oracle overview of european union pharmacovigilance directive
TRANSCRIPT

An Oracle White Paper
May 2012
An Overview of the Periodic Safety Update Report for Marketed Drugs
E2C (R2)

Overview GVP Module VII: PSURs
Executive Overview ........................................................................... 3
History of the PSUR .......................................................................... 3
Summary of GVP Module VII ......................................................... 4
Regulatory Action to Safeguard Public Health ............................... 5
Overall Content ............................................................................. 6
Preparation of PSURs ................................................................... 7
Sections ........................................................................................ 7
Reference Information ................................................................. 11
Quality System ............................................................................ 12
Transition and interim arrangements ........................................... 13
ICH E2C(R2) PBRER Guideline .................................................. 14
Changes to Summary Tabulations ............................................... 17
Conclusion ...................................................................................... 21
Overview GVP Module VII: PSURs
3
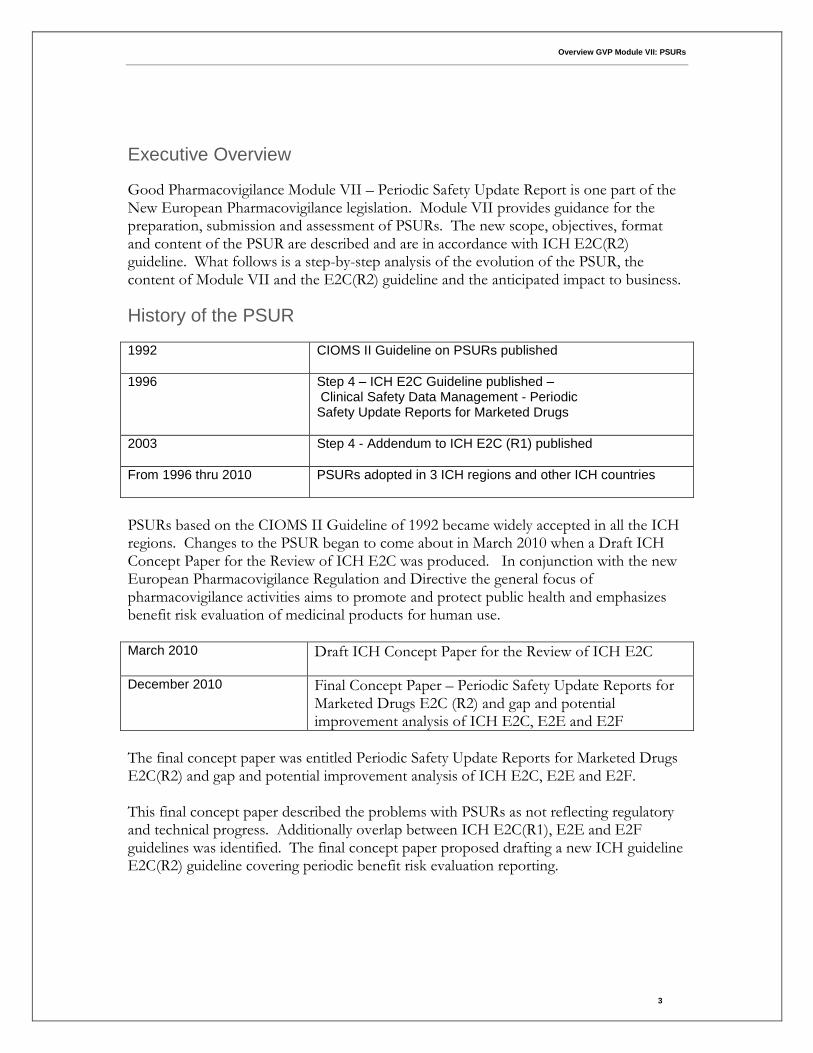
Executive Overview
Good Pharmacovigilance Module VII – Periodic Safety Update Report is one part of the New European Pharmacovigilance legislation. Module VII provides guidance for the preparation, submission and assessment of PSURs. The new scope, objectives, format and content of the PSUR are described and are in accordance with ICH E2C(R2) guideline. What follows is a step-by-step analysis of the evolution of the PSUR, the content of Module VII and the E2C(R2) guideline and the anticipated impact to business.
History of the PSUR
1992 CIOMS II Guideline on PSURs published
1996 Step 4 – ICH E2C Guideline published – Clinical Safety Data Management - Periodic Safety Update Reports for Marketed Drugs
2003 Step 4 - Addendum to ICH E2C (R1) published
From 1996 thru 2010 PSURs adopted in 3 ICH regions and other ICH countries
PSURs based on the CIOMS II Guideline of 1992 became widely accepted in all the ICH regions. Changes to the PSUR began to come about in March 2010 when a Draft ICH Concept Paper for the Review of ICH E2C was produced. In conjunction with the new European Pharmacovigilance Regulation and Directive the general focus of pharmacovigilance activities aims to promote and protect public health and emphasizes benefit risk evaluation of medicinal products for human use. March 2010 Draft ICH Concept Paper for the Review of ICH E2C
December 2010 Final Concept Paper – Periodic Safety Update Reports for Marketed Drugs E2C (R2) and gap and potential improvement analysis of ICH E2C, E2E and E2F
The final concept paper was entitled Periodic Safety Update Reports for Marketed Drugs E2C(R2) and gap and potential improvement analysis of ICH E2C, E2E and E2F. This final concept paper described the problems with PSURs as not reflecting regulatory and technical progress. Additionally overlap between ICH E2C(R1), E2E and E2F guidelines was identified. The final concept paper proposed drafting a new ICH guideline E2C(R2) guideline covering periodic benefit risk evaluation reporting.
Overview GVP Module VII: PSURs
4
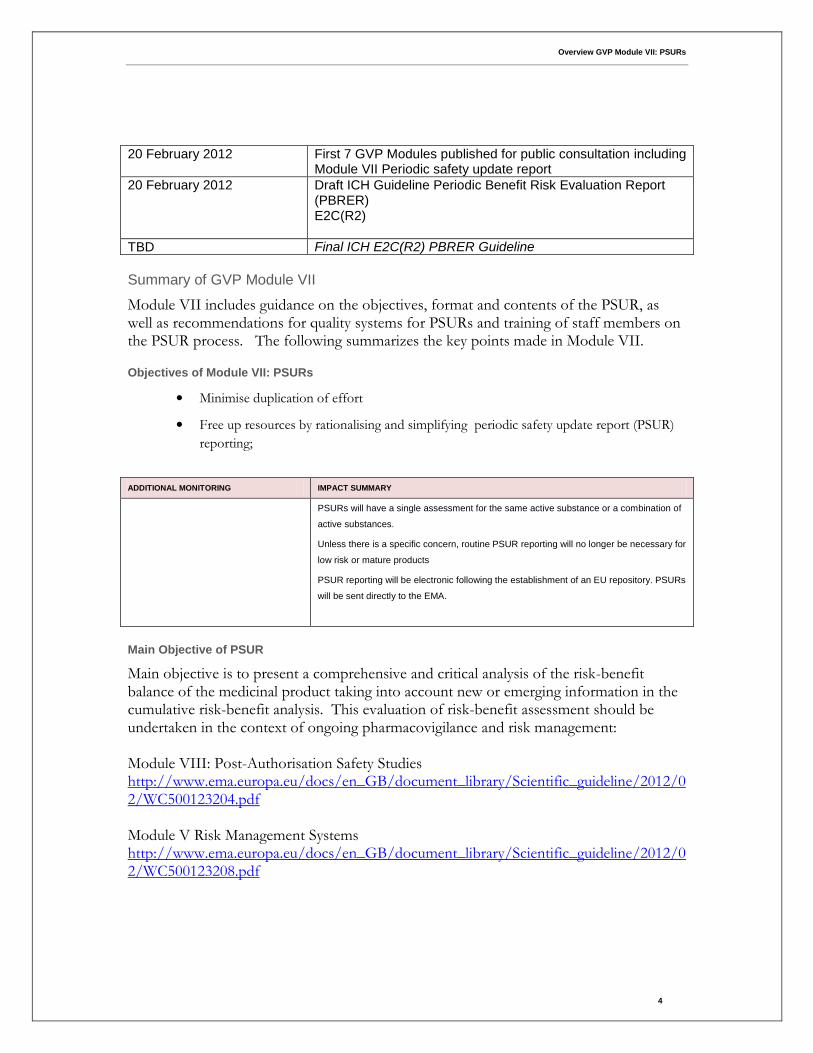
20 February 2012 First 7 GVP Modules published for public consultation including
Module VII Periodic safety update report
20 February 2012 Draft ICH Guideline Periodic Benefit Risk Evaluation Report (PBRER) E2C(R2)
TBD Final ICH E2C(R2) PBRER Guideline
Summary of GVP Module VII
Module VII includes guidance on the objectives, format and contents of the PSUR, as well as recommendations for quality systems for PSURs and training of staff members on the PSUR process. The following summarizes the key points made in Module VII.
Objectives of Module VII: PSURs
• Minimise duplication of effort
• Free up resources by rationalising and simplifying periodic safety update report (PSUR)
reporting;
ADDITIONAL MONITORING IMPACT SUMMARY
PSURs will have a single assessment for the same active substance or a combination of
active substances.
Unless there is a specific concern, routine PSUR reporting will no longer be necessary for
low risk or mature products
PSUR reporting will be electronic following the establishment of an EU repository. PSURs
will be sent directly to the EMA.
Main Objective of PSUR
Main objective is to present a comprehensive and critical analysis of the risk-benefit balance of the medicinal product taking into account new or emerging information in the cumulative risk-benefit analysis. This evaluation of risk-benefit assessment should be undertaken in the context of ongoing pharmacovigilance and risk management: Module VIII: Post-Authorisation Safety Studies http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/02/WC500123204.pdf Module V Risk Management Systems http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/02/WC500123208.pdf

