oral intoxication of mice with shiga toxin type 2a (stx2a...
TRANSCRIPT

Oral Intoxication of Mice with Shiga Toxin Type 2a (Stx2a) andProtection by Anti-Stx2a Monoclonal Antibody 11E10
L. M. Russo, A. R. Melton-Celsa, M. A. Smith, M. J. Smith, A. D. O’Brien
Department of Microbiology and Immunology, Uniformed Services University of the Health Sciences, Bethesda, Maryland, USA
Shiga toxin (Stx)-producing Escherichia coli (STEC) strains cause food-borne outbreaks of hemorrhagic colitis and, less com-monly, a serious kidney-damaging sequela called the hemolytic uremic syndrome (HUS). Stx, the primary virulence factor ex-pressed by STEC, is an AB5 toxin with two antigenically distinct forms, Stx1a and Stx2a. Although both toxins have similar bio-logical activities, Stx2a is more frequently produced by STEC strains that cause HUS than is Stx1a. Here we asked whether Stx1aand Stx2a act differently when delivered orally by gavage. We found that Stx2a had a 50% lethal dose (LD50) of 2.9 �g, but nomorbidity occurred after oral intoxication with up to 157 �g of Stx1a. We also compared several biochemical and histologicalparameters in mice intoxicated orally versus intraperitoneally with Stx2a. We discovered that both intoxication routes causedsimilar increases in serum creatinine and blood urea nitrogen, indicative of kidney damage, as well as electrolyte imbalances andweight loss in the animals. Furthermore, kidney sections from Stx2a-intoxicated mice revealed multifocal, acute tubular necrosis(ATN). Of particular note, we detected Stx2a in kidney sections from orally intoxicated mice in the same region as the epithelialcell type in which ATN was detected. Lastly, we showed reduced renal damage, as determined by renal biomarkers and histopa-thology, and full protection of orally intoxicated mice with monoclonal antibody (MAb) 11E10 directed against the toxin A sub-unit; conversely, an irrelevant MAb had no therapeutic effect. Orally intoxicated mice could be rescued by MAb 11E10 6 h butnot 24 h after Stx2a delivery.
Shiga toxin (Stx)-producing Escherichia coli (STEC) strains arefood-borne pathogens with an estimated infectious dose of
fewer than 50 organisms (1). While multiple STEC serotypes areassociated with disease, illness associated with infection by E. coliO157:H7 accounts for over 63,000 of the 113,000 total STEC caseseach year in the United States (2). Bovine and other ruminants arethe natural carriers of STEC, and contamination of meat generallyoccurs during beef processing, with up to 40% of the outbreaksoccurring from beef (3, 4). However, contaminated fresh produceis also responsible for both outbreaks and sporadic cases of STECin the United States (4, 5).
Upon STEC infection, the most common disease manifesta-tion is hemorrhagic colitis. In 5 to 15% of patients, a serious se-quela of STEC infection, the hemolytic uremic syndrome (HUS),may occur (6, 7). The HUS is characterized by a triad of symp-toms: microangiopathic hemolytic anemia, thrombocytopenia,and acute kidney failure (8). Currently, there is no vaccine toprevent or therapeutic agent to cure STEC infection, as antibioticsare contraindicated due to the potential upregulation of bacterio-phage production of Stx (9).
An STEC strain may encode Stx1a (equivalent to Stx from Shi-gella dysenteriae type 1) and/or Stx2a, two antigenically distinctbut biologically similar toxins (10, 11). Stx1a and Stx2a shareabout 57% amino acid homology, analogous crystal structures,and identical modes of action (12). Stx1a and Stx2a are AB5 tox-ins. The A subunit is responsible for the catalytic activity of thetoxin molecule, and the B subunit, a homopentamer, is requiredfor the toxin to bind to the Stx host cell receptor, globotriaosylcer-amide (Gb3) (54, 55). Once bound to its receptor, Stx undergoesretrograde transport through the cell; the enzymatically activeportion of the toxin is then released into the cytoplasm, where itdepurinates a single adenine residue from the 28S rRNA of the 60Sribosome (13, 14). This ribosomal injury results in the inhibitionof protein synthesis and, ultimately, cell death (15). Although
Stx1a and Stx2a have the same receptor and mode of action, epi-demiologic studies indicate that strains that encode Stx2a aremore likely to be associated with food-borne outbreaks and severedisease, such as the HUS, than are those that make Stx1a only orStx1a and Stx2a (16–18).
Although no one animal model recapitulates all aspects ofSTEC pathogenesis, the capacity of the Stxs to cause disease hasbeen demonstrated by either infection or intoxication models inmice, rats, pigs, baboons, and greyhounds (for reviews, see refer-ences 19 and 20). For example, oral infection with certain strainsof STEC in mouse models or injection of mice with either Stx1a orStx2a results in renal injury and death (reviewed in reference 21).Monoclonal antibody (MAb) against the toxin is able to protectthose animals from disease and death (22, 23), findings that fur-ther establish a role for Stx in pathogenesis. Only a few studies thatexamine oral intoxication by Stx in animals have been reported. Intwo such investigations, purified Stx (specific toxin type[s] un-known, [24]) and Stx2a (25) were found to be lethal after intra-gastric (i.g.) gavage of infant New Zealand White rabbits. Morerecently, Rasooly et al. demonstrated that i.g. administration of 50�g, but not 0.5 �g, of Stx2a is lethal in Swiss Webster mice (26).Here, we further defined the oral toxicity of the Stxs in mice. We
Received 8 October 2013 Returned for modification 10 November 2013Accepted 20 December 2013
Published ahead of print 30 December 2013
Editor: S. R. Blanke
Address correspondence to A. D. O’Brien, [email protected].
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01264-13.
Copyright © 2014, American Society for Microbiology. All Rights Reserved.
doi:10.1128/IAI.01264-13
March 2014 Volume 82 Number 3 Infection and Immunity p. 1213–1221 iai.asm.org 1213
on June 22, 2018 by guesthttp://iai.asm
.org/D
ownloaded from

found that although Stx1a was not lethal via the oral route, Stx2ahas an i.g. 50% lethal dose (LD50) of 2.9 �g. We further showedthat the renal pathologies caused by Stx2a by both the i.g. andintraperitoneal (i.p.) routes of intoxication were similar. Finally,we protected mice from oral Stx2a intoxication by passive anti-body transfer of MAb 11E10 against the Stx2a toxin A subunit.
(A portion of this work was presented at the 8th InternationalSymposium on Shiga Toxin [Verocytotoxin] Producing Esche-richia coli Infections, Amsterdam, The Netherlands, 6 to 9 May2012 [27]).)
MATERIALS AND METHODSBacterial strains and growth conditions. E. coli K-12 DH5� strains trans-formed with pLPSH3 (28) or pJES120 (29) encode Stx (equivalent toStx1a and called Stx1a herein for simplicity) or Stx2a, respectively. Bothstrains were grown in Luria-Bertani (LB) broth or LB agar supplementedwith 100 �g/ml ampicillin for maintenance of the recombinant plasmid.
Purification of Stx1a and Stx2a. Both toxins were purified by affinitychromatography with 5 ml AminoLink coupling resin (Thermo Scien-tific) columns.
Column preparation. MAb to the B subunit of either Stx1a or Stx2awas covalently bound to the column resin in pH 7.2 coupling buffer ac-cording to the manufacturer’s instructions. MAb 13C4 (30) was purifiedfrom hybridoma supernatant by fast protein liquid chromatography overa HiTrap protein G high-performance (HP) 5-ml column (GE Life Sci-ences, Pittsburgh, PA), desalted (HiTrap desalting column; GE Life Sci-ences) into phosphate-buffered saline (PBS), and used for purification ofStx1a (approximately 7 mg MAb/column), while MAb BC5 BB12 (31) inascites fluid, a gift from Nancy Strockbine, was diluted into PBS and usedfor Stx2a purification (approximately 13 mg MAb/column).
Cell lysate preparation. An overnight culture of the E. coli K-12 strainthat contained the plasmid that encoded the stx of interest was sedimentedby centrifugation (5,000 � g) and the pellet resuspended at 4 ml per g insonication buffer (50 mM NaPO4, 200 mM NaCl). The resuspended cellpellet was disrupted by sonication. The cell lysate was sedimented by cen-trifugation (20,000 � g), and the supernatant was filtered prior to toxinpurification.
