outer-sphere redox reactions of (n)5-macrocyclic cobalt(iii) complexes. a temperature and pressure...
TRANSCRIPT

Inorganica Chimica Acta 256 (1997) 51–59
0020-1693/97/$17.00 q 1997 Elsevier Science S.A. All rights reserved
PII S0020-1693(96)05415 -1
Journal: ICA (Inorganica Chimica Acta) Article: 5415
Outer-sphere redox reactions of (N)
5
-macrocyclic cobalt(III) complexes.
A temperature and pressure dependence kinetic study on the influence of
size and geometry of different macrocycles
Manuel Martinez
a
, Mari-Angel Pitarque
a
, Rudi van Eldik
b
a Departament de Quımica Inorganica, Facultat de Quımica, Universitat de Barcelona, Diagonal 647, E-08028 Barcelona, Spainb Institut fur Anorganische Chemie, Universitat Erlangen-Nurnberg, Egerlandstraße 1, D-91058 Erlangen, Germany
Received 23 February 1996; revised 10 June 1996
Abstract
Outer-sphere redox reactions between [Co(N)
5
H
2
O]
3q/[Co(N)
5
OH]
2q((N)
5
stetraazacycloamine ligand) and [Fe(CN)
6
]
4yhave
been studied as a function of (N)
5
, temperature and pressure. The effect of the size of the (N)
5
skeleton has been investigated to establish
possible correlations between, on the one hand, the size, geometry and charge of the cobalt(III) complex and, on the other hand, the outer-
sphere formation constant, the electron-transfer rate constant, and the thermal and baric activation parameters. The values obtained indicate
that the outer-sphere formation constants are, within experimental error, the same for all the systems studied. The electron-transfer rate
constants for the [Co(N)
5
H
2
O]
3qcomplexes increase on increasing the size of the macrocyclic ligand independently of its cis or trans
geometry (from 2.3=10
y4
s
y1
(13-membered macrocycle) to 3.4=10
y1
s
y1
(16-membered macrocycle) at Ps1 atm, 258C, Is1.0 M).
For the [Co(N)
5
OH]
2qcomplexes these differences are significantly smaller (from 1.1=10
y3
to 20=10
y3
s
y1
under the same conditions).
The values for the first-order rate constants for the hydroxo complexes are one or two orders of magnitude smaller, as found for simpler
pentaamine systems; only for the 13-membered macrocyclic [Co(N)
5
OH]
2qcomplex is the trend inverted. The thermal and pressure
activation parameters are those expected for these types of reactions and are interpreted in view of a combined effect of electrostatic and
hydrogen bonding interactions. In this respect, the highly symmetrical trans 14-membered (N)
5
macrocyclic systems show an important
increase of 10–20 cm
3
mol
y1
in the value of DV/when compared with the equivalent cis systems.
Keywords: Kinetics and mechanism; Cobalt complexes; Macrocyclic ligand complexes; Redox reactions
Scheme 1.
1. Introduction
Although simple outer-sphere redox reactions of type (1)
have been studied on several occasions as a function of
temperature and pressure [1], only a few attempts to look
III II[Co (NH ) X]q[Fe (CN) ]3 5 6
III II™ [Fe (CN) ]qCo q5NH qX (1)
6 3
charges omitted for clarity
into the effect of the size of the pentaamine skeleton as a
whole have been carried out [2]. Whereas in one study only
one of the five ammine ligands was changed for a bulkier
amine [2a], in the others the size of the complete pentaam-
mine skeleton [2b,c] was changed, as indicated in reaction
(2).
nq 4y[Co(N) (Y)] q[Fe(CN) ]
5 6
3y 2q™[Fe(CN) ] qCo q5NqY (2)
6
yNsCH NH , CH CH NH , YsH O, OH
3 2 3 2 2 2
As a continuation of our interest in the effect of steric and
electronic factors that can influence or tune the reactivity
of transition metal complexes [2–4], we have studied the
effect of replacing the {Co(RNH
2
)
5
} skeleton in
[Co(RNH
2
)
5
H
2
O]
3qby a macrocyclic system {Co(N)
5
},
where (N)
5
represents any of the ligands shown in Scheme 1.

M. Martinez et al. / Inorganica Chimica Acta 256 (1997) 51–5952
Journal: ICA (Inorganica Chimica Acta) Article: 5415
The effect of a systematic variation in the macrocycle size
and geometry (Scheme 1) has already been studied for base-
hydrolysis reactions of these types of complexes [5]. In this
paper we report the effects of such variations on the outer-
sphere redox reactions depicted in Eqs. (1) and (2). In addi-
tion, we have also varied the sixth ligand in the coordination
sphere of the cobalt(III) complex from H
2
O to OH
yby
repeating the measurements at various pH values.
This rather simple reaction was selected in order to be able
to separate the encounter complex formation constant from
the electron-transfer rate constant in terms of the mechanism
outlined in (3).
3q 4y[Co(N) H O] q[Fe(CN) ]5 2 6
Kos
3q 4y°{[Co(N) H O] ; [Fe(CN) ] }
5 2 6
3q 4y{[Co(N) H O] ; [Fe(CN) ] } (3)
5 2 6
k2q 3y™{[Co(N) H O] ; [Fe(CN) ] }
5 2 6
fast
2q 3y{[Co(N) H O] ; [Fe(CN) ] }™products
5 2 6
The nature of the final redox products depends on the size
of the different (N)
5
macrocycles. For the L
14
, L
15
and L
16
macrocycles, the final product corresponds to a {Co
III
LP
Fe
II
(CN)
6
} species that is formed via an inner-sphere oxi-
dation of the short-lived {Co
II
L} species initially produced.
For the L
13
macrocycle, the {Co
II
L} species initially formed
decomposes too fast, and no further reaction takes place. In
this case Co
2qaq
is produced, as detected by the Co/EDTA/
Fe(CN)
6
complex formation [6].
