pharmdock: a pharmcophore-based docking program user...
TRANSCRIPT

PharmDock: A Pharmcophore-Based Docking Program
User Guide
Bingjie Hu Department of Medicinal Chemistry and Molecular Pharmacology
College of Pharmacy Purdue University
575 Stadium Mall Drive West Lafayette, IN 47907 Email: [email protected]
Markus Lill Department of Medicinal Chemistry and Molecular Pharmacology
College of Pharmacy Purdue University
575 Stadium Mall Drive West Lafayette, IN 47907 Email: [email protected]

Table of Contents
Installation ................................................................................ 1
Download and Installation ................................................................. 1
PharmDock program ......................................................................... 1
Prerequisites .................................................................................................. 1
Compilers ..................................................................................................................... 1
Python with Scipy and Numpy ...................................................................................... 1
Software for conformer generation ............................................................................... 1
Open Babel with Python bindings ................................................................................. 2
Installation of PharmDock .............................................................................. 3
PharmDock plugin ............................................................................. 4
Prerequisite: PyMOL ...................................................................................... 4
Plugin: PharmDock.py.................................................................................... 4
PharmDock Usage ................................................................... 5
Generate ligand library ...................................................................... 5
Prepare and run docking ................................................................... 6
Import docking results ....................................................................... 9
Confined and Constraint Docking with PharmDock ................ 11
Docking with all the pharmacophores ............................................. 11
Docking with selected pharmacophores .......................................... 14

U S E R G U I D E
1
Installation
In this chapter we will describe the installation process of PharmDock and its PyMOL plugin.
Download and Installation
The software package consists of the PharmDock package and a PyMOL plugin. The plugin should be viewed as interface between the docking engine and the GUI PyMOL to view, prepare and analyze PharmDock docking results. The current version of our docking software and plugin is designed for Linux OS.
PharmDock program
Prerequisites
To utilize PharmDock, the following programs have to be installed:
Compilers
The compilation currently requires gcc, g++ and cmake.
Python with Scipy and Numpy
PharmDock requires the installation of Python. For the generation of protein-based pharmacophores the Python libraries Scipy and Numpy (included in standard Scipy installation) are required. PharmDock was tested with Scipy 0.7.2 with Python 2.6.6.
Software for conformer generation
Currently two different programs can be used to pre-generate ligand conformations for docking: OpenEye Omega and OpenBabel. To specify which software should be used for conformer generation, specify either
CONFORMER_PROGRAM=omega; export CONFORMER_PROGRAM
or
Chapter
1

2
CONFORMER_PROGRAM=babel; export CONFORMER_PROGRAM
O P E N E Y E O M E G A
PharmDock can use OpenEye Omega (http://www.eyesopen.com/omega) to generate multiple ligand conformations for the ligand library. PharmDock has been tested with Omega v2.2.0.[1-3]
Install Omega following OpenEye’s instructions. Currently a tar.gz file must be downloaded from OpenEye’s website after a license file has been obtained. Downloading the file to /usr/local, and unzipping and untaring it Omega will be installed under /usr/local/openeye. Please specify the environment variable:
OE_DIR=/usr/local/openeye; export OE_DIR
A license file has to be requested from OpenEye and should be placed under /usr/local/openeye. The following environment variable should be set:
OE_LICENSE =/usr/local/openeye/oe_license.txt; export OE_LICENSE.
O P E N B A B E L
PharmDock can alternatively use Open Babel for generation of ligand conformers. As Open Babel is required for ligand pharmacophore generation, we refer to the next section for installation instructions for Open Babel.
Other alternative programs to generate ligand conformations will be added in future versions of PharmDock.
Open Babel with Python bindings
For generation of ligand pharmacophores, PharmDock needs the Pybel library which is part of the Open Babel installation with Python binding. PharmDock was tested with Open Babel 2.3.2. For installation download openbabel-2.3.2.tar.gz from http://openbabel.org/wiki/Get_Open_Babel and Eigen 3.1.4 from http://eigen.tuxfamily.org to /usr/local, unzip and untar both files.
