platelets - ouhsc publications/platelets... · characterised by von willebrand factor...
TRANSCRIPT

HAEMATOLOGY
THE LANCET • Vol 355 • April 29, 2000 1531
Platelets are the smallest of blood cells, being onlyfragments of megakaryocyte cytoplasm, yet they have acritical role in normal haemostasis and are importantcontributors to thrombotic disorders. Understanding ofthe role of platelets in haemostasis and definition ofdisorders caused by abnormal platelet function have ledto important new therapies for thrombotic disease.
Platelet productionThe development of megakaryocytes and production ofplatelets are unique processes. Megakaryocytematuration involves nuclear duplication without celldivision, resulting in giant cells. Cytoplasmic organellesare organised into domains representing nascentplatelets, demarcated by a network of invaginatedplasma membranes. Within the marrow, megakaryocytesare localised next to the sinusoidal walls, whichfacilitates the exit of large segments of cytoplasm into thecirculation. The fragmentation of megakaryocytecytoplasm into individual platelets then results from theshear forces of circulating blood, perhaps largely in thepulmonary circulation.1
Thrombopoietin is the dominant hormone controllingmegakaryocyte development, but many cytokines andhormones take part, including interleukins 3, 6, and 11.2
Thrombopoietin was first identified as the ligand for areceptor on the surface membranes of megakaryocytesand platelets, termed c-mpl. This receptor is the normalproto-oncogene of the viral oncogene present in themurine myeloproliferative leukaemia virus (v-mpl).Inhibition of C-MPL expression in marrow cellsspecifically blocks megakaryocyte development. Micegenetically deficient in c-mpl have 85% fewer plateletsand megakaryocytes than normal but other haemopoieticcells are present in normal amounts; thrombopoietintherefore seems to be the major, but not the only,regulator of platelet production.3
Lancet 2000; 355: 1531–39
Hematology-Oncology Section, Department of Medicine,University of Oklahoma Health Sciences Center, PO Box 26901,Oklahoma City, OK 73190, USA (J N George MD)
(e-mail: [email protected])
Other clinical observations supporting the importanceof thrombopoietin and its receptor are the association ofmutations of C-MPL with congenital amegakaryocyticthrombocytopenia4 and the identification of auto-antibodies that neutralise the activity of thrombopoietinin a patient with acquired amegakaryocytic thrombo-cytopenia.5
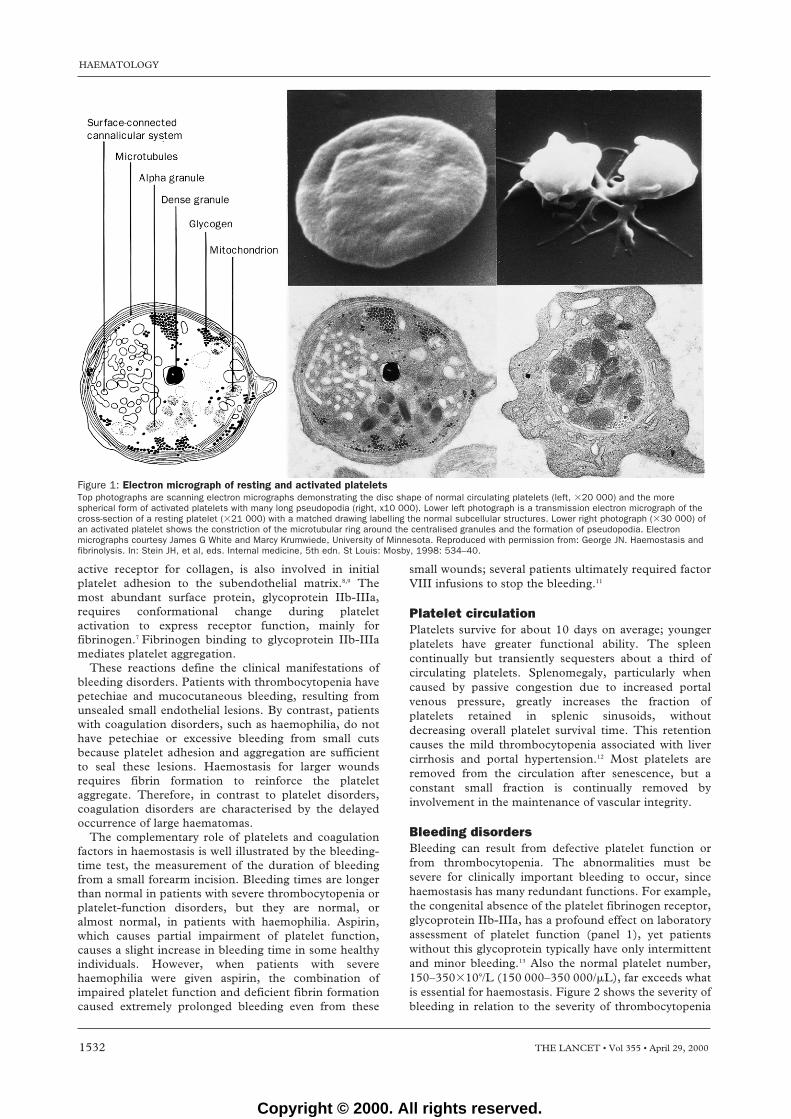
Platelet structure and functionOn activation, platelets change from the normal discshape to a compact sphere with long dendritic extensionsfacilitating adhesion (figure 1). The cytoplasm is rich inactin and myosin which bring about the change in shapeand retraction of the clot. There are two classes ofsecretory granules. The first type are dense granules thatsecrete ADP and calcium, which reinforce plateletaggregation and platelet-surface coagulation reactions.The second type are � granules, which secrete a vastarray of proteins: some, such as von Willebrand factorand platelet factor 4, are synthesised by megakaryocytes;others, such as fibrinogen, are acquired from the plasmaby receptor-mediated endocytosis; still others, such asthe abundant plasma proteins, albumin and IgG, areacquired by fluid-phase pinocytosis.6 Both secretion andendocytosis are facilitated by a network of deepinvaginations of the platelet surface membrane, thesurface-connected canalicular system, which isreminiscent of the demarcation membranes that initiallydefined platelet zones within their parent mega-karyocytes.
