studies using a Tris buffer containing Pefabloc SC forcell lysis (data not shown) Removal of DNA RNA salts andpolysaccharides from the sample was therefore not per-formed (other than the shearing of viscous DNA and RNA bysonication) and the protein extracts were sometimes difficultto focus when the sample was loaded by in-gel rehydrationLoading the sample soaked in paper strips at the anodicend of IPG strips that had already been rehydrated improvedthe reproducibility of separations significantly
Mycelium for protein extraction was harvested from cul-tures by brief centrifugation (30 s at 4000 g) at room tem-perature and immediately frozen in liquid nitrogen Typicallymycelium from 10 ml culture aliquots was collected and thetransfer time from culture flask to frozen sample was 15 minFrozen cells were thawed on ice in 5 ml of washing buffer(40 mM Tris pH 90 1 mM EGTA 1 mM EDTA) then pelletedby centrifugation (5 min at 3000 g) at 4infinC Washed cells wereresuspended in 400 ml of denaturing IEF buffer UTCHAPS[7 M urea 2 M thiourea 4 (wv) CHAPS 40 mM TrispH 90 1 mM EDTA 50 mM DTT 4 mM Pefabloc SC pro-tease inhibitor (Roche)] and disrupted by sonication (SanyoSoniprep 150 10 yen 2 s bursts at amplitude 75 mm) whilecooling in an ethanolndashice bath Cell debris was removed bycentrifugation (15 min 10 000 g 4infinC) and the protein extractwas stored frozen in aliquots at -80infinC until use For thepreparation of extracellular protein extracts washed cellswere resuspended in extraction buffer consisting of 40 mMTris pH 90 1 mM EDTA 50 mM DTT 2 SDS and 4 mMPefabloc SC protease inhibitor Cells were vortexed for 2 minbefore being pelleted by centrifugation (5 min 10 000 g4infinC) The supernatant was precipitated using the Amersham2D Clean-Up kit according to the manufacturerrsquos instructionsand the precipitated proteins were redissolved in UTCHAPSand stored at -80infinC until use
930 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
For first-dimension isoelectric focusing 18 cm IPG stripspH 4ndash7 pH 6ndash11 pH 45ndash55 or pH 55ndash67 (Amersham Bio-sciences) were rehydrated overnight in IEF buffer containing1 ampholytes according to the manufacturerrsquos instructionsusing a Phaser isoelectric focusing unit (Genomic Solutions)set at 20 V Protein samples to be separated were applied tothe rehydrated strips at the anodic end soaked in a 5ndash10 mmsection of IEF electrode strip (Amersham Biosciences)Separation was performed for 120 000 V-h with a maximumvoltage of 5000 V After isoelectric focusing IPG strips wereequilibrated for the second dimension for 15 min in IPG equil-ibration buffer (50 mM Tris pH 68 6 M urea 30 glycerol1 SDS and 001 bromophenol blue) plus 80 mM DTTthen for 10 min in IPG equilibration buffer plus 135 mMiodoacetamide Approximately 1 cm was removed from theanodic end of each equilibrated strip before applying the stripto the top of a vertical 125 SDS-PAGE gel for second-dimension separation using the Investigator 5000 systemfrom Genomic Solutions
Large-format gels (24 cm wide yen 22 cm high yen 1 mm thick)for the second-dimension separation were cast in-houseusing the system supplied by Genomic Solutions 125SDS-PAGE gels prepared using 065 NN-methylenebi-sacrylamide as cross-linking agent were used for pH 4ndash7and pH 45ndash55 IPG strips whereas pH 55ndash67 and pH 6ndash11 strips were applied to similar gels made with 08 cross-linker Electrophoresis was performed while cooling using themaximum setting (ordf 4ndash10infinC) at 20 000 mW per gel constantpower and a maximum voltage of 500 V Gels were thenstained with either colloidal Coomassie G-250 (Neuhoff et al1988) or silver nitrate (Rabilloud 1992) and scanned in aProXPRESS proteomic imaging system (Perkin-Elmer)Image analysis was performed using PHORETIX 2D version51 (NonLinear Dynamics) spot detection was optimizedautomatically using the lsquoSpot Detection Wizardrsquo and thenedited manually background subtraction was performedautomatically using the lsquoMode of Non-Spotrsquo setting imageswere then normalized to the total spot volume for quantifica-tion Spot filtering was not used
Protein identification using mass spectrometry
Protein spots of interest were excised from colloidalCoomassie-stained gels in-gel digested with trypsin andidentified by MALDI-TOF peptide mass fingerprint analysisExcised gel pieces (ordf 1-mm-diameter circles cut from 1-mm-thick gels) were washed twice with 100 ml of 50 mMammonium bicarbonate for 15 min once with 100 ml of 20acetonitrile-40 mM ammonium bicarbonate for 15 min andonce with 100 acetonitrile Washed gel pieces were allowedto air-dry for 10 min before being rehydrated in 5 ml of10 mg ml-1 trypsin (sequencing grade modified Promega) in10 mM ammonium bicarbonate Digestion (37infinC for 4 h) wasstopped by the addition of 5 ml of 5 formic acid Extractionof peptides into this solution was encouraged by a sonicwater bath treatment for 20 min Peptide extract (05 ml) wasspotted on to a thin layer of a-cyano-4-hydroxycinnamic acidapplied to a MALDI-TOF sample template and analysed byMALDI-TOF mass spectrometry (Bruker Reflex III) using anaccelerating voltage of 25 kV Samples were externally cali-brated using a standard mixture of six peptides ranging in
mass from 10465423 Da to 34946500 Da and the massaccuracy obtained was 60 ppm or better Identification ofproteins from MALDI-TOF peptide mass fingerprint data wasperformed using the lsquoMascotrsquo search engine at httpwwwmatrixsciencecom and was based on a positive resultusing their lsquoProbability Based MOWSE Scorersquo algorithm AMOWSE score of 60 or higher is significant at the 5 levelor better and proteins in this work typically gave scores gt80(frequently considerably so) In addition no identification wasaccepted unless at least five peptides representing at least20 of the protein sequence were detected in the MALDI-TOF peptide mass fingerprint
