part 7 specificity in protein protein interactions in ... · part 7 specificity in protein protein...
TRANSCRIPT

Robert Fletterick Biophysics 204
Part 7 Specificity in protein protein interactions in transcription
January 14, 2013
This lecture is focused on one type of association between proteins that seems
to be common in controlling transcription. In response to signals from hormones
binding to a receptor, protein phosphorylation or proteolysis of certain signaling
molecules, a transcription regulator protein may combine with transcriptional
activator proteins to build a transcription machine, an assembly of 30 or more
proteins to copy DNA to RNA.
Once inside the nucleus, the first step in transcription activation is by a protein-
DNA association at a particular site on the DNA, called a response element,
composed of perhaps 6 to 12 contiguous or separated base pairs.
The second and subsequent steps in activation add additional proteins to the
transcription complex to produce an active machine. The properties of the
machine are still being worked out, since in eukaryotes, the DNA is packaged
and needs to be made ready to transcribe.
We will study three similar types of helix to protein associations that are
characterized by small interfaces and weak to moderate binding. It also seems

that in these cases, the docking helix goes from disordered to ordered state
when the two proteins associate.
For the transcriptional activation, we first consider the VP16 protein. VP-16
derives from herpes virus.
VP16 is an activator, which contains a small domain that has multiple negative
charges. Based on the types of amino acids, this domain is called an acidic
activation domain. The protein binds to TAF31. Another transcriptional activator
protein involved in a checkpoint to recognize DNA damage, P53 also binds to
TAF31. The issues before the Verdine study [Uesugi M; Nyanguile O; Lu H;
Levine AJ; Verdine GL. Induced alpha helix in the VP16 activation domain upon
binding to a human TAF. Science, 1997 Aug 29, 277(5330):1310-3.] were how
the protein-protein interaction was made.
Engineering constructs showed that a 181 amino acid N-terminus of TAF 31and
a C-terminal region 452-490 were involved in the interaction. The ability to bind
was checked by making the TAF truncation 1-141, which is a conserved region in
the TAF. A segment that binds is defined by NMR [in an HSQC experiment] is
found in the figure from their paper. VP16C was labeled with amino acids
containing N15 which is NMR active. The gray bands and black bands (protons

of the sidechain) map H nuclei that change on binding TAF as measured by
NMR.
Part B shows gels of protein recovered from GST-VP16C binding assays. Lane
4 for example shows no binding for VP16 452-474. C shows the ability to
activate transcription.
NMR studies show that the VP16 domain has no regular structure in solution.

In the figure below, C shows the CB protons mapping to a face of a helix when
the amino acid sequence is drawn as a helical wheel.

Truncating the F and L to Ala reduced TAF binding and activation. Asp 486 can
be deleted without affecting binding or activation. The acidic activation domains
of VP16, P53 and NF-KB (P65) are similar in sequence as seen in the figure
below.

These three fragments of transcription factors are aligned by the FXX motif.
Purple marks the residues of VP16C perturbed by binding TAF1-140. Green
marks the residues of p53 that contact MDM2. All three contain a Phe separated
by a two-residue spacer from a hydrophobic pair. Mutations in P53 show that
these are essential for binding. Phe is required in P65.
What is the role of the acidic residues?
5 of 17 amino acids of VP16 469-485 are acidic, negatively charged. Only one
shows CB chemical shift on binding to TAF31. Mutagenesis shows that the
position of the negative charges in not important however, the number of
negatively charged side chains affects the rate of transcription. Asp 472 is not
conserved in P53 and NF-kB. Perhaps the charges are used in long range

electrostatic targeting with the hydrophobic interactions taking over as the helix
forms.
This is a mechanism where multiple weak interactions build to a strong
biochemical response.
Two hydrophobic residues are the primary motifs for the acidic activation
domains.
The p53 domain is also seen by X-ray crystallography in its interactions with
MDM2.
P53 may be one of the most important proteins in the cell. It has domains for
forming dimers, binding to DNA and attracting transcription factors. Activation of
p53 can lead to cellular growth arrest prior to entry into either S phase or mitosis
or can trigger cell death through apoptosis. The modification of p53 by multisite
phosphorylation controls its function. P53 is a tumor suppressor and is mutated
in 50 % of all human tumors. MDM2 is an oncoprotein and partner of p53. It
inhibits some functions of p53. In cancers where p53 is still functional, MDM2
may be over expressed to disable the resident p53. It binds the transactivation
domain of p53 and halts activation presumably by stopping accumulation of
accessory transcription factors. P53 can activate expression of MDM2, to further

