pre-approval inspection readiness - business … · · 2016-02-15pre-approval inspection...
TRANSCRIPT

Pre-Approval
Inspection Readiness
Presented by
Pepe Rodriguez-Perez PhD
Business Excellence Consulting Inc BEC Spain SL
wwwcalidadprcom email pepecalidadprcom
2
Agenda
US FDA Structure and Authority
Generic Drug Approval Process
ndash The PreApproval Inspection (PAI)
Pharmaceutical development
ndash QBD
ndash Process Validations
FDA Quality System Inspection Elements
Pre-Inspection Activities the Value of a Mock Audit
Interaction with FDA Inspectors
Data Integrity
3
FDA Overview US Department of Health amp Human Services
US Food and Drug Administration Centers for
ndash Food Safety and Nutrition (CFSAN)
ndash Drug Evaluation and Research (CDER)
ndash Biologics Evaluation and Research (CBER)
ndash Devices and Radiological Health (CDRH)
ndash Center for Veterinary Medicine (CVM)
ndash Center for Tobacco Products (CTP)
ndash Office of Regulatory Affairs (ORA HQ)
ndash Field Regions (SER SWR PAR NER CER) raquo District Offices
San Juan District Office (SJN-DO)
ndash SJN MRP PON USVI
14000
4
FDA Mission To ensure that
ndash Foods are safe wholesome and sanitary
ndash Human and veterinary drugs biological products
and medical devices are safe and effective
ndash Cosmetics are safe
ndash Electronic products that emit radiation are safe
Authority for Inspections
United States of America
ndash Federal Food Drugs amp
Cosmetic Act
ndash The FDA is responsible for
protecting the public health by
assuring the safety efficacy and
security of human and veterinary
drugs biological products
medical devices our nationrsquos
food supply cosmetics and
products that emit radiation 5
6
FDA Laws Overview
1906 ndash Congress enacted Food and Drug Act
1938- Federal Food Drug and Cosmetic Act
ndash To ensure that food are safe and produced under sanitary conditions
ndash Drugs and device are safe and effectives for their intended use
ndash Cosmetics are safe and made from appropriate ingredients
ndash Labeling and packaging is truthful
1963 - Fist version of GMP for Drugs
1978 cGMP for Drugs 21 CFR 211
1987 Guideline on General Principles of Process Validation
1997 ndash FDA Modernization Act (FDAMA) Fine-tuning FDA laws
1997- Electronic Records (21 CFR Part 11)
2013 ndash Combination Products
Generic Drugs The use of bioequivalence as the basis for approving generic
copies of drug products was established by the ldquoDrug Price
Competition and Patent Term Restoration Act of 1984rdquo also
known as the Waxman-Hatch Act
This Act expedites the availability of less costly generic drugs by
permitting the FDA to approve applications to market generic
versions of brand-name drugs without conducting costly and
duplicative clinical trials At the same time the brand-name
companies can apply for up to five additional years patent
protection for the new medicines they developed to make up for
time lost while their products were going through the FDArsquos
approval process Brand-name drugs are subject to the same
bioequivalence tests as generics upon reformulation 7
8
9
The FDArsquos Drug Review Process
10
The FDArsquos Drug Review Process
11
NDA vs ANDA Review Process
12
Center for Drug Evaluation amp Research Office of Generic Drugs (OGD) 13
Generic Drug Review Process
Bioequivalence Review Labeling Review Chemistry amp Micro
Review Request for Plant
Inspection
APPLICANT
ANDA
Acceptable amp
Complete
Application Review
N ChemMicro
OK
Labeling
OK
Bioequivalence
OK
PreApproval
Inspection Results
OK
Not Approvable
Letter
Approval Withheld until
Results Satisfactory
Bio Deficiency Letter
APPROVED ANDA
N N N
N
Y Y Y
Y
Y
Refuse to Receive Letter
What is the goal of a
Pre-Approval Inspection
To assure that establishments involved in the
manufacturing testing or other manipulation of
new drug dosage forms and drug substances are
evaluated for
ndash conformance with commitments in the application
ndash site cGMP compliance
ndash data authenticity reliability and accuracy
ndash adequacy of analytical methodologies
14
PAI Objectives (Program 7346832)
There are three primary inspectional objectives of
this PAI program These objectives are
1) Readiness for Commercial Manufacturing
2) Conformance to Application
3) Data Integrity Audit
15
PAI Objective 1 Readiness for Commercial Manufacturing
Determine whether the establishment has a quality
system that is designed to achieve sufficient control over
the facility and commercial manufacturing operations
a) Manufacturing and laboratory changes deviations and
trends relating to the development of new drug substance
and product manufacturing have been adequately
evaluated
b) A sound and appropriate program for sampling testing
and evaluation of components in-process materials
finished products containers and closures for the purpose
of releasing materials or products has been established
including a robust supplier qualification program
16
PAI Objective 1 Readiness for Commercial Manufacturing cont
c) The establishment has sufficient facility and equipment
controls in place to prevent contamination of and by the
application product (or API)
d) Adequate procedures exist for batch release change control
investigating failures deviations complaints and adverse
events and for reporting this information to FDA such as
field alert reporting
e) The feasibility of the proposed commercial process and
manufacturing batch record including instructions
processing parameters and process control measures are
scientifically and objectively justified This objective is
linked to the firmrsquos process validation program
17
PAI Objective 2
Conformance to Application
Verify that the formulation manufacturing or
processing methods and analytical (or
examination) methods are consistent with
descriptions contained in the CMC section of the
application for the biobatch (and other pivotal
clinical batches when applicable) the proposed
commercial scale batch and API ndash Observing processing andor testing operations
ndash Compare the biobatch manufacturing process against the
proposed commercial batch record
18
PAI Objective 3
Data Integrity Audit
Audit the raw data hardcopy or electronic to
authenticate the data submitted in the CMC
section of the application
Verify that all relevant data (eg stability
biobatch data) were submitted in the CMC section
such that CDER product reviewers can rely on the
submitted data as complete and accurate ndash Laboratory notebooks and associated chromatograms generated
during release testing of biobatch
ndash Failure to include aberrant test results in CMC section
ndash Improper invalidation of OOS results
19
Investigatorrsquos Role during PAI
Assess the following
ndash Quality Systems
ndash Manufacturing Operations
ndash Sampling Plans
ndash Laboratory
ndash Test Methods Validation
ndash Drug Product Specifications
ndash ReprocessingReworking
ndash Standard Operating Procedures
ndash Batch Records 20
The Firmrsquos Role
Make records available (as appropriate) to
conduct the pre-approval inspection
ndash Product Development Report
ndash Batch Records and Laboratory Records
ndash ProtocolsSOPs
Assure facility is cGMP compliant and ready for
an FDA inspection
ndash Once an application is submitted to Center the firm
should be considered ready for inspection
21
Product Development Report
A very useful document for both the firm
and the FDA
The data generated during product
development which defines the drug
product targets the steps in the
manufacturing process where variation is
critical to quality and thereby focuses the
subsequent process validation effort
22
Product Development Report
API Impurity Profile
ndash How is the API characterized
Excipients
Formulation Wet or Dry Granulation
ndash Solution or Suspension
ndash Sterile - Terminalaseptic conditions
ndash TabletCapsule - ImmediateModified
ReleaseExtended Release
23
Product Development Report
Describes the development of Processing
Equipment order of addition of ingredients
to the formulation mixing times and
speeds drying time and temperature
nitrogen blankets blending hold times
compression slugging filling polishing
imprinting labeling and packaging
Product Development Report may not be a
formal document
24
Batch Records
The batch records submitted in the
application must be audited as part of
the inspection to assure
ndash That the proposed production process is the
same process that was used for the
manufacture of the biostability batches
25
ReprocessingReworking
cGMP regulations require reprocessing
procedures to be in writing If firm
makes provisions for reprocessing drug
product details must be submitted as
part of the application
ndash Standard Operating Procedures
ndash QA review and approval
26
27
Different Responsibilities
28
Different Responsibilities
29
Different Responsibilities
How ICH Q8 Q9 Q10
Guidelines Are Working
Together Throughout The
Product Life Cycle
30
Nov 2005 amp Nov 2008
ICH Q8 Q9 and Q10
High level guidances
(not prescriptive)
Science and risk-based
Encourages systematic
approaches
Applicable over entire product
lifecycle
Intended to work together to
enhance pharmaceutical product
quality
31
Pharmaceutical Development -
Q8(R2)
Describes science and risk-based
approaches for pharmaceutical product and
manufacturing process development
Introduced concepts of design space and
flexible regulatory approaches
Introduced concepts of Quality by Design
(QbD) and provided examples of QbD
development approaches and design space 32
Differing Approaches to
Pharmaceutical Development
33
Q8(R2) - Example QbD
Approach
Quality Target Product Profile (QTPP)
Determine ldquopotentialrdquo critical quality attributes
Link raw material attributes and process parameters
to CQAs and perform risk assessment
Develop a design space (optional and not required)
Design and implement a control strategy
Manage product lifecycle including continual
improvement
CQArsquos
Product Profile
Risk Assessments
Design Space
Control Strategy
Continual
Improvement34
Product Lifecycle
concept
Human and veterinary
drugs
Biotech products
Finish products amp APIs
Drug constituent of a
combination product
35
Manage product lifecycle including
continual improvement
Three-stage approach to process validation
Stage 1 ndash Process Design ndash Building and capturing process knowledge and understanding
ndash Establishing a strategy for process control
Stage 2 ndash Process Qualification ndash Design a facility and qualification of utilities and equipment
ndash Process performance qualification
ndash PPQ protocol
ndash PPQ protocol execution and report
Stage 3 ndash Continued Process Verification
36
Focus on alignment with
lsquoproduct lifecyclersquo
Manufacturers should
Understand the sources of variation
Detect the presence and degree of variation
Understand the impact of variation on the process and
ultimately on product attributes
Control the variation in a manner commensurate with
the risk it represents to the process and product
37
Process Validation
38
Focus on alignment with
lsquoproduct lifecyclersquo
39
Focus on alignment with
lsquoproduct lifecyclersquo
40
Focus on alignment with
lsquoproduct lifecyclersquo
FDA Quality Systems Guidance
for Finished Pharmaceuticals
41 Inspection Readiness BEC
Inspection Readiness BEC 42
Quality System
Drug Inspection Target Includes the Quality Control Unit and all of its review
and approval duties
ndash Approval of and adherence to Procedures and associated recordkeeping systems
ndash Product Reviews
ndash Complaint Reviews
ndash Failure Evaluations
ndash Change Control
ndash Product Improvement Projects
ndash RejectsReprocessingRework
ndash Stability
ndash Validation
ndash Training
Inspection Readiness BEC 43
Facilities and Equipment
Inspection Target
Building and Facilities along with
maintenance
Equipment qualification calibration and
maintenance
Water steam compressed gas HVAC
Change control system
Investigate discrepancies
Inspection Readiness BEC 44
Materials
Inspection Target
Procedures and documentation showing adequate control of finished products in-process materials components containers closures
Qualificationvalidation and security of computerized or automated processes
Change control system
Investigation of Discrepancies
Inspection Readiness BEC 45
Production System
Drug Inspection Target
Trainingqualification of personnel
Complete batch production documentation
Process validation
Production time limits
In-process testingexamination
Change Control System
Investigation of discrepancies
Inspection Readiness BEC 46
Packaging and Labeling
Inspection Target
Training
Acceptance operations
Control of materials to prevent mix-ups
Accountability
Packaginglabeling records
Line separation
Line clearance inspection and documentation
Validation of labelingpackaging operations
Investigation of discrepancies
Inspection Readiness BEC 47
Laboratory Control
Inspection Target
Staffing and training
Adequate equipment calibration and maintenance
Reference standards
Adherence to written procedures
Validationverification analytical methods
Analytical records and raw data
Adherence to OOS procedure and timely completion of investigations
Stability testing program and reserve samples
Change control system
Before the Inspection
SOP for Inspection Readiness
Strong Internal AuditAssessment function
ndash Continuous assessment using Risk Management
Criteria
ndash Not autopilot auditing
ndash Same auditorhellip
Perform a full-system audit (Mock) at least one
month prior to inspection
ndash Be sure any necessary CA-PA has been originated 48
Before the Inspection
Educate all your employees on proper inspection
rules and etiquette
Educate your supervisory (all exempt) personnel
on FDA regulations
Inspections and external audits concern to all
employees not only those from Quality Dept
Promote the participation on the internal audit
cadre
Certify internal auditors
49
Before the Inspection
A few words about Internal Audit
Internal Audits are the most powerful tool to
avoid inspection surprises
If the inspector discovers something why your
internal auditor did not
The same auditor auditing several consecutive
years the same area is not effective
An effective internal audit function is priceless
50
During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

2
Agenda
US FDA Structure and Authority
Generic Drug Approval Process
ndash The PreApproval Inspection (PAI)
Pharmaceutical development
ndash QBD
ndash Process Validations
FDA Quality System Inspection Elements
Pre-Inspection Activities the Value of a Mock Audit
Interaction with FDA Inspectors
Data Integrity
3
FDA Overview US Department of Health amp Human Services
US Food and Drug Administration Centers for
ndash Food Safety and Nutrition (CFSAN)
ndash Drug Evaluation and Research (CDER)
ndash Biologics Evaluation and Research (CBER)
ndash Devices and Radiological Health (CDRH)
ndash Center for Veterinary Medicine (CVM)
ndash Center for Tobacco Products (CTP)
ndash Office of Regulatory Affairs (ORA HQ)
ndash Field Regions (SER SWR PAR NER CER) raquo District Offices
San Juan District Office (SJN-DO)
ndash SJN MRP PON USVI
14000
4
FDA Mission To ensure that
ndash Foods are safe wholesome and sanitary
ndash Human and veterinary drugs biological products
and medical devices are safe and effective
ndash Cosmetics are safe
ndash Electronic products that emit radiation are safe
Authority for Inspections
United States of America
ndash Federal Food Drugs amp
Cosmetic Act
ndash The FDA is responsible for
protecting the public health by
assuring the safety efficacy and
security of human and veterinary
drugs biological products
medical devices our nationrsquos
food supply cosmetics and
products that emit radiation 5
6
FDA Laws Overview
1906 ndash Congress enacted Food and Drug Act
1938- Federal Food Drug and Cosmetic Act
ndash To ensure that food are safe and produced under sanitary conditions
ndash Drugs and device are safe and effectives for their intended use
ndash Cosmetics are safe and made from appropriate ingredients
ndash Labeling and packaging is truthful
1963 - Fist version of GMP for Drugs
1978 cGMP for Drugs 21 CFR 211
1987 Guideline on General Principles of Process Validation
1997 ndash FDA Modernization Act (FDAMA) Fine-tuning FDA laws
1997- Electronic Records (21 CFR Part 11)
2013 ndash Combination Products
Generic Drugs The use of bioequivalence as the basis for approving generic
copies of drug products was established by the ldquoDrug Price
Competition and Patent Term Restoration Act of 1984rdquo also
known as the Waxman-Hatch Act
This Act expedites the availability of less costly generic drugs by
permitting the FDA to approve applications to market generic
versions of brand-name drugs without conducting costly and
duplicative clinical trials At the same time the brand-name
companies can apply for up to five additional years patent
protection for the new medicines they developed to make up for
time lost while their products were going through the FDArsquos
approval process Brand-name drugs are subject to the same
bioequivalence tests as generics upon reformulation 7
8
9
The FDArsquos Drug Review Process
10
The FDArsquos Drug Review Process
11
NDA vs ANDA Review Process
12
Center for Drug Evaluation amp Research Office of Generic Drugs (OGD) 13
Generic Drug Review Process
Bioequivalence Review Labeling Review Chemistry amp Micro
Review Request for Plant
Inspection
APPLICANT
ANDA
Acceptable amp
Complete
Application Review
N ChemMicro
OK
Labeling
OK
Bioequivalence
OK
PreApproval
Inspection Results
OK
Not Approvable
Letter
Approval Withheld until
Results Satisfactory
Bio Deficiency Letter
APPROVED ANDA
N N N
N
Y Y Y
Y
Y
Refuse to Receive Letter
What is the goal of a
Pre-Approval Inspection
To assure that establishments involved in the
manufacturing testing or other manipulation of
new drug dosage forms and drug substances are
evaluated for
ndash conformance with commitments in the application
ndash site cGMP compliance
ndash data authenticity reliability and accuracy
ndash adequacy of analytical methodologies
14
PAI Objectives (Program 7346832)
There are three primary inspectional objectives of
this PAI program These objectives are
1) Readiness for Commercial Manufacturing
2) Conformance to Application
3) Data Integrity Audit
15
PAI Objective 1 Readiness for Commercial Manufacturing
Determine whether the establishment has a quality
system that is designed to achieve sufficient control over
the facility and commercial manufacturing operations
a) Manufacturing and laboratory changes deviations and
trends relating to the development of new drug substance
and product manufacturing have been adequately
evaluated
b) A sound and appropriate program for sampling testing
and evaluation of components in-process materials
finished products containers and closures for the purpose
of releasing materials or products has been established
including a robust supplier qualification program
16
PAI Objective 1 Readiness for Commercial Manufacturing cont
c) The establishment has sufficient facility and equipment
controls in place to prevent contamination of and by the
application product (or API)
d) Adequate procedures exist for batch release change control
investigating failures deviations complaints and adverse
events and for reporting this information to FDA such as
field alert reporting
e) The feasibility of the proposed commercial process and
manufacturing batch record including instructions
processing parameters and process control measures are
scientifically and objectively justified This objective is
linked to the firmrsquos process validation program
17
PAI Objective 2
Conformance to Application
Verify that the formulation manufacturing or
processing methods and analytical (or
examination) methods are consistent with
descriptions contained in the CMC section of the
application for the biobatch (and other pivotal
clinical batches when applicable) the proposed
commercial scale batch and API ndash Observing processing andor testing operations
ndash Compare the biobatch manufacturing process against the
proposed commercial batch record
18
PAI Objective 3
Data Integrity Audit
Audit the raw data hardcopy or electronic to
authenticate the data submitted in the CMC
section of the application
Verify that all relevant data (eg stability
biobatch data) were submitted in the CMC section
such that CDER product reviewers can rely on the
submitted data as complete and accurate ndash Laboratory notebooks and associated chromatograms generated
during release testing of biobatch
ndash Failure to include aberrant test results in CMC section
ndash Improper invalidation of OOS results
19
Investigatorrsquos Role during PAI
Assess the following
ndash Quality Systems
ndash Manufacturing Operations
ndash Sampling Plans
ndash Laboratory
ndash Test Methods Validation
ndash Drug Product Specifications
ndash ReprocessingReworking
ndash Standard Operating Procedures
ndash Batch Records 20
The Firmrsquos Role
Make records available (as appropriate) to
conduct the pre-approval inspection
ndash Product Development Report
ndash Batch Records and Laboratory Records
ndash ProtocolsSOPs
Assure facility is cGMP compliant and ready for
an FDA inspection
ndash Once an application is submitted to Center the firm
should be considered ready for inspection
21
Product Development Report
A very useful document for both the firm
and the FDA
The data generated during product
development which defines the drug
product targets the steps in the
manufacturing process where variation is
critical to quality and thereby focuses the
subsequent process validation effort
22
Product Development Report
API Impurity Profile
ndash How is the API characterized
Excipients
Formulation Wet or Dry Granulation
ndash Solution or Suspension
ndash Sterile - Terminalaseptic conditions
ndash TabletCapsule - ImmediateModified
ReleaseExtended Release
23
Product Development Report
Describes the development of Processing
Equipment order of addition of ingredients
to the formulation mixing times and
speeds drying time and temperature
nitrogen blankets blending hold times
compression slugging filling polishing
imprinting labeling and packaging
Product Development Report may not be a
formal document
24
Batch Records
The batch records submitted in the
application must be audited as part of
the inspection to assure
ndash That the proposed production process is the
same process that was used for the
manufacture of the biostability batches
25
ReprocessingReworking
cGMP regulations require reprocessing
procedures to be in writing If firm
makes provisions for reprocessing drug
product details must be submitted as
part of the application
ndash Standard Operating Procedures
ndash QA review and approval
26
27
Different Responsibilities
28
Different Responsibilities
29
Different Responsibilities
How ICH Q8 Q9 Q10
Guidelines Are Working
Together Throughout The
Product Life Cycle
30
Nov 2005 amp Nov 2008
ICH Q8 Q9 and Q10
High level guidances
(not prescriptive)
Science and risk-based
Encourages systematic
approaches
Applicable over entire product
lifecycle
Intended to work together to
enhance pharmaceutical product
quality
31
Pharmaceutical Development -
Q8(R2)
Describes science and risk-based
approaches for pharmaceutical product and
manufacturing process development
Introduced concepts of design space and
flexible regulatory approaches
Introduced concepts of Quality by Design
(QbD) and provided examples of QbD
development approaches and design space 32
Differing Approaches to
Pharmaceutical Development
33
Q8(R2) - Example QbD
Approach
Quality Target Product Profile (QTPP)
Determine ldquopotentialrdquo critical quality attributes
Link raw material attributes and process parameters
to CQAs and perform risk assessment
Develop a design space (optional and not required)
Design and implement a control strategy
Manage product lifecycle including continual
improvement
CQArsquos
Product Profile
Risk Assessments
Design Space
Control Strategy
Continual
Improvement34
Product Lifecycle
concept
Human and veterinary
drugs
Biotech products
Finish products amp APIs
Drug constituent of a
combination product
35
Manage product lifecycle including
continual improvement
Three-stage approach to process validation
Stage 1 ndash Process Design ndash Building and capturing process knowledge and understanding
ndash Establishing a strategy for process control
Stage 2 ndash Process Qualification ndash Design a facility and qualification of utilities and equipment
ndash Process performance qualification
ndash PPQ protocol
ndash PPQ protocol execution and report
Stage 3 ndash Continued Process Verification
36
Focus on alignment with
lsquoproduct lifecyclersquo
Manufacturers should
Understand the sources of variation
Detect the presence and degree of variation
Understand the impact of variation on the process and
ultimately on product attributes
Control the variation in a manner commensurate with
the risk it represents to the process and product
37
Process Validation
38
Focus on alignment with
lsquoproduct lifecyclersquo
39
Focus on alignment with
lsquoproduct lifecyclersquo
40
Focus on alignment with
lsquoproduct lifecyclersquo
FDA Quality Systems Guidance
for Finished Pharmaceuticals
41 Inspection Readiness BEC
Inspection Readiness BEC 42
Quality System
Drug Inspection Target Includes the Quality Control Unit and all of its review
and approval duties
ndash Approval of and adherence to Procedures and associated recordkeeping systems
ndash Product Reviews
ndash Complaint Reviews
ndash Failure Evaluations
ndash Change Control
ndash Product Improvement Projects
ndash RejectsReprocessingRework
ndash Stability
ndash Validation
ndash Training
Inspection Readiness BEC 43
Facilities and Equipment
Inspection Target
Building and Facilities along with
maintenance
Equipment qualification calibration and
maintenance
Water steam compressed gas HVAC
Change control system
Investigate discrepancies
Inspection Readiness BEC 44
Materials
Inspection Target
Procedures and documentation showing adequate control of finished products in-process materials components containers closures
Qualificationvalidation and security of computerized or automated processes
Change control system
Investigation of Discrepancies
Inspection Readiness BEC 45
Production System
Drug Inspection Target
Trainingqualification of personnel
Complete batch production documentation
Process validation
Production time limits
In-process testingexamination
Change Control System
Investigation of discrepancies
Inspection Readiness BEC 46
Packaging and Labeling
Inspection Target
Training
Acceptance operations
Control of materials to prevent mix-ups
Accountability
Packaginglabeling records
Line separation
Line clearance inspection and documentation
Validation of labelingpackaging operations
Investigation of discrepancies
Inspection Readiness BEC 47
Laboratory Control
Inspection Target
Staffing and training
Adequate equipment calibration and maintenance
Reference standards
Adherence to written procedures
Validationverification analytical methods
Analytical records and raw data
Adherence to OOS procedure and timely completion of investigations
Stability testing program and reserve samples
Change control system
Before the Inspection
SOP for Inspection Readiness
Strong Internal AuditAssessment function
ndash Continuous assessment using Risk Management
Criteria
ndash Not autopilot auditing
ndash Same auditorhellip
Perform a full-system audit (Mock) at least one
month prior to inspection
ndash Be sure any necessary CA-PA has been originated 48
Before the Inspection
Educate all your employees on proper inspection
rules and etiquette
Educate your supervisory (all exempt) personnel
on FDA regulations
Inspections and external audits concern to all
employees not only those from Quality Dept
Promote the participation on the internal audit
cadre
Certify internal auditors
49
Before the Inspection
A few words about Internal Audit
Internal Audits are the most powerful tool to
avoid inspection surprises
If the inspector discovers something why your
internal auditor did not
The same auditor auditing several consecutive
years the same area is not effective
An effective internal audit function is priceless
50
During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

3
FDA Overview US Department of Health amp Human Services
US Food and Drug Administration Centers for
ndash Food Safety and Nutrition (CFSAN)
ndash Drug Evaluation and Research (CDER)
ndash Biologics Evaluation and Research (CBER)
ndash Devices and Radiological Health (CDRH)
ndash Center for Veterinary Medicine (CVM)
ndash Center for Tobacco Products (CTP)
ndash Office of Regulatory Affairs (ORA HQ)
ndash Field Regions (SER SWR PAR NER CER) raquo District Offices
San Juan District Office (SJN-DO)
ndash SJN MRP PON USVI
14000
4
FDA Mission To ensure that
ndash Foods are safe wholesome and sanitary
ndash Human and veterinary drugs biological products
and medical devices are safe and effective
ndash Cosmetics are safe
ndash Electronic products that emit radiation are safe
Authority for Inspections
United States of America
ndash Federal Food Drugs amp
Cosmetic Act
ndash The FDA is responsible for
protecting the public health by
assuring the safety efficacy and
security of human and veterinary
drugs biological products
medical devices our nationrsquos
food supply cosmetics and
products that emit radiation 5
6
FDA Laws Overview
1906 ndash Congress enacted Food and Drug Act
1938- Federal Food Drug and Cosmetic Act
ndash To ensure that food are safe and produced under sanitary conditions
ndash Drugs and device are safe and effectives for their intended use
ndash Cosmetics are safe and made from appropriate ingredients
ndash Labeling and packaging is truthful
1963 - Fist version of GMP for Drugs
1978 cGMP for Drugs 21 CFR 211
1987 Guideline on General Principles of Process Validation
1997 ndash FDA Modernization Act (FDAMA) Fine-tuning FDA laws
1997- Electronic Records (21 CFR Part 11)
2013 ndash Combination Products
Generic Drugs The use of bioequivalence as the basis for approving generic
copies of drug products was established by the ldquoDrug Price
Competition and Patent Term Restoration Act of 1984rdquo also
known as the Waxman-Hatch Act
This Act expedites the availability of less costly generic drugs by
permitting the FDA to approve applications to market generic
versions of brand-name drugs without conducting costly and
duplicative clinical trials At the same time the brand-name
companies can apply for up to five additional years patent
protection for the new medicines they developed to make up for
time lost while their products were going through the FDArsquos
approval process Brand-name drugs are subject to the same
bioequivalence tests as generics upon reformulation 7
8
9
The FDArsquos Drug Review Process
10
The FDArsquos Drug Review Process
11
NDA vs ANDA Review Process
12
Center for Drug Evaluation amp Research Office of Generic Drugs (OGD) 13
Generic Drug Review Process
Bioequivalence Review Labeling Review Chemistry amp Micro
Review Request for Plant
Inspection
APPLICANT
ANDA
Acceptable amp
Complete
Application Review
N ChemMicro
OK
Labeling
OK
Bioequivalence
OK
PreApproval
Inspection Results
OK
Not Approvable
Letter
Approval Withheld until
Results Satisfactory
Bio Deficiency Letter
APPROVED ANDA
N N N
N
Y Y Y
Y
Y
Refuse to Receive Letter
What is the goal of a
Pre-Approval Inspection
To assure that establishments involved in the
manufacturing testing or other manipulation of
new drug dosage forms and drug substances are
evaluated for
ndash conformance with commitments in the application
ndash site cGMP compliance
ndash data authenticity reliability and accuracy
ndash adequacy of analytical methodologies
14
PAI Objectives (Program 7346832)
There are three primary inspectional objectives of
this PAI program These objectives are
1) Readiness for Commercial Manufacturing
2) Conformance to Application
3) Data Integrity Audit
15
PAI Objective 1 Readiness for Commercial Manufacturing
Determine whether the establishment has a quality
system that is designed to achieve sufficient control over
the facility and commercial manufacturing operations
a) Manufacturing and laboratory changes deviations and
trends relating to the development of new drug substance
and product manufacturing have been adequately
evaluated
b) A sound and appropriate program for sampling testing
and evaluation of components in-process materials
finished products containers and closures for the purpose
of releasing materials or products has been established
including a robust supplier qualification program
16
PAI Objective 1 Readiness for Commercial Manufacturing cont
c) The establishment has sufficient facility and equipment
controls in place to prevent contamination of and by the
application product (or API)
d) Adequate procedures exist for batch release change control
investigating failures deviations complaints and adverse
events and for reporting this information to FDA such as
field alert reporting
e) The feasibility of the proposed commercial process and
manufacturing batch record including instructions
processing parameters and process control measures are
scientifically and objectively justified This objective is
linked to the firmrsquos process validation program
17
PAI Objective 2
Conformance to Application
Verify that the formulation manufacturing or
processing methods and analytical (or
examination) methods are consistent with
descriptions contained in the CMC section of the
application for the biobatch (and other pivotal
clinical batches when applicable) the proposed
commercial scale batch and API ndash Observing processing andor testing operations
ndash Compare the biobatch manufacturing process against the
proposed commercial batch record
18
PAI Objective 3
Data Integrity Audit
Audit the raw data hardcopy or electronic to
authenticate the data submitted in the CMC
section of the application
Verify that all relevant data (eg stability
biobatch data) were submitted in the CMC section
such that CDER product reviewers can rely on the
submitted data as complete and accurate ndash Laboratory notebooks and associated chromatograms generated
during release testing of biobatch
ndash Failure to include aberrant test results in CMC section
ndash Improper invalidation of OOS results
19
Investigatorrsquos Role during PAI
Assess the following
ndash Quality Systems
ndash Manufacturing Operations
ndash Sampling Plans
ndash Laboratory
ndash Test Methods Validation
ndash Drug Product Specifications
ndash ReprocessingReworking
ndash Standard Operating Procedures
ndash Batch Records 20
The Firmrsquos Role
Make records available (as appropriate) to
conduct the pre-approval inspection
ndash Product Development Report
ndash Batch Records and Laboratory Records
ndash ProtocolsSOPs
Assure facility is cGMP compliant and ready for
an FDA inspection
ndash Once an application is submitted to Center the firm
should be considered ready for inspection
21
Product Development Report
A very useful document for both the firm
and the FDA
The data generated during product
development which defines the drug
product targets the steps in the
manufacturing process where variation is
critical to quality and thereby focuses the
subsequent process validation effort
22
Product Development Report
API Impurity Profile
ndash How is the API characterized
Excipients
Formulation Wet or Dry Granulation
ndash Solution or Suspension
ndash Sterile - Terminalaseptic conditions
ndash TabletCapsule - ImmediateModified
ReleaseExtended Release
23
Product Development Report
Describes the development of Processing
Equipment order of addition of ingredients
to the formulation mixing times and
speeds drying time and temperature
nitrogen blankets blending hold times
compression slugging filling polishing
imprinting labeling and packaging
Product Development Report may not be a
formal document
24
Batch Records
The batch records submitted in the
application must be audited as part of
the inspection to assure
ndash That the proposed production process is the
same process that was used for the
manufacture of the biostability batches
25
ReprocessingReworking
cGMP regulations require reprocessing
procedures to be in writing If firm
makes provisions for reprocessing drug
product details must be submitted as
part of the application
ndash Standard Operating Procedures
ndash QA review and approval
26
27
Different Responsibilities
28
Different Responsibilities
29
Different Responsibilities
How ICH Q8 Q9 Q10
Guidelines Are Working
Together Throughout The
Product Life Cycle
30
Nov 2005 amp Nov 2008
ICH Q8 Q9 and Q10
High level guidances
(not prescriptive)
Science and risk-based
Encourages systematic
approaches
Applicable over entire product
lifecycle
Intended to work together to
enhance pharmaceutical product
quality
31
Pharmaceutical Development -
Q8(R2)
Describes science and risk-based
approaches for pharmaceutical product and
manufacturing process development
Introduced concepts of design space and
flexible regulatory approaches
Introduced concepts of Quality by Design
(QbD) and provided examples of QbD
development approaches and design space 32
Differing Approaches to
Pharmaceutical Development
33
Q8(R2) - Example QbD
Approach
Quality Target Product Profile (QTPP)
Determine ldquopotentialrdquo critical quality attributes
Link raw material attributes and process parameters
to CQAs and perform risk assessment
Develop a design space (optional and not required)
Design and implement a control strategy
Manage product lifecycle including continual
improvement
CQArsquos
Product Profile
Risk Assessments
Design Space
Control Strategy
Continual
Improvement34
Product Lifecycle
concept
Human and veterinary
drugs
Biotech products
Finish products amp APIs
Drug constituent of a
combination product
35
Manage product lifecycle including
continual improvement
Three-stage approach to process validation
Stage 1 ndash Process Design ndash Building and capturing process knowledge and understanding
ndash Establishing a strategy for process control
Stage 2 ndash Process Qualification ndash Design a facility and qualification of utilities and equipment
ndash Process performance qualification
ndash PPQ protocol
ndash PPQ protocol execution and report
Stage 3 ndash Continued Process Verification
36
Focus on alignment with
lsquoproduct lifecyclersquo
Manufacturers should
Understand the sources of variation
Detect the presence and degree of variation
Understand the impact of variation on the process and
ultimately on product attributes
Control the variation in a manner commensurate with
the risk it represents to the process and product
37
Process Validation
38
Focus on alignment with
lsquoproduct lifecyclersquo
39
Focus on alignment with
lsquoproduct lifecyclersquo
40
Focus on alignment with
lsquoproduct lifecyclersquo
FDA Quality Systems Guidance
for Finished Pharmaceuticals
41 Inspection Readiness BEC
Inspection Readiness BEC 42
Quality System
Drug Inspection Target Includes the Quality Control Unit and all of its review
and approval duties
ndash Approval of and adherence to Procedures and associated recordkeeping systems
ndash Product Reviews
ndash Complaint Reviews
ndash Failure Evaluations
ndash Change Control
ndash Product Improvement Projects
ndash RejectsReprocessingRework
ndash Stability
ndash Validation
ndash Training
Inspection Readiness BEC 43
Facilities and Equipment
Inspection Target
Building and Facilities along with
maintenance
Equipment qualification calibration and
maintenance
Water steam compressed gas HVAC
Change control system
Investigate discrepancies
Inspection Readiness BEC 44
Materials
Inspection Target
Procedures and documentation showing adequate control of finished products in-process materials components containers closures
Qualificationvalidation and security of computerized or automated processes
Change control system
Investigation of Discrepancies
Inspection Readiness BEC 45
Production System
Drug Inspection Target
Trainingqualification of personnel
Complete batch production documentation
Process validation
Production time limits
In-process testingexamination
Change Control System
Investigation of discrepancies
Inspection Readiness BEC 46
Packaging and Labeling
Inspection Target
Training
Acceptance operations
Control of materials to prevent mix-ups
Accountability
Packaginglabeling records
Line separation
Line clearance inspection and documentation
Validation of labelingpackaging operations
Investigation of discrepancies
Inspection Readiness BEC 47
Laboratory Control
Inspection Target
Staffing and training
Adequate equipment calibration and maintenance
Reference standards
Adherence to written procedures
Validationverification analytical methods
Analytical records and raw data
Adherence to OOS procedure and timely completion of investigations
Stability testing program and reserve samples
Change control system
Before the Inspection
SOP for Inspection Readiness
Strong Internal AuditAssessment function
ndash Continuous assessment using Risk Management
Criteria
ndash Not autopilot auditing
ndash Same auditorhellip
Perform a full-system audit (Mock) at least one
month prior to inspection
ndash Be sure any necessary CA-PA has been originated 48
Before the Inspection
Educate all your employees on proper inspection
rules and etiquette
Educate your supervisory (all exempt) personnel
on FDA regulations
Inspections and external audits concern to all
employees not only those from Quality Dept
Promote the participation on the internal audit
cadre
Certify internal auditors
49
Before the Inspection
A few words about Internal Audit
Internal Audits are the most powerful tool to
avoid inspection surprises
If the inspector discovers something why your
internal auditor did not
The same auditor auditing several consecutive
years the same area is not effective
An effective internal audit function is priceless
50
During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

