minor variation prior approval (miv-pa)...
TRANSCRIPT

MINOR VARIATION – PRIOR APPROVAL
(MiV-PA)
MALAYSIAN VARIATION GUIDELINE FOR
PHARMACEUTICAL PRODUCTS
2013
Chai Che LeongSenior Assistant Director,
Variation Section,
Centre for Post Registration,
National Pharmaceutical Control Bureau,
Tel: 03-7801 8466, fax: 03-79567151,
Email: [email protected]
Visit us: http://www.bpfk.gov.my
1
NATIONAL PHARMACEUTICAL CONTROL BUREAU
MINISTRY OF HEALTH MALAYSIA
Mission: To be a world renowned regulatory authority for medicinal products and cosmetics.
Vision: To safeguard the nation's health through scientific excellence in the regulatory control of medicinal products
and cosmetics.

Malaysian Variation Guideline for Pharmaceutical Products
Minor Variation – Prior Approval (MiV-PA)
2
Scope:
pharmaceutical drug products and drug substance (change of specifications
and test procedure only)
Definition:
Variation to a registered pharmaceutical finished product in terms of
administrative data and/or changes with minimal/no significant impact on the
aspects of efficacy, quality, and safety.
Total number of MiV-PA:
36 variations, arranged according to this order: administrative, drug substance,
drug product (including excipients).

Malaysian Variation Guideline for Pharmaceutical Products
Minor Variation – Prior Approval (MiV-PA)
3
No. MiV-PA Variation Title
1 MiV-PA1Change of drug product name
2 MiV-PA2Change of product labeling (in accordance to country specific labeling requirement)
3 MiV-PA3 Change of patient information leaflet
4 MiV-PA4Replacement of the company or party responsible for batch release
5 MiV-PA9Change of the specification of drug substance
6 MiV-PA10Change of the test procedure of non-compendial drug substance
7 MiV-PA14Change of batch size of non-sterile drug product
8 MiV-PA15Reduction or removal of overage
9 MiV-PA16Qualitative or quantitative change of excipient
10 MiV-PA17Quantitative change in coating weight of tablets or weight and/or size of capsule shell for
immediate release oral dosage form
11 MiV-PA18Change of the colouring/flavouring agent of the product [addition, deletion or replacement
of colourant(s)/flavour(s)]
12 MiV-PA19Deletion of the solvent/diluent for the drug product
Number of variations covered in this presentation: 29

Malaysian Variation Guideline for Pharmaceutical Products
Minor Variation – Prior Approval (MiV-PA)
4
No. MiV-PA Variation Title
13 MiV-PA20Change of in-process controls applied during the manufacture of the drug product
(including tightening and addition of new in-process test)
14 MiV-PA21Minor change of the manufacturing process for non-sterile product.
15 MiV-PA22Change of specifications of an excipient
16 MiV-PA23Change of a test procedure for an excipient, including replacement of an approved test
procedure by a new test procedure
17 MiV-PA24Change in the source of empty hard capsule
18 MiV-PA25Change of release and shelf-life specifications of the drug product
19 MiV-PA26Change of imprints, bossing or other markings on the tablets or printing on capsules
including addition or change of inks used for product marking
20 MiV-PA27Change of dimensions and/or shape of tablets, capsules, suppositories or pessaries
without change in qualitative and quantitative composition and mean mass
21 MiV-PA28Change in the test procedure of the drug product (including replacement or addition of a
test procedure)
22 MiV-PA29Change in primary packaging material for non-sterile product
a) Qualitative and quantitative composition and/or
b) Type of container and/or
c) Inclusion of primary packaging material
23 MiV-PA30Replacement of a manufacturer for secondary packaging
24 MiV-PA31Change of pack size/fill volume and/or change of shape or dimension of container or
closure for non-sterile product

Malaysian Variation Guideline for Pharmaceutical Products
Minor Variation – Prior Approval (MiV-PA)
5
No. MiV-PA Variation Title
25 MiV-PA32Change of outer carton pack sizes for a drug product
26 MiV-PA33Change in any part of the (primary) packaging material not in contact with the finished
product formulation (such as colour of flip-off caps, colour code rings on ampoules,
change of needle shield (different plastic used)
27 MiV-PA34Addition or replacement of measuring device for oral liquid dosage forms and other
dosage form
28 MiV-PA35Reduction of shelf-life of the drug product
29 MiV-PA36Change of storage conditions of the drug product (Increasing from the current approved
storage condition)

Malaysian Variation Guideline for Pharmaceutical Products
Minor Variation – Prior Approval (MiV-PA)
6
• Remaining variations (drug substance) will be covered by API
section later.
• 13 MiV-PA are cross – referenced with either major variation or
notification.
• The field numbers quoted in the documentation part of the following
slides refer to QUEST 2 system.
• For Quest 3 products, please submit manually.
• For discussion purpose, each document will be quoted as D1, D2,
D3, D4 etc. Not to be confused with the field for product labelling
D1,D2 or D3 of the Quest system.
• The word ‘Change’ of the variation title refers to addition,
deletion/removal and/or replacement; unless otherwise specified.

Malaysian Variation Guideline for Pharmaceutical Products
Understanding the Quest 2 system.
7
• Variation to be submitted online for Quest 2 products.
Forms tray>Variation>Product Information
• Supporting documents for certain variations are displayed but other fields
remain open to applicant.
• Upload files that are relevant to the variation. If not relevant, just upload a
blank document stating “Not Applicable”.
• Advisable to always submit the relevant supporting documents at their
respective field as well to ensure data is overwritten successfully upon
approval.
• Always bear in mind that current Quest 2 system allows only one file
attachment at anytime except part E12.
• New file will overwrite old file upon approval. Therefore please combine the
existing file with new file into a single file (preferably pdf) if existing file is to be
retained. If the file size is too big, break it down into several files. Submit first
part at the intended field and the rest at part E12.

Malaysian Variation Guideline for Pharmaceutical Products
Understanding the Quest 2 system.
8
• For part E12, please do not submit those files that have already been saved in the
system previously. You have the option to exclude those files from being submitted at
front-end. If multiple files are uploaded into part E12, please mention in the remarks box
to officer the title or content of each file briefly. It saves our time to look for the right file.
• Whenever a variation application is to be submitted, please highlight the change in
the remarks to officer. Do not just mention “For marketing purpose” or “File is uploaded
for your review”.
• Status of the application is available at Out Tray.
Variation Submitted Not Reviewed: application is submitted and currently in variation
tray at back-end.
Under Assessment: already reviewed by assistant director and now pending review by
principal assistant director who is the head of variation section.
Approved or Rejected.

