molecular dynamics study of brittle fracture in epoxy
TRANSCRIPT

lable at ScienceDirect
Composites Part B 95 (2016) 433e439
Contents lists avai
Composites Part B
journal homepage: www.elsevier .com/locate/compositesb
Molecular dynamics study of brittle fracture in epoxy-basedthermoset polymer
Bonsung Koo*, Nithya Subramanian, Aditi ChattopadhyaySchool for Engineering of Matter, Transport, and Energy, Arizona State University, Tempe, AZ, 85287, USA
a r t i c l e i n f o
Article history:Received 5 January 2016Received in revised form1 March 2016Accepted 3 April 2016Available online 13 April 2016
Keywords:A. Polymer-matrix composites (PMCs)A. Thermosetting resinB. EmbrittlementC. Computational modeling
* Corresponding author.E-mail address: [email protected] (B. Koo).
http://dx.doi.org/10.1016/j.compositesb.2016.04.0121359-8368/© 2016 Elsevier Ltd. All rights reserved.
a b s t r a c t
A novel molecular dynamics (MD) simulation methodology to capture brittle fracture in epoxy-basedthermoset polymer under mechanical loading is presented. The ductile behavior of amorphous poly-mers has been captured through traditional MD simulation methods by estimating the stress-strainresponse beyond the yield point; however, brittle fracture in highly crosslinked polymer materialssuch as epoxy thermoset has not been addressed appropriately and is the primary objective of this work.In this study, a numerically cured epoxy system comprising molecules of epoxy resin and hardener isgenerated. During the virtual deformation test, it is observed that the inherent molecular vibration dueto temperature re-equilibrates the elongated covalent bonds and this molecular vibration impedesfurther stretching the covalent bond leading to scission. In order to overcome the influence of thermalvibration, an approach that employs deformation tests at absolute zero temperature condition e aconcept borrowed from the quasi-continuum method, is developed. Bond dissociation energy ismeasured to quantify the extent of failure in the system by calculating the bond potentials during thedeformation tests. Applying zero temperature condition to the deformation test, however, requires alarge amount of computational time due to intermediate energy minimization processes. To improve thecomputational efficiency, an ultra-high strain rate (UHSRz 1013 s�1) approach is developed by which thethermal vibration is decoupled from the deformation test using a strain rate higher than molecular vi-bration frequency. Note that the deformation test performed by traditional MD simulation methods usedstrain rates ranging 108e1010 s�1. Simulation results show that the UHSR approach successfully capturesbrittle fracture in epoxy polymer due to covalent bond dissociation with high computational efficiency.
© 2016 Elsevier Ltd. All rights reserved.
1. Introduction
Although polymer matrix composites (PMCs) have drawn sig-nificant attention from the aerospace industry and their recentapplication in aircraft primary structures have shown significantweight savings and fuel efficiency, the realization of the full po-tential of these materials demands an improved understanding ofdamage initiation and progression from the material level. Theintermolecular interactions in the polymer matrix, inelasticbehavior of matrix/fiber interphase, and the effects of variability ofconstituent types (epoxy resin and hardener) on material proper-ties must be studied to develop appropriate physics-based damagemodels. Several experimental studies have been conducted toaddress the nonlinear plastic response of composites; but the
phenomena occurring at the nanoscale require further in-vestigations. Intermolecular interaction in the polymer matrixunder complex loading, in particular, needs atomistic scale inves-tigation; however, the availability of such information is limited bythe resolution/sensitivity of experimental tools.
The mechanical deformation/fracture response of epoxy matrixhas been studied using computational approach such as moleculardynamics (MD). A significant amount of research has been reportedon characterizing the elastic response of amorphous epoxy andepoxy-nanocomposites using MD simulations [1e9]. Li and Stra-chan [1] estimated thermal and mechanical properties, such as theglass transition temperature, and tensile/shear moduli of the epoxyresin DGEBF (diglycidyl ether of bisphenol f) with hardeners DETDA(diethyl toluene diamine). They also used theMDmethod to predictmechanical response under cyclic loading using DGEBF and 33DDS(3,3 diamino diphenyl sulfone) [2]. Valavala et al. [3] and Bandyo-padhyay et al. [4] studied the effect of crosslinking degree on me-chanical properties of the same epoxy matrix. However, the plastic

Fig. 1. Schematic of the MD simulation volume containing DGEBF (epoxy resin) andDETA (hardener).
B. Koo et al. / Composites Part B 95 (2016) 433e439434
deformation leading to damage initiation and propagation has notbeen captured effectively because of the implementation of clas-sical force fields. Since classical force fields were developed toanalyze system dynamics around equilibrium states, it is difficult tostudy material degradation due to covalent bond dissociationthrough classical MD simulations. Pioneering studies were under-taken by employing a special force field, finitely extensiblenonlinear elastic (FENE) [10e13], capable of capturing covalentbond dissociation by modifying the Lennard-Jones potential. Inaddition, Moller et al. [14] recently developed a new approach tocapture covalent bond scission by monitoring the covalent bondlength during mechanical deformation by employing classical forcefields. If the bond length becomes longer than a threshold, the bondis eliminated and the bond information is deleted from the systemdatabase during the virtual deformation test. While the two ap-proaches (FENE and Moller's approach) capture fracture behaviorsuccessfully, a large deformation (>100% strain) is required toobserve this failure. Such a deformation is not realistic since ma-terial failure of epoxy polymeric system appears at a small strain(<10% strain) [15e17].
