non-mendelian mitochondrial inheritance as a cause of progressive genetic sensorineural hearing loss
TRANSCRIPT

INTERNATIDMl JOURNAL OF
Pediatric
International Journal of Pediatric Otorhinolaryngology
ELSEVIER 30 (1994) 91-104
Review article
Non-Mendelian mitochondrial inheritance as a cause of progressive genetic sensorineural
hearing loss
Michael Golda3b, Isabelle Rapin* b3c
“Department of Neurology, Universiiy of South Florida College of Medicine,
Tampa. FL, USA
‘Saul R. Korey Department of Neurology, Kennedy 807. ‘Department of Pediuiric.s
and Rose F. Kennedy Center for Research in Mental Retardation and Human Development,
Albert Einsiein College of Medicine. Bron.r. NY 10461. USA
(Received 24 November 1993; revision received 22 February 1994: accepted 27 February 1994)
Abstract
Awareness of non-Mendelian mitochondrial inheritance and of its role as an agent of gene-
tic sensorineural hearing loss (SNHL) is recent. Mitochondria are passed on exclusively from the ovum to all the offspring of both sexes, a novel pattern of inheritance. Owing to the critical role of mitochondria in cellular energy metabolism, deletions or point mutations of the mito- chondrial DNA often cause progressive SNHL and a variety of disorders in other organ sys- tems (mitochondrial cytopathies). The clinical expression of mitochondrial diseases varies and depends on the proportion of mutated mitochondria in various body tissues, as well as the nature of the mutation or deletion. In order to determine how often SNHL occurs in mito- chondrial diseases and what is its presenting symptom, and also whether SNHL is a marker for particular phenotypes, we carried out a review of published case reports of patients with an established diagnosis of mitochondrial disease. The review indicates that SNHL occurs at all ages and in virtually all variants of mitochondrial diseases. It is not clear whether SNHL is a marker for a more severe and more rapid course of disease; the lower prevalence of SNHL in descriptions of live patients than of those who had died may be an artifact of case selection reported in the literature. Mitochondrial disease needs to be considered in progressive hearing loss and better longitudinal audiometric study of established cases will be required to answer these questions.
* Corresponding author.
0165-5876/94/$07.00 0 1994 Elsevier Science Ireland Ltd. All rights reserved
SSDI 0165-5876(94)01037-X

92 M. Gold, I. Rapin/lnt. J. Pediatr. Otorhinolaryngol. 30 (1994) 91-104
Keywords: Sensorineural hearing loss; Progressive hearing loss; Genetic hearing loss; Mito- chondrial cytopathy; Mitochondrial inheritance
1. Introduction
It is now well established that genetics is responsible for a large proportion of un- explained congenital and acquired hearing losses. There are many Mendelian genes for deafness, some of which are dominant, others recessive, and some X-linked [23,39,62]. The discovery of non-Mendelian mitochondrial inheritance in man is re- cent and has brought with it the realization that mitochondrial diseases are both more frequent and much more varied than previously thought. The fact that they are responsible for a significant number of cases of progressive SNHL in both child- ren and adults has attracted little attention thus far. In order to fill this gap, we car- ried out a review of published case studies of mitochondrial diseases in an attempt to determine the prevalence of hearing loss in mitochondrial diseases, how often it is their presenting symptom, whether it occurs preferentially in some syndromes, and whether it is a useful prognostic marker. Because mitochondrial diseases are still un- familiar to many professionals, we preface our review with a brief prlcis of mito- chondrial biology and mitochondrially inherited diseases.
2. Mitochondrial biology
Mitochondria are intracellular organelles thought to have evolved from pro- karyotic organisms that developed a symbiotic relationship with eukaryotic cells. Mitochondria carry out ATP synthesis and oxidative phosphorylation and thus play the central role in cellular energy metabolism. They are the only cellular organelles to carry their own genetic material in the form of a circular double stranded mole- cule of DNA with 16 569 base pairs [13,26]. Each mitochondrion contains 2-10 copies of the mitochondrial genome and each cell contains hundreds or thousands of mitochondria that replicate independently of each other and of the host cell [13,26]. Mitochondrial DNA undergoes a higher rate of spontaneous mutation than nuclear DNA because ineffectual mitochondrial DNA repair often results in an ac- cumulation of mutations and in increased susceptibility to exogenous mutagenic fac- tors [69]. Consequently, late onset and sporadic disorders ascribable to mitochondrial dysfunction are fairly common. The severity or expression of mito- chondrial diseases is variable and is a function of the proportion of mutated mito- chondria in various tissues, the energy requirements of these tissues, and the severity of the functional impairment of the electron transport chain. The high energy re- quirements of brain, muscle, retina, and the inner ear are probably responsible for the vulnerability of these tissues to mitochondrial diseases.
The evolutionary process has left mammalian mitochondria with a progressively smaller genome due to the migration of some of the genomic elements required for mitochondrial function into the cell’s nuclear genome [43]. The mitochondrial genome contains only 37 genes that code for the 2% of mitochondrial polypeptides

M. Gold, 1. Rapin / Int. J. Pediatr. Otorhinolaryngol. 30 (1994) 91-104 93
that are not coded for by the nuclear genome and are not translocated into the mito- chondria. Mitochondrial function is thus under both nuclear and mitochondrial ge- netic control. As a result, some mitochondrial disorders are inherited as classic Mendelian traits, whereas those resulting from mitochondrial DNA mutations or deletions are either maternally inherited and transmitted to both sexes or sporadic.
Cellular energy production depends on oxidative phosphorylation and the production of ATP molecules by the electron transport chain. Mitochondrial DNA codes for 13 of the enzymes required for cellular respiration and for messenger RNAs and transfer RNAs required for mitochondrial replication [13,15,64].
3. Mitochondrial diseases
The first report of a patient with a disease of mitochondrial function was pub- lished in 1962 [19,46]; the patient presented with a hypermetabolic state due to uncoupling of oxidative phosphorylation. Subsequent patients with recognized mito- chondrial diseases had myopathies or encephalomyopathies and were identified by elevations of serum and CSF lactate and pyruvate levels and mitochondrial enzyma- tic deficits in combination with abnormal muscle biopsies showing ragged-red fibers (RRF) on histochemistry.