Assignment of proteins to metabolic pathways
Proteins were assigned to primary metabolic pathwaysusing the lsquoProtein Classification Schemersquo web page ofthe annotated Streptomyces coelicolor genome sequenceat httpwwwsangeracukProjectsS_coelicolor as it ap-peared in May 2002 The annotation of the genome andtherefore the Protein Classification Scheme is likely to bemodified as more experimental data are obtained about thefunction of genes that have currently been assigned on thebasis of sequence information alone It is therefore possiblethat proteins assigned to metabolic pathways in this studymay in future be shown to perform different functions in thecell
Assignments to gene clusters encoding secondary metab-olites are based on an analysis of the genome sequencereported by Bentley et al (2002) In some cases theseassignments are based on extensive experimental data (actred reviewed by Chater and Bibb 1997 Champness 2000cda Chong et al 1998 Huang et al 2001 whiE Davis andChater 1990)
Acknowledgements
This work was funded by grants (2FGT11406 208FGT11408 and 208IGF12432) under the BBSRCrsquos Tech-nologies for Functional Genomics and Investigating GeneFunction initiatives and by a competitive strategic grant fromthe BBSRC to the John Innes Centre We thank Mike Naldrettfor his extensive contribution in establishing technology forproteome analysis at the JIC
Supplementary material
The following material is available from httpwwwblackwell-sciencecomproductsjournalssuppmatmolemole3219mmi3219smhtmSection 1 Detection of gene products assigned to individualmetabolic steps in primary metabolic pathways in ScoelicolorSection 2 Identification of proteins from individual amino acidbiosynthetic pathwaysSection 3 Detection of gene products assigned to individualmetabolic steps in secondary metabolic pathways in Scoelicolor
Proteomic analysis of S coelicolor 931
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
References
Anderson TB Brian P and Champness WC (2001)Genetic and transcriptional analysis of absA an antibioticgene cluster linked two-component system that regulatesmultiple antibiotics in Streptomyces coelicolor Mol Micro-biol 39 553ndash566
Bentley SD Chater KF Cerdeno-Tarraga A-M ChallisGL Thomson NR James KD et al (2002) Completegenome sequence of the model actinomycete Streptomy-ces coelicolor A3(2) Nature 417 141ndash147
Berdy J (1984) New ways to obtain antibiotics Chin J Anti-biot 7 272ndash290
Berggren K Chernokalskaya E Steinberg TH KemperC Lopez MF Diwu Z et al (2000) Background-freehigh sensitivity staining of proteins in one- and two-dimensional sodium dodecyl sulfate-polyacrylamide gelsusing a luminescent ruthenium complex Electrophoresis21 2509ndash2521
Blackstock WP and Weir MP (1999) Proteomics quan-titative and physical mapping of cellular proteins TrendsBiotechnol 17 121ndash127
Blattner FR Plunkett G Bloch CA Perna NT BurlandV Riley M et al (1997) The complete genome sequenceof Escherichia coli K-12 Science 277 1453ndash1462
Buttner K Bernhardt J Scharf C Schmid R MaderU Eymann C et al (2001) A comprehensive two-dimensional map of cytosolic proteins of Bacillus subtilisElectrophoresis 22 2908ndash2935
Champness W (2000) Actinomycete development antibioticproduction and phylogeny questions and challenges InProkaryotic Development Brun YV and Skimketts LJ(eds) Washington DC American Society for MicrobiologyPress pp 11ndash31
Chater KF and Bibb MJ (1997) Regulation of bacterialantibiotic production In Biotechnology Vol 7 Products ofSecondary Metabolism Kleinkauf H and von Doumlhren H(eds) Weinheim Germany VCH pp 57ndash105
Chong PP Podmore SM Kieser HM Redenbach MTurgay K Marahiel M et al (1998) Physical identificationof a chromosomal locus encoding biosynthetic genesfor the lipopeptide calcium-dependent antibiotic (CDA) ofStreptomyces coelicolor A3(2) Microbiology 144 193ndash199
de Crecy-Lagard V Servant-Moisson JV Grandvalet Cand Mazodier P (1999) Alteration of the synthesis of theClp ATP-dependent protease affects morphological andphysiological differentiation in Streptomyces Mol Microbiol32 505ndash517
Davis NK and Chater KF (1990) Spore colour inStreptomyces coelicolor A3(2) involves the developmen-tally regulated synthesis of a compound biosyntheticallyrelated to polyketide antibiotics Mol Microbiol 4 1679ndash1691
Fernandez-Moreno FA Martinez E Caballero JLIchinose K Hopwood DA and Malpartida F (1994)DNA sequence and functions of the actVI region of theactinorhodin biosynthetic gene cluster of Streptomycescoelicolor A3(2) J Biol Chem 269 24854ndash24863
Fink D Falke D Wohllheben W and Engels A (1999)Nitrogen metabolism in Streptomyces coelicolor A3(2)
modification of glutamine synthase I by an adenylyltrans-ferase Microbiology 145 2313ndash2322
Godovac-Zimmermann J Soskic V Posnanovic S andBrianza F (1999) Functional proteomics of signal trans-duction by membrane receptors Electrophoresis 20 952ndash961
Gorg A Obermaier C Boguth G Harder A Scheibe BWildgruber R and Weiss W (2000) The current state oftwo-dimensional electrophoresis with immobilized pH gra-dients Electrophoresis 21 1037ndash1053
Gramajo HC Takano E and Bibb MJ (1993) Stationary-phase production of the antibiotic actinorhodin in Strepto-myces coelicolor A3(2) is transcriptionally regulated MolMicrobiol 7 837ndash845
