plc -erk axis in vegf-induced eif4e phosphorylation and ... · plc -erk axis in vegf-induced eif4e...
TRANSCRIPT

1
PLC -Erk Axis in VEGF-induced eIF4E Phosphorylation and Protein Synthesis in
Renal Epithelial Cells
Meenalakshmi M Mariappan§, Duraisamy Senthil
§, Kavithalakshmi S. Natarajan
§,
Goutam Ghosh Choudhury§#
, and Balakuntalam S Kasinath§#
#South Texas Veterans Healthcare System, GRECC, O’Brien Kidney Research Center,
§Department of Medicine, University of Texas Health Science Center, San Antonio, TX
Running Title: VEGF-induced protein synthesis mediated by PLC and Erk
Address correspondence to: B. S. Kasinath, MD
Department of Medicine
Mail Code 7882
University of Texas Health Science Center
7703 Floyd Curl Drive
San Antonio, TX 78248
Phone: 210-567-4707
Fax: 210-567-4712
Email: [email protected]
JBC Papers in Press. Published on May 26, 2005 as Manuscript M504861200
Copyright 2005 by The American Society for Biochemistry and Molecular Biology, Inc.
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

2
PLC -ERK AXIS IN VEGF-INDUCED EIF4E PHOSPHORYLATION AND
PROTEIN SYNTHESIS IN RENAL EPITHELIAL CELLSMeenalakshmi M Mariappan
§, Duraisamy Senthil
§, Kavithalakshmi S. Natarajan
§,
Goutam Ghosh Choudhury§#
, and Balakuntalam S. Kasinath§#
#South Texas Veterans Healthcare System, GRECC, O’Brien Kidney Research Center,
§Department of Medicine, University of Texas Health Science Center, San Antonio, TX
VEGF increases protein synthesis and induces
hypertrophy in renal tubular epithelial cells
(Senthil et al., 2003). We examined the role of
Erk1/2 MAP kinase in protein synthesis induced
by VEGF. VEGF st imulated Erk
phosphorylation that was required for induction
of protein synthesis. VEGF-induced Erk
activation was not dependent on PI 3-kinase
act ivat ion but required sequent ia l
phosphorylation of type 2 VEGF receptor, PLC
and c-Src, as demonstrated by inhibitors
SU1498, U73122 and PP1, respectively. c-Src
phosphorylation was inhibited by U73122,
indicating it was downstream of PLC . Studies
with PP1/2 showed that phosphorylation of c-Src
was required for tyrosine phosphorylation of
Raf-1, an upstream regulator of Erk. VEGF also
stimulated phosphorylation of Pyk-2; VEGF-
induced phosphorylation of Pyk2, c-Src and Raf-
1 could be abolished by BAPTA-AM,
demonstrating requirement for induction of
intracellular calcium currents. We examined the
downstream events following phosphorylation of
Erk. VEGF stimulated phosphorylation of Mnk1
and eIF4E, and induced Mnk1 to shift from
cytoplasm to the nucleus upon phosphorylation.
VEGF-induced phosphorylation of Mnk1 and
eIF4E required phosphorylation of PLC , c-Src
and Erk. Expression of dominant negative Mnk1
abrogated eIF4E phosphorylation and protein
synthesis induced by VEGF. VEGF-stimulated
protein synthesis could be blocked by inhibition
of PLC by chemical inhibitor or expression of a
dominant negative construct. Our data
demonstrate that VEGF stimulated protein
synthesis is Erk-dependent and requires the
activation of VEGFR2, PLC , c-Src, Raf, Erk
pathway. VEGF also stimulates Erk-dependent
phosphorylation of Mnk1 and eIF4E, crucial
events in the initiation phase of protein
translation.
The response of a cell to injurious stimuli
includes increase in cell protein synthesis
(hypertrophy), cell division (hyperplasia) or
apoptosis. Of these processes, the mechanisms
underlying cell hypertrophy have not been well
understood. Regulation of cellular protein synthesis
occurs at the initiation phase of protein translation
(1). One of the main events in initiation phase is
phosphorylation of eukaryotic initiation factor 4E
(eIF4E) binding protein (4E-BP1, 15-20 kDa) (2).
Phosphorylation of this repressor protein results in
dissolution of eIF4E-4E-BP1 complex, releasing the
eIF4E to bind to the mRNA cap and promote
translation initiation (3). Increase in eIF4E
phosphorylation is commonly seen during the
initiation phase and has been attributed to Erk-1/-2
type MAP kinase (4). However, Erk is not the direct
kinase for eIF4E; it activates Mnk1/-2 kinase,
which, in turn, phosphorylates ser209 on eIF4E
(5,6). Agonists that induce Mnk1 phosphorylation
have not been well studied. We examined whether
VEGF regulates this important kinase.
Functional effects of VEGF as an
angiogenic agent and as a key regulator of
endothelial cell functions have been well
documented. Our recent observations have shown
that VEGF increases protein synthesis and induces
hypertrophy in renal proximal tubular epithelial
(MCT) cells, suggesting a role for VEGF in non-
angiogenic processes in non-endothelial cells (7).
Previously, we have also shown that VEGF
stimulation of protein synthesis and hypertrophy in
MCT cells recruit PI 3-kinase-Akt axis to induce
phosphorylation of 4E-BP1 (7). In that study, the
role of Erk in VEGF-induced protein synthesis was
not examined. Increase in Erk phosphorylation in
the renal cortex of mice with type 2 diabetes occurs
simultaneously with the onset of kidney
hypertrophy that mostly involves renal tubular
epithelial cells (8). Coinciding with Erk
phosphorylation and kidney hypertrophy, VEGF
expression in the kidney is increased (7), and anti-
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

3
VEGF antibody reverses kidney enlargement (9).
However, whether VEGF recruits Erk pathway in
regulation of protein synthesis and renal cell
hypertrophy is not known. Also unknown is the
identity of upstream kinases that regulate Erk
activity in response to VEGF in MCT cells. In this
study, we tested the hypothesis that PLC- , an
important kinase that binds to type 2 VEGF
receptor (VEGFR2), plays a regulatory role in
VEGF-induced Erk activation and protein synthesis.
Downstream of Erk, we also studied whether VEGF
regulated Mnk1 phosphorylation and its target,
eIF4E.
Methods
Materials. Dulbecco's Modified Eagle's Medium
(DMEM), penicillin and streptomycin were
purchased from Gibco-Invitrogen (Carlsbad, CA).
Fetal bovine serum was purchased from Hyclone
Laboratories Inc. VEGF-165 recombinant protein,
and, anti-actin antibody were purchased from
Sigma-Aldrich (St. Louis, MO). Anti-
phosphotyrosine antibody 4G10 was purchased
from Upstate Inc (Charlottesville, Virginia).
Antibody against Mnk1 and anti v-Src antibody
were bought from Santa Cruz Biotechnology (Santa
Cruz, CA) and Oncogene Research Products (San
Diego, CA), respectively. All other antibodies were
purchased from Cell Signaling (Beverly, MA). 35
S-
labeled methionine was purchased from Perkin-
Elmer (Boston, MA). ECL chemiluminescent
substrate reagent was from Pierce Biotechnology
Inc (Rockford, IL). PP1 and PP2 Analog, SU1498,
LY294002 and BAPTA/AM were purchased from
Calbiochem-EMD Biosciences (San Diego, CA).
As PP1 is no longer available we employed PP2 as
an inhibitor of c - Src. U73122 was from Biomol
International L.P. (Plymouth Meeting, PA) and
U0126 was purchased from Promega Life Sciences
(Madison, WI).
Cell culture (7 ). SV-40 immortalized murine
proximal tubular epithelial (MCT) cells (kindly
provided by Dr. Eric Neilson, Vanderbilt
University, Nashville, TN) were grown in DMEM
containing 7% fetal bovine serum, 5 mmol/L
glucose, 100 u/ml penicillin, 100 µg/ml
streptomycin and 2 mM glutamine. MCT cells
express in vivo properties of proximal tubular
epithelial cells (10). The cells were grown to 90%
confluence and then growth-arrested for 18 hours in
serum-free DMEM before experiments.
Immunoblotting (7). Equal amounts of protein
from control and VEGF-treated cells were separated
by SDS-PAGE and transferred to a nitrocellulose
membrane. After transfer overnight at 4°C, the
membrane was blocked in TBS, pH 7.2, containing
0.1% Tween 20 (TBST) for 1 hour. Membrane was
then washed and probed with primary antibody for
3 hours. After extensive washing in TBST, the
membrane was incubated with horseradish
peroxidase conjugated secondary antibody (Jackson
ImmunoResearch Laboratories,Inc, West
Grove,PA). Proteins were visualized by
chemiluminescence using ECL reagent. Images of
the bands were scanned by reflectance scanning
densitometry and the intensity of the bands was
quantified using NIH Image software.
Protein synthesis measurement (11). Serum-
starved cells were incubated with 20 ng/ml of
VEGF-165 in the presence of 10 µCi/ml of [35
S]-
methionine for 2 hours with or without the
respective inhibitors. Cells were then washed in
phosphate buffered saline (PBS) and lysed in
radioimmunoprecipitation assay (RIPA) buffer.
Equal amount of protein (20 µg) was spotted onto
the 3 mm filter paper (Whatman International,
Maidstone, UK). Filters were washed three times by
boiling for 1 minute in 10% trichloroacetic acid
(TCA) containing 0.1 g/L methionine before
determining radioactivity.
Erk1/2 MAP kinase (Erk) activity assay (12).
Equal amounts of cell lysates (200 µg) were
immunoprecipitated with Erk1/2 antibody. Protein
A-agarose beads were added and incubated at 4°C
for 1 hour. The beads were then washed and the Erk
kinase assay was performed in kinase assay buffer
in the presence of 50 µmol cold ATP, 1 µCi of
[32
P]-ATP and 5 µg of myelin basic protein (MBP)
at 30°C for 30 minutes. The reaction was arrested
by the addition of an equal volume of 2X sample
buffer. Phosphorylated bands were detected by
SDS-PAGE followed by autoradiography. In some
experiments, immunoblotting with anti-phospho-
Erk antibody was done to assess Erk
phosphorylation.
Immunofluorescence microscopy. Cells were
seeded in 8-well chamber slides. Semi-confluent
cells were serum-starved for 18 hours and treated
with VEGF (20 ng/ml) for different time periods.
Cells were washed with PBS, fixed and incubated
with rabbit anti-phospho-Mnk1 antibody followed
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

