microbial ecology crossm · the global oceans and subject to active viral infection. in this study,...
TRANSCRIPT

Genetic Diversity and Cooccurrence Patterns of MarineCyanopodoviruses and Picocyanobacteria
Yingting Sun,a,b Si Zhang,a Lijuan Long,a Junde Dong,a Feng Chen,c Sijun Huanga
aCAS Key Laboratory of Tropical Marine Bio-resources and Ecology, South China Sea Institute of Oceanology,Chinese Academy of Sciences, Guangzhou, Guangdong, China
bUniversity of Chinese Academy of Sciences, Beijing, ChinacInstitute of Marine and Environmental Technology, University of Maryland Center for Environmental Science,Baltimore, Maryland, USA
ABSTRACT Picocyanobacteria Prochlorococcus and Synechococcus are abundant inthe global oceans and subject to active viral infection. In this study, the genetic di-versity of picocyanobacteria and the genetic diversity of cyanopodoviruses were syn-chronously investigated along water columns in the equatorial Indian Ocean andover a seasonal time course in the coastal Sanya Bay, South China Sea. Using the16S-23S rRNA internal transcribed spacer (ITS)-based clone library and quantitativePCR (qPCR) analyses, the picocyanobacterial community composition and abundancewere determined. Sanya Bay was dominated by clade II Synechococcus during all theseasons, and a typical population shift from high-light-adapted Prochlorococcus tolow-light-adapted Prochlorococcus was found along the vertical profiles. Strikingly,the DNA polymerase gene sequences of cyanopodoviruses revealed a much greatergenetic diversity than we expected. Nearly one-third of the phylogenetic groupswere newly described here. No apparent seasonal pattern was observed for theSanya Bay picocyanobacterial or cyanopodoviral communities. Different dominantcyanopodovirus lineages were identified for the coastal area, upper euphotic zone,and middle-to-lower euphotic zone of the open ocean. Diversity indices of both pi-cocyanobacteria and cyanopodoviruses were highest in the middle euphotic zoneand both were lower in the upper euphotic zone, reflecting a host-virus interaction.Cyanopodoviral communities differed significantly between the upper euphotic zoneand the middle-to-lower euphotic zone, showing a vertical pattern similar to that ofpicocyanobacteria. However, in the surface waters of the open ocean, cyanopodovi-ruses exhibited no apparent biogeographic pattern, differing from picocyanobacte-ria. This study demonstrates correlated distribution patterns of picocyanobacteriaand cyanopodoviruses, as well as the complex biogeography of cyanopodoviruses.
IMPORTANCE Picocyanobacteria are highly diverse and abundant in the ocean anddisplay remarkable global biogeography and a vertical distribution pattern. However,how the diversity and distribution of picocyanobacteria affect those of the virusesthat infect them remains largely unknown. Here we synchronously analyzed thecommunity structures of cyanopodoviruses and picocyanobacteria at spatial andtemporal scales. Both spatial and temporal variations of cyanopodoviral communitiescan be linked to those of picocyanobacteria. The coastal area, upper euphotic zone,and middle-to-lower euphotic zone of the open ocean have distinct cyanopodoviralcommunities, showing horizontal and vertical variation patterns closely related tothose of picocyanobacteria. These findings emphasize the driving force of host com-munity in shaping the biogeographic structure of viruses. Our work provides impor-tant information for future assessments of the ecological roles of viruses and hostsfor each other.
Received 13 March 2018 Accepted 8 June2018
Accepted manuscript posted online 18June 2018
Citation Sun Y, Zhang S, Long L, Dong J, ChenF, Huang S. 2018. Genetic diversity andcooccurrence patterns of marinecyanopodoviruses and picocyanobacteria.Appl Environ Microbiol 84:e00591-18. https://doi.org/10.1128/AEM.00591-18.
Editor Hideaki Nojiri, University of Tokyo
Copyright © 2018 American Society forMicrobiology. All Rights Reserved.
Address correspondence to Sijun Huang,[email protected].
MICROBIAL ECOLOGY
crossm
August 2018 Volume 84 Issue 16 e00591-18 aem.asm.org 1Applied and Environmental Microbiology
on June 12, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from

KEYWORDS DNA polymerase, community composition, cyanophages,cyanopodoviruses, picocyanobacteria
Viruses are extremely abundant in the ocean (1, 2). They play key roles in the marineecosystem via modulating the abundance, diversity, and evolution of hosts, speed-
ing the recycling of nutrients and consequently influencing the global biogeochemicalcycling (1, 3). The global viral communities are of vast genetic diversity and exhibitbiogeographic structures (4–6). However, our understanding of how viral communitieschange in response to the variation of the host community is limited. Cyanophages andpicocyanobacteria have emerged to be a valuable system to investigate and demon-strate the ecologically antagonistic relationship of viruses and hosts in the ocean.Extensive studies have revealed the diversity breadth and community compositions ofpicocyanobacteria and have delineated explicit global biogeographic patterns of theirgenetic taxa. Therefore, the investigation on cooccurring cyanophage and picocyano-bacterial communities at different spatial and temporal scales could deepen ourunderstanding of the ecological interactions between viruses and hosts in the marineenvironment.
Marine unicellular picocyanobacteria of the genera Prochlorococcus and Synechoc-occus are major primary producers in the ocean. Both genera are divided into a fewclades with genetic, physiological, and ecological features. Prochlorococcus cladesdisplay remarkable vertical depth distributions, referred to as high-light (HL)-adaptedand low-light (LL)-adapted ecotypes, which are designated on the basis of their lightoptima (7–9). HL ecotypes also display horizontally latitudinal distributions, in responseto temperature (10) or iron (a trace metal element) availability (11). HL Prochlorococcusis phylogenetically more cohesive, while LL Prochlorococcus is more divergent (12).Marine Synechococcus organisms comprise three discrete subclusters, 5.1, 5.2, and 5.3,among which subcluster 5.1 is the major one and encompasses many defined clades(13, 14). The biogeographic distribution patterns of a few Synechococcus clades are clearnow. For instance, clades II and III are more likely distributed in warm oceanic waters,clades I and IV prefer high-latitude temperate waters, and clade CRD1 prefers upwellingiron-depleted waters (15–18).
Cyanophages are believed to affect their hosts in respect to their abundance,diversity, and evolution (19–23). They have been found in diverse marine environmentsand are predominantly lytic phages (24–31). All the known cyanophage isolates belongto one of the three tailed double-stranded DNA virus families, Myoviridae, Podoviridae,and Siphoviridae, on the basis of tail morphology. Cyanopodoviruses isolated thus farare genetically and morphologically similar to coliphage T7 (32–36). Metagenomicsurveys revealed that T7-like cyanopodoviruses are abundant in the sea (5, 35, 37–39).The viral DNA polymerase gene (pol) has been explored as a molecular marker toinvestigate the diversity of cyanopodoviruses. Two major phylogenetic clusters, MPP-Aand MPP-B, were identified via pol (27). The former is much less abundant than thelatter (30, 35, 38, 40, 41), in general, and the latter contains numerous definedsubclusters (30, 40, 41). However, only a few studies have delineated the diversity ofwild cyanopodoviral communities. One of our previous studies showed that cyanopo-doviral communities displayed a seasonal variation in Chesapeake Bay, a temperateestuarine ecosystem (41). Another survey showed that some ubiquitous phylogeneticgroups of cyanopodoviruses were commonly detected across distant locations in thesurface water, and the open ocean communities were less diverse than those inChesapeake Bay (40).
Many studies have revealed that cyanophage titers covary with picocyanobacterialabundances (19, 24, 25, 28, 42–44). However, only a few studies synchronously inves-tigated the genetic diversity of both picocyanobacteria and cyanophages, and theseearly studies mostly focused on cyanomyoviruses (42, 45). Picocyanobacterial ecotypesexhibit striking vertical distribution patterns and seasonal variations. Therefore, it isinteresting to further answer whether and how the change of picocyanobacterial
Sun et al. Applied and Environmental Microbiology
August 2018 Volume 84 Issue 16 e00591-18 aem.asm.org 2
on June 12, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from

communities affects the distribution of podoviruses in the ocean over spatial andtemporal scales.
In this study, we investigated the community structures of cyanopodoviruses andpicocyanobacteria along vertical profiles located in the Indian Ocean and over aseasonal time course in Sanya Bay, South China Sea. Many phylogenetic groups ofcyanopodoviruses were newly found here, suggesting a greater diversity of cyanobac-terial podoviruses exists in the marine environment. A clear vertical pattern of cya-nopodoviral community variation was observed, showing that the middle-to-lowereuphotic communities are much more diverse than the upper euphotic communities.Such a pattern was correlated with that of picocyanobacteria.
