oleary-european medical device regulations - · pdf file1 european medical device regulations...
TRANSCRIPT

1
European Medical Device Regulations— What to Expect
14th Annual Medical Device Quality Congress
March 29, 2017
Dr. Ibim Tariah, Technical Director, BSI Americas Inc.
Dan O’Leary, President, Ombu Enterprises LLC

2
Topics
• The Structure of the Presentation
• The Current Status of the EU-MDR
• Transition Time Lines
• Economic Operators
• Person Responsible for Regulatory Compliance
• Classification and Conformity Assessment
• General Safety And Performance Requirements
• Risk Management
• Clinical Evaluation
• Post-market Surveillance

Presentation Structure
• We plan to cover the topics on the previous slide
• Given the significance of the changes and the time limit, we can onlyexamine the most important elements in each topic
• We will present a few bullet points in each topic
– Make comments from the point of view of a manufacturer’simplement
– Make comments from the point of view of a Notified Body
– Invite questions from the attendees
3

Current Status of the EU-MDR
• In the EU today, there are three directives related to medicaldevices:
– Active Implantable Medical Devices (AIMD)
– In-vitro Diagnostic Directive (IVDD)
– Medical Device Directive (MDD)
• The EU intends to replace the three directives with two regulations:
– In-vitro Diagnostic Regulation (IVDR)
– Medical Device Regulation (MDR)
• The MDR includes active implantable devices
4

5
Transition Time Lines

Transition Time Lines
• Regulation transition time
– The MDR has a three-year transition from publication
– The IVDR has a five-year transition from publication
• CE Mark
– During the transition, manufacturers can use the MDD to place the CEMark on new devices
– By the end of the transition, the CE Mark must conform to the MDR
• Notified Bodies
– During the transition, Notified Bodies accredited to the MDD may stilloperate
– During the transition, Notified Bodies become accredited to the MDR
6

Transition Time Lines• EN ISO 13485:2012
– Currently, EN ISO 13485:2012 is harmonized to the MDD
– At some point, EN ISO 13485:2016 will become harmonized to the MDD
– All ISO 13485:2003 and EN ISO 13485:2012 certificates expire onMarch 1, 2019
– Anticipate EN ISO 13485:xxxx to align with the MDR
• EN ISO 14971:2012
– Currently, EN ISO 14971:2012 is harmonized to the MDD
– Anticipate EN ISO 14971:xxxx to align with the MDR
• ISO 14971:2007
– Currently, ISO 14971:2007 is the international standard
– ISO/TC 210 has a new project approved to revise the internationalstandard
7

Transition Timelines MDR (Article 120 of the Draft text Nov. 2016)
May 2017*Adoption of MDR
Entry into Force (EUOJ + 20days)(June 2017*)
NBs designation under MDR
Date of Application(June 2020*)
Last MDD/AIMDcertificates expire
(June 2024*)
MDD/AIMD certificate validity(4 years )
MDD/AIMD certificates (max 5-year expiry from issue/renewal date)
MDR certificates
Transition period3 years
* Dates are « best guess » based on our current understanding on the process/steps to be completed
Annex IV certificates expire(June 2022*)
No more « placing onthe market » of devicescovered by MDD/AIMD
certificates
8

Transitional Provisions Draft MDR
•
9

Attendees
10

11
Economic Operators

Economic Operators• Manufacturer means a natural or legal person who manufactures or fully
refurbishes a device or has a device designed, manufactured or fullyrefurbished, and markets that device under its name or trademark
• Authorized Representative means any natural or legal person establishedwithin the Union who has received and accepted a written mandate from amanufacturer, located outside the Union, to act on the manufacturer's behalfin relation to specified tasks with regard to the latter's obligations under thisRegulation
• Importer means any natural or legal person established within the Unionthat places a device from a third country on the Union market
• Distributor means any natural or legal person in the supply chain, other thanthe manufacturer or the importer, that makes a device available on themarket, up until the point of putting into service
12MDR Article 2

