new european pharmacovigilance legislation on risk management · new european pharmacovigilance...
TRANSCRIPT

New European Pharmacovigilance Legislation on Risk Management
October 22, 2013
Henri CAPLAIN, MD, MS
Associate Vice-President
Head Risk Management Center of Excellence
Global Pharmacovigilance & Epidemiology
Sanofi
1

Disclaimer
The views and opinions expressed in this
presentation are solely those of the presenter and
do not necessarily reflect those of Sanofi, or any
of its affiliated organizations.
2

Implementing Regulation520/2012
198/2013 ()
Good Pharmacovigilance Practices (GVP)
Regulatory & procedural guidance
EMA Information & Communication
EU Pharmacovigilance Legislation
Applies on 21-Jul-2012
Applies on 2-Jul-2012
Applies on 10-Jul-2012Arti.29/ 38 apply on 10 Jan 2013
Promote and protect public health by reducing burden of Adverse Drug Reactions (ADRs) and optimising the use of medicines
Clear roles and responsibilities
Robust and rapid EU decision-making
Engage patients and healthcare professionals
Science based - integrate benefit and risk
Risk based/proportionate
Increased pro activity/planning
Reduced duplication/redundancy
Increase transparency and provide better information on medicines
3
Regulation 1235/20101027/2012
Directive 2010/84/EU2012/26/EU Applies on 28-Oct-2013
Learn more on EU-PV Legislation:
Applies on 5-Jun-2013some articles apply on 4 Dec 2012
EMA website European Commission website
Applies on 31-Dec-2013.
3
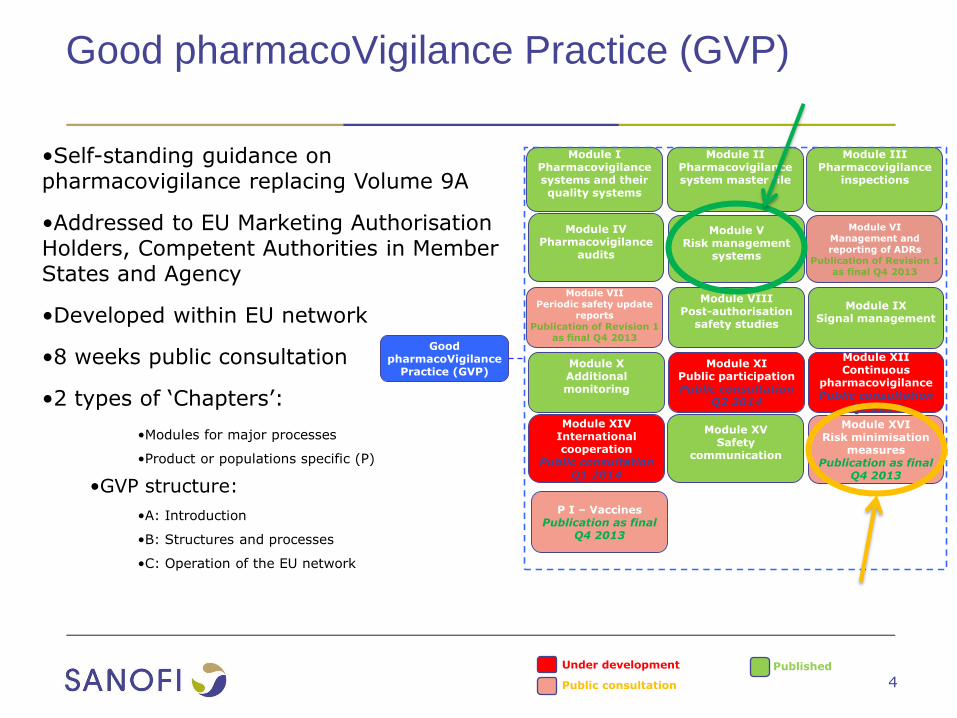
Good pharmacoVigilance Practice (GVP)
4
•Self-standing guidance on pharmacovigilance replacing Volume 9A
•Addressed to EU Marketing Authorisation Holders, Competent Authorities in Member States and Agency
•Developed within EU network
•8 weeks public consultation
•2 types of ‘Chapters’:
•Modules for major processes
•Product or populations specific (P)
•GVP structure:
•A: Introduction
•B: Structures and processes
•C: Operation of the EU network
Good pharmacoVigilance
Practice (GVP)
Module IPharmacovigilance systems and their quality systems
Module IIPharmacovigilance system master file
Module IIIPharmacovigilance
inspections
Module IVPharmacovigilance
audits
Module VRisk management
systems
Module VIManagement and reporting of ADRs
Publication of Revision 1 as final Q4 2013
Module VIIPeriodic safety update
reportsPublication of Revision 1
as final Q4 2013
Module VIIIPost-authorisation
safety studies
Module IXSignal management
Module XAdditional monitoring
Module XIPublic participationPublic consultation
Q2 2014
Module XIIContinuous
pharmacovigilancePublic consultation
Q1 2014
Module XIVInternational cooperation
Public consultation Q1 2014
Module XVSafety
communication
Under development
Public consultation
Published
P I – VaccinesPublication as final
Q4 2013
Module XVIRisk minimisation
measuresPublication as final
Q4 2013

