numerical examination of performance of some exchange-correlation functionals for molecules...
TRANSCRIPT

Numerical Examination of Performance of SomeExchange-Correlation Functionals for Molecules
Containing Heavy Elements
FAN WANG, LEMIN LIState Key Laboratory of Rare Earth Materials Chemistry and Applications,
College of Chemistry, and Molecular Engineering, Peking University,Beijing 100871, People’s Republic of China
Received 24 June 2003 ; Accepted 12 November 2003
Abstract: The performance of 17 exchange-correlation functionals for molecules containing heavy elements arenumerically examined through four-component relativistic density DFT calculations. The examined functionals showthe similar accuracy as they do for the molecules containing light elements only except for bond lengths. LDA and OP86produce good results for bond lengths and frequencies but bad bond energies. Different functionals do not show muchdifferent performance for bond energies except LDA. BP86 and GP86 produce results with average accuracy while LYPdoes not perform well. Although encouraging results are obtained with functional B97GGA-1, other heavily parame-terized and meta-GGA functionals do not produce impressive results.
© 2004 Wiley Periodicals, Inc. J Comput Chem 25: 669–677, 2004
Key words: density functional theory; exchange-correlation functional; generalized gradient approxiation (GGA);meta-GGA; four-component-relativistic DFT calculation
Introduction
Density functional theory (DFT)1 has become one of the mostpopular methods in quantum chemical studies. The exact form ofthe exchange-correlation (XC) functional is still unknown, andapproximations must be made to arrive at a practically applicableXC functional. The accuracy of the approximate DFT relies on theperformance of the XC functionals. The first approximate XCfunctional is based on local density approximation (LDA)2–4 inwhich the XC functional only depends on electronic densities. Theperformance of DFT is improved significantly over LDA when thegeneralized gradient approximation was introduced by Becke,5
Perdew,6 and others, in which besides electronic densities the XCfunctionals also depend on density gradients. Later, Becke intro-duced the hybrid functionals7 by mixing some “exact” exchangeenergy into the approximate exchange energy functionals, whichperform even better than the GGA functionals, especially fororganic molecules. A few years ago, the “meta-GGA” XC func-tionals were proposed8,9 in which the kinetic energy density is alsoinvolved in the functionals. Developing more accurate exchange-correlation functionals is still one of the most important subjects inDFT.
Besides some theoretical requirements that should be satisfiedfor an exact XC functional, the quality of an approximate XCfunctional is mainly evaluated through the results obtained by DFT
calculations using the XC functional under consideration. Mostexchange-correlation functionals used nowadays involve some pa-rameters fitted to experimental data or to results from accurate abinitio calculations related to some properties of a set of atoms ormolecules containing light elements only, for example, the G2 testset of Pople and coworkers, although a few coordination com-pounds containing the first series of d-transition elements areinvolved in some cases. Their performance in the calculations ofatoms, ions, or molecules containing light main group (up to thesecond row) elements only has been studied extensively.10,11 Al-though some of these XC functionals have also been used in thecalculations of molecules containing heavy elements by manyauthors and satisfactory results can be obtained in some cases, theperformance of the XC functionals for the systems containingheavy elements has not been systematically examined yet to ourknowledge.
It is well-known that relativistic effects12 must be taken intoaccount when the molecules containing heavy elements are studiedtheoretically. In addition, the electron correlation has to be con-
Correspondence to: Lemin Li; e-mail: [email protected]
Contract/grant sponsor: National Natural Science Foundation of China(NNSFC); contract/grant number: 20333020
Contract/grant sponsor: China Postdoctoral Science Foundation (to F.W.)
© 2004 Wiley Periodicals, Inc.

sidered also. The nonadditivity between the relativistic effects andelectron correlation makes the computation more complicated. Inthis context, density functional theory provides a good compro-mise between the computational cost and the quality of the calcu-lated results. When the large and complicated systems containingheavy elements are investigated, the DFT with relativity incorpo-rated is even more appealing, because a large number of electronshave to be explicitly treated in the calculations. Aiming at theelectronic structure calculation of atoms, molecules, and solids,DFT has been generalized to the relativistic domain on the basis ofthe no-pair Dirac–Coulomb or Dirac–Coulomb–Breit Hamilto-nian, and a few schemes to construct approximate relativisticexchange-correlation functionals have been proposed.13–19 Therelativistic DFT calculations have been done at various levels oftheory, either variationally or perturbatively. In the last years,several four-component relativistic DFT methods20–27 have beendeveloped for electronic structure calculations of molecules con-taining heavy elements. The calculation practice showed that rel-ativistic corrections to the exchange-correlation functionals, bothin LDA and GGA, affect the calculated results of the innermostorbitals indeed, but have no discernible effects on the calculatedresults of valence properties, such as the excitation energies andmolecular spectroscopic constants of molecules containing heavyand even superheavy elements.28–30 Therefore, today in currentpractical relativistic density functional calculations of moleculesand solids with the valence properties involved only, the existentforms of various nonrelativistic exchange-correlation functionalsare extensively adopted with the relativistic electronic density asthe argument. To justify this common practice further, the perfor-mance of some prevail nonrelativistic exchange-correlation func-tionals has been numerically examined by us through four-com-ponent relativistic density functional calculations of a seriesmolecules containing heavy elements with the BDF package.21–23
The results are presented and discussed in the present article.