RESULTS AND DISCUSSIONSampling environments. Water samples were collected at three sites. The vertical
stations I205 and I211 are located in the equatorial Indian Ocean (�780 km apart).Hydrologic parameters, including water temperature, density (�T), and concentrationsof macronutrients (nitrate, phosphate, and silicate) and chlorophyll a, indicate thatstations I205 and I211 represent stratified water columns of the open ocean (Fig. 1A, B,E, and F; see also Table S1 in the supplemental material). The mixed layer depths wereat 50 m and 30 m, and the deep chlorophyll maximum (DCM) depths were at 75 m and50 m for I205 and I211, respectively (Fig. 1B and F). Another coastal seasonal time serialstation, W4, is located in Sanya Bay, South China Sea. This sampling site represents atropical coastal water body without strong seasonal temperature variation (22 to 28°C)(Fig. 1I and J; Table S1).
Phylogenetic diversity and abundance of picocyanobacteria. We built clonelibraries based on the 16S-23S rRNA internal transcribed spacer (ITS) amplicons toinvestigate the genetic diversity of picocyanobacteria and used ITS-based quantitativePCR (qPCR) to quantify 14 different ecotypes of Prochlorococcus and Synechococcus. Atotal of 925 ITS environmental sequences were obtained from the 18 samples. Theobserved operational taxonomic unit (OTU) numbers dropped dramatically from usingthe OTU definition cutoff of 99% to using 95%, and leveled off below a cutoff of 95%(see Fig. S1A). Due to the highly divergent nature of ITS sequences compared with 16SrRNA sequences, we used a 90% DNA sequence similarity cutoff to cluster OTUs ratherthan the standard cutoff of 97% for 16S rRNA sequences. Rarefaction curves determinedwith the OTU definition at a cutoff of 90% (see Fig. S2A) show that the increase of OTUsreached a plateau for 10 samples, while the other 8 samples from the middle-to-lowereuphotic zone (50 to 150 m) did not. This pattern suggests that, with such a definitionof OTU, the communities in the low-light zone were not completely sampled. This isdue to the extremely high genetic diversity of LL Prochlorococcus. Nevertheless, we stillbelieve that the majority of Prochlorococcus and Synechococcus lineages have beensampled when the lineage is defined as the phylotype (see below).
We built phylogenetic trees separately for Synechococcus (see Fig. S3), HL Prochlo-rococcus (see Fig. S4), and LL Prochlorococcus (see Fig. S5). Environmental sequenceswere assigned to genetic lineages (i.e., phylotype) on the basis of the trees. At theequatorial Indian Ocean stations I205 and I211, the clade HLII Prochlorococcus clonesdominated at 5 m and 25 m, accounting for nearly 100% of the libraries (Fig. 2A). In themiddle-to-deep euphotic zone (50 to 150 m), the community became much morediverse, where sequences affiliated with LL Prochlorococcus clades and Synechococcusclade CRD1 were present in abundance. However, HLII Prochlorococcus sequences stillconstituted a large portion of the libraries at the lower euphotic layers (100 to 200 m)of I211 and at the bottom layer of I205 (200 m). This community composition variationmatches the qPCR results of picocyanobacterial ecotypes very well (Fig. 1C, D, G, and H).Prochlorococcus ecotype HLII reached the maximum concentrations, 1.0 � 108 cells · liter�1
at I205 and 2.9 � 107 cells · liter�1 at I211, in the upper euphotic waters (5 m and 25 m)and then decreased dramatically to around 104 to 105 cells · liter�1 in the mid- to lowerdepths. The abundances of LL Prochlorococcus ecotypes peaked at 50 to 75 m anddropped faster than those of HL Prochlorococcus. This may explain why HL Prochloro-
Cooccurrence of Cyanopodoviruses and Cyanobacteria Applied and Environmental Microbiology
August 2018 Volume 84 Issue 16 e00591-18 aem.asm.org 3
on June 12, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from

FIG 1 Hydrographic features (A, B, E, F, I, and J) and abundances of Prochlorococcus (C, G, and K) and Synechococcus (D, H, and L) ecotypes at IndianOcean stations I205 (A to D) and I211 (E to H) and Sanya Bay station W4 (I to L). Hydrographic parameters include concentrations of macronutrients and
(Continued on next page)
Sun et al. Applied and Environmental Microbiology
August 2018 Volume 84 Issue 16 e00591-18 aem.asm.org 4
on June 12, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from

coccus clones still dominated the libraries of deep euphotic samples. Overall, thepattern represents a typical stratified water column profile of Prochlorococcus ecotypesin the tropical open ocean (10, 46–48).
There was a large number of unassigned sequences (labeled “other LL” in Fig. 2A)in 50- to 150-m samples, constituting 2% to 49% (mean, 25%; n � 8) of sequences forone sample. These sequences formed many deeply branching nodes (Fig. S5). Previ-ously reported environmental sequences that were assigned to LL Prochlorococcusclade NC1 (12) or LLVII (49) were scattered among these nodes (Fig. S5). This phylo-genetic pattern suggests NC1 might be polyphyletic instead of forming a coherentclade. Our finding and previous knowledge (49) suggest that we do not know muchabout many extremely diverse LL-adapted Prochlorococcus organisms. These memberscould be a significant fraction and be missed during current qPCR quantification ofProchlorococcus ecotypes.
It is noteworthy that Synechococcus clades II and CRD1 exhibited depth-relateddistribution patterns (Fig. 1D and H) similar to those of HL and LL Prochlorococcusclades, respectively. Recently, the CRD1 clade of Synechococcus was found to beprevalent in the iron-depleted areas (16, 18, 50) and to be more abundant in the upperlayer than in the lower layer of water columns (16, 50). These areas are often upwellingwaters, such as the Costa Rica Dome, equatorial Pacific upwelling, and the Benguelaupwelling regions (16). Our results suggest that in the stratified water column cladeCRD1 Synechococcus may be present in low abundance in the upper layer but becomemore abundant near the DCM layer.
FIG 1 Legend (Continued)chlorophyll a, fluorescence intensity, water temperature, and water density (�T). The mixing layer depth is indicated by gray dashed lines. Abundancesof 14 Prochlorococcus and Synechococcus ecotypes and total bacteria (16S rRNA gene copies) were measured, but only the detectable ecotypes areshown. Pink dashed lines (C, D, G, H, K, and L) indicate summed ecotype abundance. Note that, in panel L, the black line representing Synechococcusclade II overlaps the pink line representing total Synechococcus.
FIG 2 Clustering of the samples on the basis of compositions of ITS (A) and pol sequences (B). OTU was defined at a cutoff of 90% DNA sequence similarityfor both ITS and pol. ANOSIM was used to test the significance of the grouping, which is indicated by light blue shading in the clustering dendrograms.Phylotype-based community composition of each sample is shown to the right of the dendrograms. Spearman correlation coefficient (Mantel test) that reflectsthe relationship between picocyanobacterial and cyanopodoviral communities is shown below the chart. “Other LL” Prochlorococcus does not represent a realclade but is comprised of other sequences that cannot be assigned to any known LL lineages.
Cooccurrence of Cyanopodoviruses and Cyanobacteria Applied and Environmental Microbiology
August 2018 Volume 84 Issue 16 e00591-18 aem.asm.org 5
on June 12, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from

In the Sanya Bay samples, only Synechococcus sequences but no Prochlorococcussequences were retrieved from the clone libraries (Fig. 2A). Synechococcus clade IIwithin the marine subcluster 5.1 was the dominant lineage across the four seasons,making up nearly 80% of sequenced clones in each library. Clade IX of subcluster 5.1and subcluster 5.2 were also present. These results are consistent with the qPCR datain which Synechococcus clade II was most abundant, outnumbering the other ecotypesby one to two orders of magnitude (Fig. 1K and L). Despite the lack of seasonal variationin diversity, picocyanobacteria in this coastal site still exhibited seasonal variation in cellabundance. Synechococcus clade II was one order of magnitude less abundant in thewinter (�108 cells · liter�1) than in other seasons (�109 cells · liter�1). HL Prochloro-coccus ecotypes were detected in much lower abundance (103 to 106 cells · liter�1) thanSynechococcus, and LL Prochlorococcus ecotypes were not detectable. Our results areconsistent with previous studies revealing that clade II is the dominant Synechococcusecotype in the warm tropical area (15–18, 51, 52).