Identification Within The Supply Chain
• Economic operators shall be able to identify the following to thecompetent authority, for 10 years in general and 15 years forimplantable devices:
(a) any economic operator to whom they have directly supplied adevice;
(b) any economic operator who has directly supplied them with adevice;
(c) any health institution or healthcare professional to which theyhave directly supplied a device.
13MDR Article 25

Manufacturers
• Determine the economic operators in your current distribution channels
• Read the MDR requirements for each kind of economic operator
• Start an information sheet for each of your economic operators so they willknow to expect
– Registration
– Post-market Surveillance
– Single registration number
• Start working on contracts and quality agreements
– EN ISO 13485:2016 requires controls on providers of outsourcedprocesses that include written quality agreements
14

Notified Body
• NB’s will ensure Economic Operator(s) must meetRequirements of the MDR [refs: Articles 11 to 16]
• Implementing Act to provide further details?
15

Chapter II, Article 4
The Commission may adopt implementing acts to ensure the uniformapplication of Annex I, to the extent necessary to resolve issues ofdivergent interpretation and practical application.
17

Attendees
17

18
Person Responsible forRegulatory Compliance

Person Responsible – Qualification• Manufacturers have at least person in the organization responsible
from regulatory compliance
– Micro and small enterprises only need a person permanently andcontinuously at their disposal
• The requisite expertise shall be demonstrated by either of thefollowing qualifications:
– a diploma, certificate or other evidence of formal qualification,awarded on completion of a university degree or an equivalentcourse of study in law, medicine, pharmacy, engineering, oranother relevant scientific discipline, and at least one year ofprofessional experience in regulatory affairs or in qualitymanagement systems relating to medical devices
– four years of professional experience in regulatory affairs or inquality management systems relating to medical devices.
19MDR Article 15

Person Responsible – Responsibilities
• The person responsible ensures:
– Device conformity is checked before release
– The technical documentation is up-to-date
– The Declaration of Conformity is up-to-date
– The company meets the PMS requirements
– The company meets the reporting requirements:
• Serious incidents
• Field safety corrective actions
• Trend reporting
• Analysis of serious incidents
• Analysis of field safety corrective actions
• Analysis of vigilance data
20MDR Article 15

Manufacturers
• Determine who in your organization meets the qualifications for a personresponsible
– It would be best to have at least two people (to provide “benchstrength”)
• Determine if your organization qualifies as an SME
• If your company is an SME and needs to use an outside person, start toidentify contractors or consultants who can provide the service
– Start working on contracts and quality agreements
– EN ISO 13485:2016 requires controls on providers of outsourcedprocesses that include written quality agreements
21

Notified Body
• NB’s will ensure “Person Responsible” Qualifications &Responsibilities meet Requirements of the MDR [Refs:
Article 15]
• Implementing Act to provide further details?
22

Attendees
23

24
Classification andConformity Assessment

Classification Rules
• The Active Implantable Medical Device directive will be retired
– Those devices will fall under the MDR
• There four device classes: I, IIa, IIb, and III
• There are about 24 classification rules
• The classification rules are in the following groups:
– Non-invasive Devices
– Invasive Devices
– Active Devices
– Special Rules
25MDR Article 51 and Annex VIII

Conformity Assessment
• Each device class has more than one conformity assessment path(in Annexes IX to XI)
• There are specific requirements for Class III implantable and ClassIIb implantable devices
• There are specific requirements for Class I devices that are sterile,reusable surgical instruments, or have a measuring function
26

Manufacturers
• For each of your existing devices, determine the class under thenew rules
• Knowing the device class, determine the possible conformityassessment paths
• For each device, select a conformity assessment path
• Develop the information needed to satisfy the elements in theselected conformity assessment path
27