4
FDA Mission To ensure that
ndash Foods are safe wholesome and sanitary
ndash Human and veterinary drugs biological products
and medical devices are safe and effective
ndash Cosmetics are safe
ndash Electronic products that emit radiation are safe
Authority for Inspections
United States of America
ndash Federal Food Drugs amp
Cosmetic Act
ndash The FDA is responsible for
protecting the public health by
assuring the safety efficacy and
security of human and veterinary
drugs biological products
medical devices our nationrsquos
food supply cosmetics and
products that emit radiation 5
6
FDA Laws Overview
1906 ndash Congress enacted Food and Drug Act
1938- Federal Food Drug and Cosmetic Act
ndash To ensure that food are safe and produced under sanitary conditions
ndash Drugs and device are safe and effectives for their intended use
ndash Cosmetics are safe and made from appropriate ingredients
ndash Labeling and packaging is truthful
1963 - Fist version of GMP for Drugs
1978 cGMP for Drugs 21 CFR 211
1987 Guideline on General Principles of Process Validation
1997 ndash FDA Modernization Act (FDAMA) Fine-tuning FDA laws
1997- Electronic Records (21 CFR Part 11)
2013 ndash Combination Products
Generic Drugs The use of bioequivalence as the basis for approving generic
copies of drug products was established by the ldquoDrug Price
Competition and Patent Term Restoration Act of 1984rdquo also
known as the Waxman-Hatch Act
This Act expedites the availability of less costly generic drugs by
permitting the FDA to approve applications to market generic
versions of brand-name drugs without conducting costly and
duplicative clinical trials At the same time the brand-name
companies can apply for up to five additional years patent
protection for the new medicines they developed to make up for
time lost while their products were going through the FDArsquos
approval process Brand-name drugs are subject to the same
bioequivalence tests as generics upon reformulation 7
8
9
The FDArsquos Drug Review Process
10
The FDArsquos Drug Review Process
11
NDA vs ANDA Review Process
12
Center for Drug Evaluation amp Research Office of Generic Drugs (OGD) 13
Generic Drug Review Process
Bioequivalence Review Labeling Review Chemistry amp Micro
Review Request for Plant
Inspection
APPLICANT
ANDA
Acceptable amp
Complete
Application Review
N ChemMicro
OK
Labeling
OK
Bioequivalence
OK
PreApproval
Inspection Results
OK
Not Approvable
Letter
Approval Withheld until
Results Satisfactory
Bio Deficiency Letter
APPROVED ANDA
N N N
N
Y Y Y
Y
Y
Refuse to Receive Letter
What is the goal of a
Pre-Approval Inspection
To assure that establishments involved in the
manufacturing testing or other manipulation of
new drug dosage forms and drug substances are
evaluated for
ndash conformance with commitments in the application
ndash site cGMP compliance
ndash data authenticity reliability and accuracy
ndash adequacy of analytical methodologies
14
PAI Objectives (Program 7346832)
There are three primary inspectional objectives of
this PAI program These objectives are
1) Readiness for Commercial Manufacturing
2) Conformance to Application
3) Data Integrity Audit
15
PAI Objective 1 Readiness for Commercial Manufacturing
Determine whether the establishment has a quality
system that is designed to achieve sufficient control over
the facility and commercial manufacturing operations
a) Manufacturing and laboratory changes deviations and
trends relating to the development of new drug substance
and product manufacturing have been adequately
evaluated
b) A sound and appropriate program for sampling testing
and evaluation of components in-process materials
finished products containers and closures for the purpose
of releasing materials or products has been established
including a robust supplier qualification program
16
PAI Objective 1 Readiness for Commercial Manufacturing cont
c) The establishment has sufficient facility and equipment
controls in place to prevent contamination of and by the
application product (or API)
d) Adequate procedures exist for batch release change control
investigating failures deviations complaints and adverse
events and for reporting this information to FDA such as
field alert reporting
e) The feasibility of the proposed commercial process and
manufacturing batch record including instructions
processing parameters and process control measures are
scientifically and objectively justified This objective is
linked to the firmrsquos process validation program
17
PAI Objective 2
Conformance to Application
Verify that the formulation manufacturing or
processing methods and analytical (or
examination) methods are consistent with
descriptions contained in the CMC section of the
application for the biobatch (and other pivotal
clinical batches when applicable) the proposed
commercial scale batch and API ndash Observing processing andor testing operations
ndash Compare the biobatch manufacturing process against the
proposed commercial batch record
18
PAI Objective 3
Data Integrity Audit
Audit the raw data hardcopy or electronic to
authenticate the data submitted in the CMC
section of the application
Verify that all relevant data (eg stability
biobatch data) were submitted in the CMC section
such that CDER product reviewers can rely on the
submitted data as complete and accurate ndash Laboratory notebooks and associated chromatograms generated
during release testing of biobatch
ndash Failure to include aberrant test results in CMC section
ndash Improper invalidation of OOS results
19
Investigatorrsquos Role during PAI
Assess the following
ndash Quality Systems
ndash Manufacturing Operations
ndash Sampling Plans
ndash Laboratory
ndash Test Methods Validation
ndash Drug Product Specifications
ndash ReprocessingReworking
ndash Standard Operating Procedures
ndash Batch Records 20
The Firmrsquos Role
Make records available (as appropriate) to
conduct the pre-approval inspection
ndash Product Development Report
ndash Batch Records and Laboratory Records
ndash ProtocolsSOPs
Assure facility is cGMP compliant and ready for
an FDA inspection
ndash Once an application is submitted to Center the firm
should be considered ready for inspection
21
Product Development Report
A very useful document for both the firm
and the FDA
The data generated during product
development which defines the drug
product targets the steps in the
manufacturing process where variation is
critical to quality and thereby focuses the
subsequent process validation effort
22
Product Development Report
API Impurity Profile
ndash How is the API characterized
Excipients
Formulation Wet or Dry Granulation
ndash Solution or Suspension
ndash Sterile - Terminalaseptic conditions
ndash TabletCapsule - ImmediateModified
ReleaseExtended Release
23
Product Development Report
Describes the development of Processing
Equipment order of addition of ingredients
to the formulation mixing times and
speeds drying time and temperature
nitrogen blankets blending hold times
compression slugging filling polishing
imprinting labeling and packaging
Product Development Report may not be a
formal document
24
Batch Records
The batch records submitted in the
application must be audited as part of
the inspection to assure
ndash That the proposed production process is the
same process that was used for the
manufacture of the biostability batches
25
ReprocessingReworking
cGMP regulations require reprocessing
procedures to be in writing If firm
makes provisions for reprocessing drug
product details must be submitted as
part of the application
ndash Standard Operating Procedures
ndash QA review and approval
26
27
Different Responsibilities
28
Different Responsibilities
29
Different Responsibilities
How ICH Q8 Q9 Q10
Guidelines Are Working
Together Throughout The
Product Life Cycle
30
Nov 2005 amp Nov 2008
ICH Q8 Q9 and Q10
High level guidances
(not prescriptive)
Science and risk-based
Encourages systematic
approaches
Applicable over entire product
lifecycle
Intended to work together to
enhance pharmaceutical product
quality
31
Pharmaceutical Development -
Q8(R2)
Describes science and risk-based
approaches for pharmaceutical product and
manufacturing process development
Introduced concepts of design space and
flexible regulatory approaches
Introduced concepts of Quality by Design
(QbD) and provided examples of QbD
development approaches and design space 32
Differing Approaches to
Pharmaceutical Development
33
Q8(R2) - Example QbD
Approach
Quality Target Product Profile (QTPP)
Determine ldquopotentialrdquo critical quality attributes
Link raw material attributes and process parameters
to CQAs and perform risk assessment
Develop a design space (optional and not required)
Design and implement a control strategy
Manage product lifecycle including continual
improvement
CQArsquos
Product Profile
Risk Assessments
Design Space
Control Strategy
Continual
Improvement34
Product Lifecycle
concept
Human and veterinary
drugs
Biotech products
Finish products amp APIs
Drug constituent of a
combination product
35
Manage product lifecycle including
continual improvement
Three-stage approach to process validation
Stage 1 ndash Process Design ndash Building and capturing process knowledge and understanding
ndash Establishing a strategy for process control
Stage 2 ndash Process Qualification ndash Design a facility and qualification of utilities and equipment
ndash Process performance qualification
ndash PPQ protocol
ndash PPQ protocol execution and report
Stage 3 ndash Continued Process Verification
36
Focus on alignment with
lsquoproduct lifecyclersquo
Manufacturers should
Understand the sources of variation
Detect the presence and degree of variation
Understand the impact of variation on the process and
ultimately on product attributes
Control the variation in a manner commensurate with
the risk it represents to the process and product
37
Process Validation
38
Focus on alignment with
lsquoproduct lifecyclersquo
39
Focus on alignment with
lsquoproduct lifecyclersquo
40
Focus on alignment with
lsquoproduct lifecyclersquo
FDA Quality Systems Guidance
for Finished Pharmaceuticals
41 Inspection Readiness BEC
Inspection Readiness BEC 42
Quality System
Drug Inspection Target Includes the Quality Control Unit and all of its review
and approval duties
ndash Approval of and adherence to Procedures and associated recordkeeping systems
ndash Product Reviews
ndash Complaint Reviews
ndash Failure Evaluations
ndash Change Control
ndash Product Improvement Projects
ndash RejectsReprocessingRework
ndash Stability
ndash Validation
ndash Training
Inspection Readiness BEC 43
Facilities and Equipment
Inspection Target
Building and Facilities along with
maintenance
Equipment qualification calibration and
maintenance
Water steam compressed gas HVAC
Change control system
Investigate discrepancies
Inspection Readiness BEC 44
Materials
Inspection Target
Procedures and documentation showing adequate control of finished products in-process materials components containers closures
Qualificationvalidation and security of computerized or automated processes
Change control system
Investigation of Discrepancies
Inspection Readiness BEC 45
Production System
Drug Inspection Target
Trainingqualification of personnel
Complete batch production documentation
Process validation
Production time limits
In-process testingexamination
Change Control System
Investigation of discrepancies
Inspection Readiness BEC 46
Packaging and Labeling
Inspection Target
Training
Acceptance operations
Control of materials to prevent mix-ups
Accountability
Packaginglabeling records
Line separation
Line clearance inspection and documentation
Validation of labelingpackaging operations
Investigation of discrepancies
Inspection Readiness BEC 47
Laboratory Control
Inspection Target
Staffing and training
Adequate equipment calibration and maintenance
Reference standards
Adherence to written procedures
Validationverification analytical methods
Analytical records and raw data
Adherence to OOS procedure and timely completion of investigations
Stability testing program and reserve samples
Change control system
Before the Inspection
SOP for Inspection Readiness
Strong Internal AuditAssessment function
ndash Continuous assessment using Risk Management
Criteria
ndash Not autopilot auditing
ndash Same auditorhellip
Perform a full-system audit (Mock) at least one
month prior to inspection
ndash Be sure any necessary CA-PA has been originated 48
Before the Inspection
Educate all your employees on proper inspection
rules and etiquette
Educate your supervisory (all exempt) personnel
on FDA regulations
Inspections and external audits concern to all
employees not only those from Quality Dept
Promote the participation on the internal audit
cadre
Certify internal auditors
49
Before the Inspection
A few words about Internal Audit
Internal Audits are the most powerful tool to
avoid inspection surprises
If the inspector discovers something why your
internal auditor did not
The same auditor auditing several consecutive
years the same area is not effective
An effective internal audit function is priceless
50
During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

Authority for Inspections
United States of America
ndash Federal Food Drugs amp
Cosmetic Act
ndash The FDA is responsible for
protecting the public health by
assuring the safety efficacy and
security of human and veterinary
drugs biological products
medical devices our nationrsquos
food supply cosmetics and
products that emit radiation 5
6
FDA Laws Overview
1906 ndash Congress enacted Food and Drug Act
1938- Federal Food Drug and Cosmetic Act
ndash To ensure that food are safe and produced under sanitary conditions
ndash Drugs and device are safe and effectives for their intended use
ndash Cosmetics are safe and made from appropriate ingredients
ndash Labeling and packaging is truthful
1963 - Fist version of GMP for Drugs
1978 cGMP for Drugs 21 CFR 211
1987 Guideline on General Principles of Process Validation
1997 ndash FDA Modernization Act (FDAMA) Fine-tuning FDA laws
1997- Electronic Records (21 CFR Part 11)
2013 ndash Combination Products
Generic Drugs The use of bioequivalence as the basis for approving generic
copies of drug products was established by the ldquoDrug Price
Competition and Patent Term Restoration Act of 1984rdquo also
known as the Waxman-Hatch Act
This Act expedites the availability of less costly generic drugs by
permitting the FDA to approve applications to market generic
versions of brand-name drugs without conducting costly and
duplicative clinical trials At the same time the brand-name
companies can apply for up to five additional years patent
protection for the new medicines they developed to make up for
time lost while their products were going through the FDArsquos
approval process Brand-name drugs are subject to the same
bioequivalence tests as generics upon reformulation 7
8
9
The FDArsquos Drug Review Process
10
The FDArsquos Drug Review Process
11
NDA vs ANDA Review Process
12
Center for Drug Evaluation amp Research Office of Generic Drugs (OGD) 13
Generic Drug Review Process
Bioequivalence Review Labeling Review Chemistry amp Micro
Review Request for Plant
Inspection
APPLICANT
ANDA
Acceptable amp
Complete
Application Review
N ChemMicro
OK
Labeling
OK
Bioequivalence
OK
PreApproval
Inspection Results
OK
Not Approvable
Letter
Approval Withheld until
Results Satisfactory
Bio Deficiency Letter
APPROVED ANDA
N N N
N
Y Y Y
Y
Y
Refuse to Receive Letter
What is the goal of a
Pre-Approval Inspection
To assure that establishments involved in the
manufacturing testing or other manipulation of
new drug dosage forms and drug substances are
evaluated for
ndash conformance with commitments in the application
ndash site cGMP compliance
ndash data authenticity reliability and accuracy
ndash adequacy of analytical methodologies
14
PAI Objectives (Program 7346832)
There are three primary inspectional objectives of
this PAI program These objectives are
1) Readiness for Commercial Manufacturing
2) Conformance to Application
3) Data Integrity Audit
15
PAI Objective 1 Readiness for Commercial Manufacturing
Determine whether the establishment has a quality
system that is designed to achieve sufficient control over
the facility and commercial manufacturing operations
a) Manufacturing and laboratory changes deviations and
trends relating to the development of new drug substance
and product manufacturing have been adequately
evaluated
b) A sound and appropriate program for sampling testing
and evaluation of components in-process materials
finished products containers and closures for the purpose
of releasing materials or products has been established
including a robust supplier qualification program
16
PAI Objective 1 Readiness for Commercial Manufacturing cont
c) The establishment has sufficient facility and equipment
controls in place to prevent contamination of and by the
application product (or API)
d) Adequate procedures exist for batch release change control
investigating failures deviations complaints and adverse
events and for reporting this information to FDA such as
field alert reporting
e) The feasibility of the proposed commercial process and
manufacturing batch record including instructions
processing parameters and process control measures are
scientifically and objectively justified This objective is
linked to the firmrsquos process validation program
17
PAI Objective 2
Conformance to Application
Verify that the formulation manufacturing or
processing methods and analytical (or
examination) methods are consistent with
descriptions contained in the CMC section of the
application for the biobatch (and other pivotal
clinical batches when applicable) the proposed
commercial scale batch and API ndash Observing processing andor testing operations
ndash Compare the biobatch manufacturing process against the
proposed commercial batch record
18
PAI Objective 3
Data Integrity Audit
Audit the raw data hardcopy or electronic to
authenticate the data submitted in the CMC
section of the application
Verify that all relevant data (eg stability
biobatch data) were submitted in the CMC section
such that CDER product reviewers can rely on the
submitted data as complete and accurate ndash Laboratory notebooks and associated chromatograms generated
during release testing of biobatch
ndash Failure to include aberrant test results in CMC section
ndash Improper invalidation of OOS results
19
Investigatorrsquos Role during PAI
Assess the following
ndash Quality Systems
ndash Manufacturing Operations
ndash Sampling Plans
ndash Laboratory
ndash Test Methods Validation
ndash Drug Product Specifications
ndash ReprocessingReworking
ndash Standard Operating Procedures
ndash Batch Records 20
The Firmrsquos Role
Make records available (as appropriate) to
conduct the pre-approval inspection
ndash Product Development Report
ndash Batch Records and Laboratory Records
ndash ProtocolsSOPs
Assure facility is cGMP compliant and ready for
an FDA inspection
ndash Once an application is submitted to Center the firm
should be considered ready for inspection
21
Product Development Report
A very useful document for both the firm
and the FDA
The data generated during product
development which defines the drug
product targets the steps in the
manufacturing process where variation is
critical to quality and thereby focuses the
subsequent process validation effort
22
Product Development Report
API Impurity Profile
ndash How is the API characterized
Excipients
Formulation Wet or Dry Granulation
ndash Solution or Suspension
ndash Sterile - Terminalaseptic conditions
ndash TabletCapsule - ImmediateModified
ReleaseExtended Release
23
Product Development Report
Describes the development of Processing
Equipment order of addition of ingredients
to the formulation mixing times and
speeds drying time and temperature
nitrogen blankets blending hold times
compression slugging filling polishing
imprinting labeling and packaging
Product Development Report may not be a
formal document
24
Batch Records
The batch records submitted in the
application must be audited as part of
the inspection to assure
ndash That the proposed production process is the
same process that was used for the
manufacture of the biostability batches
25
ReprocessingReworking
cGMP regulations require reprocessing
procedures to be in writing If firm
makes provisions for reprocessing drug
product details must be submitted as
part of the application
ndash Standard Operating Procedures
ndash QA review and approval
26
27
Different Responsibilities
28
Different Responsibilities
29
Different Responsibilities
How ICH Q8 Q9 Q10
Guidelines Are Working
Together Throughout The
Product Life Cycle
30
Nov 2005 amp Nov 2008
ICH Q8 Q9 and Q10
High level guidances
(not prescriptive)
Science and risk-based
Encourages systematic
approaches
Applicable over entire product
lifecycle
Intended to work together to
enhance pharmaceutical product
quality
31
Pharmaceutical Development -
Q8(R2)
Describes science and risk-based
approaches for pharmaceutical product and
manufacturing process development
Introduced concepts of design space and
flexible regulatory approaches
Introduced concepts of Quality by Design
(QbD) and provided examples of QbD
development approaches and design space 32
Differing Approaches to
Pharmaceutical Development
33
Q8(R2) - Example QbD
Approach
Quality Target Product Profile (QTPP)
Determine ldquopotentialrdquo critical quality attributes
Link raw material attributes and process parameters
to CQAs and perform risk assessment
Develop a design space (optional and not required)
Design and implement a control strategy
Manage product lifecycle including continual
improvement
CQArsquos
Product Profile
Risk Assessments
Design Space
Control Strategy
Continual
Improvement34
Product Lifecycle
concept
Human and veterinary
drugs
Biotech products
Finish products amp APIs
Drug constituent of a
combination product
35
Manage product lifecycle including
continual improvement
Three-stage approach to process validation
Stage 1 ndash Process Design ndash Building and capturing process knowledge and understanding
ndash Establishing a strategy for process control
Stage 2 ndash Process Qualification ndash Design a facility and qualification of utilities and equipment
ndash Process performance qualification
ndash PPQ protocol
ndash PPQ protocol execution and report
Stage 3 ndash Continued Process Verification
36
Focus on alignment with
lsquoproduct lifecyclersquo
Manufacturers should
Understand the sources of variation
Detect the presence and degree of variation
Understand the impact of variation on the process and
ultimately on product attributes
Control the variation in a manner commensurate with
the risk it represents to the process and product
37
Process Validation
38
Focus on alignment with
lsquoproduct lifecyclersquo
39
Focus on alignment with
lsquoproduct lifecyclersquo
40
Focus on alignment with
lsquoproduct lifecyclersquo
FDA Quality Systems Guidance
for Finished Pharmaceuticals
41 Inspection Readiness BEC
Inspection Readiness BEC 42
Quality System
Drug Inspection Target Includes the Quality Control Unit and all of its review
and approval duties
ndash Approval of and adherence to Procedures and associated recordkeeping systems
ndash Product Reviews
ndash Complaint Reviews
ndash Failure Evaluations
ndash Change Control
ndash Product Improvement Projects
ndash RejectsReprocessingRework
ndash Stability
ndash Validation
ndash Training
Inspection Readiness BEC 43
Facilities and Equipment
Inspection Target
Building and Facilities along with
maintenance
Equipment qualification calibration and
maintenance
Water steam compressed gas HVAC
Change control system
Investigate discrepancies
Inspection Readiness BEC 44
Materials
Inspection Target
Procedures and documentation showing adequate control of finished products in-process materials components containers closures
Qualificationvalidation and security of computerized or automated processes
Change control system
Investigation of Discrepancies
Inspection Readiness BEC 45
Production System
Drug Inspection Target
Trainingqualification of personnel
Complete batch production documentation
Process validation
Production time limits
In-process testingexamination
Change Control System
Investigation of discrepancies
Inspection Readiness BEC 46
Packaging and Labeling
Inspection Target
Training
Acceptance operations
Control of materials to prevent mix-ups
Accountability
Packaginglabeling records
Line separation
Line clearance inspection and documentation
Validation of labelingpackaging operations
Investigation of discrepancies
Inspection Readiness BEC 47
Laboratory Control
Inspection Target
Staffing and training
Adequate equipment calibration and maintenance
Reference standards
Adherence to written procedures
Validationverification analytical methods
Analytical records and raw data
Adherence to OOS procedure and timely completion of investigations
Stability testing program and reserve samples
Change control system
Before the Inspection
SOP for Inspection Readiness
Strong Internal AuditAssessment function
ndash Continuous assessment using Risk Management
Criteria
ndash Not autopilot auditing
ndash Same auditorhellip
Perform a full-system audit (Mock) at least one
month prior to inspection
ndash Be sure any necessary CA-PA has been originated 48
Before the Inspection
Educate all your employees on proper inspection
rules and etiquette
Educate your supervisory (all exempt) personnel
on FDA regulations
Inspections and external audits concern to all
employees not only those from Quality Dept
Promote the participation on the internal audit
cadre
Certify internal auditors
49
Before the Inspection
A few words about Internal Audit
Internal Audits are the most powerful tool to
avoid inspection surprises
If the inspector discovers something why your
internal auditor did not
The same auditor auditing several consecutive
years the same area is not effective
An effective internal audit function is priceless
50
During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

6
FDA Laws Overview
1906 ndash Congress enacted Food and Drug Act
1938- Federal Food Drug and Cosmetic Act
ndash To ensure that food are safe and produced under sanitary conditions
ndash Drugs and device are safe and effectives for their intended use
ndash Cosmetics are safe and made from appropriate ingredients
ndash Labeling and packaging is truthful
1963 - Fist version of GMP for Drugs
1978 cGMP for Drugs 21 CFR 211
1987 Guideline on General Principles of Process Validation
1997 ndash FDA Modernization Act (FDAMA) Fine-tuning FDA laws
1997- Electronic Records (21 CFR Part 11)
2013 ndash Combination Products
Generic Drugs The use of bioequivalence as the basis for approving generic
copies of drug products was established by the ldquoDrug Price
Competition and Patent Term Restoration Act of 1984rdquo also
known as the Waxman-Hatch Act
This Act expedites the availability of less costly generic drugs by
permitting the FDA to approve applications to market generic
versions of brand-name drugs without conducting costly and
duplicative clinical trials At the same time the brand-name
companies can apply for up to five additional years patent
protection for the new medicines they developed to make up for
time lost while their products were going through the FDArsquos
approval process Brand-name drugs are subject to the same
bioequivalence tests as generics upon reformulation 7
8
9
The FDArsquos Drug Review Process
10
The FDArsquos Drug Review Process
11
NDA vs ANDA Review Process
12
Center for Drug Evaluation amp Research Office of Generic Drugs (OGD) 13
Generic Drug Review Process
Bioequivalence Review Labeling Review Chemistry amp Micro
Review Request for Plant
Inspection
APPLICANT
ANDA
Acceptable amp
Complete
Application Review
N ChemMicro
OK
Labeling
OK
Bioequivalence
OK
PreApproval
Inspection Results
OK
Not Approvable
Letter
Approval Withheld until
Results Satisfactory
Bio Deficiency Letter
APPROVED ANDA
N N N
N
Y Y Y
Y
Y
Refuse to Receive Letter
What is the goal of a
Pre-Approval Inspection
To assure that establishments involved in the
manufacturing testing or other manipulation of
new drug dosage forms and drug substances are
evaluated for
ndash conformance with commitments in the application
ndash site cGMP compliance
ndash data authenticity reliability and accuracy
ndash adequacy of analytical methodologies
14
PAI Objectives (Program 7346832)
There are three primary inspectional objectives of
this PAI program These objectives are
1) Readiness for Commercial Manufacturing
2) Conformance to Application
3) Data Integrity Audit
15
PAI Objective 1 Readiness for Commercial Manufacturing
Determine whether the establishment has a quality
system that is designed to achieve sufficient control over
the facility and commercial manufacturing operations
a) Manufacturing and laboratory changes deviations and
trends relating to the development of new drug substance
and product manufacturing have been adequately
evaluated
b) A sound and appropriate program for sampling testing
and evaluation of components in-process materials
finished products containers and closures for the purpose
of releasing materials or products has been established
including a robust supplier qualification program
16
PAI Objective 1 Readiness for Commercial Manufacturing cont
c) The establishment has sufficient facility and equipment
controls in place to prevent contamination of and by the
application product (or API)
d) Adequate procedures exist for batch release change control
investigating failures deviations complaints and adverse
events and for reporting this information to FDA such as
field alert reporting
e) The feasibility of the proposed commercial process and
manufacturing batch record including instructions
processing parameters and process control measures are
scientifically and objectively justified This objective is
linked to the firmrsquos process validation program
17
PAI Objective 2
Conformance to Application
Verify that the formulation manufacturing or
processing methods and analytical (or
examination) methods are consistent with
descriptions contained in the CMC section of the
application for the biobatch (and other pivotal
clinical batches when applicable) the proposed
commercial scale batch and API ndash Observing processing andor testing operations
ndash Compare the biobatch manufacturing process against the
proposed commercial batch record
18
PAI Objective 3
Data Integrity Audit
Audit the raw data hardcopy or electronic to
authenticate the data submitted in the CMC
section of the application
Verify that all relevant data (eg stability
biobatch data) were submitted in the CMC section
such that CDER product reviewers can rely on the
submitted data as complete and accurate ndash Laboratory notebooks and associated chromatograms generated
during release testing of biobatch
ndash Failure to include aberrant test results in CMC section
ndash Improper invalidation of OOS results
19
Investigatorrsquos Role during PAI
Assess the following
ndash Quality Systems
ndash Manufacturing Operations
ndash Sampling Plans
ndash Laboratory
ndash Test Methods Validation
ndash Drug Product Specifications
ndash ReprocessingReworking
ndash Standard Operating Procedures
ndash Batch Records 20
The Firmrsquos Role
Make records available (as appropriate) to
conduct the pre-approval inspection
ndash Product Development Report
ndash Batch Records and Laboratory Records
ndash ProtocolsSOPs
Assure facility is cGMP compliant and ready for
an FDA inspection
ndash Once an application is submitted to Center the firm
should be considered ready for inspection
21
Product Development Report
A very useful document for both the firm
and the FDA
The data generated during product
development which defines the drug
product targets the steps in the
manufacturing process where variation is
critical to quality and thereby focuses the
subsequent process validation effort
22
Product Development Report
API Impurity Profile
ndash How is the API characterized
Excipients
Formulation Wet or Dry Granulation
ndash Solution or Suspension
ndash Sterile - Terminalaseptic conditions
ndash TabletCapsule - ImmediateModified
ReleaseExtended Release
23
Product Development Report
Describes the development of Processing
Equipment order of addition of ingredients
to the formulation mixing times and
speeds drying time and temperature
nitrogen blankets blending hold times
compression slugging filling polishing
imprinting labeling and packaging
Product Development Report may not be a
formal document
24
Batch Records
The batch records submitted in the
application must be audited as part of
the inspection to assure
ndash That the proposed production process is the
same process that was used for the
manufacture of the biostability batches
25
ReprocessingReworking
cGMP regulations require reprocessing
procedures to be in writing If firm
makes provisions for reprocessing drug
product details must be submitted as
part of the application
ndash Standard Operating Procedures
ndash QA review and approval
26
27
Different Responsibilities
28
Different Responsibilities
29
Different Responsibilities
How ICH Q8 Q9 Q10
Guidelines Are Working
Together Throughout The
Product Life Cycle
30
Nov 2005 amp Nov 2008
ICH Q8 Q9 and Q10
High level guidances
(not prescriptive)
Science and risk-based
Encourages systematic
approaches
Applicable over entire product
lifecycle
Intended to work together to
enhance pharmaceutical product
quality
31
Pharmaceutical Development -
Q8(R2)
Describes science and risk-based
approaches for pharmaceutical product and
manufacturing process development
Introduced concepts of design space and
flexible regulatory approaches
Introduced concepts of Quality by Design
(QbD) and provided examples of QbD
development approaches and design space 32
Differing Approaches to
Pharmaceutical Development
33
Q8(R2) - Example QbD
Approach
Quality Target Product Profile (QTPP)
Determine ldquopotentialrdquo critical quality attributes
Link raw material attributes and process parameters
to CQAs and perform risk assessment
Develop a design space (optional and not required)
Design and implement a control strategy
Manage product lifecycle including continual
improvement
CQArsquos
Product Profile
Risk Assessments
Design Space
Control Strategy
Continual
Improvement34
Product Lifecycle
concept
Human and veterinary
drugs
Biotech products
Finish products amp APIs
Drug constituent of a
combination product
35
Manage product lifecycle including
continual improvement
Three-stage approach to process validation
Stage 1 ndash Process Design ndash Building and capturing process knowledge and understanding
ndash Establishing a strategy for process control
Stage 2 ndash Process Qualification ndash Design a facility and qualification of utilities and equipment
ndash Process performance qualification
ndash PPQ protocol
ndash PPQ protocol execution and report
Stage 3 ndash Continued Process Verification
36
Focus on alignment with
lsquoproduct lifecyclersquo
Manufacturers should
Understand the sources of variation
Detect the presence and degree of variation
Understand the impact of variation on the process and
ultimately on product attributes
Control the variation in a manner commensurate with
the risk it represents to the process and product
37
Process Validation
38
Focus on alignment with
lsquoproduct lifecyclersquo
39
Focus on alignment with
lsquoproduct lifecyclersquo
40
Focus on alignment with
lsquoproduct lifecyclersquo
FDA Quality Systems Guidance
for Finished Pharmaceuticals
41 Inspection Readiness BEC
Inspection Readiness BEC 42
Quality System
Drug Inspection Target Includes the Quality Control Unit and all of its review
and approval duties
ndash Approval of and adherence to Procedures and associated recordkeeping systems
ndash Product Reviews
ndash Complaint Reviews
ndash Failure Evaluations
ndash Change Control
ndash Product Improvement Projects
ndash RejectsReprocessingRework
ndash Stability
ndash Validation
ndash Training
Inspection Readiness BEC 43
Facilities and Equipment
Inspection Target
Building and Facilities along with
maintenance
Equipment qualification calibration and
maintenance
Water steam compressed gas HVAC
Change control system
Investigate discrepancies
Inspection Readiness BEC 44
Materials
Inspection Target
Procedures and documentation showing adequate control of finished products in-process materials components containers closures
Qualificationvalidation and security of computerized or automated processes
Change control system
Investigation of Discrepancies
Inspection Readiness BEC 45
Production System
Drug Inspection Target
Trainingqualification of personnel
Complete batch production documentation
Process validation
Production time limits
In-process testingexamination
Change Control System
Investigation of discrepancies
Inspection Readiness BEC 46
Packaging and Labeling
Inspection Target
Training
Acceptance operations
Control of materials to prevent mix-ups
Accountability
Packaginglabeling records
Line separation
Line clearance inspection and documentation
Validation of labelingpackaging operations
Investigation of discrepancies
Inspection Readiness BEC 47
Laboratory Control
Inspection Target
Staffing and training
Adequate equipment calibration and maintenance
Reference standards
Adherence to written procedures
Validationverification analytical methods
Analytical records and raw data
Adherence to OOS procedure and timely completion of investigations
Stability testing program and reserve samples
Change control system
Before the Inspection
SOP for Inspection Readiness
Strong Internal AuditAssessment function
ndash Continuous assessment using Risk Management
Criteria
ndash Not autopilot auditing
ndash Same auditorhellip
Perform a full-system audit (Mock) at least one
month prior to inspection
ndash Be sure any necessary CA-PA has been originated 48
Before the Inspection
Educate all your employees on proper inspection
rules and etiquette
Educate your supervisory (all exempt) personnel
on FDA regulations
Inspections and external audits concern to all
employees not only those from Quality Dept
Promote the participation on the internal audit
cadre
Certify internal auditors
49
Before the Inspection
A few words about Internal Audit
Internal Audits are the most powerful tool to
avoid inspection surprises
If the inspector discovers something why your
internal auditor did not
The same auditor auditing several consecutive
years the same area is not effective
An effective internal audit function is priceless
50
During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

Generic Drugs The use of bioequivalence as the basis for approving generic
copies of drug products was established by the ldquoDrug Price
Competition and Patent Term Restoration Act of 1984rdquo also
known as the Waxman-Hatch Act
This Act expedites the availability of less costly generic drugs by
permitting the FDA to approve applications to market generic
versions of brand-name drugs without conducting costly and
duplicative clinical trials At the same time the brand-name
companies can apply for up to five additional years patent
protection for the new medicines they developed to make up for
time lost while their products were going through the FDArsquos
approval process Brand-name drugs are subject to the same
bioequivalence tests as generics upon reformulation 7
8
9
The FDArsquos Drug Review Process
10
The FDArsquos Drug Review Process
11
NDA vs ANDA Review Process
12
Center for Drug Evaluation amp Research Office of Generic Drugs (OGD) 13
Generic Drug Review Process
Bioequivalence Review Labeling Review Chemistry amp Micro
Review Request for Plant
Inspection
APPLICANT
ANDA
Acceptable amp
Complete
Application Review
N ChemMicro
OK
Labeling
OK
Bioequivalence
OK
PreApproval
Inspection Results
OK
Not Approvable
Letter
Approval Withheld until
Results Satisfactory
Bio Deficiency Letter
APPROVED ANDA
N N N
N
Y Y Y
Y
Y
Refuse to Receive Letter
What is the goal of a
Pre-Approval Inspection
To assure that establishments involved in the
manufacturing testing or other manipulation of
new drug dosage forms and drug substances are
evaluated for
ndash conformance with commitments in the application
ndash site cGMP compliance
ndash data authenticity reliability and accuracy
ndash adequacy of analytical methodologies
14
PAI Objectives (Program 7346832)
There are three primary inspectional objectives of
this PAI program These objectives are
1) Readiness for Commercial Manufacturing
2) Conformance to Application
3) Data Integrity Audit
15
PAI Objective 1 Readiness for Commercial Manufacturing
Determine whether the establishment has a quality
system that is designed to achieve sufficient control over
the facility and commercial manufacturing operations
a) Manufacturing and laboratory changes deviations and
trends relating to the development of new drug substance
and product manufacturing have been adequately
evaluated
b) A sound and appropriate program for sampling testing
and evaluation of components in-process materials
finished products containers and closures for the purpose
of releasing materials or products has been established
including a robust supplier qualification program
16
PAI Objective 1 Readiness for Commercial Manufacturing cont
c) The establishment has sufficient facility and equipment
controls in place to prevent contamination of and by the
application product (or API)
d) Adequate procedures exist for batch release change control
investigating failures deviations complaints and adverse
events and for reporting this information to FDA such as
field alert reporting
e) The feasibility of the proposed commercial process and
manufacturing batch record including instructions
processing parameters and process control measures are
scientifically and objectively justified This objective is
linked to the firmrsquos process validation program
17
PAI Objective 2
Conformance to Application
Verify that the formulation manufacturing or
processing methods and analytical (or
examination) methods are consistent with
descriptions contained in the CMC section of the
application for the biobatch (and other pivotal
clinical batches when applicable) the proposed
commercial scale batch and API ndash Observing processing andor testing operations
ndash Compare the biobatch manufacturing process against the
proposed commercial batch record
18
PAI Objective 3
Data Integrity Audit
Audit the raw data hardcopy or electronic to
authenticate the data submitted in the CMC
section of the application
Verify that all relevant data (eg stability
biobatch data) were submitted in the CMC section
such that CDER product reviewers can rely on the
submitted data as complete and accurate ndash Laboratory notebooks and associated chromatograms generated
during release testing of biobatch
ndash Failure to include aberrant test results in CMC section
ndash Improper invalidation of OOS results
19
Investigatorrsquos Role during PAI
Assess the following
ndash Quality Systems
ndash Manufacturing Operations
ndash Sampling Plans
ndash Laboratory
ndash Test Methods Validation
ndash Drug Product Specifications
ndash ReprocessingReworking
ndash Standard Operating Procedures
ndash Batch Records 20
The Firmrsquos Role
Make records available (as appropriate) to
conduct the pre-approval inspection
ndash Product Development Report
ndash Batch Records and Laboratory Records
ndash ProtocolsSOPs
Assure facility is cGMP compliant and ready for
an FDA inspection
ndash Once an application is submitted to Center the firm
should be considered ready for inspection
21
Product Development Report
A very useful document for both the firm
and the FDA
The data generated during product
development which defines the drug
product targets the steps in the
manufacturing process where variation is
critical to quality and thereby focuses the
subsequent process validation effort
22
Product Development Report
API Impurity Profile
ndash How is the API characterized
Excipients
Formulation Wet or Dry Granulation
ndash Solution or Suspension
ndash Sterile - Terminalaseptic conditions
ndash TabletCapsule - ImmediateModified
ReleaseExtended Release
23
Product Development Report
Describes the development of Processing
Equipment order of addition of ingredients
to the formulation mixing times and
speeds drying time and temperature
nitrogen blankets blending hold times
compression slugging filling polishing
imprinting labeling and packaging
Product Development Report may not be a
formal document
24
Batch Records
The batch records submitted in the
application must be audited as part of
the inspection to assure
ndash That the proposed production process is the
same process that was used for the
manufacture of the biostability batches
25
ReprocessingReworking
cGMP regulations require reprocessing
procedures to be in writing If firm
makes provisions for reprocessing drug
product details must be submitted as
part of the application
ndash Standard Operating Procedures
ndash QA review and approval
26
27
Different Responsibilities
28
Different Responsibilities
29
Different Responsibilities
How ICH Q8 Q9 Q10
Guidelines Are Working
Together Throughout The
Product Life Cycle
30
Nov 2005 amp Nov 2008
ICH Q8 Q9 and Q10
High level guidances
(not prescriptive)
Science and risk-based
Encourages systematic
approaches
Applicable over entire product
lifecycle
Intended to work together to
enhance pharmaceutical product
quality
31
Pharmaceutical Development -
Q8(R2)
Describes science and risk-based
approaches for pharmaceutical product and
manufacturing process development
Introduced concepts of design space and
flexible regulatory approaches
Introduced concepts of Quality by Design
(QbD) and provided examples of QbD
development approaches and design space 32
Differing Approaches to
Pharmaceutical Development
33
Q8(R2) - Example QbD
Approach
Quality Target Product Profile (QTPP)
Determine ldquopotentialrdquo critical quality attributes
Link raw material attributes and process parameters
to CQAs and perform risk assessment
Develop a design space (optional and not required)
Design and implement a control strategy
Manage product lifecycle including continual
improvement
CQArsquos
Product Profile
Risk Assessments
Design Space
Control Strategy
Continual
Improvement34
Product Lifecycle
concept
Human and veterinary
drugs
Biotech products
Finish products amp APIs
Drug constituent of a
combination product
35
Manage product lifecycle including
continual improvement
Three-stage approach to process validation
Stage 1 ndash Process Design ndash Building and capturing process knowledge and understanding
ndash Establishing a strategy for process control
Stage 2 ndash Process Qualification ndash Design a facility and qualification of utilities and equipment
ndash Process performance qualification
ndash PPQ protocol
ndash PPQ protocol execution and report
Stage 3 ndash Continued Process Verification
36
Focus on alignment with
lsquoproduct lifecyclersquo
Manufacturers should
Understand the sources of variation
Detect the presence and degree of variation
Understand the impact of variation on the process and
ultimately on product attributes
Control the variation in a manner commensurate with
the risk it represents to the process and product
37
Process Validation
38
Focus on alignment with
lsquoproduct lifecyclersquo
39
Focus on alignment with
lsquoproduct lifecyclersquo
40
Focus on alignment with
lsquoproduct lifecyclersquo
FDA Quality Systems Guidance
for Finished Pharmaceuticals
41 Inspection Readiness BEC
Inspection Readiness BEC 42
Quality System
Drug Inspection Target Includes the Quality Control Unit and all of its review
and approval duties
ndash Approval of and adherence to Procedures and associated recordkeeping systems
ndash Product Reviews
ndash Complaint Reviews
ndash Failure Evaluations
ndash Change Control
ndash Product Improvement Projects
ndash RejectsReprocessingRework
ndash Stability
ndash Validation
ndash Training
Inspection Readiness BEC 43
Facilities and Equipment
Inspection Target
Building and Facilities along with
maintenance
Equipment qualification calibration and
maintenance
Water steam compressed gas HVAC
Change control system
Investigate discrepancies
Inspection Readiness BEC 44
Materials
Inspection Target
Procedures and documentation showing adequate control of finished products in-process materials components containers closures
Qualificationvalidation and security of computerized or automated processes
Change control system
Investigation of Discrepancies
Inspection Readiness BEC 45
Production System
Drug Inspection Target
Trainingqualification of personnel
Complete batch production documentation
Process validation
Production time limits
In-process testingexamination
Change Control System
Investigation of discrepancies
Inspection Readiness BEC 46
Packaging and Labeling
Inspection Target
Training
Acceptance operations
Control of materials to prevent mix-ups
Accountability
Packaginglabeling records
Line separation
Line clearance inspection and documentation
Validation of labelingpackaging operations
Investigation of discrepancies
Inspection Readiness BEC 47
Laboratory Control
Inspection Target
Staffing and training
Adequate equipment calibration and maintenance
Reference standards
Adherence to written procedures
Validationverification analytical methods
Analytical records and raw data
Adherence to OOS procedure and timely completion of investigations
Stability testing program and reserve samples
Change control system
Before the Inspection
SOP for Inspection Readiness
Strong Internal AuditAssessment function
ndash Continuous assessment using Risk Management
Criteria
ndash Not autopilot auditing
ndash Same auditorhellip
Perform a full-system audit (Mock) at least one
month prior to inspection
ndash Be sure any necessary CA-PA has been originated 48
Before the Inspection
Educate all your employees on proper inspection
rules and etiquette
Educate your supervisory (all exempt) personnel
on FDA regulations
Inspections and external audits concern to all
employees not only those from Quality Dept
Promote the participation on the internal audit
cadre
Certify internal auditors
49
Before the Inspection
A few words about Internal Audit
Internal Audits are the most powerful tool to
avoid inspection surprises
If the inspector discovers something why your
internal auditor did not
The same auditor auditing several consecutive
years the same area is not effective
An effective internal audit function is priceless
50
During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

