premature translational termination and the rapidly degraded
TRANSCRIPT

Premature Translational Termination and the Rapidly Degraded Polypeptide Pathway
by
Joshua Rene Lacsina
Department of Pathology
Duke University
Date:_______________________
Approved:
___________________________
Christopher V. Nicchitta, Supervisor
___________________________
Jen‐Tsan Ashley Chi
___________________________
Salvatore V. Pizzo
___________________________
Herman F. Staats
Dissertation submitted in partial fulfillment of
the requirements for the degree of Doctor of Philosophy in the Department of
Pathology in the Graduate School
of Duke University
2011

ABSTRACT
Premature Translational Termination and the Rapidly Degraded Polypeptide Pathway
by
Joshua Rene Lacsina
Department of Pathology
Duke University
Date:_______________________
Approved:
___________________________
Christopher V. Nicchitta, Supervisor
___________________________
Jen‐Tsan Ashley Chi
___________________________
Salvatore V. Pizzo
___________________________
Herman F. Staats
An abstract of a dissertation submitted in partial
fulfillment of the requirements for the degree
of Doctor of Philosophy in the Department of
Pathology in the Graduate School
of Duke University
2011

Copyright by
Joshua Rene Lacsina
2011

iv
Abstract
Nearly thirty percent of all newly synthesized polypeptides are targeted for
rapid proteasome‐mediated degradation. These rapidly degraded polypeptides (RDPs)
are the primary source of antigenic substrates for the major histocompatibility complex
(MHC) class I presentation pathway, allowing for the immunosurveillance of newly
synthesized proteins by cytotoxic T lymphocytes. Despite the recognized role of RDPs in
MHC class I presentation, it remains unclear what molecular characteristics distinguish
RDPs from their more stable counterparts. It has been proposed that premature
translational termination products may constitute a form of RDP; indeed, in prokaryotes
translational drop‐off products are normal by‐products of protein synthesis and are
subsequently rapidly degraded.
To study the cellular fate of premature termination products, the antibiotic
puromycin was used to modulate prematurely terminated polypeptide production in
human cells. At low concentrations, puromycin doubled the fraction of rapidly
degraded polypeptides, with enhanced degradation predominantly affecting small
polypeptides, consistent with rapid degradation of truncated translation products.
Immunoprecipitation experiments using anti‐puromycin antisera demonstrated that the
majority of peptidyl‐puromycins are rapidly degraded in a proteasome‐dependent
manner. Low concentrations of puromycin increased the recovery of cell surface MHC

v
class I‐peptide complexes, indicating that prematurely terminated polypeptides can be
processed for presentation via the MHC class I pathway. In the continued presence of
puromycin, MHC class I export to the cell surface was inhibited, coincident with the
accumulation of polyubiquitinated proteins. The time‐ and dose‐dependent effects of
puromycin suggest that the pool of peptidyl‐puromycin adducts differ in their targeting
to various proteolytic pathways which, in turn, differ in the efficiency with which they
access the MHC class I presentation machinery. These studies highlight the diversity of
cellular proteolytic pathways necessary for the metabolism and immunosurveillance of
prematurely terminated polypeptides which are, by their nature, highly heterogeneous.

vi
Dedication
To my parents, Rene and Teresa Lacsina, for their faith, inspiration,
encouragement, and love. Thank you for always believing in me.

vii
Contents
Abstract ......................................................................................................................................... iv
List of Figures ............................................................................................................................... xi
Acknowledgements .................................................................................................................. xiii
1. Introduction ............................................................................................................................... 1
1.1 Overview ........................................................................................................................... 1
1.2 MHC class I presentation and the ubiquitin‐proteasome system: a short primer .. 3
1.3 The fast and the furious: a historical perspective on rapidly degraded
polypeptides (RDPs) and defective ribosomal products (DRiPs) ................................... 6
1.3.1 The defective ribosomal product hypothesis .......................................................... 6
1.3.2 The discovery and re‐discovery of rapidly degraded polypeptides .................... 7
1.3.3 Measurement of the RDP fraction: controversy and refutation .......................... 10
1.3.4 RDPs and the protein economy of cells .................................................................. 11
1.3.5 Substrate‐dependent differences in RDP degradation pathways ...................... 12
1.3.6 DRiPs and RDPs: “They are who we thought they were” .................................. 13
1.4 The search for RDPs: premature translational termination products ..................... 15
1.4.1 Premature translational termination in prokaryotes ............................................ 15
1.4.2 Does premature translational termination occur in eukaryotes? ....................... 16
1.5 Puromycin: mechanism of action and experimental applications........................... 19
1.6 Overview of Results Chapters ...................................................................................... 23
2. Materials and Methods........................................................................................................... 24
2.1 Materials .......................................................................................................................... 24

viii
2.2 Construction of the SIINFEKL Tandem Repeat reporter (TRx9) ............................. 25
2.3 Cell culture ...................................................................................................................... 27
2.4 Metabolic radiolabeling and pulse‐chase .................................................................... 27
2.5 Denaturing immunoprecipitation ................................................................................ 29
2.6 Flow cytometry ............................................................................................................... 29
2.7 MHC class I peptide stripping and recovery.............................................................. 30
2.8 Western blotting ............................................................................................................. 31
2.9 RNA interference ............................................................................................................ 33
2.10 Data analysis ................................................................................................................. 33
3. Premature translational termination products are rapidly degraded polypeptides ..... 34
3.1 Overview ......................................................................................................................... 34
3.2 Development and characterization of a model system to study the products of
premature translational termination ................................................................................. 35
3.3 Stimulating premature translational termination increases the fraction of rapidly
degraded polypeptides ........................................................................................................ 48
3.4 The products of premature translational termination are rapidly degraded
polypeptides .......................................................................................................................... 53
3.5 Summary .......................................................................................................................... 59
4. Premature translational termination promotes antigenic peptide presentation via the
major histocompatibility complex class I pathway ................................................................ 61
4.1 Overview ......................................................................................................................... 61
4.2 Effects of stimulating premature translational termination on steady state cell
surface expression of MHC class I molecules and cell death ......................................... 63

ix
4.3 Effects of stimulating premature translational termination on MHC class I export
................................................................................................................................................. 68
4.3.1 Construction of the SIINFEKL tandem repeat reporter ....................................... 68
4.3.2 Application of the SIINFEKL tandem repeat reporter to measure the recovery
of cell surface MHC class I‐peptide complexes .............................................................. 71
4.3.3 Effects of puromycin on the recovery of cell surface MHC class I‐peptide
complexes ............................................................................................................................ 73
4.3.4 Inducing premature translational termination promotes the accumulation of
polyubiquitinated proteins ............................................................................................... 79
4.4 RNA interference‐mediated knockdown of candidate RDP factors increases flux
through the RDP pathway .................................................................................................. 81
4.4.1 Pth2 .............................................................................................................................. 81
4.4.2 CHIP ............................................................................................................................ 84
4.5 Summary .......................................................................................................................... 87
5. Discussion ................................................................................................................................ 88
5.1 Summary of primary findings and overview ............................................................. 88
5.2 Rapid degradation of prematurely terminated polypeptides .................................. 89
5.3 MG132‐resistant degradation of low molecular weight RDPs ................................ 93
5.3.1 The controversies of “proteasome‐independent” degradation .......................... 94
5.3.2 Non‐proteasomal proteases ..................................................................................... 96
5.4 Relationship between premature translational termination products and MHC
class I presentation ............................................................................................................... 98
5.4.1 Models of MHC class I behavior in response to puromycin treatment ............. 99
5.4.1.1 mTORC1 model .................................................................................................. 99

x
5.4.1.2 Aggregation model .......................................................................................... 101
5.4.1.3 Substrate heterogeneity model....................................................................... 102
5.4.2 Puromycin in other studies of the MHC class I pathway .................................. 106
5.5 Candidate RDP pathway factors ................................................................................ 108
5.6 Implications for human health ................................................................................... 109
References .................................................................................................................................. 111
Biography ................................................................................................................................... 124

xi
List of Figures
Figure 1: The MHC class I antigen presentation pathway ...................................................... 4
Figure 2: Puromycin and notable structural features ............................................................ 20
Figure 3: Contrasting effects of cycloheximide and puromycin on [35S]‐methionine
incorporation during protein synthesis ................................................................................... 37
Figure 4: Contrasting effects of cycloheximide and puromycin on the profile of newly
synthesized polypeptides .......................................................................................................... 39
Figure 5: Treatment with protein synthesis inhibitors during radiolabeling has no effect
on steady state protein levels .................................................................................................... 40
Figure 6: Puromycin stimulates the production of truncated polypeptides in a
concentration‐dependent manner ............................................................................................ 46
Figure 7: Treatment with puromycin increases the fraction of rapidly degraded
polypeptides ................................................................................................................................ 50
Figure 8: Effects of puromycin on the degradation profile of newly synthesized
polypeptides. ............................................................................................................................... 52
Figure 9: Purification of premature translational termination products using puromycin‐
specific antisera ........................................................................................................................... 55
Figure 10: Rapid degradation of premature translational termination products .............. 58
Figure 11: Contrasting effects of cycloheximide and puromycin on steady‐state levels of
cell surface Kb ............................................................................................................................... 65
Figure 12: Contrasting effects of cycloheximide and puromycin on cell death ................. 67
Figure 13: Construction of the SIINFEKL tandem repeat ..................................................... 69
Figure 14: MHC class I‐peptide complex recovery assay using a fluorescent reporter
encoding antigenic peptides ...................................................................................................... 72
Figure 15: Effects of puromycin on the expression of functional fluorescent reporter
protein ........................................................................................................................................... 74

xii
Figure 16: Effects of puromycin on the recovery of cell surface Kb‐SIINFEKL complexes
....................................................................................................................................................... 75
Figure 17: Effects of puromycin on the recovery of total cell surface Kb ............................ 76
Figure 18: Dose‐dependent effects of puromycin on the recovery of cell surface MHC
class I‐peptide complexes .......................................................................................................... 78
Figure 19: Puromycin treatment leads to increased levels of polyubiquitinated proteins
....................................................................................................................................................... 80
Figure 20: Knockdown of Pth2 by RNA interference increases the fraction of RDPs ....... 82
Figure 21: Knockdown of CHIP by RNA interference increases the fraction of RDPs ..... 85
Figure 22: The mTORC1 model .............................................................................................. 100
Figure 23: The substrate heterogeneity model ...................................................................... 105

xiii
Acknowledgements
To the Monterey Bay Aquarium, who opened the heart and mind of a young boy
to the wonders of biology. I am forever in your debt.
To the dual inspiration of my high school teachers, Jack Arnold and Roz Zanides,
for instilling in me a love for molecular biology and theater respectively, the two halves
without which I am not whole. For York, that wonderful high school on the hill.
To my many, many research mentors, spanning a decade and a half across two
continents: at Hopkins Marine Station, Stephanie Clendennen, and Trish Schulte; at the
Institute for Cell Biophysics in Pushchino, Russia, Vladimir Pechatnikov, Vladislav
Dolgachev, and Natalia Dolgacheva; at Stuyvestant High School, Anne Manwell; at
Stanford, Glenn Rosen; at Harvard, Judy Lieberman, Joe Sodroski, Greg Babcock, Woj
Wojtowicz, Jason LaBonte, Christoph Grundner, and Wen Yuan.
To Chris Nicchitta, for welcoming me into his laboratory, for helping me take my
first tentative steps into the wonders of cellular immunology, and all the conversations
we have had these past few years. To all the members of the Nicchitta lab, past and
present, for teaching me about science and life. Angela Jockheck‐Clark, Lilly Zheng,
Jason Maynard, Rebecca Dodd, Sam Stephens, Brook Pyhtila, Lyuda Kadyrova, Qiang
Chen, J. Taylor Herbert, Deanna Crossman, Christine Nwosu, Mariam Totonchy, Robert
Ng, Rebecca Poliner, Helen Rankin and Ben Contrella. Special thanks to David Reid and

xiv
Sujatha Jagannathan for their contributions to my dissertation work. And a special
thanks to Xiongfei Liu, not only for his contributions to my dissertation, but also for the
privilege of being his teacher and having him as my first research student. To my
committee members, Ashley Chi, Matthias Gromeier, Sal Pizzo, and Herman Staats, for
their criticism and advice that strengthened this dissertation.
To the Duke Medical Scientist Training Program, the Duke Program in Cell and
Molecular Biology, and the Department of Pathology, for giving me the opportunity to
realize my dream of becoming a physician‐scientist. To my DukeMed, CMB, Pathology,
and especially MSTP classmates, for their constant encouragement and friendship. To
MSTP Director Emeritus Sal Pizzo, for giving me the opportunity to come to Duke, for
his enthusiasm, stories, and laughter, and for the lunches in his conference room, which
are dearly missed. To current MSTP Director Chris Kontos, for his cheerfulness,
encouragement, and understanding. And to MSTP Associate Director Dona Chikaraishi,
for her devotion to the program and its students.
To my partner and instigator in all things malaria polysome, Greg LaMonte.
Duke would never have won the 2010 National Championship if it hadn’t started with
Polysomapalooza. To our bosses, Ashley and Chris, for not pulling the plug before we
hit just the right amount of crazy to get the experiments working.
To the Chi Unit and associated acts: Greg LaMonte, “J‐Tay” Taylor Herbert,
Carolyn Sangokoya, Jeff Mito, and Jon Kotula, for long lunches and laughter.

xv
To Kelly Crace, for helping me understand my values and focus on them to
become a better person. For the invaluable advice of my advisory deans, Phil Goodman
and Mark Sebastian, in guiding me to be a better physician. For Ken Lyles, my foremost
clinical mentor and friend—it is an honor to be your Enforcer.
To the Duke Catholic Center, for offering me a faith home and a loving
community. To Fathers Joe Vetter, John McDonagh, and Mike Martin, and to Catherine
Preston and the community of Catholic graduate students at Duke.
For the Feagin Leadership Forum, who have mentored and guided me to be a
leader and teacher of physicians and scientists.
For the Marks Family, who have given new meaning to love, sacrifice, and
generosity. For my closest colleague, my fiercest advocate, my best friend, and my love,
Odessa Marks, who fights for me when I am unwilling to fight for myself.
For the unwavering support of my family around the world, but especially for
the love of my parents, Rene and Teresa Lacsina, without whom none of this would be
possible. I love you.
And most of all, to God. To you be all the praise and glory! For the opportunity
to help those in need, and for the gift you give all scientists—a chance to glimpse the
workings of your creation, and to behold its beauty in awe and wonder.
Thanks for the memories!

1
1. Introduction
1.1 Overview
A critical function of immunosurveillance is the detection and targeted
destruction of cells that have been infected with viruses or have undergone malignant
transformation. This “search and destroy” function is accomplished by cytotoxic T
lymphocytes (CTLs) bearing T cell receptors specific for peptides presented by major
histocompatibility complex class I (MHC I) molecules on the cell surface. MHC I‐peptide
complexes are primarily generated via the cytosolic degradation of proteins by the
ubiquitin‐proteasome system. The resultant peptide degradation products are
transported into the endoplasmic reticulum (ER), where peptides are loaded onto MHC
I molecules for export and presentation on the cell surface. Thus, the MHC I pathway
allows for sampling of the cellular proteome so that CTLs can detect peptides derived
from non‐self proteins, namely viral proteins or proteins bearing mutations.
Over the past decade, there has been increasing evidence that the majority of
peptides presented via the MHC I pathway do not derive from the turnover of aged,
defunct proteins (termed “retirees”) (Dolan et al. 2011a), but rather from newly
synthesized polypeptides that are rapidly degraded by the proteasome (Schubert et al.
2000; Reits et al. 2000; Princiotta et al. 2003; Qian et al. 2006a). This rapidly degraded
polypeptide (RDP) fraction has a half‐life of ~10 minutes, comprising nearly a third of all
proteins synthesized and 70% of proteasomal substrates (Schubert et al. 2000; Princiotta

2
et al. 2003; Qian et al. 2006a). A fraction of all proteins appears to be directed to the RDP
pool, even proteins that are metabolically stable. This implies that any newly
synthesized polypeptide can be directed either to the pool of stable proteins, which
display an average half‐life of 1‐2 days, or to the RDP pool (Yewdell and Nicchitta 2006;
Yewdell 2007).
This dissertation focuses on a major unanswered question about RDP biology:
what are RDPs? Specifically, it is unclear what molecular characteristics distinguish
substrates directed to the stable protein pool versus the RDP pool. The studies described
in this work test the hypothesis that the products of premature translational termination
are a source of RDPs (Yewdell et al. 1996; Dolan et al. 2011b). Support for this model
comes from studies of E. coli, where it has been estimated that nearly 25% of translation
initiation events result in the production of prematurely terminated translation products
(Manley 1978; Tsung et al. 1989; Jørgensen and Kurland 1990). While there is, as of yet,
no direct evidence for these premature termination events occurring in eukaryotes, there
are data that suggest premature termination events are a well‐conserved (and perhaps
inescapable) by‐product of translation.
In the following sections, I will outline our current knowledge about the rapidly
degraded polypeptide pathway and evidence suggesting that premature translational
termination products compose a general subclass of RDPs. I will begin by briefly
reviewing the biology of the ubiquitin‐proteasome system and the MHC class I

3
presentation pathway. Next, I will discuss the historical development of the RDP field,
starting with the emergence of the “defective ribosomal product (DRiP) hypothesis” as a
means to account for the rapidity of CTL‐mediated detection of viral infection, and the
experimental studies that have subsequently developed the theoretical framework of the
RDP model. I will then review studies of how premature translational termination
products are produced and metabolized in prokaryotes, as a touchstone for examining
truncated polypeptides as candidate RDPs in eukaryotes. In the final section, I will
introduce the antibiotic puromycin as a tool to study the biology of premature
translational termination.
1.2 MHC class I presentation and the ubiquitin-proteasome system: a short primer
Antigenic peptides presented on MHC class I molecules derive from the
degradation of cytosolic proteins by the proteasome (Fig. 1) (Rock et al. 1994; Hershko
and Ciechanover 1998; Shastri et al. 2002). To tag proteins for degradation, ubiquitin
monomers are covalently attached to exposed lysine residues on proteins by ubiquitin
ligases (Ciechanover et al. 1980). Chains of ubiquitin monomers can be joined in this
manner (Hershko et al. 1980), forming a polyubiquitin chain which is recognized as a
degradation signal and bound by the 19S regulatory subunit of the proteasome. The
substrate protein is then unfolded and translocated through the 20S core particle, a
cylindrical macromolecular machine that proteolytically degrades the substrate into
peptides. These peptides are then transported into the ER lumen in an ATP‐dependent

4
Figure 1: The MHC class I antigen presentation pathway. Polyubiquitinated proteins
are degraded by the proteasome into peptides, which are transported into the ER lumen
through TAP. The peptides are loaded onto MHC class I molecules, which then traffic
through the trans‐Golgi to the plasma membrane, where they present peptides to
cytotoxic T lymphocytes. Adapted from (Yewdell et al. 2003).

5
fashion by the transporter associated with antigen processing (TAP) (Spies et al. 1992), a
member of the ATP‐binding cassette family. In the ER, the majority of antigenic peptides
undergo further processing by ER‐resident aminopeptidases (in humans, ERAP1 and
ERAP2), which trim N‐terminal extensions to generate peptides of the appropriate
length (8‐10 amino acids) for loading onto MHC class I molecules (Saric et al. 2002;
Serwold et al. 2002; Saveanu et al. 2005). Peptide loading onto MHC class I molecules is
facilitated by the peptide loading complex, composed of TAP, 2 microglobulin,
calreticulin, ERp57, tapasin, and MHC class I. Once peptides bind empty MHC class I
molecules in the ER lumen, they are exported to the trans‐Golgi for post‐translational
modification (predominantly remodeling of N‐linked glycans) and then presented on the
cell surface for interrogation by the T cell receptors of CTLs. Recognition of a non‐self
peptide in the context of an MHC class I molecule triggers a signaling cascade through
the TCR of the CTL which triggers apoptosis in the cell bearing the offending MHC I‐
peptide complex. The MHC class I system has thus evolved to sample the cellular
proteome, allowing for CTLs to rapidly and specifically detect and delete cells infected
with viruses and cells that have undergone malignant transformation.

6
1.3 The fast and the furious: a historical perspective on rapidly degraded polypeptides (RDPs) and defective ribosomal products (DRiPs)
1.3.1 The defective ribosomal product hypothesis
While a detailed molecular picture of the MHC I presentation pathway has
emerged, in 1996, Jon Yewdell and colleagues published an article that called attention
to several unanswered questions regarding the source of degraded polypeptides that
feed the presentation pathway. First is the problem posed by the metabolic stability of
viral proteins, many of which have half‐lives on the order of days. How can such
“stable” proteins be degraded quickly enough to generate MHC class I‐peptide
complexes before the completion of the viral life cycle (as fast as 6 hours for some
positive stranded RNA viruses)? Moreover, even with the exquisite sensitivity of the T
cell receptor, how can viral proteins successfully compete for one of the scant 105 MHC
class I molecules when so dramatically outnumbered by the 3x109 cellular proteins early
in infection (Yewdell et al. 2003)? Yet despite the obstacles of metabolic stability and
scarcity, it has been observed that CTL responses can be triggered rapidly after viral
infection; a mere 45 minutes after infection with vesicular stomatitis virus, enough of the
highly stable viral nucleocapsid protein had been degraded and presented to stimulate a
CTL response specific for a nucleocapsid‐derived peptide epitope (Esquivel et al. 1992).
How could peptide presentation from a stable viral protein take place so early after
infection?

7
In response to these theoretical questions and unexplained experimental
observations, Yewdell and colleagues proposed the existence of defective ribosomal
products (DRiPs), where “defective” serves as a catch‐all descriptor of any protein that
has failed to reach its native, folded, functional state (Yewdell et al. 1996). These DRiPs
were hypothesized to comprise a fraction of all newly synthesized polypeptides which
(for whatever reason) are targeted for rapid degradation. The implication of this finding
is that proteins are directed to one of two pools, which vary markedly in stability. They
can either be directed to the (more familiar) pool of natively folded, functional proteins
or be targeted to the DRiP pool as rapidly degraded polypeptides (or RDPs).
1.3.2 The discovery and re-discovery of rapidly degraded polypeptides
In the years following the publication of the DRiP hypothesis, researchers sought
to test the predictions of the DRiP model. First, investigators proceeded to determine the
fraction of newly synthesized polypeptides degraded shortly after synthesis to estimate
the percentage of polypeptides targeted to the RDP pool (Schubert et al. 2000). This was
done by pulse‐labeling cells with [35S]‐methionine for short periods of time, followed by
a chase with unlabeled “cold” methionine to track the degradation of the labeled
population of proteins. By comparing the degradation of new proteins during the chase
between cells treated with and without a proteasome inhibitor, the investigators
determined that ~30% of newly synthesized polypeptides were targeted for rapid,
proteasome‐mediated degradation. This seemingly large fraction of RDPs was observed

8
both in cultured cell lines and in cells cultured ex vivo from mouse lymph nodes. The
addition of proteasome inhibitor rescued both cellular proteins and viral proteins from
rapid degradation. The polypeptides rescued from rapid degradation were
predominantly found in the insoluble fraction following cellular fractionation. In
support of polyubiquitinated proteins serving as a source of RDPs, the addition of
protein synthesis inhibitors led to a depletion in the cellular pool of polyubiquitinated
proteins. Finally, this study tested a major prediction of the DRiP hypothesis: that MHC
class I presentation requires ongoing protein synthesis (to produce rapidly degraded
substrates), and should therefore be highly sensitive to translational inhibitors. Indeed,
the addition of protein synthesis inhibitors led to a significant decrease in the export of
MHC I‐peptide complexes from the ER. In total, this study provided the first direct
evidence for the reliance of MHC class I presentation on ongoing protein synthesis, in
support of the DRiP hypothesis. Furthermore, the finding that nearly a third of all newly
synthesized proteins are rapidly degraded was surprising, given the inefficiency this
implies for eukaryotic translation.
Shortly after this initial study to the characterize the RDP pool was completed,
cellular immunologists realized that biochemical studies of protein turnover extending
over the previous three decades had already reported the existence of distinct “short‐“
and “long‐lived” protein pools in eukaryotic cells. In 1973, Poole and Wibo were the first
to describe a way to selectively measure proteins of long and short half‐lives in rat

9
fibroblasts (Poole and Wibo 1973). This work was extended by a series of metabolic
pulse‐chase studies conducted by Wheatley and colleagues, who demonstrated a
biphasic protein degradation profile in cells—an initial, rapid degradation phase
comprising 35% of all newly synthesized polypeptides followed by a more gradual
degradation kinetic (Wheatley et al. 1980). Wheatley also demonstrated that short
labeling times are necessary to effectively measuredly RDPs, because shortening the
time of radiolabeling led to an increase in the measured fraction of rapidly degraded
proteins. This result showed that longer radiolabeling times led to an underestimation of
the rapidly degraded protein fraction, due to ongoing protein degradation during
labeling (before the initiation of the chase). The dependence of the measured RDP
fraction on labeling time was again reported by Fuertes and colleagues during their
studies of short‐ and long‐lived protein pools in fibroblasts (Fuertes et al. 2003). In
summary, experiments spanning three decades consistently report the presence of a pool
of rapidly degraded polypeptides. Perhaps even more remarkable is the fact that the
estimate of the RDP fraction has consistently ranged from 30‐35%, even before the
advent of cell‐permeable proteasome inhibitors.
Other lines of experimental evidence for RDPs have emerged to complement the
biochemical studies of protein degradation described above. One prediction of the DRiP
hypothesis is that the flux of antigenic peptides through TAP into the ER should depend
on translational activity. To test this prediction, Reits and colleagues discovered that the

10
lateral mobility of TAP in the ER membrane is inversely proportional to the flux of
peptides through TAP (Reits et al. 2000). By employing TAP tagged with green
fluorescent protein (GFP) and measuring lateral TAP mobility in the ER membrane by
fluorescence recovery after photobleaching (FRAP), Reits and colleagues discovered that
inhibiting protein synthesis led to an increase in TAP mobility, while influenza infection
significantly decreased the mobility of TAP. These TAP mobility studies demonstrated
that peptide flux, which is fueled by protein degradation, also requires ongoing protein
synthesis.
1.3.3 Measurement of the RDP fraction: controversy and refutation
The estimate of the RDP fraction comprising 30% of protein synthesis was called
into question by a study from Vabulas and Hartl (Vabulas and Hartl 2005), which
reported that newly synthesized polypeptides are not rescued by proteasomal inhibition
except under conditions of amino acid starvation. They attribute this to their finding that
proteasomal inhibition acutely impaired the supply of amino acids for protein synthesis
during starvation. The authors interpreted this to mean that the radiolabeled proteins
rescued by proteasomal inhibition were not RDPs but rather an artifact of increased
utilization of [35S]‐Met during severe starvation for amino acids. By extension, the
authors argued that the fraction of polypeptides degraded shortly after synthesis is
relatively minor.