Toxin purification. Cell lysates were applied to the column in accor-dance with the manufacturer’s instructions for sample application exceptas noted below. All column elutions were recovered via gravity instead ofcentrifugation. The cell lysate flowthrough was reapplied to the columnfor additional toxin binding. The column contents were washed consec-utively with three column volumes each: sonication buffer, high salt buf-fer (0.5 M NaCl, 50 mM NaPO4) to further remove nonspecific contam-inants, and sonication buffer to prevent protein denaturation resultingfrom the high salt concentration. Toxin was eluted (0.1 M glycine, pH 2.8)in 1-ml fractions into 200 �l neutralization buffer (1 M Tris HCl, pH 9.5).The fractions that contained toxin were dialyzed against PBS in Slide-A-Lyzer dialysis cassettes (Thermo Scientific). Toxin activity was confirmedon Vero cells as previously described (32). The endotoxin level was lessthan 0.025 endotoxin units (EU) per �g Stx2a (data not shown) as deter-mined by the Limulus amebocyte lysate chromogenic endpoint assay (Hy-cult). When necessary, 15-ml Millipore Amicon Ultra 30K concentratorswere used in accordance with manufacturer’s instructions to concentratethe toxin preparation. The protein concentration in the toxin prepara-tions was determined with a bicinchoninic acid (BCA) assay (ThermoScientific).
Specific toxin concentration determination. To normalize for differ-ences in purity among the toxin preparations, we used densitometry anal-yses of stained gels to determine the percentage of toxin in each prepara-tion relative to the total protein in each sample as follows. Purified toxinswere separated on a NuPAGE Novex 4 to 12% bis-Tris gel(s) (Invitrogen).The gel(s) was then stained with Oriole fluorescent gel stain (Bio-Rad)and scanned with the ImageQuant LAS 4000 system (GE Healthcare). Thescanned image was analyzed with ImageQuant TL software (GE Health-
care) to determine what contribution to the total protein was made by thebands that comprised the A and B subunits of the toxins. The concentra-tion of purified toxin was then calculated by multiplying the percentage oftoxin in the preparation by the total protein concentration measured inthe BCA assay. The densitometry analyses of the individual A and B sub-unit bands suggested that the proportion of A to B subunits in each prep-aration was at or close to the expected ratio of 1:5 and that the purity of thepreparations ranged from 82 to 95% (data not shown).
Mice. All animal studies were approved by the Institutional AnimalCare and Use Committee of the Uniformed Services University of theHealth Sciences and were conducted in strict accordance with the recom-mendations of the Guide for the Care and Use of Laboratory Animals(33). Female BALB/c mice, 5 to 6 weeks old, were obtained from CharlesRiver Laboratories (Wilmington, MA) and used for all experiments. Foodand water were removed for 18 and 2 h, respectively, prior to all i.g.intoxication studies. Mice were gavaged with 0.2 ml of toxin-PBS dilu-tions or PBS as a control for oral intoxication or injected with 0.1 ml of aStx-PBS dilution for all i.p. intoxication studies. Mice were weighed dailyand monitored for morbidity and mortality for 2 weeks postintoxication.When the starting weights of mice were similar among groups, wegraphed the mean weights of animals in each group on that day. However,when the initial mean weights of the mice in groups differed by more than5%, we plotted the percent weight change per day. Two-way analysis ofvariance (ANOVA) was used to determine a significant difference inweight change among intoxication groups. The Stx2a i.g. intoxicationLD50 and 95% confidence interval (CI) values were determined by probitregression analysis with log transformation of the values.
Serum biochemistry and histopathology. Mice received Stx2a di-luted in PBS or PBS alone as a control through i.p. or i.g. administration.Three or 4 days postintoxication, mice were anesthetized with isoflurane(VetEquip Incorporated, Pleasanton, CA), and blood was collected viacardiac puncture. After the blood clotted, the serum was separated bycentrifugation at 6,000 � g for 10 min and sent to BioReliance (Rockville,MD). Serum samples were analyzed by an automated clinical chemistry an-alyzer (Cobas 600 series) and reported through a laboratory informationmanagement system (LIMS). For the initial studies, a full serum panel wasdone. However, only the kidney-specific markers showed differences fromthe expected normal range (http://www.criver.com/files/pdfs/rms/balbc/rm_rm_r_balb-c_mouse_clinical_pathology_data.aspx), so all subsequent se-rum analyses were limited to the renal panel: blood urea nitrogen (BUN),creatinine, sodium (Na), potassium (K), chloride (Cl), and albumin. One-way ANOVA was used to determine statistical significance between theexperimental and control groups.
After blood collection, the mice were euthanized and necropsied. Forsome studies, kidney, small intestine, cecum, large intestine, and/or liverwere collected, fixed in formalin, and sent to Histoserv (Germantown,MD) to be embedded in paraffin and sectioned. Sections from all organswere stained with hematoxylin and eosin (H&E). The semiquantitativeseverity modifier of a morphological diagnosis, such as mild, moderate, orsevere, was used to describe lesions. Additionally, serial sections fromsome of the kidney samples analyzed with H&E were stained with periodicacid-Schiff (PAS) stain. PAS stains polysaccharides, including basementmembranes and brush boarders of certain epithelial cells. Therefore,proximal tubules, which have a thick brush border, can be distinguishedwith the PAS stain from distal tubules that lack a prominent brush border.Slides were read by a veterinary pathologist who was blind to the studygroup identifications.
Immunofluorescence. Slides with unstained kidney or intestinal sec-tions were deparaffinized by incubation in Histoclear (National Diagnos-tics, Atlanta, GA) twice for 3 min each time. The tissue was then rehy-drated in a graded ethanol (ETOH) series as follows: three incubations for3 min in 100% ETOH, followed by 3 min in 95% ETOH, 3 min in 90%ETOH, and finally 3 min in 70% ETOH. The slides were rinsed in deion-ized water and heated in 1� citrate, pH 6, antigen retrieval buffer (Ag Plusbuffer; Novagen) at 95°C for 10 min. The slides were then rinsed in de-
Russo et al.
1214 iai.asm.org Infection and Immunity
on June 22, 2018 by guesthttp://iai.asm
.org/D
ownloaded from
ionized water and blocked overnight at 4°C in 1% goat serum diluted inantibody (Ab) diluent reagent solution (Invitrogen). Slides were incu-bated with primary Ab (polyclonal rabbit anti-Stx2a [34] diluted 1:500 inAb diluent solution) for 1 h at room temperature. Slides were washed 3times with PBS before the secondary Ab, goat anti-rabbit-Alexa-fluor 488(Invitrogen) diluted 1:500 in Ab diluent, was applied for 1 h at roomtemperature. Finally, slides were washed 3 times with PBS and the cover-slip was mounted with Vectashield (Vector Laboratories, Inc., Burlin-game, CA), which contained the counterstain, 4=,6-diamidino-2-phe-nylindole (DAPI). Slides were observed on an Olympus BX60 microscopewith a BX-FLA fluorescence attachment.
Passive Ab transfer. MAb 11E10 (BEI Resources/Hycult), directedagainst the Stx2a A subunit (22, 35), was used for all passive Ab transferstudies. TFTB1 (BEI Resources), an irrelevant Ab which recognizes thericin B subunit (36), was included as an IgG2a isotype control. Addition-ally, each experiment included a group that received Stx2a only as a pos-itive control for toxin activity. A dose of 7.5 �g Stx2a was used for all Abprotection and rescue experiments because that amount of toxin is invari-ably lethal in the oral intoxication model. Both MAbs and Stx2a werediluted in PBS for all experiments.
Ab protection. For the preliminary study, mice received 2 �g of 11E10or TFT�1 intravenously (i.v.) by tail vein 24 h prior to i.g. intoxicationwith 7.5 �g Stx2a. In all subsequent experiments, mice received 2, 4, or 40�g of Ab i.v. 1 h before intoxication.
Statistical analyses of antibody protection data. The percent weightchange values were first transformed into log ratios {log [(Y/100) � 1],where Y is weight change, to make the data more normally distributedwith more homogenous variance. Next, two-way repeated-measuresANOVA and Tukey’s post hoc tests were used to determine statistical sig-nificance of weight loss among Ab dose groups. Since repeated-measuresANOVA is an appropriate tool only for evaluation of complete data sets,analyses started on day 2. Although there was no significant group effect(P � 0.06), there was a significant interaction effect (P � 0.0002), a find-ing that indicates that the difference among groups varies over time.Tukey’s post hoc tests were used to determine statistical significance onindividual days.