Computational Details
A total of 17 prevail nonrelativistic XC functionals are examinedin the present work. Besides LDA, a part of GGA XC functionalsby combination of exchange density functionals Becke88,5 Per-dew86,31 PBE,32 revPBE,33 RPBE34 G96,35 FT97,36 and OPTX37
with correlation density functionals Perdew866 or LYP38 are in-volved. The heavily parameterized XC functionals HCTH39,40 andthe functional B97GGA�1 first proposed by Becke41 and laterreparameterized by Cohen et al.11 are included. Some meta-GGAfunctionals such as PKZB,9 VS98,42 and t-HCTH43 are also in-cluded. The exchange-correlation potential proper to each XCfunctionals was used in the SCF iterations of individual calcula-tions. It should be noted that some functionals such as OPTX,VS98 do not satisfy the requirement of homogeneous limit. Thehybrid functionals have not been involved in this work because theexact exchange potential and energy cannot be calculated withBDF program right now, and taking into account the fact that someof the afore-mentioned functionals are declared to perform as goodas or even better than the hybrid functionals.11 A set of 23closed-shell diatomic molecules containing heavy main-group el-ements were selected in the examination based on the following
consideration. (1) There are relatively definite experimental data ofbond lengths, fundamental vibrational frequencies, and bond en-ergies for these molecules. (2) In the molecules composed ofmain-group elements the chemical bonding is mainly contributedby s and p orbitals. The electronic structure of these molecules isusually simpler than that of the molecules containing d-block andf-block transitional elements, which may favor to obtain somedefinite conclusions. (3) Significantly different type of bonding,from strong polarized to covalent one, should be involved in theselected molecules. (4) Only closed-shell systems are consideredbecause there are some unsolved problems in dealing with theopen shells in the relativistic density functional calculations. Evenfor the closed-shell molecules, the calculation of bond energiesfaces the problem to calculate the energies of open-shelled atoms,which were temporarily evaluated with the “moment polarized”scheme44 in this work. (5) Only diatomic molecules are selectedbecause the computational efforts are usually quite demanding forfour-component relativistic DFT calculations with large basis setsand high numerical accuracy. The frozen-core approximation wasadopted. In our four-componemt relativistic DFT calculations withBDF program, the atom-centered and j-adapted spinor basis func-tions are used. The numerical atomic orbitals are the backbone ofbasis sets, which are obtained in advance by solving the four-component Dirac–Kohn–Sham equation for free atoms with acentral field. To describe the deformation and polarization of theelectron distribution during molecular formation, the valence spaceis further augmented, for each spinor, with several optimizedSlater-type orbitals (STOs). It has been known for a long time thatkinetic-balanced conditions have to be imposed on the smallcomponent basis sets to avoid the variational collapse. In thepresent case, the small and large components of the numericalbasis sets satisfy the exact coupling relationship. Because the smallcomponent density is localized in the vicinity of the nucleus,satisfying the kinetic-balanced condition for the valence STOs isnot a serious problem; one has some degree of freedom to approx-imate the valence STOs of the small components. Thus, in ourfour-component calculations the potential independent approxi-mate kinetic-balanced condition is adopted for the valence STOs:
Table 1. Comparison of Atomization Energies Using Different BasisSets with BLYP Functional (kcal/mol).
This work DZPa TZ2Pa cc-PVQZa
S2 104.34 101.16 104.37 105.73ClO 71.87 75.24 71.87 73.25OH 105.03 114.57 108.82 109.33SO 132.70 133.77 131.68 132.96O2 132.62 147.73 131.68 132.96HCl 104.23 103.66 103.98 104.12ClF 66.30 62.83 65.27 66.10Cl2 56.90 50.58 55.58 56.64H2 109.41 112.13 109.47 109.54F2 48.06 51.55 49.26 49.57HF 141.12 137.86 139.90 140.43
aData taken from ref. 49.
670 Wang and Li • Vol. 25, No. 5 • Journal of Computational Chemistry

�S �1
2c� � p�L,
where c is the light velocity, �S and �L are the basis functions ofthe small and large components, � and p are the vector of Paulispin matrices and the momentum operator, respectively. The j-adaptation allows one-to-one correspondence between small andlarge component basis functions, and facilitates the construction ofsymmetry-adapted basis functions according to finite doublegroups. It should be noted that for heavy elements, particularly the
