nanotechnology strategic plan
TRANSCRIPT

Experience Supporting Clinical Next Generation Sequencing
Heidi L. Rehm, PhD, FACMG Director, Laboratory for Molecular Medicine, PCPGM
Assistant Professor of Pathology, BWH, MGH, HMS

Laboratory for Molecular Medicine at PCPGM
25% of testing is from
Partners’ patients
75% is from other US and
International patients
• CLIA-certified in 2003
• LMM offers >150 tests in cardiovascular disease, cancer,
hearing loss, pharmacogenetics and genetic syndromes
• Main focus of testing is large multi-gene panels using
sequencing technologies (NGS, Sanger)
• Other technologies include TaqMan, Luminex, allele-specific
PCR, MLPA, PNAs, STRs, droplet PCR
• WGS service will launch this week

Targeted panels are enlarging and more and more labs
are launching exome and genome services.
Genomic testing is improving diagnoses, enabling
informed treatments, and defining disease risks.
With gaining community acceptance, we must now
focus on optimizing support for integrating genomics
into the practice of medicine.
Clinical Genomics is Evolving

Step 1: Improve the analytical validity of NGS
Improve variant calling accuracy
Indels, CNVs, SVs, repeat expansions
Fill in the holes
Targeted NGS panels miss a variable amount of content
depending on genes
Exome sequencing misses 5-15% of coding sequence
International Collaboration for Clinical Genomics
Medical Exome Project: Partners/Emory/CHOP

0
50
100
150
co
ve
rage
exons
AVERAGE COVERAGE PER EXON
Cardiomyopathy Test (75 bp, single read, GAII)
Courtesy of Siva Gowrisankar
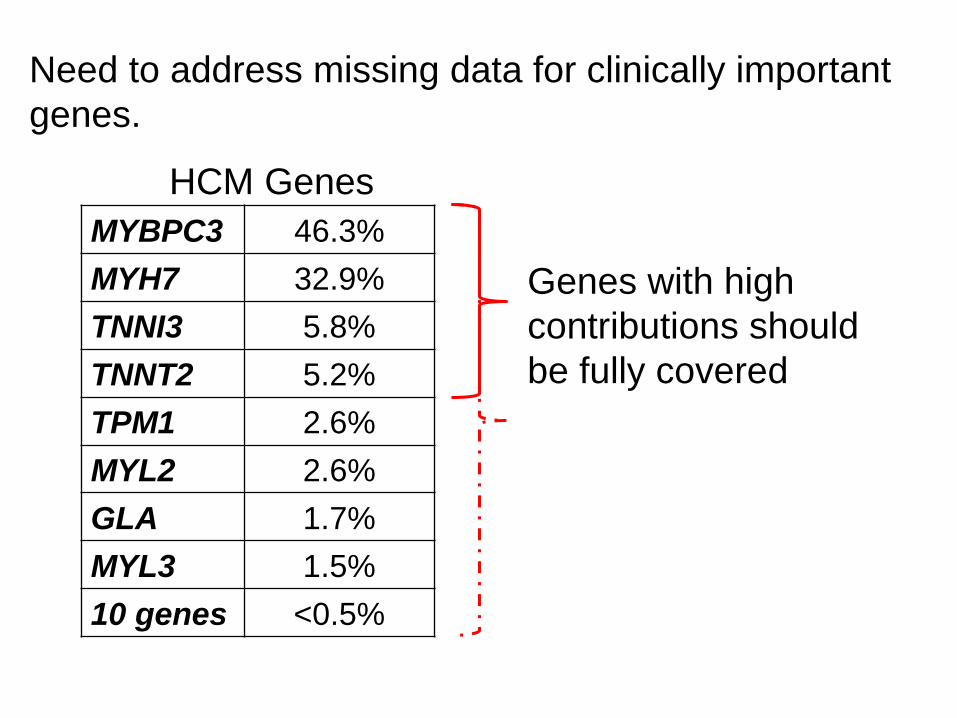
Relative Contribution of HCM Genes
MYBPC3 46.3%
MYH7 32.9%
TNNI3 5.8%
TNNT2 5.2%
TPM1 2.6%
MYL2 2.6%
GLA 1.7%
MYL3 1.5%
10 genes <0.5%
Need to address missing data for clinically important
genes.
Genes with high
contributions should
be fully covered
HCM Genes