Platelet-membrane glycoprotein receptors mediateadhesion to subendothelial tissue and subsequentaggregation to form the initial haemostatic plug (panel1).7–10 The largest glycoprotein is designated I, thesmallest IX. Letters a and b were added when bettertechniques allowed resolution of single protein bands onelectrophoresis into two separate bands (eg, glycoproteinI became glycoprotein Ia and Ib).
Glycoprotein Ib-V-IX is a constitutively activereceptor for von Willebrand factor, causing immediateplatelet attachment to exposed perivascular vonWillebrand factor.10 Glycoprotein Ia-IIa, a constitutively
Platelets
James N George
Haematology
•Platelets, derived from megakaryocte cytoplasm, have a critical role in normal haemostasis, and in thromboticdisorders.
•The development of megakaryocytes is controlled by thrombopoietin, which binds to c-mpl on the surface ofplatelets and megakaryocytes.
•Platelet membrane glycoproteins mediate binding to subendothelial tissue and aggregation into haemostaticplugs.
•Thrombocytopenia and disorders of platelet function cause petechiae and mucocutaneous bleeding.•Drugs causing specific inhibition of platelet function are important in the treatment of cardiovascular and
cerebrovascular disease.
For personal use only. Not to be reproduced without permission of The Lancet.
Copyright © 2000. All rights reserved.
active receptor for collagen, is also involved in initialplatelet adhesion to the subendothelial matrix.8,9 Themost abundant surface protein, glycoprotein IIb-IIIa,requires conformational change during plateletactivation to express receptor function, mainly forfibrinogen.7 Fibrinogen binding to glycoprotein IIb-IIIamediates platelet aggregation.
These reactions define the clinical manifestations ofbleeding disorders. Patients with thrombocytopenia havepetechiae and mucocutaneous bleeding, resulting fromunsealed small endothelial lesions. By contrast, patientswith coagulation disorders, such as haemophilia, do nothave petechiae or excessive bleeding from small cutsbecause platelet adhesion and aggregation are sufficientto seal these lesions. Haemostasis for larger woundsrequires fibrin formation to reinforce the plateletaggregate. Therefore, in contrast to platelet disorders,coagulation disorders are characterised by the delayedoccurrence of large haematomas.
The complementary role of platelets and coagulationfactors in haemostasis is well illustrated by the bleeding-time test, the measurement of the duration of bleedingfrom a small forearm incision. Bleeding times are longerthan normal in patients with severe thrombocytopenia orplatelet-function disorders, but they are normal, oralmost normal, in patients with haemophilia. Aspirin,which causes partial impairment of platelet function,causes a slight increase in bleeding time in some healthyindividuals. However, when patients with severehaemophilia were given aspirin, the combination ofimpaired platelet function and deficient fibrin formationcaused extremely prolonged bleeding even from these
small wounds; several patients ultimately required factorVIII infusions to stop the bleeding.11
Platelet circulationPlatelets survive for about 10 days on average; youngerplatelets have greater functional ability. The spleencontinually but transiently sequesters about a third ofcirculating platelets. Splenomegaly, particularly whencaused by passive congestion due to increased portalvenous pressure, greatly increases the fraction ofplatelets retained in splenic sinusoids, withoutdecreasing overall platelet survival time. This retentioncauses the mild thrombocytopenia associated with livercirrhosis and portal hypertension.12 Most platelets areremoved from the circulation after senescence, but aconstant small fraction is continually removed byinvolvement in the maintenance of vascular integrity.
Bleeding disordersBleeding can result from defective platelet function orfrom thrombocytopenia. The abnormalities must besevere for clinically important bleeding to occur, sincehaemostasis has many redundant functions. For example,the congenital absence of the platelet fibrinogen receptor,glycoprotein IIb-IIIa, has a profound effect on laboratoryassessment of platelet function (panel 1), yet patientswithout this glycoprotein typically have only intermittentand minor bleeding.13 Also the normal platelet number,150–350�109/L (150 000–350 000/�L), far exceeds whatis essential for haemostasis. Figure 2 shows the severity ofbleeding in relation to the severity of thrombocytopenia
HAEMATOLOGY
1532 THE LANCET • Vol 355 • April 29, 2000
Figure 1: Electron micrograph of resting and activated plateletsTop photographs are scanning electron micrographs demonstrating the disc shape of normal circulating platelets (left, �20 000) and the morespherical form of activated platelets with many long pseudopodia (right, x10 000). Lower left photograph is a transmission electron micrograph of thecross-section of a resting platelet (�21 000) with a matched drawing labelling the normal subcellular structures. Lower right photograph (�30 000) ofan activated platelet shows the constriction of the microtubular ring around the centralised granules and the formation of pseudopodia. Electronmicrographs courtesy James G White and Marcy Krumwiede, University of Minnesota. Reproduced with permission from: George JN. Haemostasis andfibrinolysis. In: Stein JH, et al, eds. Internal medicine, 5th edn. St Louis: Mosby, 1998: 534–40.
Copyright © 2000. All rights reserved.

in patients with idiopathic thrombocytopenic purpura:spontaneous major bleeding did not occur until theplatelet count was less than 10�109/L, and even then itwas uncommon. Also, the group of patients representedin figure 2 may be more severely affected than currentunselected patients with idiopathic thrombocytopenicpurpura, since they were treated at the University ofMichigan Medical Center, a tertiary referral centre, somay have been more severely affected; automated plateletcounts were not widely used at the time of the study andtherefore mildly affected patients were not recognised;and aspirin was not fully appreciated as a risk factor forexacerbation of bleeding.
Disorders of platelet functionAlthough hereditary disorders of platelet function arerare, they define the bleeding symptoms caused byplatelet abnormalities.13 Mucocutaneous bleeding, suchas purpura, epistaxis, gingival bleeding, andmenorrhagia, are prominent features; gastrointestinalbleeding is common; visceral haematomas,haemarthroses, and intracerebral haemorrhage rarely, ifever, occur in the absence of trauma. Even when thegenetic defect is severe, as in patients with Glanzmann’sthrombasthenia who have undetectable plateletglycoprotein IIb-IIIa, bleeding symptoms are sporadic;this observation emphasises that other platelet receptorscan compensate for the absent fibrinogen receptor.