heavy p-block elements, it is essential to construct decent bases torepresent the weak singularity of the innermost shells and the largesplitting of the 2p shells. In addition, polarization functions areadded to all atoms. Practical calculations show that such basis setsare compact and efficient and the basis set error is quite small. Allmatrix elements were evaluated by numerical quadrature. Theatomic partitioning scheme developed by Becke46 for numericalevaluation of integrals was adopted. The variant of Gauss–Cheby-shev quadrature suggested by Perez-Jorda et al.47 was used forradial integrals and Lebedev quadrature48 was used for angular
Table 2. Calculated Bond Lengths with Different XC Functionals (Å).
Ag2 AgH AgF AgCl AgBr AgI HI IF ICl IBr I2
Expt45 2.53 1.618 1.983 2.281 2.393 2.544 1.609 1.910 2.321 2.469 2.666LDA 2.493 1.592 1.950 2.234 2.352 2.509 1.630 1.925 2.328 2.477 2.677B97GGA � 1 2.581 1.616 2.009 2.302 2.422 2.582 1.614 1.930 2.336 2.488 2.693HCTH/407 2.623 1.626 2.023 2.314 2.440 2.604 1.618 1.942 2.343 2.502 2.709t-HCTH 2.572 1.619 2.004 2.305 2.425 2.585 1.616 1.932 2.344 2.497 2.702B88P86 2.569 1.615 2.001 2.295 2.417 2.579 1.632 1.956 2.366 2.521 2.726BLYP 2.608 1.625 2.021 2.326 2.452 2.620 1.638 1.978 2.401 2.560 2.771PBE 2.577 1.618 2.005 2.295 2.417 2.579 1.632 1.955 2.359 2.514 2.717revPBE 2.604 1.627 2.023 2.315 2.439 2.602 1.633 1.963 2.368 2.524 2.729P86P86 2.598 1.626 2.016 2.315 2.443 2.609 1.640 1.977 2.391 2.553 2.763RPBE 2.615 1.629 2.029 2.321 2.445 2.609 1.634 1.966 2.371 2.528 2.733GP86 2.556 1.610 1.995 2.287 2.407 2.568 1.630 1.950 2.357 2.509 2.713GLYP 2.594 1.621 2.015 2.318 2.442 2.608 1.635 1.969 2.391 2.548 2.758FT97 2.561 1.625 2.012 2.305 2.443 2.603 1.643 1.980 2.387 2.569 2.779PKZB 2.578 1.628 2.018 2.303 2.424 2.583 1.630 1.955 2.354 2.508 2.711VS98 2.586 1.626 2.022 2.327 2.451 2.616 1.618 1.946 2.363 2.526 2.738OP86 2.582 1.616 2.008 2.285 2.405 2.563 1.617 1.928 2.317 2.468 2.667OLYP 2.624 1.627 2.028 2.318 2.441 2.605 1.622 1.950 2.350 2.504 2.709Averagea 2.584 1.620 2.011 2.304 2.427 2.590 1.628 1.953 2.360 2.517 2.723Deviationb 0.054 0.002 0.028 0.023 0.034 0.046 0.019 0.043 0.039 0.048 0.057
TlH TlF TlCl TlBr TlI PbO PbS PbSe PbTe Au2 AuH Bi2
Expt.45 1.870 2.084 2.485 2.618 2.814 1.922 2.287 2.402 2.595 2.472 1.524 2.661LDA 1.869 2.076 2.460 2.599 2.786 1.913 2.285 2.401 2.594 2.456 1.526 2.641B97GGA � 1 1.894 2.110 2.517 2.663 2.857 1.919 2.294 2.414 2.611 2.501 1.523 2.664HCTH/407 1.908 2.126 2.529 2.678 2.880 1.930 2.303 2.431 2.632 2.530 1.530 2.689t-HCTH 1.893 2.105 2.514 2.660 2.854 1.922 2.298 2.418 2.615 2.501 1.527 2.668B88P86 1.900 2.122 2.520 2.665 2.860 1.941 2.318 2.440 2.638 2.514 1.536 2.690BLYP 1.919 2.145 2.553 2.696 2.903 1.958 2.343 2.469 2.671 2.550 1.544 2.728PBE 1.901 2.123 2.518 2.664 2.858 1.940 2.315 2.439 2.637 2.516 1.537 2.687revPBE 1.913 2.138 2.537 2.683 2.882 1.947 2.323 2.450 2.648 2.532 1.540 2.700P86P86 1.918 2.140 2.542 2.687 2.891 1.958 2.337 2.467 2.669 2.550 1.547 2.728RPBE 1.916 2.143 2.543 2.688 2.889 1.950 2.326 2.452 2.653 2.538 1.541 2.703GP86 1.894 2.114 2.513 2.658 2.848 1.933 2.310 2.429 2.626 2.502 1.533 2.675GLYP 1.912 2.137 2.545 2.687 2.890 1.950 2.335 2.458 2.658 2.537 1.540 2.713FT97 1.916 2.134 2.538 2.693 2.895 1.959 2.322 2.465 2.667 2.538 1.550 2.730PKZB 1.914 2.131 2.526 2.672 2.869 1.939 2.309 2.431 2.629 2.507 1.538 2.674VS98 1.911 2.133 2.548 2.695 2.905 1.938 2.316 2.443 2.646 2.533 1.531 2.701OP86 1.890 2.109 2.500 2.649 2.838 1.917 2.287 2.408 2.603 2.492 1.524 2.650OLYP 1.910 2.133 2.535 2.681 2.882 1.935 2.311 2.435 2.635 2.529 1.531 2.685Averagea 1.905 2.125 2.526 2.672 2.870 1.938 2.314 2.438 2.637 2.519 1.535 2.690Deviationb 0.035 0.041 0.041 0.054 0.056 0.016 0.027 0.036 0.042 0.047 0.011 0.029
aThe average of all values calculated with different XC functionals.bDeviation of the average from the experimental value.
Performance Exam of Exchange-Correlation Functionals 671
integrals. The numbers of the radial and angular grid points usedin the numerical integration are 100 and 80 for heavy elements, or80 and 60 for light elements, respectively. It has been checked thatthe required accuracy of numerical integration can be reached byuse of these numbers of grid points.
Results and Discussion
To check the quality of the basis sets used in this work, theatomization energies of some diatomic molecules composed of
light elements only have been calculated using BLYP functional.The result is listed in Table 1. The correspondent results calculatedby Cohen et al.49 with different basis sets are also listed forcomparison. It can be seen that the quality of the basis sets used inthis work is at least as good as that of TZ2P basis sets, and evenbetter in some cases.
The following 17 XC functionals are examined in this work:LDA (LDAX2 � VNW3), B97GGA�1,11 HCTH/407,40 t-HCTH,43
B88P86 (B885 � P866), BLYP (B885 � LYP38), PBE,32
revPBE,33, P86P86 (P8631 � P866), RPBE,34 GP86,35 GLYP,35
FT97,36 PKZB,9 VS98,42 OP86 (OPTX37 � P866), OLYP
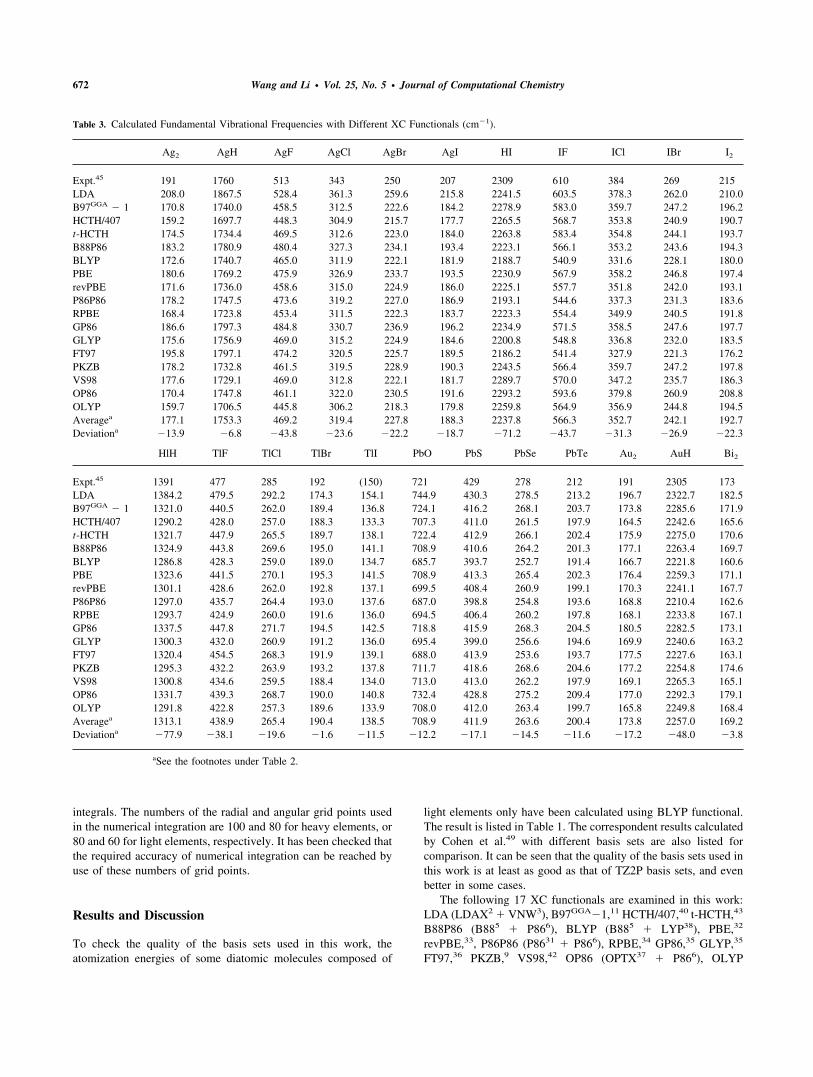
Table 3. Calculated Fundamental Vibrational Frequencies with Different XC Functionals (cm�1).