8
9
The FDArsquos Drug Review Process
10
The FDArsquos Drug Review Process
11
NDA vs ANDA Review Process
12
Center for Drug Evaluation amp Research Office of Generic Drugs (OGD) 13
Generic Drug Review Process
Bioequivalence Review Labeling Review Chemistry amp Micro
Review Request for Plant
Inspection
APPLICANT
ANDA
Acceptable amp
Complete
Application Review
N ChemMicro
OK
Labeling
OK
Bioequivalence
OK
PreApproval
Inspection Results
OK
Not Approvable
Letter
Approval Withheld until
Results Satisfactory
Bio Deficiency Letter
APPROVED ANDA
N N N
N
Y Y Y
Y
Y
Refuse to Receive Letter
What is the goal of a
Pre-Approval Inspection
To assure that establishments involved in the
manufacturing testing or other manipulation of
new drug dosage forms and drug substances are
evaluated for
ndash conformance with commitments in the application
ndash site cGMP compliance
ndash data authenticity reliability and accuracy
ndash adequacy of analytical methodologies
14
PAI Objectives (Program 7346832)
There are three primary inspectional objectives of
this PAI program These objectives are
1) Readiness for Commercial Manufacturing
2) Conformance to Application
3) Data Integrity Audit
15
PAI Objective 1 Readiness for Commercial Manufacturing
Determine whether the establishment has a quality
system that is designed to achieve sufficient control over
the facility and commercial manufacturing operations
a) Manufacturing and laboratory changes deviations and
trends relating to the development of new drug substance
and product manufacturing have been adequately
evaluated
b) A sound and appropriate program for sampling testing
and evaluation of components in-process materials
finished products containers and closures for the purpose
of releasing materials or products has been established
including a robust supplier qualification program
16
PAI Objective 1 Readiness for Commercial Manufacturing cont
c) The establishment has sufficient facility and equipment
controls in place to prevent contamination of and by the
application product (or API)
d) Adequate procedures exist for batch release change control
investigating failures deviations complaints and adverse
events and for reporting this information to FDA such as
field alert reporting
e) The feasibility of the proposed commercial process and
manufacturing batch record including instructions
processing parameters and process control measures are
scientifically and objectively justified This objective is
linked to the firmrsquos process validation program
17
PAI Objective 2
Conformance to Application
Verify that the formulation manufacturing or
processing methods and analytical (or
examination) methods are consistent with
descriptions contained in the CMC section of the
application for the biobatch (and other pivotal
clinical batches when applicable) the proposed
commercial scale batch and API ndash Observing processing andor testing operations
ndash Compare the biobatch manufacturing process against the
proposed commercial batch record
18
PAI Objective 3
Data Integrity Audit
Audit the raw data hardcopy or electronic to
authenticate the data submitted in the CMC
section of the application
Verify that all relevant data (eg stability
biobatch data) were submitted in the CMC section
such that CDER product reviewers can rely on the
submitted data as complete and accurate ndash Laboratory notebooks and associated chromatograms generated
during release testing of biobatch
ndash Failure to include aberrant test results in CMC section
ndash Improper invalidation of OOS results
19
Investigatorrsquos Role during PAI
Assess the following
ndash Quality Systems
ndash Manufacturing Operations
ndash Sampling Plans
ndash Laboratory
ndash Test Methods Validation
ndash Drug Product Specifications
ndash ReprocessingReworking
ndash Standard Operating Procedures
ndash Batch Records 20
The Firmrsquos Role
Make records available (as appropriate) to
conduct the pre-approval inspection
ndash Product Development Report
ndash Batch Records and Laboratory Records
ndash ProtocolsSOPs
Assure facility is cGMP compliant and ready for
an FDA inspection
ndash Once an application is submitted to Center the firm
should be considered ready for inspection
21
Product Development Report
A very useful document for both the firm
and the FDA
The data generated during product
development which defines the drug
product targets the steps in the
manufacturing process where variation is
critical to quality and thereby focuses the
subsequent process validation effort
22
Product Development Report
API Impurity Profile
ndash How is the API characterized
Excipients
Formulation Wet or Dry Granulation
ndash Solution or Suspension
ndash Sterile - Terminalaseptic conditions
ndash TabletCapsule - ImmediateModified
ReleaseExtended Release
23
Product Development Report
Describes the development of Processing
Equipment order of addition of ingredients
to the formulation mixing times and
speeds drying time and temperature
nitrogen blankets blending hold times
compression slugging filling polishing
imprinting labeling and packaging
Product Development Report may not be a
formal document
24
Batch Records
The batch records submitted in the
application must be audited as part of
the inspection to assure
ndash That the proposed production process is the
same process that was used for the
manufacture of the biostability batches
25
ReprocessingReworking
cGMP regulations require reprocessing
procedures to be in writing If firm
makes provisions for reprocessing drug
product details must be submitted as
part of the application
ndash Standard Operating Procedures
ndash QA review and approval
26
27
Different Responsibilities
28
Different Responsibilities
29
Different Responsibilities
How ICH Q8 Q9 Q10
Guidelines Are Working
Together Throughout The
Product Life Cycle
30
Nov 2005 amp Nov 2008
ICH Q8 Q9 and Q10
High level guidances
(not prescriptive)
Science and risk-based
Encourages systematic
approaches
Applicable over entire product
lifecycle
Intended to work together to
enhance pharmaceutical product
quality
31
Pharmaceutical Development -
Q8(R2)
Describes science and risk-based
approaches for pharmaceutical product and
manufacturing process development
Introduced concepts of design space and
flexible regulatory approaches
Introduced concepts of Quality by Design
(QbD) and provided examples of QbD
development approaches and design space 32
Differing Approaches to
Pharmaceutical Development
33
Q8(R2) - Example QbD
Approach
Quality Target Product Profile (QTPP)
Determine ldquopotentialrdquo critical quality attributes
Link raw material attributes and process parameters
to CQAs and perform risk assessment
Develop a design space (optional and not required)
Design and implement a control strategy
Manage product lifecycle including continual
improvement
CQArsquos
Product Profile
Risk Assessments
Design Space
Control Strategy
Continual
Improvement34
Product Lifecycle
concept
Human and veterinary
drugs
Biotech products
Finish products amp APIs
Drug constituent of a
combination product
35
Manage product lifecycle including
continual improvement
Three-stage approach to process validation
Stage 1 ndash Process Design ndash Building and capturing process knowledge and understanding
ndash Establishing a strategy for process control
Stage 2 ndash Process Qualification ndash Design a facility and qualification of utilities and equipment
ndash Process performance qualification
ndash PPQ protocol
ndash PPQ protocol execution and report
Stage 3 ndash Continued Process Verification
36
Focus on alignment with
lsquoproduct lifecyclersquo
Manufacturers should
Understand the sources of variation
Detect the presence and degree of variation
Understand the impact of variation on the process and
ultimately on product attributes
Control the variation in a manner commensurate with
the risk it represents to the process and product
37
Process Validation
38
Focus on alignment with
lsquoproduct lifecyclersquo
39
Focus on alignment with
lsquoproduct lifecyclersquo
40
Focus on alignment with
lsquoproduct lifecyclersquo
FDA Quality Systems Guidance
for Finished Pharmaceuticals
41 Inspection Readiness BEC
Inspection Readiness BEC 42
Quality System
Drug Inspection Target Includes the Quality Control Unit and all of its review
and approval duties
ndash Approval of and adherence to Procedures and associated recordkeeping systems
ndash Product Reviews
ndash Complaint Reviews
ndash Failure Evaluations
ndash Change Control
ndash Product Improvement Projects
ndash RejectsReprocessingRework
ndash Stability
ndash Validation
ndash Training
Inspection Readiness BEC 43
Facilities and Equipment
Inspection Target
Building and Facilities along with
maintenance
Equipment qualification calibration and
maintenance
Water steam compressed gas HVAC
Change control system
Investigate discrepancies
Inspection Readiness BEC 44
Materials
Inspection Target
Procedures and documentation showing adequate control of finished products in-process materials components containers closures
Qualificationvalidation and security of computerized or automated processes
Change control system
Investigation of Discrepancies
Inspection Readiness BEC 45
Production System
Drug Inspection Target
Trainingqualification of personnel
Complete batch production documentation
Process validation
Production time limits
In-process testingexamination
Change Control System
Investigation of discrepancies
Inspection Readiness BEC 46
Packaging and Labeling
Inspection Target
Training
Acceptance operations
Control of materials to prevent mix-ups
Accountability
Packaginglabeling records
Line separation
Line clearance inspection and documentation
Validation of labelingpackaging operations
Investigation of discrepancies
Inspection Readiness BEC 47
Laboratory Control
Inspection Target
Staffing and training
Adequate equipment calibration and maintenance
Reference standards
Adherence to written procedures
Validationverification analytical methods
Analytical records and raw data
Adherence to OOS procedure and timely completion of investigations
Stability testing program and reserve samples
Change control system
Before the Inspection
SOP for Inspection Readiness
Strong Internal AuditAssessment function
ndash Continuous assessment using Risk Management
Criteria
ndash Not autopilot auditing
ndash Same auditorhellip
Perform a full-system audit (Mock) at least one
month prior to inspection
ndash Be sure any necessary CA-PA has been originated 48
Before the Inspection
Educate all your employees on proper inspection
rules and etiquette
Educate your supervisory (all exempt) personnel
on FDA regulations
Inspections and external audits concern to all
employees not only those from Quality Dept
Promote the participation on the internal audit
cadre
Certify internal auditors
49
Before the Inspection
A few words about Internal Audit
Internal Audits are the most powerful tool to
avoid inspection surprises
If the inspector discovers something why your
internal auditor did not
The same auditor auditing several consecutive
years the same area is not effective
An effective internal audit function is priceless
50
During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

9
The FDArsquos Drug Review Process
10
The FDArsquos Drug Review Process
11
NDA vs ANDA Review Process
12
Center for Drug Evaluation amp Research Office of Generic Drugs (OGD) 13
Generic Drug Review Process
Bioequivalence Review Labeling Review Chemistry amp Micro
Review Request for Plant
Inspection
APPLICANT
ANDA
Acceptable amp
Complete
Application Review
N ChemMicro
OK
Labeling
OK
Bioequivalence
OK
PreApproval
Inspection Results
OK
Not Approvable
Letter
Approval Withheld until
Results Satisfactory
Bio Deficiency Letter
APPROVED ANDA
N N N
N
Y Y Y
Y
Y
Refuse to Receive Letter
What is the goal of a
Pre-Approval Inspection
To assure that establishments involved in the
manufacturing testing or other manipulation of
new drug dosage forms and drug substances are
evaluated for
ndash conformance with commitments in the application
ndash site cGMP compliance
ndash data authenticity reliability and accuracy
ndash adequacy of analytical methodologies
14
PAI Objectives (Program 7346832)
There are three primary inspectional objectives of
this PAI program These objectives are
1) Readiness for Commercial Manufacturing
2) Conformance to Application
3) Data Integrity Audit
15
PAI Objective 1 Readiness for Commercial Manufacturing
Determine whether the establishment has a quality
system that is designed to achieve sufficient control over
the facility and commercial manufacturing operations
a) Manufacturing and laboratory changes deviations and
trends relating to the development of new drug substance
and product manufacturing have been adequately
evaluated
b) A sound and appropriate program for sampling testing
and evaluation of components in-process materials
finished products containers and closures for the purpose
of releasing materials or products has been established
including a robust supplier qualification program
16
PAI Objective 1 Readiness for Commercial Manufacturing cont
c) The establishment has sufficient facility and equipment
controls in place to prevent contamination of and by the
application product (or API)
d) Adequate procedures exist for batch release change control
investigating failures deviations complaints and adverse
events and for reporting this information to FDA such as
field alert reporting
e) The feasibility of the proposed commercial process and
manufacturing batch record including instructions
processing parameters and process control measures are
scientifically and objectively justified This objective is
linked to the firmrsquos process validation program
17
PAI Objective 2
Conformance to Application
Verify that the formulation manufacturing or
processing methods and analytical (or
examination) methods are consistent with
descriptions contained in the CMC section of the
application for the biobatch (and other pivotal
clinical batches when applicable) the proposed
commercial scale batch and API ndash Observing processing andor testing operations
ndash Compare the biobatch manufacturing process against the
proposed commercial batch record
18
PAI Objective 3
Data Integrity Audit
Audit the raw data hardcopy or electronic to
authenticate the data submitted in the CMC
section of the application
Verify that all relevant data (eg stability
biobatch data) were submitted in the CMC section
such that CDER product reviewers can rely on the
submitted data as complete and accurate ndash Laboratory notebooks and associated chromatograms generated
during release testing of biobatch
ndash Failure to include aberrant test results in CMC section
ndash Improper invalidation of OOS results
19
Investigatorrsquos Role during PAI
Assess the following
ndash Quality Systems
ndash Manufacturing Operations
ndash Sampling Plans
ndash Laboratory
ndash Test Methods Validation
ndash Drug Product Specifications
ndash ReprocessingReworking
ndash Standard Operating Procedures
ndash Batch Records 20
The Firmrsquos Role
Make records available (as appropriate) to
conduct the pre-approval inspection
ndash Product Development Report
ndash Batch Records and Laboratory Records
ndash ProtocolsSOPs
Assure facility is cGMP compliant and ready for
an FDA inspection
ndash Once an application is submitted to Center the firm
should be considered ready for inspection
21
Product Development Report
A very useful document for both the firm
and the FDA
The data generated during product
development which defines the drug
product targets the steps in the
manufacturing process where variation is
critical to quality and thereby focuses the
subsequent process validation effort
22
Product Development Report
API Impurity Profile
ndash How is the API characterized
Excipients
Formulation Wet or Dry Granulation
ndash Solution or Suspension
ndash Sterile - Terminalaseptic conditions
ndash TabletCapsule - ImmediateModified
ReleaseExtended Release
23
Product Development Report
Describes the development of Processing
Equipment order of addition of ingredients
to the formulation mixing times and
speeds drying time and temperature
nitrogen blankets blending hold times
compression slugging filling polishing
imprinting labeling and packaging
Product Development Report may not be a
formal document
24
Batch Records
The batch records submitted in the
application must be audited as part of
the inspection to assure
ndash That the proposed production process is the
same process that was used for the
manufacture of the biostability batches
25
ReprocessingReworking
cGMP regulations require reprocessing
procedures to be in writing If firm
makes provisions for reprocessing drug
product details must be submitted as
part of the application
ndash Standard Operating Procedures
ndash QA review and approval
26
27
Different Responsibilities
28
Different Responsibilities
29
Different Responsibilities
How ICH Q8 Q9 Q10
Guidelines Are Working
Together Throughout The
Product Life Cycle
30
Nov 2005 amp Nov 2008
ICH Q8 Q9 and Q10
High level guidances
(not prescriptive)
Science and risk-based
Encourages systematic
approaches
Applicable over entire product
lifecycle
Intended to work together to
enhance pharmaceutical product
quality
31
Pharmaceutical Development -
Q8(R2)
Describes science and risk-based
approaches for pharmaceutical product and
manufacturing process development
Introduced concepts of design space and
flexible regulatory approaches
Introduced concepts of Quality by Design
(QbD) and provided examples of QbD
development approaches and design space 32
Differing Approaches to
Pharmaceutical Development
33
Q8(R2) - Example QbD
Approach
Quality Target Product Profile (QTPP)
Determine ldquopotentialrdquo critical quality attributes
Link raw material attributes and process parameters
to CQAs and perform risk assessment
Develop a design space (optional and not required)
Design and implement a control strategy
Manage product lifecycle including continual
improvement
CQArsquos
Product Profile
Risk Assessments
Design Space
Control Strategy
Continual
Improvement34
Product Lifecycle
concept
Human and veterinary
drugs
Biotech products
Finish products amp APIs
Drug constituent of a
combination product
35
Manage product lifecycle including
continual improvement
Three-stage approach to process validation
Stage 1 ndash Process Design ndash Building and capturing process knowledge and understanding
ndash Establishing a strategy for process control
Stage 2 ndash Process Qualification ndash Design a facility and qualification of utilities and equipment
ndash Process performance qualification
ndash PPQ protocol
ndash PPQ protocol execution and report
Stage 3 ndash Continued Process Verification
36
Focus on alignment with
lsquoproduct lifecyclersquo
Manufacturers should
Understand the sources of variation
Detect the presence and degree of variation
Understand the impact of variation on the process and
ultimately on product attributes
Control the variation in a manner commensurate with
the risk it represents to the process and product
37
Process Validation
38
Focus on alignment with
lsquoproduct lifecyclersquo
39
Focus on alignment with
lsquoproduct lifecyclersquo
40
Focus on alignment with
lsquoproduct lifecyclersquo
FDA Quality Systems Guidance
for Finished Pharmaceuticals
41 Inspection Readiness BEC
Inspection Readiness BEC 42
Quality System
Drug Inspection Target Includes the Quality Control Unit and all of its review
and approval duties
ndash Approval of and adherence to Procedures and associated recordkeeping systems
ndash Product Reviews
ndash Complaint Reviews
ndash Failure Evaluations
ndash Change Control
ndash Product Improvement Projects
ndash RejectsReprocessingRework
ndash Stability
ndash Validation
ndash Training
Inspection Readiness BEC 43
Facilities and Equipment
Inspection Target
Building and Facilities along with
maintenance
Equipment qualification calibration and
maintenance
Water steam compressed gas HVAC
Change control system
Investigate discrepancies
Inspection Readiness BEC 44
Materials
Inspection Target
Procedures and documentation showing adequate control of finished products in-process materials components containers closures
Qualificationvalidation and security of computerized or automated processes
Change control system
Investigation of Discrepancies
Inspection Readiness BEC 45
Production System
Drug Inspection Target
Trainingqualification of personnel
Complete batch production documentation
Process validation
Production time limits
In-process testingexamination
Change Control System
Investigation of discrepancies
Inspection Readiness BEC 46
Packaging and Labeling
Inspection Target
Training
Acceptance operations
Control of materials to prevent mix-ups
Accountability
Packaginglabeling records
Line separation
Line clearance inspection and documentation
Validation of labelingpackaging operations
Investigation of discrepancies
Inspection Readiness BEC 47
Laboratory Control
Inspection Target
Staffing and training
Adequate equipment calibration and maintenance
Reference standards
Adherence to written procedures
Validationverification analytical methods
Analytical records and raw data
Adherence to OOS procedure and timely completion of investigations
Stability testing program and reserve samples
Change control system
Before the Inspection
SOP for Inspection Readiness
Strong Internal AuditAssessment function
ndash Continuous assessment using Risk Management
Criteria
ndash Not autopilot auditing
ndash Same auditorhellip
Perform a full-system audit (Mock) at least one
month prior to inspection
ndash Be sure any necessary CA-PA has been originated 48
Before the Inspection
Educate all your employees on proper inspection
rules and etiquette
Educate your supervisory (all exempt) personnel
on FDA regulations
Inspections and external audits concern to all
employees not only those from Quality Dept
Promote the participation on the internal audit
cadre
Certify internal auditors
49
Before the Inspection
A few words about Internal Audit
Internal Audits are the most powerful tool to
avoid inspection surprises
If the inspector discovers something why your
internal auditor did not
The same auditor auditing several consecutive
years the same area is not effective
An effective internal audit function is priceless
50
During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

The FDArsquos Drug Review Process
10
The FDArsquos Drug Review Process
11
NDA vs ANDA Review Process
12
Center for Drug Evaluation amp Research Office of Generic Drugs (OGD) 13
Generic Drug Review Process
Bioequivalence Review Labeling Review Chemistry amp Micro
Review Request for Plant
Inspection
APPLICANT
ANDA
Acceptable amp
Complete
Application Review
N ChemMicro
OK
Labeling
OK
Bioequivalence
OK
PreApproval
Inspection Results
OK
Not Approvable
Letter
Approval Withheld until
Results Satisfactory
Bio Deficiency Letter
APPROVED ANDA
N N N
N
Y Y Y
Y
Y
Refuse to Receive Letter
What is the goal of a
Pre-Approval Inspection
To assure that establishments involved in the
manufacturing testing or other manipulation of
new drug dosage forms and drug substances are
evaluated for
ndash conformance with commitments in the application
ndash site cGMP compliance
ndash data authenticity reliability and accuracy
ndash adequacy of analytical methodologies
14
PAI Objectives (Program 7346832)
There are three primary inspectional objectives of
this PAI program These objectives are
1) Readiness for Commercial Manufacturing
2) Conformance to Application
3) Data Integrity Audit
15
PAI Objective 1 Readiness for Commercial Manufacturing
Determine whether the establishment has a quality
system that is designed to achieve sufficient control over
the facility and commercial manufacturing operations
a) Manufacturing and laboratory changes deviations and
trends relating to the development of new drug substance
and product manufacturing have been adequately
evaluated
b) A sound and appropriate program for sampling testing
and evaluation of components in-process materials
finished products containers and closures for the purpose
of releasing materials or products has been established
including a robust supplier qualification program
16
PAI Objective 1 Readiness for Commercial Manufacturing cont
c) The establishment has sufficient facility and equipment
controls in place to prevent contamination of and by the
application product (or API)
d) Adequate procedures exist for batch release change control
investigating failures deviations complaints and adverse
events and for reporting this information to FDA such as
field alert reporting
e) The feasibility of the proposed commercial process and
manufacturing batch record including instructions
processing parameters and process control measures are
scientifically and objectively justified This objective is
linked to the firmrsquos process validation program
17
PAI Objective 2
Conformance to Application
Verify that the formulation manufacturing or
processing methods and analytical (or
examination) methods are consistent with
descriptions contained in the CMC section of the
application for the biobatch (and other pivotal
clinical batches when applicable) the proposed
commercial scale batch and API ndash Observing processing andor testing operations
ndash Compare the biobatch manufacturing process against the
proposed commercial batch record
18
PAI Objective 3
Data Integrity Audit
Audit the raw data hardcopy or electronic to
authenticate the data submitted in the CMC
section of the application
Verify that all relevant data (eg stability
biobatch data) were submitted in the CMC section
such that CDER product reviewers can rely on the
submitted data as complete and accurate ndash Laboratory notebooks and associated chromatograms generated
during release testing of biobatch
ndash Failure to include aberrant test results in CMC section
ndash Improper invalidation of OOS results
19
Investigatorrsquos Role during PAI
Assess the following
ndash Quality Systems
ndash Manufacturing Operations
ndash Sampling Plans
ndash Laboratory
ndash Test Methods Validation
ndash Drug Product Specifications
ndash ReprocessingReworking
ndash Standard Operating Procedures
ndash Batch Records 20
The Firmrsquos Role
Make records available (as appropriate) to
conduct the pre-approval inspection
ndash Product Development Report
ndash Batch Records and Laboratory Records
ndash ProtocolsSOPs
Assure facility is cGMP compliant and ready for
an FDA inspection
ndash Once an application is submitted to Center the firm
should be considered ready for inspection
21
Product Development Report
A very useful document for both the firm
and the FDA
The data generated during product
development which defines the drug
product targets the steps in the
manufacturing process where variation is
critical to quality and thereby focuses the
subsequent process validation effort
22
Product Development Report
API Impurity Profile
ndash How is the API characterized
Excipients
Formulation Wet or Dry Granulation
ndash Solution or Suspension
ndash Sterile - Terminalaseptic conditions
ndash TabletCapsule - ImmediateModified
ReleaseExtended Release
23
Product Development Report
Describes the development of Processing
Equipment order of addition of ingredients
to the formulation mixing times and
speeds drying time and temperature
nitrogen blankets blending hold times
compression slugging filling polishing
imprinting labeling and packaging
Product Development Report may not be a
formal document
24
Batch Records
The batch records submitted in the
application must be audited as part of
the inspection to assure
ndash That the proposed production process is the
same process that was used for the
manufacture of the biostability batches
25
ReprocessingReworking
cGMP regulations require reprocessing
procedures to be in writing If firm
makes provisions for reprocessing drug
product details must be submitted as
part of the application
ndash Standard Operating Procedures
ndash QA review and approval
26
27
Different Responsibilities
28
Different Responsibilities
29
Different Responsibilities
How ICH Q8 Q9 Q10
Guidelines Are Working
Together Throughout The
Product Life Cycle
30
Nov 2005 amp Nov 2008
ICH Q8 Q9 and Q10
High level guidances
(not prescriptive)
Science and risk-based
Encourages systematic
approaches
Applicable over entire product
lifecycle
Intended to work together to
enhance pharmaceutical product
quality
31
Pharmaceutical Development -
Q8(R2)
Describes science and risk-based
approaches for pharmaceutical product and
manufacturing process development
Introduced concepts of design space and
flexible regulatory approaches
Introduced concepts of Quality by Design
(QbD) and provided examples of QbD
development approaches and design space 32
Differing Approaches to
Pharmaceutical Development
33
Q8(R2) - Example QbD
Approach
Quality Target Product Profile (QTPP)
Determine ldquopotentialrdquo critical quality attributes
Link raw material attributes and process parameters
to CQAs and perform risk assessment
Develop a design space (optional and not required)
Design and implement a control strategy
Manage product lifecycle including continual
improvement
CQArsquos
Product Profile
Risk Assessments
Design Space
Control Strategy
Continual
Improvement34
Product Lifecycle
concept
Human and veterinary
drugs
Biotech products
Finish products amp APIs
Drug constituent of a
combination product
35
Manage product lifecycle including
continual improvement
Three-stage approach to process validation
Stage 1 ndash Process Design ndash Building and capturing process knowledge and understanding
ndash Establishing a strategy for process control
Stage 2 ndash Process Qualification ndash Design a facility and qualification of utilities and equipment
ndash Process performance qualification
ndash PPQ protocol
ndash PPQ protocol execution and report
Stage 3 ndash Continued Process Verification
36
Focus on alignment with
lsquoproduct lifecyclersquo
Manufacturers should
Understand the sources of variation
Detect the presence and degree of variation
Understand the impact of variation on the process and
ultimately on product attributes
Control the variation in a manner commensurate with
the risk it represents to the process and product
37
Process Validation
38
Focus on alignment with
lsquoproduct lifecyclersquo
39
Focus on alignment with
lsquoproduct lifecyclersquo
40
Focus on alignment with
lsquoproduct lifecyclersquo
FDA Quality Systems Guidance
for Finished Pharmaceuticals
41 Inspection Readiness BEC
Inspection Readiness BEC 42
Quality System
Drug Inspection Target Includes the Quality Control Unit and all of its review
and approval duties
ndash Approval of and adherence to Procedures and associated recordkeeping systems
ndash Product Reviews
ndash Complaint Reviews
ndash Failure Evaluations
ndash Change Control
ndash Product Improvement Projects
ndash RejectsReprocessingRework
ndash Stability
ndash Validation
ndash Training
Inspection Readiness BEC 43
Facilities and Equipment
Inspection Target
Building and Facilities along with
maintenance
Equipment qualification calibration and
maintenance
Water steam compressed gas HVAC
Change control system
Investigate discrepancies
Inspection Readiness BEC 44
Materials
Inspection Target
Procedures and documentation showing adequate control of finished products in-process materials components containers closures
Qualificationvalidation and security of computerized or automated processes
Change control system
Investigation of Discrepancies
Inspection Readiness BEC 45
Production System
Drug Inspection Target
Trainingqualification of personnel
Complete batch production documentation
Process validation
Production time limits
In-process testingexamination
Change Control System
Investigation of discrepancies
Inspection Readiness BEC 46
Packaging and Labeling
Inspection Target
Training
Acceptance operations
Control of materials to prevent mix-ups
Accountability
Packaginglabeling records
Line separation
Line clearance inspection and documentation
Validation of labelingpackaging operations
Investigation of discrepancies
Inspection Readiness BEC 47
Laboratory Control
Inspection Target
Staffing and training
Adequate equipment calibration and maintenance
Reference standards
Adherence to written procedures
Validationverification analytical methods
Analytical records and raw data
Adherence to OOS procedure and timely completion of investigations
Stability testing program and reserve samples
Change control system
Before the Inspection
SOP for Inspection Readiness
Strong Internal AuditAssessment function
ndash Continuous assessment using Risk Management
Criteria
ndash Not autopilot auditing
ndash Same auditorhellip
Perform a full-system audit (Mock) at least one
month prior to inspection
ndash Be sure any necessary CA-PA has been originated 48
Before the Inspection
Educate all your employees on proper inspection
rules and etiquette
Educate your supervisory (all exempt) personnel
on FDA regulations
Inspections and external audits concern to all
employees not only those from Quality Dept
Promote the participation on the internal audit
cadre
Certify internal auditors
49
Before the Inspection
A few words about Internal Audit
Internal Audits are the most powerful tool to
avoid inspection surprises
If the inspector discovers something why your
internal auditor did not
The same auditor auditing several consecutive
years the same area is not effective
An effective internal audit function is priceless
50
During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

