molecular dissection of interactions between components of the
TRANSCRIPT

Molecular Dissection Of Interactions Between Components Of The Alternative Pathway
Of Complement And Decay Accelerating Factor (CD55)
Claire L Harris1, Rachel JM Abbott2, Richard A Smith3, B Paul Morgan1, Susan M
Lea2.
1Complement Biology Group, Department of Medical Biochemistry and Immunology, School
of Medicine, Cardiff University, Heath Park, Cardiff, UK; 2Department of Biochemistry,
Laboratory of Molecular Biophysics, Oxford University, South Parks Road, Oxford, UK;
3Adprotech Ltd., Chesterford Research Park, Little Chesterford, Essex, UK.
Corresponding Author
Claire L Harris, Complement Biology Group, Department of Medical Biochemistry and
Immunology, School of Medicine, Cardiff University, Henry Wellcome Building, Heath
Park, Cardiff, CF14 4XN, UK.
Tel: (+44) 29 20745254
Fax: (+44) 29 20744001
E-mail: [email protected]
Running Title
Interactions in the alternative pathway of complement
JBC Papers in Press. Published on November 9, 2004 as Manuscript M410179200
Copyright 2004 by The American Society for Biochemistry and Molecular Biology, Inc.
by guest on March 15, 2018
http://ww
w.jbc.org/
Dow
nloaded from

SUMMARY
The complement regulatory protein, decay accelerating factor (DAF, CD55), inhibits the
alternative complement pathway by accelerating decay of the convertase enzymes formed by
C3b and factor B. Using surface plasmon resonance we show that in the absence of Mg2+
DAF binds C3b, factor B and the Bb subunit with low affinity (KD: 14±0.1 µM, 44±10 µM
and 20±7 µM respectively). In the presence of Mg2+ DAF bound Bb or the von Willebrand
factor type A subunit of Bb with higher affinities (KD: 1.3±0.5 µM and 2.2±0.1 µM
respectively). Interaction with the proenzyme, C3bB, was investigated by flowing factor B
across a C3b-coated surface in the absence of factor D. The dissociation rate was dependent
on the time of incubation, suggesting that a time-dependant conformational transition
stabilised the C3b-factor B interaction. Activation by factor D (forming C3bBb) increased
the complex half-life, however, the enzyme became susceptible to rapid decay by DAF unlike
the proenzyme which was unaffected. A convertase assembled with cobra venom factor,
CVFBb, was decayed by DAF, albeit far less efficiently than C3bBb. DAF did not bind CVF
implying that Bb decay is accelerated, at least in part, through DAF binding this subunit. It is
likely that DAF binds the complex with higher affinity/avidity promoting a conformational
change in either or both subunits accelerating decay. Such analysis of component and
regulator interactions will inform our understanding of inhibitory mechanisms and the ways
in which regulatory proteins co-operate to control the C cascade.
by guest on March 15, 2018
http://ww
w.jbc.org/
Dow
nloaded from

INTRODUCTION
Complement (C)1 plays a central role in innate immune defence with the abilty to rapidly
opsonise or destroy microorganisms or infected cells. The alternative pathway (AP) provides
an immediate, antibody-independent means to activate the C cascade whereas the classical
pathway (CP) relies in most cases on antibody to initiate activation of the first component,
C1. A third pathway, the lectin pathway, is initiated by binding of mannan binding lectin
(MBL) to sugar residues on bacterial cell walls. Other than the initiating factor (MBL and
associated proteases) this pathway is identical to the CP. The AP provides an immediate line
of defence to potential infection, it ‘ticks-over’ continuously in plasma through low level
activation of the central component, C3, probably via hydrolysis of the internal thioester bond
(1). Tight regulation of this tickover ensures that C is not activated to excess in plasma but
provides enough active C3b to bind foreign surfaces and amplify the cascade swiftly when
appropriate (2). The AP also amplifies the CP by formation of further active convertases on
C3b deposited during early amplification steps by the CP convertase. The initiating stimulus
of the AP is nucleophilic attack on the internal thioester bond in nascent C3b by an amine or
hydroxy group present on a foreign or ‘activating’ surface (3). This results in covalent
binding of C3b and forms the nidus for C amplification. The C3 convertase is formed by
binding of factor B (fB) to C3b in an interaction that requires a Mg2+ ion. The fB changes
conformation such that the C3bB complex is capable of activating the zymogen factor D
1 Abbreviations. AP: alternative pathway; CP: classical pathway; DAF: decay accelerating factor; MCP: membrane cofactor protein; vWFA: von Willebrand factor type A; Bb: larger cleavage subunit of factor B; Ba: smaller cleavage subunit of factor B; fB: factor B; fD: factor D; SCR: short consensus repeat; CVF: cobra venom factor; sDAF: soluble decay accelerating factor comprising four short consensus repeats; SPR: surface plasmon resonance; RU: resonance unit; ligand: moiety covalently coupled to the sensor chip surface; analyte: moiety flowed across the sensor chip surface; Rmax: maximum binding capacity of ligand; KA: association equilibrium constant; KD: dissociation equilibrium constant; ka: association rate constant; kd: dissociation rate constant; HBS: HEPES buffered saline.
by guest on March 15, 2018
http://ww
w.jbc.org/
Dow
nloaded from

(fD), a serine protease present in plasma at about 2µg/ml (4). FD cleaves fB into Ba (amino-
terminal fragment) and Bb, the latter comprising an amino-terminal von Willebrand factor
type A domain (vWFA) and a carboxy terminal serine protease (SP) domain. Following
release of Ba, Bb binds with higher affinity to C3b. It is likely that conformational changes
in the vWFA domain transmit an allosteric signal to the SP domain resulting in its activation
and conferring it with the ability to cleave multiple molecules of C3 to nascent C3b thereby
amplifying the cascade (5,6).
The background tick-over of the AP and the potential for rapid amplification of all pathways
on self cells means that strict control in both biological fluids and on the membranes is
essential. Numerous C regulatory proteins (CReg) have evolved to meet the need to protect
‘self’ from the potentially destructive effects of C (7). These proteins police the body,
collaborating to control all pathways. The different CReg belong to various gene families,
those encoded in the RCA (‘Regulators of Complement Activation’) gene cluster on
chromosome 1 comprise factor H (fH), C4b binding protein (C4bp), decay accelerating factor
(DAF; CD55), membrane cofactor protein (MCP; CD46) and complement receptor 1 (CR1;
CD35) (8). All contain a structural module termed the short consensus repeat (SCR), a
domain comprising approximately 60 amino acids which confers C3b/C4b binding affinity to
the proteins and contains the regulatory function of the molecule (9). The amino-terminal
fragment of fB, Ba, is also comprised from three SCRs. The number of domains in CReg
varies from 4 (in MCP, DAF) to 37 (CR1, ‘B’ isoform). DAF is a GPI-anchored CReg
which acts to accelerate decay of the naturally labile enzymes of the AP and CP, C3bBb and
C4b2a (10,11). Other than the SCRs it comprises an elongated, heavily glycosylated ‘stalk’
by guest on March 15, 2018
http://ww
w.jbc.org/
Dow
nloaded from