11
A number of lines of experimental evidence refute the arguments of Vabulas and
Hartl, and instead support the interpretation that the radiolabeled polypeptides rescued
by proteasomal inhibition represent bona fide RDPs (Yewdell and Nicchitta 2006). First,
the early biochemical studies reporting the existence of RDPs were conducted prior to
the development of proteasome inhibitors (Poole and Wibo 1973; Wheatley et al. 1980).
Second, radiolabeled polypeptides can be recovered with proteasomal inhibitors even
without prior starvation for methionine. Third, the fact that RDPs were predominantly
recovered in the insoluble fraction indicates that RDPs have biochemical characteristics
that are distinct from bulk cellular protein. Indeed, a pulse‐chase experiment from the
Vabulas and Hartl study itself shows that 20% of newly synthesized polypeptides were
degraded during the first 30 minutes of the chase (after 10 minutes of radiolabeling).
Finally, no matter what the absolute fraction of RDPs is in cells, studies of the kinetics of
CTL responses to viral infection clearly indicate the presence of some fraction of
metabolically stable polypeptides that are degraded and presented shortly after
synthesis.
1.3.4 RDPs and the protein economy of cells
The re‐discovery of the RDP pool highlighted the need for quantitative studies to
establish the protein economy of cells—a complete accounting of protein synthesis,
protein degradation and the generation of MHC class I‐peptide complexes. For their
quantitative studies of L929 cells, Princiotta and colleagues used purified protein

12
standards to determine the number of ribosomes, proteasomes, and proteins in each cell.
In addition, they used fusion protein reporters encoding influenza nucleoprotein, the
antigenic peptide SIINFEKL, and eGFP to make sensitive measurements of the kinetics
of protein synthesis and antigen presentation by flow cytometry.
The authors demonstrated that ~45% of cellular ATP supplies are consumed by
protein synthesis. Given that 25‐30% of newly synthesized polypeptides are targeted for
rapid degradation, this implied that the synthesis of RDPs uses 11% of the energy
consumed by the cell (a seemingly large fraction of resources to synthesize products that
are immediately destroyed). The efficiency of MHC class I‐peptide complex generation
was measured to be one for every 500‐3000 degraded polypeptides. The studies also
highlighted that while RDPs are the primary source of MHC class I peptides, they are
not necessarily the most efficient source, on a peptide per protein basis; indeed, many
slowly degraded polypeptides were more efficient sources of MHC I peptide epitopes.
This study marked the first complete accounting of protein synthesis and degradation,
offering quantitative insights into the efficiency of antigen presentation and the relative
contributions of slowly versus rapidly degraded polypeptides to the MHC class I
peptide pool.
1.3.5 Substrate-dependent differences in RDP degradation pathways
Qian and colleagues performed a detailed biochemical analysis of RDPs and
their degradation characteristics (Qian et al. 2006a). Cells were fractionated using the

13
non‐ionic detergent Triton X‐100 (TX‐100) to separately characterize TX‐100‐soluble and
‐insoluble RDPs. Approximately 75% of the RDPs were TX‐100‐soluble, while the
remaining 25% were TX‐100‐insoluble. Notably, the degradation of RDPs in the TX‐100‐
insoluble fraction was insensitive to the inactivation of the E1 ubiquitin‐activating
enzyme, knockdown of the 19S regulatory subunit of the 26S proteasome, and
modulation of Hsc70 activity, whereas TX‐100‐soluble RDPs were sensitive to all these
factors. Furthermore, peptide presentation from a defined reporter protein continued
despite the inactivation of E1. These findings were interpreted to mean that TX‐100‐
insoluble RDPs represent severely misfolded polypeptides that are degraded in a
ubiquitin‐independent manner by the 20S proteasome. This study provided the first
evidence of substrate‐dependent heterogeneity in RDP degradation pathways, a theme
that will be revisited in the experiments presented in Chapter 4.
1.3.6 DRiPs and RDPs: “They are who we thought they were”
In the years following the (re)discovery of RDPs, several groups generated
evidence that antigenic peptides derived from the degradation of newly synthesized
proteins. An immunodominant epitope from the nucleoprotein of lymphocytic
choriomeningitis virus ceased to be presented after protein expression was turned off
using a tetracycline‐regulated promoter, despite the large pool of nucleoprotein in the
cell (Khan et al. 2001). In another series of studies, ‐galactosidase (‐gal) was expressed
from an inducible promoter, leading to high levels of ‐gal expression (Donohue et al.

14
2006). This correlated with the presentation of ‐gal‐derived peptides. In contrast,
peptide expression decreased after the promoter expressing ‐gal was turned off,
despite high ‐gal concentrations in the cytosol. Two groups independently
demonstrated that the presentation of peptides from Epstein‐Barr virus nuclear antigen
1 (EBNA1) requires active EBNA1 synthesis and is independent of steady state EBNA1
levels (Tellam et al. 2004; Voo et al. 2004). Tellam and colleagues extended their studies
to demonstrate that RDPs comprise a significant fraction of newly synthesized EBNA1
proteins, and that RDP production was correlated directly with EBNA1 translational
efficiency (Tellam et al. 2007). Furthermore, processing of EBNA1‐derived peptides
depended more on RDP generation than the turnover of EBNA1. In total, these studies
provided strong evidence for the dependence of peptide presentation on active synthesis
(and rapid degradation) of the antigen.
The initial investigations of DRiPs demonstrated that protein synthesis inhibitors
rapidly inhibited the export of new MHC class I molecules (Schubert et al. 2000). To
verify that this was indeed due to depletion of the supply of rapidly degraded substrates
(and not other components of the presentation pathway), Qian and colleagues treated
cells with MG132 in order to rescue a pool of DRiPs from degradation (Qian et al.
2006b). They then washed out the proteasome inhibitor to allow the DRiPs to degrade
and be presented, while simultaneously adding cycloheximide to prevent new DRiP
synthesis (CHX). This led to a burst in antigen presentation, absent protein synthesis,

15
indicating that the effects of CHX are indeed limited to inhibiting substrate supply to the
MHC class I pathway.
Measurements of DRiP‐derived peptides predominantly involved the use of
reporter fusion proteins. To test whether antigenic peptides were derived from DRiPs
when expressed from a viral protein in the context of the native virus, Dolan and
colleagues inserted the antigenic SIINFEKL peptide into the stalk of influenza A virus
neuraminidase (NA) (Dolan et al. 2010). SIINFEKL presentation was tightly correlated
with active synthesis of NA, suggesting that DRiPs are the main source of virus‐derived
antigenic peptides in the context of a natural infection.
1.4 The search for RDPs: premature translational termination products
A decade and a half after the publication of the DRiP hypothesis, the molecular
characteristics that distinguish RDPs from stable proteins still remain mysterious. In this
section, I propose that premature translational termination products are RDP pathway
substrates, and review the evidence in support of this proposal.
1.4.1 Premature translational termination in prokaryotes
Products of premature translational termination have been proposed as a source
of substrates for the RDP pathway (Yewdell et al. 1996; Dolan et al. 2011b). Support for
this model comes from studies of protein synthesis in E. coli. A study of the lacZ gene
revealed that 31% of the total ‐galactosidase monomers expressed were synthesized as
prematurely terminated polypeptide fragments, with a premature termination event

16
occurring once every 3200 codons (Manley 1978). A similar study was again conducted
on the lacZ gene, where it was determined that 24% of all initiation events resulted in the
production of a prematurely terminated polypeptide (Jørgensen and Kurland 1990).
These findings indicate that a substantial fraction of prokaryotic protein synthesis
results in the production of truncated polypeptides.
The extent to which peptidyl‐tRNA drop‐off occurs in vivo was investigated in a
series of studies using E. coli strains carrying temperature sensitive mutants of peptidyl‐
tRNA hydrolase. Peptidyl‐tRNA hydrolase catalyzes hydrolysis of the ester bond in
peptidyl‐tRNAs that have dissociated from the ribosome (Menninger et al. 1973).
Growth at the non‐permissive temperature resulted in the accumulation of peptidyl‐
tRNAs, indicating that they dissociate from ribosomes as by‐products of translation
(Menninger 1976). The rate at which peptidyl‐tRNAs dissociated from ribosomes was
estimated to be between 1 in 90 to 1 in 2600 elongation steps. Peptidyl‐tRNA
accumulation at the non‐permissive temperature led to the inhibition of translation
initiation and cell death (Atherly 1978; Menninger 1979). These findings indicate that the
premature dissociation of peptidyl‐tRNAs is a natural by‐product of prokaryotic protein
synthesis.
1.4.2 Does premature translational termination occur in eukaryotes?
There is currently no direct evidence for peptidyl‐tRNA drop‐off in eukaryotic
translation. However, there are several lines of indirect evidence that suggest premature

17
termination events are occurring. Studies of the distribution of ribosomes on mRNAs by
both polysome microarrays (Arava et al. 2003) and ribosomal footprinting (Ingolia et al.
2009) indicate relatively higher ribosomal density at the 5’ end of mRNAs. The latter
study was particularly informative in that it was able to resolve the position of
individual ribosomes at the level of single nucleotide resolution. For the first 30‐40
codons, ribosomal density was very high, followed by a decrease over the next 100‐200
codons down to a uniform density. The authors interpret these findings as reflecting
either an increase in elongation rate or premature termination events.
The presence of Pth homologs in eukaryotes (de Pereda et al. 2004, Ishii et al.
2006) suggests conservation of the mechanisms for premature translational termination
and disposal of the resulting drop‐off products, although the nature of these
degradation pathways remains to be characterized biochemically. Characterization of
the catalytic properties of Pth2 in humans indicates that Pth2 is more efficient in
catalyzing ester bond hydrolysis than the E. coli Pth enzyme (de Pereda et al. 2004).
Interestingly, the yeast homolog of Pth2 binds the ubiquitin‐like domains of Rad23 and
Dsk2, which are responsible for the delivery of polyubiquitinated proteins to the
proteasome (Ishii et al. 2006). Pth2 inhibited the interaction of Rad23 and Dsk2 with the
proteasome, thereby inhibiting the degradation of polyubiquitinated protein. The
involvement of Pth2 in the ubiquitin‐proteasome pathway suggests that the hydrolysis

18
of peptidyl‐tRNAs is functionally connected to protein degradation pathways in
eukaryotes.
Targeting of peptidyl‐tRNAs for rapid degradation may be coupled to signals
induced by ribosomal stalling. The clearance of ribosomes stalled during elongation is
regulated by the proteins Dom34 (Pelota in mammals) and Hbs1, which are paralogs of
the eukaryotic release factors (Atkinson et al. 2008). These proteins were originally
identified in the context of no‐go decay, an mRNA quality control pathway that
degrades mRNAs bearing ribosomes which have stalled due to the presence of stable
RNA stem‐loops, pseudoknots, or rare codons (Doma and Parker 2006; Passos et al.
2009). A recent structural study of the Dom34/Hbs1 complex bound to the ribosome led
the authors to propose a model in which Dom34/Hbs1 competes with elongation factors
for ribosomal binding (Becker et al. 2011). When the ribosome stalls, Dom34/Hbs1
binding is favored over the elongation factors, leading to destabilization of the mRNA‐
tRNA interaction with the ribosome and ribosomal dissociation. This mechanism
appears to be conserved regardless of the cause for the ribosomal stall. In vitro
reconstitution experiments demonstrate that Dom34/Hbs1 triggers the premature
discharge of nascent polypeptides as intact peptidyl‐tRNAs (Shoemaker et al. 2010;
Pisareva et al. 2011). Although peptidyl‐tRNA drop‐off remains to be shown in live cells,
the function of the Dom34/Hbs1 complex offers a possible mechanism through which

19
prematurely terminated polypeptides could be generated in vivo and coupled to
proteolysis.
One example has been reported for the endogenous generation of truncated
polypeptides as a DRiP source. Studies of the Epstein‐Barr virus encoded nuclear
antigen 1 (EBNA1) protein demonstrated that translation initiates normally on EBNA1
transcripts, but leads to the synthesis of truncated EBNA1 DRiPs that are efficiently
degraded for MHC class I peptide presentation (Cardinaud et al. 2010). Furthermore,
sequences within the EBNA1 mRNA auto‐inhibit the expression of the truncated
polypeptide and thereby downstream antigen presentation. Although EBNA1 may be
seen as a “special case,” it demonstrates in principle that truncated polypeptides can
serve as an efficient source substrates for the RDP pathway in human cells.
1.5 Puromycin: mechanism of action and experimental applications
I sought to use puromycin as a means to generate prematurely terminated
polypeptides for studies of the RDP pathway. As a structural mimic of tyrosyl‐tRNA
(Fig. 2), puromycin binds the ribosomal A site during elongation and binds covalently to
the C‐terminus of the nascent polypeptide. Without a tRNA to remain tethered to the
mRNA, the truncated peptidyl‐puromycin adduct then dissociates from the ribosome
(Nathans 1964; Vázquez 1979). For experimental applications, puromycin offers the
advantages of both stimulating and covalently tagging prematurely terminated
polypeptides.

20
Figure 2: Puromycin and notable structural features.
Puromycin has a well‐established history of use for studies of protein
degradation. In E. coli, proteins synthesized in the presence of puromycin showed
increased degradation (Kemshead and Hipkiss 1974), while puromycin had no effect on
proteins synthesized prior to its addition to cells (Goldberg 1972). Reticulocytes and
hepatoma cells were shown to possess a mechanism for the rapid degradation of
peptidyl‐puromycins, though this proteolytic activity was lost in cell‐free lysates
(McIlhinney and Hogan 1974). Multiple groups observed the formation of high
molecular weight aggregates following puromycin treatment in a variety of both
prokaryotic and eukaryotic cell types, leading many to suggest the aggregates
represented proteolytic intermediates (Prouty et al. 1975; Daniels et al. 1980; Klemes et

21
al. 1981). Livers from senescent mice were impaired in their ability to degrade peptidyl‐
puromycins following puromycin administration in vivo (Lavie et al. 1982).
A number of intellectual and technological advancements now make it possible
to use puromycin specifically for the study of RDPs. The previous studies were
conducted before the mechanisms of the ubiquitin‐proteasome system were understood,
and well before the development of cell permeable, small molecule inhibitors of the
proteasome which rescue RDPs from degradation (Rock et al. 1994). Reports of the
special “short‐lived” protein pool from Poole, Wibo, and Wheatley were (seemingly) not
picked up on, at the time, by investigators using puromycin.
But it was the development of anti‐puromycin antibodies that greatly expanded
the range of experimental applications for puromycin (Hansen et al. 1994). Initially,
these antibodies were used to immunoprecipitate nascent chains and identify their
bound chaperones (Hansen et al. 1994; Teter et al. 1999; McCallum et al. 2000) or
interactions with the translocon (Pariyarath et al. 2001). After the early immunologic
studies that formally tested the DRiP hypothesis, Lelouard and colleagues used
puromycin to generate DRiPs and study their behavior in dendritic cells via
immunofluorescent microscopy (Lelouard et al. 2004). Puromycin‐tagged DRiPs were
found to traffic rapidly to specialized structures known as dendritic cell aggresome‐like
induced structures (DALIS)—large cytoplasmic aggregates which function as storage
depots for polyubiquitinated proteins and protect DRiPs from being degraded.

22
The most recent technical innovation using anti‐puromycin antibodies has been
the application of puromycin to studies of protein synthesis. The underlying principle of
this family of techniques is that puromycin incorporation can be used as a proxy
measure of protein synthesis, without the need for radiolabeling. The first of these
techniques to be published was called surface sensing of translation, or SUnSET, which
takes advantage of the appearance of puromycin‐tagged polypeptides on the cell surface
(Schmidt et al. 2009). In SUnSET, puromycin‐treated cells are stained with anti‐
puromycin antibodies and then measured for cell surface puromycin expression by flow
cytometry. By this method, puromycin incorporation can be used to measure protein
synthesis with single cell resolution. SUnSET has now been used successfully applied in
vivo for studies of protein synthesis in skeletal muscle (Goodman et al. 2011). In this
study, mice were injected intraperitoneally with puromycin, followed by harvesting of
tissues for Western blotting and immunohistochemistry to measure puromycin
incorporation. In both applications of SUnSET, puromycin signal gave similar results to
radiolabeling, validating the use of this approach to measure translational activity.
Finally, anti‐puromycin antibodies have been used to detect the subcellular localization
of actively translating ribosomes, using a technique called ribopuromycylation (RPM)
(David et al. 2011). In RPM, translating ribosomes are stained by puromycin attachment
to the nascent chain. The addition of cycloheximide (CHX) arrests the nascent chain on

23
the ribosome, allowing one to stain for puromycin and characterize the subcellular
distribution of active ribosomes.
The development of anti‐puromycin antibodies has led to valuable insights into
the biology of nascent polypeptides, DRiP regulation in professional antigen presenting
cells, in vivo studies of protein synthesis, and the subcellular localization of translation.
In my studies, I propose to use puromycin to generate and track the fate of prematurely
terminated polypeptides. I hypothesize that the products of premature translational
termination are preferentially targeted to the RDP pathway. Throughout the studies in
this dissertation, I employ both quantitative biochemical analysis and assays of antigen
presentation to determine the effects of stimulating premature translational termination
on the behavior of the RDP pathway.
1.6 Overview of Results Chapters
In the following sections, I will present evidence that premature translational
termination products are rapidly degraded polypeptides. In Chapter 3, I will describe
the development of a model system that utilizes puromycin to generate and track the
fate of truncated polypeptides. I will then use this system to investigate the effects of
stimulating premature translational termination on the degradation of RDPs and to
directly measure the stability of truncated polypeptides. In Chapter 4, I will explore the
time‐ and concentration‐dependent effects of prematurely terminated polypeptides on
MHC class I presentation.

24
2. Materials and Methods
2.1 Materials
Cycloheximide (CHX), puromycin (puro) and the proteasome inhibitor MG132
were purchased from Sigma (St. Louis, MO). Mouse anti‐Kb and corresponding isotype
control antibodies were purchased from BD (Franklin Lakes, NJ). AlexaFluor 647
(AF647)‐conjugated goat anti‐mouse IgG, anti‐green fluorescent protein (GFP) rabbit
serum, and methionine/cysteine‐deficient Dulbecco’s Modified Eagle Medium (Met/Cys‐
DMEM) were purchased from Invitrogen (Carlsbad, CA). The following reagents were
also used: trichloroacetic acid (TCA, Mallinckrodt Chemicals, Phillipsburg, NJ), FK2
mouse anti‐mono‐ and polyubiquitin conjugate (Millipore, Billerica, MA), E7 mouse
anti‐‐tubulin (Developmental Studies Hybridoma Bank, University of Iowa, Iowa City,
IA), and rabbit anti‐Pth2/Bit1 (Cell Signaling, Danvers, MA). Anti‐puromycin rabbit
serum was kindly provided by Peter Walter (UCSF, San Francisco, CA). The following
were the kind gifts of Jon Yewdell (NIAID, Bethesda, MD): AF647‐conjugated 25‐D1.16,
a monoclonal mouse antibody specific for the MHC class I‐peptide complex Kb‐
SIINFEKL (Porgador et al. 1997), human embryonic kidney 293 cells stably expressing
the mouse MHC class I allele, H‐2Kb (293‐Kb) (Qian et al. 2006a), and a plasmid
containing NP‐SIINFEKL‐eGFP (NSe), composed of influenza nucleoprotein (NP) fused
to the ovalbumin antigenic peptide SIINFEKL and enhanced green fluorescent protein

25
(eGFP) (Princiotta et al. 2003). Rabbit anti‐CHIP antisera, CHIP‐specific siRNA, and non‐
targeting siRNA were the kind gift of Doug Cyr (UNC, Chapel Hill, NC).
2.2 Construction of the SIINFEKL Tandem Repeat reporter (TRx9)
The NSe construct was subcloned from pSC11 into pcDNA6B (Invitrogen) for
transfection‐based expression using the EcoRV (5’) and NotI (3’) restriction sites, making
the plasmid NSe‐pc6B. The method used to construct the tandem repeat was initially
described in (Türkel and Farabaugh 1993). Prior to constructing the tandem repeat, it
was necessary to eliminate the XbaI site in NSe‐pc6B via site‐directed mutagenesis using
the following primers (mutation underlined). Forward: 5’‐
GGCCGCTCGAGCCTAGAGGGCCC‐3’. Reverse: 5’‐
GGGCCCTCTAGGCTCGAGCGGCC‐3’. This creates a silent TC mutation. Synthetic
oligonucleotides were prepared encoding the antigenic SIINFEKL peptide flanked by 5
amino acids from the chicken ovalbumin gene sequence: LEQLESIINFEKLTEWTS. The
oligonucleotides (oligos) contain an NheI site at the 5’ end and an XbaI site at the 3’ end.
Sense: 5’‐
CTAGGTGCTAGCCTTGAGCAGCTTGAGTCGATCATCAACTTCGAAAAGCTAACT
GAATGGACCAGTTCTAGA‐3’. Antisense: 5’‐
CTAGTCTAGAACTGGTCCATTCAGTTAGCTTTTCGAAGTTGATGATCGACTCAAG
CTGCTCAAGGCTAGCAC‐3’. The oligos were annealed by mixing 2 g of the sense
and antisense strands in 1x T4 Ligase Buffer (New England Biolabs, Ipswich, MA), then

26
heating the mixture to 70 OC for 10 minutes, followed by gradual cooling to room
temperature. The oligo duplex contains sticky ends that are compatible with the NheI
restriction site. NSe‐pc6B was linearized with NheI and gel purified using a QIAquick
Gel Extraction Kit (QIAGEN, Valencia, CA). The annealed oligo was then ligated into
the NheI site of NSe‐pc6B, downstream of the existing SIINFEKL peptide between the
nucleoprotein (NP) and eGFP open reading frames. Ligation eliminated the NheI site in
the plasmid but introduced a new NheI site in the 5’ end of the ligated oligo.
Directionality of the oligo insert was verified by PCR screening.
To construct the tandem repeat, NSe‐pc6B containing the SIINFEKL oligo was
subjected to a double digest with Nhe I/Xho I or Xba I/Xho I in separate reactions. The
5’‐NheI‐XhoI‐3’ fragment and 5’‐XhoI‐XbaI‐3’ fragment (each of which contains one
SIINFEKL element). These fragments were then ligated to one another; the NheI and
XbaI sticky ends are compatible and ligate to one another, however the ligation results
in elimination of the restriction site at the junction. The product of the ligation reaction is
NSe‐pc6B containing two SIINFEKL elements (plus the SIINFEKL outside the tandem
repeat unit already present in NSe, for three total, TRx3). The tandemly repeated
SIINFEKL elements are flanked by NheI and XbaI (like the original oligo), but are joined
at the junction between the NheI and XbaI sticky ends (which no longer forms a
restriction site). Using the new construct to repeat the double digests and ligation for
two additional cycles (causing two additional rounds of duplicating the SIINFEKL