The rate law derived from this mechanism is given in (4):
4ykK [Fe(CN) ]OS 6k s (4)
obs
4y{1qK [Fe(CN) ]}
OS 6
The high charge on the complexes involved allows the
kinetic separation of the encounter complex formation con-
stant, KOS
, and the electron-transfer rate constant, k. Thus,the analysis of the [Fe(CN) ] dependence of k
obs
under
4y6
pseudo-first-order conditions as a function of temperatureand
pressure, enables us to use the obtained thermodynamic and
kinetic parameters as a source of information on the effect of
steric hindrance on outer-sphere electron transfer reactions
of significantly more complex systems.
2. Experimental
2.1. Materials
All materials were reagent grade chemicals. Na
4
[Fe-
(CN)
6
] was recrystallised twice; all other chemicals were
used without further purification.
2.2. Preparation of compounds 1
2.2.1. Cis-[Co(N)5Cl]Cl(ClO4) ((N)5sL13),cis-[Co(N)5Cl](ClO4)2 ((N)5sL14, L15),trans-[Co(N)5Cl](ClO4)2 ((N)5sL14) andtrans-[Co(N)5Cl]Cl(ClO4) ((N)5sL16)The products were prepared as described previously [5b].
Characterisation of the different compounds was done by
UV–Vis spectra (l (nm) (e (cmy1
M
y1
)): 519 (120), 459
(140), 257 (140) for (N)
5
sL
13
(cis); 525 (92), 470 (94),365 (128) for (N)
5
sL
14
(cis); 550 (79), 450 (26), 360
(87) for (N)
5
sL
14
(trans); 540 (125), 480sh (87), 368
(175) for (N)
5
sL
15
(cis); 560 (93), 390 (121) for
(N)
5
sL
16
(trans).
2.2.2. Cis-[Co(N)5(H2O)](ClO4)3 ((N)5sL13, L14, L15),trans-[Co(N5)(H2O)]3q ((N)5sL14) andtrans-[Co(N)5(H2O)]Cl(ClO4)2 ((N)5sL16)The complexes were prepared via base hydrolysis of the
corresponding chloro complexes. Thesewere dissolved in the
minimum amount of 0.05 M NaOH solution and, when the
UV–Vis spectra showed no further changes, concentrated
HClO
4
was added. On slow evaporation of the samples, the
corresponding aqua complexes were obtained. Occasionally
the isolated complexeswere contaminatedwithNaClO
4
; their
recrystallisation from diluted HClO
4
produced analytically
pure samples. The aqua complexes were characterised by
their elemental analyses,
13
C NMR and UV–Vis spectra
(Table 1), except in the case of trans-[Co(L14
)(H
2
O)]
3q
where no solid sample was obtained.
Anal. Calc. for [Co(L
13
)(H
2
O)](ClO
4
)
3
P1/2H2
O: C,
20.03; H, 4.71; N, 11.68. Found: C, 19.45; H, 4.67; N,
11.34%. Calc. for [Co(L
14
)(H
2
O)](ClO
4
)
3
P7H2
O: C,
18.23; H, 5.15; N, 9.66. Found: C, 18.57; H, 5.11; N, 9.40%.
Calc. for [Co(L
15
)(H
2
O)](ClO
4
)
3
P1/2H2
O: C, 22.96; H,
5.15; N, 11.16. Found: C, 23.02; H, 5.14; N, 11.03%. Calc.
for [Co(L
16
)(H
2
O)]Cl(ClO
4
)
2
: C, 27.41;H, 6.02;N, 12.29.
Found: C, 27.12; H, 5.86; N, 12.23%.
13
C{
1
H} NMR spectra (D
2
O, ppm referenced to TMS):
cis-[Co(L13
)(H
2
O)]
3q: 67.51, 62.85, 57.47, 57.28, 54.70,
52.67, 52.27(=2), 48.78, 20.27; cis-[Co(L14
)(H
2
O)]
3q:
77.8, 71.21, 67.63, 66.75, 66.15, 62.76, 61.72, 60.81, 59.38,
35.96, 30.16; trans-[Co(L14
)(H
2
O)]
3q: 65.58, 62.29,
53.25, 55.33, 55.74, 30.98, 20.56; cis-[Co(L15
)(H
2
O)]
3q:
66.50, 59.29, 57.06, 54.52, 52.62, 48.68, 48.49, 47.51, 45.71,
20.46, 20.07, 18.93; trans-[Co(L16
)(H
2
O)]
3q: 65.02,
63.05, 62.13, 55.05, 52.91, 51.61, 50.12, 49.69, 49.15, 26.02,
23.95, 23.58, 21.15.
In the case of trans-[Co(L14
)(H
2
O)]
3q, the very small
yield of the preparative procedure for the starting chloro com-
plex [5b] did not allow a full characterisation of the base-
hydrolysis product. Nevertheless, the
13
C NMR spectrum
(see above) and the UV–Vis spectrum (Table 1) of the
1 Caution! Special care must be taken on handling perchlorate salts of
compounds containing organic ligands. There is a high risk of explosion.

M. Martinez et al. / Inorganica Chimica Acta 256 (1997) 51–59 53
Journal: ICA (Inorganica Chimica Acta) Article: 5415
Table 1
Visible spectral data for compounds [Co(N)
5
X]
2q,3qas a function of the (N)
5
macrocyle and X (H
2
O or OH
y)
(N)
5
X
a lmax1
(nm) lmax2
(nm) lmax3
(nm)
(e) (My1
cm
y1
) (e) (My1
cm
y1
) (e) (My1
cm
y1
)
L
13
(cis) H
2
O 482(sh) 454 (126) 344 (93)
OH
y476 (157) 342 (160)
L
14
(cis) H
2
O 470(sh) 468 (103) 352 (88)
OH
y493 (169) 351 (173)
L
14
(trans) b
H
2
O 500 440 342
OH
y494 452 357
L
15
(cis) H
2
O 510 (94) 486(sh) 350 (132)
OH
y508 (90) 372 (97)
L
16
(trans) H
2
O 532 (63) 498(sh) 368 (79)
OH
y530 (157) 378 (172)
a
0.1 M HClO
4
for XsH
2
O; 0.05 M NaOH for XsOH
y.
b e not available.
obtained solution in acidic and alkaline media, as well as its
behaviour on a cation exchange column (charge q3) ena-
bled us to proceed with the study of its redox behaviour.