Malaysian Variation Guideline for Pharmaceutical Products
MiV-PA1 – Change of product name
9
MiV-PA1 Change of drug product name (A1 – apply at View/Edit Validation)
C1. There is no change to the product (formulation, release and shelf-life specifications,
manufacturing source and process) except for the product name change.
2. No confusion with another drug product either when spoken or written.
3. The new name does not (i) suggest greater safety or efficacy than supported by
clinical data (ii) imply a therapeutic use (iii) imply superiority over another similar
product and (iv) imply the presence of substance(s) not present in the product.
D1. Revised draft package insert and labeling incorporating the proposed variation.
(D1,D2,D3)
2. Updated Certificate of Pharmaceutical Product (CPP) (where applicable). (E4)
3. Official letter from product owner or product registration holder authorizing the
change of product name and committing to inform users of the relevant changes
(where applicable). (A1)
4. A declaration from the product registration holder that there is no other changes to
the product/label except for the drug product name change. (E12)
5. Trademark certificate (where applicable). (E12)

Malaysian Variation Guideline for Pharmaceutical Products
MiV-PA1 – Change of product name
10
D1: It must comply with the requirements as per Appendix 9 of current DRGD.
D3: Ensure name proposed at part A1 is same as name stated in letter of
authorization.
D3: Letter of authorization, please state current product name, proposed name
and product registration number.
D4: Change in the artwork design is allowed on labels but the content of the
product labelling should not be changed.
D5: Provide certificate of registration for the trademark ® or TM. For imported
products with shared product labelling, D5 may not be applicable.

Malaysian Variation Guideline for Pharmaceutical Products
MiV-PA2 – Change of product labeling
11
MiV-PA2 Change of product labeling (in accordance to country specific labeling
requirement)
Includes:
a) Change of the layout/artwork without altering meaning.
b) Addition/deletion/replacement of pictures, diagrams, bar code, logos and/or texts
that do not imply an unapproved indication.
c) Addition/strengthening of warnings, precautions, contraindications and/or adverse
events/effects to the approved product labeling.
d) Tightening of product’s target population.
e) Deletion of indication.
f) Change of distributor’s details.
C1. Product labeling refers to Package Insert (PI), unit carton label, inner label and/or
blister strips.
2. The change is not a MaV and does not contain promotional information. For major
change in product labeling, please refer to MaV-2.
D1. Current approved product labeling. (retrieved from the system)
2. Proposed product labeling, a clean and annotated version highlighting the changes
made. (D1,D2,D3)
3. Letter of declaration from the product registration holder stating that no other
changes on the label except for the intended change. (E12)
4. Relevant document/reference to support the changes (where applicable). (E12)

Malaysian Variation Guideline for Pharmaceutical Products
MiV-PA2 – Change of product labeling
12
Also includes:
•Editorial changes,
•Directive from Pharmacovigilance section for safety information,
•DCA directives and compulsory warnings
QR code which links to a website address (URL) is not allowed.
Removal of warning shall be submitted as MaV-2.
D2: If only the clean copy of the label is submitted, please submit at BOTH
column of “Amendment clearly marked” and “Latest amended label” so that old
label can be overwritten successfully.
D4: SmPC/approved PI from reference country or country of origin/CDS is
acceptable.

Malaysian Variation Guideline for Pharmaceutical Products
MiV-PA3 – Change of Patient Information Leaflet (PIL)
13
MiV-PA3 Change of patient information leaflet
(Processed by Pharmacovigilance section)
C1. Changes to the content (eg. Section A, C) has been approved in
the system.
D1. Proposed patient information leaflet, a clean and annotated
version highlighting the changes made. (E8,E12)

Malaysian Variation Guideline for Pharmaceutical Products
MiV-PA3 – Change of Patient Information Leaflet (PIL)
14
• Also known as Risalah Maklumat Ubat Untuk Pengguna (RiMUP) in Bahasa Melayu
(BM).
• Is compulsory for products which are self-administered by patients, including:
a. Scheduled poisons (category A),
b. Over the Counter products (category X),
c. Health supplements with high claims (disease risk reduction)
• For details, please refer to
i. Direktif Penguatkuasaan Keperluan Mengemukakan Risalah Maklumat Ubat
Untuk Pengguna (RiMUP) Bil.5 Tahun 2011 Bil(15) dlm BPFK/PPP/01/03 Jld 1
ii. Garispanduan Pelaksanaan Risalah Maklumat Ubat untuk Pengguna (RiMUP).
• The draft copy of the PIL in both English and BM shall be submitted for evaluation.
• Quest 2: online submission at part E12, state ‘Please refer to section E12’ at part
E8. Please inform the officer in-charge by email once submission is made.
• Quest 3: manual submission.
• Current officer in-charge:
Ms Rema Panickar,
Email: [email protected], Tel: 03-7883 5542

Malaysian Variation Guideline for Pharmaceutical Products
MiV-PA4 – Change of batch releaser
15
MiV-PA4 Replacement of the company or party responsible for batch release
(Propose at part E14)
C1. Only applicable for batch release.
2. The manufacturer of the drug product remains the same.
3. Method transfer from the currently approved to the proposed site or test
laboratory has been successfully completed.
D1. Revised drafts of the package insert and labeling incorporating the
proposed variation (where applicable). (D1,D2,D3)
2. Proof that the proposed site is appropriately authorized (accredited by
the authority) to be responsible for batch release such as a valid
certificate or CPP which covers the GMP certification. (E12)
3. Official letter from product owner authorizing the company/manufacturer
to be responsible for batch release (where applicable). (E12)
D2: GMP status of the proposed batch releaser must be certified by PIC/S.