Overview GVP Module VII: PSURs
5
PSUR should not be used to provide the initial notification of significant new safety information or new efficacy data or provide the means for the detection of new safety issues.
Regulatory Action to Safeguard Public Health
EMA IMPLEMENTATION MEASURES
IMPLEMENTATION DATE
Scientific committees and decision-making:
First meeting of Pharmacovigilance and Risk
Assessment Committee (PRAC)
July 2012
Revised mandate of Coordination Group for
Mutual Recognition and Decentralised
Procedures - Human (CMDh)
From September 2012
Strengthening referral procedures:
Urgent Union procedure During 2012
Implementation activities for PSURs
Public consultation on good pharmacovigilance practice (GVP) module
New procedure for centrally authorised medicines
Publication of list of Union reference dates
Focus on nationally authorised medicines
February 2012
During 2012
May 2012
After 2012
International Birth Date and Data Lock Point
The date of the first marketing approval for the medicinal product in any country in the world is the International Birth Date (IBD). Data lock point is the date designated as the cut-off for data to be included, based on IBD.
Reporting timelines
MAH should submit PSURs to EMA:
o Within 70 calendar days of the data lock point for PSURs covering intervals up to
12 months
o Within 90 calendar days of the data lock point for PSURs covering intervals in
excess of 12 months.

Overview GVP Module VII: PSURs
6
With new legal requirement for electronic submission of suspected adverse reactions to the EudraVigilance data, detailed listings of individual cases should not be included in PSURs.
Overall Content
PSURs should be linked to the Risk Management Plans (RMP) of a medicinal product and the new modular format aims to minimise duplication and improve efficiency of preparation of PSUR, development safety update reports (DSUR) and the safety specification in the RMP. PSURs should provide greater emphasis on analysis of case reports; to include:
o scientific evaluation of the benefit-risk profile
o summaries of relevant scientific/clinical data
o available sales/prescription data
Frequency of submission is specified in the Marketing Authorisation. Routine PSURs will not be required for products of ‘low risk’ or where reporting would be duplicative:
o Generics
o Well established medicinal products
o Traditional herbal medicinal products
Process of Assessment in EU Network
Competent authorities in the Member States will assess PSURs to determine whether there are new or changed risks to changes to the risk-benefit balance of the medicinal product. For medicinal products authorised in more than one Member State (Centrally authorised products, mutual recognition and decentralized procedures) a EU single assessment of all PSURs is conducted with recommendation from PRAC.
Single Assessment
A single assessment of PSURs will be performed in the European Union for different medicinal product containing the same active substance or the same combination of active substances authorised in more than one Member state. The EU single assessment can include joint assessment for medicinal products authorised through either national or centralized procedures for marketing authorisation. EMA will make PSURs available to the competent authorities in Member States, members of the Pharmacovigilance Risk Assessment Committee (PRAC), of the Committee for Medicinal Products for Human Use (CHMP) and of the Coordination Group for Mutual Recognition and Decentralised Procedures – Human (CMDh) and the European Commission by means of a PSUR repository.

Overview GVP Module VII: PSURs
7
EMA has published the list of Union reference dates and frequency of submission information which will be legally binding when Module VII becomes effective 2 July 2012.
http://www.ema.europa.eu/docs/en_GB/document_library/Other/2012/04/WC500124999.xls
Preparation of PSURs
Prepare a single PSUR for all medicinal products containing the same active substances with information on all indications, routes, forms and dosing whether authorised under different names and/or procedures. Follow the modular structure set out in the Regulation and Directive and the ICH-E2C(R2) Guideline on Periodic Benefit Risk Evaluation Report (PBRER) should also be applied.
Sections
Title Page
The title page should include the PSUR number (reports should be numbered sequentially), the name of the medicinal product(s), international birth date, reporting interval , date of the report, marketing authorisation holder details and statement of confidentiality of the information included in the PSUR. The title page shall also contain the signature.
Executive Summary
An executive summary should be placed immediately after the title page and before the table of contents. The purpose of the executive summary is to provide a concise summary of the content and the most important information in the PSUR and should contain the following information:
o introduction, report number and reporting interval;
o medicinal product(s), therapeutic class(es), mechanism(s) of action, indication(s),
pharmaceutical formulation(s), dose(s) and route(s) of administration;
o estimated cumulative clinical trials exposure;
o estimated interval and cumulative post-authorisation exposure;
o number of countries in which the medicinal product is authorised;
o summary of the overall benefit-risk analysis evaluation (based on sub-section 18.2
“benefit-risk analysis evaluation” of the PSUR);
o actions taken and proposed for safety reasons including significant changes to the
investigator brochure and post-authorisation product information or other risk
minimisation activities;