In this paper, a novel atomistic simulation methodology isdeveloped to accurately simulate the brittle behavior of epoxy-based thermoset polymer. An appropriate bond order based forcefield is necessary to capture bond dissociation during the defor-mation test. Odegard and his co-workers [18] used a bond orderbased force field to capture material degradation of an epoxypolymer system during deformation test; however, it was incon-clusive whether the material degradation is due to conformationalchange (molecular sliding) or covalent bond dissociation. In addi-tion to the selection of an appropriate force field, a method todecouple molecular thermal vibration from mechanical deforma-tion is explored in this paper. Two deformation methods are hy-pothesized to suppress the effect of thermal vibration: zero-temperature deformation and ultra-high strain rate(UHSR z 1013 s�1) deformation. The energy minimization processin the zero-temperature method maintains the system at groundstate-free of thermal effects, whereas the UHSR method allowsmolecular vibration, but deforms the simulation volume with ahigher strain rate than the bond vibration frequency of the covalentbonds. These twomethods (zero-temperature and UHSR) implicitlyproduce time-independent behavior resembling quasi-static sys-tems because the thermal activation of molecular motion isdecoupled from the virtual mechanical deformation [19]. The vir-tual mechanical deformation results show that the two deforma-tion methods with the appropriate bond order based force fieldcaptures brittle fracture in epoxy-based polymer system at a smallstrain (~10%), which correlate well with experimental results[15e17].
2. Molecular model and simulation method
Molecular structures of Di-Glycidyl Ether of Bisphenol F (DGEBF,epoxy resin) and Di-Ethylene Tri-Amine (DETA, hardener) are usedto construct a simulation volume based on the weight fraction(MDGEBF:MDETA ¼ 100:27). Table 1 contains the number of mole-cules in the simulation volume (66,560 atoms) and Fig. 1 shows aschematic of the 3D molecular structure of the volume. A classical
Table 1Components in the simulation volume (MDGEBF:MDETA ¼ 100:27).
Weight Formula Number of Molecules
DGEBF 313 g/mol C19H20O4 1120DETA 103 g/mol C4H13N3 920
force field - MMFF (Merck Molecular Force Field), is implementedto equilibrate the epoxy system and perform the curing simulation(crosslink formation) [20]. MD simulations are conducted by Large-scale Atomic Molecular Massively Parallel Simulator (LAMMPS)developed by Sandia National Laboratory [21]. A stochastic cross-link formation approach, based on a cutoff distance for covalentbond generation between active sites is adopted to simulate theepoxy curing process [22]. It is noteworthy that MD simulationswith a classical force field are very effective to study processes in asystem around its equilibrium state (e.g., curing); however, they arenot appropriate for investigating covalent bond dissociationoccurring away from the equilibrium state (e.g., deformation tests).The classical force field, MMFF shows an asymptotic increase inbond energy with elongation of bonds whereas bond order basedforce field is capable of capturing bond dissociation beyond a crit-ical bond length [23]. ReaxFF, a popular bond order based forcefield, was developed for hydrocarbon compound by van Duin andco-workers [23e27] and many parameter sets have been reportedto describe the nonlinear response of diverse materials includingzinc oxide [28], Vanadium oxide [29], fuel cell [30], Li/Si alloys [31],etc. The most suitable ReaxFF parameter set for the epoxy-basedpolymer system is determined by a bond dissociation energy(BDE) calculation of a specific CeC bond highlighted by the red boxin Fig. 2. The BDE calculation is performed by the constrained ge-ometry external force (COGEF) method developed to estimate thestrength of covalent bonds by Beyer et al. [32]. The BDEs calculatedby different ReaxFF parameter sets (Table 2) are compared with theCeC bond energy obtained by pyrolysis experiments [33]; the bondenergy was measured to be 389 kJ/mol at 298 K. Based on thevalues presented in Table 2, the force field parameters reported bySingh and co-workers [27] show the best correlation with the py-rolysis experimental result; therefore, this parameter set is used forthe virtual deformation tests performed in this study.
3. Effect of thermal vibration on potential energy underdeformation test
Temperature is directly related to the kinetic energy of themolecular system. However, temperature also causes potentialenergy variation in MD simulations. Temperature changes affectnot only the frequency but also the amplitude of molecular vibra-tion leading to changes in the potential energy. This phenomenon is
Fig. 2. Molecular structure of the resin (DGEBF) and CeC bond used for BDEcalculation.