The abnormal staining of RRF is due to alterations in intramitochondrial protein synthesis. These fibers are characteristic of some, but not all, diseases due to mito- chondrial DNA mutations and deletions and not of those due to nuclear DNA al- terations [13]. The unique features of mitochondrial biology and inheritance were elucidated by study of the pedigrees of 30 families with members affected by these diseases. Transmission was exclusively maternal in 27 of the 30 families [ 11,181. The remaining cases, in which transmission was paternal, were attributable to mutations in the nuclear encoding of the defective mitochondrial enzymes. Molecular methods now make it possible to localize the specific point mutations or deletions responsible for these biochemical defects and to classify mitochondrial diseases rationally based on whether they are inherited as mitochondrial or Mendelian traits [13].
All clinical syndromes associated with mitochondrial DNA defects are associated with enzymatic deficits of the electron transport chain. Some disorders involve only one tissue, usually muscle, or the optic nerve in the case of Leber’s hereditary optic neuropathy which is due to a mitochondrial DNA point mutation (711, while others involve multiple systems. Kearns-Sayre Syndrome (KSS) is the prototypic condition associated with large scale mitochondrial DNA deletions or, occasionally, insertions [25,37,75]. In KSS the DNA rearrangements probably take place post-fertilization because the disorder is sporadic and is not transmitted to the offspring of affected women. It starts in childhood and is characterized by hearing loss, chronic progres- sive external ophthalmoplegia (CPEO) and pigmentary retinopathy, myopathy with ragged-red fibers and cardiomyopathy - often with heart block, short stature, parathyroid deficiency, and spongy changes in the cerebral white matter. Myoclonus epilepsy with ragged-red fibers (MERRF) [24,33,45,52,67], which is associated with ataxia and sponginess of the white matter, and mitochondrial encephalopathy with lactic acidosis and stroke-like episodes (MELAS) [12,29,31,55] are both due to one

94 M. Gold, I. Rapin /Int. J. Pediatr. Otorhinolaryngol. 30 (1994) 91-104
of several mitochondrial DNA point mutations. Both may be associated with pro- gressive hearing loss and lactic acidosis. Patients with overlap syndromes [76] or with the sequential development of distinct clinically identifiable syndromes [ 11,281 in the face of a stable mitochondrial DNA deletion have been described. DeVivo [ 131 provides a lucid up-to-date review of known mitochondrial syndromes.
Mitochondrial diseases due to nuclear DNA defects, which are inherited as Mendelian traits, are associated with substrate transport and utilization deficits, in- cluding defects of fatty acid oxidation and pyruvate metabolism, but also with some deficits in the electron transport chain that overlap clinically with syndromes due to mitochondrial DNA defects.
4. Materials and Methods
We conducted a literature search on the topic of mitochondrial diseases for ar- ticles that included a case report and that were written in English and had appeared from 1984-1993. Cases reported prior to 1984 were generally excluded due to the lack of consistent clinical and biochemical evaluations and the unavailability of mitochondrial DNA analysis. Cases were accepted for this review when the diagno- sis was confirmed on the basis of deletions/mutations in the mitochondrial DNA, or of a combination of morphological evidence on muscle biopsy (ragged-red fibers) and biochemical testing of the function of the respiratory chain. In addition, the case reports had to provide adequate information on the age of onset and the ages at which prominent clinical signs or symptoms became apparent. A total of 117 case reports were sufficiently complete to be incIuded in this review of hearing loss in mi- tochondrial disease.
We attempted to estimate the prevalence of SNHL in patients with mitochondrial disorders from a series of review articles describing large collections of patients or large pedigrees. Unfortunately, many reports did not mention hearing and data on age, sex and clinical severity were often inadequate.
We presumed that if SNHL were a marker for more severe disease, patients with SNHL would have an earlier age of onset of symptoms in other organ systems than patients with normal hearing. Due to the wide range of ages of reported patients, we started by carrying out a univariate analysis in order to determine that the use of parametric statistics was appropriate. After confirming the normal distribution of ages for the overall population, a series of two tailed r-tests was carried out to com- pare the groups with and without SNHL with regard to the age at presentation of the disease and at onset of the various clinical symptoms (external ophthalmoplegia, muscle weakness, myoclonus, retinal degeneration, stroke or stroke-like episodes, and cardiac arrhythmias).
We also supposed that if SNHL functioned as a marker for more severe disease, patients with SNHL would develop more of the clinical symptoms associated with mitochondrial encephalomyopathies than those without SNHL. We tested this hy- pothesis by determining the frequency of associated clinical symptoms in patients with and without SNHL. The data were analyzed with x2 tests with Yates correc- tion. Statistical analyses were carried out using SASSTAT version 6.03. A power
M. Gold, 1. Rapin /In!. J. Pediatr. Ororhmolar~~ngoi. 30 f 1994) 91-104 9s
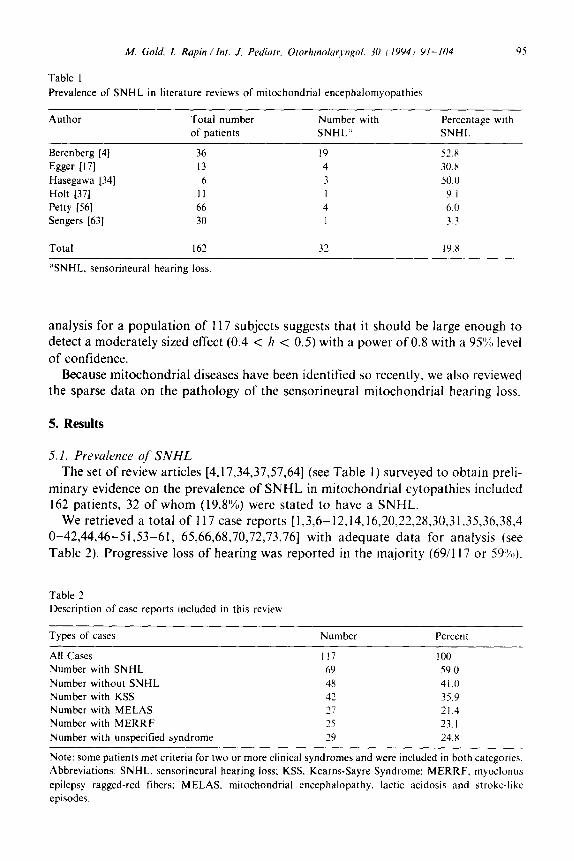
Table I Prevalence of SNHL in literature reviews of mitochondrial encephalomyopathies
Author Total number
of patients
Number with
SNHL”
Percentage with
SNHL
Berenberg [4] 36 19 52.8
Egger 1171 13 4 30.x
Hasegawa [34] 6 3 50.0
Holt [37] II 1 9.1
Petty [56] 66 4 6.0
Sengers [63] 30 I 3.3
Total I62 32 19.x
%NHL. sensorineural hearing loss
analysis for a population of 117 subjects suggests that it should be large enough to detect a moderately sized effect (0.4 < h c 0.5) with a power of 0.8 with a 95% level of confidence.