proximal to the membrane which projects the active site the correct distance from the
membrane allowing it to function efficiently (12,13).
The C components, from C1 through to C9, comprise a fascinating group of proteins whose
activities are characterised by differing conformations and presentation of continuously
changing faces or binding sites to other C components, receptors or regulators. C3 and fB are
no exception. C3 transformation starts following cleavage and release of C3a, it continues
following destruction of the internal thioester bond and moves on by presentation of binding
sites for components such as fB, Bb and eventually C5 (14,15). Along the way binding sites
for a multitude of regulators are formed, their sites may be distinct or overlapping and are
often transient as the cascade progresses or C3b becomes inactivated (16). Cleavage and
inactivation of C3b by fI results in the production of iC3b, C3c and C3dg, these inactivation
products no longer bind components such as fB and all differ in their ability to bind various C
receptors (17). FB also undergoes a multitude of conformational changes. Within the intact
protein all three domains interact with each other, possibly Ba holds the vWFA and SP
domains in a way which prevents autoactivation (5,6). FB changes conformation following
binding to C3b, once more following release of Ba and presumably again following release to
the fluid phase (18,19). The exact mechanisms of AP convertase assembly and regulation are
still being determined 50 years following discovery of this pathway (20).
Interaction of DAF with the AP convertase has largely been examined by mutagenesis
studies. These have sought to define entire domains in DAF or individual amino acids that
are crucial to decay of the convertase and protection of indicator cells (usually erythrocytes)
from lysis by C (21-23). Further information has been gained following resolution of the
structure of DAF, both in solution and in crystals (13,23,24). Various sites which extend
by guest on March 15, 2018
http://ww
w.jbc.org/
Dow
nloaded from

through SCRs 2, 3 and 4 have been proposed to be important for convertase regulation. It is
not yet clear whether DAF undergoes a conformational transition following binding to its
ligands, most likely it acts purely to induce changes in other proteins to which it binds. It is
interesting to note that DAF not only binds the convertase enzymes but also a multitude of
pathogens, both bacteria and viruses, and also the leukocyte activation antigen CD97 (25). A
potential site of interaction for DAF on fB has also been identified by mutagenesis studies
and is located distal to the C3b interaction site in vWFA domain (26). Various studies have
addressed the binding site on C3b for the RCA proteins fH, MCP and CR1 and also for fB,
but no definitive sites of interaction have been determined and that for DAF has not been
examined (27,28). In this report we examine the interactions between DAF and the AP
convertase components by surface plasmon resonance (SPR). We define the affinity of these
associations and examine in real time the assembly and decay of the active AP convertase
and its proenzyme and their interaction with DAF. This study provides new insight into these
associations and further guides our understanding of the complex mechanisms and protein
interactions intrinsic to the C cascade.
EXPERIMENTAL PROCEDURES
Preparation of complement components and regulators
C3, cobra venom factor (CVF) and factor B were prepared by classical column
chromatography using established methods (29). Factor D was purchased from Quidel
Corporation (San Diego, MA). C3b was prepared from C3 using either of two methods. In
the first, CVF was coupled to Sepharose CL-4B using the manufacturer’s protocol
(Amersham Biosciences, Chalfont St.Giles, UK). A solid-phase convertase was formed by
incubating CVF-Sepharose in normal human serum to form CVFBb. The Sepharose was
by guest on March 15, 2018
http://ww
w.jbc.org/
Dow
nloaded from

washed and incubated at 37°C with C3. C3b generated by cleavage was separated from C3a
and other minor contaminants by anion exchange on a Source Q column (Amersham
Biosciences). In the second method, C3, factor B and factor D were incubated in
complement fixation diluent (CFD; Oxoid, UK) until total C3 cleavage had occurred. C3b,
Bb and Ba were purified by anion exchange chromatography. The C3b-containing fractions
were pooled, concentrated and monomeric C3b was separated from dimeric C3b by size
exclusion on a Superose 6 column (Amersham Biosciences).
Recombinant human DAF comprising the four SCRs (soluble DAF; sDAF) was isolated and
refolded from E.coli as described previously (30). The structure and function of sDAF has
been previously studied and the purified, refolded, protein is known to consist of four
correctly folded SCR domains and demonstrates the full range of complement regulating and
pathogen binding activities associated with DAF purified from erythrocytes (13,30,31).
Human DAF-Ig fusion protein comprising four SCR domains fused to human IgG1 Fc (DAF-
Ig) was prepared as previously described (32). Recombinant vWF-A domain of human factor
B was expressed in E. coli as a fusion protein with glutathione S-transferase using published
methods (33) with the addition of a final gel filtration on a S75 column (Pharmacia) to yield
protein at a purify of >98%, as assessed by SDS PAGE electrophoeresis (data not shown).
Biosensor Analysis
All analyses were carried out on a Biacore 3000 machine (Biacore International SA,
Stevenage, UK) other than that in Figure 4c which was carried out on a Biacore 2000.
Proteins were coupled to the sensor chip surface using an amine coupling chemistry as
instructed by the manufacturers (NHS/EDC coupling kit). For all kinetic analyses a CM5
chip (carboxymethylated dextran surface) was used and data was collected at 25°C, the flow
by guest on March 15, 2018
http://ww
w.jbc.org/
Dow
nloaded from

rate was maintained at 30µl/minute and data from a reference cell was subtracted to control
for bulk refractive index changes. The Rmax was kept low and the flow rate high to eliminate
mass transfer. However, this was controlled for where appropriate by varying flow rate and
ensuring observed association rates did not vary. Samples were injected using the KINJECT
command to ensure accurate association kinetics. Interactions were analysed in HEPES
buffered saline (HBS: 10mM HEPES pH7.4, 150mM NaCl), 0.005% surfactant P20 and
either 1mM MgCl2 or 3mM EDTA as stated in the text. Data were evaluated using
Biaevaluation software (Biacore International). Concentration of analytes was assessed using
absorbance at A280, molarities were calculated using the following extinction coefficients
(molecular masses and coefficients obtained using Protean software, DNAStar): sDAF (1.3),
C3b (1.03), factor B(1.43), Bb (1.29), Ba (1.74).
RESULTS
Interaction of DAF with C3b
In order to study the interaction of DAF with the individual AP convertase components, C3
was incubated with factors B and D until formation of C3b, Bb and Ba was complete.
Individual components were separated by anion exchange chromatography (Figure 1a). This
also removed C3i from the C3b preparation. We routinely found that approximately 50% of
the C3b formed by AP activation was in a dimeric form. In order to eliminate avidity effects
from SPR analysis, C3b was further separated into monomeric and dimeric forms by size
exclusion chromatography (Figure 1b). Initial SPR studies indicated that the interaction
between DAF and C3b was of low affinity (subµM) with very fast on and off rates typical of
DAF-ligand interactions (31,34,35). The affinity of the interaction was studied under steady
state conditions by immobilising either DAF or C3b to the sensor chip surface and flowing
by guest on March 15, 2018
http://ww
w.jbc.org/
Dow
nloaded from