Phylogenetic diversity of cyanopodoviruses. A total of 1,406 pol environmentalsequences were retrieved from clone libraries for the 18 viral concentrate (VC) samples.Similar to ITS sequences, OTU numbers of pol sequences also reached a steady statebelow the definition cutoff of 95% DNA sequence similarity (Fig. S1A). With the OTUdefinition of 90%, rarefaction curves for near half of the samples are close to saturated(Fig. S2B). Although it seems that our sequences do not cover a broad diversity, at least80% of the retrieved sequences in each sample were affiliated with several majorphylotypes (see below). Therefore, by using phylotype as the taxon definition, themajority of the cyanopodovirus lineages should have been sampled.
All the pol sequences were grouped into either the MPP-A cluster (131 sequences,�10% of the total) (Fig. 3A) or the MPP-B cluster (1,275 sequences, �90% of the total)(Fig. 3B). Similar to previous studies (30, 38, 40, 41), the MPP-A sequences are much lesspresent than the MPP-B sequences. All these data support the hypothesis that MPP-Bcyanopodoviruses are more successful than the MPP-A cyanopodoviruses in the natural
FIG 3 Maximum likelihood phylogenetic trees based on the viral DNA polymerase protein sequence of cyanopo-doviruses. Trees for the MPP-A cluster (A) and the MPP-B cluster (B) were built independently. Environmentalsequences obtained in this study are labeled in the format “representative sequence identifier (ID) OTU (no. ofsequences assigned to the OTU).” Bootstrap values for 100 sampling replicates are listed only for those higher than50%. Reference sequence shown in green is from phage isolates infecting Prochlorococcus, those in blue are fromphage isolates infecting Synechococcus, and those in black are from environmental clones or from the Global OceanSampling metagenomes. Percentages of sequences assigned to each of the genotypes are listed to the right.Genotype labels are according to the numbering by Dekel-Bird and colleagues (30). Genotypes labeled in redindicate those defined in this study, and red star symbols indicate those newly detected here.
Sun et al. Applied and Environmental Microbiology
August 2018 Volume 84 Issue 16 e00591-18 aem.asm.org 6
on June 12, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from
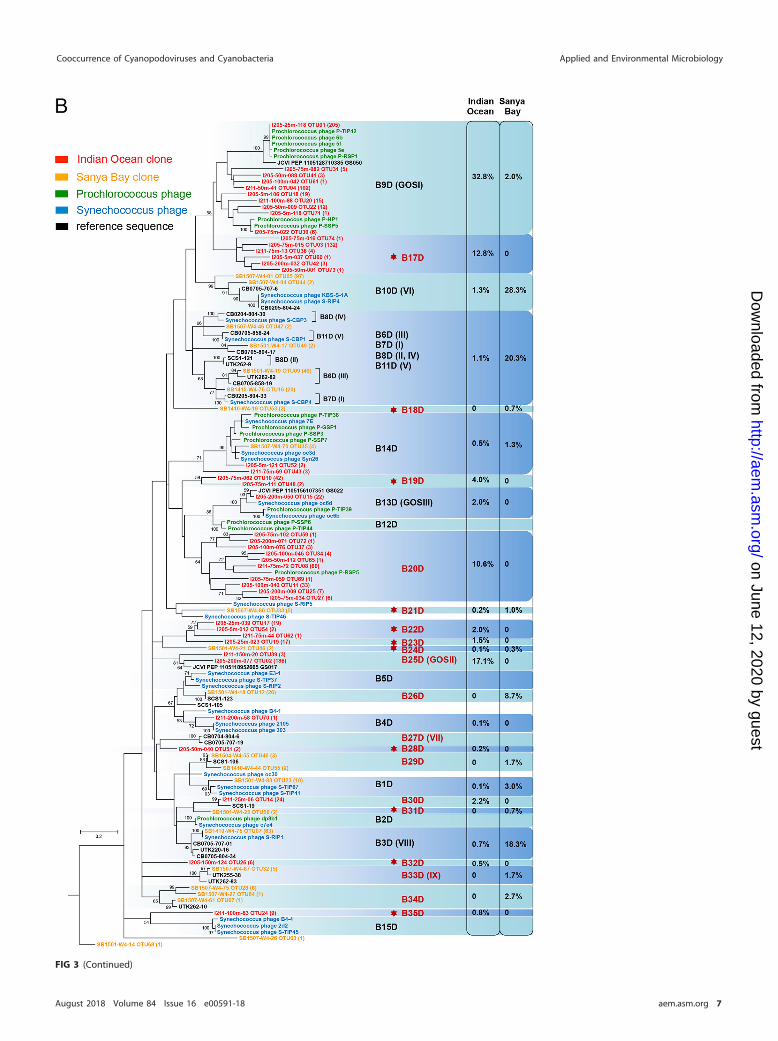
FIG 3 (Continued)
Cooccurrence of Cyanopodoviruses and Cyanobacteria Applied and Environmental Microbiology
August 2018 Volume 84 Issue 16 e00591-18 aem.asm.org 7
on June 12, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from

environments (30). Each of the two clusters can be further divided into a few subclus-ters. We use the subcluster labels established by Dekel-Bird et al. (30), while the earlierlabels with Roman numerals (40, 41) are also shown in parentheses if necessary.Compared to previous studies (30, 40, 41, 53), nearly half of the subclusters were newlydefined (labeled in red in Fig. 3), among which many were not described previously(indicated by red stars in Fig. 3). Hence, our environmental sequences greatly extendthe diversity of marine cyanopodoviruses.
Within cluster MPP-A, subclusters A7D and A9D contained a large portion ofenvironmental sequences (Fig. 3A). Most A7D sequences came from the Sanya Baysamples, while most A9D sequences came from the Indian Ocean samples (Fig. 3A).Interestingly, among the cultured MPP-B phages, only one Prochlorococcus phage,P-SSP9, fell into subcluster A9D. This phage infects an LL Prochlorococcus strain, SS-120(30, 35). Except for P-SSP9, other isolated phages in cluster MPP-A infect Synechococcus(27, 30). It will be interesting to investigate in the future whether MPP-A phages thatinfect Prochlorococcus are more common than previously thought.
Cluster MPP-B consisted of most of the retrieved environmental sequences for eachsample (Fig. 2B). In Sanya Bay, sequences of subclusters B3D (VIII), B10D (VI), and B6D(III) were dispersed in most of the samples and abundant, representing the majorgenotypes of cyanopodoviruses in this coastal environment (Fig. 3B). Previously, B3D(VIII) was also found to be a major phylogenetic cluster in Chesapeake Bay (41), as wellas in some open ocean surface waters (40). Recently, 44 cyanopodoviruses wereisolated from Synechococcus strain WH7803 from seawaters of the East Coast (RhodeIsland) and the West Coast (Washington) of North America. Most of them were groupedinto either B3D (VIII) or B10D (VI) subclusters based on the pol phylogeny (53).Synechococcus podoviruses S-RIP1 and S-RIP4 fell into B3D and B10D, respectively, asrepresentatives of the two major subclusters (53). Isolated phages within subclustersB3D and B10D thus far all infect Synechococcus (30, 53). Therefore, on the basis of ourresults and previous findings, we speculate that B3D and B10D cyanopodoviruses areprevalent in coastal waters, and they more likely infect Synechococcus.
Four major subclusters, B9D (GOSI), B17D, B20D, and B25D (GOSII), in total consti-tuted 73% of the sequences from the Indian Ocean samples (Fig. 2B and 3B). Depth-related distribution preference was observed among these lineages. B9D was relativelymore abundant in the surface mixed layer, and others became more prevalent indeeper layers (see Fig. S6). Among these major groups, B9D and B20D containedsequences from isolated Prochlorococcus phages, while B17D and B25D did not haveisolated phage representatives. Interestingly, the dominant clusters found in our studywere not the dominant ones in other surface water samples from the Atlantic andPacific Oceans (40). Instead, clusters B3D (VIII), B8D (II), and A2D (XI) made up themajority in those samples (40). Furthermore, there appears to be another discrepancythat subclusters B12D, B13D, and B14D were barely detected in this study, whilenumerous phages within these phylotypes have been isolated (30). Previous metag-enomic surveys also showed that sequences similar to that of Prochlorococcus podo-virus P-SSP7 in B14D are present at a high abundance (5, 38). This variance suggeststhat cyanopodoviral communities across the broad open oceans are likely quite vari-able.