Because mitochondrial diseases have been identified so recently, we also reviewed the sparse data on the pathology of the sensorineural mitochondrial hearing loss.
5. Results
5.1. Prevalence of SNHL
The set of review articles [4,17,34,37,57,64] (see Table 1) surveyed to obtain preli- minary evidence on the prevalence of SNHL in mitochondrial cytopathies included 162 patients, 32 of whom (19.8%) were stated to have a SNHL.
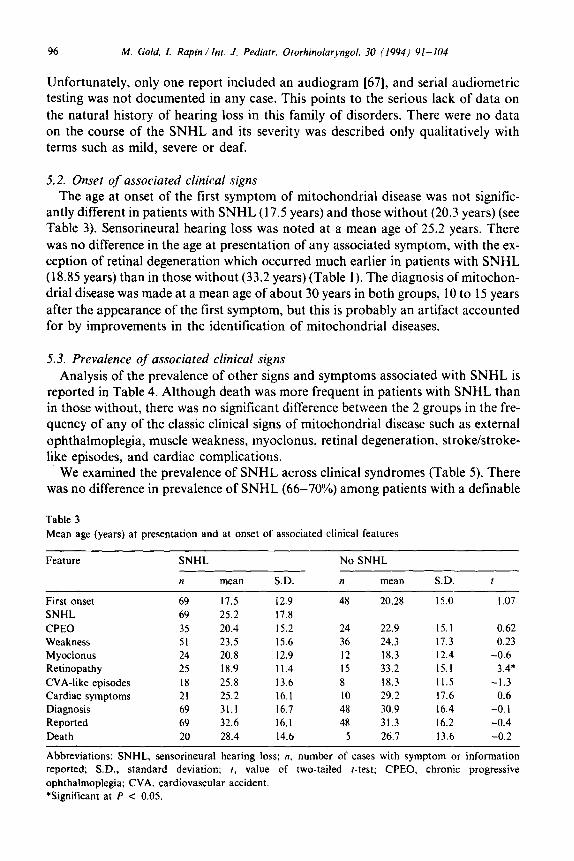
We retrieved a total of 117 case reports [ 1,3,6- 12,14,16,20,22,28,30,3 1,35,36,38,4 O-42,44,46-51,53-61, 65,66,68,70,72,73,76] with adequate data for analysis (see Table 2). Progressive loss of hearing was reported in the majority (69/l 17 or 59%).
Table 2
Description of case reports included in this review
Types of cases Number Percent
All Cases 117 100
Number with SNHL 69 59.0
Number without SNHL 48 41.0
Number with KSS 42 35.9
Number with MELAS 27 21.4
Number with MERRF 25 23. I Number with unspecified syndrome 29 24.8
Note: some patients met criteria for two or more clinical syndromes and were included in both categories.
Abbreviations: SNHL. sensorineural hearing loss; KSS, Kearns-Sayre Syndrome: MERRF. myoclonus
epilepsy ragged-red fibers: MELAS, mitochondrial encephalopathy. lactic acidosis and stroke-like
episodes.
96 M. Gold, I. Rapin/Imt. f. Pediatr. Otorhinolaryngol. 30 (1994) 91-IO4
Unfortunately, only one report included an audiogram [67], and serial audiometric testing was not documented in any case. This points to the serious lack of data on the natural history of hearing loss in this family of disorders. There were no data on the course of the SNHL and its severity was described only qualitatively with terms such as mild, severe or deaf.
5.2. Onset of associated clinical signs The age at onset of the first symptom of mitochondrial disease was not signific-
antly different in patients with SNHL (17.5 years) and those without (20.3 years) (see Table 3). Sensorineural hearing loss was noted at a mean age of 25.2 years. There was no difference in the age at presentation of any associated symptom, with the ex- ception of retinal degeneration which occurred much earlier in patients with SNHL (18.85 years) than in those without (33.2 years) (Table 1). The diagnosis of mitochon- drial disease was made at a mean age of about 30 years in both groups, 10 to 15 years after the appearance of the first symptom, but this is probably an artifact accounted for by improvements in the identification of mitochondrial diseases.
5.3. Prevalence of associated clinical signs Analysis of the prevalence of other signs and symptoms associated with SNHL is
reported in Table 4. Although death was more frequent in patients with SNHL than in those without, there was no significant difference between the 2 groups in the fre- quency of any of the classic clinical signs of mitochondrial disease such as external ophthalmoplegia, muscle weakness, myoclonus, retinal degeneration, stroke/stroke- like episodes, and cardiac complications.
We examined the prevalence of SNHL across clinical syndromes (Table 5). There was no difference in prevalence of SNHL (66-70%) among patients with a definable
Table 3 Mean age (years) at presentation and at onset of associated clinical features
Feature
First onset SNHL CPEO Weakness Myoclonus Retinopathy CVA-like episodes Cardiac symptoms Diagnosis Reported Death
SNHL No SNHL
II mean SD. n mean SD. 1
69 17.5 12.9 48 20.28 15.0 I .07 69 25.2 17.8 3s 20.4 IS.2 24 22.9 IS.1 0.62 51 23.5 IS.6 36 24.3 17.3 0.23 24 20.8 12.9 I2 18.3 12.4 -0.6 25 18.9 11.4 I5 33.2 IS.1 3.4* I8 25.8 13.6 8 18.3 II.5 -1.3 21 25.2 16.1 IO 29.2 17.6 0.6 69 31.1 16.7 48 30.9 16.4 -0.1 69 32.6 16.1 48 31.3 16.2 -0.4 20 28.4 14.6 5 26.7 13.6 -0.2
Abbreviations: SNHL, sensorineural hearing loss; n. number of cases with symptom or information reported; SD., standard deviation; 1, value of two-tailed f-test; CPEO, chronic progressive ophthalmoplegia; CVA, cardiovascular accident. *Significant at P < 0.05.