For the trans-[CoL16
H
2
O]
3qcomplex the
13
CNMR spec-
trum indicates a non-equivalence of all the carbon atoms of
the macrocycle that seems to disagree with its trans formu-lation. Nevertheless, the well-established geometry of its
chloro complex precursor [5c], the absence of isomerisation
reactions observed for the other (especially L
14
)macrocyclic
complexes, the already observed non-equivalence of carbon
atoms on trans-[Co{H(N6
)}Cl]
2q(N
6
'6,13-diamino-
6,13-dimethyl-1,4,8,11-tetraazacyclotetradecane, diammac)
complexes due to the presence of various conformers [7],
led us to relate the presence of 13
13
C signals to the existence
of the complex as a conformer not having the symmetry plane
that its chloro precursor has. In fact, even for the cis isomersof the L
13
and L
15
macrocylic complexes some of the
13
C
resonances are slightly broadened indicating the presence of
more than one conformer in solution.
2.2.3. Na[CoL15(H2O)][Fe(CN)6] and Na[L15(H2O)-Co(m-NC)Fe(CN)5]Addition of a solution of Na
4
[Fe(CN)
6
] to a solution of
[CoL
15
H
2
O](Trifl)
3
in triflic acid produces the immediate
precipitation of Na[CoL
15
(H
2
O)][Fe(CN)
6
] as character-
ised by the single band at 2044 cm
y1
in the IR spectrum [8]
and the elemental analyses. The suspension of the compound
in water at room temperature, with constant stirring, produces
a cherry red solution. Addition of ethanol to this solution
gives a precipitate of Na[L
15
(H
2
O)Co(m-NC)Fe(CN)5
] as
characterised by the two bands at 2118 and 2044 cm
y1
in the
IR spectrum and the elemental analyses. UV–Vis spectra (l
(nm) (e (cmy1
M
y1
)): 528 (285), 434 (286), 323 (374).
Anal. Calc. for Na[Co(L
15
)(H
2
O)][Fe(CN)
6
]P3H2
O:
C, 35.48; H, 6.12; N, 25.28. Found: C, 36.58; H, 6.20; N,
24.89%. Calc. for Na[Co(L
15
)(H
2
O)(m-NC)Fe(CN)5
]:C,
40.24; H, 5.44; N, 28.68. Found: C, 40.49; H, 6.48; N,
29.23%.
13
C{
1
H} NMR spectra (D
2
O, ppm referenced to TMS):
188.59(=5), 178.12, 67.05, 61.50, 58.41, 56.12, 55.00,
51.53, 49.86, 48.44, 47.17, 26.32, 21.44, 14.73.
2.3. Buffer solutions
All buffers were prepared according to well-established
procedures [9]; concentrations were chosen to provide
enough buffering for the [Fe(CN)
6
]
4ysolutions. Final pH
was set with the addition of NaOH or HClO
4
solutions to
the prepared buffers. The selected pH was 2.8–5.4
(CH
2
ClCOOH/CCH
2
ClCOO
yor CH
3
COOH/CH
3
COO
y)
for the aqua complexes and 7.8–9.0 (Tris) for the hydroxo
complexes. The effectiveness of the buffer solutions was
checked by monitoring the pH value of the final reaction
mixture.
2.4. Instruments
All UV–Vis spectra were recorded on a HP8452A instru-
ment. pH measurements were carried out with a Crison 2002
instrument equipped with an Ingold micro electrode.
13
C
NMR spectra were recorded on a GEMINI-300 instrument.
IR spectra were recorded in KBr discs with a Nicolet 520
FTIR instrument. Atmospheric pressure kinetic runs with
t1/2
)170 s were recorded on an HP8452A instrument
equipped with a thermostated multicell transport; runswithin
the 7–170 smarginwere recorded on anHP8452A instrument
equipped with a High-Tech SFA-11 Rapid Kinetics Acces-
sory; for t1/2
-7 s a Durrum D-110 stopped-flow instrument
was used. For runs at elevated pressure with t1/2
-100 s a
homemade high pressure stopped-flow system was used as
described previously [10a]; for t1/2
)800 s a previously
described pressurising system and high pressure cell were
used [10b–d].
2.5. Kinetics
All kinetic measurements were performed under pseudo-
first-order conditions with the iron complex in excess over

M. Martinez et al. / Inorganica Chimica Acta 256 (1997) 51–5954
Journal: ICA (Inorganica Chimica Acta) Article: 5415
Fig. 1. kobs
dependence on [Fe(CN) ] for the reduction of cis-[CoL15
-
4y6
(H
2
O)]
3qas a function of temperature (a), and for the reduction of cis-
[CoL
14
(OH)]
2qas a function of pressure (b), Is1.0 M (LiClO
4
).
the cobalt complex. The concentration of the cobalt(III)
complex was chosen typically in the 1–5=10
y4
M region.
For the trans-[Co(L14
)(H
2
O)]
3qcomplex the concentra-
tion was determined by using e values estimated from those
determined for the other macrocyclic cobalt(III) complexes
used in this study. Runs for the (N)
5
sL
13
system were fol-
lowed at 420 nm where the appearance of [Fe(CN)
6
]
3y
(es1023M
y1
cm
y1
) [11] can be detected; for all the other
(N)
5
systems the runs were followed at 520 nm, where the
appearance of the final products could be detected most eas-
ily. Solutions for the kinetic runs were made up by mixing
the appropriate amounts of the corresponding stock solutions
at 1.0 or 0.25 M (LiClO
4
) ionic strength. All solutions were
degassed in order to avoid anyFe(II) air oxidation andEDTA
was added to the reaction medium when (N)
5
sL
13
to pre-
vent the precipitation of the Co
2qreaction product [6,12];
for all other (N)
5
systems such precaution was not necessary
given the stability of the final redox product. The cobalt(III)
complex stock solutions were made up in water in order to
avoid any interference from anation reactions with buffer
anions during long stock, or high pressure equilibration times.