Ag2 AgH AgF AgCl AgBr AgI HI IF ICl IBr I2
Expt.45 191 1760 513 343 250 207 2309 610 384 269 215LDA 208.0 1867.5 528.4 361.3 259.6 215.8 2241.5 603.5 378.3 262.0 210.0B97GGA � 1 170.8 1740.0 458.5 312.5 222.6 184.2 2278.9 583.0 359.7 247.2 196.2HCTH/407 159.2 1697.7 448.3 304.9 215.7 177.7 2265.5 568.7 353.8 240.9 190.7t-HCTH 174.5 1734.4 469.5 312.6 223.0 184.0 2263.8 583.4 354.8 244.1 193.7B88P86 183.2 1780.9 480.4 327.3 234.1 193.4 2223.1 566.1 353.2 243.6 194.3BLYP 172.6 1740.7 465.0 311.9 222.1 181.9 2188.7 540.9 331.6 228.1 180.0PBE 180.6 1769.2 475.9 326.9 233.7 193.5 2230.9 567.9 358.2 246.8 197.4revPBE 171.6 1736.0 458.6 315.0 224.9 186.0 2225.1 557.7 351.8 242.0 193.1P86P86 178.2 1747.5 473.6 319.2 227.0 186.9 2193.1 544.6 337.3 231.3 183.6RPBE 168.4 1723.8 453.4 311.5 222.3 183.7 2223.3 554.4 349.9 240.5 191.8GP86 186.6 1797.3 484.8 330.7 236.9 196.2 2234.9 571.5 358.5 247.6 197.7GLYP 175.6 1756.9 469.0 315.2 224.9 184.6 2200.8 548.8 336.8 232.0 183.5FT97 195.8 1797.1 474.2 320.5 225.7 189.5 2186.2 541.4 327.9 221.3 176.2PKZB 178.2 1732.8 461.5 319.5 228.9 190.3 2243.5 566.4 359.7 247.2 197.8VS98 177.6 1729.1 469.0 312.8 222.1 181.7 2289.7 570.0 347.2 235.7 186.3OP86 170.4 1747.8 461.1 322.0 230.5 191.6 2293.2 593.6 379.8 260.9 208.8OLYP 159.7 1706.5 445.8 306.2 218.3 179.8 2259.8 564.9 356.9 244.8 194.5Averagea 177.1 1753.3 469.2 319.4 227.8 188.3 2237.8 566.3 352.7 242.1 192.7Deviationa �13.9 �6.8 �43.8 �23.6 �22.2 �18.7 �71.2 �43.7 �31.3 �26.9 �22.3
HlH TlF TlCl TlBr TlI PbO PbS PbSe PbTe Au2 AuH Bi2
Expt.45 1391 477 285 192 (150) 721 429 278 212 191 2305 173LDA 1384.2 479.5 292.2 174.3 154.1 744.9 430.3 278.5 213.2 196.7 2322.7 182.5B97GGA � 1 1321.0 440.5 262.0 189.4 136.8 724.1 416.2 268.1 203.7 173.8 2285.6 171.9HCTH/407 1290.2 428.0 257.0 188.3 133.3 707.3 411.0 261.5 197.9 164.5 2242.6 165.6t-HCTH 1321.7 447.9 265.5 189.7 138.1 722.4 412.9 266.1 202.4 175.9 2275.0 170.6B88P86 1324.9 443.8 269.6 195.0 141.1 708.9 410.6 264.2 201.3 177.1 2263.4 169.7BLYP 1286.8 428.3 259.0 189.0 134.7 685.7 393.7 252.7 191.4 166.7 2221.8 160.6PBE 1323.6 441.5 270.1 195.3 141.5 708.9 413.3 265.4 202.3 176.4 2259.3 171.1revPBE 1301.1 428.6 262.0 192.8 137.1 699.5 408.4 260.9 199.1 170.3 2241.1 167.7P86P86 1297.0 435.7 264.4 193.0 137.6 687.0 398.8 254.8 193.6 168.8 2210.4 162.6RPBE 1293.7 424.9 260.0 191.6 136.0 694.5 406.4 260.2 197.8 168.1 2233.8 167.1GP86 1337.5 447.8 271.7 194.5 142.5 718.8 415.9 268.3 204.5 180.5 2282.5 173.1GLYP 1300.3 432.0 260.9 191.2 136.0 695.4 399.0 256.6 194.6 169.9 2240.6 163.2FT97 1320.4 454.5 268.3 191.9 139.1 688.0 413.9 253.6 193.7 177.5 2227.6 163.1PKZB 1295.3 432.2 263.9 193.2 137.8 711.7 418.6 268.6 204.6 177.2 2254.8 174.6VS98 1300.8 434.6 259.5 188.4 134.0 713.0 413.0 262.2 197.9 169.1 2265.3 165.1OP86 1331.7 439.3 268.7 190.0 140.8 732.4 428.8 275.2 209.4 177.0 2292.3 179.1OLYP 1291.8 422.8 257.3 189.6 133.9 708.0 412.0 263.4 199.7 165.8 2249.8 168.4Averagea 1313.1 438.9 265.4 190.4 138.5 708.9 411.9 263.6 200.4 173.8 2257.0 169.2Deviationa �77.9 �38.1 �19.6 �1.6 �11.5 �12.2 �17.1 �14.5 �11.6 �17.2 �48.0 �3.8
aSee the footnotes under Table 2.