0
10
20
30
40
50
60
70
80
90
100
PM
11
-10
05
6A
_L0
05
PM
11
-10
02
5A
_L0
07
PM
11
-10
05
0A
_L0
05
PM
11
-10
06
0A
_L0
03
PM
11
-10
00
1A
_L0
01
PM
11
-10
06
5A
_L0
03
PM
11
-10
02
3A
_L0
07
PM
11
-10
02
4A
_L0
06
PM
11
-10
05
4A
_L0
05
PM
11
-10
01
9A
_L0
05
PM
11
-10
03
4A
_L0
06
PM
11
-10
00
3A
_L0
02
PM
11
-10
03
1A
_L0
06
PM
11
-10
03
0A
_L0
07
PM
11
-10
03
6A
_L0
07
PM
11
-10
00
5A
_L0
03
PM
11
-10
00
2A
_L0
05
PM
11
-10
03
1A
_L0
05
PM
11
-10
02
2A
_L0
07
PM
11
-10
00
4A
_L0
03
PM
11
-10
02
0A
_L0
06
PM
11
-10
04
7A
_L0
05
PM
11
-10
01
8A
_L0
07
PM
11
-10
02
8A
_L0
06
PM
11
-10
03
3A
_L0
05
PM
11
-10
03
3A
_L0
06
PM
11
-10
01
5A
_L0
02
PM
11
-10
04
3A
_L0
03
PM
11
-10
01
4A
_L0
02
PM
11
-10
01
3A
_L0
02
PM
11
-10
02
6A
_L0
07
PM
11
-10
03
2A
_L0
03
PM
11
-10
01
1A
_L0
02
PM
11
-10
00
7A
_L0
01
PM
11
-10
00
3A
_L0
01
PM
11
-10
02
7A
_L0
06
PM
11
-10
00
2A
_L0
06
PM
11
-10
00
5A
_L0
01
PM
11
-10
01
2A
_L0
02
PM
11
-10
00
9A
_L0
01
PM
11
-10
01
0A
_L0
01
Failed Regions
Uncallable bases
Variant Confirmation
Am
plic
on
s
Sample number
Sanger Sequencing Follow-Up for OtoGenome
Average Coverage: 876 (range 275-1643)
Average Follow-up: 27 amplicons (out of 1439)

