practical overview of genetics in childhood epilepsies...genes involves in both mild and severe...
TRANSCRIPT

Practical overview of genetics in childhood epilepsies
Dr Gaëtan LescaDepartment of Medical Genetics, Lyon
University Hospital, France

Gene discovery in Mendelian epilepsies (Helbig et al., 2016)

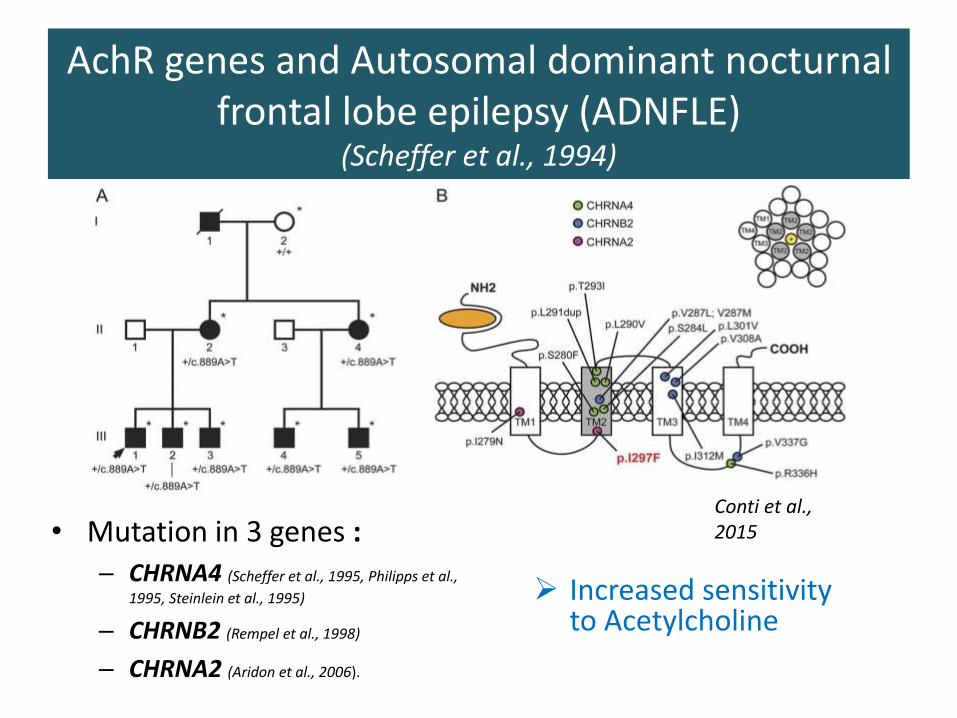
AchR genes and Autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE)
(Scheffer et al., 1994)
• Mutation in 3 genes :– CHRNA4 (Scheffer et al., 1995, Philipps et al.,
1995, Steinlein et al., 1995)
– CHRNB2 (Rempel et al., 1998)
– CHRNA2 (Aridon et al., 2006).
AchR genes and Autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE)
(Scheffer et al., 1994)
• Mutation in 3 genes :– CHRNA4 (Scheffer et al., 1995, Philipps et al.,
1995, Steinlein et al., 1995)
– CHRNB2 (Rempel et al., 1998)
– CHRNA2 (Aridon et al., 2006).
Conti et al., 2015
➢ Increased sensitivity to Acetylcholine

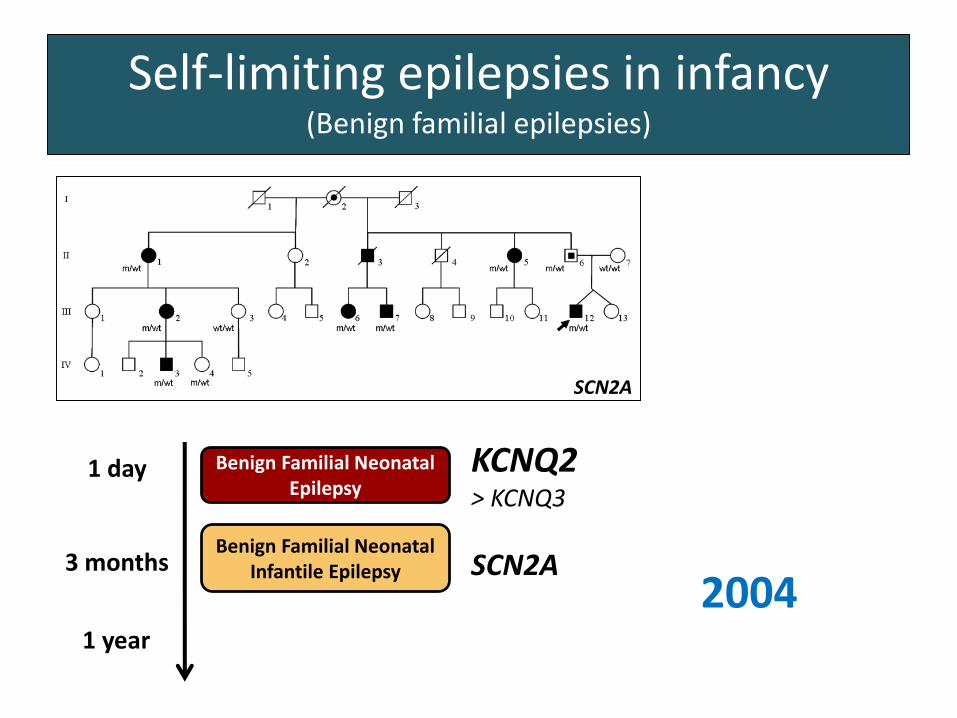
Self-limiting epilepsies in infancy(Benign familial epilepsies)
Lauxmann et al., 2013