Summary of Changes in Classification Rules
New Process in case of dispute between NB and Manufacturer
Addition of the active implantable medical devices to theclassification rules
Addition of tissue engineered products
Up-classification of devices in direct contact with the heart orthe central circulatory system
Up-classification of orthopedic devices and devices in contactwith the spinal column
Addition of Nanomaterials
Update of the wording regarding human origin material28

Classification & Conformity Assessment – Regulation
Commission Assessment
Competent Authority Assessment
Notified Body Conformity Assessment
Self-Certification
Class III
Class IIb
Risk
Class IIa
Class Im / Is / Ir
Class I
Custom Made Custom Made Class IIIImplants
Class IIa – more sampling
Class IIb Implants
Class III Implants & Class IIbactive – delivering medicines
Animal tissues, human tissues,medicinal substances, absorbable
30

Class IIb Implantable Device (Article 42.3)
Declaration of Conformity (Annex III) & CE Marking (Annex IV)Declaration of Conformity (Annex III) & CE Marking (Annex IV)
Annex VIII
QMS
Annex VIII
TechnicalDocumentation *eachGeneric Device Group
Article 42 Point 3
Annex IX
Type Examination
Annex X – Part A
ProductionQuality Assurance
Annex X – Part A
ProductionQuality Assurance
Annex X – Part B
ProductVerification
“……By way of derogation, the assessment ofthe technical documentation as specified inSection 5 of Chapter II of Annex VIII shall beapplicable for Class IIb implantable devices.Alternatively, the manufacturer may choose toapply a conformity assessment based on typeexamination as specified in Annex IX coupled witha conformity assessment based on productconformity verification as specified in Annex X”
30

Class IIb Implantable Device (Article 42.3)
Declaration of Conformity (Annex III) & CE Marking (Annex IV)Declaration of Conformity (Annex III) & CE Marking (Annex IV)
Annex VIII
QMS
Annex VIII
TechnicalDocumentation *eachGeneric Device Group
Article 42 Point 3
Exceptions: sutures, staples, dentalfillings, dental braces, tooth crowns,screws, wedges, plates, wires, pins,clips and connectors.
Article 42.3a: list above can beamended by delegated acts
31

Attendees
32

33
General Safety AndPerformance Requirements
(Annex I)

Annex I• The General Safety And Performance Requirements serve an
analogous function to the Annex I Essential Requirements in theMDD
• The are 3 major chapters
– I. General requirements
– II. Requirements regarding design and manufacture
– III. Requirements regarding the information supplied with thedevice
• Chapter II has a large number of subsections
34MDR Annex I

Harmonized Standards andCommon Specifications
• Devices in conformity with the relevant harmonized standards, or therelevant parts of those standards (published in the Official Journal)are presumed to in conformity with those requirements of thisRegulation [Article 8]
• In some cases (such as no or insufficient harmonized standard) theCommission may adopt common specifications (CS) [Article 9]
• Common specification (CS) means a set of technical and/or clinicalrequirements, other than a standard, that provides a means ofcomplying with the legal obligations applicable to a device, processor system.
35

Manufacturers• For each of your existing devices, determine the applicable
requirements in Annex I
• Determine which current harmonized standards would apply tothose requirements
– Some of them you may already used
– Identify potential gaps
• Remember that there are no Harmonized Standards for the newregulation
– The Z-annexes will have to change, but the text of the standardsprobably will not
• Remember that there are no Common Specifications for the newregulation
36

Common Specifications – Chapter II, Article 9
…where:
• no harmonized standards exist or
• where relevant harmonisedstandards are not sufficient, or
• where there is a need to addresspublic health concerns,
the Commission … may adoptcommon specifications (CS)
adopted by means of
implementing acts
Where can these apply?
• the general safety and performancerequirements set out in Annex I,
• the technical documentation set outin Annex II
• the clinical evaluation and post-market clinical follow-up set out inAnnex XIV
• the requirements regarding clinicalinvestigation set out in Annex XV.
38

