overview of us fda: drugs
TRANSCRIPT

By: Group No. 1Akshay Joshi (1)
Anjana Viswanathan (3)Mukta Asudani (4)
Nagesh Bhaykate (6)Pooja Borole (7)
Gaurav Andhansare (2)

05/01/20232 Regulatory Affairs
US FDA: Federal Agency of the US Dept. of Health & Human Services
Main responsibility is protecting the public health by assuring the safety, effectiveness, quality, and security of human and veterinary drugs, vaccines and other biological products, and medical devices
Introduction

05/01/2023Regulatory Affairs3
Organization of FDA
US FDA
Office of Commissioner
Office of Foods & Veterinary Medicine
Office of Medical Products &
Tobacco
Office of Global Regulatory
Operations and Policy

05/01/2023Regulatory Affairs4
Organization of FDA
Office of Medical Products & Tobacco
Centre for Drug Evaluation and
Research
Office of Generic Drugs Office of New Drugs
SSMRDs
Office of Prescription Drugs
Promotion(formerly DDMAC)
Centre for Biologics Evaluation and
Research
Centre for Devices and Radiological
Health
Centre for Tobacco Products
Office of Special Medical Programs

05/01/2023Regulatory Affairs5
Introduction
What does FDA regulate?
1. Animal & Veterinary
2. Cosmetics
3. Drugs
4. Food
5. Medical Devices
6. Radiation emitting products
7. Tobacco Products
8. Vaccines, Blood products and biologics
How does it regulate?

05/01/2023Regulatory Affairs6
Types of Application: IND
PURPOSE:
• Permission to ship an experimental drug across states before a marketing application for the drug has been approved.
• To propose a study of an unapproved drug, or a study of an approved product for a new indication or in a new patient population.
FDA's Role -- when the drug's sponsor has screened the new molecule for pharmacological activity and acute toxicity potential in animals and wants to test its diagnostic or therapeutic potential in humans.

05/01/2023Regulatory Affairs7
Types of Application: IND
Types:• Investigator IND
• Emergency Use IND
• Treatment IND
Classification of IND• Commercial
• Non Commercial (Research)

05/01/2023Regulatory Affairs8
05/01/2023Regulatory Affairs9
Types of Application: IND
Contents of IND Application:
• Animal Pharmacology and Toxicology Studies
• Manufacturing Information
• Clinical Protocols and Investigator Information
Once the IND is submitted, the sponsor must wait 30 days before initiating any clinical trials

05/01/2023Regulatory Affairs10


05/01/2023Regulatory Affairs12
Types of Application: NDA
• Marketing Approval of a Drug in the US
• Data gathered during the animal studies and human clinical trials of an Investigational new product become part of the NDA
• Goals of NDA
• Types of NDA
1. 505 (b)(1)
2. 505 (b)(2)
3. 505 (j)
• Once the application is submitted, the FDA has 60 days to conduct a preliminary review which will assess whether the NDA is "sufficiently complete to permit a substantive review”
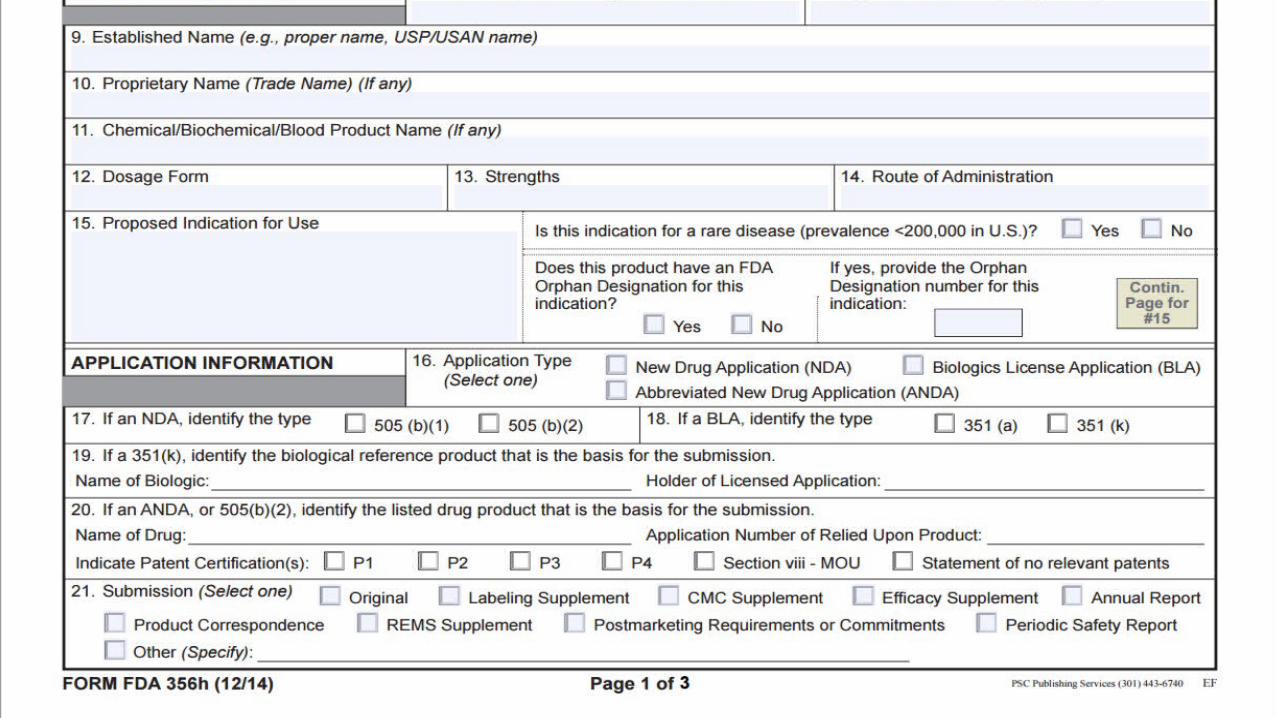
• Form FDA 356h – Application to market a New Drug.
05/01/2023Regulatory Affairs13