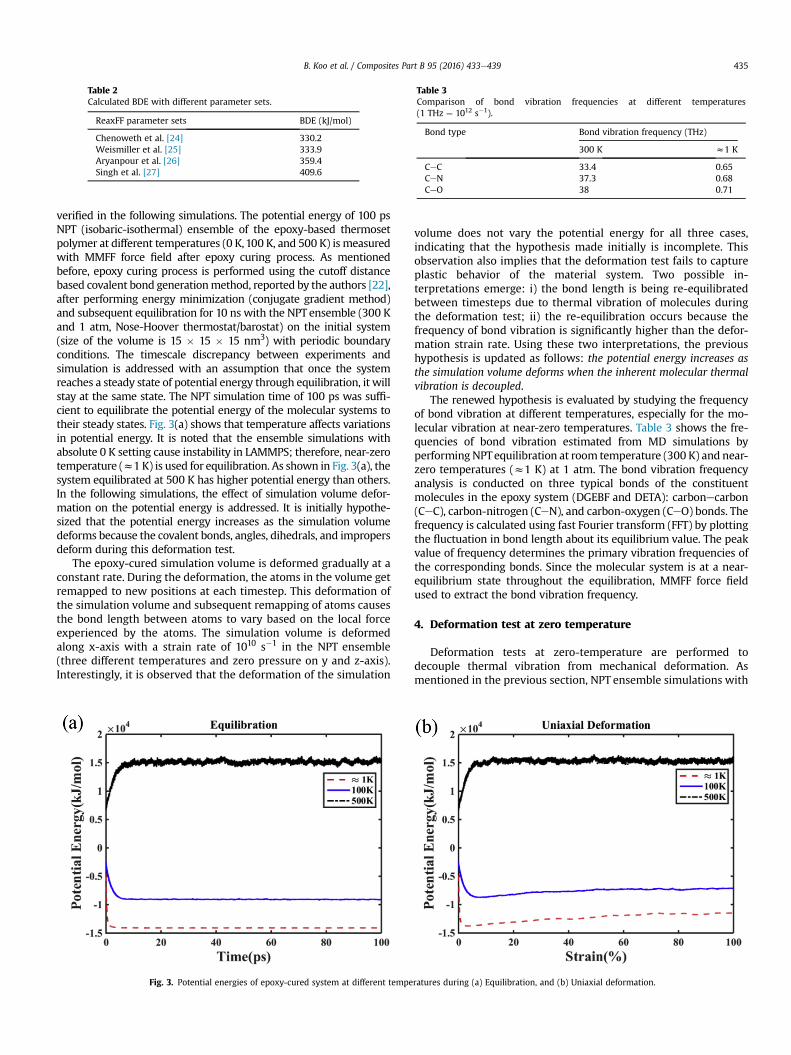
Table 2Calculated BDE with different parameter sets.
ReaxFF parameter sets BDE (kJ/mol)
Chenoweth et al. [24] 330.2Weismiller et al. [25] 333.9Aryanpour et al. [26] 359.4Singh et al. [27] 409.6
Table 3Comparison of bond vibration frequencies at different temperatures(1 THz ¼ 1012 s�1).
Bond type Bond vibration frequency (THz)
300 K z1 K
CeC 33.4 0.65CeN 37.3 0.68CeO 38 0.71
B. Koo et al. / Composites Part B 95 (2016) 433e439 435
verified in the following simulations. The potential energy of 100 psNPT (isobaric-isothermal) ensemble of the epoxy-based thermosetpolymer at different temperatures (0 K,100 K, and 500 K) is measuredwith MMFF force field after epoxy curing process. As mentionedbefore, epoxy curing process is performed using the cutoff distancebased covalent bond generationmethod, reported by the authors [22],after performing energy minimization (conjugate gradient method)and subsequent equilibration for 10 ns with the NPT ensemble (300 Kand 1 atm, Nose-Hoover thermostat/barostat) on the initial system(size of the volume is 15 � 15 � 15 nm3) with periodic boundaryconditions. The timescale discrepancy between experiments andsimulation is addressed with an assumption that once the systemreaches a steady state of potential energy through equilibration, it willstay at the same state. The NPT simulation time of 100 ps was suffi-cient to equilibrate the potential energy of the molecular systems totheir steady states. Fig. 3(a) shows that temperature affects variationsin potential energy. It is noted that the ensemble simulations withabsolute 0 K setting cause instability in LAMMPS; therefore, near-zerotemperature (z1 K) is used for equilibration. As shown in Fig. 3(a), thesystem equilibrated at 500 K has higher potential energy than others.In the following simulations, the effect of simulation volume defor-mation on the potential energy is addressed. It is initially hypothe-sized that the potential energy increases as the simulation volumedeforms because the covalent bonds, angles, dihedrals, and impropersdeform during this deformation test.
The epoxy-cured simulation volume is deformed gradually at aconstant rate. During the deformation, the atoms in the volume getremapped to new positions at each timestep. This deformation ofthe simulation volume and subsequent remapping of atoms causesthe bond length between atoms to vary based on the local forceexperienced by the atoms. The simulation volume is deformedalong x-axis with a strain rate of 1010 s�1 in the NPT ensemble(three different temperatures and zero pressure on y and z-axis).Interestingly, it is observed that the deformation of the simulation
Fig. 3. Potential energies of epoxy-cured system at different tempe
volume does not vary the potential energy for all three cases,indicating that the hypothesis made initially is incomplete. Thisobservation also implies that the deformation test fails to captureplastic behavior of the material system. Two possible in-terpretations emerge: i) the bond length is being re-equilibratedbetween timesteps due to thermal vibration of molecules duringthe deformation test; ii) the re-equilibration occurs because thefrequency of bond vibration is significantly higher than the defor-mation strain rate. Using these two interpretations, the previoushypothesis is updated as follows: the potential energy increases asthe simulation volume deforms when the inherent molecular thermalvibration is decoupled.