M. Gold, 1. Rapin /Inr. J. Pediatr. Olorhinolaryngol. 30 I19941 91-104 97
Table 4
x2 analysis of the frequency of clinical features
Feature
External ophthalmoplegia
Present
Not present Muscular weakness
Present
Not present Myoclonus
Present
Not present Retinal degeneration
Present
Not present Stroke/stroke-like episodes
Present
Not present Cardiac complications
Present
Not present Death
Yes
No
SNHL No SNHL
35 24
34 24
51 36
IH 12
24 12
45 36
25 15
44 33
18 x 51 40
21 10
48 38
20 5
49 43
xz statistic
0.0
0.0
0.85
0.13
0.96
0.x9
4.76*
Abbreviations: SNHL, sensorineural hearing loss
*Significant at P < 0.05 level.
clinical syndrome (KSS, MERRF or MELAS), whereas SNHL was much less prevalent (38%) among patients whose presentation could not be categorized.
5.4. Size of effect and power of Ihe study The 117 subjects in this study provided a sample large enough to detect a
moderately sized effect when analysing the proportion of patients presenting with the various clinical features of mitochondrial disease. An analysis of the size of the effect of proportions for the associated clinical syndromes that did not show a differ- ence between patients with and those without SNHL was performed. The power analysis revealed uniformly low size effects (h statistic) ranging from 0.02-0.381.
Table 5
x’ analysis of the frequency of SNHL within clinical groups
Clinical syndrome SNHL present SNHL absent
KSS 28 14
MERRF 17 8
MELAS 19 8
Unspecified 11 18
x2 = 8.564. d.f. = 3, P = 0.036; abbreviations: see Table 2

98 M. Gold, I. Rapin /IN. J. Pediatr. Otorhinolaryngol. 30 (1994) 91-104
Using a small size effect (h = 0.10) as a conservative estimate, a study with a power of 0.8, and a confidence level of 95% would require over 1200 subjects in order to detect a effect of such small size.
5.5. Pathology of mitochondrial SNHL Of the 20 patients with mitochondrial disease with hearing loss who died by the
time their case was reported, only 5 cases, those of Bordarier [7], Oldfors [54] and McKelvie [49] provide data on pathological changes in the auditory pathways but unfortunately they do not provide data on the cochlea. Bordarier [7] described the autopsy findings in 2 cases of KSS with SNHL. The first had demyelination of the brain-stem sparing the medial longitudinal fasciculus and the central tegmental tracts, along with severe spongiosis with loss of myelin in the cerebral white matter and brain stem, including the substantia nigra and the nuclei of cranial nerves III, VI and VIII. The second case also had severe spongiosis of the white matter of the cerebral hemispheres and brain stem, including cranial nerve VIII. In both cases, the neurons of the cranial nerve nuclei appeared normal. Oldfors [54], describing a case of KSS with SNHL, showed by electron microscopy that sponginess of the white matter was due to extracellular edema with splitting of myelin along the minor dense line. There was severe neuronal loss with gliosis in the basal ganglia, iron deposition in the globus pallidus, and a reduction in the number of cells in the inferior olivary nucleus. McKelvie 1491 describing the findings in 2 patients with MERRF, noted vacuolar changes and pallor of the myelin in the cerebrum. Brain-stem structures showed the same vacuolation in multiple cranial nerve nuclei including the vestibular nuclei. Lindsay and Hinojosa [44] provide the only data on the inner ear in a case of KSS with SNHL. They described advanced cochlear degeneration with collapse of Reisner’s membrane and degeneration of the organ of Corti, tectorial membrane, and stria vascularis. There was a 60-70”/0 reduction in the number of neurons along the entire length of the spiral ganglion.
6. Discussion
Our review of 117 adequately documented cases of mitochondrial cytopathy described over the past decade indicates that progressive SNHL may be present in almost two thirds of the cases, a figure twice as high as previously estimated. This figure may be an inadequate estimate of the true prevalence of SNHL because these disorders have only been recognized for the past 20-30 years and because of a bias towards reporting only the more severe cases. The entire spectrum of these diseases is not yet fully known and there is no information on how many cases of apparently isolated progressive SNHL will eventually declare themselves as due to mitochon- drial cytopathies.
What this review points out is the dearth of information on the character, severity, and course of mitochondrial SNHL. Information about the pathology of this disor- der, both in the cochlea and the central auditory pathway, is almost nonexistent. The fact that patients with SNHL may develop retinal degeneration earlier than those without is compatible with a selective vulnerability of certain tissues to defective

M. Gold, 1. Rapin /Int. J. Pediatr. Ororhinolaryngoi. 30 (1994) 91-104 99
energy metabolism, since energy metabolism is high both in the cochlea, whose ion pumps are continuously active, and in the retina due to rapid turnover of visual pig- ment proteins.
The dependence of the hair cells and stria vascularis on the availability of high energy compounds and oxidative phosphorylation has been investigated by exposing animal cochleas to anoxia, ischemia, and metabolic decouplers. Exposure to carbon monoxide, a model for hypoxia, led to reversible losses in compound action poten- tials, starting in the high frequency range, and to compensatory increases in blood flow [21]. Total cochlear ischemia rapidly resulted in pathological changes in the structure of the cochlea [5]. There was detectable swelling of nerve endings of inner hair cells within 15 min of the onset of ischemia that eventually led to their rupture. Outer hair cells became swollen within 30 min and showed alterations of intracellu- lar organelles, including the mitochondria. The effect of the ototoxic drug, kana- mycin, on cochlear ultrastructure was to induce mitochondrial lesions in the stria vascularis [32]. The dependence of the cochlea on high energy metabolism can be inferred from intense histochemical staining of the tegmentum vasculosum (analog structure of the stria vascularis) for Na+-K+-ATPase histochemistry in chicken cochleas which showed intense staining of the tegmentum vasculosum, especially the basal portion (high-frequency) portion of the tegmentum [63]. A central and high energy role of the stria vascularis is to maintain the ionic composition of the endo- lymph by active transport [2]. Cultured hair cells may also be more dependent on oxidative phosphorylation since they must maintain the -70 mV transmembrane potential critical to sound transduction [27].
