one pot oxidative cleavage of 1, 2-arylalkenes into...
TRANSCRIPT

One pot oxidative cleavage……… Chapter 1
37
One pot oxidative cleavage of 1,2-arylalkenes into arylketones under microwave in aqueous conditions
1.1 Introduction
The oxidative functionalization and cleavage of alkenes represents a fundamental
transformation [Lee and Chen (1991); Kuhn et al. (2004)] in organic synthesis
[Thottumkara and Vinod (2010)]. Depending upon the substitution pattern around the
double bond i.e. 1,2 or 1,1-disubstituted alkenes, the oxidative cleavage of respective
alkenes can lead to the formation of either aldehydes or ketones respectively (Scheme 1).
Scheme 1
1.2 Significance of oxidative cleavage
Oxidative cleavage has wide spread applications in synthesis of biologically important
compounds besides total synthesis of natural products [Green et al. (2008); Hirashima et al.
(2009)]. The major utility of such cleavage reactions is due to their unique ability to
truncate readily available hydrocarbon feedstocks with simultaneous introduction of
carbonyl functionality.
1.3 Reported methods for oxidative cleavage of alkenes
The oxidative cleavage of alkenes has been conventionally achieved mainly through two
principal approaches i.e. Ozonolysis [Bailey (1978); Larock (1999)] and Lemieux-Johnson
reaction [Shing et al. (1991)].
1.3.1 Conventional approaches
1.3.1.1 Ozonolysis
Ozonolysis has been used extensively in academic, research and industrial domains and it
involves the cleavage of an alkene or alkyne with O3 (ozone) to form organic compounds
R1
O
H
[O]R1
R2
R2
O
H
+
R1 R2
[O]
R1
O
R2
1,2-diarylalkene
1,1-diarylalkene
W here R 1 & R 2 = H, alkyl and Ar etc.
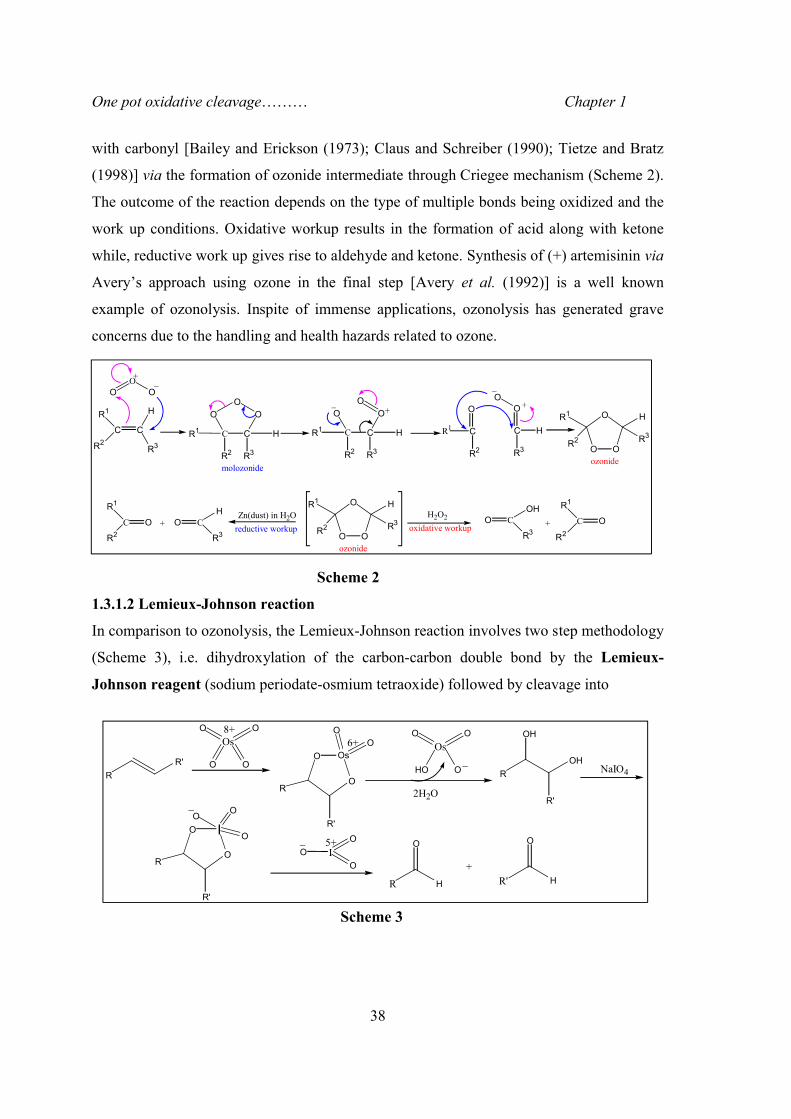
One pot oxidative cleavage……… Chapter 1
38
with carbonyl [Bailey and Erickson (1973); Claus and Schreiber (1990); Tietze and Bratz
(1998)] via the formation of ozonide intermediate through Criegee mechanism (Scheme 2).
The outcome of the reaction depends on the type of multiple bonds being oxidized and the
work up conditions. Oxidative workup results in the formation of acid along with ketone
while, reductive work up gives rise to aldehyde and ketone. Synthesis of (+) artemisinin via
Avery’s approach using ozone in the final step [Avery et al. (1992)] is a well known
example of ozonolysis. Inspite of immense applications, ozonolysis has generated grave
concerns due to the handling and health hazards related to ozone.
Scheme 2
1.3.1.2 Lemieux-Johnson reaction
In comparison to ozonolysis, the Lemieux-Johnson reaction involves two step methodology
(Scheme 3), i.e. dihydroxylation of the carbon-carbon double bond by the Lemieux-
Johnson reagent (sodium periodate-osmium tetraoxide) followed by cleavage into
Scheme 3
R'
RO
OsO
O
O
R'
R
R
R'
OH
OHNaIO4
O
IO
R'
R
O
OO
R H
O
R' H
O
2H2O
+
OsOO
OO
8+6+
IO
O
O_
_
OsOO
OHO_
5+
C C
R1
R2C C
R2 R3
HR1
O OO
OOO
H
R3
C C
R2 R3
HR1
O O
O
C
O
R2
C
O
R3
H
O
O O
O H
R3
R1
R2
O O
O H
R3
R1
R2
H2O2 C
R1
R2
OC
R1
R2
O CH
R3
O COH
R3
O
+
_+
R1
+
__
oxidative workupreductive workup
Zn(dust) in H2O+ +
ozonide
ozonide
molozonide

One pot oxidative cleavage……… Chapter 1
39
carbonyl compounds. However the strong oxidizing conditions associated with the above
protocol also enhances the probability of over oxidation into acids.
Lemieux-Johnson reaction has also been used in total synthesis of natural products like
isosteviol (Scheme 4) [Snider et al. (1998)] using sodium periodate-osmium tetraoxide.
Scheme 4
In spite of their immense utility, the ozonolysis and Lemieux-Johnson reaction are
considerable challenges in view of safety and over oxidation concerns. In particular, ozone
gas is highly toxic and its generation requires specialized equipment. Consequently, the
development of safer alternatives as well as conceptually newer olefinic cleavage
approaches has attracted increased attention.
1.3.2 Contemporary approaches for oxidative cleavage of alkenes
Travis et al. explored a mild, organometallic alternative to ozonolysis using OsO4 with
oxone as the co-oxidant in DMF, for oxidative cleavage of olefins to provide carboxylic
acids. Plausible mechanism involves the intermediacy of an osmate ester which undergoes
cleavage to provide the final oxidized product (Scheme 5) [Travis et al. (2002)].
Scheme 5
On the other hand, Yang et al. have successfully cleaved aryl olefins into aromatic
HO
O
OEt
HO
O
OEt
O
O
OH
O
OsO4, NaIO4
Isosteviol
R2R1
OsO4 (0.01 equiv.)
Oxone (4 equiv.)R1COOH + R2COOH
R2R1
OOOs
O OO O OSO3H
R1 and R2 = alkyl , aryl etc.
(DMF, 3h, RT)
intermediate
-

One pot oxidative cleavage……… Chapter 1
40
aldehydes rather than carboxylic acids, in excellent yields by using the system of RuCl3-
Oxone-NaHCO3 in CH3CN-H2O (1.5: 1) (Scheme 6) [Yang and Zhang (2001)].
Scheme 6
The current trend toward “clean and rapid” synthesis has lead to reagent immobilization to
enable easy recovery, reuse and disposal at an acceptable economic cost. In this context,
osmium tetraoxide microencapsulated in a polyurea matrix has been used as recyclable
catalyst in the dihydroxylation and the oxidative cleavage of olefins (Scheme 7) [Ley et al.
(2003)].
Scheme 7
Recently, Nicolaou et al. reported a one-pot combination of hydroxylation followed by the
addition of stoichiometric Ph(IOAc)2 to effect alkene cleavage in the presence of OsO4
(cat.) and 2,6-lutidine to yield the corresponding carbonyl compounds (Scheme 8). The
described synthetic method offers a convenient and economical alternative to the traditional
olefin cleavage methods for laboratory operations [Nicolaou et al. (2010)].
Scheme 8
R2 R3
R1
O
H R1 R2 R3
O
O
H R1 R2 R3
O
+
+
upto 90%
upto 98%
NMO (1.5 equiv), 2,6-lutidine (2.0equiv), acetone:H2O (10:1), then
PhI(OAc)2 (1.5 equiv), 25oC
PhI(OAc)2 (2.3 equiv), 2,6-lutidine(2.5 equiv), THF, 25oC
R1, R2, R3 = H, alkyl, aromatic etc.
R'
R R''
R R'
O2 mol% Os EnCat3eq. NaIO4
THF/H2O (2:1)rt, 1-8 h
Os EnCat = OsO4 microencapsulated in a polyurea matrix
(R, R', R'' = H, CH3, aryl etc.)
PhCHO
3.5 mol % RuCl31.5 eq. oxone, 4.7 eq. NaHCO3
CH3CN/H2O (1.5/1)
rt, pH 7-7.5, 0.5 h 85%Ph
Ph

