monomeric and dimericietd.inflibnet.ac.in/bitstream/10603/588/14/14_chapter9.pdfmonomeric and...
TRANSCRIPT

Monomeric and dimeric
9.1 Introduction
A s proposed in Chapter 2 and demonstrate in Chapter 4-8, our main interest
has been to study the Lewis-base linkage to CoNzS2 and NiN2S.2
chromophores formed from the three mercapto ligands mbtH, mboH and
mbmH. An obvious extension of these chromophoric species and
spectroscopicaUy interesting system is the copper (11) analogue of these
complexes bearing C U N ~ S ~ core. Eventhough there were conflicting reports
about the formation of copper (11) complexes of these ligands, we attempted
preparation of their complexes under varied conditions. The result was our
failure to get any copper (11) species. Interesting, however was the overall
nature of the reactmn anti the pmduct obtained. Eventhough the highly
insoluble yellow product obtained had 1:l (metal : ligand) composition the
reaction produced quantitative yield of the product complex only when the
metal : %and ratio employed was 1 :2. Always one equivalent of the ligand
was seen remaining uncomplexed. However this was found to be not the
original ligand but its oxidixd form. Based on these obse~vations the overall
reaction between Cuh and mbt- could be summarised as below.

LC:u2+ + Lmbt -+ 2Cu' + mbr
2Cu' + 2mbt- + 2(~umbt)k
mbr + mbr -t (mbt)~
The final organic product (mbt):! could be characterised as the disulphide of the
mercapto ligand with structure 58. The characterisation of 58 was done by
both IR, IH NMK arid molecular mass determination. Contrary to some of
the earlier reports we could confirm (by ESR and magnetic susceptibility
measurements) that the copper complex formed was in fact a Cu(1) species,
presumably with high polymeric character. No attempt was made to
characterise it further a s this copper (I) complex generated does not posses
the ability to interact with Lewis-bases.
VO is another interesting system similar to Cup) having an electronic
configuration of IAr13d1. It may be formulated as species containing V4+ and
an oxide ion. A s expected, V02+ always occur coordinated to ligating species
both in solid and in solution bringing the coordination numbers of V either
to five or six. W e have tried the interaction of VO(1V) with mbtH, mboH and
mbmH, but no characterisable product could be separated out. Some redox
reaction is seen to be operating in all the cases. However, no attempt was
made to look into the reaction mode and identify the products formed.
Among the vanous VO2+ complexes, one of the very well characterised
species is VO(acac)2 {where acac = acetylacetonato moiety). Jonesfz) assumed
a square planar arrangement of four oxygen atoms about the vanadium
atoms of acac mowties with the bond of the fifth oxygen presumably

perpendicular to the plane of the other four. Zalkin et al proposed that the
five oxygen atoms are at the comers of an approximate square pyramid, with
vanadium at the crnhe of gravity of the pyramid rather than at its base. The
crystal structure of VO(acac)z was determined by Dodge[') et a1 which showed
that the five oxygen atoms of vanadium are a t the comers of a square
pyramid wth vanad~um lie approxnnately a t its centre of symmetry.
The compelling factor for continuing interest in vanadium ion complexes of
tetradentate schifl' bases, bidentate acetylacetone and its derivatives is that
it will provide active sites capable of binding other molecules. The
interaction of solvent molecules to VO(acac)z lead to the beginning of the
study of the adducts of Lewis-bases.Is-6) After this, adduct formation between
VO(acac)z and various coordinating species have been studied by W.L.Linert
et aW) and others. Electronic, 1K and far IR spectral studies could identify
the solvatochromic hehaviour of these complexes.
A particularly interrsting study is by Nassimbeni et alm who developed a series
of substituted pvndine complexes of VO(acac)z to attempt an ordering of the
substituted pyridines and correlation against substiwent parameters utilizing
the sensitivity of vi ;,, group frequency.
The specla1 ablhtj of hldentate Lew~s-bases chosen (like bipy, azpy, pyz) in
the present studv to act a s a bndging species (through the donor centres
disposed at thr ends) rather than chelating species gave us idea to try them
for adduct formattor1 wth 'u'O(acac)~, in which case it would result in closed
dimers. To our knowledge no such strategy has been tried so far to develop
VOz+ dimers. Thr present chapter discusses our attempt to develop such
dimers and the characterisation of thr various products formed.

9.2 Experimental
9.2.1 Preparative Details
9.2.1.1 Preparation of bis(acetylacetonato)oxovanadium(IV), VO(acac),
The Vanadyl acetonate complex was synthesized by mixing a solution of
O.Olmol of vanadium sulphate in water and 0.06 mol (6g) of acetyl acetone
followed by 20 g ot urea. The reaction mixture was covered with a watch
glass and heated overnight on a steam bath. A s the urea hydrolyses to
release ammonia, the complex separates out. The crystals were washed with
water and dried in am. It was recrystafised from chlorofom.(*j
9.2.1.2 Lewis-base adducts of VO(a~ac)~
More or less a common procedure was employed for the preparation of
Lewis-base adducts of VO(acac)z. The typical method employed for the pyridine
adduct is discussed below.
A solution of pyridine (4 mmol) prepared in 10 ml of acetone was added to 2
mmol of VO(acac)2 with constant stirring. This was refluxed for about 4-5 h,
concentrated bv evaporation and cooled. The addition of about 5-10 ml of
petroleum ether resillted into the precipitation of the adduct.P) It was then
filtered, washed wth pet-ether and dried. Yield was found to be 60%.
The similar methods were made use of for the preparation of all the adducts.
But the molar ratios of VO(acac)~ and Lewis-base employed were 2:l for pyz,
bipy and azpy adducts, while 1: 1 for pyr and amp.
9.3 Results and Discussion
9.3.1 Elemental Analysis
The elemental analvsis of all the adduct~~g.~O) were canied out (Table 9.1)
which showed a 1 1 (VO(acac)z: Lewis-base) stoichiometry for pyr and amp
adduct, but 2: 1 for pvz, azpy and b1p.v adduct