Comparing picocyanobacterial and cyanopodoviral communities. To investigatethe relationship between picocyanobacterial and cyanopodoviral communities, wecompared diversity indices (alpha diversity) and composition (beta diversity) for the ITSclone libraries and the pol clone libraries. Since we analyzed both picocyanobacterialand cyanopodoviral diversities from the same sample, we referred to them as dualalpha diversity and dual beta diversity.
To gain a broader view, 14 pol clone libraries (40, 41) and 14 ITS clone libraries (52,54) from the published studies (Fig. 4A; metadata of these samples are listed in TableS2) were compared to those obtained here. These selected libraries are also from thesamples that have both pol and ITS sequence data. These additional samples include
Sun et al. Applied and Environmental Microbiology
August 2018 Volume 84 Issue 16 e00591-18 aem.asm.org 8
on June 12, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from

FIG 4 Alpha diversity and beta diversity of ITS and pol clone libraries. Previously published ITS and pol clone libraries from the Chesapeake Baysurface water samples (CB0205-858, CB0205-804, CB0205-707, CB0705-858, CB0705-804, and CB0705-707) (41, 54) and the Atlantic and Pacificsurface ocean samples (UTK202, UTK211, UTK220, UTK229, UTK240, UTK250, UTK255, and UTK262) (40, 52) were compared to those derived here.(A) Locations of the samples are shown. The map was created using Ocean Data View, version 4. Alpha diversity Chao indices (B) and Shannon
(Continued on next page)
Cooccurrence of Cyanopodoviruses and Cyanobacteria Applied and Environmental Microbiology
August 2018 Volume 84 Issue 16 e00591-18 aem.asm.org 9
on June 12, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from

Chesapeake Bay surface water (41, 54) and the surface water in the Atlantic and PacificOceans over a wide latitudinal range (40, 52).
Dual alpha diversity. At stations I205 and I211, the Chao (indicating OTU richness)and Shannon (indicating both richness and evenness) diversity indices of ITS and polclone libraries displayed similar vertical patterns, showing that both picocyanobacterialand cyanopodoviral communities in the middle-to-deep euphotic zone are generallymore diverse than those in the surface layers (Fig. 4B and C; see Fig. S7A, B, D, and E).We used the Pearson correlation test to assess the relationship between diversityindices of the viral and host communities as well as between the viral or host diversityindices and host abundances. The results (Table 1) show that the species richness (Chaoindex) of cyanopodoviruses was positively correlated with the species richness ofcooccurring picocyanobacteria but was not correlated with the abundance of them.The Shannon index did not show a significant correlation as the Chao index did.Interestingly, picocyanobacterial diversity seems to be negatively correlated with theirabundances (Table 1). Note again that the abundance of Prochlorococcus in a low-lightzone could be underestimated by our qPCR assays. At station W4, the diversity indicesof picocyanobacteria and cyanopodoviruses are both relatively low except for thecyanopodoviral community in the summer of 2015 (Fig. S7C and F).
With more samples involved in the Pearson correlation test, the diversity indices ofcyanopodoviruses were still positively correlated with those of picocyanobacteria (r �
0.593, P � 0.001 for the Chao index; r � 0.346, P � 0.052 for the Shannon index) (Fig.4B and C). Picocyanobacterial communities in the tropical and subtropical surfacewaters are commonly dominated by only a few genetic lineages, often by one majorclade (18). In the middle and lower euphotic zones, more picocyanobacterial lineagescooccurred and, especially, LL Prochlorococcus thrived, with quite genetically divergentmembers (9, 12, 48). In contrast to diversity, the abundance of picocyanobacteria oftenreaches the maximum in the upper euphotic zone and decreases toward the bottomeuphotic zone (55). Therefore, it is likely that the diversity level of cyanopodoviruses isclosely related to the diversity level of picocyanobacteria, suggesting that the hostdiversity level is a strong constraint on the diversity level of cyanopodoviruses.
Dual beta diversity. To compare the community composition of picocyanobacteriaand cyanopodoviruses, we clustered the samples on the basis of the ITS clone librariesand pol clone libraries, respectively. We also tested the significance of clustering usingan analysis of similarity (ANOSIM) and assessed the correlation between the clusteringpatterns of picocyanobacteria and cyanopodoviruses using the Mantel test. Dendro-
FIG 4 Legend (Continued)indices (C). Pearson correlation analysis results that indicate the relationship between diversity levels of picocyanobacteria and cyanopodovirusesare listed under the bar charts. Nonmetric multidimensional scaling (nMDS) plot based on compositions of ITS (D) and pol (E) OTUs. The inset plotin panel D covers all the 32 samples, and the outside plot shows the detailed relationship of a portion of 29 samples (dashed box in the insetplot). ANOSIM was used to test the significance of grouping, and the Mantel test (Spearman correlation) was used to assess the correlationbetween picocyanobacterial and cyanopodoviral communities. Coefficients (global R and �) and P values are shown.
TABLE 1 Correlation analysis for the Indian Ocean water column samples (n � 14)
Variable
Pearson test result (r/P value)
pol ITS
Chao index Shannon index Chao index Shannon index
ITSChao index 0.529/0.052Shannon index 0.382/0.177
AbundanceProchlorococcus �0.022/0.940 0.070/0.812 �0.377/0.184 �0.566/0.035Synechococcus �0.041/0.888 0.015/0.958 �0.409/0.146 �0.558/0.038Prochlorococcus and
Synechococcus�0.033/0.912 0.059/0.842 �0.383/0.177 �0.571/0.033
Sun et al. Applied and Environmental Microbiology
August 2018 Volume 84 Issue 16 e00591-18 aem.asm.org 10
on June 12, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from

grams show that the clusterings of samples (using an OTU definition cutoff of 90%) onthe basis of the two genetic markers have similar patterns (Spearman correlationcoefficient � � 0.458, P � 0.001), whereby Synechococcus-dominating waters, HLProchlorococcus-dominating waters, and LL Prochlorococcus-dominating waters wereseparated (Fig. 2). An ANOSIM showed that such clustering patterns are statisticallysignificant (global R � 0.89, P � 0.001 for ITS; global R � 0.783, P � 0.001 for pol).Additional Mantel tests (Fig. S1B) showed that most of the � coefficients calculated atOTU definition cutoffs of �86% are higher than 0.4 (P � 0.01). Our results support thateven at the microdiversity level (using a cutoff of 99%), the viral and host communitieswere still significantly correlated, reflecting a strong virus-host interaction (56). More-over, even with small library sizes for each sample (less than 100 sequences), the Manteltests using different subsets of total sequences resulted in similar correlation levels (Fig.S1C). Nevertheless, the small library size may still have an undetected impact on therobustness of the correlation analysis between the viral and host communities.
However, taking the 14 additional samples into account, the multidimensionalscaling (MDS) analysis did not show a high level of correlation between picocyanobac-terial and cyanopodoviral communities (Spearman correlation coefficient � � 0.233,P � 0.001) (Fig. 4D and E). On the MDS map, picocyanobacteria display a commonbiogeographic pattern, which separated the samples into groups as summer Chesa-peake Bay, winter Chesapeake Bay, tropical and subtropical open ocean, high-latitudeNorth Atlantic, and Sanya Bay (ANOSIM test, global R � 0.8, P � 0.001) (Fig. 4D), whilethe cyanopodoviral communities appear to be separated by individual studies (ANOSIMtest, global R � 0.814, P � 0.001) (Fig. 4E). It seems that the sampling location may playa role in shaping such a pattern. For example, samples from the Indian Ocean, SanyaBay, and Chesapeake Bay were grouped together (Fig. 4E), reflecting a local pattern.However, it was unexpected that the cyanopodoviral communities of the upper layers(5 m and 25 m) at stations I205 and I211 were not closely related with other surfaceocean samples, i.e., UTK samples (Fig. 4E). As mentioned above, the diversity ofcyanopodoviruses on the ocean surface is relatively low. Therefore, this discrepancy islikely due to the differences of dominant genetic groups of surface water samples. Thecomplex filtration procedure that concentrates viral particles and the storage of sam-ples in different studies may also impact the comparison between samples fromdifferent cruises.
Our findings suggest that environments in similar climate zones or with similarhydrographic conditions may have similar picocyanobacterial populations. In otherwords, the diversity of picocyanobacteria is predictable from the location (15, 47). Incontrast, cyanopodoviral communities are more variable and complex than their hostcommunities.