complicate controls. Deletion of the gene for p53 or MDM2 is lethal, but embryos
with both partners deleted are viable.
The structure of a 109 amino acid piece of MDM2 bound to a 15 amino acid
fragment of p53 that is the part of the trans activation domain was determined.
[Kussie PH; Gorina S; Marechal V; Elenbaas B; Moreau J; Levine AJ; Pavletich
NP. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor
transactivation domain. Science, 1996 Nov 8, 274(5289):948-53.] Mutational
analysis showed that a 12kD piece of MDM2 is conserved (70% human to frog)
and sufficient for binding.
Calorimetry of p53 with MDM2 domains showed that the Kd’s for the 15 amino
acid p53 (.6 micromolar) peptide were similar to the Kd’s for larger p53 domains.
The overview of the structure shows the helix of p53 bound between helices of
the MDM2 domain:

Description of the structure
A hydrophobic face of the p53 amphipathic helix, 2.5 turns, docks into a
hydrophobic groove of a cleft formed between two helices. Three extended
residues make weaker contacts. Four of five hydrophobic sidechains are in the
interface and solvent inaccessible.
Phe 19, Trp 23 and Leu 26 are the key players in the structure and invariant in
evolution. Mutagenesis studies have shown several to be important in p53’s
recruitment of the TAF’s.

13 amino acids are ordered. The peptide orders on binding MDM2. Thr 18 and
Asp 21 probably order the helix through sidechain to backbone interactions. Asp
21 and Lys 24 may also form a salt bridge.
This interface is unusual. MDM2 groove is lined with 14 conserved hydrophobic
residues. 1500 Å2/two proteins of surface, mostly hydrophobic atoms contribute,
are buried. Two H bonds form. The implication is that the hydrophobic side
chains are also used to attract TAF’s and this structure shows how MDM2
defeats the surveillance function of p53. Secondly, perhaps the repeat pattern of
hydrophobic sidechains may be predictive of the transactivation partner
associations.
Is this a candidate interface for drug intervention?

Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N,
Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. In vivo activation of the
p53 pathway by small-molecule antagonists of MDM2. Science. 2004 Feb
6;303(5659):844-8.
Structure and mode of binding of MDM2 inhibitors.