monomeric C3b or DAF respectively over the surface. The most accurate determination as
judged by smallest SE values for KA and Rmax was obtained by immobilising C3b and
studying the interaction with soluble DAF as this analyte was available at concentrations of
the same order of magnitude as the KDs determined (>10µM). C3b was coupled to the chip
surface either by amine coupling or through the thioester group. The latter method has the
advantage of capturing C3b on the surface in a physiologically relevant orientation. A typical
analysis is illustrated in Figure 2, reference cells were used to correct for any bulk shift
effects. Mean KDs are given in Table 1.
Interaction of DAF with fB and its cleavage products
Mutational analysis of fB has revealed two sites in the vWFA domain which are involved
with decay, whether these sites interact directly with DAF or alter the stability of the
convertase is not yet clear (26). It is likely however that there is a direct interaction of DAF
with the Bb portion of the convertase. We immobilised DAF on the chip surface and flowed
intact fB, Bb or Ba over the surface at high concentrations. A direct interaction was seen
with intact fB and the Bb subunit but not with Ba (Figure 3a). The affinity of the interaction
between DAF and intact fB was determined by immobilising either fB or DAF to the surface
via amine groups. As with the DAF/C3b interaction, the affinity was low and was analysed
at steady state. The binding profile and affinity data obtained were similar irrespective of the
protein partner immobilised (mean KD 44±10 µM in EDTA; Figure 3) and the presence or
absence of Mg2+ (not shown, mean KD 74±30 µM in 1mM Mg2+). The interaction of DAF
with intact fB was of lower affinity than with C3b (Table 1).
Whilst the interaction of fB was relatively unaffected by the presence of Mg2+, we noticed
that this cation had a marked effect on the interaction of DAF with the Bb subunit (Figure
by guest on March 15, 2018
http://ww
w.jbc.org/
Dow
nloaded from

4a). In the absence of Mg2+ the affinity of the Bb subunit for DAF and the binding profile
was similar to that of fB (data not shown, average KD 20±7 µM). However in the presence of
Mg2+ the affinity of the interaction between Bb and DAF was higher. The kinetic profile was
different to that of fB, on and off rates were slower and the affinity was calculated as 1.3 µM
(using a 1: 1 Langmuir interaction model; Figure 4b, Table 1). Further analysis using the
isolated vWFA domain demonstrated a similar binding profile (Figure 4c) and affinity of
2.2±0.1 µM for this subunit (Table 1).
Interaction of fB with C3b
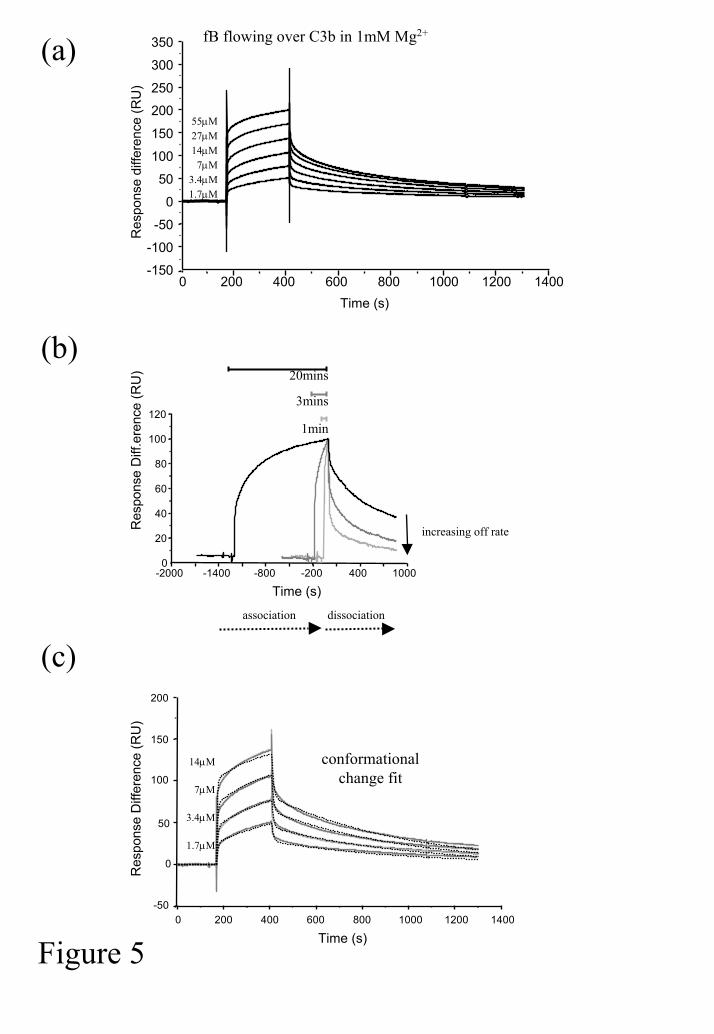
We sought to assemble the convertase on the sensor chip surface itself, the aim being to
assess the affinity of DAF for the intact enzyme in addition to individual components. Low
levels of C3b were immobilised to the chip surface and fB was flowed across in the presence
of Mg2+ (1mM). It was immediately apparent that the interaction did not fit a simple 1:1
association model (Figure 5a). Furthermore, interaction of identical concentrations of fB
with C3b for different lengths of time (1, 3 or 20 mins) demonstrated that the dissociation
became slower the longer the components interacted (Figure 5b), this can be indicative of a
conformational change which results in a tighter binding between components. Indeed,
fitting the data from Figure 5a to a conformational change model (BIAcore software) gave a
good fit, although some heterogeneity was evident at high concentrations of fB (Figure 5c).
Kinetic analysis following a 4 minute association phase gave the following information: ka1:
1.36x104M-1s-1; kd1: 0.115 s-1; ka2: 5.53x10-3M-1s-1; kd2: 1.78x10-3s-1 (χ2: 4.2). In this model
the off rate following the conformational change (kd2) is much lower.
by guest on March 15, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Formation of the AP convertase and interaction with DAF
Either the active or inactive convertase was assembled on the chip surface by flowing fB over
C3b in the presence or absence of fD. Either fB (46µg/ml) or a mixture of fB (46µg/ml) and
fD (8µg/ml) was flowed across the surface as indicated in Figure 6a. Several differences in
the two complexes were noted. Firstly, cleavage of fB by fD altered the rate of decay of the
active enzyme, prolonging its half-life. Secondly, the Mg2+ ion in the active enzyme was
'locked' in place, flowing 10mM EDTA over C3bB(Mg2+) dissociated fB from C3b whereas it
had no effect on decay of C3bBb(Mg2+).
C3b was also bound to the surface using the AP, this resulted in coupling of nascent C3b via
its thioester to hydroxy groups present on the dextran-coated surface. We used a
modification of a previously described technique in which repetitive cycles of fB/fD with C3
resulted in AP amplification on the chip surface (36). To increase efficiency of activation we
deposited the first nidus of C3b on the chip surface using a fluid phase convertase, CVFBb
(Figure 6b). Each subsequent ‘cycle’ illustrated in Figure 6b comprised incubation with fB
and fD to form active convertase followed by incubation with native C3. Following C3b
deposition, buffer was flowed across the surface for 16 hours to allow dissociation of non-
covalently bound C3b and of any active convertase. In order to study the interaction of DAF
with the convertase, factors B and D were flowed over the surface. The amount of
active/inactive convertase on the chip surface was varied by titrating the amount of fD in the
incubation (Figure 6c). In the presence of fD, the surface reached an equilibrium (Figure 6c
(E)), unlike the proenzyme which displayed complex kinetics during formation (Figures 5, 6c
(A)). When soluble DAF was flowed across the active convertase, decay of the Bb subunit
was virtually instantaneous (Figure 6c (B-E)). Decay of the enzyme was so efficient that it
was impossible to measure binding and an affinity of DAF for the intact convertase. This
by guest on March 15, 2018
http://ww
w.jbc.org/
Dow
nloaded from