Malaysian Variation Guideline for Pharmaceutical Products
MiV-PA9 – Change of specifications of drug substance
16
MiV-PA9 Change of the specification of drug substance
a) Specification limits are tightened
b) Addition of new test parameter and limits
C1. This is only applicable for drug substances which are non-compendial and generic
drug substances without European Pharmacopoeial Certificate of Suitability (CEP)
2. The change should not be the result of unexpected events arising during
manufacture or because of stability concerns.
3. Test procedures remain the same, or changes in the test procedure are minor.
4. For (b) - applicable to non-compendial method only.
5. Refer to MiV-PA13 if this change resulted in revision of CEP.
6. For widening of specification limits and deletion of test parameter and limits of drug
substance, please refer to MaV-7.
D(a) Specification limits are tightened
1. Technical justification for the change. (E12)
2. Comparative tabulated format of the currently approved and revised specification of
drug substance with changes highlighted. (E12 – tabulated differences, S4.1 -
clean copy)
3. Certificate of analysis and comparative batch analysis data of the drug substance
for all tests in the new specification for two pilot or production scale batches. (S4.4)
(b) Addition of new test parameter and limits
In addition to the above documents,
1. Description of any new analytical method (S4.2) and summary of the validation
data. (S4.3 & E12)

Malaysian Variation Guideline for Pharmaceutical Products
MiV-PA9 – Change of specifications of drug substance
17
• S4.1: Please make sure DS specs tally with CoA at S4.4. Drug product
manufacturer’s DS specs will be the main reference if they are different from
DS manufacturer’s specs.
• S4.4: CoA from each DS manufacturer (as per part S2.1) shall be combined
and uploaded in a single file. Update part S2.1 (DS manufacturer) if necessary.
Inform officer if S2.1 is not updated after last approval.
• S4.4: Name and address of DS manufacturer must be indicated clearly in
the CoA.
• S4.3: Due to technical error of the system, please upload DS validation
data at both S4.3 and E12.
• For update following a compendial change, please refer to MiV-N10.

Malaysian Variation Guideline for Pharmaceutical Products
MiV-PA10 – Change of test procedure of drug substance
18
MiV-PA10 Change of the test procedure of non-compendial drug substance
C1. Results of method validation show new test procedure to be at least equivalent to
the former procedure.
2. Refer to MiV-PA13 if this change resulted in revision of CEP.
D1. Description of the analytical methodology (S4.2), a summary of validation data
(S4.3 & E12), and comparative analytical results between the currently approved
and proposed test (where applicable) (S4.4).
2. Specification of the drug substance. (S4.1)

Malaysian Variation Guideline for Pharmaceutical Products
MiV-PA14 – Change of batch size of non-sterile drug product
19
MiV-PA14 Change of batch size of non-sterile drug product
C1. The change does not affect consistency of production.
2. Process validation scheme and/or report is available or validation of the manufacturing process
has been successfully carried out according to protocol with at least three batches at the
proposed new batch size in accordance with the ASEAN Guideline on Submission of
Manufacturing Process Validation Data For Drug Registration.
3. Release and end-of-shelf-life specifications of drug product remain unchanged.
4. This is applicable to change of batch size up to 10-fold compared to the currently registered batch
size.
5. For change of batch size for sterile products, please refer to MaV-8 and for change of batch size
more than 10-fold compared to the currently registered batch size, please refer MaV-9.
D1. Comparative tabulated format of proposed and current batch manufacturing formula. (B1.2)
2. Validation scheme and/or report of the manufacturing process as per ASEAN Guideline on
Submission of Manufacturing Process Validation Data for Drug Registration appropriate to the
proposed batch size should be provided upon submission. (P3.4)
3. Revised ACTD Section P3.1-3.4 (where applicable).
4. Release and shelf-life specifications of the drug product. (P5.1)
5. Certificate of analysis (E11) and batch analysis data (in a comparative table) of drug production a
minimum of one production batch according to currently approved and proposed batch sizes and
a letter of undertaking to submit batch data on the next full production batch. (P5.4)
6. Stability data as per ASEAN Guideline On Stability Study Of Drug Product and report if any
results fall outside shelf-life specifications (with proposed action). (P8)

Malaysian Variation Guideline for Pharmaceutical Products
MiV-PA14 – Change of batch size of non-sterile drug product
20
• Due to technical error of the system, please do not submit supporting
documents at part B1.1 but submit at their respective field.
• B1.2: Batch quantity for each ingredient should be stated clearly for all batch
sizes.
• P3.4, P5.4 and P8:
Please ensure DP specs tally with part P5.1. Combine all the relevant files if
necessary (if there are multiple batch sizes).
• P3.4 and P8: Indicate objective, the batch numbers and formula in the report
clearly.

Malaysian Variation Guideline for Pharmaceutical Products
MiV-PA15 – Reduction or removal of overage
21
MiV-PA15 Reduction or removal of overage
C1. Changes of previously approved manufacturing overages of drug substance only.
2. Release and end-of-shelf-life specifications of drug product remain unchanged.
D1. Justification for the change. (E12)
2. Comparative tabulated format of currently approved and proposed batch
manufacturing formula. (B1.2)
3. Certificate of analysis for two batches of the finished product. (E11)
4. Stability data as per ASEAN Guideline On Stability Study Of Drug Product and
report if any results fall outside shelf-life specifications (with proposed action). (P8)
• Increase in overage is considered as a new product registration.

Malaysian Variation Guideline for Pharmaceutical Products
MiV-PA16 – Qualitative & Quantitative Change of Excipient
22
MiV-PA16 Qualitative and/or quantitative change of excipient
a) For immediate release oral dosage forms
(as per Level 1, Part III Components and Composition, SUPAC guideline)
b) For other non-critical dosage forms eg. oral liquid, external preparation.
(A2.1 of View/Edit Validation)
C1. Replacement of an excipient with a comparable excipient of the same functional
characteristics.
2. The dissolution profile of the proposed product is comparable to that of the current
approved product.
3. Process validation scheme and/or report is available or validation of the
manufacturing process has been successfully carried out according to protocol with
at least three batches of the proposed product formula in accordance with the
ASEAN Guideline on Submission of Manufacturing Process Validation Data For
Drug Registration.
4. Release and shelf-life specifications of the drug product remain unchanged.
5. For qualitative or quantitative change of excipient for immediate release(Level 2
and 3 change as per SUPAC) and modified release oral dosage forms and other
critical dosage forms, please refer to MaV-11.
6. For quantitative change in coating weight of tablets or weight and/or size of capsule
shell for immediate release oral solid dosage form, please refer MiV-PA17.