Failed Regions (7 entries, 9 amplicons)Gene % uncallableUncallable/Total basesCoordinatesNGS ROI intervalSanger ConfirmationMapping Messages
CTF1 100 55/55 16:30907950-30908004CTF1_EXON_01
CTF1 48 237/492 16:30913384-30913875CTF1_EXON_03
DES 2 14/608 2:220283170-220283777DES_EXON_01
DSC2 9 50/527 18:28681851-28682377DSC2_EXON_01
DSG2 15 Nov-75 18:29078200-29078274DSG2_EXON_01
RBM20 98 217/221 10:112404198-112404418RBM20_EXON_01
TPM1 73 119/162 15:63340760-63340921TPM1_EXON_02A_(1)
Uncallable bases Gene Exon cDNA PositionReference baseVariant baseCoverage A;B breakdownB frequencyCategory ClassificationPredicted ZygosityPredicted VariantPredicted AAStrand biasSanger ConfirmationCoordinatesMapping Messages
DSP Exon 24 c.8466T T . 0 0 0 Uncallable 0 Uncallable 6:7585961-7585961
DSP Exon 24 c.8470G G . 0 0 0 Uncallable 0 Uncallable 6:7585965-7585965
DSP Exon 24 c.8471G G . 0 0 0 Uncallable 0 Uncallable 6:7585966-7585966
LDB3 Exon 12 c.1294T T . 0 0 0 Uncallable 0 Uncallable 10:88476146-88476146
LMNA Exon 10 c.1706G G . 19 19;0 0 Uncallable -10.01 Uncallable 1:156107542-156107542
LMNA Exon 10 c.1707T T . 17 17;0 0 Uncallable -10.01 Uncallable 1:156107543-156107543
TNNT2 Intron 03 c.53-8C G . 0 0 0 Uncallable 0 Uncallable 1:201341177-201341177
TNNT2 Intron 03 c.53-7T A . 0 0 0 Uncallable 0 Uncallable 1:201341176-201341176
TTN Intron 45 c.10361-6TA . 0 0 0 Uncallable 0 Uncallable 2:179616772-179616772
TTN Intron 45 c.10361-5TA . 0 0 0 Uncallable 0 Uncallable 2:179616771-179616771
Follow-up (16 entries, 16 amplicons)Gene Exon cDNA PositionReference baseVariant baseCoverage A;B breakdownB frequencyCategory ClassificationPredicted ZygosityPredicted VariantPredicted AAStrand biasSanger ConfirmationCoordinatesMapping Messages
DSC2 Intron 07 c.942+12_942+13insTTAT TTAA 524 74;450 0.86 Variant (Het) c.942+12_942+13insTTAUnknown Variant 18:28666526-28666526
LAMA4 Exon 08 c.827C G T 573 0;573 1 Variant (Hom) c.827C>A p.Ala276Glu -7610.62 Variant 6:112508770-112508770
LDB3 Intron 07 c.756-12_756-11delTCTTC T 294 133;161 0.55 Variant (Het) c.756-12_756-11delTCUnknown Variant 10:88458996-88458998
LMNA Exon 10 c.*4C C G 35 11;24 0.69 Variant (Het) c.*4C>G -10.01 Variant 1:156107559-156107559
MYH6 Exon 07 c.622G C T 809 389;420 0.52 Variant (Unclassified)(Het) c.622G>A p.Asp208Asn-6747.06 Variant 14:23873940-23873940
PKP2 Exon 09 c.1955_1956insGAAGG GCTTC 822 577;245 0.3 Variant (Het) c.1955_1956insGAAGp.Ser652ArgfsX92Unknown Variant 12:32975416-32975416
PRKAG2 Exon 05 c.700_701insGG GC 74 66;8 0.11 Variant (Het) c.700_701insGp.Ala234GlyfsX39Unknown Variant 7:151329208-151329208
RBM20 Exon 09 c.2303G G C 430 0;430 1 Variant (Unclassified)(Hom) c.2303G>Cp.Trp768Ser -7751.62 Variant 10:112572458-112572458
RBM20 Intron 12 c.3452-9G G C 561 0;561 1 Variant (Unclassified)(Hom) c.3452-9G>C -7959.5 Variant 10:112590810-112590810
RYR2 Intron 15 c.1477-11_1477-10insTA AT 358 134;224 0.63 Variant (Het) c.1477-11_1477-10insTUnknown Variant 1:237619875-237619875
RYR2 Intron 29 c.3599-9delTAT A 707 393;314 0.44 Variant (Unclassified)(Het) c.3599-9delT Unknown Variant 1:237753074-237753075
RYR2 Intron 97 c.14091-11_14091-10insTA AT 417 291;126 0.3 Variant (Het) c.14091-11_14091-10insTUnknown Variant 1:237965133-237965133
TTN Exon 43 c.10049C G A 628 308;320 0.51 Variant (Het) c.10049C>Tp.Pro3350Leu-3786.36 Variant 2:179628969-179628969
TTN Exon 44B c.10213G C T 365 0;365 1 Variant (Unclassified)(Hom) c.10213G>Ap.Ala3405Thr-3228.31 Variant 2:179621477-179621477
TTN Intron 45 c.10361-5delTGA G 349 244;105 0.3 Variant (Het) c.10361-5delT Unknown Variant 2:179616770-179616771
TTN Exon 275 c.77167C G A 846 407;439 0.52 Variant (Het) c.77167C>Tp.Arg25723Cys-5407.69 Variant 2:179425988-179425988
Common SNPs (71 entries, 66 amplicons)Gene Exon cDNA PositionReference baseVariant baseCoverage A;B breakdownB frequencyCategory ClassificationPredicted ZygosityPredicted VariantPredicted AAStrand biasSanger ConfirmationCoordinatesMapping Messages
CASQ2 Intron 03 c.420+6T A G 626 0;626 1 Variant Benign (Hom) c.420+6T>C -5986.63 Variant 1:116283343-116283343
CASQ2 Exon 11 c.1185C G A 597 299;298 0.5 Variant Benign (Het) c.1185C>Tp.Asp395Asp-4456.04 Variant 1:116243877-116243877
DSC2 Exon 15A c.2326A T C 784 361;423 0.54 Variant Benign (Het) c.2326A>Gp.Ile776Val -7024.3 Variant 18:28649042-28649042
DSC2 Exon 15A c.2393G C T 713 393;320 0.45 Variant Benign (Het) c.2393G>Ap.Arg798Gln-5346.04 Variant 18:28648975-28648975
DSG2 Exon 08 c.861C C T 797 431;366 0.46 Variant Benign (Het) c.861C>T p.Asn287Asn-6084.92 Variant 18:29104698-29104698
DSG2 Exon 14 c.2318G G A 449 256;193 0.43 Variant Benign (Het) c.2318G>Ap.Arg773Lys -871.89 Variant 18:29122799-29122799
DSG2 Exon 15 c.3321T T C 693 368;325 0.47 Variant Benign (Het) c.3321T>Cp.Val1107Val-3592.72 Variant 18:29126670-29126670
DSP Exon 24 c.7122C C T 1000 520;480 0.48 Variant Benign (Het) c.7122C>Tp.Thr2374Thr-7131.27 Variant 6:7584617-7584617
DSP Exon 24 c.8175C C A 591 290;301 0.51 Variant Benign (Het) c.8175C>Ap.Arg2725Arg -3177.6 Variant 6:7585670-7585670
FILTER OUT
• No Sanger
• No further variant assessment
• Not included on patient report
SANGER SEQUENCE
PIPELINE OUTPUT List of exons that need sanger sequencing
Courtesy of Birgit Funke

