plasma amino acids: screening, quantitation, and...
TRANSCRIPT

methods in nutrition
876The American Journal of Clinical Nutrition 24: JULY 1971, pp. 876-890. Printed in U.S.A.
Plasma amino acids: screening, quantitation,and interpretation”2
Charles R. Scriver, Carol L. Clow and Peter Lamin
Information about the steady-state concen-tration of amino acids in human plasma hasaccumulated steadily in recent years, partlyas the result of new methodology, but alsobecause of the relevance of this information
to human health and disease. There are twoareas in particular that have both served andbenefited from the growth and application ofthis knowledge. One is the field of nutrition;
the other pertains to hereditary and acquireddiseases that affect amino acid metabolism.
A number of simple qualitative or semi-quantitative methods have been developed todetect perturbation of the normal amino acidpattern in plasma. As a result, it has beenpossible to detect, with mass screening earlyin life, the few subjects with abnormal amino
acid metabolism among the many who are
normal (1 , 2). Nonetheless, abnormal pat-terns and their exact significance can berecognized only when the normal is known.
Consequently, there have been many studies,often using the newer semiautomatic elution
chromatographic methods of quantitative
analysis, to define the normal interindividual
variation in steady-state distributions of
amino acids in human plasma. When these
parameters are known, variation more subtle
than that generated by inborn errors of me-tabolism can also be identified. Hence weknow that circadian rhythms and nutritionalfactors generate intraindividual variation in
the steady-state levels of amino acids in
plasma.It is our intention to describe the methods
that have been widely used to study plasmaamino acids in man. We will also discuss the
advantages and shortcomings of the methods
with which we are familiar. The mean, stand-
ard deviation, and range of values for theprincipal amino acids in the plasma of normal
human subjects from birth to maturity arecompiled from the literature so that this re-
port may assist the clinical investigator.
The plasma free amino acid pool;
a steady state?
Measurements of plasma amino acids areusually made at a single point in time, andit is upon such measurements that we must
often decide whether a subject manifests“normal” or “abnormal” amino acid metabo-
lism, even though the dynamics of amino acid
metabolism have not been evaluated. Christ-ensen (3) has discussed the factors that regu-late the apparent steady-state levels of amino
acid in plasma; these are indicated in Fig. 1.The equilibrium of amino acids in plasma is
a reflection of their inflow (dietary intake, in-testinal absorption, release from endogenousprotein stores, and net endogenous synthesis
of nonessential amino acids), and their out-flow (uptake into liver and nonhepatic tis-sues, endogenous protein synthesis, and ca-tabolism). The fact that plasma levelsfluctuate remarkably little, indeed usuallyless than ±50% , despite intermittent dietaryinfluxes that may be tenfold greater than the
‘From the Debelle Laboratory for BiochemicalGenetics, McGill University-Montreal Children’sHospital Research Institute, Montreal 108, Quebec,Canada.
2 Supported by the Department of NationalHealth and Welfare Grant 604-7-643, Medical Re-search Council Grant MT-1085, and the NationalGenetics Foundation, New York.
by on Septem
ber 29, 2009 w
ww
.ajcn.orgD
ownloaded from

PLASMA AMINO ACIDS 877
Intestinal lumen
I
Metabolites
ILiver __ Extracellular free Nonhepatic
Free amino ‘ amino acids � free amino �-+ Metabolitesacids acids
I __ ICellular �-�- Plasma proteins *-� Cellularproteins proteins
Fio. 1. Equilibrium system for free amino acids in extracellular space, including plasma. -II- Indicatesflux across cell membranes.
amounts of amino acid present initially in theextracellular space, indicates the importanceof tissue uptake and metabolism in the con-trol of amino acid levels in plasma.
Membrane transport. Uptake of amino
acids into cells against a tissue : plasma gradi-
ent is achieved by conjugate forces in themembrane, which utilizes ionic interactionsand energy to regulate the flux of the chem-ically unmodified substrate (4, 5). Geneticevents can alter amino acid transport in man(6, 7) and this, coupled with evidence for
specificity in the selection of the substrateby the transport process, indicates that themembrane possesses transport proteins (8).
Uptake of amino acids by human cells is evi-dently constitutive and operates independ-ently of metabolic events that may affect thesize of the intracellular pool. For example,
the renal transport of an amino acid, thecatabolism of which is blocked, is no differentin the “blocked catabolic” mutant than in the
normal person. Thus, one can conclude that
transport regulates plasma steady-state levelsto some extent by allowing amino acids to
equilibrate with the intracellular pools thatare many times larger than the extracellularpool.
Periodicity of control systems. The pres-
ence of circadian periodicity produces meas-
urable intraindividual variation (9-1 1), mdi-eating that there are subtle regulatorymechanisms that control steady-state levelsof amino acids in plasma. Analysis of theperiodicity of plasma amino acid levels mdi-cates that it is partly dependent on the exoge-nous input of the amino acids themselves, but
that it is also modulated by other nutrients
and by hormonal events that influence cellu-lar uptake of amino acids. Periodicity ofenzyme activities that control entry of amino
acids into catabolic pathways further de-termines the intracellular fate of these sub-
strates. Awareness of circadian periodicitymay be important when the investigator usesthe concentration of an amino acid in plasma
to define, for example, the heterozygousphenotype of an aminoacidopathy (12).
Nutritional state. The state of nutrition
must also be considered when interpretinginterindividual and intraindividual variationin amino acid levels in plasma. Obese sub-
jects have a particular pattern of hyperamino-acidemia indicative of the insulin ineffective-
ness characteristic of obesity (13). Althoughpre- and postprandial sampling is likely towitness only subtle changes in the plasma
pattern when the normal subject is eating anormal diet (14), prolonged fasting in obese
and nonobese subjects may modulate theconcentrations of plasma amino acids to amuch greater extent. For example, in theinitial few days there may be a two- to three-
fold increase in the plasma concentration ofbranched-chain amino acids, whereas anequivalent increase in glycine and threonine
appears only after this initial period; as fast-ing continues, a dramatic fall in a-alanineoccurs. The initial rise in amino acid levelspresumably reflects a fall in plasma insulin
activity, whereas the latter changes reflect, inturn, increased peripheral release (threonineand glycine) and increased splanchnic utiliza-tion (x-alanine) of amino acids.
These observations may seem extreme andirrelevant. However, many patients withaminoacidopathies are first seen when acutelyill, often at times of diminished food intake.Under such conditions, it will be necessary
to segregate the amino acid imbalance causedby secondary illness from that associatedwith the primary disease. Furthermore, if
by on Septem
ber 29, 2009 w
ww
.ajcn.orgD
ownloaded from

878 SCRIVER ET AL.
screening programs are extended to countrieswhere malnutrition is common, as may in-deed be the case (2), the investigator willneed to distinguish the amino acid imbalancedue to primary malnutrition (17, 18) from theprimary inherited aminoacidopathy. Finally,the investigator should remember that selec-
tive modification of protein intake may affectplasma amino acids (1 4, 19); this may be-come significant, for example, when patients
with hyperammoniogenic aminoacidopathies
are treated with low protein diets. Many
additional viewpoints (20) on the influenceof protein nutrition on the steady state of freeamino acids in plasma are available for theinterested reader.
Qualitative methods of amino acid
analysis
The adaptation of simple partition chro-matographic methods to the analysis of amino
acids has provided an opportunity to evaluatethe normal amino acid pattern in plasma or
whole blood and its variations at birth andthereafter. Consequently abnormalities rep-resenting hereditary or acquired variation in
amino acid metabolism can now be easilyrecognized in mass screening programs.