4
by staining with Cy3-conjugated donkey anti-rabbit
secondary antibody (Chemicon International,
Temecula, CA). PhosphoMnk1 was visualized with
a confocal laser microscopy system (Olympus
Fluoview 500). The confocal images were analyzed
using Fluoview software to determine the
localization of phospho-Mnk.
For quantification of phosphoMnk1 nuclear
localization, cells were grown in chamber slides,
individual chambers representing one time point.
Cells were viewed under uniform magnification
(40X). For each time point, the number of cells
showing nuclear localization of phosphoMnk1 per
total number of cells in a field of fixed area
containing a minimum of 150 cells, was counted
and converted to percentage. Finally, these values
were compared with that of value at time zero
(control). The experiment was performed three
times and composite data were converted into
graphic form and analyzed by ANOVA.
Transfection studies (13). Plasmid constructs
containing dominant negative mutants of Src and
PLC (PLCz) were employed for transient
transfection in MCT cells using lipofectamine and
lipo-plus reagent (Invitrogen Life Technologies
Inc., Calrsbad, CA), as described earlier. PCR 295
expression vector containing dominant negative c-
Src was kindly provided by Dr. T. Yoneda,
University of Texas Health Science Center, San
Antonio, TX. The kinase inactivation mutation in c-
Src consisted of Lys295 to Met295. PLCz is a
dominant-negative fragment of PLC gene which
encodes the Z region (PCI region, PLC inhibitor
region) containing the SH2 and SH3 domains (aa
517-901) of this enzyme (14). The pXf/PLCz
expression was driven by SV40 early promoter.
pEBG-3X eukaryotic expression vector containing
dominant-interfering Mnk1 mutant (pEBG-T2A2
containing Thr197Ala/Thr202Ala mutants (5)) was
a kind gift from Dr J. Cooper (Fred Hutchinson
Cancer Research Center, Seattle, WA).
Statistics. All values are expressed as mean ± SE
obtained from at least three independent
experiments. Statistical analysis was performed
using one-way analysis of variance (ANOVA) for
comparison between multiple groups; p values of
p<0.05 were considered significant.
Results
VEGF-induced protein synthesis is Erk-
dependent in MCT cells
We investigated whether VEGF induced
Erk phosphorylation and if VEGF-induced protein
synthesis was Erk-dependent since Erk1/2 MAP
kinase occupies a central position in transducing
signals from extra-cellular milieu to the nucleus.
VEGF promoted Erk phosphorylation within 2 min
that lasted nearly 15 min with peak phosphorylation
at 10 min (Fig. 1, panel A). VEGF significantly
stimulated protein synthesis in MCT cells that was
abrogated by pre-incubation of cells with U0126, an
inhibitor of MEK, the direct upstream kinase of Erk
(Fig. 1, panel B). VEGF induction of protein
synthesis is, thus, dependent on stimulation of Erk.
We initiated the search for upstream
regulators of VEGF-induced Erk activation. We
started with PI 3-kinase as insulin- and IGF-1-
induced Erk activation in MCT cells is PI 3-kinase
dependent (12,13). We have previously shown that
VEGF induces PI 3-kinase activity in these cells
(7). Pre-incubation with LY294002, a selective
inhibitor of PI 3-kinase, did not affect VEGF-
induced Erk activity, although it was abolished by
U0126 (Fig. 1, panel C). These observations
suggest that VEGF-induced Erk activation is
independent of PI 3-kinase but dependent on MEK
activation.
Having excluded PI 3-kinase, we sought to
identify other kinases that could act as upstream
effectors of Erk activation by VEGF. We selected
phospholipase C (PLC ) and c-Src as potential
regulators as they are membrane-associated and
interact with VEGF receptors (15,16).
Type 2 VEGF receptor (VEGFR2) stimulates
PLC and c-Src to regulate Erk phosphorylation
Immunoblotting cell lysates with anti-
phosphotyrosine antibody following VEGF
incubation for 10 min showed increased protein
tyrosine phosphorylation of several proteins,
including those of approximate sizes 220 kD, 180
kD, 150 kD, 110 kD, 90 kD, 60 kD and 30 kD. The
180 kD, 150 kD and 60 kD bands may represent
VEGFR2, PLC and c-Src, respectively. Pre-
incubation with SU1498, a selective VEGFR2
inhibitor, abolished or markedly inhibited most of
these changes implying VEGFR2 phosphorylation
was required for these tyrosine phosphorylation
events (Fig. 2A).
VEGF stimulated tyrosine phosphorylation
of PLC and c-Src rapidly within 5 minutes (Fig. 2,
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