Ab rescue. MAb 11E10 or TFT�1 (4 �g) was delivered i.v. at 6, 24, 48,or 72 h after Stx2a i.g. intoxication. An Ab protection group, which re-ceived 4 �g 1 h prior to intoxication, was included as a positive Ab control.
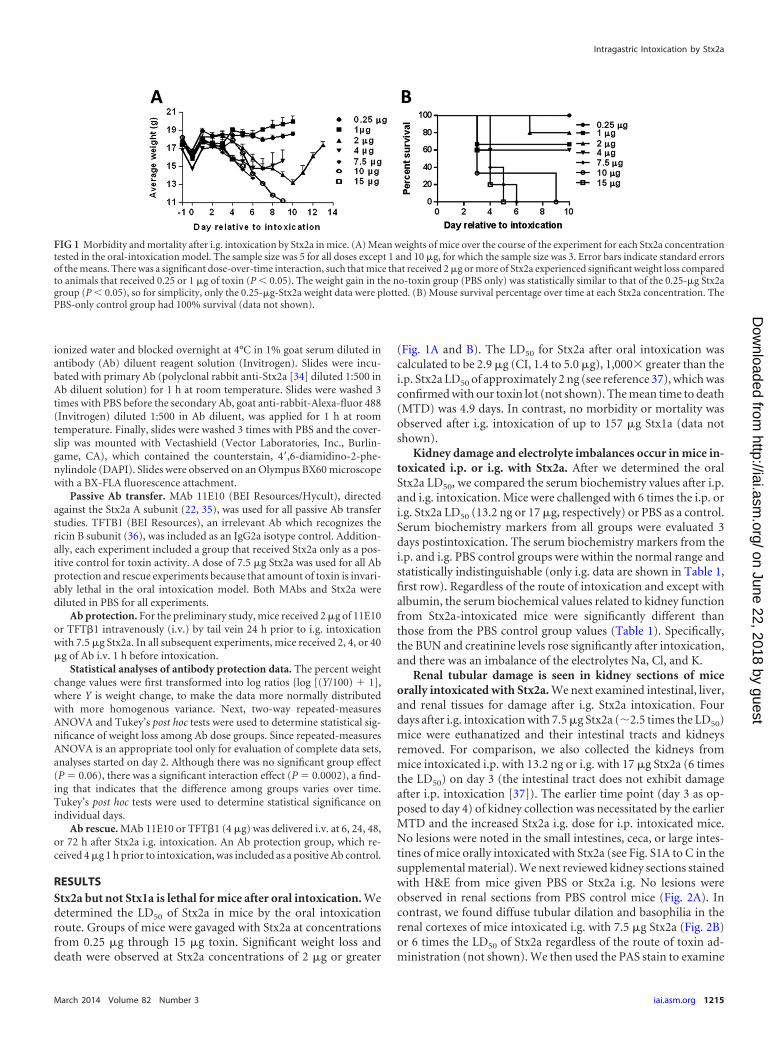
RESULTSStx2a but not Stx1a is lethal for mice after oral intoxication. Wedetermined the LD50 of Stx2a in mice by the oral intoxicationroute. Groups of mice were gavaged with Stx2a at concentrationsfrom 0.25 �g through 15 �g toxin. Significant weight loss anddeath were observed at Stx2a concentrations of 2 �g or greater
(Fig. 1A and B). The LD50 for Stx2a after oral intoxication wascalculated to be 2.9 �g (CI, 1.4 to 5.0 �g), 1,000� greater than thei.p. Stx2a LD50 of approximately 2 ng (see reference 37), which wasconfirmed with our toxin lot (not shown). The mean time to death(MTD) was 4.9 days. In contrast, no morbidity or mortality wasobserved after i.g. intoxication of up to 157 �g Stx1a (data notshown).
Kidney damage and electrolyte imbalances occur in mice in-toxicated i.p. or i.g. with Stx2a. After we determined the oralStx2a LD50, we compared the serum biochemistry values after i.p.and i.g. intoxication. Mice were challenged with 6 times the i.p. ori.g. Stx2a LD50 (13.2 ng or 17 �g, respectively) or PBS as a control.Serum biochemistry markers from all groups were evaluated 3days postintoxication. The serum biochemistry markers from thei.p. and i.g. PBS control groups were within the normal range andstatistically indistinguishable (only i.g. data are shown in Table 1,first row). Regardless of the route of intoxication and except withalbumin, the serum biochemical values related to kidney functionfrom Stx2a-intoxicated mice were significantly different thanthose from the PBS control group values (Table 1). Specifically,the BUN and creatinine levels rose significantly after intoxication,and there was an imbalance of the electrolytes Na, Cl, and K.
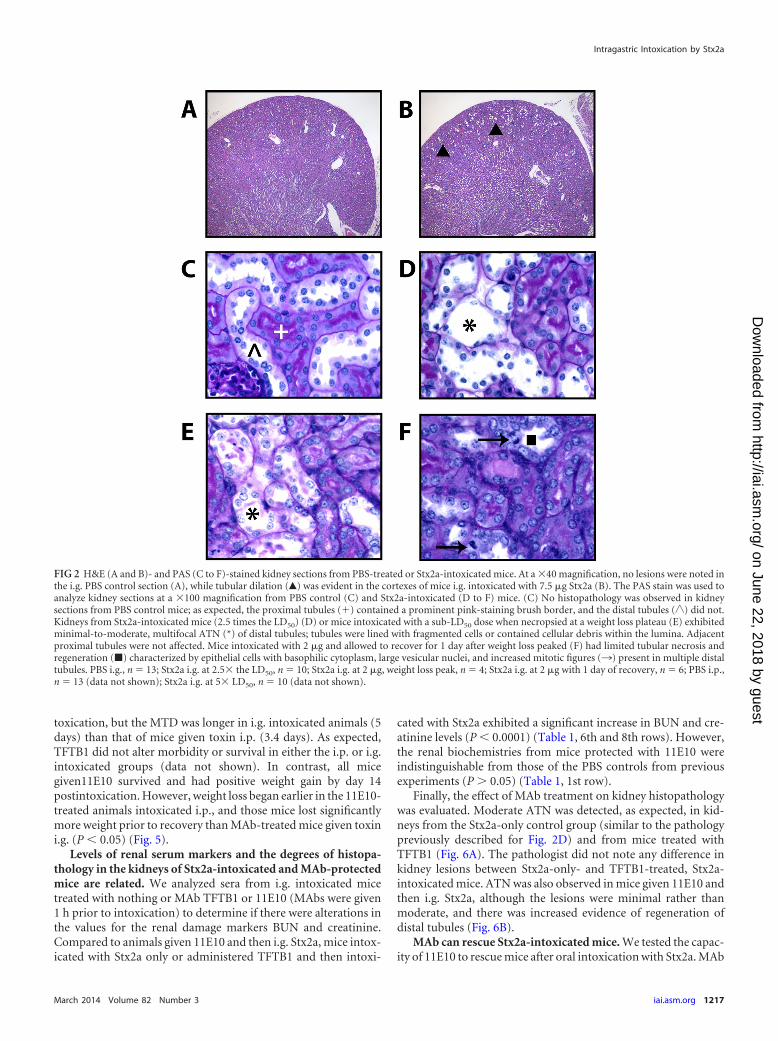
Renal tubular damage is seen in kidney sections of miceorally intoxicated with Stx2a. We next examined intestinal, liver,and renal tissues for damage after i.g. Stx2a intoxication. Fourdays after i.g. intoxication with 7.5 �g Stx2a (�2.5 times the LD50)mice were euthanatized and their intestinal tracts and kidneysremoved. For comparison, we also collected the kidneys frommice intoxicated i.p. with 13.2 ng or i.g. with 17 �g Stx2a (6 timesthe LD50) on day 3 (the intestinal tract does not exhibit damageafter i.p. intoxication [37]). The earlier time point (day 3 as op-posed to day 4) of kidney collection was necessitated by the earlierMTD and the increased Stx2a i.g. dose for i.p. intoxicated mice.No lesions were noted in the small intestines, ceca, or large intes-tines of mice orally intoxicated with Stx2a (see Fig. S1A to C in thesupplemental material). We next reviewed kidney sections stainedwith H&E from mice given PBS or Stx2a i.g. No lesions wereobserved in renal sections from PBS control mice (Fig. 2A). Incontrast, we found diffuse tubular dilation and basophilia in therenal cortexes of mice intoxicated i.g. with 7.5 �g Stx2a (Fig. 2B)or 6 times the LD50 of Stx2a regardless of the route of toxin ad-ministration (not shown). We then used the PAS stain to examine
FIG 1 Morbidity and mortality after i.g. intoxication by Stx2a in mice. (A) Mean weights of mice over the course of the experiment for each Stx2a concentrationtested in the oral-intoxication model. The sample size was 5 for all doses except 1 and 10 �g, for which the sample size was 3. Error bars indicate standard errorsof the means. There was a significant dose-over-time interaction, such that mice that received 2 �g or more of Stx2a experienced significant weight loss comparedto animals that received 0.25 or 1 �g of toxin (P 0.05). The weight gain in the no-toxin group (PBS only) was statistically similar to that of the 0.25-�g Stx2agroup (P 0.05), so for simplicity, only the 0.25-�g-Stx2a weight data were plotted. (B) Mouse survival percentage over time at each Stx2a concentration. ThePBS-only control group had 100% survival (data not shown).