Accordingly, the [Fe(CN)
6
]
4ystock solutions had to be
prepared in the corresponding buffers and with the addition
of EDTA when necessary to obtain the correct reaction con-
ditions after mixing. The stability of the cobalt(III) com-
plexes in the buffer solutions used for the study was
monitored byUV–Vis spectroscopy.No indication of anation
reactions of the aqua or hydroxo species occurring during the
reaction times was detected.
All kobs
values were derived from the obtained absorbance
versus time exponential traces using a non-linear least-
squares fitting method. All post run fittings were done by
unweighted least-squares fit to the desired equations. The
values for k and KOS
were obtained from a direct fit to Eq.
(4). Alternatively, a double reciprocal plot was also used;
the coherence of the two plots was considered as a measure
of the quality of the fit.
3. Results
All the observed pseudo-first-order rate constants, kobs
,
measured as a function of the [Fe(CN)
6
]
4yconcentrations,
(N)
5
, buffer acidity, temperature, ionic strength, andpressure
are collected in Table S1 (Section 5). All these values at
Is1.0 M LiClO
4
were fitted to Eq. (4) (or its reciprocal
form, see above) and a very good agreement between the
fitted and the experimental points was observed. Fig. 1 shows
selected plots for some of the systems studied. From these
plots the first-order electron-transfer rate constants, k, andencounter complex formation constants, K
OS
, could be cal-
culated. The errors derived for the first-order rate constants
were always in the 5–10% margin. For the cis-[Co(L
15
)(H
2
O)]
3qsystems, when I was set to 0.25 M
(LiClO
4
) the values of kobs
were found to be independent of
the [Fe(CN)
6
]
4yconcentration margin used (5–12=10
y3
M), for all the other systems this margin was reduced to the
top end. Given the large values expected for KOS
under these
conditions, rate law(4) canbe simplified to kskobs
as already
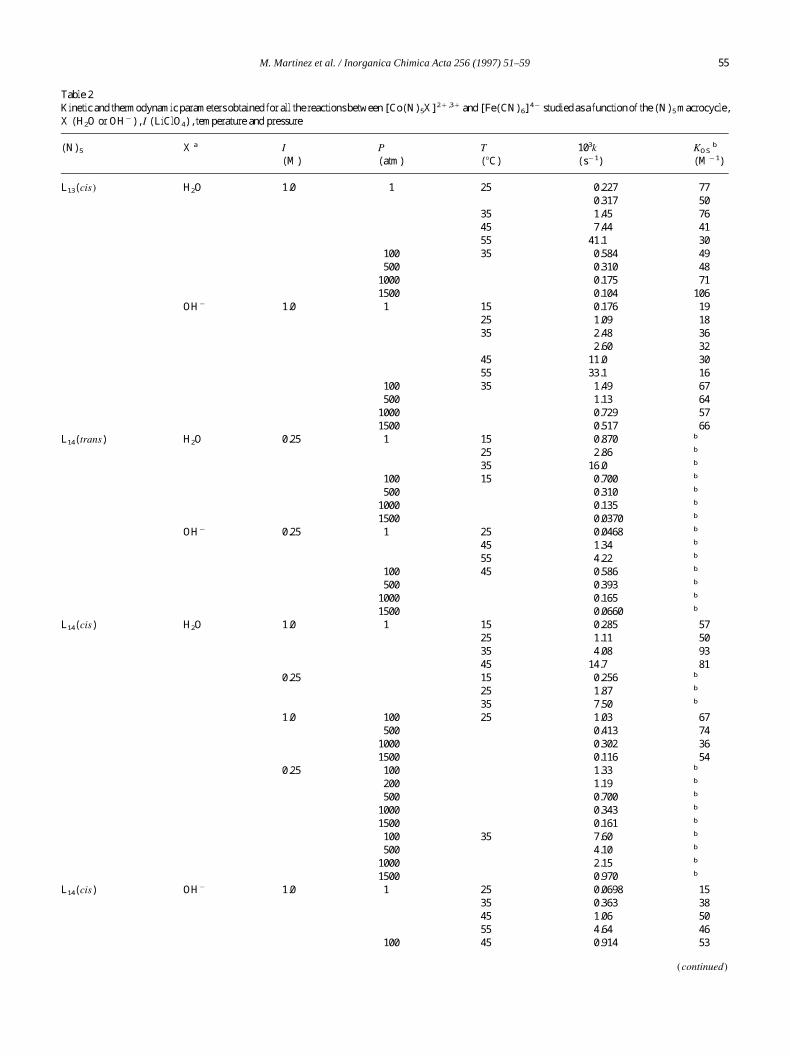
done for similar systems [2a,12]. Table 2 collects all k and
KOS
values for the systems studied as a function of themacro-
cycle (N)
5
, temperature, acidity, ionic strength and pressure.
From standard Eyring plots the thermal activationparameters
were obtained, and are summarised alongwith relevant avail-
able literature data in Table 3. Plots of ln k versus P (Fig. 2)
were used for the determination of the pressure activation
parameters; in all instances the values determined at Ps1
atmwere not used in the plots given the different instrumental
and sample manipulation required; the values determined at
atmospheric pressure correlate, in general, fairly well with
the values expected from these plots.