One pot oxidative cleavage……… Chapter 1
41
Wang et. al. have reported a palladium catalyzed protocol with O2 as the sole oxidant for
efficient oxidation of a wide range of olefins into aldehydes or ketones (Scheme 9) [Wang
and Jiang (2010)].
Scheme 9
In another report, Ho et al. utilized heterogeneous recyclable catalyst comprising ruthenium
nanoparticles grafted onto hydroxyapatite for cis-dihydroxylation and oxidative cleavage of
alkenes (Scheme 10). The utility of protocol was demonstrated both on small and larger
scale [Ho et al. (2004)].
Scheme 10
Similarly, catalytic amounts of environmentally benign organo iodine reagent 4-IB Acid (4-
iodobenzoic acid) in the presence of oxone as a co-oxidant was employed for oxidative
cleavage of different alkenes (Scheme 11) [Thottumkara and Vinod (2010)].
Scheme 11
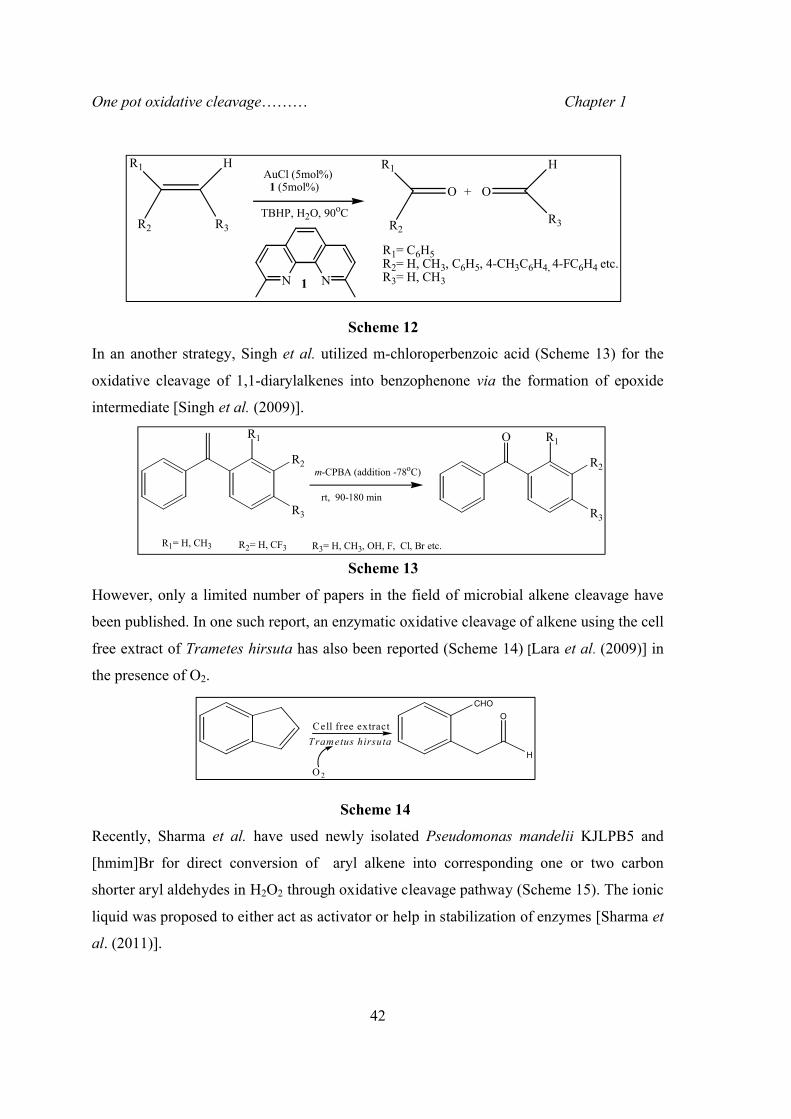
In the same vein, Xing et al. developed a unique gold catalysed homogeneous oxidation
process (Scheme 12) for cleavage of C=C bond to afford ketone or aldehyde products with
tertiary butyl hydrogen peroxide (TBHP) as the oxidant in water [Xing et. al. (2006)].
Pd(OAc)2, O2 (8 atm)
H2O, PTSACHO
24 h , 100oC
I
C OO H
I
C OO H
+OH
KHSO 4
_
R 2
R 3
H
R 1
Oxone
aq. Acetonitrile60oC aq. Acetonitrile
60oC
OxoneO
R 2
R 1
+ O
R 3
OH
R 1, R2, R 3 =H , aryl, alkyl etc.
CHO
nano RuHAP (Ru. cat.)
NaIO4/H2SO4
One pot oxidative cleavage……… Chapter 1
42
Scheme 12
In an another strategy, Singh et al. utilized m-chloroperbenzoic acid (Scheme 13) for the
oxidative cleavage of 1,1-diarylalkenes into benzophenone via the formation of epoxide
intermediate [Singh et al. (2009)].
Scheme 13
However, only a limited number of papers in the field of microbial alkene cleavage have
been published. In one such report, an enzymatic oxidative cleavage of alkene using the cell
free extract of Trametes hirsuta has also been reported (Scheme 14) [Lara et al. (2009)] in
the presence of O2.
Scheme 14
Recently, Sharma et al. have used newly isolated Pseudomonas mandelii KJLPB5 and
[hmim]Br for direct conversion of aryl alkene into corresponding one or two carbon
shorter aryl aldehydes in H2O2 through oxidative cleavage pathway (Scheme 15). The ionic
liquid was proposed to either act as activator or help in stabilization of enzymes [Sharma et
al. (2011)].
CHO
O
H
Cell free extract
Trametus hirsuta
O 2
R1
R2
R3
O R1
R2
R3
m-CPBA (addition -78oC)
rt, 90-180 min
R1= H, CH3 R2= H, CF3 R3= H, CH3, OH, F, Cl, Br etc.
R1
R2
H
R3
O
R1
R2
O
H
R3
+
AuCl (5mol%)1 (5mol%)
TBHP, H2O, 90oC
N N1
R1= C6H5R2= H, CH3, C6H5, 4-CH3C6H4, 4-FC6H4 etc.R3= H, CH3

One pot oxidative cleavage……… Chapter 1
43
Scheme 15
On the other hand, Schiaffo et al. developed a method for ozonolysis of alkenes in a
mixture of water and a miscible organic solvent for a fast, convenient and efficient one-pot
synthesis of aldehydes and ketones without the need for a separate reductive step and
further avoiding the need to isolate or decompose ozonides (Scheme 16) [Schiaffo and
Dussault (2008)].
Scheme 16
Shaikh et al. have explored inexpensive iron (III) chloride hexahydrate (FeCl3·6H2O, 5
mol%) catalyst in combination with commercially available aqueous 70% TBHP as oxidant
for the cleavage of olefins into acids. The method was suitable for a wide range of aromatic
olefins and alkynes that contain electron-donating and electron withdrawing functional
groups (Scheme 17) [Shaikh and Honga (2011)].
Scheme 17
Hart et al. described the oxidative cleavage of olefins with osmium tetraoxide using
hydrogen peroxide (H2O2) as the terminal oxidant. The protocol offered the corresponding
aldehyde and ketone products in moderate to excellent yields (Scheme 18) [Hart et al.
(2011)].
R2R1
O3/O2
5% H2O in acetone
O
R2R1
O
+
HCOOH
H2OO
R2R10oC
R1, R2 = Ph, CH3, H etc.
-
R2
R1 R1
O
HP. mandelii KJLPB5
[hmim]Br , H2O2, 37oC
Where R1= 4-OCH3, 4-Cl, 4-NO2, 2-Cl etc.R2=CH3, H etc.
RR'
NaOH, H2O, 80oC, 10h R
O
OHR R'
FeCl3.6H2O, aqueous 70% TBHP
Acidic work up
FeCl3.6H2O, aqueous 70% TBHP
Acidic work up R= arylR'=H, alkyl, aryl, allyllic
NaOH, H2O, 80oC, 10h

One pot oxidative cleavage……… Chapter 1
44
Scheme 18
Similarly, Yu et al. reported an improved procedure for oxidative cleavage of olefins by
OsO4-NaIO4. The addition of 2,6-lutidine was found to suppress the side reactions and
dramatically improved the yield of the reaction (Scheme 19) [Yu et al. (2004)].
Scheme 19
Yusubov et al. disclosed the oxidative transformations of alkenes and alkynes under the
action of diacetoxyiodobenzene (DIB) and found that non terminal alkenes behave in a
different manner. For instance oxidation of E-stilbene with the DIB-H2SO4 system (Scheme
20) gave a mixture of different products with little amount of benzophenone [Yusubov et
al. (2004)].
Scheme 20
Kim et al. elaborated a new chemoentrapment strategy for recycling osmium in the
catalytic olefin cleavage reaction. This approach utilizes KOH/i-PrOH to generate water-
R2
PhR1
PhCHO PhCOOH+
OsO4 (0.01 equiv.)50% H2O2 (4 equiv.)
DMF (20 or 0 equiv.)CH3CN (0.1M), rt
R1= H, CHO, COOH, CH2OH, COOMeR2= H, CH3
traces
Ph
Ph DIB (diacetoxyiodobenzene)-H2SO4 system
Ph
OH
O
Ph Ph
O
O
Ph
O
PhPh
Ph
O
HPhO
Ph
+
+ +Ph
-20oC, 5h
R
OsO4 (0.02 eq.), NaIO4 (4 eq.)
2,6-lutidine (2eq.), dioxane/water (3:1)RCHO
Where R =
Me
OBn
OPMB
Meor
O
O
OTBSetc.

One pot oxidative cleavage……… Chapter 1
45
soluble Os (VI) species as a recyclable metal catalyst after the oxidative cleavage reaction
(Scheme 21) [Kim et al. (2011)].
Scheme 21
It is evident from foregoing discussion that the above contemporary approaches for
oxidative cleavage have resulted in a significant improvement over classical protocols. It
would also be apparent that the substitution pattern around an olefinic bond largely
determines the nature of cleavage products. Thus, all of the above approaches cleave 1,2-
disubstituted alkenes into aldehydes while ketone cleavage products are usually obtained
only from respective 1,1-disubstituted alkenes (Scheme 1). However, a general approach
for the counterintutive one step formal scission of abundantly available 1,2-arylalkene and
diarylalkenes into one carbon shorter arylketones
instead of arylaldehydes has not yet been
disclosed although in few cases such a
transformation has been noticed as a side reaction
during oxidation of stilbenes [Yusubov et al.
(2004)] (Scheme 20). Moreover the 1,1-
diarylketones formed by the oxidative cleavage of C=C bond of 1,2-arylalkenes constitute
an important core structure of several biologically active molecules e.g. hydroxyphenstatin
(Figure 1), an anticancer benzophenone.
In this context, it would be highly desirable if an approach complementary to classical
ozonolysis & Lemieux-Johnson reaction could be devised leading to selective oxidative
cleavage of aryl- & 1,2-diarylalkenes into one-carbon shorter arylketones. Such a
methodology would provide a useful complementary tool to the prevalent approaches as it
considerably enhances the flexibility for cleaving an olefinic bond into either aldehydes or
ketones irrespective of the substitution pattern around a double bond.
RR'
OsO4
NaIO4
H2O, rt, 14 h
R H
O
R' H
OOsO4
Org.
NaIO4
NaIO3
R H
O
R' H
O
Org.
NaIO3
Aq. Aq.
OsO42-
Reducing agent
R, R' = H, Phenyl, Alkyl etc.
t-BuOH
O OH
OH
OMe
MeO
MeO
OMe
Figure 1. Hydroxyphenstatin

One pot oxidative cleavage……… Chapter 1
46
1.4 Results and Discussion
In the course of our programme towards catalytic synthesis of biologically important
phenolics [Kumar et al. (2008); Sharma et al. (2008), (2009), (2010)], we initially desired
to achieve a one step conversion of abundantly available aryl alkenes such as anethole, β-
asarone etc. into corresponding α-arylpropionic acids, which are constituents of non
steroidal anti-inflammatory drugs (NSAID’s) [Marnett and Blobaum (2007)]. In this
context, we planned to utilize our recently developed approach [Sharma et al. (2009)]
comprising direct oxidation of aryl alkenes into α-aryl aldehydes for an in situ oxidation
[Dvorak (1983); Yan and Zhang (2006)] of such aldehydes into respective α- substituted
arylalkanoic acids. Consequently, PDC (pyridinium dichromate) was chosen as the in situ
oxidizing agent due to its known ability to convert such α-substituted aryl aldehydes into
arylalkanoic acids. However,
contrary to our expectations,
the reaction of 1a (Table 1)
with N-iodosuccinimide (NIS,
1.3 eq), CTAB followed by
treatment with PDC (6 eq) in
same pot under microwave
(MW) provided a product
whose detailed GC-MS and
NMR investigations revealed it
to be one carbon shorter
acetophenone (1b, C6-C2 unit)
instead of the expected α-
arylpropionic acid (C6-C3 unit).
So the direct
oxyfunctionalization and
cleavage of even 1,2-
arylalkenes into ketones
instead of aldehydes prompted
us to further explore its scope
Entry Oxidant Amount ofoxidant (eq.)
Yield [b]
2
5
6
7
1
34
CH3
O
MeO MeOMW ( 115oC, 250W)
Same pot ii) Oxidant, Additive
Oxone
6
1.5
1.21.5
1.5
1.5
8
1.5
1.5
6
6
12
13
64
61
23
62
46
41
41
42
42
1.59
38
_
_
_
_
1a 1b
71
i) NIS, CTAB,Dioxane/Water (3:1)
PDC
PDC
PDC
PDC
PCC
Ag2O
H5IO6
TBAF
K2Cr2O7
TBHP
H2O2
15
18
14
16
17
1.5
1.5
1.5
1.5
1.5
76
78
79 (71)[c]
76
61
PDC
PDC
PDCPDC
PDC
20
19
3530PDC/H2O2
PDC/TBHP
10
11
3
0.05/6
0.05/6
1.5
Additive
CH3COOH
CH3COOHCH3COOH
CF3COOH
NaOH
L-Proline
Pyridine-2,5-dicarboxylic acid
29
traces
_
_
_
_
_
_
_
_
_
CrO3
Table 1. Optimization of conditions for oxidative cleavage of 1,2-aryl-alkenes into aryl ketone under focused microwave irradiation.[a]
[a] CEM monomode microwave. General conditions: 1a (1.35 mmol), NIS(1.75 mmol), CTAB (0.08 mmol) in dioxane (11 ml), water (3.7 ml) wereirradiated under MW (115oC, 250 W) for 15 min followed by cooling andaddition of oxidant, additive (2.0 mmol) and further MW for 15 min.[b] Based on GC-MS analysis. [c] Isolated yield of pure product.
Published in Eur. J. Org. Chem. 2010, 6025-6032.