Intragastric Intoxication by Stx2a
March 2014 Volume 82 Number 3 iai.asm.org 1215
on June 22, 2018 by guesthttp://iai.asm
.org/D
ownloaded from

kidney sections. As expected, no renal lesions were detected in thekidneys of either i.p. (not shown) or i.g. intoxicated PBS controlmice (Fig. 2C). However, we found minimal-to-moderate multi-focal acute tubular necrosis (ATN) of distal tubules, characterizedby tubules lined with degenerating, necrotic, or sloughed epithe-lial cells, independent of dose or route of intoxication (result afteradministration of 2.5 times the LD50 is shown in Fig. 2D).
Stx2a is present in the kidneys of intoxicated mice. We nextasked if we could detect Stx2a in the kidneys of mice after i.p. or i.g.intoxication. Only a minimal green fluorescent background was ob-served in kidney sections from mice given PBS i.p. (Fig. 3A) or orally(not shown). However, we were able to confirm the findings of Rutjeset al. that Stx2a can be found in kidney sections after parenteral (inour case, i.p.; in their studies, i.v.) intoxication (38) (Fig. 3B). More-over, we also detected Stx2a in kidney sections from i.g. intoxicatedmice (17 �g) (Fig. 3C). The Stx2a-positive cells appeared to be tubuleepithelial cells for both i.p. and i.g. intoxicated mice, as the Stx2astaining pattern in the tubules coincided with the histopathologicallocation of renal damage in the intoxicated mice (Fig. 2D).
Morbidity in Stx2a-intoxicated mice correlates with kidneyfunction. Because we observed significant weight loss followed byrapid weight gain in the majority of mice intoxicated i.g. with 2 �gof Stx2a, we speculated that mice given 2 �g of Stx2a i.g. mightexhibit kidney damage even though they typically recovered fromthat toxin dose (Fig. 1A and B). In a subsequent study, we foundthat mice intoxicated with 2 �g of Stx2a, a sub-LD50 dose, andsacrificed when weight loss began to plateau (day 8 or 9 in thisexperiment) exhibited serum biochemistry profiles and kidneyhistopathology similar to those of mice intoxicated with 6 timesthe LD50 (Table 1, 4th row; Fig. 2E). Stx2a was also detected byimmunofluorescence in the kidneys of those same mice (notshown). In mice given 2 �g of Stx2a and allowed to recover 1 dayafter the weight loss plateau, serum biochemistry values returnedto control levels and kidney histology improved (the ATN was lesssevere and tubules contained regenerating epithelial cells) (Table1, 5th row; Fig. 2F). Three days after the rebound in weight, nokidney lesions were noted (data not shown).
Anti-Stx2a MAb fully protects mice when given before intox-ication. We next tested whether i.g. intoxicated mice could be
protected from morbidity and/or mortality with a MAb to Stx2a.In a pilot study, mice received 2 �g of 11E10 (anti-Stx2a) orTFTB1 (isotype-matched control) i.v. 24 h prior to i.g. intoxica-tion with 7.5 �g Stx2a. A positive-control group received onlyStx2a i.g. (In this study and all subsequent experiments, MAbs andStx2a were diluted in PBS.) In this preliminary study, completemortality was observed in the Stx2a-only and Stx2a-plus-TFTB1groups, while MAb 11E10 prevented mortality though not mor-bidity (Fig. 4A, 1st row; Fig. 4B). Protected mice experienced acumulative weight loss of 1.9 g or 11% that peaked on day 7postintoxication compared to their starting weight before theyrecovered and exhibited positive weight gain by day 14 (data notshown). For our next study, in the hopes of limiting morbidity inthe toxin-treated mice, we altered the time of administration of11E10 from 24 h to 1 h so that higher levels of MAb would bepresent during the intoxication window. We found that althoughthe mice were protected from lethality, as expected, they still ex-perienced weight loss before recovery (Fig. 4A, 2nd row, and B).Therefore, in an attempt to reduce morbidity in the subsequentprotection experiment, the amount of MAb administered wasdoubled. Mice that received 4 �g 11E10 were completely pro-tected (Fig. 4A, 3rd row), and in addition, their weight loss wasreduced, although not to a statistically significant level (P � 0.055)(Fig. 4B). In a final attempt to further reduce or prevent Stx2a-mediated morbidity, 10-fold-more 11E10 was administered in thenext study. Although 11E10 again protected (Fig. 4A, 4th row), thelevel of weight loss in the intoxicated, treated animals was similarto that in the previous protection experiment in which mice re-ceived 4 �g MAb; i.e., there was no statistical difference in weightloss in mice treated with 4 or 40 �g MAb (P 0.064) (Fig. 4B).
We next compared mouse weight changes after administrationof 4 �g MAb in the i.p. and i.g. intoxication models to evaluatewhether MAb protection from morbidity might be greater in thei.p. than the i.g. model. As was done previously, an Stx2a-onlygroup served as a positive control for mortality, and MAb TFTB1was included as an isotype-matched control for 11E10. One hourafter injection of treatment groups with 4 �g MAb, mice received2.5 times the i.p. or i.g. LD50, 5.7 ng or 7.5 �g, respectively. Allmice in the Stx2a-only positive-control groups succumbed to in-
TABLE 1 Mean renal panel serum biochemistry values
Agent androute
No. ofsubjects Dose
Time of bloodcollectiona
Level (range)f
BUN(mg/dl)
Creatinine(mg/dl)
Na(mmol/liter)
K(mmol/liter)
Cl(mmol/liter)
Albumin(g/dl)
PBS i.g.b 8 0.2 ml Day 3 p.i. 33.9 (25–40) 0.24 (0.2–0.28) 135.5 (132–137) 19.9 (13.7–24.5) 105.3 (102–108) 3.3 (3.2–3.5)Stx2a i.p.c 4–9 13.2 ng Day 3 p.i. 193.9 (81–246) 0.95 (0.67–1.2) 119.5 (99–128) 30.2 (23.2–45) 84.8 (67.3–91.1) 4.1 (3.9–4.2)Stxa2 i.g.c 8–9 17 �g Day 3 p.i. 116.6 (82–245) 0.61 (0.44–1.1) 105.9 (76–121) 37.3 (25.6–67.3) 77.2 (68.9–88.8) 3.7 (1.9–4.3)Stx2a i.g. 2 2 �g Wt loss plateau 114.5 0.50 119.0 37.3 87.1 3.7Stx2a i.g. 4 2 �g After 1 day of
recovery35.8 (34–38) 0.20 (0.14–0.24) 135.8 (134–139) 25.3 (23.3–27.2) 106.6 (106–109) 3.3 (3.2–3.4)
Stx2a i.g. 2 7.5 �g Day 4 p.i. 275 1.62 NV NV NV NV�TFTB1d 5 7.5 �g Day 4 p.i. 213.2e (150–269) 1.23e (0.99–1.41) NV NV NV NV�11E1d 4 7.5 �g Day 4 p.i. 30.5e (25–40) 0.24e (0.2–0.28) NV NV NV NV
a Blood collection times were dictated by parameters for each study.b Serum biochemistry values from i.p. and i.g. PBS control mice were not statistically different (P 0.05).c The serum chemistry values were significantly different (P 0.01) from PBS control values except for the K in the Stx2a i.p. group and the albumin values in both groups.d Four micrograms of Ab.e P 0.0001, TFTB1 versus 11E10.f NV, no value returned.
Russo et al.
