neurologic phenotypes associated with col4a1 2 mutations · focal epileptiform discharges. in...
TRANSCRIPT

ARTICLE OPEN ACCESS
Neurologic phenotypes associatedwith COL4A1/2 mutationsExpanding the spectrum of disease
Sara Zagaglia, MD, Christina Selch, MD, Jelena Radic Nisevic, MD, Davide Mei, MD, Zuzanna Michalak, PhD,Laura Hernandez-Hernandez, PhD, S. Krithika, PhD, Katharina Vezyroglou, MD, Sophia M. Varadkar, MRCPI, PhD,Alexander Pepler, MBiol, Saskia Biskup, MD, PhD, Miguel Leão, MD, PhD, Jutta Gartner, MD, Andreas Merkenschlager, MD,Michaela Jaksch, MD, Rikke S. Møller, MsC, PhD, Elena Gardella, MD, PhD, Britta Schlott Kristiansen, MD,Lars Kjærsgaard Hansen, MD, Maria Stella Vari, MD, Katherine L. Helbig, MSc, Sonal Desai, MD,Constance L. Smith-Hicks, MD, PhD, Naomi Hino-Fukuyo, MD, PhD, Tiina Talvik, DrMed, Rael Laugesaar, MD,Pilvi Ilves,MD, PhD, KatrinÕunap,DrMed, IngridKorber, BSc, Till Hartlieb,MD,ManfredKudernatsch,MD, PeterWinkler,MD,Mareike Schimmel, MD, Anette Hasse, MD, Markus Knuf, MD, Jan Heinemeyer, MD, Christine Makowski, MD,Sondhya Ghedia, MBBS, FRACP, Gopinath M. Subramanian, FRACP, Pasquale Striano, MD, PhD,Rhys H. Thomas, MBChB, PhD, Caroline Micallef, FRCR, Maria Thom, FRCPath, David J. Werring, PhD, FRCP,Gerhard Josef Kluger, MD, PhD, J. Helen Cross, PhD, FRCPCH, Renzo Guerrini, MD, PhD, Simona Balestrini, MD, PhD,and Sanjay M. Sisodiya, PhD, FRCP
Neurology® 2018;91:e2078-e2088. doi:10.1212/WNL.0000000000006567
Correspondence
Dr. Sisodiya
AbstractObjectiveTo characterize the neurologic phenotypes associated with COL4A1/2 mutations and to seek genotype–phenotypecorrelation.
MethodsWe analyzed clinical, EEG, and neuroimaging data of 44 new and 55 previously reported patients with COL4A1/COL4A2mutations.
ResultsChildhood-onset focal seizures, frequently complicated by status epilepticus and resistance to antiepileptic drugs, was the mostcommon phenotype. EEG typically showed focal epileptiform discharges in the context of other abnormalities, including generalizedsharpwaves or slowing. In 46.4% of new patients with focal seizures, porencephalic cysts on brainMRI colocalizedwith the area of thefocal epileptiform discharges. In patients with porencephalic cysts, brain MRI frequently also showed extensive white matterabnormalities, consistent with the finding of diffuse cerebral disturbance on EEG. Notably, we also identified a subgroup of patientswith epilepsy as their main clinical feature, in which brain MRI showed nonspecific findings, in particular periventricular leu-koencephalopathy and ventricular asymmetry. Analysis of 15 pedigrees suggested a worsening of the severity of clinical phenotype insucceeding generations, particularly when maternally inherited. Mutations associated with epilepsy were spread across COL4A1 anda clear genotype–phenotype correlation did not emerge.
ConclusionCOL4A1/COL4A2 mutations typically cause a severe neurologic condition and a broader spectrum of milder phenotypes, inwhich epilepsy is the predominant feature. Early identification of patients carrying COL4A1/COL4A2 mutations may haveimportant clinical consequences, while for research efforts, omission from large-scale epilepsy sequencing studies of individualswith abnormalities on brain MRI may generate misleading estimates of the genetic contribution to the epilepsies overall.
From the Department of Clinical and Experimental Epilepsy (S.Z., Z.M., L.H.-H., S.K., S. Balestrini, S.M.S.) and Division of Neuropathology (Z.M., M.T.), UCL Institute of Neurology, London, UK; Clinic ofNeurology (S.Z.), Department of Experimental and Clinical Medicine, Marche Polytechnic University, Ancona, Italy; Department of Pediatric Neurology and Neurological Rehabilitation (C.S., T.H., P.W.,G.J.K.) and Neurosurgery Clinic and Clinic for Epilepsy Surgery (M.K.), Schon Klinik Vogtareuth; Department of Pediatrics (C.S.,M.S.), Children’sHospital Augsburg, Germany; UCLGreatOrmond StreetInstitute of Child Health (J.R.N., K.V., S.M.V., J.H.C.), London, UK; Paediatric Neurology and Neurogenetics Unit and Laboratories (D.M., R.G.), A. Meyer Children’s Hospital, University of Florence, Italy;Chalfont Centre for Epilepsy (Z.M., L.H.-H., S.K., S. Balestrini, S.M.S.), Chalfont-St-Peter, Buckinghamshire, UK; CeGaT–Center for Genomics and Transcriptomics (A.P., S. Biskup), Tubingen, Germany;NeurogeneticsUnit (M.L.),DepartmentofMedicalGenetics,HospitaldeSão João,Porto,Portugal;DepartmentofPediatricsandAdolescentMedicine (J.G.),UniversityMedicalCenterGottingen;Hospitalfor Children and Adolescents (A.M.), University Clinic Leipzig, Germany; Freiburg Medical Laboratory (M.J.), Dubai; The Danish Epilepsy Centre (R.S.M., E.G.), Dianalund; Institute for Regional HealthServices (R.S.M., E.G.), University of Southern Denmark, Odense; Department of Clinical Genetics (B.S.K.), Odense University Hospital; Hans Christian Andersen Children’s Hospital (L.K.H.), Odense,Denmark;PediatricNeurologyandMuscularDiseasesUnit (M.S.V., P.S.),DepartmentofNeurosciences,Rehabilitation,Ophthalmology,Genetics, andMaternalandChildHealth,UniversityofGenoa “G.Gaslini” Institute, Italy; Division of Neurology (K.L.H.), Children’s Hospital of Philadelphia, PA; Department of Neurology (S.D., C.L.S.-H.), Division of Neurogenetics, Kennedy Krieger Institute, Baltimore,MD;Center forGenomicMedicine (N.H.-F.), TohokuUniversity;Department of Pediatrics (N.H.-F.), TohokuUniversity School ofMedicine, Sendai, Japan;DepartmentofPediatrics (T.T., R.L.) and InstituteofClinicalMedicine (K.O.),UniversityofTartu;Children’sClinic (T.T.,R.L.),DepartmentofRadiology (P.I.), andDepartmentofClinicalGenetics,UnitedLaboratories (K.O.), TartuUniversityHospital, Estonia;Ludwig-Maximilians-UniversityMunich (I.K.); Departmentof PediatricNeurology (A.H.), Clinic Traunstein; Children’sHospital (M.K.), Dr. Horst Schmidt Klinik,Wiesbaden; AltonaChildren’sHospital (J.H.),Hamburg; Department of Pediatrics (C.Makowski), TechnischeUniversitatMunchen, Germany; Department of Clinical Genetics (S.G.), Royal North ShoreHospital, St Leonards; JohnHunter Children’sHospital (G.M.S.), New Lambton Heights, New South Wales, Australia; Department of Neurology (R.T.), University Hospital of Wales; Institute of Psychological Medicine and ClinicalNeurosciences (R.H.T.), Cardiff University; Division of Neuroradiology (C. Micallef), National Hospital for Neurology and Neurosurgery, London; Department of Brain Repair & Rehabilitation(D.J.W.), Stroke Research Centre, UCL Institute of Neurology, London, UK; Paracelsus Medical University (G.J.K.), Salzburg, Austria; and IRCCS Stella Maris Foundation (R.G.), Pisa, Italy.