55
Beyond 2013…what still needs to be done
Topics Activities
Literature monitoring EMA service to industry for population of EudraVigilance with
case reports of old substances.
EudraVigilance Delivery of enhanced functionalities and Information
Technology (IT) system audit results in centralised reporting for
industry
Article 57(2) data
submission and handling
Updates (variations) to the data can be submitted by industry
and data fully used to support regulation, safety and
stakeholder needs.
Periodic Safety Update
Reports
Delivery of PSUR repository and single PSUR assessment
process for National Application Procedures (NAPs) allowing
centralised reporting for industry and faster warnings for NAPs
Risk Management
System
Implement risk-based system for measuring the effectiveness
of risk minimisation
Transparency and
communication
Delivery of EU Medicines web-portal and public hearings.

EU GVP Module VRisk management systems
● GVP* are a set of measures drawn up to facilitate the performance
of pharmacovigilance (PV) in the EU
● Apply to Marketing Authorization (MA) holders, the EMA and
medicines regulatory authorities in EU Member States (MSs). They
cover medicines authorized centrally via the Agency & medicines
authorized at national level.
● Replace “Rules Governing Medicinal Products in the European Union”
(Volume 9A)
● Effective since July 2, 2012
● New format/template* required since January 10, 2013
● To be used for new submissions & RMP updates
*Guideline on good pharmacovigilance practices (GVP). Module V – Risk management systems - EMA/838713/2011
6

EU GVP Module VMajor components of the RMP (EU)● Safety Specifications: what is and is not known about safety of a drug
● Important identified and potential risks, missing information
● PV Plan = Risk Assessment Activities● Routine PV Practices
• Activities outlined in your PV System Master File (PSMF)
• Periodic Reports (e.g. PSUR)
• Enhanced PV Activities (additional monitoring)
● Additional PV Activities• Non-clinical studies
• Clinical trials
• Non-interventional studies (PASS)
● Risk Minimization Measures● Routine Measures
• Labeling (SPC, package leaflet)
• Packaging (size and design)
• Legal (prescription) status of the product
● Additional Risk Minimization Measures• Educational program / tools (checklist, brochure, poster, patient alert card)
• Controlled access program
● Other Risk Minimization Measures• Pregnancy prevention program
7