1216 iai.asm.org Infection and Immunity
on June 22, 2018 by guesthttp://iai.asm
.org/D
ownloaded from
toxication, but the MTD was longer in i.g. intoxicated animals (5days) than that of mice given toxin i.p. (3.4 days). As expected,TFTB1 did not alter morbidity or survival in either the i.p. or i.g.intoxicated groups (data not shown). In contrast, all micegiven11E10 survived and had positive weight gain by day 14postintoxication. However, weight loss began earlier in the 11E10-treated animals intoxicated i.p., and those mice lost significantlymore weight prior to recovery than MAb-treated mice given toxini.g. (P 0.05) (Fig. 5).
Levels of renal serum markers and the degrees of histopa-thology in the kidneys of Stx2a-intoxicated and MAb-protectedmice are related. We analyzed sera from i.g. intoxicated micetreated with nothing or MAb TFTB1 or 11E10 (MAbs were given1 h prior to intoxication) to determine if there were alterations inthe values for the renal damage markers BUN and creatinine.Compared to animals given 11E10 and then i.g. Stx2a, mice intox-icated with Stx2a only or administered TFTB1 and then intoxi-
cated with Stx2a exhibited a significant increase in BUN and cre-atinine levels (P 0.0001) (Table 1, 6th and 8th rows). However,the renal biochemistries from mice protected with 11E10 wereindistinguishable from those of the PBS controls from previousexperiments (P � 0.05) (Table 1, 1st row).
Finally, the effect of MAb treatment on kidney histopathologywas evaluated. Moderate ATN was detected, as expected, in kid-neys from the Stx2a-only control group (similar to the pathologypreviously described for Fig. 2D) and from mice treated withTFTB1 (Fig. 6A). The pathologist did not note any difference inkidney lesions between Stx2a-only- and TFTB1-treated, Stx2a-intoxicated mice. ATN was also observed in mice given 11E10 andthen i.g. Stx2a, although the lesions were minimal rather thanmoderate, and there was increased evidence of regeneration ofdistal tubules (Fig. 6B).
MAb can rescue Stx2a-intoxicated mice. We tested the capac-ity of 11E10 to rescue mice after oral intoxication with Stx2a. MAb
FIG 2 H&E (A and B)- and PAS (C to F)-stained kidney sections from PBS-treated or Stx2a-intoxicated mice. At a �40 magnification, no lesions were noted inthe i.g. PBS control section (A), while tubular dilation (Œ) was evident in the cortexes of mice i.g. intoxicated with 7.5 �g Stx2a (B). The PAS stain was used toanalyze kidney sections at a �100 magnification from PBS control (C) and Stx2a-intoxicated (D to F) mice. (C) No histopathology was observed in kidneysections from PBS control mice; as expected, the proximal tubules (�) contained a prominent pink-staining brush border, and the distal tubules (�) did not.Kidneys from Stx2a-intoxicated mice (2.5 times the LD50) (D) or mice intoxicated with a sub-LD50 dose when necropsied at a weight loss plateau (E) exhibitedminimal-to-moderate, multifocal ATN (*) of distal tubules; tubules were lined with fragmented cells or contained cellular debris within the lumina. Adjacentproximal tubules were not affected. Mice intoxicated with 2 �g and allowed to recover for 1 day after weight loss peaked (F) had limited tubular necrosis andregeneration (�) characterized by epithelial cells with basophilic cytoplasm, large vesicular nuclei, and increased mitotic figures (¡) present in multiple distaltubules. PBS i.g., n � 13; Stx2a i.g. at 2.5� the LD50, n � 10; Stx2a i.g. at 2 �g, weight loss peak, n � 4; Stx2a i.g. at 2 �g with 1 day of recovery, n � 6; PBS i.p.,n � 13 (data not shown); Stx2a i.g. at 5� LD50, n � 10 (data not shown).
Intragastric Intoxication by Stx2a
March 2014 Volume 82 Number 3 iai.asm.org 1217
on June 22, 2018 by guesthttp://iai.asm
.org/D
ownloaded from

11E10 or TFTB1 was administered i.v. at 6, 24, 48, or 72 h after i.g.intoxication with approximately 2.5 times the LD50 (7.5 �g) ofStx2a. None of the mice treated with TFTB1 or in the Stx2a-onlygroup survived toxin challenge. Additionally, all TFTB1-treatedand Stx2a-only groups had similar MTDs, a finding that indicatesthat the addition of fluids alone had no therapeutic effect (sincethe TFTB1 was given in PBS) (data not shown). Mice that received11E10 1 h prior (a control) or 6 h after intoxication were com-pletely protected from Stx2a. In contrast, only one mouse wasrescued when 11E10 was given at 24 h postintoxication, and nomice were rescued if the MAb was administered at 48 or 72 h after
toxin administration (Table 2). Both the 1-h and �6-h treat-ment groups also exhibited similar patterns of weight lossthroughout the experiment, and both groups displayed positiveweight gain by day 14 (data not shown).
DISCUSSION
That Stx (specific toxin type[s] unknown) alone can recapitulatesome of the symptoms of STEC disease was first reported by Pai etal. (24). These investigators found that infant New Zealand Whiterabbits orally intoxicated with Stx develop diarrhea and succumbto intoxication in a manner analogous to that of rabbits challengedwith E. coli O157:H7 (24). Similarly, Ritchie et al. showed thatrabbits orally intoxicated with Stx2a develop inflammation com-parable to that of rabbits infected with an Stx2a-producing STECstrain (25). Additionally, Rasooly et al. recently reported that oralintoxication of mice with high doses of Stx2a was lethal (26). Weextended such observations with the determination that the LD50
for Stx2a in BALB/c mice by the oral route is 2.9 �g and that boththe i.p. and i.g. Stx2a intoxication routes lead to similar systemicpathologies in those animals. Additionally, our data illustratedthat renal damage similar to that of mice inoculated with the LD50
occurred even in mice intoxicated with a sub-LD50 Stx2a dose and
FIG 3 Staining to detect Stx2a in kidney sections from PBS-treated or intox-icated mice. Immunofluorescence of kidney sections treated with rabbit poly-clonal anti-Stx2a combined with goat anti-rabbit conjugated to Alexa 488.DAPI stained the cell nuclei. (A) i.g. PBS control sections were negative forStx2a. Stx2a-positive cells adjacent to tubule lumens (bright-green fluores-cence) were detected in kidney sections from mice i.p. intoxicated with 13.2 ng(B) and i.g. intoxicated with 17 �g (C) of Stx2a. Arrows point to the lumen ofa tubule. Magnification, �400.
FIG 4 MAb 11E10 prevented mortality and limited morbidity due to Stx2a i.g. intoxication. (A) Mortality results from four independent Ab studies. Two, 4, or40 �g 11E10 protected mice from Stx2a i.g. intoxication, while TFTB1, the irrelevant MAb, had no therapeutic effect. Ab Rx, antibody administration; ND, notdone. (B) Percent weight changes from day 0 to day 14 of antibody-treated, Stx2a-intoxicated, and PBS control groups. Each group received Ab 1 h prior tointoxication. The PBS control group experienced positive weight gain over the course of the experiment, with no effect from 40 �g 11E10. Error bars indicatestandard deviations. There was a significant interaction effect (P � 0.0002), which indicates that the percentages of weight change vary over time between Abtreatment groups. The difference in percent weight change was significant between the groups given 2 or 4 �g 11E10 on days 3, 7, and 9 and between the 2-�gand 40-�g groups on days 5 to 8. There was no difference between the 4- and 40-�g groups.
FIG 5 Average weight over time in Stx2a-intoxicated mice given 11E10 (opensymbols) or no treatment (filled symbols) 1 h before toxin administration.Mice were intoxicated with 2.5 times the Stx2a LD50 i.p. (circles) or i.g.(squares). Although MAb 11E10 was protective in both intoxication models,i.p. intoxicated mice exhibited significantly more morbidity than i.g. intoxi-cated mice (P 0.05). Additionally, there was a significant difference in weightbetween the Stx2a i.p. and i.g. 11E10-protected groups on days 5, 6, 8, and 11.
Russo et al.
1218 iai.asm.org Infection and Immunity
on June 22, 2018 by guesthttp://iai.asm
.org/D
ownloaded from

that survival in those animals correlated with a return to normalserum chemistry values and tubule regeneration. Finally, we dem-onstrated protection and rescue of mice from Stx2a oral intoxica-tion through passive transfer of MAb 11E10.