Gross CA Chan C Dombroski A Gruber T Sharp MTupy J and Young B (1998) The functional and regula-tory roles of sigma factors in transcription Cold SpringHarb Symp Quant Biol 63 141ndash155
Hesketh A Sun J and Bibb MJ (2001) Induction ofppGpp synthesis in Streptomyces coelicolor A3(2) grownunder conditions of nutritional sufficiency elicits actII-ORF4transcription and actinorhodin biosynthesis Mol Microbiol39 136ndash144
Hesketh A Fink D Gust B Rexer H-U Scheel BChater K et al (2002) The PII nitrogen regulatory proteinGlnK in Streptomyces coelicolor A3(2) is modified by ade-nylylation rather than uridylylation and the GlnD and GlnKhomologues are functionally dissimilar to their nitrogenregulatory system counterparts Mol Microbiol (inpress)
Hood DW Heidstra R Swodoba UK and Hodgson DA(1992) Molecular genetic analysis of proline and tryp-tophan biosynthesis in Streptomyces coelicolor A3(2)interaction between primary and secondary metabolism- areview Gene 115 5ndash12
Hu DS-J Hood DW Heidstra R and Hodgson DA(1999) The expression of the trpD trpC and trpBA genesof Streptomyces coelicolor A3(2) is regulated by growthrate and growth phase but not by feedback repression MolMicrobiol 32 869ndash880
Huang J Lih CJ Pan KH and Cohen SN (2001)Global analysis of growth phase responsive gene expres-sion and regulation of antibiotic biosynthetic pathways inStreptomyces coelicolor using DNA microarrays GenesDev 15 3183ndash3192
Ichinose K Taguchi T Ebizuka Y and Hopwood DA(1998a) Biosynthetic gene clusters of benzoisochromane-quinone antibiotics in Streptomyces spp Identification ofgenes involved in post-PKS tailoring steps Actinomyceto-logica 12 99ndash109
Ichinose K Bedford DJ Tornus D Bechthold A BibbMJ Revill PW et al (1998b) The granaticin biosyntheticgene cluster of Streptomyces violaceoruber Tu22sequence analysis and expression in a heterologous hostChem Biol 5 647ndash659
Kelemen GH Brian P Flardh K Chamberlin L ChaterK and Buttner M (1998) Developmental regulation oftranscription of whiE a locus specifying the polyketidespore pigment in Streptomyces coelicolor A3(2) J Bacte-riol 180 2515ndash2521
Kieser T Bibb MJ Buttner MJ Chater KF and
932 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
Hopwood DA (2000) Practical Streptomyces GeneticsNorwich John Innes Foundation
Kim EJ Chung HJ Suh B Hah YC and Roe JH(1998) Expression and regulation of the sodF gene encod-ing iron- and zinc-containing superoxide dismutase inStreptomyces coelicolor Muumlller J Bacteriol 180 2014ndash2020
Kimura Y Takaoka M Tanaka S Sassa H Tanaka KPolevoda B et al (2000) Na-acetylation and proteolyticactivity of the yeast 20S proteasome J Biol Chem 2754635ndash4639
Kunst F Ogasawara N Moszer I Albertini AM AlloniG Azevedo V et al (1997) The complete genomesequence of the Gram-positive bacterium Bacillus subtilisNature 390 249ndash256
Lucchini S Thompson A and Hinton JCD (2001)Microarrays for microbiologists Microbiology 147 1403ndash1414
Mann M Hendrickson RC and Pandey A (2001) Analy-sis of proteins and proteomes by mass spectrometry AnnuRev Biochem 70 437ndash473
Miyodah S (1993) Research on antibiotic screening inJapan over the last decade a producing microorganismsapproach Actinomycetologica 7 100ndash106
Molloy MP Herbert BR Williams KL and Gooley AA(1999) Extraction of Escherichia coli proteins with organicsolvents prior to two-dimensional electrophoresis Electro-phoresis 20 701ndash704
Nagy I Tamura T Vanderleyden J Baumeister W andDe Mot R (1998) The 20S proteasome of Streptomycescoelicolor J Bacteriol 180 5448ndash5453
Neuhoff V Arold N Taube D and Ehrhardt W (1988)Improved staining of proteins in polyacrylamide gels includ-ing isoelectric focusing gels with clear background at nan-ogram sensitivity using Coomassie Brilliant Blue G-250and R-250 Electrophoresis 9 255ndash262
Nilsson CL and Davidsson P (2000) New separation toolsfor comprehensive studies of protein expression by massspectrometry Mass Spectrom Rev 19 390ndash397
Puglia AM Vohradsky J and Thompson CJ (1995)
Developmental control of the heat-shock stress regulon inStreptomyces coelicolor Mol Microbiol 17 737ndash746
Rabilloud T (1992) A comparison between low backgroundsilver diamine and silver nitrate protein stains Electro-phoresis 13 429ndash439
Robinson VL Buckler DR and Stock AM (2000) A taleof two components a novel kinase and a regulatory switchNature Struct Biol 7 626ndash633
Schena M (2000) Microarray Biochip Technology NatickMA Eaton Publishing
Spusansky O Rezuchova B Homerova D andKormanec J (2001) Expression of the gap gene encodingglyceraldehyde-3-phosphate dehydrogenase of Strepto-myces aureofaciens requires GapR a member of theAraCXylS family of transcriptional activators Microbiology147 1291ndash1301
Taguchi T Itou K Ebizuka Y Malpartida F HopwoodDA Surti CM et al (2000) Chemical characterisationof disruptants of the Streptomyces coelicolor A3(2) actVIgenes involved in actinorhodin biosynthesis J Antibiot 53144ndash152
Takano E Gramajo HC Strauch E Andres N WhiteJ and Bibb MJ (1992) Transcriptional regulation of theredD transcriptional activator gene accounts for growth-phase-dependent production of the antibotic undecylpro-digiosin in Streptomyces coelicolor A3(2) Mol Microbiol6 2797ndash2804
Tonella L Hoogland C Binz P-A Appel RDHochstrasser DF and Sanchez J-C (2001) New per-spectives in the Escherichia coli proteome investigationProteomics 1 409ndash423
Vohradsky J Li XM and Thompson CJ (1997) Identifi-cation of prokaryotic developmental stages by statisticalanalysis of two-dimensional gel patterns Electrophoresis18 1418ndash1428
Vohradsky J Li XM Dale G Folcher M Nguyen LViollier PH and Thompson CJ (2000) Developmentalcontrol of stress stimulons in Streptomyces coelicolorrevealed by statistical analyses of global gene expressionpatterns J Bacteriol 182 4979ndash4986