As found for similar systems [2b,c], neither interference
from the type and concentration of the buffer solutions, nor
from the amount of EDTA added (where necessary) was
found. Given the fact that the pK values for the aquo com-
plexes studied have not been determined, the independence
of the observed rate constants on pH,within the range studied,
was taken as an indication of the validity for the existence of
only the aqua or hydroxo species in each case. For the L
13
aqua and hydroxo systems, where EDTA had to be added to
the reacting solution in order to avoid precipitation of the
Co(II) salt of the hexacyanoferrate anions, the final spectra
correspond to those previously described [6]. For the reac-
tions run with the cobalt(III) complexes containing the L
14
,
L
15
and L
16
macrocyclic ligands, no precipitation of the
Co(II) salts was observed even in the absence of added
EDTA. In all cases, a final spectrum having two maxima at
M. Martinez et al. / Inorganica Chimica Acta 256 (1997) 51–59 55
Journal: ICA (Inorganica Chimica Acta) Article: 5415
Table 2
Kinetic and thermodynamic parameters obtained for all the reactions between [Co(N)
5
X]
2q,3qand [Fe(CN)
6
]
4ystudied as a function of the (N)
5
macrocycle,
X (H
2
O or OH
y), I (LiClO
4
), temperature and pressure
(N)
5
X
a I P T 10
3k KOS
b
(M) (atm) (8C) (s
y1
) (M
y1
)
L
13
(cis) H
2
O 1.0 1 25 0.227 77
0.317 50
35 1.45 76
45 7.44 41
55 41.1 30
100 35 0.584 49
500 0.310 48
1000 0.175 71
1500 0.104 106
OH
y1.0 1 15 0.176 19
25 1.09 18
35 2.48 36
2.60 32
45 11.0 30
55 33.1 16
100 35 1.49 67
500 1.13 64
1000 0.729 57
1500 0.517 66
L
14
(trans) H
2
O 0.25 1 15 0.870
b
25 2.86
b
35 16.0
b
100 15 0.700
b
500 0.310
b
1000 0.135
b
1500 0.0370
b
OH
y0.25 1 25 0.0468
b
45 1.34
b
55 4.22
b
100 45 0.586
b
500 0.393
b
1000 0.165
b
1500 0.0660
b
L
14
(cis) H
2
O 1.0 1 15 0.285 57
25 1.11 50
35 4.08 93
45 14.7 81
0.25 15 0.256
b
25 1.87
b
35 7.50
b
1.0 100 25 1.03 67
500 0.413 74
1000 0.302 36
1500 0.116 54
0.25 100 1.33
b
200 1.19
b
500 0.700
b
1000 0.343
b
1500 0.161
b
100 35 7.60
b
500 4.10
b
1000 2.15
b
1500 0.970
b
L
14
(cis) OH
y1.0 1 25 0.0698 15
35 0.363 38
45 1.06 50
55 4.64 46
100 45 0.914 53
(continued)

M. Martinez et al. / Inorganica Chimica Acta 256 (1997) 51–5956
Journal: ICA (Inorganica Chimica Acta) Article: 5415
Table 2 (continued)
(N)
5
X
a I P T 10
3k KOS
b
(M) (atm) (8C) (s
y1
) (M
y1
)
L
14
(cis) 500 0.569 75
1000 0.520 40
1500 0.261 55
L
15
(cis) H
2
O 1.0 1 15 4.26 21
25 14.6 44
35 32.6 70
45 144 76
0.25 15 3.14
b
25 17.6
b
35 85.1
b
1.0 100 15 2.29 35
500 0.940 52
1000 0.507 43
1500 0.219 59
0.25 100 11 1.59
b
250 1.28
b
500 0.915
b
1000 0.389
b
1500 0.198
b
L
15
(cis) OH
y1.0 1 15 0.709 46
25 1.82 64
35 5.13 55
45 15.2 59
0.25 16 0.943
b
26 3.28
b
35 6.42
b
1.0 100 25 2.95 36
500 2.27 33
1000 1.58 26
1500 0.948 31
0.25 100 2.26
b
500 1.71
b
1000 1.13
b
1500 0.620
b
L
16
(trans) H
2
O 1.0 1 15 69.2 32
25 335 25
35 1470 24
45 7380 36
0.25 15 140
b
25 398
b
35 1700
b
45 4300
b
250 25 263
b
500 166
b
750 122
b
1000 87.6
b
1250 64.9
b
OH
y1.0 15 7.07 56
25 20.3 67
35 54.7 23
45 264 47
0.25 250 34 68.0
b
500 55.0
b
750 44.5
b
1000 34.0
b
1250 29.5
b
1500 21.0
b
a
pHs2.8–5.4 (CH
2
ClCOO
y/CH
2
ClCOOH or CH
3
COO
y/CH
3
COOH) for XsH
2
O; pHs7.8–9.0 (Tris) for XsOH
y.
b
Rate law (4) becomes kobs
sk under low I conditions, see text.

