methodology for high-throughput production of soluble recombinant
TRANSCRIPT

Methodology for high-throughput production of soluble recombinant proteins in
Escherichia coli
Katrin Markland M.Sc.
The Swedish Centre for Bioprocess Technology School of Biotechnology
Royal Institute of Technology
Stockholm 2006

© Katrin Markland Stockholm, 2006 ISBN 978-91-7178-561-9 The Swedish Centre for Bioprocess Technology, CBioPT School of Biotechnology Royal Institute of Technology 106 91 STOCKHOLM SWEDEN

Katrin Markland (2006): Methodology for high-throughput production of soluble recombinant proteins in Escherichia coli. The Swedish Centre for Bioprocess Technology, CBioPT, School of Biotechnology, Royal Institute of Technology (KTH), Stockholm, Sweden ABSTRACT The aim of this work was to investigate and determine central parameters that can be used to control and increase the solubility, quality and productivity of recombinant proteins. These central parameters should be applicable under the constraints of high-throughput protein production in Escherichia coli. The present investigation shows that alternative methods exist to improve solubility, quality and productivity of the recombinant protein. The hypothesis is that by reducing the synthesis rate of the recombinant protein, a higher quality protein should be produced. The feed rate of glucose can be used to decrease the synthesis rate of the recombinant protein. The influence of feed rate on solubility and proteolysis was investigated using the lacUV5-promoter and two model proteins, Zb-MalE and Zb-MalE31. Zb-MalE31 is a mutated form of Zb-MalE that contains two different amino acids. These altered amino acids greatly affect the solubility of the protein. The soluble fraction is generally twice as high using Zb-MalE compared to Zb-MalE31. Using a low feed rate compared to high benefits the formation of the full-length soluble protein. Furthermore, by using a low feed rate, the proteolysis can be decreased. One other factor that influences the solubility is the amount of inducer used. An increase from 100 µM to 300 µM IPTG only results in more inclusion bodies being formed, the fraction of soluble protein is the same. The quality aspect of protein production was investigated for a secreted version of Zb-MalE using two different feed rates of glucose and the maltose induced promoter PmalK. It was shown that when the protein was secreted to the periplasm, the stringent response as well as the accumulation of acetic acid (even for high feed rates) was reduced. The stringent response and accumulation of acetic acid are factors that are known to affect the quality and quantity of recombinant proteins. Transporting the protein to the periplasm results in this case on a lower burden on the cell, which leads to less degradation products being formed when the protein is secreted to the periplasm. Seeing the feed rate as a critical parameter, the high-throughput production would benefit from a variation in the feed rate. However, since the fed-batch technique is technically complicated for small volumes another approach is needed. E.coli strains that have been mutated to create an internal growth limitation that simulate fed-batch were cultivated in batch and were compared to the parent strain. It was shown that the growth rate and acetic acid formation was comparable to the parent strain in fed-batch. Furthermore it was shown that a higher cell mass was reached using one of the mutants when the cells were cultivated for as long time as possible. The higher cell mass can be used to reach a higher total productivity. Key words: Escherichia coli, high-throughput methodology, recombinant protein production, solubility, productivity, quality, cultivation technology, parallel reactors

LIST OF PUBLICATIONS This thesis is based on the following papers, which in the text will be referred to by their Roman numerals: I. Sandén A M, Boström M, Markland K, Larsson G (2005) Solubility and proteolysis of the
Zb-MalE and Zb-MalE31 proteins during overproduction in Escherichia coli. Biotechnology and Bioengineering 90: 239-247
II. Boström M, Markland K, Sandén A M, Hedhammar M, Hober S, Larsson G (2005). Effect
of substrate feed rate on recombinant protein secretion, degradation and inclusion body formation in Escherichia coli. Applied Microbiology and Biotechnology 68: 82-90
III. Markland K, Johansson E, Pedersen S, Larsson G (2006). Cell engineering of Escherichia
coli allows high cell density accumulation without fed-batch process control. Manuscript

TABLE OF CONTENTS
1. INDUSTRIAL BACKGROUND_______________________________________ 1
1.1 Process technology ______________________________________________ 2 1.1.1 Cultivation techniques____________________________________________________ 2
Batch technique ___________________________________________________________ 2 Fed-batch technique________________________________________________________ 2 High cell density fed-batch cultivation__________________________________________ 3
1.1.2 Bioreactors_____________________________________________________________ 4 Microtiter plates___________________________________________________________ 4 Multi-parallel reactors______________________________________________________ 4 Conclusions on reactor systems _______________________________________________ 6
1.2 Cell and vector engineering _______________________________________ 6 1.2.1 Glucose uptake _________________________________________________________ 6 1.2.2 Quality and solubility ____________________________________________________ 7
Protein folding ____________________________________________________________ 8 Stress induced proteolysis ___________________________________________________ 9 How to improve solubility and quality _________________________________________ 15 Summary________________________________________________________________ 17
1.2.3 Specific productivity ____________________________________________________ 17 Plasmid copy number ______________________________________________________ 17 Promoters _______________________________________________________________ 18 Induction level ___________________________________________________________ 19 Translation initiation ______________________________________________________ 19 Acetic acid formation ______________________________________________________ 20 Summary________________________________________________________________ 20
2. PRESENT INVESTIGATION _______________________________________ 21
2.1 Control of solubility and proteolysis in the cytoplasm (I) _____________ 22 Strategy_________________________________________________________________ 22 Results _________________________________________________________________ 23 Conclusions _____________________________________________________________ 25
2.2 Control of stress and proteolysis in the periplasm (II) ________________ 26 Strategy_________________________________________________________________ 26 Results _________________________________________________________________ 27 Conclusions _____________________________________________________________ 30
2.3 A cellular based system for substrate uptake rate control (III)_________ 30 Strategy_________________________________________________________________ 30 Results _________________________________________________________________ 31 Conclusions _____________________________________________________________ 34
3. CONCLUSIONS__________________________________________________ 35 4. ABBREVIATIONS ________________________________________________ 36
5. ACKNOWLEDGEMENTS __________________________________________ 37
6. REFERENCES ___________________________________________________ 38


Methodology for high-throughput production of soluble recombinant proteins in E.coli - Katrin Markland
1
1. INDUSTRIAL BACKGROUND Escherichia coli, or E.coli, is a simple organism that is widely used as a host in protein production. E.coli has several advantages such as the ease of use, the low cost and the speed in which a protein can be produced. These advantages are some of the reasons why pharmaceuticals such as growth hormones, growth factors and insulin are being produced in E.coli (Schmidt 2004). The development of a new drug is a stepwise procedure and the finalization of the HUGO-project and the human genome sequencing has lead to a paradigm shift in this chain of development. As a result, a multitude of new proteins need to be produced in order to determine their structure and function for further use in this development. For pharmaceutical companies it is important that the discovery and development of new pharmaceuticals is rapid and thus that the production of e.g. target proteins does not constitute a bottleneck. The identification of new protein pharmaceuticals starts with protein production from a cDNA sequence with little or no information about the protein the sequence code for. Further, since proteins are fundamentally different from each other due to their amino acid sequence, localization, function and structure; the production is still a trial-and-error effort to find the conditions best suited for each and every protein. In order to find the optimal conditions, a high-throughput production (HTPP) methodology is often used. With this methodology, different production variables are tried simultaneously in order to find a production hit as rapidly as possible. The larger the number of process parameters, the more likely it is that the protein is produced in adequate quantities. HTPP is generally based on standard techniques that previously have been developed in successful production processes and the HTPP concept serves as a pipeline where the aim is to rapidly clone, produce, purify and validate, a preferably soluble, full-length protein. A common system in use today for production is based on forceful technology e.g. the use of strong promoters, high plasmid copy numbers and high concentration of inducer. In order to increase the number of process variables these induction systems are used in combination with a purification tag which serves both as a purification handle but also as a solubility enhancer (Berrow et al. 2006). This has given good results in several cases and presumably where the protein is relatively simple. However, the high metabolic load that such systems create has in many cases lead to the opposite result i.e. a lower production. Furthermore, the production of more difficult proteins, such as membrane proteins, is more complicated e.g. due to the hydrophobic nature of the protein and was shown to result in cell death when production was induced (Miroux and Walker 1996). Few engineering principles are used in HTPP, except for production at different temperatures, due to limitations in the control of small reactor volumes. In order to produce difficult proteins, and proteins with today unknown structure and function, there is a likely need for a larger variation in cell and vector systems as well as production techniques. With larger number of variations, in both cell and vector systems as well as in process parameters, the number of cultivations to be performed increases which increase the probability of success. To cope with the larger number of cultivations, a cultivation format is needed where multiple cultivations can be performed at the same time. To perform as many cultivations in parallel as possible, the cultivation volume needs to be relatively small so the multi-parallel format is practically feasible. However, by using a small volume the more advanced cultivation techniques, such as fed-batch, are difficult to implement due to technical difficulties of process control.