This review of published data does not support the hypothesis that SNHL is a marker for earlier presentation of mitochondrial encephalopathies or for more se- vere phenotypic expression. The seemingly higher mortality of patients with SNHL may be a sampling artifact. The similar proportion of patients with KSS, MERRF and MELAS who eventually developed SNHL suggests a common metabolic defect affecting both the cochlea and neural tissue. All 3 syndromes are associated with mitochondrial DNA defects interfering with oxidative phosphorylation and not with abnormal products of the nuclear genome.
One of the difficulties in attempting to define the natural history of SNHL in mito- chondrial diseases from published reports is the often prolonged interval between clinical evaluations of the patients. These gaps create a bias towards delayed report- ing of the onset of signs and symptoms. In fact, these gaps may have obscured any markers for earlier onset or more severe disease. The present data suggest that SNHL is not of value for predicting the eventual appearance of other clinical manifestations or the progression of the disease.
A power analysis of the data suggests that the lack of difference between patients with SNHL and those without in terms of associated clinical syndromes is not an artifact of a small sample size but is rather due to very small differences in the pro- portion of patients that are affected. Given the sparse data on the natural history of SNHL in patients with mitochondrial diseases any questions regarding clinical associations will require very large sample sizes. Conversely, better clinical and audiologic data will lead to better characterization of these two patient populations

100 M. Gold, I. Rapin /Int. J. Pediatr. Otorhinolaryngol. 30 (1994) 91-104
and should provide the answers to the questions we have raised with a more realistic number of patients.
The concept of mitochondrial inheritance is new. Physicians dealing with patients, both pediatric and adult, with an acquired SNHL need to consider the possibility of a mitochondrial disorder because of the potential involvement of other organ sys- tems and of the unusual pattern of inheritance. They should not only inquire specifically about progressive hearing loss in the family but also about other symp- toms suggestive of a mitochondrial disorder among maternal relatives. If the history is suspicious, they should examine living relatives, especially maternal ones, for clini- cal markers of mitochondrial disease. The determination of maternal inheritance is crucial for genetic counselling in affected patients and their offspring.
Blood tests of mitochondrial function and molecular approaches to diagnosis using blood, skin tibroblasts, and muscle samples provide an opportunity for early and specific diagnosis [74]. Some of these disorders may respond to future therapies directed at replacing the defective enzyme or components thereof. The development of enzymatic replacement therapies depends on identifying the exact deficiency and establishing a correlation between the defective enzyme and particular functional deficits.
Note added in proof
Two more mitochondrial syndromes with hearing loss have been identified, one autosomal recessive with gastrointestinal pseudo-obstruction and encephalomyo- pathy (Hirano et al., 1994) the other maternally transmitted with insulin-dependent diabetes mellitus (Kadowaki et al., 1994).
Hirano, M., Silvestri, G., Blake, D.M., et al. (1994) Mitochondrial neurogastrointestinal encephalomyo- pathy (MNGIE): Clinical, biochemical, and genetic features of an autosomal recessive mitochondrial disorder. Neurology 44, 721-727.
Kadowaki, T., Kadowaki, H., Mori, Y. et al. (1994) A subtype of diabetes mellitus associated with a mutation of mitochondrial DNA. N. Engl. J. Med. 330, 962-968.
References
I Angelini, C., Bresolin, N., Pegolo, G., Bet, L., Rinaldo, P., Trevisan. C. and Vergani, L. (1986) Childhood encephalomyopathy with cytochrome C oxidase deficiency, ataxia, muscle wasting and mental impairment. Neurology 36, lO48- 1052.
2 Anniko, M. and Wroblewski, R. (1986) Ionic environment of cochlear hair cells. Hear. Res. 22, 279-293.
3 Bendahan, D., Desnuelle, C., Vanuxem, D., Confort-Gouny, S., Figarella-Branger, D.. Pellisier, J.F., Kozak-Ribbens, G., Pouget, J., Serratrice, G. and Cozzone, P.J. (1982) 3’P NMR spec- troscopy and ergometer exercise test as evidence for oxidative performance improvement with co- enzyme Q in mitochondrial myopathies. Neurology 42, 1203-1208.
4 Berenberg, R.A., Pellock, J.M., DiMauro, S., Schotland, D.S., Bonilla, E., Eastwood, A., Hays, A., Vicale, CT., Behrens, M., Chutorian, A. and Rowland, L.P. (1977) Lumping or splitting? “Ophthalmoplegia plus” or Kearns-Sayre Syndrome? Ann. Neurol. I, 37-54.
5 Billett, T.E., Thorne, P.R. and Gavin, J.B. (1989) The nature and progression of injury in the organ of corti during ischemia. Hear. Res. 41, 198-197.

M. Gold. 1. Rapin / Inr. J. Pediarr. Otorhinolaryn~ol. 30 ( 1994) 91-104 101
6 Bindoff. L.A., Desnuelle, C., Birch-Machin, M.A.. Peilissier, J.F.. Serratrice, G., Dravet, C..
Bureau, M., Howell, N. and Turnbull, D.M. (1991) Multiple defects of the mitochondrial respiratory
chain in a mitochondrial encephalopathy (MERRF): a clinical. biochemical and molecular study.
J. Neurol. Sci. 102. 17-24.
7 Bordarier, C., Duyckaerts, C., Robain. 0.. Ponsot. G. and Laplane. D. (1990) Kearns-Sayre Syn-
drome: two clinico-pathological cases. Neuropediatrics 21. 106-109.