A Genetic Sequencing Test is Not One Test
DNA
PCR
Hundreds of assays per sample
Sequencing
Hundreds of bases per exon
Failed exons/bases
One OtoGenome Test is actually ~380,000 tests with an infinite
number of possible results. After the NGS process, Sanger
follow-up begins:

VARIANT TYPE # FN SENSITIVITY 95%CI
All variants 400 2 99.50% 98.2-100
Substitutions 335 0 100.00% 98.9-100
Indels (all) 65 2 96.92% 98.8-99.2
>10 bp 14 1 92.86% 70.2-98.8
?10 bp 51 1 98.04% 89.9-99.7
5-10 bp 3 0 100.00% ND
3-5 bp 19 1 94.74% 76.4-99.1
1-2 bp 29 0 100.00% 88.3-100Pan Cardiomyopathy+ Hearing Loss Panel data FN = False Negative
VARIANT TYPE # FN SENSITIVITY 95%CI
All variants 400 2 99.50% 98.2-100
Substitutions 335 0 100.00% 98.9-100
Indels (all) 65 2 96.92% 98.8-99.2
>10 bp 14 1 92.86% 70.2-98.8
?10 bp 51 1 98.04% 89.9-99.7
5-10 bp 3 0 100.00% ND
3-5 bp 19 1 94.74% 76.4-99.1
1-2 bp 29 0 100.00% 88.3-100
VARIANT TYPE # FN SENSITIVITY 95%CI
All variants 400 2 99.50% 98.2-100
Substitutions 335 0 100.00% 98.9-100
Indels (all) 65 2 96.92% 98.8-99.2
>10 bp 14 1 92.86% 70.2-98.8
?10 bp 51 1 98.04% 89.9-99.7
5-10 bp 3 0 100.00% ND
3-5 bp 19 1 94.74% 76.4-99.1
1-2 bp 29 0 100.00% 88.3-100CNVs 16 0 100.00%
Modified from Birgit Funke
% FN SENS 95% CI % FP SPEC 95% CI
Substitutions * 0% 100% 99.3 - 100 0.08% 92.0% 99.6 - 99.9
In/dels 4.29% 95.70% 89.8 - 99.2 15.00% 85.0% 73.9 - 91.9
CNVs ? ?
Other structural ? ?
*non-repetitive regions
% FN SENS 95% CI % FP SPEC 95% CI
Substitutions * 0% 100% 99.3 - 100 0.08% 92.0% 99.6 - 99.9
In/dels 4.29% 95.70% 89.8 - 99.2 15.00% 85.0% 73.9 - 91.9
CNVs ? ?
Other structural ? ?
*non-repetitive regions
NGS Validation
99.9%