Two qualitative methods in particular (21,22) have been widely used to survey the hu-man plasma amino acid profile; these areindicated in Fig. 2. Both techniques combinedeproteinization of the sample directly on thefilter paper, with development of the chro-matogram in an n-butanol, acetic acid, watersolvent (1 2 : 3 : 5) (23); the use of various
location reagents increases the sensitivity withwhich various amino acids can be detected.The ease with which samples in large num-
bers may be processed with these filter paper
methods, makes them preferable to thin-layermethods for purposes of mass screening.
Plasma method. A sample of whole bloodis drawn from a skin puncture into heparmn-ized microcapillary tubes (75 mm x 1.4 mm,
outer diameter) (21). After sealing one endof the tube with plasticine, the plasma isseparated from the cells by centrifugation inan International microcapillary centrifuge(Model MB). A lO-�d aliquot of plasma3 is
applied directly to a line drawn 2.5 cm fromone edge of a 25.4-cm square, corner-
punched Whatman No. 3 MM ifiter paper(A pattern). Eleven samples can be appliedat about 2-cm intervals to one sheet of filter
paper. Transfer of plasma is simplified if acut segment of tube, the length of which isequivalent to 10 �d plasma, is held in thevertical position against the paper and theplasma is allowed to flow onto the paper in asingle application. The mean error in obtain-
ing a 1 � volume by this method is ± 3%
(24). Approximately 1 mm is required toprocess one sample completely. The filter
papers are then mounted on “universal”frames with spacers (25). They are then de-veloped overnight by ascending partitionchromatography in a freshly prepared n-buta-nol, acetic acid, water mixture (12 : 3 : 5). Inthe morning, the papers are removed fromthe solvent, dried for 1 hr at room tempera-ture in a stream of air, then removed from
the frame and stained in a mixture of ninhy-drin (0.25% w/v), isatin (0.01 % w/v), and
2 , 6-lutidine (1 % v/v) in acetone. The chro-matograms are dried at 80 C for 1 5 mm andthen viewed with combined transmitted andreflected light upon a lighted X-ray viewingscreen. Significant elevation or diminution of
amino acid content is readily visible, and thesensitivity of the method has been evaluated(2 1). The upper three-fourths of the paper(above the glutamic acid spot) may then beoverstained with Ehrlich’s reagent (p-di-
methylaminobenzaldehyde) to identify hy-droxyproline, citrulline, and homocitrulline.If the lower portion of the chromatogram isstained with Pauly’s reagent (diazotized sulfa-
nilic acid), the histidine spot will be selec-tively identified.
Whole blood method. Whole blood istransferred from a skin puncture ontoSchleicher and Schuell (Keene, New Hamp-shire) paper No. 903 ; the paper must be
soaked through on both surfaces (22). Theair-dried sample is then dry-autoclaved at
250 F for 3 mm. A 3/16-inch punch (equiva-
lent to about 7 �l of blood) or #{188}-inch punch
of the blood spot is then withdrawn and in-
serted into holes of equivalent size in a
Aliquots of 20 �l applied in two lots can alsobe analyzed by this method without sacrificing
resolution. The sensitivity of the method can thus
be increased.
by on Septem
ber 29, 2009 w
ww
.ajcn.orgD
ownloaded from

to
SIre.’
i �8’
�d.
, I I I �. � 1*
Leucune (70)
Isolouclse)67)
() Pttenytoioouse (60)
� Volt’. (SI)\-J MetNuo,iuse (501
Trypteytloce 150)Tyrosine)45)
0 �-AmuooboIy’uc ocud (40)
‘S�q ‘� ,
I
2 1 Z I I I#{174} SttctlOfl 3 uRCNII
879
g- LEUCINE
- ISOLEUCINE
- PP4(NYLALANINE
p VALPNEO-. METPIIONIN(
�. TIYPTOPNAN
9- TYROSINE
-�-AMINO-N-SUTYI1C ACID
i- PROLINE
- ALANINE
TNIEONIP4EGLUTAMIC ACID
� IIVD*OXYPROLINE
- GLYCINE, SER!NE
CLUTAMINE, CITRULLIP4E
*. ARGJNINOSUCC1NIC ACID
a- P4ISTIDINE
- L’VSINE, ARGININE, ORNITHINE
#{149}CYSTATPIIONIP4E
ORIGIN (PROTEIN SPOT)
3 Proluwe (34) Yellow
- AlOeuee(30)
CTj�, Glotemuc ocud (28)Tltreonet,(26)
Situ’. (22t � Glycue. (23)
Glotowu,e(l Cut’uiiu,te)18(- T0o,uuW(2O�/�H�t0h1Pi0l�tt1 (22) Yellow
r Aqe�)
�
0 0 0 (�l C,,’ ‘e (5)
STANDARDSTANDARD AMINO ACID SPOTS 3/16
RI ) Soetple Discs
Fio. 2. Examples of Partition chromatographic methods for screening on plasma amino acids (see text for
details of development and staining). A) Map of the spots for “plasma method” (21). B) Examples of actualsamples examined by “plasma method” showing, from left to right: 1) normal plasma; 2) hyperphenylalaninemia(in PKU); 3) hypermethioninemia; 4) transient neonatal hypertyrosinemia; 5) hypercitrullinemia (incitrulline-mia); 6) hyperhistidinemia (in histidinemia). The next channel (6’) shows Pauly overstain of sample 6 and the
last channel (5’) shows Ehrlich overstain to confirm citrulline. (Reproduced from Scriver (52).) C) Examplesof actual samples examined by “whole blood method” (22) showing from left to right: 1) “homocystinuria;2) “maple syrup urine disease;” 3) transient hypermethioninemia; 4) “phenylketonuria;” 5) “neonatal tyro-sinemia;” 6) hypertyrosinemia; 7) normal plasma; and 8) standard amino acid mixture. (Reproduced fromLevy et al. (29.) D) Map of the spots for whole blood method. (Taken from Efron et al. (22).)
by on Septem
ber 29, 2009 w
ww
.ajcn.orgD
ownloaded from

880 SCRIVER ET AL.
“carrier” sheet of filter paper (Whatman No.3 MM); the discs should be handled with
forceps to avoid contamination (26). Frictionholds the discs in place satisfactorily. The
carrier sheet, which may be charged with 25discs set 2.5 cm from one edge, is developedovernight by ascending chromatography(paper size, 63 x 30 cm, cut from 30-cmwidth rolls) or by descending chromatog-raphy (paper size, 57 x 33 cm with a wick,57 x 7-6 cm, sewn in with zigzag stitching(22)), in the n-butanol, acetic acid, watersolvent (12 : 3 : 5). Staining and location of
amino acids is performed as described above.This method is advantageous in its ease of
collection and storage of samples. However,streaking of amino acid spots on the devel-
oped chromatogram, because of hemoglobin
tailing, may be a problem. Two modifications
avoid this problem:1) The “tandem chromatography tech-
nique.” Szeinberg and colleagues (27) use25-cm square Whatman No. 3 MM filter
paper (see plasma method) as the carriersheet for the 3A6-inch Schleicher and Schuelldiscs containing the whole blood sample. Thechromatogram is developed first by ascendingchromatography for 6 hr in an isopropanol,water mixture (3 : 1). The paper is then driedfor 1 hr at room temperature in a stream ofair, after which the discs, which retain the
hemoglobin, are pushed out. The chromato-gram is then replaced in the same direction,in a second solvent mixture of n-butanol,acetic acid, water (1 2 : 3 : 5) solvent and de-veloped overnight; the developed chromato-gram is dried and stained as described above.