The FDArsquos Drug Review Process
11
NDA vs ANDA Review Process
12
Center for Drug Evaluation amp Research Office of Generic Drugs (OGD) 13
Generic Drug Review Process
Bioequivalence Review Labeling Review Chemistry amp Micro
Review Request for Plant
Inspection
APPLICANT
ANDA
Acceptable amp
Complete
Application Review
N ChemMicro
OK
Labeling
OK
Bioequivalence
OK
PreApproval
Inspection Results
OK
Not Approvable
Letter
Approval Withheld until
Results Satisfactory
Bio Deficiency Letter
APPROVED ANDA
N N N
N
Y Y Y
Y
Y
Refuse to Receive Letter
What is the goal of a
Pre-Approval Inspection
To assure that establishments involved in the
manufacturing testing or other manipulation of
new drug dosage forms and drug substances are
evaluated for
ndash conformance with commitments in the application
ndash site cGMP compliance
ndash data authenticity reliability and accuracy
ndash adequacy of analytical methodologies
14
PAI Objectives (Program 7346832)
There are three primary inspectional objectives of
this PAI program These objectives are
1) Readiness for Commercial Manufacturing
2) Conformance to Application
3) Data Integrity Audit
15
PAI Objective 1 Readiness for Commercial Manufacturing
Determine whether the establishment has a quality
system that is designed to achieve sufficient control over
the facility and commercial manufacturing operations
a) Manufacturing and laboratory changes deviations and
trends relating to the development of new drug substance
and product manufacturing have been adequately
evaluated
b) A sound and appropriate program for sampling testing
and evaluation of components in-process materials
finished products containers and closures for the purpose
of releasing materials or products has been established
including a robust supplier qualification program
16
PAI Objective 1 Readiness for Commercial Manufacturing cont
c) The establishment has sufficient facility and equipment
controls in place to prevent contamination of and by the
application product (or API)
d) Adequate procedures exist for batch release change control
investigating failures deviations complaints and adverse
events and for reporting this information to FDA such as
field alert reporting
e) The feasibility of the proposed commercial process and
manufacturing batch record including instructions
processing parameters and process control measures are
scientifically and objectively justified This objective is
linked to the firmrsquos process validation program
17
PAI Objective 2
Conformance to Application
Verify that the formulation manufacturing or
processing methods and analytical (or
examination) methods are consistent with
descriptions contained in the CMC section of the
application for the biobatch (and other pivotal
clinical batches when applicable) the proposed
commercial scale batch and API ndash Observing processing andor testing operations
ndash Compare the biobatch manufacturing process against the
proposed commercial batch record
18
PAI Objective 3
Data Integrity Audit
Audit the raw data hardcopy or electronic to
authenticate the data submitted in the CMC
section of the application
Verify that all relevant data (eg stability
biobatch data) were submitted in the CMC section
such that CDER product reviewers can rely on the
submitted data as complete and accurate ndash Laboratory notebooks and associated chromatograms generated
during release testing of biobatch
ndash Failure to include aberrant test results in CMC section
ndash Improper invalidation of OOS results
19
Investigatorrsquos Role during PAI
Assess the following
ndash Quality Systems
ndash Manufacturing Operations
ndash Sampling Plans
ndash Laboratory
ndash Test Methods Validation
ndash Drug Product Specifications
ndash ReprocessingReworking
ndash Standard Operating Procedures
ndash Batch Records 20
The Firmrsquos Role
Make records available (as appropriate) to
conduct the pre-approval inspection
ndash Product Development Report
ndash Batch Records and Laboratory Records
ndash ProtocolsSOPs
Assure facility is cGMP compliant and ready for
an FDA inspection
ndash Once an application is submitted to Center the firm
should be considered ready for inspection
21
Product Development Report
A very useful document for both the firm
and the FDA
The data generated during product
development which defines the drug
product targets the steps in the
manufacturing process where variation is
critical to quality and thereby focuses the
subsequent process validation effort
22
Product Development Report
API Impurity Profile
ndash How is the API characterized
Excipients
Formulation Wet or Dry Granulation
ndash Solution or Suspension
ndash Sterile - Terminalaseptic conditions
ndash TabletCapsule - ImmediateModified
ReleaseExtended Release
23
Product Development Report
Describes the development of Processing
Equipment order of addition of ingredients
to the formulation mixing times and
speeds drying time and temperature
nitrogen blankets blending hold times
compression slugging filling polishing
imprinting labeling and packaging
Product Development Report may not be a
formal document
24
Batch Records
The batch records submitted in the
application must be audited as part of
the inspection to assure
ndash That the proposed production process is the
same process that was used for the
manufacture of the biostability batches
25
ReprocessingReworking
cGMP regulations require reprocessing
procedures to be in writing If firm
makes provisions for reprocessing drug
product details must be submitted as
part of the application
ndash Standard Operating Procedures
ndash QA review and approval
26
27
Different Responsibilities
28
Different Responsibilities
29
Different Responsibilities
How ICH Q8 Q9 Q10
Guidelines Are Working
Together Throughout The
Product Life Cycle
30
Nov 2005 amp Nov 2008
ICH Q8 Q9 and Q10
High level guidances
(not prescriptive)
Science and risk-based
Encourages systematic
approaches
Applicable over entire product
lifecycle
Intended to work together to
enhance pharmaceutical product
quality
31
Pharmaceutical Development -
Q8(R2)
Describes science and risk-based
approaches for pharmaceutical product and
manufacturing process development
Introduced concepts of design space and
flexible regulatory approaches
Introduced concepts of Quality by Design
(QbD) and provided examples of QbD
development approaches and design space 32
Differing Approaches to
Pharmaceutical Development
33
Q8(R2) - Example QbD
Approach
Quality Target Product Profile (QTPP)
Determine ldquopotentialrdquo critical quality attributes
Link raw material attributes and process parameters
to CQAs and perform risk assessment
Develop a design space (optional and not required)
Design and implement a control strategy
Manage product lifecycle including continual
improvement
CQArsquos
Product Profile
Risk Assessments
Design Space
Control Strategy
Continual
Improvement34
Product Lifecycle
concept
Human and veterinary
drugs
Biotech products
Finish products amp APIs
Drug constituent of a
combination product
35
Manage product lifecycle including
continual improvement
Three-stage approach to process validation
Stage 1 ndash Process Design ndash Building and capturing process knowledge and understanding
ndash Establishing a strategy for process control
Stage 2 ndash Process Qualification ndash Design a facility and qualification of utilities and equipment
ndash Process performance qualification
ndash PPQ protocol
ndash PPQ protocol execution and report
Stage 3 ndash Continued Process Verification
36
Focus on alignment with
lsquoproduct lifecyclersquo
Manufacturers should
Understand the sources of variation
Detect the presence and degree of variation
Understand the impact of variation on the process and
ultimately on product attributes
Control the variation in a manner commensurate with
the risk it represents to the process and product
37
Process Validation
38
Focus on alignment with
lsquoproduct lifecyclersquo
39
Focus on alignment with
lsquoproduct lifecyclersquo
40
Focus on alignment with
lsquoproduct lifecyclersquo
FDA Quality Systems Guidance
for Finished Pharmaceuticals
41 Inspection Readiness BEC
Inspection Readiness BEC 42
Quality System
Drug Inspection Target Includes the Quality Control Unit and all of its review
and approval duties
ndash Approval of and adherence to Procedures and associated recordkeeping systems
ndash Product Reviews
ndash Complaint Reviews
ndash Failure Evaluations
ndash Change Control
ndash Product Improvement Projects
ndash RejectsReprocessingRework
ndash Stability
ndash Validation
ndash Training
Inspection Readiness BEC 43
Facilities and Equipment
Inspection Target
Building and Facilities along with
maintenance
Equipment qualification calibration and
maintenance
Water steam compressed gas HVAC
Change control system
Investigate discrepancies
Inspection Readiness BEC 44
Materials
Inspection Target
Procedures and documentation showing adequate control of finished products in-process materials components containers closures
Qualificationvalidation and security of computerized or automated processes
Change control system
Investigation of Discrepancies
Inspection Readiness BEC 45
Production System
Drug Inspection Target
Trainingqualification of personnel
Complete batch production documentation
Process validation
Production time limits
In-process testingexamination
Change Control System
Investigation of discrepancies
Inspection Readiness BEC 46
Packaging and Labeling
Inspection Target
Training
Acceptance operations
Control of materials to prevent mix-ups
Accountability
Packaginglabeling records
Line separation
Line clearance inspection and documentation
Validation of labelingpackaging operations
Investigation of discrepancies
Inspection Readiness BEC 47
Laboratory Control
Inspection Target
Staffing and training
Adequate equipment calibration and maintenance
Reference standards
Adherence to written procedures
Validationverification analytical methods
Analytical records and raw data
Adherence to OOS procedure and timely completion of investigations
Stability testing program and reserve samples
Change control system
Before the Inspection
SOP for Inspection Readiness
Strong Internal AuditAssessment function
ndash Continuous assessment using Risk Management
Criteria
ndash Not autopilot auditing
ndash Same auditorhellip
Perform a full-system audit (Mock) at least one
month prior to inspection
ndash Be sure any necessary CA-PA has been originated 48
Before the Inspection
Educate all your employees on proper inspection
rules and etiquette
Educate your supervisory (all exempt) personnel
on FDA regulations
Inspections and external audits concern to all
employees not only those from Quality Dept
Promote the participation on the internal audit
cadre
Certify internal auditors
49
Before the Inspection
A few words about Internal Audit
Internal Audits are the most powerful tool to
avoid inspection surprises
If the inspector discovers something why your
internal auditor did not
The same auditor auditing several consecutive
years the same area is not effective
An effective internal audit function is priceless
50
During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

NDA vs ANDA Review Process
12
Center for Drug Evaluation amp Research Office of Generic Drugs (OGD) 13
Generic Drug Review Process
Bioequivalence Review Labeling Review Chemistry amp Micro
Review Request for Plant
Inspection
APPLICANT
ANDA
Acceptable amp
Complete
Application Review
N ChemMicro
OK
Labeling
OK
Bioequivalence
OK
PreApproval
Inspection Results
OK
Not Approvable
Letter
Approval Withheld until
Results Satisfactory
Bio Deficiency Letter
APPROVED ANDA
N N N
N
Y Y Y
Y
Y
Refuse to Receive Letter
What is the goal of a
Pre-Approval Inspection
To assure that establishments involved in the
manufacturing testing or other manipulation of
new drug dosage forms and drug substances are
evaluated for
ndash conformance with commitments in the application
ndash site cGMP compliance
ndash data authenticity reliability and accuracy
ndash adequacy of analytical methodologies
14
PAI Objectives (Program 7346832)
There are three primary inspectional objectives of
this PAI program These objectives are
1) Readiness for Commercial Manufacturing
2) Conformance to Application
3) Data Integrity Audit
15
PAI Objective 1 Readiness for Commercial Manufacturing
Determine whether the establishment has a quality
system that is designed to achieve sufficient control over
the facility and commercial manufacturing operations
a) Manufacturing and laboratory changes deviations and
trends relating to the development of new drug substance
and product manufacturing have been adequately
evaluated
b) A sound and appropriate program for sampling testing
and evaluation of components in-process materials
finished products containers and closures for the purpose
of releasing materials or products has been established
including a robust supplier qualification program
16
PAI Objective 1 Readiness for Commercial Manufacturing cont
c) The establishment has sufficient facility and equipment
controls in place to prevent contamination of and by the
application product (or API)
d) Adequate procedures exist for batch release change control
investigating failures deviations complaints and adverse
events and for reporting this information to FDA such as
field alert reporting
e) The feasibility of the proposed commercial process and
manufacturing batch record including instructions
processing parameters and process control measures are
scientifically and objectively justified This objective is
linked to the firmrsquos process validation program
17
PAI Objective 2
Conformance to Application
Verify that the formulation manufacturing or
processing methods and analytical (or
examination) methods are consistent with
descriptions contained in the CMC section of the
application for the biobatch (and other pivotal
clinical batches when applicable) the proposed
commercial scale batch and API ndash Observing processing andor testing operations
ndash Compare the biobatch manufacturing process against the
proposed commercial batch record
18
PAI Objective 3
Data Integrity Audit
Audit the raw data hardcopy or electronic to
authenticate the data submitted in the CMC
section of the application
Verify that all relevant data (eg stability
biobatch data) were submitted in the CMC section
such that CDER product reviewers can rely on the
submitted data as complete and accurate ndash Laboratory notebooks and associated chromatograms generated
during release testing of biobatch
ndash Failure to include aberrant test results in CMC section
ndash Improper invalidation of OOS results
19
Investigatorrsquos Role during PAI
Assess the following
ndash Quality Systems
ndash Manufacturing Operations
ndash Sampling Plans
ndash Laboratory
ndash Test Methods Validation
ndash Drug Product Specifications
ndash ReprocessingReworking
ndash Standard Operating Procedures
ndash Batch Records 20
The Firmrsquos Role
Make records available (as appropriate) to
conduct the pre-approval inspection
ndash Product Development Report
ndash Batch Records and Laboratory Records
ndash ProtocolsSOPs
Assure facility is cGMP compliant and ready for
an FDA inspection
ndash Once an application is submitted to Center the firm
should be considered ready for inspection
21
Product Development Report
A very useful document for both the firm
and the FDA
The data generated during product
development which defines the drug
product targets the steps in the
manufacturing process where variation is
critical to quality and thereby focuses the
subsequent process validation effort
22
Product Development Report
API Impurity Profile
ndash How is the API characterized
Excipients
Formulation Wet or Dry Granulation
ndash Solution or Suspension
ndash Sterile - Terminalaseptic conditions
ndash TabletCapsule - ImmediateModified
ReleaseExtended Release
23
Product Development Report
Describes the development of Processing
Equipment order of addition of ingredients
to the formulation mixing times and
speeds drying time and temperature
nitrogen blankets blending hold times
compression slugging filling polishing
imprinting labeling and packaging
Product Development Report may not be a
formal document
24
Batch Records
The batch records submitted in the
application must be audited as part of
the inspection to assure
ndash That the proposed production process is the
same process that was used for the
manufacture of the biostability batches
25
ReprocessingReworking
cGMP regulations require reprocessing
procedures to be in writing If firm
makes provisions for reprocessing drug
product details must be submitted as
part of the application
ndash Standard Operating Procedures
ndash QA review and approval
26
27
Different Responsibilities
28
Different Responsibilities
29
Different Responsibilities
How ICH Q8 Q9 Q10
Guidelines Are Working
Together Throughout The
Product Life Cycle
30
Nov 2005 amp Nov 2008
ICH Q8 Q9 and Q10
High level guidances
(not prescriptive)
Science and risk-based
Encourages systematic
approaches
Applicable over entire product
lifecycle
Intended to work together to
enhance pharmaceutical product
quality
31
Pharmaceutical Development -
Q8(R2)
Describes science and risk-based
approaches for pharmaceutical product and
manufacturing process development
Introduced concepts of design space and
flexible regulatory approaches
Introduced concepts of Quality by Design
(QbD) and provided examples of QbD
development approaches and design space 32
Differing Approaches to
Pharmaceutical Development
33
Q8(R2) - Example QbD
Approach
Quality Target Product Profile (QTPP)
Determine ldquopotentialrdquo critical quality attributes
Link raw material attributes and process parameters
to CQAs and perform risk assessment
Develop a design space (optional and not required)
Design and implement a control strategy
Manage product lifecycle including continual
improvement
CQArsquos
Product Profile
Risk Assessments
Design Space
Control Strategy
Continual
Improvement34
Product Lifecycle
concept
Human and veterinary
drugs
Biotech products
Finish products amp APIs
Drug constituent of a
combination product
35
Manage product lifecycle including
continual improvement
Three-stage approach to process validation
Stage 1 ndash Process Design ndash Building and capturing process knowledge and understanding
ndash Establishing a strategy for process control
Stage 2 ndash Process Qualification ndash Design a facility and qualification of utilities and equipment
ndash Process performance qualification
ndash PPQ protocol
ndash PPQ protocol execution and report
Stage 3 ndash Continued Process Verification
36
Focus on alignment with
lsquoproduct lifecyclersquo
Manufacturers should
Understand the sources of variation
Detect the presence and degree of variation
Understand the impact of variation on the process and
ultimately on product attributes
Control the variation in a manner commensurate with
the risk it represents to the process and product
37
Process Validation
38
Focus on alignment with
lsquoproduct lifecyclersquo
39
Focus on alignment with
lsquoproduct lifecyclersquo
40
Focus on alignment with
lsquoproduct lifecyclersquo
FDA Quality Systems Guidance
for Finished Pharmaceuticals
41 Inspection Readiness BEC
Inspection Readiness BEC 42
Quality System
Drug Inspection Target Includes the Quality Control Unit and all of its review
and approval duties
ndash Approval of and adherence to Procedures and associated recordkeeping systems
ndash Product Reviews
ndash Complaint Reviews
ndash Failure Evaluations
ndash Change Control
ndash Product Improvement Projects
ndash RejectsReprocessingRework
ndash Stability
ndash Validation
ndash Training
Inspection Readiness BEC 43
Facilities and Equipment
Inspection Target
Building and Facilities along with
maintenance
Equipment qualification calibration and
maintenance
Water steam compressed gas HVAC
Change control system
Investigate discrepancies
Inspection Readiness BEC 44
Materials
Inspection Target
Procedures and documentation showing adequate control of finished products in-process materials components containers closures
Qualificationvalidation and security of computerized or automated processes
Change control system
Investigation of Discrepancies
Inspection Readiness BEC 45
Production System
Drug Inspection Target
Trainingqualification of personnel
Complete batch production documentation
Process validation
Production time limits
In-process testingexamination
Change Control System
Investigation of discrepancies
Inspection Readiness BEC 46
Packaging and Labeling
Inspection Target
Training
Acceptance operations
Control of materials to prevent mix-ups
Accountability
Packaginglabeling records
Line separation
Line clearance inspection and documentation
Validation of labelingpackaging operations
Investigation of discrepancies
Inspection Readiness BEC 47
Laboratory Control
Inspection Target
Staffing and training
Adequate equipment calibration and maintenance
Reference standards
Adherence to written procedures
Validationverification analytical methods
Analytical records and raw data
Adherence to OOS procedure and timely completion of investigations
Stability testing program and reserve samples
Change control system
Before the Inspection
SOP for Inspection Readiness
Strong Internal AuditAssessment function
ndash Continuous assessment using Risk Management
Criteria
ndash Not autopilot auditing
ndash Same auditorhellip
Perform a full-system audit (Mock) at least one
month prior to inspection
ndash Be sure any necessary CA-PA has been originated 48
Before the Inspection
Educate all your employees on proper inspection
rules and etiquette
Educate your supervisory (all exempt) personnel
on FDA regulations
Inspections and external audits concern to all
employees not only those from Quality Dept
Promote the participation on the internal audit
cadre
Certify internal auditors
49
Before the Inspection
A few words about Internal Audit
Internal Audits are the most powerful tool to
avoid inspection surprises
If the inspector discovers something why your
internal auditor did not
The same auditor auditing several consecutive
years the same area is not effective
An effective internal audit function is priceless
50
During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62
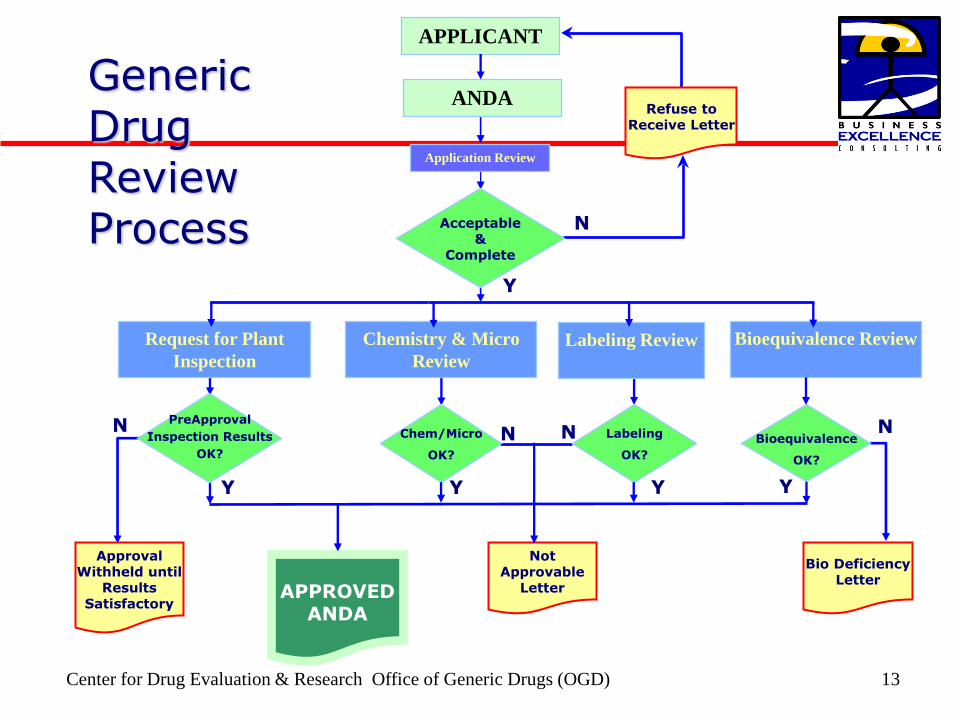
Center for Drug Evaluation amp Research Office of Generic Drugs (OGD) 13
Generic Drug Review Process
Bioequivalence Review Labeling Review Chemistry amp Micro
Review Request for Plant
Inspection
APPLICANT
ANDA
Acceptable amp
Complete
Application Review
N ChemMicro
OK
Labeling
OK
Bioequivalence
OK
PreApproval
Inspection Results
OK
Not Approvable
Letter
Approval Withheld until
Results Satisfactory
Bio Deficiency Letter
APPROVED ANDA
N N N
N
Y Y Y
Y
Y
Refuse to Receive Letter
What is the goal of a
Pre-Approval Inspection
To assure that establishments involved in the
manufacturing testing or other manipulation of
new drug dosage forms and drug substances are
evaluated for
ndash conformance with commitments in the application
ndash site cGMP compliance
ndash data authenticity reliability and accuracy
ndash adequacy of analytical methodologies
14
PAI Objectives (Program 7346832)
There are three primary inspectional objectives of
this PAI program These objectives are
1) Readiness for Commercial Manufacturing
2) Conformance to Application
3) Data Integrity Audit
15
PAI Objective 1 Readiness for Commercial Manufacturing
Determine whether the establishment has a quality
system that is designed to achieve sufficient control over
the facility and commercial manufacturing operations
a) Manufacturing and laboratory changes deviations and
trends relating to the development of new drug substance
and product manufacturing have been adequately
evaluated
b) A sound and appropriate program for sampling testing
and evaluation of components in-process materials
finished products containers and closures for the purpose
of releasing materials or products has been established
including a robust supplier qualification program
16
PAI Objective 1 Readiness for Commercial Manufacturing cont
c) The establishment has sufficient facility and equipment
controls in place to prevent contamination of and by the
application product (or API)
d) Adequate procedures exist for batch release change control
investigating failures deviations complaints and adverse
events and for reporting this information to FDA such as
field alert reporting
e) The feasibility of the proposed commercial process and
manufacturing batch record including instructions
processing parameters and process control measures are
scientifically and objectively justified This objective is
linked to the firmrsquos process validation program
17
PAI Objective 2
Conformance to Application
Verify that the formulation manufacturing or
processing methods and analytical (or
examination) methods are consistent with
descriptions contained in the CMC section of the
application for the biobatch (and other pivotal
clinical batches when applicable) the proposed
commercial scale batch and API ndash Observing processing andor testing operations
ndash Compare the biobatch manufacturing process against the
proposed commercial batch record
18
PAI Objective 3
Data Integrity Audit
Audit the raw data hardcopy or electronic to
authenticate the data submitted in the CMC
section of the application
Verify that all relevant data (eg stability
biobatch data) were submitted in the CMC section
such that CDER product reviewers can rely on the
submitted data as complete and accurate ndash Laboratory notebooks and associated chromatograms generated
during release testing of biobatch
ndash Failure to include aberrant test results in CMC section
ndash Improper invalidation of OOS results
19
Investigatorrsquos Role during PAI
Assess the following
ndash Quality Systems
ndash Manufacturing Operations
ndash Sampling Plans
ndash Laboratory
ndash Test Methods Validation
ndash Drug Product Specifications
ndash ReprocessingReworking
ndash Standard Operating Procedures
ndash Batch Records 20
The Firmrsquos Role
Make records available (as appropriate) to
conduct the pre-approval inspection
ndash Product Development Report
ndash Batch Records and Laboratory Records
ndash ProtocolsSOPs
Assure facility is cGMP compliant and ready for
an FDA inspection
ndash Once an application is submitted to Center the firm
should be considered ready for inspection
21
Product Development Report
A very useful document for both the firm
and the FDA
The data generated during product
development which defines the drug
product targets the steps in the
manufacturing process where variation is
critical to quality and thereby focuses the
subsequent process validation effort
22
Product Development Report
API Impurity Profile
ndash How is the API characterized
Excipients
Formulation Wet or Dry Granulation
ndash Solution or Suspension
ndash Sterile - Terminalaseptic conditions
ndash TabletCapsule - ImmediateModified
ReleaseExtended Release
23
Product Development Report
Describes the development of Processing
Equipment order of addition of ingredients
to the formulation mixing times and
speeds drying time and temperature
nitrogen blankets blending hold times
compression slugging filling polishing
imprinting labeling and packaging
Product Development Report may not be a
formal document
24
Batch Records
The batch records submitted in the
application must be audited as part of
the inspection to assure
ndash That the proposed production process is the
same process that was used for the
manufacture of the biostability batches
25
ReprocessingReworking
cGMP regulations require reprocessing
procedures to be in writing If firm
makes provisions for reprocessing drug
product details must be submitted as
part of the application
ndash Standard Operating Procedures
ndash QA review and approval
26
27
Different Responsibilities
28
Different Responsibilities
29
Different Responsibilities
How ICH Q8 Q9 Q10
Guidelines Are Working
Together Throughout The
Product Life Cycle
30
Nov 2005 amp Nov 2008
ICH Q8 Q9 and Q10
High level guidances
(not prescriptive)
Science and risk-based
Encourages systematic
approaches
Applicable over entire product
lifecycle
Intended to work together to
enhance pharmaceutical product
quality
31
Pharmaceutical Development -
Q8(R2)
Describes science and risk-based
approaches for pharmaceutical product and
manufacturing process development
Introduced concepts of design space and
flexible regulatory approaches
Introduced concepts of Quality by Design
(QbD) and provided examples of QbD
development approaches and design space 32
Differing Approaches to
Pharmaceutical Development
33
Q8(R2) - Example QbD
Approach
Quality Target Product Profile (QTPP)
Determine ldquopotentialrdquo critical quality attributes
Link raw material attributes and process parameters
to CQAs and perform risk assessment
Develop a design space (optional and not required)
Design and implement a control strategy
Manage product lifecycle including continual
improvement
CQArsquos
Product Profile
Risk Assessments
Design Space
Control Strategy
Continual
Improvement34
Product Lifecycle
concept
Human and veterinary
drugs
Biotech products
Finish products amp APIs
Drug constituent of a
combination product
35
Manage product lifecycle including
continual improvement
Three-stage approach to process validation
Stage 1 ndash Process Design ndash Building and capturing process knowledge and understanding
ndash Establishing a strategy for process control
Stage 2 ndash Process Qualification ndash Design a facility and qualification of utilities and equipment
ndash Process performance qualification
ndash PPQ protocol
ndash PPQ protocol execution and report
Stage 3 ndash Continued Process Verification
36
Focus on alignment with
lsquoproduct lifecyclersquo
Manufacturers should
Understand the sources of variation
Detect the presence and degree of variation
Understand the impact of variation on the process and
ultimately on product attributes
Control the variation in a manner commensurate with
the risk it represents to the process and product
37
Process Validation
38
Focus on alignment with
lsquoproduct lifecyclersquo
39
Focus on alignment with
lsquoproduct lifecyclersquo
40
Focus on alignment with
lsquoproduct lifecyclersquo
FDA Quality Systems Guidance
for Finished Pharmaceuticals
41 Inspection Readiness BEC
Inspection Readiness BEC 42
Quality System
Drug Inspection Target Includes the Quality Control Unit and all of its review
and approval duties
ndash Approval of and adherence to Procedures and associated recordkeeping systems
ndash Product Reviews
ndash Complaint Reviews
ndash Failure Evaluations
ndash Change Control
ndash Product Improvement Projects
ndash RejectsReprocessingRework
ndash Stability
ndash Validation
ndash Training
Inspection Readiness BEC 43
Facilities and Equipment
Inspection Target
Building and Facilities along with
maintenance
Equipment qualification calibration and
maintenance
Water steam compressed gas HVAC
Change control system
Investigate discrepancies
Inspection Readiness BEC 44
Materials
Inspection Target
Procedures and documentation showing adequate control of finished products in-process materials components containers closures
Qualificationvalidation and security of computerized or automated processes
Change control system
Investigation of Discrepancies
Inspection Readiness BEC 45
Production System
Drug Inspection Target
Trainingqualification of personnel
Complete batch production documentation
Process validation
Production time limits
In-process testingexamination
Change Control System
Investigation of discrepancies
Inspection Readiness BEC 46
Packaging and Labeling
Inspection Target
Training
Acceptance operations
Control of materials to prevent mix-ups
Accountability
Packaginglabeling records
Line separation
Line clearance inspection and documentation
Validation of labelingpackaging operations
Investigation of discrepancies
Inspection Readiness BEC 47
Laboratory Control
Inspection Target
Staffing and training
Adequate equipment calibration and maintenance
Reference standards
Adherence to written procedures
Validationverification analytical methods
Analytical records and raw data
Adherence to OOS procedure and timely completion of investigations
Stability testing program and reserve samples
Change control system
Before the Inspection
SOP for Inspection Readiness
Strong Internal AuditAssessment function
ndash Continuous assessment using Risk Management
Criteria
ndash Not autopilot auditing
ndash Same auditorhellip
Perform a full-system audit (Mock) at least one
month prior to inspection
ndash Be sure any necessary CA-PA has been originated 48
Before the Inspection
Educate all your employees on proper inspection
rules and etiquette
Educate your supervisory (all exempt) personnel
on FDA regulations
Inspections and external audits concern to all
employees not only those from Quality Dept
Promote the participation on the internal audit
cadre
Certify internal auditors
49
Before the Inspection
A few words about Internal Audit
Internal Audits are the most powerful tool to
avoid inspection surprises
If the inspector discovers something why your
internal auditor did not
The same auditor auditing several consecutive
years the same area is not effective
An effective internal audit function is priceless
50
During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

What is the goal of a
Pre-Approval Inspection
To assure that establishments involved in the
manufacturing testing or other manipulation of
new drug dosage forms and drug substances are
evaluated for
ndash conformance with commitments in the application
ndash site cGMP compliance
ndash data authenticity reliability and accuracy
ndash adequacy of analytical methodologies
14
PAI Objectives (Program 7346832)
There are three primary inspectional objectives of
this PAI program These objectives are
1) Readiness for Commercial Manufacturing
2) Conformance to Application
3) Data Integrity Audit
15
PAI Objective 1 Readiness for Commercial Manufacturing
Determine whether the establishment has a quality
system that is designed to achieve sufficient control over
the facility and commercial manufacturing operations
a) Manufacturing and laboratory changes deviations and
trends relating to the development of new drug substance
and product manufacturing have been adequately
evaluated
b) A sound and appropriate program for sampling testing
and evaluation of components in-process materials
finished products containers and closures for the purpose
of releasing materials or products has been established
including a robust supplier qualification program
16
PAI Objective 1 Readiness for Commercial Manufacturing cont
c) The establishment has sufficient facility and equipment
controls in place to prevent contamination of and by the
application product (or API)
d) Adequate procedures exist for batch release change control
investigating failures deviations complaints and adverse
events and for reporting this information to FDA such as
field alert reporting
e) The feasibility of the proposed commercial process and
manufacturing batch record including instructions
processing parameters and process control measures are
scientifically and objectively justified This objective is
linked to the firmrsquos process validation program
17
PAI Objective 2
Conformance to Application
Verify that the formulation manufacturing or
processing methods and analytical (or
examination) methods are consistent with
descriptions contained in the CMC section of the
application for the biobatch (and other pivotal
clinical batches when applicable) the proposed
commercial scale batch and API ndash Observing processing andor testing operations
ndash Compare the biobatch manufacturing process against the
proposed commercial batch record
18
PAI Objective 3
Data Integrity Audit
Audit the raw data hardcopy or electronic to
authenticate the data submitted in the CMC
section of the application
Verify that all relevant data (eg stability
biobatch data) were submitted in the CMC section
such that CDER product reviewers can rely on the
submitted data as complete and accurate ndash Laboratory notebooks and associated chromatograms generated
during release testing of biobatch
ndash Failure to include aberrant test results in CMC section
ndash Improper invalidation of OOS results
19
Investigatorrsquos Role during PAI
Assess the following
ndash Quality Systems
ndash Manufacturing Operations
ndash Sampling Plans
ndash Laboratory
ndash Test Methods Validation
ndash Drug Product Specifications
ndash ReprocessingReworking
ndash Standard Operating Procedures
ndash Batch Records 20
The Firmrsquos Role
Make records available (as appropriate) to
conduct the pre-approval inspection
ndash Product Development Report
ndash Batch Records and Laboratory Records
ndash ProtocolsSOPs
Assure facility is cGMP compliant and ready for
an FDA inspection
ndash Once an application is submitted to Center the firm
should be considered ready for inspection
21
Product Development Report
A very useful document for both the firm
and the FDA
The data generated during product
development which defines the drug
product targets the steps in the
manufacturing process where variation is
critical to quality and thereby focuses the
subsequent process validation effort
22
Product Development Report
API Impurity Profile
ndash How is the API characterized
Excipients
Formulation Wet or Dry Granulation
ndash Solution or Suspension
ndash Sterile - Terminalaseptic conditions
ndash TabletCapsule - ImmediateModified
ReleaseExtended Release
23
Product Development Report
Describes the development of Processing
Equipment order of addition of ingredients
to the formulation mixing times and
speeds drying time and temperature
nitrogen blankets blending hold times
compression slugging filling polishing
imprinting labeling and packaging
Product Development Report may not be a
formal document
24
Batch Records
The batch records submitted in the
application must be audited as part of
the inspection to assure
ndash That the proposed production process is the
same process that was used for the
manufacture of the biostability batches
25
ReprocessingReworking
cGMP regulations require reprocessing
procedures to be in writing If firm
makes provisions for reprocessing drug
product details must be submitted as
part of the application
ndash Standard Operating Procedures
ndash QA review and approval
26
27
Different Responsibilities
28
Different Responsibilities
29
Different Responsibilities
How ICH Q8 Q9 Q10
Guidelines Are Working
Together Throughout The
Product Life Cycle
30
Nov 2005 amp Nov 2008
ICH Q8 Q9 and Q10
High level guidances
(not prescriptive)
Science and risk-based
Encourages systematic
approaches
Applicable over entire product
lifecycle
Intended to work together to
enhance pharmaceutical product
quality
31
Pharmaceutical Development -
Q8(R2)
Describes science and risk-based
approaches for pharmaceutical product and
manufacturing process development
Introduced concepts of design space and
flexible regulatory approaches
Introduced concepts of Quality by Design
(QbD) and provided examples of QbD
development approaches and design space 32
Differing Approaches to
Pharmaceutical Development
33
Q8(R2) - Example QbD
Approach
Quality Target Product Profile (QTPP)
Determine ldquopotentialrdquo critical quality attributes
Link raw material attributes and process parameters
to CQAs and perform risk assessment
Develop a design space (optional and not required)
Design and implement a control strategy
Manage product lifecycle including continual
improvement
CQArsquos
Product Profile
Risk Assessments
Design Space
Control Strategy
Continual
Improvement34
Product Lifecycle
concept
Human and veterinary
drugs
Biotech products
Finish products amp APIs
Drug constituent of a
combination product
35
Manage product lifecycle including
continual improvement
Three-stage approach to process validation
Stage 1 ndash Process Design ndash Building and capturing process knowledge and understanding
ndash Establishing a strategy for process control
Stage 2 ndash Process Qualification ndash Design a facility and qualification of utilities and equipment
ndash Process performance qualification
ndash PPQ protocol
ndash PPQ protocol execution and report
Stage 3 ndash Continued Process Verification
36
Focus on alignment with
lsquoproduct lifecyclersquo
Manufacturers should
Understand the sources of variation
Detect the presence and degree of variation
Understand the impact of variation on the process and
ultimately on product attributes
Control the variation in a manner commensurate with
the risk it represents to the process and product
37
Process Validation
38
Focus on alignment with
lsquoproduct lifecyclersquo
39
Focus on alignment with
lsquoproduct lifecyclersquo
40
Focus on alignment with
lsquoproduct lifecyclersquo
FDA Quality Systems Guidance
for Finished Pharmaceuticals
41 Inspection Readiness BEC
Inspection Readiness BEC 42
Quality System
Drug Inspection Target Includes the Quality Control Unit and all of its review
and approval duties
ndash Approval of and adherence to Procedures and associated recordkeeping systems
ndash Product Reviews
ndash Complaint Reviews
ndash Failure Evaluations
ndash Change Control
ndash Product Improvement Projects
ndash RejectsReprocessingRework
ndash Stability
ndash Validation
ndash Training
Inspection Readiness BEC 43
Facilities and Equipment
Inspection Target
Building and Facilities along with
maintenance
Equipment qualification calibration and
maintenance
Water steam compressed gas HVAC
Change control system
Investigate discrepancies
Inspection Readiness BEC 44
Materials
Inspection Target
Procedures and documentation showing adequate control of finished products in-process materials components containers closures
Qualificationvalidation and security of computerized or automated processes
Change control system
Investigation of Discrepancies
Inspection Readiness BEC 45
Production System
Drug Inspection Target
Trainingqualification of personnel
Complete batch production documentation
Process validation
Production time limits
In-process testingexamination
Change Control System
Investigation of discrepancies
Inspection Readiness BEC 46
Packaging and Labeling
Inspection Target
Training
Acceptance operations
Control of materials to prevent mix-ups
Accountability
Packaginglabeling records
Line separation
Line clearance inspection and documentation
Validation of labelingpackaging operations
Investigation of discrepancies
Inspection Readiness BEC 47
Laboratory Control
Inspection Target
Staffing and training
Adequate equipment calibration and maintenance
Reference standards
Adherence to written procedures
Validationverification analytical methods
Analytical records and raw data
Adherence to OOS procedure and timely completion of investigations
Stability testing program and reserve samples
Change control system
Before the Inspection
SOP for Inspection Readiness
Strong Internal AuditAssessment function
ndash Continuous assessment using Risk Management
Criteria
ndash Not autopilot auditing
ndash Same auditorhellip
Perform a full-system audit (Mock) at least one
month prior to inspection
ndash Be sure any necessary CA-PA has been originated 48
Before the Inspection
Educate all your employees on proper inspection
rules and etiquette
Educate your supervisory (all exempt) personnel
on FDA regulations
Inspections and external audits concern to all
employees not only those from Quality Dept
Promote the participation on the internal audit
cadre
Certify internal auditors
49
Before the Inspection
A few words about Internal Audit
Internal Audits are the most powerful tool to
avoid inspection surprises
If the inspector discovers something why your
internal auditor did not
The same auditor auditing several consecutive
years the same area is not effective
An effective internal audit function is priceless
50
During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