M. Martinez et al. / Inorganica Chimica Acta 256 (1997) 51–59 57
Journal: ICA (Inorganica Chimica Acta) Article: 5415
Table 3
Kinetic, thermal and pressure activation parameters for all the reactions between [Co(N)
5
X]
2q,3qand [Fe(CN)
6
]
4ystudied as a function of the (N)
5
macrocyle, X (H
2
O or OH
y) and I (LiClO
4
). Available data for similar systems in the literature are also included
(N)
5
X
a I 10
3k298 H/D S/D V /(T)D
(M) (s
y1
) (kJ mol
y1
) (J K
y1
mol
y1
) (cm
3
mol
y1
) (K)
L
13
(cis) H
2
O 1.0 0.23"0.02
0.32"0.06 132"4 131"14 31"2 (308)
OH
y1.0 1.1"0.2 98"5 25"15 20"1 (308)
L
14
(trans) H
2
O 0.25 2.9
b
105"11 59"38 49"3 (288)
OH
y0.25 0.039 121"7 79"22 42"3 (318)
L
14
(cis) H
2
O 1.0 1.1"0.1 98"1 27"3 36"5 (298)
0.25 1.9
b
122"1 110"4 37"1 (308)
38"1 (298)
OH
y1.0 0.070"0.004 109"4 42"13 22"4 (318)
L
15
(cis) H
2
O 1.0 15"1 84"7 1"23 39"3 (288)
0.25 17
b
115"2 106"8 36"1 (284)
OH
y1.0 1.8"0.1 76"3 y43"10 20"2 (298)
0.25 3.3
b
75"3 y43"9 22"1 (298)
L
16
(trans) H
2
O 1.0 340"10 116"3 134"10
0.25 400
b
95"2 68"8 34"1 (298)
OH
y1.0 20"2 88"8 17"24
0.25 23"1 (307)
(NH
3
)
5
H
2
O
c
0.50 1.3 102"5 79"15 26.5"2.4 (298)
OH
y d
1.0 0.081 109"10 84"30
(MeNH
2
)
5
H
2
O
d
1.0 93 79"7 39"22 29.4"1.6 (298)
OH
y d
1.0 2.0 83"3 21"9 32.9"1.3 (298)
(EtNH
2
)
5
H
2
O
d
1.0 350 84"6 65"21 33.1"2.0 (298)
OH
y d
1.0 6.4 102"1 95"2 30.6"2.8 (308)
a
pHs2.8–5.4 (CH
2
ClCOO
y/CH
2
ClCOOH or CH
3
COO
y/CH
3
COOH) for XsH
2
O; pHs7.8–9.0 (Tris) for XsOH
y.
b
Rate law (4) becomes kobs
sk under low I conditions, see text.c
Ref. [2a].
d
Ref. [2b].
Fig. 2. Plots of ln k vs. P for some of the systems studied: d cis-[CoL15
-
(H
2
O)]
3q, 118C, Is0.25 M (LiClO
4
); s cis-[CoL15
(H
2
O)]
3q, 158C,
Is1.0 (LiClO
4
); j cis-[CoL15
(OH)]
2q, 258C, Is0.25 M (LiClO
4
); h
cis-[CoL15
(OH)]
2q, 258C, Is1.0 (LiClO
4
).
;508 and 442 nm and no signal at 420 nm, corresponding
to [Fe(CN)
6
]
3y, was found. From these solutions a cherry
red compound with an IR spectrum showing the bands
of a {m-(CN)Fe(CN)5
} moiety can be precipitated. No
substitution reaction of H
2
O by [Fe(CN)
6
]
3yin the
[Co(N)
5
(H
2
O)]
3qcompounds, either in acidic or alkaline
medium, was detected under the experimental conditions of
this study indicating that a possible substitution reaction of
[Co(N)
5
(H
2
O)]
3qby [Fe(CN)
6
]
4ycan be ruled out. The
extremely slow acid aquation reaction of [Co(N)
5
Cl]
2qions
(kf6=10
y5
s
y1
at [H
q]s0.2 M and 508C) confirms the
previous assumption. On the other hand, the final cherry
red redox final compound can also be obtained from a
direct reaction of [Co(N)
5
(H
2
O)](O
3
SCF
3
)
3
with
Na
4
[Fe(CN)
6
]. On mixing equimolar amounts of the two
species in highly acidic medium immediate precipitation of
the compound Na[Co(N)
5
(H
2
O)][Fe(CN)
6
] occurs, as
proved by elemental analysis and IR spectroscopy; on stand-
ing, the product slowly dissolves to produce an intense cherry
red solution with UV–Vis maxima at 508, 442 and 322 nm
that analyses with the same C:N ratio, but now showing an
IR signal at 2118 cm
y1
indicative of a m-CN group not
present in the initial product, aswell as an extra
13
C resonance
in theNMR spectrum. The stability of the final {Co
II
(N)
5
}
2q
redox products, their lability, and subsequent inner-sphere
redox reaction, can be considered to be responsible for these
observations.
Table 2 clearly indicates a lack of meaningful differences
in the values of KOS
determined at Is1.0 M, although
changes in ionic strength do affect the KOS
values signifi-
cantly. On the other hand, no pressure dependence of these
values has also been detected. As seen in Tables 2 and 3, the
data obtained for the reduction of [Co(N)
5
(H
2
O)]
3qand

M. Martinez et al. / Inorganica Chimica Acta 256 (1997) 51–5958
Journal: ICA (Inorganica Chimica Acta) Article: 5415
[Co(N)
5
(OH)]
2qagree extremely well with the trends
already observed for similar systems [2b]. The differences
in the first-order electron transfer rate constants on changing
the size of the macrocycle (Table 2) are quite significant for
the aqua complexes; the value of k increases by up to three
orders of magnitude on increasing the size of themacrocyclic
(N)
5
ligand. The difference between the L
14
(trans) and
L
14
(cis) systems is significantly smaller and for the trans-L
16
complex the trend is maintained. The observed effect is
significantly different for the hydroxo species. In this case
the L
14
cis and trans systems show a minimum in the rate
constant, whereas the values for L
13
and L
15
systems are very
similar and an increase of one order of magnitude is observed
for the trans-L16
complex.
The activation enthalpy values do not show any trend.
There is a large scatter in the S/values, whereas the V/D D
values clearly show that there is a significant decrease on
going from the aqua to the hydroxo species. Changes in the
macrocyclic ligand only seem to affect in an important way
the trans-L14
system. Changes in ionic strength only affect
the KOS
value and the differences observed for the thermal
and pressure activation parameters are within the intrinsic
experimental error limits. The values found for the cis-L14
and cis-L15
systems studied at Is0.25M seem to show larger
values of H/and S/
. The reverse trend is obtained forD D
the trans-L16
systems, and the trans-L14
has not been deter-
mined at Is1.0 M. Consequently, given the fact that the
activation volumes are the same within experimental error, it
is very difficult to establish this fact as a proper trend.