Biacore- A concentration series of each compound incubated with MDM2 and
injected onto a chip with p53 protein
(B) MDM2 with the Nutlin-2 in p53 pocket.
(C) Nutlin-2 (carbon white, nitrogen blue, oxygen red, and bromine brown) with
side chains of Phe19, Trp23, and Leu26
(D) Surface of p53 binding pocket of MDM2 (buried regions, green; exposed
portions, red) showing one bromophenyl group buried deep in the Trp pocket.
A related example is for CREB and CPB, but here phosphorylation controls
assembly.
Radhakrishnan I; Perez-Alvarado GC; Parker D; Dyson HJ; Montminy MR;
Wright PE. Solution structure of the KIX domain of CBP bound to the
transactivation domain of CREB: a model for activator:coactivator interactions.
Cell, 1997 Dec 12, 91(6):741-52.
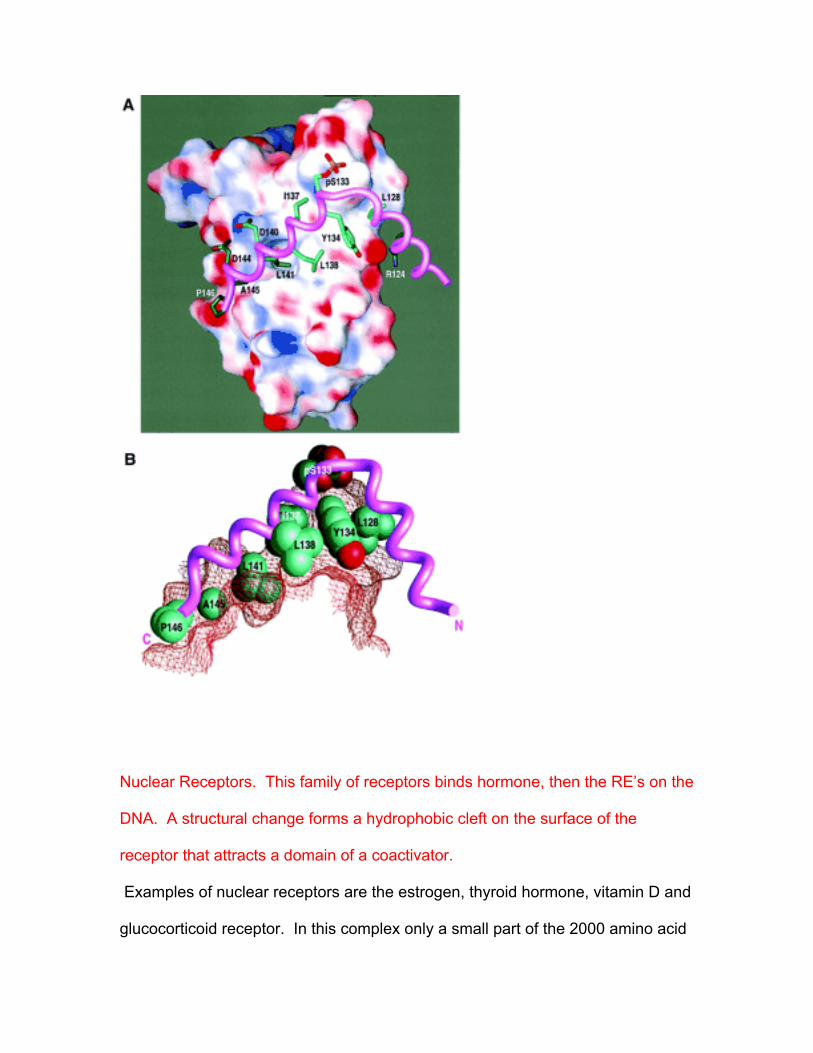
Signals at the cell surface get into the nucleus to activate transcription. One of
the first described activation processes was for the cAMP system. In this
response to increased levels of the second messenger, the nuclear protein factor
CREB (cAMP response element Binding) activates transcription of target genes.
CREB is phosphorylated on Ser 133 by cAMP kinase, PKA, and binds to the

appropriate spot on DNA. This interaction is not fully understood, but occurs in
part through direct interactions with a second general transcription factor or
coactivator called CBP for CREB Binding Protein. Only a small part of CPB and
of CREB participates in the interaction, this is the KIX domain of the CBP.
Further, the CREB-CBP(KIX) binds only when the Ser is phosphorylated.
NMR was used to determine the details of the interaction. The solution structure
of the complex formed by the phosphorylated kinase-inducible domain (pKID) of
CREB with KIX was determined. NMR analysis for the free and complex reveals
that pKID undergoes a coil to α-helix folding transition when it binds to KIX. In
this process, two α−helices form.
The amphipathic helix αB of pKID binds to a hydrophobic groove. In this case as
in the other examples in this lecture, the groove is shallow, here defined by
helices α−1 and α−3 of KIX. The other pKID helix, α−A, contacts a different face
of α3.
Phosphorylation is required to form the complex. The phosphate group of the
critical phosphoserine might be expected to bind to Arg or Lys sidechains and
require a conformational change. But the phosphoryl group of pKID forms a
hydrogen bond to the side chain of the uncharged Tyr-658 of KIX.