Malaysian Variation Guideline for Pharmaceutical Products
23
D1. Revised drafts of the package insert and labeling incorporating the proposed variation (where
applicable). (D1,D2,D3)
2. A declaration that the new excipient does not interfere with the drug product release and shelf-life
specifications test method (where applicable). (E12)
3. Justification for the change must be given by appropriate development of pharmaceutics. (E12)
4. Comparative tabulated format of the current and revised product formulation with calculated
changes highlighted (please state changes in the percentage of the proposed excipient out of the
total target dosage form weight, where applicable). (E12)
5. Comparative dissolution profile data of at least one representative pilot/production batch of the
drug product between the currently approved and proposed solid dosage forms formulation. (E12)
6. Revised batch manufacturing formula. (B1.2)
7. Validation scheme and/or report of the manufacturing process as per ASEAN Guideline on
Submission of Manufacturing Process Validation Data for Drug Registration appropriate to the
proposed change in product formula should be provided upon submission (where applicable).
(P3.4)
8. Revised ACTD Section P3.1-3.4 (where applicable).
9. Specifications of the proposed excipient. (P4.1)
10. For proposed excipients made of ruminants source, Transmitting Animal Spongiform
Encephalopathy (TSE)-free certificate or Bovine Spongiform Encephalopathy (BSE)-free cert
issued from relevant veterinary authority of the issuing country (where applicable). (E12)
11. Release and shelf-life specifications. (P5.1)
12. Certificate of analysis (E11) and batch analysis data (in a comparative tabulated format) of drug
product of at least two production (or one production batch and two pilot batches) according to
currently approved and proposed product formula. (P5.4)
13. Stability data as per ASEAN Guideline On Stability Study Of Drug Product and report if any
results fall outside shelf-life specifications (with proposed action). (P8)
14. Justification for not submitting a new bioequivalence study according to the ASEAN Guidelines
For The Conduct of Bioavailability and Bioequivalence Studies. (E12)
MiV-PA16 – Qualitative & Quantitative Change of Excipient

Malaysian Variation Guideline for Pharmaceutical Products
24
No. Ingredient Type/Function Quantity
Unit Per
Tablet
(Registered)
Quantity Unit
Per Tablet
(Proposed)
% Difference /
registered quantity
Absolute
additive
effect (%)
1. Folic Acid Active Ingredient 5mg 5mg nil
2. Lactose
Monohydrate
Excipient:
Bulking agent
70mg 72mg |72 – 70| x100%
105
= 1.90%
1.90
3. Corn Starch Excipient:
Disintegrant
30mg 29mg |29 – 30| x100%
105
= 0.95%
0.95
4. Magnesium
Stearate
Excipient:
Lubricant
- 2mg |2 – 0| x100%
105
= 1.90%
1.90
Total weight 105mg 108mg 4.75%
(Level 1)
Example of SUPAC Level 1 calculation:
• Total absolute additive effect should be NMT5%.
MiV-PA16 – Qualitative & Quantitative Change of Excipient

Malaysian Variation Guideline for Pharmaceutical Products
25
Indicate function of the excipient clearly.
C4: Quantitative change may result in change of DP specs, e.g. Weight of the
tablet. Revised DP specs have to be submitted as per D11.
Comparative dissolution profile as per ASEAN BA/BE Guideline
Innovator – 1pH
Generic – 3 pH
Minimum 3 time points,
f2 value between 50-100 suggests similarity between the two dissolution
profiles.
Acceptable if more than 85% of the drug dissolved within 15 minutes.
MiV-PA16 – Qualitative & Quantitative Change of Excipient

Malaysian Variation Guideline for Pharmaceutical Products
MiV-PA17 – Quantitative change in coating weight of tablets and/or size of capsule
26
MiV-PA17 Quantitative change in coating weight of tablets or weight and/or size of capsule shell for
immediate release oral solid dosage form
(A2.1 of View/Edit Validation)
C1. The dissolution profile of the proposed product is comparable to that of the current approved
product.
2. The product release and end-of-shelf-life specifications of the drug product remain unchanged
except for the weight and/or size.
3. For quantitative change in coating weight of tablets or weight and/or size of capsule shell for
modified release oral solid dosage forms please refer to MaV-12.
D1. Revised draft of product label incorporating the proposed change (where applicable). (D1,D2,D3)
2. A declaration from product registration holder that the change does not interfere with the drug
product release and shelf-life specifications test method. (E12)
3. Comparative tabulated format of current and proposed product and batch manufacturing formula.
(B1.2)
4. Comparative dissolution profile data of at least one pilot/production batch of the drug product
between the currently approved and proposed composition. (E12)
5. Revised release and shelf-life specifications of the drug product. (P5.1)
6. Certificate of analysis (E11) and batch analysis data (P5.4) for two production/pilot scale batches
of the drug product.
7. Stability data as per ASEAN Guideline On Stability Study Of Drug Product and report if any
results fall outside shelf-life specifications (with proposed action). Except for the change in weight
and/or size of capsule shell, a letter of declaration from the applicant that the relevant stability
studies of the drug product in accordance with ASEAN Guideline on Stability Study of Drug
Product have been started will suffice. (P8)
8. Justification for not submitting a new bioequivalence study according to the ASEAN Guidelines
For The Conduct of Bioavailability and Bioequivalence Studies (where applicable). (E12)

Malaysian Variation Guideline for Pharmaceutical Products
MiV-PA17 – Quantitative change in coating weight of tablets and/or size of capsule
27
• MiV-PA16 is applicable for the excipients of the core tablets and other non-
sterile immediate release dosage forms.
• MiV-PA17 is applicable for the excipients used in the coat of the tablets.

Malaysian Variation Guideline for Pharmaceutical Products
MiV-PA18 – Change of the colouring/flavouring agent
28
MiV-PA18 Change of the colouring/flavouring agent of the product [addition, deletion or replacement of
colourant(s)/flavour(s)]
(A2.1 of View/Edit Validation)
C1. Same functional characteristics, no change in dissolution profile for solid oral dosage forms.
2. The proposed colouring/flavouring agents must not have been rejected for pharmaceutical use.
3. The release and shelf-life specifications of the drug product remain unchanged except for the
change in colour/flavour.
D1. Revised drafts of the package insert and labeling incorporating the proposed variation (where
applicable). (D1,D2,D3)
2. A declaration from product registration holder that the change does not interfere with the drug
product release and shelf-life specifications test method. (E12)
3. A letter of commitment from product owner or product registration holder to inform users of the
relevant change (where applicable). (E12)
4. Revised product formulation and batch manufacturing formula. (B1.2)
5. Qualitative and quantitative information of the current and proposed colouring/flavouring agent in
a comparative table. (E12)
6. For proposed excipients made of ruminants source, Transmitting Animal Spongiform
Encephalopathy (TSE)-free certificate or Bovine Spongiform Encephalopathy (BSE)-free
certificate issued from relevant veterinary authority of the issuing country (where applicable).
(E12)
7. Revised release and shelf-life specifications of the drug product. (P5.1)
8. Certificate of analysis (E11) and batch analysis data (P5.4) for two production/pilot scale batches
of the drug product.
9. Stability data as per ASEAN Guideline On Stability Study Of Drug Product and report if any
results fall outside shelf-life specifications (with proposed action). (P8)

Malaysian Variation Guideline for Pharmaceutical Products
MiV-PA18 – Change of the colouring/flavouring agent
29
• No PV data is required, making it different from MiV-PA16 or MiV-PA17.