PAI Objectives (Program 7346832)
There are three primary inspectional objectives of
this PAI program These objectives are
1) Readiness for Commercial Manufacturing
2) Conformance to Application
3) Data Integrity Audit
15
PAI Objective 1 Readiness for Commercial Manufacturing
Determine whether the establishment has a quality
system that is designed to achieve sufficient control over
the facility and commercial manufacturing operations
a) Manufacturing and laboratory changes deviations and
trends relating to the development of new drug substance
and product manufacturing have been adequately
evaluated
b) A sound and appropriate program for sampling testing
and evaluation of components in-process materials
finished products containers and closures for the purpose
of releasing materials or products has been established
including a robust supplier qualification program
16
PAI Objective 1 Readiness for Commercial Manufacturing cont
c) The establishment has sufficient facility and equipment
controls in place to prevent contamination of and by the
application product (or API)
d) Adequate procedures exist for batch release change control
investigating failures deviations complaints and adverse
events and for reporting this information to FDA such as
field alert reporting
e) The feasibility of the proposed commercial process and
manufacturing batch record including instructions
processing parameters and process control measures are
scientifically and objectively justified This objective is
linked to the firmrsquos process validation program
17
PAI Objective 2
Conformance to Application
Verify that the formulation manufacturing or
processing methods and analytical (or
examination) methods are consistent with
descriptions contained in the CMC section of the
application for the biobatch (and other pivotal
clinical batches when applicable) the proposed
commercial scale batch and API ndash Observing processing andor testing operations
ndash Compare the biobatch manufacturing process against the
proposed commercial batch record
18
PAI Objective 3
Data Integrity Audit
Audit the raw data hardcopy or electronic to
authenticate the data submitted in the CMC
section of the application
Verify that all relevant data (eg stability
biobatch data) were submitted in the CMC section
such that CDER product reviewers can rely on the
submitted data as complete and accurate ndash Laboratory notebooks and associated chromatograms generated
during release testing of biobatch
ndash Failure to include aberrant test results in CMC section
ndash Improper invalidation of OOS results
19
Investigatorrsquos Role during PAI
Assess the following
ndash Quality Systems
ndash Manufacturing Operations
ndash Sampling Plans
ndash Laboratory
ndash Test Methods Validation
ndash Drug Product Specifications
ndash ReprocessingReworking
ndash Standard Operating Procedures
ndash Batch Records 20
The Firmrsquos Role
Make records available (as appropriate) to
conduct the pre-approval inspection
ndash Product Development Report
ndash Batch Records and Laboratory Records
ndash ProtocolsSOPs
Assure facility is cGMP compliant and ready for
an FDA inspection
ndash Once an application is submitted to Center the firm
should be considered ready for inspection
21
Product Development Report
A very useful document for both the firm
and the FDA
The data generated during product
development which defines the drug
product targets the steps in the
manufacturing process where variation is
critical to quality and thereby focuses the
subsequent process validation effort
22
Product Development Report
API Impurity Profile
ndash How is the API characterized
Excipients
Formulation Wet or Dry Granulation
ndash Solution or Suspension
ndash Sterile - Terminalaseptic conditions
ndash TabletCapsule - ImmediateModified
ReleaseExtended Release
23
Product Development Report
Describes the development of Processing
Equipment order of addition of ingredients
to the formulation mixing times and
speeds drying time and temperature
nitrogen blankets blending hold times
compression slugging filling polishing
imprinting labeling and packaging
Product Development Report may not be a
formal document
24
Batch Records
The batch records submitted in the
application must be audited as part of
the inspection to assure
ndash That the proposed production process is the
same process that was used for the
manufacture of the biostability batches
25
ReprocessingReworking
cGMP regulations require reprocessing
procedures to be in writing If firm
makes provisions for reprocessing drug
product details must be submitted as
part of the application
ndash Standard Operating Procedures
ndash QA review and approval
26
27
Different Responsibilities
28
Different Responsibilities
29
Different Responsibilities
How ICH Q8 Q9 Q10
Guidelines Are Working
Together Throughout The
Product Life Cycle
30
Nov 2005 amp Nov 2008
ICH Q8 Q9 and Q10
High level guidances
(not prescriptive)
Science and risk-based
Encourages systematic
approaches
Applicable over entire product
lifecycle
Intended to work together to
enhance pharmaceutical product
quality
31
Pharmaceutical Development -
Q8(R2)
Describes science and risk-based
approaches for pharmaceutical product and
manufacturing process development
Introduced concepts of design space and
flexible regulatory approaches
Introduced concepts of Quality by Design
(QbD) and provided examples of QbD
development approaches and design space 32
Differing Approaches to
Pharmaceutical Development
33
Q8(R2) - Example QbD
Approach
Quality Target Product Profile (QTPP)
Determine ldquopotentialrdquo critical quality attributes
Link raw material attributes and process parameters
to CQAs and perform risk assessment
Develop a design space (optional and not required)
Design and implement a control strategy
Manage product lifecycle including continual
improvement
CQArsquos
Product Profile
Risk Assessments
Design Space
Control Strategy
Continual
Improvement34
Product Lifecycle
concept
Human and veterinary
drugs
Biotech products
Finish products amp APIs
Drug constituent of a
combination product
35
Manage product lifecycle including
continual improvement
Three-stage approach to process validation
Stage 1 ndash Process Design ndash Building and capturing process knowledge and understanding
ndash Establishing a strategy for process control
Stage 2 ndash Process Qualification ndash Design a facility and qualification of utilities and equipment
ndash Process performance qualification
ndash PPQ protocol
ndash PPQ protocol execution and report
Stage 3 ndash Continued Process Verification
36
Focus on alignment with
lsquoproduct lifecyclersquo
Manufacturers should
Understand the sources of variation
Detect the presence and degree of variation
Understand the impact of variation on the process and
ultimately on product attributes
Control the variation in a manner commensurate with
the risk it represents to the process and product
37
Process Validation
38
Focus on alignment with
lsquoproduct lifecyclersquo
39
Focus on alignment with
lsquoproduct lifecyclersquo
40
Focus on alignment with
lsquoproduct lifecyclersquo
FDA Quality Systems Guidance
for Finished Pharmaceuticals
41 Inspection Readiness BEC
Inspection Readiness BEC 42
Quality System
Drug Inspection Target Includes the Quality Control Unit and all of its review
and approval duties
ndash Approval of and adherence to Procedures and associated recordkeeping systems
ndash Product Reviews
ndash Complaint Reviews
ndash Failure Evaluations
ndash Change Control
ndash Product Improvement Projects
ndash RejectsReprocessingRework
ndash Stability
ndash Validation
ndash Training
Inspection Readiness BEC 43
Facilities and Equipment
Inspection Target
Building and Facilities along with
maintenance
Equipment qualification calibration and
maintenance
Water steam compressed gas HVAC
Change control system
Investigate discrepancies
Inspection Readiness BEC 44
Materials
Inspection Target
Procedures and documentation showing adequate control of finished products in-process materials components containers closures
Qualificationvalidation and security of computerized or automated processes
Change control system
Investigation of Discrepancies
Inspection Readiness BEC 45
Production System
Drug Inspection Target
Trainingqualification of personnel
Complete batch production documentation
Process validation
Production time limits
In-process testingexamination
Change Control System
Investigation of discrepancies
Inspection Readiness BEC 46
Packaging and Labeling
Inspection Target
Training
Acceptance operations
Control of materials to prevent mix-ups
Accountability
Packaginglabeling records
Line separation
Line clearance inspection and documentation
Validation of labelingpackaging operations
Investigation of discrepancies
Inspection Readiness BEC 47
Laboratory Control
Inspection Target
Staffing and training
Adequate equipment calibration and maintenance
Reference standards
Adherence to written procedures
Validationverification analytical methods
Analytical records and raw data
Adherence to OOS procedure and timely completion of investigations
Stability testing program and reserve samples
Change control system
Before the Inspection
SOP for Inspection Readiness
Strong Internal AuditAssessment function
ndash Continuous assessment using Risk Management
Criteria
ndash Not autopilot auditing
ndash Same auditorhellip
Perform a full-system audit (Mock) at least one
month prior to inspection
ndash Be sure any necessary CA-PA has been originated 48
Before the Inspection
Educate all your employees on proper inspection
rules and etiquette
Educate your supervisory (all exempt) personnel
on FDA regulations
Inspections and external audits concern to all
employees not only those from Quality Dept
Promote the participation on the internal audit
cadre
Certify internal auditors
49
Before the Inspection
A few words about Internal Audit
Internal Audits are the most powerful tool to
avoid inspection surprises
If the inspector discovers something why your
internal auditor did not
The same auditor auditing several consecutive
years the same area is not effective
An effective internal audit function is priceless
50
During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

PAI Objective 1 Readiness for Commercial Manufacturing
Determine whether the establishment has a quality
system that is designed to achieve sufficient control over
the facility and commercial manufacturing operations
a) Manufacturing and laboratory changes deviations and
trends relating to the development of new drug substance
and product manufacturing have been adequately
evaluated
b) A sound and appropriate program for sampling testing
and evaluation of components in-process materials
finished products containers and closures for the purpose
of releasing materials or products has been established
including a robust supplier qualification program
16
PAI Objective 1 Readiness for Commercial Manufacturing cont
c) The establishment has sufficient facility and equipment
controls in place to prevent contamination of and by the
application product (or API)
d) Adequate procedures exist for batch release change control
investigating failures deviations complaints and adverse
events and for reporting this information to FDA such as
field alert reporting
e) The feasibility of the proposed commercial process and
manufacturing batch record including instructions
processing parameters and process control measures are
scientifically and objectively justified This objective is
linked to the firmrsquos process validation program
17
PAI Objective 2
Conformance to Application
Verify that the formulation manufacturing or
processing methods and analytical (or
examination) methods are consistent with
descriptions contained in the CMC section of the
application for the biobatch (and other pivotal
clinical batches when applicable) the proposed
commercial scale batch and API ndash Observing processing andor testing operations
ndash Compare the biobatch manufacturing process against the
proposed commercial batch record
18
PAI Objective 3
Data Integrity Audit
Audit the raw data hardcopy or electronic to
authenticate the data submitted in the CMC
section of the application
Verify that all relevant data (eg stability
biobatch data) were submitted in the CMC section
such that CDER product reviewers can rely on the
submitted data as complete and accurate ndash Laboratory notebooks and associated chromatograms generated
during release testing of biobatch
ndash Failure to include aberrant test results in CMC section
ndash Improper invalidation of OOS results
19
Investigatorrsquos Role during PAI
Assess the following
ndash Quality Systems
ndash Manufacturing Operations
ndash Sampling Plans
ndash Laboratory
ndash Test Methods Validation
ndash Drug Product Specifications
ndash ReprocessingReworking
ndash Standard Operating Procedures
ndash Batch Records 20
The Firmrsquos Role
Make records available (as appropriate) to
conduct the pre-approval inspection
ndash Product Development Report
ndash Batch Records and Laboratory Records
ndash ProtocolsSOPs
Assure facility is cGMP compliant and ready for
an FDA inspection
ndash Once an application is submitted to Center the firm
should be considered ready for inspection
21
Product Development Report
A very useful document for both the firm
and the FDA
The data generated during product
development which defines the drug
product targets the steps in the
manufacturing process where variation is
critical to quality and thereby focuses the
subsequent process validation effort
22
Product Development Report
API Impurity Profile
ndash How is the API characterized
Excipients
Formulation Wet or Dry Granulation
ndash Solution or Suspension
ndash Sterile - Terminalaseptic conditions
ndash TabletCapsule - ImmediateModified
ReleaseExtended Release
23
Product Development Report
Describes the development of Processing
Equipment order of addition of ingredients
to the formulation mixing times and
speeds drying time and temperature
nitrogen blankets blending hold times
compression slugging filling polishing
imprinting labeling and packaging
Product Development Report may not be a
formal document
24
Batch Records
The batch records submitted in the
application must be audited as part of
the inspection to assure
ndash That the proposed production process is the
same process that was used for the
manufacture of the biostability batches
25
ReprocessingReworking
cGMP regulations require reprocessing
procedures to be in writing If firm
makes provisions for reprocessing drug
product details must be submitted as
part of the application
ndash Standard Operating Procedures
ndash QA review and approval
26
27
Different Responsibilities
28
Different Responsibilities
29
Different Responsibilities
How ICH Q8 Q9 Q10
Guidelines Are Working
Together Throughout The
Product Life Cycle
30
Nov 2005 amp Nov 2008
ICH Q8 Q9 and Q10
High level guidances
(not prescriptive)
Science and risk-based
Encourages systematic
approaches
Applicable over entire product
lifecycle
Intended to work together to
enhance pharmaceutical product
quality
31
Pharmaceutical Development -
Q8(R2)
Describes science and risk-based
approaches for pharmaceutical product and
manufacturing process development
Introduced concepts of design space and
flexible regulatory approaches
Introduced concepts of Quality by Design
(QbD) and provided examples of QbD
development approaches and design space 32
Differing Approaches to
Pharmaceutical Development
33
Q8(R2) - Example QbD
Approach
Quality Target Product Profile (QTPP)
Determine ldquopotentialrdquo critical quality attributes
Link raw material attributes and process parameters
to CQAs and perform risk assessment
Develop a design space (optional and not required)
Design and implement a control strategy
Manage product lifecycle including continual
improvement
CQArsquos
Product Profile
Risk Assessments
Design Space
Control Strategy
Continual
Improvement34
Product Lifecycle
concept
Human and veterinary
drugs
Biotech products
Finish products amp APIs
Drug constituent of a
combination product
35
Manage product lifecycle including
continual improvement
Three-stage approach to process validation
Stage 1 ndash Process Design ndash Building and capturing process knowledge and understanding
ndash Establishing a strategy for process control
Stage 2 ndash Process Qualification ndash Design a facility and qualification of utilities and equipment
ndash Process performance qualification
ndash PPQ protocol
ndash PPQ protocol execution and report
Stage 3 ndash Continued Process Verification
36
Focus on alignment with
lsquoproduct lifecyclersquo
Manufacturers should
Understand the sources of variation
Detect the presence and degree of variation
Understand the impact of variation on the process and
ultimately on product attributes
Control the variation in a manner commensurate with
the risk it represents to the process and product
37
Process Validation
38
Focus on alignment with
lsquoproduct lifecyclersquo
39
Focus on alignment with
lsquoproduct lifecyclersquo
40
Focus on alignment with
lsquoproduct lifecyclersquo
FDA Quality Systems Guidance
for Finished Pharmaceuticals
41 Inspection Readiness BEC
Inspection Readiness BEC 42
Quality System
Drug Inspection Target Includes the Quality Control Unit and all of its review
and approval duties
ndash Approval of and adherence to Procedures and associated recordkeeping systems
ndash Product Reviews
ndash Complaint Reviews
ndash Failure Evaluations
ndash Change Control
ndash Product Improvement Projects
ndash RejectsReprocessingRework
ndash Stability
ndash Validation
ndash Training
Inspection Readiness BEC 43
Facilities and Equipment
Inspection Target
Building and Facilities along with
maintenance
Equipment qualification calibration and
maintenance
Water steam compressed gas HVAC
Change control system
Investigate discrepancies
Inspection Readiness BEC 44
Materials
Inspection Target
Procedures and documentation showing adequate control of finished products in-process materials components containers closures
Qualificationvalidation and security of computerized or automated processes
Change control system
Investigation of Discrepancies
Inspection Readiness BEC 45
Production System
Drug Inspection Target
Trainingqualification of personnel
Complete batch production documentation
Process validation
Production time limits
In-process testingexamination
Change Control System
Investigation of discrepancies
Inspection Readiness BEC 46
Packaging and Labeling
Inspection Target
Training
Acceptance operations
Control of materials to prevent mix-ups
Accountability
Packaginglabeling records
Line separation
Line clearance inspection and documentation
Validation of labelingpackaging operations
Investigation of discrepancies
Inspection Readiness BEC 47
Laboratory Control
Inspection Target
Staffing and training
Adequate equipment calibration and maintenance
Reference standards
Adherence to written procedures
Validationverification analytical methods
Analytical records and raw data
Adherence to OOS procedure and timely completion of investigations
Stability testing program and reserve samples
Change control system
Before the Inspection
SOP for Inspection Readiness
Strong Internal AuditAssessment function
ndash Continuous assessment using Risk Management
Criteria
ndash Not autopilot auditing
ndash Same auditorhellip
Perform a full-system audit (Mock) at least one
month prior to inspection
ndash Be sure any necessary CA-PA has been originated 48
Before the Inspection
Educate all your employees on proper inspection
rules and etiquette
Educate your supervisory (all exempt) personnel
on FDA regulations
Inspections and external audits concern to all
employees not only those from Quality Dept
Promote the participation on the internal audit
cadre
Certify internal auditors
49
Before the Inspection
A few words about Internal Audit
Internal Audits are the most powerful tool to
avoid inspection surprises
If the inspector discovers something why your
internal auditor did not
The same auditor auditing several consecutive
years the same area is not effective
An effective internal audit function is priceless
50
During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

PAI Objective 1 Readiness for Commercial Manufacturing cont
c) The establishment has sufficient facility and equipment
controls in place to prevent contamination of and by the
application product (or API)
d) Adequate procedures exist for batch release change control
investigating failures deviations complaints and adverse
events and for reporting this information to FDA such as
field alert reporting
e) The feasibility of the proposed commercial process and
manufacturing batch record including instructions
processing parameters and process control measures are
scientifically and objectively justified This objective is
linked to the firmrsquos process validation program
17
PAI Objective 2
Conformance to Application
Verify that the formulation manufacturing or
processing methods and analytical (or
examination) methods are consistent with
descriptions contained in the CMC section of the
application for the biobatch (and other pivotal
clinical batches when applicable) the proposed
commercial scale batch and API ndash Observing processing andor testing operations
ndash Compare the biobatch manufacturing process against the
proposed commercial batch record
18
PAI Objective 3
Data Integrity Audit
Audit the raw data hardcopy or electronic to
authenticate the data submitted in the CMC
section of the application
Verify that all relevant data (eg stability
biobatch data) were submitted in the CMC section
such that CDER product reviewers can rely on the
submitted data as complete and accurate ndash Laboratory notebooks and associated chromatograms generated
during release testing of biobatch
ndash Failure to include aberrant test results in CMC section
ndash Improper invalidation of OOS results
19
Investigatorrsquos Role during PAI
Assess the following
ndash Quality Systems
ndash Manufacturing Operations
ndash Sampling Plans
ndash Laboratory
ndash Test Methods Validation
ndash Drug Product Specifications
ndash ReprocessingReworking
ndash Standard Operating Procedures
ndash Batch Records 20
The Firmrsquos Role
Make records available (as appropriate) to
conduct the pre-approval inspection
ndash Product Development Report
ndash Batch Records and Laboratory Records
ndash ProtocolsSOPs
Assure facility is cGMP compliant and ready for
an FDA inspection
ndash Once an application is submitted to Center the firm
should be considered ready for inspection
21
Product Development Report
A very useful document for both the firm
and the FDA
The data generated during product
development which defines the drug
product targets the steps in the
manufacturing process where variation is
critical to quality and thereby focuses the
subsequent process validation effort
22
Product Development Report
API Impurity Profile
ndash How is the API characterized
Excipients
Formulation Wet or Dry Granulation
ndash Solution or Suspension
ndash Sterile - Terminalaseptic conditions
ndash TabletCapsule - ImmediateModified
ReleaseExtended Release
23
Product Development Report
Describes the development of Processing
Equipment order of addition of ingredients
to the formulation mixing times and
speeds drying time and temperature
nitrogen blankets blending hold times
compression slugging filling polishing
imprinting labeling and packaging
Product Development Report may not be a
formal document
24
Batch Records
The batch records submitted in the
application must be audited as part of
the inspection to assure
ndash That the proposed production process is the
same process that was used for the
manufacture of the biostability batches
25
ReprocessingReworking
cGMP regulations require reprocessing
procedures to be in writing If firm
makes provisions for reprocessing drug
product details must be submitted as
part of the application
ndash Standard Operating Procedures
ndash QA review and approval
26
27
Different Responsibilities
28
Different Responsibilities
29
Different Responsibilities
How ICH Q8 Q9 Q10
Guidelines Are Working
Together Throughout The
Product Life Cycle
30
Nov 2005 amp Nov 2008
ICH Q8 Q9 and Q10
High level guidances
(not prescriptive)
Science and risk-based
Encourages systematic
approaches
Applicable over entire product
lifecycle
Intended to work together to
enhance pharmaceutical product
quality
31
Pharmaceutical Development -
Q8(R2)
Describes science and risk-based
approaches for pharmaceutical product and
manufacturing process development
Introduced concepts of design space and
flexible regulatory approaches
Introduced concepts of Quality by Design
(QbD) and provided examples of QbD
development approaches and design space 32
Differing Approaches to
Pharmaceutical Development
33
Q8(R2) - Example QbD
Approach
Quality Target Product Profile (QTPP)
Determine ldquopotentialrdquo critical quality attributes
Link raw material attributes and process parameters
to CQAs and perform risk assessment
Develop a design space (optional and not required)
Design and implement a control strategy
Manage product lifecycle including continual
improvement
CQArsquos
Product Profile
Risk Assessments
Design Space
Control Strategy
Continual
Improvement34
Product Lifecycle
concept
Human and veterinary
drugs
Biotech products
Finish products amp APIs
Drug constituent of a
combination product
35
Manage product lifecycle including
continual improvement
Three-stage approach to process validation
Stage 1 ndash Process Design ndash Building and capturing process knowledge and understanding
ndash Establishing a strategy for process control
Stage 2 ndash Process Qualification ndash Design a facility and qualification of utilities and equipment
ndash Process performance qualification
ndash PPQ protocol
ndash PPQ protocol execution and report
Stage 3 ndash Continued Process Verification
36
Focus on alignment with
lsquoproduct lifecyclersquo
Manufacturers should
Understand the sources of variation
Detect the presence and degree of variation
Understand the impact of variation on the process and
ultimately on product attributes
Control the variation in a manner commensurate with
the risk it represents to the process and product
37
Process Validation
38
Focus on alignment with
lsquoproduct lifecyclersquo
39
Focus on alignment with
lsquoproduct lifecyclersquo
40
Focus on alignment with
lsquoproduct lifecyclersquo
FDA Quality Systems Guidance
for Finished Pharmaceuticals
41 Inspection Readiness BEC
Inspection Readiness BEC 42
Quality System
Drug Inspection Target Includes the Quality Control Unit and all of its review
and approval duties
ndash Approval of and adherence to Procedures and associated recordkeeping systems
ndash Product Reviews
ndash Complaint Reviews
ndash Failure Evaluations
ndash Change Control
ndash Product Improvement Projects
ndash RejectsReprocessingRework
ndash Stability
ndash Validation
ndash Training
Inspection Readiness BEC 43
Facilities and Equipment
Inspection Target
Building and Facilities along with
maintenance
Equipment qualification calibration and
maintenance
Water steam compressed gas HVAC
Change control system
Investigate discrepancies
Inspection Readiness BEC 44
Materials
Inspection Target
Procedures and documentation showing adequate control of finished products in-process materials components containers closures
Qualificationvalidation and security of computerized or automated processes
Change control system
Investigation of Discrepancies
Inspection Readiness BEC 45
Production System
Drug Inspection Target
Trainingqualification of personnel
Complete batch production documentation
Process validation
Production time limits
In-process testingexamination
Change Control System
Investigation of discrepancies
Inspection Readiness BEC 46
Packaging and Labeling
Inspection Target
Training
Acceptance operations
Control of materials to prevent mix-ups
Accountability
Packaginglabeling records
Line separation
Line clearance inspection and documentation
Validation of labelingpackaging operations
Investigation of discrepancies
Inspection Readiness BEC 47
Laboratory Control
Inspection Target
Staffing and training
Adequate equipment calibration and maintenance
Reference standards
Adherence to written procedures
Validationverification analytical methods
Analytical records and raw data
Adherence to OOS procedure and timely completion of investigations
Stability testing program and reserve samples
Change control system
Before the Inspection
SOP for Inspection Readiness
Strong Internal AuditAssessment function
ndash Continuous assessment using Risk Management
Criteria
ndash Not autopilot auditing
ndash Same auditorhellip
Perform a full-system audit (Mock) at least one
month prior to inspection
ndash Be sure any necessary CA-PA has been originated 48
Before the Inspection
Educate all your employees on proper inspection
rules and etiquette
Educate your supervisory (all exempt) personnel
on FDA regulations
Inspections and external audits concern to all
employees not only those from Quality Dept
Promote the participation on the internal audit
cadre
Certify internal auditors
49
Before the Inspection
A few words about Internal Audit
Internal Audits are the most powerful tool to
avoid inspection surprises
If the inspector discovers something why your
internal auditor did not
The same auditor auditing several consecutive
years the same area is not effective
An effective internal audit function is priceless
50
During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

PAI Objective 2
Conformance to Application
Verify that the formulation manufacturing or
processing methods and analytical (or
examination) methods are consistent with
descriptions contained in the CMC section of the
application for the biobatch (and other pivotal
clinical batches when applicable) the proposed
commercial scale batch and API ndash Observing processing andor testing operations
ndash Compare the biobatch manufacturing process against the
proposed commercial batch record
18
PAI Objective 3
Data Integrity Audit
Audit the raw data hardcopy or electronic to
authenticate the data submitted in the CMC
section of the application
Verify that all relevant data (eg stability
biobatch data) were submitted in the CMC section
such that CDER product reviewers can rely on the
submitted data as complete and accurate ndash Laboratory notebooks and associated chromatograms generated
during release testing of biobatch
ndash Failure to include aberrant test results in CMC section
ndash Improper invalidation of OOS results
19
Investigatorrsquos Role during PAI
Assess the following
ndash Quality Systems
ndash Manufacturing Operations
ndash Sampling Plans
ndash Laboratory
ndash Test Methods Validation
ndash Drug Product Specifications
ndash ReprocessingReworking
ndash Standard Operating Procedures
ndash Batch Records 20
The Firmrsquos Role
Make records available (as appropriate) to
conduct the pre-approval inspection
ndash Product Development Report
ndash Batch Records and Laboratory Records
ndash ProtocolsSOPs
Assure facility is cGMP compliant and ready for
an FDA inspection
ndash Once an application is submitted to Center the firm
should be considered ready for inspection
21
Product Development Report
A very useful document for both the firm
and the FDA
The data generated during product
development which defines the drug
product targets the steps in the
manufacturing process where variation is
critical to quality and thereby focuses the
subsequent process validation effort
22
Product Development Report
API Impurity Profile
ndash How is the API characterized
Excipients
Formulation Wet or Dry Granulation
ndash Solution or Suspension
ndash Sterile - Terminalaseptic conditions
ndash TabletCapsule - ImmediateModified
ReleaseExtended Release
23
Product Development Report
Describes the development of Processing
Equipment order of addition of ingredients
to the formulation mixing times and
speeds drying time and temperature
nitrogen blankets blending hold times
compression slugging filling polishing
imprinting labeling and packaging
Product Development Report may not be a
formal document
24
Batch Records
The batch records submitted in the
application must be audited as part of
the inspection to assure
ndash That the proposed production process is the
same process that was used for the
manufacture of the biostability batches
25
ReprocessingReworking
cGMP regulations require reprocessing
procedures to be in writing If firm
makes provisions for reprocessing drug
product details must be submitted as
part of the application
ndash Standard Operating Procedures
ndash QA review and approval
26
27
Different Responsibilities
28
Different Responsibilities
29
Different Responsibilities
How ICH Q8 Q9 Q10
Guidelines Are Working
Together Throughout The
Product Life Cycle
30
Nov 2005 amp Nov 2008
ICH Q8 Q9 and Q10
High level guidances
(not prescriptive)
Science and risk-based
Encourages systematic
approaches
Applicable over entire product
lifecycle
Intended to work together to
enhance pharmaceutical product
quality
31
Pharmaceutical Development -
Q8(R2)
Describes science and risk-based
approaches for pharmaceutical product and
manufacturing process development
Introduced concepts of design space and
flexible regulatory approaches
Introduced concepts of Quality by Design
(QbD) and provided examples of QbD
development approaches and design space 32
Differing Approaches to
Pharmaceutical Development
33
Q8(R2) - Example QbD
Approach
Quality Target Product Profile (QTPP)
Determine ldquopotentialrdquo critical quality attributes
Link raw material attributes and process parameters
to CQAs and perform risk assessment
Develop a design space (optional and not required)
Design and implement a control strategy
Manage product lifecycle including continual
improvement
CQArsquos
Product Profile
Risk Assessments
Design Space
Control Strategy
Continual
Improvement34
Product Lifecycle
concept
Human and veterinary
drugs
Biotech products
Finish products amp APIs
Drug constituent of a
combination product
35
Manage product lifecycle including
continual improvement
Three-stage approach to process validation
Stage 1 ndash Process Design ndash Building and capturing process knowledge and understanding
ndash Establishing a strategy for process control
Stage 2 ndash Process Qualification ndash Design a facility and qualification of utilities and equipment
ndash Process performance qualification
ndash PPQ protocol
ndash PPQ protocol execution and report
Stage 3 ndash Continued Process Verification
36
Focus on alignment with
lsquoproduct lifecyclersquo
Manufacturers should
Understand the sources of variation
Detect the presence and degree of variation
Understand the impact of variation on the process and
ultimately on product attributes
Control the variation in a manner commensurate with
the risk it represents to the process and product
37
Process Validation
38
Focus on alignment with
lsquoproduct lifecyclersquo
39
Focus on alignment with
lsquoproduct lifecyclersquo
40
Focus on alignment with
lsquoproduct lifecyclersquo
FDA Quality Systems Guidance
for Finished Pharmaceuticals
41 Inspection Readiness BEC
Inspection Readiness BEC 42
Quality System
Drug Inspection Target Includes the Quality Control Unit and all of its review
and approval duties
ndash Approval of and adherence to Procedures and associated recordkeeping systems
ndash Product Reviews
ndash Complaint Reviews
ndash Failure Evaluations
ndash Change Control
ndash Product Improvement Projects
ndash RejectsReprocessingRework
ndash Stability
ndash Validation
ndash Training
Inspection Readiness BEC 43
Facilities and Equipment
Inspection Target
Building and Facilities along with
maintenance
Equipment qualification calibration and
maintenance
Water steam compressed gas HVAC
Change control system
Investigate discrepancies
Inspection Readiness BEC 44
Materials
Inspection Target
Procedures and documentation showing adequate control of finished products in-process materials components containers closures
Qualificationvalidation and security of computerized or automated processes
Change control system
Investigation of Discrepancies
Inspection Readiness BEC 45
Production System
Drug Inspection Target
Trainingqualification of personnel
Complete batch production documentation
Process validation
Production time limits
In-process testingexamination
Change Control System
Investigation of discrepancies
Inspection Readiness BEC 46
Packaging and Labeling
Inspection Target
Training
Acceptance operations
Control of materials to prevent mix-ups
Accountability
Packaginglabeling records
Line separation
Line clearance inspection and documentation
Validation of labelingpackaging operations
Investigation of discrepancies
Inspection Readiness BEC 47
Laboratory Control
Inspection Target
Staffing and training
Adequate equipment calibration and maintenance
Reference standards
Adherence to written procedures
Validationverification analytical methods
Analytical records and raw data
Adherence to OOS procedure and timely completion of investigations
Stability testing program and reserve samples
Change control system
Before the Inspection
SOP for Inspection Readiness
Strong Internal AuditAssessment function
ndash Continuous assessment using Risk Management
Criteria
ndash Not autopilot auditing
ndash Same auditorhellip
Perform a full-system audit (Mock) at least one
month prior to inspection
ndash Be sure any necessary CA-PA has been originated 48
Before the Inspection
Educate all your employees on proper inspection
rules and etiquette
Educate your supervisory (all exempt) personnel
on FDA regulations
Inspections and external audits concern to all
employees not only those from Quality Dept
Promote the participation on the internal audit
cadre
Certify internal auditors
49
Before the Inspection
A few words about Internal Audit
Internal Audits are the most powerful tool to
avoid inspection surprises
If the inspector discovers something why your
internal auditor did not
The same auditor auditing several consecutive
years the same area is not effective
An effective internal audit function is priceless
50
During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

PAI Objective 3
Data Integrity Audit
Audit the raw data hardcopy or electronic to
authenticate the data submitted in the CMC
section of the application
Verify that all relevant data (eg stability
biobatch data) were submitted in the CMC section
such that CDER product reviewers can rely on the
submitted data as complete and accurate ndash Laboratory notebooks and associated chromatograms generated
during release testing of biobatch
ndash Failure to include aberrant test results in CMC section
ndash Improper invalidation of OOS results
19
Investigatorrsquos Role during PAI
Assess the following
ndash Quality Systems
ndash Manufacturing Operations
ndash Sampling Plans
ndash Laboratory
ndash Test Methods Validation
ndash Drug Product Specifications
ndash ReprocessingReworking
ndash Standard Operating Procedures
ndash Batch Records 20
The Firmrsquos Role
Make records available (as appropriate) to
conduct the pre-approval inspection
ndash Product Development Report
ndash Batch Records and Laboratory Records
ndash ProtocolsSOPs
Assure facility is cGMP compliant and ready for
an FDA inspection
ndash Once an application is submitted to Center the firm
should be considered ready for inspection
21
Product Development Report
A very useful document for both the firm
and the FDA
The data generated during product
development which defines the drug
product targets the steps in the
manufacturing process where variation is
critical to quality and thereby focuses the
subsequent process validation effort
22
Product Development Report
API Impurity Profile
ndash How is the API characterized
Excipients
Formulation Wet or Dry Granulation
ndash Solution or Suspension
ndash Sterile - Terminalaseptic conditions
ndash TabletCapsule - ImmediateModified
ReleaseExtended Release
23
Product Development Report
Describes the development of Processing
Equipment order of addition of ingredients
to the formulation mixing times and
speeds drying time and temperature
nitrogen blankets blending hold times
compression slugging filling polishing
imprinting labeling and packaging
Product Development Report may not be a
formal document
24
Batch Records
The batch records submitted in the
application must be audited as part of
the inspection to assure
ndash That the proposed production process is the
same process that was used for the
manufacture of the biostability batches
25
ReprocessingReworking
cGMP regulations require reprocessing
procedures to be in writing If firm
makes provisions for reprocessing drug
product details must be submitted as
part of the application
ndash Standard Operating Procedures
ndash QA review and approval
26
27
Different Responsibilities
28
Different Responsibilities
29
Different Responsibilities
How ICH Q8 Q9 Q10
Guidelines Are Working
Together Throughout The
Product Life Cycle
30
Nov 2005 amp Nov 2008
ICH Q8 Q9 and Q10
High level guidances
(not prescriptive)
Science and risk-based
Encourages systematic
approaches
Applicable over entire product
lifecycle
Intended to work together to
enhance pharmaceutical product
quality
31
Pharmaceutical Development -
Q8(R2)
Describes science and risk-based
approaches for pharmaceutical product and
manufacturing process development
Introduced concepts of design space and
flexible regulatory approaches
Introduced concepts of Quality by Design
(QbD) and provided examples of QbD
development approaches and design space 32
Differing Approaches to
Pharmaceutical Development
33
Q8(R2) - Example QbD
Approach
Quality Target Product Profile (QTPP)
Determine ldquopotentialrdquo critical quality attributes
Link raw material attributes and process parameters
to CQAs and perform risk assessment
Develop a design space (optional and not required)
Design and implement a control strategy
Manage product lifecycle including continual
improvement
CQArsquos
Product Profile
Risk Assessments
Design Space
Control Strategy
Continual
Improvement34
Product Lifecycle
concept
Human and veterinary
drugs
Biotech products
Finish products amp APIs
Drug constituent of a
combination product
35
Manage product lifecycle including
continual improvement
Three-stage approach to process validation
Stage 1 ndash Process Design ndash Building and capturing process knowledge and understanding
ndash Establishing a strategy for process control
Stage 2 ndash Process Qualification ndash Design a facility and qualification of utilities and equipment
ndash Process performance qualification
ndash PPQ protocol
ndash PPQ protocol execution and report
Stage 3 ndash Continued Process Verification
36
Focus on alignment with
lsquoproduct lifecyclersquo
Manufacturers should
Understand the sources of variation
Detect the presence and degree of variation
Understand the impact of variation on the process and
ultimately on product attributes
Control the variation in a manner commensurate with
the risk it represents to the process and product
37
Process Validation
38
Focus on alignment with
lsquoproduct lifecyclersquo
39
Focus on alignment with
lsquoproduct lifecyclersquo
40
Focus on alignment with
lsquoproduct lifecyclersquo
FDA Quality Systems Guidance
for Finished Pharmaceuticals
41 Inspection Readiness BEC
Inspection Readiness BEC 42
Quality System
Drug Inspection Target Includes the Quality Control Unit and all of its review
and approval duties
ndash Approval of and adherence to Procedures and associated recordkeeping systems
ndash Product Reviews
ndash Complaint Reviews
ndash Failure Evaluations
ndash Change Control
ndash Product Improvement Projects
ndash RejectsReprocessingRework
ndash Stability
ndash Validation
ndash Training
Inspection Readiness BEC 43
Facilities and Equipment
Inspection Target
Building and Facilities along with
maintenance
Equipment qualification calibration and
maintenance
Water steam compressed gas HVAC
Change control system
Investigate discrepancies
Inspection Readiness BEC 44
Materials
Inspection Target
Procedures and documentation showing adequate control of finished products in-process materials components containers closures
Qualificationvalidation and security of computerized or automated processes
Change control system
Investigation of Discrepancies
Inspection Readiness BEC 45
Production System
Drug Inspection Target
Trainingqualification of personnel
Complete batch production documentation
Process validation
Production time limits
In-process testingexamination
Change Control System
Investigation of discrepancies
Inspection Readiness BEC 46
Packaging and Labeling
Inspection Target
Training
Acceptance operations
Control of materials to prevent mix-ups
Accountability
Packaginglabeling records
Line separation
Line clearance inspection and documentation
Validation of labelingpackaging operations
Investigation of discrepancies
Inspection Readiness BEC 47
Laboratory Control
Inspection Target
Staffing and training
Adequate equipment calibration and maintenance
Reference standards
Adherence to written procedures
Validationverification analytical methods
Analytical records and raw data
Adherence to OOS procedure and timely completion of investigations
Stability testing program and reserve samples
Change control system
Before the Inspection
SOP for Inspection Readiness
Strong Internal AuditAssessment function
ndash Continuous assessment using Risk Management
Criteria
ndash Not autopilot auditing
ndash Same auditorhellip
Perform a full-system audit (Mock) at least one
month prior to inspection
ndash Be sure any necessary CA-PA has been originated 48
Before the Inspection
Educate all your employees on proper inspection
rules and etiquette
Educate your supervisory (all exempt) personnel
on FDA regulations
Inspections and external audits concern to all
employees not only those from Quality Dept
Promote the participation on the internal audit
cadre
Certify internal auditors
49
Before the Inspection
A few words about Internal Audit
Internal Audits are the most powerful tool to
avoid inspection surprises
If the inspector discovers something why your
internal auditor did not
The same auditor auditing several consecutive
years the same area is not effective
An effective internal audit function is priceless
50
During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