5
panels B and C). VEGFR2 is believed to mediate
most of the biological effects of VEGF. Pre-
treatment of cells with SU-1498 abolished VEGF-
induced PLC and c-Src phosphorylation (Fig. 2,
panels D and E), suggesting involvement of
VEGFR2. Next, we examined the sequence of
phosphorylation of PLC and c-Src by VEGF.
U73122, a PLC inhibitor, blocked phosphorylation
of c-Src, demonstrating that the latter is
downstream of PLC in VEGFR2 signaling events
in MCT cells (Fig. 2, panel F).
We investigated whether PLC plays a role
in regulating VEGF-induced Erk activation. VEGF-
stimulated Erk activation was inhibited by U-
73122, the PLC inhibitor (Fig. 3, panel A). This
was further confirmed by expressing dominant
negative constructs of PLC (PLCz). VEGF-
stimulated Erk activation was abolished in cells
transfected with PLCz, but not in control cells
transfected with vector alone (Fig. 3, panel B).
Although it appears as though expression of PLCz
reduced Erk activity only upon addition of VEGF,
densitometric analysis of bands from 3 experiments
did not reveal a significant difference in band
intensity between VEGF+PLCz and untreated
control groups (bars 1 Vs 4 in Fig. 3, panel B).
We next examined the role of c-Src in
VEGF induction of Erk. PP1, a c-Src inhibitor,
abolished VEGF induced Erk phosphorylation (Fig.
4, panel A). Further confirmation was obtained by
employing a kinase-inactive dominant negative
construct of c-Src (Fig. 4, panel B). Raf is an
upstream kinase in Erk pathway (17,18). VEGF
stimulated Raf phosphorylation at Tyr340, 341
within 5 min that lasted for nearly 15 min (Fig. 4,
panel C). This was completely inhibited by PP2, a
c-Src inhibitor (Fig. 4, panel D), indicating
Tyr340/341 phosphorylation of Raf was c-Src
dependent. The Src inhibitor, at this concentration
at this concentration, did not affect VEGF-induced
PLC phosphorylation, which is VEGFR2-
dependent (See Supplementary fig. S1).
VEGF induces phosphorylation of Pyk2, c-Src
and Erk in a Ca-dependent manner
VEGF increases intracellular calcium transients
through VEGFR2 (19) that correlate with
phosphorylation of PLC (20). Activation of PLC
has been linked to the production of inositol 1,4,5,
tris-phosphate and diacylglycerol, leading to
increased intracellular calcium mobilization and
PKC activation, respectively. A read-out of Ca
signaling is the phosphorylation of Pyk2, a proline-
rich, Ca++
-dependent non-receptor tyrosine kinase.
Pyk2 phosphorylation was studied using a phospho-
specific antibody that detects phosphorylation on
tyr402. Incubation with VEGF rapidly induced
phosphorylation of Pyk2 that lasted for nearly 30
min (Fig. 5, panel A). We next examined the role of
Ca++
in Erk activation induced by VEGF. Chelation
of intracellular Ca++
by BAPTA/AM abolished
VEGF-induced phosphorylation of Pyk2, Src and
Erk (Fig. 5, panels B, C and D), confirming that
Ca++
transients are required for VEGF-induced
phosphorylation of Erk and its upstream regulators.
The data described above established the
upstream regulators of VEGF-induced Erk
activation along the lines of VEGFR2, PLC ,
Pyk2/c-Src, Raf, Erk. We next sought to gain
insight into the possible mechanism downstream of
Erk activation related to events of regulatory
importance in protein synthesis.
VEGF induces phosphorylation of Mnk1
As discussed before, phosphorylation of 4E-BP1
releases eIF4E from the dimeric complex. 4E-BP-1
phosphorylation is required for VEGF-induced
protein synthesis and hypertrophy in MCT cells;
this process is PI 3-kinase- and Akt-dependent (7).
Following dissolution of eIF4E-4E-BP1 complex,
free eIF4E binds to a scaffolding protein eIF4G and
is phosphorylated by MAP kinase integrating
kinases (Mnk1/2) that also bind eIF4G at the C-
terminus (21). Mnk1, a 52 kDa protein, is activated
by phosphorylation at Thr197 by Erk1/2. As Erk
regulation of protein synthesis could involve Mnk
and eIF4E, we examined whether VEGF regulates
their phosphorylation. VEGF rapidly stimulated
phosphorylation of Mnk1 within 5 min and the
effect lasted for nearly 30 min (Fig. 6, panel A).
These data suggest that VEGF activates a
downstream target of Erk. Mnk1 shuttles between
the cytoplasm and the nucleus (22); however,
agonists that stimulate nuclear shift of Mnk1 have
not been extensively identified. Therefore, we
examined if VEGF regulates shift of Mnk1 into the
nucleus. As seen in Fig. 6, panel B, we observed
increased immunofluorescence corresponding to
Mnk1 phosphorylation in the cytoplasm within 2
minutes with perinuclear localization at 5 min.
Nuclear translocation was observed at 15 min,
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

6
coinciding with maximum phosphorylation of the
protein (Fig. 6, panel A). Figure 6B shows the
scoring for nuclear localization of phosphoMnk1.
There was a progressive increase in nuclear
immunofluorescence that peaked at 15 min
following treatment of the cells with VEGF (p<0.01
at 5 and 15 min). To our knowledge, this is the first
demonstration of VEGF regulation of Mnk1
phosphorylation and its localization in the cell. We
examined the upstream regulators of VEGF-
induced Mnk1 phosphorylation. U73122, the PLC
inhibitor, PP2, the c-Src inhibitor, and U0126, the
MEK inhibitor, all abolished VEGF-stimulated
Mnk1 phosphorylation (Fig. 7, panels A, B and C),
identifying PLC, c-Src and MEK (immediately
upstream of Erk) as kinases that control Mnk1
phosphorylation.
VEGF induces phosphorylation of eIF4E.
Our next aim was to test whether VEGF induced
phosphorylation of eIF4E, the Mnk1 substrate.
VEGF stimulated phosphorylation of eIF4E
robustly within 5 min and the effect lasted for about
30 min (Fig. 8, panel A). U73122, PP2, and U0126
completely inhibited VEGF-induced
phosphorylation of eIF4E (Fig. 8, panels B, C and
D). Thus, VEGF-induced phosphorylation of eIF4E
is regulated via the PLC , c-Src and Erk axis.
DN-Mnk1 expression inhibits eIF4E
phosphorylation and protein synthesis induced
by VEGF
We next investigated the involvement of Mnk1 in
VEGF-stimulated protein synthesis by expressing
dominant negative Mnk1 in which the Thr197 and
202 are mutated to Ala. Both VEGF-induced eIF4E
phosphorylation and protein synthesis were
abrogated by expression of mutant Mnk1 (Fig. 9,
panels A and B). These data demonstrate that
VEGF-induced protein synthesis requires Mnk1
phosphorylation.
PLC activation is required for VEGF-induced
enhanced protein synthesis. We have established
the role of PLC in activation of Erk to increase
phosphorylation of eIF4E, a regulator of protein
synthesis initiation. We directly examined whether
PLC activation, the proximal event in Erk
activation, is required for VEGF induction of
protein synthesis. VEGF-induced protein synthesis
in MCT cells was completely abrogated when the
cells were pre-treated with U73122 (Fig. 10, panel
A). Further confirmation was obtained employing
cells transfected to express PLCz, the dominant
negative construct of PLC (Fig. 10, panel B).
Fig.11 summarizes the proposed sequence of
signaling events induced by VEGF leading to
protein synthesis in MCT cells.
Discussion
In this report we demonstrate that VEGF
augments protein synthesis in an Erk
phosphorylation-dependent manner. Our data show
that VEGF-induced Erk phosphorylation is
regulated by sequential phosphorylation of
VEGFR2, PLC , and c-Src. We report for the first
time that VEGF stimulation of Erk phosphorylation,
in turn, results in phosphorylation of Mnk1 and its
substrate eIF4E. Phosphorylation of Mnk1 is
associated with its shift from the cytoplasm to the
nucleus. Our results provide new evidence for a role
for PLC in VEGF-induced eIF4E phosphorylation
and protein synthesis.
VEGF stimulation of protein synthesis in MCT
cells is mediated via activation of VEGFR2 (7).
Pathways regulating phosphorylation of 4E-BP1
and eIF4E seem to diverge downstream of
VEGFR2, the former being dependent on PI 3-
kinase-Akt activation (7) whereas, as reported here,
e IF4E phosphory la t ion requi res Erk
phosphorylation. As Erk phosphorylation was
independent of PI 3-kinase, our observations
suggest distinct roles for PI 3-kinase and Erk
pathways in regulation of crucial events that control
initiation phase of protein translation. PLC
phosphorylation by VEGFR2 is the most proximal
event identified in eIF4E phosphorylation in this
study. PLC binds to tyr1175 in the cytoplasmic
domain of VEGFR2 (23). Its role in regulation of
protein translation has not been studied in depth.
U73122, an inhibitor of PLC, has been shown to
abolish cardiac myocyte hypertrophy induced by
norepinephrine (24), although it appears not
required in angiotensin II-induction of protein
synthesis in cardiac fibroblasts (25). Thus,
involvement of PLC in protein synthesis appears to
be cell- and agonist-specific. In several previous
studies, involvement of PLC was not directly
studied but suspected by the use of its inhibitor, and
changes in eIF4E were not addressed. Our study
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