Self-limiting epilepsies in infancy(Benign familial epilepsies)
Benign Familial Neonatal Epilepsy
1 day
3 months
1 year
KCNQ2> KCNQ3
SCN2A
1998
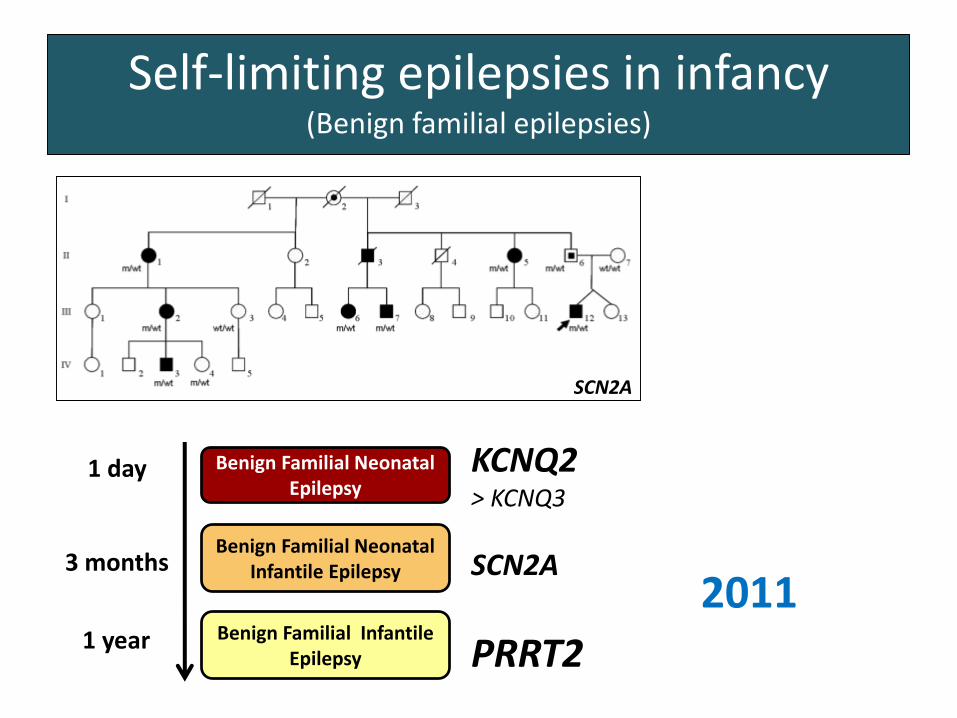
Self-limiting epilepsies in infancy(Benign familial epilepsies)
Benign Familial Neonatal Epilepsy
Benign Familial Neonatal Infantile Epilepsy
1 day
3 months
1 year
KCNQ2> KCNQ3
SCN2A
SCN2A
2004
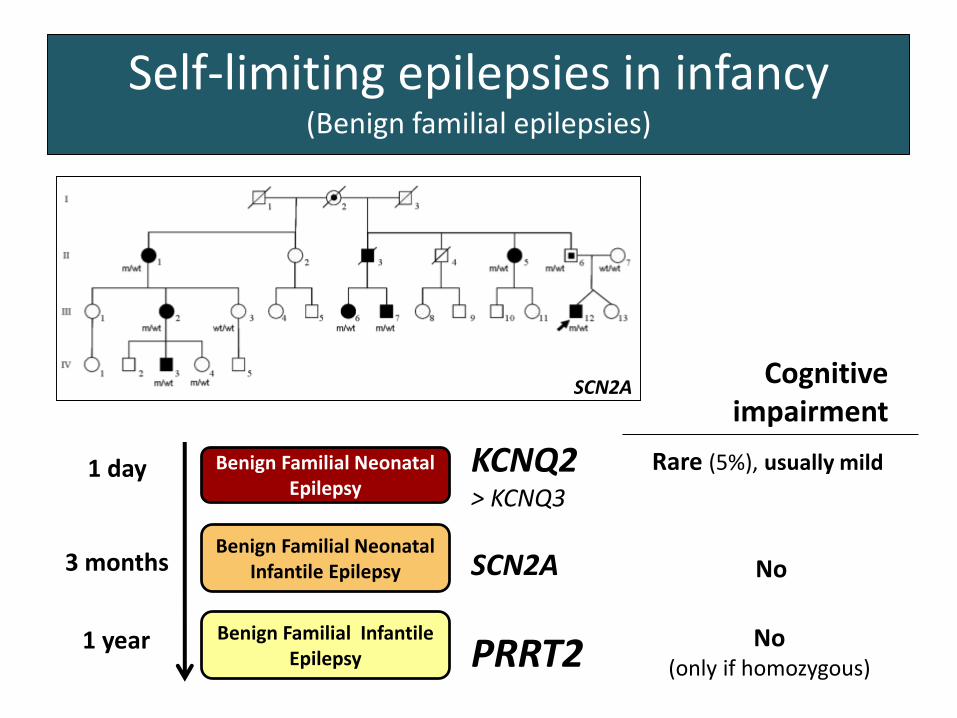
Self-limiting epilepsies in infancy(Benign familial epilepsies)
Benign Familial Neonatal Epilepsy
Benign Familial Neonatal Infantile Epilepsy
Benign Familial Infantile Epilepsy
1 day
3 months
1 year
KCNQ2> KCNQ3
SCN2A
PRRT2
SCN2A
2011
Self-limiting epilepsies in infancy(Benign familial epilepsies)
Benign Familial Neonatal Epilepsy
Benign Familial Neonatal Infantile Epilepsy
Benign Familial Infantile Epilepsy
1 day
3 months
1 year
KCNQ2> KCNQ3
SCN2A
PRRT2
Cognitive impairment
No(only if homozygous)
No
Rare (5%), usually mild
SCN2A

Loss-of-function mutations
Missense mutations
KCNQ2-related epilepsies(Millichap et al., 2016)
Self-limiting epilepsy(BFNE > BFNIE, BFIE)

KCNQ2-related epilepsies(Millichap et al., 2016)
Self-limiting epilepsy(BFNE > BFNIE, BFIE)
Early-onset epileptic encephalopathy (EOEE)
Loss-of-function mutations
Missense mutations Missense mutations with dominant-negative effect

SCN2A-related disorders (Sanders et al., 2018)
Lauxmann et al., 2013
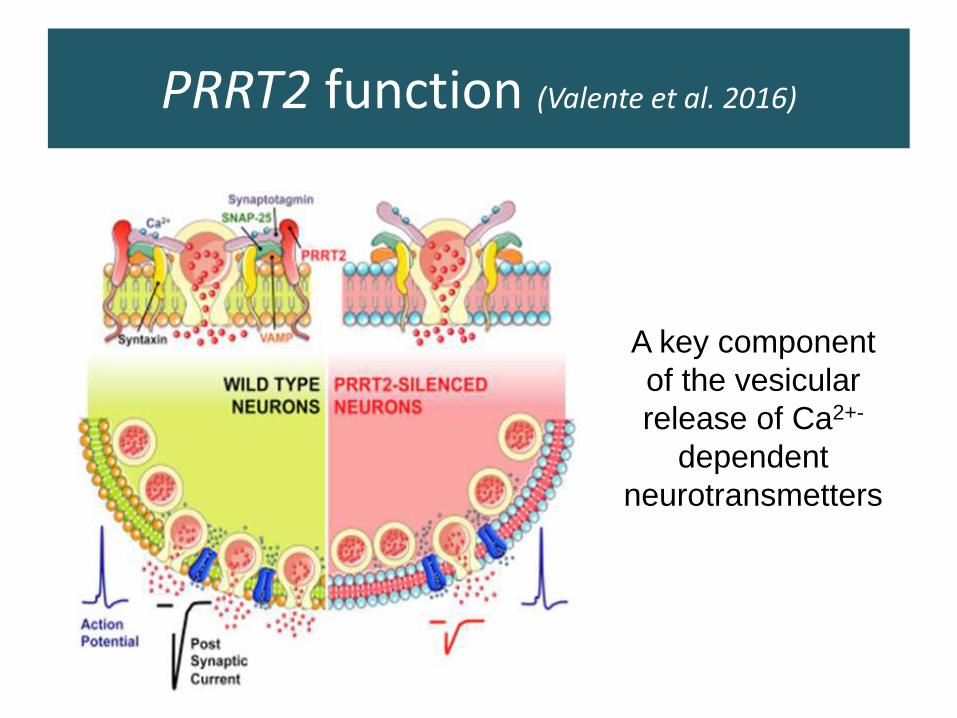
PRRT2 function (Valente et al. 2016)
A key component
of the vesicular
release of Ca2+-
dependent
neurotransmetters
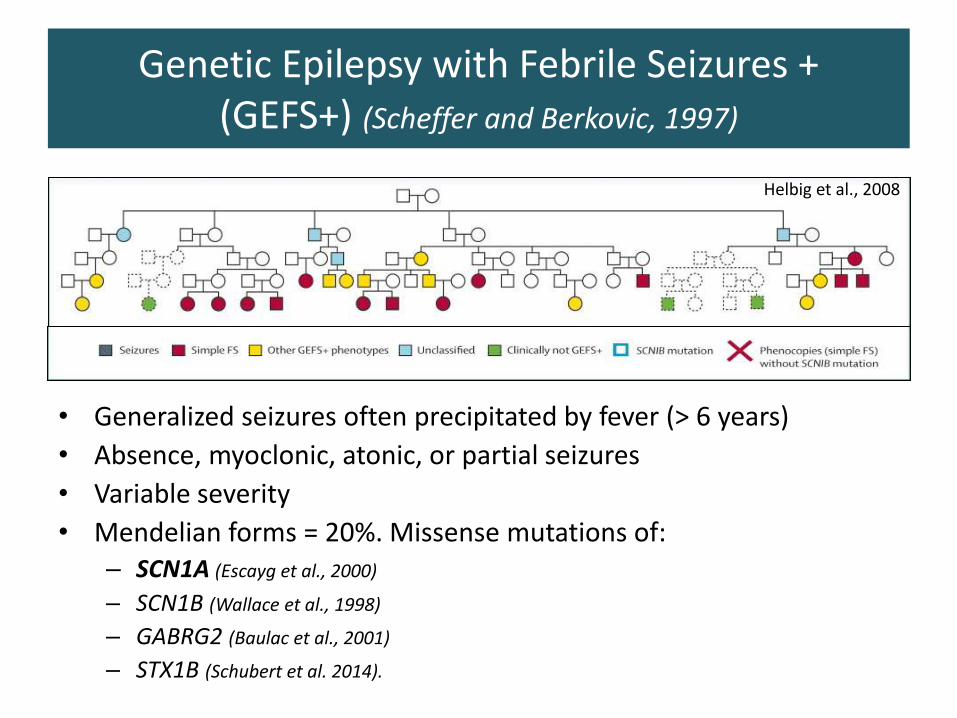
• Generalized seizures often precipitated by fever (> 6 years)
• Absence, myoclonic, atonic, or partial seizures
• Variable severity
• Mendelian forms = 20%. Missense mutations of:– SCN1A (Escayg et al., 2000)
– SCN1B (Wallace et al., 1998)
– GABRG2 (Baulac et al., 2001)
– STX1B (Schubert et al. 2014).
Helbig et al., 2008
Genetic Epilepsy with Febrile Seizures +(GEFS+) (Scheffer and Berkovic, 1997)