Acquired platelet function defects are mild andubiquitous, considering, for example, the number ofpeople who take aspirin regularly and who therefore haveimpaired platelet function caused by irreversibleinhibition of cyclo-oxygenase-dependent thromboxaneformation. More than 100 other drugs, foods, spices,and vitamins have been reported to impair plateletfunction.14 For almost all agents, the data are limited todescriptions of abnormal in-vitro platelet aggregationtests or a long bleeding time, which may have no clinicalimportance. Only for aspirin has an increased risk ofbleeding been clearly documented, and that proof camefrom a study of 22 071 physicians followed up for 5years, to assess the efficacy of aspirin (325 mg onalternate days) for the primary prevention of myocardial
infarction. 27% of the physicians taking aspirin reportedbleeding symptoms consistent with impaired plateletfunction; however, perhaps more important was theobservation that 20% of those taking placebo alsoreported “excessive” bleeding.15 These data emphasisethe simple, familiar facts that bleeding symptoms occurin normal people and that excessive bleeding is difficultto define. Negligible or no excessive bleeding may beexpected in acquired platelet-function disorders, sincethe impairment of platelet function is much less severethan in Glanzmann’s thrombasthenia, and since patientswith thrombasthenia may have no bleeding for manyyears.13 However, aspirin, and presumably other causesof abnormal platelet function such as chronic renalfailure and cardiac surgery,14 can profoundly exacerbatebleeding in patients who already have compromisedhaemostasis from any cause, such as a coagulationdisorder11 or anticoagulant therapy.
HAEMATOLOGY
THE LANCET • Vol 355 • April 29, 2000 1533
Panel 1: Major platelet membrane glycoprotein receptors
Receptor Structure Function Polymorphisms DisordersGlycoprotein Integrin family of On platelet activation, Leu33/Pro33 creates platelet- Deficiency results in Glanzmann’sIIb-IIIa receptors (aIIb�3); becomes a receptor for specific antigens, HPA-1a/1b, thrombasthenia, characterised by absent
80 000 molecules fibrinogen, also for the important antigens for aggregation in response to all physiologicalper platelet von Willebrand factor, neonatal alloimmune agonists and absent clot retraction;
fibronectin, vitronectin, thrombocytopenia platelet number and morphology are normaland thrombospondin
Glycoprotein Integrin family of Constitutively active C807T silent dimorphism affects Deficiency, with absent collagen-inducedIa-IIa receptors (�2�1); receptor for collagen �2�1 surface density and collagen aggregation, may result in mildly increased
900–2300 molecules receptor activity; may influence bleedingper platelet occurrence of coronary
thrombosis and stroke
Glycoprotein Complex of four gene Constitutively active None are clinically important Deficiency results in Bernard-SoulierIb-V-IX products, each receptor for insoluble syndrome, characterised by
characterised by von Willebrand factor thrombocytopenia, giant platelets, and leucine-rich repeats; in the perivascular matrix lack of binding of von Willebrand factorcovalent heterodimerof Ib�–Ib� and non- Mutations with increased function resultcovalent association in platelet-type von Willebrand’s disease;with glycoproteins characterised by spontaneous binding ofV and IX; 25 000 von Willebrand factor causing itsmolecules per platelet depletion from plasma
4
Ble
edin
g se
verit
y
3
2
1
010 20 40 50 60
Platelet count (�109/L)80 100 120 140
Figure 2: Bleeding severity in relation to the platelet count inpatients with idiopathic thrombocytopenic purpuraBleeding symptoms: O=no bleeding or purpura; 1=minor bruising withtrauma; 2=spontaneous but self-limited bleeding (eg, purpura, epistaxis);3=spontaneous bleeding requiring medical attention (eg, epistaxisrequiring nasal packing); 4=major bleeding requiring emergencytreatment. Reproduced and modifed with permission from Lacey andPenner, Seminars in Thrombosis and Hemostasis 1977; 3: 3.
Copyright © 2000. All rights reserved.

The low risk of major bleeding in patients withhereditary and acquired disorders of platelet functionhas been exploited in the use of aspirin and plateletglycoprotein IIb-IIIa blockers as antithrombotic agents.
Thrombocytopenia occurring as an isolatedabnormalityThrombocytopenia in an otherwise healthy personThrombocytopenia may be suspected from bleedingsymptoms, or may be discovered by a routine blood countin a person without symptoms. The incidental discoveryof thrombocytopenia, which has occurred since plateletcounting became routine, has shown the existence ofpseudothrombocytopenia and expanded the clinical rangeof thrombocytopenic disorders to include manysymptom-free patients. The investigation andmanagement of patients with isolated thrombocytopeniais illustrated by the algorithm in figure 3 (the algorithmdoes not include other disorders associated withthrombocytopenia, such as chronic liver disease withhypersplenism, and autoimmune, lymphoproliferative,and infectious diseases, because they are generallyaccompanied by signs and symptoms suggesting systemicdisease). Isolated thrombocytopenia initially diagnosed asidiopathic thrombocytopenic purpura may subsequentlybe diagnosed as congenital thrombocytopenia ormyelodysplasia. Congenital thrombocytopenias are rarebut their recognition is critical for avoiding unnecessarytreatment; commonly they manifest giant platelets, butplatelet size may be normal or small.
PseudothrombocytopeniaMany large studies have shown that falsely low plateletcounts, due in most cases to platelet agglutinationcaused by the anticoagulant edetic acid (EDTA), usedfor routine blood counts (figure 4), occurs in about oneperson in 1000, irrespective of the presence or absenceof any disease. Therefore the diagnosis ofthrombocytopenia must always be confirmed byexamination of the peripheral-blood smear. In patientswith edetic-acid-induced pseudothrombocytopenia, anaturally occurring antibody causes plateletagglutination by binding to normally concealed epitopeson glycoprotein IIb, which are revealed when edetic acidchelates calcium and thereby alters the glycoprotein IIb-IIIa molecule.16 Other anticoagulants, such as sodiumcitrate used for coagulation assays, generally do notperturb glycoprotein IIb-IIIa and therefore do not leadto platelet agglutination in these individuals. Edetic-acidagglutinins are not clinically important,17 except for therisk of a mistaken diagnosis of thrombocytopenia andresulting inappropriate treatments.