Overview GVP Module VII: PSURs
8
o conclusions
Table of Contents
1. Introduction –introduce product so PSUR stands alone and also put in
perspective of previous PSURs include:
IBD, reporting interval and sequential number of report
medicinal product(s), therapeutic class(es), mechanism(s) of action,
authorised indication(s), pharmaceutical form(s), dose(s) and route(s)
of administration;
brief description of population(s) treated and studied;
brief description and explanation of any information that has not
been included in the PSUR.
2. Worldwide Marketing Approval Status – provide cumulative information and
brief narrative including date of first authorisation worldwide, indications,
authorised dose(s), and where authorised if applicable.
3. Actions Taken in the Reporting Interval for Safety Reasons – include
description of significant actions related to safety that have been taken
during the reporting interval, related to either investigation or marketing
experience. Module VII includes many details and examples.
4. Changes to Reference Safety Information – List any significant changes made
to the reference safety information within the reporting interval. Also provide
information on any final and ongoing changes to the national/local
authorized product information.
5. Estimated Exposure and Use Patterns – provide accurate estimation of
population exposed including all data on volume of sales and prescriptions.
Module VII includes many details
5.1 Cumulative Subject Exposure in Clinical Trials - This section should
include the following information on patients studied in clinical trials
presented in tabular format:
Cumulative numbers of subjects from ongoing and completed
clinical trials exposed to the investigational medicinal product, placebo, and/or active comparator(s) since the DIBD. It is recognized that for older products, detailed data might not be available;
More detailed cumulative subject exposure in clinical trials should be presented if available (e.g. sub-grouped by age, sex, and racial group for the entire development program);

Overview GVP Module VII: PSURs
9
• Important differences among trials in dose, routes of administration, or patient populations can be noted in the tables, if applicable, or separate tables can be considered;
• If clinical trials have been or are being performed in special populations (e.g. pregnant women; patients with renal, hepatic, or cardiac impairment; or patients with relevant genetic polymorphisms), exposure data should be provided, as appropriate;
• When there are substantial differences in time of exposure between subjects randomized to the investigational medicinal
product or comparator(s), or disparities in length of exposure between clinical trials, it can be useful to express exposure in subject-time (subject-days, months or years.)
• Investigational drug exposure in healthy volunteers might be less relevant to the overall safety profile, depending on the type of adverse reaction, particularly when subjects are exposed to a single
dose. Such data can be presented separately with an explanation as appropriate. • If the serious adverse events from clinical trials are presented by indication in the summary tabulations, the patient exposure should also be presented by indication, where available.
• For individual trials of particular importance, demographic characteristics should be provided separately.
5.2 Cumulative and Interval Patient Exposure from Marketing Experience
– Separate estimations should be provided for cumulative exposure
(since the IBD) and interval exposure (since the data lock point of the
previous PSUR).
6. Data in Summary Tabulations
6.1 Reference Information – Specify version of MedDRA used for analysis
and Preferred Term (PT) level and system organ class (SOC) should be
presented in the summary tabulations.
6.2 Cumulative Summary Tabulations of Serious Adverse Events from
Clinical Trials – Summary tabulation of serious adverse events
reported in clinical trials from IBD to data lock point. Tabulation
should be organized by MedDRA SOC for investigational drug.
Comparator arm(s), placebo. Presented by trials, indication, route or
other variables.
6.3 Cumulative and Interval Summary Tabulations from Post-marketing
Data Sources – Provide cumulative and interval summary tabulations
of adverse reactions, from IBD to data lock. Serious and non-serious

Overview GVP Module VII: PSURs
10
reactions presented in a single table with interval and cumulative
data presented side-by-side.
7. Summaries of Significant Findings from Clinical Trials in the Reporting Interval
– include listing of trials with data categorized by sex and age, indication,
dose, region.
7.1 Completed Clinical Trials – brief summary of efficacy and safety
findings from clinical trials completed. Present as narrative or
synopsis.
7.2 Ongoing Clinical Trials – brief summary of clinically important
information arising from ongoing clinical trials or information which
supports or refutes previous safety concerns as well as new safety
signals
7.3 Long-term Follow-up – information from long-term follow-up of
subjects.
7.4 Other Therapeutic Use of Medicinal Product – clinically important
safety information from other programs (expanded access,
compassionate use, etc.)
7.5 New Safety Data Related to Fixed Combination Therapies –
summarize important safety information arising from fixed
combination product.
8. Findings from Non-interventional Studies – summarize safety information
from non-interventional studies.
9. Information from Other Clinical Trials and Sources – summarize information
from other clinical trial/study sources (pool analysis or meta-analysis of
randomized clinical trials, etc.)
10. Non-clinical Data – summarize information from non clinical in vivo and in
vitro studies
11. Literature – summary of new and significant safety findings from wide
literature search including studies reporting safety outcomes.
12. Other Periodic Reports – applies for fixed combination products, multiple
indications or formulations were multiple PSURs are prepared. Summarize
findings from other PSURs.
13. Lack of Efficacy in Controlled Clinical Trials – summarize clinical trials
demonstrating lack of efficacy.
14. Late-Breaking Information – summarize findings which arise after data lock
but during preparation of PSUR.
15. Overview of Signals: New, Ongoing or Closed – provide overview of signals
detected, under review and evaluated during reporting interval.