Attendees
38

39
Risk Management System

Risk Management System
• Establish a risk management system with the following elements:
– A Risk Management Plan for each device
– Identify and analyze known and foreseeable hazards
– Estimate and evaluate the risks during intended use
– Eliminate or control the risks following the risk control measures
– Evaluate information from production and the PMS system
– Amend controls as necessary based on the evaluation
40MDR Annex I, Section 3

Risk Control Measures
• Risk control measures conform to safety principles taking intoaccount the generally acknowledged state of the art
• Manage risk so the residual risk of each hazard and the overallresidual risk are acceptable
• Adopt risk control measures in the priority order:
– Safe design and manufacture
– Adequate protection measures including alarms if necessary
– Information for safety and, where appropriate, user training
• Inform users of any residual risk
41MDR Annex I, Section 4

Manufacturers
• Ensure your implementation of EN ISO 14971:2012
– Recognize that there will be a new version for the MDR
• Notice the change in the risk control priority order from the MDD
– Many people misread MDD Content Deviation #7, believing thatinformation for safety is not a risk control measure
– The MDR fixes the source of the misunderstanding
42

Risk Management
... much more than analysis
Risk analysis
Intended use/intended purposeidentification of characteristics related tothe safety of the medical device
Identification of hazards Estimation of the risks for each hazardous
situation
Risk evaluation
Risk control
Risk control option analysis Implementation of risk control measure(s) Residual risk evaluation Risk/benefit analysis Risks arising from risk control measures Completeness of risk control
Evaluation of overall residualrisk acceptability
Risk management report
Production & post productioninformation
43

Attendees
44

45
Clinical Evaluation

Clinical Evaluation
• Clinical data provides clinical evidence to support:
– Conformity with Annex I
– Evaluation of undesirable side-effects
– Acceptability of the benefit-risk ratio
• Manufacturers plan, conduct, and document a clinical evaluation following adefined and methodologically sound procedure that includes:
– Evaluation of data on an equivalent device
– Evaluation of the results of clinical investigations
– Consideration of alternate treatment options
• Update the clinical evaluation through the product lifecycle using the PMCFand post-market surveillance (PMS)
46MDR Article 61

Clinical Evaluation
• Develop a Clinical Evaluation Plan
– There are 8 required elements including a Clinical Development Plan
• Identify available clinical data
• Appraise all relevant clinical data
• Generate, through clinical investigations following the Clinical DevelopmentPlan, any needed additional clinical data
• Analyze all relevant clinical data
• Document the results in a Clinical Evaluation Report
• Base the clinical evaluation on clinical data from an equivalent device:
– Technical
– Biological
– Clinical
47MDR Annex XIV Part A

Manufacturers• The MDD requires Clinical Evaluation
• In June 2016, the EU published MedDev 2.7/1 Rev. 4 ClinicalEvaluation: A Guide For Manufacturers And Notified Bodies UnderDirectives 93/42/EEC and 90/385/EEC which made significantchanges to Clinical Evaluation process
• Review MedDev 2.7/1 Rev. 4 and make a plan to implement it
– Expect a new version of MedDev 2.7/1 for the MDR
– The changes will probably be additive – not requiring you toundo anything
• Review MedDev 2.12/2 Rev. 2 Post Market Clinical Follow-upStudies – A Guide For Manufacturers And Notified Bodies
– Expect a new version of MedDev 2.12/2 for the MDR
48

Clinical Evidence – MedDev 2.7.1 & MDR
Clinical Evidence
Clinical Evaluation
Clinical Data
• the clinical data and clinical evaluationreport pertaining to a device
• sufficient amount and quality to allow aqualified assessment of whether thedevice achieves the intended clinicalbenefit(s) and safety, when used asintended by the manufacturer
• a methodologically sound / systematic andplanned process to continuously generate,collect, analyse and assess the clinical datapertaining to a device
• to verify the safety and performance, includingclinical benefits, of the device when used asintended by the manufacturer
• clinical investigation on the device concerned
• clinical investigation reported in the scientificliterature, of a similar device for which equivalence tothe device in question can be demonstrated
• peer reviewed scientific literature on other clinicalexperience of either the device in question or a similardevice for which equivalence can be demonstrated
• data from the manufacturer’s post-marketsurveillance system, in particular post-market clinicalfollow-up
50