7
demonstrates for the first time, a link between PLC
phosphorylation and eIF4E phosphorylation for
inducing protein synthesis in cells stimulated with
VEGF.
Phosphorylation of PLC is followed by
generation of inositol 1,4,5, tris-phosphate and
diacylglycerol, which are known to stimulate
mobilization of intracellular calcium and protein
kinase C, respectively. Ca++
transients appear to be
important for VEGF induction of Erk
phosphorylation as BAPTA/AM abolished this
effect. In MCT cells, VEGF stimulation of c-Src
requires activation of PLC . c-Src has been
identified as a mediator of angiotensin II-
stimulation of protein synthesis in smooth muscle
cells grown from resistance arteries from
hypertensive patients (26). Additionally, c-Src is
involved in phosphorylation of RNA binding
protein hnRNP-K that regulates translation of select
mRNAs (27). Our observations identify c-Src as
one of the upstream regulators of VEGF-induced
phosphorylation of Mnk1 and eIF4E. Pyk2, a
proline-rich non-receptor tyrosine kinase, is
activated in a Ca++
-dependent manner, resulting in
its interaction with activated c-Src (28).
Phosphorylation of tyr402 of Pyk2 leads to binding
and activation of c-Src in PC12 cells (29). Pyk2
phosphorylation is required for angiotensin II-
induced eIF4E phosphorylation and protein
synthesis in vascular smooth muscle cells (30,31).
Pyk2 may serve as a bridge between PLC -c-Src
axis and the canonical Raf-MEK-Erk pathway (29).
Activated Raf is phosphorylated on Ser338 (32).
However, for full activation of Raf-1, c-Src
mediated phosphorylation of Tyr340/341 is
necessary (33,34). Our data suggest that VEGF
stimulation of Erk pathway involves Raf-1 tyrosine
phosphorylation by c-Src. It is well known that Raf-
1 directly phosphorylates and activates the protein
kinase MEK.
eIF4E regulates the initiation phase of protein
synthesis that is regarded as the rate-limiting step
(35). In general, translation of most capped mRNAs
in eukaryotes is facilitated by eIF4E, the cap
binding protein (36). In response to signals that
promote protein synthesis, 4E-BP1 is
phosphorylated which results in dissolution of its
heterodimeric complex with eIF4E (37). Once freed
from 4E-BP1, eIF4E associates with eIF4G and
eIF4A to form the eIF4F complex and binds to the
m7 cap of the mRNA. Erk has been implicated in
eIF4E phosphorylation on Ser209; the
physiologically important site (38) although it is not
the direct kinase as eIF4E lacks the proline residues
around Ser209 involved in Erk recognition. It is
now established that Erk phosphorylates Mnk1,
which serves as the kinase for eIF4E (5,6). Binding
studies have shown that Mnk1 associates with C
terminus of eIF4G, the scaffolding protein, which
has binding sites for eIF4E and eIF4A, and forms
the anchor for eIF4F complex. Bulk of
phosphorylation of eIF4E by Mnk1 occurs
following their binding to eIF4G (21). Mnk1 carries
a basic amino acid-rich nuclear import signal in its
N-terminus that determines its shift from the
cytoplasm to the nucleus (22). Our observations
show that while phosphorylation of Mnk1 following
VEGF incubation starts while the protein is in the
cytoplasm, the peak in phosphorylation appears to
occur when the protein has shifted to the nucleus.
The relevance of Mnk1 nuclear shift to cellular
protein synthesis needs to be elucidated.
The functional significance of eIF4E
phosphorylation is also unclear. Initially, in vitro
studies showed that phosphorylation of eIF4E
augmented the avidity of the molecule for the
mRNA cap and promoted the efficiency of its
translation (39). In Drosophila, expression of an
eIF4E phosphorylation mutant resulted in stunted
development, suggesting phosphorylation of eIF4E
facilitated cell protein synthesis (40). However,
opposing views have recently been expressed.
Phosphorylation of eIF4E is thought to cause
electrostatic repulsion from phosphate groups on
the mRNA cap and inhibit its cap binding (41,42).
The precise timing of eIF4E phosphorylation
relative to mRNA cap binding is not known. If
phosphorylation of eIF4E were to occur after
binding of the factor to the mRNA cap, anionic
repulsion could serve to disengage eIF4E from the
mRNA. The association and dissociation reactions
between eIF4E and mRNA are known to be rapid in
the range of 0.1 sec (43) and could be facilitated by
eIF4E phosphorylation. Although present in
cytoplasm, a significant fraction of cellular eIF4E is
present in the nucleus and may participate in
nuclear protein translation (44) and export of select
mRNAs e.g., cyclin D1, from the nucleus (45,46).
Role of eIF4E phosphorylation in these processes is
not yet known.
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

8
Stimulation of protein synthesis in renal
epithelial (MCT) cells by VEGF has implications
for kidney hypertrophy, which may occur
physiologically, after removal of one kidney (9), or,
pathologically, in early phase of diabetes (7,47).
Neutralizing antibodies against VEGF ameliorate
early renal dysfunction in experimental diabetes
including renal hypertrophy (9,48). These data
implicate VEGF in the pathogenesis of kidney
hypertrophy in diabetes that mostly involves renal
epithelial cells (49). VEGF could have constructive
effects on the kidney in a different setting. In
thrombotic glomerular disease, VEGF
administration helps restore renal architecture (50).
Knowledge of signaling intermediaries that regulate
VEGF-induction of protein synthesis in renal cells
should help facilitate modulation of this process to
advantage in renal disease states.
Acknowledgements: These studies were supported
by grants from the NIH (BSK, GGC-DK55815 &
DK50190), American Diabetes Association (BSK),
Veterans Administration (BSK, GGC), the Juvenile
Diabetes Research Foundation (GGC), and the
National Kidney Foundation of South and Central
Texas (MMM). We acknowledge the staff at the
Cellular and Structural Biology Department at
UTHSC San Antonio for providing the confocal
microscopy instruments. GGC is a recipient of the
Veterans Administration Career Scientist Award.
Abbreviations used are: IGF-1, insulin-like
growth factor 1; PAGE, polyacrylamide gel
electrophoresis; PP1, 4-Amino-1-tert-butyl-3-(1¢-
naphthyl)pyrazolo[3,4-d]pyrimidine; PP2, 4-amino-
5-(4-chlorophenyl)-7-(t-
butyl)pyrazolo[3,4,d]pyrimidine; PCI domain,
phospholipase C inhibitory domain; PI 3-kinase,
phosphoinositide-3-kinase; PLC- 1, phospholipase
C-gamma one; SH2, SH3, Src homology 2 and 3;
VEGF, vascular endothelial growth factor;
VEGFR2, vascular endothelial growth factor
receptor 2
References
1. Miron, M., and Sonenberg, N. (2001) J Nutr 131, 2988S-2993S
2. Lin, T. A., Kong, X., Haystead, T. A., Pause, A., Belsham, G., Sonenberg, N., and Lawrence, J. C.,
Jr. (1994) Science 266, 653-656
3. Thornton, S., Anand, N., Purcell, D., and Lee, J. (2003) J Mol Med 81, 536-548
4. Wang, X., Flynn, A., Waskiewicz, A. J., Webb, B. L., Vries, R. G., Baines, I. A., Cooper, J. A., and
Proud, C. G. (1998) J Biol Chem 273, 9373-9377
5. Waskiewicz, A. J., Flynn, A., Proud, C. G., and Cooper, J. A. (1997) Embo J 16, 1909-1920
6. Waskiewicz, A. J., Johnson, J. C., Penn, B., Mahalingam, M., Kimball, S. R., and Cooper, J. A.
(1999) Mol Cell Biol 19, 1871-1880
7. Senthil, D., Choudhury, G. G., McLaurin, C., and Kasinath, B. S. (2003) Kidney Int 64, 468-479
8. Feliers, D., Duraisamy, S., Faulkner, J. L., Duch, J., Lee, A. V., Abboud, H. E., Choudhury, G. G.,
and Kasinath, B. S. (2001) Kidney Int 60, 495-504
9. Flyvbjerg, A., Dagnaes-Hansen, F., De Vriese, A. S., Schrijvers, B. F., Tilton, R. G., and Rasch, R.
(2002) Diabetes 51, 3090-3094
10. Haverty, T. P., Kelly, C. J., Hines, W. H., Amenta, P. S., Watanabe, M., Harper, R. A., Kefalides,
N. A., and Neilson, E. G. (1988) J Cell Biol 107, 1359-1368
11. Senthil, D., Choudhury, G. G., Abboud, H. E., Sonenberg, N., and Kasinath, B. S. (2002) Am J
Physiol Renal Physiol 283, F1226-1236
12. Bhandari, B. K., Feliers, D., Duraisamy, S., Stewart, J. L., Gingras, A. C., Abboud, H. E.,
Choudhury, G. G., Sonenberg, N., and Kasinath, B. S. (2001) Kidney Int 59, 866-875
13. Senthil, D., Faulkner, J. L., Choudhury, G. G., Abboud, H. E., and Kasinath, B. S. (2001) Biochem
J 360, 87-95
14. Homma, Y., and Takenawa, T. (1992) J Biol Chem 267, 21844-21849
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