Overview GVP Module VII: PSURs
11
16. Signal and Risk Evaluation
16.1 Summaries of Safety Concerns – baseline summary of safety concerns
16.2 Signal Evaluation – summary of results of evaluation of safety signals
closed during reporting interval
16.3 Evaluation of Risks and New Information – critical appraisal of new
information on new or previously detected risks
16.4 Characterisation of Risks – describe important identified risks
16.5 Effectiveness of Risk Minimisation (if applicable) – summarize results
of risk minimization activities.
17. Benefit Evaluation
17.1 Important Baseline Efficacy and Effectiveness Information –
summarize baseline efficacy and effectiveness information.
17.2 Newly Identified information on Efficacy and Effectiveness –
summarize additional information on efficacy and effectiveness
available during the reporting interval.
17.3 Characterisation of Benefits – integration of baseline benefits and
new benefit information available during the reporting interval.
18. Integrated Benefit-risk Analysis for Authorised Indications
18.1 Benefit-risk Context–Medical Need and Important Alternatives –brief
description of medical need for medicinal product
18.2 Benefit-risk Analysis Evaluation – benefit risk profile specific to
indication and population.
19. Conclusions –implications of any new information based on evaluation of
cumulative safety data and befit-risk analysis, assess need for changes to
CCDS/CCSI. Include preliminary proposal to optimize or further evaluation
benefit-risk balance which should be incorporated into RMP.
20. Appendices to the PSUR – include appropriate appendices to comply with
national or regional requirements.
Reference Information
The latest Core Company Data Sheet (CCDS) in effect at the end of the reporting interval should be used as reference for both the benefit and risk sections. Any changes to CCDS needed during the reporting interval should be described in section 4 “Changes to the reference safety information” and/or section 16 “Signal and risk evaluation”. The CCDS should be dated, version controlled and note the version of the coding dictionary used.

Overview GVP Module VII: PSURs
12
Quality System
Marketing Authorisation Holders must check the list of EU reference dates and frequency of submission to ensure compliance. MAH must have systems in place to produce PSURs according to:
• List of EU reference dates and frequency of PSURs
• Conditions in the marketing authorisation
• Standard PSUR submission schedule established for products authorised before
July 2012
• Ad hoc requests for PSURs by regulatory authority

Overview GVP Module VII: PSURs
13
The Qualified Person for Pharmacovigilance (QPPV) is responsible for the establishment and maintenance of the pharmacovigilance system and with the requirements for the production and submission of PSURs. Additional responsibilities for the QPPV in relation to PSURs include:
• Ensuring quality, correctness and completeness of data submitted in PSURs
• Ensuring full response according to timelines (next PSUR)
• Awareness of PSUR and assessment report conclusions, PRAC recommendations,
CMDh positions and EC decisions to ensure appropriate actions takes place.
EMA’s quality system should support compliance by EMA for fulfilling its tasks and responsibilities for the management of PSUR procedures and EU single assessments. Each competent authority in the Member States should have a pharmacovigilance system for surveillance of medicinal products and for receipt and evaluation of all pharmacovigilance data including PSURs. National competent authorities should implement a quality system for operating tasks relating to PSURs and for monitoring MAH for compliance.
Transition and interim arrangements
The EMA with the competent authorities in Member States and the European Commission will set up and maintain a repository for PSURs and the corresponding assessment reports so they are fully and permanently accessible to EU, competent authorities, PRAC, CHMP and CMDh. Until the repository is established PSURs must be submitted to all competent authorities in Members States where the medicinal products are authorised. PSURs should also be sent to the EMA. From 12 months after the functionalities of the repository have been established, MAH will submit PSURs electronically. Once the ePSUR format and content per ICH-E2C(R2) becomes available MAH will submit PSURs and related documents automatically via an electronic gateway. Until the repository is in place, the following documents should be circulated through a dedicated mailbox:
• Preliminary assessment report created by Rapporteur
• Comments submitted by the marketing authorisation holders and members of
• Updated Rapporteur’s assessment report
• CHMP opinion with annexes and appendices to the European Commission, MAH
and competent authorities in Member States.