Change into directory /usr/local
cd /usr/local
and generate a ‘build’ directory:
mkdir build cd build
Configure the build system:

3
cmake ../openbabel-2.3.2 -DPYTHON_BINDINGS=ON –DEIGEN3_INCLUDE_DIR=/usr/local/eigen-eigen-version_number
Compile:
make –j2
Install:
sudo make install
Specify the path to pybel.py, openbabel.py and _openbabel.so which will be /usr/local/lib for the standard installation of openbabel:
OPENBABELLIB=’/usr/local/lib’; export OPENBABELLIB
Installation of PharmDock
The PharmDock package PharmDock.tar.gz needs to be downloaded (default to /usr/local) and extracted using
tar –zxf PharmDock.tar.gz
Please specify the environmental variable
$PHARMDOCKHOME
e.g. for bash shell:
PHARMDOCKHOME=/usr/local/PharmDock export PHARMDOCKHOME
If PharmDock is installed under a different directory, the environment variable should be modified accordingly.
Next, go to src folder:
cd $PHARMDOCKHOME/src
and compile and install program:
make

4
PharmDock plugin
Prerequisite: PyMOL
Before downloading and installing the plugin, please download PyMOL from http://pymol.org and install it on your computer. The location of PyMOL will be noted as
$PyMOL (here: /usr/local/pymol)
in the following.
To prevent potential conflicts of standard Python installation and Python libraries as part of PyMOL for running the Python utilities of PharmDock, we installed PyMOL from source code. The following packages (including development libraries for most) need to be installed: OpenGL, (free)glut, glew, libpng, tkinter, pmw, freetype2. The package manager can be used to install those packages for most standard Linux distributions.
PyMOL is then installed using (write premission to installation location required):
python setup.py build install --home=/usr/local/pymol
Add /usr/local/pymol/bin to PATH, e.g.
PATH=$PATH:/usr/local/pymol/bin
Plugin: PharmDock.py
The plugin to perform docking using PharmDock can be downloaded here:
PharmDock.py