Malaysian Variation Guideline for Pharmaceutical Products
MiV-PA19 – Deletion of the solvent/diluent
30
MiV-PA19 Deletion of the solvent/diluent for the drug product
C1. The proposed change does not result in any change in the dosage form,
regimen, indication, method of administration of the product.
D1. Revised drafts of the package insert and labeling incorporating the
proposed variation (where applicable). (D1,D2,D3)
2. Justification for the deletion of the solvent/diluent, including a statement
regarding alternative means to obtain the solvent/diluent. (E12)
3. Amended relevant ACTD Section P (where applicable).

Malaysian Variation Guideline for Pharmaceutical Products
MiV-PA19 – Deletion of the solvent/diluent
31
Addition of solvent/diluent is a MaV.
Applicable to the following scenario:
• Lyophilized powder vial packed together with solvent/diluent.
• Updating of package insert by removing one of the solvents/diluents.
Example:
Currently in PI:
Product A lyophilized powder should be reconstituted with water for injection,
normal saline or bacteriostatic water for injection.
Proposed:
Product A lyophilized powder should be reconstituted with water for injection,
normal saline or bacteriostatic water for injection.

Malaysian Variation Guideline for Pharmaceutical Products
MiV-PA20 – Change of in-process controls
32
MiV-PA20 Change of in-process controls applied during the manufacture of the
drug product (including tightening and addition of new in-process test)
C1. Release and shelf-life specifications of drug product remain unchanged.
2. The change does not result from unexpected events arising during
manufacture e.g. new unqualified impurity; change in total impurity limits.
3. Any new test method does not concern a novel non-standard technique
or a standard technique used in a novel way.
D1. Comparative tabulated format of currently approved and proposed in-
process controls. (P3.3)
2. A description of the analytical methodology (E9) and summary of
validation data (E10) must be provided for all new analytical methods
(where applicable).
3. Revised in-process specifications together with justification and relevant
process validation data. (P3.3)
4. Certificate of analysis (E11) and comparative batch analysis data (P5.4)
of drug product of at least two production/pilot batches.

Malaysian Variation Guideline for Pharmaceutical Products
MiV-PA21 – Minor change of the manufacturing process
33
MiV-PA21 Minor change of the manufacturing process for non-sterile product
C1. The same currently approved manufacturing site.
2. The overall manufacturing principle remains the same.
3. The change does not cause negative impact on the quality, safety and efficacy of the drug product.
4. The dissolution profile of the proposed product is comparable to that of the current approved product.
5. Release and end-of-shelf-life specifications of drug product remain unchanged.
6. For major change in the manufacturing process for drug product, please refer to MaV-10.
D1. For solid oral dosage forms, comparative dissolution profile data of at least one representative production
batch of the drug product between the currently approved and proposed solid oral dosage forms
formulation. (E12)
2. Description of the new manufacturing process and technical justification for the change. (P2.4, P3.2,
P3.2.1)
3. Comparative tabulated format of present and proposed process with changes highlighted. (E12)
4. For semi solid and suspension products, validation scheme and/or report of the manufacturing process as
per ASEAN Guideline on Submission of Manufacturing Process Validation Data for Drug Registration
should be provided upon submission. (P3.4)
5. Copy of currently approved release and shelf-life specifications. Or, alternately, copy of revised release
and shelf-life specifications that supports that the new process must lead to an identical or better product
regarding all aspects of quality, safety and efficacy. (P5.1)
6. Certificate of analysis (E11) and batch analysis data (P5.4) (in a comparative tabulated format) of drug
product on a minimum of one batch manufactured to both the currently approved and the proposed
process; batch data on the next two full production batches should be made available upon request.
7. A declaration from the product registration holder that the relevant stability studies of the drug product in
accordance with the ASEAN Guideline on Stability Study of Drug Product have been started and that the
relevant stability studies will be finalized; data should be provided only if outside specification (with
proposed action). (P8)
8. Justification for not submitting a new bioequivalence study according to the current Bioavailability and
Bioequivalence guidance (where applicable). (E12)

Malaysian Variation Guideline for Pharmaceutical Products
MiV-PA21 – Minor change of the manufacturing process
34
• Overall manufacturing principle remains the same.
• Please declare if it also involves in equipment change and justify how the
manufacturing principle remains unchanged.
• D4: PV data is not required for oral solid dosage forms but for semi solid and
suspension products. However, CDP is required as per D1.
• Declare site where such process takes place and batch size affected.

Malaysian Variation Guideline for Pharmaceutical Products
MiV-PA22 – Change of specifications of an excipient
35
MiV-PA22 Change of specifications of an excipient
a) Specification limits are tightened
b) Addition of new test parameter and limits
C1. Release and end-of-shelf-life specifications of drug product remain unchanged.
2. The change should not be the result of unexpected events arising during
manufacture or because of stability concerns.
3. Applicable to non compendial excipients. For compendial excipients, please refer to
MiV-N10.
D1. Description of new method (P4.2) and summary of analytical validation (P4.3)
(applicable for addition of new parameter).
2. Comparative tabulated format of the current and revised specification of the
excipient with changes highlighted. (P4.1)
3. Batch analysis data of the excipient for all tests in the new specification. (E12)

Malaysian Variation Guideline for Pharmaceutical Products
MiV-PA23 – Change of test procedure for an excipient
36
MiV-PA23 Change of a test procedure for an excipient, including replacement of
an approved test procedure by a new test procedure
C1. Appropriate method validation studies have been performed in
accordance with the ASEAN Guidelines For Validation of Analytical
Procedures.
2. Results of method validation show new test procedure to be at least
equivalent to the former procedure.
3. There have been no changes of the total impurity limits.
4. Only applicable to the currently approved test parameters.
5. No new unqualified impurities are detected.
6. This applies for non-compendial excipient.
D1. Description of the analytical methodology with a comparative tabulation
of the changes. (P4.2)
2. For quantitative test change, comparative analytical validation results
showing that the current and proposed tests are equivalent. (P4.3)