Go to Neurology.org/N for full disclosures. Funding information and disclosures deemed relevant by the authors, if any, are provided at the end of the article.
The Article Processing Charge was funded by the Wellcome Trust.
This is an open access article distributed under the terms of the Creative Commons Attribution License 4.0 (CC BY), which permits unrestricted use, distribution, and reproduction in anymedium, provided the original work is properly cited.
e2078 Copyright © 2018 The Author(s). Published by Wolters Kluwer Health, Inc. on behalf of the American Academy of Neurology.

COL4A1 and COL4A2 encode α1 and α2 chains of type IVcollagen, respectively, and share a common locus at 13q34.One α2 and 2 α1 chains assemble into a heterotrimer of typeIV collagen, a structural component of basement membranes.α-Chains are composed of 3 domains: the amino-terminalregion (7S), the carboxy-terminal region (NC1), which ini-tiates heterotrimer assembly, and the collagenous part of themolecule, the triple helix region (THR). The THR is com-posed of amino acid triplet repeats (Gly-Xaa-Yaa), the firstbeing glycine (Gly) and the other 2 any amino acid.1 Mostpathogenic COL4A1/2 mutations are missense and lead tosubstitution of a glycine with a different amino acid.2 In 2005,semi-dominant Col4a1 mutations were demonstrated to in-duce perinatal cerebral hemorrhages and predispose to por-encephaly in an animal model, with COL4A1 mutationssegregating with human familial porencephaly.3 Sub-sequently, it has been recognized that autosomal dominantCOL4A1 and COL4A2 mutations cause a broad spectrum ofcerebrovascular disease, whose onset occurs from fetal lifeonward and whose severity may range from small-vessel dis-ease to fatal intraparenchymal hemorrhage.4–8 While epilepsyis known to be a clinical feature of porencephaly,3 the epilepsyphenotypes associated with mutations in COL4A1 and CO-L4A2 have not yet been detailed. We hypothesized that epi-lepsy could be a manifestation of disease even in patients inwhom porencephaly is not evident and aimed to characterizethe phenotypes associated with COL4A1/COL4A2 muta-tions, seeking genotype–phenotype correlation.
MethodsStandard protocol approvals, registrations,and patient consentsThis research was approved by the institutional ethics com-mittees of the participating centers. Informed consent wasobtained from all participants, or from parents or legalguardians of minors or individuals with intellectual disability.A bespoke questionnaire was used to collect clinical and ge-netic data.
Data were collected from published and new patients. Pub-lished cases were sought using COL4A1 and COL4A2 askeywords on PubMed/PubMed Gene and selected if theyprovided sufficient clinical details: 31 articles werereviewed.8–38
New patients were gathered through informal links andcontact with established consortia (EuroEPINOMICS RESand Deciphering Developmental Disorders). They were in-cluded if their variants were considered pathogenic, judged as
follows: nonsynonymous, splice-site altering, or truncatingchanges; present less than 2 times in >120,000 controls in theGenome Aggregation Database (gnomAD) browser and denovo, inherited from an affected parent, or found in affectedsiblings; or found in patients with MRI findings resemblingthe previously known COL4A1/COL4A2 phenotype (e.g.,with porencephaly). The following clinical variables wereassessed for all new patients: maternal complications duringpregnancy, antenatal and perinatal history, neuro-psychological delay and cognitive disturbances, and seizurehistory (age at seizure onset, seizure types, seizure frequency,history of status epilepticus, antiepileptic drug history).Seizures were classified according to the 2017 InternationalLeague Against Epilepsy (ILAE) classification and terminol-ogy.39 Drug-resistant epilepsy was defined according to theILAE Consensus.40 Available EEG recordings and brain MRIscans were evaluated. COL4A1 and COL4A2 mutations wereidentified through various methods (table 1, doi.org/10.5061/dryad.gj58t0v). The same data were sought from pub-lished cases, though were not always available.
Statistical analysisData were tested for normal distribution. We applied the χ2
test to estimate the significance of the differences in perinatalcomplications and Fisher exact test to assess the significanceof differences in prenatal evidence of brain pathology in 2groups (maternal or paternal inheritance). We applied theWilcoxon matched-pairs signed-rank test to assess the dif-ference in disease severity across generations in families withestablished disease. Data were analyzed using Stata/IC 11.1(StataCorp, College Station, TX).
ImmunohistochemistryImmunohistochemistry was performed from consented sur-plus resected tissue from case 1 and compared with 3 controlcases (additional methods, doi.org/10.5061/dryad.gj58t0v).
Data availabilityData not published within the article are available in a publicrepository (doi.org/10.5061/dryad.gj58t0v) and anonymizeddata will be shared by request from any qualified investigator.
ResultsGeneral description of previouslypublished patientsAltogether, 123 patients, from 73 different families, and 69different mutations (63 COL4A1 and 6 COL4A2) wereidentified.8–38 Epilepsy was reported in 55 patients, all ana-lyzed in this study, associated with 44 different mutations (42
GlossaryGly = glycine; HELLP = hemolysis–elevated liver enzymes–low platelet; ILAE = International League Against Epilepsy;MCD = malformations of cortical development; THR = triple helix region.
Neurology.org/N Neurology | Volume 91, Number 22 | November 27, 2018 e2079

for COL4A1 and 2 for COL4A2).8–29 Among published caseswith epilepsy, there were 12 of maternal origin, 11 of paternalorigin, 8 de novo mutations, and 24 with unknown in-heritance. Genetic and clinical details are summarized in dataavailable from Dryad (table 2, doi.org/10.5061/dryad.gj58t0v).
Demographic characteristics, mode ofinheritance, andprenatal andperinatal historyin new patientsData are available from Dryad (table 3a/b, doi.org/10.5061/dryad.gj58t0v). There were 46 new patients (24 male) in 9countries: Germany (n = 14, 2 from the same family), UnitedKingdom (n = 12), Italy (n = 10, 5 from the same family),Denmark (n = 3), Australia (n = 2), United States (n = 2),Estonia (n = 1), Japan (n = 1), and Portugal (n = 1). In thiscohort, 2 families were included (nos. 33a, 33b, 33c, 33d, and33e and 23a and 23b), in which at least one participant (nos.33b and 33d and 23a) had epilepsy. In 2 cases (nos. 26 and28), epilepsy was not found after evaluation in specializedcenters, but these cases were retained in the current studybecause they carried novel mutations and a compatible neu-rologic phenotype, described below separately. Two caseswere excluded from further analysis due to uncertainty aboutmutation pathogenicity.
In the final group of 44 new patients, mean age at last follow-up was 9.7 years (SD ± 13.4): 7 patients were adults (meanage 35.6 years; SD ± 15.4) and 37 individuals were children(mean age 4.9 years; SD ± 3.7).
De novo mutations were identified in 24 patients; maternalinheritance was found in 5 patients (including 2 sibling pairs),paternal in 6 (2 of whom were siblings). In one family, theparents tested negative, but both the proband (no. 26) and hissister (not included in the study) carried the same mutation;parental mosaicism is assumed but not proven. In 8 cases,inheritance was unknown.
Natural delivery was reported in 28 patients. Delivery wassurgical in 15 patients, due to the following complications:prenatal ventriculomegaly (nos. 4 and 10), severe intrauterinegrowth retardation (no. 24), polyhydramnios (nos. 26 and37), fetal arrhythmia (no. 2), intrauterine growth retardationin the other fetus (not included in the cohort) (no. 27), fetalmicrocephaly and mild renal pelvic dilation (no. 5), placentaprevia and intraventricular hemorrhage in utero detected byfetal MRI (no. 17), mild maternal abdominal trauma 3 weeksbefore due delivery date and subsequent failure to thrive andpathologic cardiotocographic recording (no. 13),hemolysis–elevated liver enzymes–low platelet (HELLP)syndrome (no. 22), prolonged labor (no. 19/a), and sus-pected hydrocephalus (no. 32); in 2, the reasons were un-known (nos. 20 and 34). Prenatal evidence of vascularcerebral insult was reported in 7 patients (nos. 4, 10, 17, 19/b,29, 32, and 37). All patients with prenatal evidence of a cere-bral vascular event or a prenatal complication requiring
surgical delivery developed severe intellectual disability andabnormal neurologic signs.