It needs to be saved to $PyMOL/lib64/python/pmg_tk/startup/ where $PyMOL is the top-directory containing your local PyMOL installation.

5
PharmDock Usage
In this chapter we will describe how to use PharmDock through the PyMOL plugin.
We will demonstrate the use of the docking engine PharmDock in conjunction with the PyMOL plugin PharmDock.py for docking several inhibitors to the Retinoic Acid Receptor gamma (RXRgamma, PDB-code: 1FCX [4]). We will first generate a small ligand library, setup the protein system, and then perform docking calculations. All necessary file are located under $PHARMDOCKHOME/examples/RXRgamma.
Generate ligand library
The general procedure to generate a ligand library is to generate ligands as individual PyMOL objects. Alternatively ligands can be imported as individual files (e.g. as pdb or mol2 files). Make sure that the topology (including bond strength) and hydration state is correctly assigned to each ligand. Please note the net charge of each ligand.
We will start with a series of pre-generated Retinoic Acid Receptor gamma (RXRgamma) inhibitors from the PDBbind dataset [5,6].
Change to subdirectory $PHARMDOCKHOME/examples/RXRgamma/Ligands
cd $PHARMDOCKHOME/examples/RXRgamma/Ligands
and then open the mol2 files in PyMOL by typing:
pymol *.mol2
Set-up ligand library for PharmDock using the menu item:
PharmDock → Export ligand library
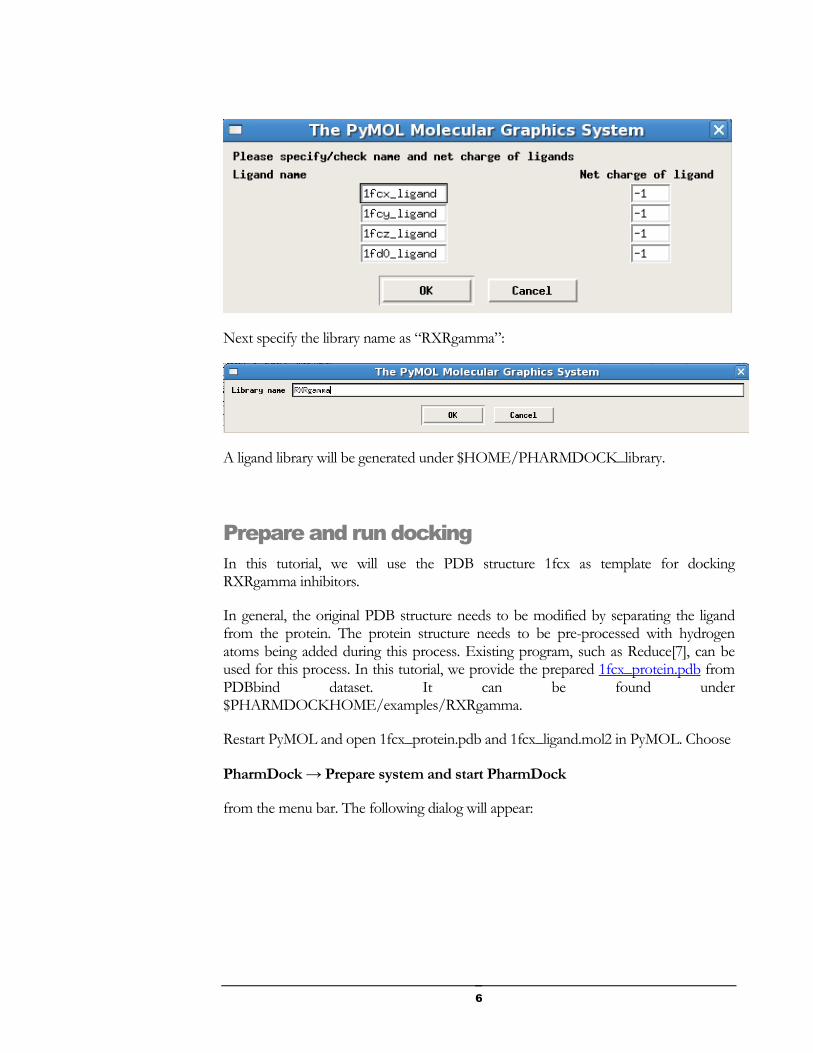
Specify ligand name and net charge of ligand:
Chapter
2
6
Next specify the library name as “RXRgamma”:
A ligand library will be generated under $HOME/PHARMDOCK_library.
Prepare and run docking
In this tutorial, we will use the PDB structure 1fcx as template for docking RXRgamma inhibitors.
In general, the original PDB structure needs to be modified by separating the ligand from the protein. The protein structure needs to be pre-processed with hydrogen atoms being added during this process. Existing program, such as Reduce[7], can be used for this process. In this tutorial, we provide the prepared 1fcx_protein.pdb from PDBbind dataset. It can be found under $PHARMDOCKHOME/examples/RXRgamma.