Relative sequence coverage is reproducible and comparable across exons and samples
Base c
overa
ge p
rofile
s fro
m 1
0 P
anC
ard
iom
yopath
y
tests
MYH7 exons
0-206
0-249
0-272
0-270
0-250
0-240
0-274
0-280
0-247
0-272
Trevor Pugh

Detection of full and partial gene deletions
through NGS: VisCap
Lo
g2
ratio
sa
mp
le/b
atc
h m
ed
ian
USH2A heterozygous
exon 10 deletion
All exons, sorted by genome position USH2A exons (3’→5’)
OTOF deletion
47 exons
Trevor Pugh
Need to balance FN and FP Rates
CNVs are confirmed by ddPCR

WGS Case 1: Non-syndromic Hearing Loss
• Mild-moderate “Cookie-bite” hearing loss
• Inheritance could be recessive or dominant with reduced penetrance
• Three affected children sequenced by WGS
• ~ 3.6 million variants identified
• No obvious candidates through initial analysis

Analyzed Hearing Loss Case by OtoGenome
STRC locus
All exons, sorted by genome position

Chromosome 15: STRC gene
Francey et al., 2011
high homology to pSTRC
STRC pSTRC
STRC pSTRC
Hom deletion of STRC
pSTRC
pSTRC
3 2 1 0 -1 -2 -3

2 1 1
2
4
16
47
0
5
10
15
20
25
30
35
40
45
50
0% 1-24% 25-49% 50-74% 75-89% 90-98% >98%
Exome Coverage of 73 Hearing Loss Genes
miRNAs
PTPRQ:
Reference
genome
assembly
problem
STRC:
Pseudogene
Common CNV

100 kb deletion
(43.89 Mb to 43.99 Mb)

WGS Case 1: Nonsyndromic Hearing Loss
Males with this deletion
will be infertile due to
deletion of the adjacent
CATSPER gene

Take Home Message
Targeted gene panels is still the recommended approach
when clinical sensitivity is high and until the cost of
exome/genome sequencing drops below targeted panels
such that one could start with exome/genome and then
reflex to targeted if negative.

Case 2: Distal Arthrogryposis Type 5
Disease is known to be AD and to occur de novo
Skeletal Spine stiffness, Hunched anteverted shoulders, Pectus excavatum, Limited forearm rotation and
wrist extension, Bilateral club feet, Congenital finger contractures, Long fingers, Absent phalangeal
creases, Poorly formed palmar creases, Camptodactyly, Dimples over large joints
Muscle Decreased muscle mass (especially in lower limbs), Firm muscles
Face Triangular face, Decreased facial expression
Ears Prominent ears
Eyes Ophthalmoplegia, Deep-set eyes, Epicanthal folds, Ptosis, Duane anomaly, Keratoglobus,
Keratoconus, Macular retinal folds, Strabismus, Astigmatism, Abnormal electroretinogram, Abnormal
retinal pigmentation
Clinical features:

Case 2: Distal Arthrogryposis Type 5
Previous DA phenotypes associated with MYBPC1,
MYH3, TPM2, MYH8, FBN2, TNNI2, TNNT3 were negative for variants
Two de novo mutations in exonic sequence:
ACSM4 – acyl-CoA synthetase medium-chain family member 4
5 nonsense variants identified in ESP; 1 with 6.4% MAF; 4 occur once
PIEZO2: mechanosensitive
ion channel component 2
Great candidate, but how to we prove causality for a
novel gene-disease association?

Project under discussion with Ada Hamosh to
deploy the PhenoDB system as a means to
connect labs with candidate gene identifications

A New Paradigm in Clinical Genomics
Patient/Provider
Researcher Clinical Lab
Research Discoveries
Clinical Lab Patient Care
Traditional Paradigm
New Paradigm

What is the biggest
bottleneck in clinical
sequencing?