Hereditary thrombocytopeniasAlthough rare, many types of hereditarythrombocytopenia have been described. Some areautosomal dominant traits, such as the May-Hegglinanomaly (characterised by giant platelets and neutrophilinclusions) and type 2B von Willebrand’s disease.Others are X-linked, such as the Wiskott-Aldrichsyndrome (characterised by small platelets and
HAEMATOLOGY
1534 THE LANCET • Vol 355 • April 29, 2000
Assess-peripheral blood smear
Platelet clumps,pseudothrombocytopenia
True thrombocytopenia
Normal platelet morphology
ITP: defined as isolatedthrombocytopenia withno clinically apparentassociated disordersor other causes ofthrombocytopenia
Giant platelets,suspect hereditarythrombocytopenia
Drug-inducedthrombocytopenia
Hereditarythrombocytopenia
Pregnancy
Hypertension,proteinuria:thrombocytopeniadue to eclampsia
Normal bloodpressure,no proteinuria
Early-mid pregnancy,severe thrombocytopenia:suspect ITP
Late pregnancy, mildthrombocytopenia:suspect gestationalthrombocytopenia
Figure 3: Algorithm for investigation of isolated thrombocytopenia in an otherwise healthy person
Copyright © 2000. All rights reserved.

immunodeficiency). And yet others are autosomalrecessive traits, such as the Bernard-Soulier syndrome(also characterised by giant platelets; figure 5). Mosthereditary thrombocytopenias are apparent in infancy,though some are symptomless and not detected untiladulthood. The important issue is to distinguish thesedisorders from idiopathic thrombocytopenic purpuraand to avoid inappropriate treatment. A hereditarydisorder should be considered in patients who have adiagnosis of idiopathic thrombocytopenic purpura withpersistent thrombocytopenia, particularly children andadolescents in whom chronic idiopathicthrombocytopenic purpura is uncommon.18 Hereditarythrombocytopenias should also be considered whengiant platelets are present. Although some patients withidiopathic thrombocytopenic purpura have platelets thatare somewhat larger than normal, truly giant plateletsapproaching the diameter of red cells (figures 3 and 5)occur only in congenital disorders.
Drug-induced thrombocytopeniaIn patients with unexpected, isolated thrombocytopenia,a drug-induced mechanism must be considered. Thebest-described mechanism is platelet destruction bydrug-dependent antibodies against platelets formedwhen binding of the drug to a platelet-membrane
molecule exposes a normally concealed aminoacid orcarbohydrate sequence that becomes antigenic(neoepitope). In affected patients the offending drugmay bind only weakly and reversibly, if at all, to eitherthe antibody or platelets, but it can promote high-affinitybinding of the antibody to the platelet antigen. Drug-dependent antibody-binding sites (the neoepitopes) arehighly specific and are restricted to small domains onglycoprotein IX, glycoprotein Ib, and less commonly onglycoprotein IIb-IIIa.19,20
The suspicion of drug-induced thrombocytopenia isconfirmed by recovery from thrombocytopenia after thesuspected drug is withdrawn, which is predictable within5–7 days.21 Laboratory tests for drug-dependentantibodies are not routinely and promptly available. Toaddress the issue of which drugs are most likely to causethrombocytopenia, a systematic review analysed allpublished case reports with defined levels of evidence todocument the causal relation between the drug andthrombocytopenia.21 In that review, case reportsdescribing 98 drugs provided definite or probableevidence that the drug caused the thrombocytopenia.The most commonly reported drugs with definiteevidence were quinidine, quinine, rifampicin, andtrimethoprim-sulphamethoxazole.21 The databaseestablished by this systematic review, including the fulllist of drugs and articles and data on clinical course andseverity of bleeding, is updated annually and is availableat the website: http://moon.ouhsc.edu/jgeorge.
Heparin-induced thrombocytopenia needs specificemphasis because of its frequency and the variability ofits clinical manifestations, which include thromboticcomplications. Antibodies are specific for heparin boundto platelet factor 4, a positively charged platelet-secretedprotein that binds tightly to the negatively chargedheparin. The complex of these three molecules can causeplatelet activation; activated platelets then increase therisk of further thrombosis.22 Obviously, since thesepatients are being treated with heparin, they alreadyhave, or are at risk of, thrombosis. In most patients thethrombocytopenia is mild and transient, occurringseveral days after the occurrence of thrombosis whenheparin can safely be discontinued. Severethrombocytopenia with thrombosis is much lesscommon, and its frequency is further decreasing withshorter durations of heparin treatment and increased useof low-molecular-weight heparins, which are lesscommonly associated with thrombocytopenia.22
Idiopathic (or immune) thrombocytopenic purpuraIdiopathic thrombocytopenic purpura is defined asisolated thrombocytopenia with no clinically apparentassociated disorders (eg, HIV infection, systemic lupuserythematosus). No specific criteria establish thediagnosis of idiopathic thrombocytopenic purpura; thediagnosis relies on the exclusion of other causes ofthrombocytopenia.23 Idiopathic thrombocytopenicpurpura is caused by autoantibody destruction ofplatelets, but tests for antiplatelet antibodies have not yetbeen shown to be important for diagnosis andmanagement.23 If the history, physical examination, andinitial blood counts with examination of the blood smearare compatible with the diagnosis and do not includeatypical findings that are uncommon in idiopathicthrombocytopenic purpura or suggest other causes forthe thrombocytopenia, further diagnostic tests are
HAEMATOLOGY
THE LANCET • Vol 355 • April 29, 2000 1535
Figure 4: Blood film showing platelet clumps, made fromblood anticoagulated with edetic acidReproduced with permission from: George JN. Thrombocytopenia. In:Beutler E, Lichtman MA, Coller BS, Kipps TJ, eds. Hematology, 5th edn.New York: McGraw-Hill, 1995: 1356.
Figure 5: Blood film showing giant platelets, made from apatient with Bernard-Soulier diseaseReproduced with permission from: Hoffbrand AV, Pettit JE. Sandoz slideatlas of clinical haematology. London: Gower Medical Publishers, 1990.
Copyright © 2000. All rights reserved.