One pot oxidative cleavage……… Chapter 1
47
and mechanistic pathway.
Consequently, a detailed optimization study (Table 1) was conducted to evaluate the effect
of various oxidizing agents and additives. Interestingly, the use of other well known
oxidizing agents like oxone, TBHP, H2O2 etc. didn’t proved beneficial for oxidative
cleavage of arylalkenes (Table 1, entries 6, 12-13). However, addition of a reduced amount
of PDC (1.5 eq) led to a significantly increased yield of 1b (71%, Table 1, Entry 3).
Amongst the various acidic/basic additives, the use of acetic acid led to further
improvement in reaction performance besides helping in work up of the reaction mixture
(Table 1, Entry 14).
The apparently counterintuitive cleavage of even 1, 2-disubstituted arylalkenes into aryl
ketones instead of aryl aldehydes implied a oxygenation-rearrangement-oxidative cleavage
pathway. Thus, arylalkene (a) is initially converted to corresponding α-arylaldehyde in the
presence of NIS/H2O (Figure 2, a'). Subsequently, the oxidation of this α-arylaldehyde into
one carbon shorter ketone can proceed through a number of intermediates [Perlman et al.
(1994); Clennan and Pan (2003); Bryant et al. (2004); Velosa et al. (2007)] including α-
arylacid [Rozner et al. (2007); Das et al. (2008)] and enol [Heathcock et al. (1985);
Perlman et al. (1994)] etc. Thus, the incipient α-arylaldehyde (a′) can undergo oxidation
into corresponding α-arylacid which is further converted into respective ketone through
oxidative decarboxylation (Figure 2). In order to evaluate the above possibility, the standard
acids i.e. Flurbiprofen (2-(2-fluorobiphenyl-4-yl) propanoic acid) or Ibuprofen (2-(4-(2-
methyl propyl)phenyl)propionic acid) were treated with PDC under identical conditions,
however, the corresponding ketones were not detected. In another alternative pathway, the
incipient α-arylaldehyde undergoes acid catalyzed enolization [Heathcock et al. (1985);
Perlman et al. (1994)] followed by PDC assisted oxidative cleavage into corresponding
RR
O
I
O-H
a a' b
NIS/H2O
O
CO2
O
CHO
COOH
R
R
R
O
RHO
Figure 2. Plausible mechanistic pathways for one pot oxidative cleavage of 1, 2-disubstituted arylalkenesinto one carbon shorter ketones (Published in Eur. J. Org. Chem. 2010, 6025-6032).

One pot oxidative cleavage……… Chapter 1
48
ketone (b) (Figure 2) [Rozner et al. (2007); Das et al. (2008)].
In order to further confirm the above mechanistic rationale, arylalkene (1a) was treated with
PDC/acetic acid alone in dioxane/water (3:1) under MW. Interestingly, the above reaction
afforded only methoxybenzaldehyde, thereby, clearly highlighting the critical role of
cascade pathway (Figure 2) in providing an exclusive access to ketone cleavage products.
Further, the role of MW [Kappe (2004)] was also evaluated by reaction of 1a under thermal
heating at similar temp., however, 1b was obtained in comparatively lower yield (55%)
with longer reaction times (6 h). It is pertinent to mention here that such a cleavage of
incipient α-arylaldehyde into respective one carbon shorter ketones has earlier been
observed using reagents like O2 (in combination with polyoxometalates/zeolite), ruthenium-
oxo complexes, peroxidises and PhI(OAc)2 etc.
The utility of above optimized protocol for cleavage of C=C double bonds was
subsequently ascertained. As would be evident from Table 2, various aromatic and
polyaromatic arylalkenes bearing electron donating/withdrawing groups (EDG’s/EWG’s)
underwent facile oxidative cleavage into corresponding aryl ketones instead of the aryl
aldehydes. Further, the arylalkene with elongated side chain (C6-C5 unit) was also found
Entry Substrate(a)
Product(b)
H3COH3CO
O
i) NIS, CTAB,
MW (115oC, 250W)R
R'R'
O
H3CO
H3CO
O
O
1
H3CO
OCH3
OCH3
H3CO
OCH3
OCH3
H3CO H3CO
O
Yield[b] (%)
1-15a 1-15b
O
OO
H3CO
H3CO
O
3
4
O
5
71
78
51
86
45[c]
2
OCH3 OCH3
Same pot ii) PDC, CH3COOH R = H, OMe, OH, OCH2O, Cl,C6H5, OC4H9 , OC6H13 etc.R' = CH3, CH2CH2CH3 etc.
Dioxane /Water (3:1)
R
Table 2. Oxidative cleavage of 1,2-arylalkenes into arylketones instead of arylaldehydesunder focused microwave irradiation.[a]
H3CO H3CO
O
6
67

One pot oxidative cleavage……… Chapter 1
49
to be compatible (Table 2, Entry 3) with the developed methodology. On the other hand,
the olefin possessing free phenolic group (Table 2, Entry 11) provided lower yield of
respective aryl ketone owing to probable polymerization besides formation of some side
products. Similarly, the relatively unactivated substrates like 1,1-disubstituted (Table 2,
Entry 14) and aliphatic alkenes (Table 2, Entry 15) also led to low or no reaction
performance presumably due to the reduced formation of corresponding α-substituted aryl
aldehydes in the first step as shown in figure 2.
Having successfully realized the oxidative cleavage approach, it was envisaged to further
explore the one pot halogenation-oxidative cleavage sequence. However, the in situ
reaction of 2b (obtained from oxidative cleavage of 2a) with NIS [Yadav et al. (2004)]
C4H9O
OCH3
O
C4H9O
OCH3
HO
OCH3
O
HO
OCH3
11
12
14[c]
76
O
C6H13O
OCH3
C6H13O
OCH3
13
O
O
14
15
48
12[c]
nd[c-d]
O
[a] CEM monomode microwave. General conditions: Substrate (0.9 mmol), NIS (1.18 mmol),CTAB (0.08 mmol) in dioxane (11 ml), water (3.7 ml) were irradiated under MW (115oC, 250W)for 15 min followed by cooling and addition of PDC (1.36 mmol), CH3COOH (1.36 mmol) andfurther MW for 15 min. [b] Yield of pure isolated product (single run). The structure of all productswas confirmed by NMR (1H & 13C) and HRMS analysis. [c] Based on GC-MS. [d] Not detected.
O
O
O
8
9
23
37
Cl10
30[c]
O
Cl
O
7 54
Published in Eur. J. Org. Chem. 2010, 6025-6032.

One pot oxidative cleavage……… Chapter 1
50
didn’t afford the expected 2-iodoacetophenone (2b′) (Scheme 22). Surprisingly, an
alternative approach proved fruitful, wherein, initial treatment of 2a with excess NIS
[Sharma et al. (2009)] followed by oxidative cleavage with PDC provided the expected
iodo derivative 2b′,
thereby, demonst-
rating the success-
ful incorporation of
both oxygen and
iodo groups in a
single operational
step (Scheme 22). Scheme 22. One-pot halogenation-oxidative cleavage of arylalkene
The success with 1,2-arylalkenes (Table 2) motivated us to explore an analogous
oxidative cleavage of 1,2-diaryl alkenes into corresponding diarylketones (benzophenones).
However, the developed protocol didn’t provide expected result as stilbenes (16-18a) were
selectively cleaved into benzaldehydes (Scheme 23).
Scheme 23. Selective oxidative cleavage of symmetrical and unsymmetrical 1,2- diarylalkenes into arylaldehydes
In the view of above result as well as the well known tendency of such diarylalkenes
towards cleavage into arylaldehydes, a reduced amount of PDC (0.8 eq.) was also tried but
to no avail. On the other hand, the use of other oxidizing agents like H2O2, TBHP, K2Cr2O7
etc. did provide desired benzophenone (19b, Table 3) but in low yields. Further, some
unreacted starting 1,2-diaryl alkene was also obtained using above reaction conditions.
Thereafter, detailed studies were conducted for identifying an oxidation system compatible
with such 1,2-diaryl alkenes. Interestingly, a combination of catalytic PDC (0.05 eq.),
i) NIS, CTAB,
MW (115oC, 250W)
Dioxane/ Water (3:1)
16-18c
Same pot ii) PDC, CH3COOH16-18a
CHOR2
R1
R3
R2
i) 16c, 69 % yieldii) 17c, 78 % yieldiii) 18c, 45 % yield + 5-Iodovanillin; (41% yield)
R1i) 16a , R1, R2, R3 = Hii) 17a, R1 = H, R2 = R3 = OMeiii) 18a, R1
, R3 = OMe, R2 = OH
R2
R1
O
R3
16-18b
NIS (1.3 eq.)PDC (1.5 eq.)
(i)
(ii)
NIS (4 eq.)
MeO
MeO
MeO
MeO
CHO
I
MeO
MeO
O
MeO
MeO
O
PDC (2.5 eq.)
I
XNIS (2.0 eq.)
2a
2b
2b' (24% overall yieldfrom two steps)
Same pot
Same pot

One pot oxidative cleavage……… Chapter 1
51
TBHP (6 eq.) along with an increased amount of NIS (2 eq. in place of 1.3 eq., Table 2)
and acetic acid (at the start) was required to overcome the initial sluggish conversion of
such diaryl substituted alkenes into corresponding iodohydrins (Figure 2) which finally
afforded the desired 19b (Table 3, Entry 19) in enhanced 49% yield. Subsequently, above
optimum reaction conditions were also found to be applicable on various unsubstituted,
electron rich and electron deficient 1,2-diaryl alkenes (Table 3, Entries 19-22 & 25-26)
Entry Substrate(a)
Product(b)
i) NIS, CTAB, CH3COOH
MW (115oC, 250W)
H3CO
H3CO
19-26b
21
20
OCH3
Same pot ii) Catal. PDC/TBHP
OCH3
OCH3
49
60
R1
R2
19
O
O
H3CO OCH3
O
H3CO
OCH3
OCH3
63
Yield[b] (%)
O
Dioxane/Water (3:1), MW
R1, R2 = H, OMe, OH , Cl etc.
R2R1
Table 3. Oxidative cleavage of 1,2-diarylalkenes into benzophenones instead ofbenzaldehydes under focused microwave irradiation[a]
O
O
OCH3
22
O
O
OCH3
67
H3CO
OCH3
H3CO
O
2329
OCH3
24
O
OCH3
H3CO
ClO
H3CO Cl
HO
OCH3
OCH3
HO
OCH3
O
OCH3
31
65
15[c]
25
O
26
OCH3
[a] CEM monomode microwave. General conditions: Substrate (0.79 mmol), NIS (1.5 mmol),CH3COOH (1.16 mmol), CTAB (0.08 mmol) in dioxane (11 ml), water (3.7 ml) were irradiatedunder MW (115oC, 250W) for 15 min followed by cooling and addition of PDC (0.04mmol)/TBHP (4.7 mmol), and further MW for 15 min. [b] Yield of pure isolated product (singlerun). The structure of all products was confirmed by NMR (1H & 13C) and HRMS analysis. [c]Based on GC-MS.(Published in Eur. J. Org. Chem. 2010, 6025-6032).