Maternal complications during pregnancy included gesta-tional diabetes (no. 38), placenta previa (no. 17), bleedingduring the first trimester treated with progesterone togetherwith detection of a single umbilical artery (no. 1), and HELLPsyndrome (no. 22). None of the mothers with pregnancycomplications carried themutation found in the affected child.
There were 6 late preterm births (nos. 13, 15, 18, 19/a, 20,and 27). Head circumference at birth was known for 20patients: 15 (nos. 1, 2, 4, 5, 9, 14, 19/a, 20, 21, 22, 24, 25, 29,31, and 32) had microcephaly.
Seizure semiology, EEG features, and anatomo-electroclinical correlationsPatients without epilepsy (nos. 23b, 26, 28, 33a, 33c, and 33e)were excluded from this analysis.
Seizure types included focal-onset seizures, epileptic spasms,and generalized tonic-clonic seizures without known focalonset. Mean age at seizure onset was 15.4 (SD ± 26.4)months. Focal-onset seizures, defined by seizure semiologyand interictal or ictal EEG findings, occurred in 28/38 patients(73.7%), 10 of whom showed multifocal changes on ictal orinterictal EEG. Ictal EEG was available in 5 patients. Video-EEG was not available. Among these 28, impairment ofawareness during seizures was described in 13 patients; evo-lution to bilateral tonic-clonic seizures occurred in 11 patients.Status epilepticus or prolonged seizures (lasting >5 minutes)occurred in 15/38 patients (39.5%) (nos. 1, 2, 3, 9, 10, 13, 19/a, 21, 23a, 29, 30, 31, 32, 34, and 36). Status epilepticus wasthe presenting symptom in 4 patients (nos. 3, 19a, 23a, and34). In 18/28 (64.3%) patients with focal seizures, EEGshowed diffuse abnormalities (spike-wave activity or gener-alized slowing) and brain MRI revealed widespread whitematter alterations (periventricular leukoencephalopathy,supratentorial white matter loss, and thinning of corpus cal-losum). In 13/28 patients (46.4%), a porencephalic cyst ora malformation of cortical development localized to the samearea as the identified seizure onset zone, with additionalwidespread white matter abnormalities. In 15/28 (53.6%)patients with focal seizures but no porencephaly, we founddiffuse abnormalities on brain MRI, including ventricularenlargement and asymmetry or periventricular leukoence-phalopathy and extensive white matter loss (nos. 3, 6, 7, 8, 9,11, 15, 16, 19a, 19b, 27, 30, 33b, 33d, and 34).
Nine patients had epileptic spasms (nos. 12, 13, 17, 18, 20, 25,32, 35, and 37). EEG was not available for patients 12 and 32.In the other 7 patients, focal onset of spasms was demon-strated on EEG and in 5 patients (nos. 17, 18, 20, 35, and 37)an association was found between EEG localizing features anda structural abnormality on brain MRI. One patient hadgeneralized tonic-clonic seizures only; EEG was not availableand it was not possible to exclude a focal onset (no. 29).
e2080 Neurology | Volume 91, Number 22 | November 27, 2018 Neurology.org/N

Drug resistance was reported in 24/36 (66.6%); 8 patients(22.2%) had a “good response” to treatment. No single drugstood out for efficacy data (table 3a/b, doi.org/10.5061/dryad.gj58t0v).
Three patients had surgical treatment for epilepsy. One pa-tient (no. 27) with drug-resistant focal seizures underwentcorpus callosotomy at 6 years of age, with significant re-duction in seizure frequency, with seizures currently every 6–8weeks.41 No complications due to anesthetic or surgery werereported. Patient 24, diagnosed with West syndrome at 6months, underwent corpus callosotomy at 20 months.42 After1 month of reduced seizure frequency, drug-resistant seizuresreturned and psychomotor delay became evident. Functionalhemispherectomy was then performed, leading to seizurefreedom and subsequent improved head control and eyecontact. No surgical complications were reported. Patient 1had surgery to remove a left temporo-occipital dysplasia at 21months: the pathology is reported below. He remainedseizure-free at the latest follow-up, 1 year after surgery.
Of the 55 published patients with reported epilepsy, de-scription of epilepsy phenotypes was provided in 16. Focalseizures were reported in 11 patients: 5 had porencephaly onMRI; 6 had periventricular leukoencephalopathy and irregularenlargement of the lateral ventricles. Four patients with focalepilepsy had EEG records reported, 2 showing a focal ab-normality and generalized slowing and spike-wave activity,with extensive hemispheric white matter loss and right-sidedporencephalic cyst on MRI. In one patient, EEG showeda slow background and generalized spike-wave discharges,with periventricular leukoencephalopathy and calcificationson MRI. One patient had generalized tonic-clonic seizuresand a right-sided porencephalic cyst, 1 had epileptic spasmswith good response to vigabatrin and extensive periventricularwhite matter changes, 1 had epileptic encephalopathy, and 2had neonatal seizures.
In a subgroup of the new patients (5/38 [13%]) (nos. 3, 7, 8,33/d, and 34) and 4/55 published cases12,17,21,28 (7%), epi-lepsy was the presenting clinical problem.
Neuropsychological development andneurologic examination in patientswith epilepsyIntellectual impairment was found in 39/55 previously pub-lished cases and in 36/38 new patients.
Neurologic examination showed a wide spectrum of motorabnormalities: pyramidal signs and spasticity were reported in21 new patients, dystonic features in 7, and hypotonia at birthin 12. Four new patients (2 children [nos. 3 and 34] and 2adults [nos. 33/d and 8]) had normal neurologic examinationat the mean age of 22.7 years at observation (SD ± 18.9 years):notably, patients 8, 34, and 33/d had epilepsy onset after thefirst year of life, at 11, 5, and 6 years, respectively. In publishedpatients, neurologic examination was abnormal in all but one.
Extra-CNS involvement in patientswith epilepsyOcular defects, reported in 16/55 published patients and 19/38 new patients, were the most frequent extra-CNS signs andcomprised congenital cataract, retinal vessel tortuosity, andanterior chamber dysgenesis. Increased serum creatine kinaseor muscle cramps were documented in 6 new and 7 publishedpatients. Kidney abnormalities (hematuria, hydronephrosis,renal agenesis, and polycystic kidneys) were found in 3 pub-lished and 3 new patients. Cardiac disease, reported in 3 newpatients and in 2 published patients, comprised mitral valveprolapse, ventricular septal defect, tricuspid regurgitation, andpatent foramen ovale. The extra-CNS signs were alreadypresent at the time of onset of epilepsy; the timing of onset ofthe increased serum creatine kinase could not be establishedfrom the histories and records available for review (tables 2and 3a/b, doi.org/10.5061/dryad.gj58t0v).
Brain MRI findings in patients with epilepsyA wide spectrum of abnormalities, summarized in figure 1,was observed on brain MRI. In 29 cases, the brain MRI wasperformed at epilepsy onset. Porencephaly (figure 1F) wasfound in 31/55 (56%) published patients and in 15/38(39.5%) new patients. All patients with porencephaly hada complex syndromic presentation, with severe de-velopmental delay, abnormalities on neurologic examination,and early-onset, drug-resistant seizures. Malformations ofcortical development (MCD) (figure 1D), including schi-zencephaly, polymicrogyria, focal cortical dysplasia, andnodular heterotopia, were identified in 11 new (28.9%) and 7(11%) published patients. Where present, MCD were alwaysassociated with signs of white matter vascular insult(i.e., periventricular leukoencephalopathy, ventricular dys-morphisms, or white matter thinning).
Periventricular leukoencephalopathy (figure 1, B and C) wasreported in 11/55 (20%) published and 16/38 (42.1%) newpatients. Asymmetry of the lateral ventricles or basal ganglia(figure 1, A and E) was reported in 9/55 (16.4%) publishedand 22/38 (57.8%) new patients. Posterior fossa abnormali-ties were reported in 6 new (15.8%) (nos. 2, 9, 17, 18, 29, and38) and 7 (12.8%) published patients. In one new patient (no.31), MRI angiography showed reduced development of leftmedial and posterior cerebral arteries.