2) The “elution technique.” Blood-soakeddiscs (12-mm diameter, Schleicher andSchuell filter paper No. 2992 containing
about 40 � whole blood) are eluted overnightin 100 �l of an ethanol and water mixture(60 :40) in capped conical microcentrifugetubes (0.4-mi capacity) (28). Forty micro-liters of the hemoglobin-free eluate is thenspotted, using several applications, ontoWhatman No. 3 MM paper (25 x 25 cm)
and developed by ascending chromatographyas described above (21). An additional spot,probably glutathione, is found between the“dibasic” amino acids and the origin. The
sensitivity of this modification is equivalent
to the plasma method (21). The size of thedisc and the volume of the alcohol mixture
could probably be reduced to diminish thenumber of transfers of eluate to the ifiter
paper.Performance. Both basic methods have
also been used satisfactorily for semiquanti-tative analysis. As a result, monitoring ofplasma amino acid concentrations in patients
who are being treated for an aminoacidopa-thy is greatly facilitated. Present experi-
ence indicates that this relaxed approach iscompatible with good clinical control of mostaminoacidopathies.
Large field trials have been performed
with both methods. Mass screening of new-born infants has been reported on about1 50,000 subjects (24, 29-3 1 ) and it is evi-dent that the methods perform well for this
purpose. A wide variety of normal transient
patterns of hyperaminoacidemia has been ob-served (24, 29-3 1). Many of the aminoacid-opathies associated with a specific hyper-
aminoacidemia have also been recognized inthe newborn period with these methods (seeTable 1). Partington (32) has evaluated the
sensitivity and specificity of the plasmamethod (21) for the specific detection ofhyperphenylalaninemia; he found it to beequivalent to the inhibition assay of Guthrieand Susi in reliability.
Ninhydrin-positive aminoacidopathies that
are presently known and could be detectedby chromatographic screening of plasma andwhole blood are listed in Table 1 ; aminoacid-
opathies still unknown might also be discov-ered by these methods in the future. The
above-mentioned methods can be used forpreventive community screening, if personnelare trained to interpret the chromatogramsand to be aware of the significance of theamino acid patterns that they observe. Theremust also be facilities to locate, retest, andfollow all subjects with abnormal initial tests(24).
Quantitative methods of amino acid
analysis
A little over a decade ago, very few in-
vestigators were able to perform more than
one complete analysis per week, i.e., of the
free amino acids in physiological fluids.
by on Septem
ber 29, 2009 w
ww
.ajcn.orgD
ownloaded from

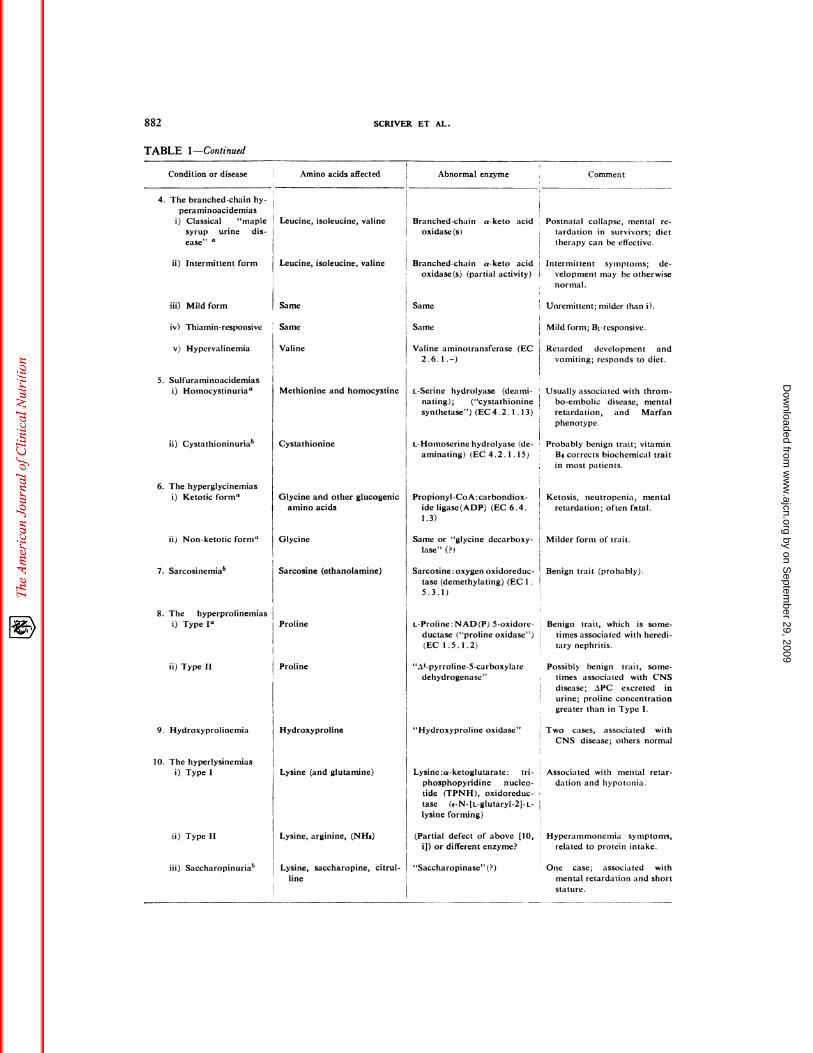
Amino acids affected Abnormal enzyme Comment
Phenylalanine hydroxylating
system (?)
Phenylalanine
Tyrosine
Methionine
Histidine
Phenylalaninc
p-Hydroxyphenyl pyruvic acid Benign; responds to ascorbic
hydroxylase (EC 1.14.2.2) acid and reduced protein in-
take.
5-adeno- Benign; usually found with
(EC 2.5. high protein intake.
phenylke- Phenylalanine
iii ( Transient phenylke- Phenylalanine
tonuria
iv ) Benign hyperphe- Phenylalanine
nylalaninemia a
Tyrosine
Tyrosine
tyrosin- Tyrosine (and methionine)
ii) “5uper tyrosinemia”
(Oregon)”
L-Tyrosine: pyruvate amino- One case known; myasthenia
transferase (?) (EC 2.6. 1 . gravis, probably incidental
20) fInding.
ii ( Variant form Histidine
TABLE 1
PLASMA AMINO ACIDS
Aminoacidopathies that can be detected by plasma or whole blood screening
881
Condition or disease
Group I: Common perinatal
(adaptive) traits
Neonatal hyperphenylalanin-
emia#{176}
Neonatal tyrosinemia#{176}
Hvpermethioninemia’
Hyperhistidinemia a
Group II: Inherited amino-
acidopathies
1. The hyperphenylala-
ninemias
i) Classical phenylke-
tonuria
ii) Atypical
tonuria a
2. The hypertyrosinemias
i) Tyrosinosis (Medes)
iii) Hereditary
emia”
3. The hyperhistidinemias
i) Classical form#{176} Histidine (alanine in some
cases
ATP: L-methioninesyltransferase(?)
l.6
i-Histidine ammonia lyase (?)
(EC 4.3.1.3)
i-Phenylalanine tetrahydrop-
teridine: oxygen oxidore-
ductase (4-hydroxylating)
(EC 1.14.3.1)
L-Phcnylalaninetetrahydrop-
teridine: oxygen oxidore-
ductase (4-hydroxylating)
(EC 1.14.3.1)
i-Phenylalanine tetrahydrop-
teridine: oxygen oxidore-
ductase (4-hydroxylating)
(EC 1.14.3.1) (?)
i-Phenylalaninc tetrahydrop-
teridine: oxygen oxidore-
ductase (4-hydroxylating)
(EC 1.14.3.1)
Soluble (cytoplasmic) iso-zyme of tyrosine amino-
transferase
p-HHPA hydroxylase (EC 1.
14.2.2) Primary or second-
ary defect?
L-HiStidine ammonia lyase
(EC 4. 3. 1 .3) (liver and
skin)
L-HiStidine ammonia lyase
(EC4.3.1.3) (liver only)
Benign; may respond to folic
acid; often occurs with tyro-
sinemia.