27
element) produces NSe‐pc6B containing 9 total SIINFEKL elements (TRx9). All
sequences were verified by the Duke University Comprehensive Cancer DNA
Sequencing Facility (Durham, NC).
2.3 Cell culture
293‐Kb cells were cultured in DMEM with 10% fetal bovine serum at 37 OC and
5% CO2. Unless otherwise indicated, cell monolayers were used at 85% confluency.
2.4 Metabolic radiolabeling and pulse-chase
Radiolabeling and pulse‐chase conditions to measure RDPs were adapted from
(Qian et al. 2005). 293‐Kb cells were harvested and resuspended at a concentration of 107
cells/ml in methionine‐deficient (Met/Cys‐) DMEM prewarmed to 37 OC supplemented
with 1 mM glutamine, 1 mM sodium pyruvate and 25 mM HEPES. Cells were labeled
without prior methionine starvation with 300 Ci/ml 35S‐methionine/cysteine (EasyTag
Express Protein Labeling Mix, Perkin Elmer, Waltham, MA) at 37 OC. To terminate
labeling and precipitate polypeptides, TCA was added to a final concentration of 10%
w/v and samples were incubated on ice for 10 min. Precipitates were washed twice in
acetone, air‐dried, resuspended in solubilization buffer (5% SDS, 0.5 M Tris) and heated
at 95 OC for 10 min to fully solubilize polypeptides. Radiolabeled polypeptides were
measured by liquid scintillation counting using a Packard Liquid Scintillation Analyzer
Tri‐Carb 2100TR. Alternatively, radiolabeled polypeptides were separated on either 10%
or 16%/6% (total/crosslinker) polyacrylamide gels by tricine SDS‐PAGE (Schägger 2006)

28
to resolve low molecular weight polypeptides or on 7.5% polyacrylamide Laemmli gels
by standard SDS‐PAGE to resolve high molecular weight polypeptides. Gels were
stained for total protein with Coomassie blue, dried, and exposed to a PhosphorImager
plate overnight at room temperature or to HyBlot CL Autoradiography Film (Denville
Scientific, Inc., Metuchen, NJ) at ‐80 OC for 4 days. PhosphorImager plates were scanned
using a Typhoon 9400 (GE Healthcare) and quantified using ImageQuant TL version 7.0
(GE Healthcare) or ImageJ (NIH).
To generate lane intensity plots, custom Python scripts developed by David Reid
(Duke University) were used to measure the sum of pixel intensities at each vertical
position in a lane, with the sum plotted as a function of vertical position in the gel. To
calculate the total signal in a specified region of a lane, the sum of pixel intensities was
integrated over the given range.
For pulse‐chase experiments, cells were radiolabeled at a concentration of 107
cells/ml for 5 minutes, as previously described. Pulse labeling was terminated by the
addition of >10‐fold excess volume of ice‐cold chase solution (Dulbecco’s phosphate
buffered saline (DPBS) with 1% bovine serum albumin (BSA), 10 mM unlabeled
methionine, 200 M CHX) and placing the samples on ice. Cells were quickly pelleted by
centrifugation for 30 s at 3400 xg, then washed twice with 1 ml chase solution and
resuspended in chase media at a concentration of 2.2x106 cells/ml (Met/Cys‐ DMEM with
10 mM methionine and 200 M CHX). For the chase, cells were incubated at 37 OC in a

29
water bath; the chase was terminated at specific time points by the addition of TCA to a
final concentration of 10% v/v and placing samples on ice, as described above.
2.5 Denaturing immunoprecipitation
Solubilized TCA precipitates from radiolabeled cells were used at a
concentration of 4x104 cell equivalents per 10 l of solubilization buffer, unless otherwise
indicated. For each immunoprecipitation (IP) reaction, 10l of solubilized TCA
precipitate was diluted into 990 l of IP buffer (1% Triton X‐100, 25 mM HEPES, 150 mM
NaCl, 1 mM EDTA) and precleared with 15 l of Pansorbin cells (EMD, Gibbstown, NJ)
for 30 minutes at room temperature. To precipitate peptidyl‐puromycins, 1 l of anti‐
puromycin serum was incubated with the precleared lysate for 1 hour at room
temperature with gentle mixing. Immune complexes were captured by adding 15 l of a
50% slurry of Pierce protein A/G agarose beads (Thermo Fisher, Rockford, IL) and
incubating for 1 hour at room temperature with gentle mixing. Beads were washed four
times with 1 ml IP buffer, then mixed with 22 l sample buffer (300 mM Tris, pH 6.8,
36% glycerol, 10% SDS, 0.012% bromophenol blue) with 50 mM dithiothreitol (DTT) and
heated to 95 OC for 5 minutes. Beads were pelleted and the supernatants were used for
liquid scintillation counting or tricine SDS‐PAGE.
2.6 Flow cytometry
Twenty‐four hours prior to drug treatment, 293‐Kb cells were seeded into a 6‐
well plate at a density of 2x105 cells/well. Cells were treated for 12 hours with various

30
drugs, then harvested and stained with 0.5 g of either isotype control or anti‐Kb
antibody in 100 l of FACS buffer (DPBS with 1% BSA and 0.02% sodium azide) on ice
for 45 minutes. Cells were washed twice with FACS buffer, and then stained with 1 g of
AF647‐goat anti‐mouse IgG in 100 l of FACS buffer on ice for 45 minutes. Cells were
washed twice more with FACS buffer, then resuspended in 300 l FACS buffer with 2
g/ml propidium iodide (PI, Sigma‐Aldrich, St. Louis, MO) on ice for 30 minutes. A
similar procedure was used to measure cell surface Kb‐SIINFEKL complexes using the
AF647‐conjugated 25‐D1.16 monoclonal antibody (1:500 dilution) except for the
exclusion of a secondary antibody staining step. Samples were analyzed immediately
using an LSRII flow cytometer (BD Biosciences, San Jose, CA). PI‐positive cells were
excluded from analyses of cell surface antibody staining. For each fluorescence channel,
the minimum and maximum values of geometric mean fluorescence intensity (MFI)
were standardized between trials. All flow cytometry data were analyzed using FlowJo
version 8.6.1 (Treestar, Ashland, OR).
2.7 MHC class I peptide stripping and recovery
Twenty‐four hours prior to reporter plasmid transfection, 293‐Kb cells were
seeded into 10‐cm plates at a density of 106 cells/plate. For transfection, 54 g of
polyethylenimine (PEI, 25 kDa, linear, Polysciences, Warrington, PA) was complexed
with 18 g of reporter plasmid DNA in Opti‐MEM (Invitrogen) and incubated with cells
for 7 hours. Media containing PEI‐DNA complexes was exchanged for fresh, prewarmed

31
media, and transfected cells were incubated for an additional 17 hours. Cells were then
harvested and stripped of MHC I peptides as described in (Sugawara et al. 1987). Briefly,
cell pellets were resuspended in peptide stripping buffer (0.131 M citric acid, 0.66 M
Na2HPO4, 1% BSA, pH 3) and incubated on ice for 2 minutes. The pH was neutralized to
7.4 and the cells were resuspended at a concentration of 105 cells/ml in standard media,
then seeded into a 12‐well plate with varying concentrations of protein synthesis
inhibitors. Cells were incubated for up to 4 hours at 37 OC to allow for the recovery of
cell surface MHC class I‐peptide complexes. The cells were harvested at the indicated
time points, stained for cell surface Kb and Kb‐SIINFEKL complexes, and analyzed by
flow cytometry as described above.
2.8 Western blotting
293‐Kb cells were harvested and lysed on ice in IP buffer with 1 mM
phenylmethylsulfonyl fluoride (PMSF) for 10 minutes. Lysates were clarified by
centrifugation at 20000 xg for 10 minutes at 4 OC. Cleared lysates were mixed 1:1 with
sample buffer and 20 mM DTT, heated to 95 OC for 5 minutes, and separated by SDS‐
PAGE on standard 10% or 12.5% Laemmli gels, using 3x105 cell equivalents/lane.
Proteins were transferred via overnight wet transfer in Towbin buffer (25 mM Tris, 192
mM glycine, 20% methanol) onto a polyvinylidene fluoride (PVDF) membrane.
For detection of ‐tubulin, membranes were blocked with 10% milk in Tris‐
buffered saline and 0.05% Tween 20 (TBS‐T). E7 (anti‐‐tubulin, 1:3000) was diluted in

32
1% milk in TBS‐T then incubated with the membranes for 1 hour at room temperature
with gentle rocking. Membranes were washed three times with 1% milk in TBS‐T, then
probed with the secondary antibody HRP‐goat anti‐mouse IgG (1:2500 in TBS‐T) to
detect E7 for 30 minutes.
For detection of polyubiquitinated proteins, membranes were blocked with 1%
BSA in TBS‐T, then probed with FK2 antibody (1:1000 in 1% BSA TBS‐T). Membranes
were washed three times in 1% BSA TBS‐T, then FK2 was detected using HRP‐goat anti‐
mouse (1:2500 in 1% BSA TBS‐T).
To detect peptidyl‐tRNA hydrolase 2 (Pth2), membranes were blocked with 5%
BSA in TBS‐T, then probed with anti‐Pth2 antibody (1:500 in 5% BSA TBS‐T) overnight
at 4 OC with gentle rocking. Membranes were washed 3 times in 5% BSA TBS‐T, then
probed with mouse anti‐rabbit IgG (1:1000 in 5% BSA TBS‐T) secondary antibody.
Membranes were washed 3 additional times in 5% BSA TBS‐T, then probed with HRP‐
goat anti‐mouse IgG (1:2500 in 5% BSA TBS‐T) tertiary antibody for 30 minutes at room
temperature with gentle rocking.
To detect CHIP, membranes were blocked with 10% milk in TBS‐T, then probed
with 1:250 rabbit anti‐CHIP antisera (diluted in 1% milk TBS‐T) overnight at 4 OC with
gentle rocking. Membranes were washed 3 times in 1% milk TBS‐T, then probed with
HRP‐goat anti‐rabbit IgG (1:2500 in 1% milk TBS‐T) secondary antibody for 30 minutes
at room temperature with gentle rocking.

33
For all Western blots, just prior to the addition of HRP substrate, blots were
washed 3 times in TBS‐T, then 2 additional times in TBS. HRP‐antibody conjugates were
detected by incubating blots for 5 minutes with the SuperSignal West Pico
Chemiluminescent Substrate (Thermo Scientific), then imaging the chemiluminescence
on HyBlot CL Autoradiography Film.
2.9 RNA interference
293‐Kb cells were seeded at 50% confluence in 12‐well plates 24 hours prior to
transfection, then transfected with 100 nM Pth2 SMARTpool siRNA (Dharmacon,
Lafayette, CO), 100 nM CHIP siRNA or 100 nM non‐targeting siRNA complexed with 2
l of Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer’s
instructions. After 7 hours, media on the cells was changed to fresh, prewarmed media.
Seventy‐two hours after transfection, cells were harvested for Western blotting, pulse‐
chase analysis, or flow cytometry as described above.
2.10 Data analysis
Statistical analyses were conducted using Microsoft Excel 2010. Graphs were
generated using either Microsoft Excel 2010 or GraphPad Prism 4.0 (GraphPad Software,
San Diego, CA). Images were assembled in Adobe InDesign CS4, Adobe Illustrator CS4,
and Adobe Photoshop CS4 (Adobe Systems Inc., San Jose, CA). Schematics were
generated in Microsoft PowerPoint 2010.

34
3. Premature translational termination products are rapidly degraded polypeptides
3.1 Overview
Nearly a third of all newly synthesized polypeptides are directed to the RDP
pool, which comprises 70% of proteasomal substrates (Schubert et al. 2000; Princiotta et
al. 2003; Qian et al. 2006a). However, the molecular characteristics of RDP pathway
substrates remain mysterious. From the initial formulation of the DRiP hypothesis
fifteen years ago to the present day, premature translational termination products have
been proposed to be a source of RDP pathway substrates, although this has not yet been
tested experimentally in eukaryotic cells (Yewdell 1996; Dolan et al. 2011b). Studies in E.
coli have demonstrated that the drop‐off of nascent polypeptides occurs as a normal by‐
product of protein synthesis (Menninger 1976), with nearly 25% of translation initiation
events resulting in the production of truncated polypeptides which are rapidly
degraded (Manley 1978, Tsung et al. 1989, Jorgensen and Kurland 1990).
To determine whether prematurely terminated polypeptides can serve as RDP
pathway substrates, I used puromycin to stimulate premature translational drop‐off of
nascent chains. In this chapter, I describe the development of a model system employing
puromycin to track the fate of prematurely terminated polypeptides in mammalian cells.
Based on my biochemical studies with puromycin, I report four primary findings: 1)
Puromycin treatment doubles the fraction of polypeptides targeted for rapid
degradation in cells. 2) During puromycin treatment, a fraction of polypeptides is

35
rapidly degraded via a mechanism resistant to MG132, a proteasome inhibitor. 3) Low
molecular weight polypeptides show increased degradation in puromycin‐treated cells,
even in the presence of MG132. 4) Prematurely terminated peptidyl‐puromycins are
targeted for rapid degradation, a fraction of which occurs in an MG132‐resistant
manner. These studies provide biochemical evidence to support the proposal that
prematurely terminated polypeptides are preferentially targeted to the RDP pathway,
suggesting that truncated polypeptides compose a general subclass of RDPs.
3.2 Development and characterization of a model system to study the products of premature translational termination
To test the hypothesis that prematurely terminated polypeptides are RDPs, I
sought to develop a model system in mammalian cells in which the generation of
prematurely terminated polypeptides could be precisely controlled. To accomplish this,
I exploited the properties of the antibiotic puromycin, a structural mimic of tyrosyl‐
tRNA. Puromycin covalently incorporates into the C‐terminus of nascent polypeptides,
leading to their premature termination and dissociation from the ribosome (Vázquez
1979). I postulated that the production of truncated polypeptides could be predictably
controlled by varying the concentration of puromycin. Furthermore, with the
development of anti‐puromycin antisera (McCallum et al. 2000), puromycin serves as a
covalent tag that can be used to selectively purify and track the fate of prematurely
terminated peptidyl‐puromycin adducts.

36
First, I sought to identify a puromycin concentration range that promotes the
production of prematurely terminated polypeptides while maintaining a substantial
level of protein synthesis. To accomplish this, 293‐Kb cells were radiolabeled with [35S]‐
Met in the presence of varying concentrations of puromycin ranging from 0.02‐200 M.
Radiolabel incorporation was then determined by liquid scintillation counting to
measure total cellular protein synthesis. For comparison, I used cycloheximide (CHX),
an antibiotic that arrests elongating ribosomes without prematurely discharging the
nascent chain (Vázquez 1979). CHX acts by binding the E‐site of the large ribosomal
subunit (60S) and preventing translocation (Schneider‐Poetsch et al. 2010).
The data in Fig. 3 illustrate the protein synthesis dose inhibition profiles for
puromycin and CHX. CHX elicited a biphasic inhibition of [35S] incorporation, with
~70% inhibition over the first 2 log order increase in concentration then ~20% inhibition
over the remaining 2 log order increase. The results correspond to the linear portion and
tail of a sigmoidal inhibitory dose‐response profile, based on comparison to a similar
experiment done in HeLa cells (Schneider‐Poetsch et al. 2010).

37
Figure 3: Contrasting effects of cycloheximide and puromycin on [35S]‐methionine
incorporation during protein synthesis. 293‐Kb cells were radiolabeled with [35S]‐Met
for 10 minutes in the presence of varying concentrations (M) of CHX and puro,
followed by precipitation of polypeptides with trichloroacetic acid (TCA). TCA
precipitates were solubilized and radiolabel incorporation was measured by liquid
scintillation counting. [35S] signal was normalized to untreated controls (n = 3; mean ±
s.e.m.)
Puromycin treatment yields a very different dose‐response profile. In contrast to
CHX, treatment with 0.2 to 2 M puromycin resulted in a small but significant increase
in [35S] labeling compared to untreated controls (p = 0.01 and p = 0.07 at 0.2 and 2 M,
respectively), with ~90% inhibition occurring over the remaining 2 log order increase in
concentration. These results demonstrate that inducing premature termination with low
concentrations of puromycin allows the majority of total protein synthesis to be
maintained (20 M) or causes it to slightly increase (0.2‐2 M).
The stimulation of protein synthesis observed at low puromycin concentrations
is consistent with a study reporting that moderate accumulation of misfolded

38
proteins induces a small increase in protein synthesis (Qian et al. 2010). In that study, the
increase in misfolded proteins was found to decrease chaperone availability, thereby
triggering signaling through the mammalian target of rapamycin complex 1 (mTORC1),
which in turn, phosphorylated p70 S6 kinase 1 (S6K1). Phosphorylation of the ribosomal
S6 protein by S6K stimulated protein synthesis. This feedback loop is proposed to
regulate protein quantity depending on protein quality; the increase in translation likely
represents an acute response to replenish cellular proteins lost to misfolding. In my
system, it is reasonable to suggest that puromycin concentrations from 0.2 to 2 M
stimulated the production of truncated polypeptides (many of which are likely to be
misfolded or unfolded) which triggered the feedback loop to increase protein synthesis.
To examine the molecular characteristics of puromycin‐elicited drop‐off
products, lysates from cells radiolabeled with the same concentrations of puromycin or
CHX used in Fig. 3 were separated by tricine SDS‐PAGE, which is better able to resolve
low molecular weight proteins than standard Laemmli SDS‐PAGE (Schägger and von
Jagow 1987; Schägger 2006). [35S]‐labeled proteins were visualized by phosphor imaging.

39
Figure 4: Contrasting effects of cycloheximide and puromycin on the profile of newly
synthesized polypeptides. 293‐Kb cells were radiolabeled with [35S]‐Met for 10 minutes
in the presence of varying concentrations (M) of CHX and puro, followed by TCA
precipitation of polypeptides. (A) TCA precipitates were solubilized and separated by
tricine SDS‐PAGE in a 10% gel (left panel) or 16%/6% gel (total/crosslinker, lower right
panel) or by Laemmli SDS‐PAGE in a 7.5% gel (upper right panel). Gels were dried and
exposed to a PhosphorImager plate overnight. (B) and (C) Pixel intensities were
determined using ImageJ and plotted for polypeptides at the top of the gel in the
stacking/separating gel interface (High MW), resolved proteins in the separating gel
(Bulk), and unresolved small polypeptides in the dye front (Low MW). The sum of all
three signals was also plotted. All values for CHX (B) and puromycin‐treated (C) cells
were normalized to untreated samples. Results are representative of three independent
experiments.

40
Figure 5: Treatment with protein synthesis inhibitors during radiolabeling has no
effect on steady state protein levels. The gels from Fig. 4A were stained with Coomassie
blue and scanned. Results are representative of three independent experiments.
The size distribution of [35S]‐labeled polypeptides showed considerable
concentration‐dependent differences between CHX and puromycin‐treated cells (Fig.
4A). This was most evident in the populations of poorly resolved polypeptides at the top
and bottom of the gel. To distinguish between the behavior of poorly resolved high and
low molecular weight polypeptides, as well the bulk of proteins resolved in the
separating gel, ImageJ was used to determine the pixel intensities for each of the three
polypeptide populations across all samples (Figs. 4B and 4C). I will discuss these
populations, in turn, and compare their behavior to total 35S signal in the lane (Figs. 4B
and 4C, Sum) to compare the effects of CHX and puromycin on the profile of
polypeptides of different molecular weights.

41
High molecular polypeptides (>260 kDa) accumulated as a single band at the
interface of the stacking and separating gel layers in 10% tricine gels. In an attempt to
resolve these high molecular weight species, lysates from CHX and puromycin‐treated
cells were resolved on 7.5% Laemmli gels (Fig. 4A, upper right panel). Despite better
resolution of polypeptides between 95 and 260 kDa, the population at the
stacking/separating gel interface persisted. While the exact identity of this population is
unclear, it has been observed previously in the context of studying RDPs (Schubert et al.
2000) in which it was suggested that these represent polyubiquitinated proteins, though
this was not demonstrated explicitly. This explanation is plausible, given that
polyubiquitin chains branch off the primary polypeptide chain, and polyubiquitin
chains can be branched themselves. Thus, even when maximally solvated and unfolded
by SDS, proteins conjugated to sufficiently long and branched polyubiquitin moieties
would be poorly able to migrate through the polyacrylamide pores of the separating gel,
and instead get trapped at the stacking/separating gel interface. Alternatively, this
population may simply represent high molecular weight proteins or SDS‐resistant
protein aggregates. Small, unresolved polypeptides at the dye front of the 10% tricine
gel were successfully resolved on a 16%/6% (total/crosslinker) tricine gel (Fig. 4A, lower
right panel).
Increasing concentrations of CHX resulted in progressive decreases in [35S] signal
that uniformly affected the high molecular weight (High MW) and resolved (Bulk)

42
polypeptide populations (Fig 4B), suggesting that these two populations are inhibited by
CHX in the same manner. Unexpectedly, the production of low molecular weight (Low
MW) polypeptides increased slightly at 0.02 M CHX, and was less potently inhibited
by CHX from 0.02‐20 M, but was more potently inhibited by 200 M CHX. The reason
for this behavior is unclear; the data would be consistent with ribosomes being more
sensitive to CHX in later rounds of elongation than earlier (and this could even explain
the biphasic inhibition profile of CHX seen in Fig. 3), but there is no reason to expect this
based on CHX’s mechanism of action (Schneider‐Poetsch et al. 2010).
In contrast to CHX, at puromycin concentrations from 0.02 to 2 M, the profile of
resolved [35S]‐labeled bands was of slightly higher intensity relative to untreated
controls (Fig. 4A), with an increase in 35S signal of 12‐16% (Fig. 4C, Bulk). This increase is
consistent with the [35S] incorporation data showing a small increase in protein synthesis
over this low range of puromycin concentrations (Fig. 3). Polypeptides at the top of the
gel (Fig. 4C, High MW) showed no change in signal from 0.02 to 2 M puromycin, while
smaller polypeptides (Low MW) increased in a concentration‐dependent manner.
The profile shifted markedly at 20 M puromycin, where radiolabel
incorporation into the prominent resolved bands was reduced, with a clearly discernible
increase in background radioactivity throughout the lane, consistent with the
appearance of highly heterogeneous, radiolabeled species. While the high molecular
weight and resolved proteins showed a ~30% decrease in signal relative to untreated

43
controls (Fig. 4C, High MW and Bulk, respectively), there was a >250% signal increase in
unresolved polypeptides at the dye front of the 10% tricine gel (Fig. 4A, left panel).
These small polypeptides were successfully resolved as a heterogeneous population
extending down to 4.6 kDa on a 16%/6% (total/crosslinker) tricine gel (Fig. 4A, lower
right panel). The low molecular weight polypeptides whose accumulation was robustly
stimulated by 20 M puromycin are, by definition, premature translational termination
products.
At 20 M puromycin, there is a ~30% inhibition of total 35S incorporation (Figs. 3
and 4C). Because of puromycin’s mechanism of action as a premature chain terminator,
it is instructive to explore possible mechanisms by which puromycin decreases 35S
labeling to better understand its effects in this experimental system. First, truncation of
polypeptides will lead to a decrease in the number of radiolabeled amino acids per
polypeptide (assuming methionine residues are randomly distributed along the length
of any given protein), meaning part of the decrease in 35S signal reflects the decrease in
average polypeptide length, rather than a decrease in initiation events. Second,
saturation of either the translational termination or initiation machinery by the high
number of premature termination events would result in a net decrease in protein
synthesis. (I favor saturation of initiation, since it is the rate‐limiting step of translation
(Kapp and Lorsch 2004; Gebauer and Hentze 2004)). Third, the increase in the cellular
load of misfolded, truncated proteins could cause a massive drop in chaperone

44
availability. Studies of the mTORC1 pathway demonstrate that sizable depletions in
available chaperones lead to the failure of mTORC1 assembly and downstream
translational inhibition (Qian et al. 2010). Finally, (and most significantly for this study),
the drop in 35S incorporation likely reflects the rapid degradation of prematurely
terminated peptidyl‐puromycin chains during radiolabeling. One or more of these
factors is likely responsible for the partial inhibition of 35S incorporation observed
following treatment with 20 M puromycin.
At 200 M puromycin, there was a nearly complete loss of [35S]‐labeled high
molecular weight and resolved polypeptide species, while there were still 25% more low
molecular weight peptides in this sample than untreated controls (Figs. 4A and 4C).
These data indicate that at such a high concentration of puromycin, nearly all
translational initiation events in the cell are followed shortly by the covalent attachment
of puromycin to nascent chains in one of the first few elongation steps, exclusively
yielding short peptidyl‐puromycins.
One potential concern is that the changes in 35S‐labeled polypeptide profiles are
not due to changes in protein synthesis, but rather to off‐target effects of the inhibitors
that globally perturb protein homeostasis. To address this concern, the gels from the
radiolabeling studies (Fig. 4) were stained with Coomassie blue to assess changes to the
steady state protein profile. As seen in Fig. 5, treatment with either protein synthesis
inhibitor caused no discernible change in steady state cellular protein concentration or

45
composition across the entire concentration range, indicating that the concentration‐
specific effects of the protein synthesis inhibitors result from their effects on translation.
To more precisely control the production of truncated polypeptides, I sought to
obtain a more detailed picture of the dose‐response relationship between puromycin
and the polypeptide profile. To accomplish this, cells were radiolabeled with [35S]‐Met
over a linear range of puromycin concentrations from 0 to 20 M, and the labeled
polypeptide composition was analyzed by tricine SDS‐PAGE (Fig. 6A). The profiles of
radiolabeled polypeptides showed a puromycin‐dependent shift in the average intensity
of [35S] signals from higher to lower molecular weight polypeptides. There was also a
puromycin‐dependent increase in signal for unresolved low molecular weight
polypeptides at the dye front. The gel data were plotted in Fig. 6B to visualize lane
intensity profiles. Additionally, the effects of varying puromycin concentration on [35S]
signal were determined for polypeptides of different molecular weights (Regions 1‐4
throughout Fig. 6, plotted in Fig. 6C). For the gel regions analyzed, puromycin treatment
resulted both in the progressive loss of polypeptides from ~60‐85 kDa in size (Region 1)
and in the accumulation of polypeptides ~10‐12 kDa (Region 3) and <10 kDa (Region 4)
in size, reflecting increased production of truncated polypeptides. Notably, signal for
polypeptides ~15‐17 kDa in size (Region 2) showed minimal variation with puromycin
concentration.