unnecessary in most patients. Bone-marrow examinationmay be appropriate in older patients, to excludemyelodysplasia (which can present as isolatedthrombocytopenia), and in patients with severethrombocytopenia in whom initial treatment isunsuccesful and in whom splenectomy is considered.23 Anormal bone marrow is consistent with the diagnosis ofidiopathic thrombocytopenic purpura.
In adults, idiopathic thrombocytopenic purpuratypically has an insidious onset and a chronic course.The incidence of the disorder is increasing, especiallyamong older adults, with the routine reporting of plateletcounts;24 currently 30–40% of adult patients aresymptom-free and the diagnosis is made incidentally.Major bleeding is not an important risk unless theplatelet count is less than 10�109/L (figure 2).Intracerebral haemorrhage and death occur, but toorarely for an incidence to be calculated.23 Adult patientswith severe and symptomatic thrombocytopeniaare treated initially with prednisone 1 mg/kgdaily; however, when symptomless thrombocytopeniais incidentally discovered and the platelet count isover 30�109/L, patients can safely be observed with nospecific treatment.23 The aim of treatment is not cure ofthe idiopathic thrombocytopenic purpura, butprevention of bleeding. Splenectomy is the conventionaltreatment for adults who continue to have severe andsymptomatic thrombocytopenia despite treatment withprednisone. Although long-term remissions, with normalplatelet counts on no further treatment, occur in onlyabout 50% of patients, the results are better than withother available treatments. For patients who continue tohave severe and symptomatic thrombocytopenia aftersplenectomy, various immunosuppressive regimens havebeen used, all with some reports of success, but mostpatients have incomplete responses.
Emergency treatment for life-threatening bleedingconsists of platelet transfusions, high doses ofglucocorticoids given intravenously (eg, 1000 mgmethylprednisolone ), and intravenous immunoglobulin.
In children younger than 10 years, idiopathicthrombocytopenic purpura typically has an acute onsetand resolves spontaneously within 6–12 months.Therefore initial management is typically even moreconservative than that in adults. Some paediatrichaematologists recommend no specific treatment evenfor children with severe thrombocytopenia, in theabsence of clinically important bleeding.23,25,26 However,although the clinical benefit is uncertain, others treatchildren who have severe thrombocytopenia withintravenous immunoglobulin or glucocorticoids, even inthe absence of bleeding symptoms.25,27 This variation inpractice must be resolved by a randomised clinical trial,documenting the efficacy (or lack of efficacy) oftreatment on prevention of major bleeding as well as thecomplications of the treatment itself. Persistentthrombocytopenia is uncommon in children, andsplenectomy is rarely done, because most childreneventually have a spontaneous remission.25
Thrombocytopenia in a patient with acutesystemic illnessThrombocytopenia caused by infectionThe most common causes of thrombocytopenia areinfections. Thrombocytopenia can occur in infections
caused by viruses (eg, HIV, cytomegalovirus, Epstein-Barr virus, hantavirus), mycoplasmas, bacteria,mycobacteria, rickettsiae, or protozoal parasites (eg,malaria). In most cases the mechanism is decreasedplatelet production, though hypersplenism cancontribute. In HIV infection, thrombocytopenia iscaused by infection of marrow stromal cells that facilitatehaemopoiesis.28 Thrombocytopenia is common incritically ill patients with sepsis, in whom the dominantcause is platelet phagocytosis mediated by increasedconcentrations of macrophage colony-stimulatingfactor.29
Thrombotic thrombocytopenic purpura-haemolyticuraemic syndrome (TTP-HUS)Before effective treatment with plasma exchange becameavailable in the 1970s, the mortality from TTP-HUSwas 90%.30 At that time, when the clinical course waslong and fatal in most cases, the diagnosis was made bythe presence of five cardinal clinical features:thrombocytopenia, microangiopathic haemolyticanaemia, renal and neurological abnormalities, andfever, as well as the characteristic arteriolar hyalinethrombi on histopathology.30 Now that effective plasmaexchange treatment is available, lowering mortality toabout 20%, establishment of the diagnosis is urgent;therefore the stringency of diagnostic criteria hasinevitably decreased.31 In a case series reported in 1991,only the presence of thrombocytopenia and micro-angiopathic haemolytic anaemia, without a clinicallyapparent cause, were required for initiation of treatment.32
Earlier diagnosis and use of fewer diagnostic criteriahave, in turn, resulted in a large increase in the numberof patients treated for TTP-HUS33 and a wider clinicalrange of disease. TTP and HUS were initially describedas different syndromes (with TTP having more severeneurological abnormalities and HUS having more severerenal abnormalities) but a clear distinction is typicallynot apparent. Even the distinction between TTP-HUSand other diseases with acute thrombocytopenia andanaemia (eg, viral, bacterial, or rickettsial sepsis) isinitially unclear in many cases.31 This overlap of clinicalsyndromes is understandable, since the characteristicabnormality of thrombotic microangiopathy is notspecific for TTP-HUS, but is also seen in otherdisorders with distinct causes and outcomes, such asmalignant hypertension, acute scleroderma, anti-phospholipid antibody syndrome, and renal allograftrejection.34 Currently TTP-HUS can best be describedas a syndrome, not a specific disease, which can resultfrom many causes and can be associated with a variety ofclinical disorders (panel 2). As more specific causes forTTP-HUS are documented, such as hypersensitivity toquinine and ticlopidine,35,36 the proportion of patients inthe idiopathic category will decrease.
Endothelial-cell damage seems to be a central featurein the pathogenesis of the TTP-HUS syndromes. Plasmafrom patients diagnosed as having TTP or sporadicHUS, but not HUS associated with childhood epidemicdiarrhoea (panel 2), can cause apoptosis ofmicrovascular endothelial cells isolated from organstypically affected by TTP-HUS.37 Endothelial cellssynthesise and secrete large von Willebrand factormolecules that are later decreased in size by a plasmaprotease. A deficiency of the protease that cleaves vonWillebrand factor may allow unusually large multimers
HAEMATOLOGY
1536 THE LANCET • Vol 355 • April 29, 2000
Copyright © 2000. All rights reserved.

