parallel computing—a route to complexity and reality in material simulations shiwu gao department...
TRANSCRIPT

Parallel Computing—a route to complexity and reality in material simulations
Shiwu Gao
Department of Applied Physics
Chalmers/Göteborg University

Parallel computing and materials simulations
Water-metal interface
Dynamics of electron excitation/transfer
BiomembraneAquaporin water channel in membraneK. Murata et al, Nature, 407, 599 (2002)

Macroscopic(meter, hour)
Mesoscopic
Kinetics
Energetics
Atomic
Electronic(Å, fs)
Bottom-up approach
Theoretical approach based on:
1) Fundamental laws of physics
2) Computer modeling and simulations

Density Functional Theory based simulations
2
22
)()(
)()()()(2
Fi
i
iiiXCCoulomb
n
nvnm
rr
rr
Solving the Kohn-Sham Equations for all electorns
Full-potential and pseudopotential methods -Ful-potential methods (FP-LAPW, FP-LMTO) accurate and slow
-Pseudo-potential methods (VASP, CPMD, PWSCF) fast but with uncertainty in pseudopotentials

Outline • Parallelization of WIEN package
FP-(L)APW method
• Applications - Hydrogen bonding by CH group
- Pressure melting of confined water films

WIEN97 (T. U. Vienna)
Typical timing (s)
H/Cu(100) p(3x3) 3+2+5layers 29 atoms
Potential
Eigenproblem
Density
Core Electron
Mixing in/out data
+ Accurate + Versatile -- Slow-- larger RAM

Timing in LAPW1
0
10
20
30
40
50
60
70
80
90
H S Hns Solver
Exact
Iterative
- Large memory needed for H,S
RAM ~ M2
- Time-consuming
H |Ψk>= εkS |Ψk> t ~ M3
For large systems (>30 atoms)
- more than 90 % CPU time
- severval GB RAM

Parallelizing the eigenproblem (LAPW1)
2. Parallelizing the eigensolver
-Incorporating PQR
-Writting an iterative parallel solver
Myid = 0 1 2 3 0 1 2 3 0
1. Distributing and parallel setting H and S
PQR:X.B. Chi, Inst. Software, Chinese Academy of Sciences,Beijing

Further Parallelizations
+ LAPW1 Distributing H S setting and parallelizing the eigensolver -Incorporating PQR
-Writting an iterative parallel solver
+ LAPW2 and LAPW0
Distributing the calculation atom-wise
+ Implemeting the new APW+lo basis, E. Sjöstedt, Nordström, and Singh, Solid State Commun 114, 15 (2001)
S. Gao, Comput. Phys. Commun. (to be published)

Test example: C2H4+O2/Ag(110) coadsorption
- 100 surface atoms -Ag(110) 3x4x7=84
-(C2H4+o2)x2=16
- 6 layer vacuum- 21x23x35 au3
- Dual basis -Ag(110) LAPW -molecules, APW+lo
- 1-k point- 9 Ry cut-off structure- 13 -16 Ry in energy- 12 min/SCF 24 SGI3k- 12-15 Ionic steps/day

Scaling on IBM SP3 (PDC, KTH)
Tested up to48 CPUs
+ Nearly linear-scaling
M=14400

Scaling on Seth---Linux cluster at HPC2N
Up to 128 CPUs
Seth and SP3: 1) comparable scaling, 2) different in speed

Summary on scaling and performance
Timing consuming parts Acceleration on p CPUs
Setting H and S 0.98—1.0 p
HNS 0.79—0.9 p
Eigensolver--PQR 0.91–-0.94 p
Iterative Diag. 0.7— 0.8 p
Charge (LAPW2) ~ Na (or no acc.)
Potential (LAPW0) ~ Na (or no acc.)
Applicable to large systems, as PW-PP methods

Hydrogen bonding by CH group C2H4+O2/Ag(110)
Expt: J. R. Hahn, W. Ho, UCITheory: S. W. Gao, Chalmers

Why hydrogen bond with CH group
• H-bond is ubiquetous in biomolecules and organics
• Also of interest for fundamental studies (Ionic, covelency, vdW?)
• Usually with FH (VII), OH (VI), and NH(V) due to the large affinity, favoring ionic coupling
• H-bond with CH, weak—controversial EHB < 1 kcal/mol (c.a. 43 meV)

Building artificial complex with organics

Questions:
- Structure and orientations
- Interaction between ethylene oxygen
- IE-STS