On one hand, cyanopodoviral communities in estuarine, coastal, and open oceanwaters differ from each other, and so do the communities in the upper euphotic zoneand in the lower euphotic zone, showing a niche adaptation. Depth was considered tobe a driver of viral community structure in the whole water column, where communi-ties in the photic zone differ significantly from those in the aphotic zone (4, 57).However, it was also shown that the total viral communities displayed minimal variationat different euphotic depths (6). Here, we found that the cyanopodoviral communitiesare structured by depth within the euphotic zone. Similar depth-related communityvariation was also observed for cyanomyoviruses (58). We speculate that hosts alsomediate niche adaptation and play a key role in shaping the regional and verticalcommunity structures of cyanophages. This is also supported by a previous study whichsuggested that both cyanopodoviral communities and cyanomyoviral communitiesdiffer between the Synechococcus-dominating and Prochlorococcus-dominating envi-ronments (38). Cyanopodoviruses are commonly host strain specific, while many cya-nomyoviruses have broader host ranges. Despite having different host specificities,cyanomyoviral diversity was also shown to be correlated with that of cooccurringSynechococcus in a seasonal time course study (42). Recently, the host range phenotypeof cyanomyoviruses was found to be a factor that contributes to their local and regional
Cooccurrence of Cyanopodoviruses and Cyanobacteria Applied and Environmental Microbiology
August 2018 Volume 84 Issue 16 e00591-18 aem.asm.org 11
on June 12, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from

distribution patterns (53). It is well known that infectious cyanophages are abundant inthe field and quantitatively covary with their hosts (44, 59). The virioplankton abun-dance was found to be highly synchronous with Prochlorococcus distribution, and thissuggested that cyanophages contributed to a significant fraction of viruses in thesubtropical Atlantic gyre (60). Altogether, these findings imply a tight relationshipbetween wild cyanophages and cyanobacteria, with respect to both abundance andcommunity structure.
On the other hand, one host strain can be infected by diverse podoviruses that fallinto distinct phylogenetic groups (30, 41). This could in part explain the randomness ofthe cyanopodoviral community structures over the broad surface waters of the openocean. Moreover, the Prochlorococcus community is composed of numerous coexistingsubpopulations with a large degree of genomic variation (61). The rapid change ofthese subpopulations may contribute to a poor correlation between the cyanopodo-viral and picocyanobacterial communities across the ocean surface.
Conclusion. This study demonstrates the highly diverse and variable cyanopodo-viral communities in different marine regions and at different depths. Specific phylo-genetic lineages of cyanopodoviruses were found to occupy the near-shore waterswhere Synechococcus organisms are abundant, and other different lineages thrive in theProchlorococcus-dominating open ocean. Cyanopodoviral communities also changedramatically along the vertical scale in the open ocean, and the changing pattern iscorrelated to that of the picocyanobacterial community in the water column. Ourfindings further support that host availability (6, 38, 53, 62) and coevolutionary inter-actions (63, 64) are the main driving forces of viral diversity and distribution.
MATERIALS AND METHODSSampling and parameter measuring. Seawater samples were collected from Sanya Bay station W4,
located in the northern South China Sea, and the Indian Ocean stations I205 and I211. The details ofsampling locations, dates, and parameters are shown in Table S1 in the supplemental material. Tem-perature and salinity data were extracted from the CTD sensor (Sea-Bird Scientific) or the YSI sensor (YSIInc.) data set. Chlorophyll a concentrations were measured using Turner Designs Trilogy fluorometer(Turner Designs Inc.) according to a protocol described previously (65). N (nitrate), soluble reactivephosphate (SRP), and Si (silicate) were measured with a Seal AA3 autoanalyzer (Bran-Luebbe, GmbH)using long-path spectrophotometry (66, 67) with detection limits of 0.02 �mol · liter�1, 0.02 �mol ·liter�1, and 0.03 �mol · liter�1, respectively.
To collect viral concentrates (VCs), 20 liters of seawater was filtered through 0.22-�m-pore-sizepolycarbonate filter membranes (Millipore). A tangential flow filtration system was used to concentratethe filtrate to a final volume of 200 ml, with a 30-kDa molecular weight cutoff filter (Pall). VCs were storedat 4°C in the dark. The 0.22-�m-pore-size membranes were stored at �80°C until DNA extraction.
DNA extraction, PCR, cloning, and sequencing. DNA was extracted from filter membranes usingthe MoBio Powersoil DNA isolation kit according to the manufacturer’s protocol. The amplification ofpicocyanobacterial 16S-23S rRNA internal transcribed spacer (ITS) sequences from the extracted DNA wasperformed using the primers Picocya16S-F and Picocya23S-R, according to the PCR program describedpreviously (54). A partial sequence of the cyanopodovirus DNA polymerase gene was amplified using 2�l VC as the template and the primers CP-DNAP-349F, CP-DNAP-533Ra, and CP-DNAP-533Rb, asdescribed previously (40). The PCR products were purified using the TaKaRa agarose gel DNA purificationkit (TaKaRa) and cloned using the TaKaRa pMD18-T vector cloning kit (TaKaRa), according to themanufacturer’s instructions. Randomly picked clones were sequenced on an ABI 3730 genetic analyzer(Applied Biosystems) at Major Bio-tech Co., Ltd., Shanghai, China.
Phylogenetic diversity analysis. Raw Sanger sequences were examined and trimmed using BioEdit,and those of low quality were discarded. For the pol sequences, only those that were successfullytranslated to amino acid sequences were retained. ITS DNA sequences and pol amino acid sequenceswere aligned using Clustal X2 (68). Chimeras were discarded via a manual examination of the alignment.After quality control, 925 ITS sequences and 1,406 pol sequences were obtained. To reduce the sequenceredundancy in the phylogenetic analysis, all the sequences were binned into OTUs using mothur (69) atthe cutoff of 90% similarity of DNA sequences for ITS and at 80% for pol. A cutoff of 90% sequencesimilarity for pol was also tested, while it resulted in a very high degree of sequence redundancy atphylogenetic tree nodes. Thus, the results for the cutoff of 80% are shown. As a result, 149 ITS OTUs and74 pol OTUs were obtained, and representative sequences for each of the OTUs were imported to buildthe tree. Note that we used the translated amino acid sequences for constructing the Pol phylogeny.Phylogenetic analysis was performed with MEGA 7 (70). The maximum likelihood (ML) method based onthe Jukes-Cantor model and the ML method based on the JTT matrix-based model were used forestimating the ITS phylogeny and the Pol phylogeny, respectively. A discrete gamma distribution wasused to model the evolutionary rate differences among sites. The rate variation model enabled somesites to be evolutionarily invariable. The robustness of the phylogeny was tested using a bootstrap
Sun et al. Applied and Environmental Microbiology
August 2018 Volume 84 Issue 16 e00591-18 aem.asm.org 12
on June 12, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from

method with 100 bootstrap replications. Rarefaction curves were calculated using DOTUR (71) at a 90%DNA sequence similarity cutoff for both ITS and pol.
Previously, we investigated the cyanopodoviral communities (40, 41) and picocyanobacterialcommunities (52, 54) for the same bulk of seawater samples (see Table S2 for the metadata).However, the viral and host communities were independently assessed in different studies. Tocompare with those already published data, whole clone libraries for ITS and pol sequences wereretrieved from the NCBI GenBank. These retrieved DNA sequences were aligned together with thoseobtained in this study and the resulting alignment was imported into mothur to calculate thediversity indices of Chao and Shannon at a cutoff of 90% DNA sequence similarity for both ITS andpol sequences. A “shared file” created by the mothur command line “make.shared” was loaded intothe software PRIMER 5 (72) to calculate the square-root-transformed Bray-Curtis similarities amongclone libraries and to further plot the multidimensional scaling (MDS) map. The clone libraries for polsequences and for ITS sequences obtained in this study were also analyzed, and a “shared file” wasgenerated and loaded into PRIMER 5 to cluster the samples on the basis of the square-root-transformed Bray-Curtis similarity matrix, using the CLUSTER module and the “complete linkage”mode. ANOSIM implemented in PRIMER 5 was used to test the significance of grouping amongsamples during the MDS and CLUSTER analyses. The Spearman correlation-based Mantel test(RELATE function in PRIMER 5) was used to assess the correlation between picocyanobacterial andcyanopodoviral communities. The Pearson correlation between diversity indices of picocyanobac-teria and cyanopodoviruses was tested using SPSS version 13.