Variant Assessment and Reporting
NVAs
Fellows/Residents
Draft Reports
Genetic Counselors
Signout
Geneticists
Clinical Correlation
Pathologists
Somatic Cancer
~15,000 variants
interpreted in patient
reports to date
Average
22 min
25 min
120 min
Variant Assessment Type
Variant with no data
Variant with dbSNP/ESP data only
Variant with publications

0
50
100
150
200
250
300
350
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32
HCM Gene Mutations – 3000 cases tested
>500 clinically significant mutations identified
66% of clinically significant mutations are seen in
only one family
Number of probands
Nu
mb
er
of va
ria
nts
MYBPC3 E258K
MYBPC3 MYH7 R502W W792fs R663H

0
50
100
150
200
250
300
350
400
450
1 3 5 7 9 11 13 15 17 19 21 23 25 27 29 31 33 35 37 39 41 43 45 47 49 51 53 55 57 59 61 63 65 67 69 71 73 75 77 79 81 83 85 87 89 91
Hearing Loss Gene Mutations – 2000 Cases Tested
Number of probands
Num
ber
of variants
GJB2
35delG
GJB2
V37I
GJB2
M34T
USH2A
2299delG
SLC26A4
81% (423/523) of clinically significant variants have been seen in only one family

100 Healthy Patients (10 PCPs)
100 HCM Patients (10 cardiologists)
Cardiomyopathy
Report
Cardiac Risk
Report
General Genome
Report
50 50
Project 2 Workflow
Whole Genome
Sequencing
MedSeq – WGS Pilot Clinical Trial
Standard of
Care
with
Family
History
50 50
Standard of
Care
with
Family
History Cardiac
Risk
Report
General
Genome
Report
Compare Outcomes Compare Outcomes

The General Genome Report
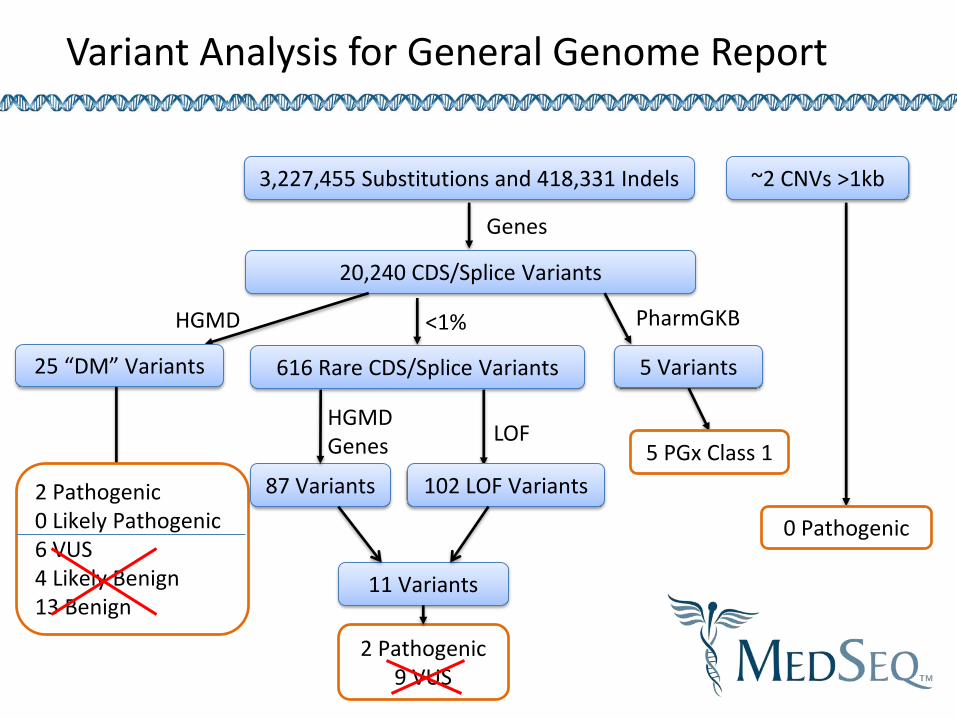
Variant Analysis for General Genome Report
3,227,455 Substitutions and 418,331 Indels
20,240 CDS/Splice Variants
25 “DM” Variants
HGMD
Genes
5 PGx Class 1
5 Variants
PharmGKB
2 Pathogenic 0 Likely Pathogenic 6 VUS 4 Likely Benign 13 Benign
<1%
616 Rare CDS/Splice Variants
87 Variants 102 LOF Variants
HGMD Genes
LOF
2 Pathogenic 9 VUS
11 Variants
~2 CNVs >1kb
0 Pathogenic

Fetus with US finding: ↑NT
PTPN11 p.Ile309Val Published as “pathogenic” for Noonan syndrome
Patient contacted author of paper who said he later found the variant in 7% of AJ controls; now feels the variant is benign
Courtesy Sherri Bale
Noonan Syndrome Case
?
LMM Case