Table 9.1 Elemental analysis data
9.3.2 Infra-red Spectra
The IR spectra of VO(acac):! have several interesting features. Eventhough it
has a VOs chromophore, all the ligating O atoms are not similar. While ~ 0 ~ '
has a double bond character between V and 0 the other four oxygen atoms
of the two acac moieties coordinated to the metal have different bonding
mode. Several diagnostic peaks have been identified for the VO(acac)z. This
include the W=O of the vanadyl moiety. vv-0's resulting h m V-acac bonding, vc,o
and vcZc of the amr: moiety and the h v u . The assignment of these peaks have
been satisfactonly done by several authom.~11~7) In the solid state IR spectra of
VO(acac)~, the w =G has been found to occur at 999 cm-I, VC=O at 1590 cm-I, vc,c
at 1550 cm~l, w,'s at 488 and 61 1 cm and 60.~0 at 425 cm-'. Some interesting
changes are seen in these band positions on adduct formation of VO(acac)z with
the Lewis-bases which could be used to gauge their donor characteristics and
also to assess the overall electronic modulation.
Along with all the characteristic peaks of VO(acac):! the vibrational peaks due
to the Lewis-base moieties are also seen in all the adducts prepared. Some
of the important and characteristic peaks of the adducts are tabulated in
Table 9.2.
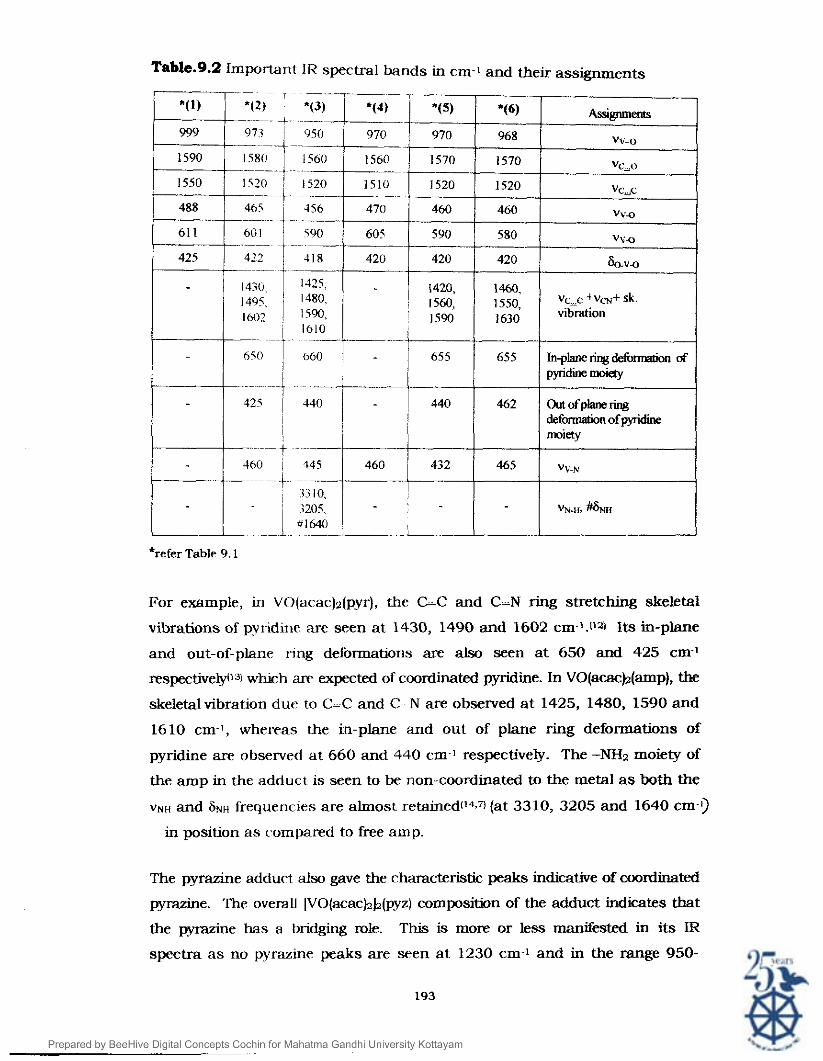
Table.9.2 Important IR spectral bands in cm-I and their assignments
*refer Table 9.1
For example, in VO(aca~)~(pyr), the L C and C-N ring stretching skeletal
vibrations of pyridine are seen at 1430, 1490 and 1602 cm-l.t12) Its in-plane
and out-of-plane ring deformations are also seen at 650 and 425 cm-'
respectivelylla w k h arr expected of coordinated pyridine. In VO(acac)2(amp), the
skeletal vibrafion due to C-C and C N are observed a t 1425, 1480, 1590 and
1610 cm l , whereas the in-plane and out of plane ring deformations of
pyridine are observed at 660 and 440 cm-I respectively. The -NH2 moiety of
the amp in the adduct is seen to be non-coordinated to the metal a s both the
VNH and ~ N H frequeric~es are almost retained(l4.7, (at 3310, 3205 and 1640 cm-l)
in position as compared to free amp.
The pyrazine adduct also gave the characteristic peaks indicative of coordinated
pyrazine. The overall [VO(ac.ac)zb(pyz) composition of the adduct indicates that
the pyrazine has a bridging role. This is more or less manifested in its IR
spectra a s no pyrazine peaks are seen at 1230 cm-1 and in the range 950-

1000 cm ' rrp'cted from . I O ~ O Coordinated (non-bfidged) mrme However orl ~ ~ ~ f i . ~ ' u ~ analysis a vev weak abmmtion at 695 cm-i is
o f m i m ~ h p ~ r i t y of mono c o o r d b m pyrezine adduct.
The IR swctra 'J*' ivo(acacJiJ2(biw) m d ~O(acacj2b(w) dso showed
the pw*nce of br*~mg irwis-br mokties m them. me cbcw& ~c a L N skelew vibrations of the irulebssp
m n at 1420, 1560 and 1590 cm-' in the b i p ~ adduct and at 14 10 1550 and 1630 cm.' h its erpy a d d ~ c t ~ ~ ~ l .
The correspondirlg m-plslr~e ring deformation is seen a t 655 cm-1 in both the
cases and the okrt of plane rurg deformation at 440 and 465 cm-l - -
respectively.
Like in the Case of the adducts of CoN2S2 and ~ f i ~ ~ ~ formed by hipy
coordination (discussed in Chapters 4 and 6) the ~ ~ ( a c a c ) ~ ] ~ ~ i p y ) also has
been checked for the characteristic. IR absorptions to monitor the mode of
bonding by the bipv moiety in the vanadyl adduct. If the bipy is acting a s a
monodentate species c,oordi.nating through only one of the pyridine moieties,
six characteristic peaks of bipy are expected at 800, 1070, 1215, 1402, 1483 and
1589 cm-l.il9l If coordination through both the pyridyl moieties exists the three
M d s at 1215, 1402 and 1589 cm~' are expected to be absent in the resulting
bipy bridged adducts in tale caw of (VO(acac)a]z (bipy) complex we
the abwncr ot above three absorptions indicating that the bipy is
actmg a s a bridgmg rnokety between two VO(acac)z species. The other three
characteristic peaks (out of the 6 peaks) are seen to occur at 820, 1065 and 1460 cm 1 for th~s adduct Further we could find a low intense peak at
1210 cm-1 whlrh could be assigned to an in-plane vibration indicating that -
the two pyridyl moietit.s are riot coplanar but tilted.
In all the Lews-base complexes a new band in the range 460-432 cm 1 is
seen which could be ass~gned to v i N as a result of the coordinated pyridyl
moiety.
Since all the Lewis-bases studied art mostly a-donor species coordinating
through the lone-pair based on a s p hybrid orbital of l the centrd V atom
of VO(a~ac)~ is expected to gain conside* electron density on the adduct
f t i o n . Since the metal atom is electmpsitire and the basal acac