Malaysian Variation Guideline for Pharmaceutical Products
MiV-PA24 – Change in the source of empty hard capsule
37
MiV-PA24 Change in the source of empty hard capsule
C1. From TSE-risk material to vegetable-sourced or synthetic empty hard capsules or
vice versa.
2. No change in the formulation and manufacturing process of drug product.
3. Not applicable to change from hard capsule to soft gel.
4. Excipient and finished product release and end of shelf-life specifications remain
unchanged.
D1. A letter of declaration from the manufacturer or the product registration holder
stating that the material is purely of vegetable, animal or synthetic origin. (E12)
2. Technical specifications and composition of the empty hard capsule of the new
source. (E12)
3. For empty hard capsule made of ruminants source, Transmitting Animal Spongiform
Encephalopathy (TSE)-free certificate or Bovine Spongiform Encephalopathy
(BSE)-free cert issued by a competent authority of the issuing country. (E12)
4. Comparative dissolution profile data of one batch representative of pilot/production
batch of the drug product using hard capsule between the two sources (where
applicable). (E12)
5. Certificate of analysis of the empty hard capsule of the proposed new source. (A3.2
& E12)
6. Certificate of analysis (E11) and batch analysis data (P5.4) for two production/pilot
scale batches of the drug product.
7. Stability data as per ASEAN Guideline On Stability Study Of Drug Product and
report if any results fall outside shelf-life specifications (with proposed action). (P8)

Malaysian Variation Guideline for Pharmaceutical Products
MiV-PA24 – Change in the source of empty hard capsule
38
Important!
Please also submit revised drafts of package insert and labeling at D1, D2 or
D3.
C3: Change of hard capsule to softgel is considered as change in the dosage
form which leads to a new product registration.

Malaysian Variation Guideline for Pharmaceutical Products
MiV-PA25 – Change of specifications of drug product
39
MiV-PA25 Change of release and shelf-life specifications of the drug product
a) Specification limits are tightened
b) Addition of new test parameter and limits
C1. Applicable to non-compendial method.
2. The change should not be the result of unexpected events arising during
manufacture or because of stability concerns.
3. The test methods remain the same or changes in the test methods are minor.
4. If there are changes to the test procedure, MiV-PA28 is also applicable.
5. For widening of specification limits and deletion of test parameter and limits of drug
product, please refer to MaV-7.
D(a) Specification limits are tightened
1. Technical justification for the change. (E12)
2. Comparative tabulated format of the current and revised release and shelf-life
specifications of the drug product with changes highlighted. (P5.1)
3. Certificate of analysis (E11) and comparative batch analysis data (P5.4) of the drug
product for all tests in the new specification of at least two batches.
(b) Addition of new test parameter and limits
In addition to the above documents:
1. Description of any new method (E9 & P5.2) and summary of analytical validation
data (E10 & P5.3) for non-compendial method.
2. Stability data as per ASEAN Guideline On Stability Study Of Drug Product and
report if any results fall outside shelf-life specifications (with proposed action).
(where applicable). (P8)

Malaysian Variation Guideline for Pharmaceutical Products
MiV-PA26 – Change of imprints, bossing or markings on tablets or printing on capsule
40
MiV-PA26 Change of imprints, bossing or other markings on tablets or printing on capsules including
addition or change of inks used for product marking
C(a) Except score/break-line
1. New markings do not cause confusion with other registered products.
2. Any ink proposed must comply to relevant pharmaceutical legislation or of food grade and not a
listed banned substance.
3. Release and shelf-life specifications of the drug product remain unchanged except for
appearance.
(b) On score/break-line
In addition to the above conditions,
4. Score/break-line is not meant for cosmetic purpose.
5. Applicable to addition or removal of score/break-line.
D(a) Except score/break-line
1. Revised drafts of the package insert and labeling incorporating the proposed variation (where
applicable). (D1,D2,D3)
2. A letter of commitment from product owner or product registration holder to inform users of the
relevant change (where applicable). (E12)
3. Details and specifications of the proposed new inks (where applicable). (E12)
4. Detailed drawing or written description of the current and proposed imprint/bossing/markings.
(E12)
5. Certificate of analysis of ink/printing material (pharmaceutical grade and of
food grade) (where applicable). (E12)
6. Release and shelf-life specifications of the drug product with the new product description. (P5.1)
(b) On score/break-line
In addition to the above documents,
7. Justification for the change (i.e. change in dosing regimen). (E12)
8. Data on test of uniformity of the subdivided parts of the tablets at release should be submitted.
(E11 & P5.4)
9. Certificate of analysis (E11) and batch analysis data (P5.4) of two production/pilot scale batches.

Malaysian Variation Guideline for Pharmaceutical Products
MiV-PA26 – Change of imprints, bossing or markings on tablets or printing on capsule
41
• D8: Depending on the weight and ratio of drug substance in a dosage unit,
uniformity test is either weight variation or uniformity of content. Test
methods and acceptance criteria are described in lead pharmacopoeia.
• For uncoated tablets, film coated tablets and hard capsules containing 25
mg or more of a drug substance comprising 25% or more by weight of the
dosage unit, weight variation on subdivided parts should be conducted. If the
weight of drug substance is less than 25 mg or the ratio is less than 25%,
content uniformity shall be conducted.
• Otherwise, content uniformity is applicable for other forms of tablets and
capsules.
• Please refer pharmacopoeia for further details.

Malaysian Variation Guideline for Pharmaceutical Products
MiV-PA27 – Change of dimensions and/or shape of dosage forms
42
MiV-PA27 Change of dimensions and/or shape of tablets, capsules, suppositories or pessaries
without change in qualitative and quantitative composition and mean mass
a) Immediate release oral solid dosage form, suppositories and pessaries
b) Other than immediate release oral solid dosage forms, suppositories and pessaries.
C1. If appropriate, the dissolution profile of the proposed product is comparable to that of the
current approved product.
2. Release and shelf-life specifications of the drug product remain unchanged except for
dimension and/or shape.
D(a) Immediate release oral solid dosage form, suppositories and pessaries
1. Revised drafts of the package insert and labeling incorporating the proposed variation
(where applicable). (D1,D2,D3)
2. Detailed drawing or written description of the current and proposed appearance. (E12)
3. Comparative dissolution data on at least one pilot/production batch of the currently
approved and proposed dimensions. (E12)
4. Data on test of uniformity of the subdivided parts of tablets at release as conformed to
compendial requirement should be submitted (only applicable for drug product with
score/break-line). (E11 & P5.4)
5. Release and shelf-life specifications of the drug product. (P5.1)
(b) Other than immediate release oral solid dosage forms, suppositories and pessaries
In addition to the above condition,
6. Justification for not submitting a new bioequivalence study according to the ASEAN
Guidelines For The Conduct of Bioavailability and Bioequivalence Studies (where
applicable). (E12)

