pathogen informatics 21 st nov 2014 pathogen sequencing informatics jacqui keane pathogen...
TRANSCRIPT

Pathogen Informatics 21st Nov 2014
Pathogen Sequencing Informatics
Jacqui KeanePathogen Informatics

Pathogen Informatics 21st Nov 2014
Pathogen Informatics Team
▸ Team of 9 software developers and bioinformaticians
CarlaCummins

Pathogen Informatics 21st Nov 2014
Role of Pathogen Informatics
▸ Informatics support to pathogen variation programme
▸ Dougan, Lawley, Parkhill, Berriman, Thomson & Kellam faculty teams
▸ Researchers, visiting workers, collaborators
▸ Approx. 120 people
▸ Applications and systems to support research activities
▸ Automated pipelines for sequence tracking and analysis
▸ Ad-hoc bioinformatics support and training
Pathogen Informatics 21st Nov 2014
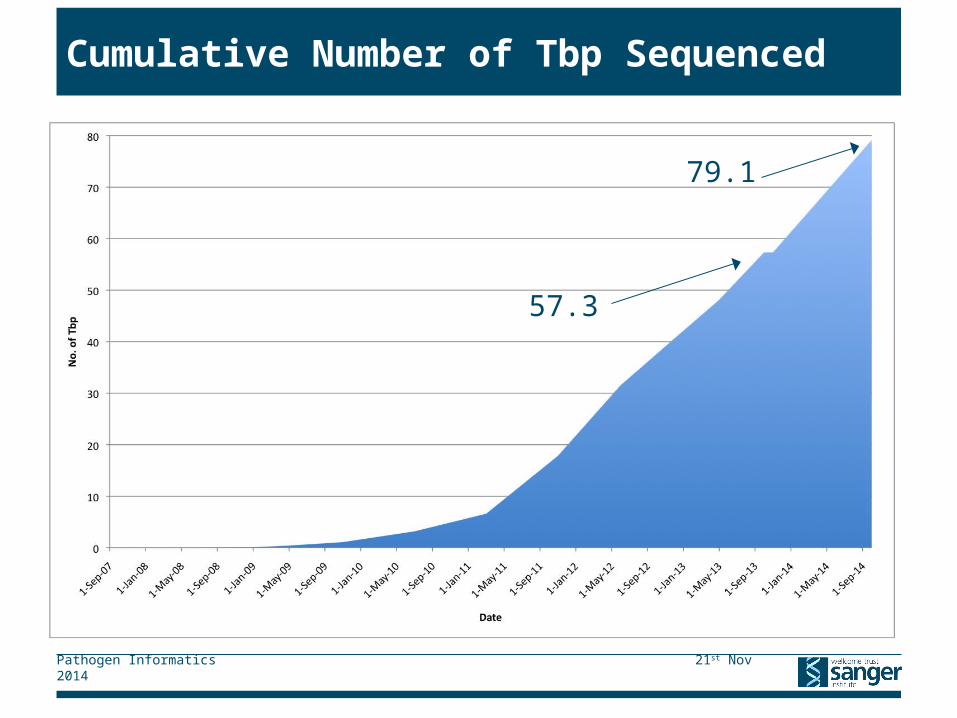
Cumulative Number of Tbp Sequenced
79.1
57.3
Pathogen Informatics 21st Nov 2014
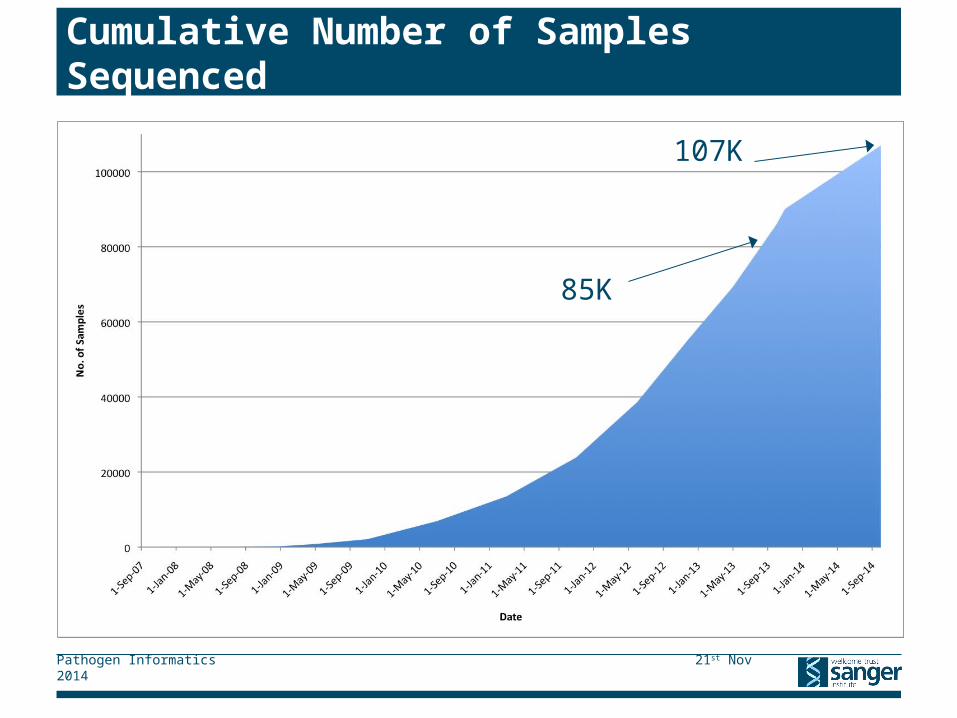
Cumulative Number of Samples Sequenced
107K
85K