The renewed hypothesis is evaluated by studying the frequencyof bond vibration at different temperatures, especially for the mo-lecular vibration at near-zero temperatures. Table 3 shows the fre-quencies of bond vibration estimated from MD simulations byperforming NPTequilibration at room temperature (300 K) and near-zero temperatures (z1 K) at 1 atm. The bond vibration frequencyanalysis is conducted on three typical bonds of the constituentmolecules in the epoxy system (DGEBF and DETA): carbonecarbon(CeC), carbon-nitrogen (CeN), and carbon-oxygen (CeO) bonds. Thefrequency is calculated using fast Fourier transform (FFT) by plottingthe fluctuation in bond length about its equilibrium value. The peakvalue of frequency determines the primary vibration frequencies ofthe corresponding bonds. Since the molecular system is at a near-equilibrium state throughout the equilibration, MMFF force fieldused to extract the bond vibration frequency.
4. Deformation test at zero temperature
Deformation tests at zero-temperature are performed todecouple thermal vibration from mechanical deformation. Asmentioned in the previous section, NPT ensemble simulations with
ratures during (a) Equilibration, and (b) Uniaxial deformation.
B. Koo et al. / Composites Part B 95 (2016) 433e439436
absolute 0 K setting makes the system unstable in LAMMPS.Introducing an energy minimization step into the virtual defor-mation test maintains the system temperature at zero. This idea isborrowed from the quasi-continuummethod, which was originallydeveloped as a multiscale modeling method to bridge molecularscale to continuum scale by decoupling temperature effect [34]. Thesimulation volume is deformed by gradually varying strain usingReaxFF and the atom coordinates in the volume are remapped froma previously stored box shape to a new deformed one based on anaffine transformation followed by energy minimization [35,36].Through this simulation method, the absolute zero temperaturecondition successfully allows to decouple thermal vibration fromMD simulations and plastic deformation induced by covalent bondbreakage is observed. However, the energy minimization con-ducted at every small strain deformation makes the methodcomputationally expensive. This method is applied to the epoxy-cured system (66,560 atoms) for 1000 tensile strain steps result-ing in 50% final strain. This simulation is performed on a computingnode with Intel(R) Xeon(R) CPU E5-2640 v2 @ 2.00 GHz processorsusing eight CPU cores and the total CPU hours for a single simula-tion were well over 1000 h. The prohibitively large computationaltime associated with energy minimization warrants the develop-ment of a numerical framework that can simulate fracture inepoxy-based thermoset polymer reliably and efficiently.
5. Deformation test with ultra-high strain rate
The feasibility of performing deformation tests at UHSRs(>>1010 s�1) is explored as an alternative to the computationally