One pot oxidative cleavage……… Chapter 1
52
along with the substrates having aromatic or polyaromatic cores (Table 3, Entry 23 & 24).
It is pertinent to mention that the oxidative cleavage of olefins into benzophenones has
been earlier reported using only 1,1-diaryl substituted C=C double bonds. In this context,
the developed methodology affords the first direct scission of even 1,2-diaryl olefins into
corresponding benzophenones.
Having developed a new approach for oxidative cleavage of diverse arylalkenes, we were
intrigued to evaluate further applications of the developed methodology. In this context, we
were attracted by several reports of total synthesis of natural products, wherein, a two step
sequence of oxidative cleavage followed by condensation [Gwaltney II et al. (1996); Hayes
et al. (2006); Somu and Johnson (2006)] has comprised an important strategy. Consequently,
the realization of a one pot protocol for tandem oxidative cleavage-condensation would be
beneficial as it would eliminate isolation of intermediates [Somu and Johnson (2006)]. As a
proof of concept, 1a was subjected to developed oxidation protocol (Table 2) till formation of
1b (Table 2), and thereafter a base such as NaOH (10 %) and 2,4,5-trimethoxybenzaldehyde
Entry Substrate(a)
Benzaldehyde
i) NIS, CTAB, Dioxane/Water (3:1)
MW (115oC, 250W)
H3CO
29
27
28
O
O
30
Yield[b] (%)
H3CO
OCH3
OCH3
OCH3
31
Product(b)
O
H3CO Cl
O
H3CO Cl
Cl
O
H3CO Cl
OCH3
O
H3CO OCH3
OCH3
OCH3
O
O
O
Cl
R1 = R2 = H, OMe, Cl etc.
O
R1 Same pot iii) Benzaldehyde, NaOH, MeOH
ii) PDC, CH3COOH
27-37b R2R1
60
56
34
52
68
1a
(1a)
(2a)
(4a)
ClCl
CHO
H3CO
CHO
Cl
CHO
CHO
Cl
CHO
Cl
Table 4. One pot oxidative cleavage-condensation of arylalkenes with benzaldehydesunder focused microwave irradiation[a]
1a
(a)

One pot oxidative cleavage……… Chapter 1
53
(1.3 eq) was added to the same pot and stirred for 2 h (Table 4). Gratifyingly, this one pot
approach proved useful to directly obtain the condensation product (27b) from corresponding
arylalkene (1a) in 56% overall yield (Table 4, Entry 27). Later on, the above one pot strategy
was also successfully applied for hitherto unknown route to one pot oxidation-condensation
of arylalkenes (Table 4). Further various condensation products (Table 4, 28-37) along with
some heteroaromatic moieties (Entries 32-37) have also been synthesized.
1.5 Conclusion
In conclusion, a new oxidative cleavage approach has been developed which affords for the
first time a selective scission of 1,2-disubstituted alkenes into one carbon shorter ketones
using NIS and PDC/TBHP as co-oxidants. The methodology considerably enhances the
flexibility for cleaving a 1,2-disubstituted olefinic bond into either ketones or aldehydes.
Moreover, the reaction showed wide substrate scope as diverse 1,2-arylalkenes as well as
O
H3CO
OCH3
N33 14
H
2a
O
O
O
N BrNBr
CHO
[a] CEM monomode microwave. General conditions: Substrate (1.35 mmol), NIS (1.75mmol), CTAB (0.08 mmol), dioxane (11 ml), water (3.7 ml) were irradiated under MW(250W, 115oC) for 15 min followed by cooling and additon of PDC (2.0 mmol). CH3COOH(2.0 mmol) and further MW for 15 min thereafter added 10% NaOH (5-8 ml) andmethanolic solution (2-3 ml) of benzaldehyde (1.48 mmol) and stirred for 2 h.[b] Overall yield of pure isolated product from arylalkene (single run).
35 51
37
36
O
H3COS CHO
S
49
50
O
H3CO
O
O CHO
4a
1a
1a
O
CHO
34 524a
O
O
O
O
HN
CHO
32
O
H3CO
OCH3
48S CHO
S2a
Published in Eur. J. Org. Chem. 2010, 6025-6032.

One pot oxidative cleavage……… Chapter 1
54
1,2-diaryl alkenes bearing electron donating or withdrawing groups underwent facile
cleavage into corresponding arylketones. Significantly, the protocol also paved way for the
synthesis of chalcones and some hetroaromatic chalcones through valuable one pot
oxidative cleavage-condensation reaction which has widespread utility in total synthesis of
natural products.
1.6 Experimental Section
1.6.1 General
β-asarone was obtained from natural Acorus calamus oil following our earlier reported
procedure [Sinha et al. (2002)]. The naphthyl and stilbene derivatives were prepared
through a previously reported Grignard–dehydration [Kumar et al. (2008)] or Heck
approach [Plevyak and Heck (1978)] respectively. The rest of the arylalkenes and NIS (N-
iodosuccinimide) were reagent grade (purchased from Merck and Aldrich). Glacial acetic
acid (99-100 % for synthesis), PDC (pyridinium dichromate) and other oxidants were
purchased from either Merck or Aldrich and used as supplied. The solvents used for
isolation/purification of compounds were obtained from commercial sources (Merck) and
used without further purification. Column chromatography was performed using silica gel
(Merck, 60-120 mesh size). 1H (300 MHz) and 13C (75.4 MHz) NMR spectra were
recorded on a Bruker Avance-300 spectrometer. The following abbreviations were used to
designate chemical shift multiplicities: s = singlet, d = doublet, dd = double doublet, t =
triplet, m = multiplet, q = quartet. HRMS-ESI spectra were determined using micromass Q-
TOF ultima spectrometer. GC-MS analysis was carried out on Shimadzu MS-QP-2010
system equipped with a stationary phase DB-5MS column (Agilent Technologies, U.S.A.).
CEM Discover© focused microwave (2450 MHz, 300W) was used wherever mentioned.
The temperature of reactions in microwave heating experiments was measured by an inbuilt
infrared temperature probe that determined the temperature on the surface of reaction flask.
The sensor is attached in a feedback loop with an on-board microprocessor to control the
temperature rise rate. In the case of conventional heating in oil bath, the temperature of
reaction mixture was monitored by an inner thermometer.

One pot oxidative cleavage……… Chapter 1
55
1.6.2 Optimization of reaction conditions
1.6.2.1 One pot cascade oxidative cleavage of p-methoxyphenylpropene (1a) into one
carbon shorter 4′-methoxyacetophenone using NIS/PDC as an oxidizing agent (Table
1, Entry 1)
The p-methoxyphenylpropene (1a, 1.35 mmol) was treated with NIS (1.75 mmol), CTAB
(0.08 mmol) in dioxane (11ml), water (3.7 ml) was conducted under focused microwave
system (250W, 115oC) for 15 min followed by treatment with PDC (6 eq) in same pot
under microwave (250W, 115oC) for 15 min. The above mixture was cooled, washed with
saturated aq. Na2S2O3 solution (1x10 ml) and extracted with ethyl acetate (3x20 ml). The
combined organic layer was washed with brine (1x10 ml), dried over Na2SO4 and vacuum
evaporated. The obtained residue was subsequently purified by column chromatography on
silica gel (60-120 mesh size) using hexane:ethylacetate (9.3:0.7) to give a product (light
yellow solid, 41% yield) whose detailed GC-MS and NMR investigations revealed it to be
4′-methoxyacetophenone (1b) instead of the expected α-arylpropionic acid.
4′-methoxyacetophenone (1b, Table 1)
Light yellow solid, m.p. 35-39°C (lit. m.p. 36-38°C) [Ohno et al. (2004)], 1H NMR (300
MHz, CDCl3); (ppm) 7.93 (2H, d, J = 7.8 Hz), 6.93 (2H, d, J = 8.2 Hz), 3.84 (3H, s),
2.53 (3H, s); 13C NMR (75.4 MHz, CDCl3); (ppm) 195.6, 162.7, 130.6, 130.3, 113.7, 56.4
and 28.1.
1.6.2.2 One pot cascade oxidative cleavage of p-methoxyphenylpropene (1a) into one
carbon shorter 4′-methoxyacetophenone using varying amounts of PDC (Table 1,
Entries 2-4) as an oxidizing agent
To a stirred mixture of p-methoxyphenylpropene (1a, 0.2 g, 0.9 mmol) and dioxane (11ml),
water (3.7 ml), CTAB (0.03g, 0.08 mmol) and NIS (0.26 g, 1.18 mmol) were added and the
mixture was allowed to stir for 5 min at room temperature. Subsequently, the flask was
irradiated under focused microwave system (250W, 115oC) for 15 min. Thereafter, the
above reaction flask was cooled and varying amounts of PDC (3 or 1.5 or 1.2 equiv.
H3CO
O

One pot oxidative cleavage……… Chapter 1
56
respectively) were added and further irradiated under microwave (250W, 115oC) for 15
min. The above mixture was cooled, washed with saturated aq. Na2S2O3 solution (1x10 ml)
and extracted with ethyl acetate (3x20 ml). The combined organic layer was washed with
brine (1x10 ml), dried over Na2SO4 and vacuum evaporated and obtained residue subjected
to GCMS analysis. The above experiments provided 1b in 64, 71 and 61% yields in the
case of 3, 1.5 and 1.2 eq of PDC respectively. Thus, 1.5 equiv. of PDC was found to be the
appropriate.
1.6.2.3 One pot cascade oxidative cleavage of 1a into 1b using different oxidizing agent
(Table 1, Entries 5-13)
The reactions were performed in the same manner as given in section 1.6.2.2 with PCC,
oxone, Ag2O, H5IO6, TBAF, CrO3 and K2Cr2O7 replacing PDC in equal molar amounts in
each case, while in case of TBHP and H2O2, 6 equivalents of each were utilized. The above
reactions upon completion followed by work up as given in section 1.6.2.2 and followed by
GCMS analysis provided 1b in 42, 38, 42, 29, traces, 23, 62, 46 and 41% yields in the each
case respectively.
Thus, PDC (Table 1, Entry 3) was found to be the optimum oxidizing agent and used in all
the subsequent experiments.
1.6.2.4 One pot cascade oxidative cleavage of 1a to 1b using PDC and different acidic
and basic additives (Table 1, Entries 14-18)
The reactions were performed in the same manner as given in section 1.6.2.2 except that
(2.0 mmol) of CH3COOH, CF3COOH, NaOH, L-Proline or pyridine-2,5-dicarboxylic acid
were added in the second step of each case. The above reactions upon completion followed
by work up and GCMS analysis as given in section 1.6.2.2 provided 1b in 79 (71% after
column purification), 76, 61, 78 and 76% yields in each case respectively. Thus,
CH3COOH was found to be the effective additive and used in all the subsequent
experiments.
1.6.2.5 One pot cascade oxidative cleavage of 1a to 1b using H2O2 and TBHP as co-
oxidant with PDC using CH3COOH as additive (Table 1, Entries 19 & 20)
The reactions were performed in the same manner as given in section 1.6.2.2 except that
PDC (0.05 equiv.) was used along with H2O2 and TBHP (6 equiv. of each) as co-oxidants