moieties are full? conjugated, it is highly probable that the accumulated
electron density on the vanadium atom gets partly dissipated in the acac
framework. This would result in noticeable shift in some of the characteristic
acac vibrations. Further, the VO moiety has a double bond character
between V and O and any change in the electron density on the V is
expected to alter the VV=O stretching frequency also.
For various adducts prepared from the substituted pyridines, we could find
that the vv-o vibration is undergoing a downward shift from 999 cm-1 (in
VO(acac)z) to 975-950 cm-I. This effect could be expected, since the V=O bond in
vanadyl complexes is a multiple covalent bond consisting of pi-& donation of
electrons by the oxygen ta the vanadium, superimposed upon the o-bond. The
electron accepting capacity of vanadium (rv) which has one half-faed and
four emptv d-orbitals should be markedly affected by any coordinating
ligand frame work. The ligands (acac moieties) which are attached to the
metal (V02') donate their electron pairs which increase the electron density
on the metal d-orbitals and consequently the px-dn donation from oxygen to
vanadium is expectrd to be reduced to an extent which depends upon the
donor ability of the sixth ligand. A s a result there will be a lowering of V=O
bond orded71 in all the Lewis-base adducts.
In order to understand the Ixwis-base effects on the overall bonding and
electronic tuning in the various adducts prepared in the present study, we
have looked into the changes that occur in the four diagnostic peaks of
VO(acac)~ moietv. These an: vv-0, vv o, 60-"-0 and the combination peak of w-o
+~C.CH~. Among these the peak that is most sensitive to Lewis-base adduct
formation is seen to tw VV=O. While in the parent VO(acac)z species this peak
appears at 999 cm I , in the various Lewis-base adducts it gets shifted even
to the extent of 950 c m ~ l . Noticeable shifts are seen in other three peaks
also on the adduct formation. What is, however, interesting is that while in
the amp adduct the . I~V=O (shift in VV-0) was to the extent of 40 cm-I, in the
rest of the adducts it was only marginal (29cm~1).(7) Since we could not offer
any straight forward answer to this observation we have gone through the
literature and picket1 up the IR details of the various Lewis-base adducts of
VO(acac)z with substituted pyridines. Based on the trend observed for the

four diagnostic peaks of VO(acac)a moiety mentioned above we could class@
the adducts into two categories. These values are tabulated in Table 9.3
dong with those observed for our present complexes. In type I species the
vv-o shift was marked (with Aw-o in the range 40 k 4 cm-1). The substituent
on the pyridine moiety in this class of compound is seen to be having only
negligible effect on the vv.0 shift. Whether the substituent groups are either
electron donating or withdrawing, or the substituent position is varied, there
is seen to be no effect of them on the vv-o shift. Similar trend is seen in the
v ~ o , 60.~~0 and v ~ ~ ~ , + v ~ C H ~ peaks. In the second categoly of adducts (type 11)
their vv-o shift was seen to be only to the extent of 29M cm-1. Similarly the
vv-0, 60-v~o and I:V o+vr:. C H , shifts are also found to be only marginal. But in
this class of compounds, pyridine substituent is seen to be showing dominant
role. For example, while in 4-cyano pyridine adduct the w-o was 974 cm-1 in the
3,4-dimethyl pyridine adduct it was seen at 965 cm-1. The electron donating
groups on the pvridine moiety is found to be effecting vv-o more than
electron withdrawing groups. Na~simbeni(~) et a1 have analysed some of the
pyxidiie adducts of the VC)(acac)z species. They have stated that a simple
mass effect (due to pyridine moiety) can account for a shift of vv-o to the
extent of onlv 6 cm . So in both type 1 and type I1 classes some additional
factors should be involved. They have attributed the changes in these two
categories as due to the structural differences. There are two structural
isomers possible for the adducts which could be depicted a s below (Fig.9.1).
One of them is cis-isomer and the other is tran~-isomer.[~)
It has been stated that the cis-isomers are the ones that undergo maximum
shifts in vv-o on addurt fonnation. So among the various adducts tabulated
in Table 9.3, thr type 1 can be considered to be cis-and type I1 the trans.
Based on t h ~ ~ categotisation wr attribute a trans-structure (type IZ) to
[VO(acacbl@yr). IVO(acac)zb(wz), PO(acacblz(azpy) and IVO(acachlz@iW) and
cis-structure (tyw 1) to [VO(a~ac)~j(amp).

Table 9.3 IR data (in cm~l) of substituted pyridine complexes of VO(acac)z Substituted pyridinr is represented by R-CsH4N
I Twe 11 I
-- .- 601
- ~ ~ ~ . 42 1 462
Present work
(trans-) type 11
Fig.9. lThe cis-trans isomers possible for Lewis- base addurts of VO(acac)2

A trans-structure tcl the bipy, azpy and pyz adduct is tughly acceptable as they
are essenldly 2: 1 [VO(acac):2:lewis-b;lse] 'dimeric' species. The bridging ability of
the Lewis-base in these adducts is possible only when if the Lewis-base is
trans to V=O bond(becausc: of sterk reasons). The unusual cis-structure for
the 4-aminopyridine adduct may be essentially due to the high o- donor
ability of the Lewis-base for which the preferred site of cooxiination could be
cis to V=O. As discussed earlier amp cannot acts as a bridging ligand because
the inability of NH2 groups to take part in coordination. The shifts observed in
the other three diagnostic peaks also show the same trend.
The increase in electron density on the metal ion on adduct formation is also
expected to affect the vclr:, and vc c stretching frequencies of coordinated
acac (Table 9.2). 'fhe decrease in the IR absorptions of C - 0 and C C
frequencies (from 1 590 to 1510- 1520cmi assignable to VC-o and from 1550
to 1520 cm~ ' in the case of vc,~) is due to n-back donation of electron from
vanadium to oxygen which weakens the C-C and C-0 bonds.121221
9.3.3 Electronic Spectra
The energy level scheme for vanadyl moiety has been satisfactorily well explained
by Jorgensenr23! and fi'UrlaniR41, using simple crystal field model. Fulrani's
calculation conside~d only t.he C,V symmehy of bare V@+ alone, and, themfore,
cannot hope to account for all the obse~ed levels in its complexes. By
considering V(Y' in aqueous solution as tetragonal D.'O(H&)$+ molecule ion,
with axial destabilization, Jorgenseni231 obtained a level scheme which
qualitatively accounted for the crystal field part of the spectrum. Later,
Ballhausen1251 and <;ray developed the electrostatic model for the hydrated
vanadyl ion consisting V4+ situated in a tetragonal electric field caused by
the oxide ion and five water dipoles. The crystal field energy level diagram
for such a situation is given in Fig.9.2.

Ng. 9.2 Energy levels in crystal fields of Ob and compressed C ,, svmmel ry
If the tetragonal perturbation results in axial compression, the axial a,(W)
orbital is less stable than b,(dx2-yJ), but the ordering of e and & orbitals
depends on the relatlve values of splitting parameters. The ground state
configuraQon of thr V 0 2 + involves placing the one electron in the b2 orbital.
The predicted transitions are b+e, &+br and b~+al as shown in Fig.9.2.
The bz+b, transiuon can be considered to be corresponding to 10Dq.125)
The spectrum of VOS04.5H20 in aqueous medium shows two crystal field
bands at 13000 cm and 16000 cm which can be assigned (fhm the Fig.9.2)
to M e and b.+bI transitions respectively. The transition is expected at
higher energy but it is often indistmgmshable, presumably being covered by the
broad charge-transfer- band, which often appears around 30000 cm-1.
For VO(acac)~, the above mentioned transitions are observed at
40650(LMCT), 34015(b+a~), 16750 (b2+b1) and 14410 c m - l ( M ) Fable 9.4).
The adducts of VO(ac:ac:)? prepared in the present study show three transitions in
the range 13880- 14590 cm~l, 16520-17605 cm-I and 33220-34130 cm-1
respectively (Fig. 9. :$a and Fig.9.3b).