Fig. 4. Effect of temperature on potential energy at different strain rates: (a) 1 � 1010 s�1,
A. Compatibility between zero-temperature and UHSR methods
intensive zero-temperature method. The average bond vibrationfrequency (0.68 THz ¼ 0.68 � 1012 s�1) is calculated to be higherthan deformation strain rates, 108e1010 s�1, which are normallyused in literature pertaining to MD simulations [1e4]. Hence, it isexpected that these UHSRs, higher than the average vibration fre-quency of the bonds present in the molecular system of interest,deform the simulation volume faster than bond vibrations. There-fore, elongated bond lengths overcome thermally-induced re-equilibration during the deformation test. Here, the term UHSR isused to differentiate from the ‘high strain rate’ used in the currentMD research field [1e4].
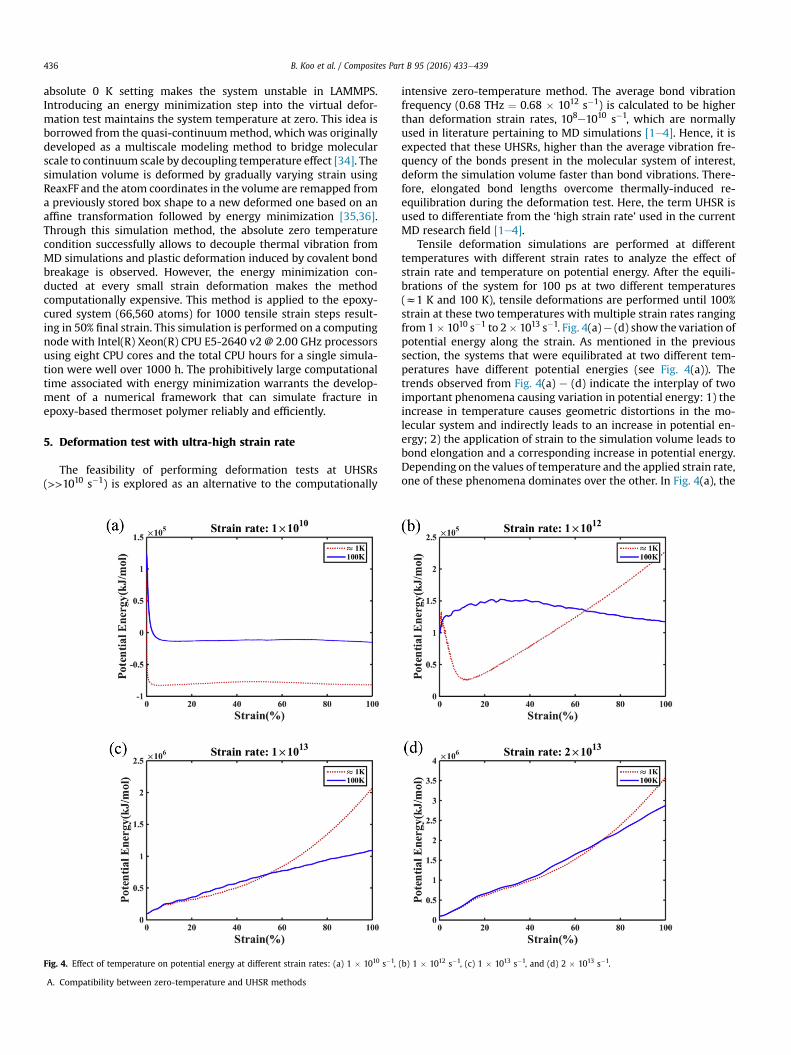
Tensile deformation simulations are performed at differenttemperatures with different strain rates to analyze the effect ofstrain rate and temperature on potential energy. After the equili-brations of the system for 100 ps at two different temperatures(z1 K and 100 K), tensile deformations are performed until 100%strain at these two temperatures with multiple strain rates rangingfrom1� 1010 s�1 to 2� 1013 s�1. Fig. 4(a)e (d) show the variation ofpotential energy along the strain. As mentioned in the previoussection, the systems that were equilibrated at two different tem-peratures have different potential energies (see Fig. 4(a)). Thetrends observed from Fig. 4(a) e (d) indicate the interplay of twoimportant phenomena causing variation in potential energy: 1) theincrease in temperature causes geometric distortions in the mo-lecular system and indirectly leads to an increase in potential en-ergy; 2) the application of strain to the simulation volume leads tobond elongation and a corresponding increase in potential energy.Depending on the values of temperature and the applied strain rate,one of these phenomena dominates over the other. In Fig. 4(a), the
(b) 1 � 1012 s�1, (c) 1 � 1013 s�1, and (d) 2 � 1013 s�1.
B. Koo et al. / Composites Part B 95 (2016) 433e439 437
applied strain rate is less than the value of bond vibration fre-quencies at z1 K and 100 K; therefore, case (1) dominates in thisrange of strain rate. In Fig. 4(b), the applied strain rate is higher thanbond vibration frequencies at z1 K but lower than the bond vi-bration frequencies at 100 K. This indicates that case (2) is domi-nant in the z1 K simulations whereas case (1) plays a moreimportant role in the 100 K simulation. At higher strain rates(z1013 s�1), as shown in Fig. 4(c) and (d), the influence of tem-perature is reduced significantly because the applied strain rate ishigher than the bond vibrational frequencies in both the z1 K andthe 100 K simulations. If the strain rate of the deformation is fasterthan the bond stretching vibration, the effect of re-equilibration ofthe stretched bond due to thermal vibration is reduced. Therefore,the applied strain is