Benign; related to high protein
intake.
i) Plasma phenylalanine >16
mg/lOO ml; causes mentalretardation. When treated,
i-phenylalanine tolerancein diet is 250-500 mg phe
day.
ii) Plasma phenylalanine > l6
mg/lOO ml; similar to i) butdietary tolerance for L-phe-
nylalanine is >500 mg/day
iii) Plasma phenylalanine >16
mg/l0O ml; change in sta-tus to iv), or normal, sev-
eral months or years after
birth.
iv) Plasma phenylalanine <16
mg ‘100 ml; on normal
diet. Benign trait.
One case; associated mentalretardation.
Hepat.ic cirrhosis, and renal
tubular failure, eventually
fatal; responds to tyrosine
restriction.
Usually associated with mental
retardation and speech de-
feet.
As above.
by on Septem
ber 29, 2009 w
ww
.ajcn.orgD
ownloaded from
Branched-chain a-keto acid
oxidase(s) (partial activity)
Leucine, isoleucine, valine
Leucine, isoleucine, valine
Same
Same
Valine
Methionine and homocystine
Cystathionine
Milder form of trait.
ii) Type II
Glycine
Sarcosine (ethanolamine)
Proline
Proline
Hydroxyproline
Lysine (and glutamine)
Lysine, arginine, (NHs)ii) Type II
iii) Saccharopinunia�’
882 SCRIVER ET AL.
Lysine, saccharopine, citrul-
line
TABLE 1-Continued
Condition or disease Amino acids affected Abnormal enzyme Comment
4. The branched-chain hy-
peraminoacidemias
I) Classical “maplesyrup urine dis-
ease” a
ii) Intermittent form
iii) Mild form
iv) Thiamin-respoissive
v) Hypervalinemia
5. Sulfuraminoacidemias
i) Homocystinuria5
ii) Cystathioninuriab
6. The hyperglycinemias
i) Ketotic form”
ii) Non-ketotic form”
7. Sarcosinemiab
8. The hyperprolinemias
i) Type I”
9. Hydroxyprolinemia
10. The hyperlysinemias
1) Type I
Glycine and other glucogenic
amino acids
Branched-chain a-keto acid
oxidase(s)
Same
Same
Valine aminotranaferase (EC
2.6. 1.-)
i-Serine hydrolyase (deami-
nating); (“cystathionine
synthetase”) (EC4.2. I .13)
L-Homoserine hydrolyase (de-
aminating) (EC 4.2. 1.15)
Propionyl-CoA :carbondiox-ide ligase(ADP) (EC 6.4.
1.3)
Same or “glycine decarboxy-
lase” (?)
Sarcosine: oxygen oxidoreduc-
tase (demethylating) (EC 1.5.3.1)
L-Proline:NAD(P) 5-oxidore-
ductase (“proline oxidase”)(EC 1.5.1.2)
“i�-pyrroline-5-carboxylate
dehydrogenase”
“Hydroxyproline oxidase”
Lysine:a-ketoglutarate: tn-
phosphopynidine nucleo-
tide (TPNH), oxidoreduc-
tase (e-N-[L-glutaryl-2]-L-
lysine forming)
(Partial defect of above [10,
i]) or different enzyme?
“Saccharopinase” (?)
Postnatal collapse, mental re-
‘ tardation in survivors; diet
therapy can be effective.
Intermittent symptoms; de-� velopment may be otherwise
normal.
� Unremittent; milder than i)
� Mild form; Bi-responsive.
Retarded development and
vomiting; responds to diet.
� Usually associated with throm.� bo-embolic disease, mental
� retardation, and Marfan
� phenotype.
� Probably benign trait; vitamin‘ Bt corrects biochemical trait
in most patients.
Ketosis, neutropenia, mental
retardation; often fatal.
Benign trait (probably).
Benign trait, which is some-
times associated with heredi-
tary nephritis.
Possibly benign trait, some-
times associated with CNS
disease; .XPC excreted in
urine; proline concentration
greater than in Type I.
Two cases, associated with
CNS disease; others normal
Associated with mental retar-
dation and hypotonia.
Hyperammonemia symptoms,
related to protein intake.
One case; associated with
mental retardation and short
stature.
by on Septem
ber 29, 2009 w
ww
.ajcn.orgD
ownloaded from

Condition or disease
11. Pipecolicacidemia1’
Amino acids affected Abnormal enzyme
“Pipecolate oxidase” (?)Pipecolicacd
Glycine, glutamine
Glutamine
Ornithine
Citrulline
ASA
ii) Type II
iii) Hyperornithinemia
iv) Citrullinentia”
v) Argininosuccinicaci-
duniat
Group Ill. Nutritional andother diseaseswhich may affect
amino acids inplasma
Protein-calorie malnutrition
Prolonged fasting
Obesity
Hepatitis
See text.
I;
a These diseases have been detected in newborn patients by the screening tests described in this paper.
b These traits may not be detectable by screening tests because plasma elevation is too small; since the
renal clearance of the relevant substances is high, urine testing would be preferable. Other traits in thisclass are phosphoethanolaminuria (in the hypophosphatasias) , hyper-�-alaninemia, and fl-aminoiso-butyricacidemia.
PLASMA AMINO ACIDS 883
TABLE 1-Continued
12. The hyperammonemias
i) Type I ATP: carbamate phospho-
transferase (EC 2.7.2.2)
Carbamoylphosphate: L-or-
nithine carbamoyltransfer-
ase (EC2.l.3.3)
Ornithine - ketoacid amino-
transferase
L-Citrulline: 1-aspartase ligase
(AMP) (EC6.3.4.5)
1-Argininosuccinate arginine-
lyase (EC4.3.2.l)
Comment
Hepatomegaly and mental re-
tardation.
A group of diseases that shows
ammonia intoxication, pro-
tein intolerance, hepato-
megaly, vomiting, et cetera.
ASAuria also has trichor-
rhexis nodosa.
vi) (See 10, Type II)
Tryptophan,’leucine/isoleu-
cine/valine �; Tyrosine/
glycine/proline I
Alanine � ; threonine;
glycine I
Leucine,. isoleucine/valine,
phenylalanine/tyrosine
Glycine I
Methionine/tyrosine I
Severity of change related to
severity of malnutrition.
Reflects insulin insensitivity.
Reflects severity of liver dis-
ease.
Moreover, a large sample was often requiredto obtain reliable results. It is now possible,by virtue of better resins and instrumentation,
to perform (by elution chromatography onion exchange resins) several analyses dailyof very small samples of plasma or serum. We
and others (33-35) have found that attentionpaid to details in handling the samples isimportant (Table 2), and when certain arti-facts are avoided the investigator can obtainaccurate analysis of the free amino acids inplasma samples.
1) The effect of venipuncture. The con-centration of taurine and glutamic acid inplasma falls about 30 mm after venipuncture
and returns to normal in about 1 hr (36).This artifact should be distinguished from
physiological changes in studies requiring
multiple sequential venipunctures.2) Plasma versus serum, and choice of
anticoagulant. In general, plasma is easier to
prepare for application to the resin column.Plasma may be deproteinized immediately
after the blood cells are separated by centrif-ugation, whereas serum may experiencechanges in its amino acid composition uponstanding at room temperature during clotretraction.