Dravet syndrome(Dravet et al., 1992)
• Onset < 1 year
• Seizures often triggered by fever
• Prolonged seizures, status epilepticus
• Drug refractory
• Progressive motor and cognitive delay, ataxia
➢Loss-of-function heterozygous de novo mutations of SCN1A in 80% of patients (Sugawara et al., 2002)

The unified loss-of-function hypothesis for Nav1.1 genetic epilepsies (Catteral et al., 2010)
Nav1.1 expressed in GABAergicinhibitoryneurons(including
Purkinje cells)
SCN1A mutations
Clinical continuum
Dravet

CDKL5 encephalopathy (Kalscheuer et al., 2003)
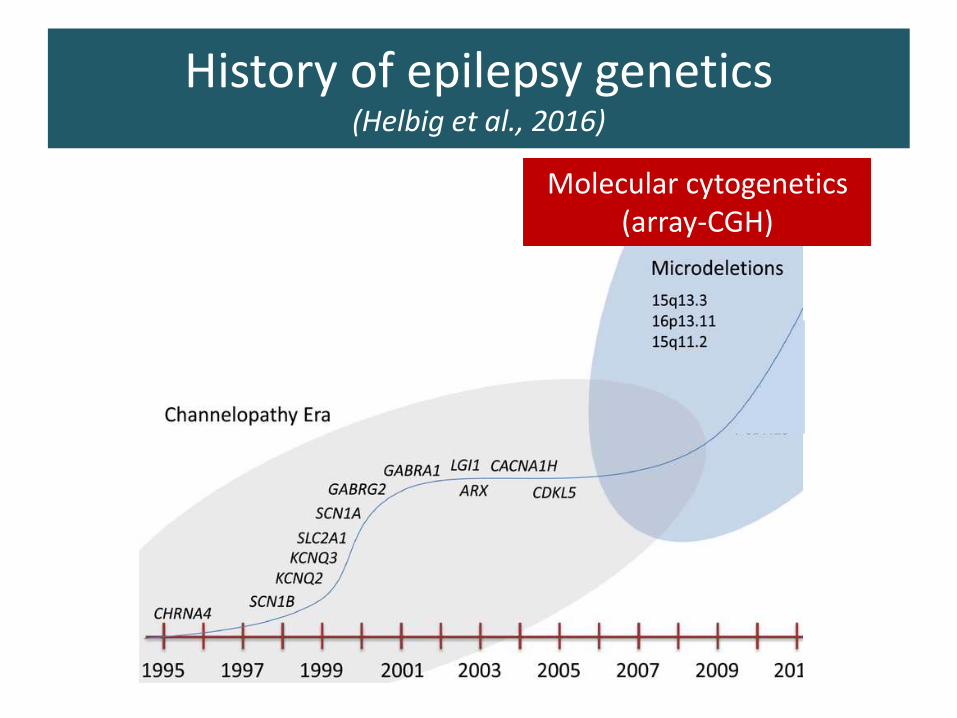
History of epilepsy genetics(Helbig et al., 2016)
Molecular cytogenetics(array-CGH)
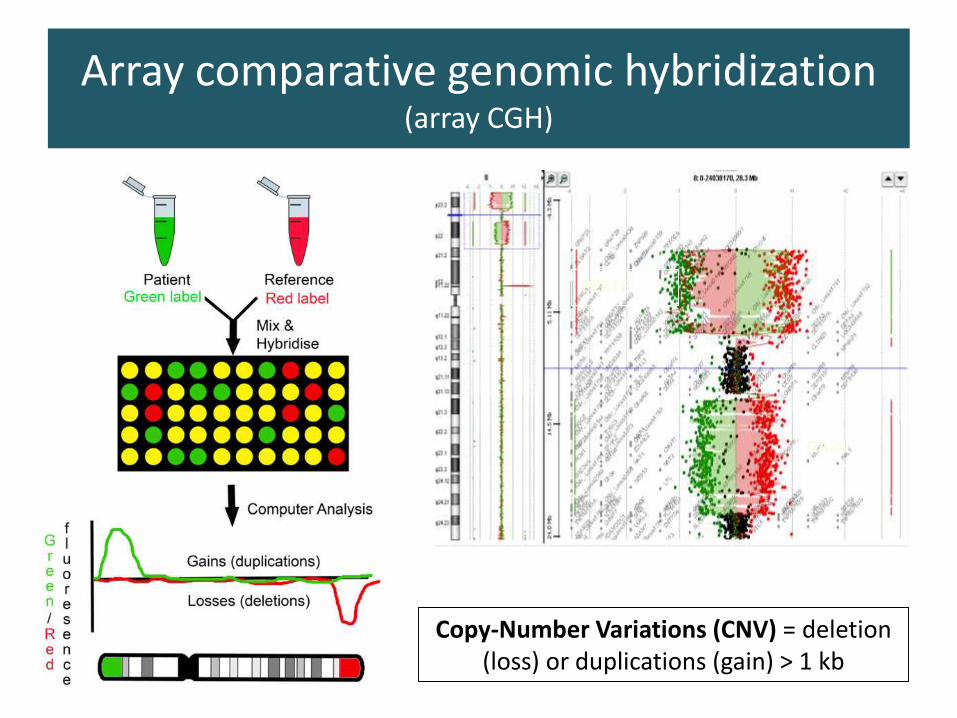
Array comparative genomic hybridization (array CGH)
PERTE
GAIN
Copy-Number Variations (CNV) = deletion(loss) or duplications (gain) > 1 kb

CNVs and epilepsy
1. Rare microdeletion syndromes:
– Angelman, del 1p36…
– Many other very rare ones
2. Recurrent CNVs associated with idiopathic epilepsies
3. CNVs that led to gene identification: STXBP1, PCDH19, GRIN2A…

•15q11.2 (de Kovel et al.,
2010)
•15q13.3 (Helbig et al.,
2009; Dibbens et al., Genet 2009)
•16p13.11 (de Kovel et al.,
2010; Heinzen et al., 2010)
Found in 3% of IGE
CNVs and idiopathic generalized epilepsy

Recurrent CNVs in IGE
• Female, 2 years: absence epilepsy with falls -> remission (stop valproate)
• 6 years: childhood absence epilepsy -> remission (stop valproate)• 13 years: myoclonia in the morning, 2 TCGS, no cognitive
impairment• No family history of epilepsy

Phenotypical variability associated with the recurrent 15q13.3 deletion
Intellectual deficiency, autism, generalized
epilepsy(0.3% patients,
0% controls)
Schizophrenia(0.2% patients,
0,01% controls)
Generalized epilepsy
(1% patients, <0,02%
controls)
Odds ratio : 68 [29 – 181]
Often inherited from an unaffected parent
= risk factor
15q13.3

Two major categories of genetic factors
Strong influence of a major gene
Mendelian epilepsies
--------
Mutation = high risk of disease

Two major categories of genetic factors
Strong influence of a major gene
Mendelian epilepsies
--------
Mutation = high risk of disease
Limited influence of a given gene
Multifactorial epilepsies
--------
Risk factors, neither necessary nor sufficient

• Mendelian inheritance rare (< 5%) -> polygenic ?
• Association studies in common epilepsies (IGE, FNLE) :
– Candidate genes or genome-wide asociation studies
– Non reproductible results: false positive, underpowered ?
• Large cohort studies :
– Epi4K/EPGP. Lancet neurol 2017: enrichment in ultrarare variants of some known genes (SCN1A, KCNQ2...)
– May et al. Lancet Neurol 2018: Enrichment in rare variants in GABAr genes
– EPi25K : ongoing
Genetic basis for common epilepsies ?