930 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
For first-dimension isoelectric focusing 18 cm IPG stripspH 4ndash7 pH 6ndash11 pH 45ndash55 or pH 55ndash67 (Amersham Bio-sciences) were rehydrated overnight in IEF buffer containing1 ampholytes according to the manufacturerrsquos instructionsusing a Phaser isoelectric focusing unit (Genomic Solutions)set at 20 V Protein samples to be separated were applied tothe rehydrated strips at the anodic end soaked in a 5ndash10 mmsection of IEF electrode strip (Amersham Biosciences)Separation was performed for 120 000 V-h with a maximumvoltage of 5000 V After isoelectric focusing IPG strips wereequilibrated for the second dimension for 15 min in IPG equil-ibration buffer (50 mM Tris pH 68 6 M urea 30 glycerol1 SDS and 001 bromophenol blue) plus 80 mM DTTthen for 10 min in IPG equilibration buffer plus 135 mMiodoacetamide Approximately 1 cm was removed from theanodic end of each equilibrated strip before applying the stripto the top of a vertical 125 SDS-PAGE gel for second-dimension separation using the Investigator 5000 systemfrom Genomic Solutions
Large-format gels (24 cm wide yen 22 cm high yen 1 mm thick)for the second-dimension separation were cast in-houseusing the system supplied by Genomic Solutions 125SDS-PAGE gels prepared using 065 NN-methylenebi-sacrylamide as cross-linking agent were used for pH 4ndash7and pH 45ndash55 IPG strips whereas pH 55ndash67 and pH 6ndash11 strips were applied to similar gels made with 08 cross-linker Electrophoresis was performed while cooling using themaximum setting (ordf 4ndash10infinC) at 20 000 mW per gel constantpower and a maximum voltage of 500 V Gels were thenstained with either colloidal Coomassie G-250 (Neuhoff et al1988) or silver nitrate (Rabilloud 1992) and scanned in aProXPRESS proteomic imaging system (Perkin-Elmer)Image analysis was performed using PHORETIX 2D version51 (NonLinear Dynamics) spot detection was optimizedautomatically using the lsquoSpot Detection Wizardrsquo and thenedited manually background subtraction was performedautomatically using the lsquoMode of Non-Spotrsquo setting imageswere then normalized to the total spot volume for quantifica-tion Spot filtering was not used
Protein identification using mass spectrometry
Protein spots of interest were excised from colloidalCoomassie-stained gels in-gel digested with trypsin andidentified by MALDI-TOF peptide mass fingerprint analysisExcised gel pieces (ordf 1-mm-diameter circles cut from 1-mm-thick gels) were washed twice with 100 ml of 50 mMammonium bicarbonate for 15 min once with 100 ml of 20acetonitrile-40 mM ammonium bicarbonate for 15 min andonce with 100 acetonitrile Washed gel pieces were allowedto air-dry for 10 min before being rehydrated in 5 ml of10 mg ml-1 trypsin (sequencing grade modified Promega) in10 mM ammonium bicarbonate Digestion (37infinC for 4 h) wasstopped by the addition of 5 ml of 5 formic acid Extractionof peptides into this solution was encouraged by a sonicwater bath treatment for 20 min Peptide extract (05 ml) wasspotted on to a thin layer of a-cyano-4-hydroxycinnamic acidapplied to a MALDI-TOF sample template and analysed byMALDI-TOF mass spectrometry (Bruker Reflex III) using anaccelerating voltage of 25 kV Samples were externally cali-brated using a standard mixture of six peptides ranging in
mass from 10465423 Da to 34946500 Da and the massaccuracy obtained was 60 ppm or better Identification ofproteins from MALDI-TOF peptide mass fingerprint data wasperformed using the lsquoMascotrsquo search engine at httpwwwmatrixsciencecom and was based on a positive resultusing their lsquoProbability Based MOWSE Scorersquo algorithm AMOWSE score of 60 or higher is significant at the 5 levelor better and proteins in this work typically gave scores gt80(frequently considerably so) In addition no identification wasaccepted unless at least five peptides representing at least20 of the protein sequence were detected in the MALDI-TOF peptide mass fingerprint
Assignment of proteins to metabolic pathways
Proteins were assigned to primary metabolic pathwaysusing the lsquoProtein Classification Schemersquo web page ofthe annotated Streptomyces coelicolor genome sequenceat httpwwwsangeracukProjectsS_coelicolor as it ap-peared in May 2002 The annotation of the genome andtherefore the Protein Classification Scheme is likely to bemodified as more experimental data are obtained about thefunction of genes that have currently been assigned on thebasis of sequence information alone It is therefore possiblethat proteins assigned to metabolic pathways in this studymay in future be shown to perform different functions in thecell
Assignments to gene clusters encoding secondary metab-olites are based on an analysis of the genome sequencereported by Bentley et al (2002) In some cases theseassignments are based on extensive experimental data (actred reviewed by Chater and Bibb 1997 Champness 2000cda Chong et al 1998 Huang et al 2001 whiE Davis andChater 1990)
Acknowledgements
This work was funded by grants (2FGT11406 208FGT11408 and 208IGF12432) under the BBSRCrsquos Tech-nologies for Functional Genomics and Investigating GeneFunction initiatives and by a competitive strategic grant fromthe BBSRC to the John Innes Centre We thank Mike Naldrettfor his extensive contribution in establishing technology forproteome analysis at the JIC
Supplementary material