One pot oxidative cleavage……… Chapter 1
57
and acetic acid (2.0 mmol) as additive. The above reactions upon completion followed by
work up and GCMS analysis as given in section 1.6.2.2 provided 1b in 30 and 35% yields
respectively.
1.6.2.6 Optimized procedure for one pot cascade oxidative cleavage of 1a to 1b using
PDC as oxidising agent with CH3COOH as additive (Table 2)
To a stirred mixture of p-methoxyphenylpropene (Table 2, Entry 1, 0.2 g, 0.9 mmol) and
dioxane (11ml), water (3.7 ml), CTAB (0.03g, 0.08 mmol) and NIS (0.26 g, 1.18 mmol)
were added and the mixture was allowed to stir for 5 min at room temperature.
Subsequently, the flask was irradiated under focused microwave system (250W, 115oC) for
15 min. Thereafter, the above reaction flask was cooled then acetic acid (0.08 ml, 1.36
mmol), PDC (0.51 g, 1.36 mmol) were added and further irradiated under microwave
(250W, 115oC) for 15 min. The above mixture was cooled, washed with saturated aq.
Na2S2O3 solution (1x10 ml) and extracted with ethyl acetate (3x20 ml). The combined
organic layer was washed with brine (1x10 ml), dried over Na2SO4 and vacuum evaporated.
The obtained residue was subsequently purified by column chromatography on silica gel
(60-120 mesh size) using hexane: ethylacetate (9.3:0.7) to give 1-(4-
methoxyphenyl)ethanone (1b) as light yellow solid in 71% yield, m. p. 35-39°C.
The above procedure was also followed for oxidative cleavage of all the other
arylalkenes (Table 2, 2-14a) into corresponding product (Table 2, 2-14b)
3', 4'- Dimethoxyacetophenone (2b, Table 2)
White solid, m.p. 48-50°C (lit. m.p. 49-51°C) [Jiang and Ragauskas (2007)], 1H NMR (300
MHz, CDCl3); (ppm) 7.56 (1H, d, J = 8.6 Hz), 7.49 (1H, s), 6.87 (1H, d, J = 8.6 Hz),
3.92 (6H, s), 2.54 (3H, s); 13C NMR (75.4 MHz, CDCl3); (ppm) 197.2, 153.8, 149.6,
131.1, 123.7, 110.7, 56.5 and 26.6.
H3CO
H3CO
O

One pot oxidative cleavage……… Chapter 1
58
1-(3, 4-Dimethoxyphenyl)- 1-butanone (3b, Table 2)
White solid, m.p. 65-68°C, 1H NMR (300 MHz, CDCl3); (ppm) 7.60 (2H, t, J = 9.2 Hz),
6.90 (1H, s), 3.94 (6H, s), 2.92 (2H, t, J = 7.1 Hz), 1.80-1.72 (2H, m), 1.03 (3H, t, J = 7.9
Hz); 13C NMR (75.4 MHz, CDCl3); (ppm) 199.4, 153.5, 149.4, 130.8, 123.0, 110.6,
56.4, 40.4, 18.5 and 14.6.
3', 4'- (Dioxymethylene) acetophenone (4b, Table 2)
White solid, m.p. 84-87°C (lit. m.p. 86–89°C) [Giddens et al.(2005)] ; IR (KBr): 1663 cm-1
(C=O); 1H NMR (300 MHz, CDCl3); (ppm) 7.57 (1H, d, J = 8.2 Hz), 7.43 (1H, s), 6.86
(1H, d, J = 8.2 Hz), 6.05 (2H, s), 2.54 (3H, s); 13C NMR (75.4 MHz, CDCl3); (ppm)
197.1, 152.7, 149.1, 133.1, 125.7, 108.9, 108.8, 102.8, and 27.4.
1-(6'-Methoxy-2'-naphthyl)ethanone (6b, Table 2)
White solid, m.p. 63-66°C, 1H NMR (300 MHz, CDCl3); (ppm) 8.37 (1H, s), 8.01 (1H,
d, J = 8.4), 7.84 (1H, d, J = 8.7), 7.76 (1H, d, J = 8.7), 7.21 (1H, d, J = 8.7), 7.14 (1H, s),
3.92 (3H, s), 2.69 (3H, s) ; 13C NMR (75.4 MHz, CDCl3); (ppm) 198.2, 160.2, 137.7,
133.0, 131.5, 130.1, 128.2, 127.5, 125.0, 120.3, 106.5, 55.8 and 26.9. HRMS-ESI: m/z
[M+H]+ for C13H12O2, calculated 201.0894; observed 201.0882.
1-Naphthyl methyl ketone (7b Table 2)
H3CO
O
OCH3
O
H3CO
O
O
O
O

One pot oxidative cleavage……… Chapter 1
59
Light yellow liquid, [Yao et al. (2003)], 1H NMR (300 MHz, CDCl3); (ppm) 8.78 (1H, d,
J = 8.7), 8.00-7.83 (3H, m), 7.63-7.46 (3H, m), 2.74 (3H, s); 13C NMR (75.4 MHz, CDCl3);
(ppm) 202.2, 135.8, 134.4, 133.4, 130.6, 129.1, 128.8, 128.4, 126.8, 126.4, 124.7 and
30.3.
4-Acetylbiphenyl (8b Table 2)
Creamish solid, m.p. 116-119°C (lit. m.p. 117-120°C), [Balasankar et al. (2005)], 1H NMR
(300 MHz, CDCl3); (ppm) 8.26 (2H, t, J = 8.8 Hz), 7.96-7.92 (4H, m), 7.74 (3H, t, J =
8.8 Hz), 2.90 (3H, s) ; 13C NMR (75.4 MHz, CDCl3); (ppm) 197.8, 145.8, 139.9, 135.9,
129.0, 128.3, 127.3 and 14.2.
Acetophenone (9b, Table 2)
Colourless liquid, 1H NMR (300 MHz, CDCl3); (ppm) 8.14 (2H, d, J = 8.0 Hz), 7.74
(1H, dd, J = 6.0, 8.0), 7.64 (2H, dd, J = 7.8, 6.7), 2.75 (3H, s) ; 13C NMR (75.4 MHz,
CDCl3); (ppm) 197.5, 137.1, 132.7, 128.2, 127.9 and 26.1.
4'-Butoxy-3'-methoxyacetophenone (12b, Table 2)
Creamish solid, m.p. 35-37°C; IR (KBr): 1674 cm-1 (C=O), 1H NMR (300 MHz, CDCl3);
(ppm) 7.66 (2H, t, J = 6.5 Hz), 7.08 (1H, s), 4.30 (2H, t, J = 7.1 Hz), 4.07 (3H, s), 2.78
(3H, s), 2.12-2.04 (2H, m), 1.79-1.67 (2H, m), 1.23 (3H, t, J = 6.7Hz); 13C NMR (75.4
MHz, CDCl3); (ppm) 197.0, 153.1, 149.4, 130.4, 123.4, 111.2, 110.6, 68.9, 56.2, 31.1,
26.3, 19.3 and 14.0. HRMS-ESI: m/z [M+H]+ for C13H18O3, calculated 223.1302; observed
O
O
O
OCH 3
O

One pot oxidative cleavage……… Chapter 1
60
223.1315.
4'-Hexoxy-3'-methoxyacetophenone (13b, Table 2 )
Transparent viscous liquid, 1H NMR (300 MHz, CDCl3); (ppm) 7.62 (2H, d, J = 7.6 Hz),
6.98 (1H, t, J = 9.5 Hz), 4.17 (2H, d, J = 6.9 Hz), 4.01 (3H, s), 2.65 (3H, s), 1.94 (2H, s),
1.54-1.35 (6H, m), 0.97 (3H, s); 13C NMR (75.4 MHz, CDCl3); (ppm) 197.5, 153.7,
150.0, 130.9, 123.9, 111.8, 111.2, 69.8, 56.7, 32.2, 29.6, 26.0, 25.4, 23.2 and 14.7. HRMS-
ESI: m/z [M+H]+ for C15H22O3, calculated 251.1614; observed 251.1628.
1.6.2.7 Representative procedure for one pot halogenation-oxidative cleavage of 3,4-
dimethoxyphenylpropene into 1-(2-Iodo-4,5-dimethoxyphenyl) ethanone (2b', Scheme
22) :
To a stirred mixture of 3,4-dimethoxyphenylpropene (2a, 0.2 g, 1.1 mmol) in dioxane
(11ml), water (3.7 ml) added CTAB (0.03 g, 0.08 mmol) and NIS (1.0 g, 4.44 mmol) and
the reaction mixture was allowed to stir for 5 min at room temperature. Subsequently the
flask was irradiated under focused microwave system (250W, 115oC) for 15 min.
Thereafter, reaction flask was cooled, acetic acid (0.096 ml, 1.66 mmol), PDC (1.05g, 2.79
mmol) were added and further irradiated under microwave (250W, 115oC) for 15 min. The
above mixture was cooled, washed with saturated aq. Na2S2O3 solution (1x10 ml) and
extracted with ethyl acetate (3x20 ml). The combined organic layer was washed with brine
(1x10 ml), dried over Na2SO4 and vacuum evaporated. The residue was purified by column
chromatography on silica gel (60-120 mesh size) using hexane: ethylacetate (9.3:0.7) to
give 1-(2-Iodo-4, 5-dimethoxyphenyl) ethanone (2b', 0.082 g, 24 % yield).
1-(2-Iodo-4, 5-dimethoxyphenyl) ethanone (2b', Scheme 22)
O
H3CO
OCH3
I
O
O
OCH 3

One pot oxidative cleavage……… Chapter 1
61
Yellow viscous liquid, 1H NMR (300 MHz, CDCl3); (ppm) 7.34 (1H, s), 7.12 (1H, s),
3.93 (6H, s), 2.57 (3H, s); 13C NMR (75.4 MHz, CDCl3); (ppm) 200.0, 151.8, 149.3,
135.9, 123.9, 112.9, 81.9, 56.7 and 55.8. HRMS-ESI: m/z [M+H]+ for C10H11O3I,
calculated 306.9879; observed 306.9826.
1.6.2.8 Representative procedure for selective oxidative cleavage of 1,2-diaryl alkenes
(16a, Scheme 23) into corresponding benzaldehydes
To a stirred mixture of trans- stilbene (16a, 0.2 g, 1.1 mmol) and dioxane (11 ml), water
(3.7 ml), CTAB (0.03 g, 0.08 mmol ), NIS (0.5g, 2.2 mmol) and acetic acid (0.095 ml, 1.66
mmol) were added and the reaction mixture was allowed to stir for 3-4 hr at room
temperature. Subsequently the flask was irradiated under focused microwave system
(250W, 115oC) for 15 min. Thereafter, reaction flask was cooled, PDC (0.63g, 1.67 mmol)
was added and further irradiated under microwave (250W, 115oC) for 15 min. The above
mixture was cooled, washed with saturated aq. Na2S2O3 solution (1x10 ml) and extracted
with ethyl acetate (3x20 ml). The combined organic layer was washed with brine (1x10
ml), dried over Na2SO4 and vacuum evaporated. The GCMS analysis of residue revealed in
it 69% benzaldehyde (16c, Scheme 23).
The above procedure was also applied on other symmetrical and unsymmetrical 1, 2
diaryl alkenes (Scheme 23, 17a –18a) and products (17c-18c) confirmed through
GCMS analysis.
1.6.2.9 Representative procedure for cascade oxidative cleavage of trans-stilbene into
corresponding benzophenones
To a stirred mixture of trans-stilbene (19a, 0.2 g, 0.79 mmol) and dioxane (11 ml), water
(3.7 ml), CTAB (0.03 g, 0.08 mmol), NIS (0. 35 g, 1.5 mmol) and acetic acid (0.07 ml,
1.16 mmol) were added and the reaction mixture was allowed to stir for 3-4 hr at room
temperature. Subsequently, the flask was irradiated under focused microwave system
(250W, 115oC) for 15 min. Thereafter, the reaction flask was cooled, PDC (0.015g, 0.04
mmol), TBHP (0.45ml, 4.7 mmol) were added and further irradiated under microwave
(250W, 115oC) for 15 min. The above mixture was cooled, washed with saturated aq.
Na2S2O3 solution (1x10 ml) and extracted with ethyl acetate (3x20 ml). The combined