Fig. 9.3a Electronic spectra of Lewis-base adducts of VO(acac)z
Fig.9.3b Electronic spectrum of [VO(acac)a]z(bipy)
These bands could be assigned to bz-te, bz+b~ and b+al respectively which
are typical of octahedral (tetragonally distorted) VO2+ cornplexes.@5.'0) The
lODq values of these adducts were also calculated form bz-tbl transition

which corresponds to 10Ilq The electronic transitions, their assignments and
Dq values of the various Lewis-base adducts of VO(acac)z prepared are given in
Table 9.4.
Table 9.4. Ligand field transitions VO(acac)2 and its adducts
It was found that from the 'Table 9.4: the Dq values of these adducts decrease in
the order [VO(acac)~]~(bipy)~[VO(a~ac~]~(azpy)~pO(acac)z](amp) [VO(acac)z]@yrJ>
[VO(acac)~]z(pvz). This change in Llq values could be explained in terms of
the donor abilities of the Lewis-bases. Since amp is more basic than pyr, the
ligand field effect of amp could be more than that of pyr.(26) The greater Dq
value observed for- [VO(acac)2](amp) a s compared to [VO(acac)~](pyr) and
[VO(acac)2]2(pyz) is in agreement with this. Among the various Lewis-bases
selected for the study, pyz, pyr, and amp can be considered to be pure o-
donors. The donor ability of acac moiety and these Lewis-bases would be to
interact strongly aith dx2-yZ and dz2 orbitals of the metal and push them to
higher in rnergv. Greater the donor ability, greater will be the
destabilisation of dx2-y2 and dzr. Consequently, Dq will increase with
increase in the destabilisation of these axjally oriented orbitals. This is
depicted in Fig.9.4(b) (as compared to Fig. 9.4(a). The observed trend in Dq
values in the order ~IVO(acac)2](amp)> [VO(acac)~Jz@yr)> [VO(acac)2Jz(pyz) is in
perfect agreement with the donor abilities of the bases. It can be seen from
Fig. 9.4(c) that the lODq values can be enhanced if the lower tz, orbitals are
i Assignments nrn(cm-') .- Compound
294 597 694
294 590 698 (3401 5) (1 6949) (14326)
589 747
750 712
-.+ 1 334 296 562 73 7
(17793) (13568)
568 736
Dq (cm-' )
167s
1695
1698
1689
1779
1760
P (a (BM)
1.74
1.81
1.81
179
1.80
1.79

pushed down. This can happen if the Lewis-base in question has got some
n-acceptor property. 'The dxz and dvz orbitals of the tz, set are ideally suited
to interact with the rr* orbitals of the Lewis-bases.
We expect that awpyridine and bipyridine, because of their extensive
conjugation have energetically favorable x* MO's which would stabilize the
metal x-acceptor orbitals. The result of both the G-donor and this x-acceptor
property of bipv and azpy would be cumulative and result in larger lODq
separation. The observed higher Dq values for both azpy and bipy adducts
could be attributed to this synergic interaction.
A
energy
Fig. 9.4 Relative ordering of Dq in a) moderatively strong cr-donor Ligands b) veIy stmngly donating ligand firlds c) ligands with synergic interactions (a-donation and n-back donation)
9.3.4 Magnetic Susceptibility Measurements
The room temperature magnetic susceptibiliQ measurements were canied
out and values evaluated for all the adducts. The magnetic moment
values for the adducts are found to be in the range 1.74-1.81 BM (Table 9.4).
These values correspond to one unpaired spin per vanadium atom
demonstrating the presence of vanadium (IV) in these complexes and the
absence of direct metal-metal interactions between atoms. The magnetic
moment values are found to be slightly higher than the spin only value of

1.73 BM due to rhr possible mixing of vandium 3d~-* , 3dn and 4s orbitals.
These results also suggest a distorted octahedral geometry for the
ad duct^.^^^,^^^ Eventhough we could identify some ordering of the Lewis-base
adducts in terms of their Ilq-values, the observed b~ values did not give any
indication of ordering among the adducts. The perceptible change, however,
was that while the pentacoordinated VO(acac):! has a pea value of 1.74BM,
the rest of the adducts had their pee values around 1.80BM. We believe that
a temperature dependent magnetic measurement (especially a t low
temperature) would give a. clear ordering of these adducts. However in the
present study no attempt was made in this direction.
9.3.5 Electron Spin Resonance Spectra (ESR)
Electron spin resonance studies can yield useful information about the overall
bonding in vanadyl complexes (V02').@81 The V4' ion has a 3d1configuration and
since the orbital angular momentum gets quenched by crystal fields, the
paramagnetisn~ of the vanadyl ion arises form a single unpaired spin. The ESR
spectrum of V" is, therefore, quite analogous to that of C U ~ + which has a 3d1
hole configuration.fa 321
Looking at the overall geometry of the VO(acac)z molecule the complex can be
considered to have a &V symmetry with almost coplanar bonds between the V
ion and each of the four O atoms of the acac moieties. The vanadyl oxygen is
attached axially above the vanadium, ie. along the z-axis. The sixth ligand which
is a Lewis-base is expected to lie opposite to vanadyl oxygen, ie. on the z-axis.
It is worthwhile to consider the relevant molecular orbitals as developed by
Ballhausen and G r a p before we discussed the ESR in d e t d The MO's can be
considered as follows

The d's represent vanadium 3d orbitals, so-the vanadium 4s orbital, c, refers
to an sp ligand orbital on the ith ligand directed towards the vanadium
nucleus; the p's are &and 2p orbitals directed along the ith molecular axis
and subscript. 5 indicates the vanadyl oxygen. The p, a and E'S are coefficients
of the ion wave functions whereas the p' d, E' and E"S are coefficients of the
kand wave functions. AU. the molecular orbitals represented are bonding
orbitals. The general geometry is indicated in Fig. 9.5.
Ng.9.5 Structure of adducts ofVO(1V) complexes (0-orbitals)
A The appromate spln Hamiltonian Hcan be obtained by the same method
a s we use in Cu2+ complexes and it ran be given a s
where (2, ) and (x, y , 3 refers to the directions -el and perpendicular
respectively, to the vanadyl V - 0 bonds. S and I(vi refers to the electron spin
and the vanadium nuclear spin respectively. H is the applied magnetic field
and is the Bohr Magneton. An axial symmetry is assumed for the above
Hamiltonian
The electron spin resonance spectra of all the adducts were recorded in the
solid state and or in solutio~i (either rn toluene or in toluene + methanol (1: 1)

mixture) at LNT 'The solid state ESI? spectra of all the adducts gave a very broad
featureless spectra at room temperature. The spectra for the VO(acac)a(amp) is
shown in Fig.9.6
Fig.9.6 The solid state ESR spectrum of VO(acac)~(amp) a t LNT
In frozen solution at liquid nitrogen temperature all the compounds showed
anholmpic features. A s expected of 100% abundant 5lV isotope with I = 7/2 the
complexes gave typical &line spectra with two sets of resonance peaks, one
each due to parallel and perpendicular ~ornponentsc3~~*1. The spectra are
reproduced in Fig.9.7 to 9.10.
Table 9.5 ESK parameters of the Lewis-base adducts of VO(acac)~
compound
Ivq-hl @ur) -
- J.
.
The splitting pattern is also shown in the spectra. Both the parallel and
perpendicular components of g (gi and gi) is evaluated using the equation.
gpH=hv The g(av) is calculated by

Pig. 9.7 ESK spectrum of VO(acac)&m) in frozen solution at LNT
I l l I r I I I I I I I I I I
I 3300 4300
Field (G)
Flg. 9.8 ESR specbmm of VO(acac)g(amp) in frozen solution at LNT

I I I I I I I I I I I I I I I I
I 2300 3300 4300
Field (G)
Fig. 9.9 ESK spectrum of W(acachjz(pyz) in ! h e n solution at LNT
Field (G)
Fig. 9.10 ESK spectrum of w(acach]z(azpy) in h z n solution at LNT
207