Pathogen Informatics 21st Nov 2014
Sequence Analysis Pipelines
Assembly
Annotation
Mapping
Variant Calling
RNA-Seq Expression
QC
Sequence Tracking
Pathogen Informatics 21st Nov 2014
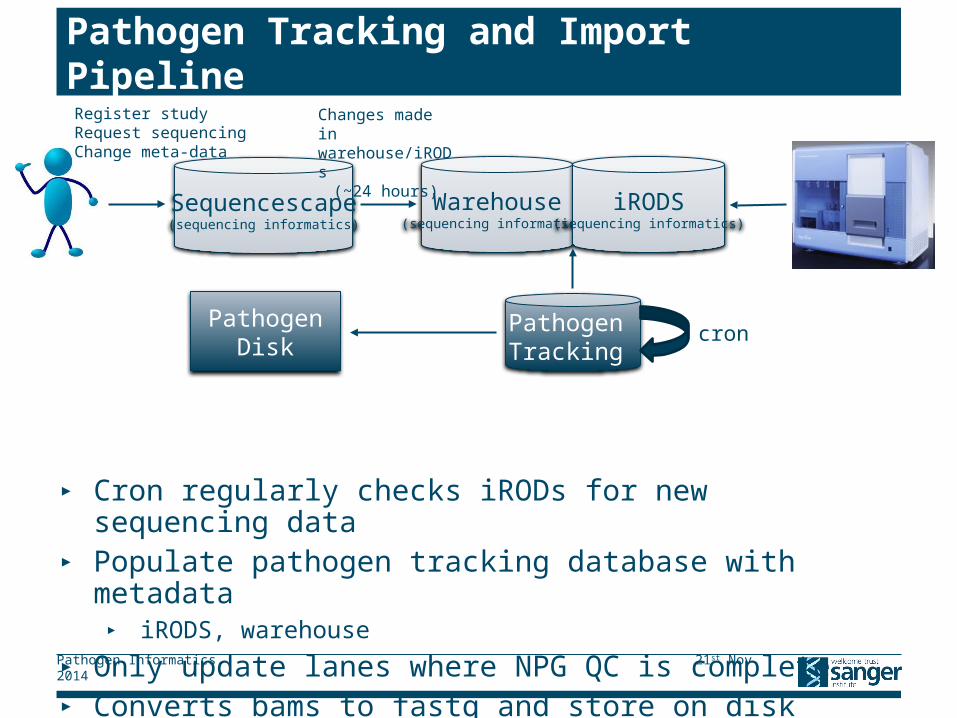
Pathogen Tracking and Import Pipeline
▸ Cron regularly checks iRODs for new sequencing data▸ Populate pathogen tracking database with metadata
▸ iRODS, warehouse
▸ Only update lanes where NPG QC is complete▸ Converts bams to fastq and store on disk▸ Convert bax5 files to fastq and store on disk
Warehouse(sequencing informatics)
iRODS(sequencing informatics)
Pathogen Tracking
PathogenDisk
cron
Sequencescape(sequencing informatics)
Changes made in warehouse/iRODs
(~24 hours)
Register studyRequest sequencingChange meta-data