Investigatorrsquos Role during PAI
Assess the following
ndash Quality Systems
ndash Manufacturing Operations
ndash Sampling Plans
ndash Laboratory
ndash Test Methods Validation
ndash Drug Product Specifications
ndash ReprocessingReworking
ndash Standard Operating Procedures
ndash Batch Records 20
The Firmrsquos Role
Make records available (as appropriate) to
conduct the pre-approval inspection
ndash Product Development Report
ndash Batch Records and Laboratory Records
ndash ProtocolsSOPs
Assure facility is cGMP compliant and ready for
an FDA inspection
ndash Once an application is submitted to Center the firm
should be considered ready for inspection
21
Product Development Report
A very useful document for both the firm
and the FDA
The data generated during product
development which defines the drug
product targets the steps in the
manufacturing process where variation is
critical to quality and thereby focuses the
subsequent process validation effort
22
Product Development Report
API Impurity Profile
ndash How is the API characterized
Excipients
Formulation Wet or Dry Granulation
ndash Solution or Suspension
ndash Sterile - Terminalaseptic conditions
ndash TabletCapsule - ImmediateModified
ReleaseExtended Release
23
Product Development Report
Describes the development of Processing
Equipment order of addition of ingredients
to the formulation mixing times and
speeds drying time and temperature
nitrogen blankets blending hold times
compression slugging filling polishing
imprinting labeling and packaging
Product Development Report may not be a
formal document
24
Batch Records
The batch records submitted in the
application must be audited as part of
the inspection to assure
ndash That the proposed production process is the
same process that was used for the
manufacture of the biostability batches
25
ReprocessingReworking
cGMP regulations require reprocessing
procedures to be in writing If firm
makes provisions for reprocessing drug
product details must be submitted as
part of the application
ndash Standard Operating Procedures
ndash QA review and approval
26
27
Different Responsibilities
28
Different Responsibilities
29
Different Responsibilities
How ICH Q8 Q9 Q10
Guidelines Are Working
Together Throughout The
Product Life Cycle
30
Nov 2005 amp Nov 2008
ICH Q8 Q9 and Q10
High level guidances
(not prescriptive)
Science and risk-based
Encourages systematic
approaches
Applicable over entire product
lifecycle
Intended to work together to
enhance pharmaceutical product
quality
31
Pharmaceutical Development -
Q8(R2)
Describes science and risk-based
approaches for pharmaceutical product and
manufacturing process development
Introduced concepts of design space and
flexible regulatory approaches
Introduced concepts of Quality by Design
(QbD) and provided examples of QbD
development approaches and design space 32
Differing Approaches to
Pharmaceutical Development
33
Q8(R2) - Example QbD
Approach
Quality Target Product Profile (QTPP)
Determine ldquopotentialrdquo critical quality attributes
Link raw material attributes and process parameters
to CQAs and perform risk assessment
Develop a design space (optional and not required)
Design and implement a control strategy
Manage product lifecycle including continual
improvement
CQArsquos
Product Profile
Risk Assessments
Design Space
Control Strategy
Continual
Improvement34
Product Lifecycle
concept
Human and veterinary
drugs
Biotech products
Finish products amp APIs
Drug constituent of a
combination product
35
Manage product lifecycle including
continual improvement
Three-stage approach to process validation
Stage 1 ndash Process Design ndash Building and capturing process knowledge and understanding
ndash Establishing a strategy for process control
Stage 2 ndash Process Qualification ndash Design a facility and qualification of utilities and equipment
ndash Process performance qualification
ndash PPQ protocol
ndash PPQ protocol execution and report
Stage 3 ndash Continued Process Verification
36
Focus on alignment with
lsquoproduct lifecyclersquo
Manufacturers should
Understand the sources of variation
Detect the presence and degree of variation
Understand the impact of variation on the process and
ultimately on product attributes
Control the variation in a manner commensurate with
the risk it represents to the process and product
37
Process Validation
38
Focus on alignment with
lsquoproduct lifecyclersquo
39
Focus on alignment with
lsquoproduct lifecyclersquo
40
Focus on alignment with
lsquoproduct lifecyclersquo
FDA Quality Systems Guidance
for Finished Pharmaceuticals
41 Inspection Readiness BEC
Inspection Readiness BEC 42
Quality System
Drug Inspection Target Includes the Quality Control Unit and all of its review
and approval duties
ndash Approval of and adherence to Procedures and associated recordkeeping systems
ndash Product Reviews
ndash Complaint Reviews
ndash Failure Evaluations
ndash Change Control
ndash Product Improvement Projects
ndash RejectsReprocessingRework
ndash Stability
ndash Validation
ndash Training
Inspection Readiness BEC 43
Facilities and Equipment
Inspection Target
Building and Facilities along with
maintenance
Equipment qualification calibration and
maintenance
Water steam compressed gas HVAC
Change control system
Investigate discrepancies
Inspection Readiness BEC 44
Materials
Inspection Target
Procedures and documentation showing adequate control of finished products in-process materials components containers closures
Qualificationvalidation and security of computerized or automated processes
Change control system
Investigation of Discrepancies
Inspection Readiness BEC 45
Production System
Drug Inspection Target
Trainingqualification of personnel
Complete batch production documentation
Process validation
Production time limits
In-process testingexamination
Change Control System
Investigation of discrepancies
Inspection Readiness BEC 46
Packaging and Labeling
Inspection Target
Training
Acceptance operations
Control of materials to prevent mix-ups
Accountability
Packaginglabeling records
Line separation
Line clearance inspection and documentation
Validation of labelingpackaging operations
Investigation of discrepancies
Inspection Readiness BEC 47
Laboratory Control
Inspection Target
Staffing and training
Adequate equipment calibration and maintenance
Reference standards
Adherence to written procedures
Validationverification analytical methods
Analytical records and raw data
Adherence to OOS procedure and timely completion of investigations
Stability testing program and reserve samples
Change control system
Before the Inspection
SOP for Inspection Readiness
Strong Internal AuditAssessment function
ndash Continuous assessment using Risk Management
Criteria
ndash Not autopilot auditing
ndash Same auditorhellip
Perform a full-system audit (Mock) at least one
month prior to inspection
ndash Be sure any necessary CA-PA has been originated 48
Before the Inspection
Educate all your employees on proper inspection
rules and etiquette
Educate your supervisory (all exempt) personnel
on FDA regulations
Inspections and external audits concern to all
employees not only those from Quality Dept
Promote the participation on the internal audit
cadre
Certify internal auditors
49
Before the Inspection
A few words about Internal Audit
Internal Audits are the most powerful tool to
avoid inspection surprises
If the inspector discovers something why your
internal auditor did not
The same auditor auditing several consecutive
years the same area is not effective
An effective internal audit function is priceless
50
During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

The Firmrsquos Role
Make records available (as appropriate) to
conduct the pre-approval inspection
ndash Product Development Report
ndash Batch Records and Laboratory Records
ndash ProtocolsSOPs
Assure facility is cGMP compliant and ready for
an FDA inspection
ndash Once an application is submitted to Center the firm
should be considered ready for inspection
21
Product Development Report
A very useful document for both the firm
and the FDA
The data generated during product
development which defines the drug
product targets the steps in the
manufacturing process where variation is
critical to quality and thereby focuses the
subsequent process validation effort
22
Product Development Report
API Impurity Profile
ndash How is the API characterized
Excipients
Formulation Wet or Dry Granulation
ndash Solution or Suspension
ndash Sterile - Terminalaseptic conditions
ndash TabletCapsule - ImmediateModified
ReleaseExtended Release
23
Product Development Report
Describes the development of Processing
Equipment order of addition of ingredients
to the formulation mixing times and
speeds drying time and temperature
nitrogen blankets blending hold times
compression slugging filling polishing
imprinting labeling and packaging
Product Development Report may not be a
formal document
24
Batch Records
The batch records submitted in the
application must be audited as part of
the inspection to assure
ndash That the proposed production process is the
same process that was used for the
manufacture of the biostability batches
25
ReprocessingReworking
cGMP regulations require reprocessing
procedures to be in writing If firm
makes provisions for reprocessing drug
product details must be submitted as
part of the application
ndash Standard Operating Procedures
ndash QA review and approval
26
27
Different Responsibilities
28
Different Responsibilities
29
Different Responsibilities
How ICH Q8 Q9 Q10
Guidelines Are Working
Together Throughout The
Product Life Cycle
30
Nov 2005 amp Nov 2008
ICH Q8 Q9 and Q10
High level guidances
(not prescriptive)
Science and risk-based
Encourages systematic
approaches
Applicable over entire product
lifecycle
Intended to work together to
enhance pharmaceutical product
quality
31
Pharmaceutical Development -
Q8(R2)
Describes science and risk-based
approaches for pharmaceutical product and
manufacturing process development
Introduced concepts of design space and
flexible regulatory approaches
Introduced concepts of Quality by Design
(QbD) and provided examples of QbD
development approaches and design space 32
Differing Approaches to
Pharmaceutical Development
33
Q8(R2) - Example QbD
Approach
Quality Target Product Profile (QTPP)
Determine ldquopotentialrdquo critical quality attributes
Link raw material attributes and process parameters
to CQAs and perform risk assessment
Develop a design space (optional and not required)
Design and implement a control strategy
Manage product lifecycle including continual
improvement
CQArsquos
Product Profile
Risk Assessments
Design Space
Control Strategy
Continual
Improvement34
Product Lifecycle
concept
Human and veterinary
drugs
Biotech products
Finish products amp APIs
Drug constituent of a
combination product
35
Manage product lifecycle including
continual improvement
Three-stage approach to process validation
Stage 1 ndash Process Design ndash Building and capturing process knowledge and understanding
ndash Establishing a strategy for process control
Stage 2 ndash Process Qualification ndash Design a facility and qualification of utilities and equipment
ndash Process performance qualification
ndash PPQ protocol
ndash PPQ protocol execution and report
Stage 3 ndash Continued Process Verification
36
Focus on alignment with
lsquoproduct lifecyclersquo
Manufacturers should
Understand the sources of variation
Detect the presence and degree of variation
Understand the impact of variation on the process and
ultimately on product attributes
Control the variation in a manner commensurate with
the risk it represents to the process and product
37
Process Validation
38
Focus on alignment with
lsquoproduct lifecyclersquo
39
Focus on alignment with
lsquoproduct lifecyclersquo
40
Focus on alignment with
lsquoproduct lifecyclersquo
FDA Quality Systems Guidance
for Finished Pharmaceuticals
41 Inspection Readiness BEC
Inspection Readiness BEC 42
Quality System
Drug Inspection Target Includes the Quality Control Unit and all of its review
and approval duties
ndash Approval of and adherence to Procedures and associated recordkeeping systems
ndash Product Reviews
ndash Complaint Reviews
ndash Failure Evaluations
ndash Change Control
ndash Product Improvement Projects
ndash RejectsReprocessingRework
ndash Stability
ndash Validation
ndash Training
Inspection Readiness BEC 43
Facilities and Equipment
Inspection Target
Building and Facilities along with
maintenance
Equipment qualification calibration and
maintenance
Water steam compressed gas HVAC
Change control system
Investigate discrepancies
Inspection Readiness BEC 44
Materials
Inspection Target
Procedures and documentation showing adequate control of finished products in-process materials components containers closures
Qualificationvalidation and security of computerized or automated processes
Change control system
Investigation of Discrepancies
Inspection Readiness BEC 45
Production System
Drug Inspection Target
Trainingqualification of personnel
Complete batch production documentation
Process validation
Production time limits
In-process testingexamination
Change Control System
Investigation of discrepancies
Inspection Readiness BEC 46
Packaging and Labeling
Inspection Target
Training
Acceptance operations
Control of materials to prevent mix-ups
Accountability
Packaginglabeling records
Line separation
Line clearance inspection and documentation
Validation of labelingpackaging operations
Investigation of discrepancies
Inspection Readiness BEC 47
Laboratory Control
Inspection Target
Staffing and training
Adequate equipment calibration and maintenance
Reference standards
Adherence to written procedures
Validationverification analytical methods
Analytical records and raw data
Adherence to OOS procedure and timely completion of investigations
Stability testing program and reserve samples
Change control system
Before the Inspection
SOP for Inspection Readiness
Strong Internal AuditAssessment function
ndash Continuous assessment using Risk Management
Criteria
ndash Not autopilot auditing
ndash Same auditorhellip
Perform a full-system audit (Mock) at least one
month prior to inspection
ndash Be sure any necessary CA-PA has been originated 48
Before the Inspection
Educate all your employees on proper inspection
rules and etiquette
Educate your supervisory (all exempt) personnel
on FDA regulations
Inspections and external audits concern to all
employees not only those from Quality Dept
Promote the participation on the internal audit
cadre
Certify internal auditors
49
Before the Inspection
A few words about Internal Audit
Internal Audits are the most powerful tool to
avoid inspection surprises
If the inspector discovers something why your
internal auditor did not
The same auditor auditing several consecutive
years the same area is not effective
An effective internal audit function is priceless
50
During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

Product Development Report
A very useful document for both the firm
and the FDA
The data generated during product
development which defines the drug
product targets the steps in the
manufacturing process where variation is
critical to quality and thereby focuses the
subsequent process validation effort
22
Product Development Report
API Impurity Profile
ndash How is the API characterized
Excipients
Formulation Wet or Dry Granulation
ndash Solution or Suspension
ndash Sterile - Terminalaseptic conditions
ndash TabletCapsule - ImmediateModified
ReleaseExtended Release
23
Product Development Report
Describes the development of Processing
Equipment order of addition of ingredients
to the formulation mixing times and
speeds drying time and temperature
nitrogen blankets blending hold times
compression slugging filling polishing
imprinting labeling and packaging
Product Development Report may not be a
formal document
24
Batch Records
The batch records submitted in the
application must be audited as part of
the inspection to assure
ndash That the proposed production process is the
same process that was used for the
manufacture of the biostability batches
25
ReprocessingReworking
cGMP regulations require reprocessing
procedures to be in writing If firm
makes provisions for reprocessing drug
product details must be submitted as
part of the application
ndash Standard Operating Procedures
ndash QA review and approval
26
27
Different Responsibilities
28
Different Responsibilities
29
Different Responsibilities
How ICH Q8 Q9 Q10
Guidelines Are Working
Together Throughout The
Product Life Cycle
30
Nov 2005 amp Nov 2008
ICH Q8 Q9 and Q10
High level guidances
(not prescriptive)
Science and risk-based
Encourages systematic
approaches
Applicable over entire product
lifecycle
Intended to work together to
enhance pharmaceutical product
quality
31
Pharmaceutical Development -
Q8(R2)
Describes science and risk-based
approaches for pharmaceutical product and
manufacturing process development
Introduced concepts of design space and
flexible regulatory approaches
Introduced concepts of Quality by Design
(QbD) and provided examples of QbD
development approaches and design space 32
Differing Approaches to
Pharmaceutical Development
33
Q8(R2) - Example QbD
Approach
Quality Target Product Profile (QTPP)
Determine ldquopotentialrdquo critical quality attributes
Link raw material attributes and process parameters
to CQAs and perform risk assessment
Develop a design space (optional and not required)
Design and implement a control strategy
Manage product lifecycle including continual
improvement
CQArsquos
Product Profile
Risk Assessments
Design Space
Control Strategy
Continual
Improvement34
Product Lifecycle
concept
Human and veterinary
drugs
Biotech products
Finish products amp APIs
Drug constituent of a
combination product
35
Manage product lifecycle including
continual improvement
Three-stage approach to process validation
Stage 1 ndash Process Design ndash Building and capturing process knowledge and understanding
ndash Establishing a strategy for process control
Stage 2 ndash Process Qualification ndash Design a facility and qualification of utilities and equipment
ndash Process performance qualification
ndash PPQ protocol
ndash PPQ protocol execution and report
Stage 3 ndash Continued Process Verification
36
Focus on alignment with
lsquoproduct lifecyclersquo
Manufacturers should
Understand the sources of variation
Detect the presence and degree of variation
Understand the impact of variation on the process and
ultimately on product attributes
Control the variation in a manner commensurate with
the risk it represents to the process and product
37
Process Validation
38
Focus on alignment with
lsquoproduct lifecyclersquo
39
Focus on alignment with
lsquoproduct lifecyclersquo
40
Focus on alignment with
lsquoproduct lifecyclersquo
FDA Quality Systems Guidance
for Finished Pharmaceuticals
41 Inspection Readiness BEC
Inspection Readiness BEC 42
Quality System
Drug Inspection Target Includes the Quality Control Unit and all of its review
and approval duties
ndash Approval of and adherence to Procedures and associated recordkeeping systems
ndash Product Reviews
ndash Complaint Reviews
ndash Failure Evaluations
ndash Change Control
ndash Product Improvement Projects
ndash RejectsReprocessingRework
ndash Stability
ndash Validation
ndash Training
Inspection Readiness BEC 43
Facilities and Equipment
Inspection Target
Building and Facilities along with
maintenance
Equipment qualification calibration and
maintenance
Water steam compressed gas HVAC
Change control system
Investigate discrepancies
Inspection Readiness BEC 44
Materials
Inspection Target
Procedures and documentation showing adequate control of finished products in-process materials components containers closures
Qualificationvalidation and security of computerized or automated processes
Change control system
Investigation of Discrepancies
Inspection Readiness BEC 45
Production System
Drug Inspection Target
Trainingqualification of personnel
Complete batch production documentation
Process validation
Production time limits
In-process testingexamination
Change Control System
Investigation of discrepancies
Inspection Readiness BEC 46
Packaging and Labeling
Inspection Target
Training
Acceptance operations
Control of materials to prevent mix-ups
Accountability
Packaginglabeling records
Line separation
Line clearance inspection and documentation
Validation of labelingpackaging operations
Investigation of discrepancies
Inspection Readiness BEC 47
Laboratory Control
Inspection Target
Staffing and training
Adequate equipment calibration and maintenance
Reference standards
Adherence to written procedures
Validationverification analytical methods
Analytical records and raw data
Adherence to OOS procedure and timely completion of investigations
Stability testing program and reserve samples
Change control system
Before the Inspection
SOP for Inspection Readiness
Strong Internal AuditAssessment function
ndash Continuous assessment using Risk Management
Criteria
ndash Not autopilot auditing
ndash Same auditorhellip
Perform a full-system audit (Mock) at least one
month prior to inspection
ndash Be sure any necessary CA-PA has been originated 48
Before the Inspection
Educate all your employees on proper inspection
rules and etiquette
Educate your supervisory (all exempt) personnel
on FDA regulations
Inspections and external audits concern to all
employees not only those from Quality Dept
Promote the participation on the internal audit
cadre
Certify internal auditors
49
Before the Inspection
A few words about Internal Audit
Internal Audits are the most powerful tool to
avoid inspection surprises
If the inspector discovers something why your
internal auditor did not
The same auditor auditing several consecutive
years the same area is not effective
An effective internal audit function is priceless
50
During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

Product Development Report
API Impurity Profile
ndash How is the API characterized
Excipients
Formulation Wet or Dry Granulation
ndash Solution or Suspension
ndash Sterile - Terminalaseptic conditions
ndash TabletCapsule - ImmediateModified
ReleaseExtended Release
23
Product Development Report
Describes the development of Processing
Equipment order of addition of ingredients
to the formulation mixing times and
speeds drying time and temperature
nitrogen blankets blending hold times
compression slugging filling polishing
imprinting labeling and packaging
Product Development Report may not be a
formal document
24
Batch Records
The batch records submitted in the
application must be audited as part of
the inspection to assure
ndash That the proposed production process is the
same process that was used for the
manufacture of the biostability batches
25
ReprocessingReworking
cGMP regulations require reprocessing
procedures to be in writing If firm
makes provisions for reprocessing drug
product details must be submitted as
part of the application
ndash Standard Operating Procedures
ndash QA review and approval
26
27
Different Responsibilities
28
Different Responsibilities
29
Different Responsibilities
How ICH Q8 Q9 Q10
Guidelines Are Working
Together Throughout The
Product Life Cycle
30
Nov 2005 amp Nov 2008
ICH Q8 Q9 and Q10
High level guidances
(not prescriptive)
Science and risk-based
Encourages systematic
approaches
Applicable over entire product
lifecycle
Intended to work together to
enhance pharmaceutical product
quality
31
Pharmaceutical Development -
Q8(R2)
Describes science and risk-based
approaches for pharmaceutical product and
manufacturing process development
Introduced concepts of design space and
flexible regulatory approaches
Introduced concepts of Quality by Design
(QbD) and provided examples of QbD
development approaches and design space 32
Differing Approaches to
Pharmaceutical Development
33
Q8(R2) - Example QbD
Approach
Quality Target Product Profile (QTPP)
Determine ldquopotentialrdquo critical quality attributes
Link raw material attributes and process parameters
to CQAs and perform risk assessment
Develop a design space (optional and not required)
Design and implement a control strategy
Manage product lifecycle including continual
improvement
CQArsquos
Product Profile
Risk Assessments
Design Space
Control Strategy
Continual
Improvement34
Product Lifecycle
concept
Human and veterinary
drugs
Biotech products
Finish products amp APIs
Drug constituent of a
combination product
35
Manage product lifecycle including
continual improvement
Three-stage approach to process validation
Stage 1 ndash Process Design ndash Building and capturing process knowledge and understanding
ndash Establishing a strategy for process control
Stage 2 ndash Process Qualification ndash Design a facility and qualification of utilities and equipment
ndash Process performance qualification
ndash PPQ protocol
ndash PPQ protocol execution and report
Stage 3 ndash Continued Process Verification
36
Focus on alignment with
lsquoproduct lifecyclersquo
Manufacturers should
Understand the sources of variation
Detect the presence and degree of variation
Understand the impact of variation on the process and
ultimately on product attributes
Control the variation in a manner commensurate with
the risk it represents to the process and product
37
Process Validation
38
Focus on alignment with
lsquoproduct lifecyclersquo
39
Focus on alignment with
lsquoproduct lifecyclersquo
40
Focus on alignment with
lsquoproduct lifecyclersquo
FDA Quality Systems Guidance
for Finished Pharmaceuticals
41 Inspection Readiness BEC
Inspection Readiness BEC 42
Quality System
Drug Inspection Target Includes the Quality Control Unit and all of its review
and approval duties
ndash Approval of and adherence to Procedures and associated recordkeeping systems
ndash Product Reviews
ndash Complaint Reviews
ndash Failure Evaluations
ndash Change Control
ndash Product Improvement Projects
ndash RejectsReprocessingRework
ndash Stability
ndash Validation
ndash Training
Inspection Readiness BEC 43
Facilities and Equipment
Inspection Target
Building and Facilities along with
maintenance
Equipment qualification calibration and
maintenance
Water steam compressed gas HVAC
Change control system
Investigate discrepancies
Inspection Readiness BEC 44
Materials
Inspection Target
Procedures and documentation showing adequate control of finished products in-process materials components containers closures
Qualificationvalidation and security of computerized or automated processes
Change control system
Investigation of Discrepancies
Inspection Readiness BEC 45
Production System
Drug Inspection Target
Trainingqualification of personnel
Complete batch production documentation
Process validation
Production time limits
In-process testingexamination
Change Control System
Investigation of discrepancies
Inspection Readiness BEC 46
Packaging and Labeling
Inspection Target
Training
Acceptance operations
Control of materials to prevent mix-ups
Accountability
Packaginglabeling records
Line separation
Line clearance inspection and documentation
Validation of labelingpackaging operations
Investigation of discrepancies
Inspection Readiness BEC 47
Laboratory Control
Inspection Target
Staffing and training
Adequate equipment calibration and maintenance
Reference standards
Adherence to written procedures
Validationverification analytical methods
Analytical records and raw data
Adherence to OOS procedure and timely completion of investigations
Stability testing program and reserve samples
Change control system
Before the Inspection
SOP for Inspection Readiness
Strong Internal AuditAssessment function
ndash Continuous assessment using Risk Management
Criteria
ndash Not autopilot auditing
ndash Same auditorhellip
Perform a full-system audit (Mock) at least one
month prior to inspection
ndash Be sure any necessary CA-PA has been originated 48
Before the Inspection
Educate all your employees on proper inspection
rules and etiquette
Educate your supervisory (all exempt) personnel
on FDA regulations
Inspections and external audits concern to all
employees not only those from Quality Dept
Promote the participation on the internal audit
cadre
Certify internal auditors
49
Before the Inspection
A few words about Internal Audit
Internal Audits are the most powerful tool to
avoid inspection surprises
If the inspector discovers something why your
internal auditor did not
The same auditor auditing several consecutive
years the same area is not effective
An effective internal audit function is priceless
50
During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

Product Development Report
Describes the development of Processing
Equipment order of addition of ingredients
to the formulation mixing times and
speeds drying time and temperature
nitrogen blankets blending hold times
compression slugging filling polishing
imprinting labeling and packaging
Product Development Report may not be a
formal document
24
Batch Records
The batch records submitted in the
application must be audited as part of
the inspection to assure
ndash That the proposed production process is the
same process that was used for the
manufacture of the biostability batches
25
ReprocessingReworking
cGMP regulations require reprocessing
procedures to be in writing If firm
makes provisions for reprocessing drug
product details must be submitted as
part of the application
ndash Standard Operating Procedures
ndash QA review and approval
26
27
Different Responsibilities
28
Different Responsibilities
29
Different Responsibilities
How ICH Q8 Q9 Q10
Guidelines Are Working
Together Throughout The
Product Life Cycle
30
Nov 2005 amp Nov 2008
ICH Q8 Q9 and Q10
High level guidances
(not prescriptive)
Science and risk-based
Encourages systematic
approaches
Applicable over entire product
lifecycle
Intended to work together to
enhance pharmaceutical product
quality
31
Pharmaceutical Development -
Q8(R2)
Describes science and risk-based
approaches for pharmaceutical product and
manufacturing process development
Introduced concepts of design space and
flexible regulatory approaches
Introduced concepts of Quality by Design
(QbD) and provided examples of QbD
development approaches and design space 32
Differing Approaches to
Pharmaceutical Development
33
Q8(R2) - Example QbD
Approach
Quality Target Product Profile (QTPP)
Determine ldquopotentialrdquo critical quality attributes
Link raw material attributes and process parameters
to CQAs and perform risk assessment
Develop a design space (optional and not required)
Design and implement a control strategy
Manage product lifecycle including continual
improvement
CQArsquos
Product Profile
Risk Assessments
Design Space
Control Strategy
Continual
Improvement34
Product Lifecycle
concept
Human and veterinary
drugs
Biotech products
Finish products amp APIs
Drug constituent of a
combination product
35
Manage product lifecycle including
continual improvement
Three-stage approach to process validation
Stage 1 ndash Process Design ndash Building and capturing process knowledge and understanding
ndash Establishing a strategy for process control
Stage 2 ndash Process Qualification ndash Design a facility and qualification of utilities and equipment
ndash Process performance qualification
ndash PPQ protocol
ndash PPQ protocol execution and report
Stage 3 ndash Continued Process Verification
36
Focus on alignment with
lsquoproduct lifecyclersquo
Manufacturers should
Understand the sources of variation
Detect the presence and degree of variation
Understand the impact of variation on the process and
ultimately on product attributes
Control the variation in a manner commensurate with
the risk it represents to the process and product
37
Process Validation
38
Focus on alignment with
lsquoproduct lifecyclersquo
39
Focus on alignment with
lsquoproduct lifecyclersquo
40
Focus on alignment with
lsquoproduct lifecyclersquo
FDA Quality Systems Guidance
for Finished Pharmaceuticals
41 Inspection Readiness BEC
Inspection Readiness BEC 42
Quality System
Drug Inspection Target Includes the Quality Control Unit and all of its review
and approval duties
ndash Approval of and adherence to Procedures and associated recordkeeping systems
ndash Product Reviews
ndash Complaint Reviews
ndash Failure Evaluations
ndash Change Control
ndash Product Improvement Projects
ndash RejectsReprocessingRework
ndash Stability
ndash Validation
ndash Training
Inspection Readiness BEC 43
Facilities and Equipment
Inspection Target
Building and Facilities along with
maintenance
Equipment qualification calibration and
maintenance
Water steam compressed gas HVAC
Change control system
Investigate discrepancies
Inspection Readiness BEC 44
Materials
Inspection Target
Procedures and documentation showing adequate control of finished products in-process materials components containers closures
Qualificationvalidation and security of computerized or automated processes
Change control system
Investigation of Discrepancies
Inspection Readiness BEC 45
Production System
Drug Inspection Target
Trainingqualification of personnel
Complete batch production documentation
Process validation
Production time limits
In-process testingexamination
Change Control System
Investigation of discrepancies
Inspection Readiness BEC 46
Packaging and Labeling
Inspection Target
Training
Acceptance operations
Control of materials to prevent mix-ups
Accountability
Packaginglabeling records
Line separation
Line clearance inspection and documentation
Validation of labelingpackaging operations
Investigation of discrepancies
Inspection Readiness BEC 47
Laboratory Control
Inspection Target
Staffing and training
Adequate equipment calibration and maintenance
Reference standards
Adherence to written procedures
Validationverification analytical methods
Analytical records and raw data
Adherence to OOS procedure and timely completion of investigations
Stability testing program and reserve samples
Change control system
Before the Inspection
SOP for Inspection Readiness
Strong Internal AuditAssessment function
ndash Continuous assessment using Risk Management
Criteria
ndash Not autopilot auditing
ndash Same auditorhellip
Perform a full-system audit (Mock) at least one
month prior to inspection
ndash Be sure any necessary CA-PA has been originated 48
Before the Inspection
Educate all your employees on proper inspection
rules and etiquette
Educate your supervisory (all exempt) personnel
on FDA regulations
Inspections and external audits concern to all
employees not only those from Quality Dept
Promote the participation on the internal audit
cadre
Certify internal auditors
49
Before the Inspection
A few words about Internal Audit
Internal Audits are the most powerful tool to
avoid inspection surprises
If the inspector discovers something why your
internal auditor did not
The same auditor auditing several consecutive
years the same area is not effective
An effective internal audit function is priceless
50
During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

Batch Records
The batch records submitted in the
application must be audited as part of
the inspection to assure
ndash That the proposed production process is the
same process that was used for the
manufacture of the biostability batches
25
ReprocessingReworking
cGMP regulations require reprocessing
procedures to be in writing If firm
makes provisions for reprocessing drug
product details must be submitted as
part of the application
ndash Standard Operating Procedures
ndash QA review and approval
26
27
Different Responsibilities
28
Different Responsibilities
29
Different Responsibilities
How ICH Q8 Q9 Q10
Guidelines Are Working
Together Throughout The
Product Life Cycle
30
Nov 2005 amp Nov 2008
ICH Q8 Q9 and Q10
High level guidances
(not prescriptive)
Science and risk-based
Encourages systematic
approaches
Applicable over entire product
lifecycle
Intended to work together to
enhance pharmaceutical product
quality
31
Pharmaceutical Development -
Q8(R2)
Describes science and risk-based
approaches for pharmaceutical product and
manufacturing process development
Introduced concepts of design space and
flexible regulatory approaches
Introduced concepts of Quality by Design
(QbD) and provided examples of QbD
development approaches and design space 32
Differing Approaches to
Pharmaceutical Development
33
Q8(R2) - Example QbD
Approach
Quality Target Product Profile (QTPP)
Determine ldquopotentialrdquo critical quality attributes
Link raw material attributes and process parameters
to CQAs and perform risk assessment
Develop a design space (optional and not required)
Design and implement a control strategy
Manage product lifecycle including continual
improvement
CQArsquos
Product Profile
Risk Assessments
Design Space
Control Strategy
Continual
Improvement34
Product Lifecycle
concept
Human and veterinary
drugs
Biotech products
Finish products amp APIs
Drug constituent of a
combination product
35
Manage product lifecycle including
continual improvement
Three-stage approach to process validation
Stage 1 ndash Process Design ndash Building and capturing process knowledge and understanding
ndash Establishing a strategy for process control
Stage 2 ndash Process Qualification ndash Design a facility and qualification of utilities and equipment
ndash Process performance qualification
ndash PPQ protocol
ndash PPQ protocol execution and report
Stage 3 ndash Continued Process Verification
36
Focus on alignment with
lsquoproduct lifecyclersquo
Manufacturers should
Understand the sources of variation
Detect the presence and degree of variation
Understand the impact of variation on the process and
ultimately on product attributes
Control the variation in a manner commensurate with
the risk it represents to the process and product
37
Process Validation
38
Focus on alignment with
lsquoproduct lifecyclersquo
39
Focus on alignment with
lsquoproduct lifecyclersquo
40
Focus on alignment with
lsquoproduct lifecyclersquo
FDA Quality Systems Guidance
for Finished Pharmaceuticals
41 Inspection Readiness BEC
Inspection Readiness BEC 42
Quality System
Drug Inspection Target Includes the Quality Control Unit and all of its review
and approval duties
ndash Approval of and adherence to Procedures and associated recordkeeping systems
ndash Product Reviews
ndash Complaint Reviews
ndash Failure Evaluations
ndash Change Control
ndash Product Improvement Projects
ndash RejectsReprocessingRework
ndash Stability
ndash Validation
ndash Training
Inspection Readiness BEC 43
Facilities and Equipment
Inspection Target
Building and Facilities along with
maintenance
Equipment qualification calibration and
maintenance
Water steam compressed gas HVAC
Change control system
Investigate discrepancies
Inspection Readiness BEC 44
Materials
Inspection Target
Procedures and documentation showing adequate control of finished products in-process materials components containers closures
Qualificationvalidation and security of computerized or automated processes
Change control system
Investigation of Discrepancies
Inspection Readiness BEC 45
Production System
Drug Inspection Target
Trainingqualification of personnel
Complete batch production documentation
Process validation
Production time limits
In-process testingexamination
Change Control System
Investigation of discrepancies
Inspection Readiness BEC 46
Packaging and Labeling
Inspection Target
Training
Acceptance operations
Control of materials to prevent mix-ups
Accountability
Packaginglabeling records
Line separation
Line clearance inspection and documentation
Validation of labelingpackaging operations
Investigation of discrepancies
Inspection Readiness BEC 47
Laboratory Control
Inspection Target
Staffing and training
Adequate equipment calibration and maintenance
Reference standards
Adherence to written procedures
Validationverification analytical methods
Analytical records and raw data
Adherence to OOS procedure and timely completion of investigations
Stability testing program and reserve samples
Change control system
Before the Inspection
SOP for Inspection Readiness
Strong Internal AuditAssessment function
ndash Continuous assessment using Risk Management
Criteria
ndash Not autopilot auditing
ndash Same auditorhellip
Perform a full-system audit (Mock) at least one
month prior to inspection
ndash Be sure any necessary CA-PA has been originated 48
Before the Inspection
Educate all your employees on proper inspection
rules and etiquette
Educate your supervisory (all exempt) personnel
on FDA regulations
Inspections and external audits concern to all
employees not only those from Quality Dept
Promote the participation on the internal audit
cadre
Certify internal auditors
49
Before the Inspection
A few words about Internal Audit
Internal Audits are the most powerful tool to
avoid inspection surprises
If the inspector discovers something why your
internal auditor did not
The same auditor auditing several consecutive
years the same area is not effective
An effective internal audit function is priceless
50
During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

ReprocessingReworking
cGMP regulations require reprocessing
procedures to be in writing If firm
makes provisions for reprocessing drug
product details must be submitted as
part of the application
ndash Standard Operating Procedures
ndash QA review and approval
26
27
Different Responsibilities
28
Different Responsibilities
29
Different Responsibilities
How ICH Q8 Q9 Q10
Guidelines Are Working
Together Throughout The
Product Life Cycle
30
Nov 2005 amp Nov 2008
ICH Q8 Q9 and Q10
High level guidances
(not prescriptive)
Science and risk-based
Encourages systematic
approaches
Applicable over entire product
lifecycle
Intended to work together to
enhance pharmaceutical product
quality
31
Pharmaceutical Development -
Q8(R2)
Describes science and risk-based
approaches for pharmaceutical product and
manufacturing process development
Introduced concepts of design space and
flexible regulatory approaches
Introduced concepts of Quality by Design
(QbD) and provided examples of QbD
development approaches and design space 32
Differing Approaches to
Pharmaceutical Development
33
Q8(R2) - Example QbD
Approach
Quality Target Product Profile (QTPP)
Determine ldquopotentialrdquo critical quality attributes
Link raw material attributes and process parameters
to CQAs and perform risk assessment
Develop a design space (optional and not required)
Design and implement a control strategy
Manage product lifecycle including continual
improvement
CQArsquos
Product Profile
Risk Assessments
Design Space
Control Strategy
Continual
Improvement34
Product Lifecycle
concept
Human and veterinary
drugs
Biotech products
Finish products amp APIs
Drug constituent of a
combination product
35
Manage product lifecycle including
continual improvement
Three-stage approach to process validation
Stage 1 ndash Process Design ndash Building and capturing process knowledge and understanding
ndash Establishing a strategy for process control
Stage 2 ndash Process Qualification ndash Design a facility and qualification of utilities and equipment
ndash Process performance qualification
ndash PPQ protocol
ndash PPQ protocol execution and report
Stage 3 ndash Continued Process Verification
36
Focus on alignment with
lsquoproduct lifecyclersquo
Manufacturers should
Understand the sources of variation
Detect the presence and degree of variation
Understand the impact of variation on the process and
ultimately on product attributes
Control the variation in a manner commensurate with
the risk it represents to the process and product
37
Process Validation
38
Focus on alignment with
lsquoproduct lifecyclersquo
39
Focus on alignment with
lsquoproduct lifecyclersquo
40
Focus on alignment with
lsquoproduct lifecyclersquo
FDA Quality Systems Guidance
for Finished Pharmaceuticals
41 Inspection Readiness BEC
Inspection Readiness BEC 42
Quality System
Drug Inspection Target Includes the Quality Control Unit and all of its review
and approval duties
ndash Approval of and adherence to Procedures and associated recordkeeping systems
ndash Product Reviews
ndash Complaint Reviews
ndash Failure Evaluations
ndash Change Control
ndash Product Improvement Projects
ndash RejectsReprocessingRework
ndash Stability
ndash Validation
ndash Training
Inspection Readiness BEC 43
Facilities and Equipment
Inspection Target
Building and Facilities along with
maintenance
Equipment qualification calibration and
maintenance
Water steam compressed gas HVAC
Change control system
Investigate discrepancies
Inspection Readiness BEC 44
Materials
Inspection Target
Procedures and documentation showing adequate control of finished products in-process materials components containers closures
Qualificationvalidation and security of computerized or automated processes
Change control system
Investigation of Discrepancies
Inspection Readiness BEC 45
Production System
Drug Inspection Target
Trainingqualification of personnel
Complete batch production documentation
Process validation
Production time limits
In-process testingexamination
Change Control System
Investigation of discrepancies
Inspection Readiness BEC 46
Packaging and Labeling
Inspection Target
Training
Acceptance operations
Control of materials to prevent mix-ups
Accountability
Packaginglabeling records
Line separation
Line clearance inspection and documentation
Validation of labelingpackaging operations
Investigation of discrepancies
Inspection Readiness BEC 47
Laboratory Control
Inspection Target
Staffing and training
Adequate equipment calibration and maintenance
Reference standards
Adherence to written procedures
Validationverification analytical methods
Analytical records and raw data
Adherence to OOS procedure and timely completion of investigations
Stability testing program and reserve samples
Change control system
Before the Inspection
SOP for Inspection Readiness
Strong Internal AuditAssessment function
ndash Continuous assessment using Risk Management
Criteria
ndash Not autopilot auditing
ndash Same auditorhellip
Perform a full-system audit (Mock) at least one
month prior to inspection
ndash Be sure any necessary CA-PA has been originated 48
Before the Inspection
Educate all your employees on proper inspection
rules and etiquette
Educate your supervisory (all exempt) personnel
on FDA regulations
Inspections and external audits concern to all
employees not only those from Quality Dept
Promote the participation on the internal audit
cadre
Certify internal auditors
49
Before the Inspection
A few words about Internal Audit
Internal Audits are the most powerful tool to
avoid inspection surprises
If the inspector discovers something why your
internal auditor did not
The same auditor auditing several consecutive
years the same area is not effective
An effective internal audit function is priceless
50
During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