The hypehe constants A , AL and A(av) are also evaluated. The various spin
Hamiltonian paramc-ters evaluated m-e tabulated in Table 9.5.
The parameter (PZ*)~ wM:h is a measure of the in-plane R-bonding was
evaluated using the equation~351
where, p x 10' = 128, K is the isotropic contact term, is related to the
amount of unpaired electron densip at vanadium nucleus, which is taken as
0.8 in our calculations.
The g!, g~ and g(av) values for all the adducts are in the expected range for V4+
in a tehgonal@ distorted octahedral field.(%) In 51V spectra the difference
g~ - g!, is generally considered to be indicative of the donor strength of the
basal ligand. In the adducts of VO(acac)2 the donor strengths of basal acac
moiety gets largely modulated by the axially ligating Lewis-bases. Greater
the donor ahilitri of the Lewis-base lesser will be the ligating strength of the
acac moiety. The Table 9.5 shows that g i - g p parameter decmases in the order
[ v O ( a c a c ) ~ j ( a m p ) ~ p O ( a c a c h ~ ( p y r ) ~ ~ O ( a ~ [ v O ( a c a c ~ ~ ( p y z ) . This trend
is in perfect agreement with the donor ability of the Lewis-bases. The amp is
a strong 0-donor and hence on coordination a t the sixth axial position it
would increase the electron densitv on the metal. This would in turn
decrease the ligating strength of the (acac) moiety. The highest value for
gi-g; agrees well with this. Similarly pyz, which is a poor donor would
coordinate only weakly and hence the ligating ability of acac would be
disturbed only shghtly which is evident from the 0.0364 value for the @%\I.
The values obtained for (Pz" )~ (36) are in the range 1.0917-1.09876 which is
expected for tetragonallq distorted octahedral VOW comple~es(~~1. Both Ari, AL
and A(av) values are also in the range of such complexes. It is interesting the
note the trend oh%rved for the A values for the complexes, which is in the order
[ v O ( a c a c ) 2 ( p y r ) ~ [ v O ( a c a c ~ p ) ~ [ v O ( a c a c ) z . This indicate
that the electron spin density gets more delocalised in the VO(aca+Jz(pyz)
adduct than in pO(acacM(amp) and VO(acac)~](pyrl.

On careful analvsis of the ESR spectra we could find a set of low intensity peaks
lying adjacent to the main peaks with similar characteristics. We attribute this to
the occurrence of' a dissociative reaction of the adduct in solution condition as
given below yielding two vanadyl complexes.
The extent of dissocmtion, however, may be quite independent on the nature of
Lewis-base and is likely to be o m marginal. We have seen that the ESR
spectrum of ~Olacach](amp) does not show such multispecies features
indicating that the complex is resistant to dissociation(").
Based on the spectral data (IR, electronic, ESR) and magnetic moment values the
structure of the various adducts could be suggested as shown in Fig.9.11.
Fig.9.11The possible geometries of various Lewis-base adducts of VO(acac)2
(a) VO(acacb(amp) (b) VO(acacb(pyr) (c) IVO(acac)zbL ( b w z , a*, biw)
9.3.6 Thermal Analysis
The thermal decomposition of the adducts were studied in N2 atmosphere at a
heating rate of 1 WC/ mm, in a Seiko-.5 Thermal Analyser. The phenomenological,

kinetic and thermodynamic parameters of the decomposition are discussed
below.
9.3.6.1 Phenomenological aspects
The thennogram obtained for this compound is given in Fig.9.12. The adduct is
stable upto 115% indicated by a weU-defmed horizontal line in the TG curve.
It shows a large mass loss of about 73.04'/0 in the temperature range
115-2950C. The peak temperature during this change was found to be
233.470C. The second stage of decomposition is found to starting at 2950C (Ti)
itself and ending at 4570C (Tf) with peak temperature 353.23OC. The mass loss
for this was found to be only 2.8% and the mass of the residue being 24.06%
afler the two decompositions. Above this temperature a mass gain of about
1.25% was observed in the thermograrn. The loss in mass data tabulated is
given in Table 9.6.
Table 9.6. Thermal decomposition features of VO(acac)z pyr
~ . ~ -
Compound I i.ci Trs(*.'C) -1 obsvd P r O b a b l r /
V02(residue)
~p
V20s(stable residue)
From the chemical analysis, spectroscopic and magnetic susceptibility data,
the adduct was found to be a monomeric 1: 1 species. The formation of the
adduct can be such that the pyridyl N gets attached to the V(W) from the
direction which is trans to vanadyl oxygen. During heating the expulsion of
the pyridine molecule coonlinated to the metal ion was expected first, before the
decomposition of VO(acac:)z. But a single stage decompsition is seen in this
case. I t indicates the loss of both m d i n e and the (acac) units simuhmeoudy
from the complex. The calculated value was also agreeing well with this. The
second stage can be expected to be due to the decomposition of some

intermediates formed. After the second stage of decomposition, the mass of
the residue is seen Lo he 24.06%. This could be due to the formation of VOz
(24.12%). But abovr 460°C the weight was gradually increasing and reached
to 26.40%. This could he due to the formation of VzOs from V&.1371 The
calculated valut. of 26.45% agrees well with this.
Fig. 9.12 The thermogram of VO(acac)z(pyr)
The thermogram obtained for VO(acac)~(amp) is given in Fig.9.13. It shows
an initial decomposition starting at 1150C 6) and ending a t 2600C vtj with a
peak temperature (T.) of 221.860C. The loss in mass corresponding to this
decomposition stage amounted to 68.98%. The second stage of decomposition is
seen in the range 26 I - 484oC with Ts of 278.32%. The loss in mass o b s e ~ e d
in this stage was a b u t 6.f3,%, anti the total mass loss was 75.81% with a
residual mass of 24. 18O/0. Above 485OC, it is seen that the mass of the
residue is increasing to an extent of 1.62% which then becomes constant.
The mass loss and other features are given in Table 9.7.

Table 9.7. Thermal decomposition features of VO(acac)2(amp)
- . -- .
Mass loss in % Probable reaction
! 1
- I 4buve5001 .- / - 25.78 1 25.33 1 VzO,(stable residue)
Eventhough amp has two donor sites (amino N and pyridyl N) the coodination of
amp can take place only through its pyridyl N atom. Thus 1: 1 (VO(acac)z:amp)
adduct can be expected to have a distorted octahedral (tetragonal) structure.
The thermal decompositiorl features of such type of adducts can be expected
to be very interesting, because of the possible release of pyridine moieties
from the adduct without any major structural change in its parent complex.
But the decomposition of this compound is seen to be not in the expected
manner. The total weight loss observed during the first and second stages of
decompositions indicate t.he loss of both (acac) moieties in addition to
4-aminopyridine. The mass of the residue observed (24.18%) is in good
0 200 400 600 860 (Temperature, OC)
Fig. 9.13 The thermogram of VO(acac)~(amp)