The following material is available from httpwwwblackwell-sciencecomproductsjournalssuppmatmolemole3219mmi3219smhtmSection 1 Detection of gene products assigned to individualmetabolic steps in primary metabolic pathways in ScoelicolorSection 2 Identification of proteins from individual amino acidbiosynthetic pathwaysSection 3 Detection of gene products assigned to individualmetabolic steps in secondary metabolic pathways in Scoelicolor
Proteomic analysis of S coelicolor 931
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
References
Anderson TB Brian P and Champness WC (2001)Genetic and transcriptional analysis of absA an antibioticgene cluster linked two-component system that regulatesmultiple antibiotics in Streptomyces coelicolor Mol Micro-biol 39 553ndash566
Bentley SD Chater KF Cerdeno-Tarraga A-M ChallisGL Thomson NR James KD et al (2002) Completegenome sequence of the model actinomycete Streptomy-ces coelicolor A3(2) Nature 417 141ndash147
Berdy J (1984) New ways to obtain antibiotics Chin J Anti-biot 7 272ndash290
Berggren K Chernokalskaya E Steinberg TH KemperC Lopez MF Diwu Z et al (2000) Background-freehigh sensitivity staining of proteins in one- and two-dimensional sodium dodecyl sulfate-polyacrylamide gelsusing a luminescent ruthenium complex Electrophoresis21 2509ndash2521
Blackstock WP and Weir MP (1999) Proteomics quan-titative and physical mapping of cellular proteins TrendsBiotechnol 17 121ndash127
Blattner FR Plunkett G Bloch CA Perna NT BurlandV Riley M et al (1997) The complete genome sequenceof Escherichia coli K-12 Science 277 1453ndash1462
Buttner K Bernhardt J Scharf C Schmid R MaderU Eymann C et al (2001) A comprehensive two-dimensional map of cytosolic proteins of Bacillus subtilisElectrophoresis 22 2908ndash2935
Champness W (2000) Actinomycete development antibioticproduction and phylogeny questions and challenges InProkaryotic Development Brun YV and Skimketts LJ(eds) Washington DC American Society for MicrobiologyPress pp 11ndash31
Chater KF and Bibb MJ (1997) Regulation of bacterialantibiotic production In Biotechnology Vol 7 Products ofSecondary Metabolism Kleinkauf H and von Doumlhren H(eds) Weinheim Germany VCH pp 57ndash105
Chong PP Podmore SM Kieser HM Redenbach MTurgay K Marahiel M et al (1998) Physical identificationof a chromosomal locus encoding biosynthetic genesfor the lipopeptide calcium-dependent antibiotic (CDA) ofStreptomyces coelicolor A3(2) Microbiology 144 193ndash199
de Crecy-Lagard V Servant-Moisson JV Grandvalet Cand Mazodier P (1999) Alteration of the synthesis of theClp ATP-dependent protease affects morphological andphysiological differentiation in Streptomyces Mol Microbiol32 505ndash517
Davis NK and Chater KF (1990) Spore colour inStreptomyces coelicolor A3(2) involves the developmen-tally regulated synthesis of a compound biosyntheticallyrelated to polyketide antibiotics Mol Microbiol 4 1679ndash1691
Fernandez-Moreno FA Martinez E Caballero JLIchinose K Hopwood DA and Malpartida F (1994)DNA sequence and functions of the actVI region of theactinorhodin biosynthetic gene cluster of Streptomycescoelicolor A3(2) J Biol Chem 269 24854ndash24863
Fink D Falke D Wohllheben W and Engels A (1999)Nitrogen metabolism in Streptomyces coelicolor A3(2)
modification of glutamine synthase I by an adenylyltrans-ferase Microbiology 145 2313ndash2322
Godovac-Zimmermann J Soskic V Posnanovic S andBrianza F (1999) Functional proteomics of signal trans-duction by membrane receptors Electrophoresis 20 952ndash961
Gorg A Obermaier C Boguth G Harder A Scheibe BWildgruber R and Weiss W (2000) The current state oftwo-dimensional electrophoresis with immobilized pH gra-dients Electrophoresis 21 1037ndash1053
Gramajo HC Takano E and Bibb MJ (1993) Stationary-phase production of the antibiotic actinorhodin in Strepto-myces coelicolor A3(2) is transcriptionally regulated MolMicrobiol 7 837ndash845
Gross CA Chan C Dombroski A Gruber T Sharp MTupy J and Young B (1998) The functional and regula-tory roles of sigma factors in transcription Cold SpringHarb Symp Quant Biol 63 141ndash155
Hesketh A Sun J and Bibb MJ (2001) Induction ofppGpp synthesis in Streptomyces coelicolor A3(2) grownunder conditions of nutritional sufficiency elicits actII-ORF4transcription and actinorhodin biosynthesis Mol Microbiol39 136ndash144
Hesketh A Fink D Gust B Rexer H-U Scheel BChater K et al (2002) The PII nitrogen regulatory proteinGlnK in Streptomyces coelicolor A3(2) is modified by ade-nylylation rather than uridylylation and the GlnD and GlnKhomologues are functionally dissimilar to their nitrogenregulatory system counterparts Mol Microbiol (inpress)
Hood DW Heidstra R Swodoba UK and Hodgson DA(1992) Molecular genetic analysis of proline and tryp-tophan biosynthesis in Streptomyces coelicolor A3(2)interaction between primary and secondary metabolism- areview Gene 115 5ndash12
Hu DS-J Hood DW Heidstra R and Hodgson DA(1999) The expression of the trpD trpC and trpBA genesof Streptomyces coelicolor A3(2) is regulated by growthrate and growth phase but not by feedback repression MolMicrobiol 32 869ndash880
Huang J Lih CJ Pan KH and Cohen SN (2001)Global analysis of growth phase responsive gene expres-sion and regulation of antibiotic biosynthetic pathways inStreptomyces coelicolor using DNA microarrays GenesDev 15 3183ndash3192
Ichinose K Taguchi T Ebizuka Y and Hopwood DA(1998a) Biosynthetic gene clusters of benzoisochromane-quinone antibiotics in Streptomyces spp Identification ofgenes involved in post-PKS tailoring steps Actinomyceto-logica 12 99ndash109
Ichinose K Bedford DJ Tornus D Bechthold A BibbMJ Revill PW et al (1998b) The granaticin biosyntheticgene cluster of Streptomyces violaceoruber Tu22sequence analysis and expression in a heterologous hostChem Biol 5 647ndash659