9
15. Guo, D., Jia, Q., Song, H. Y., Warren, R. S., and Donner, D. B. (1995) J Biol Chem 270, 6729-6733
16. Cunningham, S. A., Arrate, M. P., Brock, T. A., and Waxham, M. N. (1997) Biochem Biophys Res
Commun 240, 635-639
17. Pearson, G., Robinson, F., Beers Gibson, T., Xu, B. E., Karandikar, M., Berman, K., and Cobb,
M. H. (2001) Endocr Rev 22, 153-183
18. von Gise, A., Lorenz, P., Wellbrock, C., Hemmings, B., Berberich-Siebelt, F., Rapp, U. R., and
Troppmair, J. (2001) Mol Cell Biol 21, 2324-2336
19. Cunningham, S. A., Tran, T. M., Arrate, M. P., Bjercke, R., and Brock, T. A. (1999) Am J Physiol
276, C176-181
20. McLaughlin, A. P., and De Vries, G. W. (2001) Am J Physiol Cell Physiol 281, C1448-1456
21. Pyronnet, S., Imataka, H., Gingras, A. C., Fukunaga, R., Hunter, T., and Sonenberg, N. (1999)
Embo J 18, 270-279
22. Parra-Palau, J. L., Scheper, G. C., Wilson, M. L., and Proud, C. G. (2003) J Biol Chem 278,
44197-44204
23. Takahashi, T., Yamaguchi, S., Chida, K., and Shibuya, M. (2001) Embo J 20, 2768-2778
24. Singal, T., Dhalla, N. S., and Tappia, P. S. (2004) Biochem Biophys Res Commun 320, 1015-1019
25. Hou, M., Pantev, E., Moller, S., Erlinge, D., and Edvinsson, L. (2000) Acta Physiol Scand 168, 301-
309
26. Touyz, R. M., He, G., Wu, X. H., Park, J. B., Mabrouk, M. E., and Schiffrin, E. L. (2001)
Hypertension 38, 56-64
27. Ostareck-Lederer, A., Ostareck, D. H., Cans, C., Neubauer, G., Bomsztyk, K., Superti-Furga, G.,
and Hentze, M. W. (2002) Mol Cell Biol 22, 4535-4543
28. Sabri, A., Govindarajan, G., Griffin, T. M., Byron, K. L., Samarel, A. M., and Lucchesi, P. A.
(1998) Circ Res 83, 841-851
29. Dikic, I., Tokiwa, G., Lev, S., Courtneidge, S. A., and Schlessinger, J. (1996) Nature 383, 547-550
30. Rocic, P., Govindarajan, G., Sabri, A., and Lucchesi, P. A. (2001) Am J Physiol Cell Physiol 280,
C90-99
31. Rocic, P., Jo, H., and Lucchesi, P. A. (2003) Am J Physiol Cell Physiol 285, C1437-1444
32. Mason, C. S., Springer, C. J., Cooper, R. G., Superti-Furga, G., Marshall, C. J., and Marais, R.
(1999) Embo J 18, 2137-2148
33. Fabian, J. R., Daar, I. O., and Morrison, D. K. (1993) Mol Cell Biol 13, 7170-7179
34. Marais, R., Light, Y., Paterson, H. F., and Marshall, C. J. (1995) Embo J 14, 3136-3145
35. Sonenberg, N., and Dever, T. E. (2003) Curr Opin Struct Biol 13, 56-63
36. von der Haar, T., Gross, J. D., Wagner, G., and McCarthy, J. E. (2004) Nat Struct Mol Biol 11,
503-511
37. Pause, A., Belsham, G. J., Gingras, A. C., Donze, O., Lin, T. A., Lawrence, J. C., Jr., and
Sonenberg, N. (1994) Nature 371, 762-767
38. Flynn, A., and Proud, C. G. (1995) J Biol Chem 270, 21684-21688
39. Minich, W. B., Balasta, M. L., Goss, D. J., and Rhoads, R. E. (1994) Proc Natl Acad Sci U S A 91,
7668-7672
40. Lachance, P. E., Miron, M., Raught, B., Sonenberg, N., and Lasko, P. (2002) Mol Cell Biol 22,
1656-1663
41. Scheper, G. C., and Proud, C. G. (2002) Eur J Biochem 269, 5350-5359
42. Zuberek, J., Wyslouch-Cieszynska, A., Niedzwiecka, A., Dadlez, M., Stepinski, J., Augustyniak,
W., Gingras, A. C., Zhang, Z., Burley, S. K., Sonenberg, N., Stolarski, R., and Darzynkiewicz, E.
(2003) Rna 9, 52-61
43. Scheper, G. C., van Kollenburg, B., Hu, J., Luo, Y., Goss, D. J., and Proud, C. G. (2002) J Biol
Chem 277, 3303-3309
44. Iborra, F. J., Jackson, D. A., and Cook, P. R. (2001) Science 293, 1139-1142
45. Cohen, N., Sharma, M., Kentsis, A., Perez, J. M., Strudwick, S., and Borden, K. L. (2001) Embo J
20, 4547-4559
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

10
46. Topisirovic, I., Culjkovic, B., Cohen, N., Perez, J. M., Skrabanek, L., and Borden, K. L. (2003)
Embo J 22, 689-703
47. Cha, D. R., Kang, Y. S., Han, S. Y., Jee, Y. H., Han, K. H., Han, J. Y., Kim, Y. S., and Kim, N. H.
(2004) J Endocrinol 183, 183-194
48. de Vriese, A. S., Tilton, R. G., Elger, M., Stephan, C. C., Kriz, W., and Lameire, N. H. (2001) J Am
Soc Nephrol 12, 993-1000
49. Schrijvers, B. F., Flyvbjerg, A., and De Vriese, A. S. (2004) Kidney Int 65, 2003-2017
50. Kim, Y. G., Suga, S. I., Kang, D. H., Jefferson, J. A., Mazzali, M., Gordon, K. L., Matsui, K.,
Breiteneder-Geleff, S., Shankland, S. J., Hughes, J., Kerjaschki, D., Schreiner, G. F., and Johnson,
R. J. (2000) Kidney Int 58, 2390-2399
Figure legends
Fig 1.
Panel A. VEGF stimulates Erk phosphorylation. Serum-deprived cells were incubated with VEGF (20 ng/ml).
Equal amounts of protein from the cell lysates were fractionated on a 12.5% gel. Immunoblotting was performed
using 1:2000 dilution of an antibody that specifically detects phospho-Erk. The blots were stripped and re-probed
with an Erk antibody to assess loading. A representative blot from more than 3 experiments is shown.
Densitometric quantification of data from 3 experiments is shown in the graph *p<0.01 VEGF Vs control.
Panel B. VEGF stimulation of protein synthesis is Erk-dependent. Quiescent MCT cells were incubated with
or without VEGF (20 ng/ml) simultaneously in the presence of [35
S]-methionine for 2 hours with or without pre-
incubation with 25 µM U0126, a MEK inhibitor, for 30 min. Incorporation of radiolabel into TCA-precipitable
protein was taken as a measure of protein synthesis and expressed as percentage of control. Composite data from 3
experiments are shown in a graph; *p<0.001 VEGF Vs control, **p<0.001 for VEGF+U0126 Vs VEGF, by
ANOVA.
Panel C. VEGF-induced Erk activation is independent of PI 3-kinase activation. MCT cells were incubated
with or without VEGF (20 ng/ml) for 10 min with or without pre-incubation with LY294002 (25 µM) or U0126
(25 µM) for 30 min. Equal amounts of cell lysates (100 µg) were immunoprecipitated with an anti-Erk-1/-2-type
MAP kinase antibody. Erk kinase activity was determined in the immunoprecipitates by an in vitro kinase assay
using myelin basic protein (MBP) as the substrate. The samples were fractionated by 15% SDS-PAGE and
phosphorylation of MBP visualized by autoradiography. Immunoblotting with total Erk antibody was done to
assess loading and band intensities were measured by densitometry. A representative blot from 3 individual
experiments is shown. The graph presents composite densitometric data from 3 experiments with controls
expressed as 100%; *p<0.001 VEGF or VEGF+LY294002 Vs control, **p<0.001 VEGF+U0126 Vs VEGF, by
ANOVA.
Fig 2.
Panel A. VEGF-induced tyrosine phosphorylation of proteins is dependent on VEGFR2 tyrosine
phosphorylation. Serum-starved MCT cells were incubated with VEGF (20 ng/ml) for 10 min with or without
pre-incubation with SU1498, the selective VEGFR2 inhibitor. Equal amounts of cell lysates were resolved on
SDS-PAGE and immunoblotted with phospho-tyrosine antibody. A representative blot from 3 independent
experiments is shown. The bottom panel shows immunoblot analysis of the same samples with anti-actin antibody.
Panels B, C. VEGF stimulates phosphorylation of phospholipase C (PLC ) (B) and c-Src kinase (C).
Serum-starved MCT cells were incubated with VEGF (20 ng/ml) for the time periods indicated. Equal amounts of
cell lysates were resolved on SDS-PAGE and immunoblotted with phospho-specific antibodies for PLC and c-
Src; loading was assessed by immunoblotting with antibodies against PLC and Src, respectively. A representative
blot from 3 independent experiments for each kinase is shown.
Panels D, E. VEGF-induced phosphorylation of PLC and c-Src is mediated through VEGFR2. Cells were
incubated with or without VEGF for 10 min after pre-incubation for 30 min with or without 10 µM SU1498, a
VEGFR2-selective inhibitor. Equal amounts of cell lysates were fractionated on SDS-PAGE and immunoblotted
with the phospho-specific antibodies for PLC and c-Src; loading was assessed by immunoblotting with antibodies
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