Methodology for high-throughput production of soluble recombinant proteins in E.coli - Katrin Markland
2
The aim of this work was to investigate and determine central parameters that can be used to control and increase the solubility, quality and productivity of recombinant proteins. These central parameters should be applicable under the constraints of high-throughput production i.e. the multi-parallel format at restricted volume. This implies the investigation of two central areas of technology. The first is reactor and cultivation technology and the second area is cell and vector engineering. 1.1 Process technology The first part of a successful high-throughput methodology is knowledge about cultivation techniques and the multi-parallel bioreactors. These tools will be a first step on the way to a high cell density. With a high cell density, the concentration of the recombinant protein may be increased and this will enable further studies on the active and soluble protein.
1.1.1 Cultivation techniques Batch technique Screening for a new protein is often done in small scale and the easiest cultivation method to use in small scale is the batch technique. All necessary components (salts, trace elements and substrate) are added in concentrations to make the reaction rate un-restricted. This means that the specific growth rate (µ) will be at its highest until for example the cells have consumed the added substrate, exhausted all oxygen or inhibitory metabolic by-products have formed. During this time the cells will at first grow exponentially but when nutrients are depleted or inhibitory by-products are formed, the growth rate will decrease and the cells will stop growing. The only additives to a batch process during cultivation are air, a base or acid to control the pH and an antifoam agent if necessary. The cell growth during ideal conditions can be described by: dX/dt = µX, where dX/dt is the change in cell mass over time, µ is the specific growth rate (h-1) and X is the cell mass (g/L). Fed-batch technique Another cultivation technique is the fed-batch and this is the only way possible to reach high cell densities. The difference between batch and fed-batch is that a substrate feed is continuously added to the bioreactor during the cultivation making it possible to use different feed profiles to control growth. The fed-batch can start as a batch cultivation and when the initial substrate is depleted a feed is started. But in industry the feed is often started directly after inoculation of the bioreactor. The feed substrate is most often glucose in a high concentration, ≥ 600 g/L, to minimize the volume increase. The condition for growth rate limitation is: F/VSi < qs,maxX(t) where F is the feed rate of the limiting substrate (L/h), V is the cultivation volume (L), Si is the concentration of the substrate (g/L), qs,max is the maximum substrate consumption rate (g/g,h) and X(t) is the biomass concentration changing with time (g/L). The feed rate can be designed in multiple ways. A constant feed rate results in a decreasing growth rate as each cell receives less and less glucose. A exponential feed on the other hand results in a constant growth rate since the feed is increased in the same exponential pattern that the cells are growing in. Every cell receives the same amount of glucose for the duration of the cultivation. There are two advantages with the fed-batch technique compared to batch. The first is the reaction rate control and the second is the metabolic control.

Methodology for high-throughput production of soluble recombinant proteins in E.coli - Katrin Markland
3
The reaction rate control limits the growth rate. The bacteria will increase their cell mass with the same rate as the substrate being fed into the reactor. As long as there is enough glucose and the yield of biomass on consumed substrate,YX/S, is constant, the growth rate will also be constant. When the reaction rate is controlled, problems with cooling capacity and oxygen transfer can be avoided. Through the metabolic control, catabolite repression and sugar over-flow metabolism can be avoided. By reducing the over-flow metabolism, the accumulation of the by-product acetic acid may be reduced. However, one disadvantage with fed-batch cultivation is the need for more technically trained personnel as well as more advanced equipment that can handle e.g. feed-profiles. The difference between batch and fed-batch is illustrated in Figure1. Figure 1: The difference between batch and fed-batch cultivation techniques. F = feed rate of limiting substrate (L/h), Si = concentration of the substrate (g/L) High cell density fed-batch cultivation Strategies to improve cultivations have developed gradually since the 1920’s when 2 patent applications were submitted in Denmark and Germany. It had been discovered that high initial substrate and nutrient concentrations could inhibit the growth and could thus not be added all at once. Metabolic control In order to avoid the growth inhibition caused by high substrate and nutrient concentrations and also to control the metabolism, medium components are added gradually in a feed. When all glucose is added at the same time, the cell directs some of the glucose to the formation of acetic acid. Furthermore, if other medium components such as salts are added in to high concentrations, growth is also inhibited (Risenberg 1991). On the other hand, nutrient limitation can affect protein yields, plasmid copy number and plasmid stability. The plasmid copy number increased from 50-400 during phosphate limitation (Horn et al. 1990) and it resulted in an enhanced plasmid instability (Jones and Melling 1984). It has however, also been shown that the protein yield increased two-fold when cells are grown on glucose after the depletion of phosphate (Jensen and Carlsen 1990). A balance between too much and too little nutrients needs to be found to reach the best conditions for the cells. Limiting factors In high-cell density cultivation the oxygen demand increases with the growth rate. A higher growth rate requires more oxygen than a lower growth rate. Different methods are used to increase the oxygen supply. Examples are increasing the aeration rate, use of O2-enriched air or pure oxygen, decreasing the temperature or cultivating under pressure (Risenberg 1991). If only the supply of oxygen is considered, the theoretical maximum cell density is about 350 g/L dry cell weight with
Air in
Gas out
Air in
Gas out
F, Si

Methodology for high-throughput production of soluble recombinant proteins in E.coli - Katrin Markland
4
pure oxygen (Risenberg 1991). However, this is not possible in practice due to a high viscosity making it difficult to stir the cultivation as well as the volume of the cells. Another calculation considers the void volume in between the cells and the density of the cells. In 1 liter of culture the maximum wet weight is 800 g of E.coli cells. Since the dry weight is approximately 20-25% of the wet weight, this corresponds to about 200 g/L dry weight (Märkl et al. 1993). This is consistent with the maximum cell density practically reached, 190 g/l (Fuchs et al. 2002; Nakano et al. 1997).
1.1.2 Bioreactors Several types of bioreactors exist that are possible to use for high throughput production. Important aspects that need to be considered for all bioreactors are the sterility and the possibility to control and regulate the cultivation. Parameters that need to be controlled and regulated are for example the temperature, pH and oxygen tension. Furthermore, the screening of a protein is often done in a parallel format where several constructs are tried at the same time, so it is preferable with a reactor where parallel cultivations can be performed. Microtiter plates A microtiter plate offers a possibility to perform many identical and parallel reactions in a very small scale. This is the reason why microtiter plates have been used as miniature bioreactors for cell cultivation. The cultivation volume is 25 µL to 5 mL. The plates are most often made in plastic and the number of wells can be 6, 12, 24, 96, 384 and even as many as 1536 or 3456 (Mere et al. 1999). The wells can have a rectangular or a cylindrical shape and they are deep or shallow. Oxygen transfer and mixing is accomplished by orbital shaking of the entire plate and the rectangular shape enhances the mixing and oxygen transfer through their baffle-like function (Duetz et al. 2000). The plate is placed on a heated block that at the same time controls the temperature (Betts and Baganz 2006). The oxygen transfer rate can to a certain amount be increased by an increased shaking speed. With a modified shaker it has been shown to reach a maximum KLa of 175 h-1. After that higher shaking speeds only accomplishes that medium is spilled into other wells (Hermann et al. 2002). A solution is to have a cover on the plate to minimize the contamination risk between wells. This cover must at the same time allow the wells to be aerated through contact with air. To accomplish this, a membrane can be used (Duetz et al. 2000). At the same time, the membrane also helps to avoid medium evaporation which otherwise may be a problem during slow growing cultivations (Kumar et al. 2004). Other reports have shown that KLa values normally are in the range of 100-130 h-1 for microtiter plates (Kumar et al. 2004). These relatively low values depend on the fact that microtiter plates are shaken systems where the surface aeration is the sole source of oxygen. One difficulty with microtiter plates has been pH and DOT measurement and control. During the last years, plates with integrated fluorescent sensors for both DOT and pH have been developed. These plates can be used with a fluorescent reader equipped with a shaking device (John et al. 2003a; John et al. 2003b). Since it is difficult to control the pH, a medium with high buffer capacity is required. Multi-parallel reactors Multi-parallel reactors are similar to conventional fermenters in that temperature, pH and dissolved oxygen can be monitored and maintained at desired levels. They have a greater potential for monitoring and control than for example microtiter plates. The reactors are usually made of borosilicate glass, different types of plastics or stainless steel (Betts and Baganz 2006; Kumar et al. 2004).