Kelemen GH Brian P Flardh K Chamberlin L ChaterK and Buttner M (1998) Developmental regulation oftranscription of whiE a locus specifying the polyketidespore pigment in Streptomyces coelicolor A3(2) J Bacte-riol 180 2515ndash2521
Kieser T Bibb MJ Buttner MJ Chater KF and
932 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
Hopwood DA (2000) Practical Streptomyces GeneticsNorwich John Innes Foundation
Kim EJ Chung HJ Suh B Hah YC and Roe JH(1998) Expression and regulation of the sodF gene encod-ing iron- and zinc-containing superoxide dismutase inStreptomyces coelicolor Muumlller J Bacteriol 180 2014ndash2020
Kimura Y Takaoka M Tanaka S Sassa H Tanaka KPolevoda B et al (2000) Na-acetylation and proteolyticactivity of the yeast 20S proteasome J Biol Chem 2754635ndash4639
Kunst F Ogasawara N Moszer I Albertini AM AlloniG Azevedo V et al (1997) The complete genomesequence of the Gram-positive bacterium Bacillus subtilisNature 390 249ndash256
Lucchini S Thompson A and Hinton JCD (2001)Microarrays for microbiologists Microbiology 147 1403ndash1414
Mann M Hendrickson RC and Pandey A (2001) Analy-sis of proteins and proteomes by mass spectrometry AnnuRev Biochem 70 437ndash473
Miyodah S (1993) Research on antibiotic screening inJapan over the last decade a producing microorganismsapproach Actinomycetologica 7 100ndash106
Molloy MP Herbert BR Williams KL and Gooley AA(1999) Extraction of Escherichia coli proteins with organicsolvents prior to two-dimensional electrophoresis Electro-phoresis 20 701ndash704
Nagy I Tamura T Vanderleyden J Baumeister W andDe Mot R (1998) The 20S proteasome of Streptomycescoelicolor J Bacteriol 180 5448ndash5453
Neuhoff V Arold N Taube D and Ehrhardt W (1988)Improved staining of proteins in polyacrylamide gels includ-ing isoelectric focusing gels with clear background at nan-ogram sensitivity using Coomassie Brilliant Blue G-250and R-250 Electrophoresis 9 255ndash262
Nilsson CL and Davidsson P (2000) New separation toolsfor comprehensive studies of protein expression by massspectrometry Mass Spectrom Rev 19 390ndash397
Puglia AM Vohradsky J and Thompson CJ (1995)
Developmental control of the heat-shock stress regulon inStreptomyces coelicolor Mol Microbiol 17 737ndash746
Rabilloud T (1992) A comparison between low backgroundsilver diamine and silver nitrate protein stains Electro-phoresis 13 429ndash439
Robinson VL Buckler DR and Stock AM (2000) A taleof two components a novel kinase and a regulatory switchNature Struct Biol 7 626ndash633
Schena M (2000) Microarray Biochip Technology NatickMA Eaton Publishing
Spusansky O Rezuchova B Homerova D andKormanec J (2001) Expression of the gap gene encodingglyceraldehyde-3-phosphate dehydrogenase of Strepto-myces aureofaciens requires GapR a member of theAraCXylS family of transcriptional activators Microbiology147 1291ndash1301
Taguchi T Itou K Ebizuka Y Malpartida F HopwoodDA Surti CM et al (2000) Chemical characterisationof disruptants of the Streptomyces coelicolor A3(2) actVIgenes involved in actinorhodin biosynthesis J Antibiot 53144ndash152
Takano E Gramajo HC Strauch E Andres N WhiteJ and Bibb MJ (1992) Transcriptional regulation of theredD transcriptional activator gene accounts for growth-phase-dependent production of the antibotic undecylpro-digiosin in Streptomyces coelicolor A3(2) Mol Microbiol6 2797ndash2804
Tonella L Hoogland C Binz P-A Appel RDHochstrasser DF and Sanchez J-C (2001) New per-spectives in the Escherichia coli proteome investigationProteomics 1 409ndash423
Vohradsky J Li XM and Thompson CJ (1997) Identifi-cation of prokaryotic developmental stages by statisticalanalysis of two-dimensional gel patterns Electrophoresis18 1418ndash1428
Vohradsky J Li XM Dale G Folcher M Nguyen LViollier PH and Thompson CJ (2000) Developmentalcontrol of stress stimulons in Streptomyces coelicolorrevealed by statistical analyses of global gene expressionpatterns J Bacteriol 182 4979ndash4986

Proteomic analysis of S coelicolor 931
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
References
Anderson TB Brian P and Champness WC (2001)Genetic and transcriptional analysis of absA an antibioticgene cluster linked two-component system that regulatesmultiple antibiotics in Streptomyces coelicolor Mol Micro-biol 39 553ndash566
Bentley SD Chater KF Cerdeno-Tarraga A-M ChallisGL Thomson NR James KD et al (2002) Completegenome sequence of the model actinomycete Streptomy-ces coelicolor A3(2) Nature 417 141ndash147
Berdy J (1984) New ways to obtain antibiotics Chin J Anti-biot 7 272ndash290
Berggren K Chernokalskaya E Steinberg TH KemperC Lopez MF Diwu Z et al (2000) Background-freehigh sensitivity staining of proteins in one- and two-dimensional sodium dodecyl sulfate-polyacrylamide gelsusing a luminescent ruthenium complex Electrophoresis21 2509ndash2521
Blackstock WP and Weir MP (1999) Proteomics quan-titative and physical mapping of cellular proteins TrendsBiotechnol 17 121ndash127
Blattner FR Plunkett G Bloch CA Perna NT BurlandV Riley M et al (1997) The complete genome sequenceof Escherichia coli K-12 Science 277 1453ndash1462
Buttner K Bernhardt J Scharf C Schmid R MaderU Eymann C et al (2001) A comprehensive two-dimensional map of cytosolic proteins of Bacillus subtilisElectrophoresis 22 2908ndash2935
Champness W (2000) Actinomycete development antibioticproduction and phylogeny questions and challenges InProkaryotic Development Brun YV and Skimketts LJ(eds) Washington DC American Society for MicrobiologyPress pp 11ndash31
Chater KF and Bibb MJ (1997) Regulation of bacterialantibiotic production In Biotechnology Vol 7 Products ofSecondary Metabolism Kleinkauf H and von Doumlhren H(eds) Weinheim Germany VCH pp 57ndash105