Risk Management Plan Template: New format (EU)
Part I Product(s) overview
Part II Safety specification
• SI Epidemiology of the indication(s) and target population(s)
• SII Non-clinical part of the safety specification
• SIII Clinical trial exposure
• SIV Populations not studied in clinical trials
• SV Post-authorisation experience
• SVI Additional EU requirements for the safety specification
• SVII Identified and potential risks
• SVIII Summary of the safety concerns
Part III Pharmacovigilance plan
Part IV Plans for post-authorisation efficacy studies
Part V Risk minimisation measures (including evaluation of the effectiveness of
risk minimisation measures)
Part VI Summary of the risk management plan
Part VII Annexes
8
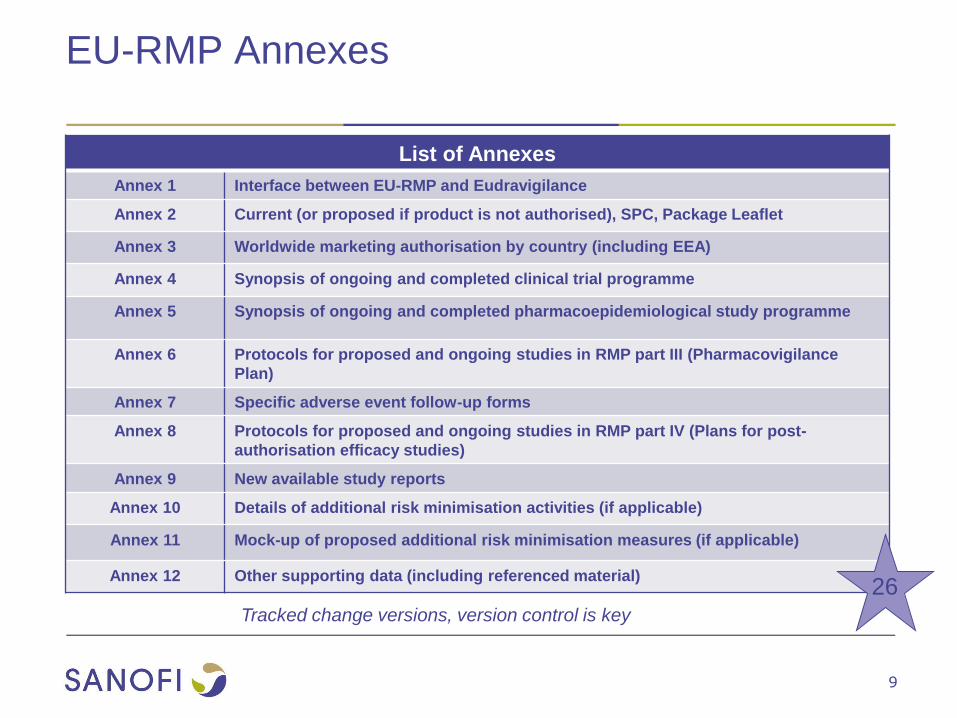
EU-RMP Annexes
List of Annexes
Annex 1 Interface between EU-RMP and Eudravigilance
Annex 2 Current (or proposed if product is not authorised), SPC, Package Leaflet
Annex 3 Worldwide marketing authorisation by country (including EEA)
Annex 4 Synopsis of ongoing and completed clinical trial programme
Annex 5 Synopsis of ongoing and completed pharmacoepidemiological study programme
Annex 6 Protocols for proposed and ongoing studies in RMP part III (Pharmacovigilance
Plan)
Annex 7 Specific adverse event follow-up forms
Annex 8 Protocols for proposed and ongoing studies in RMP part IV (Plans for post-
authorisation efficacy studies)
Annex 9 New available study reports
Annex 10 Details of additional risk minimisation activities (if applicable)
Annex 11 Mock-up of proposed additional risk minimisation measures (if applicable)
Annex 12 Other supporting data (including referenced material)26
Tracked change versions, version control is key
9

New Template
Part I - Product(s) overview
Part II - Safety specification
SI: Epidemiology of the indication(s) and target
population(s)
SII: Non-clinical part of the Safety Specification
SIII: Clinical trial exposure
SIV: Populations not studied in clinical trials
SV: Post-Authorisation Experience
SVI: Additional EU requirements for the Safety
Specification
SVII: Identified and potential risks
SVIII: Summary of the safety concerns
Part III - Pharmacovigilance plan
Part IV - Plans for post-authorisation efficacy
studies
Part V - Risk minimization measures
Part VI - Summary of the RMP
Part VII - Annexes
Past and present EU-RMP
Previous Template
Product information
Safety specification
Non-clinical
Limitations of the human safety database
Populations not studied in the pre-authorisation
phase
Post Marketing Experience
Adverse Events/Adverse Reactions
Identified and potential interactions including food-
drug and drug-drug interactions
Epidemiology of the indication and safety concerns
Pharmacological class effects
Additional EU requirements
Summary on ongoing safety concerns
Pharmacovigilance plan
Evaluation of the need for risk minimisation activities
Risk minimisation plan
Summary of the EU-RMP
Annexes
Part I
Part II
Part IV - Plans for post-authorisation efficacy
studies
10

GVP Module VRequirement for an EU-RMP
● For all new marketing applications
● For applications involving a significant change to an existing
marketing authorisation (MA)
● New dosage form, route of administration, manufacturing process of a
biotechnologically-derived product
● Paediatric indication
● Other significant change in indication (e.g. new disease area, age group
or a more severe disease to a less severely affected population).
● At the request of the Agency (EMA) or national Competent Authority
(NCA) when there is a concern about the risk-benefit balance
● At the time of the renewal of the MA if the product has an existing
RMP
● An updated EU-RMP should always be submitted if there is a
significant change to the benefit-risk balance of one or more medicinal
products included in the RMP
11