Pathogen Informatics 21st Nov 2014
Finding Data
Script:
pathfind
Examples:
▸ Where is the FASTQ for a lane:pathfind -t –id 1234_5
▸ Make a symlink to FASTQpathfind -t –id 1234_5 –symlink
▸ Find all FASTQs for a species:pathfind -t species -i Staph
▸ Output lane stats to a .csv file:pathfind -t species -i Staph –results out.csv
▸ Get all the options:pathfind -h

Pathogen Informatics 21st Nov 2014
Sequence Analysis Pipelines
Assembly
Annotation
Mapping
Variant Calling
RNA-Seq Expression
Sequence Tracking
QC

Pathogen Informatics 21st Nov 2014
QC Pipeline
▸ Align 100MB to reference with bwa▸ Generate QC stats
▸ Basic statistics on fastq e.g. yield
▸ Percent reads/bases mapped
▸ Percent genome covered
▸ Error rate
▸ Create QC Plots▸ GC plot vs. reference GC
▸ Insert size distribution
▸ Base quality
▸ Coverage
▸ Run Kraken ▸ assigns taxonomic labels to short DNA sequences
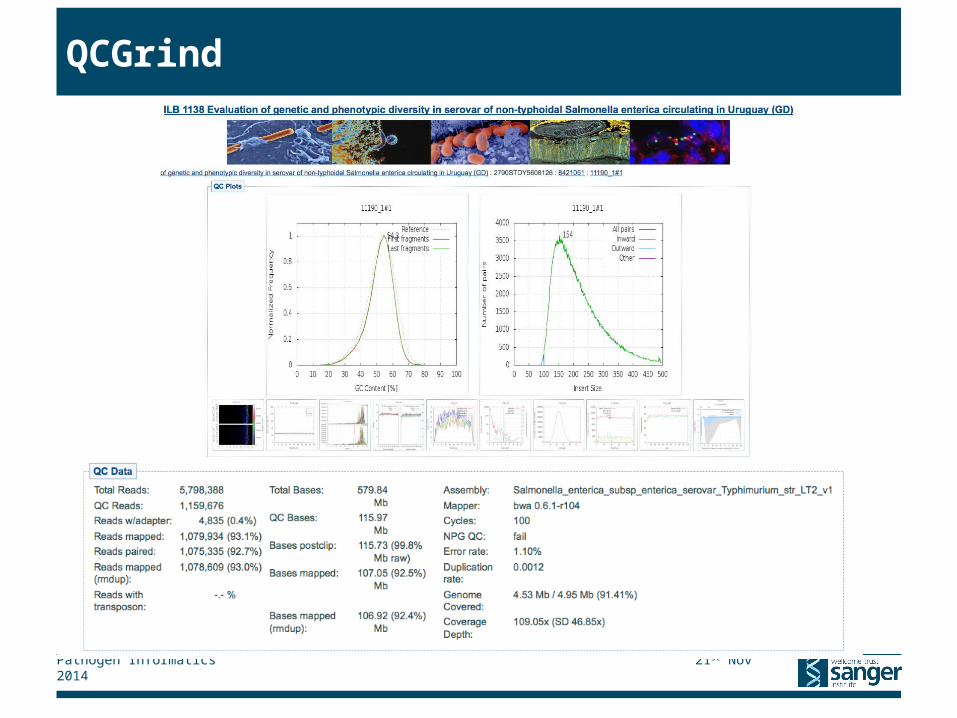
▸ Results presented through QCGrind web interface
Pathogen Informatics 21st Nov 2014
QCGrind
Pathogen Informatics 21st Nov 2014
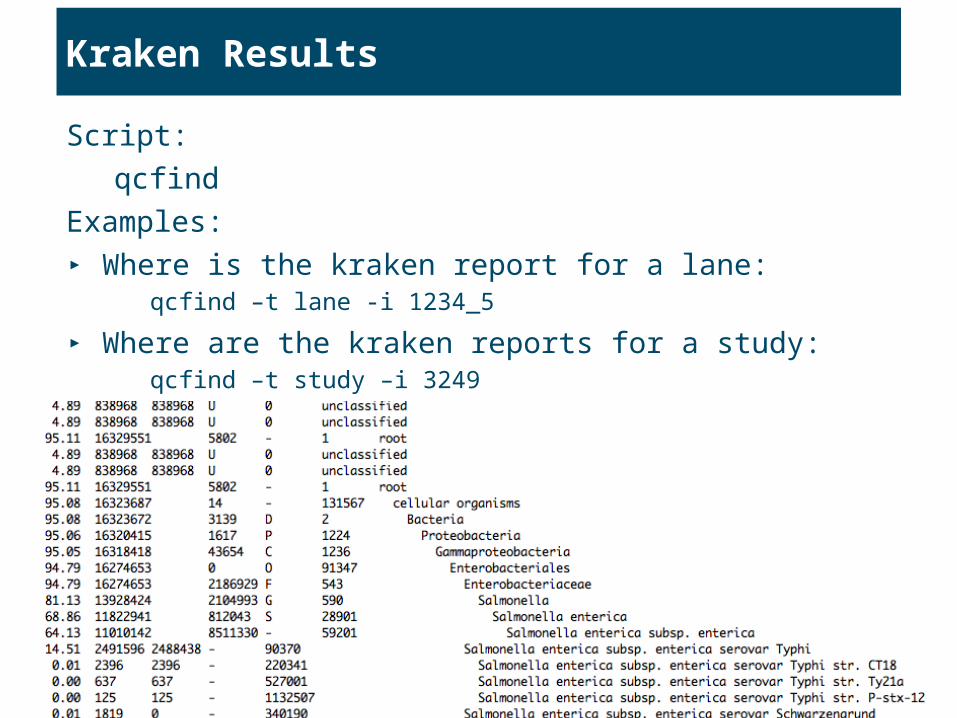
Kraken Results
Script:
qcfind
Examples:
▸ Where is the kraken report for a lane:qcfind –t lane -i 1234_5
▸ Where are the kraken reports for a study:qcfind –t study –i 3249