The choice of anticoagulant can be im-portant. Most investigators use heparin; how-
by on Septem
ber 29, 2009 w
ww
.ajcn.orgD
ownloaded from

884 SCRIVER ET AL.
TABLE 2Artifacts in plasma amino acid composition related to technique in handling samples
Procedure
Effect on particular amino acids
-
Concentration in sample decreases Concentration in sample increases
1) Repeated venipuncture Taurine, glutamic acid
2) Clotting of serum standingroom temperature
at Glutamine, asparagine Taurine, aspartic acid, glu-tamic acid
3) Delay in deproteinization Cystine, homocystine, and mixeddisulfides
Picric acid method” Citrulline, homocitrulline, trypto-phan
4) ContaminationPlatelets and WBC Taurine, aspartic acid, glu-
tamic acid
RBC (hemolysis) Arginine, cystine Glutathione, ornithine
“Fingerprints” (sweat) Many amino acids
5) StorageTemperature above -68 C Glutamine, asparagine, tryptophan Glutamic acid, aspartic
acid
Handling and elution at tem-perature >40 C
Glutamine, asparagine Glutamic acid, asparticacid
“Losses occur during step when picric acid is removed on Dowex 1 or 2 resin.
ever, an excess of this anticoagulant maycause hemolysis leading to artifacts relatedto constituents of the red blood cells. Im-purities in some batches of ethylenediamine-tetraacetic acid (EDTA), which is sometimesused as an anticoagulant, can produce nm-
hydrin-positive peaks in the elution chro-
matogram (33).3) Deproteinization. Many investigators
prefer to deproteinize plasma with 3 % sulfo-salicylic acid (35) (1 vol plasma: 5 vol 3%sulfosalicylic acid), or with direct addition of30 mg sulfosalicylic acid powder/mi plasma.After high speed centrifugation (21 ,000 x g)for 10 mm, the supernatant may be stored orapplied directly to the ion exchange column.
The other popular method of deproteiniza-tion (37) uses picric acid, which because ofits yellow color, must be removed on Dowex
1 or 2 resin before the deproteinized samplecan be analyzed for taurine and urea. Thesulfosalicylic acid method of deproteinizationalso prevents substantial losses of tryptophan,
citrulline, and homocitrulline that occur whenpicric acid is removed from deproteinized
samples (35). The arguments are numerous,both for and against sulfosalicylic acid versus
picric acid as the agent of choice for de-
proteinization in various situations, and the
numerous discussions on this matter (33, 35,
38, 39) should be consulted by the reader.Immediate removal of protein from the
sample is important so as to avoid significant
losses of disulfide amino acids that will bind
to plasma proteins on standing at room
temperature or in the refrigerator (33, 35,
37, 40). Rapid deproteinization is necessaryin field studies, or in those situations in which
shipment of samples is required,4 or when the
treatment of patients with disorders of sulfur
amino acid metabolism is being monitored.
4 Homocystine was not found in plasma samplesobtained from the original cases of homocystinuria(41). A delay in deproteinization accounted forthis artifact.
by on Septem
ber 29, 2009 w
ww
.ajcn.orgD
ownloaded from

PLASMA AMINO ACIDS 885
4) Contamination. Hamilton (26) has
mentioned that contamination of glasswarewith sweat from a fingertip can jeopardize the
reliability of analyses obtained by some highsensitivity methods.
Platelets and leukocytes contain largeamounts of taurine and dicarboxylic aminoacids (33, 36, 42). Therefore, plasma orserum samples can be contaminated withthese substances released from the cellularconstituents in the buffy coat. Plasma should
be drawn off from the cell-plasma interfacewith care to avoid disturbing the buffy coat.
Hemolysis of red blood cells may also dis-tort the apparent plasma pattern. Glutathi-one (reduced and oxidized) is present in red
cells (36); release of arginase from erythro-cytes may diminish arginine and increaseornithine in plasma; and plasma cystine maybe lowered, either by dilution with an intra-cellular pool that is low in cystine, or bybinding to protein in the hemolysate. Other
amino acids are not significantly altered, astheir concentration in mature red cells is
similar to that of plasma (42, 43).
5) Storage. The concentrations of glutamicacid and aspartic acid rise slowly, and gluta-mine and asparagine fall equivalently insamples stored for long periods at -20 C.These changes are minimized by storage at
-68 C and enhanced at -4 C, or at roomtemperature (35, 38). Evaporating and dry-ing of samples or elution of amino acidsfrom ion exchange columns at temperaturesabove 40 C will reduce the glutamine andasparagine content of plasma samples.
Methods of quantitative analysis. Thereare now many reports in the literature thatdescribe technical modifications in the basicmethods for semiautomated elution chroma-
tography of amino acids on ion exchangeresin columns. These modifications, in gen-eral, provide improved speed, resolution, and
automation of analysis; a later article in thisJournal will discuss these developments. We
will mention only two reports, which provide
the investigator with a portfolio of simple
operating programs, and which will allow him
to investigate specific aminoacidopathies
without sacrificing column time.
Shih and colleagues (44) described a series
of protocols for operation of the single col-
umn (45, 46) type of analyzer. They were
able to obtain within 1 to 2 hr quantitativeanalysis of particular groups of amino acidfrom short resin columns. The protocols arerelevant for the investigation of many of theaminoacidopathies listed in Table 1.
Our group (47) described methods forrapid operation of the two-column analyzer
system (48) as well as the simultaneous use oftwo columns. Recoveries and reproducibilityof the methods compare well with the parent
techniques (48).Experience with these modified techniques
has demonstrated their value in monitoring
patients under treatment for hereditaryaminoacidopathies and in performing investi-.gations that require analysis of many samples
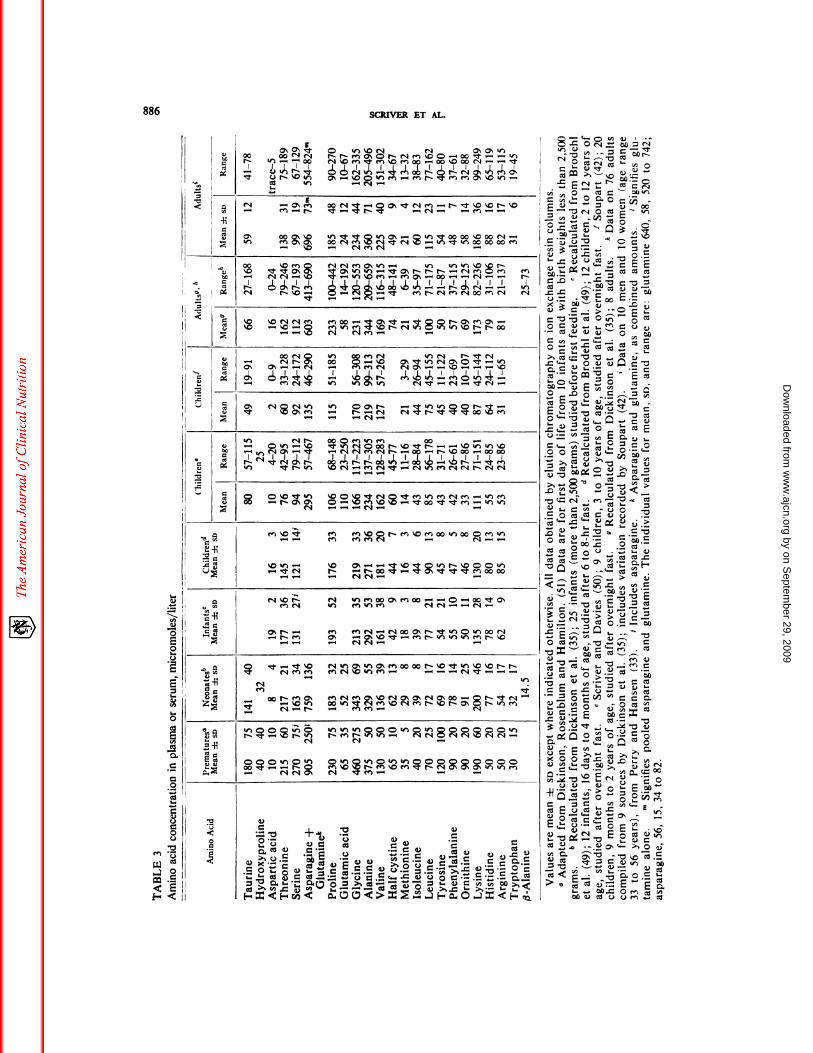
for a particular amino acid.Normal values. Values for amino acid
concentration in plasma or serum of healthyhuman subjects are presented in Table 3.These data represent many studies performed
by investigators using reasonable precautionsfor the handling and analysis of the samples
of plasma or serum.5 Several different tech-niques for amino acid analysis by elutionchromatography on ion exchange resin wereused to obtain the results presented in Table
3 and yet the data are reasonably homogene-ous. One observes that essential amino acids
in plasma are lower during the period ofrapid somatic growth (49, 50) despite thehigher protein intakes on the basis of bodyweight at such times. Dickinson et al. (51)showed that amino acid concentrations inplasma fall after the first day of life, with par-
ticular reference to nonessential amino acids.Nonetheless, despite undoubted variation indiet of the subjects of the various reports, the
interindividual variation in human subjectsis relatively modest, reflecting presumably the
remarkable control exerted upon the steady-state free amino acid pattern in plasma.