Methodology for high-throughput production of soluble recombinant proteins in E.coli - Katrin Markland
5
A number of research groups are developing different reactor-prototypes in a multi-parallel format. Professor Weuster-Botz at the Technical University at München has developed a set consisting of 48 reactors (volume 8-15 mL) where pH, DOT and OD can be measured, pH can be controlled and fed-batch cultivation is possible. These reactors have a maximum KLa of 700 h-1 and cell dry weights of 20 g/L has been accomplished (Puskeiler et al. 2005; Weuster-Botz et al. 2005). This system seems to be highly efficient due to both the cultivation control and the high KLa values reached. Another system that has been developed is a set of miniaturized bubble columns. By using a 48-well microplate with porous frits in the bottom of each well to allow an upward flow of gas, a microplate with 48 bubble columns each 2 mL in volume was created. The maximum kLa is 220 h-1 and the dry cell weight has reached 10 g/L in this bubble column (Doig et al. 2005). A comparison of a set of commercial parallel fermentation systems can be seen in Table 1. The list is not intended to be complete but shows some examples. Some of these systems are equipped to handle fed-batch cultivations but requires then a larger number of tubes and wires for additions and measurements. The working volume can be as low as 35 mL in the Cellstation from Fluorometrix (Massachusetts, US) and up to 1000 mL in the Greta (Figure 2) from Belach Bioteknik (Stockholm, Sweden) (Hedrén et al. 2006). Manufacturer Volume
mL Stirrer Measured Controlled Cultivation
technology KLa h-1
Number of units
Cellstation (Fluorometrix)
≥ 35 Dual paddle impeller
pH, DOT, OD pH, DOT Batch NR 12
Fed-batch pro (Dasgip)
50-500 Orbital shaker
pH, DOT pH, DOT Batch, Fed-batch
1100 16
Sixfors (Infors) 200-500 Impeller pH, DOT pH, DOT Batch, Fed-batch
300 6
Greta (Belach) 200-1000
Impeller pH, DOT, OD pH, DOT Batch, Fed-batch
800 6-24
Table 1: Overview of some commercial multi-parallel bioreactors. NR = not reported. KLa values achieved from companies (personal communication) One drawback with these systems is the relatively high cost compared to microtiter plates. But if high biomass is required, these reactors have the advantage of higher KLa values. Since the working volume is relatively large, samples can be taken and more information can be gained during cultivation. However, this is generally more important in process development than in high-throughput production. Figure 2: The multi-parallel fermentation system Greta manufactured by Belach Bioteknik, Sweden

Methodology for high-throughput production of soluble recombinant proteins in E.coli - Katrin Markland
6
Conclusions on reactor systems For an efficient high-throughput system, i.e. a system where many parallel cultivations are rapidly processed at the same time, the microtiter plate is the reactor of choice due to its ability to run many parallel cultivations. However, the only cultivation technique possible in microtiter plates is the batch technique. This system is best suited for high yield proteins i.e. proteins produced to a high percentage of the total protein content. For other proteins, bioreactors are preferred. First for their larger cultivation volumes, second for the possibility to use fed-batch cultivation and third for their higher KLa values, which enable a higher biomass hence, possibilities for a higher total productivity.
1.2 Cell and vector engineering The goal in high-throughput protein production is to receive a high total productivity of the recombinant protein. The total productivity depends on the cultivation volume, the cell density and the specific productivity, i.e. Q = V*X*qp. To increase the total productivity a higher cell density, a larger cultivation volume or a higher specific productivity is required. But the total productivity is not the only important parameter. Since the recombinant protein is intended for structure and function determinations, it also needs to be active and of full-length. Finally since it is also important that the production of new proteins is rapid, a soluble protein is preferred. The cell is influenced by a lot of different factors or stimuli. These can be external due to changing environmental conditions such as altered pH or nutrient depletion. They can also be internal for example the situation when the recombinant protein production is induced. In order to produce a soluble, full-length and active protein with a high total productivity, the external and internal stimuli needs to be controlled so that the cell machinery functions properly and quality, solubility and productivity of the recombinant protein can be maintained.
1.2.1 Glucose uptake The cell uses glucose as a carbon source for cell growth, product formation, respiration and also by-product formation. In order to utilize the glucose, it needs to be transported from the medium and into the cell. The proteins OmpC and OmpF transport glucose into the periplasm during high glucose concentrations (Nikaido and Vaara 1985) whereas the protein LamB is responsible during glucose limitation (Death et al. 1993). When the glucose is present in the periplasm, it is actively transported into the cytoplasm by the phosphoenolpyruvate: phosphotransferase system (PTS). PTS transports and phosphorylates several sugars. The PTS consist of Enzyme I (EI) and a phosphohistidine carrier protein (HPr), which are both soluble and non-sugar specific. These proteins transfer a phosphate group from phosphoenolpyruvate to the sugar specific enzymes IIA and IIB. Furthermore, an integral membrane protein (IIC and sometimes IID) transports the sugars across the cytoplasmic membrane where the sugar is phosphorylated by enzyme IIB. Glucose is transported by the specific glucose enzyme, IIGlc as well as by the enzyme for mannose, IIMan (Curtis and Epstein 1975). The specific glucose enzyme, IIGlc consist of the soluble enzyme IIAGlc and the membrane permease IICBGlc. These two proteins are encoded by crr and ptsG respectively. The enzyme IIMan also consists of two parts, IIABMan and IICDMan. IIABMan is a homodimer enzyme coded by the gene manX and IICDMan is a membrane permease encoded by manYZ. From the literature it is known that the regulation of the glucose- and mannose-PTS systems is under the control of a repressor protein called Mlc (Kimata et al. 1998; Plumbridge 1998). This protein binds to the un-phosphorylated form of the enzyme IIBGlc when glucose is present in the