Pathogen Informatics 21st Nov 2014
Sequence Analysis Pipelines
QC
Annotation
Mapping
Variant Calling
RNA-Seq Expression
Sequence Tracking
Assembly

Pathogen Informatics 21st Nov 2014
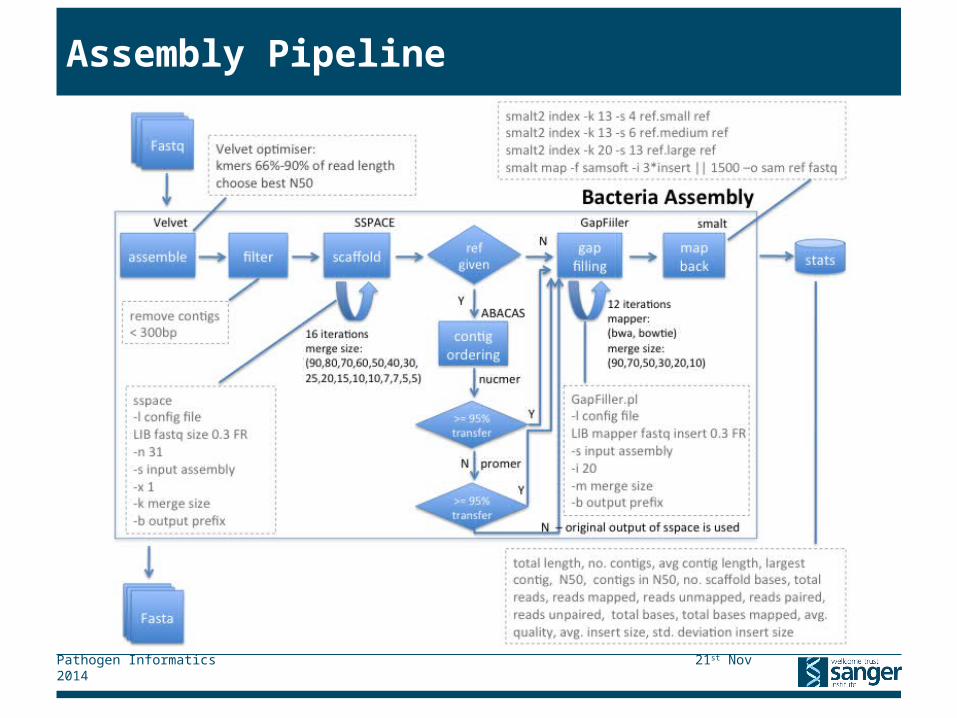
Assembly Pipeline
▸ Bacteria samples are assembled automatically
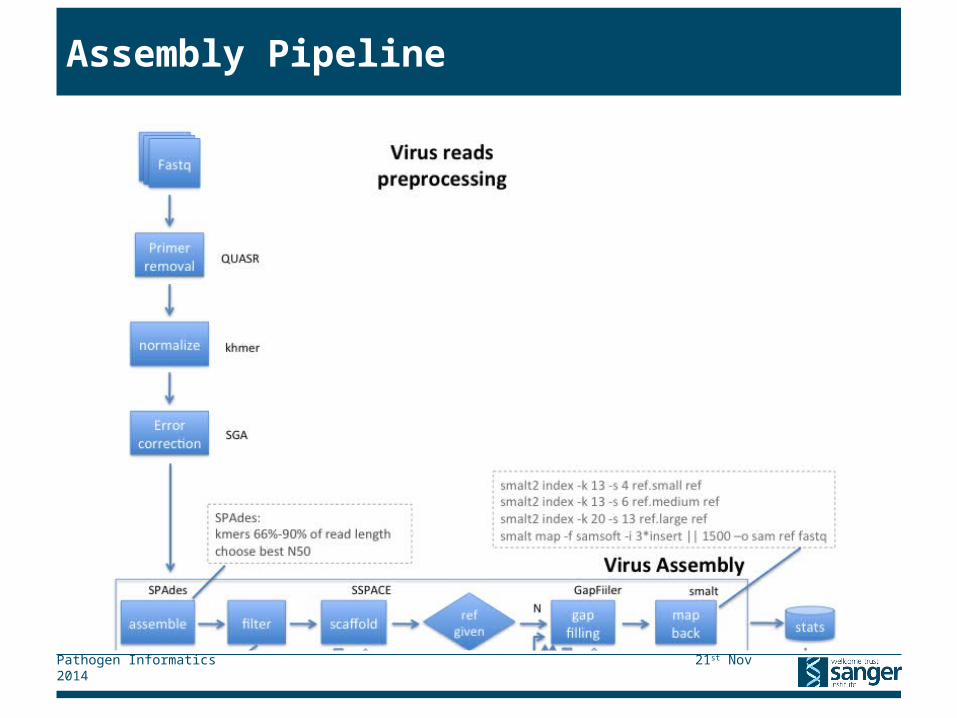
▸ Virus samples are assembled automatically on a study by study basis
▸ Eukaryote samples are assembled on a per request basis
▸ PacBio samples are assembled automatically using HGAP
Pathogen Informatics 21st Nov 2014
Assembly Pipeline
Pathogen Informatics 21st Nov 2014
Assembly Pipeline