Longitudinal MRI data were available only for patients 1, 2, 5,8, 9, 10, and 16: subsequent MRIs were performed within 3years from the first one, except for patients 5 and 31, with 5and 12 years follow-up, respectively. In all cases, consecutivebrain MRI findings were stable.
Phenotypes of patients 26 and 28Patient 26 (COL4A1 p. G1169S), aged 17 at last follow-up,had moderate learning difficulties and left hemiparesis. EEGwas normal and brain MRI, stable after 2 years, showed bi-lateral fronto-parietal polymicrogyria and schizencephaly,periventricular nodular heterotopia, and white matter loss.
Neurology.org/N Neurology | Volume 91, Number 22 | November 27, 2018 e2081

Patient 28 (COL4A1 p.G1207V), aged 16 years at last ob-servation, had severe language impairment with dysarthria,language automatism, and left spastic hemiparesis. Brain MRIshowed right fronto-parietal schizencephaly. No seizures werereported. He had agenesis of the right kidney and a severe
ocular dysmorphism with bilateral ptosis, hemangioma of theleft superior eyelid, right cataract, and bilateral retinal atrophy.
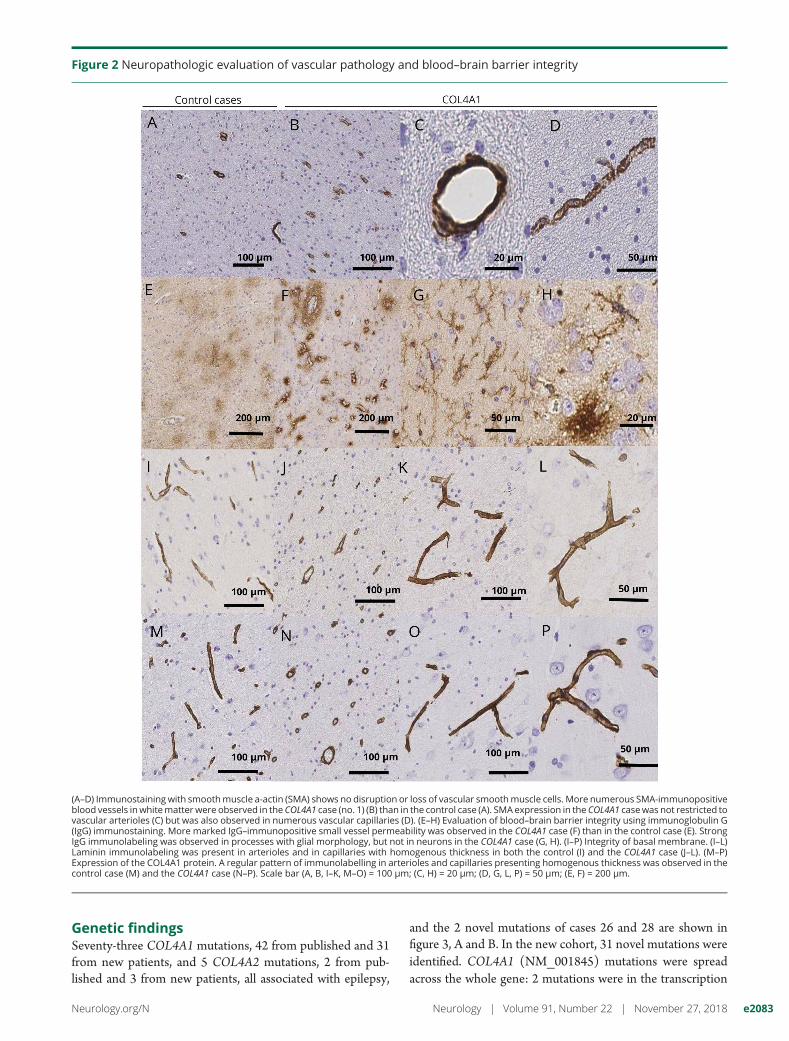
PathologyPathology results are detailed in figure 2.
Figure 1 The spectrum of imaging abnormalities observed with COL4A1 mutations
(A) Ventricular enlargement (arrows) anddysmorphism (dotted arrow), thinning ofcorpus callosum (*), white matter loss (pa-tient 1). (B) Periventricular leukoencephal-opathy (arrows) (patient 33/a). (C) Acutegerminal matrix hemorrhage on fetal brainMRI (arrows) and consequent extensive leu-koencephalopathy on postnatal brain MRI (*)at 8 days of life (patient 33/c). (D) Malforma-tions of cortical development: porencephalywith schizencephalic cleft (dotted lines) andpolymicrogyria (arrows) (patient 17). (E) Dys-morphism and asymmetry of basal ganglia(patient 30). (F) Porencephaly (patient 17).
e2082 Neurology | Volume 91, Number 22 | November 27, 2018 Neurology.org/N
Genetic findingsSeventy-three COL4A1 mutations, 42 from published and 31from new patients, and 5 COL4A2 mutations, 2 from pub-lished and 3 from new patients, all associated with epilepsy,
and the 2 novel mutations of cases 26 and 28 are shown infigure 3, A and B. In the new cohort, 31 novel mutations wereidentified. COL4A1 (NM_001845) mutations were spreadacross the whole gene: 2 mutations were in the transcription
Figure 2 Neuropathologic evaluation of vascular pathology and blood–brain barrier integrity
(A–D) Immunostaining with smoothmuscle a-actin (SMA) shows no disruption or loss of vascular smoothmuscle cells. More numerous SMA-immunopositiveblood vessels inwhitematter were observed in the COL4A1 case (no. 1) (B) than in the control case (A). SMA expression in the COL4A1 casewas not restricted tovascular arterioles (C) but was also observed in numerous vascular capillaries (D). (E–H) Evaluation of blood–brain barrier integrity using immunoglobulin G(IgG) immunostaining. More marked IgG–immunopositive small vessel permeability was observed in the COL4A1 case (F) than in the control case (E). StrongIgG immunolabeling was observed in processes with glial morphology, but not in neurons in the COL4A1 case (G, H). (I–P) Integrity of basal membrane. (I–L)Laminin immunolabeling was present in arterioles and in capillaries with homogenous thickness in both the control (I) and the COL4A1 case (J–L). (M–P)Expression of the COL4A1 protein. A regular pattern of immunolabelling in arterioles and capillaries presenting homogenous thickness was observed in thecontrol case (M) and the COL4A1 case (N–P). Scale bar (A, B, I–K, M–O) = 100 μm; (C, H) = 20 μm; (D, G, L, P) = 50 μm; (E, F) = 200 μm.
Neurology.org/N Neurology | Volume 91, Number 22 | November 27, 2018 e2083

initiation site, 68 in the THR, and 5 localized to theC-terminal region. The 2 mutations localized in the initial partof the gene (nos. 1 and 2) were associated with a severeclinical phenotype, with onset of epilepsy at 3 months anda history of status epilepticus. THR mutations comprised 9splice-site and frameshift mutations, 1 substitution leading toprotein truncation, and 58 missense mutations leading toglycine substitutions in Gly-Xaa-Yaa motifs. No obviouscorrelation between the position of the mutation and theseverity of the associated phenotype was observed in the THRregion. The 5 mutations in the C-terminal domain were allmissense and were all associated with a severe syndromicpicture, except the variant p.C1551Y, found in patient 34, withfocal epilepsy and behavioral problems, normal neurologicexamination, nonspecific white matter lesions, and an arach-noid cyst on MRI.
The COL4A1 p. G601S variant is newly described, identifiedin 2 new patients (nos. 8 and 9). Patient 8 had developmentaldelay, moderate cognitive impairment, autism, and normalneurologic examination. Focal-onset drug-resistant seizuresstarted at 11 years of age. Brain MRI showed extensivesupratentorial white matter loss and abnormalities, originallyinterpreted as perinatal infection. Patient 9 had onset of focal
drug-resistant seizures at age 10 months; neurologic exami-nation showed microcephaly and hypotonia at birth. MRIshowed periventricular white matter loss, thinning of thecorpus callosum, and cerebellar atrophy.