agreement with the theoretical valur of 23.10?/0, due to the formation of VO2.
But, at above 450°C:. the increase in mass of the residue to the extent of 1.60%
could be attributed to the formation of VZOS fmm VOz. The mass of the final
residue 25.7% is in close agreement with the calculated value of 25.3%.
The thermogram of this compound is given in Fig.9.14. This compound was
seen to be comparativeiy unstable. It shows an initial decomposition in the range
75-1460C. The loss in mass was found to be about 9.95%. The second stage
of decomposition is seen initiating at 146°C and pmgressing tiu 4130C. The
mass loss corresponding to this stage was found to be 64.27% leaving behind a
residual mass of 25.78O/0. Above 4150C, there is a gradual constant increase in
the mass. These data are tabulated iri Table 9.8.
Table 9.8 Thermal decomposition features of [VO(acac)z]~@yz)
i- - 1 Probable reaction
Compound iUc I
The mass loss obsewed in the first stage of decomposition in the temperature
range 75-1460C m y be due to the breaking of pyrazine N-M bond which helps
the expulsion of pvrazine moieties. Rut the mass )ass calculated for the change
(13.11%) is not agreeing well with the observed value of 9.95%. This may be
due to the decomposition of the mkrmediate (may be VO(acac)z) formed
before completing the first stage of decomposition. During the second stage
this intermediate was converted into V02. The residual mass of 25.78 is
agreeing well with this. But above 4500C, it was found that the mass of ihe
residue was graduallv increasing due to the oxidation of VOz to V205. The
calculated value of 27.19% is agreeing well with the observed value (26.70?!).
- I
d too
P 1 1 1 i
560 i 600 Temperature, OC
Fig.9.14 The thermogram of [VO(acac)z]~(pyz)
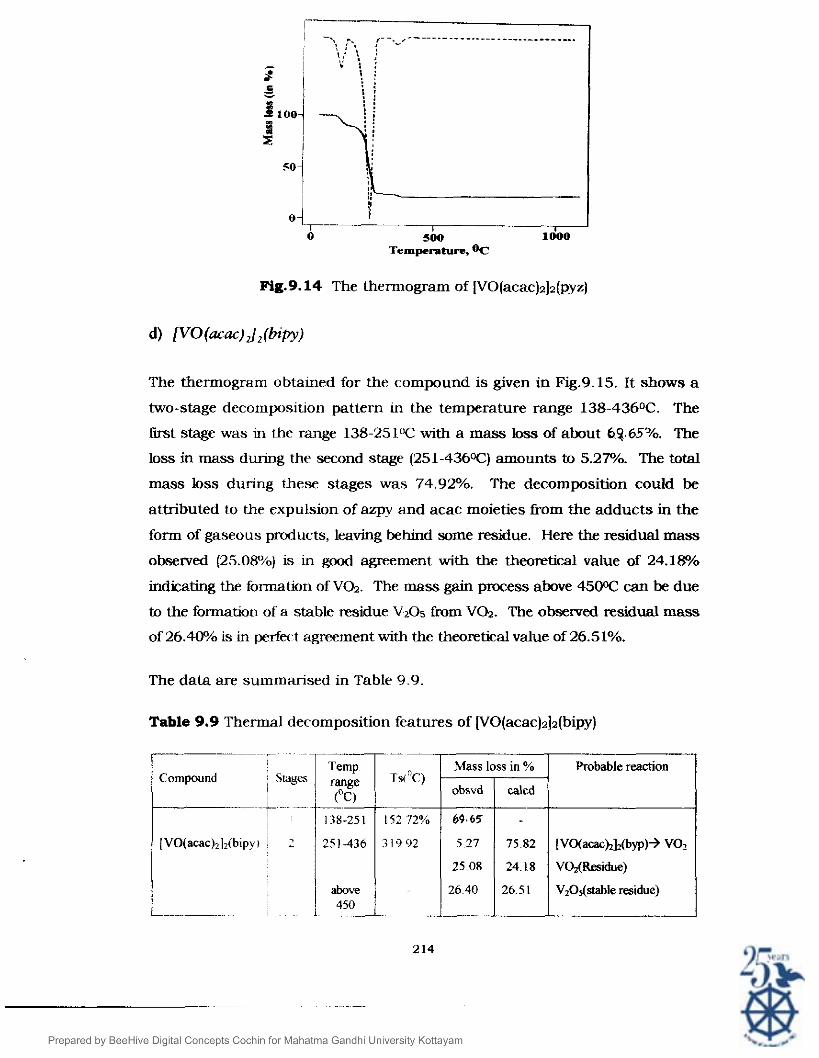
The thermogram obtained for the compound is given in Fig.9.15. It shows a
two-stage decomposition pattern in the temperature range 138-4360C. The
fust stage was in the range 138-251" with a mass loss of about 62.65"/0. The
loss in mass during the second stage (251-4360C) amounts to 5.27%. The total
mass loss during these stages was 74.92%. The decomposition could be
attributed to the expulsion of azpy and acac moieties from the adducts in the
form of gaseous products, leaving behind some residue. Here the residual mass
observed (25.08%) is in good agreement with the theomtical value of 24.1%
indicating the formation of V G . The mass gain pmcess above 4500C can be due
to the formation of a stable residue V205 from V G . The obsemed residual mass
of 26.40% is in perfect agreement with the theoretical value of 26.51%.
The data are summarised in Table 9.9.
Table 9.9 Thermal decomposition features of [VO(acac)a]z(bipy)
Probable reaction
[Vo(aca~~]~(byp)+ V02
V@(Residue)
V,O&table residue)
i Mass loss in % Compound 1 Stages
i obsvd
[VO(acac)~]2(bipyi 5.27
25.08
above 26.40
~ ~ - -
caled
75.82
24.18
26.5 1
10.0 1, I 40 200 360 520 680 760
(Temperature, O C )
Fig. 9.15 The thermogram of [VO(acac)z]~(bipy)
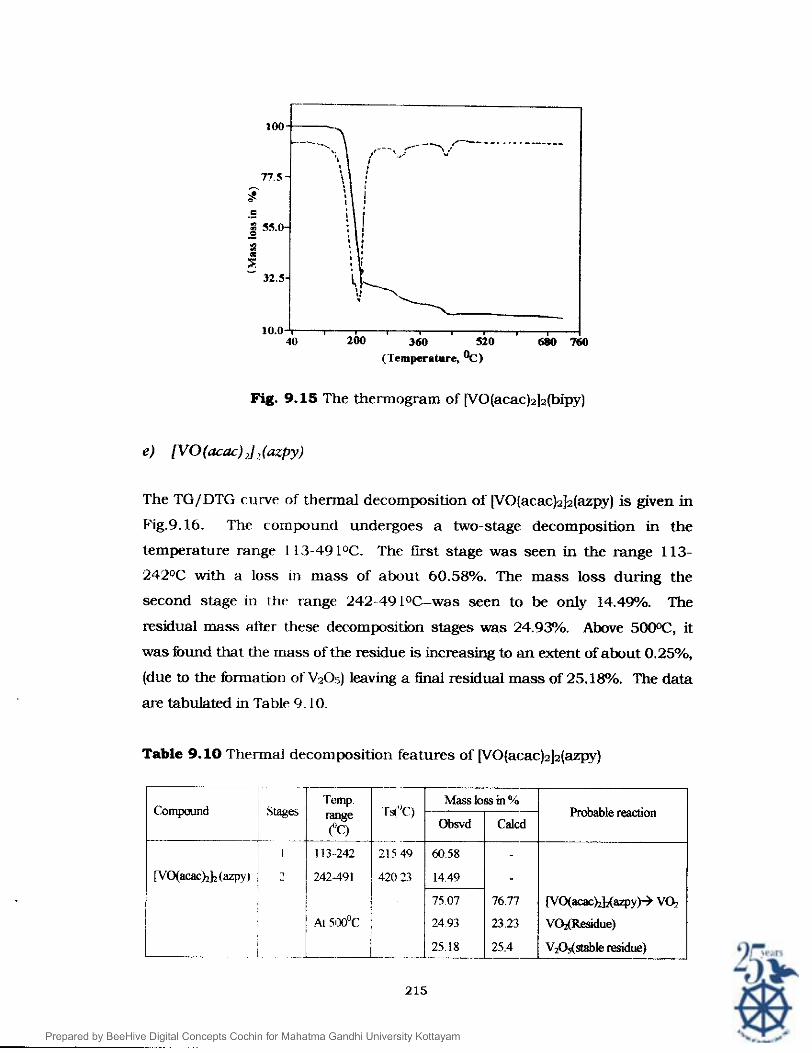
The TG/DTG curve of thermal decomposition of [VO(acac)~]z(azpy) is given in
Fig.9.16. The cornpound undergoes a two-stage decomposition in the
temperature range 1 13-49 10C. The first stage was seen in the range 113-
242oC with a loss in mass of about 60.58%. The mass loss during the
second stage in thr range 242-4910C-was seen to be only 14.49%. The
residual mass after these decomposition stages was 24.93%. Above 5000C, it
was found that the mass of the residue is increasing to an extent of about 0.2S0o,
(due to the formation of VZOS) leaving a final residual mass of 25.18%. The data
are tabulated in Table 9.10.
Table 9.10 Thermal decomposition features of [VO(acac)z]z(azpy)
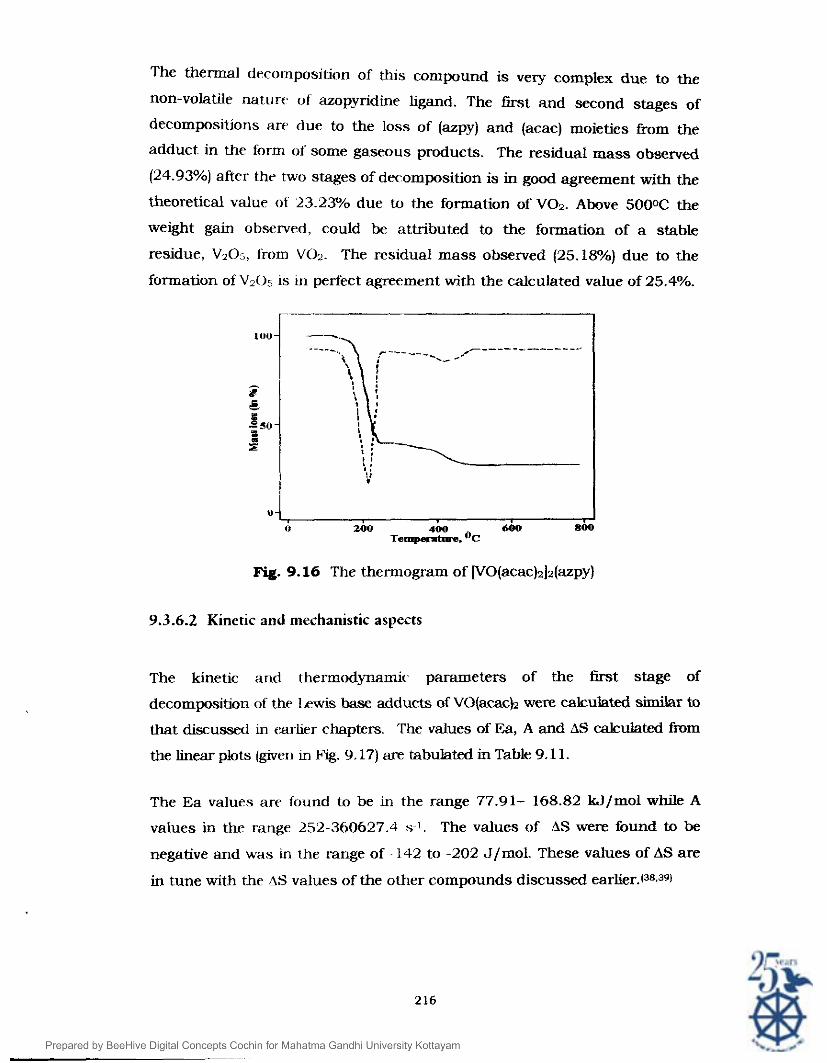
The thermal decomposition of this compound is very complex due to the
non-volatile nature of' azopyridine ligand. The first and second stages of
decompositions arr due to the loss of (azpy) and (acac) moieties from the
adduct in the form of some gaseous products. The residual mass observed
(24.93%) after the two stages of dec:omposition is in good agreement with the
theoretical value of 23.23?/0 due to the formation of V02. Above 500°C the
weight gain observed, could be attributed to the formation of a stable
residue, V205, from VOz. The residual mass observed (25.18%) due to the
formation of V205 is in perfect agreement with the calculated value of 25.4%.
Fig. 9.16 The thennogram of [VO(acac)~]z(azpy)
9.3.6.2 Kinetic and mechanistic aspects
The kinetic ant1 thermodynamic. parameters of the first stage of
decomposition of the l ~ w i s base adducts of VO(aca+ were calculated similar to
that discussed in carlier chapters. The values of Ea, A and A S calculated from
the linear plots (given in Fig. 9.17) are tabulated in Table 9.11.
The Ea values arc found to be in the range 77.91- 168.82 kJ/mol while A
values in the range 252-360627.4 s 1 . The values of A S were found to be
negative and was in the range of 142 to -202 J/mol. These values of A S are
in tune with the A S values of the other compounds discussed earlier.(38.39)
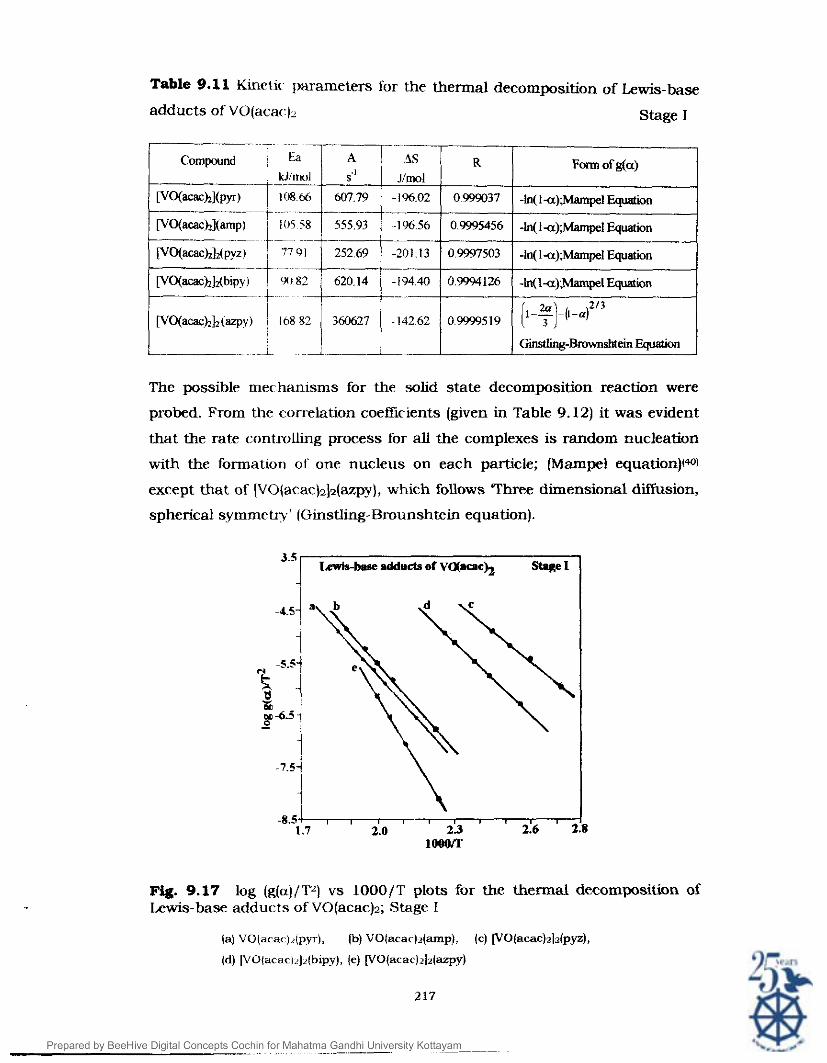
Table 9.11 Klnef parameters for the thermal decomposition of Lewis-base
adducts of VO(acac~~ Stage I
R Form of g(a) J/ml -
0.999037 -h(la)Mampel Equataon -~
0.9995456 -In(la);ml -on
0.9997503 -~la)$hmpel Eguation
0.9994126 -ln(la);Mampel EEquation - ~. ~ - ~
The possible mechanisms for the solid state decomposition reaction were
probed. From the correlation coefficients (given in Table 9.12) it was evident
that the rate controlling process for all the complexes is random nucleation
with the formation of one nucleus on each particle; (Mampel equation)(wl
except that of (VO(acac)2j~(azpy), which follows Three dimensional diffusion,
spherical symmetry' (Ginstling-Brounshtein equation).
Fig. 9.17 log (g(a)/'lVA) vs 1000/T plots for the thermal decomposition of Lewis-base adducts of VO(acac)z; Stage I