Update: Changes to RMPs (August 2013)
● There is no longer an automatic requirement to update RMPs on a
fixed-time basis. The Agency and the NCAs are now adopting a
risk-based approach to RMP updates.
● An updated RMP should now be submitted:
● at the request of the Agency or an NCA;
● whenever the risk-management system is modified, especially as the
result of new information being received that may lead to a significant
change to the benefit-risk profile or as a result of an important
pharmacovigilance or risk-minimisation milestone being reached.
● When justified by risk, the competent authority may still specify a date
for submission of the next RMP as a condition of the marketing
authorisation in exceptional cases.
● If the date for the submission of a periodic safety update report
(PSUR) and the need to update a RMP coincide, both can be
submitted at the same time.
12

Update: Changes to RMPs (August 2013)
● Changes to 'important missing information‘: The word 'important' has
been removed from the phrase 'important missing information' within
risk-management documents defining what constitutes a safety concern in
an RMP.
● Safety concerns are now classified as:
● important identified risks;
● important potential risks;
● missing information.
● Previously, International Conference on Harmonisation (ICH) of
Technical Requirements for Registration of Pharmaceuticals for
Human Use E2E and all EU risk-management documents used the
terms 'important identified risks', 'important potential risks' and'
important missing information' to define safety concerns in RMPs.
● The concept of a safety concern has not changed; it is still missing
information that could be clinically important and that needs to be
captured. The only change is the way that this concept is expressed.
13
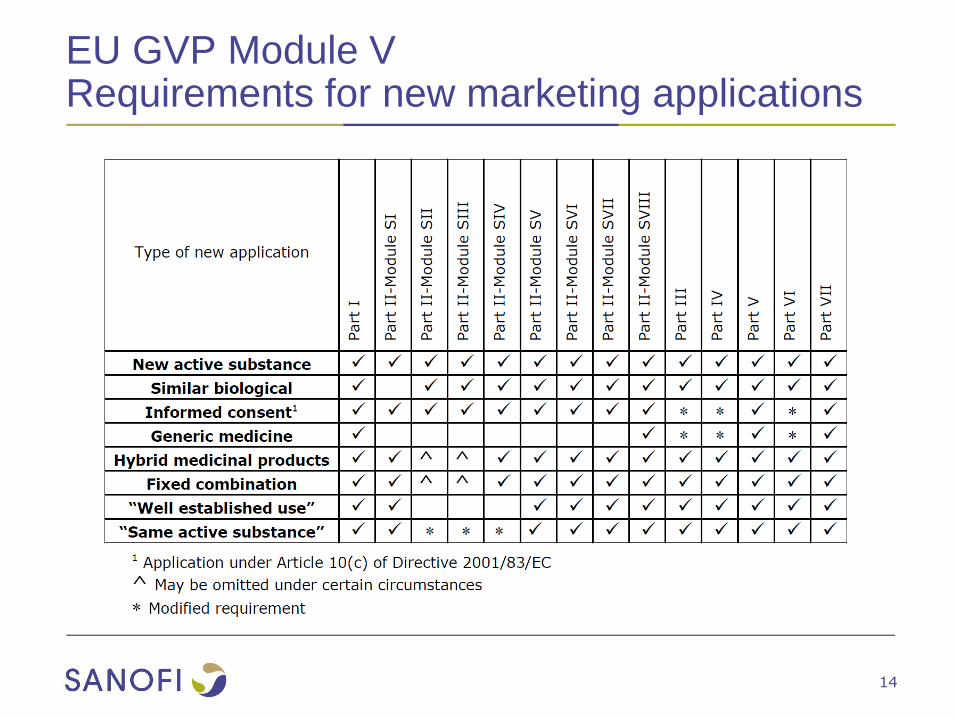
EU GVP Module VRequirements for new marketing applications
14

Common Sections Between the New EU-RMP and PSUR / PBRER
● The PSUR and RMP modular format is intended to minimize duplication by enabling common
(sections of) modules to be utilized interchangeably across both reports
15

PRAC (PV Risk Assessment Committee) Highlights
● EMA 7th scientific Committee (Replacing the PhVWP)
• Composed of EU National Agencies representatives, 6 independent scientific experts,
Healthcare Professionals and Patient associations representatives (2 each)
• Key role in PV assessments: mandatory or consultative role
• Referrals (Art 20, 31, 107i)
• PSUR/PBRER
• PASS protocols and results
• Signal detection
• Safety communication
• EURD lists
• Safety concerns
• Rapid risks evaluation in the context of the identified benefits
● Final responsibility for opinion on the risk-benefit remains with CHMP (centralized products) or CMDh(other products)
Additional monitoring
RMPs
Renewals & annual assessment
Type II safety variations
PV audits, inspections requests & results
16