05/01/2023Regulatory Affairs14

05/01/2023Regulatory Affairs15
505(b)(1) 505(b)(2) 505(j)
Type of product Drug molecule not approved before by the FDA
Approved drug by the FDA but which is either a NCE/NME or with new changes
(not just duplication)
Duplication of previously approved drug with identical specifications (API, strength, dosage
forms, etc.)
Type of Information required
Complete reports of Investigations of safety and effectiveness
Complete reports of Investigations of safety and effectiveness but some of the information required for approval comes
from studies not conducted by the applicant
To show that the proposed product is identical in active ingredients, dosage form, strength,
route of administration, quality to a previously approved product.
Only Bioequivalence need to be established
Data ownership Applicant is the Owner of the DataApplicant does not have a right of
reference or right of use of data (or part of it) submitted
Data from Agency of NDA is relied upon
Purpose of Application
Approval of a new drug (for clinical use) whose active ingredient has
not previously been approved
Approval of a new drug that relies, at least in part, on data not developed by the
applicant
Approval of a “generic” version of a drug that has already been approved
User Fees Yes Yes No
Patent Period / Market Exclusivity
20 years from date of filingEffectively 8-10 years*
3 years (Formulation/Indication)5 years (NCE)
7 years (Orphan)6 months if FTF
Average FDA Review Period Approx. 10-12 months Approx. 12 months Approx. 27 months

05/01/2023Regulatory Affairs16
Types of Application: OTC
Drugs that are safe and effective for use by the general public without a
prescription.
Play an increasingly vital role in health care system.
Are considered safe if warnings and directions are followed.
Labelling is regulated by FDA
Advertising is regulated by Federal Trade Commission(FTC)

05/01/2023Regulatory Affairs17
Types of Application: OTC
• OTC Drug Categories:
Antacids Antiemetic
Antihistamine
Cough Medicine DecongestantLaxatives
Vitamins Pain Killers
Herbal Products

05/01/2023Regulatory Affairs18
Types of Application: OTC
Drug Monograph Process
FDA's review of OTC drugs is primarily handled by CDER's Office of Drug Evaluation IV.
Advisory panel review
• Category I: generally recognized as safe and effective for the claimed therapeutic indication
• Category II: not generally recognized as safe and effective or unacceptable indications
• Category III: insufficient data available to permit final classification
Creation of tentative Monograph (agency’s review)
Publication of Final monograph
FDA approval

05/01/2023Regulatory Affairs19
Types of Application: OTC

05/01/2023Regulatory Affairs20
Types of Application: OTC
• Two Regulatory pathways exist for the legal marketing of such product
• NDA process
• OTC Monographs
NDA Process OTC Monographs
• Pre-marketing approval• Confidential filling• Drug product are specify• May require a user fee• Potential for marketing exclusivity • Mandated FDA review timelines• May require clinical studies
• No Pre-marketing approval• Public process• Active ingredient –specific• No user fees• No marketing exclusivity• No mandated time lines• May require clinical studies

05/01/2023Regulatory Affairs21
IND NDA BLA ANDA OTC
Covered under FDC Act FDC Act PHS ActFDC Act,
Hatch-Waxman Act, BPCIA
FDC Act
Reviewed by CDER/CBER CDER CBER CDER, OGD CDER (ODE IV) and FTC
Listed in - Orange Book Purple Book Orange BookOTC Monograph
and Federal Register
Type of application Research Marketing Marketing Marketing Marketing
Data available/required
Preclinical,CMC,
Proposed CT protocol
Clinical, CMC, proposed labelling
Clinical, CMC, proposed labelling BA/BE
Additional safety studies, labelling
info
Types
Investigator initiated
EmergencyTreatment
505(b)(1)505(b)(2) 351(a) Generic – 505(j)
Biosimilar – 351(k) -
Comparison between IND, NDA, ANDA, BLA and OTC

05/01/2023Regulatory Affairs22
FAERS: FDA Adverse Event Reporting System (in accordance with ICH E2B)
• Who can report?
• How can one report?
• Is it made public?
• Any Limitations?
Safety

05/01/2023Regulatory Affairs23
Drug Recalls
By: Pharmaceutical firm, FDA request, other statutory body ordering FDA
Classification: Class I: Reasonable probability of serious adverse health consequences or death
Class II: May cause temporary or medically reversible adverse health consequence or remote probability of serious adverse health consequences
Class III: Not likely to cause adverse health consequences
Market withdrawal: Minor violation which may not qualify for legal action by FDA
Medical device safety alert
Information made public through website
Safety