The NMR structure shows again that KID is unstructured before it interacts.
Then, the two helices of the CREB KID domain interact by a primarily
hydrophobic surface on an amphipathic alpha helix, Pro 132 to Asp 144. Three
turns bind. A second helix at 90 degrees to the first makes fewer interactions.
Here 1200 Å2 are buried. The interactions are with Y 134, I 137, L 138 and L
141. Also, Asp 140 forms a pair with K of KIX. The phosphoserine binds to a Tyr
and is near a Lys.
What is the role of the Ser-phosphate?
Since it is not interacting strongly with the KIX domain, the charges are likely
used to stabilize the KID helix by initiation of the helix 2 through interactions with
the backbone NH’s.
Other factors contact the CBP KIX have a motif Glu Ph X X Ph Ph, similar to the
KID motif.
Nuclear Receptors. This family of receptors binds hormone, then the RE’s on the
DNA. A structural change forms a hydrophobic cleft on the surface of the
receptor that attracts a domain of a coactivator.
Examples of nuclear receptors are the estrogen, thyroid hormone, vitamin D and
glucocorticoid receptor. In this complex only a small part of the 2000 amino acid

protein binds to the receptor. The remaining domains of the coactivator contact
DNA, bind other proteins, such as CBP, and have acetyl transferase activity. A
helix of the transcriptional coactivator of the SRC-1 or GRIP-1 family form from
13-15 amino acids that contain the LXXLL motif. The coactivators may have two
or three copies of the motif that is called the NR box. This motif is a strikingly
small and bland!
Robert Nolte et al: Ligand binding and co-activator assembly of the peroxisome
proliferator-activated receptor- Nature 395, 137-143 (1998)
The figure below shows familiar interactions and concepts.
Figure 3 SRC-1 interactions with PPAR-gamma, A sigma-weighted 2Fo-Fc omit
electron-density map is shown contoured at 1.0 for the area surrounding the
rosiglitazone ligand. b, A ribbons drawing of the PPAR- LBD dimer and SRC-1,
including the ligand rosiglitazone. The two PPAR- monomers are blue and green
and the two SRC-1 interacting helices are yellow. The structure of SRC-1 was
determined from amino acids 628-640 and 684-703 and was crystallographically
refined. Very weak electron density from residues 670 to 684 was visible but was
not crystallographically refined and is shown as a dashed line. SRC-1 amino
acids 642-669 were disordered and not structurally determined. The diagram
shows how one SRC-1 molecule, with two interacting domains, forms a complex

with a PPAR- homodimer. The dashed line connecting the two structurally
determined domains of SRC-1 is the proposed connection between these two
domains. c, The binding of SRC-1 (amino acids 628-642) to the LXXLL-binding
site of PPAR-. SRC-1 is colored: yellow, carbon; blue, nitrogen; red, oxygen. The
ribbon backbone of the PPAR- LBD is in green. PPAR- amino acids binding to
the LXXLL helix are also shown in green. d, Residues H631-T640 of SRC-1 are
colored as in c, with an electrostatic surface of PPAR- showing the coactivator-
binding site. E471 and K301 side chains result in the red (negative) and blue
(positive) charges on the surface of the coactivator-binding site at the N and C
termini of the SRC-1 helix, respectively. e, Residues H687-E696 of SRC-1 are
colored as in c, with an electrostatic surface of PPAR- showing the coactivator-
binding site. f, Amino acids L465-K474 of the PPAR- AF-2 helix of one monomer
in the apo structure are shown in: green, carbon; blue, nitrogen; red, oxygen, with
an electrostatic surface of PPAR- showing the coactivator-binding site. E471 and
K301 side chains result in the red (negative) and blue (positive) charges on the
surface at the N and C terminus of the other PPAR- monomer. This figure shows
how one monomer in the apo crystal structure orientates its AF-2 helix into the
coactivator-binding site of another crystallographically related monomer.

The PPAR γ structure is known with RXR on DNA.

This is discussion paper for the last lecture in this series.
Nature , 350-356 20 November 2008
Structure of the intact PPAR-big gamma–RXR-alpha nuclear receptor complex
on DNA
Vikas Chandra1,4, Pengxiang Huang1,4, Yoshitomo Hamuro2, Srilatha
Raghuram1, Yongjun Wang3, Thomas P. Burris3 & Fraydoon Rastinejad1