672 Wang and Li • Vol. 25, No. 5 • Journal of Computational Chemistry

(OPTX37 � LYP3), where the expressions for individual approx-imate XC functionals are specified with the related references. Thecalculated results of bond lengths, vibrational frequencies for theexamined set of molecules using different XC functionals arelisted in Tables 2 and 3, respectively. The deviation statistics of thecalculated results by different XC functionals are listed in Table 4.It can be seen that BLYP, P86P86, FT97, GLYP, and RPBEfunctionals predict too long bond lengths and correspondentlypredict too low fundamental vibrational frequencies, while OP86,LDA, B97GGA �1, t-HCTH, and GP86 give satisfactory results. Itis interesting to note that LDA gives quite good results for bondlengths and fundamental vibrational frequencies. Handy et al.found that LDA is the best DFT model for some selected diatomicmolecules containing second-row elements,50 and pointed out thatit predicts better geometry parameters than other many GGAfunctionals.11 This result is in accordance with the observationknown to the nonrelativistic DFT calculations of molecules con-taining light elements of main groups only that the performance ofGGA functionals usually is even worse in predicting bond lengthsand vibrational frequencies than that of LDA. This may be due tothe fact that the bonding is substantially contributed by the p ands orbitals in both cases. However, the combination of OPTXexchange with Perdew86 correlation functional (OP86) gives verygood prediction on bond lengths and vibrational frequencies. Thismay mean that satisfying the homogeneous limit requirement maynot be necessary for getting satisfactory results in the calculationsof molecular systems, at least for the calculation of bond lengthsand vibrational frequencies.
The calculated bond energies are listed in Table 5. The requiredenergies of the component atoms are calculated with the spheri-
cally averaging “moment-polarized” electronic density of atomsbecause the ground states of atoms are usually degenerated. It isvery well known that the energy of an open-shell atom in itsground state cannot be definitely determined with current approx-imate functionals, because it is not invariant with respect to dif-ferent choices of ground state electronic densities.51 For the timebeing, there are two schemes to evaluate the energy of the open-shell atoms in ground states: to calculate the energy with thespherical averaging ground state electron density, or to calculatethe lowest energy by variation of the atomic electron density. Theatomic energy calculated with spherically averaging “moment-polarized” electronic density is not the lowest energy. In this work,the bond energies evaluated with respect to the lowest ground stateatomic energies are also calculated and given in Table 6 forcomparison. The differences between the bond energies evaluatedin these two ways are remarkable only for F, Cl, O, and Selements; thus, only the bond energies of those molecules contain-ing these elements are presented in Table 6. The deviation statisticsof the bond energies calculated with different approximate func-tionals are listed in Table 7. Obviously, the bond energies evalu-ated with respect to the lowest energies of component atoms inground states are indeed improved to some extent, although thecomputational efforts increase to a certain extent also. It can beseen from the presented data that except the bond lengths theexamined functionals do not show much different performance,variation of the standard deviation is 0.042 � 0.014 Å for bondlengths, 35.5 � 7.8 cm�1 for fundamental vibrational frequencies,and 0.232 � 0.026 or 0.197 � 0.030 eV for bond energies (withLDA excluded). As a whole they show the similar accuracy as theydo for the molecules containing light elements only except that in
Table 4. Deviation Statistics for the Calculated Bond Lengths and Fundamental Vibrational Frequencies withDifferent XC Functionals.a
Deviation statistics forbond lengths (BL)
Deviation statistics forvibrational frequencies (�)
� BLabs �BL �� BLmax �� BLmax � �abs �� �� �max �� �max
LDA 0.018 0.023 0.021 �0.047 14.9 28.1 107.5 �67.5B97GGA � 1 0.022 0.026 0.051 �0.002 22.8 27.3 0.0 �70.0HCTH/407 0.037 0.043 0.093 �0.000 34.3 40.6 0.0 �100.8t-HCTH 0.023 0.026 0.042 �0.000 23.8 27.8 0.0 �69.3B88P86 0.034 0.037 0.060 �0.003 24.3 31.1 20.9 �85.9BLYP 0.063 0.067 0.105 �0.000 40.0 49.2 0.0 �120.3PBE 0.033 0.035 0.051 �0.000 23.2 30.1 9.2 �78.1revPBE 0.047 0.049 0.074 �0.000 31.5 38.9 0.8 �89.9P86P86 0.057 0.061 0.097 �0.000 36.0 46.2 1.0 �115.9RPBE 0.051 0.054 0.085 �0.000 34.5 42.1 0.0 �97.3GP86 0.026 0.028 0.047 �0.008 20.2 26.4 37.3 �74.1GLYP 0.053 0.057 0.092 �0.000 34.4 42.9 0.0 �108.2FT97 0.055 0.060 0.113 �0.000 33.6 44.0 37.1 �122.8PKZB 0.034 0.036 0.055 �0.000 26.2 34.4 1.6 �95.7VS98 0.047 0.051 0.091 �0.000 27.8 32.7 0.0 �90.2OP86 0.013 0.018 0.052 �0.011 15.4 21.5 6.1 �59.3OLYP 0.041 0.045 0.094 �0.000 33.0 39.6 0.0 �99.2
a�Qabs, mean absolute deviation of Q; �Q, standard deviation of Q; �� Qmax (��Qmax), the maximal positive(negative) deviation of Q from experiments; where Q denotes bond length or vibrational frequency.
Performance Exam of Exchange-Correlation Functionals 673

the calculation of bond lengths, for example, the mean absolutedeviation of bond lengths, fundamental vibrational frequencies,and atomization energies of 32 small molecules calculated withBLYP functional by Pople et al.10 are 0.020 Å, 45 cm�1 and 5.6kcal/mol (0.243 eV), while the correspondent values of this workare 0.063 Å, 40.0 cm�1 and 0.168 eV (0.153 eV); those calculatedwith the combination of Becke 88 and VWN functionals are 0.018Å, 45 cm�1 and 4.4 kcal/mol (0.191 eV), while this work gives0.034 Å, 24.3 cm�1 and 0.174 (0.137) eV, respectively. However,there are some points different. In contrast to the calculations of
organic molecules, here the correlation functional LYP does notperform well almost in all cases. Functionals B88P86 and GP86only produce results with average accuracy for bond lengths,fundamental vibrational frequencies, and bond energies. For theheavily parameterized XC functionals, B97GGA�1 gives encour-aging results. HCTH/407 predicts fair good bond energies butgives disappointing bond lengths and vibrational frequencies. Themeta-GGA functionals such as VS98, t-HCTH and PKZB do notsignificantly and systematically improve the calculated results overthe GGA functionals even though the functional VS98 is heavily
Table 5. Calculated Bond Energies with Respect to the Energies of Constituent Atoms with Average OpenShell Density by Different XC Functionals (eV).