The COL4A1 p.G720D variant was previously described in 2families. In the first family,30 5 individuals had malformationsof the anterior chamber of the eye and cerebral vasculopathy(one member had infantile-onset hemiparesis). No epilepsywas reported in this family. In the second family,24 2 memberswere affected. The proband had intraventricular hemorrhageresulting in porencephaly and developed “generalized epi-lepsy” in the first year of life. He also had optic coloboma andcataract. His father had bilateral congenital cataracts, mi-graine, and recurrent TIA.
We found 5 recurrent COL4A1 mutations: p.G601S,p.G720D, p.G749S, p.G1044R, and p.G1239R. As detailedbelow, they were usually associated with phenotypicvariability.
The COL4A1 p.G1044R variant was described as a de novomutation in a patient with low birthweight, congenital bi-lateral cataracts, microcephaly, and porencephaly.19 Among
Figure 3 The distribution of mutations in the genes
The upper half of each figure depicts missense mutations, the lower half frameshift and splice-site mutations. (A) Distribution of COL4A1 mutations. (B)Distribution of COL4A2 mutations.
e2084 Neurology | Volume 91, Number 22 | November 27, 2018 Neurology.org/N
the new patients, we found a similar phenotype in a child whodied at 6 years of age (no. 24) and had bilateral porencephaly,intractable epilepsy, profound global developmental delay,microphthalmia, and congenital cataracts.
The COL4A1 p.G749S variant was described in an Italianfamily11: 2 siblings had spastic quadriparesis and focal epi-lepsy; their father had normal intellect and mild left hemi-paresis. The same variant was found in a patient with prenatalultrasound evidence of massive brain parenchymal hemor-rhage and neonatal seizures.27 His father, who had the mu-tation, only had minor white matter abnormalities onbrain MRI.
The COL4A1 p.G1239R variant was first reported as a pater-nally inherited mutation in a child with intracranial hemor-rhage identified on prenatal screening and subsequent leftporencephaly and progressive hemolytic anemia.23 The fatherhad features of hereditary angiopathy with nephropathy,aneurysms, and muscle cramps (HANAC) syndrome. In ourseries of new cases, the same mutation was found de novo inan affected 3-year-old girl. Surgical delivery was performedbecause prenatal hydrocephalus was suspected. The childdeveloped microcephaly, severe cognitive impairment, anddrug-resistant epileptic spasms. Of note, her paternal grand-mother died of a ruptured cerebral aneurysm (no otherclinical details were known).
The 5 COL4A2 (NM_001846) mutations were all missensemutations and localized to the THR domain.
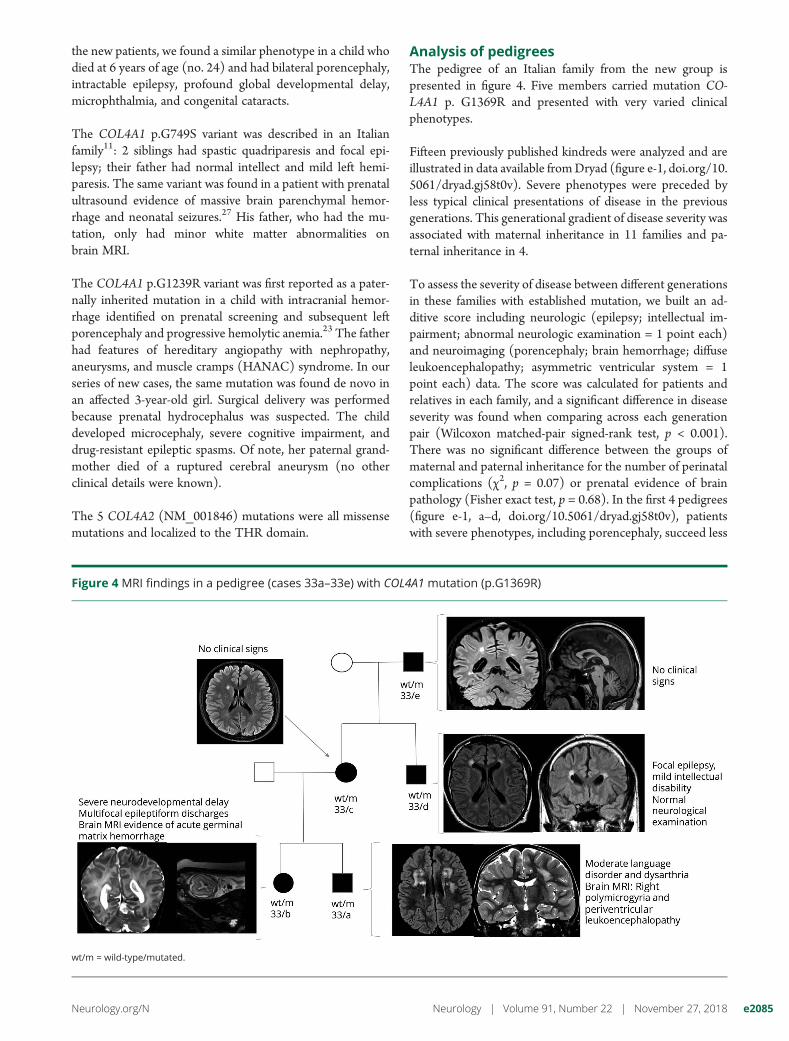
Analysis of pedigreesThe pedigree of an Italian family from the new group ispresented in figure 4. Five members carried mutation CO-L4A1 p. G1369R and presented with very varied clinicalphenotypes.
Fifteen previously published kindreds were analyzed and areillustrated in data available fromDryad (figure e-1, doi.org/10.5061/dryad.gj58t0v). Severe phenotypes were preceded byless typical clinical presentations of disease in the previousgenerations. This generational gradient of disease severity wasassociated with maternal inheritance in 11 families and pa-ternal inheritance in 4.
To assess the severity of disease between different generationsin these families with established mutation, we built an ad-ditive score including neurologic (epilepsy; intellectual im-pairment; abnormal neurologic examination = 1 point each)and neuroimaging (porencephaly; brain hemorrhage; diffuseleukoencephalopathy; asymmetric ventricular system = 1point each) data. The score was calculated for patients andrelatives in each family, and a significant difference in diseaseseverity was found when comparing across each generationpair (Wilcoxon matched-pair signed-rank test, p < 0.001).There was no significant difference between the groups ofmaternal and paternal inheritance for the number of perinatalcomplications (χ2, p = 0.07) or prenatal evidence of brainpathology (Fisher exact test, p = 0.68). In the first 4 pedigrees(figure e-1, a–d, doi.org/10.5061/dryad.gj58t0v), patientswith severe phenotypes, including porencephaly, succeed less
Figure 4 MRI findings in a pedigree (cases 33a–33e) with COL4A1 mutation (p.G1369R)
wt/m = wild-type/mutated.
Neurology.org/N Neurology | Volume 91, Number 22 | November 27, 2018 e2085

severe phenotypes having epilepsy as their main clinical fea-ture. Of note, in the less severely affected patients, brain MRIshowed nonspecific findings (in particular periventricularleukoencephalopathy and asymmetric enlargement of theventricular system) that would not have suggested the geneticdiagnosis until a more typical and severe phenotype appearedin the family.
DiscussionIn our series of new and published patients, the neurologicpatterns associated with COL4A1/COL4A2 mutations com-prised a typical severe presentation and a spectrum of lesscommon phenotypes, in which epilepsy can be the pre-dominant feature. In the present study, we retained the term“porencephaly,” notwithstanding its lack of specificity, as it isin common clinical usage. The typical severe phenotype wascharacterized by porencephalic cysts on brain MRI, andclinically defined by severe developmental delay, intellectualand behavioral difficulties, microcephaly, and motor abnor-malities on neurologic examination, with involvement of bothpyramidal and extrapyramidal systems.