We found that Stx1a was not lethal by the intragastric route, incontrast to the oral lethality of Stx2a. We speculate that if, as withStx2a, the i.g. LD50 for Stx1a is 1,000 times greater than the i.p.LD50 (as it was for Stx2a), more than 500 �g Stx1a per mousewould be necessary to result in lethality. However, it is not prac-tical to purify such large quantities of toxin to test the hypothesisthat Stx1 is potentially lethal by the oral route. Our finding thatStx1a is not lethal by the oral route as far as we can test may not besurprising, since STEC strains that produce only Stx1a are notlethal in mouse models of oral infection (39).
We did not observe lesions in any section of the intestinal tractsfrom Stx2a i.g. intoxicated mice, a finding that suggests that thetoxin reached systemic circulation without injury to the intestinalepithelial cell lining. We also examined the liver for damage be-cause we thought that that organ had the potential to absorb alarge concentration of Stx2a as the toxin traveled through thehepatic portal system. However, we did not observe lesions in thelivers of orally intoxicated mice, and we did not detect alterationsin the hepatic serum biochemistry values (data not shown).Therefore, we concluded that the liver is not a target site for Stx2ain mice, a contention supported by the observation that Gb3 con-centrations are low in that organ (40).
We found that once sufficient Stx2a entered systemic circula-tion, regardless of the route of intoxication (i.p. or i.g.), similarelevations in renal serum biochemistry values occurred. The ele-vated BUN and creatinine values suggest acute renal failure, whichis characterized by a reduced glomerular filtration rate (GFR).Although the pathophysiologic mechanism of the GFR is not fullyunderstood, it likely involves reduction in blood flow to or withinthe kidney. The apparently reduced GFR is not related to prob-lems with the glomerulus in these mice. In addition, the significantreduction in sodium and chloride levels due to Stx2a exposureindicate renal tubular malfunction (41, 42). We do not believe thatthe changes in serum biochemistry values after Stx2a intoxicationare a result of hemoconcentration due to dehydration because theexperimental serum albumin levels were equivalent to albuminlevels from control mice.
Our immunofluorescence data suggest that once Stx2a reachedthe kidney, it targeted tubular epithelial cells because the toxinstaining was in the cells adjacent to the renal tubule lumen. The
toxin molecule likely killed those cells, events that would lead toATN. ATN causes loss of renal function, which, in turn, can resultin mortality. ATN is the same pathology that occurs in STECmouse infection models. Overall, these results demonstrate thatoral intoxication with Stx2a alone recapitulates disease noted afterinfection of mice with STEC in various models (21).
The specificity of the PAS stain revealed approximately equiv-alent proportions of distal and proximal tubules in both controland experimental mice (data not shown). The PAS stain furtherindicated that the Stx2a-mediated lesions were in distal ratherthan proximal tubules (as we had previously believed [37]), be-cause of the absence of a brush boarder in the affected tubules. Thehypothesis that distal rather than proximal tubules are more af-fected by Stx2a is supported by a study that showed that distaltubules have a higher concentration of Gb3 receptors on the cellsurface than do proximal tubules (43). We were surprised to findthat the renal pathology and serum biochemistry values due to asub-LD50 oral Stx2a dose were similar to those found after i.g.delivery of 6 times the LD50 of toxin if the mice were necropsiedwhen weight loss reached its nadir. In contrast, once the sub-LD50-intoxicated animals began to gain weight, they had serumbiochemistry values similar to those of control mice and exhibitedless severe kidney histopathology. We believe that although thetypes of histopathology observed in the sub-LD50 and 6-fold-LD50
Stx2a groups were similar when assessed at the point of lowestweight for the sub-LD50 group, the magnitude of damage wasgreater in the higher-dose group. We further speculate that thosemice that received a sub-LD50 dose survived if they were able toregenerate tubular epithelial cells (observed in Fig. 2F) and restorea minimum threshold of kidney function. This study also demon-strated the importance of timing of sample collection after intox-ication; we note that damage can occur yet go undetected becauseof subsequent repair if samples are taken too long after toxin in-sult.
We protected all mice from 2.5 times the LD50 of Stx2a with asingle passive transfer of 4 �g 11E10. Although there was a reduc-tion in weight loss at the 4-�g compared to the 2-�g 11E10 dose,a 10-fold increase to 40 �g of the MAb did not result in a furtherdecrease in morbidity. Thus, there appears to be a threshold forlevels of antibody that can decrease morbidity in intoxicated ani-mals. We also found that mice treated with 11E10 and then givenStx2a had normal kidney function 4 days postintoxication, as in-dicated by serum BUN and creatinine levels. Although minimalrenal ATN was observed in the MAb-treated Stx2a-intoxicatedmice, there were increased instances of tubule regeneration com-pared to that in animals that received Stx2a only. Our study sug-gests that 11E10 pretreatment reduced kidney damage in Stx2a-intoxicated mice. Finally, since we observed no protective effect by
FIG 6 PAS-stained kidney sections from the MAb protection/Stx2a i.g. intox-ication study. (A) Moderate ATN (}) of distal tubules was observed in thekidneys of mice in Stx2a-plus-TFTB1 groups. (B) Minimal ATN (●) and in-creased regeneration of distal tubules were seen in Stx2a- and 11E10-treatedmice.
TABLE 2 Rescue by 11E10 of Stx2a i.g. intoxicated mice
Time of 11E10 treatmentrelative to i.g. Stx2a (h)
No. of deadmice/total MTD (days)
1 0/8�6 0/10�24 9/10 4.4�48 10/10 4.4�72 8/8 4.3NAa (Stx2a only) 8/8 4.4a NA, not applicable.
Intragastric Intoxication by Stx2a
March 2014 Volume 82 Number 3 iai.asm.org 1219
on June 22, 2018 by guesthttp://iai.asm
.org/D
ownloaded from

irrelevant MAb TFTB1, an IgG2a isotype control, we concludethat fluid alone was not protective.
We also observed that in mice treated with 11E10, greater mor-bidity occurred after i.p. than i.g. Stx2a intoxication. The i.p.-intoxicated animals exhibited a shorter time to death of approxi-mately 1.5 days than mice given toxin i.g. The reduced MTD in i.p.intoxicated mice suggests that Stx2a reached the target site of thekidneys earlier when it was delivered to the peritoneum than whenit was delivered to the gastrointestinal tract. Although ATN wasprobably present at increased levels in i.p. compared to i.g. intox-icated mice, kidney function was not lost and the mice were able torecover.
We demonstrated rescue of mice with 4 �g of 11E10 at 6 butnot 24 h after oral intoxication with Stx2a. A recent study in whichmice were orally intoxicated with botulinum neurotoxin serotypeA (BotA) and then treated by passive Ab transfer demonstratedthat differences of just 1 to 2 h in antibody administration timepoints resulted in the loss of the capacity to rescue intoxicatedanimals (44). We speculate, therefore, that as with BotA, onceStx2a is sequestered in target tissues (the kidneys), passive Abtransfer is not able to rescue intoxicated animals. However, therescue time frame may be increased if a larger MAb dose is pro-vided or if additional doses are delivered. In support of the latterhypothesis, another group was able to rescue baboons 24 h afterintravenous intoxication with Stx2a by providing a therapeuticdose of TVP, an acetylated tetravalent peptide, daily until day 4(45). We believe that passive Ab transfer is a viable therapeuticoption for STEC infections. Furthermore, for someone infectedwith STEC, the rescue time frame is likely extended compared tothat of the oral intoxication model, as Stx would be delivered at acontinual low dose rather than in a single large bolus. Previousresearch in our lab demonstrated that low levels of active Stx2acould be found in the feces of mice with an intact commensal floraup to 72 h postinfection with an Stx2a-producing O157:H7 strain(46).
Finally, we found that an oral Stx2a dose of 2 �g (�0.1 mg/kgof body weight) was sufficient to cause weight loss and renal injurybut not death in mice. If mice and humans are equivalently sus-ceptible to Stx, then we calculate that 1.8 mg Stx2a would be suf-ficient to cause some disease in an 18-kg child. However, we be-lieve that humans are more susceptible to Stx than are micebecause, unlike mice, people have Gb3 in their glomeruli in addi-tion to in their tubules (37, 47, 48). Therefore, we believe that theoral toxicity of Stx2a is potentially relevant as a public health issue.There is a possibility that preformed Stx occurs in contaminatedfood, which might contribute to morbidity (49–51) and possiblyenhance colonization by the organism, as Stx2 has been shown toincrease the adherence of O157:H7 to tissue culture cells and inanimal models (52, 53).
ACKNOWLEDGMENTS
We thank Farhang Alem and Stephen Darnell for assistance with animalwork and Cara Olsen for facilitation of statistical analyses.