8 Bresolin. N., Moggio, M., Bet, L., Gallanti, A., Prelle. A., Nobile-Orazio, E.. Adobbati. L.. Fer-
rante, C., Pellegrini. G. and Scarlato. G. (1987) Progressive cytochrome C oxidase deficiency in a
case of KSS: morphological, immunological and biochemical studies in muscle biopsies and autopsy
tissues. Ann. Neurol. 21. 564-572.
9 Bresolin, N., Martinelli, P., Barbiroli, B., Zaniol, P.. Ausenda. C.. Montagna. P.. Gallant]. A..
Comi, G.P.. Scarlato, G. and Lugaresi. E. (1991) Muscle mitochondrial DNA deletion and “P
NMR spectroscopy alterations in a migraine patient. J. Neurol. Sci. 104. 182-189.
IO Byrne. E.. Marzuki, S., Sattayasai. N., Dennet. X. and Trounce. I. (1987) Mitochondrial studies m
Kearns-Sayre Syndrome: normal respiratory chain function with absence of a mitochondrial transla-
tion product. Neurology 37, 1530-1534.
I I Byrne, E., Trounce, I., Dennett. X.. Gilligan, B.. Morley, J.B. and Marzuki. S. (1988) Progression
from MERRF to MELAS phenotype in a patient with combined respiratory complex I and IV defi-
ciencies. J. Neurol. Sci. 88, 327-337.
I2 Ciafaloni, E., Ricci, E., Servidei, S., Shanske, S.. Silvestri. G., Manfredi, G., Schon. E.A. and
DiMauro, S. (1991) Widespread tissue distribution of a tRNA Leu’U”R’ mutation in the mitochon-
drial DNA of a patient with MELAS syndrome. Neurology 41. 1663-1665.
I3 DeVivo, D.C. (1993) The expanding clinical spectrum of mitochondrial disease. Brain Dev. 15. I -22.
I4 DeQuick. M., Lammens, M., Dom, R. and Carlton, H. (1991) MELAS: a family with paternal In-
heritance. Ann. Neurol. 29, 456-457.
I5 DiMauro, S., Bonilla. E., Zeviani. M.. Nakagawa. M. and DeVivo. D.C. (1985) Mitochondrial
myopathies. Ann. Neurol. 17, 521-538.
I6 Doriguzzi. C., Palmucci, L., Mongini, T., Bresolin. N.. Bet, L.. Comi, G. and Lala. R. (1989) Endo-
crine involvement in mitochondrial encephalomyopathy with partial cytochrome C oxidase deficien-
cy. J. Neurol. Neurosurg. Psych. 52. 122-125.
17 Egger, J., Lake, B.D. and Wilson, J. (1981) Mitochondrial cytopathy: a multi-system disorder with
ragged-red fibers on muscle biopsy. Arch. Dis. Child. 56. 741-752.
18 Egger. J. and Wilson. J. (1983) Mitochondrial inheritance in a mitochondrially mediated disease. N.
Eng. J. Med. 309, 142-146.
19 Enster. L., Ikkos, D. and Luft, R. (19.59) Enzymic activittes of human muscle mitochondria: a tool
in clinical metabolic research. Nature 184. 1851-1854.
20 Eviatar. L.. Shanske, S., Gauthier. B., Abrams. C.. Maytal. J.. Slavin. M.. Valderrama, E. and
DiMauro. S. (1990) Kearns-Sayre Syndrome presenting as renal tubular acidosis. Neurology 40.
1761-1763.
21 Fechter. L.D.. Thorne. P.R. and Nuttall. A.L. (1987) Effects of carbon monoxide on cochlear elec-
trophysiology and blood flow. Hear. Res. 27. 37-45.
22 Frackowiak. R.S.J.. Herold, S., Petty, R.K.H. and Morgan-Hughes. J.A. (1988) The cerebral merab-
olism of glucose and oxygen measured with positron emission tomography in patients with mtto-
chondrial diseases. Brain I I I. 1009-1024.
23 Fraser, G.F. (1976) The Causes of Profound Deafness in Childhood. Johns Hopkins University
Press, Baltimore.
24 Fukahara. N.. Tokiguchi. S., Shirakawa, S. and Tsubaki, T. (1980) Myoclonus epilepsy assoctated
with ragged-red fibers: disease entity or syndrome? Light and electron microscopy studies of two
cases and a review of the literature. J. Neurol. Sci. 47, 117-133.
25 Gerbitz, K.D., Obermaier-Kusser. B., Zierz. S.. Pongratz. D., Muller-Hacker. J. and Lesttene, P.
(1990) Mitochondrial myopathies: divergences of genetic deletions. biochemical defects and the clim-
cal syndromes. J. Neurol. 237, 5-10.
26 Giles, R.E.. Blanc, H.. Cann, H.M. and Wallace, D.C. (1980) Maternal inherttance of human mtto-
chondrial DNA. Proc. Natl. Acad Sci. 77. 6715-6719.