Pathogen Informatics 21st Nov 2014
Assembly: get results
Script:assemblyfind
Examples:
▸ Create symlinks to all the final assemblies in the given studyassemblyfind -t study -id "My study" –symlink
▸ Find an assembly for a given laneassemblyfind -t lane -id 1234_5#6
▸ Make a .csv file of assembly stats for a given species:assemblyfind -t species -i "Leishmania donovani” -stats
▸ Get all the options:assemblyfind -h

Pathogen Informatics 21st Nov 2014
Sequence Analysis Pipelines
QC
Assembly
Mapping
Variant Calling
RNA-Seq Expression
Sequence Tracking
Annotation

Pathogen Informatics 21st Nov 2014
Annotation Pipeline
▸ Run automatically on all Bacteria denovo assemblies (also works for Viruses)
▸ Can be run in standalone modeannotate_bacteria
▸ Annotation ready for submission to EMBL/Genbank
▸ Pipeline Steps▸ Genes predicted with Prodigal
▸ RNA predicted with Infernal
▸ The databases are searched in the following order:▸ Genus specific RefSeq databases
▸ UniprotKB – bacteria/virus databases
▸ Conserved domain database
▸ pfam (A)
▸ rfam

Pathogen Informatics 21st Nov 2014
Annotation: get results
Script:annotationfind
Examples:
▸ To get annotation for all samples in study 123:annotationfind -t study –id 123
▸ Find annotation for a given lane:annotationfind -t lane -id 1234_5#6
▸ Create a multi fasta file of all of the gryA genes for Staph:annotationfind -t species -i “Staph” –g gryA
▸ Get all the options:annotationfind -h