One pot oxidative cleavage……… Chapter 1
62
organic layer was washed with brine (1x10 ml), dried over Na2SO4 and vacuum evaporated.
The obtained residue was subsequently purified by column chromatography on silica gel
(60-120 mesh size) using hexane: ethylacetate (9.3:0.7) to give benzophenone (19b, Table
3) (49% yield).
Benzophenone (19b, Table 3)
White solid, m.p. 46-48°C (lit. m.p. 47-48°C) [Rao et al. (2009)]; IR (KBr): 1651 cm-1
(C=O), 1H NMR (300 MHz, CDCl3); (ppm) 8.02 (4H, d, J = 6.2 Hz), 7.80-7.68 (6H, m); 13C NMR (75.4 MHz, CDCl3); (ppm) 197.5, 138.4, 133.2, 130.8 and 129.0.
The above procedure was also used for oxidative cleavage of all the other 1,2-diaryl
alkenes (Table 3, Entries 20-26)
4, 4'-Dimethoxybenzophenone (20b, Table 3)
White solid, m.p. 135-137°C, [Rao et al. (2009)], IR (KBr): 1636 cm-1 (C=O); 1H NMR
(300 MHz, CDCl3); (ppm) 7.80 (4H, d, J = 9.5 Hz), 6.98 (4H, d, J = 7.5 Hz), 3.89 (6H,
s); 13C NMR (75.4 MHz, CDCl3); (ppm) 194.8, 163.2, 132.6, 131.3, 113.9 and 55.8.
HRMS-ESI: m/z [M+H]+ for C15H14O3, calculated 243.1016; observed 243.1026.
3, 4, 4'-Trimethoxybenzophenone (21b, Table 3)
White solid, m.p. 57-60°C, 1H NMR (300 MHz, CDCl3); (ppm) 7.81 (2H, d, J = 8.7
Hz), 7.44 (1H, s), 7.38 (1H, d, J = 9.1 Hz), 6.98 (3H, m), 3.96 (3H, s), 3.94 (3H, s), 3.89
(3H, s); 13C NMR (75.4 MHz, CDCl3); (ppm) 194.5, 162.9, 152.7, 149.0, 132.3, 130.2,
124.9, 114.3, 112.4, 109.9, 56.1 and 55.4. HRMS-ESI: m/z [M+H]+ for C16H16O4,
calculated 273.1121; observed 273.1132.
O
OCH3H3CO
H3CO
O
OCH3H3CO
O

One pot oxidative cleavage……… Chapter 1
63
3-4-Dioxymethylene-4'-methoxybenzophenone (22b, Table 3)
White solid, m.p. 96-98°C, 1H NMR (300 MHz, CDCl3); (ppm) 7.80 (2H, d, J = 9.7 Hz),
7.33 (2H, d, J = 9.3 Hz), 7.00 (2H, d, J = 9.7 Hz), 6.95 (1H, d, J = 8.0 Hz), 6.07 (2H, s),
3.89 (3H, s); 13C NMR (75.4 MHz, CDCl3); (ppm) 194.4, 163.3, 151.5, 148.2, 132.9,
132.6, 131.0, 126.6, 113.9, 110.3, 108.0, 102.1 and 55.9. HRMS-ESI: m/z [M+H]+ for
C15H12O4, calculated 257.0808 observed 257.0816.
(6- Methoxy-2-naphthalen-2yl) (4′-methoxyphenyl) methanone (23b, Table 3)
White solid, m.p. 128-131°C; IR (KBr): 1645cm-1 (C=O), 1H NMR (300 MHz, CDCl3);
(ppm) 8.20 (1H, s), 7.89-7.81 (5H, m), 7.23 (2H, d, J = 8.4 Hz), 7.01 (2H, d, J = 7.6 Hz),
3.98 (3H, s), 3.97 (3H, s); 13C NMR (75.4 MHz, CDCl3); (ppm) 195.8, 163.4, 159.9,
137.1, 133.7, 132.8, 131.6, 131.2, 128.0, 127.3, 127.0, 120.0, 113.9, 106.1, 60.8 and
55.8. HRMS-ESI: m/z [M+H]+ for C19H16O3, calculated 293.1310; observed 293.1382.
1-(2-Naphthyl )-4′-methoxyphenylmethanone (24b, Table 3 )
Yellow viscous liquid, 1H NMR (300 MHz, CDCl3); (ppm) 8.30 (1H, d, J = 9.7 Hz),
8.00-7.80 (5H, m), 7.61-7.52 (2H, m), 7.26-7.19 (3H, m), 3.96 (3H, s); 13C NMR (75.4
MHz, CDCl3); (ppm) 198.5, 160.3, 139.1, 138.9, 132.8, 131.7, 130.6, 128.9, 127.6,
127.2, 120.4, 106.4 and 56.1. HRMS-ESI: m/z [M+H]+ for C18H14O2, calculated
263.1066; observed 263.1065.
O
OCH3
O
O
O
OCH3H3CO
O
OCH3

One pot oxidative cleavage……… Chapter 1
64
4-Chloro- 4'- methoxybenzophenone (25b Table 3 )
White solid, m.p. 111-115°C, [Rao et al. (2009)], IR (KBr): 1637 cm-1 (C=O), 1H NMR
(300 MHz, CDCl3); (ppm) 7.80 (2H, d, J = 8.9 Hz), 7.71 (2H, d, J = 8.6 Hz), 7.45 (2H,
d, J = 8.9 Hz), 6.97 (2H, d, J = 9.1 Hz), 3.89 (3H, s); 13C NMR (75.4 MHz, CDCl3);
(ppm) 194.2, 163.5, 138.3, 136.7, 132.5, 131.2, 130.0, 128.6, 113.8 and 55.6. HRMS-ESI:
m/z [M+H]+ for C14H11ClO2,calculated 247.0520 observed 247.0519.
1.6.2.10 Representative procedure for one pot oxidative cleavage-condensation of 4-
(methoxyphenyl)propene with 2,4,5-trimethoxybenzaldehyde (Table 4, 27b)
To a stirred mixture of 1a (0.2 g, 1.35 mmol) and dioxane (11ml), water (3.7 ml), CTAB
(0.03 g, 0.08 mmol), NIS (0.395 g, 1.75 mmol) were added and the reaction mixture was
allowed to stir for 5 min at room temperature. Subsequently, the flask was irradiated under
MW (250W, 115oC) for 15 min followed by addition of acetic acid (0.12 ml, 2.0 mmol),
PDC (2.0 mmol) and further MW (250W, 115oC) for 15 min. Thereafter, the reaction
mixture was filtered and 5-8 ml of 10% sodium hydroxide (till basic conditions) along with
a methanolic solution (2-3 ml) of 2,4,5-trimethoxybenzaldehyde (0.29 g, 1.48 mmol) were
added. The reaction mixture was allowed to stir for 2 hr after which it was washed with
saturated aq. Na2S2O3 solution (1x10 ml) and extracted with ethyl acetate (3x20 ml). The
combined organic layer was washed with brine (1x10 ml), dried over Na2SO4 and vacuum
evaporated to obtain a crude mixture which upon addition of methanol, led to precipitation
of condensation product (27b, 0.24 g, 56% yield).
1-(4′-Methoxyphenyl)-3-(2,4,5-trimethoxyphenyl)- 2-propen-1-ones (Table 4, 27b)
O
OCH3Cl
O
H3CO
OCH3
OCH3
OCH3

One pot oxidative cleavage……… Chapter 1
65
Light yellow solid, m.p. 107-110°C, 1H NMR (300 MHz, CDCl3); (ppm) 8.10-8.00 (3H,
m), 7.50 (1H, d, J = 16.6 Hz), 7.12 (1H, s), 6.96 (1H, d, J = 8.0 Hz), 6.50 (1H, s), 3.87 (3H,
s), 3.85 (6H, s), 3.84 (3H, s); 13C NMR (75.4 MHz, CDCl3); (ppm) 189.6, 163.5, 154.9,
151.8, 143.1, 139.6, 132.0, 130.8, 120.5, 116.5, 114.1, 112.0, 97.1, 57.3, 57.0, 56.3 and
56.2. HRMS-ESI: m/z [M+H]+ for C19H20O5, calculated 329.1383; observed 329.1387.
The above procedure was also used for the one pot oxidative cleavage-condensation of
all the other arylalkenes (Table 4, 28-31b)
3-(4-Chlorophenyl)-1-(4-methoxyphenyl)prop-2-en-1-one, [Dong et al. (2008)] (Table
4, 28b)
Creamish solid, m.p. 128-131°C, 1H NMR (300 MHz, CDCl3); (ppm) 7.97 (2H, d, J = 9.3
Hz), 7.69 (1H, d, J = 16.2 Hz), 7.50 (2H, d, J = 8.1 Hz), 7.46 (1H, d, J = 16.2 Hz), 7.31
(2H, d, J = 8.1 Hz), 6.92 (2H, d, J = 8.7 Hz), 3.81 (3H, s); 13C NMR (75.4 MHz, CDCl3);
(ppm) 188.4, 163.5, 142.4, 136.1, 133.6, 130.8, 129.5, 129.2, 122.3, 113.9 and 55.5.
3-(2,4-Dichlorophenyl)-1-(4-methoxyphenyl)prop-2-en-1-one, [Liu et al. (2001)] (Table
4, 29b)
White solid, m.p. 134-137°C, 1H NMR (300 MHz, CDCl3); (ppm) 8.09-8.00 (3H, m),
7.67 (1H, d, J = 8.0 Hz), 7.49 (1H, d, J = 15.8 Hz), 7.43 (1H, s), 7.28 (1H, d, J = 8.4 Hz),
6.98 (2H, d, J = 8.2 Hz), 3.88 (3H, s); 13C NMR (75.4 MHz, CDCl3); (ppm) 188.4, 164.0,
138.6, 136.5, 136.2, 132.4, 131.3, 131.0, 130.4, 128.8, 127.8, 125.2, 114.3 and 55.8.
3-(4-dichlorophenyl)-1-(3, 4-dimethoxyphenyl)prop-2-en-1-one (Table 4, 30b)
O
H3CO Cl
OCH3
O
H3CO Cl
O
H3CO ClCl

One pot oxidative cleavage……… Chapter 1
66
White solid, m.p. 104-106°C, 1H NMR (300 MHz, CDCl3); (ppm) 7.79 (1H, d, J = 15.2
Hz), 7.71 (1H, d, J = 8.9 Hz), 7.64-7.51 (4H, m), 7.42 (1H, d, J = 8.0Hz), 6.96 (1H, d, J =
8.2 Hz), 3.99 (6H, s); 13C NMR (75.4 MHz, CDCl3); (ppm) 188.6, 153.8, 149.8, 142.8,
136.6, 134.0, 131.6, 129.9, 129.6, 123.4, 122.5, 111.2, 110.4 and 56.5.
1-(4′-Chlorophenyl)-3-(3,4-dioxymethylenephenyl)- 2-propen-1-ones (Table 4, 31b)
White solid, m.p. 149-151°C, 1H NMR (300 MHz, CDCl3); (ppm) 7.68 (1H, d, J = 15.7
Hz), 7.57 (1H, d, J = 8.0 Hz), 7.49-7.44 (3H, m), 7.40 (1H, d, J = 15.7 Hz), 7.31 (2H, d, J
= 8.4 Hz), 6.82 (1H, d, J = 8.0 Hz), 5.98 (2H, s); 13C NMR (75.4 MHz, CDCl3); (ppm)
187.9, 151.8, 148.3, 142.7, 136.2, 133.5, 132.8, 129.5, 129.2, 124.7, 122.1, 108.4, 107.9
and 101.9. HRMS-ESI: m/z [M+H]+ for C16H11ClO3, calculated 287.0469; observed
287.0467.
The above procedure was also successfully followed for the synthesis of
heteroaromatic based chalcones (32b-37b) (Table 4).
1-(3,4-dimethoxyphenyl)-3-(thiophen-2-yl)prop-2-en-1-one (Table 4, 32b)
Light yellow solid, m.p. 97-98 °C, 1H NMR (300 MHz, CDCl3); (ppm) 7.97 (1H, d, J=
15.3 Hz), 7.68 (1H, d, J= 8.4 Hz), 7.62 (1H, s) 7.41-7.33 (3H, m), 7.01 (1H, t, J= 4.2 Hz),
6.94 (1H, d, J= 8.3 Hz), 3.97 (6H, s); 13C NMR (75.4 MHz, CDCl3); (ppm) 188.3, 153.7,
149.7, 141.0, 136.7, 132.1, 131.7, 128.8, 128.7, 123.3, 120.9, 111.2, 110.4 and 56.4.
HRMS-ESI: m/z [M+H]+ for C15H14SO3, calculated 275.0736; observed 275.0719.
1-(3,4-dimethoxyphenyl)-3-(1H-indol-3-yl)prop-2-en-1-one (Table 4, 33b)
O
O
O
Cl
O
S
MeO
OMe
O
H3CO
OCH3
NH