4. Discussion
The studied reactions merit special attention with respect
to the final reaction products. The results clearly indicate that
the final m-CN species detected for the L
14
, L
15
and L
16
systems cannot appear via a substitution process on the initial
[CoL(H
2
O)]
3qcomplex, given its extreme inert substitu-
tion nature in acidic medium. Nevertheless, the overall proc-
ess is very similar to the set of reactions occurring in the
Co
II,III
/EDTA/Fe
II,III
(CN)
6
redox processes [6,12]. In our
case the [CoL(H
2
O)]
2qspecies formed after reduction
probably undergo a macrocyclic pendant arm dechelation to
produce a [CoL(H
2
O)
2
]
2qspecies. The new redox potential
probably allows an inner-sphere oxidation process with the
produced [Fe(CN)
6
]
3ycomplex, to yield the final detected
[Co
III
L(H
2
O)(m-CN)FeII(CN)5
]
ycomplex. The non-
reversible cyclic voltammograms of the Co(III) complexes
indicate that the Co(II) species are very labile. The fact that
the final cyano bridged product exists for the macrocyclic
system that has the largest cavity, L
16
, even though itsCo(III)
complex is the less stable one, and the fact that the smallest
cavity, L
13
, does not form this final compound, are in agree-
ment with the overall picture. This means that the Co(II)
species initially formed is only long-lived enough to undergo
the inner-sphere oxidation when the macrocyclic cavity
allows the presence of the Co(II) ion in a relaxedenvironment.
The values collected forKOS
are very similar within exper-
imental error limits, indicating that, unlike for some other
systems [2b], outer-sphere formation is the same, regardless
of the complexes involved for the reactions studied. Simple
electrostatic arguments cannot account for the obtained val-
ues. The effect of ionic strength is such that electrostatic
interaction seems to be mainly responsible for the overall
precursor complex formation. Nevertheless, hydrogen bond-
ing interactions must also be taken into account, as already
found for species involving oxoanionic species where charge
and hydrogen bonding factors can easily compensate effects
[2c,d]. In fact, on the basis of (KOS
) data, the solventD
separated nature of outer-sphere complexes, havingan impor-
tant contribution of hydrogen bonding, has already been con-
cluded [1a,d]. The lack of significant differences between
the kinetic and activation parameters determined at Is1.0
and 0.25 M must be related to the above-mentioned facts.
Although charge redistribution in the transition state is impor-
tant enough as to be affected by the ionic strength, the values
obtained for S/and V/
do not show the differencesD D
expected due to the directly related electrostriction terms
[13], suggesting that hydrogen bonding must in some way
compensate the purely electrostatic ionic strength effects.The
lack of the expected pressure dependence of the values deter-
mined for KOS
also indicates that hydrogen bonding must
play a very important role in the precursor complex; its exis-
tence justifies a strong interactionwhile keeping the separated
ionic nature of the reactants.
The size of the different macrocyclic ligands does affect
in a very important manner the value of k for the reduction
of the aqua complexes. The acceleration observed on increas-
ing the size of the ligand, and the metal–ligand bond length,
is in line with that already observed for the simple linear
amine systems. As pointed out [14], the larger the N
5
O
cavity, the easier the reduction of Co
III
to Co
II
occurs as
expected on the basis of their respective sizes; in our case the
average Co–N distance for the [Co(N)
5
Cl]
2qseries of com-
plexes increases on going from the L
13
to the L
16
macrocycles
[5b,c] and an acceleration should be present on going along
the series. This effect is, however, not observed for the cor-
responding L
13
–hydroxo complex; the small size of the
ligand, that could favour specific O∆H interactions within
the deprotonated complex encapsulating the cobalt centre
even more, could be responsible for this observation.
Differences in geometry do not seem to be very important
as seen from a comparison of the values for the two L
14
systems. This seems to rule out special encounter complex
geometries that could also induce the concept of a dead-end
complex as indicated for similar systems [2c,14]. Even so,
it is difficult to believe that the reducing species
[Fe(CN)
6
]
4ysees the cobalt complex as a perfect spherical
entity [15]. Although a facial approach that could be com-
mon to both cis and trans geometries (Scheme 1) could bea better possibility to understand the results obtained [16],

M. Martinez et al. / Inorganica Chimica Acta 256 (1997) 51–59 59
Journal: ICA (Inorganica Chimica Acta) Article: 5415
the smaller deformation capacity of the trans isomer that caneasily compensate the larger cavity size, can be easily be held
responsible for the observed facts. In this respect, it is impor-
tant to point out the possibility of the existence of several
conformers for the L
16
system, somehow increasing the flex-
ibility of the complex and enabling a faster electron transfer
reaction (larger S/values).D
As for the thermal and pressure activation parameters col-
lected in Table 3, the only real trends appear for the V/D
values. All the values are very positive,which is characteristic
for these types of reactions, where the V/values arise fromD
changes in electrostriction associated with charge neutralis-
ation (especially [Fe(CN)
6
]
4yto [Fe(CN)
6
]
3y) and vol-
ume increase on reduction from Co
III
to Co
II
[1,2]. The
difference in V/between the aquo and hydroxo speciesD
does not correspond to that observed for the simple linear
amine analogous complexes as seen in Table 3 [2b]. For the
trans-L14
complex the obtained values are extremely large
and for all systems the value obtained for the hydroxo com-
plex is consistently lower than that of its aquo counterpart.
Given the above-mentioned facts, the lower V/found forD
the hydroxo systems have to be related to the absence of
contribution arising from the reduction of Co
III
to Co
II
, both
due to charge differences (absence of a 3q cation) and bond
distances (shorter Co–O bond), that is the increased com-
pactness of the L cage.
Recent studies carried out on the reduction of oxoanionic
pentaa(m)minecobalt(III) complexes [2c,d] also show the
same trend; the effects have also been related to hydrogen
bonding that seems to dominate electrostatic interaction. The
differences observed for the trans-L14
complex can partly be
related to its geometry, but also to the size of themacrocycle;
the trans-L16
complex does not show such a difference. If
hydrogen bonding is a key aspect in these processes, the
electrostriction effects that dominate the V/values willD
produce an important electrostriction volume increase for the
extremely symmetric trans-L14
complex (see Section 2),
when the charge on the metal centre is reduced.