Pathogen Informatics 21st Nov 2014
Sequence Analysis Pipelines
QC
Assembly
Variant Calling
Sequence Tracking
Mapping
Annotation
RNA-Seq Expression
Pathogen Informatics 21st Nov 2014
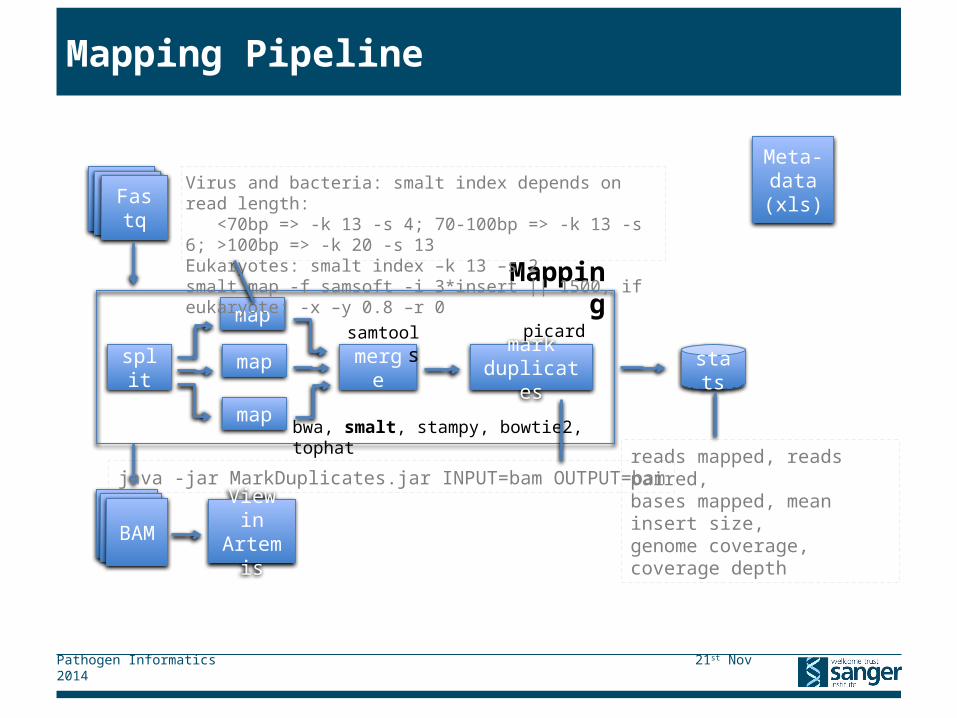
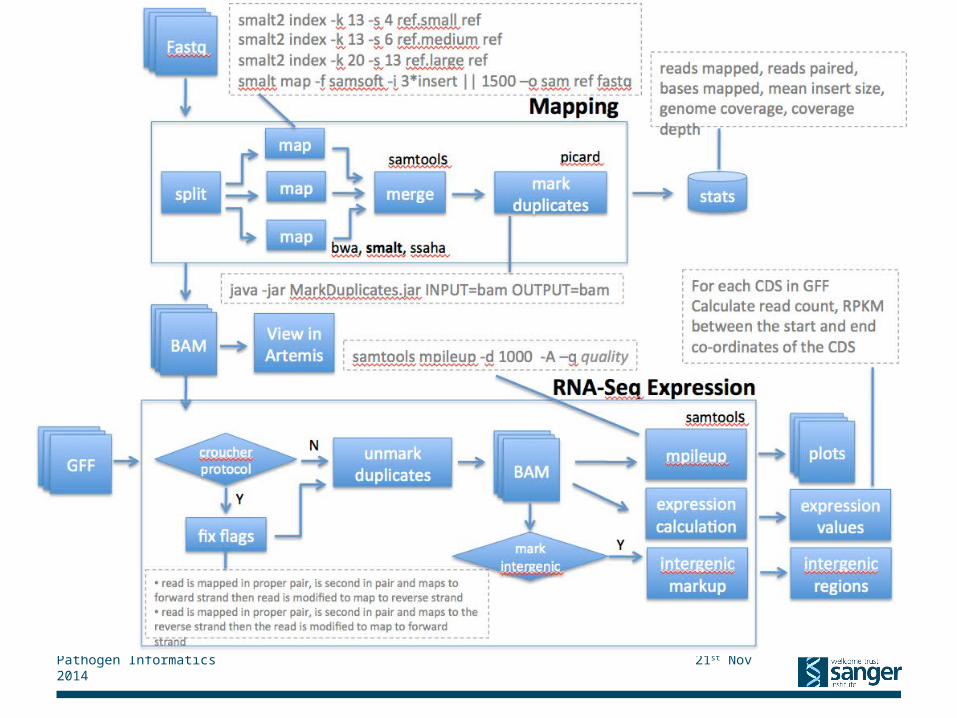
Mapping Pipeline
Mapping
FastqFastqFastq
split
map
merge
bwa, smalt, stampy, bowtie2, tophat
markduplicates
map
map
stats
picard
BAM View in Artemis
BAMBAM
Virus and bacteria: smalt index depends on read length: <70bp => -k 13 -s 4; 70-100bp => -k 13 -s 6; >100bp => -k 20 -s 13Eukaryotes: smalt index –k 13 –s 2smalt map -f samsoft -i 3*insert || 1500, if eukaryote: -x –y 0.8 –r 0
reads mapped, reads paired, bases mapped, mean insert size,genome coverage, coverage depth
java -jar MarkDuplicates.jar INPUT=bam OUTPUT=bam
samtools
Meta-data(xls)

Pathogen Informatics 21st Nov 2014
Mapping: get results
Script:
mapfind
Examples:
▸ Where is the BAM for a lane:mapfind –t lane -i 1234_5
▸ Make a symlink to BAM (and its index file)mapfind -t lane -id 1234_5 –symlink
▸ Find all BAMs for a species:mapfind -t species -i Staph
▸ Output mapping stats to a .csv file:mapfind -t species -i Staph –results out.csv
▸ Get all the options:mapfind -h