To improve our knowledge of DNA
variation will require a massive
effort in data sharing

GeneInsight Laboratory Network
BWH
MGH
Intermount
ain Health
Litwin
Center
Michigan
More to
come
MGH
Pathology
LMM
Bioreference
Genetic
Data
Hub
Patient
Care
Providers
Illumina
CLIA Lab
ARUP
GeneInsight
Network
GeneInsight Clinics
GeneInsight Labs
Mt Sinai
NY
Genome
Center

Alert Delivery
3
4
This screenshot was taken from a demonstration system – the content of this screen should
not be used for any clinical purpose

Alerts are Common
~4% of case per year
received medium or high
alerts (.33% per month)

ICCG Project Goals To raise the quality of patient care by:
• Standardizing testing platforms and result interpretation related to structural and sequence-level variation
• Creating a centralized database of clinically relevant variant annotations to share data for clinical and research purposes
• Implementing a QC and expert consensus process for curating data submitted across laboratories and developing evidence based classifications

www.ncbi.nlm.nih.gov/clinvar
ClinVar
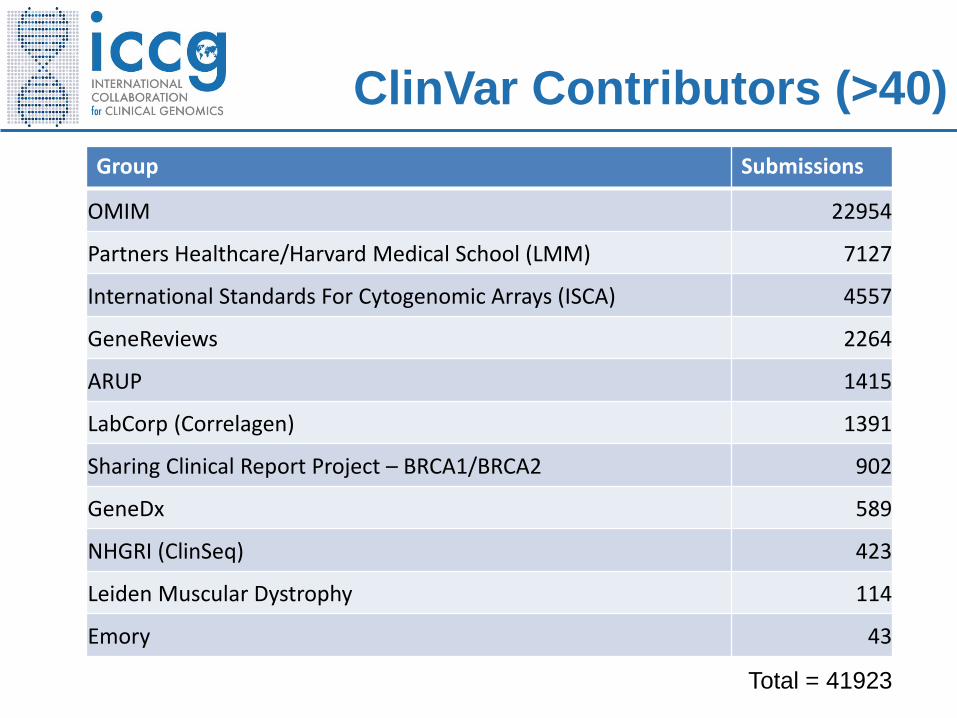
ClinVar Contributors (>40)
Group Submissions
OMIM 22954
Partners Healthcare/Harvard Medical School (LMM) 7127
International Standards For Cytogenomic Arrays (ISCA) 4557
GeneReviews 2264
ARUP 1415
LabCorp (Correlagen) 1391
Sharing Clinical Report Project – BRCA1/BRCA2 902
GeneDx 589
NHGRI (ClinSeq) 423
Leiden Muscular Dystrophy 114
Emory 43
Total = 41923