Methodology for high-throughput production of soluble recombinant proteins in E.coli - Katrin Markland
7
medium resulting in the de-repression of for example the genes ptsHI, ptsG and manXYZ (Kimata et al. 1998; Plumbridge 1998; Plumbridge 1999). During conditions without glucose in the medium, the enzyme IIBGlc~P is unable to bind the Mlc protein, which results in the repression of the above mentioned genes (Plumbridge 2002). Furthermore, it has also been shown that the transcription of ptsG is under the control of cAMP-CRP (Kimata et al. 1997).
1.2.2 Quality and solubility When producing proteins the quality and solubility are two important factors. In this thesis the definition of a high quality is the production of a full-length protein with correct amino acid sequence and which is correctly folded and active. The solubility is important when it comes to the time aspect of the production process. The purification of the protein will be more rapid if the protein is soluble than if inclusion bodies are formed and needs to be re-solubilized and reactivated. A relatively new method to monitor the solubility of a recombinant protein in the cytoplasm is by fusing it to the green fluorescent protein (GFP). When GFP is properly folded it is fluorescent and can be used as a solubility marker. This means that when the recombinant protein is correctly folded, GFP is fluorescent whereas it is not fluorescent if the recombinant protein has folded incorrectly (Waldo et al. 1999). However, all recombinant proteins are not possible to produce in the cytoplasm. These proteins can benefit from being secreted to the periplasm where the environment is completely different. The protein quality is affected by the amino acid sequence that can be influenced by the cDNA quality or by errors in the transcription or translation processes. In the transcription process, RNA polymerase can sometimes select and insert the wrong ribonucleotide. This can result in the incorporation of an incorrect amino acid during translation due to the faulty codon. In translation, missense errors can occur which is the substitution of one amino acid for another. This results from either that a tRNA is charged with the wrong amino acid or an anticodon-codon mismatch on the ribosome (Parker 1989). Other errors include the premature termination of translation because the ribosome is stalled or the addition of extra amino acids due to the fact that the termination codon is not recognized (Parker 1989). Frameshift errors can also occur. If the reading frame shifts, a termination codon may be formed before the intended termination codon resulting in a too short protein (Parker 1989). Errors also occur from post-translational modifications. One example is the production of human growth hormone. The correct form is 191 amino acids and the protein contains two intramolecular disulphide bridges. The hormone can be altered to exist in a methylated form where one or several amino groups have been lost. It can also receive one or two additional oxygen atoms giving an oxidized form (Gellerfors et al. 1990) The average number of translation errors is 10-4, which means that for every 10000 codons translated, one error occurs (Parker 1989). For example, the misreading of arginine resulted in incorporation of cysteine in flagellin, the subunit of bacterial flagella (Edelmann and Gallant 1977). Another report showed that norleucine was translated instead of methionine (Tsai et al. 1988). As much as 19 % of the methionine was replaced by norleucine when interleukin-2 was overexpressed in minimal medium, whereas the level of norleucine was only 3% in rich media. During starvation for certain amino acids, the translational errors increase to the extent that it is visible on a two-dimensional polyacrylamide gel. After 25 minutes of starvation for histidine, the misincorporation of amino acids leads to an altered charge of the protein. This charge shift leads to the formation of

Methodology for high-throughput production of soluble recombinant proteins in E.coli - Katrin Markland
8
several spots on a 2D-PAGE, with the same mobility in the SDS-PAGE but differing in the isoelectric focusing (Parker et al. 1978). The errors are often due to the fact that the codons used in a foreign gene are not the same as the codons used by the host to synthesis other highly expressed proteins. Therefore the rarely used amino acids will be depleted and the ribosome will stall while waiting for the correct amino acid. This increases the probability that an incorrect amino acid will be inserted instead of the limiting amino acid (Glick 1995). Protein folding During the last 20 years there has been a lot of work done in the area of protein folding to understand the mechanisms behind folding and misfolding. An important group of proteins in this process are the chaperones. They bind to exposed hydrophobic regions on proteins thus preventing the proteins from aggregating into insoluble and nonfunctional inclusion bodies and instead reach their native state (Wickner et al. 1999). There are three classes of chaperones that are important, folding chaperones (DnaK, DnaJ, GrpE, GroEL, GroES), holding chaperones (IbpB, Hsp31, Hsp33) and disaggregating chaperones (ClpB) (Baneyx 2004). Other important groups of molecules are foldases and proteases. Rate-limiting steps in the folding pathway are catalyzed by foldases for example peptidyl-prolyl cis/trans isomerases (PPIases) that accelerate trans to cis isomerization of peptidyl-prolyl bonds and thiol-disulfide oxidoreductases (Dsbs) that are involved in the formation of disulfide bridges (Baneyx 2004). If a protein is too damaged to be rescued by any other pathway it will be degraded by proteases (Baneyx 2004). When a cytoplasmic nascent polypeptide exits the ribosome it is either properly folded or it needs assistance to fold correctly. If aid is required the protein can either interact with a trigger factor, TF or with the chaperone DnaK and its co-chaperone DnaJ (Deuerling et al. 2003). Once released from the chaperones by a GrpE-catalyzed ADP/ATP exchange, three possibilities exist for the protein. The first possibility is that the protein is in a native form. The second is that it will interact with DnaK until it folds or thirdly it can be transferred to the GroEL-GroES system (Baneyx 1999; Baneyx and Palumbo 2003). During environmental stress native proteins may be unfolded and then it is the holding chaperones (IbpA/B, Hsp31 and Hsp33) that stabilize the protein until DnaK and GroEL can once again fold the protein (Veinger et al. 1998; Baneyx and Mujacic 2004). During severe or prolonged stress, the holding chaperones are unable to stabilize the protein and it will aggregate until the disaggregating chaperone (ClpB) can solubilize the protein (Baneyx and Mujacic 2004). Sometimes it is not possible to solubilize the protein and the aggregates will remain insoluble as inclusion bodies. See Figure 3 for the folding pathway. When inclusion bodies do form they are highly stable and resistant to proteases in vivo (Fahnert et al. 2004). But inclusion body formation is a result of an unbalance between protein precipitation and cell-mediated refolding and solubilisation and as a result the inclusion bodies may be dissolved (Mar Carrio and Villaverde 2001). Proteins that are toxic, unstable or easy to refold may benefit from being produced as inclusion bodies. But since there is no guarantee that the inclusion bodies will regain activity or result in a high yield of the refolded protein (Baneyx 1999), it is not optimal to produce the protein as inclusion bodies. For every protein, the specific conditions during refolding have to be optimized in respect to buffer composition, temperature, and protein concentration (Lilie et al. 1998). Furthermore, it is more expensive and time-consuming to purify and refold inclusion bodies than to purify soluble proteins (Sorensen and Mortensen 2005). The production of inclusion bodies is thus not an interesting system for high-throughput production due to the increased time for these processes.

Methodology for high-throughput production of soluble recombinant proteins in E.coli - Katrin Markland
9
Figure 3: A nascent polypeptide is first bound to TF or DnaK-DnaJ. When released, the folding intermediate may reach a native conformation or it will be transferred to GroEL-GroES for help in the folding process. Native proteins may also be stabilized by holding chaperones (IbpA/B, Hsp31, Hsp33) until it either is folded or aggregates. When the stress is reduced the disaggregating chaperones refolds the aggregated proteins. 1 Stress induced proteolysis In order to receive an active protein, it is vital that the protein is not degraded by proteases. During overexpression of proteins, there are several stress response systems that are activated by misfolded proteins. When these stress response systems are activated, they produce, among other proteins, proteases that may degrade the desired protein. The heat-shock response is induced due to overproduction of proteins in the cytoplasm (Gross 1996) and the envelope stress response is induced when misfolded proteins are detected in the periplasm (Mecsas et al. 1993). It has also been shown that the stringent response induces expression of heat-shock proteins (Grossman et al. 1985). The heat-shock response The heat-shock response is induced by a variety of stress conditions for example temperature shifts, harmful substances such as ethanol and the antibiotics nalidixic acid and trimetoprim (Booth 1999; Laskovska et al. 2003) and unfolded proteins (Allen et al. 1992; Gross 1996). When the heat-shock response is induced, nearly 50 heat-shock induced proteins (HSPs) are synthesized. Many of the HSPs are chaperones and proteases. It is important that these chaperones and proteases have a high affinity and folds the appropriate proteins at a high rate. The immediate stress is followed by an adaptation period after which the HSP synthesis rates have decreased to their normal levels (Arsène et al. 2000).
1 Reprinted by permission from Macmillan Publishers Ltd: [Nature Biotechnology], (Francois Baneyx and Mirna Mujacic, 2004, Recombinant protein folding and misfolding in Escherichia coli, Nature Biotechnology, Vol. 22, No 11, p 1399-1408), Copyright (2004).