11
against PLC and Src, respectively. A representative blot from 3 independent experiments for each kinase is
shown.
Panel F. c-Src phosphorylation is PLC -dependent. Cells were pre-treated with or without U73122 (5 µM), a
PLC inhibitor, prior to incubation for 10 min with or without VEGF 20 ng/ml. Western blot analysis was
performed with the cell lysates using phospho-Src antibody. The lower panel represents immunoblot analyses of
the same samples with Src antibody to assess loading. A representative blot from 3 independent experiments is
shown.
Fig 3.
VEGF-stimulated Erk activation is PLC -dependent in MCT cells
Panel A. Cell lysates prepared from cells, treated with or without VEGF (20 ng/ml) after pre-treatment with or
without U73122 (5 µM), a PLC inhibitor, were run on SDS-PAGE, transferred and immunoblotted with phospho-
Erk antibody. Loading was assessed by stripping the blot and re-probing with an Erk antibody. A representative
blot from 4 independent experiments is shown. The graph shows quantification of results from 4 different
experiments. *p<0.01 VEGF Vs control, **p<0.01 VEGF Vs VEGF+U73122 by ANOVA.
Panel B. MCT cells were transfected with 1 µg of PLCz (a dominant negative construct of PLC ) per well or an
empty vector without the construct as control and allowed to grow for 48 hours. Cells were treated with or without
VEGF (20 ng/ml) for 10 min and Erk phosphorylation was assessed as described in panel A. A representative blot
from 3 experiments is shown. Densitometric analysis of the bands is presented in a graph. *p<0.01 VEGF Vs
control, **p<0.01 VEGF Vs VEGF+PLCz by ANOVA.
Fig 4.
VEGF-induced Erk phosphorylation is dependent on c-Src
Panel A. Cells were treated with or without PP1, a Src inhibitor, for 30 min prior to incubation with or without
VEGF (20 ng/ml). Erk phosphorylation was examined as described in panel A. A representative blot from 4
experiments is shown. The graph shows quantification of composite data from 3 experiments. *p<0.01 VEGF Vs
control, **p<0.01 VEGF Vs VEGF+PP1 by ANOVA.
Panel B. Cells were transfected with a plasmid carrying a c-Src dominant negative (DN-Src) construct or an
empty vector before incubation with or without VEGF for 10 min. Cell lysates were fractionated on SDS-PAGE
and Erk phosphorylation was assessed as described in panel A. A representative blot from 3 experiments is
shown. The graph shows quantification of composite data from 3 experiments. *p<0.01 VEGF Vs control,
**p<0.01 VEGF Vs VEGF+DN Src by ANOVA.
Panel C. VEGF stimulates Raf-1 phosphorylation that is c-Src-dependent. MCT cells were incubated with
or without VEGF for 10 min. Western blot analysis was done with phospho-specific antibodies for Tyr340/341
of Raf. The blots were stripped and re-probed with Raf-1 antibody (bottom panel) to assess loading. A
representative blot from three experiments is shown.
Panel D. MCT cells were pre-treated with or without PP2 analog, a Src inhibitor, prior to incubation with or
without VEGF (20ng/ml) for 10 min. Cell lysates were fractionated by SDS-PAGE gel and immunoblotted with
phospho-specific Raf antibody. Blots were stripped and re-probed with Raf-1 antibody to assess loading (bottom
panel). A representative blot from 3 experiments is shown.
Fig 5.
VEGF induces tyrosine phosphorylation of Pyk2, and, Ca++
-dependence of VEGF activation of kinases.
Panels A. MCT cells were incubated with or without VEGF for 10 min. Immunoblot analysis was done with anti-
phospho-specific antibodies for Pyk2. The blots were stripped and re-probed with Pyk2 antibody (bottom panel) to
assess loading. A representative blot from three experiments is shown.
Panels B, C, and D. Serum-starved MCT cells were pre-treated with BAPTA/AM (100 µM) for 30 min followed
by incubation with or without VEGF (20 ng/ml). Equal amounts of cell lysates were fractionated by SDS PAGE
and immunoblotted with phospho-specific antibodies for Pyk2 (B), c-Src (C) and Erk (D). The lower panel in each
section shows immunoblotting with antibodies against Pyk2, c-Src and Erk, respectively, done to assess loading.
Representative blots from three experiments are shown for each kinase. The graph shows quantification of
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