Malaysian Variation Guideline for Pharmaceutical Products
MiV-PA28 – Change in the test procedure of drug product
43
MiV-PA28 Change in the test procedure of the drug product (including
replacement or addition of a test procedure)
C1. Drug product specifications are not adversely affected unless the
specifications are tightened.
2. Results of method verification/validation show new test procedure to be
at least equivalent to the former procedure.
3. The change should not be the result of unexpected events arising during
manufacture or because of stability concerns.
D1. Justification for the proposed change. (E12)
2. Comparative tabulated format of the currently approved and proposed
release and shelf-life specifications of the drug product. (P5.1)
3. Description of the analytical methodology. (P5.2 & E9)
4. Appropriate verification/validation data and comparative analytical results
between the currently approved and proposed test. (P5.3 & E10)
5. Certificate of analysis and batch analysis data of the finished product of
two production batches when made available. (E11 & P5.4)

Malaysian Variation Guideline for Pharmaceutical Products
MiV-PA29 – Change in primary packaging material for non-sterile product
44
MiV-PA29 Change in primary packaging material for non-sterile product (Apply at part C)
a) Qualitative and quantitative composition and/or
b) Type of container and/or
c) Inclusion of primary packaging material
C1. The proposed packaging material must be at least equivalent to or better than the
approved material in respect of its relevant properties.
2. The change includes the same packaging type (for example from blister to blister,
from transparent glass bottle to amber glass bottle).
3. Release and end-of-shelf-life specifications of drug product remain unchanged.
4. For change in the primary packaging material for sterile drug product, please refer
to MaV-13.
D1. Revised drafts of the package insert incorporating the proposed variation (where
applicable). (D1,D2,D3)
2. Justification for the change in packaging material and appropriate scientific studies
on the new packaging. (E12)
3. For semi-solid and liquid dosage forms, proof must be provided that no interaction
between the content and the packaging material occurs (e.g. no migration of
components of the proposed material into the content and no loss of components of
the product into the pack). (E12)
4. Comparative tabulated format of the currently approved and proposed
specifications of the primary packaging material (where applicable). (P7)
5. Stability data as per ASEAN Guideline On Stability Study Of Drug Product and
report if any results fall outside shelf-life specifications (with proposed action). (P8)

Malaysian Variation Guideline for Pharmaceutical Products
MiV-PA29 – Change in primary packaging material for non-sterile product
45
• Example of blister to blister change:
Alu/PVC blister to Alu/Alu blister.
• Please also retain current approved pack sizes in the proposed data. Remove
any pack size that is no longer marketed.
• Inform officer if data is not updated upon approval. Please retrieve the variation
approval record from your end and email to officer.
• The “Container Type Description” part must be filled in with details of the
primary packaging material.
• Combine stability report of the drug product packed in new packaging material
with current approved stability in a single file.
• Please submit supporting document at their respective field as well.
Malaysian Variation Guideline for Pharmaceutical Products
MiV-PA30 – Replacement of a secondary packer
46
MiV-
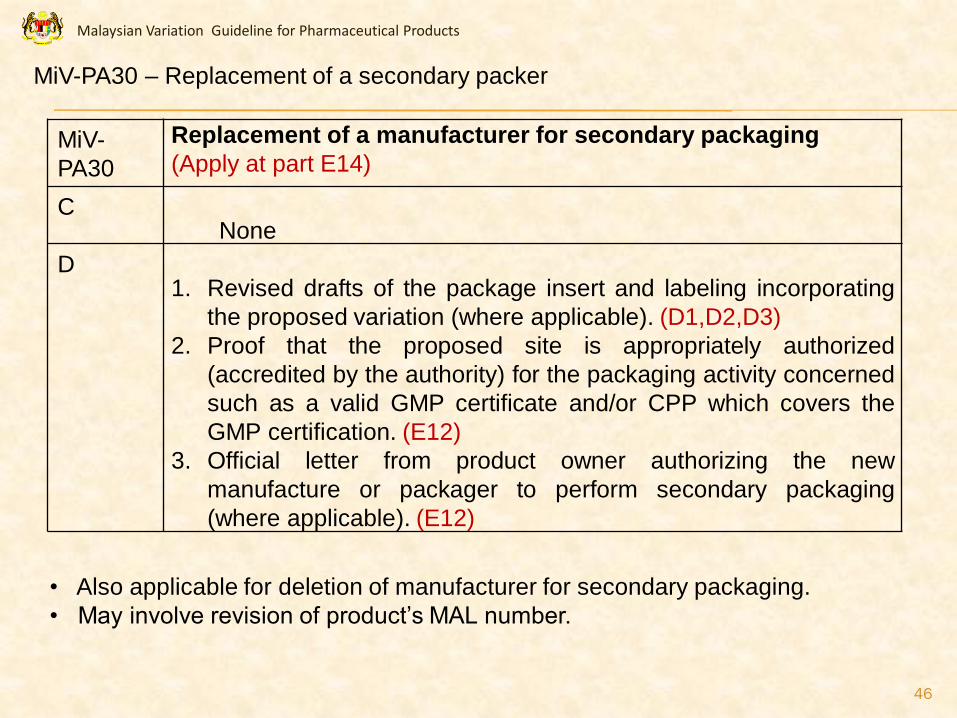
PA30
Replacement of a manufacturer for secondary packaging
(Apply at part E14)
CNone
D1. Revised drafts of the package insert and labeling incorporating
the proposed variation (where applicable). (D1,D2,D3)
2. Proof that the proposed site is appropriately authorized
(accredited by the authority) for the packaging activity concerned
such as a valid GMP certificate and/or CPP which covers the
GMP certification. (E12)
3. Official letter from product owner authorizing the new
manufacture or packager to perform secondary packaging
(where applicable). (E12)
• Also applicable for deletion of manufacturer for secondary packaging.
• May involve revision of product’s MAL number.

Malaysian Variation Guideline for Pharmaceutical Products
MiV-PA31 – Change of pack size/fill volume or shape or dimension of container
47
MiV-PA31 Change of pack size/fill volume and/or change of shape or dimension
of container or closure for non-sterile product (Apply at part C)
C1. The change only concerns the same packaging type and material.
2. The new size is consistent with the dosage regimen and duration of use
as approved in the package insert.
3. Change in the dimension of the primary packaging material (where
applicable).
4. Release and shelf-life specifications of the drug product remain
unchanged.
5. For change of pack size/fill volume and/or change of shape or dimension
of container or closure for sterile solid and liquid drug product, please
refer to MaV-14.
D1. Revised drafts of the package insert and labeling incorporating the
proposed variation (where applicable). (D1,D2,D3)
2. Justification for the proposed pack size. (E12)
3. A declaration from the product registration holder that the relevant
stability studies of the drug product in accordance with the ASEAN
Guideline on Stability Study of Drug Product have been started and that
the relevant stability studies will be finalized; data should be provided
only if outside specification (with proposed action). (E12)

Malaysian Variation Guideline for Pharmaceutical Products
MiV-PA31 – Change of pack size/fill volume or shape or dimension of container
48
• Applicable for oral solid dosage forms, liquids, semi-solid, topical preparations.
• Example:
Current approved pack size:
10 capsules packed in a Alu/PVC blister, 2x10’s/box, 3x10’s/box, 10x10’s/box
Proposed pack size:
10 capsules packed in a Alu/PVC blister, 2x10’s/box, 3x10’s/box, 10x10’s/box
30 capsules packed in a Alu/PVC blister, 1x30’s/box, 4x30’s/box
• D3 for the proposed pack size shall be ‘where applicable’ provided that current
approved pack size has already been supported with established stability data.