Quantifying ecotypes of Prochlorococcus and Synechococcus by qPCR. Real-time PCR was used toquantify a total of 14 ecotypes of Prochlorococcus and Synechococcus. We used the published primers andqPCR programs to quantify Prochlorococcus ecotypes eMED4 (HLI) (73), eMIT9312 (HLII) (73), eNATL2A(LLI) (73), eSS120 (LLII) (46), and eMIT9313 (LLIV) (73). Note that the reverse primer for eMED4 is5=-GAAGCTAGATTCGCTCAGAGC-3= (Z. Johnson, personal communication). The primers and qPCR pro-grams for quantifying Synechococcus clades I, II, III, IV, X, XV, XVI, CRD1, and CRD2 were according to thestudy by Ahlgren and Rocap (74). The gene copies of 16S rRNA genes were also quantified using a qPCRassay with the primers EUB338 and EUB518 (75).
To prepare qPCR standards, nested PCR was performed. First, ITS sequences amplified from allthe field samples using primers Picocya16S-F and Picocya23S-R were pooled. Second, using thepooled ITS PCR products as the template, ecotype-specific PCR products were generated. Then eachof the secondary PCR products was cloned, and randomly picked clones were sequenced to makesure that they indeed represent the corresponding ecotypes. Plasmid DNA was isolated and servedas the template to amplify a fragment with the universal M13-47/RV-M primers. Finally, thesegel-purified PCR products served as the qPCR standards. Their concentrations were determined withPicoGreen double-stranded DNA quantification reagent (Yeasen Biotech) using 2-fold serially dilutedlambda DNA (350 ng/�l; TaKaRa) as the standards. A multimode plate reader (EnSight; PerkinElmer)was used to read the fluorescence intensity. The preparation of standards for 16S rRNA genequantification was according to the same procedure. The first-round PCR was carried out with theprimers EUB338 and EUB518.
Standard curves were generated from triplicate reactions in 25-�l volumes, using the TaKaRa SYBRII agents and the Bio-Rad CFX Connect machine (Bio-Rad Life Science). Theoretical amplification effi-ciencies for quantifying picocyanobacterial ecotypes ranged from 82% to 96%, consistent with a previousreport (73). The amplification efficiency for quantifying 16S rRNA genes was 98%. qPCR assays wereperformed with 1 �l of template DNA extracted from filter membranes described above.
Accession number(s). Environmental sequences obtained in this study were deposited in the NCBIGenBank under accession numbers MG427080 to MG427884 and MH034887 to MH035487 for the viralDNA polymerase gene sequences and accession numbers MG427885 to MG428399 and MH034477 toMH034886 for the picocyanobacterial 16S-23S rRNA ITS sequences.
SUPPLEMENTAL MATERIAL
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.00591-18.
SUPPLEMENTAL FILE 1, PDF file, 1.4 MB.
ACKNOWLEDGMENTSWe thank the staff of Tropical Marine Biological Research Station in Hainan (CAS)
and the crew of the R.V. Shiyan I for assistance in sampling and the Xu Jie Lab and LiQian Lab at South China Sea Institute of Oceanology for help in measuring chlorophylland macronutrients.
This study was supported by the NSFC grants 41576126 (S.H.) and 41230962 (S.Z.),the Natural Science Foundation of Guangdong Province grant 2017A030306020 (S.H.),the Youth Innovation Promotion Association CAS (S.H.), and the Strategic PriorityResearch Program of the Chinese Academy of Sciences, grant no. XDA13020301 (S.H.).
Cooccurrence of Cyanopodoviruses and Cyanobacteria Applied and Environmental Microbiology
August 2018 Volume 84 Issue 16 e00591-18 aem.asm.org 13
on June 12, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from

REFERENCES1. Fuhrman JA. 1999. Marine viruses and their biogeochemical and eco-
logical effects. Nature 399:541–548. https://doi.org/10.1038/21119.2. Suttle CA. 2005. Viruses in the sea. Nature 437:356 –361. https://doi.org/
10.1038/nature04160.3. Suttle CA. 2007. Marine viruses–major players in the global ecosystem.
Nat Rev Microbiol 5:801– 812. https://doi.org/10.1038/nrmicro1750.4. Hurwitz BL, Brum JR, Sullivan MB. 2015. Depth-stratified functional and
taxonomic niche specialization in the ‘core’ and ‘flexible’ Pacific Oceanvirome. ISME J 9:472– 484. https://doi.org/10.1038/ismej.2014.143.
5. Angly FE, Felts B, Breitbart M, Salamon P, Edwards RA, Carlson C, ChanAM, Haynes M, Kelley S, Liu H. 2006. The marine viromes of four oceanicregions. PLoS Biol 4:e368. https://doi.org/10.1371/journal.pbio.0040368.
6. Brum JR, Ignacio-Espinoza JC, Roux S, Doulcier G, Acinas SG, AlbertiA, Chaffron S, Cruaud C, de Vargas C, Gasol JM, Gorsky G, Gregory AC,Guidi L, Hingamp P, Iudicone D, Not F, Ogata H, Pesant S, Poulos BT,Schwenck SM, Speich S, Dimier C, Kandels-Lewis S, Picheral M, Sear-son S, Tara Oceans Coordinators, Bork P, Bowler C, Sunagawa S,Wincker P, Karsenti E, Sullivan MB. 2015. Ocean plankton. Patternsand ecological drivers of ocean viral communities. Science 348:1261498. https://doi.org/10.1126/science.1261498.
7. Moore LR, Chisholm SW. 1999. Photophysiology of the marine cyano-bacterium Prochlorococcus: ecotypic differences among cultured iso-lates. Limnol Oceanogr 44:628 – 638. https://doi.org/10.4319/lo.1999.44.3.0628.
8. Moore LR, Rocap G, Chisholm SW. 1998. Physiology and molecularphylogeny of coexisting Prochlorococcus ecotypes. Nature 393:464 – 467.https://doi.org/10.1038/30965.
9. Rocap G, Distel DL, Waterbury JB, Chisholm SW. 2002. Resolution ofProchlorococcus and Synechococcus ecotypes by using 16S-23S ribo-somal DNA internal transcribed spacer sequences. Appl Environ Micro-biol 68:1180 –1191. https://doi.org/10.1128/AEM.68.3.1180-1191.2002.
10. Johnson ZI, Zinser ER, Coe A, McNulty NP, Woodward EM, Chisholm SW.2006. Niche partitioning among Prochlorococcus ecotypes along ocean-scale environmental gradients. Science 311:1737–1740. https://doi.org/10.1126/science.1118052.
11. Rusch DB, Martiny AC, Dupont CL, Halpern AL, Venter JC. 2010. Charac-terization of Prochlorococcus clades from iron-depleted oceanic regions.Proc Natl Acad Sci U S A 107:16184 –16189. https://doi.org/10.1073/pnas.1009513107.
12. Martiny AC, Tai AP, Veneziano D, Primeau F, Chisholm SW. 2009. Taxo-nomic resolution, ecotypes and the biogeography of Prochlorococcus.Environ Microbiol 11:823– 832. https://doi.org/10.1111/j.1462-2920.2008.01803.x.
13. Fuller NJ, Marie D, Partensky F, Vaulot D, Post AF, Scanlan DJ. 2003.Clade-specific 16S ribosomal DNA oligonucleotides reveal the predom-inance of a single marine Synechococcus clade throughout a stratifiedwater column in the Red Sea. Appl Environ Microbiol 69:2430 –2443.https://doi.org/10.1128/AEM.69.5.2430-2443.2003.
14. Penno S, Lindell D, Post AF. 2006. Diversity of Synechococcus and Pro-chlorococcus populations determined from DNA sequences of theN-regulatory gene ntcA. Environ Microbiol 8:1200 –1211. https://doi.org/10.1111/j.1462-2920.2006.01010.x.
15. Zwirglmaier K, Jardillier L, Ostrowski M, Mazard S, Garczarek L, Vaulot D,Not F, Massana R, Ulloa O, Scanlan DJ. 2008. Global phylogeography ofmarine Synechococcus and Prochlorococcus reveals a distinct partitioningof lineages among oceanic biomes. Environ Microbiol 10:147–161.
16. Sohm JA, Ahlgren NA, Thomson ZJ, Williams C, Moffett JW, Saito MA,Webb EA, Rocap G. 2016. Co-occurring Synechococcus ecotypes occupyfour major oceanic regimes defined by temperature, macronutrients andiron. ISME J 10:333–345. https://doi.org/10.1038/ismej.2015.115.
17. Zwirglmaier K, Heywood JL, Chamberlain K, Woodward EM, Zubkov MV,Scanlan DJ. 2007. Basin-scale distribution patterns of picocyanobacteriallineages in the Atlantic Ocean. Environ Microbiol 9:1278 –1290. https://doi.org/10.1111/j.1462-2920.2007.01246.x.