We identified a subgroup of new and published patients inwhich epilepsy was the main feature leading to medical at-tention, associated with mild to moderate intellectual or be-havioral difficulties, while neurologic examination showedslight and insidiously developing motor abnormalities. Twoadult patients with epilepsy had normal neurologic examina-tion: patient 33/d was diagnosed after a severe phenotype ofdisease appeared in the family. Patient 8 was diagnosed 25years after epilepsy onset after specialist review. Milder pre-sentations are likely underdiagnosed, due to limited awarenessof the full spectrum of neurologic presentations, such thatclinicians (in particular those seeing adults) may suspectCOL4A1/2 etiology only in the most severe cases. The di-agnosis in milder cases with new-onset epilepsy is challengingbecause of the nonspecific brain MRI features(i.e., asymmetric ventricular enlargement, diffuse periven-tricular leukoencephalopathy, white matter thinning), whosecausation is frequently attributed to traumatic or hypoxic-ischemic birth injury and intrauterine infections. For instance,new patient 8 was initially diagnosed with epilepsy secondaryto unidentified perinatal infection. A similar misdiagnosis waspreviously described.22 Notably, the involvement of otherorgans (especially eyes, kidneys, andmuscles) was found to bealready present at the time of onset of epilepsy and can pro-vide a diagnostic clue.
We also observed a generational gradient of disease severity,especially with maternal inheritance. COL4A1/2mutations inthe fetus induce susceptibility to intrauterine environmentstressors that increase the risk of intraventricular hemor-rhage.7 Since COL4A1/COL4A2 are among the maternalsusceptibility genes for preeclampsia,43 we hypothesized thata mutation expressed in the maternal uterus may further
increase the risk of prenatal brain complications and, conse-quently, the severity of disease. Our analysis did not showa significant difference in prenatal and perinatal complicationsbetween maternally and paternally inherited cases. However,we suggest this hypothesis as one explanation, for testing infuture studies as the low numbers and potential selectionbiases may have limited the conclusions of the present study.The pedigree in figure 4 also highlights that asymptomaticcarriers (nos. 33/c and 33/e) can precede severe phenotypes.This observation suggests a reduced penetrance of COL4A1/COL4A2 mutations that may partly contribute to the gener-ational gradient of disease severity.
COL4A1/COL4A2 mutation-related seizures typically hadfocal onset, also in cases with epileptic spasms. EEG record-ings showed focal or multifocal epileptiform discharges andgeneralized, frequently asymmetric, abnormalities (general-ized spike-waves or diffuse slowing). Focal epileptiform dis-charges were related to a specific lesion (in particularporencephalic cysts, schizencephaly, or polymicrogyria) in46.4% of patients, while in the remaining patients, less specificEEG abnormalities were described, without a clear correlationwith a focal lesion. This complex anatomo-electroclinicalpicture suggests different pathogenic associations. The mosttypical is through predisposition to hemorrhagic and ischemicinsults, as demonstrated by mouse models.3,4 MCD were alsoassociated with Col4a1 mutations, as a result of defects ofcortical lamination.44 Here, we found a notably high preva-lence (28.9% of new cases) of polymicrogyria, schizencephaly,or focal cortical dysplasia.
De novo mutations seemed more common in the newlyidentified patients. One possible explanation of this discrep-ancy is that growing evidence of de novo variants in epilepsycausation has led to an increasing search for such variants, anda slight move away from familial studies. However, thenumbers in this study are modest overall and for some pub-lished cases data on inheritance were unavailable; thus, wecannot draw secure conclusions on this aspect.
An increased awareness of COL4A1/2-related epilepsy phe-notypes has clinical and research implications. One is forfollow-up. COL4A1/COL4A2 mutations are establishedmonogenic causes of stroke and can present for the first timein adult life with features of cerebral small-vessel disease, in-cluding subcortical hemorrhage and ischemic stroke, withlacunar infarcts, leukoaraiosis, and cerebral microbleeds onMRI,6 suggesting a dynamic evolution of COL4A1/2 leu-koencephalopathy. Although our subset of longitudinal MRIdata did not demonstrate progressive increase in the burdenof cerebrovascular disease, important limitations (young age,low numbers, short follow-up) hamper definitive statements.Although data on the risk from COL4A1/2 mutations forfuture intracerebral hemorrhage or ischemic stroke remainlimited, these mutations might increase the intracranialhemorrhagic risk in anticoagulated patients: one patient car-rying COL4A1 mutation p.G562E died at age 40 after
e2086 Neurology | Volume 91, Number 22 | November 27, 2018 Neurology.org/N

a spontaneous cerebral hemorrhage while on oralanticoagulation.25,26 The intracranial bleeding risk during IVthrombolysis in patients carrying COL4A1/COL4A2 muta-tions also needs consideration. The presence of cerebralmicrobleeds on brain MRI might help to identify those withCOL4A1/COL4A2 mutations at highest risk of intracranialhemorrhage prior to anticoagulation or thrombolysis.45,46
Epilepsy surgery, including both functional surgical procedures(like corpus callosotomy) and focal resections, was performedin 3 patients. To our knowledge, patient 1 is the first witha known COL4A1 mutation to have undergone a resection ofMCD, resulting in complete seizure control. Although we areaware of only 3 patients with this genetic condition treatedsurgically, notably the outcomes have been successful, in termsof both safety and effectiveness. There is rising interest in therole of genetic diagnostics during presurgical evaluation.47 Thegenetic epilepsies are heterogeneous and for some (e.g., focalcortical dysplasia due to mutations in MTOR pathway genes),surgery may be appropriate, while for other genetic conditionssurgerymay not be effective.47 It is therefore desirable that eachcausation is considered gene by gene in a multidisciplinaryteam, with, wherever possible, decisions based on un-derstanding of the underlying mechanisms of disease. Theevidence so far, although limited, suggests that surgery may bea valid option for drug-resistant COL4A1/2-associated epi-lepsy. The presurgical evaluation should consider other organinvolvement (which may contribute to an increased perioper-ative risk). Broadening the spectrum of clinical phenotypesassociated with COL4A1/COL4A2 mutations may help ourunderstanding of the genetic architecture of the epilepsies.Many large-scale genetic research efforts tend to exclude peoplewith structural changes on MRI, including cysts and periven-tricular leukoencephalopathy. The epilepsy phenotypes asso-ciated with COL4A1/COL4A2mutations suggest that this maynot be the most comprehensive strategy to determine the fulleffect of genetic variation in the causation and biology of theepilepsies, or to best apply genetically driven precision medi-cine approaches.48
There are certain phenotypic pointers to considering CO-L4A1/2mutations in individual patients, with implications forindividual patient management and for our understanding ofepilepsy genetics.
Author contributionsStudy concept: S.M.S., S. Balestrini, S.Z. Data acquisition:S.Z., C.S., J.R.N., D.M., Z.M., L.H.-H., K.S., K.V., S.M.V., A.P.,S. Biskup, M.L., J.G., A.M., M.J., R.S.M., E.G., B.S.K., L.K.H.,M.S.V., K.L.H., S.D., C.L.S.-H., N.H.-F., T.T., R.L., P.I., K.O.,I.K., T.H., M.K., P.W., M.S., A.H., M.K., J.H., C. Makowski,S.G., G.S., P.S., R.H.T., C. Micallef, M.T., D.J.W., G.J.K.,J.H.C., R.G. Data analysis and interpretation: S.Z., P.S., M.T.,D.J.W., G.J.K., J.H.C., R.G., S. Balestrini, S.M.S. Drafting ofthe manuscript: S.Z. Study supervision and critical revision:S.M.S., S. Balestrini. All authors critically reviewed and ap-proved the manuscript.
AcknowledgmentThe authors thank the patients and their families forparticipation in this study; Natascha Schneider for help withthe figures; David Goldstein, Slave Petrovski, and ErinHeinzen (Columbia University, New York, New York) forexome sequencing and variant analysis of one case; and ZaneJaunmuktane for help in pathology data interpretation.