The thermal parameters of the second stage decomposition of
(VO(acac)~]~(p~j were also calculated. The Ea, A and A S values were found to
be 212.50 kJ/mol, 1.43:,:107 s~l and -112.31 J/mol-I respectively. The
possible mechan~sm of this step is also seen to be involving 'Ginstling-
Brounshtein equation' with a correlation coefficient of 0.9997323.
AU the Lews-base adducts of VO(acac)~ show a single stage decomposition in
the range 115 140°C' except (VO(acac)23(pyz), whose decomposition is seen a
starting at 7S°C itself So this compound is thermally unstable compared to
the other adducts ~ndicating the weak bonding nature of pyrazine moiety.
This compound shows two distinct decomposition stages, fxst assignable to
the loss of pyz molecule (75-146OC) and the second to the decomposition of
VO(acac)~ to V02
Table 9.12 Correlation coefficients calculated using nine forms of g(a) for the Lewis-base adducts of VO(acac)~. Stage I
References
1. R.P. Dodge, 11. H . Templeton and A.Zalldn, J. ChemPhys., 35 (1% 1) 55.
2. M.M.Jones, J A m C h a S o c , 76 (1954) 5995.
3. N.M Atherton, P.J.Gihhn and M. Candida B.Shohoji, J. Chem Soc.,
Dalton's 7?-am.. ( 1982) 2289.