Chong PP Podmore SM Kieser HM Redenbach MTurgay K Marahiel M et al (1998) Physical identificationof a chromosomal locus encoding biosynthetic genesfor the lipopeptide calcium-dependent antibiotic (CDA) ofStreptomyces coelicolor A3(2) Microbiology 144 193ndash199
de Crecy-Lagard V Servant-Moisson JV Grandvalet Cand Mazodier P (1999) Alteration of the synthesis of theClp ATP-dependent protease affects morphological andphysiological differentiation in Streptomyces Mol Microbiol32 505ndash517
Davis NK and Chater KF (1990) Spore colour inStreptomyces coelicolor A3(2) involves the developmen-tally regulated synthesis of a compound biosyntheticallyrelated to polyketide antibiotics Mol Microbiol 4 1679ndash1691
Fernandez-Moreno FA Martinez E Caballero JLIchinose K Hopwood DA and Malpartida F (1994)DNA sequence and functions of the actVI region of theactinorhodin biosynthetic gene cluster of Streptomycescoelicolor A3(2) J Biol Chem 269 24854ndash24863
Fink D Falke D Wohllheben W and Engels A (1999)Nitrogen metabolism in Streptomyces coelicolor A3(2)
modification of glutamine synthase I by an adenylyltrans-ferase Microbiology 145 2313ndash2322
Godovac-Zimmermann J Soskic V Posnanovic S andBrianza F (1999) Functional proteomics of signal trans-duction by membrane receptors Electrophoresis 20 952ndash961
Gorg A Obermaier C Boguth G Harder A Scheibe BWildgruber R and Weiss W (2000) The current state oftwo-dimensional electrophoresis with immobilized pH gra-dients Electrophoresis 21 1037ndash1053
Gramajo HC Takano E and Bibb MJ (1993) Stationary-phase production of the antibiotic actinorhodin in Strepto-myces coelicolor A3(2) is transcriptionally regulated MolMicrobiol 7 837ndash845
Gross CA Chan C Dombroski A Gruber T Sharp MTupy J and Young B (1998) The functional and regula-tory roles of sigma factors in transcription Cold SpringHarb Symp Quant Biol 63 141ndash155
Hesketh A Sun J and Bibb MJ (2001) Induction ofppGpp synthesis in Streptomyces coelicolor A3(2) grownunder conditions of nutritional sufficiency elicits actII-ORF4transcription and actinorhodin biosynthesis Mol Microbiol39 136ndash144
Hesketh A Fink D Gust B Rexer H-U Scheel BChater K et al (2002) The PII nitrogen regulatory proteinGlnK in Streptomyces coelicolor A3(2) is modified by ade-nylylation rather than uridylylation and the GlnD and GlnKhomologues are functionally dissimilar to their nitrogenregulatory system counterparts Mol Microbiol (inpress)
Hood DW Heidstra R Swodoba UK and Hodgson DA(1992) Molecular genetic analysis of proline and tryp-tophan biosynthesis in Streptomyces coelicolor A3(2)interaction between primary and secondary metabolism- areview Gene 115 5ndash12
Hu DS-J Hood DW Heidstra R and Hodgson DA(1999) The expression of the trpD trpC and trpBA genesof Streptomyces coelicolor A3(2) is regulated by growthrate and growth phase but not by feedback repression MolMicrobiol 32 869ndash880
Huang J Lih CJ Pan KH and Cohen SN (2001)Global analysis of growth phase responsive gene expres-sion and regulation of antibiotic biosynthetic pathways inStreptomyces coelicolor using DNA microarrays GenesDev 15 3183ndash3192
Ichinose K Taguchi T Ebizuka Y and Hopwood DA(1998a) Biosynthetic gene clusters of benzoisochromane-quinone antibiotics in Streptomyces spp Identification ofgenes involved in post-PKS tailoring steps Actinomyceto-logica 12 99ndash109
Ichinose K Bedford DJ Tornus D Bechthold A BibbMJ Revill PW et al (1998b) The granaticin biosyntheticgene cluster of Streptomyces violaceoruber Tu22sequence analysis and expression in a heterologous hostChem Biol 5 647ndash659
Kelemen GH Brian P Flardh K Chamberlin L ChaterK and Buttner M (1998) Developmental regulation oftranscription of whiE a locus specifying the polyketidespore pigment in Streptomyces coelicolor A3(2) J Bacte-riol 180 2515ndash2521
Kieser T Bibb MJ Buttner MJ Chater KF and
932 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
Hopwood DA (2000) Practical Streptomyces GeneticsNorwich John Innes Foundation
Kim EJ Chung HJ Suh B Hah YC and Roe JH(1998) Expression and regulation of the sodF gene encod-ing iron- and zinc-containing superoxide dismutase inStreptomyces coelicolor Muumlller J Bacteriol 180 2014ndash2020
Kimura Y Takaoka M Tanaka S Sassa H Tanaka KPolevoda B et al (2000) Na-acetylation and proteolyticactivity of the yeast 20S proteasome J Biol Chem 2754635ndash4639
Kunst F Ogasawara N Moszer I Albertini AM AlloniG Azevedo V et al (1997) The complete genomesequence of the Gram-positive bacterium Bacillus subtilisNature 390 249ndash256
Lucchini S Thompson A and Hinton JCD (2001)Microarrays for microbiologists Microbiology 147 1403ndash1414
Mann M Hendrickson RC and Pandey A (2001) Analy-sis of proteins and proteomes by mass spectrometry AnnuRev Biochem 70 437ndash473
Miyodah S (1993) Research on antibiotic screening inJapan over the last decade a producing microorganismsapproach Actinomycetologica 7 100ndash106
Molloy MP Herbert BR Williams KL and Gooley AA(1999) Extraction of Escherichia coli proteins with organicsolvents prior to two-dimensional electrophoresis Electro-phoresis 20 701ndash704
Nagy I Tamura T Vanderleyden J Baumeister W andDe Mot R (1998) The 20S proteasome of Streptomycescoelicolor J Bacteriol 180 5448ndash5453
Neuhoff V Arold N Taube D and Ehrhardt W (1988)Improved staining of proteins in polyacrylamide gels includ-ing isoelectric focusing gels with clear background at nan-ogram sensitivity using Coomassie Brilliant Blue G-250and R-250 Electrophoresis 9 255ndash262