of von Willebrand factor to be present in plasma, whichcould agglutinate circulating platelets, causing arteriolarthrombi.38,39
Epidemic HUS in children caused by shiga toxinsfrom enterohaemorrhagic infections, mostly caused byEscherichia coli O157:H7,40 is clinically distinct amongthe disorders in panel 2. Acute renal failure is thedominant abnormality in these children, and mostsurvive with only supportive care, without plasmaexchange. The other disorders in panel 2 occur mainly inadults; all have similar clinical presentations; all arepotentially fatal without plasma-exchange treatment;therefore all are treated with plasma exchange, evenhaemorrhagic colitis due to E coli O157:H7 in adults.Although the basis for the efficacy of plasma exchangeremains unknown, this treatment has had a remarkableeffect on the clinical course of patients with TTP-HUS.The long-term risk of recurrent episodes has nowbecome apparent,41 but recurrence was almost unknownin the era before plasma exchange.30 These recurrencesseem to be restricted to adult patients with idiopathicTTP-HUS; they are unlikely to occur among patientswho have haemorrhagic colitis or drug-induced TTP-HUS (unless the drug is taken again, of course).
Thrombocytopenia associated with pregnancyThe occurrence of thrombocytopenia during pregnancyraises important diagnostic and management issues.23
Mild, symptomless thrombocytopenia (gestationalthrombocytopenia), is common at term, occurring in 5%of women.42 If thrombocytopenia is more severe (plateletcounts less than 70�109/L) or occurs early in pregnancy,idiopathic thrombocytopenic purpura is diagnosed. Thisdistinction is not precise but nor is it important formanagement of the woman; severe, symptomaticthrombocytopenia during pregnancy is treated in thesame way as that occurring at other times, andobservation without treatment is appropriate for mild,symptomless thrombocytopenia. The distinction isimportant for the infant, however, because fetalthrombocytopenia does not occur with gestationalthrombocytopenia but infants born to mothers withidiopathic thrombocytopenic purpura have a 4–10% riskof having severe thrombocytopenia at birth or during thefirst week of life.23,42 Gestational thrombocytopenia maybe simply a mild, transient manifestation of idiopathicthrombocytopenic purpura. This idea is supported byseveral observations. First, platelet counts in somewomen with an established diagnosis of idiopathic
thrombocytopenic purpura decrease during pregnancyand recover after delivery. Second, concentrations ofantibodies against platelets are increased in bothidiopathic thrombocytopenic purpura and gestationalthrombocytopenia.43 Finally, severe neonatalthrombocytopenia is more common in infants born tomothers with more severe idiopathic thrombocytopenicpurpura.44 Follow-up studies are needed to find out thelong-term clinical outcomes of women who have beendiagnosed as having gestational thrombocytopenia.
Pre-eclampsia, defined by hypertension andproteinuria, occurs in 5–10% of all pregnancies, andthrombocytopenia occurs in about 15% of women withpre-eclampsia.42 In addition, some women with severepre-eclampsia have microangiopathic haemolysis andliver dysfunction (a syndrome described by the acronym,HELLP), and neurological abnormalities, such as hyper-reflexia and visual disturbances. Characteristically, allmanifestations of pre-eclampsia resolve promptly afterdelivery, but some women continue to be affected forsome time post partum.45 Severe pre-eclampsia/HELLPsyndrome can be indistinguishable from TTP-HUS, andintervention with plasma exchange may be appropriate.This similarity explains why case series of TTP-HUSdescribe frequent occurrences during pregnancy andpost partum.30,31
ThrombocythaemiaEssential thrombocythaemia is a clonal disorder thatoriginates from a multipotent stem cell and ischaracterised by an isolated, persistently high plateletcount, typically greater than 600�109/L, without thepresence of features diagnostic for othermyeloproliferative disorders or clinical evidence forreactive thrombocytosis. Many patients have nosymptoms, and the diagnosis is made incidentally.Although patients with essential thrombocythaemia donot have the Philadelphia chromosome t(9;22), whichdefines chronic myelocytic leukaemia, the chimericBCR-ABL transcript mRNA from this translocation hasbeen identified in patients with clinically typical essentialthrombocythaemia.46 The important management issueis whether to start treatment, either to lower the plateletcount or to inhibit platelet aggregation, or to observe thepatient without specific treatment.
Current options for lowering the platelet count arehydroxyurea, an alkylating agent, and anagrelide, whichselectively inhibits platelet production. The potentialbenefit of treatment is prevention of thromboticcomplications such as stroke and myocardial infarction.However, these agents do not permanently controlthrombocytosis, so they must be given indefinitely. Animportant concern is that long-term or lifetime exposureof young patients to an akylating agent such ashydroxyurea may increase the risk of acute leukaemia.47
Anagrelide is associated with side-effects ofvasodilatation (eg, headache, cardiac failure). Aspirincan prevent thrombotic complications, but it alsoincreases the risk of bleeding, especially in patients withvery high platelet counts. There are no datadocumenting a clinical benefit from treatment withaspirin or anagrelide. Standard practice is to usehydroxyurea to lower the platelet count to less than600�109/L if any of the following criteria apply: age over60 years, platelet count over 1500�109/L, or a history ofthrombosis.47,48
HAEMATOLOGY
THE LANCET • Vol 355 • April 29, 2000 1537
Panel 2: TTP-HUS: a classification of clinicalpresentations and associated conditions
Childhood epidemic HUSAssociation with haemorrhagic colitis (typically E coli O157:H7infection)
Adult TTP-HUS syndromesIdiopathicAssociations with other disorders:
Haemorrhagic colitisDrug-induced TTP-HUS
Hypersensitivity reactions (quinine, ticlopidine)Cumulative dose-related toxicity (mitomycin A)Pregnancy
Autoimmune diseases (systemic lupus erythematosus,antiphospholipid antibody syndrome, scleroderma)
Allogeneic bone-marrow transplantation
Copyright © 2000. All rights reserved.