Comment
It has been several years since the merits
of mass screening for hereditary metabolicdiseases (52) became a popular topic for dis-
cussion. Although the rate of discovery of
“new” aminoacidopathies has continued to
Artifacts, with regard to glutamine and gluta-mate, are noticeable in some of the older studies.
by on Septem
ber 29, 2009 w
ww
.ajcn.orgD
ownloaded from
ScCa
a
0 ‘ft “0 r�.i � O\ C�s *11
t- N �t O\ 0 r- r�i r� ‘.0 � - 00 � �‘
o� - 9 � � ret - �i p.. r��i r� O� “0 tni -
a
Ca
Co e.i � - 0 a� � e�i �i - r- � ‘.0 �C r-. �� _c�4_ -f�.’--
,,� � �. 0 in Q�, ,.a © �1�i � � 00 ‘.0 00 �
00 � (� �O (�4 � e�4 ‘.0 - ‘(� � “� 00 00 �,- e�1r��ir�l - -
ii� � Sc.e� �� 1”� -�--
< ‘�a�
i?�: ��
� -
00 ‘.0 r�i 0so �
e�i r� - �O
rL �N r-. � �
r�t r� r�t O� ‘r � .,�, i,� .,� ‘.0 ‘0 r-��� - ‘ft �,O r”� - c�s C’� - 00 � � N - -
�,_ - ,_ � � �. (� r-. � r�s e� � � r’�
e�i r� r�‘.0 ‘ �C - �
� �� �- (eir.1�.
r� 00 - ‘�1� a� � - ‘�i� 0 0 r- a� � � -� � � � ‘,� s-.. c’.i “rnt � I1�t In �O I’.. N 00
; ; �,ia ��1�1 ‘n�O�v�t
:�5’
�
e-1�N’ri�. s.o��rei
-
“Vt � �
r--r-i �- - t,1 -
‘�:��0
�C.�
4’te
��
©Lr)�e�i as ‘
rL�i4r�ia‘ft �‘:rN’rt
‘� t�’1 � r�i trn.- r�. � 00 ‘ 00 00IJI�I4�
�- - -
C
‘�O%O”t’ft
00 - N � g� �c � ‘i� 00 � “1� r’�i � U’S ‘ft
a�C’�
r�i ‘.0 ��
�, 00 ‘ft 00 0 n�i ‘itCe) � -
�+.
�C.C ‘�
� c��.O ‘ft
��.O O� - - � ‘.0 � C ‘ft N. � � 0 ‘ii
�:
a‘ v a‘
c�-i�.oR(“�tr�4
r�ilit r�s’ftr�t
,�C‘ eg
�
O\N-
�
ret �
�
a.%u� 0 ‘�t - � ‘so� e.1cr)r��) r�i ‘ft O� ‘ft C� ret co 00 t��- �C “�2� ‘ft �O �O 1-.. F-.�e,1�.C�rtce)� �
�4CC
8 ‘az
�
c-I -ret
00 F-� � a�- - � Ifs- c-I - F-.
‘1�.e� (�-1 f’�i O� �.O c�.1 a� o� c-i 0” 00 ‘-‘ � N. ‘�I� �00 ‘fS � e-� r� �o c’� r� t-. � � � � �. ‘i�- m r�t .-
a �� ‘a�1
N. � ‘.0 r-. ‘�r�iI�- F�t N ‘ft �f) - e-i c-i � �i N “0 (‘1 C�-1 -
c�9 -
11
5)
0E0‘a0
.�
‘a
�0
0
.E
;�‘u �
� � � �v �aD,t� .� � � � 5) �
5)�’.9._ 0ot� ���)5) ._s)�c�� �.� 0� � s� � � � �) o.� �.E.;;r��4: � �
t�>%C#�.�5)C#) �F-��<�-in< �
‘ft
‘�0‘ft
5)
00
886 S�RIVER ET AL.
00 ooNr�tr- �-‘-‘oo�a� �r-�-
o� 000\\O��.0
by on Septem
ber 29, 2009 w
ww
.ajcn.orgD
ownloaded from

PLASMA AMINO ACIDS 887
burgeon (53), a clearer interphase has de-
veloped between the use of mass screeningmethods and the recognition and the under-standing of many acquired and inheriteddiseases of amino acid metabolism. There islittle doubt that the proper use of screeningmethods, such as those discussed in this arti-cle, benefits the student of health and diseaseand his patient. Yet, as more use is madeof mass screening programs, there must beequally wide awareness of the limitationsinherent in these methods and in the interpre-
tation of their findings. Today’s advances indiagnosis will further burden those facilitiesthat must provide medical care to patientswith disordered amino acid metabolism; it isclear that the methods described here canalso be used to facilitate the investigationand treatment of patients with such problems.It must be a matter of some gratification forthe pioneers of partition and elution chroma-tography to see their little bits of filter paper
and their batches of resin performing yeomanservice in the mainstream of human biologytoday.
Addendum
An unfortunate delay between receipt ofthis solicited manuscript by the editors andits publication by the Journal can be offset tosome extent with the following selected ma-
terial:
Transport and metabolism
Brodehi and colleagues (54) studied tubu-lar reabsorption of phenylalanine in classical
phenylketonuric patients. Transport is nor-mal even though endogenous oxidation of thesubstrate is completely blocked. It is nowknown that phenylalanine is oxidized to a
significant extent in mammalian kidney (55,
56). Similar observations on the independ-ence of transport of a substrate from its intra-cellular fate were obtained by Glorieux et al.(57), studying sarcosine transport in normal
and sarcosinemic subjects.
Circadian variation
G#{252}ttler, Olesen and Wamberg (58) ob-
served a reversal of the normal circadian
rhythm for plasma phenylalanine in phenyl-
ketonuric patients. This finding further em-phasizes the need to standardize conditionsfor discrimination of normal subjects and
heterozygotes for phenylketonuria. Burghen
et al. (59) discovered that chloramphenicoltherapy altered normal periodicity and theinitial steady-state concentration of amino
acids in plasma.
Nutrition and plasma amino acids
Relationships between feeding and plasma
amino acids continue to be of interest. Adibiand colleagues (60) showed that starvationleading to changes in plasma insulin couldnot solely explain changes in branched chain
amino acid levels. Alanine was clearly sen-sitive to endogenous carbohydrate metabo-
lism.Synderman’s group continues to publish
data on the effect of protein intake on theplasma amino acids (61). Restricted protein
intake (2 g/kg per day) in the premature in-
fant depresses plasma ‘lysine and elevatesglycine. High protein intakes (9 g/kg per day)
increase proline, phenylalanine, tyrosine,methionine, valine, leucine, and isoleucine inplasma. The RBC : plasma distribution ratiofor most free amino acids is not significantly
altered by widely differing protein intakes.Leucine feeding depresses isoleucine andvaline in plasma (20, 62), presumably by in-fluencing catabolism of the affected amino
acid specifically. Whitehead’s group inUganda (63) reported that fasting serumamino acids provide a useful monitor of theprogress of kwashiorkor and its treatment.