Methodology for high-throughput production of soluble recombinant proteins in E.coli - Katrin Markland
10
The heat-shock response is controlled at the transcriptional level by the rpoH gene product, σ32
(Straus et al. 1987). When the σ32 –levels increase, the heat-shock response is induced (Arsène et al. 2000). The first mechanism is at the transcriptional level of rpoH. Four promoters, three σ70-dependent (P1, P4, P5) and one σE-dependent (P3), transcribe the rpoH gene (Fig 4). The use of each promoter seems to be temperature-dependent (Arsène et al. 2000). The second mechanism, affects translation of rpoH mRNA (Fig 4). During normal growth temperatures, cis-acting mRNA sites within the 5’ region of the rpoH messenger form secondary structures that block the ribosome-binding site. These secondary structures are temperature sensitive. At elevated temperatures, the structures melt thus enabling a more efficient translation of the rpoH mRNA (Moat et al. 2002). In Figure 4, the regulatory mechanism of the heat-shock response is illustrated. σ32 bind to RNA-polymerase and start the production of heat-shock proteins for example DnaK, DnaJ and GrpE. These proteins regulate the heat-shock response. During unstressful conditions when the amount of misfolded or unfolded proteins in the cell is low, the chaperones bind to σ32. This binding has a negative effect on stability and translation of σ32. During temperature up-shifts, the amount of misfolded proteins will increase thereby removing the chaperones from σ32. The sigma factor is free to start the production of HSPs. When the stress has decreased and the amount of misfolded proteins is lower, the chaperones will once again bind σ32 and repress its activities (Gross 1996). Figure 4: The regulatory mechanism of the heat-shock response. The chaperones DnaK, DnaJ and GrpE bind to σ32 when there are no proteins to fold. During heat-shock the level of misfolded proteins increase, forcing the chaperones to release σ32 and to help other proteins in their folding process. σ32 start the production of HSPs. When the heat-shock is released, the chaperones have no misfolded proteins to bind; hence they will bind σ32, thereby repressing it. Adapted from (Arsène et al. 2000; Gross 1996) Some of these 50 heat shock proteins produced are located within the σ32 regulon and function as chaperones (DnaK, DnaJ, ClpB, GroEL, GroES), proteases (Lon, ClpP, ClpX) or nucleotide exchanging factors (GrpE). Other heat shock proteins are located in the σE regulon for example Deg P, which can function both as a chaperone and a protease. The envelope stress response The cytoplasm and the periplasm in E.coli are two different environments. The periplasmic space is more viscous, it is devoid of ATP and it has oxidizing conditions to favour disulfide bond formation. It is therefore not surprising that there are specific systems to cope with stress in the periplasm. E.coli has two different stress response mechanisms in the periplasm. They are called the σE – and the Cpx- pathway.
rpoH
mRNA
σ32
Eσ32
Promoter Hsp genes
DnaK, DnaJ, GrpE (50 proteins)
Activity
Translation
Stability
σ70
P1 P3 P4 P5
σE

Methodology for high-throughput production of soluble recombinant proteins in E.coli - Katrin Markland
11
The σE-pathway is induced by heat, ethanol and overexpression of outer membrane proteins (Grossman et al. 1984; Mecsas et al. 1993; Rouvière et al. 1995). The pathway was discovered when the gene degP was studied. This gene codes for a periplasmic protease that is essential for survival at temperatures over 42°C (Lipinska et al. 1989). In response to heat-shock the levels of DegP is increased but it is not dependent on the sigma factor for cytoplasmic heat-shock σ32. Instead the degP gene is transcribed by another sigma factor, σE, which is a new heat-stable form of RNA-polymerase (Erickson and Gross 1989; Wang and Kaguni 1989). The operon that codes for σE also codes for the proteins RseA, RseB and RseC that are the key-players in the σE-pathway (De Las Penas et al. 1997; Missiakas et al. 1997). RseA is a protein that spans the membrane. The N-terminal of RseA is localized in the cytoplasm where it interacts with σE and the C-terminal faces the periplasm and interacts with RseB, a periplasmic protein (De Las Penas et al. 1997; Missiakas et al. 1997). As shown in Figure 5, during normal growth conditions envelope proteins are folded and correctly localized and the negative regulators RseA and RseB interact with σE (De Las Penas et al. 1997; Missiakas et al. 1997). But during stressful conditions, RseA is degraded by DegS, an inner membrane serine-protease, in the periplasm and by YaeL, a metallo-protease, in the cytoplasm thus releasing σE which is then free to bind to RNA-polymerase (Alba and Gross 2004; Alba et al. 2002; Kanehara et al. 2002). This is the start of the transcription of folding factors (SurA, FkpA), chaperones (Skp) and proteases (DegP) (Dartigalongue et al. 2001).
σE
σE
Figure 5: The σE pathway. During normal growth conditions the negative regulators RseA and RseB interact with σE. But when heat, ethanol or OMP’s are expressed, RseA is degraded by DegS on the periplasmic side and by YaeL on the cytoplasmic side of the membrane thus releasing σE that is free to bind to RNA-polymerase and start the transcription of folding factors (SurA, FkpA), chaperones (Skp) and proteases (DegP). Modified from (Raivio and Silhavy 2001)
OM
IM
Heat EtOH
OMPs RseB
RseB RseB
N
C
RseA
β -barrel proteins
Normal conditions Stressful conditions
RNAP
surA, degP, rpoH, rpoE, rseA, rseB, rseC
Misfolded envelope proteins
1
2
DegS
YaeL

Methodology for high-throughput production of soluble recombinant proteins in E.coli - Katrin Markland
12
The second envelope stress response is a two-component system consisting of CpxA and CpxR. It is induced by an alkaline pH (Danese and Silhavy 1998), misfolded P pilin subunits (Jones et al. 1997), overproduction of a rare outer membrane lipoprotein E, NlpE (Snyder et al. 1995) and by major alterations in membrane phospholipid composition (Mileykovskaya and Dowhan 1996). CpxA is an integral membrane protein with both a cytoplasmic and a periplasmic domain and it function as a sensor-protein (Weber and Silverman 1988). CpxR on the other hand function as a cytoplasmic response regulator (Dong et al. 1993). During non-stressful conditions, the sensing domain of CpxA interacts with a small protein called CpxP, which results in the inactivation of the Cpx-system, as shown in Figure 6 (Raivio et al. 1999). When the Cpx-pathway is induced, CpxP is removed from CpxA, which in turn is auto-phosphorylated. CpxR is then activated by phosphorylation when the phosphate is transferred from CpxA to CpxR (Raivio et al. 1999). The phosphorylated form of CpxR is then free to bind upstream of multiple promoters thereby activating transcription of folding factors, chaperones etc (Raivio and Silhavy 2001). Figure 6: The Cpx-pathway. During non-stressful conditions, CpxA interacts with CpxP which results in the inactivation of the Cpx-system. When the Cpx-pathway is induced by an alkaline pH, misfolded P pilin subunits or overproduction of NlpE, CpxP is titrated away from CpxA.CpxA is autophosphorylatedand CpxR is activated by phosphorylation when thephosphate is transferred fromCpxA to CpxR. The phosphorylated form of CpxR is then free to bindupstream of multiple promoters thereby activating transcription of folding factors, chaperones etc. (Raivio and Silhavy 2001)2
2 Reprinted, with permission, from the Annual Review of Microbiology, Volume 55 © 2001 by Annual Reviews, www.annualreviews.org
Outer membrane
periplasm
CpxA ATP ADP
autophosphorylation
kinase Phosphatase (ATP)
Inner membrane
CpxP
CpxP
>H
CpxP
STRESS
CpxP
CpxA >H-P
CpxR >D CpxR >D CpxR >D-P
Pi
degP, dsbA, ppiA, cpxP, cpxR, pap, pili
Folded protein
Misfolded protein