contrasted with flow of DAF across the inactive enzyme in which rapid and total decay was
not apparent (Figure 6b (A)). Uncleaved fB was dissociated from the surface at the end of
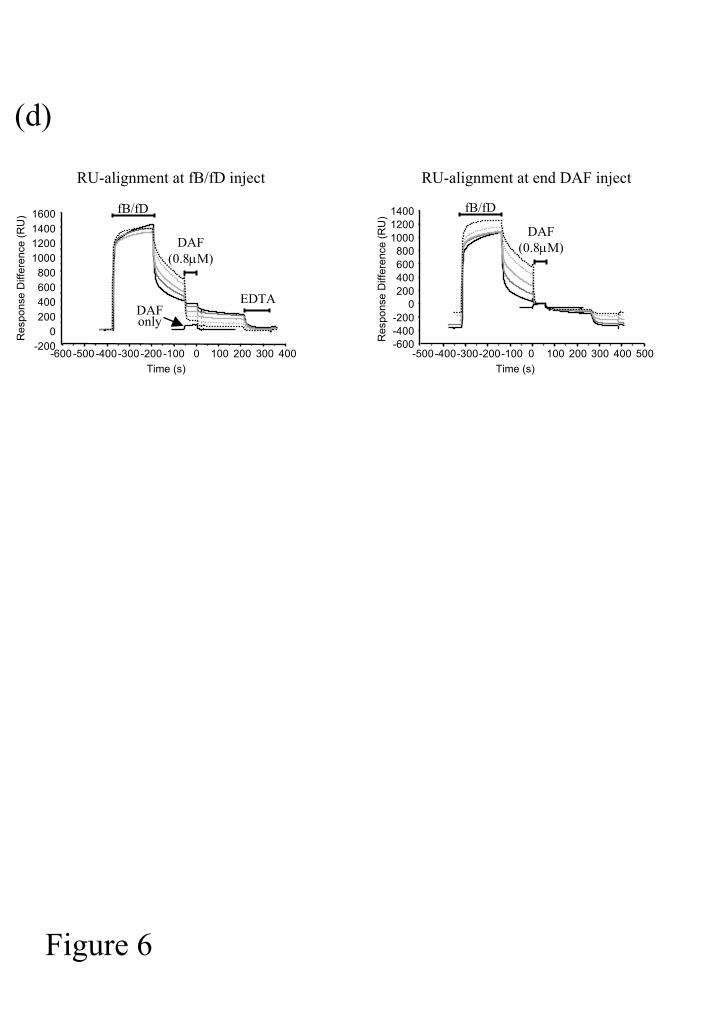
the incubation using EDTA as shown in Figure 6. Alignment of individual sensorgrams at
either the fB/fD inject or DAF inject demonstrates the variation in decay and identical
binding of DAF to C3b on the chip surface (Figure 6d).
Formation of CVFBb and interaction with DAF
We have demonstrated that DAF interacts with both subunits of the active enzyme and that
decay is rapid and efficient. The convertase enzymes formed from CVF (C3b-like molecule
found in cobra venom) and fB are very stable and are assumed to be resistant to decay by
human RCA proteins. To analyse the interaction with DAF, CVF was immobilised on the
sensor chip surface and DAF was flowed across, no interaction between CVF and DAF was
evident even at high concentrations (47µM) (Figure 7a). To assess whether DAF could decay
the CVF-containing convertase, fB and fD were flowed sequentially across the CVF surface
and a C3b-coated surface (Figure 7b). In the absence of fD the CVFB complex dissociated
rapidly and the surface could be regenerated with EDTA. Inclusion of fD resulted in an
EDTA-stable surface which efficiently cleaved C3 to C3b (data not shown). Formation of
the active enzyme was less efficient than with human C3b but that which did form was very
stable (Figure 7b). When DAF was flowed across the surface at concentrations (15µM) much
higher than those which brought about immediate decay of C3bBb, it was apparent that it
could indeed accelerate decay of CVFBb. However, this was far less efficient than with the
native, human enzyme. The large concentration of DAF used in this experiment is apparent
in Figure 7b where binding to the C3b-coated surface can be seen during the DAF inject
(indicated in the figure).
by guest on March 15, 2018
http://ww
w.jbc.org/
Dow
nloaded from

DISCUSSION
We have used SPR technology to analyse the interaction of DAF with individual components
of the AP convertase and also with the intact (pro)enzyme and to visualise assembly and
decay of the convertase in real time. We demonstrate that DAF interacts with the individual
components C3b and fB with low affinity. The equilibrium dissociation constant (KD) for the
interaction of DAF with fB is 44±10µM demonstrating that this is a significantly weaker
interaction than that of DAF with C3b where the interaction with amine-coupled C3b is
characterised by a KD of ~14±1µM and with thioester-coupled C3b with a KD of ~7±1µM
(Table 1 and Figures 2 and 3). The small differences between the KDs for DAF interactions
with differently coupled C3b probably reflect the more optimal presentation of binding sites
in the natively coupled C3b as compared to the amine coupled protein where steric hindrance
is more likely to occur.
DAF did not interact with the Ba subunit of fB but did interact with the Bb subunit. The
affinities of DAF for fB and Bb in EDTA were comparable (44±10 µM and 20±7 µM
respectively). However, the affinity of DAF for the Bb subunit was increased >10-fold to
1.3µM (KD) in the presence of Mg2+; a similar affinity was obtained for the isolated vWFA
domain (2.2µM ) suggesting that this domain mediates the contact with DAF (Figure 4).
Indeed, a previous report has defined two surface patches on opposing surfaces of the vWFA
domain that may be involved in the decay of C3bBb by DAF; one of these may mediate
binding to C3b whilst the other may be involved in binding DAF (26). It is interesting that
DAF has a higher affinity for Mg2+-bound Bb. It is known that the Mg2+ coordination site of
fB is in the active site cleft of the vWFA domain, a site that is involved in the binding to C3b,
and that the structure of this domain and of Bb is more stable in the presence of the cation. A
by guest on March 15, 2018
http://ww
w.jbc.org/
Dow
nloaded from

conformational dependence of the vWFA domain on the presence of a metal ion has been
demonstrated by various spectroscopic techniques (6), in particular the α-helix A7 is
conformationally mobile between the metal-free and metal-bound forms of the vWFA
domain. Our data show that DAF may bind distinct conformational states of this molecule
with different affinities. Whilst we have demonstrated a direct interaction of DAF with the
isolated Bb fragment, the physiologically relevant complex is Bb in association with C3b. Bb
has little or no affinity for C3b following dissociation indicating that fluid phase Bb differs in
conformation from C3b-bound Bb ((18) and our unpublished SPR data). It is known that the
vWFA domain in Bb, in common with similar domains in other molecules such as CR3 or
von Willebrand factor, exists in high affinity and low affinity conformations (37). Following
cleavage of fB by fD, Bb binds to C3b with higher affinity. The vWFA domain likely
transmits an allosteric signal resulting in proteolytic activity in the SP domain of Bb (6). We
cannot determine whether the affinity of DAF that we demonstrate here is for a low or high
affinity conformation of vWFA domain.
The initial binding of fB to surface bound C3b is complex and clearly does not follow a
simple 1:1 Langmuir interaction (Figure 5). The data fit a model in which a conformational
change occurs resulting in a higher affinity interaction with a slower dissociation rate (kd2).
This model can only be applied when the change is slow and occurs over the period of the
association phase, extremely rapid changes are more likely to fit a simple 1:1 interaction. It
is known that fB changes conformation upon binding C3b allowing it to induce a
proteolytically active conformation of fD (4). However inclusion of fD in the incubation
resulted in a very different binding profile (Figure 6), the binding rapidly reached a plateau
and an equilibrium was obtained. Interestingly, the resulting complex was resistant to EDTA
(Figure 6a), this is in agreement with previous work which has shown that a transition in Bb
by guest on March 15, 2018
http://ww
w.jbc.org/
Dow
nloaded from