102 M. Gold, I. Rapin /Int. J. Pediatr. Otorhinolaryngol. 30 (1994) 91-104
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43 44
45
46
41
Gitter, A.H., Zenner, H.P. and Fromter, E. (1986) Membrane potential and ion channels in isolated outer hair cells of guinea pig cochlea. J. Otorhinolaryngol. Rel. Spec. 48, 68-75. Gold. M., Rapin, 1. and Shanske, S. (1991) Mitochondrial inheritance of acquired deafness. Ann. N.Y. Acad. Sci. 630, 301-302. Goto, Y., Nonaka, I. and Horai, S. (1990) A mutation in the tRNA Leu(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature 348, 651-653. Goto, Y., Itami, N., Kajii, N., Tochimaru, H., Endo, M. and Horai, S. (1990) Renal tubular involve- ment mimicking Bartter Syndrome in a patient with Kearns-Sayre Syndrome. J. Pediatr. 116, 904-910. Goto, Y., Horai, S., Matsuoka, T., Koga, Y., Nihei, K., Kobayashi, M. and Nonaka, 1. (1992) Mito- chondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS): a correla- tive study of the clinical features and mitochondrial DNA mutation, Neurology 42, 545-550. Gratacap, B., Charachon, R. and Stoebner, P. (1985) Results of an ultrastructural study comparing stria vascularis with organ of corti in guinea pigs treated with kanamycin. Acta Otolaryngol. 99, 339-342. Hammans, S.R., Sweeney, M.G., Brockington, M., Lennox. G.G., Lawton, N.F., Kennedy, C.R., Morgan-Hughes, J.A. and Harding, A.E. (1993) The mitochondrial DNA transfer RNALYS A - G(s3”) mutation and the syndrome of myoclonic epilepsy with ragged red fibers. Brain 116, 617-632. Hasegawa, H., Matsuoka, T., Goto, Y. and Nonaka, 1. (1991) Strongly succinate dehydrogenase- reactive blood vessels in muscles from patients with mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes. Ann. Neural. 29, 601-605. Hayes, D.J., Lecky, B.R.F., Landon, D.N., Morgan-Hughes, J.A. and Clark, J.B. (1984) A new mi- tochondrial myopathy: biochemical studies revealing a deficiency in the cytochrome B-C, complex (complex III) of the respiratory chain. Brain 107, 1165-l 177. Herzberg, N.H., van Schooneveld, M.J., Bleeker-Wagemakers, E.M., Zwart, R., Cremers, F.M.P., van der Knapp, M.D., Bolhuis, P.A. and de Visser, M. (1993) Kearns-Sayre Syndrome with a phenocopy of choroidemia instead of pigmentary retinopathy. Neurology 43, 218-221. Holt, LJ., Harding, A.E., Cooper, J.M., Schapira, A.H.V.. Toscano, A., Clark, J.B. and Morgan- Hughes, J.A. (1989) Mitochondrial myopathies: clinical and biochemical features of 30 patients with major deletions of muscle mitochondrial DNA. Ann. Neurol. 26. 699-708. Koga, Y., Nonaka, I., Sunohara, N., Yamanaka, R. and Kumagai, K. (1988) Variability in the activ- ity of respiratory chain enzymes in mitochondrial myopathies. Acta Neuropathol. 76, 135-141. Konigsmark, B.W. and Gorlin, R.J. (1976) Genetic and Metabolic Deafness, Saunders, Philadelphia. Kuriyama, M., Suehara, M., Marume, N., Osame, M. and Igata, A. (1984) High CSF lactate and pyruvate content in Kearns-Sayre Syndrome. Neurology (Cleveland) 34, 253-255. Kuriyama, M., Umezaki, H., Fukuda, Y., Osame, M., Koike, K., Tateishi. J. and Igata. A. (1984) Mitochondrial encephalomyopathy with lactate-pyruvate elevation and brain infarctions. Neurology (Cleveland) 34, 72-77. Larsson. N.G., Holme, E., Kristiansson, B., Oldfors, A. and Tulinius, M. (1990) Progressive increase of the mutated mitochondrial DNA fraction in Kearns-Sayre Syndrome. Pediatr. Res. 28, 131-I 36. Leving, C.S. and Brown, G.G. (1989) Molecular biology of plant mitochondria. Cell 56, 171-179. Lindsay, J.R. and Hinojosa, R. (1976) Histopathologic features of the inner ear associated with Kearns-Sayre Syndrome. Arch. Otolaryngol. 102, 747-752. Lombes, A., Mendell, J.R., Nakase, H., Barohn, R.J., Bonilla, E., Zeviani. M., Yates, A.J., Omerza, J., Gales, T.L., Nakahara, K., Rizutto, R., Engel, W.K. and DiMauro. S. (1989) Myoclonic epilepsy and ragged-red fibers with cytochrome oxidase deficiency: neuropathology, biochemistry and molec- ular genetics. Ann. Neurol. 26, 20-33. Luft, R., Ikkos, D., Palmieri, G., Ernster, L. and Afzelius, B.A. (1962) A case of severe hypermetabolism of non-thyroid origin with a defect in the maintenance of mitochondrial control: a correlated clinical, biochemical and morphological study. J. Clin. Invest. 41, 1776-1804. Macmillan, C., Lath, B. and Shoubridge, E.A. (1993) Variable distribution of mutant mitochondrial DNAs (tRNALEU(3243’) in tissues of symptomatic relatives with MELAS: the role of mitotic segre- gation. Neurology 43, 1586- 1590.

M. Gold, I. Rapin/lnr. J. Pediatr. Otorhinolaryngol. 30 (1994) 91-104 103
48 Matthews, P.M., Tampieri, D. and Berkovic, SF. (1991) Magnetic resonance imaging shows specific
abnormalities in the MELAS syndrome. Neurology 41. 1043-1046.
49 McKelvie, P.A., Morley, J.B.. Byrne. E. and Marzuki, S. (1991) Mitochondrial encephalomyo-
pathies: a correlation between neuropathological findings and defects in mitochondrial DNA. J.
Neurol. Sci. 102. 51-60.
50 Montagna, P.. Gallassi, R.. Medori. R.. Govoni, E.. Zeviani. M., DiMauro. S.. Lugaresi. E. and
Anderman, F. (1988) MELAS Syndrome: characteristic migrainous end epileptic features and mater-
nal transmission. Neurology 38, 75 I-754.
51 Mori, K.. Narahara, K., Ninomiya, S., Goto. Y. and Nonaka, 1. (1991) Renal and skin involvement
in a patient with the complete Kearns-Sayre syndrome. Am. J. Med. Genet. 38. 583-587.
52 Noer. A.S., Sudoyo. P.. Lertrit, P.. Thyagarajan, D., Utthanapol. P.. Kapsa. R.. Byrne, E. and Mar-
zuki. S. (1991) A tRNAtLy’) mutation in the mtDNA is the causal genetic lesion underlying the
myoclonic epilepsy and ragged-red fiber (MERRF) syndrome. Am. J. Hum. Genet. 49. 715-722.
53 Okamoto. T., Mizuno, K., lida. M., Sobue. 1. and Mukoyama, M. (1981) Opthalmoplegia plus: it’s
occurrence with periventricular diffuse low density on computed tomographic scan. Arch. Neurol.
38, 423-426.
54 Oldfors, A., Fyhr, I.M., Holme, E.. Larsson. N.G. and Tulinius. M. (1990) Neuropathology in
Kearns-Sayre Syndrome. Acta Neuropathol. 80. 541-546.