transferred completely to the atomic bonds andcase (2) dominates over case (1). Fig. 4(d) shows that the potentialenergy values almost converge in spite of the difference in tem-perature, indicating that the potential energy is invariant to tem-perature at UHSRs. Thus, the effects of temperature on thedeformation test are reduced at UHSRs, and the variation in po-tential energy can be fully attributed to the increase in strain in thesystem causing bond elongation and bond dissociation.
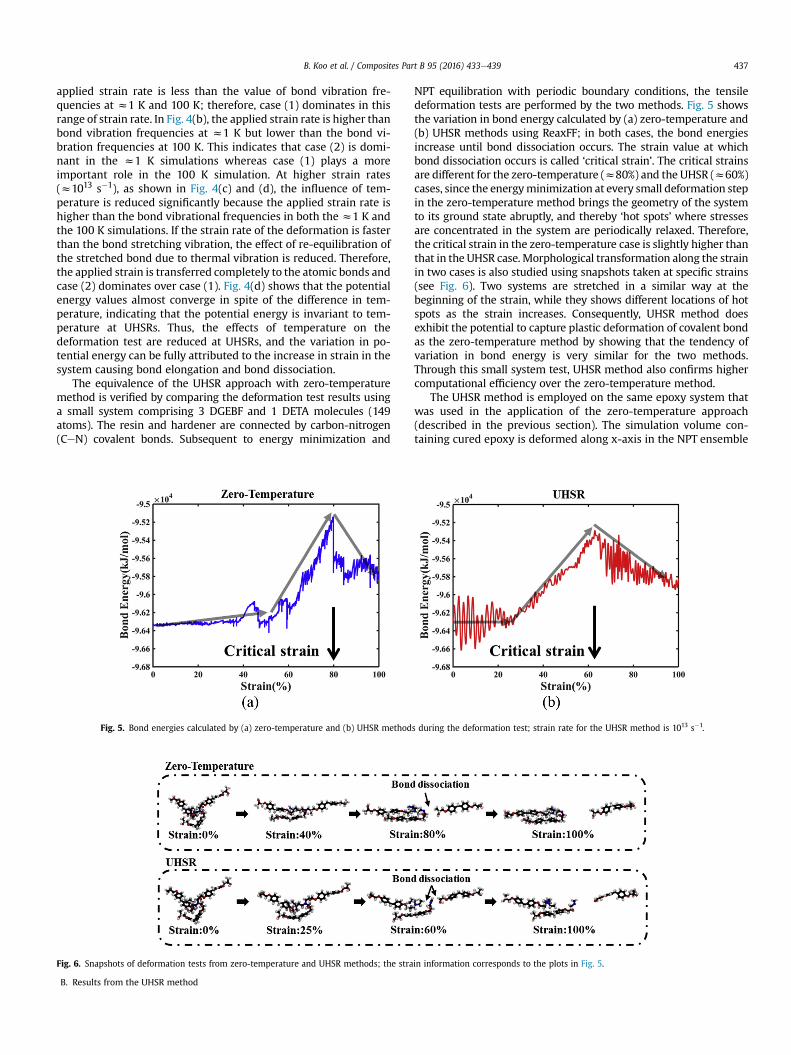
The equivalence of the UHSR approach with zero-temperaturemethod is verified by comparing the deformation test results usinga small system comprising 3 DGEBF and 1 DETA molecules (149atoms). The resin and hardener are connected by carbon-nitrogen(CeN) covalent bonds. Subsequent to energy minimization and
Fig. 5. Bond energies calculated by (a) zero-temperature and (b) UHSR method
Fig. 6. Snapshots of deformation tests from zero-temperature and UHSR methods; the stra
B. Results from the UHSR method
NPT equilibration with periodic boundary conditions, the tensiledeformation tests are performed by the two methods. Fig. 5 showsthe variation in bond energy calculated by (a) zero-temperature and(b) UHSR methods using ReaxFF; in both cases, the bond energiesincrease until bond dissociation occurs. The strain value at whichbond dissociation occurs is called ‘critical strain’. The critical strainsare different for the zero-temperature (z80%) and the UHSR (z60%)cases, since the energyminimization at every small deformation stepin the zero-temperature method brings the geometry of the systemto its ground state abruptly, and thereby ‘hot spots’ where stressesare concentrated in the system are periodically relaxed. Therefore,the critical strain in the zero-temperature case is slightly higher thanthat in the UHSR case. Morphological transformation along the strainin two cases is also studied using snapshots taken at specific strains(see Fig. 6). Two systems are stretched in a similar way at thebeginning of the strain, while they shows different locations of hotspots as the strain increases. Consequently, UHSR method doesexhibit the potential to capture plastic deformation of covalent bondas the zero-temperature method by showing that the tendency ofvariation in bond energy is very similar for the two methods.Through this small system test, UHSR method also confirms highercomputational efficiency over the zero-temperature method.
The UHSR method is employed on the same epoxy system thatwas used in the application of the zero-temperature approach(described in the previous section). The simulation volume con-taining cured epoxy is deformed along x-axis in the NPT ensemble
s during the deformation test; strain rate for the UHSR method is 1013 s�1.
in information corresponds to the plots in Fig. 5.
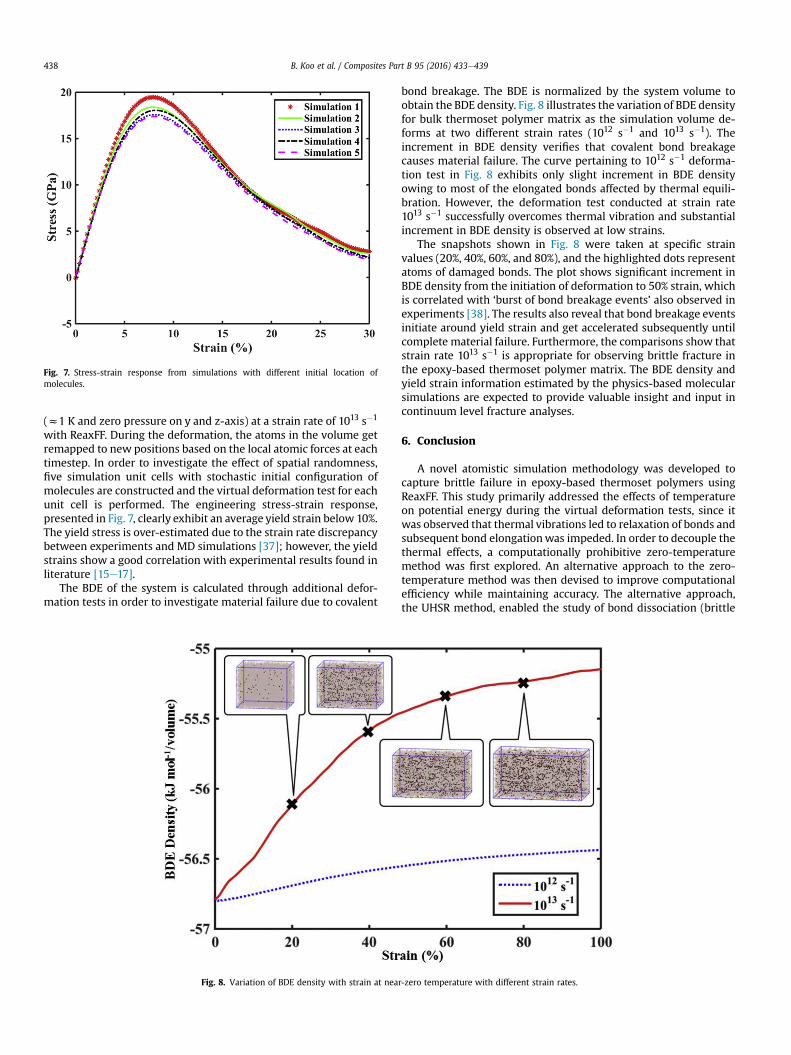
Fig. 7. Stress-strain response from simulations with different initial location ofmolecules.