Pharmacological inhibition of plateletfunction to prevent thrombosisSince the demonstration that aspirin is effective in theprimary prevention of myocardial infarction,15 theprophylactic use of aspirin for thrombotic disorders hasincreased enormously. Aspirin has also become astandard treatment for patients with both cardiovascularand cerebrovascular diseases. However, the use ofangioplasty and stent placement to open obstructedcoronary arteries has necessitated even more effectiveantithrombotic agents to prevent restenosis. Ticlopidine,which blocks the platelet ADP receptor, has become astandard agent in addition to aspirin for patients withcoronary-artery stents, and it shows greater efficacy thanaspirin for prevention of recurrent stroke.49 Howeverticlopidine has greater risks than aspirin, with thepotential for causing neutropenia and TTP-HUS.36,49 Anewer analogue of ticlopidine, clopidogrel, is now usedfor these indications; its safety compared with that ofticlopidine is not yet certain.49
The most effective antithrombotic agents forcoronary-artery disease are those that block the plateletfibrinogen receptor, glycoprotein IIb-IIIa.50 These drugshave been approved for use only within the past 3 yearsbut already are used in most coronary-angioplasty andstent procedures. All currently approved glycoproteinIIb-IIIa blockers, including monoclonal antibodies,peptides, and other small molecules, are givenintravenously, but oral agents for long-term use are inclinical trials.50 An adverse effect of these agents is theoccurrence of profound thrombocytopenia in about 1%of patients.51 Thrombocytopenia can occur immediatelyafter the initial exposure, distinct from typical drug-induced thrombocytopenia, which requires sensitisationby repeated administration. A possible mechanism forthe acute thrombocytopenia after initial exposure to aglycoprotein IIb-IIIa blocker is that “naturallyoccurring” antibodies to glycoprotein IIb-IIIa (describedabove as causing platelet agglutination in edetic-acid-anticoagulated blood samples and resulting inpseudothrombocytopenia) react with platelets in whichthe glycoprotein IIb-IIIa is perturbed by interaction withthe blocking agent.
References1 Behnke O, Forer A. From megakaryocytes to platelets: platelet
morphogenesis takes place in the bloodstream. Eur J Haematol 1998;60: 3–23.
2 Kaushansky K. Thrombopoietin: the primary regulator of plateletproduction. Blood 1995; 86: 419–31.
3 Gurney AL, Carver-Moore K, De Sauvage FJ, Moore MW.Thrombocytopenia in c-mpl-deficient mice. Science 1994; 265:1445–47.
4 Ihara K, Ishii E, Eguchi M, et al. Identification of mutations in thec-mpl gene in congenital amegakaryocytic thrombocytopenia. ProcNatl Acad Sci USA 1999; 96: 3132–36.
5 Shiozaki H, Miyawaki S, Kuwaki T, Hagiwara T, Kato T,Miyazaki H. Autoantibodies neutralizing thrombopoietin in a patientwith megakaryocytic thrombocytopenic purpura. Blood 2000; 95:2187–88.
6 George JN. Platelet immunoglobulin G: its significance for theevaluation of thrombocytopenia and for understanding the origin ofalpha-granule proteins. Blood 1990; 76: 859.
7 Shattil SJ, Kashiwagi H, Pampori N. Integrin signaling: the plateletparadigm. Blood 1998; 91: 2645–57.
8 Santoro SA, Zutter MM. The alpha2beta1 integrin: a collagenreceptor on platelets and other cells. Thromb Haemost 1995; 74:813–21.
9 Santoso S, Kunicki TJ, Kroll H, Haberbosch W, Gardemann A.Association of the platelet glycoprotein Ia C807 T gene
polymorphism with nonfatal myocardial infarction in youngerpatients. Blood 1999; 93: 2449–53.
10 Lopez JA, Andrews RK, Afshar-Kharghan V, Berndt MC. Bernard-Soulier syndrome. Blood 1998; 91: 4397–418.
11 Kaneshiro MM, Mielke CHJr, Kasper CK, Rapaport SI. Bleedingtime after aspirin in disorders of intrinsic clotting. N Engl J Med1969; 281: 1039–42.
12 Aster RH. Pooling of platelets in the spleen: role in the pathogenesisof “hypersplenic” thrombocytopenia. J Clin Invest 1966; 45: 645-57.
13 George JN, Caen JP, Nurden AT. Glanzmann’s thrombasthenia: thespectrum of clinical disease. Blood 1990; 75: 1383–95.
14 George JN, Shattil SJ. The clinical importance of acquiredabnormalities of platelet function. N Engl J Med 1991; 324: 27–39.
15 Steering Committee of the Physicians’ Health Study ResearchGroup. Final report of the aspirin component of the ongoingPhysicians’ Health Study. N Engl J Med 1989; 321: 129–35.
16 Fiorin F, Steffan A, Pradella P, Bizzaro N, Potenza R, De Angelis V.IgG platelet antibodies in EDTA-dependentpseudothrombocytopenia bind to platelet membrane glycoproteinIIb. Am J Clin Pathol 1998; 110: 178–83.
17 Bizzaro N. EDTA-dependent pseudothrombocytopenia: a clinicaland epidemiological study of 112 cases, with 10-year follow-up.Am J Hematol 1995; 50: 103–09.
18 Najean Y, Lecompte T. Hereditary thrombocytopenias in childhood.Semin Thromb Hemost 1995; 21: 294–304.
19 Gentilini G, Curtis BR, Aster RH. An antibody from a patient withranitidine-induced thrombocytopenia recognizes a site onglycoprotein IX that is a favored target for drug-induced antibodies.Blood 1998; 92: 2359–65.
20 Burgess JK, Lopez JA, Berndt MC, Dawes I, Chesterman CN,Chong BH. Quinine-dependent antibodies bind a restricted set ofepitopes on the glycoprotein Ib-IX complex: characterization of theepitopes. Blood 1998; 92: 2366–73.
21 George JN, Raskob GE, Shah SR, et al. Drug-inducedthrombocytopenia: a systematic review of published case reports.Ann Intern Med 1998; 129: 886–90.
22 Warkentin TE, Chong BH, Greinacher A. Heparin-inducedthrombocytopenia: toward consensus. Thromb Haemost 1998; 79:1–7.
23 George JN, Woolf SH, Raskob GE, et al. Idiopathicthromboctyopenic purpura: a practice guideline developed byexplicit methods for the American Society of Hematology. Blood1996; 88: 3–40.
24 Frederiksen H, Schmidt K. The incidence of ITP in adults increaseswith age. Blood 1999; 94: 909–13.
25 Bolton-Maggs PHB, Moon I. Assessment of UK practice formanagement of acute childhood idiopathic thrombocytopenicpurpura against published guidelines. Lancet 1997; 350: 620–23.