12
composite data from 3 experiments on BAPTA/AM abolition of Erk activation by VEGF in panel D. *p<0.01
VEGF Vs Control, **p<0.01 VEGF Vs VEGF+BAPTA/AM by ANOVA.
Fig 6.
VEGF induces phosphorylation of (Mnk1) and promotes its shift from cytoplasm into nucleus.
Panel A. Serum-deprived MCT cells were stimulated with VEGF 20 ng/ml. The lysates were fractionated by SDS
PAGE and immunoblotted with phospho-specific antibodies for Mnk1. Probing with Mnk1 antibody (lower panel)
was performed to assess loading. A representative blot from 3 experiments is shown.
Panel B. Quiescent MCT cells were immunostained with anti-phospho Mnk1 antibody with or without incubation
with VEGF (20ng/ml), for various time durations. Immunofluorescent staining was performed as described in
Methods and the images were analyzed by confocal microscopy. Control cells showed little staining for
phosphorylated Mnk1. After incubation with VEGF for 2 min, intense staining for phospho Mnk1 in the
cytoplasm is seen followed by peri-nuclear staining at 5 min. The nuclear staining is maximal at 15 min and gets
dispersed by about 30 min. The graph represents percent of cells that showed nuclear localization of phospho-
Mnk1 at individual time points following VEGF treatment. Cells at time zero served as control. Equal numbers of
cells were plated in 8-chambered slides; after treatment with VEGF, cells were viewed at uniform magnification
of 40X and counted for phospho-Mnk1 nuclear localization. Composite data from 3 experiments are shown in a
graph (p<0.01 time zero Vs 5 min and 15 min, by ANOVA).
Fig 7.
Panels A, B, and C. VEGF-induced Mnk1 phosphorylation is dependent on PLC , c-Src and Erk. Cells
were serum-starved overnight and subjected to pre-treatment with or without U73122 (5 µM), a PLC inhibitor (A),
PP2 (10 µM), a Src inhibitor (B), and U0126 (25 µM), a MEK inhibitor (C), prior to incubation with or without
VEGF 20 ng/ml for 10 min. Equal amounts of cell lysates were run on a SDS-PAGE and immunoblotted with
phospho-specific antibody for Mnk1. The lower panels of A, B and C show the blots probed with antibody against
Mnk1 to assess loading. Representative blots from 3 experiments with each kinase inhibitor are shown.
Fig 8.
VEGF stimulates eIF4E phosphorylation that is dependent on PLC , c-Src and Erk.
Panel A. Serum-deprived MCT cells were stimulated with VEGF 20 ng/ml. The lysates were fractionated by SDS
PAGE and immunoblotted with a phospho-specific antibody for eIF4E. Loading was assessed by probing with
eIF4E antibody (lower panel). A representative blot from 3 experiments is shown for each kinase.
Panels B, C, and D. Quiescent cells were subjected to pre-treatment with or without U73122 (5 µM), a PLC
inhibitor (B), PP2 analog (10 µM), a Src inhibitor (C) and U0126 (25 µM), a MEK inhibitor (D), prior to
incubation with or without VEGF 20 ng/ml for 10 min. Equal amounts of cell lysates were run on a SDS-PAGE
and immunoblotted with phospho-specific antibody for eIF4E. The lower panels of B, C and D show the blots
probed with eIF4E antibody to assess loading. Representative blots from 3 experiments with each kinase inhibitor
are shown.
Fig. 9.
Mnk1 mediates eIF4E phosphorylation and is needed for VEGF-induced protein synthesis
Panel A. MCT cells were transfected with a plasmid carrying DN-Mnk1 (pEBG-T2/A2) or empty vector and
allowed to grow for 48 hours. Phosphorylation of eIF4E was assessed by immunoblotting as described in Fig. 8A.
A representative blot from 3 experiments is shown.
Panel B. MCT cells were transfected with dominant negative Mnk1 or empty vector as described in panel A, and
treated with or without VEGF prior to addition of [35
S]-methionine and incubated for a total of 2 hours. Equal
amount of protein from the lysates were taken for the estimation of [35
S]-label incorporation into TCA-precipitable
protein and expressed as percentage of control (Mean±SE). Data from 3 experiments are shown in a graph;
*p<0.01 for VEGF Vs vector-transfected control, **p<0.01 for VEGF Vs VEGF-treated DN-Mnk1 expressing
cells, by ANOVA.
Fig. 10.
VEGF-stimulated protein synthesis is dependent on phosphorylation of PLC .
Panel A. Quiescent cells were stimulated with VEGF simultaneously in the presence of [35
S]-methionine for 2
hours with or without pre-incubation with 5 µM U73122, a PLC inhibitor, for 30 min. Incorporation of radiolabel
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

13
into TCA-precipitable protein was taken as a measure of protein synthesis and expressed as percentage of control
(Mean ± SE). Composite data from 3 experiments are shown in a graph; *p<0.01 VEGF Vs control, **p<0.01 for
VEGF+U73122 Vs VEGF, by ANOVA.
Panel B. MCT cells transfected with empty plasmid vector or with PLCz, a plasmid carrying a dominant negative
construct of PLC , were incubated with VEGF in the presence of [35
S]-methionine for 2 hours. Protein synthesis
was measured as in Panel A. The values are expressed as percentage of control from an average of 3 individual
experiments; *p<0.01 VEGF Vs control, **p<0.01 for VEGF+U73122 Vs VEGF, by ANOVA.
Fig 11.
Erk-1/-2 MAP kinase mediated signaling mechanism involved in VEGF-induced protein synthesis.
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

Figure 1
0 2 5 10 15 30 60050100150200250300350
Time in minutes
P E
rk/T
ota
l E
rk
(% o
f C
on
tro
l)
A
0 2 5 10 15 30 60
Erk
VEGF(20 ng/ml)
Phospho-Erk
0
50
100
150
[35S
] -M
et.
Inco
rp[
% o
f co
ntr
ol]
VEGF
U0126
+ – +–
B
VEGF (20 ng/ml) + – + – +–
– – – – + +– – + + – –LY (25 µM)
U0126 (25 µM)
MBP
Erk
C
0
50
100
150
200
Erk
kin
ase
act
ivit
y (%
of
con
tro
l)
VEGF
LY294002 U0126
+ – + – +–
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

Figure 2
A B
Phospho-PLCγ
VEGF(20 ng/ml)Time in minutes
PLCγ
0 5 10 15 30 60
VEGF (20 ng/ml) U73122 (5 µM)
Phospho-Src
Src
_ + _ +_ _ ++
F
_ + _ +_ _ ++
E
Phospho-Src
Src
VEGF(20 ng/ml)
SU1498 (10 µM)
D
Phospho-PLCγ
PLCγ
VEGF(20 ng/ml)_ + _ +_ _ ++SU1498 (10 µM)
220kD
97kD
66kD
46kD
VEGF(20 ng/ml) _ + _ +_ _ ++SU1498 (10 µM)
Actin
C
Phospho-Src
Src
0 5 10 15 30 60VEGF(20 ng/ml)Time in minutes
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from
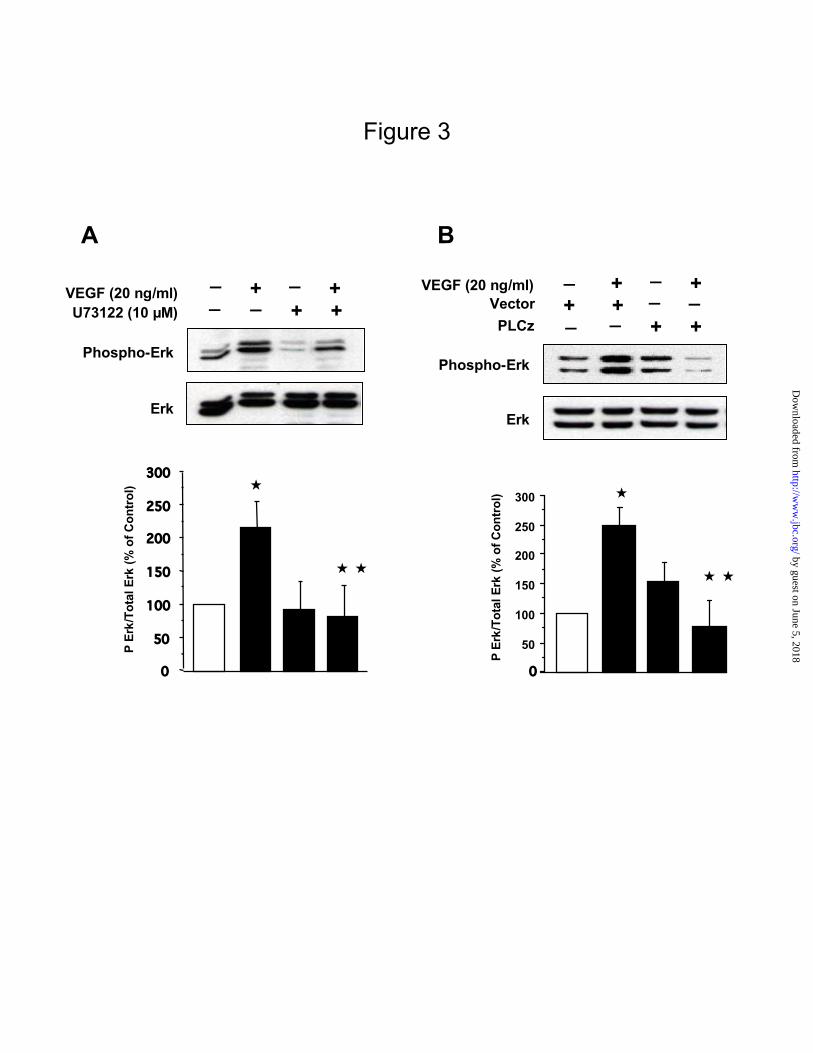
Figure 3
Phospho-Erk
VEGF (20 ng/ml)
Erk
U73122 (10 µM)
_ + _ +_ _ ++
A
0
50
100
150
200
250
300
P E
rk/T
ota
l E
rk (
% o
f C
on
tro
l)
B
Phospho-Erk
VEGF (20 ng/ml)
Erk
_ + _ +
_ _ ++
__++
PLCz
Vector
50
100
150
200
250
300 P
Erk
/To
tal
Erk
(%
of
Co
ntr
ol)
0
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