Methodology for high-throughput production of soluble recombinant proteins in E.coli - Katrin Markland
13
The stringent response The stringent response has been shown to trigger the induction of certain heat-shock proteins such as DnaK, GroEL and Lon (Grossman et al. 1985). Lon is a protease with a broad specificity and could increase the proteolysis of the recombinant protein. One hypothesis is that during overproduction of proteins, certain amino acids may be depleted leading to amino acid limitation, which is inducing the stringent response. The stringent response adjusts the cells’ macromolecular composition to the actual growth rate. During slow growth, the cells are small, have few ribosomes and less DNA, RNA, proteins, phospholipids and cell wall material compared to cells with a higher growth rate that are also larger and have more ribosomes (Lengeler and Postma 1999). When the cells suffer from carbon starvation or from a lack of charged tRNAs, the protein synthesis is blocked by stalled ribosomes. This stimulates the cell, and an alarmone called guanosine tetraphosphate (ppGpp) is produced. The alarmone ppGpp affects transcription by inhibiting DNA initiation. Translation is affected by the inhibition of tRNA synthesis, rRNA synthesis as well as the protein chain elongation. On the protein level, ppGpp is responsible for the degradation of ribosomal proteins as well as the production of universal stress proteins. Furthermore, the amino acid biosynthesis and the induction of the general stress response regulated by σs are stimulated by ppGpp as can be seen in Figure 7 (Cashel et al. 1996; Neidhardt et al. 1990). Figure 7: The effects of stringent response. Both positive and negative effects exist. Negative effects are inhibition of stable RNA, ribosomal proteins, elongation factors, fatty acid synthesis, lipid synthesis and DNA replication. Positive effects are induction of universal stress proteins, amino acid biosynthesis and induction of the general stress response controlled by rpoS, which activates many survival genes.3
3 Reprinted from Trends in Microbiology, Vol.13 No. 5, Magnusson, Farewell and Nyström, ppGpp: a global regulator in Escherichia coli, 236-242., Copyright (2005), with permission from Elsevier
Stable RNA
Ribosomal proteins
Elongation factors
Fatty acid Lipids
Cell wall
DNA replication
rpoS Universal stress
proteins
Amino acid biosynthesis Proteolysis
σs
Glycolysis Stasis survival genes
Oxidative stress
survival genes
Osmotic stress
survival genes
+ + + +
+ + + - - - -
RNAP
pppGpp
GTP + ATP
GDP + PPi
SpoT Stress and starvation
L11 RelA
Lack of charged tRNA
ppGpp

Methodology for high-throughput production of soluble recombinant proteins in E.coli - Katrin Markland
14
Mechanisms of the stringent response There are two different mechanisms in which a cell can produce the alarmone ppGpp. The first is the RelA-dependent pathway and the second is SpoT-dependent as also can be seen in Figure 7 (Magnusson et al. 2005). Both pathways use GTP and ATP to form first pppGpp and then ppGpp. During stringent response when the RelA pathway is induced, a pool of uncharged tRNAs has been generated. The uncharged tRNA will bind to the ribosomal A-site and stall the ribosome (Fig 8a); relA will then detect the stalled ribosome and bind to it (Fig 8b). After that RelA converts ATP and GTP to (p)ppGpp and AMP. RelA but not the uncharged tRNA in the ribosomal A-site will then be released (Fig 8c). When RelA is released from the ribosome it jumps to the next stalled ribosome to repeat the ppGpp synthesis (Fig 8d). Once the stress is lowered, a charged tRNA replaces the uncharged on the ribosome due to the higher affinity of the charged tRNA as can be seen in Figure 8e (Wendrich et al. 2002). Figure 8: The mechanism of the RelA pathway4 To restore its’ growth, the cell needs amino acids to “unstall” the ribosomes thus restarting the protein synthesis. When the stringent response has been activated and ppGpp has been formed (Fig 9a), the alarmone plays a crucial role in the balance between the two enzymes polyP kinase (PPK) and exopolyphosphatase (PPX). The enzyme polyP kinase (PPK) transfers a phosphate group from ATP to a short-chain of a linear polyphosphate (polyP) thus creating longer polyP-chains whereas PPX degrades polyP to phosphate groups (Fig 9b). PPX is inhibited by (p)ppGpp so during stringent response when (p)ppGpp builds up in the cell, the concentration of polyP increases (Fig 9c). PolyP forms a complex with four molecules of the protease Lon (Fig 9d). This complex binds to free ribosomal proteins and degrade them to amino acids (Fig 9e). The cell incorporates these amino acids into essential proteins that are required for survival (Kuroda 2006).
4 Reprinted from Molecular Cell, Vol. 10, Wendrich TM, Blaha G, Wilson DN, Marahiel MA, Nierhaus KH, Dissection of the mechanism for the stringent factor RelA, p 779-788, Copyright (2002) with permission from Elsevier.
E P A
Amino acid starvation
E P A E P A
RelA
RelA
ATP + GTP
(p)ppGpp +
AMP
E P A E P A
RelA
Stringent response
RelA detects blocked ribosome
RelA hops to the next blocked ribosome (p)ppGpp synthesis releases RelA
post stress: aminoacyl-tRNA has higher affinity
for A site
a b
c d
e