Ag2 AgH AgF AgCl AgBr AgI HI IF ICl IBr I2
Expt.45 1.66 2.39 3.67 3.24 3.05 2.6 3.20 2.92 2.18 1.83 1.56LDA 2.268 3.078 4.321 3.684 3.257 2.843 3.592 3.875 2.864 2.422 2.068B97GGA � 1 1.835 2.686 3.920 3.273 2.850 2.393 3.209 3.155 2.202 1.781 1.443HCTH/407 1.675 2.466 3.854 3.131 2.665 2.216 3.108 3.200 2.198 1.739 1.396t-HCTH 1.976 2.637 4.118 3.388 2.961 2.506 3.153 3.328 2.296 1.878 1.540B88P86 1.701 2.568 3.787 3.140 2.711 2.295 3.251 3.317 2.351 1.920 1.591BLYP 1.662 2.326 3.732 2.950 2.515 2.097 3.063 3.191 2.118 1.688 1.359PBE 1.753 2.374 3.857 3.229 2.796 2.384 3.153 3.381 2.445 2.012 1.685revPBE 1.546 2.274 3.679 3.059 2.626 2.209 3.082 3.184 2.276 1.848 1.527P86P86 1.825 2.474 3.832 3.104 2.660 2.264 3.183 3.323 2.293 1.852 1.533RPBE 1.539 2.242 3.673 3.047 2.612 2.194 3.072 3.166 2.258 1.829 1.509GP86 1.577 2.617 3.704 3.095 2.669 2.247 3.257 3.259 2.323 1.896 1.568GLYP 1.543 2.364 3.650 2.902 2.470 2.047 3.068 3.132 2.089 1.662 1.335FT97 1.729 2.646 3.605 3.010 2.583 2.233 3.221 3.096 2.176 1.766 1.502PKZB 1.505 2.565 3.662 3.110 2.684 2.261 3.308 3.153 2.296 1.872 1.549VS98 2.069 2.417 4.155 3.409 2.906 2.429 3.193 3.280 2.205 1.724 1.383OP86 1.400 2.639 3.724 3.180 2.740 2.292 3.372 3.271 2.428 1.984 1.640OLYP 1.379 2.375 3.663 2.985 2.533 2.080 3.176 3.128 2.173 1.726 1.383Averagea 1.670 2.479 3.788 3.126 2.686 2.259 3.179 3.223 2.258 1.824 1.496Deviationa 0.010 0.089 0.118 �0.114 �0.364 �0.341 �0.021 0.303 0.078 �0.006 �0.064
TlH TlF TlCl TlBr TlI PbO PbS PbSe PbTe Au2 AuH Bi2
Expt.45 2.06 4.60 3.83 3.43 2.77 3.87 3.46 3.06 2.57 2.31 3.36 2.03LDA 2.392 5.522 4.330 3.798 3.218 5.356 4.459 3.946 3.237 2.998 3.838 3.024B97GGA � 1 2.279 5.087 3.973 3.460 2.860 4.421 3.699 3.160 2.467 2.214 3.325 2.346HCTH/407 2.104 5.088 3.891 3.325 2.729 4.415 3.610 3.037 2.350 2.072 3.149 2.188t-HCTH 2.244 5.241 4.051 3.536 2.940 4.477 3.688 3.221 2.547 2.315 3.267 2.423B88P86 2.165 5.033 3.898 3.367 2.792 4.701 3.866 3.332 2.647 2.327 3.389 2.439BLYP 2.067 4.976 3.723 3.182 2.605 4.497 3.569 3.061 2.397 2.199 3.253 2.167PBE 2.044 5.100 3.980 3.445 2.875 4.817 4.000 3.438 2.752 2.380 3.262 2.537revPBE 2.011 4.930 3.841 3.308 2.740 4.600 3.831 3.253 2.566 2.145 3.151 2.336P86P86 2.148 5.077 3.867 3.313 2.753 4.705 3.756 3.251 2.613 2.377 3.356 2.407RPBE 2.008 4.921 3.830 3.295 2.727 4.573 3.807 3.221 2.534 2.117 3.132 2.304GP86 2.161 4.964 3.868 3.344 2.763 4.646 3.857 3.332 2.634 2.256 3.376 2.419GLYP 2.060 4.906 3.690 3.156 2.574 4.436 3.557 3.055 2.379 2.126 3.238 2.145FT97 2.292 4.884 3.827 3.301 2.789 4.331 3.572 3.534 3.074 2.596 3.548 2.753PKZB 2.247 4.910 3.886 3.361 2.789 4.600 3.982 3.383 2.667 2.171 3.351 2.411VS98 2.182 5.339 4.120 3.513 2.877 4.582 3.817 3.157 2.479 2.480 3.362 2.374OP86 2.204 4.988 3.955 3.418 2.825 4.677 4.037 3.378 2.631 2.029 3.301 2.402OLYP 2.102 4.923 3.775 3.224 2.626 4.448 3.713 3.084 2.359 1.910 3.170 2.103Averagea 2.145 5.023 3.886 3.347 2.767 4.558 3.773 3.244 2.569 2.232 3.289 2.360Deviationa 0.085 0.423 0.056 �0.083 �0.003 0.688 0.313 0.184 �0.001 �0.078 �0.071 0.330
aSee the footnotes under Table 2.
674 Wang and Li • Vol. 25, No. 5 • Journal of Computational Chemistry

parameterized. The heavily parameterized t-HCTH performs bet-ter, but it is still inferior to B97GGA�1. It may be of meaning tocompare the results of PKZB with those of PBE. These twofunctionals were both developed by Perdew et al. in a similar way,
while theoretically PKZB satisfies more rigorous conditions thanPBE does. Handy et al. came to a similar conclusion, that themeta-GGA functionals do not significantly go beyond the GGAfunctionals, and did not consider PKZB better than PBE from the
Table 6. Calculated Bond Energies with Respect to the Lowest Energies of the Constituent Atoms byDifferent XC Functionals (eV).