0 5 10 15 30 60
Phospho-Raf
VEGF (20 ng/ml)
Raf-1
C
Figure 4
D
_ + _ +_ _ ++
Phospho-Raf
VEGF (20 ng/ml)
Raf-1
PP2(10 µM)
A
VEGF(20 ng/ml)
Phospho-Erk
Erk
_ + _ +_ _ ++PP1 (10 µM)
0
50100150
200250300
350400
P Er
k/To
tal E
rk (
% o
f Co
ntro
l)
B
Phospho-Erk
VEGF (20 ng/ml)
Erk
_ +_
+_ _ ++
__++DN-SrcVector
0
50
100
150
200
250
300
350
P Er
k/To
tal E
rk (
% o
f Co
ntro
l)
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

Figure 5
B
Phospho-Pyk2
Pyk2
VEGF (20 ng/ml)
BAPTA/AM (100 µM)
_ + _ +_ _ ++A
VEGF (20 ng/ml)
Phospho-Pyk2
Pyk2
0 5 10 15 30 60
BAPTA/AM (100 µM)
Phospho-Src
Src
VEGF (20 ng/ml)
BAPTA/AM (100 µM)
_ + _ +_ _ ++
C
D
0
50
100
150
200
250
300
350
400
P Er
k/To
tal E
rk (
% o
f Co
ntro
l)
Phospho-Erk
Erk
VEGF (20 ng/ml) _ + _ +_ _ ++
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

AFigure 6
Phospho-Mnk
VEGF (20 ng/ml)Time in minutes 0 5 10 15 30 60
Total Mnk
2 min 5 min
15 min 30 min
0 minB
0 2 5 15 30 0
5
10
15
20
25
30
35
Num
ber
of c
ells
. % o
f co
ntro
l
VEGF (20 ng/ml)
Time in minutes
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

A
Phospho-Mnk
U73122 (5 µM) VEGF(20 ng/ml) _ + _ +
_ _ ++
C
Phospho-Mnk
VEGF(20 ng/ml) U0126 (25 µM)
_ + _ +_ _ ++
Figure 7
B
Phospho-Mnk
PP2 (10 µM)VEGF(20 ng/ml) _ + _ +
_ _ ++
Total Mnk
Total Mnk
Total Mnk
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from
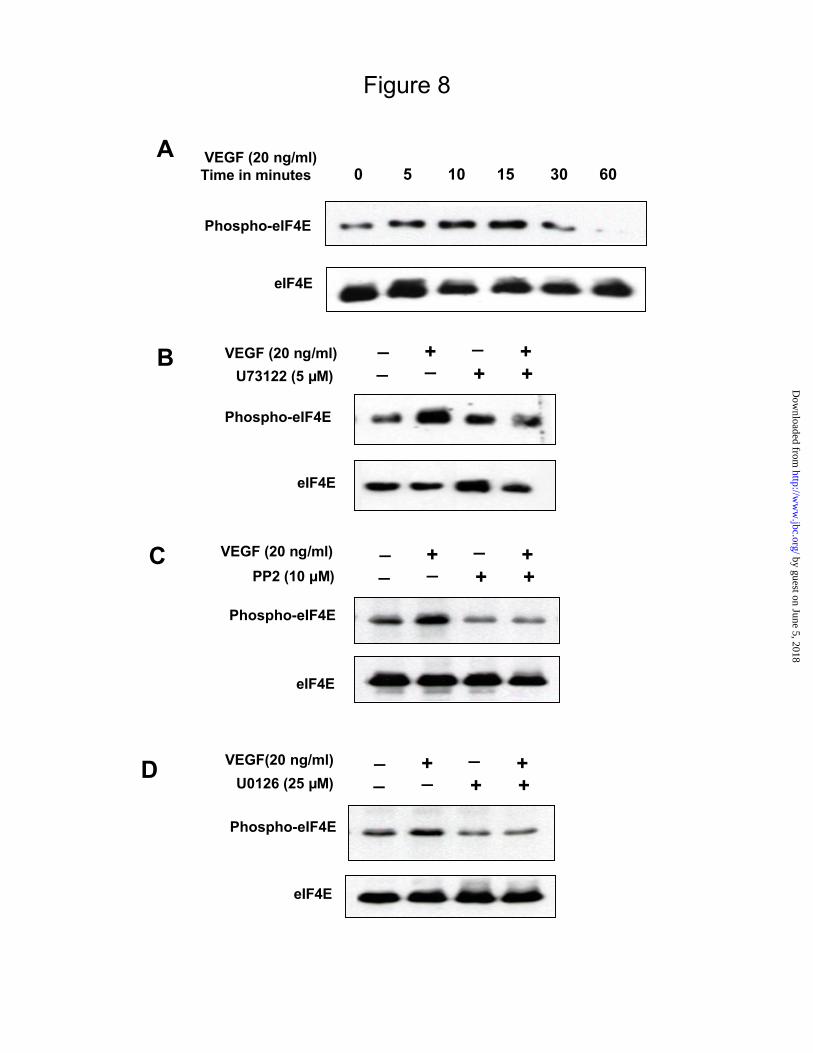
Figure 8
B VEGF (20 ng/ml)
Phospho-eIF4E
eIF4E
U73122 (5 µM)
_ + _ +_ _ ++
D VEGF(20 ng/ml)
Phospho-eIF4E
eIF4E
U0126 (25 µM)
_ + _ +_ _ ++
C VEGF (20 ng/ml)
eIF4E
Phospho-eIF4E
PP2 (10 µM)
_ + _ +_ _ ++
Phospho-eIF4E
eIF4E
VEGF (20 ng/ml)Time in minutes 0 5 10 15 30 60
A
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

Figure 9
VEGF (20 ng/ml)
DN-MnkVector
_ _ + ++ +
_ _++_ _
Phospho-eIF4E
eIF4E
A
B
VEGF (20 ng/ml)
DN-MnkVector
_ _ + ++ +
_ _++_ _0
20
40
60
80
100
120
140
160
180
[35S
] -M
et.
Inco
rp. %
of
con
tro
l
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

Figure 10
A
0
20
40
60
80
100
120
140
160
_ VEGF (20 ng/ml)
U73122 (5 µM)+ _ +
_ _ + +
[35S
] -M
et.
Inco
rp. %
of
con
tro
l
B
0
50
100
150
200
250
300
350
400
VEGF (20 ng/ml)
PLCzVector
+_+
_ _ + +
_
+ + _ _
[35S
] -M
et.
Inco
rp. %
of
con
tro
l
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

Figure 11
Pyk2
c-Src PP2Src-DN
Raf MEK
Erk-1/-2
Mnk-1/-2
eIF4E
Protein synthesis
U0126
VEGF + VEGF RECEPTOR 2
PLCγ
SU1498
U73122 PLCz
BAPTA/AM
Mnk-DN
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from

PP2(10 µM) - - +VEGF (20ng/ml) - + +
Phospho-PLCγ
PLCγ
Supplementary figure 1. (S1)
Cells were incubated with or without VEGF for 10 min after pre-incubation with PP2 for 30 min. Equal amounts of cell lysates were
fractionated on SDS PAGE and immunoblotted with phospho-specific antibody against PLCγ. Loading was assessed by immunoblotting
with an antibody against PLCγ. PP2 did not inhibit VEGF-induced PLCγ phosphorylation.
by guest on June 5, 2018 http://www.jbc.org/ Downloaded from

Goutam Ghosh Choudhury and Balakuntalam S. KasinathMeenalakshmi Malini Mariappan, Duraisamy Senthil, Kavithalakshmi S. Natarajan,
renal epithelial cells-Erk axis in VEGF-induced eIF4E phosphorylation and protein synthesis inγPLC
published online May 26, 2005J. Biol. Chem.
10.1074/jbc.M504861200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
Supplemental material:
http://www.jbc.org/content/suppl/2005/06/14/M504861200.DC1
by guest on June 5, 2018http://w
ww
.jbc.org/D
ownloaded from