Sequencing Laboratories Which Have Agreed to Share Data
Ackerman Lab, Mayo Alfred I Dupont Hospital for Children All Children's Hospital St. Petersburg Ambry Laboratories ARUP Athena Diagnostics Baylor Medical Genetic Laboratories Boston Children's Hospital Boston University Children's Hospital of Philadelphia Children's Mercy Hospital, Kansas City Cincinnati Children's Hospital City of Hope Molecular Diagnostic Lab CureCMD Denver Genetic Laboratories Detroit Medical Center Emory University Fullerton Genetics Laboratory GeneDx Cleveland Clinic Greenwood Genetics Harvard-Partners Lab for Molec. Medicine Henry Ford Hospital Huntington Medical Research Institutes
Illumina Clinical Services Lab Indiana University/Perdue University InSiGHT LabCorp / Integrated Genetics / Correlagen Masonic Medical Research Laboratory Mayo Clinic Mt. Sinai School of Medicine Nationwide Children's Hospital Nemours Biomolecular Core, Jefferson Medical Oregon Health Sciences University Providence Sacred Heart Medical Center Quest Diagnostics SickKids Molecular Genetic Laboratory Transgenomics University of Chicago University of Michigan University of Nebraska Medical Center University of Oklahoma University of Penn University of Sydney University of Washington Women and Children's Hospital Wayne State University School of Medicine Yale University

SCRP: The Sharing Clinical Report Project: BRCA1/2
Robert Nussbaum, Dawn Lee, Heidi Rehm, George Riley, Peter Kolchinsky, Breast Cancer Clinics
445
19
255
62
119 Pathogenic
ProbablyPathogenic
VUS
Likely Benign
Benign
1981 variants (962 unique) from 25 clinics

Large variant
datasets
Intra-laboratory
Evidence-based review
Practice guidelines
Expert Curation
Single-Source Curation
Uncurated
Multi-Source Curation
Guideline
Inter-laboratory
dbSNP/dbVar
ClinVar
877
39004
1
ISCA
Maintaining Quality and Supporting Multiple Uses in ClinVar

N = 4,432 genes – not yet curated, still contain “false positives”
HGMD
OMIM
ClinVar
847
484
432
1897
108
91
31 Cosmic
11
CNV genes
7
CHOP
98 Animal
Models
278
Orpha
net
282
GeneTests
56
Defining the Medical Exome
Lead Groups: Partners, Emory, CHOP

1. Technical challenges • High GC • Pseudogenes/homologies • Repeat expansions • Common sites of structural variation
2. Variant types (denote common vs rare types) • Sequence variants (substitutions, small indels)
• Loss-of-function • Gain-of-function
• CNV – haploinsufficient • CNV – triplosensitive • Other structural changes (translocations, inversions, etc) • Imprinted loci • Repeat expansions
3. Medically relevant transcripts 4. Gene regions of pathogenic relevance 5. Patterns of inheritance (dominant, recessive, X-linked, mitochondrial, de novo, etc) 6. Phenotypes and evidence base for phenotype associations 7. Available approaches to define variant pathogenicity (assays, tools, etc) 8. Clinical utility measures 9. Clinical decision support opportunities
Community Efforts to Curate Genes and Loci in the Medical Exome/Genome

Annual Meeting
May 9-10, 2013
Bethesda, MD
Visit www.iccg.org for more details

Acknowledgements
NextGen Team
Birgit Funke
Lisa Farwell
Beth Duffy
Shangtao
Trevor Pugh
Siva Gowrisankar
Alison Brown
Natalie Boutin
Kevin Embree
Ana Holzbach
Nanda Kishore
Tom McLaughlin
Christine Poandl
Neeta Rathi
Brent Richter
Matilde Vickers
WGS Team
Matt Lebo
Robert Green – MedSeq
Sandy Aronson
Eugene Clark
Siva Gowrisankar
Amy Hernandez
Mike Murray
Shamil Sunyaev
GeneInsight Team
Sandy Aronson
Eugene Clark
Larry Babb
Frank Russell
Matt Varugheese
Tom Venman
Matt Lebo
Diana Toledo
Ashesh Patel
Fei Wang
Bob Hurley
Maureen Denning
Mike Oats
Gary Dombrowski
Juhan Sonin
Shane Thomas
Sharma Addepalli
Trung Do
Mike Band
Nancy Lugn
LMM Staff
ICCG/U41
Christa Martin
David Ledbetter
Robert Nussbaum
Joyce Mitchell
Andy Faucett
Erin Riggs
Erin Kaminsky
Sherri Bale
Madhuri Hegde
Patrick Willems
David Miller
Donna Maglott
Deanna Church
Justin Paschall
And many more……