CNVs and epilepsy
1. Rare microdeletion syndromes:
– Angelman, del 1p36…
– Many other very rare ones
2. Recurrent CNVs associated with idiopathic epilepsies
3. CNVs that led to gene identification: STXBP1, PCDH19, GRIN2A…
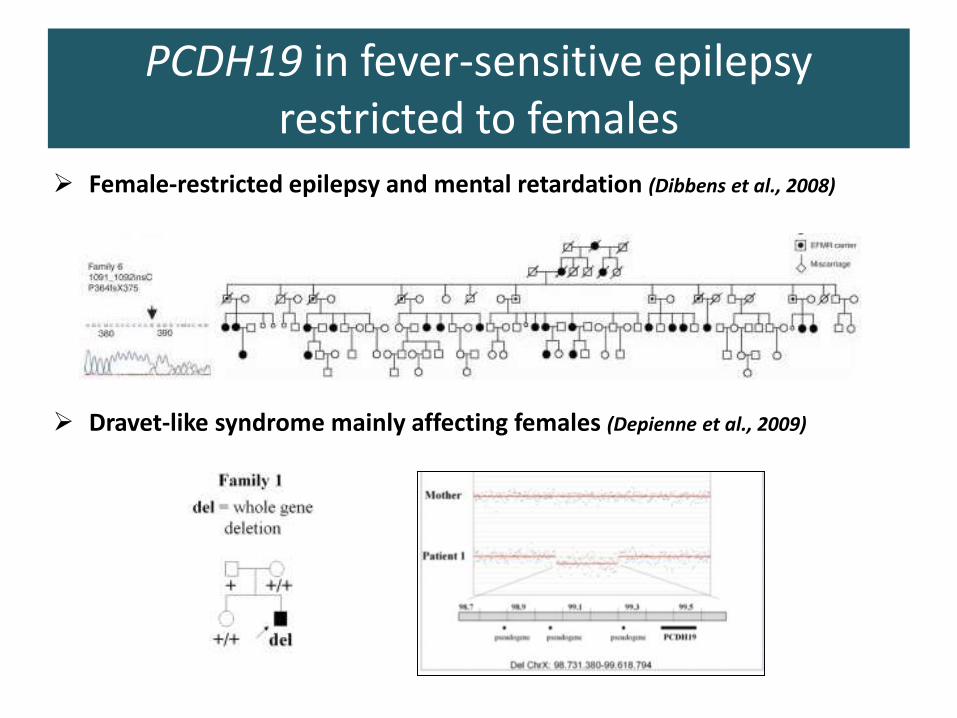
PCDH19 in fever-sensitive epilepsy restricted to females
➢ Female-restricted epilepsy and mental retardation (Dibbens et al., 2008)
➢ Dravet-like syndrome mainly affecting females (Depienne et al., 2009)
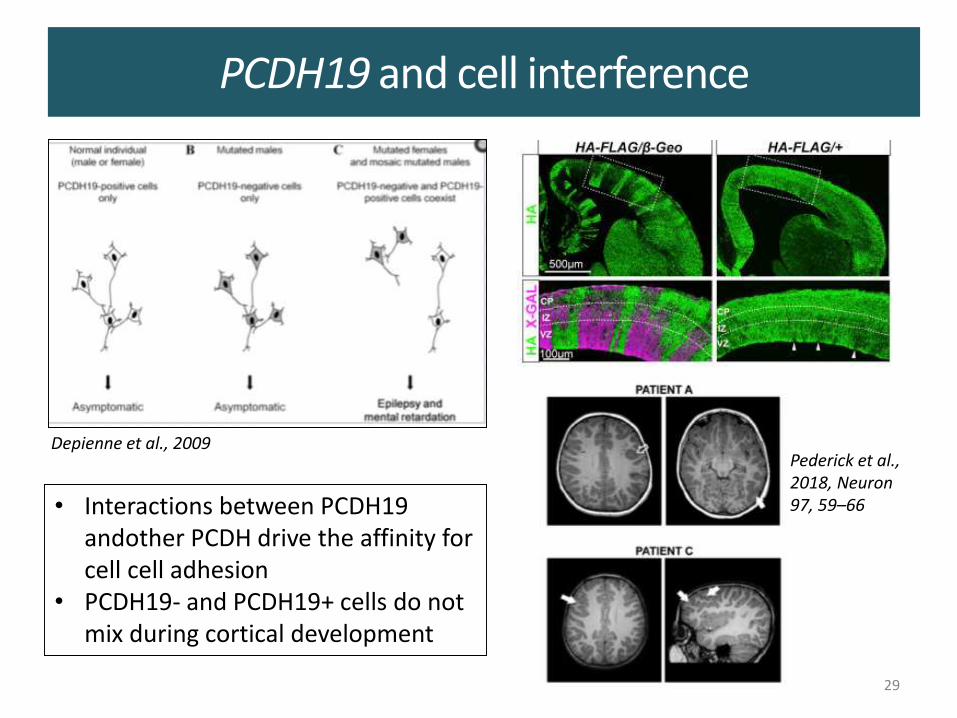
PCDH19 and cell interference
29
Depienne et al., 2009Pederick et al., 2018, Neuron 97, 59–66• Interactions between PCDH19
andother PCDH drive the affinity for cell cell adhesion
• PCDH19- and PCDH19+ cells do not mix during cortical development
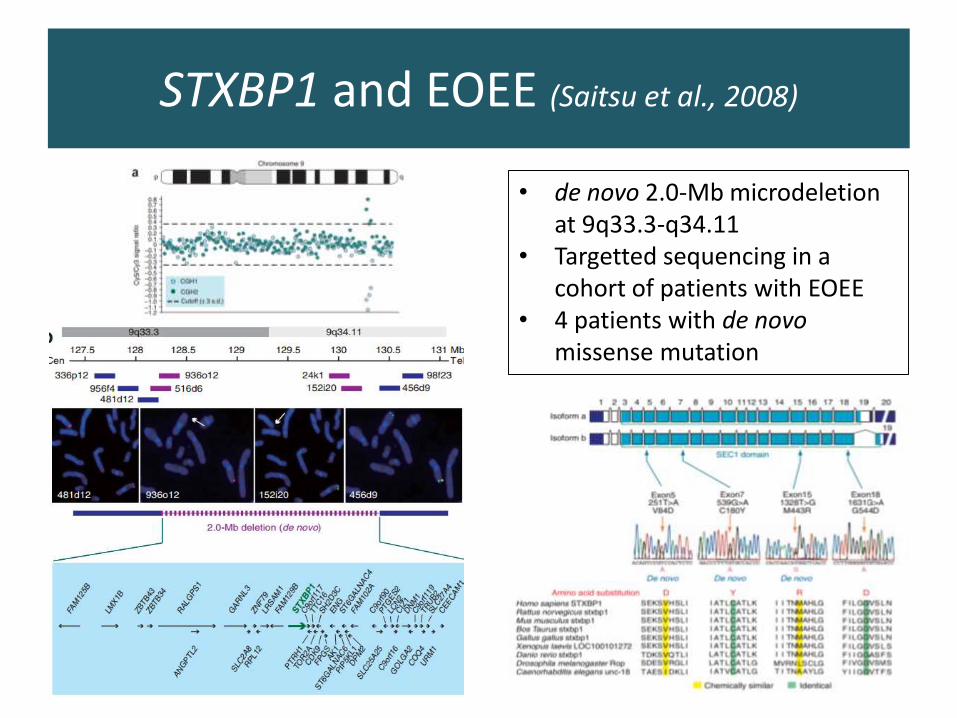
STXBP1 and EOEE (Saitsu et al., 2008)
• de novo 2.0-Mb microdeletion at 9q33.3-q34.11
• Targetted sequencing in a cohort of patients with EOEE
• 4 patients with de novomissense mutation