46
Figure 6: Puromycin stimulates the production of truncated polypeptides in a
concentration‐dependent manner. (A) 293‐Kb cells were radiolabeled with [35S]‐Met for
10 minutes in the presence of a linear range of puromycin concentrations from 0 to 20
M, followed by TCA precipitation of polypeptides. TCA precipitates were solubilized
and separated by tricine SDS‐PAGE in a 10% gel. In the later panels, the [35S] signals
from the four regions indicated to the right of the gel are analyzed. (B) PhosphorImager
signal intensities (arbitrary units) from selected lanes in (A). The left side of the graph
corresponds to the top of the gel while the right side of the graph corresponds to the
bottom of the gel at the dye front. The highlighted regions correspond to the parts of the
gel indicated in (A). (C) The effects of puromycin concentration on [35S] signal for each of
the highlighted gel regions in (A) and (B). Results are representative of three
independent experiments. The data presented in this figure was contributed by David
Reid and Joshua Lacsina, in experiments designed by Joshua Lacsina.
Interestingly, the concentration‐dependent effects of puromycin on the size
profile of polypeptides are not consistent with a simple model in which puromycin

47
causes progressively shorter truncation of polypeptides at higher concentrations. If such
a model were true, we would have expected to see an increase in signal in the 15‐17 kDa
range, followed by a decrease over the course of the titration. Instead, we see the
progressive loss of high molecular weight polypeptides (as we expect), but only the
accumulation of polypeptides <15 kDa in size.
What can account for this size bias in puromycin‐truncated polypeptides? I
suggest two possible contributing factors. First, high‐resolution studies of ribosomal
position indicate that there is a high density of ribosomes in the first 30‐40 codons of an
mRNA, which relaxes to a uniform density in a transitional zone of 100‐200 additional
codons (Ingolia et al. 2009). Assuming an average molecular weight of 110 Da per amino
acid, this would correspond to a higher density of ribosomes with nascent chains
ranging from 3.3 to 4.4 kDa, followed by ribosomes in the transitional zone with nascent
chains 15.4 to 26.4 kDa in size. The 5’‐skewed distribution of ribosomes along transcripts
would bias puromycin (which we assume acts stochastically) to discharge polypeptides
in the size ranges listed, consistent with my experimental observations.
Second, ribosomes that dissociate as a result of puromycin‐induced termination
would be recycled for a subsequent round of initiation. As more elongation events are
prematurely terminated, this would shift the equilibrium away from ribosomes
participating in elongation and towards ribosomes undergoing initiation (assuming the
translational initiation machinery is not saturated). The net result of this shift in

48
ribosomal distribution to the 5’ end of transcripts for initiation would result in
puromycin‐induced discharge predominantly of short polypeptides.
For the purposes of studying prematurely terminated polypeptides in the RDP
pathway, the major finding from this series of studies was that treatment with 20 M
puromycin markedly enhanced the production of premature termination products while
maintaining a substantial fraction (~70%) of total protein synthesis (Figs. 3 and 4C).
These experimental conditions were used in the studies described below to study the
cellular fate of prematurely terminated polypeptides.
3.3 Stimulating premature translational termination increases the fraction of rapidly degraded polypeptides
In the previous section, I identified experimental conditions that promote the
robust production of premature translational termination products. In the following
series of studies, I explore how inducing premature termination affects the flux of
polypeptides through the RDP pathway. Quantitative studies of RDPs were made
possible by the development of proteasome inhibitors permeable to live cells (Rock et al.
1994; Kisselev and Goldberg 2001). Measurement of RDPs involves short pulse
radiolabeling of cells, followed by a chase with excess unlabeled amino acid to track the
degradation of the radiolabeled proteins. Conducting the pulse‐chase in the presence or
absence of proteasome inhibitors allows measurement of both the rate of proteasome‐
dependent RDP degradation and the fraction of RDPs (Qian et al. 2005). A short pulse
labeling period has been shown to be critical for measuring the RDP pool, because RDPs

49
are degraded during the duration of labeling, which leads to an underestimation of the
RDP fraction (Wheatley et al. 1980; Fuertes et al. 2003). Because RDPs have a
characteristic half‐life of ~10 minutes (Schubert et al. 2000; Qian et al. 2005; Qian et al.
2006a), they are almost completely degraded after the first 50 minutes of the chase. For
this reason, I term the “RDP fraction” as the fraction of polypeptides degraded after 50
minutes.
To determine the effects of inducing premature termination on the fraction of
polypeptides targeted for rapid degradation, cells were pulse‐labeled for 5 minutes in
the presence or absence of 20 M puromycin, then chased for up to 50 minutes without
puromycin in the presence or absence of the proteasome inhibitor, MG132 (Fig. 7). In
control cells without puromycin, 20% of the radiolabeled proteins were degraded by the
proteasome 5 minutes into the chase in an initial, rapid degradation phase. This was
followed by a more gradual loss in signal with a total loss of 25% of the radiolabel by the
end of the chase relative to MG132‐treated controls. These results are consistent with
previous reports of the biphasic degradation kinetic and measures of the fraction of
RDPs in mammalian cells (Princiotta et al. 2003; Qian et al. 2006a).
In contrast, in puromycin‐treated cells, 25% of the radiolabeled polypeptides had
already been lost by the beginning of the chase in a proteasome‐dependent manner,
indicating that they were degraded during pulse labeling. This is similar to what I
suggested previously to explain the partial inhibition of 35S incorporation at 20 M

50
Figure 7: Treatment with puromycin increases the fraction of rapidly degraded
polypeptides. 293‐Kb cells were pulse labeled with [35S]‐Met +/‐ 20 M puro and +/‐ 20 M MG132 for 5 minutes, then chased from 0 to 50 minutes in the presence of excess
cold methionine, CHX and +/‐ 20 M MG132. DMSO is a solvent control for MG132. The
chase was terminated at the indicated time points by the addition of TCA to cell
suspensions to precipitate polypeptides. TCA precipitates were solubilized and [35S] was
measured by liquid scintillation counting (n ≥ 4; mean ± s.e.m.)
puromycin (Fig. 3). Rapid loss of radiolabeled protein continued up to 20 minutes into
the chase, compared to the 5 minutes of rapid degradation seen in the absence of
puromycin. I speculate that puromycin treatment prolongs the rapid degradation phase
of the decay curve by saturating the proteasome with substrates. By the end of the 50
min chase in puromycin‐treated cells, 49% of the radiolabeled polypeptides were
degraded relative to MG132‐treated controls, representing a doubling of the RDP
fraction. These findings indicate that the premature termination products induced by
puromycin treatment were targeted for rapid degradation.

51
Interestingly, in puromycin‐treated cells there was a trend towards increased
degradation despite the presence of proteasome inhibitor. Comparing MG132‐treated
cells in the presence or absence of puromycin, there was an initial rapid loss of
radiolabeled polypeptide in puromycin‐treated cells, with the eventual loss of 20% of the
radiolabel by the end of the chase. These results indicate that a fraction of prematurely
terminated polypeptides are degraded in an MG132‐resistant manner.
Because puromycin treatment increased the fraction of polypeptides targeted for
rapid degradation, I wanted to determine whether polypeptides displayed size‐
dependent differences in their sensitivity to this effect. Lysates from cells subjected to
pulse‐chase protocols similar to those described in Fig. 7 were separated by tricine SDS‐
PAGE and radiolabeled polypeptides were visualized by phosphor imaging. In control
cells pulse‐labeled without puromycin (Fig. 8A), MG132 treatment led to a modest
recovery of [35S] signal that showed no bias for molecular weight. For cells pulse‐labeled
with puromycin (Fig. 8B), there was a substantial loss in signal over the course of the
chase in the absence of proteasome inhibitor. This loss was especially pronounced for
low molecular weight polypeptides. The addition of MG132 to puromycin‐treated cells
rescued [35S] signal for polypeptides of all sizes. Notably, even in the presence of
proteasome inhibitor, there was a marked loss of low molecular weight polypeptides in
puromycin‐treated cells over the course of the chase.

52
Figure 8: Effects of puromycin on the degradation profile of newly synthesized
polypeptides. (A) and (B) 293‐Kb cells were pulse labeled with [35S]‐Met +/‐ 20 M puro and +/‐ 20 M MG132 for 5 minutes, then chased from 0 to 100 minutes in the presence
of excess cold methionine, CHX and +/‐ 20 M MG132. The chase was terminated at 0, 5,
10, 20, 50, and 100 minutes by the addition of TCA to cell suspensions to precipitate
polypeptides. Solubilized TCA precipitates from cells radiolabeled in the absence (A) or
presence (B) of 20 M puro were separated by tricine SDS‐PAGE on 10% gels. Gels were
dried and exposed to a PhosphorImager plate overnight. Note that for (B), the darkness
of the image has been enhanced in order to see the contrast in degradation rates between
(A) and (B) more clearly. Results are representative of three independent experiments.

53
Based on these pulse‐chase studies, I report the following findings. 1)
Stimulating premature termination with puromycin doubles the fraction of RDPs in
cells. 2) Low molecular weight polypeptides representing premature termination
products are particularly unstable in puromycin‐treated cells. 3) Puromycin treatment
results in the MG132‐resistant degradation of a fraction of polypeptides. 4) Even in the
presence of MG132, low molecular weight polypeptides are rapidly degraded in
puromycin‐treated cells. Taken together, the pulse‐chase data strongly suggest that the
prematurely terminated polypeptides induced by puromycin treatment are rapidly
degraded. The data also suggest the presence of an alternative, MG132‐resistant RDP
pathway that acts on small, truncated polypeptides. Having examined the effects of
puromycin on the degradation of total cellular protein, in the next section, I describe my
efforts to specifically track the fate of prematurely terminated polypeptides.
3.4 The products of premature translational termination are rapidly degraded polypeptides
As a structural mimic of tyrosyl‐tRNA, puromycin acts by covalently
incorporating into the C‐terminus of nascent polypeptides, leading to their premature
termination and dissociation from the ribosome (Vázquez 1979). Because of its covalent
attachment to the dissociated nascent chain, puromycin can serve as an affinity tag for
the specific detection and purification of premature termination products, which was
made possible by the development of anti‐puromycin antibodies (McCallum et al. 2000).
Puromycin has been used to generate DRiPs and study their trafficking by

54
immunofluorescence microscopy in a variety of cell types (Lelouard et al. 2004; Szeto et
al. 2006). To date, however, anti‐puromycin antibodies have not been applied to study
prematurely terminated polypeptides in the context of the RDP pathway. In this section,
I describe the development and optimization of an immunoprecipitation (IP)‐based
method to purify and track the fate of truncated polypeptides.
To specifically analyze prematurely terminated polypeptides, I performed
denaturing IPs using anti‐puromycin antisera to capture peptidyl‐puromycin
termination products. Denaturing immunoprecipitation entails solubilization of proteins
in SDS‐containing buffer, to dissociate protein‐protein interactions. This ensures that the
IP step directly precipitates peptidyl‐puromycins, and not the proteins bound to them.
SDS in the lysate is then diluted in non‐ionic detergent (in these experiments, Triton X‐
100) to a concentration that forms mixed micelles which do not disrupt antibody
structure or antibody‐antigen binding interactions (Dimitriadis 1979).
To establish the profile of immunoprecipitated peptidyl‐puromycins, cells were
radiolabeled with [35S]‐Met in the presence of 20 M puromycin, followed by TCA
precipitation of polypeptides. TCA precipitates were solubilized in SDS‐containing
buffer, then diluted in Triton X‐100 for the anti‐puromycin IP. The results are shown in
Fig. 9A. While non‐specific rabbit sera precipitated no appreciable signal, anti‐
puromycin sera precipitated a heterogeneous smear with a signal peak of 19 kDa. The
immunoprecipitated population has a higher average molecular weight than the

55
Figure 9: Purification of premature translational termination products using
puromycin‐specific antisera. (A) Denaturing IP of peptidyl‐puromycins. 293‐Kb cells
were radiolabeled for 30 minutes with [35S]‐Met, 20 M puromycin, and 20 M MG132.
Polypeptides were precipitated with TCA, solubilized, then subjected to a denaturing IP
using either non‐specific rabbit serum (negative IP control) or anti‐puromycin serum.
Immunoprecipitates were separated by tricine SDS‐PAGE on 10% gels. Gels were dried
and exposed to film for 72 hours at ‐80 OC. Results are representative of three
independent experiments. (B) Optimization of anti‐puromycin immunoprecipitation.
293‐Kb cells were radiolabeled for 5 minutes with [35S]‐Met, 20 M puromycin, and 20
M MG132. Varying cell equivalents (half‐log serial dilutions) of solubilized TCA
precipitate were used as inputs for IPs using anti‐puromycin antisera. Radioactivity in
the immunoprecipitates was measured by liquid scintillation counting and normalized
to the maximal radioactivity recovered. (C) Proteasomal sensitivity of newly synthesized
peptidyl‐puromycins. 293‐Kb cells were radiolabeled for varying periods of time with
[35S]‐Met and 20 M puromycin in the presence or absence of 20 M MG132, then
subjected to TCA precipitation and denaturing anti‐puromycin IP. Radioactivity in the
immunoprecipitates was measured by liquid scintillation counting and normalized to
radiolabel incorporation in MG132‐treated cells at the final (5 minute) time point.

56
puromycin‐induced polypeptide profile observed previously (Fig. 6A), though this
likely reflects the longer labeling time for the IP experiment (30 minutes) versus the
puromycin titration (10 minutes). The relative concentration of puromycin would be
expected to decrease gradually over the course of treatment as puromycin is utilized
(since it is used at a non‐saturating concentration), leading to the production of longer
polypeptides, on average. This result demonstrates that treatment with 20 M
puromycin produces a heterogeneous population of peptidyl‐puromycins spanning a
wide range of sizes, but is predominantly composed of low molecular weight, truncated
products.
To establish experimental conditions for quantitative anti‐puromycin
immunoprecipitation, cells were labeled with [35S]‐Met and 20 M puromycin, followed
by TCA precipitation and solubilization of polypeptides. The solubilized precipitates
were serially diluted in half‐log steps to determine the binding isotherm of peptidyl‐
puromycins to the anti‐puromycin antibody (Fig. 9B). The peptidyl‐puromycin binding
profile fit a one‐site binding hyperbolic curve (R2 = 0.999). Binding was approximately
linear up to 9x104 cell equivalents, followed by an inflection and binding curve with a
more shallow slope from 9x104 to 3.2x105 cell equivalents. To achieve a balance between
sensitivity of detection and dynamic range, 4x104 cell equivalents were used as the input
for the anti‐puromycin IPs described in the experiments below; this input corresponds to
~30% of the maximal [35S] signal observed.

57
To characterize the proteasomal sensitivity of peptidyl‐puromycins, a pilot study
was conducted in which cells were labeled with [35S]‐Met and 20 M puromycin for
varying periods of time in the presence or absence of proteasome inhibitor, followed by
purification of peptidyl‐puromycins via denaturing IP (Fig. 9C). In the absence of
proteasome inhibitor, radiolabel incorporation increased linearly with time. Under
conditions of proteasomal inhibition, radiolabel incorporation was similar to DMSO‐
treated controls for the first minute of labeling. This was followed by a sharp increase in
peptidyl‐puromycin recovery over the next four minutes of labeling, up to a 3‐fold
increase over cells without proteasome inhibitors. This initial finding strongly suggested
that peptidyl‐puromycins are rapidly degraded in a proteasome‐dependent manner.
With conditions established for the quantitative measurement of peptidyl‐
puromycins, I sought to directly test the stability of prematurely terminated
polypeptides using the same pulse‐chase protocol I employed previously to track the
degradation of RDPs (Figs. 7 and 8). Cells were pulse‐labeled for 5 min with [35S]‐Met
and 20 M puromycin, then chased for up to 50 minutes in the presence or absence of
MG132. Denaturing anti‐puromycin IPs were performed on all samples to recover and
measure peptidyl‐puromycins by liquid scintillation counting (Fig. 10).
In the absence of MG132, 21% of the peptidyl‐puromycins were degraded by the
start of the chase. Peptidyl‐puromycins displayed a biphasic decay kinetic, with 30%
being degraded during the first 5 minutes of the chase, followed by a more gradual

58
Figure 10: Rapid degradation of premature translational termination products. 293‐Kb
cells were pulse labeled with [35S]‐Met, 20 M puro and +/‐ 20 M MG132 for 5 minutes,
then chased from 0 to 50 minutes in the presence of excess cold methionine, CHX and +/‐
20 M MG132. The chase was terminated at the indicated time points by TCA
precipitation. Solubilized TCA precipitates were subjected to denaturing
immunoprecipitation using anti‐puromycin serum. [35S] in the anti‐puromycin
immunoprecipitates was measured by liquid scintillation counting. (n = 4; mean ±
s.e.m.) The data presented in this figure was contributed by Odessa Marks and Joshua
Lacsina, in experiments designed by Joshua Lacsina.
degradation profile over the final 45 minutes. By the end of the chase, 62% of the
peptidyl‐puromycin signal was lost relative to MG132‐treated cells. These results
confirm that peptidyl‐puromycins are subject to rapid degradation, with a nearly 2.5‐
fold higher fraction of RDPs compared to the RDP fraction of proteins from non‐
puromycin treated cells (Fig. 7). Notably, in MG132‐treated cells, 20% of the

59
immunoprecipitated peptidyl‐puromycin signal was still lost by the end of the chase
(Fig. 10). This result mirrors the MG132‐resistant degradation profile of total protein
from puromycin‐treated cells (Fig. 7), providing additional evidence for MG132‐
sensitive and ‐insensitive peptidyl‐puromycin degradation pathways.
In this section, I described the development and optimization of a quantitative IP
to specifically measure prematurely terminated polypeptides. Using this approach, I
uncovered direct evidence to support the hypothesis that premature translational
termination products are predominantly targeted to the RDP pathway.
3.5 Summary
In this chapter, I tested the hypothesis that premature translational termination
products are substrates for the RDP pathway. To accomplish this, I employed
puromycin to precisely control the production of prematurely terminated polypeptides.
Through titration experiments, I identified a range of puromycin concentrations that
promote the robust production of prematurely terminated polypeptides (Figs. 4 and 6)
while minimizing inhibition of protein synthesis (Fig. 3). Stimulating premature
termination doubled the fraction of cellular proteins targeted for rapid, proteasome‐
mediated degradation (Fig. 7). In puromycin‐treated cells, ~20% of newly synthesized
polypeptides were degraded via an MG132‐resistant RDP pathway (Fig. 7) which
predominantly affects low molecular weight polypeptides (Fig. 8). Following the
optimization of a quantitative assay to specifically measure premature termination

60
products (Fig. 9), I determined that prematurely terminated polypeptides are
predominantly targeted for rapid degradation, a subset of which occurs via an MG132‐
resistant mechanism (Fig. 10). These studies provide direct evidence for the hypothesis
that premature translational termination products can serve as RDP pathway substrates.
Based on these findings, I speculate that prematurely terminated polypeptides may
compose a general subclass of substrates for the RDP pathway in mammalian cells.

61
4. Premature translational termination promotes antigenic peptide presentation via the major histocompatibility complex class I pathway
4.1 Overview
RDPs are the primary source of antigenic peptides presented on MHC class I
molecules (Schubert et al. 2000; Reits et al. 2000; Qian et al. 2006b; Dolan et al. 2011a).
MHC I molecules are thereby able to sample and present peptides derived from the
rapid degradation of newly synthesized polypeptides. By directing a fraction (~30%) of
all translation products to the RDP pool (Schubert et al. 2000; Princiotta et al. 2003), the
MHC I pathway can sample peptides derived from even the most metabolically stable
proteins, including viral proteins. Selectively sampling RDPs also prevents the ~3x109
cellular proteins from outcompeting the vastly outnumbered viral proteins for
presentation on the ~105 MHC class I molecules per cell, especially early in infection
(Princiotta et al. 2003; Yewdell et al. 2003). Coupling protein synthesis to
immunosurveillance thus allows the rapid detection and elimination of virally infected
cells by CTLs. This is critical to prevent the dissemination of viruses, some of which
have life cycles as short as four hours (Yewdell et al. 2003).
In the previous chapter, I reported that puromycin‐induced premature
translational termination products are substrates for the RDP pathway. Because
puromycin treatment increases the net production of RDPs, in this chapter, I test the
hypothesis that inducing premature translational termination stimulates peptide

62
presentation via the MHC class I pathway. For these studies, I constructed a reporter
(modified from Antón et al. 1999) encoding tandem repeats of the antigenic peptide
SIINFEKL, derived from chicken ovalbumin (Rötzschke et al. 1991). The SIINFEKL
tandem repeats are fused to eGFP, allowing functional reporter protein and MHC I
peptides derived from reporter degradation to be measured sensitively and
simultaneously by flow cytometry. I utilized the reporter to examine the effects of
stimulating truncated polypeptide production on peptide generation from a model
antigen.
From these studies, I report three primary findings: 1) Treatment with low
concentrations of puromycin or over a short duration increases MHC class I
presentation. 2) Extended treatments with puromycin inhibit the MHC class I pathway.
3) Impairment of MHC class I presentation correlates with the increased production of
prematurely terminated polypeptides and the accumulation of polyubiquitinated
proteins, consistent with saturation and inhibition of the proteasome. The time‐ and
dose‐dependent effects of puromycin suggest that prematurely terminated polypeptides
differ in their targeting to various proteolytic pathways which, in turn, differ in the
efficiency with which they access the MHC class I presentation machinery, an emerging
theme in the field of RDP immunology (Princiotta et al. 2003; Dolan et al. 2011a; Dolan et
al. 2011b).