and tight binding of the vWFA active site cleft to C3b protects the Mg ion from chelation by
EDTA (19,38). The binding profile of fB in the presence of fD was more typical of a simple
interaction implying that if conformational changes have occurred they were rapid and an
'end-point' was swiftly reached. Others have previously reported that fB can bind C3b (and
CVF) and form an active enzyme in the absence of fD (39-41). It is possible that the
conformational change evident in the absence of fD in Figure 5 represents a slow transition to
an active conformation in which the SP domain of fB can cleave C3b, fD-mediated cleavage
of fB to Bb may act to expedite this transition.
We have shown that DAF binds individual components of the AP convertase with low
affinity, the crucial question is how does DAF bind the intact convertase? DAF must
'recycle' in order to protect self cells from C attack. Clearly if DAF bound to C3b with the
same affinity as to the convertase it would rapidly become saturated with C3b at a site of C
activation and would not be available for decay of the active convertase. In 1986, Pangburn
examined the ability of DAF and other CReg to decay the zymosan-bound convertases in the
presence of fluid phase competitors such as C3b, Bb, C3bB and C3bBb (42). Apparent
association constants (appKA) of DAF with C3b and Bb were 0.045µM-1 and 0.067µM-1
respectively. The affinity of DAF for C3bB or C3bBb was >10 fold higher (0.71µM-1,
0.91µM-1) suggesting that following decay of the enzyme DAF would be 'released' enabling it
to bind the next convertase with high affinity. Despite the fact that DAF appeared to bind
C3bB almost as well as C3bBb in this study, it was not clear whether DAF decayed both the
active convertase and the proenzyme. Early studies using DAF incorporated into erythrocyte
membrane showed that DAF did not prevent fB binding surface bound C3b or C4b, but
rapidly decayed the activated fragments Bb (43). More recently it has been demonstrated
using an ELISA-based decay assay that DAF decays C3bBb(Ni2+) but not C3bB(Ni2+) (44),
by guest on March 15, 2018
http://ww
w.jbc.org/
Dow
nloaded from

this selective decay may be related to the >10-fold higher affinity of DAF for Bb compared to
fB in the presence of Mg2+. Our data support the suggestion that DAF only decays the active
enzyme. In order to monitor the decay of the convertase using SPR, we formed the
convertase on the sensor chip surface in the presence or absence of fD (Figure 6). DAF-
mediated decay of the active convertase, C3bBb, was visualised in real time. It was
immediately apparent that dissociation of Bb from C3b was virtually instantaneous and it was
not possible to measure an affinity of DAF for the active convertase directly. In support of
the ELISA-based assay it was evident that DAF decayed the active enzyme and had little
effect on the inactive convertase. By titrating the amount of active convertase on the chip
surface it was possible to see this differential decay (Figure 6c). In the absence of any fD a
small amount of DAF-mediated decay was evident (Figure 6c(A)), we do not know what this
represents but it is possible that a portion of C3bB has undergone a suitable transition such
that it is in an ‘active’ conformation and the affinity for DAF is enhanced as discussed above
rather than this being the observation of decay of the inactive convertase. When DAF was
flowed across the C3b-coated surface in the absence of any fB or fD an increase in RU was
evident (Figure 6d, alignment at DAF inject). This represented binding of DAF to C3b and
decreased again rapidly after the DAF inject was completed, the off rate for this interaction is
extrememly rapid as illustrated in Figure 2. Following an identical inject of DAF over the
C3bB or C3bBb coated surface the same decrease in RU was evident at the end of the inject
and dissociation was rapid (Figure 6d). The dissociation of DAF from the surface was
evident above the background decay of fB from C3b which remained unaltered. These
preliminary data imply that in our assay system the affinity of DAF for C3b was unaffected
by the presence or absence of proteolytically inactive fB, we are currently investigating the
kinetics of this interaction further. Attempts to stabilise the C3bBb surface using chemical
crosslinking in order to measure the affinity of DAF for the active enzyme were not
by guest on March 15, 2018
http://ww
w.jbc.org/
Dow
nloaded from

successful. Whilst the release of Bb from the surface was prevented, an interaction with DAF
could not be visualised. Either the crosslinking reagent destroyed the interaction sites with
the convertase or DAF did indeed decay the components but they remained loosely tethered
to each other via the crosslinker without any physical protein/protein interaction. There are
various reasons why DAF may decay the active but not the inactive convertase. Firstly the
SCR-containing subunit of fB, Ba, may directly compete for the same binding site on C3b.
Secondly, the presence of Ba may stabilise the C3bB complex, a direct interaction of Ba with
C3b has been demonstrated ((45) and our unpublished SPR data), several points of contact
will increase the avidity of the interaction. Thirdly, as suggested above Bb may adopt a
conformation in the absence of Ba which binds DAF with a higher affinity. Finally, it is also
possible that the Ba domain partially blocks the binding site on Bb for DAF, it has been
shown previously that Ba contacts both the vWFA and SP domains in inactive fB (5).
It is interesting that DAF accelerates decay of the CVFBb convertase, albeit with much lower
efficiency than decay of C3bBb (Figure 7b). DAF had no affinity for the isolated C3b-like
component of the enzyme, CVF (Figure 7a). In the study described above Pangburn
demonstrated that the affinity of DAF for CVFBb was identical to that of DAF for Bb (42).
Although we show here that the accelerated decay of CVFBb mediated by DAF was very
inefficient, it was the only way in which we could regenerate the surface of the chip, neither
EDTA nor another powerful CReg, soluble recombinant CR1 (sCR1), could regenerate the
surface back to uncomplexed CVF. The inefficiency of this decay implies that either the
conformational change in Bb resulting in its release is slowed by the presence of CVF, or that
DAF works by also inducing a transition in C3b (not seen with CVF) that accelerates decay
of the enzyme.
by guest on March 15, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Current concepts regarding AP convertase assembly and decay are as follows. Ba mediates
initial binding of fB to C3b, the avidity of binding may be enhanced by sites on the Bb
subunit which interact with C3b. If sites in Bb interact with C3b the conformation differs
from that of the dissociated subunit, Bb, which has much decreased affinity for C3b.
Following binding of fB a conformational change occurs such that the complex activates the
zymogen fD, which in turn cleaves fB into Bb and Ba. Ba is released from the complex
(although this interaction is reversible). Release of Ba results in a higher affinity complex
between C3b and Bb, the Mg2+ ion in the active site cleft of the vWFA domain is 'locked' into
the enzyme through association with C3b, the complex becomes more stable with a longer
half-life and the SP domain in Bb is activated such that it can proteolytically cleave C3.
However, this complex is subject to decay by DAF. We show that DAF binds individual
components of the convertase in the following order of affinity: Bb>C3b>fB. Presumably it
associates weakly with C3b deposited covalently on a cell surface. However, in order to
selectively bind and decay active convertase, binding to C3bBb must be greater than that to
C3b. A simple explanation would be that a second point of contact with the Bb subunit
increases the avidity of the interaction, we have noted that apparent affinities of bivalent
interactions can be 10-fold that seen with the same monovalent interaction (unpublished
data). It is also possible that complexing of C3b and Bb favours a particular conformation in
one or both of the components that bind DAF with higher affinity. Increased avidity of DAF
binding to the multi-molecular complex would in effect stabilise the complex further and
prevent decay/dissociation. It is likely therefore that DAF promotes a further change in
conformation resulting in dissociation of Bb from C3b, possibly by inducing the 'low affinity'
conformation of the vWFA domain. Decay of the complex results in a low (subµM)
interaction with the individual components allowing DAF to recycle to other active
convertase. It is of note that the AP convertase is stabilised in vivo by binding of properdin
by guest on March 15, 2018
http://ww
w.jbc.org/
Dow
nloaded from