Study fundingPart of this work was undertaken at University College LondonHospitals, which received a proportion of funding from theNIHR Biomedical Research Centres funding scheme. The workwas also supported by a Wellcome Trust Strategic Award(WT104033AIA), byMuirMaxwell Trust and Epilepsy Society,by Epilepsy Research UK (P1104) (to R.H.T.), by a grant fromthe EU Seventh Framework Programme FP7 under the projectDESIRE (grant agreement no 602531) (to R.G.), and by PUT(148) grant of the Estonian Research Council.
The DDD study presents independent research commis-sioned by the Health Innovation Challenge Fund (grantnumber HICF-1009-003), a parallel funding partnership be-tween Wellcome and the Department of Health, and theWellcome Sanger Institute (grant number WT098051). Theviews expressed in this publication are those of the author(s)and not necessarily those of Wellcome or the Department ofHealth. The study has UK Research Ethics Committee ap-proval (10/H0305/83, granted by the Cambridge SouthREC, and GEN/284/12 granted by the Republic of IrelandREC). The research team acknowledges the support of theNational Institute for Health Research, through the Com-prehensive Clinical Research Network. This study makes useof DECIPHER, which is funded by the Wellcome Trust.
DisclosureS. Zagaglia, C. Selch, J. Radic Nisevic, D. Mei, Z. Michalak, L.Hernandez-Hernandez, S. Krithika, K. Vezyroglou, S. Varadkar,A. Pepler, S. Biskup, M. Leão, J. Gartner, A. Merkenschlager, M.Jaksch, R. Møller, E. Gardella, B. Schlott Kristiansen, L.Kjærsgaard Hansen, M. Vari, K. Helbig, S. Desai, C. Smith-Hicks, N. Hino-Fukuyo, T. Talvik, R. Laugesaar, P. Ilves, K.Õunap, I. Korber, T. Hartlieb, M. Kundernatsch, P.Winkler, M.Schimmel, A. Hasse, M. Knuf, J. Heinemeyer, C. Makowski, S.Ghedia, G. Subramanian, and P. Striano report no disclosuresrelevant to themanuscript. R.H. Thomas has received honorariafrom Eiiai, UCB Pharma, and Sanofi. C. Micallef, M. Thom, D.Werring, G. Kluger, J. Cross, R. Guerrini, and S. Balestrinireport no disclosures relevant to the manuscript. S. Sisodiya hasreceived research funding or personal/institutional honorariafrom UCB, GSK, and Eisai Inc. and research support fromUCB; has an academic collaboration with Congenica; andserves on the editorial boards of Epileptic Disorders and PracticalNeurology. Go to Neurology.org/N for full disclosures.
Publication historyReceived by Neurology January 12, 2018. Accepted in finalform August 17, 2018.
Neurology.org/N Neurology | Volume 91, Number 22 | November 27, 2018 e2087

References1. Ricard-Blum S. The collagen family. Cold Spring Harb Perspect Biol 2011;3:a004978.2. Jeanne M, Gould DB. Genotype–phenotype correlations in pathology caused by
collagen type IV alpha 1 and 2 mutations. Matrix Biol 2017;57-58:29–44.3. Gould DB, Phalan FC, Breedveld GJ, et al. Mutations in COL4A1 cause perinatal
cerebral hemorrhage and porencephaly. Science 2005;308:1167–1171.4. Gould DB, Phalan FC, van Mil SE, et al. Role of Col4a1 in small-vessel disease and
hemorrhagic stroke. N Engl J Med 2006;354:1489–1496.5. Vahedi K, Alamowitch S. Clinical spectrum of type IV collagen (COL4A1)mutations:
a novel genetic multisystem disease. Curr Opin Neurol 2011;24:63–68.6. Lanfranconi S, Markus H.S. COL4A1 mutations as a monogenic cause of cerebral
small vessel disease: a systematic review. Stroke 2010;41:e513–518.7. Ment LR, Aden U, Lin A, et al. Gene Targets for IVH Study Group: gene-
environment interactions in severe intraventricular hemorrhage of preterm neonates.Pediatr Res 2014;75:241–250.
8. Meuwissen ME, Halley DJ, Smit LS, et al. The expanding phenotype of COL4A1 andCOL4A2 mutations: clinical data on 13 newly identified families and a review of theliterature. Genet Med 2015;17:843–853.
9. Aguglia U, Gambardella A, Breedveld GJ, et al. Suggestive evidence for linkage tochromosome 13qter for autosomal dominant type 1 porencephaly. Neurology 2004;62:1613–1615.
10. Breedveld G, de Coo IF, Lequin MH, et al. Novel mutations in three families confirma major role of COL4A1 in hereditary porencephaly. J Med Genet 2006;43:490–495.
11. Gasparini S, Qualtieri A, Ferlazzo E, et al. Normal immunofluorescence pattern of skinbasement membranes in a family with porencephaly due to COL4A1 G749S muta-tion. Neurol Sci 2016;37:459–463.
12. Giorgio E, Vaula G, Bosco G, et al. Two families with novel missense mutations inCOL4A1: when diagnosis can be missed. J Neurol Sci 2015;352:99–104.
13. Ha TT, Sadleir LG, Mandelstam SA, et al. A mutation in COL4A2 causes autosomaldominant porencephaly with cataracts. Am J Med Genet A 2016;170A:1059–1063.
14. van der KnaapMS, Smit LM, Barkhof F, et al. Neonatal porencephaly and adult strokerelated to mutations in collagen IV A1. Ann Neurol 2006;59:504–511.
15. Lemmens R, Maugeri A, Niessen HW, et al. Novel COL4A1 mutations cause cerebralsmall vessel disease by haploinsufficiency. Hum Mol Genet 2013;22:391–397.
16. Leung M, Lewis E, Humphreys P, et al. COL4A1 mutation in a pediatric patientpresenting with post-ictal hemiparesis. Can J Neurol Sci 2012;39:654–657.
17. Livingston J, Doherty D, Orcesi S, et al. COL4A1 mutations associated with a char-acteristic pattern of intracranial calcification. Neuropediatrics 2011;42:227–233.
18. Mancini GM, de Coo IF, Lequin MH, Arts WF. Hereditary porencephaly: clinical andMRI findings in two Dutch families. Eur J Paediatr Neurol 2004;8:45–54.
19. Meuwissen ME, de Vries LS, Verbeek HA, et al. Sporadic COL4A1 mutations withextensive prenatal porencephaly resembling hydranencephaly. Neurology 2011;76:844–846.
20. Rødahl E, Knappskog PM, Majewski J, et al. Variants of anterior segment dysgenesisand cerebral involvement in a large family with a novel COL4A1 mutation. Am JOphthalmol 2013;155:946–953.
21. Shah S, Ellard S, Kneen R, et al. Childhood presentation of COL4A1 mutations. DevMed Child Neurol 2012;54:569–574.
22. Smigiel R, Cabala M, Jakubiak A, et al. Novel COL4A1 mutation in an infant withsevere dysmorphic syndrome with schizencephaly, periventricular calcifications, andcataract resembling congenital infection. Birth Defects Res A Clin Mol Teratol 2016;106:304–307.
23. Takenouchi T, Ohyagi M, Torii C, Kosaki R, Takahashi T, Kosaki K. Porencephaly ina fetus and HANAC in her father: variable expression of COL4A1 mutation. Am JMed Genet A 2015;167A:156–158.
24. Tonduti D, Pichiecchio A, La Piana R, et al. COL4A1-related disease: raised creatinekinase and cerebral calcification as useful pointers. Neuropediatrics 2012;43:283–288.
25. Vahedi K, Massin P, Guichard JP, et al. Hereditary infantile hemiparesis, retinalarteriolar tortuosity, and leukoencephalopathy. Neurology 2003;60:57–63.
26. Vahedi K, Boukobza M, Massin P, Gould DB, Tournier-Lasserve E, Bousser MG.Clinical and brain MRI follow-up study of a family with COL4A1 mutation. Neu-rology 2007;69:1564–1568.