Nevertheless, the thermodynamics of the electron transfer
for these systems can also be held responsible for the effects
observed. As stated before, the non-reversibility of the cyclic
voltammograms of these species does not allow the necessary
accurate measure of the corresponding reduction potentials
for the complexes.
5. Supplementary material
Table S1 is available from the authors on request.
Acknowledgements
Helpful comments from Dr G.A. Lawrance are highly
appreciated. Financial support from the German–Spanish
cooperation projects, DGICYT, and the Direccio General de
Universitats de la Generalitat de Catalunya is gratefully
acknowledged.
References
[1] (a) R. van Eldik and H. Kelm, Inorg. Chim. Acta, 73 (1983) 91; (b)
I. Krack and R. van Eldik, Inorg. Chem., 25 (1986) 1743; (c) Y.
Sasaki, K. Endo, A. Nagasawa and K. Saito, Inorg. Chem., 25 (1986)4845; (d) I. Krack and R. van Eldik, Inorg. Chem., 28 (1989) 85.
[2] (a) I. Krack and R. van Eldik, Inorg. Chem., 29 (1990) 1700; (b) M.
Martinez, M.A. Pitarque and R. van Eldik, J. Chem. Soc., DaltonTrans., (1994) 3159; (c) M. Martinez and M.A. Pitarque, J. Chem.Soc., Dalton Trans., (1995) 4107; (d) M. Martinez, M.A. Pitarque
and R. van Eldik, J. Chem. Soc., Dalton Trans., (1996) 2665.[3] (a) M. Martinez and G. Muller, J. Chem. Soc., Dalton Trans., (1989)
1669; (b)M. Ferrer, G. Gonzalez andM.Martinez, Inorg. Chim. Acta,188 (1991) 211; (c) G. Gonzalez, M. Martinez, X. Solans and M.
Font-Bardıa, Inorg. Chim. Acta, 203 (1993) 229; (d) G. Gonzalez
andM.Martinez, Inorg. Chim. Acta, 230 (1995) 67; (e) G. Gonzalez,M. Martinez, A.E. Merbach and B. Moullet, Inorg. Chem., 33 (1994)2330; (f) G. Gonzalez, M. Martinez and E. Rodriguez, J. Chem. Soc.,Dalton Trans., (1995) 891.
[4] (a) R. van Eldik, Inorganic High Pressure Chemistry: Kinetics andMechanisms, Elsevier, Amsterdam, 1986; (b) M. Kotowski and R.
van Eldik, Coord. Chem. Rev., 93 (1989) 19.
[5] (a) G.A. Lawrance, M. Martinez, B.W. Skelton and A.H. White, J.Chem. Soc., Dalton Trans., (1992) 823; (b) G.A. Lawrance, M.
Martinez, T.M. Manning, M. Maeder, M.A. O’Leary, W.C.
Patalinghug, B.W. Skelton and A.H. White, J. Chem. Soc., DaltonTrans., (1992) 1635; (c) G.A. Lawrance, T.W. Hambley, M.
Martinez, B.W. Skelton and A.H.White, J. Chem. Soc., Dalton Trans.,(1992) 1643; (d) G.A. Lawrance, M. Martinez, B.W. Skelton and
A.H. White, J. Chem. Soc., Dalton Trans., (1992) 1649.[6] D.H. Huchital and R.G. Wilkins, Inorg. Chem., 6 (1967) 1022.
[7] (a) P.V. Bernhardt, G.A. Lawrance and T.W.Hambley, J. Chem. Soc.,Dalton Trans., (1989) 1059; (b) P.V. Bernhardt, P. Comba, N.F.
Curtis, T.W. Hambley, G.A. Lawrance,M.Maeder andA. Sirwardena,
Inorg. Chem., 29 (1990) 3208.
[8] (a) K. Nakamoto, Infrared Spectra of Inorganic and CoordinationCompounds, Wiley, New York, 3rd edn., 1978; (b) P. Forlano, L.M.
Baraldo, J.A. Olabe and C.O. Della Vedova, Inorg. Chim. Acta, 223(1994) 37; (c) V.G. Poulopoulou, Z.-W. Li and H. Taube, Inorg.Chim. Acta, 225 (1994) 173.
[9] D.D. Perrin, Aust. J. Chem., 16 (1963) 372.
[10] (a) R. van Eldik, W. Gaede, S. Wieland, J. Kraft, M. Spitzer and D.A.
Palmer, Rev. Sci. Instrum., 64 (1993) 1355; (b) R. van Eldik, D.A.
Palmer, R. Schmidt and H. Kelm, Inorg. Chim. Acta, 50 (1981) 131;
(c) M. Spitzer, F. Gartig and R. van Eldik, Rev. Sci. Instrum., 59(1988) 2092; (d) F.K. Fleischman, E.G. Conze, D.R. Stranks and H.
Kelm, Rev. Sci. Instrum., 99 (1977) 1427.
[11] A.J. Miralles, R.E. Armstrong and A. Haim, J. Am. Chem. Soc., 99(1977) 1416.
[12] D. Gaswick and A. Haim, J. Am. Chem. Soc., 93 (1971) 7347.
[13] (a) J. Burgess, Ions in Solution, Ellis Horwood, Chichester, UK, 1988;(b) T.W. Swaddle, Inorg. Chem., 29 (1990) 5017.
[14] A.G. Lappin, Redox Mechanisms in Inorganic Chemistry, Ellis
Horwood, Chichester, UK, 1994.
[15] M.R. Grace, H. Takagi and T.W. Swaddle, Inorg. Chem., 33 (1994)
1915.
[16] J. Berglund and L.I. Elding, Inorg. Chem., 34 (1995) 513.