63
Towards the end of the chapter, I present my early studies on identifying the role
of two candidate factors in the RDP pathway: Pth2 and CHIP. Interestingly, knockdown
of these factors by RNA interference increases flux through the RDP pathway,
suggesting they may play a role in rescuing defective proteins from rapid degradation
or in directing them to alternative degradation pathways.
4.2 Effects of stimulating premature translational termination on steady state cell surface expression of MHC class I molecules and cell death
Because puromycin stimulates RDP production, I sought to test the effects of
puromycin treatment on peptide presentation via the MHC class I pathway. As in my
initial biochemical studies, I used cycloheximide for comparison because CHX prevents
nascent chain release from the ribosome and thereby inhibits the flux of substrates into
the RDP pathway (Schubert et al. 2000). In the experiments described below (and in the
previous chapter), I used human embryonic kidney 293 (HEK293) cells stably expressing
the mouse MHC class I allele, Kb (293‐Kb) (Qian et al. 2006a). Kb presents the antigenic
peptide SIINFEKL, to which it binds with high affinity (Rötzschke et al. 1991); Kb‐
SIINFEKL has been used extensively as a model system to study MHC class I
presentation, leading to the development of highly sensitive and specific reagents to
detect cell surface Kb‐SIINFEKL complexes (detailed in the next section). I therefore
chose to study the effects of inducing premature termination in 293‐Kb cells due to their
amenability for studies of the MHC class I pathway.

64
293‐Kb cells were treated with varying concentrations of CHX and puromycin for
12 hours. Cell surface levels of Kb were then measured by flow cytometry, with dead
cells excluded from the analysis by propidium iodide (PI) staining (Fig. 11A). CHX
exhibited a sigmoidal dose response profile, with decreasing cell surface Kb from 0.06 to
20 M, up to a maximal inhibition of ~65%. The CHX profile represents a standard
inhibitory dose response; the remaining 35% of Kb signal at the maximum CHX
concentration likely represents pre‐existing cell surface Kb that failed to turn over during
the 12 hour treatment window.
In contrast, puromycin treatment caused a trend towards increased Kb up to 0.6
M, where a 25% increase in Kb surface expression was observed relative to untreated
cells. This was followed by a 72% drop in Kb expression over the next two log step
increases in puromycin, down to 35% Kb expression, as observed for CHX‐treated cells.
The latter finding suggests that 35% Kb expression represents maximal inhibition of
MHC I presentation within the parameters of this experiment. The puromycin dose
response profile suggests that low levels of truncated polypeptide production stimulate
MHC I presentation. In contrast, further increases in the production of premature
termination products inhibit the MHC I pathway. (At 200 M puromycin, the truncated
polypeptides produced are likely too short to be efficient MHC I pathway substrates,
and thus result in maximal inhibition of Kb expression.)

65
Figure 11: Contrasting effects of cycloheximide and puromycin on steady‐state levels
of cell surface Kb. (A) 293‐Kb cells were treated with the indicated concentrations of
CHX and puro for 12 hours. Cells were harvested and stained live with propidium
iodide (PI) and anti‐Kb antibody. Mean fluorescence intensity (MFI) for cell surface Kb
was measured by flow cytometry and normalized to untreated cells. PI‐positive cells
were excluded from the analysis. (B) MHC class I machinery is functional in the
presence of protein synthesis inhibitors. MFI is of anti‐Kb signal, normalized to untreated
sample. “C200” and “P200” refer to cells treated with 200 M CHX or puro, respectively
(red bars). “+S” indicates cells treated with both 200 M protein synthesis inhibitor and 5 M SIINFEKL peptide (blue bars) (n = 3; mean ± s.e.m.) The data presented in this figure
was contributed by Xiongfei Liu, in experiments designed by Joshua Lacsina and
Xiongfei Liu.

66
A potential concern with using protein synthesis inhibitors in these experiments
is that they may disrupt the expression of factors needed for peptide generation or the
formation and transport of MHC class I‐peptide complexes (raised previously and
addressed over a shorter timescale in Qian et al. 2006b). To test this possibility,
exogenous SIINFEKL peptide was added to cells treated with protein synthesis
inhibitors to determine whether cell surface Kb levels could be rescued (Minami et al.
2010). Rescue can occur via two distinct mechanisms, both of which depend on the
functionality of the MHC I machinery. First, peptide can complex directly with and
stabilize peptide‐receptive, “empty” MHC class I molecules on the cell surface, which
would otherwise be recycled shortly after arrival at the plasma membrane (Rock et al.
1991). Second, peptides can be taken up by cells and transported in a retrograde fashion
to the endoplasmic reticulum (ER), where they are loaded onto MHC class I molecules
and exported to the cell surface as MHC I‐peptide complexes via the standard pathway
(Day et al. 1997). The addition of SIINFEKL peptide rescued cell surface Kb to levels
comparable to cells that were not treated with protein synthesis inhibitors (Fig. 11B).
These data indicate that the MHC class I machinery remains functional in cells treated
with protein synthesis inhibitors, and that cell surface MHC class I levels accurately
reflect the flux of substrates through the presentation pathway.
An incidental observation during these initial flow cytometric studies was that
samples treated with 20 M puromycin consistently had higher numbers of rounded or

67
Figure 12: Contrasting effects of cycloheximide and puromycin on cell death. 293‐Kb
cells were treated with the indicated concentrations of CHX and puro for 12 hours. Cells
were harvested and stained live with propidium iodide (PI). The data shown are the
percentage of PI‐positive cells, subtracting the percentage of PI‐positive cells in the
untreated sample as a baseline. (n = 3; mean ± s.e.m.) The data presented in this figure
was contributed by Xiongfei Liu, in experiments designed by Joshua Lacsina and
Xiongfei Liu.
floating cells when viewed by light microscopy, even more so than the highest
concentration of puromycin (200 M). To extend this observation, flow cytometry was
used to determine the effects of varying concentrations of CHX and puromycin on the
percentage of PI‐positive cells. Strikingly, cells treated with 20 M puromycin showed
the highest percentage of PI‐stained cells, even compared to cells treated with 200 M
puromycin (Fig. 12). In contrast, cells treated with CHX showed no significant increase
in PI staining over baseline. This experiment demonstrates an association between high
levels of truncated polypeptide production and cell death.

68
These studies suggested that low levels of prematurely terminated polypeptides
stimulate MHC class I presentation, while higher levels impair the presentation
pathway. Indeed, higher levels of truncated polypeptide production were associated
with increased cell death.
4.3 Effects of stimulating premature translational termination on MHC class I export
Because the studies described in the previous section were conducted after
twelve hours of treatment, I next sought to establish the time‐ and concentration‐
dependent dynamics of MHC class I responses to truncated polypeptide production. I
postulated that puromycin might exert time‐dependent changes to the MHC I pathway
if cells possess adaptive mechanisms to respond to the prolonged production of
truncated polypeptides
4.3.1 Construction of the SIINFEKL tandem repeat reporter
For my studies on MHC I presentation dynamics, I sought to complement my
measurements of total cell surface Kb levels with experiments to track the presentation of
peptides derived from the degradation of a model antigen. To accomplish this, I
synthesized a reporter construct encoding a fusion protein consisting of influenza
nucleoprotein (NP), nine tandem repeats of the SIINFEKL peptide (each flanked by its
native amino acid sequence for proper processing), and eGFP (Fig. 14A). The reporter,
called Tandem Repeat x9 (TRx9), can be readily utilized via flow cytometry for the

69
Figure 13: Construction of the SIINFEKL tandem repeat. The strategy relies on flanking
the repeat element with two different restriction sites which have complementary sticky
ends, but which eliminate the restriction site when joined. The NheI/XbaI pair was used
to build the reporter for these studies, TRx9. The repeat element can be amplified
exponentially with each cycle of the procedure.

70
simultaneous measurement of both native, functional reporter protein by eGFP
fluorescence, and antigenic peptides derived from reporter protein degradation, in the
form of cell surface Kb‐SIINFEKL complexes. Cell surface Kb‐SIINFEKL was measured
with the use of the monoclonal antibody, 25‐D1.16, which binds Kb‐SIINFEKL complexes
with the sensitivity and specificity of a T cell receptor (Porgador et al. 1997).
TRx9 was constructed using a previously published NP‐SIINFEKL‐eGFP
reporter as a starting point (Antón et al. 1999; Princiotta et al. 2003; Berglund et al. 2007).
The original reporter was designed for expression in vaccinia, which achieves
substantially higher expression than transfected plasmids. To increase the sensitivity
with which changes in SIINFEKL presentation from the reporter could be detected, I
introduced additional SIINFEKL peptides to the reporter as tandemly repeated elements
using a cloning strategy described in (Türkel and Farabaugh 1993) (Fig. 13).
To build TRx9, the SIINFEKL element was flanked with two different restriction
sites which yield compatible sticky ends, but which eliminate the original restriction
sites when ligated together. In each cloning step, restriction fragments were generated
with separate double digests of either the 5’ or 3’ restriction site flanking the repeat
element, plus an additional restriction site (using a third enzyme common to both
digestion reactions) outside the repeat element. The restriction fragments containing the
repeat element were purified from each of the two digestion reactions and ligated
together. The resulting product contained a duplication of the repeat element, with no

71
changes in the rest of the plasmid. Also, the duplicated region was now flanked by the
restriction sites which flanked the original element, meaning the number of repeat
elements can be increased exponentially with each cloning step.
4.3.2 Application of the SIINFEKL tandem repeat reporter to measure the recovery of cell surface MHC class I-peptide complexes
It is difficult to measure rapid, dynamic changes in antigen presentation by
measuring steady state levels of MHC I, given the high background of pre‐existing cell
surface MHC I molecules. To facilitate sensitive detection of newly exported MHC I‐
peptide complexes (derived from RDP degradation), cells transfected with the TRx9
reporter plasmid were stripped of MHC I‐bound peptides by a short acid treatment
(Sugawara et al. 1987). The recovery of cell surface MHC I‐peptide complexes was then
tracked by flow cytometry to measure changes in antigen presentation.
As shown in Fig. 14B, prior to acid stripping, cells transfected with the TRx9
reporter (left panel) showed robust expression of the fluorescent reporter protein (eGFP,
x‐axis) and Kb‐SIINFEKL presentation (y‐axis). Acid‐stripped cells showed complete loss
of Kb‐SIINFEKL expression while eGFP fluorescence was maintained (middle panel).
After a 4 hour incubation at 37 OC, Kb‐SIINFEKL expression partially recovered,
reflecting the export of new MHC I‐peptide complexes to the cell surface (right panel).
Over this period, eGFP fluorescence also increased (Fig. 14C), due to the continued
synthesis of fluorescent reporter proteins. These findings demonstrate successful

72
Figure 14: MHC class I‐peptide complex recovery assay using a fluorescent reporter
encoding antigenic peptides. (A) Schematic of the modified NP‐SIINFEKL‐eGFP
reporter (adapted from Princiotta et al. 2003) containing eight additional tandem repeats
of SIINFEKL (nine total) and its five flanking amino acids from the native ovalbumin
sequence, NP‐[SIINFEKL]9‐eGFP (Tandem Repeat x9 or TRx9). (B) Validation of MHC
class I peptide stripping and recovery in TRx9‐expressing 293‐Kb cells. Biexponential
scatter plots show single cell profiles of the MFI for eGFP on the x‐axis and 25‐D1.16 on
the y‐axis. Plots show fluorescence profiles immediately pre‐ (left) and post‐ (middle)
peptide stripping, and after a 4 hour recovery (right). (C) Graph of MFI data from (B)
normalized to pre‐stripping MFI. Results are representative of three independent
experiments.

73
expression of the TRx9 reporter and its application to measure the generation of MHC I‐
peptide complexes.
4.3.3 Effects of puromycin on the recovery of cell surface MHC class I-peptide complexes
Using the acid stripping protocol outlined above, I proceeded to test the effects of
puromycin on MHC I‐peptide complex recovery in TRx9‐expressing cells. Cells
transfected with the TRx9 plasmid were stripped of MHC I peptides and allowed to
recover in the presence of varying concentrations of puromycin from 60 to 180 minutes.
Sixty minutes was chosen as the initial time point for measurement during the recovery,
as this is the approximate timeframe for protein degradation, peptide loading, and Kb‐
SIINFEKL export to the cell surface (Qian et al. 2006b). Flow cytometry was used to
measure functional reporter protein (by eGFP fluorescence, Fig. 15) as well as cell
surface Kb‐SIINFEKL complexes (Fig. 16) and total Kb (Fig. 17).
Reporter eGFP signal in untreated cells increased steadily throughout the
recovery, reflecting the continued synthesis of reporter protein during this interval (Fig.
15). Ninety minutes into the recovery, cells treated with either 2 M or 20 M puromycin
showed decreased eGFP signal relative to untreated controls, causing the signal to
plateau over the final 90 minutes. This plateau in eGFP signal could reflect: 1)
production of truncated, non‐fluorescent reporter protein, 2) increased reporter
degradation, or 3) impaired protein synthesis. All of these interpretations would be
consistent with the effects of puromycin.

74
Figure 15: Effects of puromycin on the expression of functional fluorescent reporter
protein. 293‐Kb cells expressing the TRx9 reporter were stripped of cell surface MHC I
peptides as in Fig. 12. Recovery of cell surface MHC class I‐peptide complexes was
conducted in the presence of varying concentrations of puro from 0 to 180 minutes. Flow
cytometry was used to quantitate reporter eGFP fluorescence as a proxy measure of
natively folded, functional reporter protein (n = 5; mean ± s.e.m.) The data presented in
this figure was contributed by Odessa Marks and Joshua Lacsina, in experiments
designed by Joshua Lacsina.
All samples showed substantial generation of new Kb‐SIINFEKL complexes
during the recovery, decelerating as the recovery progressed. To more clearly ascertain
the effects of puromycin Kb‐SIINFEKL recovery, the raw data (Fig. 16A) for the
puromycin‐treated cells was normalized to control (untreated) cells (Fig. 16B) as
described in (Qian et al. 2006b). Treatment with 2 M puromycin caused no significant
change in the recovery of cell surface Kb‐SIINFEKL complexes. In contrast, treatment
with 20 M puromycin elicited an initial increase in Kb‐SIINFEKL levels at 60 minutes,

75
Figure 16: Effects of puromycin on the recovery of cell surface Kb‐SIINFEKL
complexes. (A) 293‐Kb cells expressing the TRx9 reporter were stripped of cell surface
MHC I peptides and allowed to recover in the presence of varying concentrations of
puro from 0 to 180 minutes. Cells were stained with 25‐D1.16 to measure cell surface Kb‐
SIINFEKL complexes by flow cytometry. (B) To quantitate differences in the kinetics of
MHC class I‐peptide complex recovery, the MFI values of puro‐treated cells were
normalized to untreated cells as described in (Qian et al. 2006b) (n = 5; mean ± s.e.m.)
* p < 0.05 versus untreated controls. The data presented in this figure was contributed by
Odessa Marks and Joshua Lacsina, in experiments designed by Joshua Lacsina.
followed by a constant decline in the rate of Kb‐SIINFEKL presentation. These findings
suggest that the initial burst of puromycin‐induced drop‐off products caused an increase
in MHC I presentation, but that sustained production of truncated polypeptides
inhibited the flux of reporter‐derived peptides into the MHC I presentation pathway.
To generalize these findings to MHC I‐peptide complexes other than Kb‐
SIINFEKL, a similar experiment was conducted to measure total cell surface Kb recovery
in the presence of varying concentrations of puromycin (Fig. 17A). Data from
puromycin‐treated cells were normalized to the untreated samples, as before (Fig. 17B).

76
Figure 17: Effects of puromycin on the recovery of total cell surface Kb. (A) 293‐Kb cells
expressing the TRx9 reporter were stripped of cell surface MHC I peptides and allowed
to recover in the presence of varying concentrations of puro from 0 to 180 minutes. Cells
were stained with anti‐Kb antibody and analyzed by flow cytometry. (B) MFI values of
puro‐treated cells were normalized to untreated cells as in Figure 16B (n = 5; mean ±
s.e.m.) The data presented in this figure was contributed by Odessa Marks and Joshua
Lacsina, in experiments designed by Joshua Lacsina.
After acid stripping, cells displayed a time‐dependent loss of cell surface Kb molecules
(Fig. 17A). Because MHC I molecules lacking bound peptide show decreased cell surface
stability (Leonhardt et al. 2010), the loss of cell surface Kb after acid stripping reflects the
shedding, unfolding, and internalization of destabilized Kb molecules, which has been
observed previously (Qian et al. 2006b). Puromycin treatment had no discernible effect
on Kb surface presentation during the first 90 minutes of recovery. Treatment with 2 M
puromycin led to accelerated delivery of Kb to the cell surface from 120 to 180 minutes, a
trend reaching statistical significance at the 180 minute time point (Fig. 17B). Cells
treated with 20 M puromycin showed a trend towards impaired Kb surface

77
presentation from 120 minutes onwards, though this did not reach statistical
significance. The Kb recovery studies indicate that translational drop‐off products
induced by low concentrations of puromycin stimulate MHC class I presentation,
whereas at higher concentrations, puromycin treatment impairs the MHC class I
pathway. I speculate that the contrasting effects of puromycin on Kb‐SIINFEKL
compared to Kb recovery can be attributed to differences in the efficiency of epitope
processing from truncated products of the TRx9 reporter versus the global population of
precursors for all Kb‐binding peptides.
A consistent observation in the analysis of cell surface presentation of Kb‐
SIINFEKL and Kb is that higher concentrations of puromycin elicited a decrease in the
rate of cell surface MHC class I presentation, particularly late in the recovery period. To
extend these observations, a similar acid stripping/recovery experiment was performed
in the presence of increasing concentrations of puromycin. After 240 minutes of
recovery, eGFP, Kb‐SIINFEKL, and Kb were measured by flow cytometry (Fig. 18).
Reporter eGFP fluorescence was unchanged up to 0.2 M puromycin, followed by a
dose‐dependent decrease over the remaining concentration range. Similar to the data in
Fig. 15, the eGFP profile is consistent with dose‐dependent production of truncated,
non‐fluorescent reporter protein, increased reporter degradation, or decreased reporter
synthesis.
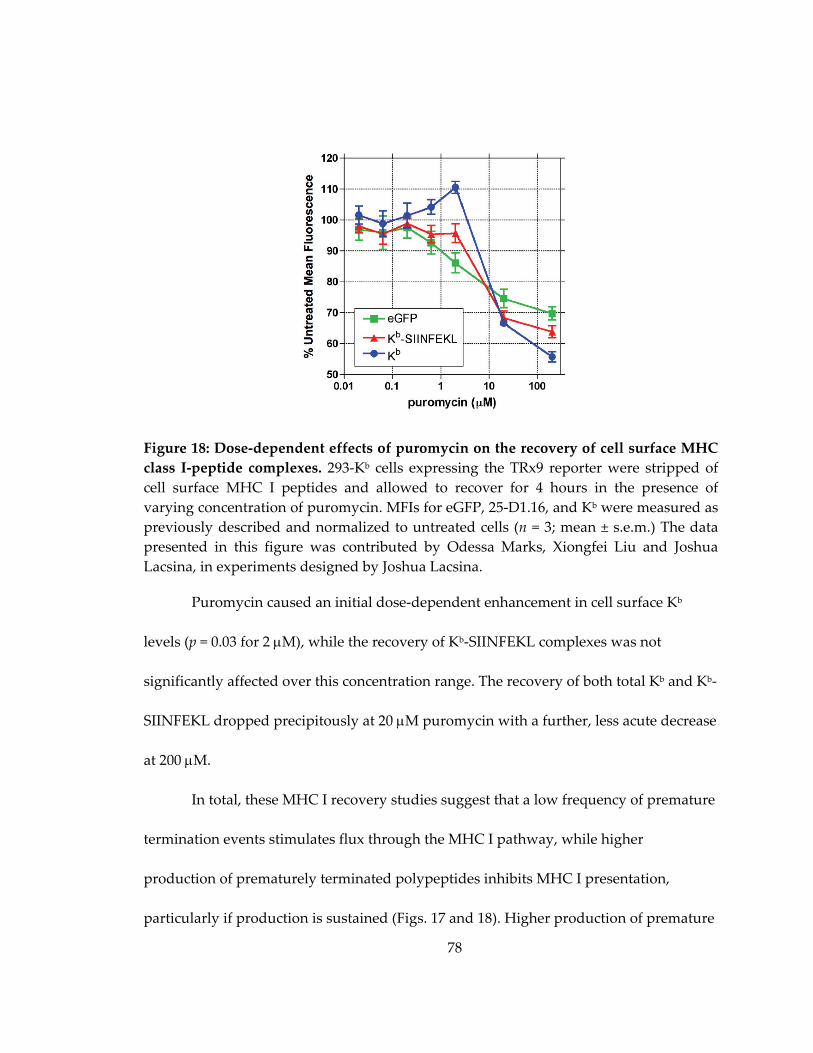
78
Figure 18: Dose‐dependent effects of puromycin on the recovery of cell surface MHC
class I‐peptide complexes. 293‐Kb cells expressing the TRx9 reporter were stripped of
cell surface MHC I peptides and allowed to recover for 4 hours in the presence of
varying concentration of puromycin. MFIs for eGFP, 25‐D1.16, and Kb were measured as
previously described and normalized to untreated cells (n = 3; mean ± s.e.m.) The data
presented in this figure was contributed by Odessa Marks, Xiongfei Liu and Joshua
Lacsina, in experiments designed by Joshua Lacsina.
Puromycin caused an initial dose‐dependent enhancement in cell surface Kb
levels (p = 0.03 for 2 M), while the recovery of Kb‐SIINFEKL complexes was not
significantly affected over this concentration range. The recovery of both total Kb and Kb‐
SIINFEKL dropped precipitously at 20 M puromycin with a further, less acute decrease
at 200 M.
In total, these MHC I recovery studies suggest that a low frequency of premature
termination events stimulates flux through the MHC I pathway, while higher
production of prematurely terminated polypeptides inhibits MHC I presentation,
particularly if production is sustained (Figs. 17 and 18). Higher production of premature

79
termination products at 20 M puromycin only stimulated MHC I presentation at the
earliest time point after acid stripping, followed by progressive inhibition of the MHC I
pathway over time (Fig. 16).
4.3.4 Inducing premature translational termination promotes the accumulation of polyubiquitinated proteins
If RDPs are the primary source of MHC class I peptides, why is MHC class I
presentation inhibited under experimental conditions that produce high levels of
prematurely terminated polypeptides? One possibility is that the increase in truncated
polypeptides saturates the proteasome with RDP pathway substrates. To test this, cells
were treated with varying concentrations of puromycin and the amount of
polyubiquitinated protein was determined by Western blotting using the FK2
monoclonal antibody, which is specific for mono‐ and polyubiquitin‐conjugated proteins
(Fujimuro et al. 1994). CHX was again used for comparison.
As a positive control, inhibition of the proteasome with MG132 caused a
dramatic increase in polyubiquitinated proteins (Fig. 19), which migrated as a high
molecular weight smear by Western blotting, as observed previously (Schubert et al.
2000). (The lack of signal in the molecular weight range >260 kDa reflects the poor ability
of the highest molecular weight polypeptides to transfer from the gel to the membrane.)
Treatment with CHX resulted in a dose‐dependent decrease in polyubiquitinated
proteins, due to CHX preventing nascent polypeptide dissociation from the ribosome.

80
Figure 19: Puromycin treatment leads to increased levels of polyubiquitinated
proteins. 293‐Kb cells were treated for 4 hours with media alone, 20 M MG132, or the
indicated concentrations of CHX and puro (M). Lysates were subjected to Western
blotting with FK2 (upper panel), a monoclonal antibody specific for mono‐ and
polyubiquitinated protein conjugates. ‐tubulin was probed as a loading control (lower
panel). Results are representative of three independent experiments. The data presented
in this figure was contributed by Xiongfei Liu, in experiments designed by Joshua
Lacsina and Xiongfei Liu.
In contrast, treating cells with 0.02 to 20 M puromycin resulted in the accumulation of
polyubiquitinated proteins in a dose‐dependent manner, consistent with proteasomal
saturation. In further support of this interpretation, this increase was not observed
following treatment with 200 M puromycin, which produces premature termination
products that are too short (only a few amino acids long) to serve as proteasomal
substrates. These results highlight a correlation between increased production of
prematurely terminated polypeptides, proteasomal inhibition, and impaired MHC I
presentation.