AgF AgCl IF ICl TlF TlCl PbO PbS
Expt.45 3.67 3.24 2.92 2.18 4.60 3.83 3.87 3.46LDA 4.328 3.695 3.882 2.894 5.531 4.341 5.302 4.459B97GGA � 1 3.696 3.194 2.931 2.118 4.858 3.894 4.122 3.618HCTH/407 3.652 3.041 3.003 2.101 4.885 3.801 4.136 3.522t-HCTH 3.825 3.282 3.038 2.185 4.946 3.945 4.107 3.593B88P86 3.537 3.126 3.073 2.276 4.782 3.823 4.349 3.752BLYP 3.517 2.909 2.983 2.085 4.761 3.682 4.224 3.516PBE 3.636 3.160 3.167 2.375 4.879 3.911 4.496 3.885revPBE 3.418 3.042 2.929 2.179 4.668 3.750 4.234 3.687P86P86 3.577 3.087 3.073 2.221 4.822 3.788 4.356 3.657RPBE 3.405 2.952 2.904 2.157 4.653 3.734 4.205 3.660GP86 3.445 3.017 3.007 2.244 4.705 3.789 4.272 3.725GLYP 3.427 2.859 2.919 2.052 4.684 3.646 4.141 3.485FT97 3.410 2.971 2.906 2.186 4.690 3.788 4.056 3.572PKZB 3.335 3.099 2.830 2.179 4.583 3.777 4.199 3.821VS98 3.811 3.269 2.940 2.053 4.996 3.980 4.207 3.718OP86 3.480 3.075 3.033 2.305 4.744 3.850 4.335 3.867OLYP 3.456 2.916 2.927 2.095 4.715 3.706 4.187 3.607Averagea 3.539 3.062 2.979 2.176 4.773 3.804 4.227 3.668Deviationa �0.131 �0.178 0.059 �0.004 0.173 �0.026 0.357 0.208
aSee the footnotes under Table 2.
Table 7. Deviation Statistics for the Calculated Bond Energies (BE) with Different XC Functionals.a
Deviation statistics for thebond energies in Table 5
Deviation statistics for thebond energies in Table 6
� BEabs �BE (�� BE)max (�� BE)max � BEabs �BE (�� BE)max (�� BE)max
LDA 0.617 0.659 0.999 �0.000 0.625 0.666 0.999 �0.000B97GGA � 1 0.155 0.193 0.487 �0.207 0.118 0.149 0.316 �0.216HCTH/407 0.158 0.199 0.488 �0.384 0.135 0.172 0.285 �0.394t-HCTH 0.187 0.244 0.641 �0.094 0.126 0.165 0.393 �0.104B88P86 0.174 0.225 0.433 �0.309 0.137 0.176 0.409 �0.313BLYP 0.168 0.217 0.376 �0.505 0.153 0.202 0.161 �0.504PBE 0.210 0.272 0.540 �0.224 0.171 0.218 0.507 �0.235revPBE 0.165 0.210 0.371 �0.394 0.143 0.184 0.306 �0.414P86P86 0.162 0.215 0.477 �0.360 0.127 0.167 0.377 �0.364RPBE 0.166 0.208 0.347 �0.408 0.154 0.193 0.274 �0.431GP86 0.169 0.219 0.397 �0.353 0.144 0.186 0.389 �0.361GLYP 0.180 0.231 0.306 �0.553 0.172 0.227 0.115 �0.550FT97 0.205 0.275 0.723 �0.437 0.198 0.269 0.723 �0.407PKZB 0.182 0.234 0.522 �0.339 0.158 0.207 0.381 �0.368VS98 0.205 0.271 0.739 �0.177 0.148 0.185 0.409 �0.185OP86 0.221 0.268 0.577 �0.308 0.184 0.220 0.407 �0.341OLYP 0.182 0.234 0.323 �0.520 0.179 0.230 0.147 �0.537
a�Qabs, mean absolute deviation of Q; �Q, standard deviation of Q; �� Qmax (��Qmax), the maximal positive(negative) deviation of Q from experiments; where Q denotes bond energy.
Performance Exam of Exchange-Correlation Functionals 675

assessment of some XC functionals with a set of atoms, molecules,and ions containing light elements only.11 These functionals per-form pretty well for molecules containing light elements only, buttheir performance is not impressive when the molecules containingheavy elements are considered. It may not be a good way tooptimize the approximate XC functionals through heavily param-eterization for reaching generally accurate functionals, becausemany chemical interesting systems may not always be included inthe training set.
Scrutinizing the deviations of the calculated results from theexperimental data along the series of F, Cl, Br, I or O, S, Se, Te,it can be found that the deviations change regularly. They increasegradually for the calculated bond lengths, while they change in aninverted order for the calculated fundamental vibrational frequen-cies in most cases. For the calculated bond energies they alsochange regularly. This fact may show implicitly some commondefects of the current approximate XC functionals, and it is worthwhile to study further.
Of course, it should be noted that the number of moleculesinvolved in our examined set is limited and only a part of molec-ular properties is considered in the present work. Further studies onmore molecules, particularly those containing d-block and f-blockelements, and involving more molecular properties are needed forobtaining more definite conclusion about the performance of dif-ferent approximate XC functionals in the calculations of moleculescontaining heavy elements. Further work is in progress.
Conclusion
In the present work, the performance of 17 current approximateexchange-correlation functionals for the molecules containingheavy elements are examined through four-componenet relativisticdensity functional calculations of a set of 23 diatomic moleculescontaining heavy main-group atoms. It is found that for the mol-ecules containing heavy elements in which chemical bonding iscontributed mainly by s and p orbitals, the examined approximateXC functionals show the similar accuracy as they do in thecalculation of molecules containing light elements only, except forthe calculation of bond lengths. LDA and OP86 functionals predictquite good bond lengths and vibrational frequencies, while allother approximate XC functionals predict too long bond lengthsand too low fundamental vibrational frequencies. Different XCfunctionals do not show much difference in performance for cal-culation of bond energies except LDA which predicts too large abond energy as usual. The bond energies evaluated with respect tothe lowest energies of component atoms in ground states areslightly improved compared to those calculated with the averagingground electron density. B88P86 and GP86 produce results withaverage accuracy in all aspects. In contrast to the calculation oforganic molecules, the correlation functional LYP does not showexcellent performance in all cases. The heavily parameterized XCfunctionals and meta-GGA XC functionals do not produce signif-icantly better results than other GGA functionals, although encour-aging results are produced with the functional B97GGA�1. It isfound that the deviations vary regularly with the change of elec-tronegativity of the elements combined to the heavy atoms forhalide and chalcogenide, which needs further study. It seems that
LDA can be used to reliably predict bond lengths, while B88P86and GP86 may give relatively more stable results in the calcula-tions of molecules containing heavy main group elements with anaverage accuracy in all aspects with moderate computational ef-forts.