Restart PyMOL and open 1fcx_protein.pdb and 1fcx_ligand.mol2 in PyMOL. Choose
PharmDock → Prepare system and start PharmDock
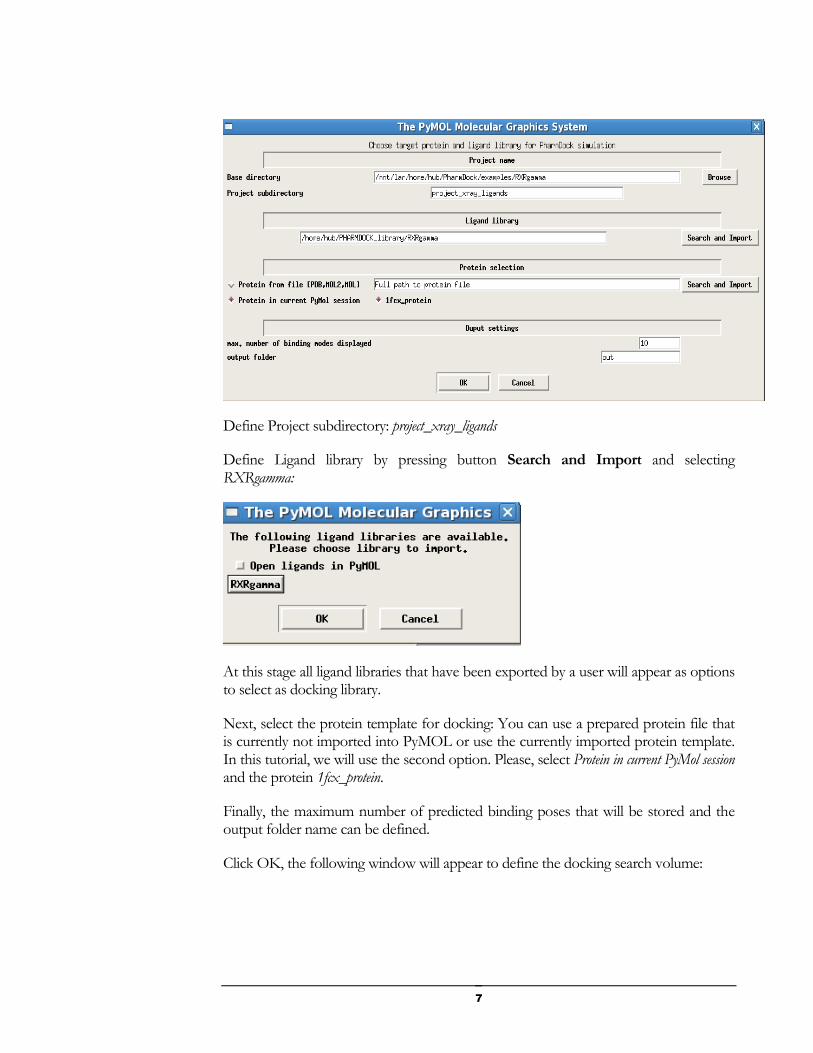
from the menu bar. The following dialog will appear:
7
Define Project subdirectory: project_xray_ligands
Define Ligand library by pressing button Search and Import and selecting RXRgamma:
At this stage all ligand libraries that have been exported by a user will appear as options to select as docking library.
Next, select the protein template for docking: You can use a prepared protein file that is currently not imported into PyMOL or use the currently imported protein template. In this tutorial, we will use the second option. Please, select Protein in current PyMol session and the protein 1fcx_protein.
Finally, the maximum number of predicted binding poses that will be stored and the output folder name can be defined.
Click OK, the following window will appear to define the docking search volume:

8
The search volume should cover the binding pocket of fXa. Two options are suggested:
i. Display only x-ray ligand (or several x-ray ligands if applicable) and manually define box to cover space of ligand plus some extra space. Use the sliders to define center and box size in every direction (see figure above).
ii. Select the x-ray ligand (or several ligands) in the PyMOL Viewer (selection (sele) has to show up as object/selection in the right column of the PyMOL Viewer). Press button Determine new box coordinates, and the center of the box will be shifted to the center of mass of all atoms in the selection (sele). [Can also be used by selection several amino acid residues in the binding site without use of ligand data.]
Press button Determine new box dimensions and the box will be rescaled to fit all atoms in the selection (sele) plus a user-defined extra space in every direction using Radius around selection (has to be set before pressing button).