to the C3bBb complex (46). We are currently examining the effect of properdin on DAF
binding to the AP convertase and to individual components. Whilst the exact mechanisms of
assembly and decay of the AP convertase are still unclear, we have come a long way in
deciphering the ways in which C is activated and regulated. Data presented here and similar
analyses of other C regulators will provide a more informed picture of their co-operation in
inhibiting the complement activation pathways and the mechanisms responsible for release
from the convertase and ‘recycling’ of the regulatory proteins.
REFERENCES
1. Pangburn, M. K., Schreiber, R. D., and Muller-Eberhard, H. J. (1981) J Exp Med 154,
856-867
2. Nicol, P. A., and Lachmann, P. J. (1973) Immunol 24, 259-275
3. Law, S. K., and Levine, R. P. (1977) Proc Natl Acad Sci USA 74, 2701-2705
4. Volanakis, J. E., and Narayana, S. V. (1996) Protein Sci 5, 553-564.
5. Hinshelwood, J., and Perkins, S. J. (2000) J Mol Biol 301, 1267-1285.
6. Hinshelwood, J., and Perkins, S. J. (2000) J Mol Biol 298, 135-147.
7. Morgan, B. P., and Harris, C. L. (1999) Complement Regulatory Proteins, Academic
Press, London
8. Holers, V. M., Cole, J. L., Lublin, D. M., Seya, T., and Atkinson, J. P. (1985)
Immunol Today 6, 188-192
9. Reid, K. B., and Day, A. J. (1989) Immunol Today 10, 177-180
10. Nicholson-Weller, A. (1992) Curr Top Microbiol 178, 7-30
11. Kuttner-Kondo, L., Medof, M. E., Brodbeck, W., and Shoham, M. (1996) Protein Eng
9, 1143-1149
by guest on March 15, 2018
http://ww
w.jbc.org/
Dow
nloaded from

12. Coyne, K. E., Hall, S. E., Thompson, S., Arce, M. A., Kinoshita, T., Fujita, T.,
Anstee, D. J., Rosse, W., and Lublin, D. M. (1992) J Immunol 149, 2906-2913
13. Lukacik, P., Roversi, P., White, J., Esser, D., Smith, G. P., Billington, J., Williams, P.
A., Rudd, P. M., Wormald, M. R., Harvey, D. J., Crispin, M. D., Radcliffe, C. M.,
Dwek, R. A., Evans, D. J., Morgan, B. P., Smith, R. A., and Lea, S. M. (2004) Proc
Natl Acad Sci U S A 101, 1279-1284.
14. Isenman, D. E., and Cooper, N. R. (1981) Mol Immunol 18, 331-339.
15. Isenman, D. E., Kells, D. I., Cooper, N. R., Muller-Eberhard, H. J., and Pangburn, M.
K. (1981) Biochemistry 20, 4458-4467.
16. Lambris, J. D., and Muller-Eberhard, H. J. (1986) Mol Immunol 23, 1237-1242
17. Harrison, R. A., and Lachmann, P. J. (1980) Mol Immunol 17, 9-20
18. Fishelson, Z., and Muller-Eberhard, H. J. (1984) J Immunol 132, 1425-1429
19. Tuckwell, D. S., Xu, Y., Newham, P., Humphries, M. J., and Volanakis, J. E. (1997)
Biochemistry 36, 6605-6613
20. Pillemer, L., Blum, L., Lepow, I.H., Ross, O.A., Todd, E.W., Wardlaw, A.C. (1954)
Science 120, 279-285
21. Brodbeck, W. G., Kuttner-Kondo, L., Mold, C., and Medof, M. E. (2000) Immunol
101, 104-111.
22. Kuttner-Kondo, L. A., Mitchell, L., Hourcade, D. E., and Medof, M. E. (2001) J
Immunol 167, 2164-2171.
23. Williams, P., Chaudhry, Y., Goodfellow, I. G., Billington, J., Powell, R., Spiller, O.
B., Evans, D. J., and Lea, S. (2003) J Biol Chem 278, 10691-10696.
24. Uhrinova, S., Lin, F., Ball, G., Bromek, K., Uhrin, D., Medof, M. E., and Barlow, P.
N. (2003) Proc Natl Acad Sci U S A 100, 4718-4723.
25. Lea, S. (2002) Biochem Soc Trans 30, 1014-1019.
by guest on March 15, 2018
http://ww
w.jbc.org/
Dow
nloaded from

26. Hourcade, D. E., Mitchell, L., Kuttner-Kondo, L. A., Atkinson, J. P., and Medof, M.
E. (2002) J Biol Chem 277, 1107-1112. Epub 2001 Nov 1102.
27. Farries, T. C., Seya, T., Harrison, R. A., and Atkinson, J. P. (1990) Complement &
Inflammation 7, 30-41
28. Lambris, J. D., Lao, Z., Oglesby, T. J., Atkinson, J. P., Hack, C. E., and Becherer, J.
D. (1996) J Immunol 156, 4821-4832
29. Harrison, R. A. (1996) in Weir's handbook of experimental immunology, Volume II.
Cell surface and messenger molecules of the immune system. (Herzenberg, L. A.,
Weir, D. M., Herzenberg, L. A., and Blackwell, C., eds), pp. 75.71-75.50, Blackwell
Science, Cambridge, MA
30. White, J., Lukacik, P., Esser, D., Steward, M., Giddings, N., Bright, J., <organ, B. P.,
Lea, S. M., Smith, G. P., and Smith, R. A. G. (2004) Protein Sci In press,
31. Anderson, K. L., Billington, J., Pettigrew, D., Cola, E., Roversi, P., Simpson, P.,
Chen, H. A., Urvil, P., du Merle, L., Barlow, P., Medof, E., Smith, R. A. G., Nowicki,
B., Le Bouguenec, Lea, S. M., and Matthews, S. (2004) Mol Cell In press,
32. Harris, C. L., Lublin, D. M., and Morgan, B. P. (2002) J Immunol Methods 268, 245-
258.
33. Williams, S. C., Hinshelwood, J., Perkins, S. J., and Sim, R. B. (1999) Biochem J 342,
625-632.
34. Lea, S. M., Powell, R. M., McKee, T., Evans, D. J., Brown, D., Stuart, D. I., and van
der Merwe, P. A. (1998) J Biol Chem 273, 30443-30447.
35. Lin, H. H., Stacey, M., Saxby, C., Knott, V., Chaudhry, Y., Evans, D., Gordon, S.,
McKnight, A. J., Handford, P., and Lea, S. (2001) J Biol Chem 276, 24160-24169.
36. Jokiranta, T. S., Westin, J., Nilsson, U. R., Nilsson, B., Hellwage, J., Lofas, S.,
Gordon, D. L., Ekdahl, K. N., and Meri, S. (2001) Int Immunopharmacol 1, 495-506.
by guest on March 15, 2018
http://ww
w.jbc.org/
Dow
nloaded from