References
1. Parr, R. G.; Yang, W. Density-Functional Theory of Atoms andMolecules; Clarendon: Oxford, 1989.
2. Kohn, W.; Sham, L. J. Phys Rev A 1965, 140, 1133.3. Vosko, S. H.; Wilk, L.; Nusair, M. Can J Phys 1980, 58, 1200.4. Perdew, J. P.; Wang, Y. Phys Rev B 1992, 45, 13244.5. Becke, A. D. Phys Rev A 1988, 38, 3098.6. Perdew, J. P. Phys Rev B 1986, 33, 8822.7. Becke, A. D. J Chem Phys 1993, 98, 5648.8. Becke, A. D.; Roussel, M. R. Phys Rev A 1989, 39, 3761.9. Perdew, J. P.; Kurth, S.; Zupan, A.; Blaha, P. Phys Rev Lett 1999, 82,
2544.10. Johnson, B. G.; Gill, P. M. W.; Pople, J. A. J Chem Phys 1993, 98,
5612.11. Cohen, A. J.; Handy, N.C. Chem Phys Lett 2000, 316, 160.12. Pyykko, P. Chem Rev 1988, 88, 563.13. Ellis, D. E. J Phys B 1977, 10, 1.14. Rajagopal, A. K. J Phys C 1978, 11, L943.15. MacDonald A. H.; Vosko S. H. J Phys C 1979, 12, 2977.16. Engel, E.; Keller, S.; Dreizler, R. M. In Electronic Density Functional
Theory: Recent Progress and New Directions; Dobson, J. F.; Vignale,G.; Das, M. P., Eds.; Plenum: New York, 1997, p. 149.
17. Engel, E.; Keller, S.; Bonetti, A. F.; Muller, H.; Dreizler, R. M. PhysRev A 1995, 52, 2750.
18. Engel, E.; Keller, S.; Dreizler, R. M. Phys Rev A 1996, 53, 1367.19. Engel, E.; Bonetti, A. F.; Keller, S.; Andrejkovics, I.; Dreizler, R. M.
Phys Rev A 1998, 58, 964.20. Rosen, A.; Ellis, D. E. J Chem Phys 1975, 62, 3039.21. Liu, W.; Hong, G.; Li, L.; Xu, G. Chin Sci Bull 1996, 41, 651.22. Liu, W.; Hong, G.; Dai, D.; Li, L.; Dolg, M. Theor Chem Acc 1997,
96, 75.23. Liu, W.; Wang, F.; Li, L. J Theor Comput Chem 2003, 2, 257.24. Varga, S.; Fricke, B.; Nakamatsu, H., Mukoyama, T.; Anton, J.;
Geschke, D.; Heitmann, A.; Engel, E.; Bastug, T. J Chem Phys 2000,112, 3499.
25. Yanai, T.; Iikura, H.; Nakajima, T.; Ishikawa, Y.; Hirao, K. J ChemPhys 2001, 115, 8267.
26. Saue, T.; Helgaker, T. J Comput Chem 2001, 23, 814.27. Quiney, H. M.; Belanzoni, P. J Chem Phys 2002, 117, 5550.28. Mayer, M.; Haberlen, O. D.; Rosh, N. Phys Rev A 1996, 54, 4775.29. Liu, W.; Kuchle, W.; Dolg, M. Phys Rev A 1998, 58, 1103.30. Liu W.; van Wullen, C. J Chem Phys 1999, 110, 3730.31. Perdew, J. P.; Wang, Y. Phys Rev B 1986, 33, 8800.32. Perdew, J. P.; Burke, K.; Ernzehof, M. Phys Rev Lett 1996, 77, 3865.33. Zhang, Y.; Yang, W. Phys Rev Lett 1999, 80, 890.34. Hammer, B.; Hansen, L. B.; Norskov, J. K. Phys Rev B 1999, 59,
7413.35. Gilla, P. M. W. Mol Phys 1996, 89, 433.36. Filatov, M.; Thiel, W. Mol Phys 1997, 91, 847.37. Handy, N. C.; Cohen, A. J. Mol Phys 2001, 99, 403.38. Lee, C.; Yang, W.; Parr, R. G. Phys Rev B 1988, 37, 785.39. Hamprecht, F. A.; Cohen, A. J.; Tozer, D. J.; Handy, N. C. J Chem
Phys 1998, 109, 6264.
676 Wang and Li • Vol. 25, No. 5 • Journal of Computational Chemistry

40. Boese, A. D.; Handy, N. C. J Chem Phys 2001, 114, 5497.41. Becke, A. D. J Chem. Phys 1997, 107, 8554.42. Voorhis, T. V.; Scuseria, G. E. J Chem Phys 1998, 109, 400.43. Boese, A. D.; Handy, N. C. J Chem Phys 2002, 116, 9559.44. Ellis, D. E.; Goodman, G. L. Int J Quantum Chem 1984, 25, 185.45. Huber, K. P.; Herzberg, G. Molecular Spectra and Molecular Structure
IV: Constants of Diatomic Molecules; Van Nostrand Reihold Co.:New York, 1979.
46. Becke, A. D. J Chem Phys 1988, 88, 2547.47. Perez–Jorda, J. M.; San-Fabian, E.; Moscardo, F. Comput Phys Com-
mun 1992, 70, 271.48. Lebedev, V. I. Zh Vychisi Mat Mat Fiz 1975, 15, 48.49. Cohen, A. J.; Handy, N. C. J Chem Phys 2002, 117, 1470.50. Neumann, R.; Nobes, R. H.; Handy, N. C. Mol Phys 1996, 87, 1.51. Baerends, E. J.; Branchadell, V.; Sodupe, M. Chem Phys Lett 1997,
265, 481.
Performance Exam of Exchange-Correlation Functionals 677