Membership of PRAC
Appointed by
each Member
State:
Appointed by
European
Commission:
1 member + alternate
28 + EEA countries non
voting members
6 members - relevant expertiseincluding clinical pharmacology
and pharmacoepidemiology
1 member/alternate representing patient organizations
1 member/alternate representing healthcare professionals
17
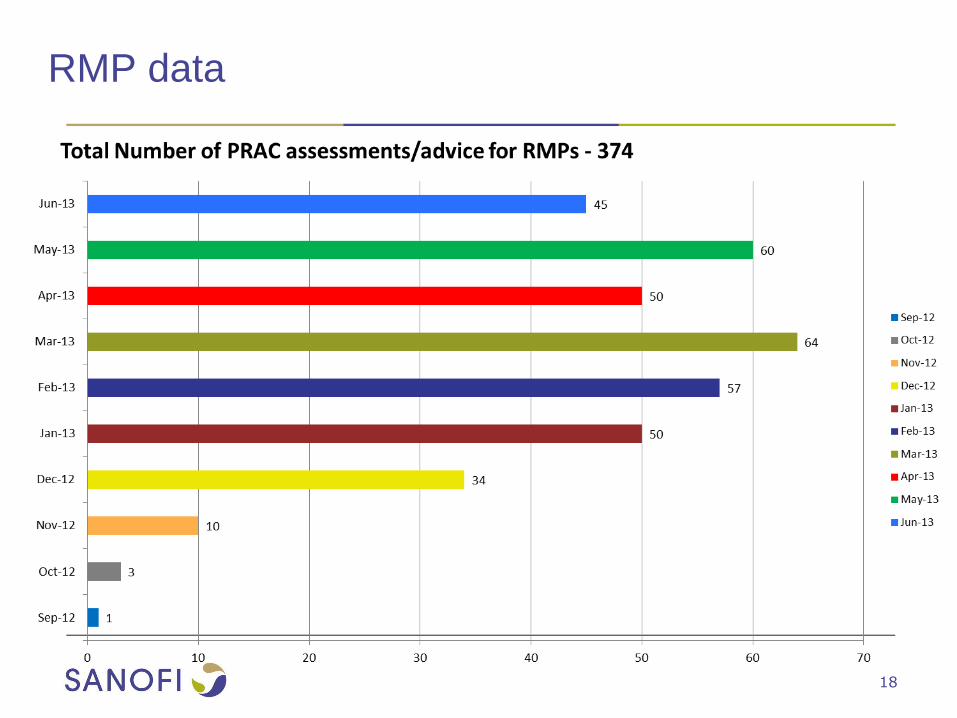
RMP data
18

RMP Data
19

● Views of patient,
consumer and healthcare
professional
organisations were
pivotal to choice of
inverted black triangle as
symbol
● Additional monitoring list
now published from April
2013
▼ This medicine is subject
to additional monitoring. This will allow quick identification of new safety information
You can help by reporting any side effects you may get
See the end of section 4 for how to report side effects
Additional Monitoring
20

Additional monitoring list
Monthly review by
PRAC of proposals
for additions to the
list
Communications
campaign starting
1 October 2012
21

Further legislation…
22

Publication of RMP summaries
●Pilot phase to be initiated in October 2013.
•Summary (Part VI.2 of RMP) to be published at the time of the EPAR
publication.
•Summary to be updated in case of important changes to RMP.
•Summary is aligned with other information (European Public
Assessment Reports (EPAR) summary, product information).
•For all newly - authorized Centralized Application Procedures (CAPs);
•For other (not newly authorised) CAPs, RMP summary to be published
when RMP is updated.
23

● VI.2.4: Summary of safety concerns
● Safety concerns in lay language
Important identified risk
Risk What is known Preventability
Risk 1 Frequency and severity
Important potential risk
Risk What is known
Risk 1 Reason why it is thought to be a potential risk along with
uncertainties
Missing information
Risk What is known
Risk 1 State risk has not been studied, the relevance to the target
population and what the recommendations are
Part VI.2: Elements for a public summary
24

Conclusion
Risk management is fluid and continuous
Global responsibility
Benefit/Risk
● Pharmacovigilance legislation is
continually evolving and a work-in-
progress
● Safety principles remain relatively
constant
● Main change in the RMP – Addition
of benefit/risk evaluation
● More details about planned
efficacy studies
● Opportunity to design efficacy /
safety studies in post-marketing
setting
25