26 Lilleyman JS. Management of childhood idiopathicthrombocytopenic purpura. Br J Haematol 1999; 105: 871–75.
27 Vesely S, Buchanan GR, George JN, Raskob GE, Cohen A. Self-reported diagnostic and management strategies in childhoodidiopathic thrombocytopenic purpura: results of a survey ofpracticing pediatric hematology/oncology specialists. Am J PediatrHematol Oncol 2000; 22: 55–61.
28 Bahner I, Kearns K, Coutinho S, Leonard EH, Kohn DB. Infectionof human marrow stroma by human immunodeficiency virus-1(HIV-1) is both required and sufficient for HIV-1-inducedhematopoietic suppression in vitro: demonstration by genemodification of primary human stroma. Blood 1997; 90: 1787–98.
29 Francois B, Trimoreau F, Vignon P, Fixe P, Praloran V,Gastinne H. Thrombocytopenia in the sepsis syndrome: role ofhemophagocytosis and macrophage colony-stimulating factor.Am J Med 1997; 103: 114-20.
30 Amorosi EL, Ultmann JE. Thrombotic thrombocytopenic purpura:report of 16 cases and review of the literature. Medicine 1966; 45:139–59.
31 George JN, Gilcher RO, Smith JW, Chandler L, Duvall D, Ellis C.Thrombotic thrombocytopenia purpura-hemolytic uremic syndrome:diagnosis and management. J Clin Apheresis 1998; 13: 120–25.
32 Rock GA, Shumak KH, Buskard NA, et al. Comparison of plasmaexchange with plasma infusion in the treatment of thromboticthrombocytopenic purpura. N Engl J Med 1991; 325: 393–97.
33 Clark WF, Rock GA, Buskard N, et al. Therapeutic plasmaexchange: an update from the Canadian Apheresis Group.Ann Intern Med 1999; 131: 453–62.
34 Laszik Z, Silva F. Hemolytic-uremic syndrome, thromboticthrombocytopenia purpura, and systemic sclerosis (systemicscleroderma). In: Jennett JC, Olson JL, Schwartz MM, Silva FG,eds. Heptinstall’s pathology of the kidney, 5th edn. Philadelphia:Lippincott-Raven, 1998: 1003–57.
35 Gottschall JL, Neahring B, McFarland JG, Wu G-G, Weitekamp LA,Aster RH. Quinine-induced immune thrombocytopenia with
HAEMATOLOGY
1538 THE LANCET • Vol 355 • April 29, 2000
Copyright © 2000. All rights reserved.

hemolytic uremic syndrome: clinical and serological findings in ninepatients and review of literature. Am J Hematol 1994; 47: 283–89.
36 Bennett CL, Weinberg PD, Rozenberg-Ben-Dror K, Yarnold PR,Kwaan HC, Green D. Thrombotic thrombocytopenic purpuraassociated with ticlopidine: a review of 60 cases. Ann Intern Med1998; 128: 541–44.
37 Dang CT, Magid MS, Weksler B, Chadburn A, Laurence J.Enhanced endothelial cell apoptosis in splenic tissues of patientswith thrombotic thrombocytopenic purpura. Blood 1999; 93:1264–70.
38 Furlan M, Robles R, Galbusera M, et al. Von Willebrand factor-cleaving protease in thrombotic thrombocytopenic purpura and thehemolytic-uremic syndrome. N Engl J Med 1998; 339: 1578–84.
39 Tsai H-M, Lian ECY. Antibodies to von-Willebrand factor-cleavingprotease in acute thrombotic thrombocytopenic purpura. N Engl JMed 1998; 339: 1585–94.
40 Mead PS, Griffin PM. Escherichia coli O157:H7. Lancet 1998; 352:1207–12.
41 Shumak KH, Rock GA, Nair RC, Canadian Apheresis Group. Laterelapses in patients successfully treated for thromboticthrombocytopenic purpura. Ann Intern Med 1995; 122: 569–72.
42 Burrows RF, Kelton JG. Fetal thrombocytopenia and its relation tomaternal thrombocytopenia. N Engl J Med 1993; 329: 1463–66.
43 Lescale KB, Eddleman KA, Cines DB, et al. Antiplatelet antibodytesting in thrombocytopenic pregnant women. Am J Obstet Gynecol1996; 174: 1014–18.
44 Valat AS, Caulier MT, Devos P, et al. Relationships between severeneonatal thrombocytopenia and maternal characteristics inpregnancies associated with autoimmune thrombocytopenia. Br JHaematol 1998; 103: 397–401.
45 Martin JN Jr, Files JC, Blake PG, Perry KG Jr, Morrison JC,Norman PH. Postpartum plasma exchange for atypicalpreeclampsia-eclampsia as HELLP (hemolysis, elevated liverenzymes, and low platelets) syndrome. Am J Obstet Gynecol 1995;172: 1107–27.
46 Blickstein D, Aviram A, Luboshitz J, et al. BCR-ABL transcripts in bonemarrow aspirates of Philadelphia-negative essential thrombocythemiapatients: clinical presentations. Blood 1997; 90: 2768–71.
47 Cortelazzo S, Finazzi G, Ruggeri M, et al. Hydroxyurea for patientswith essential thrombocytopenia and a high risk of thrombosis.N Engl J Med 1995; 332: 1132–36.
48 Ruggeri M, Finazzi G, Tosetto A, Riva S, Rodeghiero F, Barbui T.No treatment for low-risk thrombocythaemia: results from aprospective study. Br J Haematol 1998; 103: 772–77.
49 Sharis PJ, Cannon CP, Loscalzo J. The antiplatelet effects ofticlopidine and clopidogrel. Ann Intern Med 1998; 129: 394–405.
50 Topol EJ, Byzova TV, Plow EF. Platelet GPIIb-IIIa blockers. Lancet1999; 353: 227–31.
51 Berkowitz SD, Sane DC, Sigmon KN, et al. Occurrence and clinicalsignificance of thrombocytopenia in a population undergoing high-risk percutaneous coronary revascularization. J Am Coll Cardiol1998; 32: 311–19.
HAEMATOLOGY
THE LANCET • Vol 355 • April 29, 2000 1539
Copyright © 2000. All rights reserved.