Stegink and Baker (64) reported relationshipsbetween malnutrition and plasma amino acidlevels. Eskimo and Caucasian children hadsimilar postprandial amino acid levels in se-
rum despite different diets.The so-called “alanine cycle” in man has
received considerable attention (65). Alanineflux from muscle to liver serves gluconeo-
genesis in the latter organ, assuming net al-anine synthesis from pyruvate in muscle. Thecycle might also serve urea synthesis in the
transfer and removal of ammonia from pe-ripheral tissues. Formation and release of
alanine in muscle may be the rate limiting
by on Septem
ber 29, 2009 w
ww
.ajcn.orgD
ownloaded from

888 SCRIVER ET AL.
step in gluconeogenesis in human starvation.Hypoalaninemia may play a pivotal role inthe pathogenesis of ketotic hypoglycemia ofchildhood (66). Cahill’s group (67) hasshown that glutamate is taken up by muscle,whereas glutamine is released; the latter is
taken up across the splanchnic bed in man.
It is possible that this other “shunt” may be
an important source of urinary ammonia!!
We wish to thank Mrs. Eveline Zifkin for herassistance in the preparation of this manuscript.
References
1. WILSON, J. M. G., AND G. Jui��onit. The Princi-pies and Practice of Screening for Disease.World Health Organ. Public Health PapersNo. 34, Geneva, 1968.
2. Report of a WHO Scientific Group. Screeningfor Inborn Errors of Metabolism. World HealthOrgan. Tech. Rept. Ser. No. 401. Geneva, 1968.
3. CHRISTENSEN, H. N. Some transport lessionstaught by the organic solute. Perspectives Biol.Med. 10: 471, 1967.
4. CHRISTENSEN, H. N. Free amino acids and
peptides in tissues. In: Mammalian ProteinMetabolism, edited by H. N. Munro and J. B.Allison. New York: Academic, vol. 1, 1964,p. 105-124.
5. SCHULTZ, S. G. Mechanisms of absorption. In:Biological Membranes, edited by R. M. Dow-ben. Boston: Little, Brown, 1969, p. 59-108.
6. SCRIVER, C. R. The human biochemical genet-ics of amino acid transport. Pediatrics 44: 348,
1969.7. SCRIVER, C. R., AND P. HECHThIAN. Human ge-
netics of membrane transport with emphasison amino acids. In: Advan. Human Genet., ed-ited by H. Harris and K. Hirschhorn. NewYork: Plenum, 1970, p. 211-274.
8. PARDEE, A. B. Membrane transport proteins.Science 162: 632, 1968.
9. FEIGIN, R. D., A. S. KLAINER AND W. R. BEISEL.
Circadian periodicity of blood amino-acids inadult men. Nature 215: 512, 1967.
10. FEIGIN, R. D., A. S. KLAINER AND W. R. BEISEL.
Factors affecting circadian periodicity of bloodamino acids in man. Metab. Clin. Exptl. 17:764, 1968.
11. WURTMAN, R. J., C. M. ROSE, C. CHOU AND
F. F. LARIN. Daily rhythms in the concentra-tions of various amino acids in human plasma.New Engi. I. Med. 279: 171, 1968.
12. ROSENBLATF, D., AND C. R. SCRIVER. Heter-ogeneity in genetic control of phenylalaninemetabolism in man. Nature 218: 677, 1968.
13. FELIG, P., E. MARLISS AND G. F. CAUHU., JR.
Plasma amino acid levels and insulin secretionin obesity. New Engl. I. Med. 281: 811, 1969.
14. WELLER, L. A., S. MARGEN AND D. H. CALLO-
WAY. Variation in fasting and postprandialamino acids of men fed adequate or protein-free diets. Am. I. Cliii. Nutr. 22: 1577, 1969.
15. FELIG, P., D. E. OWEN, J. WAHREN AND G. F.CAHILL, JR. Ammo acid metabolism during pro-longed starvation. I. C/in. Invest. 48: 584, 1969.
16. ADIBI, S. A. Influence of dietary deprivationson plasma concentration of man. I. App!.
Physiol. 25: 52, 1968.
17. HOLT, L. E., JR., S. E. SNYDERMAN, P. M.NORTON, E. ROITMAN AND J. FINCH. Plasmaaminogram in kwashiorkor. Lancel 2: 1343,1963.
18. SAUNDERS, S. J., A. S. TRUSWELL, G. 0. BARBE-
ZAT, W. WITTMAN AND J. D. L. HANSEN. Plasmafree aminoacid pattern in protein-calorie mal-nutrition. Reappraisal of its diagnostic value.Lancet 2: 795, 1967.
19. Sr�.wDERMAN, S. E., L. E. HOLT, JR., P. M.NORTON, E. ROITMAN AND S. V. PHANSALKAR.
The plasma aminogram. I. Influence of thelevel of protein intake and a comparison ofwhole protein and amino acid diets. Pediat.
Res. 2: 131, 1968.
20. LEATHEM, J. H. (editor). Protein Nutrition and
Free Amino Acid Patterns. Twentieth Ann.Conf. Bureau Biol. Res. February 1965. NewBrunswick, N.J.: Rutgers Univ. Press, 1968,227 p.
21. SCRIVER, C. R., E. DAVIES AND A. M. CULLEN.
Application of a simple method to the screeningof plasma for a variety of aminoacidopathies.Lancet 2: 230, 1964.
22. EFRON, M. L., D. YOUNG, H. W. MOSER AND
R. A. MACCREADY. A simple chromatographic
screening test for the detection of disorders ofamino acid metabolism: a technique usingwhite blood cells or urine collected on filterpaper. New Eng!. J. Med. 270: 1378, 1964.
23. CULLEY, W. J., E. T. MERTZ, M. W. LUCE,
J. M. CALANDRO AND D. H. JOLLY. Paperchromatographic estimation of phenylalanineand tyrosine using finger-tip blood. Clin. Chem.8: 266, 1962.
24. CLow, C. L., C. R. SCRIVER AND E. DAVIES.
Results of mass screening for hyperamino-acidemias in the newborn infant. Am. I.Diseases Children 1 17: 48, 1969.
25. DATTA, S. P., C. E. DENT AND H. HARRIS. Anapparatus for the simultaneous production ofmany two-dimensional paper chromatograms.Science 112: 621, 1950.
26. HAMILTON, P. B. Amino-acids on hands. Nature
205: 284, 1965.
27. SZEINBERG, A., B. SZEINBERG AND B. E. COHEN.
Screening method for detection of specificaminoacidemias. Clin. Chim. Acta 23: 93, 1969.
28. ADRIAENSSENS, K., R. VANHEULE AND M. VAN
BELLE. A new simple screening method for de-tecting pathological aminoacidemias with col-iection of blood on paper. Clin. C/tim. Acta
15: 362, 1967.29. LEvy, H. L., V. E. SmH, P. M. MADIGAN, V.
KAROLKEWICZ AND R. A. MACCREADY. Results
\
by on Septem
ber 29, 2009 w
ww
.ajcn.orgD
ownloaded from

PLASMA AMINO ACIDS 889
of a screening method for free amino acids. I.Whole blood. Clin. Biochem. 1: 200, 1968.
30. KOMROWER, G. M., B. FOWLER, M. J. GRW-FITHS AND A. M. LAMBERT. A prospective com-munity survey for aminoacidemias. Proc. Roy.Soc. Med. 61: 294, 1968.
3 1 . KELLY, S., AND H. SWIFT. Amino acid abnor-malities in a mentally retarded population. Am.I. Epidemiol. 85: 250, 1967.