STXBP1 encephalopathy
• De novo mutations of STXBP1 = frequent cause of:
– EOEE with or without suppression-burst
– Infantile spasms/West, Lennox-Gastaut
• Moderate to severe intellectual disability
• Behavior disorders, autistic features
• Movement disorders: ataxia, dyskinesia,choreoathetosis

Deletions of 16p13 including GRIN2A in patients with intellectual disability, various dysmorphic features, and seizure disorders of the Rolandic region
(Reutlingen et al., Epilepsia 2010).
Microdeletions involving GRIN2A
Lesca et al., 2013

GRIN2A and epilepsia-aphasia(Lesca et al., 2013; Carvill et al., 2013; Lemke e al., 2013)
NTD = N-terminal domain, S1-2 = ligand binding sites, CTD = C-terminal domain,
LBD = ligand-binding domain
Subunit 2A of the glutamate
NMDA receptor

GRIN2A-related disorders: genotype and functional consequence predict phenotype (Strehlow et al., 2019)
Truncating variants
Missense variants

Massive parrarelsequencing era
History of epilepsy genetics(Helbig et al., 2016)

Massive parrarelsequencing era
History of epilepsy genetics(Helbig et al., 2016)
Epileptic encephalopathies
> 100 genes

Massive parallel sequencing(Next-generation sequencing)
37
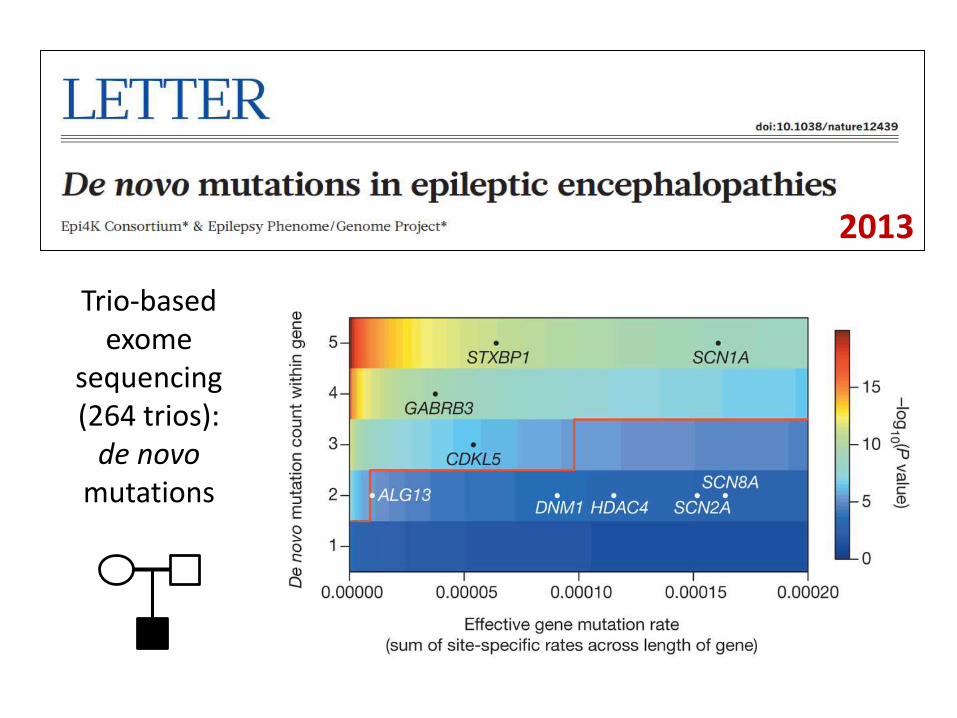
Trio-based exome
sequencing(264 trios):
de novo mutations
2013

Trio-based exome
sequencing(264 trios):
de novo mutations
2013

SCN8A developmental and epileptic encephalopathy(Gardella et al., 2018)
Gain of function (Meisler et al., 2016)
• Median age at onset : 4 months
• Prolonged focal seizures,
cyanosis, vegetative semiology
followed by clonias
• Developmental slowing,
pyramidal/extrapyramidal signs,
movement disorders, cortical
blindness
• EEG: background deterioration,
epileptiform abnormalities
• Sodium blockers more efficient

SCN8A-related EE: two modes of onset(Denis et al., 2019)
Sudden
• Onset < 6 months
• Frequent seizures: tonic seizures or spasms
• Normal inter-ictal EEG (no supression burst)
• Evolve to GTCS and tonic seizures
Progressive
• Progressive onset or unclear
• Rare and subltle seizures: jerks, myoclonia or tremor
• Normal interictal EEG at onset
• Developmental slowing or arrest

Benign infantile seizures and paroxysmal dyskinesia caused by an SCN8A mutation
(Gardella et al., 2016)
• Tonic or GTCS
• May persist >2y
• Normal psychomotor development
• Drug-sensitive or dependant

Spectrum of SCN8A related epilepsy
➢ Efficiency of sodium blockers ?
Severe early onset DEE, high
mortality
Mild phenotype, seizures in
childhood normal cognition
Moderate DEE, often drug-
resistant seizures
Treatable seizures, mild ID
Loss of function
Gain of function
Adapted from Johannesen et al., 2019

Genes involves in both mild and severe epilepsy phenotypes
Gene Mild phenotype Severe phenotype
SCN1A GEFS+ Dravet
SCN1B GEFS+ Dravet (homozygous variants)
SCN2A Benign Familial neonatal-infantile Epilepsy EOEE
SCN8A Benign familial infantile epilepsy and paroxysmal choreoathetosis
EOEE
KCNQ2 Benign familial neonatal epilepsy EOEE
SLC2A1 Refractory generalized or focal epilepsy (early absence epilepsy)
De Vivo
GABRA1 Idiopathic generalized epilepsy EOEE
GABRG2 GEFS+ EOEE
GRIN2A Epilepsy-aphasia syndrome EOEE
Modified from Helbig and Abou Tayoum, 2016