Pathogen Informatics 21st Nov 2014
Sequence Analysis Pipelines
QC
Assembly
Mapping
RNA-Seq Expression
Sequence Tracking
Variant Calling
Annotation
Pathogen Informatics 21st Nov 2014
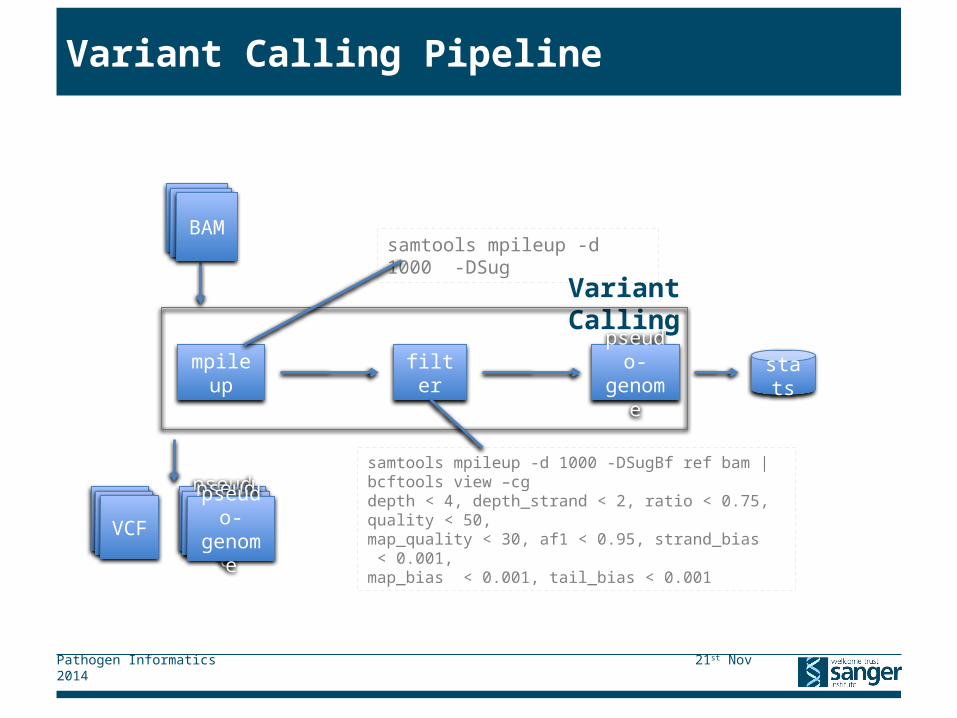
Variant Calling Pipeline
Variant Calling
VCF pseudo-genomeVCF pseudo-genomeVCF pseudo-genome
mpileup filter pseudo-genome stats
samtools mpileup -d 1000 -DSugBf ref bam | bcftools view –cg depth < 4, depth_strand < 2, ratio < 0.75, quality < 50, map_quality < 30, af1 < 0.95, strand_bias < 0.001, map_bias < 0.001, tail_bias < 0.001
samtools mpileup -d 1000 -DSug BAMBAMBAM

Pathogen Informatics 21st Nov 2014
Variant Calling: get results
Script:snpfind
Examples:
▸ Find vcf file for a lane:snpfind -t lane -i 1234_5
▸ Make symlink to vcf file (and its index) for a lane:snpfind -t lane -i 1234_5 -symlink
▸ Get single file with multifasta alignment of pseudogenomes from a file of lanes:
snpfind -t file -i filename –p
▸ Read the usage:snpfind -h

Pathogen Informatics 21st Nov 2014
Sequence Analysis Pipelines
QC
Assembly
Annotation
Mapping
Variant Calling
Sequence Tracking
RNA-Seq Expression
Pathogen Informatics 21st Nov 2014

Pathogen Informatics 21st Nov 2014
RNASeq Expression: get results
Script:rnaseqfind
Examples:
▸ All directories with RNASeq results for study 1234:rnaseqfind -t study -i 1234
▸ All spreadsheets for study 1234:rnaseqfind -t study -i 1234 -f spreadsheet
▸ Coverage plots:rnaseqfind -t study -i 1234 -f coverage
▸ Standalone script: rna_seq_expression -h

Pathogen Informatics 21st Nov 2014
Pathogen Informatics Training
▸ New starters induction▸ Getting started, basic UNIX, compute and storage
▸ Sequencing pipelines
▸ Support services provided by Pathogen Informatics▸ Queries about location of sequencing data
▸ External software applications
▸ Small scale bespoke analysis
▸ Queries about how to use pcs/farm
▸ To arrange induction if/when join pathogen team▸ Email [email protected]