B. Koo et al. / Composites Part B 95 (2016) 433e439438
(z1 K and zero pressure on y and z-axis) at a strain rate of 1013 s�1
with ReaxFF. During the deformation, the atoms in the volume getremapped to new positions based on the local atomic forces at eachtimestep. In order to investigate the effect of spatial randomness,five simulation unit cells with stochastic initial configuration ofmolecules are constructed and the virtual deformation test for eachunit cell is performed. The engineering stress-strain response,presented in Fig. 7, clearly exhibit an average yield strain below 10%.The yield stress is over-estimated due to the strain rate discrepancybetween experiments and MD simulations [37]; however, the yieldstrains show a good correlation with experimental results found inliterature [15e17].
The BDE of the system is calculated through additional defor-mation tests in order to investigate material failure due to covalent
Fig. 8. Variation of BDE density with strain at nea
bond breakage. The BDE is normalized by the system volume toobtain the BDE density. Fig. 8 illustrates the variation of BDE densityfor bulk thermoset polymer matrix as the simulation volume de-forms at two different strain rates (1012 s�1 and 1013 s�1). Theincrement in BDE density verifies that covalent bond breakagecauses material failure. The curve pertaining to 1012 s�1 deforma-tion test in Fig. 8 exhibits only slight increment in BDE densityowing to most of the elongated bonds affected by thermal equili-bration. However, the deformation test conducted at strain rate1013 s�1 successfully overcomes thermal vibration and substantialincrement in BDE density is observed at low strains.
The snapshots shown in Fig. 8 were taken at specific strainvalues (20%, 40%, 60%, and 80%), and the highlighted dots representatoms of damaged bonds. The plot shows significant increment inBDE density from the initiation of deformation to 50% strain, whichis correlated with ‘burst of bond breakage events’ also observed inexperiments [38]. The results also reveal that bond breakage eventsinitiate around yield strain and get accelerated subsequently untilcomplete material failure. Furthermore, the comparisons show thatstrain rate 1013 s�1 is appropriate for observing brittle fracture inthe epoxy-based thermoset polymer matrix. The BDE density andyield strain information estimated by the physics-based molecularsimulations are expected to provide valuable insight and input incontinuum level fracture analyses.
6. Conclusion
A novel atomistic simulation methodology was developed tocapture brittle failure in epoxy-based thermoset polymers usingReaxFF. This study primarily addressed the effects of temperatureon potential energy during the virtual deformation tests, since itwas observed that thermal vibrations led to relaxation of bonds andsubsequent bond elongationwas impeded. In order to decouple thethermal effects, a computationally prohibitive zero-temperaturemethod was first explored. An alternative approach to the zero-temperature method was then devised to improve computationalefficiency while maintaining accuracy. The alternative approach,the UHSR method, enabled the study of bond dissociation (brittle
r-zero temperature with different strain rates.

B. Koo et al. / Composites Part B 95 (2016) 433e439 439
behavior) of bulk polymer system (large system size) using ultra-high strain rates (z1013 s�1) which are higher than the averagevibration frequency of the bonds present in themolecular system ofinterest. The UHSR method produced very similar results as thezero-temperature method, but with significant computationalbenefits using a combination of low temperature (z1 K) and ultra-high strain rate. The UHSR method also provides a platform toquantify both the BDE and BDE density in a bulk polymer system;these parameters are capable of describing the extent of damage incontinuum level fracture analysis of thermoset polymer systems.
Acknowledgments
This research is supported in parts by the U.S. Army ResearchOffice, technical monitor: Dr. David Stepp (Agreement Number:W911NF-15-1-0072) and the Office of Naval Research, technicalmonitor: Mr. William Nickerson (Agreement Number: N00014-14-1-0068).
References
[1] Li C, Strachan A. Molecular dynamics predictions of thermal and mechanicalproperties of thermoset polymer EPON862/DETDA. Polymer 2011;52(13):2920e8.
[2] Li C, Jaramillo E, Strachan A. Molecular dynamics simulations on cyclicdeformation of an epoxy thermoset. Polymer 2013;54(2):881e90.
[3] Valavala P, Odegard G, Aifantis E. Influence of representative volume elementsize on predicted elastic properties of polymer materials. Model Simul MaterSci Eng 2009;17(4):045004.
[4] Bandyopadhyay A, Valavala PK, Clancy TC. Molecular modeling of crosslinkedepoxy polymers: the effect of crosslink density on thermomechanical prop-erties. Polymer 2011;52(11):2445e52.
[5] Shiu S, Tsai J. Characterizing thermal and mechanical properties of graphene/epoxy nanocomposites. Compos Part B Eng 2014;56:691e7.
[6] Shin H, Chang S, Yang S. Statistical multiscale homogenization approach foranalyzing polymer nanocomposites that include model inherent uncertaintiesof molecular dynamics simulations. Compos Part B Eng 2016;87:120e31.
[7] Rahman R, Haque A. Molecular modeling of crosslinked grapheneeepoxynanocomposites for characterization of elastic constants and interfacialproperties. Compos Part B Eng 2013;54:353e64.
[8] Ionita M. Multiscale molecular modeling of SWCNTs/epoxy resin compositesmechanical behaviour. Compos Part B Eng 2012;43(8):3491e6.