MedDev 2.7.1 Rev 4 / MDR – Equivalence
Technical
• be of similar design• used under similar conditions of
use• have similar specifications and
properties (e.g. physicochemicalproperties such as intensity ofenergy, tensile strength, viscosity,surface characteristics,wavelength, software algorithms,porosity, particle size,nanotechnology, specific mass,atomic inclusions –nitrocarburising, oxidability)
• use similar deployment methods(if relevant)
• have similar principles ofoperation and criticalperformance requirements
Biological
• use same materials orsubstances in contact with thesame human tissues or bodyfluids
• for a similar kind andduration of contact and similarrelease characteristics ofsubstances
• including degradationproducts and leachables
• Exceptions can be foreseen for devices incontact with intact skin and minorcomponents; in these cases risk analysisresults may allow the use of similar materialstaking into account the role and nature of thesimilar material. Evaluators should considerbiological safety (e.g. ISO 10993) as well asother aspects necessary for a comprehensivedemonstration of equivalence. A justificationexplaining the situation should be provided forany difference.
Clinical
• used for the same clinicalcondition or intendedpurpose (including similarseverity and stage ofdisease, medical indication)
• at the same site in the body• in a similar population
(including age, gender,anatomy, physiology)
• have same kind of user• not foreseen to deliver
significantly differentperformances
• have similar relevant criticalperformance according to theexpected clinical effect for aspecific intended purpose
51

Attendees
51

52
Post-market Surveillance

PMS System• As part of the Quality Management System, QMS, develop a PMS System.
– The PMS system elements include planning, establishing, documenting,implementing , maintaining, and updating
• Data from the PMS system:
– Updates the benefit/risk determination and improves risk management
– Updates design, manufacturing, IFU and labeling
– Updates Clinical Evaluation
– Updates the Summary of Safety and Clinical Performance
– Identifies the need to corrective action, preventive action, or filed safetycorrective action
– Identifies options to improve the usability, performance, or safety
– Contributes to post-market surveillance of other devices
– Detects and reports trends
53MDR Article 83

PMS System Components
• The components of the PMS system include:
– A PMS Plan {Article 84, Annex III, Section 1.1}
– A PMS report
• For Class I device a PMS Report {Article 85}
• For Class IIa, IIb, and III devices a Periodic Safety UpdateReport, PSUR {Article 86}
• Annex III includes the requirements for the Technical Documentationon Post-market Surveillance
• For Class III and implantable devices, a Summary of Safety andClinical Performance {Article 32}
54

PMCF
• Post-market Clinical Follow-up, PMCF, means a continuous process thatupdates the clinical evaluation. PMCF is addressed in the manufacturer'spost-market surveillance (PMS) plan.
• PMCF proactively collects and evaluates clinical data to:
– Confirm safety and performance throughout the lifetime
– Ensure the identified risks remain acceptable
– Detecting emerging risks
• Develop a PMCF plan
– There are 8 required element including linkage to the Clinical EvaluationReport and Risk Management
• Implement the PMCF plan
• Document the results in a PMCF Evaluation Report
– Use the results to update Clinical Evaluation and Risk Management
55MDR Annex XIV Part B