Distance-dependent interaction
-27.4 meV
-90.4 meV
-6.6 meV
In the gas phase: the interaction is negligible ~ + 10 meV

Mechanism of H-bond formation --adsorption induced electron transfer

Pressure Melting of Confined Water from ab initio Molecular Dynamics Simulation

Background and Motivation
• Special phenomena in confined water• Bio-membrane fusion: role of thin water films• Pressure:
-phase control-material synthesis-mechanical stimuli in biology
• Ice-skating and lubrication• How to characterize confined liquid water from computer simulations

• New water phases in confined water
• Existence of solid-liquid critical points
K.Koga et al.,Nature 412, 802 (2001)

Bio-membrane Fusion
Science 297, 1817 & 1878 (2002)

Phase Diagram of Water
Science 297, 1288 (2002)

Simulation Method
• VASP—Veinna ab intio simulation package (better adapted to MD simulations)
.• Slab representation in a supercell
geometry: up to 48 Pt atoms and 32 H2O molecules

Applying the pressure
Pt
Water
Pt
ΔZ

Kinetic Energy vs. ΔZ
Transition atvolume change 6.6 %
Bulk ice (expt.) 6.4 %

Layer-resolved Ek~ΔZ
4th Layer3rd Layer2nd Layer1st Layer

Estimating the pressure
P = F / S

Trajectories of a molecule from the simulation
Before meltingAfter melting
--------
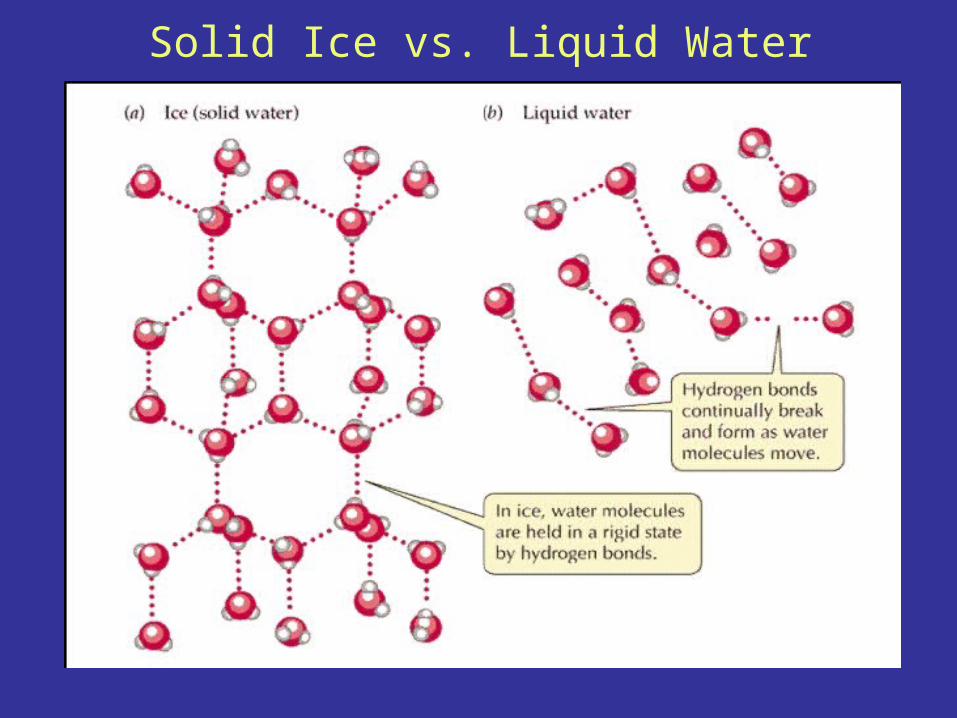
Solid Ice vs. Liquid Water

Hydrogen Bond Dynamics

Pair Correlation Function

Summary
• Parallel WIEN for large-scale ab initio electron structure calculations
• Applications in material simulations1. Hydrogen bonding mechanism induced by
adsorption 2. Pressure induced phase transitions of water
films

Route to complexityParallelcomputing
Single CPU processing

Acknowledgements
• Sheng Meng
• Supported by VR
• E. G. Wang’s (IOP, Beijing), B. Kasemo, Chalmers • P. Blaha (TU Vienna)
• E. Sjöstedt, L. Nordström (Uppsala)
• Technical support from (Ulf Andersson , Niclas Andersson, Torgny Faxen)