Methodology for high-throughput production of soluble recombinant proteins in E.coli - Katrin Markland
15
Figure 9: Amino acid formation during stringent response. Modified from reference (Kuroda 2006)
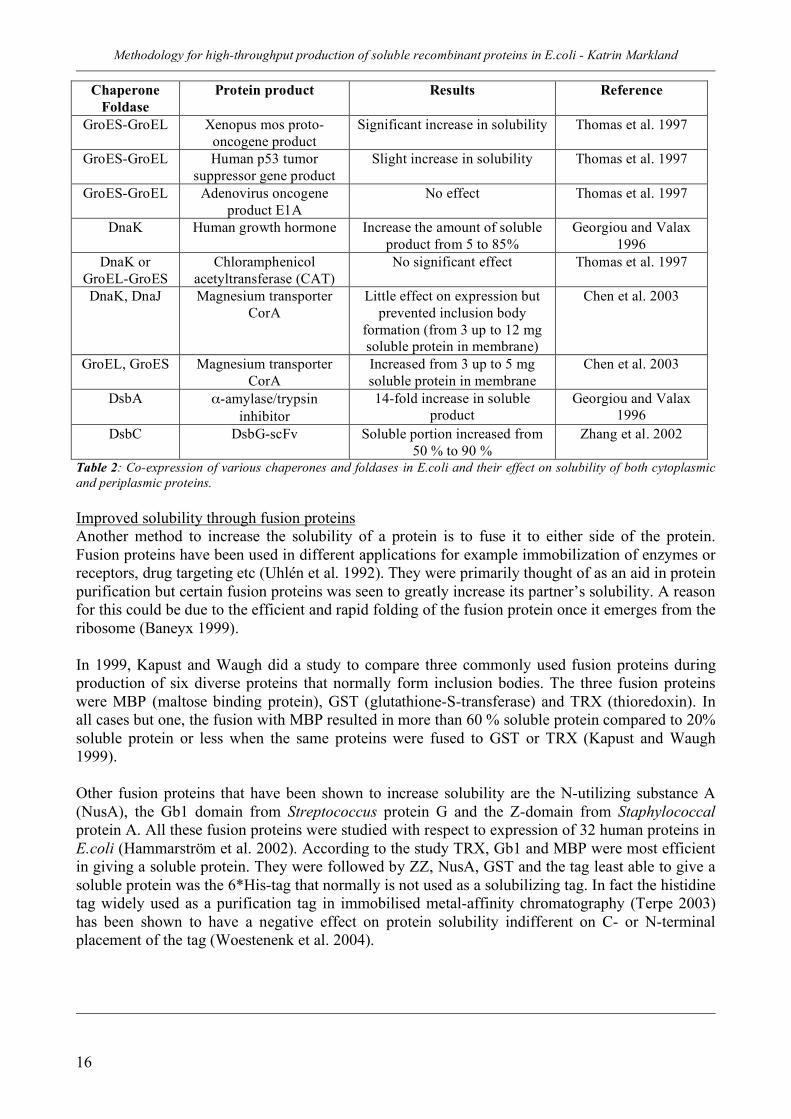
When ppGpp is accumulated during most stresses and nutrient limitations except for amino acid starvation, the SpoT-pathway is the responsible mechanism although not that much is known about the exact mechanism (Magnusson et al. 2005). One way to minimize the effect of the stress response on protein production is to use bacterial strains that lack the stringent response. It has been shown that chloramphenicol acetyltransferase (CAT) cultivated in batch has a 5-fold increase in activity when a strain lacking spoT and relA was grown in LB-medium (Dedhia et al. 1996). How to improve solubility and quality Since solubility and quality are important aspects of protein production, different methods have been developed that can be used to increase both solubility and quality. Co-expression of chaperones to increase solubility It was first shown in 1989 that co-expression of chaperones facilitated the solubility of a heterologous protein. Since then chaperone co-expression have been utilized to increase the solubility of many proteins. Examples of co-expression in E.coli can be seen in Table 2, and as can be seen, co-expression of chaperones is not an instant method to success. It is an trial-and-error effort to find the correct chaperone that interacts with the specific protein of interest. The optimal level of co-expression also needs to be found since overexpression of chaperones have drawbacks like cell filamentation (Georgiou and Valax 1996). Furthermore, co-expression of one chaperone may be insufficient to increase solubility since different types of chaperones act together (Langer et al. 1992). Adding 3% ethanol together with chaperone co-expression increased the production of preS2-S’-β-galactosidase probably due to induction of the heat-shock response in combination with the co-expression (Thomas and Baneyx 1997). The periplasmic production of single-chain Fv antibodies was improved markedly by not only overexpressing DsbC but also by fusing the antibody to DsbG (Zhang et al. 2002).
(p)ppGpp Ribosomal proteins
Amino acids
Lon-PolyP complex
Degradation
Lon
Stringent response
Inhibition
PPX
PPK
ATP + Pi
Ribosome
PolyP
P P P P
P P P
P P
a
b
c
d
e
Methodology for high-throughput production of soluble recombinant proteins in E.coli - Katrin Markland
16
Chaperone Foldase
Protein product Results Reference
GroES-GroEL Xenopus mos proto-oncogene product
Significant increase in solubility Thomas et al. 1997
GroES-GroEL Human p53 tumor suppressor gene product
Slight increase in solubility Thomas et al. 1997
GroES-GroEL Adenovirus oncogene product E1A
No effect Thomas et al. 1997
DnaK Human growth hormone Increase the amount of soluble product from 5 to 85%
Georgiou and Valax 1996
DnaK or GroEL-GroES
Chloramphenicol acetyltransferase (CAT)
No significant effect Thomas et al. 1997
DnaK, DnaJ Magnesium transporter CorA
Little effect on expression but prevented inclusion body
formation (from 3 up to 12 mg soluble protein in membrane)
Chen et al. 2003
GroEL, GroES Magnesium transporter CorA
Increased from 3 up to 5 mg soluble protein in membrane
Chen et al. 2003
DsbA α-amylase/trypsin inhibitor
14-fold increase in soluble product
Georgiou and Valax 1996
DsbC DsbG-scFv Soluble portion increased from 50 % to 90 %
Zhang et al. 2002
Table 2: Co-expression of various chaperones and foldases in E.coli and their effect on solubility of both cytoplasmic and periplasmic proteins. Improved solubility through fusion proteins Another method to increase the solubility of a protein is to fuse it to either side of the protein. Fusion proteins have been used in different applications for example immobilization of enzymes or receptors, drug targeting etc (Uhlén et al. 1992). They were primarily thought of as an aid in protein purification but certain fusion proteins was seen to greatly increase its partner’s solubility. A reason for this could be due to the efficient and rapid folding of the fusion protein once it emerges from the ribosome (Baneyx 1999). In 1999, Kapust and Waugh did a study to compare three commonly used fusion proteins during production of six diverse proteins that normally form inclusion bodies. The three fusion proteins were MBP (maltose binding protein), GST (glutathione-S-transferase) and TRX (thioredoxin). In all cases but one, the fusion with MBP resulted in more than 60 % soluble protein compared to 20% soluble protein or less when the same proteins were fused to GST or TRX (Kapust and Waugh 1999). Other fusion proteins that have been shown to increase solubility are the N-utilizing substance A (NusA), the Gb1 domain from Streptococcus protein G and the Z-domain from Staphylococcal protein A. All these fusion proteins were studied with respect to expression of 32 human proteins in E.coli (Hammarström et al. 2002). According to the study TRX, Gb1 and MBP were most efficient in giving a soluble protein. They were followed by ZZ, NusA, GST and the tag least able to give a soluble protein was the 6*His-tag that normally is not used as a solubilizing tag. In fact the histidine tag widely used as a purification tag in immobilised metal-affinity chromatography (Terpe 2003) has been shown to have a negative effect on protein solubility indifferent on C- or N-terminal placement of the tag (Woestenenk et al. 2004).