[9] Zhang J, Koo B, Subramanian N. An optimized cross-linked network model tosimulate the linear elastic material response of a smart polymer. J Intell MaterSyst Struct 2015. http://dx.doi.org/10.1177/1045389X15595292.
[10] Panico M, Narayanan S, Brinson L. Simulations of tensile failure in glassypolymers: effect of cross-link density. Model Simul Mater Sci Eng 2010;18(5):055005.
[11] Sliozberg YR, Hoy RS, Mrozek RA. Role of entanglements and bond scission inhigh strain-rate deformation of polymer gels. Polymer 2014;55(10):2543e51.
[12] Hoy RS, Robbins MO. Strain hardening in polymer glasses: limitations ofnetwork models. Phys Rev Lett 2007;99(11):117801.
[13] Rottler J, Robbins MO. Growth, microstructure, and failure of crazes in glassypolymers. Phys Rev E 2003;68(1):011801.
[14] Moller J, Barr S, Schultz E. Simulation of fracture nucleation in cross-linkedpolymer networks. Jom 2013;65(2):147e67.
[15] Kinloch AJ. Fracture behaviour of polymers. Springer Science & BusinessMedia; 2013.
[16] Cook WD, Mayr AE, Edward GH. Yielding behaviour in model epoxy ther-mosetsdII. Temperature dependence. Polymer 1998;39(16):3725e33.
[17] Dlubek G, Pointeck J, Shaikh MQ. Free volume of an Oligomeric epoxy resinand its relation to structural relaxation: Evidence from Positron Lifetime andpressure-volume-temperature experiments. Phys Rev E 2007;75(2):021802.
[18] Odegard GM, Jensen BD, Gowtham S. Predicting mechanical response ofcrosslinked epoxy using ReaxFF. Chem Phys Lett 2014;591:175e8.
[19] Mott P, Argon A, Suter U. Atomistic modelling of plastic deformation of glassypolymers. Philos Mag A 1993;67(4):931e78.
[20] Halgren TA. Merck molecular force field. I. Basis, form, scope, parameteriza-tion, and performance of MMFF94. J Comput Chem 1996;17(5e6):490e519.
[21] Plimpton S. Fast parallel algorithms for short-range molecular dynamics.J Comput Phys 1995;117(1):1e19.
[22] Koo B, Liu Y, Zou J. Study of glass transition temperature (Tg) of novel stress-sensitive composites using molecular dynamic simulation. Model Simul MaterSci Eng 2014;22(6):065018.
[23] Van Duin AC, Dasgupta S, Lorant F. ReaxFF: a Reactive Force Field for hydro-carbons,”. J Phys Chem A 2001;105(41):9396e409.
[24] Chenoweth K, Cheung S, Van Duin AC. Simulations on the thermal decom-position of a poly (dimethylsiloxane) polymer using the ReaxFF Reactive forcefield. J Am Chem Soc 2005;127(19):7192e202.
[25] Weismiller MR, Duin A C v, Lee J. ReaxFF Reactive Force Field developmentand applications for molecular dynamics simulations of ammonia boranedehydrogenation and combustion. J Phys Chem A 2010;114(17):5485e92.
[26] Aryanpour M, van Duin AC, Kubicki JD. Development of a Reactive Force Fieldfor ironeoxyhydroxide systems. J Phys Chem A 2010;114(21):6298e307.
[27] Singh SK, Srinivasan SG, Neek-Amal M. Thermal properties of fluorinatedgraphene. Prb 2013;87(10):104114.
[28] Raymand D, van Duin AC, Baudin M. A Reactive Force Field (ReaxFF) for zincoxide,”. Surf Sci 2008;602(5):1020e31.
[29] Chenoweth K, Duin A C v, Persson P. Development and application of a ReaxFFReactive Force Field for oxidative dehydrogenation on vanadium oxide cata-lysts. J Phys Chem C 2008;112(37):14645e54.
[30] van Duin AC, Merinov BV, Jang SS. ReaxFF Reactive Force Field for solid oxidefuel cell systems with application to oxygen ion transport in yttria-stabilizedzirconia. J Phys Chem A 2008;112(14):3133e40.
[31] Fan F, Huang S, Yang H. Mechanical properties of amorphous LixSi alloys: aReactive Force Field Study. Model Simul Mater Sci Eng 2013;21(7):074002.
[32] Beyer MK. The mechanical strength of a covalent bond calculated by densityfunctional theory. J Chem Phys 2000;112(17):7307e12.
[33] Dean JA. Lange's handbook of chemistry. New York, NY: McGraw Hill BookCo.; 1985.
[34] Tadmor EB, Ortiz M, Phillips R. Quasicontinuum analysis of defects in solids.Philos Mag A 1996;73(6):1529e63.
[35] Sommer J, Lay S. Topological structure and nonaffine swelling of bimodalpolymer networks. Macromolecules 2002;35(26):9832e43.
[36] Basu A, Wen Q, Mao X. Nonaffine displacements in flexible polymer networks.Macromolecules 2011;44(6):1671e9.
[37] Fratzl P. Collagen: structure and mechanics, an introduction. Springer; 2008.[38] Zhurkov S, Zakrevskyi V, Korsukov V. Mechanism of submicrocrack generation
in stressed polymers. J Polym Sci Part A-2 Polym Phys 1972;10(8):1509e20.