In this tutorial, we used the 1fcx_ligand.mol2 and a radius of 3.0 Å to define the binding site. Click 1fcx_ligand under PyMOL viewer and press Determine new box coordinates and Determine new box dimensions. The search volume is displayed and always updated as yellow box in the PyMOL Viewer:

9
After pressing OK, the docking calculation will start in the background. An information dialog will appear:
Docking per ligand on a single core takes about 1-5 minutes on current architecture. PyMOL can be closed while docking simulations are running in the background.
Import docking results
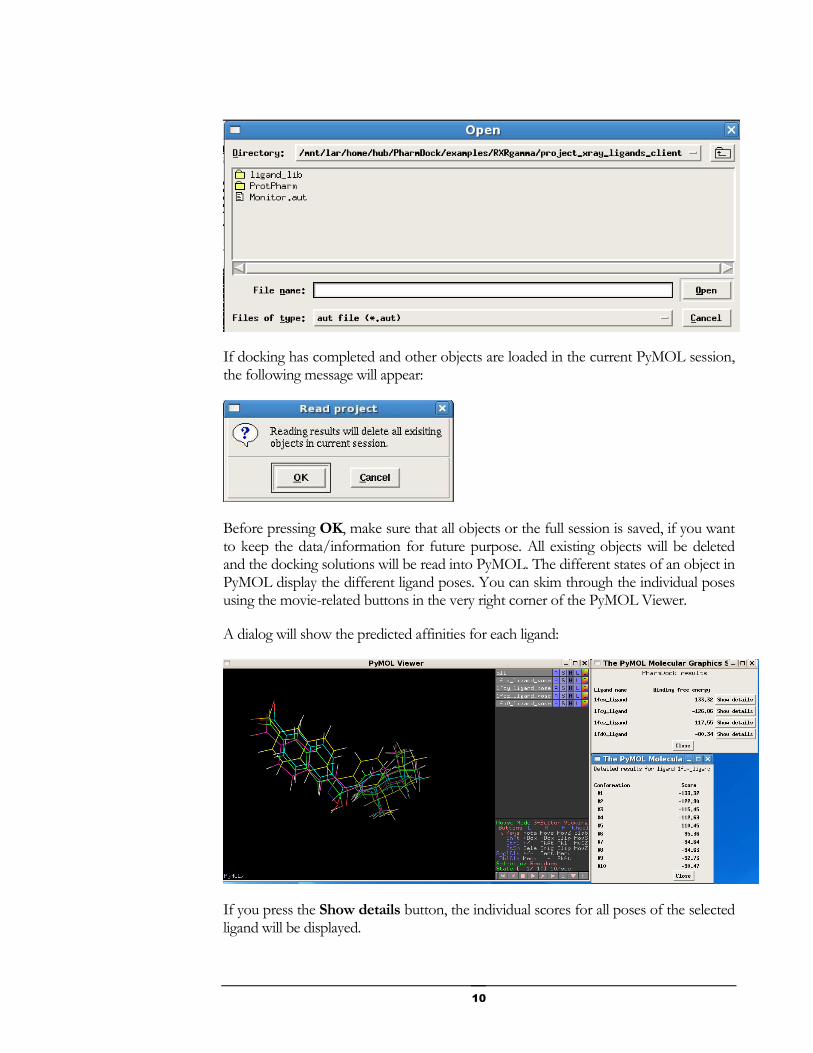
You can check if the simulation has been completed and import the results with PharmDock Import Results:
PharmDock → Import Results
Select Monitor.aut from the folder project_xray_ligands_client.
10
If docking has completed and other objects are loaded in the current PyMOL session, the following message will appear:
Before pressing OK, make sure that all objects or the full session is saved, if you want to keep the data/information for future purpose. All existing objects will be deleted and the docking solutions will be read into PyMOL. The different states of an object in PyMOL display the different ligand poses. You can skim through the individual poses using the movie-related buttons in the very right corner of the PyMOL Viewer.
A dialog will show the predicted affinities for each ligand:
If you press the Show details button, the individual scores for all poses of the selected ligand will be displayed.

11
Confined and Constraint
Docking with PharmDock
In this chapter we will describe an advanced setting of PharmDock that allows docking with selected protein pharmacophores.