One pot oxidative cleavage……… Chapter 1
67
Dark orange solid, m.p. 73-75 °C, 1H NMR (300 MHz, CDCl3); (ppm) 8.85 (1H, s), 8.35
(1H, s), 8.16 (1H, d, J= 15.5 Hz), 7.87 (1H, s), 7.78-7.62 (2H, m) 7.36-7.28 (4H, m), 7.00
(1H, d, J= 8.3 Hz), 3.99 (6H, s); 13C NMR (75.4 MHz, CDCl3); (ppm) 185.6, 153.3,
149.6, 138.6, 135.9, 132.4, 130.5, 125.8, 124.8, 123.9, 122.1, 121.0, 117.9, 114.9, 112.4,
111.3, 110.5 and 56.5. HRMS-ESI: m/z [M+H]+ for C19H17NO3, calculated 308.1281;
observed 308.1267.
1-(2H-1,3-benzodioxol-5-yl)-3-(furan-3-yl)prop-2-en-1-one (Table 4, 34b)
Brown solid, m.p. 95-97 °C, 1H NMR (300 MHz, CDCl3); (ppm) 7.67 (1H, d, J= 8.1 Hz),
7.60 (1H, d, J= 15.3Hz), 7.53-7.52 (2H, m), 7.43 (1H, d, J= 15.3 Hz), 6.89 (1H, d, J= 8.1
Hz), 6.70 (1H, s), 6.50 (1H, s), 6.05 (2H, s); 13C NMR (75.4 MHz, CDCl3); (ppm) 187.9,
152.2, 152.1, 148.7, 145.2, 133.4, 130.5, 125.0, 119.5, 116.2, 113.0, 108.7, 108.3 and
102.2. HRMS-ESI: m/z [M+H]+ for C14H10O4, calculated 243.0651; observed 243.0662.
1-(2H-1,3-benzodioxol-5-yl)-3-(6-bromopyridin-3-yl)prop-2-en-1-one (Table 4, 35b)
White solid, m.p. 146-148 °C, 1H NMR (300 MHz, CDCl3); (ppm) 8.56 (1H, s), 7.89
(1H, s), 7.64-7.51 (5H, m), 6.90 (1H, s), 6.07 (2H, s); 13C NMR (75.4 MHz, CDCl3);
(ppm) 180.3, 159.8, 149.7, 148.2, 138.2, 136.7, 133.1, 131.7, 128.8, 124.7, 123.3, 107.9,
107.6 and 101.7. HRMS-ESI: m/z [M+H]+ for C15H10BrNO3, calculated 331.9916;
observed 331.9959.
1-(4-methoxyphenyl)-3-(thiophen-2-yl)prop-2-en-1-one (Table 4, 36b)
Brown solid, m.p. 93-94 °C, 1H NMR (300 MHz, CDCl3); (ppm) 8.06 (2H, d, J= 4.5 Hz),
7.97 (1H, d, J= 15.3 Hz), 7.42-7.33 (3H, m), 7.10-7.07 (1H, m), 7.01 (2H, d, J= 4.5 Hz),
O
O
O
O
O
O
O
N Br
O
H3CO
S

One pot oxidative cleavage……… Chapter 1
68
3.89 (3H, s); 13C NMR (75.4 MHz, CDCl3); (ppm) 188.4, 163.8, 140.9, 136.8, 132.1,
131.4, 131.1, 128.8, 128.7, 121.1, 114.2 and 55.9. HRMS-ESI: m/z [M+H]+ for C14H12 SO2,
calculated 245.0630; observed 245.0611.
3-(Furan-2-yl)-1-(4-methoxyphenyl)prop-2-en-1-one (Table 4, 37b)
Brown solid, m.p. 68-70°C, 1H NMR (300 MHz, CDCl3); (ppm) 8.05 (2H, d, J= 8.8 Hz),
7.60-7.43 (3H, m), 6.97 (2H, d, J= 8.8 Hz), 6.69 (1H, s), 6.50 (1H, s), 3.85 (3H, s); 13C
NMR (75.4 MHz, CDCl3); (ppm) 188.4, 163.8, 152.2, 145.1, 131.4, 131.1, 130.3, 119.6,
116.1, 114.2, 113.0 and 55.8. HRMS-ESI: m/z [M+H]+ for C14H12O3, calculated 229.0859;
observed 229.0831.
1.7 References
Avery, M. A., Chong, W. K. M. and White, C. J. (1992). Stereoselective total synthesis of
(+)-artemisinin, the antimalarial constituent of Artemisia annua L. Journal of American
Chemical Society 114: 974-79.
Bailey, P. S. and Erickson, R. E. (1973). Diphenaldehyde. Collective Volume 5: 489.
Bailey, P. S. (1978). In ozonization in organic chemistry. Academic Press: New York.
Balasankar, T., Gopalakrishnan, M. and Nagarajan, S. (2005). Synthesis and antibacterial
activity of some 5-(4-biphenylyl)-7-aryl[3,4-d][1,2,3]-benzothiadiazoles. European
Journal of Medicinal Chemistry 40: 728-31.
Bryant, J. R., Matsuo, T. and Mayer, J. M. (2004). Cumene oxidation by cis-[Ru-
IV(bPY)(2)(py)(O)](2+), revisited. Inorganic Chemistry 43: 1587-92.
Claus, R. E. and Schreiber, S. L. (1990). Ozonolytic cleavage of cyclohexene to terminally
differentiated products. Collective Volume 7: 168.
Clennan, E. L. and Pan, G. I. (2003). Zeolite-promoted oxidations of 1,1-diarylethylenes.
Organic Letters 5: 4979-82.
Das, S., Brudvig, G. W. and Crabtree, R. H. (2008). High turnover remote catalytic
oxygenation of alkyl groups: How steric exclusion of unbound substrate contributes to
O
H3CO
O

One pot oxidative cleavage……… Chapter 1
69
high molecular recognition selectivity. Journal of American Chemical Society 130:
1628-37.
Dong, F., Jian, C., Zhenghao, F., Kai, G. and Zuliang, L. (2008). The increased amount of
PDC was presumably required due to the comparatively lower reactivity of halogenated
α-aryl aldehyde towards oxidative cleavage. Catalysis communications 9: 1924-27.
Dvorak, C. A. (1983). Process for the preparation of d,1-2-(6-methoxy-2-
naphthyl)propionic acid. U S Patent 4395571.6
Giddens, A. C., Boshoff, H. I. M., Franzblau, S. G., Barry, C. E. and Coppa, B. R. (2005).
Antimycobacterial natural products: Synthesis and preliminary biological evaluation of
the oxazole-containing alkaloid texaline. Tetrahedron Letters 46: 7355-57.
Green, M. E., Rech, J. C. and Floreancig, P. E. (2008). Total synthesis of theopederin.
Angewandte Chemie International Edition 47: 7317-20.
Gwaltney II, S. L., Sakata, S. T. and Shea, K. J. (1996). Bridged to fused ring interchange
methodology for the construction of fused cycloheptanes and cyclooctanes. Total
syntheses of ledol, ledene, and compressanolide. Journal of Organic Chemistry 61:
7438-51.
Hart, S. R., Whitehead, D. C., Travis, B. R. and Borhan, B. (2011). Catalytic oxidative
cleavage of olefins promoted by osmium tetroxide and hydrogen peroxide. Organic and
Biomolecular Chemistry 9: 4741–44.
Hayes, C. J., Bradley, D. M. and Thomson, N. M. (2006). An efficient enantioselective
synthesis of (2R)-hydroxymethyl glutamic acid and an approach to the (2R)-
hydroxymethyl-substituted sphingofungins. Journal of Organic Chemistry 71: 2661-
65.
Heathcock, C. H., Young, S. D., Hagen, J. P., Pilli, R. and Badertscher, U. (1985). Acyclic
stereoselection. 25. Stereoselective synthesis of the C-1 to C-7 moiety of erythronolide-
A. Journal of Organic Chemistry 50: 2095-2105.
Hirashima, S., Kudo, Y., Nobuta, T., Tada, N. and Itoh, A. (2009). Aerobic photo-oxidative
cleavage of the C–C double bonds of styrenes. Tetrahedron Letters 50: 4328-30.
Ho, C. M., Yu, W. Y. and Che, C. M. (2004). Ruthenium nanoparticles supported on
hydroxyapatite as an efficient and recyclable catalyst for cis-dihydroxylation and
oxidative cleavage of alkenes. Angewandte Chemie International Edition 43: 3303-07.

One pot oxidative cleavage……… Chapter 1
70
Jiang, N. and Ragauskas, A. (2007). Selective aerobic oxidation of activated alcohols into
acids or aldehydes in ionic liquids. Journal of Organic Chemistry 72: 7030-33.
Kappe, C. O. (2004). Controlled microwave heating in modern organic synthesis.
Angewandte Chemie International Edition 43: 6250-84.
Kim, S., Chung, J. and Kim, B. M. (2011). Recycling of osmium catalyst in oxidative
olefin cleavage: A chemoentrapment approach. Tetrahedron Letters 52: 1363-67.
Kuhn, F. E., Fischer, R. W., Herrmann, W. A. and Weskamp, T. (2004). Transition Metals
for Organic Synthesis. pp 427. Wiley-VCH: Weinheim, Germany.
Kumar, R., Sharma, A., Kumar, V. and Sinha, A. K. (2008). Neutral ionic liquid [hmim]Br
as a green reagent and solvent for the mild and efficient dehydration of benzyl alcohols
into (E)-arylalkenes under microwave irradiation. European Journal of Organic
Chemistry 5577-82.
Larock, R. C. (1999). Comprehensive Organic Transformations, 2nd ed. pp 1213-15.
Wiley-VCH: New York.
Lara, M., Mutti, F. G., Glueck, S. M. and Kroutil, W. (2009). Oxidative enzymatic alkene
cleavage: Indications for a nonclassical enzyme mechanism. Journal of American
Chemical Society 131: 5368-69.
Lee, D. G. and Chen, T. (1991). Comprehensive organic synthesis. pp 541-91. Pergamon
Press, Oxford.
Ley, S. V., Ramarao, C., Lee, A., Ostergaard, N., Smith, S. C. and Shirley, I. M. (2003).
Microencapsulation of osmium tetroxide in polyurea. Organic Letters 5: 185-87.
Liu, M., Wilairat, P. and Go, M. L. (2001). Antimalarial alkoxylated and hydroxylated
chalcones: Structure-activity relationship analysis. Journal of Medicinal Chemistry 44:
4443-52.
Marnett, L. J. and Blobaum, A. L. (2007). Structural and functional basis of
cyclooxygenase inhibition. Journal of Medicinal Chemistry 50: 1425-41.
Nicolaou, K. C., Adsool, V. A. and Hale, C. R. H. (2010). An expedient procedure for the
oxidative cleavage of olefinic bonds with PhI(OAc)2, NMO, and catalytic OsO4.
Organic Letters 12: 1552-55.