Overview GVP Module VII: PSURs
14
ICH E2C(R2) PBRER Guideline
The new ICH E2C(R2) guideline encourages a critical analysis of new and emerging safety/benefit-risk information such that PBRER reports support safety evaluation of all relevant available information. http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E2C/E2C_R2__Step2.pdf
ICH published the new Step 2 ICH-E2C (R2) Periodic Benefit-Risk Evaluation Report (PBRER) guideline for public consultant in February 2012. Step 3 Consultation Deadline for Comments EMA 21 May 2012 FDA 11 May 2012 MHLW 1 May 2012 The final approved Step 4 version of the ICH-E2C (R2) PBRER guideline is anticipated by the end of 2012.
The emphasis for the PBRER is on thoughtful analysis of the benefit/risk profile of the medicinal products for human use. The objective is to optimise the evaluation of benefits and risks and consequently to enable the optimisation of risk minimization and of benefit maximization through product labeling and other risk minimization and health promotion activities. Summary Bridging Reports and Addendum Reports, introduced in ICH guideline E2C(R1), will no longer be accepted. The line listings will no longer be required. A new modular format is proposed to facilitate synchronization between the PBRER and the DSUR whenever a drug undergoes further clinical studies after obtaining marketing authorisation. . Each report should include interval data for the period covered, as well as cumulative data presented side-by-side for ease of analysis. The PBRER will emphasize cumulative information as well as a focus on new information. The level of detail within the PBRER will vary and depend on the extent of the information which is known or emerging during the reporting interval. Ad hoc PBRERs may be required by some authorities when new risks emerge or when risks have changed, when efficacy or effectiveness has change or when there are changes to the benefit-risk profile. Ad hoc PBRERs will most likely focus on specific concerns and the authority will specify the duration of interval data needed.
Overview GVP Module VII: PSURs
15
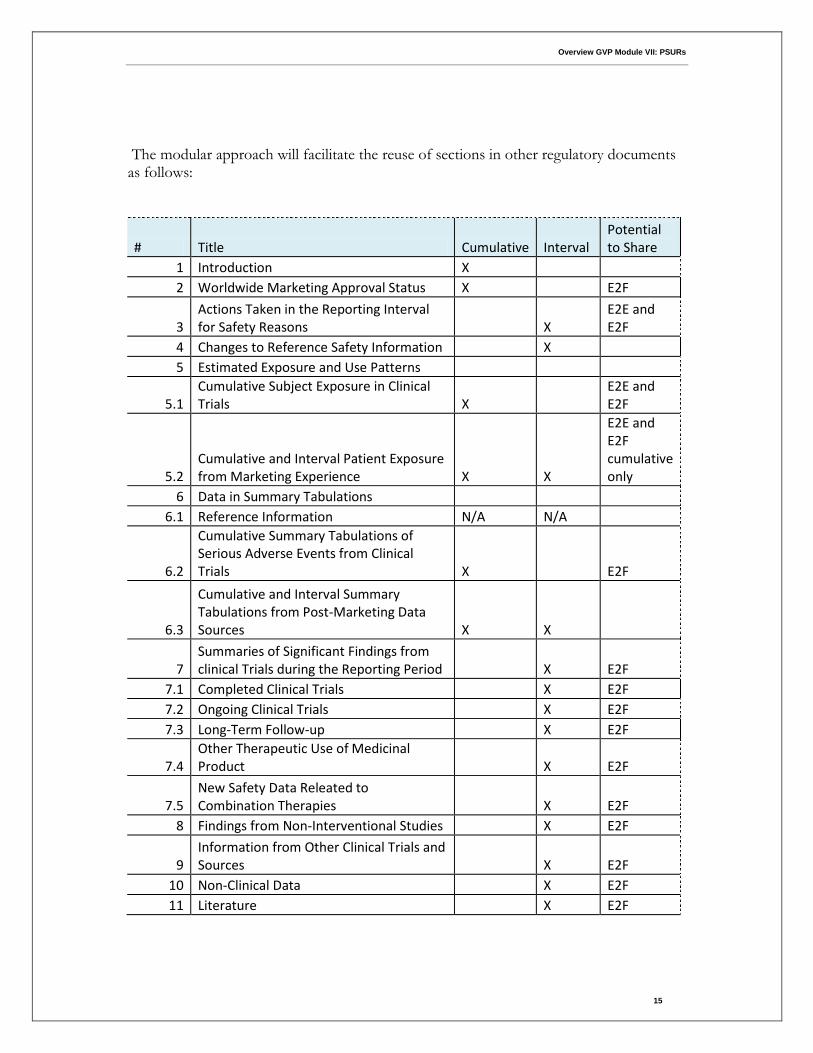
The modular approach will facilitate the reuse of sections in other regulatory documents as follows:
# Title Cumulative Interval Potential to Share
1 Introduction X 2 Worldwide Marketing Approval Status X
E2F
3 Actions Taken in the Reporting Interval for Safety Reasons
X
E2E and E2F
4 Changes to Reference Safety Information
X 5 Estimated Exposure and Use Patterns
5.1 Cumulative Subject Exposure in Clinical Trials X
E2E and E2F
5.2 Cumulative and Interval Patient Exposure from Marketing Experience X X
E2E and E2F cumulative only
6 Data in Summary Tabulations 6.1 Reference Information N/A N/A
6.2
Cumulative Summary Tabulations of Serious Adverse Events from Clinical Trials X
E2F
6.3
Cumulative and Interval Summary Tabulations from Post-Marketing Data Sources X X
7 Summaries of Significant Findings from clinical Trials during the Reporting Period
X E2F
7.1 Completed Clinical Trials
X E2F
7.2 Ongoing Clinical Trials
X E2F
7.3 Long-Term Follow-up
X E2F
7.4 Other Therapeutic Use of Medicinal Product
X E2F
7.5 New Safety Data Releated to Combination Therapies
X E2F
8 Findings from Non-Interventional Studies
X E2F
9 Information from Other Clinical Trials and Sources
X E2F
10 Non-Clinical Data
X E2F
11 Literature
X E2F
Overview GVP Module VII: PSURs
16
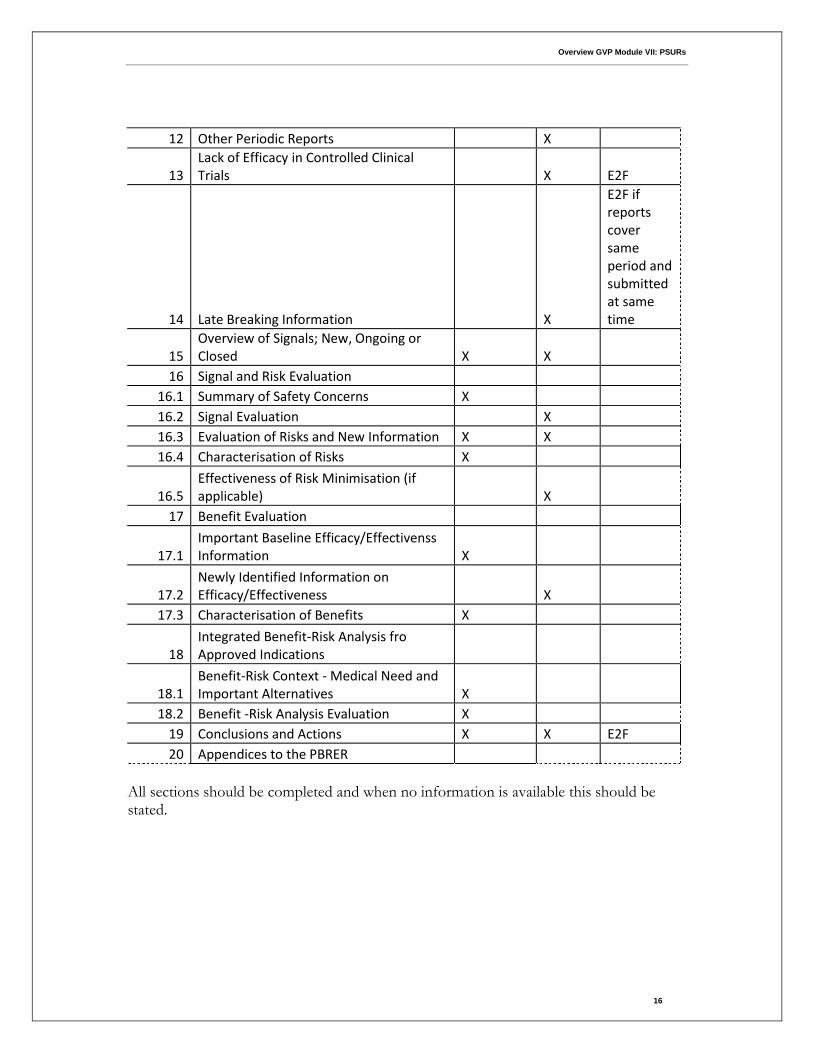
12 Other Periodic Reports
X
13 Lack of Efficacy in Controlled Clinical Trials
X E2F
14 Late Breaking Information
X
E2F if reports cover same period and submitted at same time
15 Overview of Signals; New, Ongoing or Closed X X
16 Signal and Risk Evaluation 16.1 Summary of Safety Concerns X
16.2 Signal Evaluation
X 16.3 Evaluation of Risks and New Information X X 16.4 Characterisation of Risks X
16.5 Effectiveness of Risk Minimisation (if applicable)
X
17 Benefit Evaluation
17.1 Important Baseline Efficacy/Effectivenss Information X
17.2 Newly Identified Information on Efficacy/Effectiveness
X
17.3 Characterisation of Benefits X
18 Integrated Benefit-Risk Analysis fro Approved Indications
18.1 Benefit-Risk Context - Medical Need and Important Alternatives X
18.2 Benefit -Risk Analysis Evaluation X 19 Conclusions and Actions X X E2F
20 Appendices to the PBRER
All sections should be completed and when no information is available this should be stated.