81
4.4 RNA interference-mediated knockdown of candidate RDP factors increases flux through the RDP pathway
In this section, I present my early studies using RNA interference to test the
putative roles for two candidate factors in the RDP pathway: Pth2 and CHIP.
4.4.1 Pth2
The physiologic correlates of puromycin‐induced premature termination
products are peptidyl‐tRNAs, which dissociate prematurely from the ribosome during
elongation. In E. coli, peptidyl‐tRNAs are produced as a normal by‐product of protein
synthesis and are metabolized by the enzyme, peptidyl‐tRNA hydrolase (Pth), which
cleaves the carboxylic ester bond between the peptide moiety and the tRNA (Menninger
1976). Deletion of Pth results in the toxic accumulation of peptidyl‐tRNAs, with similar
peptidyl‐tRNA hydrolase activity being reported in archaeal species from a structurally
unrelated protein family, Pth2 (Rosas‐Sandoval et al. 2002; Fromant et al. 2003). While
peptidyl‐tRNA drop‐off has not been reported in eukaryotes, homologs of both Pth and
Pth2 family members are conserved in humans, with human Pth2 showing greater
catalytic activity than Pth (de Pereda et al. 2004).
The studies described above suggested that Pth2 could play a role in the
metabolism of prematurely terminated polypeptides in the RDP pathway. To test this, I
examined the effects of siRNA‐mediated knockdown of Pth2 on the RDP pathway.
Treatment of cells with Pth2‐targeted siRNAs produced a ~90% knockdown in Pth2
protein expression (Fig. 20A). I then sought to determine the effects of Pth2 knockdown

82
Figure 20: Knockdown of Pth2 by RNA interference increases the fraction of RDPs.
293‐Kb cells were transfected with either a non‐targeting control siRNA or Pth2‐specific
siRNA, and assayed 72 hours later. (A) Western blot for Pth2 (upper panel) and ‐tubulin as a loading control (lower panel). Knockdown efficacy was measured with ImageJ.
Results are representative of three independent experiments. (B) Control and Pth2
knockdown cells were pulse labeled for 5 min with [35S]‐Met and 20 M puro +/‐ 20 M MG132, then chased from 0 to 50 minutes in the presence of excess cold methionine,
CHX and +/‐ 20 M MG132. The chase was terminated at the indicated time points by
the addition of TCA to cell suspensions to precipitate polypeptides. TCA precipitates
were solubilized and [35S] incorporation was measured by liquid scintillation counting (n
= 3; mean ± s.e.m.) (C) Control and Pth2 knockdown cells were stained for cell surface Kb
molecules and fluorescence was measured by flow cytometry. Isotype control (dashed
line), control siRNA (thin line) and Pth2 siRNA (thick line) are shown. Results are
representative of five independent experiments.

83
on the metabolism of prematurely terminated polypeptides. Control and Pth2
knockdown cells were subjected to a pulse‐chase with [35S]‐Met in the presence or
absence of MG132 in order to measure the RDP fraction (Qian et al. 2005). During the
pulse, cells were treated with 20 M puromycin to induce the production of truncated
polypeptides.
In control siRNA‐transfected cells, there was minimal loss of radiolabel by the
beginning of the chase. (This differs from the findings reported in Fig. 7, in which
puromycin treatment resulted in 25% loss of radiolabel by the start of the chase. The
different degradation profiles observed in the following experiments may be due to the
non‐specific effects of transfection or siRNAs, such as low level activation of stress
pathways (Szeto et al. 2006).) In contrast, in Pth2 knockdown cells, 29% of the [35S] signal
was already lost by the beginning of the chase. The control siRNA samples showed a
biphasic RDP degradation profile, with a sharp decrease in signal from 0 to 10 minutes,
followed by a shallower second decay phase. Pth2 knockdown cells lacked this sharp
decay phase during the chase (as it occurred during pulse labeling), and showed a near‐
linear degradation profile similar to the slower rate of loss seen in controls. By the end of
the chase, the Pth2 knockdown samples exhibited a 39% increase in the proteasome‐
sensitive fraction relative to controls. Both ctrl and Pth2 knockdown cells exhibited
similar MG132‐resistant degradation profiles, indicating that the MG132‐resistant

84
degradation pathway observed previously (Fig. 7) is unaffected by Pth2 function. These
findings are consistent with Pth2 inhibiting flux through the RDP pathway.
If Pth2 indeed antagonizes the RDP pathway, one prediction of this model is that
Pth2 knockdown should increase peptide presentation via the MHC class I pathway. To
test this prediction, I compared the level of cell surface Kb molecules on control and Pth2
siRNA‐transfected 293‐Kb cells by flow cytometry (Fig. 20C). Pth2 knockdown resulted
in a 16% increase in Kb signal relative to control siRNA‐transfected cells, supporting the
idea that Pth2 inhibits flux through the RDP pathway.
4.4.2 CHIP
CHIP (Carboxyl terminus of Hsp70‐Interacting Protein) is a key regulator of
cellular proteostasis, such that CHIP knockout mice show marked reductions in lifespan,
accelerated aging, accelerated cellular senescence, and impairments in protein quality
control (Ballinger et al. 1999; Min et al. 2008). CHIP is unique in that it functions as a
chaperone, cochaperone, and E3 ubiquitin ligase (Rosser et al. 2007), and acts by
recognizing and directly ubiquitinating unfolded chaperone substrates (Connell et al.
2001; Meacham et al. 2001; Petrucelli et al. 2004; Qian et al. 2006c). The ability of CHIP to
directly recognize and ubiquitinate unfolded polypeptides suggests that CHIP could
play a role in the metabolism of RDPs, particularly prematurely terminated
polypeptides (which are likely to be unfolded).

85
Figure 21: Knockdown of CHIP by RNA interference increases the fraction of RDPs.
293‐Kb cells were transfected with either a non‐targeting control siRNA or CHIP‐specific
siRNA, and assayed 72 hours later. (A) Western blot for CHIP (upper panel) and ‐tubulin as a loading control (lower panel). Results are representative of three independent
experiments. (B) Control and CHIP knockdown cells were subjected to pulse‐chase as
described in Fig. 20. [35S] incorporation was measured by liquid scintillation counting (n
= 3; mean ± s.e.m.) (C) Control and CHIP knockdown cells were stained for cell surface
Kb molecules and fluorescence was measured by flow cytometry. Isotype control
(dashed line), control siRNA (thin line) and CHIP siRNA (thick line) are shown. Results
are representative of three independent experiments.

86
I tested the effects of CHIP knockdown on the RDP pathway in puromycin‐
treated cells. Transfection of cells with CHIP‐specific siRNAs resulted in an 85%
decrease in CHIP protein (Fig. 21A). RDP degradation was then compared in
puromycin‐treated CHIP knockdown and control cells using the same pulse‐chase
conditions as in Fig. 20 (Fig. 21B). In CHIP knockdown cells, 18% of radiolabeled
polypeptides were degraded in a proteasome‐dependent manner by the start of the
chase, indicating increased degradation of RDP pathway substrates. The rapid
degradation kinetic in CHIP knockdown cells was not as steep as in control cells, but
was prolonged an extra 10 minutes, suggesting saturation of the degradation pathway.
By the end of the chase, CHIP knockdown resulted in a 26% increase in the proteasome‐
dependent RDP fraction. CHIP knockdown cells also showed a trend towards delayed
MG132‐resistant degradation, though the fraction of polypeptides degraded by this
pathway was similar to control cells by the end of the chase. These data suggest that
CHIP antagonizes flux through the RDP pathway. To determine whether CHIP
knockdown affects downstream MHC class I presentation, the levels of cell surface Kb on
control and CHIP knockdown cells were compared by flow cytometry (Fig. 21C). CHIP
knockdown resulted in a 25% increase in Kb surface expression, indicating that CHIP has
an inhibitory effect on the MHC class I pathway.

87
4.5 Summary
In this chapter, I examined the effects of stimulating premature translational
termination on the MHC class I presentation pathway. Using an antigenic reporter to
measure both functional protein expression and peptide presentation (Fig. 14A), I report
time‐ and concentration‐dependent effects of puromycin on the generation of MHC class
I‐peptide complexes (Figs. 16‐18). Low concentrations of puromycin stimulated MHC
class I presentation. At concentrations shown in the previous chapter to cause robust
production of RDPs, puromycin initially stimulated, then progressively inhibited the
export of MHC class I molecules presenting reporter peptide. Overall, higher levels of
premature termination products were associated with inhibition of the MHC class I
pathway, particularly when truncated polypeptide production was sustained.
Furthermore, I report an association between impaired MHC class I presentation, high
levels of truncated polypeptides, increases in polyubiquitinated proteins (Fig. 19), and
cell death (Fig. 12). Together, these data suggest diverse responses of the MHC class I
pathway to premature translational termination products, reflecting the heterogeneous
characteristics of truncated polypeptides. The results also suggest the presence of
homeostatic mechanisms in cells to cope with prolonged production of premature
translational termination products. Finally, I report preliminary evidence that the
putative RDP pathway factors Pth2 and CHIP inhibit flux through the RDP pathway
(Figs. 20‐22).

88
5. Discussion
5.1 Summary of primary findings and overview
In my studies, I have shown that premature translational termination products
induced by puromycin treatment are substrates for the rapidly degraded polypeptide
pathway. In Chapter 3, I described the biochemical experiments demonstrating the rapid
degradation of prematurely terminated polypeptides. These findings support the
proposal that prematurely terminated polypeptides represent a general subclass of RDP
pathway substrates, an idea put forth in the original publication of the DRiP hypothesis
fifteen years ago (Yewdell et al. 1996) and revisited recently (Dolan et al. 2011b). I also
reported that after puromycin treatment, a fraction of RDPs were degraded via an
MG132‐resistant mechanism which predominantly affected small polypeptides. This
finding highlights the importance of previously unappreciated RDP degradation
pathways which are insensitive to small molecule inhibitors of the proteasome.
Because RDPs are the primary source of MHC class I peptides (Schubert et al.
2000; Reits et al. 2000; Qian et al. 2006b), in Chapter 4, I investigated the effects of
stimulating premature translational termination on the MHC class I presentation
pathway. I reported that MHC I presentation was modestly stimulated by conditions
producing low concentrations of prematurely terminated polypeptides, as originally
predicted. Interestingly, sustained production of prematurely terminated polypeptides
correlated with the inhibition of MHC I presentation, the accumulation of

89
polyubiquitinated proteins and increased cell death. These results indicate that the
relationship between RDP production and MHC I presentation is not monotonic,
thereby highlighting heterogeneity in the efficiency which with RDP pathway substrates
can access the MHC class I pathway.
In this chapter, I will discuss the following: 1) Potential mechanisms for the rapid
degradation of prematurely terminated polypeptides. 2) Possible mechanisms for the
MG132‐resistant degradation of low molecular weight RDPs. 3) Models for the time‐ and
concentration‐dependent effects of stimulating premature termination on MHC class I
presentation. 4) Candidate factors for RDP metabolism. I will conclude with some brief
thoughts on the significance of this work to studies of human health.
5.2 Rapid degradation of prematurely terminated polypeptides
While premature translational termination has not been reported in eukaryotes,
prematurely terminated polypeptides are generated as highly unstable, normal by‐
products of protein synthesis in prokaryotic cells (Menninger 1976; Manley 1978; Tsung
et al. 1989; Jørgensen and Kurland 1990). In Chapter 3, I used puromycin to induce the
premature dissociation of nascent polypeptides from ribosomes as peptidyl‐puromycin
adducts. Stimulating premature termination with puromycin led to a doubling of the
fraction of rapidly degraded polypeptides (Fig. 7). By purifying peptidyl‐puromycin
truncation products using anti‐puromycin antisera (Fig. 9A) (McCallum et al. 2000), I
determined that 70% of the prematurely terminated polypeptides were targeted for

90
rapid degradation (Fig. 10). Truncated polypeptide degradation was largely
proteasome‐dependent, although ~35% of the degradation occurred via an MG132‐
resistant mechanism (discussed in the next section). While the truncated polypeptides
produced in my system are likely to be highly defective, both in conformation and
function, the exact molecular signals that target peptidyl‐puromycins for rapid
degradation are unknown.
Since RDPs have a characteristic half‐life of ~10 minutes (Princiotta et al. 2003),
RDPs would be expected to be almost completely degraded by the end of a 50 minute
chase. If this is the case, what are the 30% of peptidyl‐puromycins that remain at the end
of the chase (Fig. 10)? Immunoprecipitation of the peptidyl‐puromycins revealed a
heterogeneous profile of polypeptides (Fig. 9A). Given the diversity in the peptidyl‐
puromycin pool, it is likely that a subpopulation of truncated products possesses the
characteristics to be directed to the stable protein pool. In puromycin‐treated cells, small
polypeptides (representing premature termination products) were particularly sensitive
to rapid degradation, as determined by SDS‐PAGE (Fig. 8). Based on these findings, I
propose that the peptidyl‐puromycins which resist rapid degradation are a population
of relatively longer truncated products.
The absolute length of the truncated polypeptide is unlikely to be the
determining factor for triage between the RDP and stable protein pools. Rather, the
observed “size dependence” likely reflects the higher probability for longer

91
polypeptides to contain folded domains, thereby decreasing the exposure of
hydrophobic regions that would predispose the protein to degradation (Gilon et al. 1998;
Johnson et al. 1998; Arteaga et al. 2006). This model is supported by a recent study in
which a hydrophobic stretch of amino acids was inserted into a model antigenic protein.
The hydrophobic region functioned as a dose‐dependent degradation motif which
stimulated the presentation of antigen‐derived peptides on MHC I (Huang et al. 2011b).
Similarly, I propose that exposed hydrophobic regions in the truncated polypeptides
serve to target them for rapid degradation.
It is formally possible that the puromycin moiety itself serves as a rapid
degradation signal, though it is unclear what cellular mechanisms would have evolved
to recognize puromycin. Accessibility of the puromycin molecule may vary between
substrates, depending on the folding state of the truncated polypeptide. This would be
consistent with longer polypeptides containing more folded domains being able to
sterically conceal the puromycin moiety and escape the RDP pathway. (As I shall
discuss in a later section, peptidyl‐tRNA hydrolase 2, which might be expected to
recognize peptidyl‐puromycins and target them for rapid degradation, was found to
exert the opposite effect.)
A related question is the extent to which peptidyl‐puromycin degradation
depends on polyubiquitination. While the immunoprecipitated peptidyl‐puromycins
had an average molecular weight of 19 kDa (Fig. 9), the average weight of

92
polyubiquitinated proteins that accumulated in response to puromycin treatment was
~190 kDa (Fig. 19). This suggests that the majority of peptidyl‐puromycins we observed
were not directly polyubiquitinated, raising the possibility that the prematurely
terminated products were degraded by the 20S proteasome via a ubiquitin‐independent
mechanism. This is consistent with a previous report demonstrating that 25% of RDPs
(~7% of all proteins synthesized) are degraded in a ubiquitin‐independent manner (Qian
et al. 2006a). These ubiquitin‐independent RDPs are insoluble in the non‐ionic detergent
Triton X‐100 and insensitive to the cytosolic chaperone Hsc70, suggesting this RDP pool
is composed of severely misfolded polypeptides, as would be expected for a substantial
fraction (if not the majority) of peptidyl‐puromycins.
A different series of experiments using a dominant negative mutant of ubiquitin
lacking lysine residues demonstrated that MHC class I peptides can be processed and
presented from cytosolic antigens independently of ubiquitin (Huang et al. 2011a). The
authors of the latter study suggest that cytosolic RDPs derive from nascent polypeptides
that fail to be intercepted by the cellular folding machinery, and are instead immediately
directed to the 20S proteasome for ubiquitin‐independent degradation (Eisenlohr et al.
2007). By this model, the puromycin‐stimulated production of truncated polypeptides
likely leads to acute saturation of the cellular folding machinery. The excess truncated
polypeptides would then be targeted for rapid degradation in 20S proteasomes.

93
In summary, the peptidyl‐puromycin pool generated in my studies is
heterogeneous. While the majority of these truncated products are RDPs, a minority,
likely representing partially or fully folded polypeptides, is targeted to the stable protein
pool. I propose that hydrophobicity or the puromycin moiety may serve as signals to
target peptidyl‐puromycins to the RDP pathway. Finally, I suggest that peptidyl‐
puromycin degradation occurs in a largely ubiquitin‐independent manner via the 20S
proteasome, which has been proposed to degrade severely misfolded or unchaperoned
RDPs.
5.3 MG132-resistant degradation of low molecular weight RDPs
The ubiquitin‐proteasome system is the main pathway responsible for cytosolic
proteolysis (Hochstrasser 1996; Hershko and Ciechanover 1998; Pickart and Cohen
2004). For this reason, studies of the RDP pathway have largely focused on proteasome‐
dependent degradation (Schubert et al. 2000; Qian et al. 2005; Qian et al. 2006a), which
was measured using small molecule inhibitors of the proteasome (Rock et al. 1994).
Comparatively little attention has been paid to non‐proteasomal pathways for RDP
degradation. In my studies of truncated polypeptide degradation, I observed 20% of
peptidyl‐puromycins were degraded in the presence of the proteasome inhibitor,
MG132. (Fig. 10) This accounted for 35% of the total fraction of peptidyl‐puromycins
targeted for rapid degradation. Examining the polypeptide profiles of puromycin‐
treated cells revealed that low molecular weight polypeptides (<17 kDa) were

94
particularly susceptible to rapid degradation, even in the presence of MG132 (Fig. 8B).
Some earlier studies of RDPs show rapid polypeptide degradation in MG132‐treated
cells (Qian et al. 2005), though this was not noted or commented on. Do these results
indicate that small, truncated polypeptides are degraded by a proteasome‐independent
RDP pathway? And do these alternative degradation mechanisms provide substrates for
the MHC class I pathway?
5.3.1 The controversies of “proteasome-independent” degradation
Following the initial application of small molecule proteasome inhibitors to
studies of the MHC class I pathway, evidence emerged for the continued presentation of
a subset of MHC I‐peptide complexes in cells treated with these inhibitors. The MHC
class I alleles HLA‐A3, HLA‐A11, and HLA‐B35 were found to load peptides and
undergo normal maturation in cells subjected to very high inhibitor concentrations
(Benham et al. 1998). Two different studies demonstrated that treatment with
proteasome inhibitors actually stimulated the presentation of peptides derived from
influenza A virus proteins (Anton et al. 1998; Luckey et al. 1998). Finally, relatively
efficient MHC class I loading was observed in cells adapted to long‐term growth in the
presence of covalent inhibitors of the proteasome (Glas et al. 1998). This body of
evidence was interpreted as signifying the presence of a proteasome‐independent
mechanism to degrade substrates for MHC class I presentation.

95
A re‐examination of the evidence suggests alternative explanations for the
maintenance of MHC class I presentation in the presence of “maximal” proteasomal
inhibition (van Endert 2011). The common proteasome inhibitors epoxomicin and
MG132 (with MG132 being used in my studies) primarily inhibit the chymotrypsin‐like
activity of the proteasome, while leaving the trypsin‐like and caspase‐like activities
intact. The consequence of this altered cleavage specificity is a shift in the profile of
proteasomal degradation products, leading to the generation of new MHC class I
epitopes and the destruction of others (Gavioli et al. 2002; Wherry et al. 2006), mirroring
the findings cited as evidence of proteasome‐independent antigen processing .
Additionally, long‐term treatment with proteasome inhibitors has been shown to
activate compensatory pathways that increase the expression of proteasomal subunits in
a variety of cellular contexts (Naujokat et al. 2007; Mitsiades et al. 2002; Meiners et al.
2003; Rückrich et al. 2009). Indeed, it was later shown that the originally described
proteasome inhibitor‐adapted cells actually expressed functional proteasomes as a result
of this compensatory pathway (Princiotta et al. 2001).
With these caveats about the interpretation of experiments using proteasome
inhibitors, I propose that the MG132‐resistant degradation of peptidyl‐puromycins may
reflect 1) residual proteasomal activity or 2) MG132‐induced alterations in proteasomal
substrate specificity. It is notable that MG132‐resistant degradation was only apparent in
puromycin‐treated cells, and predominantly affected small polypeptides. This suggests

96
that MG132 binding results in increased selectivity for small polypeptides, perhaps due
to partial steric occlusion of the interior chamber of the proteasome.
5.3.2 Non-proteasomal proteases
Although MG132 does not completely inactivate the proteasome, the size
selectivity of MG132‐resistant degradation prompted me to identify non‐proteasomal
proteases that may contribute to the observed degradation of peptidyl‐puromycins.
One well‐known non‐proteasomal protease is tripeptidyl peptidase II, which was
originally identified as an enzyme that compensates for loss of proteasome function in
cells treated with proteasome inhibitors for extended periods (Geier et al. 1999). TPP II
cleaves tripeptides from the amino terminus of proteins with little sequence specificity,
and also exhibits weak trypsin‐like endoprotease activity. The lack of sequence
specificity may allow TPP II to metabolize a range of substrates that are highly
heterogeneous in composition and characteristics, such as prematurely terminated
polypeptides. The endogenous substrates for TPP II remain unknown (van Endert 2011).
While TPP II was originally suggested to generate MHC class I‐binding peptides (Geier
et al. 1999), more recent evidence from mouse models indicates that TPP II deficiency
leads to increased MHC class I presentation (Firat et al. 2007; Kawahara et al. 2009),
indicating that the net effect of TPP II is to destroy potential epitopes in vivo.
Insulin‐degrading enzyme (IDE) is a promising candidate protease for peptidyl‐
puromycin degradation, because of its specificity for substrates <12 kDa in size (in

97
addition to insulin) (Duckworth et al. 1998). IDE was found to generate a proteasome‐
independent HLA‐A1‐restricted peptide derived from the MAGE‐A3 human tumor
protein (Parmentier et al. 2010). Interestingly, IDE is specific for substrates bearing
amyloid‐prone beta sheet conformations, rather than targeting specific amino acid
sequences (Kurochkin 2001). While it is unclear whether peptidyl‐puromycins tend to
fold into amyloidogenic structures, it is known that puromycin treatment results in the
formation of structures called ALIS, which resemble aggresomes (Szeto et al. 2006).
Thus, IDE has a number of characteristics that make it well‐suited to metabolize
peptidyl‐puromycins and prevent them from accumulating as toxic aggregates.
Finally, a study of the HLA‐B27‐binding peptide repertoire suggested the
existence of a proteasome‐independent degradation pathway (Marcilla et al. 2007).
Using quantitative mass spectrometry, the investigators demonstrated continued
production of one‐third of the original repertoire of HLA‐B27‐peptide complexes in the
presence of epoxomicin. Interestingly, these peptides were specifically derived from
small (6‐16.5 kDa), basic polypeptides, and were unlikely to be generated from the
residual caspase‐like activity of the epoxomicin‐bound proteasome. The degradation
mechanism exhibiting this restricted substrate profile remains to be identified. The
activity may represent residual proteasome function or a yet‐unidentified protease. In
either case, a proteolytic pathway targeting small polypeptides is consistent with the
MG132‐resistant degradation of truncated polypeptides observed in my studies.