Manufacturers• Look at the PMS System requirements
• Develop a plan to implement them including:
– Risk Management
– Clinical Evaluation
– PMCF
– Clinical Investigation, if applicable
• Develop a plan to include the information in the:
– Annex II Technical documentation
– Annex III Technical documentation on post-market surveillance
56
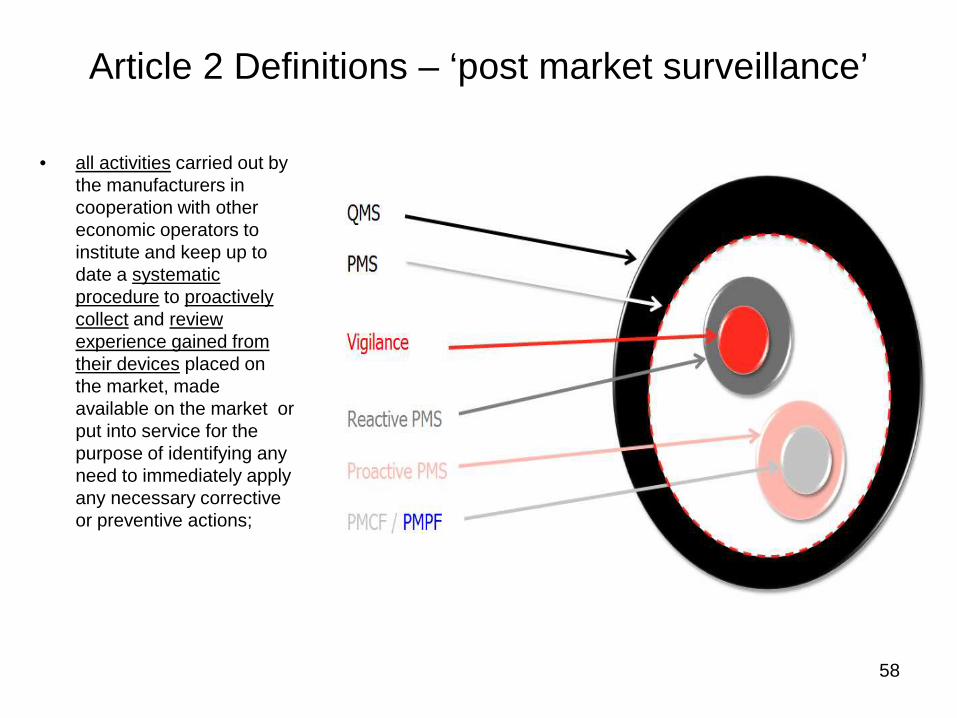
Article 2 Definitions – ‘post market surveillance’
• all activities carried out bythe manufacturers incooperation with othereconomic operators toinstitute and keep up todate a systematicprocedure to proactivelycollect and reviewexperience gained fromtheir devices placed onthe market, madeavailable on the market orput into service for thepurpose of identifying anyneed to immediately applyany necessary correctiveor preventive actions;
58

ANNEX XIV – CLINICAL EVALUATION AND POST-MARKET CLINICAL FOLLOW-UPPART B: POST-MARKET CLINICAL FOLLOW-UP
5. PMCF shall be understood to be a continuousprocess that updates the clinical evaluation referredto in Article 61 and Part A of this Annex and shall beaddressed in the manufacturer's post-marketsurveillance plan.
• When conducting PMCF, the manufacturer shallproactively collect and evaluate clinical data fromthe use in or on humans of a device, with the aim ofconfirming the safety and performance throughoutthe expected lifetime of the device, of ensuring thecontinued acceptability of identified risks and ofdetecting emerging risks on the basis of factualevidence.
6. PMCF shall be performed pursuant to adocumented method laid down in a PMCF plan.
59

ANNEX XIV – CLINICAL EVALUATION AND POST-MARKET CLINICAL FOLLOW-UP
PART B: POST-MARKET CLINICAL FOLLOW-UP
• The PMCF plan shall specify the methods and procedures for proactively collecting andevaluating clinical data with the aim of:
confirming the safety andperformance of the devicethroughout its expected lifetime
identifying previously unknown side-effects and monitoring the identifiedside-effects and contraindications
identifying and analysing emergentrisks on the basis of factualevidence
ensuring the continued acceptabilityof the risk-benefit ratio (Annex I)
identifying possible systematicmisuse or off-label use of thedevice, with a view to verifying thatthe intended purpose is correct
60

60
Questions