Overview GVP Module VII: PSURs
17
Changes to Summary Tabulations
Summary tabulations will include cumulative and interval data side-by-side. Appendix B of the PBRER guideline includes seven (7) examples of the Summary Tabulations in table format.
TABLE 1 ESTIMATED CUMULATIVE SUBJECT EXPOSURE FROM CLINICAL TRIALS
Includes cumulative SAEs from completed clinical trials and enrollment/randomization for ongoing trials
Treatment Number of Subjects
Medicinal product
Comparator
Placebo
TABLE 2 CUMULATIVE SUBJECT EXPOSURE TO INVESTIGATIONAL DRUG FROM COMPLETED CLINICAL TRIALS BY AGE AND
SEX
Includes data from completed trials as of (date)
Age Range Number of Subjects Total
Male Female
TABLE 3 CUMULATIVE SUBJECT EXPOSURE TO INVESTIGATIONAL DRUG FROM COMPLETED CLINICAL TRIALS BY RACIAL
GROUP
Includes data from completed trials as of (date)
Racial Group Number of subjects
Asian
Black
Caucasian
Other
Unknown
Total
Overview GVP Module VII: PSURs
18
TABLE 4 CUMULATIVE EXPOSURE FROM MARKETING EXPERIENCE
Includes cumulative data obtained from month/day/year through month/day/year, where available.
Indication Sex Age Dose Formulation Region
Male Female 2-16
>16 to 65
>65
<40 40 Unknown Oral IV EU Japan Mexico US CA Other
Depression
Migraine
TABLE 5 INTERVAL EXPOSURE FROM MARKETING EXPERIENCE
Includes interval data obtained from month/day/year through month/day/year, where available.
Indication Sex Age Dose Formulation Region
Male Female 2-16
>16 to 65
>65
<40 40 Unknown Oral IV EU Japan Mexico US CA Other
Depression
Migraine
Overview GVP Module VII: PSURs
19
TABLE 6 CUMULATIVE TABULATIONS OF SERIOUS ADVERSE EVENTS FROM CLINICAL
TRIALS
System Organ Class Preferred Term
Medicinal Product
Blinded Active Comparator
Placebo
Investigations N N N N
Alanine aminotransferase Increased
N N N N
Aspartate aminotransferase increased
N N N N
Nervous System Disorders
N N N N
Syncope N N N N
Headache N N N N
Overview GVP Module VII: PSURs
20
TABLE 7 NUMBERS OF ADVERSE DRUG REACTIONS BY TERM FROM POST-MARKETING SOURCES
Includes serious and non-serious reactions from all post-marketing sources, includes PASS, consumer reports, cumulative and interval data side-by-side
SOC
Spontaneous, including regulatory authority and literature
Non-interventional post-marketing study Total
Serious Non-serious Serious Non-serious Cumulative, all
Interval Cumulative Interval Cumulative Interval Cumulative Interval Cumulative
SOC 1 MedDRA PT MedDRA PT MedDRA PT SOC 2 MedDRA PT MedDRA Pt