27
Different Responsibilities
28
Different Responsibilities
29
Different Responsibilities
How ICH Q8 Q9 Q10
Guidelines Are Working
Together Throughout The
Product Life Cycle
30
Nov 2005 amp Nov 2008
ICH Q8 Q9 and Q10
High level guidances
(not prescriptive)
Science and risk-based
Encourages systematic
approaches
Applicable over entire product
lifecycle
Intended to work together to
enhance pharmaceutical product
quality
31
Pharmaceutical Development -
Q8(R2)
Describes science and risk-based
approaches for pharmaceutical product and
manufacturing process development
Introduced concepts of design space and
flexible regulatory approaches
Introduced concepts of Quality by Design
(QbD) and provided examples of QbD
development approaches and design space 32
Differing Approaches to
Pharmaceutical Development
33
Q8(R2) - Example QbD
Approach
Quality Target Product Profile (QTPP)
Determine ldquopotentialrdquo critical quality attributes
Link raw material attributes and process parameters
to CQAs and perform risk assessment
Develop a design space (optional and not required)
Design and implement a control strategy
Manage product lifecycle including continual
improvement
CQArsquos
Product Profile
Risk Assessments
Design Space
Control Strategy
Continual
Improvement34
Product Lifecycle
concept
Human and veterinary
drugs
Biotech products
Finish products amp APIs
Drug constituent of a
combination product
35
Manage product lifecycle including
continual improvement
Three-stage approach to process validation
Stage 1 ndash Process Design ndash Building and capturing process knowledge and understanding
ndash Establishing a strategy for process control
Stage 2 ndash Process Qualification ndash Design a facility and qualification of utilities and equipment
ndash Process performance qualification
ndash PPQ protocol
ndash PPQ protocol execution and report
Stage 3 ndash Continued Process Verification
36
Focus on alignment with
lsquoproduct lifecyclersquo
Manufacturers should
Understand the sources of variation
Detect the presence and degree of variation
Understand the impact of variation on the process and
ultimately on product attributes
Control the variation in a manner commensurate with
the risk it represents to the process and product
37
Process Validation
38
Focus on alignment with
lsquoproduct lifecyclersquo
39
Focus on alignment with
lsquoproduct lifecyclersquo
40
Focus on alignment with
lsquoproduct lifecyclersquo
FDA Quality Systems Guidance
for Finished Pharmaceuticals
41 Inspection Readiness BEC
Inspection Readiness BEC 42
Quality System
Drug Inspection Target Includes the Quality Control Unit and all of its review
and approval duties
ndash Approval of and adherence to Procedures and associated recordkeeping systems
ndash Product Reviews
ndash Complaint Reviews
ndash Failure Evaluations
ndash Change Control
ndash Product Improvement Projects
ndash RejectsReprocessingRework
ndash Stability
ndash Validation
ndash Training
Inspection Readiness BEC 43
Facilities and Equipment
Inspection Target
Building and Facilities along with
maintenance
Equipment qualification calibration and
maintenance
Water steam compressed gas HVAC
Change control system
Investigate discrepancies
Inspection Readiness BEC 44
Materials
Inspection Target
Procedures and documentation showing adequate control of finished products in-process materials components containers closures
Qualificationvalidation and security of computerized or automated processes
Change control system
Investigation of Discrepancies
Inspection Readiness BEC 45
Production System
Drug Inspection Target
Trainingqualification of personnel
Complete batch production documentation
Process validation
Production time limits
In-process testingexamination
Change Control System
Investigation of discrepancies
Inspection Readiness BEC 46
Packaging and Labeling
Inspection Target
Training
Acceptance operations
Control of materials to prevent mix-ups
Accountability
Packaginglabeling records
Line separation
Line clearance inspection and documentation
Validation of labelingpackaging operations
Investigation of discrepancies
Inspection Readiness BEC 47
Laboratory Control
Inspection Target
Staffing and training
Adequate equipment calibration and maintenance
Reference standards
Adherence to written procedures
Validationverification analytical methods
Analytical records and raw data
Adherence to OOS procedure and timely completion of investigations
Stability testing program and reserve samples
Change control system
Before the Inspection
SOP for Inspection Readiness
Strong Internal AuditAssessment function
ndash Continuous assessment using Risk Management
Criteria
ndash Not autopilot auditing
ndash Same auditorhellip
Perform a full-system audit (Mock) at least one
month prior to inspection
ndash Be sure any necessary CA-PA has been originated 48
Before the Inspection
Educate all your employees on proper inspection
rules and etiquette
Educate your supervisory (all exempt) personnel
on FDA regulations
Inspections and external audits concern to all
employees not only those from Quality Dept
Promote the participation on the internal audit
cadre
Certify internal auditors
49
Before the Inspection
A few words about Internal Audit
Internal Audits are the most powerful tool to
avoid inspection surprises
If the inspector discovers something why your
internal auditor did not
The same auditor auditing several consecutive
years the same area is not effective
An effective internal audit function is priceless
50
During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

28
Different Responsibilities
29
Different Responsibilities
How ICH Q8 Q9 Q10
Guidelines Are Working
Together Throughout The
Product Life Cycle
30
Nov 2005 amp Nov 2008
ICH Q8 Q9 and Q10
High level guidances
(not prescriptive)
Science and risk-based
Encourages systematic
approaches
Applicable over entire product
lifecycle
Intended to work together to
enhance pharmaceutical product
quality
31
Pharmaceutical Development -
Q8(R2)
Describes science and risk-based
approaches for pharmaceutical product and
manufacturing process development
Introduced concepts of design space and
flexible regulatory approaches
Introduced concepts of Quality by Design
(QbD) and provided examples of QbD
development approaches and design space 32
Differing Approaches to
Pharmaceutical Development
33
Q8(R2) - Example QbD
Approach
Quality Target Product Profile (QTPP)
Determine ldquopotentialrdquo critical quality attributes
Link raw material attributes and process parameters
to CQAs and perform risk assessment
Develop a design space (optional and not required)
Design and implement a control strategy
Manage product lifecycle including continual
improvement
CQArsquos
Product Profile
Risk Assessments
Design Space
Control Strategy
Continual
Improvement34
Product Lifecycle
concept
Human and veterinary
drugs
Biotech products
Finish products amp APIs
Drug constituent of a
combination product
35
Manage product lifecycle including
continual improvement
Three-stage approach to process validation
Stage 1 ndash Process Design ndash Building and capturing process knowledge and understanding
ndash Establishing a strategy for process control
Stage 2 ndash Process Qualification ndash Design a facility and qualification of utilities and equipment
ndash Process performance qualification
ndash PPQ protocol
ndash PPQ protocol execution and report
Stage 3 ndash Continued Process Verification
36
Focus on alignment with
lsquoproduct lifecyclersquo
Manufacturers should
Understand the sources of variation
Detect the presence and degree of variation
Understand the impact of variation on the process and
ultimately on product attributes
Control the variation in a manner commensurate with
the risk it represents to the process and product
37
Process Validation
38
Focus on alignment with
lsquoproduct lifecyclersquo
39
Focus on alignment with
lsquoproduct lifecyclersquo
40
Focus on alignment with
lsquoproduct lifecyclersquo
FDA Quality Systems Guidance
for Finished Pharmaceuticals
41 Inspection Readiness BEC
Inspection Readiness BEC 42
Quality System
Drug Inspection Target Includes the Quality Control Unit and all of its review
and approval duties
ndash Approval of and adherence to Procedures and associated recordkeeping systems
ndash Product Reviews
ndash Complaint Reviews
ndash Failure Evaluations
ndash Change Control
ndash Product Improvement Projects
ndash RejectsReprocessingRework
ndash Stability
ndash Validation
ndash Training
Inspection Readiness BEC 43
Facilities and Equipment
Inspection Target
Building and Facilities along with
maintenance
Equipment qualification calibration and
maintenance
Water steam compressed gas HVAC
Change control system
Investigate discrepancies
Inspection Readiness BEC 44
Materials
Inspection Target
Procedures and documentation showing adequate control of finished products in-process materials components containers closures
Qualificationvalidation and security of computerized or automated processes
Change control system
Investigation of Discrepancies
Inspection Readiness BEC 45
Production System
Drug Inspection Target
Trainingqualification of personnel
Complete batch production documentation
Process validation
Production time limits
In-process testingexamination
Change Control System
Investigation of discrepancies
Inspection Readiness BEC 46
Packaging and Labeling
Inspection Target
Training
Acceptance operations
Control of materials to prevent mix-ups
Accountability
Packaginglabeling records
Line separation
Line clearance inspection and documentation
Validation of labelingpackaging operations
Investigation of discrepancies
Inspection Readiness BEC 47
Laboratory Control
Inspection Target
Staffing and training
Adequate equipment calibration and maintenance
Reference standards
Adherence to written procedures
Validationverification analytical methods
Analytical records and raw data
Adherence to OOS procedure and timely completion of investigations
Stability testing program and reserve samples
Change control system
Before the Inspection
SOP for Inspection Readiness
Strong Internal AuditAssessment function
ndash Continuous assessment using Risk Management
Criteria
ndash Not autopilot auditing
ndash Same auditorhellip
Perform a full-system audit (Mock) at least one
month prior to inspection
ndash Be sure any necessary CA-PA has been originated 48
Before the Inspection
Educate all your employees on proper inspection
rules and etiquette
Educate your supervisory (all exempt) personnel
on FDA regulations
Inspections and external audits concern to all
employees not only those from Quality Dept
Promote the participation on the internal audit
cadre
Certify internal auditors
49
Before the Inspection
A few words about Internal Audit
Internal Audits are the most powerful tool to
avoid inspection surprises
If the inspector discovers something why your
internal auditor did not
The same auditor auditing several consecutive
years the same area is not effective
An effective internal audit function is priceless
50
During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

29
Different Responsibilities
How ICH Q8 Q9 Q10
Guidelines Are Working
Together Throughout The
Product Life Cycle
30
Nov 2005 amp Nov 2008
ICH Q8 Q9 and Q10
High level guidances
(not prescriptive)
Science and risk-based
Encourages systematic
approaches
Applicable over entire product
lifecycle
Intended to work together to
enhance pharmaceutical product
quality
31
Pharmaceutical Development -
Q8(R2)
Describes science and risk-based
approaches for pharmaceutical product and
manufacturing process development
Introduced concepts of design space and
flexible regulatory approaches
Introduced concepts of Quality by Design
(QbD) and provided examples of QbD
development approaches and design space 32
Differing Approaches to
Pharmaceutical Development
33
Q8(R2) - Example QbD
Approach
Quality Target Product Profile (QTPP)
Determine ldquopotentialrdquo critical quality attributes
Link raw material attributes and process parameters
to CQAs and perform risk assessment
Develop a design space (optional and not required)
Design and implement a control strategy
Manage product lifecycle including continual
improvement
CQArsquos
Product Profile
Risk Assessments
Design Space
Control Strategy
Continual
Improvement34
Product Lifecycle
concept
Human and veterinary
drugs
Biotech products
Finish products amp APIs
Drug constituent of a
combination product
35
Manage product lifecycle including
continual improvement
Three-stage approach to process validation
Stage 1 ndash Process Design ndash Building and capturing process knowledge and understanding
ndash Establishing a strategy for process control
Stage 2 ndash Process Qualification ndash Design a facility and qualification of utilities and equipment
ndash Process performance qualification
ndash PPQ protocol
ndash PPQ protocol execution and report
Stage 3 ndash Continued Process Verification
36
Focus on alignment with
lsquoproduct lifecyclersquo
Manufacturers should
Understand the sources of variation
Detect the presence and degree of variation
Understand the impact of variation on the process and
ultimately on product attributes
Control the variation in a manner commensurate with
the risk it represents to the process and product
37
Process Validation
38
Focus on alignment with
lsquoproduct lifecyclersquo
39
Focus on alignment with
lsquoproduct lifecyclersquo
40
Focus on alignment with
lsquoproduct lifecyclersquo
FDA Quality Systems Guidance
for Finished Pharmaceuticals
41 Inspection Readiness BEC
Inspection Readiness BEC 42
Quality System
Drug Inspection Target Includes the Quality Control Unit and all of its review
and approval duties
ndash Approval of and adherence to Procedures and associated recordkeeping systems
ndash Product Reviews
ndash Complaint Reviews
ndash Failure Evaluations
ndash Change Control
ndash Product Improvement Projects
ndash RejectsReprocessingRework
ndash Stability
ndash Validation
ndash Training
Inspection Readiness BEC 43
Facilities and Equipment
Inspection Target
Building and Facilities along with
maintenance
Equipment qualification calibration and
maintenance
Water steam compressed gas HVAC
Change control system
Investigate discrepancies
Inspection Readiness BEC 44
Materials
Inspection Target
Procedures and documentation showing adequate control of finished products in-process materials components containers closures
Qualificationvalidation and security of computerized or automated processes
Change control system
Investigation of Discrepancies
Inspection Readiness BEC 45
Production System
Drug Inspection Target
Trainingqualification of personnel
Complete batch production documentation
Process validation
Production time limits
In-process testingexamination
Change Control System
Investigation of discrepancies
Inspection Readiness BEC 46
Packaging and Labeling
Inspection Target
Training
Acceptance operations
Control of materials to prevent mix-ups
Accountability
Packaginglabeling records
Line separation
Line clearance inspection and documentation
Validation of labelingpackaging operations
Investigation of discrepancies
Inspection Readiness BEC 47
Laboratory Control
Inspection Target
Staffing and training
Adequate equipment calibration and maintenance
Reference standards
Adherence to written procedures
Validationverification analytical methods
Analytical records and raw data
Adherence to OOS procedure and timely completion of investigations
Stability testing program and reserve samples
Change control system
Before the Inspection
SOP for Inspection Readiness
Strong Internal AuditAssessment function
ndash Continuous assessment using Risk Management
Criteria
ndash Not autopilot auditing
ndash Same auditorhellip
Perform a full-system audit (Mock) at least one
month prior to inspection
ndash Be sure any necessary CA-PA has been originated 48
Before the Inspection
Educate all your employees on proper inspection
rules and etiquette
Educate your supervisory (all exempt) personnel
on FDA regulations
Inspections and external audits concern to all
employees not only those from Quality Dept
Promote the participation on the internal audit
cadre
Certify internal auditors
49
Before the Inspection
A few words about Internal Audit
Internal Audits are the most powerful tool to
avoid inspection surprises
If the inspector discovers something why your
internal auditor did not
The same auditor auditing several consecutive
years the same area is not effective
An effective internal audit function is priceless
50
During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

How ICH Q8 Q9 Q10
Guidelines Are Working
Together Throughout The
Product Life Cycle
30
Nov 2005 amp Nov 2008
ICH Q8 Q9 and Q10
High level guidances
(not prescriptive)
Science and risk-based
Encourages systematic
approaches
Applicable over entire product
lifecycle
Intended to work together to
enhance pharmaceutical product
quality
31
Pharmaceutical Development -
Q8(R2)
Describes science and risk-based
approaches for pharmaceutical product and
manufacturing process development
Introduced concepts of design space and
flexible regulatory approaches
Introduced concepts of Quality by Design
(QbD) and provided examples of QbD
development approaches and design space 32
Differing Approaches to
Pharmaceutical Development
33
Q8(R2) - Example QbD
Approach
Quality Target Product Profile (QTPP)
Determine ldquopotentialrdquo critical quality attributes
Link raw material attributes and process parameters
to CQAs and perform risk assessment
Develop a design space (optional and not required)
Design and implement a control strategy
Manage product lifecycle including continual
improvement
CQArsquos
Product Profile
Risk Assessments
Design Space
Control Strategy
Continual
Improvement34
Product Lifecycle
concept
Human and veterinary
drugs
Biotech products
Finish products amp APIs
Drug constituent of a
combination product
35
Manage product lifecycle including
continual improvement
Three-stage approach to process validation
Stage 1 ndash Process Design ndash Building and capturing process knowledge and understanding
ndash Establishing a strategy for process control
Stage 2 ndash Process Qualification ndash Design a facility and qualification of utilities and equipment
ndash Process performance qualification
ndash PPQ protocol
ndash PPQ protocol execution and report
Stage 3 ndash Continued Process Verification
36
Focus on alignment with
lsquoproduct lifecyclersquo
Manufacturers should
Understand the sources of variation
Detect the presence and degree of variation
Understand the impact of variation on the process and
ultimately on product attributes
Control the variation in a manner commensurate with
the risk it represents to the process and product
37
Process Validation
38
Focus on alignment with
lsquoproduct lifecyclersquo
39
Focus on alignment with
lsquoproduct lifecyclersquo
40
Focus on alignment with
lsquoproduct lifecyclersquo
FDA Quality Systems Guidance
for Finished Pharmaceuticals
41 Inspection Readiness BEC
Inspection Readiness BEC 42
Quality System
Drug Inspection Target Includes the Quality Control Unit and all of its review
and approval duties
ndash Approval of and adherence to Procedures and associated recordkeeping systems
ndash Product Reviews
ndash Complaint Reviews
ndash Failure Evaluations
ndash Change Control
ndash Product Improvement Projects
ndash RejectsReprocessingRework
ndash Stability
ndash Validation
ndash Training
Inspection Readiness BEC 43
Facilities and Equipment
Inspection Target
Building and Facilities along with
maintenance
Equipment qualification calibration and
maintenance
Water steam compressed gas HVAC
Change control system
Investigate discrepancies
Inspection Readiness BEC 44
Materials
Inspection Target
Procedures and documentation showing adequate control of finished products in-process materials components containers closures
Qualificationvalidation and security of computerized or automated processes
Change control system
Investigation of Discrepancies
Inspection Readiness BEC 45
Production System
Drug Inspection Target
Trainingqualification of personnel
Complete batch production documentation
Process validation
Production time limits
In-process testingexamination
Change Control System
Investigation of discrepancies
Inspection Readiness BEC 46
Packaging and Labeling
Inspection Target
Training
Acceptance operations
Control of materials to prevent mix-ups
Accountability
Packaginglabeling records
Line separation
Line clearance inspection and documentation
Validation of labelingpackaging operations
Investigation of discrepancies
Inspection Readiness BEC 47
Laboratory Control
Inspection Target
Staffing and training
Adequate equipment calibration and maintenance
Reference standards
Adherence to written procedures
Validationverification analytical methods
Analytical records and raw data
Adherence to OOS procedure and timely completion of investigations
Stability testing program and reserve samples
Change control system
Before the Inspection
SOP for Inspection Readiness
Strong Internal AuditAssessment function
ndash Continuous assessment using Risk Management
Criteria
ndash Not autopilot auditing
ndash Same auditorhellip
Perform a full-system audit (Mock) at least one
month prior to inspection
ndash Be sure any necessary CA-PA has been originated 48
Before the Inspection
Educate all your employees on proper inspection
rules and etiquette
Educate your supervisory (all exempt) personnel
on FDA regulations
Inspections and external audits concern to all
employees not only those from Quality Dept
Promote the participation on the internal audit
cadre
Certify internal auditors
49
Before the Inspection
A few words about Internal Audit
Internal Audits are the most powerful tool to
avoid inspection surprises
If the inspector discovers something why your
internal auditor did not
The same auditor auditing several consecutive
years the same area is not effective
An effective internal audit function is priceless
50
During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

Nov 2005 amp Nov 2008
ICH Q8 Q9 and Q10
High level guidances
(not prescriptive)
Science and risk-based
Encourages systematic
approaches
Applicable over entire product
lifecycle
Intended to work together to
enhance pharmaceutical product
quality
31
Pharmaceutical Development -
Q8(R2)
Describes science and risk-based
approaches for pharmaceutical product and
manufacturing process development
Introduced concepts of design space and
flexible regulatory approaches
Introduced concepts of Quality by Design
(QbD) and provided examples of QbD
development approaches and design space 32
Differing Approaches to
Pharmaceutical Development
33
Q8(R2) - Example QbD
Approach
Quality Target Product Profile (QTPP)
Determine ldquopotentialrdquo critical quality attributes
Link raw material attributes and process parameters
to CQAs and perform risk assessment
Develop a design space (optional and not required)
Design and implement a control strategy
Manage product lifecycle including continual
improvement
CQArsquos
Product Profile
Risk Assessments
Design Space
Control Strategy
Continual
Improvement34
Product Lifecycle
concept
Human and veterinary
drugs
Biotech products
Finish products amp APIs
Drug constituent of a
combination product
35
Manage product lifecycle including
continual improvement
Three-stage approach to process validation
Stage 1 ndash Process Design ndash Building and capturing process knowledge and understanding
ndash Establishing a strategy for process control
Stage 2 ndash Process Qualification ndash Design a facility and qualification of utilities and equipment
ndash Process performance qualification
ndash PPQ protocol
ndash PPQ protocol execution and report
Stage 3 ndash Continued Process Verification
36
Focus on alignment with
lsquoproduct lifecyclersquo
Manufacturers should
Understand the sources of variation
Detect the presence and degree of variation
Understand the impact of variation on the process and
ultimately on product attributes
Control the variation in a manner commensurate with
the risk it represents to the process and product
37
Process Validation
38
Focus on alignment with
lsquoproduct lifecyclersquo
39
Focus on alignment with
lsquoproduct lifecyclersquo
40
Focus on alignment with
lsquoproduct lifecyclersquo
FDA Quality Systems Guidance
for Finished Pharmaceuticals
41 Inspection Readiness BEC
Inspection Readiness BEC 42
Quality System
Drug Inspection Target Includes the Quality Control Unit and all of its review
and approval duties
ndash Approval of and adherence to Procedures and associated recordkeeping systems
ndash Product Reviews
ndash Complaint Reviews
ndash Failure Evaluations
ndash Change Control
ndash Product Improvement Projects
ndash RejectsReprocessingRework
ndash Stability
ndash Validation
ndash Training
Inspection Readiness BEC 43
Facilities and Equipment
Inspection Target
Building and Facilities along with
maintenance
Equipment qualification calibration and
maintenance
Water steam compressed gas HVAC
Change control system
Investigate discrepancies
Inspection Readiness BEC 44
Materials
Inspection Target
Procedures and documentation showing adequate control of finished products in-process materials components containers closures
Qualificationvalidation and security of computerized or automated processes
Change control system
Investigation of Discrepancies
Inspection Readiness BEC 45
Production System
Drug Inspection Target
Trainingqualification of personnel
Complete batch production documentation
Process validation
Production time limits
In-process testingexamination
Change Control System
Investigation of discrepancies
Inspection Readiness BEC 46
Packaging and Labeling
Inspection Target
Training
Acceptance operations
Control of materials to prevent mix-ups
Accountability
Packaginglabeling records
Line separation
Line clearance inspection and documentation
Validation of labelingpackaging operations
Investigation of discrepancies
Inspection Readiness BEC 47
Laboratory Control
Inspection Target
Staffing and training
Adequate equipment calibration and maintenance
Reference standards
Adherence to written procedures
Validationverification analytical methods
Analytical records and raw data
Adherence to OOS procedure and timely completion of investigations
Stability testing program and reserve samples
Change control system
Before the Inspection
SOP for Inspection Readiness
Strong Internal AuditAssessment function
ndash Continuous assessment using Risk Management
Criteria
ndash Not autopilot auditing
ndash Same auditorhellip
Perform a full-system audit (Mock) at least one
month prior to inspection
ndash Be sure any necessary CA-PA has been originated 48
Before the Inspection
Educate all your employees on proper inspection
rules and etiquette
Educate your supervisory (all exempt) personnel
on FDA regulations
Inspections and external audits concern to all
employees not only those from Quality Dept
Promote the participation on the internal audit
cadre
Certify internal auditors
49
Before the Inspection
A few words about Internal Audit
Internal Audits are the most powerful tool to
avoid inspection surprises
If the inspector discovers something why your
internal auditor did not
The same auditor auditing several consecutive
years the same area is not effective
An effective internal audit function is priceless
50
During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

Pharmaceutical Development -
Q8(R2)
Describes science and risk-based
approaches for pharmaceutical product and
manufacturing process development
Introduced concepts of design space and
flexible regulatory approaches
Introduced concepts of Quality by Design
(QbD) and provided examples of QbD
development approaches and design space 32
Differing Approaches to
Pharmaceutical Development
33
Q8(R2) - Example QbD
Approach
Quality Target Product Profile (QTPP)
Determine ldquopotentialrdquo critical quality attributes
Link raw material attributes and process parameters
to CQAs and perform risk assessment
Develop a design space (optional and not required)
Design and implement a control strategy
Manage product lifecycle including continual
improvement
CQArsquos
Product Profile
Risk Assessments
Design Space
Control Strategy
Continual
Improvement34
Product Lifecycle
concept
Human and veterinary
drugs
Biotech products
Finish products amp APIs
Drug constituent of a
combination product
35
Manage product lifecycle including
continual improvement
Three-stage approach to process validation
Stage 1 ndash Process Design ndash Building and capturing process knowledge and understanding
ndash Establishing a strategy for process control
Stage 2 ndash Process Qualification ndash Design a facility and qualification of utilities and equipment
ndash Process performance qualification
ndash PPQ protocol
ndash PPQ protocol execution and report
Stage 3 ndash Continued Process Verification
36
Focus on alignment with
lsquoproduct lifecyclersquo
Manufacturers should
Understand the sources of variation
Detect the presence and degree of variation
Understand the impact of variation on the process and
ultimately on product attributes
Control the variation in a manner commensurate with
the risk it represents to the process and product
37
Process Validation
38
Focus on alignment with
lsquoproduct lifecyclersquo
39
Focus on alignment with
lsquoproduct lifecyclersquo
40
Focus on alignment with
lsquoproduct lifecyclersquo
FDA Quality Systems Guidance
for Finished Pharmaceuticals
41 Inspection Readiness BEC
Inspection Readiness BEC 42
Quality System
Drug Inspection Target Includes the Quality Control Unit and all of its review
and approval duties
ndash Approval of and adherence to Procedures and associated recordkeeping systems
ndash Product Reviews
ndash Complaint Reviews
ndash Failure Evaluations
ndash Change Control
ndash Product Improvement Projects
ndash RejectsReprocessingRework
ndash Stability
ndash Validation
ndash Training
Inspection Readiness BEC 43
Facilities and Equipment
Inspection Target
Building and Facilities along with
maintenance
Equipment qualification calibration and
maintenance
Water steam compressed gas HVAC
Change control system
Investigate discrepancies
Inspection Readiness BEC 44
Materials
Inspection Target
Procedures and documentation showing adequate control of finished products in-process materials components containers closures
Qualificationvalidation and security of computerized or automated processes
Change control system
Investigation of Discrepancies
Inspection Readiness BEC 45
Production System
Drug Inspection Target
Trainingqualification of personnel
Complete batch production documentation
Process validation
Production time limits
In-process testingexamination
Change Control System
Investigation of discrepancies
Inspection Readiness BEC 46
Packaging and Labeling
Inspection Target
Training
Acceptance operations
Control of materials to prevent mix-ups
Accountability
Packaginglabeling records
Line separation
Line clearance inspection and documentation
Validation of labelingpackaging operations
Investigation of discrepancies
Inspection Readiness BEC 47
Laboratory Control
Inspection Target
Staffing and training
Adequate equipment calibration and maintenance
Reference standards
Adherence to written procedures
Validationverification analytical methods
Analytical records and raw data
Adherence to OOS procedure and timely completion of investigations
Stability testing program and reserve samples
Change control system
Before the Inspection
SOP for Inspection Readiness
Strong Internal AuditAssessment function
ndash Continuous assessment using Risk Management
Criteria
ndash Not autopilot auditing
ndash Same auditorhellip
Perform a full-system audit (Mock) at least one
month prior to inspection
ndash Be sure any necessary CA-PA has been originated 48
Before the Inspection
Educate all your employees on proper inspection
rules and etiquette
Educate your supervisory (all exempt) personnel
on FDA regulations
Inspections and external audits concern to all
employees not only those from Quality Dept
Promote the participation on the internal audit
cadre
Certify internal auditors
49
Before the Inspection
A few words about Internal Audit
Internal Audits are the most powerful tool to
avoid inspection surprises
If the inspector discovers something why your
internal auditor did not
The same auditor auditing several consecutive
years the same area is not effective
An effective internal audit function is priceless
50
During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

Differing Approaches to
Pharmaceutical Development
33
Q8(R2) - Example QbD
Approach
Quality Target Product Profile (QTPP)
Determine ldquopotentialrdquo critical quality attributes
Link raw material attributes and process parameters
to CQAs and perform risk assessment
Develop a design space (optional and not required)
Design and implement a control strategy
Manage product lifecycle including continual
improvement
CQArsquos
Product Profile
Risk Assessments
Design Space
Control Strategy
Continual
Improvement34
Product Lifecycle
concept
Human and veterinary
drugs
Biotech products
Finish products amp APIs
Drug constituent of a
combination product
35
Manage product lifecycle including
continual improvement
Three-stage approach to process validation
Stage 1 ndash Process Design ndash Building and capturing process knowledge and understanding
ndash Establishing a strategy for process control
Stage 2 ndash Process Qualification ndash Design a facility and qualification of utilities and equipment
ndash Process performance qualification
ndash PPQ protocol
ndash PPQ protocol execution and report
Stage 3 ndash Continued Process Verification
36
Focus on alignment with
lsquoproduct lifecyclersquo
Manufacturers should
Understand the sources of variation
Detect the presence and degree of variation
Understand the impact of variation on the process and
ultimately on product attributes
Control the variation in a manner commensurate with
the risk it represents to the process and product
37
Process Validation
38
Focus on alignment with
lsquoproduct lifecyclersquo
39
Focus on alignment with
lsquoproduct lifecyclersquo
40
Focus on alignment with
lsquoproduct lifecyclersquo
FDA Quality Systems Guidance
for Finished Pharmaceuticals
41 Inspection Readiness BEC
Inspection Readiness BEC 42
Quality System
Drug Inspection Target Includes the Quality Control Unit and all of its review
and approval duties
ndash Approval of and adherence to Procedures and associated recordkeeping systems
ndash Product Reviews
ndash Complaint Reviews
ndash Failure Evaluations
ndash Change Control
ndash Product Improvement Projects
ndash RejectsReprocessingRework
ndash Stability
ndash Validation
ndash Training
Inspection Readiness BEC 43
Facilities and Equipment
Inspection Target
Building and Facilities along with
maintenance
Equipment qualification calibration and
maintenance
Water steam compressed gas HVAC
Change control system
Investigate discrepancies
Inspection Readiness BEC 44
Materials
Inspection Target
Procedures and documentation showing adequate control of finished products in-process materials components containers closures
Qualificationvalidation and security of computerized or automated processes
Change control system
Investigation of Discrepancies
Inspection Readiness BEC 45
Production System
Drug Inspection Target
Trainingqualification of personnel
Complete batch production documentation
Process validation
Production time limits
In-process testingexamination
Change Control System
Investigation of discrepancies
Inspection Readiness BEC 46
Packaging and Labeling
Inspection Target
Training
Acceptance operations
Control of materials to prevent mix-ups
Accountability
Packaginglabeling records
Line separation
Line clearance inspection and documentation
Validation of labelingpackaging operations
Investigation of discrepancies
Inspection Readiness BEC 47
Laboratory Control
Inspection Target
Staffing and training
Adequate equipment calibration and maintenance
Reference standards
Adherence to written procedures
Validationverification analytical methods
Analytical records and raw data
Adherence to OOS procedure and timely completion of investigations
Stability testing program and reserve samples
Change control system
Before the Inspection
SOP for Inspection Readiness
Strong Internal AuditAssessment function
ndash Continuous assessment using Risk Management
Criteria
ndash Not autopilot auditing
ndash Same auditorhellip
Perform a full-system audit (Mock) at least one
month prior to inspection
ndash Be sure any necessary CA-PA has been originated 48
Before the Inspection
Educate all your employees on proper inspection
rules and etiquette
Educate your supervisory (all exempt) personnel
on FDA regulations
Inspections and external audits concern to all
employees not only those from Quality Dept
Promote the participation on the internal audit
cadre
Certify internal auditors
49
Before the Inspection
A few words about Internal Audit
Internal Audits are the most powerful tool to
avoid inspection surprises
If the inspector discovers something why your
internal auditor did not
The same auditor auditing several consecutive
years the same area is not effective
An effective internal audit function is priceless
50
During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62
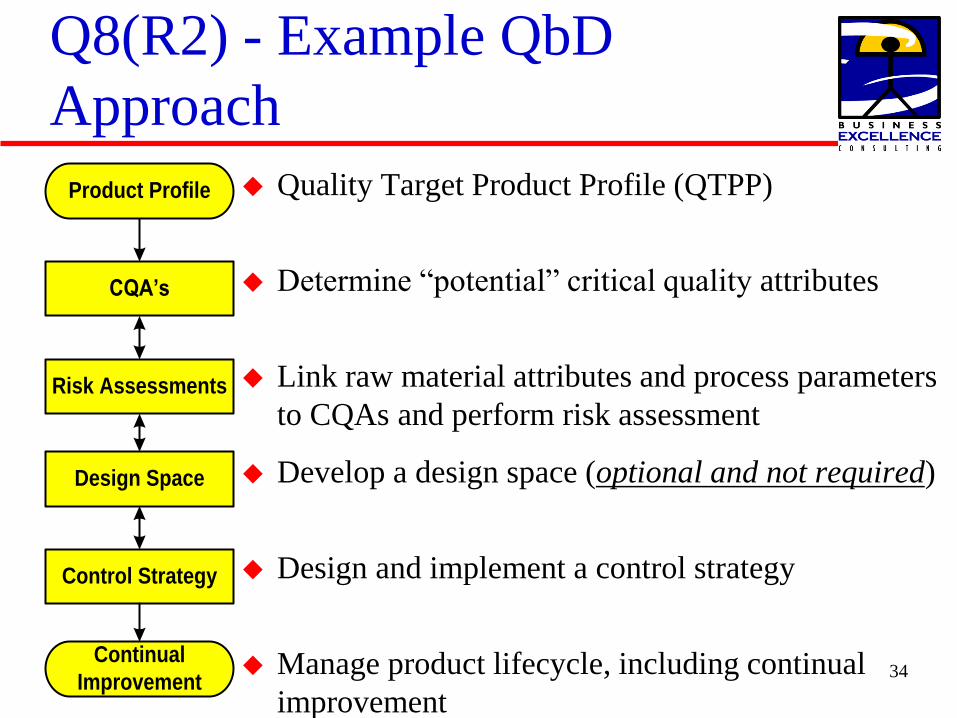
Q8(R2) - Example QbD
Approach
Quality Target Product Profile (QTPP)
Determine ldquopotentialrdquo critical quality attributes
Link raw material attributes and process parameters
to CQAs and perform risk assessment
Develop a design space (optional and not required)
Design and implement a control strategy
Manage product lifecycle including continual
improvement
CQArsquos
Product Profile
Risk Assessments
Design Space
Control Strategy
Continual
Improvement34
Product Lifecycle
concept
Human and veterinary
drugs
Biotech products
Finish products amp APIs
Drug constituent of a
combination product
35
Manage product lifecycle including
continual improvement
Three-stage approach to process validation
Stage 1 ndash Process Design ndash Building and capturing process knowledge and understanding
ndash Establishing a strategy for process control
Stage 2 ndash Process Qualification ndash Design a facility and qualification of utilities and equipment
ndash Process performance qualification
ndash PPQ protocol
ndash PPQ protocol execution and report
Stage 3 ndash Continued Process Verification
36
Focus on alignment with
lsquoproduct lifecyclersquo
Manufacturers should
Understand the sources of variation
Detect the presence and degree of variation
Understand the impact of variation on the process and
ultimately on product attributes
Control the variation in a manner commensurate with
the risk it represents to the process and product
37
Process Validation
38
Focus on alignment with
lsquoproduct lifecyclersquo
39
Focus on alignment with
lsquoproduct lifecyclersquo
40
Focus on alignment with
lsquoproduct lifecyclersquo
FDA Quality Systems Guidance
for Finished Pharmaceuticals
41 Inspection Readiness BEC
Inspection Readiness BEC 42
Quality System
Drug Inspection Target Includes the Quality Control Unit and all of its review
and approval duties
ndash Approval of and adherence to Procedures and associated recordkeeping systems
ndash Product Reviews
ndash Complaint Reviews
ndash Failure Evaluations
ndash Change Control
ndash Product Improvement Projects
ndash RejectsReprocessingRework
ndash Stability
ndash Validation
ndash Training
Inspection Readiness BEC 43
Facilities and Equipment
Inspection Target
Building and Facilities along with
maintenance
Equipment qualification calibration and
maintenance
Water steam compressed gas HVAC
Change control system
Investigate discrepancies
Inspection Readiness BEC 44
Materials
Inspection Target
Procedures and documentation showing adequate control of finished products in-process materials components containers closures
Qualificationvalidation and security of computerized or automated processes
Change control system
Investigation of Discrepancies
Inspection Readiness BEC 45
Production System
Drug Inspection Target
Trainingqualification of personnel
Complete batch production documentation
Process validation
Production time limits
In-process testingexamination
Change Control System
Investigation of discrepancies
Inspection Readiness BEC 46
Packaging and Labeling
Inspection Target
Training
Acceptance operations
Control of materials to prevent mix-ups
Accountability
Packaginglabeling records
Line separation
Line clearance inspection and documentation
Validation of labelingpackaging operations
Investigation of discrepancies
Inspection Readiness BEC 47
Laboratory Control
Inspection Target
Staffing and training
Adequate equipment calibration and maintenance
Reference standards
Adherence to written procedures
Validationverification analytical methods
Analytical records and raw data
Adherence to OOS procedure and timely completion of investigations
Stability testing program and reserve samples
Change control system
Before the Inspection
SOP for Inspection Readiness
Strong Internal AuditAssessment function
ndash Continuous assessment using Risk Management
Criteria
ndash Not autopilot auditing
ndash Same auditorhellip
Perform a full-system audit (Mock) at least one
month prior to inspection
ndash Be sure any necessary CA-PA has been originated 48
Before the Inspection
Educate all your employees on proper inspection
rules and etiquette
Educate your supervisory (all exempt) personnel
on FDA regulations
Inspections and external audits concern to all
employees not only those from Quality Dept
Promote the participation on the internal audit
cadre
Certify internal auditors
49
Before the Inspection
A few words about Internal Audit
Internal Audits are the most powerful tool to
avoid inspection surprises
If the inspector discovers something why your
internal auditor did not
The same auditor auditing several consecutive
years the same area is not effective
An effective internal audit function is priceless
50
During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