37. Loftus, J. C., and Liddington, R. C. (1997) J Clin Invest 100, S77-81.
38. Fishelson, Z., Pangburn, M. K., and Muller-Eberhard, H. J. (1983) J Biol Chem 258,
7411-7415.
39. Vogt, W., Dames, W., Schmidt, G., and Dieminger, L. (1977) Immunochemistry 14,
201-205
40. Cooper, N. R. (1973) J Exp Med 137, 451-460.
41. Vogel, C. W., and Muller-Eberhard, H. J. (1982) J Biol Chem 257, 8292-8299.
42. Pangburn, M. K. (1986) J Immunol 136, 2216-2221
43. Fujita, T., Inoue, T., Ogawa, K., Iida, K., and Tamura, N. (1987) J Exp Med 166,
1221-1228
44. Hourcade, D. E., Mitchell, L. M., and Medof, M. E. (1999) Immunopharmacology 42,
167-173.
45. Pryzdial, E. L., and Isenman, D. E. (1987) J Biol Chem 262, 1519-1525.
46. Medicus, R. G., Gotze, O., Muller-Eberhard, H. J. (1976) J Exp Med 144 1076-1093.
ACKNOWLEDGEMENTS
This work was supported by the Wellcome Trust (grant reference 068823/Z). We
acknowledge the support of the MRC (studentship to RJMA).
by guest on March 15, 2018
http://ww
w.jbc.org/
Dow
nloaded from

FIGURE LEGENDS
Figure 1. Isolation of activated C components. C3 was incubated with fB and fD in CFD.
Activated components were separated by anion exchange (a), C3b monomers and dimers
were further fractionated by size exclusion (b).
Figure 2. Interaction between C3b and DAF. (a) C3b (monomeric) was immobilised on
the chip surface via amine coupling. Interaction with sDAF was analysed in HBS-EDTA at
the indicated concentrations and sensorgram traces were obtained (grey/black lines are
duplicates and virtually overlie each other). The affinity of the interaction was analysed by
steady state analysis (see inset). Values for KD and Rmax and standard errors associated with
the fit illustrated here are shown.
Figure 3. Interaction between DAF and factor B, Bb and Ba. (a) Differing amounts of
DAF (either sDAF or DAF-Ig) were immobilised on the chip surface via amine groups.
Factor B, Bb or Ba was flowed at the indicated concentrations in HBS-EDTA and
sensorgram traces were obtained. (b) sDAF was immobilised on the chip surface via amine
groups. Interaction with factor B was analysed in HBS-EDTA at the indicated concentrations
and sensorgram traces were obtained. The affinity of the interaction was revealed by steady
state analysis to yield the values shown. (c) FB was immobilised on the chip surface via
amine groups and interaction with sDAF was analysed in HBS-EDTA at the indicated
concentrations.
Figure 4. Interaction between DAF, Bb and vWFA domain. sDAF was immobilised on
the chip surface via amine groups. FB or Bb in either EDTA or 1mM Mg2+ was flowed
by guest on March 15, 2018
http://ww
w.jbc.org/
Dow
nloaded from

across the surface (a). Further kinetic analysis of either (b) Bb or (c) vWFA domain was
performed at the indicated concentrations in HBS-Mg2+ and sensorgram traces were obtained.
The affinity of the interactions were analysised by 1:1 Langmuir binding. Black traces
represent the actual data and grey traces the modelled data. Values derived from and errors
associated with these fits are also shown.
Figure 5. Interaction between C3b and factor B. (a) C3b was immobilised on the chip
surface via amine coupling. Interaction with factor B was analysed at the indicated
concentrations in HBS-Mg2+ allowing association for 4 minutes. (b) Factor B was flowed
over C3b for different lengths of time (1, 3 and 20 minutes), data were normalised using
BIAcore evaluation software and sensorgrams overlaid at the start of the dissociation curve.
(c) The affinity of the interaction was analysed using the ‘conformational change’ model;
dashed lines are modelled data, χ2=4.2; grey lines are actual data.
Figure 6. Assembly and decay of the AP C3 convertase in real time. (a) C3b was amine-
coupled to the chip surface. fB was flowed across in HBS-Mg2+ as indicated, incubation in
10mM EDTA decayed the complex. In contrast C3bBb was not decayed by 10mM EDTA.
(b) C3b was deposited on to the acceptor surface using a method similar to that already
described (36). The AP was amplified by repeated cycling (in HBS-1mM Ni2+) at 30ºC of C3
followed by fB and fD. Convertases and non-covalently bound C3b was allowed to decay for
16 hours before using the surface. (c) Different amounts of active convertase were assembled
on the chip surface by varying the concentration of factor D in the incubation. As the
quantity of active convertase increased, the ability of sDAF to decay the components was
greater. FB, fD, sDAF and EDTA were injected at the indicated timepoints. Decay mediated
by (0.8µM) sDAF is indicated by a double-headed arrow. FB was injected at 450µg/ml and
by guest on March 15, 2018
http://ww
w.jbc.org/
Dow
nloaded from

fD at 0, 2.5, 5, 10 and 20ng/ml (numbered A-E respectively). (d) Sensorgrams from (c) are
aligned at the start of the fB/fD inject to allow for comparison of convertase assembly or at
the end of the DAF inject to allow comparison of DAF-mediated decay.
Figure 7. Interaction of DAF with cobra venom factor (CVF). CVF was immobilised on
the chip surface via amine groups. (a) CVF did not bind sDAF although it formed a C3
convertase when factor B and factor D were flowed across the chip surface (b). sDAF was
injected as indicated during which time the rate of decay of the convertase increased. The
C3bBb sensorgram is indicated by the dashed line, that of CVFBb by the solid black line.
CVF convertase was assembled in HBS-Mg2+.
by guest on March 15, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Table 1. Parameters measured that define DAF-ligand interactions. Values given are
mean ± SD of multiple experiments (n) other than Bb (1mM Mg2+) where individual datasets
are illustrated. Concentration ranges of analytes are as indicated in previous figures. ND:
not determined.
Protein interacting with DAF
ka
(mol-1
s-1
)
kd
(ms-1
)
KD
(µM)
χ2
C3b monomer amine coupled (n=5) ND ND 14 ± 1 1.2 ± 0.7
C3b dimer amine coupled (n=3) ND ND 13 ± 1 2.5 ± 1.3
Thioester coupled C3b (n=4) ND ND 7 ± 1 6.1 ± 6.4
fB (no Mg2+) (n=4) ND ND 44 ± 10 1.2 ± 0.6
fB (1mM Mg2+) (n=4) ND ND 74 ± 30 2.3 ± 3.2
Bb (no Mg2+) (n=4) ND ND 20 ± 7 2.3 ± 0.8
Bb (1mM Mg2+) (n=2) 824, 1420
1.4, 1.2 1.7, 0.8
mean 1.3
3.7, 1.8
vWF-A (1mM Mg2+) (n=6) 830 ± 30 1.8 ± 0.03 2.2 ± 0.1 0.4 ± 0.3
by guest on March 15, 2018
http://ww
w.jbc.org/
Dow
nloaded from