Overview GVP Module VII: PSURs
21
Conclusion
The definition and scope of the Periodic Safety Update Report (PSUR) is changing. The overlap of the content of the ICH Guidelines E2C(R1), E2E and E2F has been analyzed and a new ICH-E2C(R2) guideline has been released for public consultation. The format and content will now emphasis a thoughtful analysis of risk-benefit balance of a medicinal product for submission by the marketing authorisation holder at defined time points during the post-authorisation phase. In the European Union the periodic safety update reports should follow the format of the periodic benefit-risk evaluation report (PBRER) in accordance with the ICH-E2C(R2) guideline. The ICH-E2C(R2) guideline is referenced in the new European Pharmacovigilance legislation which becomes legally binding in July 2012. The final Step 4 version of the ICH-E2C(R2) guideline to incorporate public comments is anticipated by the end of 2012 and it will include examples of potential sources of information to be used in preparation of the PBRER.

Overview EU PV Directive
May 2012
Oracle Corporation
World Headquarters
500 Oracle Parkway
Redwood Shores, CA 94065
U.S.A.
Worldwide Inquiries:
Phone: +1.650.506.7000
Fax: +1.650.506.7200
oracle.com
Copyright © 2012, Oracle and/or its affiliates. All rights reserved. This document is provided for information purposes only and the
contents hereof are subject to change without notice. This document is not warranted to be error-free, nor subject to any other
warranties or conditions, whether expressed orally or implied in law, including implied warranties and conditions of merchantability or
fitness for a particular purpose. We specifically disclaim any liability with respect to this document and no contractual obligations are
formed either directly or indirectly by this document. This document may not be reproduced or transmitted in any form or by any
means, electronic or mechanical, for any purpose, without our prior written permission.
Oracle and Java are registered trademarks of Oracle and/or its affiliates. Other names may be trademarks of their respective owners.
Intel and Intel Xeon are trademarks or registered trademarks of Intel Corporation. All SPARC trademarks are used under license and
are trademarks or registered trademarks of SPARC International, Inc. AMD, Opteron, the AMD logo, and the AMD Opteron logo are
trademarks or registered trademarks of Advanced Micro Devices. UNIX is a registered trademark licensed through X/Open
Company, Ltd. 0112