18. Farrant GK, Dore H, Cornejo-Castillo FM, Partensky F, Ratin M, OstrowskiM, Pitt FD, Wincker P, Scanlan DJ, Iudicone D, Acinas SG, Garczarek L.2016. Delineating ecologically significant taxonomic units from globalpatterns of marine picocyanobacteria. Proc Natl Acad Sci U S A 113:E3365–E3374. https://doi.org/10.1073/pnas.1524865113.
19. Waterbury JB, Valois FW. 1993. Resistance to co-occurring phages en-
ables marine Synechococcus communities to coexist with cyanophagesabundant in seawater. Appl Environ Microbiol 59:3393–3399.
20. Suttle CA, Chan AM. 1994. Dynamics and distribution of cyanophagesand their effect on marine Synechococcus spp. Appl Environ Microbiol60:3167–3174.
21. Lindell D, Sullivan MB, Johnson ZI, Tolonen AC, Rohwer F, Chisholm SW.2004. Transfer of photosynthesis genes to and from Prochlorococcusviruses. Proc Natl Acad Sci U S A 101:11013–11018. https://doi.org/10.1073/pnas.0401526101.
22. Avrani S, Lindell D. 2015. Convergent evolution toward an improvedgrowth rate and a reduced resistance range in Prochlorococcus strainsresistant to phage. Proc Natl Acad Sci U S A 112:E2191–E2200. https://doi.org/10.1073/pnas.1420347112.
23. Avrani S, Wurtzel O, Sharon I, Sorek R, Lindell D. 2011. Genomic islandvariability facilitates Prochlorococcus-virus coexistence. Nature 474:604 – 608. https://doi.org/10.1038/nature10172.
24. Suttle CA, Chan AM. 1993. Marine cyanophages infecting oceanic andcoastal strains of Synechococcus: abundance, morphology, cross-infectivity and growth characteristics. Mar Ecol Prog Ser 92:99 –109.https://doi.org/10.3354/meps092099.
25. Sullivan MB, Waterbury JB, Chisholm SW. 2003. Cyanophages infectingthe oceanic cyanobacterium Prochlorococcus. Nature 424:1047–1051.https://doi.org/10.1038/nature01929.
26. Waterbury JB, Watson SW, Valois FW, Franks DG. 1986. Biological andecological characterization of the marine unicellular cyanobacteriumSynechococcus. Can Bull Fish Aquat Sci 214:71–120.
27. Wang K, Chen F. 2008. Prevalence of highly host-specific cyanophages inthe estuarine environment. Environ Microbiol 10:300 –312. https://doi.org/10.1111/j.1462-2920.2007.01452.x.
28. Marston MF, Sallee JL. 2003. Genetic diversity and temporal variation inthe cyanophage community infecting marine Synechococcus species inRhode Island’s coastal waters. Appl Environ Microbiol 69:4639 – 4647.https://doi.org/10.1128/AEM.69.8.4639-4647.2003.
29. Dekel-Bird NP, Sabehi G, Mosevitzky B, Lindell D. 2015. Host-dependentdifferences in abundance, composition and host range of cyanophagesfrom the Red Sea. Environ Microbiol 17:1286 –1299. https://doi.org/10.1111/1462-2920.12569.
30. Dekel-Bird NP, Avrani S, Sabehi G, Pekarsky I, Marston MF, Kirzner S,Lindell D. 2013. Diversity and evolutionary relationships of T7-like podo-viruses infecting marine cyanobacteria. Environ Microbiol 15:1476 –1491.https://doi.org/10.1111/1462-2920.12103.
31. Wilson WH, Joint IR, Carr NG, Mann NH. 1993. Isolation and molecularcharacterization of five marine cyanophages propagated on Synechoc-occus sp. strain WH7803. Appl Environ Microbiol 59:3736 –3743.
32. Sullivan MB, Coleman ML, Weigele P, Rohwer F, Chisholm SW. 2005.Three Prochlorococcus cyanophage genomes: signature features andecological interpretations. PLoS Biol 3:e144. https://doi.org/10.1371/journal.pbio.0030144.
33. Chen F, Lu J. 2002. Genomic sequence and evolution of marinecyanophage P60: a new insight on lytic and lysogenic phages. ApplEnviron Microbiol 68:2589 –2594. https://doi.org/10.1128/AEM.68.5.2589-2594.2002.
34. Pope WH, Weigele PR, Chang J, Pedulla ML, Ford ME, Houtz JM, Jiang W,Chiu W, Hatfull GF, Hendrix RW, King J. 2007. Genome sequence, struc-tural proteins, and capsid organization of the cyanophage Syn5: a“horned” bacteriophage of marine Synechococcus. J Mol Biol 368:966 –981. https://doi.org/10.1016/j.jmb.2007.02.046.
35. Labrie SJ, Frois-Moniz K, Osburne MS, Kelly L, Roggensack SE, SullivanMB, Gearin G, Zeng Q, Fitzgerald M, Henn MR, Chisholm SW. 2013.Genomes of marine cyanopodoviruses reveal multiple origins of diver-sity. Environ Microbiol 15:1356 –1376. https://doi.org/10.1111/1462-2920.12053.
36. Huang SJ, Zhang S, Jiao NZ, Chen F. 2015. Comparative genomic andphylogenomic analyses reveal a conserved core genome shared byestuarine and oceanic cyanopodoviruses. PLoS One 10:e0142962. https://doi.org/10.1371/journal.pone.0142962.
37. Bench SR, Hanson TE, Williamson KE, Ghosh D, Radosovich M, Wang K,Wommack KE. 2007. Metagenomic characterization of Chesapeake Bayvirioplankton. Appl Environ Microbiol 73:7629 –7641. https://doi.org/10.1128/AEM.00938-07.
38. Huang S, Zhang S, Jiao N, Chen F. 2015. Marine cyanophages demon-
Sun et al. Applied and Environmental Microbiology
August 2018 Volume 84 Issue 16 e00591-18 aem.asm.org 14
on June 12, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from

strate biogeographic patterns throughout the global ocean. Appl Envi-ron Microbiol 81:441– 452. https://doi.org/10.1128/AEM.02483-14.
39. Williamson SJ, Rusch DB, Yooseph S, Halpern AL, Heidelberg KB, Glass JI,Andrews-Pfannkoch C, Fadrosh D, Miller CS, Sutton G, Frazier M, VenterJC. 2008. The Sorcerer II Global Ocean Sampling Expedition: metag-enomic characterization of viruses within aquatic microbial samples.PLoS One 3:e1456. https://doi.org/10.1371/journal.pone.0001456.
40. Huang S, Wilhelm SW, Jiao N, Chen F. 2010. Ubiquitous cyanobacterialpodoviruses in the global oceans unveiled through viral DNA polymer-ase gene sequences. ISME J 4:1243–1251. https://doi.org/10.1038/ismej.2010.56.
41. Chen F, Wang K, Huang S, Cai H, Zhao M, Jiao N, Wommack KE. 2009.Diverse and dynamic populations of cyanobacterial podoviruses inthe Chesapeake Bay unveiled through DNA polymerase gene se-quences. Environ Microbiol 11:2884 –2892. https://doi.org/10.1111/j.1462-2920.2009.02033.x.
42. Mühling M, Fuller NJ, Millard A, Somerfield PJ, Marie D, Wilson WH,Scanlan DJ, Post AF, Joint I, Mann NH. 2005. Genetic diversity of marineSynechococcus and co-occurring cyanophage communities: evidence forviral control of phytoplankton. Environ Microbiol 7:499 –508. https://doi.org/10.1111/j.1462-2920.2005.00713.x.
43. Millard AD, Mann NH. 2006. A temporal and spatial investigation ofcyanophage abundance in the Gulf of Aqaba, Red Sea. J Mar Biol AssocU.K. 86:507–515. https://doi.org/10.1017/S0025315406013415.
44. Wang K, Wommack KE, Chen F. 2011. Abundance and distribution ofSynechococcus spp. and cyanophages in the Chesapeake Bay. ApplEnviron Microbiol 77:7459 –7468. https://doi.org/10.1128/AEM.00267-11.
45. Jameson E, Mann NH, Joint I, Sambles C, Muhling M. 2011. The diversityof cyanomyovirus populations along a North-South Atlantic Ocean tran-sect. ISME J 5:1713–1721. https://doi.org/10.1038/ismej.2011.54.