The opinions or assertions contained herein are the private ones of theauthors and are not to be construed as official or reflecting the views of theDepartment of Defense, the Uniformed Services University of the HealthSciences, or the National Institutes of Health.
This work was supported by National Institutes of Health grants R37AI020148 to A.D.O. and U54 AI057168 to Myron Levine (subaward toA.D.O.).
REFERENCES1. Tilden J, Jr, Young W, McNamara AM, Custer C, Boesel B, Lambert-
Fair MA, Majkowski J, Vugia D, Werner SB, Hollingsworth J, MorrisJG, Jr. 1996. A new route of transmission for Escherichia coli: infectionfrom dry fermented salami. Am. J. Public Health 86:1142–1145. http://dx.doi.org/10.2105/AJPH.86.8_Pt_1.1142.
2. Scallan E, Hoekstra RM, Angulo FJ, Tauxe RV, Widdowson MA, RoySL, Jones JL, Griffin PM. 2011. Foodborne illness acquired in the UnitedStates—major pathogens. Emerg. Infect. Dis. 17:7–15. http://dx.doi.org/10.3201/eid1701.09-1101p1.
3. Hancock DD, Besser TE, Rice DH, Tarr PI. 1998. Ecology of Escherichiacoli O157:H7 in cattle and impact of management practices, p 85–91. InKaper JB, O’Brien AD (ed), Escherichia coli O157:H7 and other Shigatoxin-producing E. coli strains. American Society for Microbiology,Washington, DC.
4. Painter JA, Hoekstra RM, Ayers T, Tauxe RV, Braden CR, Angulo FJ,Griffin PM. 2013. Attribution of foodborne illnesses, hospitalizations,and deaths to food commodities by using outbreak data, United States,1998 –2008. Emerg. Infect. Dis. 19:407– 415. http://dx.doi.org/10.3201/eid1903.111866.
5. Rangel JM, Sparling PH, Crowe C, Griffin PM, Swerdlow DL. 2005.Epidemiology of Escherichia coli O157:H7 outbreaks, United States,1982–2002. Emerg. Infect. Dis. 11:603– 609. http://dx.doi.org/10.3201/eid1104.040739.
6. Karmali MA, Petric M, Lim C, Fleming PC, Arbus GS, Lior H. 1985.The association between idiopathic hemolytic uremic syndrome and in-fection by verotoxin-producing Escherichia coli. J. Infect. Dis. 151:775–782. http://dx.doi.org/10.1093/infdis/151.5.775.
7. Mayer CL, Leibowitz CS, Kurosawa S, Stearns-Kurosawa DJ. 2012.Shiga toxins and the pathophysiology of hemolytic uremic syndrome inhumans and animals. Toxins 4:1261–1287. http://dx.doi.org/10.3390/toxins4111261.
8. Kaplan BS, Cleary TG, Obrig TG. 1990. Recent advances in understand-ing the pathogenesis of the hemolytic uremic syndromes. Pediatr. Neph-rol. 4:276 –283. http://dx.doi.org/10.1007/BF00857676.
9. Wong CS, Jelacic S, Habeeb RL, Watkins SL, Tarr PI. 2000. The risk ofthe hemolytic-uremic syndrome after antibiotic treatment of Escherichiacoli O157:H7 infections. N. Engl. J. Med. 342:1930 –1936. http://dx.doi.org/10.1056/NEJM200006293422601.
10. Wen SX, Teel LD, Judge NA, O’Brien AD. 2006. Genetic toxoids of Shiga toxintypes 1 and 2 protect mice against homologous but not heterologous toxin chal-lenge. Vaccine 24:1142–1148. http://dx.doi.org/10.1016/j.vaccine.2005.08.094.
11. O’Brien AD, Holmes RK. 1987. Shiga and Shiga-like toxins. Microbiol.Rev. 51:206 –220.
12. Melton-Celsa AR, Smith MJ, O’Brien AD. 2005. Shiga toxins: potentpoisons, pathogenicity determinants, and pharmacological agents.EcoSal Plus http://dx.doi.org/10.1128/ecosalplus.8.7.8.
13. Endo Y, Tsurugi K, Yutsudo T, Takeda Y, Ogasawara T, Igarashi K.1988. Site of action of a Vero toxin (VT2) from Escherichia coli O157:H7and of Shiga toxin on eukaryotic ribosomes. RNA N-glycosidase activity ofthe toxins. Eur. J. Biochem. 171:45–50.
14. Saxena SK, O’Brien AD, Ackerman EJ. 1989. Shiga toxin, Shiga-liketoxin II variant, and ricin are all single-site RNA N-glycosidases of 28 SRNA when microinjected into Xenopus oocytes. J. Biol. Chem. 264:596 –601.
15. Obrig TG. 1997. Shiga toxin mode of action in E. coli O157:H7 disease.Front. Biosci. 2:d635– d642.
16. Boerlin P, McEwen SA, Boerlin-Petzold F, Wilson JB, Johnson RP,Gyles CL. 1999. Associations between virulence factors of Shiga toxin-producing Escherichia coli and disease in humans. J. Clin. Microbiol. 37:497–503.
17. Ostroff SM, Tarr PI, Neill MA, Lewis JH, Hargrett-Bean N, KobayashiJM. 1989. Toxin genotypes and plasmid profiles as determinants of sys-temic sequelae in Escherichia coli O157:H7 infections. J. Infect. Dis. 160:994 –998. http://dx.doi.org/10.1093/infdis/160.6.994.
18. Scotland SM, Willshaw GA, Smith HR, Rowe B. 1987. Properties ofstrains of Escherichia coli belonging to serogroup O157 with special ref-erence to production of Vero cytotoxins VT1 and VT2. Epidemiol. Infect.99:613– 624. http://dx.doi.org/10.1017/S0950268800066462.
19. Moxley RA, Francis DH. 1998. Overview of animal models, p 249 –260. InKaper JB, O’Brien AD (ed), Escherichia coli O157:H7 and other Shigatoxin-producing E. coli strains. ASM Press, Washington, DC.
Russo et al.
1220 iai.asm.org Infection and Immunity
on June 22, 2018 by guesthttp://iai.asm
.org/D
ownloaded from

20. Melton-Celsa AR, O’Brien AD. 2003. Animal models for STEC-mediateddisease. Methods Mol. Med. 73:291–305.
21. Mohawk KL, O’Brien AD. 2011. Mouse models of Escherichia coliO157:H7 infection and Shiga toxin injection. J. Biomed. Biotechnol. 2011:258185. http://dx.doi.org/10.1155/2011/258185.
22. Smith MJ, Melton-Celsa AR, Sinclair JF, Carvalho HM, Robinson CM,O’Brien AD. 2009. Monoclonal antibody 11E10, which neutralizes Shigatoxin type 2 (Stx2), recognizes three regions on the Stx2 A subunit, blocksthe enzymatic action of the toxin in vitro, and alters the overall cellulardistribution of the toxin. Infect. Immun. 77:2730 –2740. http://dx.doi.org/10.1128/IAI.00005-09.
23. Edwards AC, Melton-Celsa AR, Arbuthnott K, Stinson JR, Schmitt CK,Wong HC, O’Brien AD. 1998. Vero cell neutralization and mouse pro-tective efficacy of humanized monoclonal antibodies against Escherichiacoli toxins Stx1 and Stx2, p 388 –392. In Kaper JB, O’Brien AD (ed), Esch-erichia coli O157:H7 and other Shiga toxin-producing E. coli strains. ASMPress, Washington, DC.
24. Pai CH, Kelly JK, Meyers GL. 1986. Experimental infection of infantrabbits with verotoxin-producing Escherichia coli. Infect. Immun. 51:16 –23.
25. Ritchie JM, Thorpe CM, Rogers AB, Waldor MK. 2003. Critical roles forstx2, eae, and tir in enterohemorrhagic Escherichia coli-induced diarrheaand intestinal inflammation in infant rabbits. Infect. Immun. 71:7129 –7139. http://dx.doi.org/10.1128/IAI.71.12.7129-7139.2003.
26. Rasooly R, Do PM, Griffey SM, Vilches-Moure JG, Friedman M. 2010.Ingested Shiga toxin 2 (Stx2) causes histopathological changes in kidney,spleen, and thymus tissues and mortality in mice. J. Agric. Food Chem.58:9281–9286. http://dx.doi.org/10.1021/jf101744z.