Malaysian Variation Guideline for Pharmaceutical Products
MiV-PA32 – Change of outer carton pack sizes
49
MiV-PA32 Change of outer carton pack sizes for a drug product
C1. Primary packaging materials remain unchanged.
2. No other changes except for the change of outer carton pack sizes for a
drug product.
3. The remaining pack sizes are adequate to accommodate the dosing
regimen as per the approved product labeling.
D1. Revised drafts of the package insert and labeling incorporating the
proposed variation (where applicable). (D1,D2,D3)
2. Letter of declaration from the product registration holder stating that no
other changes except for the change of outer carton pack sizes for a
drug product. (E12)

Malaysian Variation Guideline for Pharmaceutical Products
MiV-PA32 – Change of outer carton pack sizes
50
Covers change in the number of units packed in an outer carton.
Example:
Current approved pack sizes:
5 vials of 1mL in a box,
12 vials of 1mL in a box.
Proposed pack sizes:
5 vials of 1mL in a box,
10 vials of 1mL in a box.

Malaysian Variation Guideline for Pharmaceutical Products
MiV-PA33 – Change in any part of the primary packaging not in contact with
finished product formulation
51
MiV-PA33 Change in any part of the (primary)packaging material not in contact
with the finished product formulation such as colour of flip-off caps,
colour code rings on ampoules, change of needle shield (different
plastic used)
C1. The change does not concern a part of the packaging material, which
affects the delivery, use, safety or stability of the finished product.
D
1. Amendment of the relevant section(s) of the dossier (presented in the
ACTD format), including revised product labeling as appropriate.

Malaysian Variation Guideline for Pharmaceutical Products
MiV-PA34 – Addition or replacement of measuring device
52
MiV-PA34 Addition or replacement of measuring device for oral liquid dosage
forms and other dosage forms
C1. The size and where applicable, the accuracy of the proposed measuring
device must be compatible with the approved posology.
2. The new device is compatible with the drug product.
D1. Revised draft of the package insert and labeling incorporating the
proposed variation (where applicable). (D1,D2,D3)
2. Description of the device (including a drawing; where applicable). (E12)
3. The composition of the device material. Where applicable the materials
should comply with the pharmacopoeia. (E12)
4. Justification that size and accuracy of the device are adequate for the
posology as approved in the product labeling. (E12)

Malaysian Variation Guideline for Pharmaceutical Products
MiV-PA35 – Reduction of shelf-life of drug product
53
MiV-PA35 Reduction of shelf-life of the drug product
a) As a package for sale and/or
b) After first opening and/or
c) After dilution/reconstitution
C1. For (a) & (b) - The studies must show conformance to the currently approved shelf-
life specification.
2. For (c) – The studies must show conformance to the currently approved shelf-life
specification for the reconstituted product.
3. For extension of shelf-life, please refer to MaV-16.
D1. Revised drafts of the package insert and labeling incorporating the proposed
variation (where applicable). (D1,D2,D3)
2. Justification letter for the change of shelf-life of the drug product (where applicable).
(E12)
3. A letter of commitment from product owner or product registration holder to inform
users of the relevant change (where applicable). (E12)
4. Results of appropriate real time stability studies covering the duration of proposed
shelf-life of at least two pilot/production scale batches of the product in the
authorized packaging material
a) as a package for sale and/or
b) after first opening and/or
c) after the dilution/reconstitution
in accordance with the ASEAN Guidelines on Stability Study of Drug
Product; results of appropriate microbiological testing should be included
(where appropriate). (P8)

Malaysian Variation Guideline for Pharmaceutical Products
MiV-PA35 – Reduction of shelf-life of drug product
54
• Apply at part A16.
• Reduction of shelf life could be due to:
Degradation of the product, making it separated from MaV.
Company’s strategy to standardize the shelf life.
• Depending on the case, full real time stability data covering the proposed
shelf life is required.
• Stability report as per Annex 5.2 of ASEAN Stability Guideline should be
provided.

Malaysian Variation Guideline for Pharmaceutical Products
MiV-PA36 – Increasing of storage condition
55
MiV-PA36 Change of storage conditions of the drug product (Increasing from the
current approved storage condition)
a) As a package for sale and/or
b) After first opening and/or
c) After dilution/reconstitution
C1. For (a) & (b) - The studies must show conformance to the currently
approved shelf-life specification.
2. For (c) – The studies must show conformance to the currently approved
shelf-life specification for the reconstituted product.
3. For change of storage condition (lowering from the current approved
storage condition), please refer to MaV-17.
D1. Revised drafts of the package insert and labeling incorporating the
proposed variation (where applicable). (D1,D2,D3)
2. Technical justification for the change of storage condition. (E12)
3. Results of appropriate real time stability studies covering the duration of
currently approved shelf-life (at proposed storage condition) of at least
two pilot/production scale batches of the product and in the authorized
packaging material in accordance with the ASEAN Guidelines on
Stability Study of Drug Product. (P8)

Malaysian Variation Guideline for Pharmaceutical Products
MiV-PA36 – Increasing of storage condition
56
• Apply at part A15.
• Example:
To comply to Zone IVb requirement. Storage condition is increased from 25oC to
30oC.
• Full real time stability data covering the shelf life of the product at the
proposed storage condition must be submitted.

NATIONAL PHARMACEUTICAL CONTROL BUREAU
MINISTRY OF HEALTH MALAYSIA
Mission: To be a world renowned regulatory authority for medicinal products and cosmetics.
Vision: To safeguard the nation's health through scientific excellence in the regulatory control of medicinal products
and cosmetics.
THANK YOU
Chai Che LeongSenior Assistant Director,
Variation Section,
Centre for Post Registration,
National Pharmaceutical Control Bureau,
Tel: 03-7801 8466, fax: 03-79567151,
Email: [email protected]
Visit us: http://www.bpfk.gov.my
57