46. Malmstrom RR, Coe A, Kettler GC, Martiny AC, Frias-Lopez J, Zinser ER,Chisholm SW. 2010. Temporal dynamics of Prochlorococcus ecotypes inthe Atlantic and Pacific oceans. ISME J 4:1252–1264. https://doi.org/10.1038/ismej.2010.60.
47. Zinser ER, Johnson ZI, Coe A, Karaca E, Veneziano D, Chisholm SW. 2007.Influence of light and temperature on Prochlorococcus ecotype distribu-tions in the Atlantic Ocean. Limnol Oceanogr 52:2205–2220. https://doi.org/10.4319/lo.2007.52.5.2205.
48. Zinser ER, Coe A, Johnson ZI, Martiny AC, Fuller NJ, Scanlan DJ, ChisholmSW. 2006. Prochlorococcus ecotype abundances in the North AtlanticOcean as revealed by an improved quantitative PCR method. ApplEnviron Microbiol 72:723–732. https://doi.org/10.1128/AEM.72.1.723-732.2006.
49. Biller SJ, Berube PM, Lindell D, Chisholm SW. 2015. Prochlorococcus: thestructure and function of collective diversity. Nat Rev Microbiol 13:13–27. https://doi.org/10.1038/nrmicro3378.
50. Ahlgren NA, Noble A, Patton AP, Roache-Johnson K, Jackson L, RobinsonD, McKay C, Moore LR, Saito MA, Rocap G. 2014. The unique trace metaland mixed layer conditions of the Costa Rica upwelling dome support adistinct and dense community of Synechococcus. Limnol Oceanogr 59:2166 –2184. https://doi.org/10.4319/lo.2014.59.6.2166.
51. Mazard S, Ostrowski M, Partensky F, Scanlan DJ. 2012. Multi-locus se-quence analysis, taxonomic resolution and biogeography of marineSynechococcus. Environ Microbiol 14:372–386. https://doi.org/10.1111/j.1462-2920.2011.02514.x.
52. Huang S, Wilhelm S, Harvey H, Taylor K, Jiao N, Chen F. 2012. Novellineages of Prochlorococcus and Synechococcus in the global oceans.ISME J 6:285–297. https://doi.org/10.1038/ismej.2011.106.
53. Hanson CA, Marston MF, Martiny JBH. 2016. Biogeographic variation inhost range phenotypes and taxonomic composition of marine cya-nophage isolates. Front Microbiol 7:983. https://doi.org/10.3389/fmicb.2016.00983.
54. Cai H, Wang K, Huang S, Jiao N, Chen F. 2010. Distinct patterns ofpicocyanobacterial communities in winter and summer in the Chesa-peake Bay. Appl Environ Microbiol 76:2955–2960. https://doi.org/10.1128/AEM.02868-09.
55. Partensky F, Hess WR, Vaulot D. 1999. Prochlorococcus, a marine photo-synthetic prokaryote of global significance. Microbiol Mol Biol Rev 63:106 –127.
56. Needham DM, Sachdeva R, Fuhrman JA. 2017. Ecological dynamics andco-occurrence among marine phytoplankton, bacteria and myovirusesshows microdiversity matters. ISME J 11:1614 –1629. https://doi.org/10.1038/ismej.2017.29.
57. Hurwitz BL, Westveld AH, Brum JR, Sullivan MB. 2014. Modeling ecolog-
ical drivers in marine viral communities using comparative metagenom-ics and network analyses. Proc Natl Acad Sci U S A 111:10714 –10719.https://doi.org/10.1073/pnas.1319778111.
58. Frederickson CM, Short SM, Suttle CA. 2003. The physical environmentaffects cyanophage communities in British Columbia inlets. Microb Ecol46:348 –357. https://doi.org/10.1007/s00248-003-1010-2.
59. Sandaa RA, Larsen A. 2006. Seasonal variations in virus-host populationsin Norwegian coastal waters: focusing on the cyanophage communityinfecting marine Synechococcus spp. Appl Environ Microbiol 72:4610 – 4618. https://doi.org/10.1128/AEM.00168-06.
60. Parsons RJ, Breitbart M, Lomas MW, Carlson CA. 2012. Ocean time-seriesreveals recurring seasonal patterns of virioplankton dynamics in thenorthwestern Sargasso Sea. ISME J 6:273–284. https://doi.org/10.1038/ismej.2011.101.
61. Kashtan N, Roggensack SE, Rodrigue S, Thompson JW, Biller SJ, Coe A,Ding H, Marttinen P, Malmstrom RR, Stocker R, Follows MJ, StepanauskasR, Chisholm SW. 2014. Single-cell genomics reveals hundreds of coex-isting subpopulations in wild Prochlorococcus. Science 344:416 – 420.https://doi.org/10.1126/science.1248575.
62. Chow CET, Kim DY, Sachdeva R, Caron DA, Fuhrman JA. 2014. Top-downcontrols on bacterial community structure: microbial network analysis ofbacteria, T4-like viruses and protists. ISME J 8:816 – 829. https://doi.org/10.1038/ismej.2013.199.
63. Schwartz DA, Lindell D. 2017. Genetic hurdles limit the arms race be-tween Prochlorococcus and the T7-like podoviruses infecting them. ISMEJ 11:1836 –1851. https://doi.org/10.1038/ismej.2017.47.
64. Marston MF, Pierciey FJ, Shepard A, Gearin G, Qi J, Yandava C, SchusterSC, Henn MR, Martiny JBH. 2012. Rapid diversification of coevolvingmarine Synechococcus and a virus. Proc Natl Acad Sci U S A 109:4544 – 4549. https://doi.org/10.1073/pnas.1120310109.
65. Knap AH, Michaels AF, Close AR, Ducklow HW, Dickson AG. 1996. Pro-tocols for the Joint Global Ocean Flux Study (JGOFS) core measure-ments. JGOFS report no. 19. Reprint of Intergovernmental OceanicCommission manuals and guides, no. 29. UNESCO, Bergen, Norway.
66. Li QP, Hansell DA, Zhang JZ. 2008. Underway monitoring of nanomolarnitrate plus nitrite and phosphate in oligotrophic seawater. LimnolOceanogr Methods 6:319 –326. https://doi.org/10.4319/lom.2008.6.319.
67. Li QP, Hansell DA. 2008. Nutrient distributions in baroclinic eddies of theoligotrophic North Atlantic and inferred impacts on biology. Deep SeaRes Part 2 Top Stud Oceanogr 55:1291–1299. https://doi.org/10.1016/j.dsr2.2008.01.009.
68. Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliamH, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, HigginsDG. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948.https://doi.org/10.1093/bioinformatics/btm404.
69. Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB,Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B,Thallinger GG, Van Horn DJ, Weber CF. 2009. Introducing mothur: open-source, platform-independent, community-supported software for de-scribing and comparing microbial communities. Appl Environ Microbiol75:7537–7541. https://doi.org/10.1128/AEM.01541-09.
70. Kumar S, Stecher G, Tamura K. 2016. MEGA7: Molecular EvolutionaryGenetics Analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870 –1874. https://doi.org/10.1093/molbev/msw054.
71. Schloss PD, Handelsman J. 2005. Introducing DOTUR, a computer pro-gram for defining operational taxonomic units and estimating speciesrichness. Appl Environ Microbiol 71:1501–1506. https://doi.org/10.1128/AEM.71.3.1501-1506.2005.
72. Clarke KR. 1993. Nonparametric multivariate analyses of changes incommunity structure. Aust J Ecol 18:117–143. https://doi.org/10.1111/j.1442-9993.1993.tb00438.x.
73. Ahlgren NA, Rocap G, Chisholm SW. 2006. Measurement of Prochloro-coccus ecotypes using real-time polymerase chain reaction reveals dif-ferent abundances of genotypes with similar light physiologies. EnvironMicrobiol 8:441– 454. https://doi.org/10.1111/j.1462-2920.2005.00910.x.
74. Ahlgren NA, Rocap G. 2012. Diversity and distribution of marineSynechococcus: multiple gene phylogenies for consensus classificationand development of qPCR assays for sensitive measurement of clades inthe ocean. Front Microbiol 3:213. https://doi.org/10.3389/fmicb.2012.00213.
75. Fierer N, Jackson JA, Vilgalys R, Jackson RB. 2005. Assessment of soilmicrobial community structure by use of taxon-specific quantitative PCRassays. Appl Environ Microbiol 71:4117– 4120. https://doi.org/10.1128/AEM.71.7.4117-4120.2005.
Cooccurrence of Cyanopodoviruses and Cyanobacteria Applied and Environmental Microbiology
August 2018 Volume 84 Issue 16 e00591-18 aem.asm.org 15
on June 12, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from