27. Russo LM, Melton-Celsa AR, Smith MJ, O’Brien AD. 2012. Analysis ofchimeric shiga toxins (Stxs) to determine the contribution of individualStx subunits to cytotoxicity and mouse lethality, abstr D2-3. Abstr. 8th Int.Symp. Shiga Toxin Producing E. coli Infect. VTEC 2012, 6 to 9 May 2012,Amsterdam, The Netherlands.
28. Tesh VL, Samuel JE, Perera LP, Sharefkin JB, O’Brien AD. 1991.Evaluation of the role of Shiga and Shiga-like toxins in mediating directdamage to human vascular endothelial cells. J. Infect. Dis. 164:344 –352.http://dx.doi.org/10.1093/infdis/164.2.344.
29. Lindgren SW, Melton AR, O’Brien AD. 1993. Virulence of enterohem-orrhagic Escherichia coli O91:H21 clinical isolates in an orally infectedmouse model. Infect. Immun. 61:3832–3842.
30. Strockbine NA, Marques LR, Holmes RK, O’Brien AD. 1985. Charac-terization of monoclonal antibodies against Shiga-like toxin from Esche-richia coli. Infect. Immun. 50:695–700.
31. Downes FP, Barrett TJ, Green JH, Aloisio CH, Spika JS, Strockbine NA,Wachsmuth IK. 1988. Affinity purification and characterization of Shiga-like toxin II and production of toxin-specific monoclonal antibodies. In-fect. Immun. 56:1926 –1933.
32. Gentry MK, Dalrymple JM. 1980. Quantitative microtiter cytotoxicityassay for Shigella toxin. J. Clin. Microbiol. 12:361–366.
33. U.S. National Research Council, Committee for the Update of theGuide for the Care and Use of Laboratory Animals, Institute forLaboratory Animal Research. 2011. Guide for the care and use of labo-ratory animals, 8th ed. National Academies Press, Washington, DC.
34. Kokai-Kun JF, Melton-Celsa AR, O’Brien AD. 2000. Elastase in intesti-nal mucus enhances the cytotoxicity of Shiga toxin type 2d. J. Biol. Chem.275:3713–3721. http://dx.doi.org/10.1074/jbc.275.5.3713.
35. Perera LP, Marques LR, O’Brien AD. 1988. Isolation and characteriza-tion of monoclonal antibodies to Shiga-like toxin II of enterohemorrhagicEscherichia coli and use of the monoclonal antibodies in a colony enzyme-linked immunosorbent assay. J. Clin. Microbiol. 26:2127–2131.
36. Fulton RJ, Uhr JW, Vitetta ES. 1986. The effect of antibody valency andlysosomotropic amines on the synergy between ricin A chain- and ricin Bchain-containing immunotoxins. J. Immunol. 136:3103–3109.
37. Tesh VL, Burris JA, Owens JW, Gordon VM, Wadolkowski EA, O’BrienAD, Samuel JE. 1993. Comparison of the relative toxicities of Shiga-liketoxins type I and type II for mice. Infect. Immun. 61:3392–3402.
38. Rutjes NW, Binnington BA, Smith CR, Maloney MD, Lingwood CA.2002. Differential tissue targeting and pathogenesis of verotoxins 1 and 2in the mouse animal model. Kidney Int. 62:832– 845. http://dx.doi.org/10.1046/j.1523-1755.2002.00502.x.
39. Wadolkowski EA, Sung LM, Burris JA, Samuel JE, O’Brien AD. 1990.Acute renal tubular necrosis and death of mice orally infected with Esch-erichia coli strains that produce Shiga-like toxin type II. Infect. Immun.58:3959 –3965.
40. Fujii Y, Numata S, Nakamura Y, Honda T, Furukawa K, Urano T, WielsJ, Uchikawa M, Ozaki N, Matsuo S, Sugiura Y, Furukawa K. 2005.Murine glycosyltransferases responsible for the expression of globo-seriesglycolipids: cDNA structures, mRNA expression, and distribution of theirproducts. Glycobiology 15:1257–1267. http://dx.doi.org/10.1093/glycob/cwj015.
41. Lopez-Novoa JM, Rodriguez-Pena AB, Ortiz A, Martinez-Salgado C,Lopez Hernandez FJ. 2011. Etiopathology of chronic tubular, glomerularand renovascular nephropathies: clinical implications. J. Transl. Med.9:13. http://dx.doi.org/10.1186/1479-5876-9-13.
42. Tarr PI, Gordon CA, Chandler WL. 2005. Shiga-toxin-producing Esch-erichia coli and haemolytic uraemic syndrome. Lancet 365:1073–1086.http://dx.doi.org/10.1016/S0140-6736(05)71144-2.
43. Kashiwamura M, Kurohane K, Tanikawa T, Deguchi A, Miyamoto D,Imai Y. 2009. Shiga toxin kills epithelial cells isolated from distal but notproximal part of mouse colon. Biol. Pharm. Bull. 32:1614 –1617. http://dx.doi.org/10.1248/bpb.32.1614.
44. Cheng LW, Stanker LH, Henderson TD, II, Lou J, Marks JD. 2009.Antibody protection against botulinum neurotoxin intoxication in mice.Infect. Immun. 77:4305– 4313. http://dx.doi.org/10.1128/IAI.00405-09.
45. Stearns-Kurosawa DJ, Collins V, Freeman S, Debord D, Nishikawa K,Oh SY, Leibowitz CS, Kurosawa S. 2011. Rescue from lethal Shiga toxin2-induced renal failure with a cell-permeable peptide. Pediatr. Nephrol.26:2031–2039. http://dx.doi.org/10.1007/s00467-011-1913-y.
46. Mohawk KL, Melton-Celsa AR, Zangari T, Carroll EE, O’Brien AD.2010. Pathogenesis of Escherichia coli O157:H7 strain 86-24 followingoral infection of BALB/c mice with an intact commensal flora. Microb.Pathog. 48:131–142. http://dx.doi.org/10.1016/j.micpath.2010.01.003.
47. Richardson SE, Karmali MA, Becker LE, Smith CR. 1988. The histopa-thology of the hemolytic uremic syndrome associated with verocytotoxin-producing Escherichia coli infections. Hum. Pathol. 19:1102–1108. http://dx.doi.org/10.1016/S0046-8177(88)80093-5.
48. Boyd B, Lingwood C. 1989. Verotoxin receptor glycolipid in human renaltissue. Nephron 51:207–210. http://dx.doi.org/10.1159/000185286.
49. Weeratna RD, Doyle MP. 1991. Detection and production of verotoxin 1of Escherichia coli O157:H7 in food. Appl. Environ. Microbiol. 57:2951–2955.
50. Rasooly R, Do PM. 2010. Shiga toxin Stx2 is heat-stable and not inacti-vated by pasteurization. Int. J. Food Microbiol. 136:290 –294. http://dx.doi.org/10.1016/j.ijfoodmicro.2009.10.005.
51. Harris SM, Yue WF, Olsen SA, Hu J, Means WJ, McCormick RJ, Du M,Zhu MJ. 2012. Salt at concentrations relevant to meat processing en-hances Shiga toxin 2 production in Escherichia coli O157:H7. Int. J. FoodMicrobiol. 159:186 –192. http://dx.doi.org/10.1016/j.ijfoodmicro.2012.09.007.
52. Mohawk KL, Melton-Celsa AR, Robinson CM, O’Brien AD. 2010.Neutralizing antibodies to Shiga toxin type 2 (Stx2) reduce colonization ofmice by Stx2-expressing Escherichia coli O157:H7. Vaccine 28:4777–4785. http://dx.doi.org/10.1016/j.vaccine.2010.04.099.
53. Robinson CM, Sinclair JF, Smith MJ, O’Brien AD. 2006. Shiga toxin ofenterohemorrhagic Escherichia coli type O157:H7 promotes intestinalcolonization. Proc. Natl. Acad. Sci. U. S. A. 103:9667–9672. http://dx.doi.org/10.1073/pnas.0602359103.
54. Lingwood CA. 1993. Verotoxins and their glycolipid receptors. Adv. LipidRes. 25:189 –211.
55. Lingwood CA. 1996. Role of verotoxin receptors in pathogenesis. TrendsMicrobiol. 4:147–153. http://dx.doi.org/10.1016/0966-842X(96)10017-2.
Intragastric Intoxication by Stx2a
March 2014 Volume 82 Number 3 iai.asm.org 1221
on June 22, 2018 by guesthttp://iai.asm
.org/D
ownloaded from