32. PARTINGTON, M. W. Case finding in phenyl-ketonuria. III. One-way paper chromatographyof the amino acids in blood. Can. Med. Assoc.I. 99: 638, 1968.
33. PERRY, T. L., AND S. HANSEN. Technical pitfallsleading to errors in the quantitation of plasmaamino acids. C/in. C/tim. Acta 25: 53, 1969.
34. Beckman-Spinco Manual A-IM-3, 1965. SpincoDivision, Beckman Instruments Inc., Palo Alto,California 94304.
35. DICKINSON, J. C., H. ROSENBLUM AND P. B.HAMILTON. Ion exchange chromatography ofthe free amino acids in the plasma of the new-born infant. Pediatrics 36: 2, 1965.
36. ROUSER, G., B. JELINEK, A. J. SAMUELS AND
K. KINUGASA. Free amino acids in the bloodof man and animals. I. Method of study andthe effects of venipuncture and food intake onblood free amino acids. In: Amino Acid Pools,
edited by J. T. Holden. New York: Elsevier,1962, p. 350-372.
37. MOORE, S., AND W. H. STEIN. Procedures for
the chromatographic determination of aminoacids on four percent cross-linked sulfonatedpolystyrene resins. J. Biol. Chem. 21 1: 893,1954.
38. DEWOLFE, M. S., S. BASKURT AND W. A.COCHRA.NE. Automatic amino acid analysis ofblood serum and plasma. C/in. Bioc/zem. 1: 75,1967.
39. KNIPFEL, J. E., D. A. CHRISTENSEN AND B. D.OWEN. Effect of deproteinating agents (picricacid and sulfosalicylic acid) on analysis for freeamino acids in swine blood and tissue. I. As-soc. Offic. Agr. Chemists 52: 981, 1969.
40. CRAWHALL, J. C., C. J. THOMPSON AND K. H.BRADLEY. Separation of cystine, penicillamine
disulfide and cysteine-penicillamine mixed di-sulfide by automatic amino acid analysis. Anal.Biochem. 14: 405, 1966.
41. CARSON, N. A. J., D. C. CUSWORTH, C. E.DENT, C. M. B. FIELD, D W. NEILL AND R. G.WESTALL. Homocystinuria: a new inborn errorof metabolism associated with mental de-ficiency. Arch. Disease Childhood 38: 425,1963.
42. Sour’ART, P.: In: Amino Acid Pools, edited byJ. T. Holden. New York: Elsevier, 1962, p.220-262.
43. WINTER, C. G., AND H. N. CHRISTENSEN. Mi-gration of amino acids across the membraneof the human erythrocyte. I. Biol. Chem. 239:872, 1964.
44. SHIH, V., M. L. EFRON AND G. L. MECHANIC.
Rapid short column chromatography of amino
acids: a method for blood and urine specimensin the diagnosis and treatment of metabolic
diseases. Anal. Biochem. 20: 299, 1967.45. PIEZ, K. A., AND L. MoRRis. A modified pro-
cedure for the automatic analysis of aminoacids. Anal. Biochem. 1: 187, 1960.
46. HAMILTON, P. B. Ion exchange chromatographyof amino acids: a single column high resolving,fully automatic procedure. Anal. Chem. 35:2055, 1963.
47. SCRIVER, C. R., E. DAVIES AND P. LAMM. Ac-celerated selective short column chromatog-raphy of neutral and acidic amino acids on aBeckman-Spinco analyzer, modified for simul-taneous analysis of two samples. Clin. Biochem.1: 179, 1968.
48. SPACKMAN, D. H., W. H. STEIN AND S. MOORE.
Automatic recording apparatus for use in thechromatography of amino acids. Anal. Chem.30: 1190, 1958.
49. BRODEHL, J., AND K. GELLISSEN. Endogenous
renal transport of free amino acids in infancyand childhood. Pediatrics 42: 395, 1968.
so. ScRIvrR, C. R., AND E. DAVIES. Endogenous
renal clearance rates of free amino acids inpre-pubertal children. Pediatrics 32: 592, 1965.
51. DICKINSON, J. C., H. ROSENBLUM AND P. B.HAMILTON. Ion exchange chromatography of
the free amino acids in the plasma of infantsunder 2,500 grams at birth. Pediatrics 45: 606,1970.
52. SCRIvER, C. R. Screening newborns for heredi-tary metabolic disease. Pediat. Clin. N. Am.12: 807, 1965.
53. SCRIVER, C. R. Inborn errors of amino acidmetabolism. Brit. Med. Bull. 25: 35, 1969.
54. BRODEHL, J., K. GELLISEN AND W. P. KAAS. Therenal transport of amino acids in untreated in-fants with phenylketonuria. A cta Paediat.Scand. 59: 241-248, 1970.
55. TOURIAN, A., J. GODDARD AND T. T. PUCK.Phenylalanine hydroxlyase activity in mamma-han cells. I. Cell. Physiol. 73: 159, 1969.
56. MURTHY, L. I., AND H. K. BERRY. Characteris-tics of phenylalanine hydroxylase. FederationProc. 30: 460, 1971 (abstr.).
57. GLORIEUX, F. H., C. R. SCRIvER, E. DELVIN
AND F. MOHYUDDIN. Transport and metabo-lism of sarcosine in hypersarcosinemia and nor-mai phenotypes. I. Clin. Invest. In press.
58. GUi-rLER, F., S. OLESEN AND E. W�tM�r.Ro.Diurnal variations of serum phenylaianine inphenylketonuric children on low phenylalaninediet. Am. I. C/in. Nutr. 22: 1568, 1969.
59. BURGHEN, G. A., W. R. BEISEL AND P. J. BAR-TELLONI. Influences of chloramphenicol admin-istration on whole blood amino acids in man.C/in. Med. 77: 26, 1970.
60. ADIBI, S. A., A. L. DRASH AND E. D. LIvi.
Hormone and amino acid levels in altered nu-
tritional states. I. Lab. C/in. Med. 76: 722,
1970.61. SNYDERMAN, S. E., L. E. HOLT, JR., P. M.
NORTON AND S. V. PHANSALKAR. Protein re-
by on Septem
ber 29, 2009 w
ww
.ajcn.orgD
ownloaded from

890 SCRIVER ET AL.
quirement of the premature infant. II. Influenceof protein intake on free amino acid content ofplasma and red blood cells. Am. I. Clin. Nutr.23: 890, 1970.
62. PHANSALKAR, S. V., P. M. NORTON, L. E. HOLT,JR. AND S. E. SNYDERMAN. Amino acid inter-relationships: the effect of a load of leucine onthe metabolism of isoleucine. Proc. Soc. Exptl.Biol. Med. 134: 262, 1970.
63. GRIMBLE, R. F., AND R. G. WmTEHE&n. Fast-ing serum-amino acid patterns in Kwashiorkorand after administration of different levels ofprotein. Lancet 1: 918, 1970.
64. STEGINK, L. D., AND G. L. BAKER. Serum amino
acid levels of northern Alaskan Eskimo infantsand children. Am. I. C/in. Nutr. 23: 1642, 1970.
65. FELIG, P., T. POZEFSKY, E. M�niISS AND G. F.CAHILL, JR. Alanine: key role in gluconeogene-sis. Science 167: 1003, 1970.
66. PAGLIARA, A., I. Ki�ni, D. DEvivo, R. FEIGIN
AND D. KIPNIS. Hypoalaninemia: the cause ofketotic hypoglycemia of childhood. I. C/in.invest. 50: 73a, 1971.
67. M�uu.iss, E. B., T. T. Aoiu, T. POZEFSKY, A. S.MOST AND G. F. CAHILL, JR. Muscle andsplanchnic glutamine and glutamate metabo-lism in postabsorptive and starved man. J. C/in.
invest. 50: 814, 1970.
by on Septem
ber 29, 2009 w
ww
.ajcn.orgD
ownloaded from