Nilsson CL and Davidsson P (2000) New separation toolsfor comprehensive studies of protein expression by massspectrometry Mass Spectrom Rev 19 390ndash397
Puglia AM Vohradsky J and Thompson CJ (1995)
Developmental control of the heat-shock stress regulon inStreptomyces coelicolor Mol Microbiol 17 737ndash746
Rabilloud T (1992) A comparison between low backgroundsilver diamine and silver nitrate protein stains Electro-phoresis 13 429ndash439
Robinson VL Buckler DR and Stock AM (2000) A taleof two components a novel kinase and a regulatory switchNature Struct Biol 7 626ndash633
Schena M (2000) Microarray Biochip Technology NatickMA Eaton Publishing
Spusansky O Rezuchova B Homerova D andKormanec J (2001) Expression of the gap gene encodingglyceraldehyde-3-phosphate dehydrogenase of Strepto-myces aureofaciens requires GapR a member of theAraCXylS family of transcriptional activators Microbiology147 1291ndash1301
Taguchi T Itou K Ebizuka Y Malpartida F HopwoodDA Surti CM et al (2000) Chemical characterisationof disruptants of the Streptomyces coelicolor A3(2) actVIgenes involved in actinorhodin biosynthesis J Antibiot 53144ndash152
Takano E Gramajo HC Strauch E Andres N WhiteJ and Bibb MJ (1992) Transcriptional regulation of theredD transcriptional activator gene accounts for growth-phase-dependent production of the antibotic undecylpro-digiosin in Streptomyces coelicolor A3(2) Mol Microbiol6 2797ndash2804
Tonella L Hoogland C Binz P-A Appel RDHochstrasser DF and Sanchez J-C (2001) New per-spectives in the Escherichia coli proteome investigationProteomics 1 409ndash423
Vohradsky J Li XM and Thompson CJ (1997) Identifi-cation of prokaryotic developmental stages by statisticalanalysis of two-dimensional gel patterns Electrophoresis18 1418ndash1428
Vohradsky J Li XM Dale G Folcher M Nguyen LViollier PH and Thompson CJ (2000) Developmentalcontrol of stress stimulons in Streptomyces coelicolorrevealed by statistical analyses of global gene expressionpatterns J Bacteriol 182 4979ndash4986

932 A R Hesketh et al
copy 2002 Blackwell Publishing Ltd Molecular Microbiology 46 917ndash932
Hopwood DA (2000) Practical Streptomyces GeneticsNorwich John Innes Foundation
Kim EJ Chung HJ Suh B Hah YC and Roe JH(1998) Expression and regulation of the sodF gene encod-ing iron- and zinc-containing superoxide dismutase inStreptomyces coelicolor Muumlller J Bacteriol 180 2014ndash2020
Kimura Y Takaoka M Tanaka S Sassa H Tanaka KPolevoda B et al (2000) Na-acetylation and proteolyticactivity of the yeast 20S proteasome J Biol Chem 2754635ndash4639
Kunst F Ogasawara N Moszer I Albertini AM AlloniG Azevedo V et al (1997) The complete genomesequence of the Gram-positive bacterium Bacillus subtilisNature 390 249ndash256
Lucchini S Thompson A and Hinton JCD (2001)Microarrays for microbiologists Microbiology 147 1403ndash1414
Mann M Hendrickson RC and Pandey A (2001) Analy-sis of proteins and proteomes by mass spectrometry AnnuRev Biochem 70 437ndash473
Miyodah S (1993) Research on antibiotic screening inJapan over the last decade a producing microorganismsapproach Actinomycetologica 7 100ndash106
Molloy MP Herbert BR Williams KL and Gooley AA(1999) Extraction of Escherichia coli proteins with organicsolvents prior to two-dimensional electrophoresis Electro-phoresis 20 701ndash704
Nagy I Tamura T Vanderleyden J Baumeister W andDe Mot R (1998) The 20S proteasome of Streptomycescoelicolor J Bacteriol 180 5448ndash5453
Neuhoff V Arold N Taube D and Ehrhardt W (1988)Improved staining of proteins in polyacrylamide gels includ-ing isoelectric focusing gels with clear background at nan-ogram sensitivity using Coomassie Brilliant Blue G-250and R-250 Electrophoresis 9 255ndash262
Nilsson CL and Davidsson P (2000) New separation toolsfor comprehensive studies of protein expression by massspectrometry Mass Spectrom Rev 19 390ndash397
Puglia AM Vohradsky J and Thompson CJ (1995)
Developmental control of the heat-shock stress regulon inStreptomyces coelicolor Mol Microbiol 17 737ndash746
Rabilloud T (1992) A comparison between low backgroundsilver diamine and silver nitrate protein stains Electro-phoresis 13 429ndash439
Robinson VL Buckler DR and Stock AM (2000) A taleof two components a novel kinase and a regulatory switchNature Struct Biol 7 626ndash633
Schena M (2000) Microarray Biochip Technology NatickMA Eaton Publishing
Spusansky O Rezuchova B Homerova D andKormanec J (2001) Expression of the gap gene encodingglyceraldehyde-3-phosphate dehydrogenase of Strepto-myces aureofaciens requires GapR a member of theAraCXylS family of transcriptional activators Microbiology147 1291ndash1301
Taguchi T Itou K Ebizuka Y Malpartida F HopwoodDA Surti CM et al (2000) Chemical characterisationof disruptants of the Streptomyces coelicolor A3(2) actVIgenes involved in actinorhodin biosynthesis J Antibiot 53144ndash152
Takano E Gramajo HC Strauch E Andres N WhiteJ and Bibb MJ (1992) Transcriptional regulation of theredD transcriptional activator gene accounts for growth-phase-dependent production of the antibotic undecylpro-digiosin in Streptomyces coelicolor A3(2) Mol Microbiol6 2797ndash2804
Tonella L Hoogland C Binz P-A Appel RDHochstrasser DF and Sanchez J-C (2001) New per-spectives in the Escherichia coli proteome investigationProteomics 1 409ndash423
Vohradsky J Li XM and Thompson CJ (1997) Identifi-cation of prokaryotic developmental stages by statisticalanalysis of two-dimensional gel patterns Electrophoresis18 1418ndash1428
Vohradsky J Li XM Dale G Folcher M Nguyen LViollier PH and Thompson CJ (2000) Developmentalcontrol of stress stimulons in Streptomyces coelicolorrevealed by statistical analyses of global gene expressionpatterns J Bacteriol 182 4979ndash4986