27. Vermeulen RJ, Peeters-Scholte C, Van Vugt JJ, et al. Fetal origin of brain damage in 2infants with a COL4A1mutation: fetal and neonatalMRI.Neuropediatrics 2011;42:1–3.
28. Yoneda Y, Haginoya K, Arai H, et al. De novo and inherited mutations in COL4A2,encoding the type IV collagen α2 chain cause porencephaly. Am J Hum Genet 2012;90:86–90.
29. Yoneda Y, Haginoya K, Kato M, et al. Phenotypic spectrum of COL4A1 mutations:porencephaly to schizencephaly. Ann Neurol 2013;73:48–57.
30. Sibon I, Coupry I, Menegon P, et al. COL4A1 mutation in Axenfeld-Rieger anomalywith leukoencephalopathy and stroke. Ann Neurol 2007;62:177–184.
31. Zenteno JC, Crespı J, Buentello-Volante B, et al. Next generation sequencinguncovers a missense mutation in COL4A1 as the cause of familial retinal arteriolartortuosity. Graefes Arch Clin Exp Ophthalmol 2014;252:1789–1794.
32. Bilguvar KL, DiLuna ML, Bizzarro MJ, et al. COL4A1 mutation in preterm in-traventricular hemorrhage. J Pediatr 2009;155:743–745.
33. de Vries LS, Koopman C, Groenendaal F, et al. COL4A1 mutation in two pretermsiblings with antenatal onset of parenchymal hemorrhage. Ann Neurol 2009;65:12–18.
34. Alamowitch S, Plaisier E, Favrole P, et al. Cerebrovascular disease related to COL4A1mutations in HANAC syndrome. Neurology 2009;73:1873–1882.
35. Plancher JM, Hufnagel RB, Vagal A, Peariso K, Saal HM, Broderick JP. Case of smallvessel disease associated with COL4A1 mutations following trauma. Case Rep Neurol2015;7:142–147.
36. Lichtenbelt KD, Pistorius LR, De Tollenaer SM, Mancini GM, De Vries LS. Prenatalgenetic confirmation of a COL4A1 mutation presenting with sonographic fetal in-tracranial hemorrhage. Ultrasound Obstet Gynecol 2012;39:726–727.
37. Durrani-Kolarik S, Manickam K, Chen B. COL4A1 mutation in a neonate withintrauterine stroke and anterior segment dysgenesis. Pediatr Neurol 2017;66:100–103.
38. Verbeek E, Meuwissen ME, Verheijen FW, et al. COL4A2 mutation associatedwith familial porencephaly and small-vessel disease. Eur J Hum Genet 2012;20:844–851.
39. Fisher RS, Cross JH, French JA, et al. Operational classification of seizure types by theInternational League Against Epilepsy: position paper of the ILAE commission forclassification and terminology. Epilepsia 2017;58:522–530.
40. Kwan P, Arzimanoglou A, Berg AT, et al. Definition of drug resistant epilepsy:consensus proposal by the ad hoc Task Force of the ILAE Commission on Thera-peutic Strategies. Epilepsia 2010;51:1069–1077.
41. Papandreou A, Tisdall MM, Chong WK, Cross JH, Harkness WF, Varadkar SM.COL4A1 mutations should not be a contraindication for epilepsy surgery. ChildsNerv Syst 2014;30:1467–1469.
42. Hino-Fukuyo N, Kikuchi A, Iwasaki M, et al. Dramatic response after functionalhemispherectomy in a patient with epileptic encephalopathy carrying a de novoCOL4A1 mutation. Brain Dev 2016;39:337–340.
43. Yong HE, Murthi P, Borg A, et al. Increased decidual mRNA expression levels ofcandidate maternal pre-eclampsia susceptibility genes are associated with clinicalseverity. Placenta 2014;35:117–124.
44. Labelle-Dumais C, Dilworth DJ, Harrington EP, et al. COL4A1 mutations causeocular dysgenesis, neuronal localization defects, and myopathy in mice and Walker-Warburg syndrome in humans. PLoS Genet 2011;7:e1002062.
45. Charidimou A, Turc G, Oppenheim C, et al. Microbleeds, cerebral hemorrhage, andfunctional outcome after stroke thrombolysis: individual patient data meta-analysis.Stroke 2017;48:e332.
46. Wilson D, Charidimou A, Ambler G, et al. Recurrent stroke risk and cerebralmicrobleed burden in ischemic stroke and TIA: a meta-analysis. Neurology 2016;87:1501–1510.
47. Stevelink R, Sanders MW, Tuinman MP, et al. Epilepsy surgery for patients withgenetic refractory epilepsy: a systematic review. Epileptic Disord 2018;20:99–115.
48. Thomas RH, Berkovic SF. The hidden genetics of epilepsy: a clinically important newparadigm. Nat Rev Neurol 2014;10:283–292.
e2088 Neurology | Volume 91, Number 22 | November 27, 2018 Neurology.org/N

DOI 10.1212/WNL.00000000000065672018;91;e2078-e2088 Published Online before print November 9, 2018Neurology Sara Zagaglia, Christina Selch, Jelena Radic Nisevic, et al.
of disease mutations: Expanding the spectrum2/COL4A1Neurologic phenotypes associated with
This information is current as of November 9, 2018
ISSN: 0028-3878. Online ISSN: 1526-632X.Wolters Kluwer Health, Inc. on behalf of the American Academy of Neurology.. All rights reserved. Print1951, it is now a weekly with 48 issues per year. Copyright Copyright © 2018 The Author(s). Published by
® is the official journal of the American Academy of Neurology. Published continuously sinceNeurology

ServicesUpdated Information &
http://n.neurology.org/content/91/22/e2078.fullincluding high resolution figures, can be found at:
References http://n.neurology.org/content/91/22/e2078.full#ref-list-1
This article cites 48 articles, 11 of which you can access for free at:
Subspecialty Collections
http://n.neurology.org/cgi/collection/partial_seizuresPartial seizures
http://n.neurology.org/cgi/collection/infarctionInfarction
http://n.neurology.org/cgi/collection/epilepsy_surgery_Epilepsy surgery
http://n.neurology.org/cgi/collection/all_geneticsAll Genetics e
http://n.neurology.org/cgi/collection/all_cerebrovascular_disease_strokAll Cerebrovascular disease/Strokefollowing collection(s): This article, along with others on similar topics, appears in the
Errata
/content/early/2020/01/23/WNL.0000000000008787.full.pdf or: page
nextAn erratum has been published regarding this article. Please see
Permissions & Licensing
http://www.neurology.org/about/about_the_journal#permissionsits entirety can be found online at:Information about reproducing this article in parts (figures,tables) or in
Reprints
http://n.neurology.org/subscribers/advertiseInformation about ordering reprints can be found online:
ISSN: 0028-3878. Online ISSN: 1526-632X.Wolters Kluwer Health, Inc. on behalf of the American Academy of Neurology.. All rights reserved. Print1951, it is now a weekly with 48 issues per year. Copyright Copyright © 2018 The Author(s). Published by
® is the official journal of the American Academy of Neurology. Published continuously sinceNeurology

CORRECTION
Neurologic phenotypes associated with COL4A1/2 mutationsExpanding the spectrum of diseaseNeurology® 2019;00:1. doi:10.1212/WNL.0000000000008787
In the article “Neurologic phenotypes associated with COL4A1/2 mutations: Expanding thespectrum of disease” by Zagaglia et al.,1 the text for “3/M, 5 years” under the “Mutation Gene”column in supplementary table 3a should read “c.607G>A; p. G203R//paternal.” The authorsregret the error.
Reference1. Zagaglia S, Selch C, Nisevic JR, et al. Neurologic phenotypes associated with COL4A1/2mutations: expanding the spectrum of disease.
Neurology 2019;91:e2078–e2088.
Copyright © 2020 American Academy of Neurology 1
Copyright © 2020 American Academy of Neurology. Unauthorized reproduction of this article is prohibited.
Published Ahead of Print on January 24, 2020 as 10.1212/WNL.0000000000008787