4. W. Linert, I?. tlerlmnger and Peer Margl. J. Coord Chem, 28 (1993) 1.
5. R.C. Amdwal and D.S.S. Narayana, Indian J. Chem, (1982) 318.
6. R.C. Aggrawal and B. Ebsad, Indian J. Chem, @ (1975) 728.
7. M.R.Caira, J . M . Haigh and 1,.R Nassimbeni, Inorg. Nucl. Chem, 34 (1972) 317 1
8. A. Rowe arid M.Jones, Inorgam Synthesis, 5 (1957) 114.
9. N.H.Fuman, "Standard Methods of Chemical Analysis" 6th Ed., Vo1.l
(1962) 1.2 1 I .
10. R.C. Maurya, U D Mlshra and V.Plllai, Synth React. Inorg. Met-Org. Chem,
m, (1995) 1 127
11. K.Nakamoto, Y Morunoto and A.E. Martell, J.Am ChemSoc, 83 (196 1) 453.<
12. Silverstein. Bassler arid Moml. "Spedroswpic Ident@don of Organic
Compounds", S'hEXl., John-Wiley 8a Sons. Inc.
13. K. Nakamoto, "Inf7-a-red and Raman w a s w p y of Inorganic and
Coordinakon Compounds", 4'h Ed., John-Wiley 86 Sons, New York, (1986).
14. A.M.Dom, J Them A d , 39 (1993) 323.
15. A.B.P. Lever, .J Lewis and RS.Nvhom, Nature, Lond., 58 (196 1) 189.
16. AB.P Lever, J.Ixu?s and R.S.Nyhom, J .ChemSx, (1962) 1235.
17. kB.P Lever. J.IxwLs and R.S. Nyhom, J.Chem Soc, (1963) 3156.
18. A.B.P Lever, .J. lxwis ard R.S. Nvhom, J. Chem. Soc., (1963) 5042.
19. J.Metz, 0. Schneider and M.Hanack, Spectrochirn A d a , (1982) 1265.
20. W.De W Hom:ks, D.H.Templeton and A.Zallrin, Inorg.Chem., 7 (1968) 1552.
21. M.Mohan, P.Sharnia and N.K. Jha, Imrg. Chim Acta, 106 (1985) 11.
22. M. Dudeja, K. Malhotra, M.P. Gupta and K.S. Dhindsa, Synth React.
Inorg. Met-Org. <:hem., (1996) 925.
23. C. K. Jorgenser~, Acta. Chem. Scand., 11 (1957) 73.
24. C. Furlanil Rlcerca Sci., 27 (1957) 1 141.
25. Ballhausen and H.G. Harry, Inorg. Chem., 1 (1962) 11 1.
26. J.A. Dean.. 'Lange's Hand Book of Chemistry', 13th Ed. McGraw Hill
Book Cornpan,~, New York (1972) 5.18-5.60.
27. C.P. Stewart and A.L. Po&, J . C M Soc, Datton's IIZans., (1972) 1661.
28. D. Kivelson and S.K. Lee, J. C h a Phys., 41 No.7 (1964) 1896.
29. A.H. Maki and B.K. Mc. Garvev, ,I. Chem Phys., 29 (1958) 31.

30. D. Kivelson and K Neiman, J. Chem. Phys., 35 (196 1) 149.
31. S.E. Harrison atid J.M. Assour, J. Chem. Phys., 40 (1964) 365.
32. H.R. Gensmann and J.D. Swalen, J. Chem Phys., 36 (1962) 3221.
33. K. Wiithrich, Helv. Chim Acta. 48 (1965) 779.
34. J. Selbin, Coord. Chem Rev., 1 (1966) 308.
35. L.J. Boucher, E .C. Tynan and T. F. Yen, In 'Electron Spin Resonance Spectra
ofMetal Complexes: T.F. Yen Ed., Plenum Press, New York (1969).
36. K. B. Pandeya and Divya Khare, Synth React. Inorg. Met- Org.Chem.,
(1992) 52 1 .
37. Agarwal and Singh, Synth React Inorg. Met- Org. C h , (1986) 1183.
38. Y.M. Gorbechev, ,J. Them Anal., 8 (1975) 349.
39. A.A.Froast and R.G. Pearson, 'Kinetics and Mechanism', Wiley New
York , (19611