98
In summary, I present a series of different models that explain the MG132‐
resistant degradation of peptidyl‐puromycins: 1) MG132 biases the substrate specificity
of the proteasome towards the degradation of small polypeptides. 2) Peptidyl‐
puromycins are degraded via TPP II, which has a broad range of potential substrates. 3)
Peptidyl‐puromycins are degraded by IDE, which is specific for small, amyloidogenic
polypeptides. 4) Peptidyl‐puromycins are degraded by a yet uncharacterized protease
specific for small, basic proteins that is involved in the generation of HLA‐B27‐binding
epitopes. Interactions between different pathways are also possible; for example,
cleavage by non‐proteasomal proteases may require the altered activity of the MG132‐
bound proteasome, or vice‐versa. Distinguishing between these models will be
important for understanding the mechanisms by which small prematurely terminated
polypeptides are degraded.
5.4 Relationship between premature translational termination products and MHC class I presentation
RDPs are the primary source of peptides presented on MHC class I molecules
(Schubert et al. 2000; Reits et al. 2000; Qian et al. 2006b). As described in Chapter 3, I
found that prematurely terminated peptidyl‐puromycins are predominantly targeted to
the RDP pool. In Chapter 4, I proceeded to examine the effects of puromycin‐induced
premature translational termination on MHC class I presentation. Puromycin
concentrations inducing low levels of truncated polypeptide production stimulated
MHC class I presentation. In contrast, puromycin concentrations inducing high levels of

99
premature termination products inhibited the MHC class I pathway, particularly after
extended time periods. Given that RDP degradation is so tightly coupled to MHC I
presentation, why do rapidly degraded peptidyl‐puromycins stimulate MHC I
presentation under some conditions and inhibit presentation under others? In this
section, I will introduce three models to account for the time‐ and concentration‐
dependent effects of puromycin on the MHC class I pathway. I will also review my
findings in the context of previous studies using puromycin to study the MHC class I
pathway.
5.4.1 Models of MHC class I behavior in response to puromycin treatment
5.4.1.1 mTORC1 model
The first model discusses how the MHC class I pathway responds to regulatory
feedback between protein quality control and protein synthesis. Qian and colleagues
reported a mechanism by which mTORC1 senses changes in protein quality via the
occupancy of chaperones (Qian et al. 2010). I adapted their model of this regulatory
pathway to explain my findings as follows (Fig. 22).
Low concentrations of puromycin stimulate the production of small quantities of
prematurely terminated polypeptides of a diverse range of lengths, many of which are
long enough to contain one or more folded domains. This profile of truncated

100
Figure 22: The mTORC1 model. Low concentrations of peptidyl‐puromycins cause
moderate decreases in chaperone availability, resulting in mTORC1 activation and
promoting MHC class I presentation. High levels of severely truncated peptidyl‐
puromycins cause acute depletion of chaperones, resulting in mTORC1 inhibition by
aggregation. Downstream MHC class I levels are impaired.
polypeptides exerts a mild stress on the protein folding machinery, leading to moderate
reductions in chaperone availability. The modest decrease in available chaperones
activates mTORC1 via Hsp90‐mediated assembly of the full mTORC1 complex.
Activated mTORC1 phosphorylates S6K1, which in turn, phosphorylates the ribosomal
S6 protein, leading to the upregulation of both protein synthesis (Fig. 3) and the
downstream supply of substrates to the MHC class I pathway.
High concentrations of puromycin stimulate the production of short, truncated
polypeptides which are predominantly unfolded. The flood of unfolded polypeptides
depletes the supply of available chaperones. Shortly after puromycin treatment,
mTORC1 is briefly activated, leading to the initial burst in Kb‐SIINFEKL presentation
(Fig. 16B). However, the sequestration of Hsp90 by truncated polypeptides eventually
causes mTOR to aggregate and fail to form functional mTORC1. This results in

101
decreased protein synthesis and the observed impairment in MHC class I presentation at
high puromycin concentrations and later time points.
This model highlights the potential importance of protein quality‐mediated
regulation of translation as a determinant of MHC class I presentation. In that context,
puromycin allows me to manipulate protein quality and quantity to explore the
dynamic range of responses to different protein quality environments.
5.4.1.2 Aggregation model
The second model focuses on the potential for prematurely terminated
polypeptides to aggregate and thereby inhibit the proteasome. Low concentrations of
puromycin produce a small amount of truncated polypeptides of diverse sizes, many of
which will contain folded domains to prevent aggregation. These truncated
polypeptides are targeted for rapid degradation, leading to the stimulation of MHC class
I presentation. In contrast, high concentrations of puromycin produce a large number of
short, unfolded, prematurely terminated polypeptides. These are also rapidly degraded,
leading to the observed early burst in Kb‐SIINFEKL presentation (Fig. 16B). However,
the high volume of truncated polypeptides saturates the proteasome, which has little
excess capacity (Gidalevitz et al. 2006), resulting in the accumulation of
polyubiquitinated proteins (Fig. 19). Proteasomal saturation results in the accumulation
of largely unfolded peptidyl‐puromycins, leading to the formation of aggregates that are

102
toxic to cells over time (Fig. 12). Aggregate accumulation inhibits proteasomal activity
(Bence et al. 2001; Bennett et al. 2005), thereby decreasing MHC class I presentation.
This model highlights the consequences of prematurely terminated peptides on
protein homeostasis and the time‐dependent effects of disruptions to proteostasis on
degradation pathways, antigen presentation, and cell viability.
5.4.1.3 Substrate heterogeneity model
The final model focuses on the intrinsic heterogeneity of the prematurely
terminated polypeptide pool, and proposes a mechanism for how truncations of
different lengths affect the efficiency of MHC class I presentation.
Lower concentrations of puromycin stimulated the premature termination of a
relatively higher amount of near full‐length polypeptides; the finding that these
puromycin conditions are associated with increased MHC class I presentation is
consistent with reports from other systems that newly synthesized, full‐length or near
full‐length defective ribosomal products are the most efficient source of peptides
presented on MHC class I molecules (Schubert et al. 2000; Princiotta et al. 2003; Dolan et
al. 2011a). In contrast, higher concentrations of puromycin stimulated the production of
polypeptides truncated earlier in their synthesis. These conditions were associated with
progressive impairment of the MHC class I pathway.
Based on these data, I propose that near full‐length truncated polypeptides are a
more efficient source of MHC class I substrates than polypeptides truncated early in

103
their synthesis. This is consistent with the finding that proteasomal substrates (including
RDPs) show wide variation in the efficiency with which they access the MHC class I
pathway (Princiotta et al. 2003). The difference in presentation efficiency likely cannot be
attributed to substrate hydrophobicity (if anything, this should favor presentation from
the poorly folded, shorter polypeptide population, based on Huang et al. 2011b). So how
can near full‐length peptidyl‐puromycins be distinguished from their shorter
counterparts?
One possibility lies in the organization of eukaryotic messenger
ribonucleoproteins (mRNPs), which form a closed loop structure juxtaposing the 5’ and
3’ ends of the transcript (Amrani et al. 2008). One of the proteins that mediates bridging
interactions to maintain the closed loop is eukaryotic initiation factor 3 (eIF3).
Interestingly, eIF3 was shown to form a complex with the 26S proteasome, suggesting
that proteasomes localize to the joined 5’ and 3’ ends of mRNPs (Sha et al. 2009). With
~8x105 functional proteasomes per cell (Princiotta et al. 2003), this is more than enough
to tether 1‐2 proteasomes per translating mRNP (given ~3.8x105 mRNAs per cell, which
is the upper limit on the number of translating mRNPs) (Qiagen). The proximity of the
eIF3‐bound proteasome near the end of the open reading frame positions the
proteasome to selectively receive full‐length and near full‐length substrates. The eIF3‐
bound proteasome could then “channel” its degradation products specifically for
presentation on MHC class I molecules (perhaps by directly binding TAP or via an

104
accessory tethering protein). This is consistent with the finding that MHC class I
substrate processing is compartmentalized for high efficiency (Lev et al. 2010).
Compartmentalization (via an unknown mechanism) favors the loading of MHC class I
molecules with peptides derived from the proteasomal degradation of newly
synthesized RDPs and prevents competition from “retiree” proteins that have reached
the end of their lifespan. In contrast, shorter peptidyl‐puromycins are released distally
from the eIF3‐bound proteasome, and are instead rapidly degraded by cytosolic
proteasomes that are not channeled for presentation (Dolan et al. 2011b).
With this model, I can propose a speculative accounting for the fates of different
classes of peptidyl‐puromycins (Fig. 23). Short peptides produced by premature
termination should degrade in seconds, due to the activity of cytosolic peptidases (Reits
et al. 2003). Because peptide degradation is more efficient than peptide translocation into
the ER (van Endert 2011), peptide‐length premature termination products would be
predicted to be highly inefficient sources of MHC class I peptides. The majority of
peptidyl‐puromycins truncated early or in the middle of their synthesis (such as those
induced by 20 M puromycin) are rapidly degraded by cytosolic proteasomes (20S for
severely misfolded substrates, 26S for moderately misfolded substrates) (Qian et al.
2006a). A minority of peptidyl‐puromycins, (predominantly smaller ones) are degraded
by non‐proteasomal proteases, such as IDE and TPP II. These smaller truncated
polypeptides are not degraded by proteasomes channeled for antigen presentation

105
Figure 23: The substrate heterogeneity model. Indicates proposed degradation
pathways for prematurely terminated substrates at varying points of truncation. Only
the eIF3‐bound 26S proteasome is compartmentalized for efficient MHC class I
presentation. At the bottom, the profiles of polypeptides produced under different
puromycin treatment conditions are indicated. Note that the mRNP forms a closed loop
with the 5’ and 3’ ends juxtaposed by bridging proteins (like eIF4G) but is shown here in
extended form for simplicity.
(being too distant from the eIF3‐bound proteasome), and are thus poor MHC class I
pathway substrates, despite being RDPs. Finally, near‐full length truncated polypeptides
(such as those induced by 2 M puromycin) are degraded by eIF3‐bound proteasomes
channeled for antigen presentation.
A key feature of this model is the spatial and functional separation of truncated
polypeptide metabolism from proteolytic pathways channeled for MHC class I
presentation. Separation of these pathways could be desirable to prevent N‐terminally
biased sampling of the proteome for MHC class I‐binding peptides. Thus, rapid
clearance of premature termination products could be accomplished without biasing the
antigenic peptide repertoire. These issues highlight the need for bioinformatics‐based

106
surveys of MHC I peptides. Such studies would help determine the positional bias of
peptide epitopes within ORFs and thereby provide a preliminary estimate of the relative
contribution of prematurely terminated polypeptides to the MHC I peptidome (or lack
thereof).
In summary, this model highlights the heterogeneity of prematurely terminated
polypeptides, and the corresponding heterogeneity in degradation pathways for their
disposal. The diversity of degradation mechanisms was highlighted by a recent study
suggesting that distinct biochemical pathways are involved in the processing of newly
synthesized DRiPs versus old proteins that have reached the end of their lifespan
(“retirees”) (Dolan et al. 2011a). I propose to extend this analogy, and suggest that there
exist diverse pathways for RDP degradation, each of which metabolizes specific
subclasses of RDPs, including prematurely terminated polypeptides. Identifying the
distinctive features of these proteolytic pathways, the basis for their substrate selectivity
and the mechanisms that govern their access to the MHC class I machinery are
important future avenues for making sense of the complex calculus behind RDP
metabolism.
5.4.2 Puromycin in other studies of the MHC class I pathway
Two previous studies reported conflicting findings on the effects of puromycin
on MHC class I presentation (Gileadi et al. 1999; Golovina et al. 2005). In the first study,
cells were treated with 2000 M puromycin for 30 minutes during infection with a

107
vaccinia vector expressing the matrix protein of influenza virus (Gileadi et al. 1999).
Puromycin treatment resulted in increased lysis of antigen‐expressing cells by CTLs
specific for a matrix‐derived epitope. In the second study, cells were infected with
vaccinia vectors expressing a model NP‐SIINFEKL antigen (Golovina et al. 2005). One
hour after infection, cells were treated with 200 M puromycin for 20 minutes, which
resulted in either no change or a slight decrease in the presentation of peptides derived
from the model antigen.
Both studies used puromycin with the goal of stimulating truncated polypeptide
production. However, it is clear from my puromycin titration experiments that the
production of truncated polypeptides is (expectedly) concentration‐dependent, peaking
at 20 M (in 293‐Kb cells), and that few translation products are produced at puromycin
concentrations of 200 M or higher (Figs. 3 and 4). I suggest that the high concentrations
of puromycin used in prior studies may lead to an initial discharge of nascent chains
from elongating ribosomes, but predominantly results in the production of methionyl‐
puromycins or peptidyl‐puromycins only a few amino acids in length, both of which are
poor substrates for presentation on MHC I molecules.
MHC class I recovery exhibited both puromycin‐mediated stimulation and
inhibition, depending on the concentration and duration of puromycin treatment. This
may be responsible for the seemingly opposite effects of puromycin on the MHC class I
pathway observed by previous investigators. By establishing the dynamic range of time‐

108
and concentration‐dependent effects of puromycin, I was able to observe previously
unappreciated complexities in the behavior of prematurely terminated polypeptides and
their processing by the MHC class I pathway.
5.5 Candidate RDP pathway factors
Despite ongoing study of RDPs for over a decade (and reports of the RDP pool
extending almost three decades before that), identification of RDP pathway factors has
proven elusive. Towards the end of Chapter 4, I reported my preliminary investigations
into the role of two different candidate factors in RDP metabolism: Pth2 and CHIP.
Peptidyl‐tRNA hydrolase 2 (Pth2) metabolizes peptidyl‐tRNAs by hydrolyzing
the ester bond between the peptide and tRNA (de Pereda et al. 2004). Since peptidyl‐
tRNAs are the closest endogenous correlate of peptidyl‐puromycins, I tested the effects
of Pth2 knockdown on RDP degradation and antigen presentation (Fig. 20). Pth2
knockdown increased the cellular fraction of RDPs and stimulated MHC class I
presentation, suggesting that Pth2 inhibits flux through the RDP pathway. One possible
explanation for this is that Pth2 functions to direct peptidyl‐tRNAs to alternative
degradation pathways, such as cytosolic proteases. In yeast, Pth2 has been shown to
bind the ubiquitin‐like (UBL) domains of Rad23 and Dsk2, two proteins that deliver
polyubiquitinated substrates to the proteasome (Ishii et al. 2006). Pth2 binding to these
delivery proteins inhibited their interaction with polyubiquitin receptors, resulting in a
decrease in proteasomal degradation consistent with my observations.

109
In total, these findings suggest that Pth2 inhibits the delivery of substrates to the
proteasome. Because prematurely terminated polypeptides have a propensity to
aggregate, Pth2 may serve a protective function by directing truncated polypeptides
away from the proteasome towards less critical proteolytic pathways. By shunting
prematurely terminated substrates away from the proteasome, Pth2 may also serve an
immunologic function by helping to prevent N‐terminal biasing of the MHC class I
peptide repertoire.
CHIP is uniquely well‐suited to serve as an RDP pathway factor, since it
functions as a chaperone, cochaperone, and E3 ubiquitin ligase (Rosser et al. 2007). The
chaperone activity of CHIP would allow it to recognize misfolded substrates, while its
intrinsic E3 ligase activity could facilitate the ubiquitination and destruction of RDPs.
Knockdown of CHIP increased the fraction of RDPs and stimulated MHC class I
presentation (Fig. 21). These findings suggest that the net effect of CHIP is to inhibit flux
through the RDP pathway, possibly by directly chaperoning substrates that would
otherwise have been targeted for rapid degradation.
5.6 Implications for human health
My studies on the fate of prematurely terminated polypeptides highlight the
diversity of proteolytic pathways necessary for the rapid clearance of defective proteins.
The extent to which disruptions in the various proteolytic pathways contribute to
human disease is a topic of great clinical interest. From cardiomyopathy to

110
neurodegenerative disorders, disruptions in proteostasis have been associated with an
ever‐growing number of diverse disease states (Powers et al. 2009), with small molecules
that modulate the proteostasis network showing promise in clinical trials (Balch et al.
2008). Furthermore, the mechanisms that regulate the efficiency with which different
proteolytic pathways access the MHC class I machinery remain unknown (Dolan et al.
2011b). Characterizing these mechanisms has the potential to offer us new insights into
antiviral immunity and facilitate the design of more effective CTL vaccines.

111
References
Amrani N, Ghosh S, Mangus DA and Jacobson A. (2008) Translation factors promote the
formation of two states of the closed‐loop mRNP. Nature. 453(7199): 1276‐80.
Antón LC, Snyder HL, Bennink JR, Vinitsky A, Orlowski M, Porgador A and Yewdell
JW. (1998) Dissociation of proteasomal degradation of biosynthesized viral
proteins from generation of MHC class I‐associated antigenic peptides. J
Immunol. 160(10): 4859‐68.
Antón LC, Schubert U, Bacík I, Princiotta MF, Wearsch PA, Gibbs J, Day PM, Realini C,
Rechsteiner MC, Bennink JR and Yewdell JW. (1999) Intracellular localization of
proteasomal degradation of a viral antigen. J Cell Biol. 146(1): 113‐24.
Arava Y, Wang Y, Storey JD, Liu CL, Brown PO and Herschlag D. (2003) Genome‐wide
analysis of mRNA translation profiles in Saccharomyces cerevisiae. Proc Natl
Acad Sci U S A. 100(7): 3889‐94.
Arteaga MF, Wang L, Ravid T, Hochstrasser M and Canessa CM. (2006) An amphipathic
helix targets serum and glucocorticoid‐induced kinase 1 to the endoplasmic
reticulum‐associated ubiquitin‐conjugation machinery. Proc Natl Acad Sci U S A.
103(30): 11178‐83.
Atherly AG. (1978) Peptidyl‐transfer RNA hydrolase prevents inhibition of protein
synthesis initiation. Nature. 275(5682): 769.
Atkinson GC, Baldauf SL and Hauryliuk V (2008) Evolution of nonstop, no‐go and
nonsense‐mediated mRNA decay and their termination factor‐derived
components. BMC Evol Biol. 8: 290.
Balch WE, Morimoto RI, Dillin A and Kelly JW. (2008) Adapting proteostasis for disease
intervention. Science. 319(5865): 916‐9.
Ballinger CA, Connell P, Wu Y, Hu Z, Thompson LJ, Yin LY and Patterson C. (1999)
Identification of CHIP, a novel tetratricopeptide repeat‐containing protein that
interacts with heat shock proteins and negatively regulates chaperone functions.
Mol Cell Biol. 19(6): 4535‐45.
Banerji J, Sands J, Strominger JL and Spies T. (1990) A gene pair from the human major
histocompatibility complex encodes large proline‐rich proteins with multiple

112
repeated motifs and a single ubiquitin‐like domain. Proc Natl Acad Sci U S A.
87(6): 2374‐8.
Becker T, Armache JP, Jarasch A, Anger AM, Villa E, Sieber H, Motaal BA, Mielke T,
Berninghausen O and Beckmann R. (2011) Structure of the no‐go mRNA decay
complex Dom34‐Hbs1 bound to a stalled 80S ribosome. Nat Struct Mol Biol. 18(6):
715‐20.
Bence NF, Sampat RM and Kopito RR. (2001) Impairment of the ubiquitin‐proteasome
system by protein aggregation. Science. 292(5521): 1552‐5.
Benham AM, Grommé M and Neefjes J. (1998) Allelic differences in the relationship
between proteasome activity and MHC class I peptide loading. J Immunol.
161(1): 83‐9.
Bennett EJ, Bence NF, Jayakumar R and Kopito RR. (2005) Global impairment of the
ubiquitin‐proteasome system by nuclear or cytoplasmic protein aggregates
precedes inclusion body formation. Mol Cell. 17(3): 351‐65.
Berglund P, Finzi D, Bennink JR and Yewdell JW. (2007) Viral alteration of cellular
translational machinery increases defective ribosomal products. J Virol. 81(13):
7220‐9.
Cardinaud S, Starck SR, Chandra P and Shastri N. (2010) The synthesis of truncated
polypeptides for immune surveillance and viral evasion. PLoS One. 5(1): e8692.
Cascio P, Hilton C, Kisselev AF, Rock KL and Goldberg AL. (2001) 26S proteasomes and
immunoproteasomes produce mainly N‐extended versions of an antigenic
peptide. EMBO J. 20(10): 2357‐66.
Ciechanover A, Heller H, Elias S, Haas AL and Hershko A. (1980) ATP‐dependent
conjugation of reticulocyte proteins with the polypeptide required for protein
degradation. Proc Natl Acad Sci U S A. 77(3): 1365‐8.
Connell P, Ballinger CA, Jiang J, Wu Y, Thompson LJ, Höhfeld J and Patterson C. (2001)
The co‐chaperone CHIP regulates protein triage decisions mediated by heat‐
shock proteins. Nat Cell Biol. 3(1): 93‐6.
Daniels RS, Worthington VC, Atkinson EM and Hipkiss AR. (1980) Proteolysis of
puromycin‐peptides in rabbit reticulocytes: detection of a high molecular weight
oligopeptide proteolytic substrate. FEBS Lett. 113(2): 245‐8.

113
David A, Netzer N, Strader MB, Das SR, Chen CY, Gibbs J, Pierre P, Bennink JR and
Yewdell JW. (2011) RNA binding targets aminoacyl‐tRNA synthetases to
translating ribosomes. J Biol Chem. 286(23): 20688‐700.
Day PM, Yewdell JW, Porgador A, Germain RN and Bennink JR. (1997) Direct delivery
of exogenous MHC class I molecule‐binding oligopeptides to the endoplasmic
reticulum of viable cells. Proc Natl Acad Sci U S A. 94(15): 8064‐9.
de Pereda JM, Waas WF, Jan Y, Ruoslahti E, Schimmel P and Pascual J. (2004) Crystal
structure of a human peptidyl‐tRNA hydrolase reveals a new fold and suggests
basis for a bifunctional activity. J Biol Chem. 279(9): 8111‐5.
Desmots F, Russell HR, Lee Y, Boyd K and McKinnon PJ. (2005) The reaper‐binding
protein scythe modulates apoptosis and proliferation during mammalian
development. Mol Cell Biol. 25(23): 10329‐37.
Dimitriadis GJ. (1979) Effect of detergents on antibody‐antigen interaction. Anal
Biochem.98(2): 445‐51.
Dolan BP, Li L, Takeda K, Bennink JR and Yewdell JW. (2010) Defective ribosomal
products are the major source of antigenic peptides endogenously generated
from influenza A virus neuraminidase. J Immunol. 184(3): 1419‐24.
Dolan BP, Li L, Veltri CA, Ireland CM, Bennink JR and Yewdell JW. (2011a) Distinct
pathways generate peptides from defective ribosomal products for CD8+ T cell
immunosurveillance. J Immunol. 186(4): 2065‐72.
Dolan BP, Bennink JR and Yewdell JW. (2011b) Translating DRiPs: progress in
understanding viral and cellular sources of MHC class I peptide ligands. Cell Mol
Life Sci. 68(9): 1481‐9.
Doma MK and Parker R. (2006) Endonucleolytic cleavage of eukaryotic mRNAs with
stalls in translation elongation. Nature. 440(7083): 561‐4.
Donohue KB, Grant JM, Tewalt EF, Palmer DC, Theoret MR, Restifo NP and Norbury
CC. (2006) Cross‐priming utilizes antigen not available to the direct presentation
pathway. Immunology. 119(1): 63‐73.
Duckworth WC, Bennett RG and Hamel FG. (1998) Insulin degradation: progress and
potential. Endocr Rev. 19(5): 608‐24.

114
Eisenlohr LC, Huang L and Golovina TN. (2007) Rethinking peptide supply to MHC
class I molecules. Nat Rev Immunol. 7(5): 403‐10.
Esquivel F, Yewdell J and Bennink J. (1992) RMA/S cells present endogenously
synthesized cytosolic proteins to class I‐restricted cytotoxic T lymphocytes. J Exp
Med. 175(1): 163‐8.
Firat E, Huai J, Saveanu L, Gaedicke S, Aichele P, Eichmann K, van Endert P and
Niedermann G. (2007) Analysis of direct and cross‐presentation of antigens in
TPPII knockout mice. J Immunol. 179(12): 8137‐45.
Fromant M, Ferri‐Fioni ML, Plateau P and Blanquet S. (2003) Peptidyl‐tRNA hydrolase
from Sulfolobus solfataricus. Nucleic Acids Res. 31(12): 3227‐35.
Fuertes G, Villarroya A and Knecht E. (2003) Role of proteasomes in the degradation of
short‐lived proteins in human fibroblasts under various growth conditions. Int J
Biochem Cell Biol. 35(5): 651‐64.
Fujimuro M, Sawada H and Yokosawa H. (1994) Production and characterization of
monoclonal antibodies specific to multi‐ubiquitin chains of polyubiquitinated
proteins. FEBS Lett. 349(2): 173‐80.
Fujimuro M and Yokosawa H. (2005) Production of antipolyubiquitin monoclonal
antibodies and their use for characterization and isolation of polyubiquitinated
proteins. Methods Enzymol. 399: 75‐86.
Gavioli R, Vertuani S and Masucci MG. (2002) Proteasome inhibitors reconstitute the
presentation of cytotoxic T‐cell epitopes in Epstein‐Barr virus‐associated tumors.
Int J Cancer. 101(6): 532‐8.
Gebauer F and Hentze MW. (2004) Molecular mechanisms of translational control.
(2004) Nat Rev Mol Cell Biol. 5(10): 827‐35.
Geier E, Pfeifer G, Wilm M, Lucchiari‐Hartz M, Baumeister W, Eichmann K and
Niedermann G. (1999) A giant protease with potential to substitute for some
functions of the proteasome. Science. 283(5404): 978‐81.
Gidalevitz T, Ben‐Zvi A, Ho KH, Brignull HR and Morimoto RI. (2006) Progressive
disruption of cellular protein folding in models of polyglutamine diseases.
Science. 311(5766): 1471‐4.