Genetic heterogeneity in epileptic syndromes

Genetic heterogeneity in epileptic syndromes
15-20% with exome sequencing!
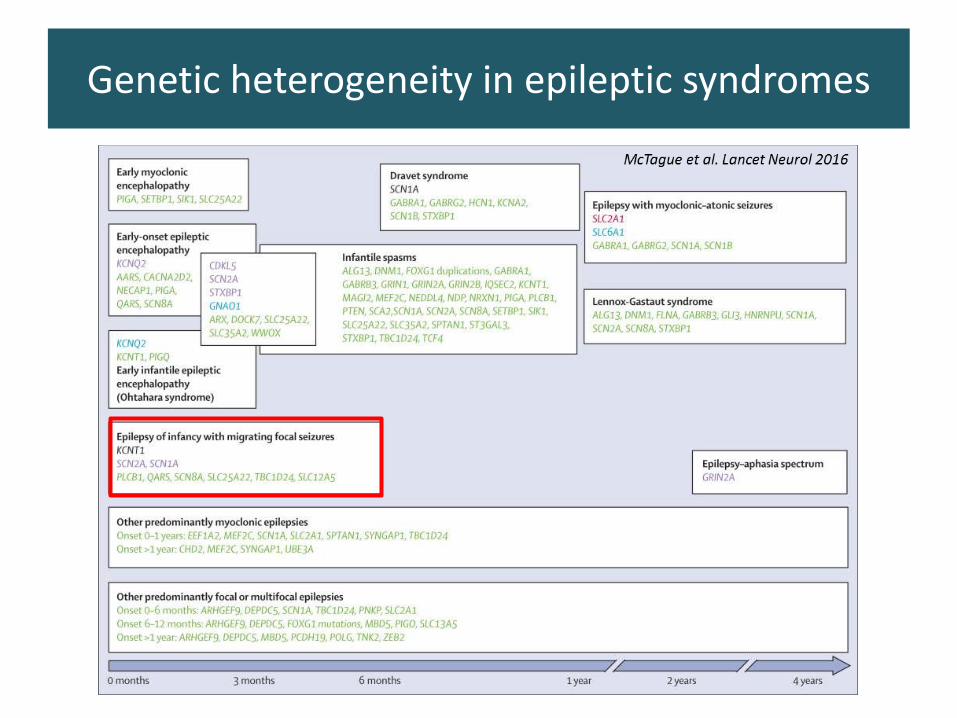
Genetic heterogeneity in epileptic syndromes
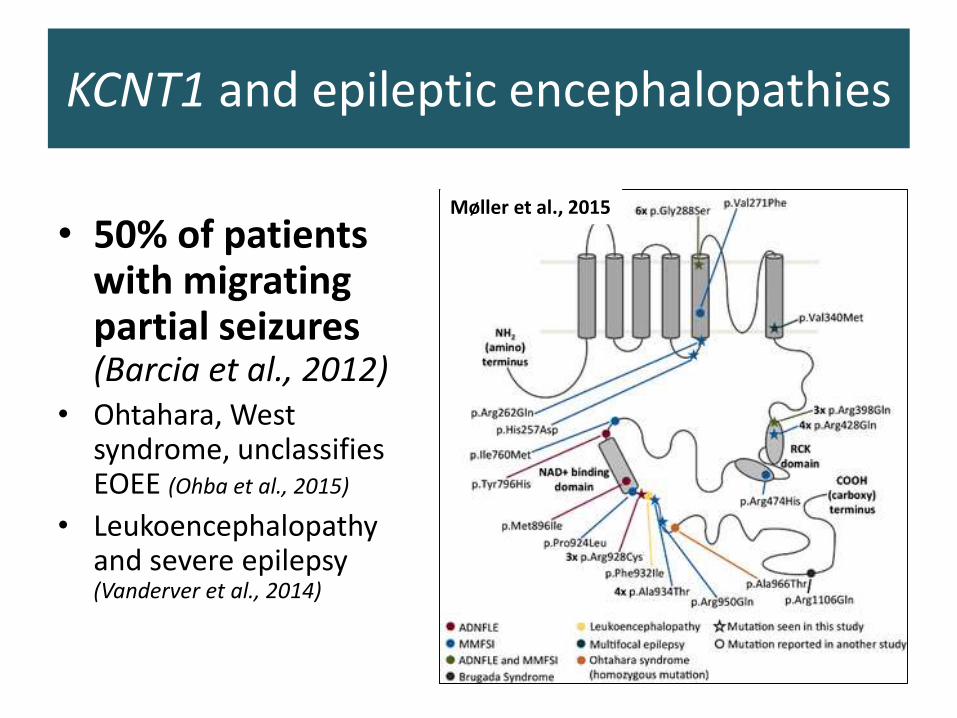
KCNT1 and epileptic encephalopathies
• 50% of patients with migrating partial seizures (Barcia et al., 2012)
• Ohtahara, West syndrome, unclassifiesEOEE (Ohba et al., 2015)
• Leukoencephalopathy and severe epilepsy (Vanderver et al., 2014)
Møller et al., 2015

Delineation of new epileptic syndromes
• Rare disorders with few patients in each center
• Initial description in a few patients on a rather biased manner
• Genetic findings as the common denominator
• Improve delineating :
– The electro-clinical profile(s) relateto a given gene
– Some novel electro-clinical syndromes
• Towards a gene centered classification of the epilepsies ?
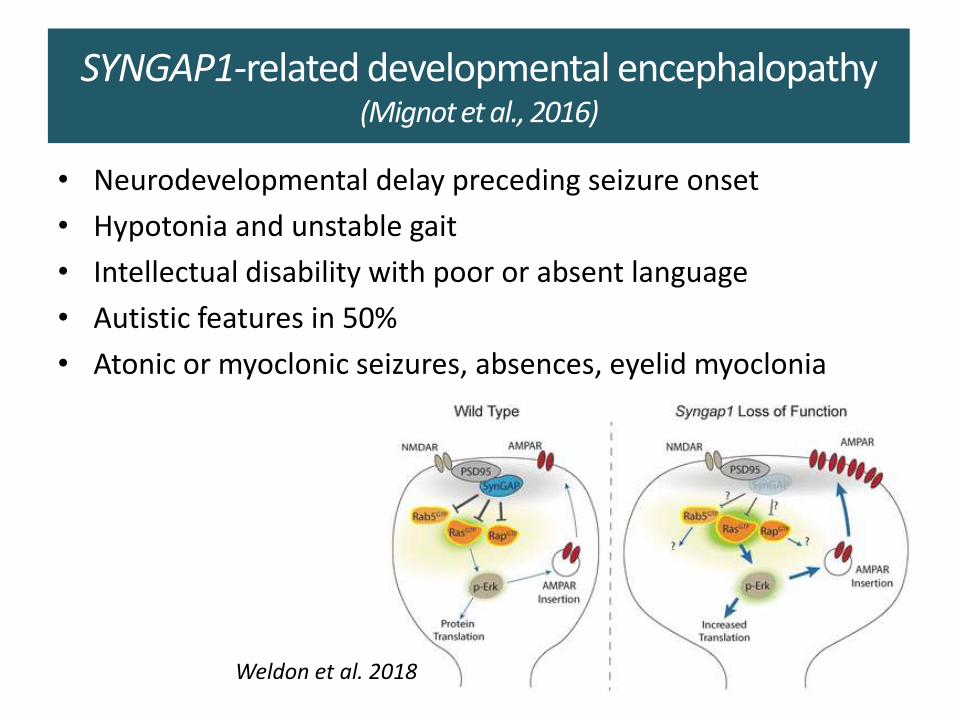
SYNGAP1-related developmental encephalopathy (Mignot et al., 2016)
• Neurodevelopmental delay preceding seizure onset
• Hypotonia and unstable gait
• Intellectual disability with poor or absent language
• Autistic features in 50%
• Atonic or myoclonic seizures, absences, eyelid myoclonia
Weldon et al. 2018
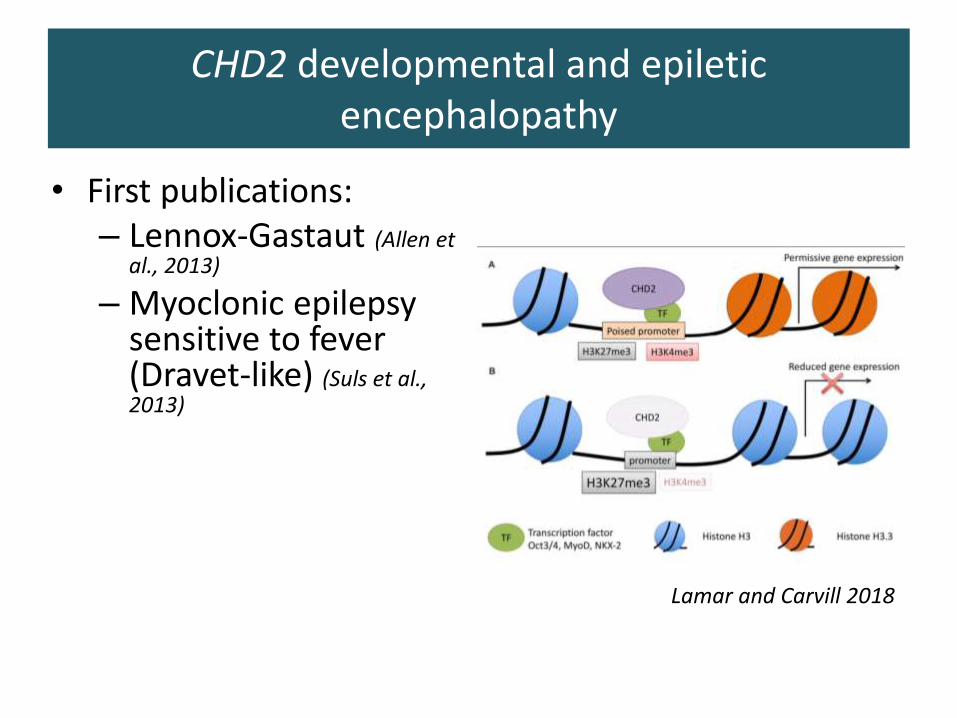
• First publications:– Lennox-Gastaut (Allen et
al., 2013)
– Myoclonic epilepsy sensitive to fever (Dravet-like) (Suls et al., 2013)
Lamar and Carvill 2018
CHD2 developmental and epiletic encephalopathy