It is possible that researchers would like to use previously acquired empirical knowledge about the target and identify compounds that can form specific interactions with certain region or specific residues of the binding pocket. To allow for inclusion of such information, we provide two options to perform docking with PharmDock focusing on a set of selected protein pharmacophore elements. For a complete motivation and description of this advanced setting, please refer to our paper [Journal of Cheminformatics, submitted]. We will demonstrate the confined docking with the enzyme human factor Xa (fXa, PDB: 1MQ6[8]) in this tutorial.
Docking with all the pharmacophores
Change to subdirectory $PHARMDOCKHOME/examples/fXa/ and then open the 1mq6_ligand.mol2 file by typing:
pymol 1mq6_ligand.mol2
Set-up ligand library for PharmDock as instructed in Chapter 2:
PharmDock → Export ligand library
Specify the library name as “fXa”:
Chapter
3
12
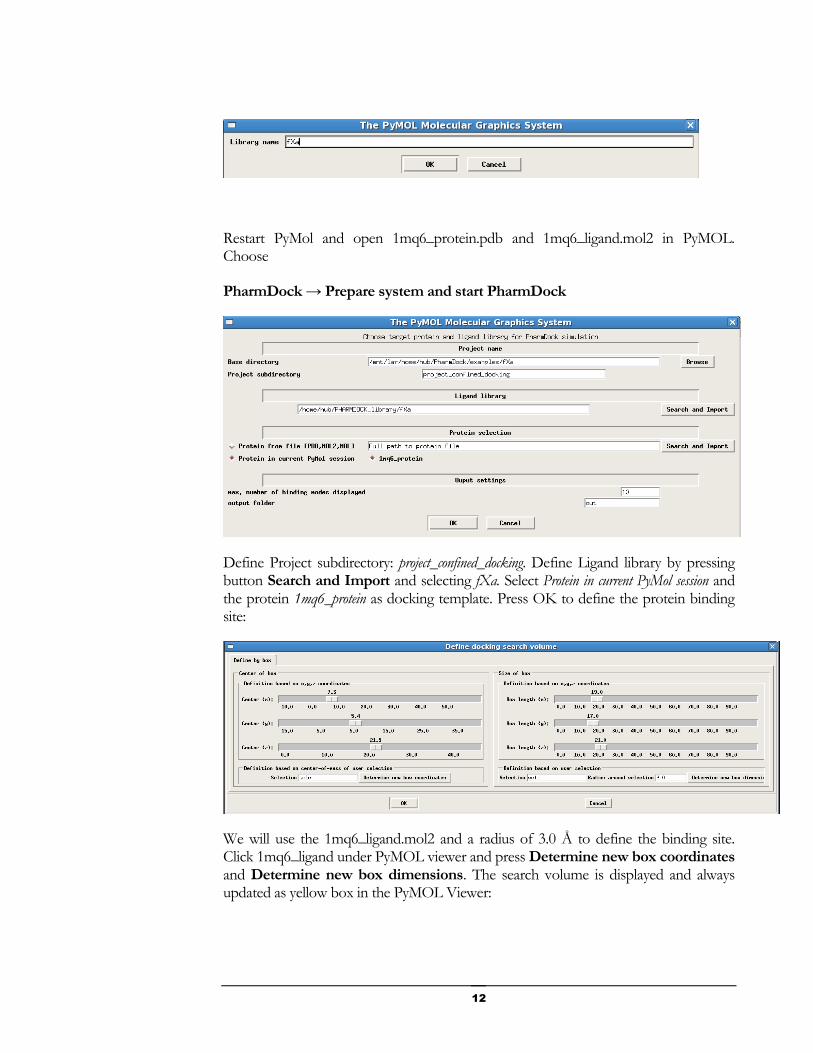
Restart PyMol and open 1mq6_protein.pdb and 1mq6_ligand.mol2 in PyMOL. Choose
PharmDock → Prepare system and start PharmDock
Define Project subdirectory: project_confined_docking. Define Ligand library by pressing button Search and Import and selecting fXa. Select Protein in current PyMol session and the protein 1mq6_protein as docking template. Press OK to define the protein binding site:
We will use the 1mq6_ligand.mol2 and a radius of 3.0 Å to define the binding site. Click 1mq6_ligand under PyMOL viewer and press Determine new box coordinates and Determine new box dimensions. The search volume is displayed and always updated as yellow box in the PyMOL Viewer:

13
Press OK to submit the docking simulation.
When the docking simulation finished, imported the results into PyMOL:
Load the 1mq6_ligand.mol2 into PyMOL to observe that there is a large discrepancy between the top predicted binding pose (green) and the original X-ray ligand pose (purple).
14
Docking with selected pharmacophores
Now let’s try to confine the docking search volume to certain region of the protein binding site. Under the project directory project_confined_docking_client, restart PyMOL and choose
PharmDock → Rerun PharmDock with selected protein pharmacophores
from the menu bar. The following dialog will appear:
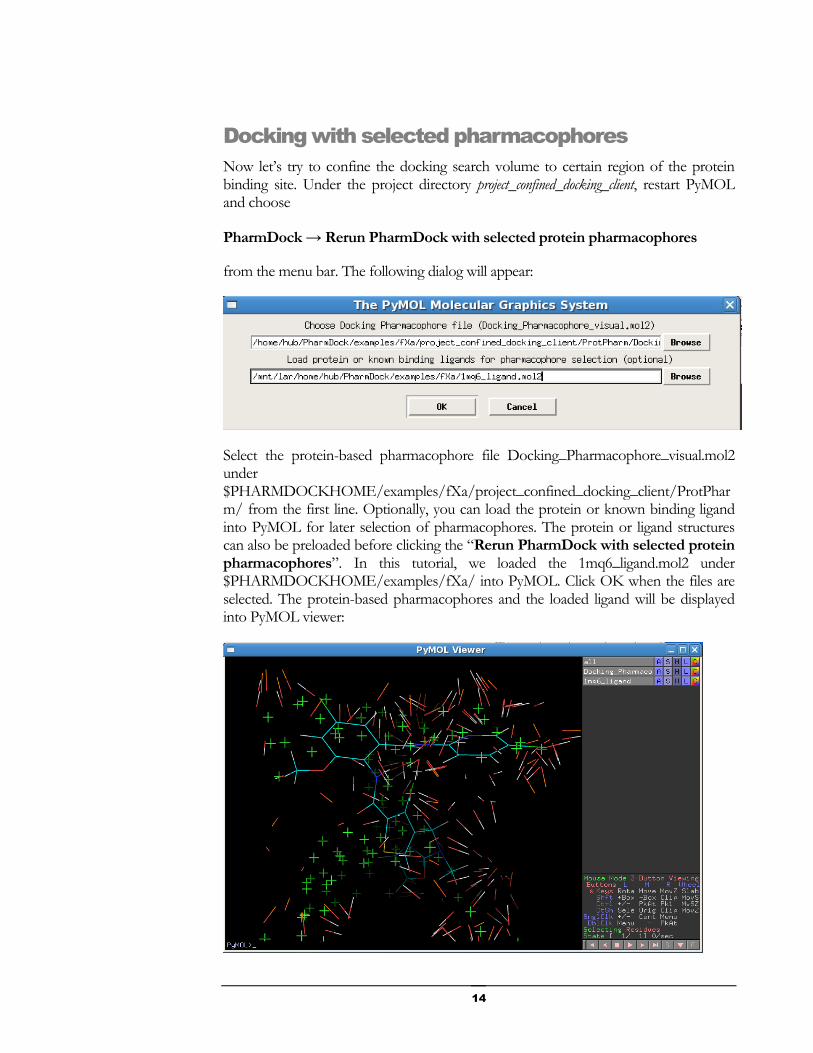
Select the protein-based pharmacophore file Docking_Pharmacophore_visual.mol2 under $PHARMDOCKHOME/examples/fXa/project_confined_docking_client/ProtPharm/ from the first line. Optionally, you can load the protein or known binding ligand into PyMOL for later selection of pharmacophores. The protein or ligand structures can also be preloaded before clicking the “Rerun PharmDock with selected protein pharmacophores”. In this tutorial, we loaded the 1mq6_ligand.mol2 under $PHARMDOCKHOME/examples/fXa/ into PyMOL. Click OK when the files are selected. The protein-based pharmacophores and the loaded ligand will be displayed into PyMOL viewer:

15
The following dialog will also appear:
First, select the protein pharmacophores for the rerun in the PyMOL Viewer window by clicking on individual pharmacophores using the left mouse button. As default, a selection “sele” will appear as selection in the right column of the PyMOL Viewer. You can also change the “sele” to other selection names you defined in the PyMOL Viewer. To make the selection of pharmacophore easier, the “select” command in PyMOL is recommended as will be shown below in our example.
Next, select the rerun docking settings. Two options are available to guide docking towards the selected pharmacophores:
1. Confined docking: The search volume will be confined to include only the selected pharmacophores. In this case, please select all the pharmacophores that you would like to use to define the search volume. A minimum of three pharmacophores are required for PharmDock to perform the docking simulation.
2. Constraint docking: The generated docking poses must match at least one of the selected pharmacophores but the search volume is unmodified compared to the original unbiased docking (i.e. all the pharmacophore loaded in the PyMOL viewer will be used in defining the search volume). In this case, please select at least one pharmacophore that you would like the ligand to match with.