Product Lifecycle
concept
Human and veterinary
drugs
Biotech products
Finish products amp APIs
Drug constituent of a
combination product
35
Manage product lifecycle including
continual improvement
Three-stage approach to process validation
Stage 1 ndash Process Design ndash Building and capturing process knowledge and understanding
ndash Establishing a strategy for process control
Stage 2 ndash Process Qualification ndash Design a facility and qualification of utilities and equipment
ndash Process performance qualification
ndash PPQ protocol
ndash PPQ protocol execution and report
Stage 3 ndash Continued Process Verification
36
Focus on alignment with
lsquoproduct lifecyclersquo
Manufacturers should
Understand the sources of variation
Detect the presence and degree of variation
Understand the impact of variation on the process and
ultimately on product attributes
Control the variation in a manner commensurate with
the risk it represents to the process and product
37
Process Validation
38
Focus on alignment with
lsquoproduct lifecyclersquo
39
Focus on alignment with
lsquoproduct lifecyclersquo
40
Focus on alignment with
lsquoproduct lifecyclersquo
FDA Quality Systems Guidance
for Finished Pharmaceuticals
41 Inspection Readiness BEC
Inspection Readiness BEC 42
Quality System
Drug Inspection Target Includes the Quality Control Unit and all of its review
and approval duties
ndash Approval of and adherence to Procedures and associated recordkeeping systems
ndash Product Reviews
ndash Complaint Reviews
ndash Failure Evaluations
ndash Change Control
ndash Product Improvement Projects
ndash RejectsReprocessingRework
ndash Stability
ndash Validation
ndash Training
Inspection Readiness BEC 43
Facilities and Equipment
Inspection Target
Building and Facilities along with
maintenance
Equipment qualification calibration and
maintenance
Water steam compressed gas HVAC
Change control system
Investigate discrepancies
Inspection Readiness BEC 44
Materials
Inspection Target
Procedures and documentation showing adequate control of finished products in-process materials components containers closures
Qualificationvalidation and security of computerized or automated processes
Change control system
Investigation of Discrepancies
Inspection Readiness BEC 45
Production System
Drug Inspection Target
Trainingqualification of personnel
Complete batch production documentation
Process validation
Production time limits
In-process testingexamination
Change Control System
Investigation of discrepancies
Inspection Readiness BEC 46
Packaging and Labeling
Inspection Target
Training
Acceptance operations
Control of materials to prevent mix-ups
Accountability
Packaginglabeling records
Line separation
Line clearance inspection and documentation
Validation of labelingpackaging operations
Investigation of discrepancies
Inspection Readiness BEC 47
Laboratory Control
Inspection Target
Staffing and training
Adequate equipment calibration and maintenance
Reference standards
Adherence to written procedures
Validationverification analytical methods
Analytical records and raw data
Adherence to OOS procedure and timely completion of investigations
Stability testing program and reserve samples
Change control system
Before the Inspection
SOP for Inspection Readiness
Strong Internal AuditAssessment function
ndash Continuous assessment using Risk Management
Criteria
ndash Not autopilot auditing
ndash Same auditorhellip
Perform a full-system audit (Mock) at least one
month prior to inspection
ndash Be sure any necessary CA-PA has been originated 48
Before the Inspection
Educate all your employees on proper inspection
rules and etiquette
Educate your supervisory (all exempt) personnel
on FDA regulations
Inspections and external audits concern to all
employees not only those from Quality Dept
Promote the participation on the internal audit
cadre
Certify internal auditors
49
Before the Inspection
A few words about Internal Audit
Internal Audits are the most powerful tool to
avoid inspection surprises
If the inspector discovers something why your
internal auditor did not
The same auditor auditing several consecutive
years the same area is not effective
An effective internal audit function is priceless
50
During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

Three-stage approach to process validation
Stage 1 ndash Process Design ndash Building and capturing process knowledge and understanding
ndash Establishing a strategy for process control
Stage 2 ndash Process Qualification ndash Design a facility and qualification of utilities and equipment
ndash Process performance qualification
ndash PPQ protocol
ndash PPQ protocol execution and report
Stage 3 ndash Continued Process Verification
36
Focus on alignment with
lsquoproduct lifecyclersquo
Manufacturers should
Understand the sources of variation
Detect the presence and degree of variation
Understand the impact of variation on the process and
ultimately on product attributes
Control the variation in a manner commensurate with
the risk it represents to the process and product
37
Process Validation
38
Focus on alignment with
lsquoproduct lifecyclersquo
39
Focus on alignment with
lsquoproduct lifecyclersquo
40
Focus on alignment with
lsquoproduct lifecyclersquo
FDA Quality Systems Guidance
for Finished Pharmaceuticals
41 Inspection Readiness BEC
Inspection Readiness BEC 42
Quality System
Drug Inspection Target Includes the Quality Control Unit and all of its review
and approval duties
ndash Approval of and adherence to Procedures and associated recordkeeping systems
ndash Product Reviews
ndash Complaint Reviews
ndash Failure Evaluations
ndash Change Control
ndash Product Improvement Projects
ndash RejectsReprocessingRework
ndash Stability
ndash Validation
ndash Training
Inspection Readiness BEC 43
Facilities and Equipment
Inspection Target
Building and Facilities along with
maintenance
Equipment qualification calibration and
maintenance
Water steam compressed gas HVAC
Change control system
Investigate discrepancies
Inspection Readiness BEC 44
Materials
Inspection Target
Procedures and documentation showing adequate control of finished products in-process materials components containers closures
Qualificationvalidation and security of computerized or automated processes
Change control system
Investigation of Discrepancies
Inspection Readiness BEC 45
Production System
Drug Inspection Target
Trainingqualification of personnel
Complete batch production documentation
Process validation
Production time limits
In-process testingexamination
Change Control System
Investigation of discrepancies
Inspection Readiness BEC 46
Packaging and Labeling
Inspection Target
Training
Acceptance operations
Control of materials to prevent mix-ups
Accountability
Packaginglabeling records
Line separation
Line clearance inspection and documentation
Validation of labelingpackaging operations
Investigation of discrepancies
Inspection Readiness BEC 47
Laboratory Control
Inspection Target
Staffing and training
Adequate equipment calibration and maintenance
Reference standards
Adherence to written procedures
Validationverification analytical methods
Analytical records and raw data
Adherence to OOS procedure and timely completion of investigations
Stability testing program and reserve samples
Change control system
Before the Inspection
SOP for Inspection Readiness
Strong Internal AuditAssessment function
ndash Continuous assessment using Risk Management
Criteria
ndash Not autopilot auditing
ndash Same auditorhellip
Perform a full-system audit (Mock) at least one
month prior to inspection
ndash Be sure any necessary CA-PA has been originated 48
Before the Inspection
Educate all your employees on proper inspection
rules and etiquette
Educate your supervisory (all exempt) personnel
on FDA regulations
Inspections and external audits concern to all
employees not only those from Quality Dept
Promote the participation on the internal audit
cadre
Certify internal auditors
49
Before the Inspection
A few words about Internal Audit
Internal Audits are the most powerful tool to
avoid inspection surprises
If the inspector discovers something why your
internal auditor did not
The same auditor auditing several consecutive
years the same area is not effective
An effective internal audit function is priceless
50
During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

Manufacturers should
Understand the sources of variation
Detect the presence and degree of variation
Understand the impact of variation on the process and
ultimately on product attributes
Control the variation in a manner commensurate with
the risk it represents to the process and product
37
Process Validation
38
Focus on alignment with
lsquoproduct lifecyclersquo
39
Focus on alignment with
lsquoproduct lifecyclersquo
40
Focus on alignment with
lsquoproduct lifecyclersquo
FDA Quality Systems Guidance
for Finished Pharmaceuticals
41 Inspection Readiness BEC
Inspection Readiness BEC 42
Quality System
Drug Inspection Target Includes the Quality Control Unit and all of its review
and approval duties
ndash Approval of and adherence to Procedures and associated recordkeeping systems
ndash Product Reviews
ndash Complaint Reviews
ndash Failure Evaluations
ndash Change Control
ndash Product Improvement Projects
ndash RejectsReprocessingRework
ndash Stability
ndash Validation
ndash Training
Inspection Readiness BEC 43
Facilities and Equipment
Inspection Target
Building and Facilities along with
maintenance
Equipment qualification calibration and
maintenance
Water steam compressed gas HVAC
Change control system
Investigate discrepancies
Inspection Readiness BEC 44
Materials
Inspection Target
Procedures and documentation showing adequate control of finished products in-process materials components containers closures
Qualificationvalidation and security of computerized or automated processes
Change control system
Investigation of Discrepancies
Inspection Readiness BEC 45
Production System
Drug Inspection Target
Trainingqualification of personnel
Complete batch production documentation
Process validation
Production time limits
In-process testingexamination
Change Control System
Investigation of discrepancies
Inspection Readiness BEC 46
Packaging and Labeling
Inspection Target
Training
Acceptance operations
Control of materials to prevent mix-ups
Accountability
Packaginglabeling records
Line separation
Line clearance inspection and documentation
Validation of labelingpackaging operations
Investigation of discrepancies
Inspection Readiness BEC 47
Laboratory Control
Inspection Target
Staffing and training
Adequate equipment calibration and maintenance
Reference standards
Adherence to written procedures
Validationverification analytical methods
Analytical records and raw data
Adherence to OOS procedure and timely completion of investigations
Stability testing program and reserve samples
Change control system
Before the Inspection
SOP for Inspection Readiness
Strong Internal AuditAssessment function
ndash Continuous assessment using Risk Management
Criteria
ndash Not autopilot auditing
ndash Same auditorhellip
Perform a full-system audit (Mock) at least one
month prior to inspection
ndash Be sure any necessary CA-PA has been originated 48
Before the Inspection
Educate all your employees on proper inspection
rules and etiquette
Educate your supervisory (all exempt) personnel
on FDA regulations
Inspections and external audits concern to all
employees not only those from Quality Dept
Promote the participation on the internal audit
cadre
Certify internal auditors
49
Before the Inspection
A few words about Internal Audit
Internal Audits are the most powerful tool to
avoid inspection surprises
If the inspector discovers something why your
internal auditor did not
The same auditor auditing several consecutive
years the same area is not effective
An effective internal audit function is priceless
50
During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

38
Focus on alignment with
lsquoproduct lifecyclersquo
39
Focus on alignment with
lsquoproduct lifecyclersquo
40
Focus on alignment with
lsquoproduct lifecyclersquo
FDA Quality Systems Guidance
for Finished Pharmaceuticals
41 Inspection Readiness BEC
Inspection Readiness BEC 42
Quality System
Drug Inspection Target Includes the Quality Control Unit and all of its review
and approval duties
ndash Approval of and adherence to Procedures and associated recordkeeping systems
ndash Product Reviews
ndash Complaint Reviews
ndash Failure Evaluations
ndash Change Control
ndash Product Improvement Projects
ndash RejectsReprocessingRework
ndash Stability
ndash Validation
ndash Training
Inspection Readiness BEC 43
Facilities and Equipment
Inspection Target
Building and Facilities along with
maintenance
Equipment qualification calibration and
maintenance
Water steam compressed gas HVAC
Change control system
Investigate discrepancies
Inspection Readiness BEC 44
Materials
Inspection Target
Procedures and documentation showing adequate control of finished products in-process materials components containers closures
Qualificationvalidation and security of computerized or automated processes
Change control system
Investigation of Discrepancies
Inspection Readiness BEC 45
Production System
Drug Inspection Target
Trainingqualification of personnel
Complete batch production documentation
Process validation
Production time limits
In-process testingexamination
Change Control System
Investigation of discrepancies
Inspection Readiness BEC 46
Packaging and Labeling
Inspection Target
Training
Acceptance operations
Control of materials to prevent mix-ups
Accountability
Packaginglabeling records
Line separation
Line clearance inspection and documentation
Validation of labelingpackaging operations
Investigation of discrepancies
Inspection Readiness BEC 47
Laboratory Control
Inspection Target
Staffing and training
Adequate equipment calibration and maintenance
Reference standards
Adherence to written procedures
Validationverification analytical methods
Analytical records and raw data
Adherence to OOS procedure and timely completion of investigations
Stability testing program and reserve samples
Change control system
Before the Inspection
SOP for Inspection Readiness
Strong Internal AuditAssessment function
ndash Continuous assessment using Risk Management
Criteria
ndash Not autopilot auditing
ndash Same auditorhellip
Perform a full-system audit (Mock) at least one
month prior to inspection
ndash Be sure any necessary CA-PA has been originated 48
Before the Inspection
Educate all your employees on proper inspection
rules and etiquette
Educate your supervisory (all exempt) personnel
on FDA regulations
Inspections and external audits concern to all
employees not only those from Quality Dept
Promote the participation on the internal audit
cadre
Certify internal auditors
49
Before the Inspection
A few words about Internal Audit
Internal Audits are the most powerful tool to
avoid inspection surprises
If the inspector discovers something why your
internal auditor did not
The same auditor auditing several consecutive
years the same area is not effective
An effective internal audit function is priceless
50
During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

39
Focus on alignment with
lsquoproduct lifecyclersquo
40
Focus on alignment with
lsquoproduct lifecyclersquo
FDA Quality Systems Guidance
for Finished Pharmaceuticals
41 Inspection Readiness BEC
Inspection Readiness BEC 42
Quality System
Drug Inspection Target Includes the Quality Control Unit and all of its review
and approval duties
ndash Approval of and adherence to Procedures and associated recordkeeping systems
ndash Product Reviews
ndash Complaint Reviews
ndash Failure Evaluations
ndash Change Control
ndash Product Improvement Projects
ndash RejectsReprocessingRework
ndash Stability
ndash Validation
ndash Training
Inspection Readiness BEC 43
Facilities and Equipment
Inspection Target
Building and Facilities along with
maintenance
Equipment qualification calibration and
maintenance
Water steam compressed gas HVAC
Change control system
Investigate discrepancies
Inspection Readiness BEC 44
Materials
Inspection Target
Procedures and documentation showing adequate control of finished products in-process materials components containers closures
Qualificationvalidation and security of computerized or automated processes
Change control system
Investigation of Discrepancies
Inspection Readiness BEC 45
Production System
Drug Inspection Target
Trainingqualification of personnel
Complete batch production documentation
Process validation
Production time limits
In-process testingexamination
Change Control System
Investigation of discrepancies
Inspection Readiness BEC 46
Packaging and Labeling
Inspection Target
Training
Acceptance operations
Control of materials to prevent mix-ups
Accountability
Packaginglabeling records
Line separation
Line clearance inspection and documentation
Validation of labelingpackaging operations
Investigation of discrepancies
Inspection Readiness BEC 47
Laboratory Control
Inspection Target
Staffing and training
Adequate equipment calibration and maintenance
Reference standards
Adherence to written procedures
Validationverification analytical methods
Analytical records and raw data
Adherence to OOS procedure and timely completion of investigations
Stability testing program and reserve samples
Change control system
Before the Inspection
SOP for Inspection Readiness
Strong Internal AuditAssessment function
ndash Continuous assessment using Risk Management
Criteria
ndash Not autopilot auditing
ndash Same auditorhellip
Perform a full-system audit (Mock) at least one
month prior to inspection
ndash Be sure any necessary CA-PA has been originated 48
Before the Inspection
Educate all your employees on proper inspection
rules and etiquette
Educate your supervisory (all exempt) personnel
on FDA regulations
Inspections and external audits concern to all
employees not only those from Quality Dept
Promote the participation on the internal audit
cadre
Certify internal auditors
49
Before the Inspection
A few words about Internal Audit
Internal Audits are the most powerful tool to
avoid inspection surprises
If the inspector discovers something why your
internal auditor did not
The same auditor auditing several consecutive
years the same area is not effective
An effective internal audit function is priceless
50
During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

40
Focus on alignment with
lsquoproduct lifecyclersquo
FDA Quality Systems Guidance
for Finished Pharmaceuticals
41 Inspection Readiness BEC
Inspection Readiness BEC 42
Quality System
Drug Inspection Target Includes the Quality Control Unit and all of its review
and approval duties
ndash Approval of and adherence to Procedures and associated recordkeeping systems
ndash Product Reviews
ndash Complaint Reviews
ndash Failure Evaluations
ndash Change Control
ndash Product Improvement Projects
ndash RejectsReprocessingRework
ndash Stability
ndash Validation
ndash Training
Inspection Readiness BEC 43
Facilities and Equipment
Inspection Target
Building and Facilities along with
maintenance
Equipment qualification calibration and
maintenance
Water steam compressed gas HVAC
Change control system
Investigate discrepancies
Inspection Readiness BEC 44
Materials
Inspection Target
Procedures and documentation showing adequate control of finished products in-process materials components containers closures
Qualificationvalidation and security of computerized or automated processes
Change control system
Investigation of Discrepancies
Inspection Readiness BEC 45
Production System
Drug Inspection Target
Trainingqualification of personnel
Complete batch production documentation
Process validation
Production time limits
In-process testingexamination
Change Control System
Investigation of discrepancies
Inspection Readiness BEC 46
Packaging and Labeling
Inspection Target
Training
Acceptance operations
Control of materials to prevent mix-ups
Accountability
Packaginglabeling records
Line separation
Line clearance inspection and documentation
Validation of labelingpackaging operations
Investigation of discrepancies
Inspection Readiness BEC 47
Laboratory Control
Inspection Target
Staffing and training
Adequate equipment calibration and maintenance
Reference standards
Adherence to written procedures
Validationverification analytical methods
Analytical records and raw data
Adherence to OOS procedure and timely completion of investigations
Stability testing program and reserve samples
Change control system
Before the Inspection
SOP for Inspection Readiness
Strong Internal AuditAssessment function
ndash Continuous assessment using Risk Management
Criteria
ndash Not autopilot auditing
ndash Same auditorhellip
Perform a full-system audit (Mock) at least one
month prior to inspection
ndash Be sure any necessary CA-PA has been originated 48
Before the Inspection
Educate all your employees on proper inspection
rules and etiquette
Educate your supervisory (all exempt) personnel
on FDA regulations
Inspections and external audits concern to all
employees not only those from Quality Dept
Promote the participation on the internal audit
cadre
Certify internal auditors
49
Before the Inspection
A few words about Internal Audit
Internal Audits are the most powerful tool to
avoid inspection surprises
If the inspector discovers something why your
internal auditor did not
The same auditor auditing several consecutive
years the same area is not effective
An effective internal audit function is priceless
50
During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

FDA Quality Systems Guidance
for Finished Pharmaceuticals
41 Inspection Readiness BEC
Inspection Readiness BEC 42
Quality System
Drug Inspection Target Includes the Quality Control Unit and all of its review
and approval duties
ndash Approval of and adherence to Procedures and associated recordkeeping systems
ndash Product Reviews
ndash Complaint Reviews
ndash Failure Evaluations
ndash Change Control
ndash Product Improvement Projects
ndash RejectsReprocessingRework
ndash Stability
ndash Validation
ndash Training
Inspection Readiness BEC 43
Facilities and Equipment
Inspection Target
Building and Facilities along with
maintenance
Equipment qualification calibration and
maintenance
Water steam compressed gas HVAC
Change control system
Investigate discrepancies
Inspection Readiness BEC 44
Materials
Inspection Target
Procedures and documentation showing adequate control of finished products in-process materials components containers closures
Qualificationvalidation and security of computerized or automated processes
Change control system
Investigation of Discrepancies
Inspection Readiness BEC 45
Production System
Drug Inspection Target
Trainingqualification of personnel
Complete batch production documentation
Process validation
Production time limits
In-process testingexamination
Change Control System
Investigation of discrepancies
Inspection Readiness BEC 46
Packaging and Labeling
Inspection Target
Training
Acceptance operations
Control of materials to prevent mix-ups
Accountability
Packaginglabeling records
Line separation
Line clearance inspection and documentation
Validation of labelingpackaging operations
Investigation of discrepancies
Inspection Readiness BEC 47
Laboratory Control
Inspection Target
Staffing and training
Adequate equipment calibration and maintenance
Reference standards
Adherence to written procedures
Validationverification analytical methods
Analytical records and raw data
Adherence to OOS procedure and timely completion of investigations
Stability testing program and reserve samples
Change control system
Before the Inspection
SOP for Inspection Readiness
Strong Internal AuditAssessment function
ndash Continuous assessment using Risk Management
Criteria
ndash Not autopilot auditing
ndash Same auditorhellip
Perform a full-system audit (Mock) at least one
month prior to inspection
ndash Be sure any necessary CA-PA has been originated 48
Before the Inspection
Educate all your employees on proper inspection
rules and etiquette
Educate your supervisory (all exempt) personnel
on FDA regulations
Inspections and external audits concern to all
employees not only those from Quality Dept
Promote the participation on the internal audit
cadre
Certify internal auditors
49
Before the Inspection
A few words about Internal Audit
Internal Audits are the most powerful tool to
avoid inspection surprises
If the inspector discovers something why your
internal auditor did not
The same auditor auditing several consecutive
years the same area is not effective
An effective internal audit function is priceless
50
During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

Inspection Readiness BEC 42
Quality System
Drug Inspection Target Includes the Quality Control Unit and all of its review
and approval duties
ndash Approval of and adherence to Procedures and associated recordkeeping systems
ndash Product Reviews
ndash Complaint Reviews
ndash Failure Evaluations
ndash Change Control
ndash Product Improvement Projects
ndash RejectsReprocessingRework
ndash Stability
ndash Validation
ndash Training
Inspection Readiness BEC 43
Facilities and Equipment
Inspection Target
Building and Facilities along with
maintenance
Equipment qualification calibration and
maintenance
Water steam compressed gas HVAC
Change control system
Investigate discrepancies
Inspection Readiness BEC 44
Materials
Inspection Target
Procedures and documentation showing adequate control of finished products in-process materials components containers closures
Qualificationvalidation and security of computerized or automated processes
Change control system
Investigation of Discrepancies
Inspection Readiness BEC 45
Production System
Drug Inspection Target
Trainingqualification of personnel
Complete batch production documentation
Process validation
Production time limits
In-process testingexamination
Change Control System
Investigation of discrepancies
Inspection Readiness BEC 46
Packaging and Labeling
Inspection Target
Training
Acceptance operations
Control of materials to prevent mix-ups
Accountability
Packaginglabeling records
Line separation
Line clearance inspection and documentation
Validation of labelingpackaging operations
Investigation of discrepancies
Inspection Readiness BEC 47
Laboratory Control
Inspection Target
Staffing and training
Adequate equipment calibration and maintenance
Reference standards
Adherence to written procedures
Validationverification analytical methods
Analytical records and raw data
Adherence to OOS procedure and timely completion of investigations
Stability testing program and reserve samples
Change control system
Before the Inspection
SOP for Inspection Readiness
Strong Internal AuditAssessment function
ndash Continuous assessment using Risk Management
Criteria
ndash Not autopilot auditing
ndash Same auditorhellip
Perform a full-system audit (Mock) at least one
month prior to inspection
ndash Be sure any necessary CA-PA has been originated 48
Before the Inspection
Educate all your employees on proper inspection
rules and etiquette
Educate your supervisory (all exempt) personnel
on FDA regulations
Inspections and external audits concern to all
employees not only those from Quality Dept
Promote the participation on the internal audit
cadre
Certify internal auditors
49
Before the Inspection
A few words about Internal Audit
Internal Audits are the most powerful tool to
avoid inspection surprises
If the inspector discovers something why your
internal auditor did not
The same auditor auditing several consecutive
years the same area is not effective
An effective internal audit function is priceless
50
During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

Inspection Readiness BEC 43
Facilities and Equipment
Inspection Target
Building and Facilities along with
maintenance
Equipment qualification calibration and
maintenance
Water steam compressed gas HVAC
Change control system
Investigate discrepancies
Inspection Readiness BEC 44
Materials
Inspection Target
Procedures and documentation showing adequate control of finished products in-process materials components containers closures
Qualificationvalidation and security of computerized or automated processes
Change control system
Investigation of Discrepancies
Inspection Readiness BEC 45
Production System
Drug Inspection Target
Trainingqualification of personnel
Complete batch production documentation
Process validation
Production time limits
In-process testingexamination
Change Control System
Investigation of discrepancies
Inspection Readiness BEC 46
Packaging and Labeling
Inspection Target
Training
Acceptance operations
Control of materials to prevent mix-ups
Accountability
Packaginglabeling records
Line separation
Line clearance inspection and documentation
Validation of labelingpackaging operations
Investigation of discrepancies
Inspection Readiness BEC 47
Laboratory Control
Inspection Target
Staffing and training
Adequate equipment calibration and maintenance
Reference standards
Adherence to written procedures
Validationverification analytical methods
Analytical records and raw data
Adherence to OOS procedure and timely completion of investigations
Stability testing program and reserve samples
Change control system
Before the Inspection
SOP for Inspection Readiness
Strong Internal AuditAssessment function
ndash Continuous assessment using Risk Management
Criteria
ndash Not autopilot auditing
ndash Same auditorhellip
Perform a full-system audit (Mock) at least one
month prior to inspection
ndash Be sure any necessary CA-PA has been originated 48
Before the Inspection
Educate all your employees on proper inspection
rules and etiquette
Educate your supervisory (all exempt) personnel
on FDA regulations
Inspections and external audits concern to all
employees not only those from Quality Dept
Promote the participation on the internal audit
cadre
Certify internal auditors
49
Before the Inspection
A few words about Internal Audit
Internal Audits are the most powerful tool to
avoid inspection surprises
If the inspector discovers something why your
internal auditor did not
The same auditor auditing several consecutive
years the same area is not effective
An effective internal audit function is priceless
50
During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

Inspection Readiness BEC 44
Materials
Inspection Target
Procedures and documentation showing adequate control of finished products in-process materials components containers closures
Qualificationvalidation and security of computerized or automated processes
Change control system
Investigation of Discrepancies
Inspection Readiness BEC 45
Production System
Drug Inspection Target
Trainingqualification of personnel
Complete batch production documentation
Process validation
Production time limits
In-process testingexamination
Change Control System
Investigation of discrepancies
Inspection Readiness BEC 46
Packaging and Labeling
Inspection Target
Training
Acceptance operations
Control of materials to prevent mix-ups
Accountability
Packaginglabeling records
Line separation
Line clearance inspection and documentation
Validation of labelingpackaging operations
Investigation of discrepancies
Inspection Readiness BEC 47
Laboratory Control
Inspection Target
Staffing and training
Adequate equipment calibration and maintenance
Reference standards
Adherence to written procedures
Validationverification analytical methods
Analytical records and raw data
Adherence to OOS procedure and timely completion of investigations
Stability testing program and reserve samples
Change control system
Before the Inspection
SOP for Inspection Readiness
Strong Internal AuditAssessment function
ndash Continuous assessment using Risk Management
Criteria
ndash Not autopilot auditing
ndash Same auditorhellip
Perform a full-system audit (Mock) at least one
month prior to inspection
ndash Be sure any necessary CA-PA has been originated 48
Before the Inspection
Educate all your employees on proper inspection
rules and etiquette
Educate your supervisory (all exempt) personnel
on FDA regulations
Inspections and external audits concern to all
employees not only those from Quality Dept
Promote the participation on the internal audit
cadre
Certify internal auditors
49
Before the Inspection
A few words about Internal Audit
Internal Audits are the most powerful tool to
avoid inspection surprises
If the inspector discovers something why your
internal auditor did not
The same auditor auditing several consecutive
years the same area is not effective
An effective internal audit function is priceless
50
During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

Inspection Readiness BEC 45
Production System
Drug Inspection Target
Trainingqualification of personnel
Complete batch production documentation
Process validation
Production time limits
In-process testingexamination
Change Control System
Investigation of discrepancies
Inspection Readiness BEC 46
Packaging and Labeling
Inspection Target
Training
Acceptance operations
Control of materials to prevent mix-ups
Accountability
Packaginglabeling records
Line separation
Line clearance inspection and documentation
Validation of labelingpackaging operations
Investigation of discrepancies
Inspection Readiness BEC 47
Laboratory Control
Inspection Target
Staffing and training
Adequate equipment calibration and maintenance
Reference standards
Adherence to written procedures
Validationverification analytical methods
Analytical records and raw data
Adherence to OOS procedure and timely completion of investigations
Stability testing program and reserve samples
Change control system
Before the Inspection
SOP for Inspection Readiness
Strong Internal AuditAssessment function
ndash Continuous assessment using Risk Management
Criteria
ndash Not autopilot auditing
ndash Same auditorhellip
Perform a full-system audit (Mock) at least one
month prior to inspection
ndash Be sure any necessary CA-PA has been originated 48
Before the Inspection
Educate all your employees on proper inspection
rules and etiquette
Educate your supervisory (all exempt) personnel
on FDA regulations
Inspections and external audits concern to all
employees not only those from Quality Dept
Promote the participation on the internal audit
cadre
Certify internal auditors
49
Before the Inspection
A few words about Internal Audit
Internal Audits are the most powerful tool to
avoid inspection surprises
If the inspector discovers something why your
internal auditor did not
The same auditor auditing several consecutive
years the same area is not effective
An effective internal audit function is priceless
50
During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

Inspection Readiness BEC 46
Packaging and Labeling
Inspection Target
Training
Acceptance operations
Control of materials to prevent mix-ups
Accountability
Packaginglabeling records
Line separation
Line clearance inspection and documentation
Validation of labelingpackaging operations
Investigation of discrepancies
Inspection Readiness BEC 47
Laboratory Control
Inspection Target
Staffing and training
Adequate equipment calibration and maintenance
Reference standards
Adherence to written procedures
Validationverification analytical methods
Analytical records and raw data
Adherence to OOS procedure and timely completion of investigations
Stability testing program and reserve samples
Change control system
Before the Inspection
SOP for Inspection Readiness
Strong Internal AuditAssessment function
ndash Continuous assessment using Risk Management
Criteria
ndash Not autopilot auditing
ndash Same auditorhellip
Perform a full-system audit (Mock) at least one
month prior to inspection
ndash Be sure any necessary CA-PA has been originated 48
Before the Inspection
Educate all your employees on proper inspection
rules and etiquette
Educate your supervisory (all exempt) personnel
on FDA regulations
Inspections and external audits concern to all
employees not only those from Quality Dept
Promote the participation on the internal audit
cadre
Certify internal auditors
49
Before the Inspection
A few words about Internal Audit
Internal Audits are the most powerful tool to
avoid inspection surprises
If the inspector discovers something why your
internal auditor did not
The same auditor auditing several consecutive
years the same area is not effective
An effective internal audit function is priceless
50
During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

Inspection Readiness BEC 47
Laboratory Control
Inspection Target
Staffing and training
Adequate equipment calibration and maintenance
Reference standards
Adherence to written procedures
Validationverification analytical methods
Analytical records and raw data
Adherence to OOS procedure and timely completion of investigations
Stability testing program and reserve samples
Change control system
Before the Inspection
SOP for Inspection Readiness
Strong Internal AuditAssessment function
ndash Continuous assessment using Risk Management
Criteria
ndash Not autopilot auditing
ndash Same auditorhellip
Perform a full-system audit (Mock) at least one
month prior to inspection
ndash Be sure any necessary CA-PA has been originated 48
Before the Inspection
Educate all your employees on proper inspection
rules and etiquette
Educate your supervisory (all exempt) personnel
on FDA regulations
Inspections and external audits concern to all
employees not only those from Quality Dept
Promote the participation on the internal audit
cadre
Certify internal auditors
49
Before the Inspection
A few words about Internal Audit
Internal Audits are the most powerful tool to
avoid inspection surprises
If the inspector discovers something why your
internal auditor did not
The same auditor auditing several consecutive
years the same area is not effective
An effective internal audit function is priceless
50
During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

Before the Inspection
SOP for Inspection Readiness
Strong Internal AuditAssessment function
ndash Continuous assessment using Risk Management
Criteria
ndash Not autopilot auditing
ndash Same auditorhellip
Perform a full-system audit (Mock) at least one
month prior to inspection
ndash Be sure any necessary CA-PA has been originated 48
Before the Inspection
Educate all your employees on proper inspection
rules and etiquette
Educate your supervisory (all exempt) personnel
on FDA regulations
Inspections and external audits concern to all
employees not only those from Quality Dept
Promote the participation on the internal audit
cadre
Certify internal auditors
49
Before the Inspection
A few words about Internal Audit
Internal Audits are the most powerful tool to
avoid inspection surprises
If the inspector discovers something why your
internal auditor did not
The same auditor auditing several consecutive
years the same area is not effective
An effective internal audit function is priceless
50
During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

Before the Inspection
Educate all your employees on proper inspection
rules and etiquette
Educate your supervisory (all exempt) personnel
on FDA regulations
Inspections and external audits concern to all
employees not only those from Quality Dept
Promote the participation on the internal audit
cadre
Certify internal auditors
49
Before the Inspection
A few words about Internal Audit
Internal Audits are the most powerful tool to
avoid inspection surprises
If the inspector discovers something why your
internal auditor did not
The same auditor auditing several consecutive
years the same area is not effective
An effective internal audit function is priceless
50
During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

Before the Inspection
A few words about Internal Audit
Internal Audits are the most powerful tool to
avoid inspection surprises
If the inspector discovers something why your
internal auditor did not
The same auditor auditing several consecutive
years the same area is not effective
An effective internal audit function is priceless
50
During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

During the Inspection
Use your best resources to assist during
inspection
War room
Managing documents
Photographs etc
Opening meeting
Daily wrap up
Close-our meeting
51
During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

During the Inspection
Answering Inspectorrsquos Questions
Think before your answer
Answer questions accurately and truthfully
Donrsquot be intimidated or defensive
Know your work and be confident
Be professional
If you donrsquot know the answer it is
acceptable to reply that donrsquot know but you
can find out
52
At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

At the end of the Inspection
Do not hesitate to challenge (be polite) the
inspector to explain the basis of citations
Many companies are unwilling to challenge
the inspector out of fear to provoking some
sort of retaliatory response
However FDA may interpret silence as
agreement
Inspection Readiness BEC 53
Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

Data integrity data manipulation and fraud appears to be increasing
Itrsquos occuring in early stages of drug development (ie clinical studies) during commercial manufacturing and in various FDA regulated products
The FDampC Act is a strict liability statute
FDA takes the position that corporations act through the actions of individuals
Part of an FDA Investigatorrsquos job is to document individual responsibility for violations noted during inspections
54
Data Integrity Information that is accurate complete and truthful
Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

Data Integrity and Quality
FDA needs to be able to verify the quality and
integrity of the data during inspections
ndash Data needs to meet ALCOA elements of quality
ndash Attributable ndash data are identified with a specific subject and a specific
observer and recorder (Password audit trail and e-signature)
ndash Legible ndash data are readable and understandable by humans (reports tables
and listings)
ndash Contemporaneous - data are recorded at the time they are generated or
observed (Time stamps and time-limited entry)
ndash Original ndash data are recorded for the first time (Source data)
ndash Accurate ndash data are correct (Calculations algorithms analyses)
55
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62
56
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62
57
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62
58
Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

Train employees on proper data handling
and reporting
Assure the reliability of data reported in
applications and manufacturing records
Emphasize that everyone in the company is
responsible for data integrity 59
What Can Industry Do
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62
FDA ONLINE Resources httpwwwfdagov
60
FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

FDA ONLINE Resources
wwwfdagovora
61
Gracias
wwwcalidadprcom
wwwbecspainslcom
62

Gracias
wwwcalidadprcom
wwwbecspainslcom
62