0
200
400
600
800
1000
1200
0 50 100 150 200 250 300 350
(a)
volume
A28
0 (m
AU
)anion exchange
BbBa
C3b
C3b aggregatesC3i
(b)
-20
30
80
130
180
230
280
0 5 10 15 20 25volume
A28
0 (m
AU
)
size exclusion; C3b
C3bmonomer
C3bdimer
Figure 1
by guest on March 15, 2018
http://ww
w.jbc.org/
Dow
nloaded from

-100
-50
0
50
100
150
200
100 150 200 250 300 350Time (s)
Res
pons
e D
iffer
ence
(RU
)
1.28µM
40.8µM20.4µM10.2µM5.11µM2.55µM
Amine coupling Req
10
30
50
70
90
110
concentration DAF (µM)0 5 10 15 20 25 30 35 40 45
KA = 7.2x104± 2.4x103 M-1
Rmax =135 ± 2KD = 13.9µΜχ2 = 1.3
Figure 2
by guest on March 15, 2018
http://ww
w.jbc.org/
Dow
nloaded from

(a)
-100
-50
0
50
100
150
200
250
300
350
2200 2400 2600 2800 3000 3200 3400 3600
Time (s)
Res
pons
e D
iffer
ence
(RU
)
Ba at 7.5µMfB at 3.2µM
Bb at 5.0µMLigand:
CD55-Ig low RUCD55-Ig high RUsDAF
(b) (c)
350 400
Time (s)
39.5µM
1.2µM2.5µM4.9µM9.9µM
19.8µM
100 150 200 250 300 350-40
-200
2040
6080
100120
140
Res
pons
e D
iffer
ence
(RU
)
-100
-50
0
50
100
150
200
250
100 150 200 250
Time (s)
Res
pons
e D
iffer
ence
(RU
)
300
13.7µM
0.9µM1.7µM3.4µM6.9µM
27.4µM
FB flowing over CD55 CD55 flowing over fB
KA = 1.8x104± 0.8x103 M-1
Rmax =413 ± 13KD = 53µMχ2 = 0.8
KA = 3.0x104± 2.4x103 M-1
Rmax =140 ± 6KD = 33µMχ2 = 1.8
Figure 3
by guest on March 15, 2018
http://ww
w.jbc.org/
Dow
nloaded from

(a)R
espo
nse
Diff
eren
ce (R
U)
-40-20
020406080
100
Time
fB (11µM)EDTA
Bb1mM Mg2+
Bb (17µM)EDTA
fB1mM Mg2+
(b) Bb flowing over DAF in 1mM Mg2+
(c)
-100
-50
0
50
100
150
200
-500 -200 100 400 700 1000 1300
Res
pons
e D
iffer
ence
(RU
)
Time (s)
ka = 824 ± 5 M-1s-1
kd = 1.4x10-3 ± 4x10-6s-1
Rmax =155 ± 0.6KD = 1.7µMχ2 = 3.7
7.9µM
4.0µM2.0µM1.0µM
0.25µM0.5µM
vWFA flowing over DAF in 1mM Mg2+
1600 1900 2200 2500
7.2µM
4.5µM
2.8µM
1.8µM1.1µM
0.43µM0.27µM0.68µM
-200 100 400 700 1000 1300 1600 1900 2200 2500-40
-20
0
20
40
60
-500
Res
pons
e (R
U)
ka = 851 ± 1 M-1s-1
kd = 1.9x10-3 ± 1x10-6s-1
Rmax =106 ± 0.1KD = 2.2µMχ2 = 0.2
140
120
100
80
Time (s)
Figure 4
by guest on March 15, 2018
http://ww
w.jbc.org/
Dow
nloaded from
(a)
-150
-100
-50
0
50
100
150
200
250
300
350
0 200 400 600 800 1000 1200 1400Time (s)
Res
pons
e di
ffere
nce
(RU
)55µM27µM14µM
7µM3.4µM1.7µM
fB flowing over C3b in 1mM Mg2+
(b)
0
20
40
60
80
100
120
-2000 -1400 -800 -200 400 1000
Res
pons
e D
iff.e
renc
e(R
U)
1min
3mins
20mins
increasing off rate
Time (s)
association dissociation
(c)
-50
0
50
100
0
Res
pons
e D
iffer
ence
(RU
200 400 600 800 1000 1200
Time (s)
14µM
7µM
3.4µM
1.7µM
Figure 5
conformational change fit
200
)
150
1400
by guest on March 15, 2018
http://ww
w.jbc.org/
Dow
nloaded from

(a)
-1000
100200300400500600700
500 1000 1500 2000 2500 3000
Time
Res
pons
e D
iffer
ence fB
10mMEDTA
fB+fD10mMEDTA
Decayof fB
18000
20000
22000
24000
26000
28000
30000
32000
34000
4000 5000 6000 7000 8000 9000 10000 11000 12000 13000 14000
Time (s)
Res
pons
e (R
U)
CVFBb+ C3
fB+fD
C3
Cycle 1
Cycle 2Cycle 3
Cycle 4
(b)
(c)increasing [fD]
-2000
200400600800
1000120014001600
0 500 1000 1500 2000 2500
Res
pons
e D
iffer
ence
(RU
)
EDTADAF
(A) (B) (C)
fB/fD
D
E
D
E
fB/fD
fB
3000 3500
(D)
D
E
fB/fD
4000 4500
(E)
D
E
fB/fD
5000Time (s)
Figure 6
by guest on March 15, 2018
http://ww
w.jbc.org/
Dow
nloaded from
(d)
Res
pons
e D
iffer
ence
(RU
)
Time (s)
RU-alignment at fB/fD inject
-600-500-400-300-200-100 0 100 200 300 400-200
200400600800
1000120014001600
0
fB/fD
DAF(0.8µM)
EDTADAFonly
RU-alignment at end DAF inject
-400-300-200-100 0 100 200 300 400 500-600-400-200
200400600800
100012001400
-500
0
Res
pons
e D
iffer
ence
(RU
)Time (s)
fB/fD
DAF(0.8µM)
Figure 6
by guest on March 15, 2018
http://ww
w.jbc.org/
Dow
nloaded from

DAF flowing across CVF
(a)
-100
-80
-60
-40
-20
0
20
40
2060 2080 2100 2120 2140 2160 2180 2200 2220 2240
Res
pons
e D
iffer
ence
(RU
)
Time (s)
DAF (47µM)
Figure 7
(b)enzyme
formation enzyme
-100
0
100
200
300
400
500
500
Res
pons
e D
iffer
ence
(RU
)
DAF (15µM)
C3bBb
CVFBbDAF
bindingC3b
accelerated decay of CVFBb
decay
DAFdecay of C3bBb
1000 1500 2000 2500 3000 3500
Time (s)
by guest on March 15, 2018
http://ww
w.jbc.org/
Dow
nloaded from

LeaClaire L. Harris, Rachel J. M. Abbott, Richard A. Smith, B. Paul Morgan and Susan M.
pathway of complement and decay accelerating factor (CD55)Molecular dissection of interactions between components of the alternative
published online November 9, 2004J. Biol. Chem.
10.1074/jbc.M410179200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
by guest on March 15, 2018
http://ww
w.jbc.org/
Dow
nloaded from