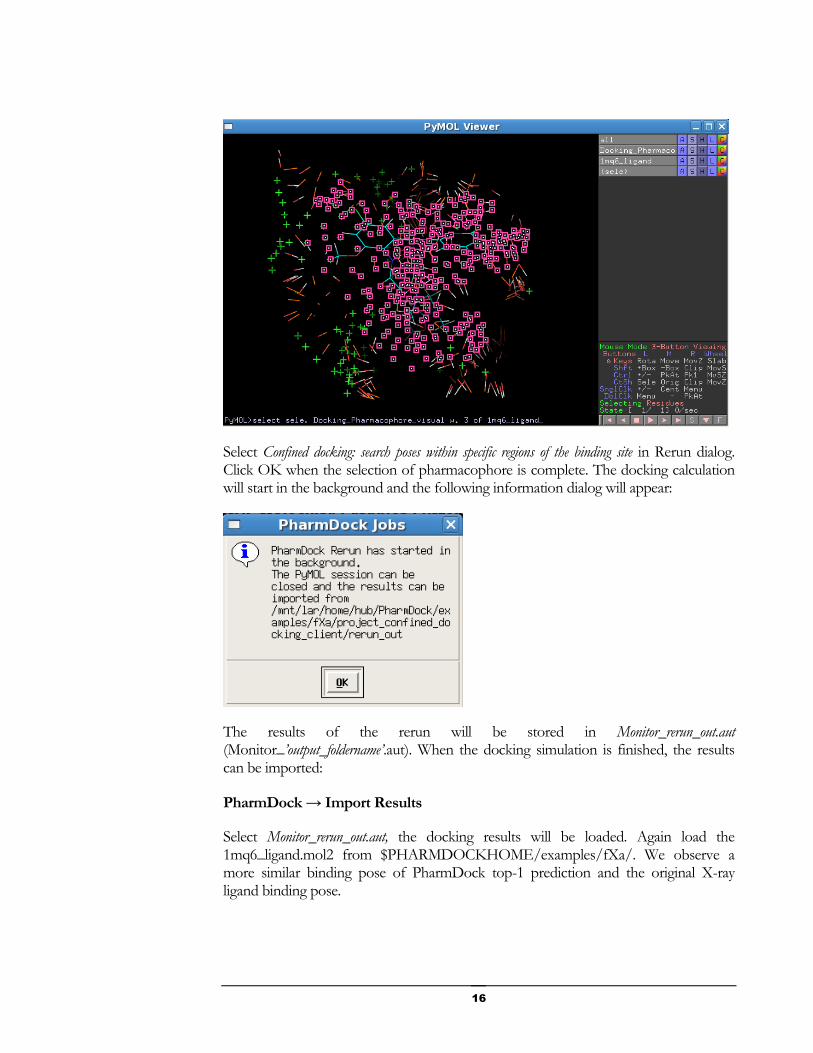
In our fXa example, we will demonstrate the confined docking setting. We select all the pharmacophore elements that are within 3Å to the loaded ligand 1mq6_ligand.mol2. In the bottom left of PyMOL viewer, type:
select sele, Docking_Pharmacophore_visual w. 2 of 1mq6_ligand
hit Enter, the pharmacophores within 2Å to the loaded ligand 1mq6_ligand.mol2 will be selected as shown below:
16
Select Confined docking: search poses within specific regions of the binding site in Rerun dialog. Click OK when the selection of pharmacophore is complete. The docking calculation will start in the background and the following information dialog will appear:
The results of the rerun will be stored in Monitor_rerun_out.aut (Monitor_’output_foldername’.aut). When the docking simulation is finished, the results can be imported:
PharmDock → Import Results
Select Monitor_rerun_out.aut, the docking results will be loaded. Again load the 1mq6_ligand.mol2 from $PHARMDOCKHOME/examples/fXa/. We observe a more similar binding pose of PharmDock top-1 prediction and the original X-ray ligand binding pose.

17
References
1. OpenEye Scientific Software: Santa Fe N, USA, www.eyesopen.com. OMEGA: version 2.2.0 2. Hawkins PCD, Skillman AG, Warren GL, Ellingson BA, Stahl MT (2010) Conformer generation with OMEGA: algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J Chem Inf Model 50 (4):572-584 3. Hawkins PC, Nicholls A (2012) Conformer Generation with OMEGA: learning from the data set and the analysis of failures. J Chem Inf Model 52 (11):2919-2936 4. Klaholz BP, Mitschler A, Moras D (2000) Structural basis for isotype selectivity of the human retinoic acid nuclear receptor. J Mol Biol 302 (1):155-170 5. Wang R, Fang X, Lu Y, Wang S (2004) The PDBbind database: collection of binding affinities for protein-ligand complexes with known three-dimensional structures. J Med Chem 47 (12):2977-2980. doi:10.1021/jm030580l 6. Wang R, Fang X, Lu Y, Yang CY, Wang S (2005) The PDBbind database: methodologies and updates. J Med Chem 48 (12):4111-4119 7. Word J, Lovell S, Richardson J, Richardson D (1999) Asparagine and glutamine: using hydrogen atom contacts in the choice of side-chain amide orientation1. J Mol Biol 285 (4):1735-1747 8. Adler M, Kochanny MJ, Ye B, Rumennik G, David R, Biancalana S, Whitlow M (2002) Crystal structures of two potent nonamidine inhibitors bound to factor Xa. Biochemistry (Mosc) 41 (52):15514-15523