One pot oxidative cleavage……… Chapter 1
71
Ohno, K., Kataoka, Y. and Mashima, K. (2004). Asymmetric transfer hydrogenation of
aryl ketones catalyzed by salt-free two samarium centers supported by a chiral
multidentate alkoxy ligand. Organic Letters 6: 4695-97.
Perlman, K. L., Darwish, H. and DeLuca, M. H. F. (1994). 20-oxopregnacalciferols -
vitamin-D compounds that bind the progesterone-receptor. Tetrahedron Letters 35:
2295-98.
Plevyak, J. E. and Heck, R. R. F. (1978). Palladium-catalyzed arylation of ethylene.
Journal of Organic Chemistry 43: 2454-56.
Rao, M. L. N., Jadhav, D. N. and Venkatesh, V. (2009). Pd(0)/C-catalyzed cross-couplings
of acyl chlorides with triarylbismuths as atom-efficient sub-stoichiometric multi-
coupling reagents. Tetrahedron Letters 50: 4268-71.
Rozner, D. S., Neimann, K. and Neumann, R. (2007). Aerobic oxidation of aldehydes
catalyzed by Keggin type polyoxometalates [Mo12VO39 (μ2-OH)10H2{XII(H2O)3}4] (X =
Ni, Co, Mn and Cu) as heterogeneous catalysts. Journal of Molecular Catalysis A 262:
109-13.
Schiaffo, C. E. and Dussault, P. H. (2008). Ozonolysis in solvent/water mixtures: Direct
conversion of alkenes to aldehydes and ketones. Journal of Organic Chemistry 73:
4688-90.
Shaikh, T. M. and Honga, F. E. (2011). Iron-catalyzed oxidative cleavage of olefins and
alkynes to carboxylic acids with aqueous tert-butyl hydroperoxide. Advanced Synthesis
and Catalysis 353: 1491-96.
Sharma, A., Kumar, R., Sharma, N., Kumar, V. and Sinha, A. K. (2008). Unique versatility
of ionic liquids as clean decarboxylation catalyst cum solvent: A metal- and quinoline-
free paradigm towards synthesis of indoles, styrenes, stilbenes and arene derivatives
under microwave irradiation in aqueous conditions. Advanced Synthesis and Catalysis
350: 909-18.
Sharma, A., Sharma, N., Kumar, R., Sharma, U. and Sinha, A. K. (2009). Water-promoted
cascade synthesis of alpha-arylaldehydes from arylalkenes using N-halosuccinimides:
An avenue for asymmetric oxidation using Cinchona organocatalysis. Chemical
Communication 5299-5301.

One pot oxidative cleavage……… Chapter 1
72
Sharma, A., Sharma, N., Kumar, R., Shard, A. and Sinha, A. K. (2010). Direct olefination
of benzaldehydes into hydroxy functionalized oligo (p-phenylenevinylene)s via Pd-
catalyzed heterodomino Knoevenagel-decarboxylation-Heck sequence and its
application for fluoride sensing pi-conjugated units. Chemical Communication 46:
3283-85.
Sharma, N., Sharma, U. K., Salwan, R., Kasana, R. C. and Sinha. A. K. (2011). A synergic
blend of newly isolated Pseudomonas mandelii KJLPB5 and [hmim]Br for
chemoselective 2o aryl alcohol oxidation in H2O2: Synthesis of aryl ketone or aldehydes
via sequential dehydration-oxidative C=C cleavage. Catalysis Letters 141: 616-22.
Shing, T. K. M. (1991). Comprehensive organic synthesis. pp 703-16. Pergamon Press,
Oxford.
Sinha, A. K., Acharya, R. and Joshi, B. P. (2002). A mild and convenient procedure for the
conversion of toxic beta-asarone into rare phenylpropanoids: 2,4,5-
trimethoxycinnamaldehyde and gamma-asarone. Journal of Natural Products 65: 764-
65.
Singh, F. V., Milagre, H. M. S., Eberlin, M. N. and Stefani, H. A. (2009). Synthesis of
benzophenones from geminal biaryl ethenes using m-chloroperbenzoic acid.
Tetrahedron Letters 50: 2312-16.
Snider, B. B., Kiselgof, J. Y. and Foxman, B. M. (1998). Total syntheses of (±)-Isosteviol
and (±)-beyer-15-ene-3-β,19-diol by manganese(III)-based oxidative quadruple free-
radical cyclization. Journal of Organic Chemistry 63 : 7945-52.
Somu, R. V. and Johnson, R. L. (2005). Synthesis of pipecolic acid-based spiro bicyclic
lactam scaffolds as ß-turn mimics. Journal of Organic Chemistry 70: 5954-63.
Thottumkara, P. and Vinod, T. K. (2010). Oxidative cleavage of alkenes using an in situ
generated iodonium ion with oxone as a terminal oxidant. Organic Letters 24: 5640-43.
Tietze, L. F. and Bratz, M. (1998). Dialkyl mesoxalates by ozonolysis of dialkyl
benzalmalonates. Collective Volume 9: 314.
Travis, B. R., Narayan, R. S. and Borhan, B. (2002). Osmium tetroxide-promoted catalytic
oxidative cleavage of olefins: An organometallic ozonolysis. Journal of American
Chemical Society 124: 3824-25.

One pot oxidative cleavage……… Chapter 1
73
Velosa, A. C., Baader, W. J., Stevani, C. V., Mano, C. M. and Bechara, E. J. H. (2007).
1,3-Diene probes for detection of triplet carbonyls in biological systems. Chemical
Research in Toxicology 20: 1162-69.
Wang, A. and Jiang, H. (2010). Palladium-catalyzed direct oxidation of alkenes with
molecular oxygen: General and practical methods for the preparation of 1,2-diols,
aldehydes, and ketones. Journal of Organic Chemistry 75: 2321-26.
Xing, D., Guan, B., Cai, G., Fang, Z., Yang, L. and Shi, Z. (2006). Gold (I)-catalyzed
oxidative cleavage of a C=C double bond in water. Organic Letters 8: 693-96.
Yadav, J. S., Reddy, B. V. S., Reddy, P. S. R., Basak, A. K. and Narsaiah, A. V. (2004).
Efficient halogenation of aromatic systems using N-halosuccinimides in ionic liquids.
Advanced Synthesis and Catalysis 346: 77-82.
Yang, D. and Zhang, C. (2001). Ruthenium-catalyzed oxidative cleavage of olefins to
aldehydes. Journal of Organic Chemistry 66: 4814-18.
Yan, Y. and Zhang, X. (2006). A hybrid phosphorus ligand for highly enantioselective
asymmetric hydroformylation. Journal of American Chemical Society 128: 7198-7202.
Yao, S., Meng, J. C., Siuzdak, G. and Finn, M. G. (2003). New catalysts for the asymmetric
hydrosilylation of ketones discovered by mass spectrometry screening. Journal of
Organic Chemistry 68: 2540-46.
Yu, W., Mei, Y., Kang, Y., Hua, Z. and Jin, Z. (2004). Improved procedure for the
oxidative cleavage of olefins by OsO4-NaIO4. Organic Letters 6: 3217-19.
Yusubov, M. S., Zholobova, G. A., Filimonova, I. L. and Ki, W. C. (2004). New oxidative
transformations of alkenes and alkynes under the action of diacetoxyiodobenzene.
Russian Chemical Bulletin International Edition 53: 1735-42.

One pot oxidative cleavage……… Chapter 1
74
Spectral figures of some representative compounds
1H NMR (in CDCl3) spectrum of 3',4'-Dimethoxyacetophenone (2b, Table 2)
13C NMR (in CDCl3) spectrum of 3',4'-Dimethoxyacetophenone (2b, Table 2)
H3CO
H3CO
O

One pot oxidative cleavage……… Chapter 1
75
1H NMR (in CDCl3) spectrum of 4-Chloro-4'-methoxybenzophenone (25b, Table 3)
13C NMR (in CDCl3) spectrum of 4-Chloro-4'-methoxybenzophenone (25b, Table 3)
O
OCH3Cl

One pot oxidative cleavage……… Chapter 1
76
9 8 7 6 5 4 3 2 1 ppm
Prep ByNPP DIVISIONIHBT (CSIR)SHIV
m/z100 110 120 130 140 150 160 170 180 190 200 210 220 230 240 250 260 270 280 290
%
0
100
AKB6H 11 (0.194) Cm (7:13) TOF MS ES+ 1.36e4247.0519
102.1694
212.0410153.0847
174.1484 195.1440
249.0606
HRMS spectrum of 4-Chloro-4'-methoxybenzophenone (25b, Table 3)
1H NMR (in CDCl3) spectrum of (2E)-3-(furan-2-yl)-1-(4-methoxyphenyl)prop-2-en-1- one (37b, Table 4)
O
H3CO
O
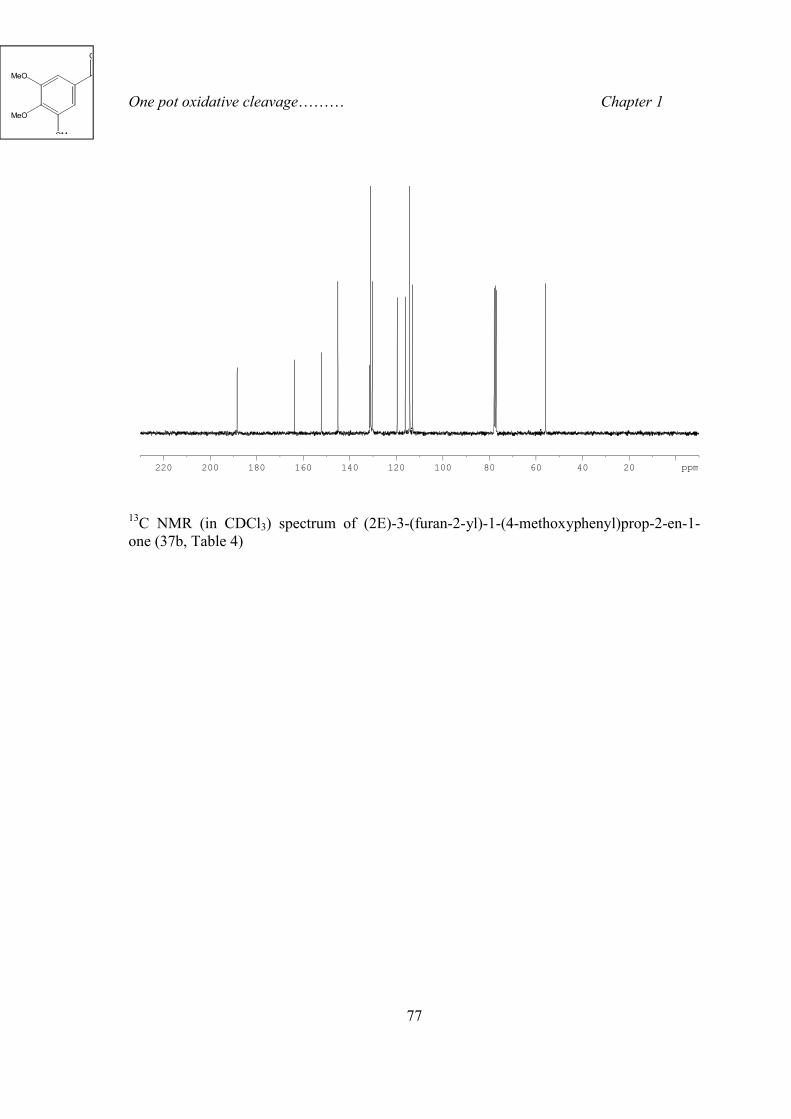
One pot oxidative cleavage……… Chapter 1
77
220 200 180 160 140 120 100 80 60 40 20 ppm
13C NMR (in CDCl3) spectrum of (2E)-3-(furan-2-yl)-1-(4-methoxyphenyl)prop-2-en-1- one (37b, Table 4)
O
MeO
MeO
OMe