• First publications:– Lennox-Gastaut (Allen et
al., 2013)
– Myoclonic epilepsy sensitive to fever (Dravet-like) (Suls et al., 2013)
• Emerging phenotype: – Febrile seizures– Absences, myoclonias,
tonic clonic seizures– Mild to moderate ID– Pyschotic features
Lamar and Carvill 2018
CHD2 developmental and epiletic encephalopathy

SLC6A13p25.3 microdeletion of GABA transporters SLC6A1 and SLC6A11 results in intellectual disability, epilepsy and
stereotypic behaviour (Dikow et al., 2014)
• Reuptake of GABA from the inhibitory synapses
• Mutation -> reduction of GABA synaptic signalling

SLC6A1-related DEE(Johannesen et al., 2018)
• Mild to moderate ID
• Epilepsy > 90%
• Ataxia and psychitaric troubles in 20%
• Mean age at seizure onset: 3.7y
• Atypical absences, atonic, myoclonia, TCGS
• Rythmic activity 3-4 Hz, occipital predominance
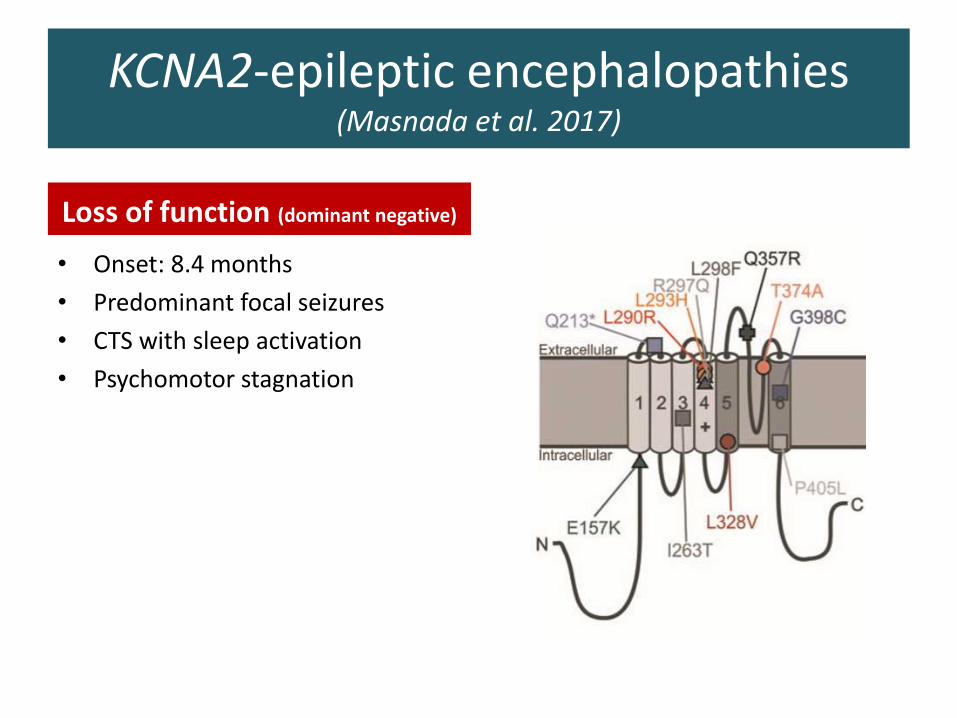
KCNA2-epileptic encephalopathies (Masnada et al. 2017)
Loss of function (dominant negative)
• Onset: 8.4 months
• Predominant focal seizures
• CTS with sleep activation
• Psychomotor stagnation

KCNA2-epileptic encephalopathies (Masnada et al. 2017)
Loss of function (dominant negative)
• Onset: 8.4 months
• Predominant focal seizures
• CTS with sleep activation
• Psychomotor stagnation
Gain of function
• Onset: 8.7 months
• Febrile seizures
• Predominant generalized seizures
• More severe epilepsy
• Cognitive impairment, movement disorders