115
Gileadi U, Moins‐Teisserenc HT, Correa I, Booth BL Jr, Dunbar PR, Sewell AK,
Trowsdale J, Phillips RE and Cerundolo V. (1999) Generation of an
immunodominant CTL epitope is affected by proteasome subunit composition
and stability of the antigenic protein. J Immunol. 163(11): 6045‐52.
Gilon T, Chomsky O and Kulka RG. (1998) Degradation signals for ubiquitin system
proteolysis in Saccharomyces cerevisiae. EMBO J. 17(10): 2759‐66.
Glas R, Bogyo M, McMaster JS, Gaczynska M and Ploegh HL. (1998) A proteolytic
system that compensates for loss of proteasome function. Nature. 392(6676): 618‐
22.
Goldberg AL. (1972) Degradation of abnormal proteins in Escherichia coli. Proc Natl Acad
Sci U S A. 69(2): 422‐6.
Golovina TN, Morrison SE and Eisenlohr LC. (2005) The impact of misfolding versus
targeted degradation on the efficiency of the MHC class I‐restricted antigen
processing. J Immunol. 174(5): 2763‐9.
Goodman CA, Mabrey DM, Frey JW, Miu MH, Schmidt EK, Pierre P and Hornberger
TA. (2011) Novel insights into the regulation of skeletal muscle protein synthesis
as revealed by a new nonradioactive in vivo technique. FASEB J. 25(3): 1028‐39.
Hansen WJ, Lingappa VR and Welch WJ. (1994) Complex environment of nascent
polypeptide chains. J Biol Chem. 269(43): 26610‐3.
Hershko A, Ciechanover A, Heller H, Haas AL and Rose IA. (1980) Proposed role of
ATP in protein breakdown: conjugation of protein with multiple chains of the
polypeptide of ATP‐dependent proteolysis. Proc Natl Acad Sci U S A. 77(4): 1783‐
6.
Hershko A and Ciechanover A. (1998) The ubiquitin system. Annu Rev Biochem. 67: 425‐
79.
Hochstrasser M. (1996) Ubiquitin‐dependent protein degradation. Annu Rev Genet. 30:
405‐39.
Huang L, Marvin JM, Tatsis N and Eisenlohr LC. (2011a) Selective role of ubiquitin in
MHC class I antigen presentation. J Immunol. 186(4): 1904‐8.
Huang L, Kuhls MC and Eisenlohr LC. (2011b) Hydrophobicity as a driver of MHC class
I antigen processing. EMBO J. 30(8): 1634‐44.

116
Ingolia NT, Ghaemmaghami S, Newman JR and Weissman JS. (2009) Genome‐wide
analysis in vivo of translation with nucleotide resolution using ribosome
profiling. Science. 324(5924): 218‐23.
Ishii T, Funakoshi M and Kobayashi H. (2006) Yeast Pth2 is a UBL domain‐binding
protein that participates in the ubiquitin‐proteasome pathway. EMBO J. 25(23):
5492‐503.
Johnson PR, Swanson R, Rakhilina L and Hochstrasser M. (1998) Degradation signal
masking by heterodimerization of MATalpha2 and MATa1 blocks their mutual
destruction by the ubiquitin‐proteasome pathway. Cell. 94(2): 217‐27.
Jørgensen F and Kurland CG. (1990) Processivity errors of gene expression in
Escherichia coli. J Mol Biol. 215: 511‐21.
Kapp LD and Lorsch JR. (2004) The molecular mechanics of eukaryotic translation.
Annu Rev Biochem. 73: 657‐704.
Kawahara M, York IA, Hearn A, Farfan D and Rock KL. (2009) Analysis of the role of
tripeptidyl peptidase II in MHC class I antigen presentation in vivo. J Immunol.
183(10): 6069‐77.
Kemshead JT and Hipkiss AR. (1974) Degradation of abnormal proteins in Escherichia
coli: relative susceptibility of canavanyl proteins and puromycin peptides to
proteolysis in vitro. Eur J Biochem. 45(2): 535‐40.
Khan S, de Giuli R, Schmidtke G, Bruns M, Buchmeier M, van den Broek M and
Groettrup M. (2001) Cutting edge: neosynthesis is required for the presentation
of a T cell epitope from a long‐lived viral protein. J Immunol. 167(9): 4801‐4.
Kisselev AF and Goldberg AL. (2001) Proteasome inhibitors: from research tools to drug
candidates. Chem Biol. 8(8): 739‐58.
Klemes Y, Etlinger JD and Goldberg AL. (1981) Properties of abnormal proteins
degraded rapidly in reticulocytes. Intracellular aggregation of the globin
molecules prior to hydrolysis. J Biol Chem. 256(16): 8436‐44.
Kurochkin IV. (2001) Insulin‐degrading enzyme: embarking on amyloid destruction.
Trends Biochem Sci. 26(7): 421‐5.
Lavie L, Reznick AZ and Gershon D. (1982) Decreased protein and puromycinyl‐peptide
degradation in livers of senescent mice. Biochem J. 202(1): 47‐51.

117
Lelouard H, Ferrand V, Marguet D, Bania J, Camosseto V, David A, Gatti E and Pierre P.
(2004) Dendritic cell aggresome‐like induced structures are dedicated areas for
ubiquitination and storage of newly synthesized defective proteins. J Cell Biol.
164(5): 667‐75.
Leonhardt RM, Fiegl D, Rufer E, Karger A, Bettin B and Knittler MR. (2010) Post‐
endoplasmic reticulum rescue of unstable MHC class I requires proprotein
convertase PC7. J Immunol. 184: 2985‐98.
Lev A, Princiotta MF, Zanker D, Takeda K, Gibbs JS, Kumagai C, Waffarn E, Dolan BP,
Burgevin A, Van Endert P, Chen W, Bennink JR and Yewdell JW. (2010)
Compartmentalized MHC class I antigen processing enhances
immunosurveillance by circumventing the law of mass action. Proc Natl Acad Sci
U S A. 107(15): 6964‐9.
Luckey CJ, King GM, Marto JA, Venketeswaran S, Maier BF, Crotzer VL, Colella TA,
Shabanowitz J, Hunt DF and Engelhard VH. (1998) Proteasomes can either
generate or destroy MHC class I epitopes: evidence for nonproteasomal epitope
generation in the cytosol. J Immunol. 161(1): 112‐21.
Manley JL. (1978) Synthesis and degradation of termination and premature‐termination
fragments of beta‐galactosidase in vitro and in vivo. J Mol Biol. 125: 407‐32.
Marcilla M, Cragnolini JJ and López de Castro JA. (2007) Proteasome‐independent HLA‐
B27 ligands arise mainly from small basic proteins. Mol Cell Proteomics. 6(5): 923‐
38.
McCallum CD, Do H, Johnson AE and Frydman J. (2000) The interaction of the
chaperonin tailless complex polypeptide 1 (TCP1) ring complex (TRiC) with
ribosome‐bound nascent chains examined using photo‐cross‐linking. J Cell Biol.
149: 591‐602.
McIlhinney A and Hogan BL. (1974) Rapid degradation of puromycyl peptides in
hepatoma cells and reticulocytes. FEBS Lett. 40(2): 297‐301.
Meacham GC, Patterson C, Zhang W, Younger JM and Cyr DM. (2001) The Hsc70 co‐
chaperone CHIP targets immature CFTR for proteasomal degradation. Nat Cell
Biol. 3(1): 100‐5.
Meiners S, Heyken D, Weller A, Ludwig A, Stangl K, Kloetzel PM and Krüger E. (2003)
Inhibition of proteasome activity induces concerted expression of proteasome

118
genes and de novo formation of mammalian proteasomes. J Biol Chem. 278(24):
21517‐25.
Menninger JR, Walker C and Tan PF. (1973) Studies on the metabolic role of peptidyl‐
tRNA hydrolase. I. Properties of a mutant E. coli with temperature‐sensitive
peptidyl‐tRNA hydrolase. Mol Gen Genet. 121(4): 307‐24.
Menninger JR. (1976) Peptidyl transfer RNA dissociates during protein synthesis from
ribosomes of Escherichia coli. J Biol Chem. 251(11): 3392‐8.
Menninger JR. (1979) Accumulation of peptidyl tRNA is lethal to Escherichia coli. J
Bacteriol. 137(1): 694‐6.
Min JN, Whaley RA, Sharpless NE, Lockyer P, Portbury AL and Patterson C. (2008)
CHIP deficiency decreases longevity, with accelerated aging phenotypes
accompanied by altered protein quality control. Mol Cell Biol. 28(12): 4018‐25.
Minami R, Hayakawa A, Kagawa H, Yanagi Y, Yokosawa H and Kawahara H. (2010)
BAG‐6 is essential for selective elimination of defective proteasomal substrates. J
Cell Biol. 190(4): 637‐50.
Mitsiades N, Mitsiades CS, Poulaki V, Chauhan D, Fanourakis G, Gu X, Bailey C, Joseph
M, Libermann TA, Treon SP, Munshi NC, Richardson PG, Hideshima T and
Anderson KC. (2002) Molecular sequelae of proteasome inhibition in human
multiple myeloma cells. Proc Natl Acad Sci U S A. 99(22): 14374‐9.
Nathans D. (1964) Puromycin inhibition of protein synthesis: incorporation of
puromycin into peptide chains. Proc Natl Acad Sci U S A. 51: 585‐92.
Naujokat C, Fuchs D and Berges C. (2007) Adaptive modification and flexibility of the
proteasome system in response to proteasome inhibition. Biochim Biophys Acta.
1773(9): 1389‐97.
Pariyarath R, Wang H, Aitchison JD, Ginsberg HN, Welch WJ, Johnson AE and Fisher
EA. (2001) Co‐translational interactions of apoprotein B with the ribosome and
translocon during lipoprotein assembly or targeting to the proteasome. J Biol
Chem. 276(1): 541‐50.
Parmentier N, Stroobant V, Colau D, de Diesbach P, Morel S, Chapiro J, van Endert P
and Van den Eynde BJ. (2010) Production of an antigenic peptide by insulin‐
degrading enzyme. Nat Immunol. 11(5): 449‐54.

119
Passos DO, Doma MK, Shoemaker CJ, Muhlrad D, Green R, Weissman J, Hollien J and
Parker R. (2009) Analysis of Dom34 and its function in no‐go decay. Mol Biol Cell.
20(13): 3025‐32.
Petrucelli L, Dickson D, Kehoe K, Taylor J, Snyder H, Grover A, De Lucia M, McGowan
E, Lewis J, Prihar G, Kim J, Dillmann WH, Browne SE, Hall A, Voellmy R, Tsuboi
Y, Dawson TM, Wolozin B, Hardy J and Hutton M. (2004) CHIP and Hsp70
regulate tau ubiquitination, degradation and aggregation. Hum Mol Genet. 13(7):
703‐14.
Pickart CM and Cohen RE. (2004) Proteasomes and their kin: proteases in the machine
age. Nat Rev Mol Cell Biol. 5(3): 177‐87.
Pisareva VP, Skabkin MA, Hellen CU, Pestova TV and Pisarev AV. (2011) Dissociation
by Pelota, Hbs1 and ABCE1 of mammalian vacant 80S ribosomes and stalled
elongation complexes. EMBO J. 30(9): 1804‐17.
Poole B and Wibo M. (1973) Protein degradation in cultured cells. The effect of fresh
medium, fluoride, and iodoacetate on the digestion of cellular protein of rat
fibroblasts. J Biol Chem. 248(17): 6221‐6.
Porgador A, Yewdell JW, Deng Y, Bennink JR and Germain RN. (1997) Localization,
quantitation, and in situ detection of specific peptide‐MHC class I complexes
using a monoclonal antibody. Immunity. 6(6): 715‐26.
Powers ET, Morimoto RI, Dillin A, Kelly JW and Balch WE. (2009) Biological and
chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem. 78:
959‐91.
Princiotta MF, Schubert U, Chen W, Bennink JR, Myung J, Crews CM and Yewdell JW.
(2001) Cells adapted to the proteasome inhibitor 4‐hydroxy‐ 5‐iodo‐3‐
nitrophenylacetyl‐Leu‐Leu‐leucinal‐vinyl sulfone require enzymatically active
proteasomes for continued survival. Proc Natl Acad Sci U S A. 98(2): 513‐8.
Princiotta MF, Finzi D, Qian SB, Gibbs J, Schuchmann S, Buttgereit F, Bennink JR and
Yewdell JW. (2003) Quantitating protein synthesis, degradation, and endogenous
antigen processing. Immunity. 18(3): 343‐54.
Prouty WF, Karnovsky MJ and Goldberg AL. (1975) Degradation of abnormal proteins
in Escherichia coli. Formation of protein inclusions in cells exposed to amino acid
analogs. J Biol Chem. 250(3): 1112‐22.

120
Qiagen. “Nucleic Acids and Proteins: Mammalian Cells and Tissues.” Technical
Information. (2011) August 24, 2011
<http://www.qiagen.com/resources/info/nucleic_acids_and_proteins_mammalia
n_cells_and_tissues.aspx#1>.
Qian SB, Bennink JR and Yewdell JW. (2005) Quantitating defective ribosome products.
Methods Mol Biol. 301: 271‐81.
Qian SB, Princiotta MF, Bennink JR and Yewdell JW. (2006a) Characterization of rapidly
degraded polypeptides in mammalian cells reveals a novel layer of nascent
protein quality control. J Biol Chem. 281(1): 392‐400.
Qian SB, Reits E, Neefjes J, Deslich JM, Bennink JR and Yewdell JW. (2006b) Tight
linkage between translation and MHC class I peptide ligand generation implies
specialized antigen processing for defective ribosomal products. J Immunol.
177(1): 227‐33.
Qian SB, McDonough H, Boellmann F, Cyr DM and Patterson C. (2006c) CHIP‐mediated
stress recovery by sequential ubiquitination of substrates and Hsp70. Nature.
440(7083): 551‐5.
Qian SB, Zhang X, Sun J, Bennink JR, Yewdell JW and Patterson C. (2010) mTORC1 links
protein quality and quantity control by sensing chaperone availability. J Biol
Chem. 285(35): 27385‐95.
Reits EA, Vos JC, Grommé M and Neefjes J. (2000) The major substrates for TAP in vivo
are derived from newly synthesized proteins. Nature. 404(6779): 774‐8.
Rock KL, Gamble S, Rothstein L, Gramm C and Benacerraf B. (1991) Dissociation of beta
2‐microglobulin leads to the accumulation of a substantial pool of inactive class I
MHC heavy chains on the cell surface. Cell. 65(4): 611‐20.
Rock KL, Gramm C, Rothstein L, Clark K, Stein R, Dick L, Hwang D and Goldberg AL.
(1994) Inhibitors of the proteasome block the degradation of most cell proteins
and the generation of peptides presented on MHC class I molecules. Cell.
78(5):761‐71.
Rosas‐Sandoval G, Ambrogelly A, Rinehart J, Wei D, Cruz‐Vera LR, Graham DE, Stetter
KO, Guarneros G and Söll D. (2002) Orthologs of a novel archaeal and of the
bacterial peptidyl‐tRNA hydrolase are nonessential in yeast. Proc Natl Acad Sci U
S A. 99(26): 16707‐12.

121
Rosser MF, Washburn E, Muchowski PJ, Patterson C and Cyr DM. (2007) Chaperone
functions of the E3 ubiquitin ligase CHIP. J Biol Chem. 282(31): 22267‐77.
Rötzschke O, Falk K, Stevanović S, Jung G, Walden P and Rammensee HG. (1991) Exact
prediction of a natural T cell epitope. Eur J Immunol. 21(11): 2891‐4.
Rückrich T, Kraus M, Gogel J, Beck A, Ovaa H, Verdoes M, Overkleeft HS, Kalbacher H
and Driessen C. (2009) Characterization of the ubiquitin‐proteasome system in
bortezomib‐adapted cells. Leukemia. 23(6): 1098‐105.
Saric T, Chang SC, Hattori A, York IA, Markant S, Rock KL, Tsujimoto M and Goldberg
AL. (2002) An IFN‐gamma‐induced aminopeptidase in the ER, ERAP1, trims
precursors to MHC class I‐presented peptides. Nat Immunol. 3(12):1169‐76.
Saveanu L, Carroll O, Lindo V, Del Val M, Lopez D, Lepelletier Y, Greer F, Schomburg
L, Fruci D, Niedermann G and van Endert PM. (2005) Concerted peptide
trimming by human ERAP1 and ERAP2 aminopeptidase complexes in the
endoplasmic reticulum. Nat Immunol. 6(7): 689‐97.
Schägger H and von Jagow G. (1987) Tricine‐sodium dodecyl sulfate‐polyacrylamide gel
electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal
Biochem. 166(2): 368‐79.
Schägger H. (2006) Tricine‐SDS‐PAGE. Nat Protoc. 1: 16‐22.
Schmidt EK, Clavarino G, Ceppi M and Pierre P. (2009) SUnSET, a nonradioactive
method to monitor protein synthesis. Nat Methods. 6(4): 275‐7.
Schneider‐Poetsch T, Ju J, Eyler DE, Dang Y, Bhat S, Merrick WC, Green R, Shen B and
Liu JO. (2010) Inhibition of eukaryotic translation elongation by cycloheximide
and lactimidomycin. Nat Chem Biol. 6(3): 209‐217.
Schubert U, Antón LC, Gibbs J, Norbury CC, Yewdell JW and Bennink JR. (2000) Rapid
degradation of a large fraction of newly synthesized proteins by proteasomes.
Nature. 404(6779): 770‐4.
Serwold T, Gonzalez F, Kim J, Jacob R and Shastri N. (2002) ERAAP customizes peptides
for MHC class I molecules in the endoplasmic reticulum. Nature. 419(6906): 480‐3.
Sha Z, Brill LM, Cabrera R, Kleifeld O, Scheliga JS, Glickman MH, Chang EC and Wolf
DA. (2009) The eIF3 interactome reveals the translasome, a supercomplex linking
protein synthesis and degradation machineries. Mol Cell. 36(1): 141‐52.

122
Shastri N, Schwab S and Serwold T. (2002) Producing natureʹs gene‐chips: the
generation of peptides for display by MHC class I molecules. Annu Rev Immunol.
20: 463‐93.
Shoemaker CJ, Eyler DE and Green R. (2010) Dom34:Hbs1 promotes subunit
dissociation and peptidyl‐tRNA drop‐off to initiate no‐go decay. Science.
330(6002): 369‐72.
Spies T, Cerundolo V, Colonna M, Cresswell P, Townsend A and DeMars R. (1992)
Presentation of viral antigen by MHC class I molecules is dependent on a
putative peptide transporter heterodimer. Nature. 355(6361): 644‐6.
Sugawara S, Abo T and Kumagai K. (1987) A simple method to eliminate the
antigenicity of surface class I MHC molecules from the membrane of viable cells
by acid treatment at pH 3. J Immunol Methods. 100: 83‐90.
Szeto J, Kaniuk NA, Canadien V, Nisman R, Mizushima N, Yoshimori T, Bazett‐Jones
DP and Brumell JH. (2006) ALIS are stress‐induced protein storage
compartments for substrates of the proteasome and autophagy. Autophagy.
2(3):189‐99.
Tellam J, Connolly G, Green KJ, Miles JJ, Moss DJ, Burrows SR and Khanna R. (2004)
Endogenous presentation of CD8+ T cell epitopes from Epstein‐Barr virus‐
encoded nuclear antigen 1. J Exp Med. 199(10): 1421‐31.
Tellam J, Fogg MH, Rist M, Connolly G, Tscharke D, Webb N, Heslop L, Wang F and
Khanna R. (2007) Influence of translation efficiency of homologous viral proteins
on the endogenous presentation of CD8+ T cell epitopes. J Exp Med. 204(3): 525‐
32.
Teter SA, Houry WA, Ang D, Tradler T, Rockabrand D, Fischer G, Blum P,
Georgopoulos C and Hartl FU. (1999) Polypeptide flux through bacterial Hsp70:
DnaK cooperates with trigger factor in chaperoning nascent chains. Cell. 97(6):
755‐65.
Tsung K, Inouye S and Inouye M. (1989) Factors affecting the efficiency of protein
synthesis in Escherichia coli. Production of a polypeptide of more than 6000
amino acid residues. J Biol Chem. 264: 4428‐33.
Türkel S and Farabaugh PJ. (1993) Interspersion of an unusual GCN4 activation site with
a complex transcriptional repression site in Ty2 elements of Saccharomyces
cerevisiae. Mol Cell Biol. 13(4): 2091‐103.

123
Vabulas RM and Hartl FU. (2005) Protein synthesis upon acute nutrient restriction relies
on proteasome function. Science. 310(5756): 1960‐3.
van Endert P. (2011) Post‐proteasomal and proteasome‐independent generation of MHC
class I ligands. Cell Mol Life Sci. 68(9): 1553‐67.
Vázquez D. (1979) Inhibitors of protein biosynthesis. Mol Biol Biochem Biophys. 30: i‐x, 1‐
312.
Voo KS, Fu T, Wang HY, Tellam J, Heslop HE, Brenner MK, Rooney CM and Wang RF.
(2004) Evidence for the presentation of major histocompatibility complex class I‐
restricted Epstein‐Barr virus nuclear antigen 1 peptides to CD8+ T lymphocytes. J
Exp Med. 199(4): 459‐70.
Wheatley DN, Giddings MR and Inglis MS. (1980) Kinetics of degradation of ʺshort‐ʺ
and ʺlong‐livedʺ proteins in cultured mammalian cells. Cell Biol Int Rep. 4(12):
1081‐90.
Wherry EJ, Golovina TN, Morrison SE, Sinnathamby G, McElhaugh MJ, Shockey DC
and Eisenlohr LC. (2006) Re‐evaluating the generation of a ʺproteasome‐
independentʺ MHC class I‐restricted CD8 T cell epitope. J Immunol. 176(4): 2249‐
61.
Yewdell JW, Antón LC and Bennink JR. (1996) Defective ribosomal products (DRiPs): a
major source of antigenic peptides for MHC class I molecules? J Immunol. 157(5):
1823‐6.
Yewdell JW, Reits E and Neefjes J. (2003) Making sense of mass destruction: quantitating
MHC class I antigen presentation. Nat Rev Immunol. 3(12): 952‐61.
Yewdell JW and Nicchitta CV. (2006) The DRiP hypothesis decennial: support,
controversy, refinement and extension. Trends Immunol. 27: 368‐73.
Yewdell JW. (2007) Plumbing the sources of endogenous MHC class I peptide ligands.
Curr Opin Immunol 19: 79‐86.

124
Biography
Joshua Rene Lacsina was born on July 24, 1982 in Denver, Colorado to Rene
Quiambao Lacsina and Teresa Anden Lacsina. After Josh’s early years in Denver and
Rochester Hills, Michigan, the Lacsina family moved in 1989 to Salinas, California,
where Josh enjoyed a happy childhood. In 2003, Josh graduated magna cum laude from
Harvard University with an A.B. in Biochemical Sciences. Later that year, he began his
studies as an MD‐PhD student in the Medical Scientist Training Program at Duke
University. Josh met and started dating his long‐time girlfriend and professional
collaborator, Odessa Marks, during his first lab rotation in 2005 through the Duke
Program in Cell and Molecular Biology. The following year, Josh joined the lab of Dr.
Christopher Nicchitta through the Department of Pathology to study cellular
immunology and protein quality control. In 2004, Josh received the Award for Academic
Excellence and Achievement from the American Society for Clinical Pathology. In 2006,
he was elected to Alpha Omega Alpha, the medical honor society. In 2008, he was
elected to Sigma Xi, the scientific honor society and was awarded an NIH Kirschstein
Predoctoral Fellowship. In 2010, he received an Award for Outstanding Promise in
Research during Duke Research Career Day and was selected to the inaugural class of
Feagin Medical Scholars, through the John A. Feagin, Jr., MD, International Leadership
Endowment. He currently resides in Durham, North Carolina, and plans to pursue a
residency in internal medicine and a fellowship in infectious diseases.

125
Lacsina JR, Marks OA, Liu X, Reid DW, Jagannathan S and Nicchitta CV. Premature
translational termination products are rapidly degraded substrates for MHC
class I presentation. In revision.
LaMonte G, Philip N, Reardon J, Lacsina JR, Majoros W, Chapman L, Telen MJ, Ohler U,
Nicchitta CV, Haystead T and Chi JT. Erythrocytic microRNAs are genetic
determinants of malaria resistance in sickle cell diseases. In review.
Lampson BL, Prinz JA, Lacsina JR, Nicchitta CV, MacAlpine DA and Counter CM. Rare
codons limit KRas tumorigenic activity. Manuscript in preparation.
Lacsina JR, LaMonte G, Nicchitta CV and Chi JT. (2011) Polysome profiling of the
malaria parasite Plasmodium falciparum. Mol Biochem Parasitol. 179(1): 42‐6.
Yewdell JW, Lacsina JR, Rechsteiner MC and Nicchitta CV. (2011) Out with the old, in
with the new? Comparing methods for measuring protein degradation. Cell Biol
Int. 35(5): 457‐62.