55 Pavlakis, SC.. Phillips, P.C., DiMauro, S.. DeVivo, D.C. and Rowland, L.P. (1984) Mitochondrial
myopathy, encephalopathy, lactic acidosis and stroke-like episodes: a distinctive clinical syndrome
Ann. Neural. 16, 481-488.
56
57
58
59
60
61
62
63
64
65
66
67
Penn, A.M.W., Lee, J.W.K., Thuiller. P., Wagner, M., Maclure, K.M.. Menard. M.R.. Hall. L.D.
and Kennaway, N.G. (1992) MELAS syndrome with mitochondrial tRNALeu(uuR) mutation: cor-
relation of clinical state, nerve conduction. and muscle “P MRS during treatment with
nicotinamide and riboflavin. Neurology 42. 2147-2 152.
Petty, R.K.H., Harding, A.E. and Morgan-Hughes, J.A. (1986) The clinical features of mitochon-
drial myopathy. Brain 109. 915-938.
Remes, A.M.. Majamaa, K., Herva, R. and Hassinen. I.E. (1993) Adult onset diabetes mellitus and
neurosensory hearing loss in maternal relatives of MELAS patients in a family with the
tRNALEUtUUR’ mutation. Neurology 43. 1015-1020.
Riggs. J.E.. Schochet, S.S., Fakadej, A.V., Papadimitriou. A.. DiMauro, S., Crosby, T.W., Gut-
mann, L. and Moxley, R.T. (1984) Mitochondrial encephalomyopathy with decreased succinate-
cytochrome C reductase activity. Neurology (Cleveland) 34. 48-53.
Rosing. H.S.. Hopkins. L.C.. Wallace, D.C., Epstein, C.M. and Weidenheim. K. (1985) Maternally
inherited mitochondrial myopathy and myoclonic epilepsy. Ann. Neurol. 17. 228-237.
Rowland. L.P., Hausmanowa-Petrusewicz. 1.. Badurska, B.. Warburton, D.. Nibroj-Dobosz. I..
Pallai, M. and Johnson, W.G. (1988) Kearns-Sayre Syndrome in twins: lethal mutation or acquired
disease? Neurology 38, 1399-1402.
Ruben, R.J.. van der Water, T.R. and Steel, K.P. (Eds). (1991) Genetics of Hearing Impairment,
Vol. 630. Ann. N.Y. Acad. Sci.. New York.
Schneider, M.E., Cotanche. D.A., Fambrough. D.M.. Saunders. J.C. and Matschinsky, F.M. (1987)
lmmunocytochemical and quantitative studies of the Na+-K’-ATPase distribution in the develop-
ing chick cochlea. Hear. Res. 31, 39-53.
Sengers, R.C.A., Stadhouers, A.M. and Trijbels. J.M.F. (1984) Mitochondrial myopathies: clinical.
morphological and biochemical aspects. Eur. J. Pediatr. 141. 192-207.
Servidei, S., Zeviani, M., Manfredi. G.. Ricci. E., Silvestri. G.. Bertini, E.. Gellera, C., DiMauro.
S., Di Donato, S. and Tonali, P. (1991) Dominantly inherited mitochondrial myopathy with multiple
deletion of mitochondrial DNA: clinical, morphologic, and biochemical studies. Neurology 41.
1053-1059.
Shimoizumi, H., Momoi, M.Y., Ohta, S., Kagawa, Y.. Momoi, T. and Yanagisawa. M. (1989)
Cytochrome c oxidase-deficient myogenic cell lines in mitochondrial myopathy. Ann. Neurol. 25,
615-621.
Shoffner, J.M., Lott, M.T., Lezza, A.M., Seibel, P.. Ballinger. S.W. and Wallace. D.C. (1990)
Myoclonic epilepsy with ragged-red fibers is associated with a mitochondrial DNA tRNAtLY’)
mutation. Cell 61, 931-937.

104 M. Gold. I. Rapin/lnt. J. Pediatr. Otorhinolaryngol. 30 (1994) 91-104
68 Swift, A.C. and Singh, S.D. (1988) Hearing impairment and the Kearns-Sayre Syndrome. J. Laryngol. Otol. 102, 626-627.
69 Trischler, H.J. and Medori, R. (1993) Mitochondrial DNA alterations as a source of human dis- orders Neurology 43, 280-288.
70 Tveskov, C. and Angelo-Nielsen, K. (1990) Kearns-Sayre Syndrome and dilated cardiomyopathy. Neurology 40. 553-554.
71 Wallace, D.C., Singh, G., Lott, M.T., Hodge. J.A.. Schurr. T.G.. Lezza, A.M., Elsas. L.J. and Nikoskelanien, E.K. (1988) Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science 242. 1427- 1430.
72 Yamamoto, M., Sato, T., Anno, M.. Ujike, 1. and Takemoto. M. (1987) Mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes with recurrent abdominal symptoms and coenzyme QIO administration. J. Neurol. Neurosurg. Psych. 50. 1475-1481.
73 Yoda, S., Terauchi, A.. Kitahara. F. and Akabane. T. (1984) Neurologic deterioration with progres- sive CT changes in a child with Kearns-Sayre Syndrome. Brain Dev. 6. 323-327.
74 Zeviani. M., Moraes, C., DiMauro, S., Nakase. H.. Bonilla. E.. Schon, E.A. and Rowland, L.P. (1988) Deletions in mitochondrial DNA in Kearns-Sayre Syndrome. Neurology 38. 1339-1346.
75 Zeviani, M., Amati. P., Bresolin. N., Antonzzi. C.. Picollo, G., Toscano, A. and DiDonato, S. (1991) Rapid detection of the A-G (8344) mutation in Italian families with myoclonus epilepsy and ragged- red fibers (MERRF). Am. J. Hum. Genet. 48, 203-21 I.
76 Zupanc, M.L., Moraes, C.T.. Shanske, S.. Langman, B.C.. Ciafaloni, E. and DiMauro. S. (1991) Deletion of mitochondrial DNA in patients with combined features of Kearns-Sayre and MELAS syndromes. Ann. Neurol. 29. 680-683.