KCNA2-epileptic encephalopathies (Masnada et al. 2017)
Loss of function (dominant negative)
• Onset: 8.4 months
• Predominant focal seizures
• CTS with sleep activation
• Psychomotor stagnation
Gain of function
• Onset: 8.7 months
• Febrile seizures
• Predominant generalized seizures
• More severe epilepsy
• Cognitive impairment, movement disorders
Gain and loss of function
• Onset: 2.1 months
• Generalized and focal seizures
• More severe developmental impairment
• Cerebellar atrophy
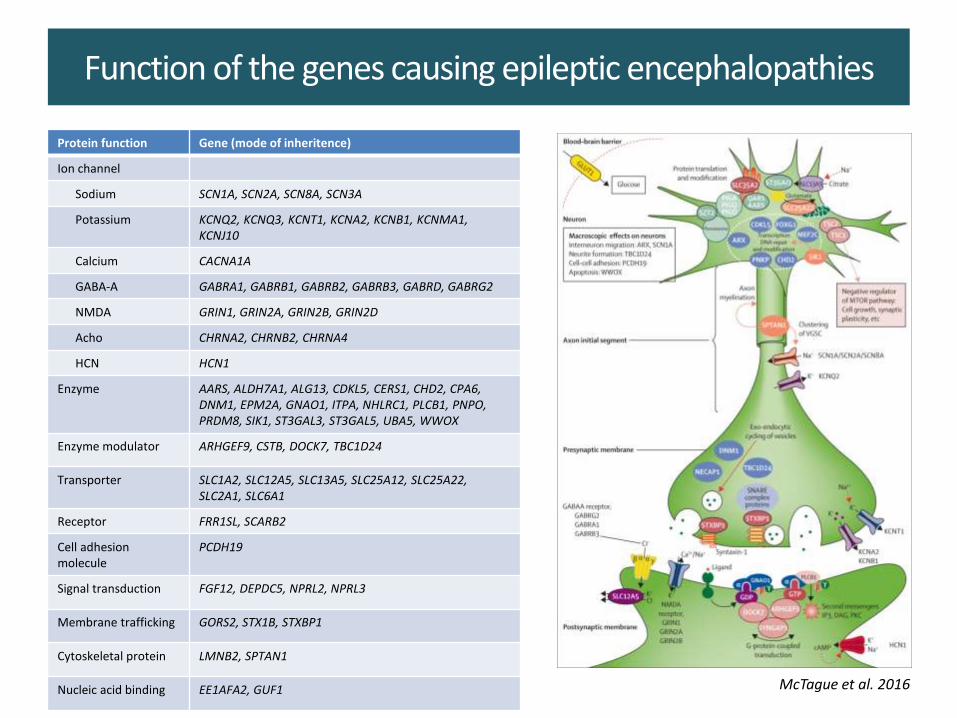
Function of the genes causing epileptic encephalopathies
Protein function Gene (mode of inheritence)
Ion channel
Sodium SCN1A, SCN2A, SCN8A, SCN3A
Potassium KCNQ2, KCNQ3, KCNT1, KCNA2, KCNB1, KCNMA1, KCNJ10
Calcium CACNA1A
GABA-A GABRA1, GABRB1, GABRB2, GABRB3, GABRD, GABRG2
NMDA GRIN1, GRIN2A, GRIN2B, GRIN2D
Acho CHRNA2, CHRNB2, CHRNA4
HCN HCN1
Enzyme AARS, ALDH7A1, ALG13, CDKL5, CERS1, CHD2, CPA6, DNM1, EPM2A, GNAO1, ITPA, NHLRC1, PLCB1, PNPO, PRDM8, SIK1, ST3GAL3, ST3GAL5, UBA5, WWOX
Enzyme modulator ARHGEF9, CSTB, DOCK7, TBC1D24
Transporter SLC1A2, SLC12A5, SLC13A5, SLC25A12, SLC25A22, SLC2A1, SLC6A1
Receptor FRR1SL, SCARB2
Cell adhesion molecule
PCDH19
Signal transduction FGF12, DEPDC5, NPRL2, NPRL3
Membrane trafficking GORS2, STX1B, STXBP1
Cytoskeletal protein LMNB2, SPTAN1
Nucleic acid binding EE1AFA2, GUF1 McTague et al. 2016
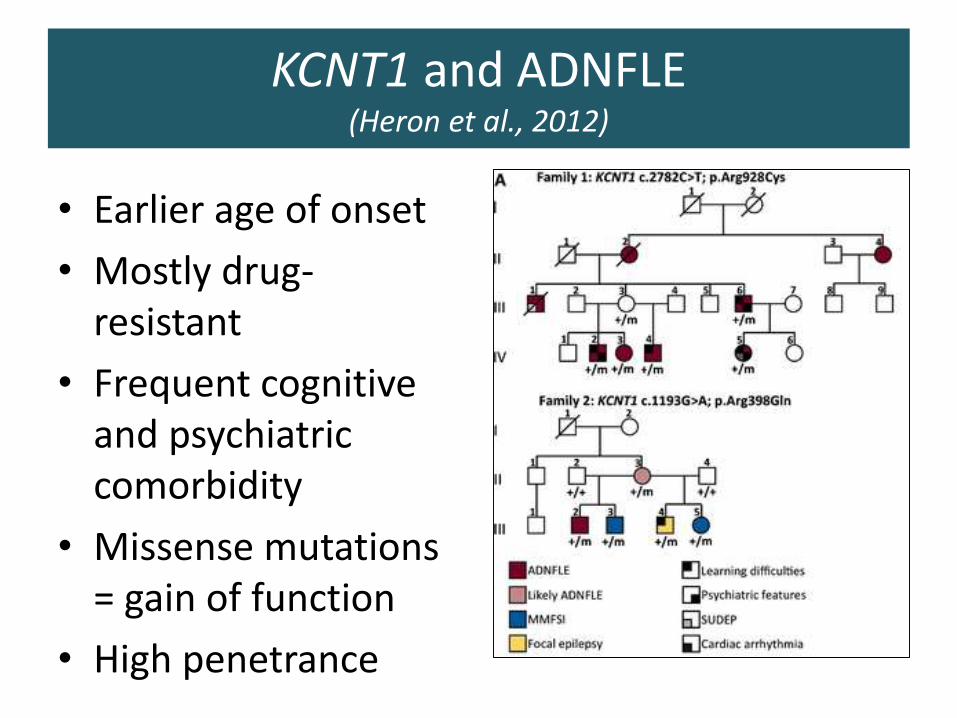
KCNT1 and ADNFLE(Heron et al., 2012)
• Earlier age of onset
• Mostly drug-resistant
• Frequent cognitive and psychiatric comorbidity
• Missense mutations = gain of function
• High penetrance

Familial focal epilepsy with variable foci (FFEVF) (Dibbens et al.; 2013, Ishida et al., 2013)
• Variable age of onset
• +/- cognitive and psychiatric involvement
• Intra familial varability
• Loss of function mutations in: DEPDC5, NPRL2, or NPRL3
• Low penetrance (60%)

Epilepsy and mutations of gene belonging to the GATOR1 Complex (Ricos et al., 2015)
GATOR1 = GTPase-activating protein Activity Towards RAGs complex 1)

Germline DEPDC5 mutations and lesionnal focal epilepsies
• FCD type II• Heterotopia• Megalencephaly• 2nd hit = somatic
mutation in dysplastic tissue ? (Baulac et al., 2015)
Scheffer et al., 2014
Role of Somatic mutations in focal epilepsies ?
Koh and Lee, 2018

Role of Somatic mutations in focal epilepsies ?
Koh and Lee, 2018

Role of Somatic mutations in focal epilepsies ?
Koh and Lee, 2018
100-800x
> 2500x
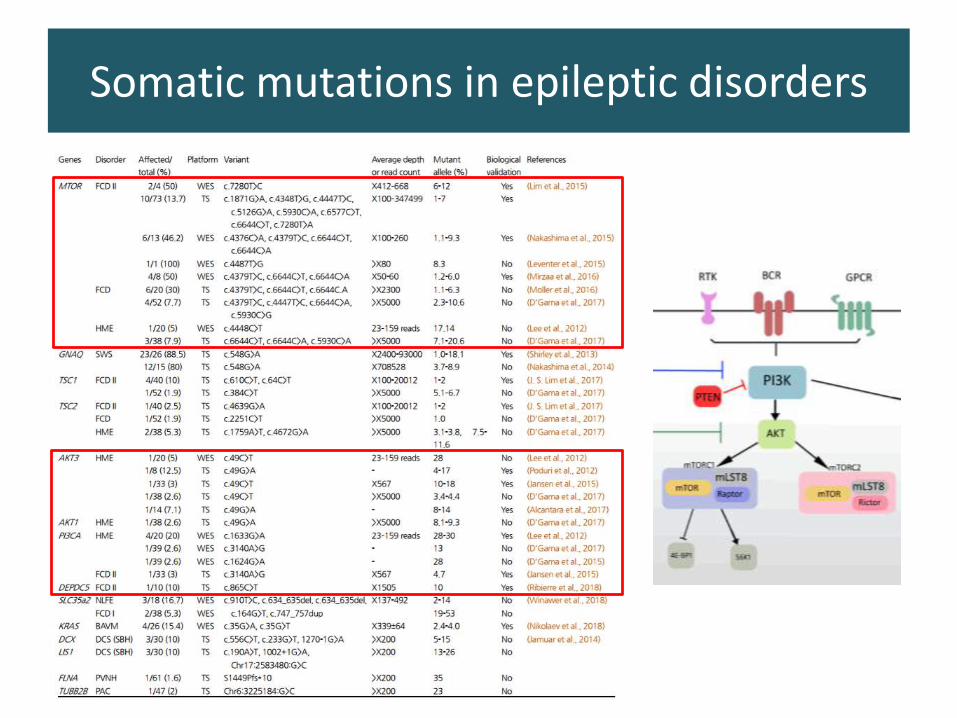
Somatic mutations in epileptic disorders

Department of medical genetics,team for molecular diagnosis of
epileptic disorders
Pediatric neurology departmentDorothée Ville
Anne-Lise PoulatIsabelle Sabatier
Maryline CarneiroLaurence Lion-François
Vincent des Portes
Pediatric epilepsy departmentJulitta de BellesciseEleni PanagiotakakiPascale Keo-KosalJoseph ToulouseKarine Ostrowsky
Alexis Arzimanoglou