Methodology for high-throughput production of soluble recombinant proteins in E.coli - Katrin Markland
17
One disadvantage with the use of fusion proteins is that the solubility may decrease when the fusion partner is removed. Furthermore, the cleavage of the target protein from the fusion protein takes time and it is also expensive. Protein engineering to decrease proteolysis If a proteolytically sensitive site is found in a region of a protein that is not important for the activity, folding or other functional properties protein-engineering can be used to improve the solubility (Murby et al. 1996). It may be possible to delete or substitute amino acids in the sensitive site and obtain a more stable product. Even small changes in the primary structure can have large effects on protein solubility and stability (Schein 1990). Furthermore, many hydrophobic residues on the surface of the protein can lead to an insoluble protein (Sorensen and Mortensen 2005). In 1989 Hellebust et al showed that by exchanging a linker region where 2 basic amino acids contributed to the degradation of a fusion between Staphylococcus protein A (SpA) and E.coli β-galactosidase, both IgG-binding and β-galactosidase activity were retained (Hellebust et al. 1989). Another example is from the expression of a 101-amino acid long fragment from the human respiratory syncytial virus (RSV) major glycoprotein G where 4 phenylalanine residues and 2 cysteins were replaced by serines. This engineering increased the fraction of soluble protein from 27 to 75 % (Murby et al. 1995). It has also been shown that if surface-exposed hydrophobic residues are exchanged for positively charged residues the protein solubility can be significantly improved. A designed protein, 4ANK was only soluble between pH 3 to 4.25 but when six leucines were replaced by arginines to increase the net positive charge, the protein was soluble between pH 4 and 7 (Mosavi and Peng 2003). Summary During overproduction of a recombinant protein, the conditions during cultivation act as stimuli on the cell. The quality of the recombinant protein can be influenced by the different stress response systems that exist in the cell. The heat shock and envelope stress responses are for example induced by misfolded proteins and respond with the production of proteases that may degrade the recombinant product. Furthermore, carbon depletion caused by too little glucose may induce the stringent response, which puts the cell in a survival mode greatly affecting the production of the recombinant protein.
1.2.3 Specific productivity During high-throughput protein production it is important to get a high total product concentration. Amounts of 5-100 mg soluble protein are necessary for structural studies. A high total productivity is accomplished by either a scale-up of the cultivation volume, an increased biomass or an increased specific productivity. Due to the constraints of HTPP, the volume of the production process is fixed, as is the biomass possible to reach due to the limited oxygen transfer. The only possible way to increase the total productivity is to increase the specific productivity, that is the productivity per cell, which can be influenced by the rate of the protein synthesis. Plasmid copy number Multicopy plasmids have been used for protein overexpression during a long time and especially high copy number plasmids are used. They have several advantages, they are small (about 5 kbp), easy to manipulate and generally result in good expression (Jones et al. 2000). But there are disadvantages as well, the plasmid may become structurally unstable and it may shift the normal metabolism in the host also increasing the risk of instability (Jones et al. 2000). The plasmid copy number influences the productivity. There is a gene dosage effect, i.e. that the amount of product
Methodology for high-throughput production of soluble recombinant proteins in E.coli - Katrin Markland
18
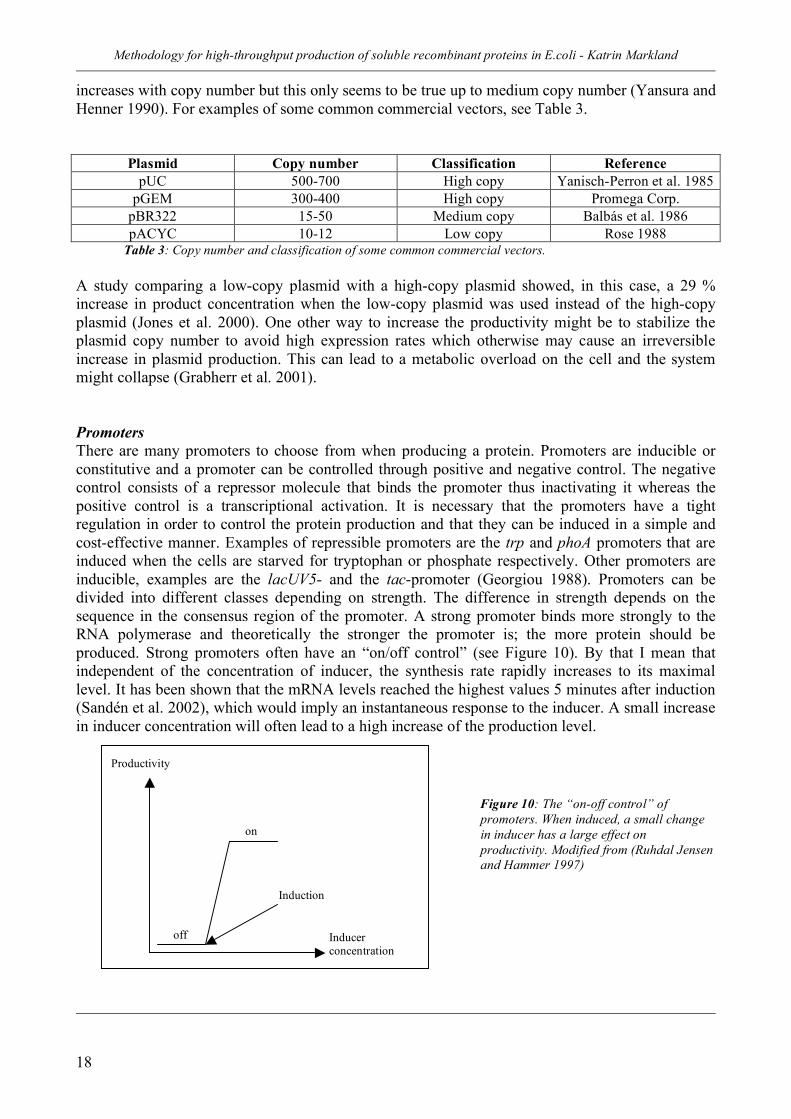
increases with copy number but this only seems to be true up to medium copy number (Yansura and Henner 1990). For examples of some common commercial vectors, see Table 3.
Plasmid Copy number Classification Reference pUC 500-700 High copy Yanisch-Perron et al. 1985
pGEM 300-400 High copy Promega Corp. pBR322 15-50 Medium copy Balbás et al. 1986 pACYC 10-12 Low copy Rose 1988
Table 3: Copy number and classification of some common commercial vectors. A study comparing a low-copy plasmid with a high-copy plasmid showed, in this case, a 29 % increase in product concentration when the low-copy plasmid was used instead of the high-copy plasmid (Jones et al. 2000). One other way to increase the productivity might be to stabilize the plasmid copy number to avoid high expression rates which otherwise may cause an irreversible increase in plasmid production. This can lead to a metabolic overload on the cell and the system might collapse (Grabherr et al. 2001). Promoters There are many promoters to choose from when producing a protein. Promoters are inducible or constitutive and a promoter can be controlled through positive and negative control. The negative control consists of a repressor molecule that binds the promoter thus inactivating it whereas the positive control is a transcriptional activation. It is necessary that the promoters have a tight regulation in order to control the protein production and that they can be induced in a simple and cost-effective manner. Examples of repressible promoters are the trp and phoA promoters that are induced when the cells are starved for tryptophan or phosphate respectively. Other promoters are inducible, examples are the lacUV5- and the tac-promoter (Georgiou 1988). Promoters can be divided into different classes depending on strength. The difference in strength depends on the sequence in the consensus region of the promoter. A strong promoter binds more strongly to the RNA polymerase and theoretically the stronger the promoter is; the more protein should be produced. Strong promoters often have an “on/off control” (see Figure 10). By that I mean that independent of the concentration of inducer, the synthesis rate rapidly increases to its maximal level. It has been shown that the mRNA levels reached the highest values 5 minutes after induction (Sandén et al. 2002), which would imply an instantaneous response to the inducer. A small increase in inducer concentration will often lead to a high increase of the production level.
Figure 10: The “on-off control” of promoters. When induced, a small change in inducer has a large effect on productivity. Modified from (Ruhdal Jensen and Hammer 1997)
Induction
Productivity
Inducer concentration
on
off

Methodology for high-throughput production of soluble recombinant proteins in E.coli - Katrin Markland
19
Induction level In high-throughput production a constitutive production might be a good choice. A constitutive production system has a promoter that is constantly active which means that no extra additions need to be made to the bioreactor. Auto-induction is another possibility where cells for example are growing on a mixture on glucose and lactose. Due to catabolite repression the cells will favour the uptake of glucose and when the glucose is exhausted, lactose will be utilized and will lead to induction of the lac-promoter. But a high expression level can cause a metabolic burden on the cell leading to decreasing growth rates (Glick 1995). Therefore an induced production of the recombinant protein can be a better option if the system is not overinduced. When using the lac-promoter, an optimal inducer concentration needs to be chosen to balance the decreasing yields of cells with increasing levels of target protein. When too high inducer concentration is used, in this example 3.4 mM isopropyl-β-D-thiogalactopyranoside (IPTG), the reduction in specific growth rate may be so severe that it will lead to cells entering stationary phase and even cell death (Bentley et al. 1991). Even an induction with 0.1 mM IPTG can lead to a 23% lower biomass than an un-induced culture (Andersson et al. 1996). Apart from reducing the growth rate of the bacteria, if too high concentrations of IPTG is used it will lead to a high production which causes stress responses in the cell. Heat-shock proteins such as DnaK, GroEL and GroES were observed 30 minutes after induction with 0.5 mM IPTG (Kosinski et al. 1992). One can also take advantage of the effects of IPTG. IPTG causes a high production rate, which in turn might lead to an increased permeability of the outer membrane, thus releasing 90 % of the produced periplasmic protein to the medium (Georgiou et al. 1988). This simplifies the purification process of the product since the medium contains fewer proteins that need to be removed compared to if the product were still in the cells. The productivity may be increased through a reduction in the IPTG concentration. When decreased from 1 mM to 0.01 mM, the concentration of functional and secreted Fab fragment was increased from below 90 ng/mL culture to almost 1700 ng/mL culture (Shibui and Nagahari 1992). Translation initiation The main elements of translation initiation region (TIR) are a Shine-Dalgarno sequence (SD), an initiation codon and a downstream region (DR). The SD is located 5-9 bases upstream of the initiation codon and it is important in the binding of mRNA to the 30S complex of the ribosome. SD base pairs to a region located at 3’ end of 16S rRNA that locates the ribosome to the proper initiation codon. In 91 % of the translation initiation regions in E.coli that have been sequenced, the initiation codon used is AUG, followed by GUG (8 %) and UUG (1 %) (Gualerzi and Pon 1990). The downstream region (DR) consists of 5-10 codons where the first codon, the +2 codon, has a strong influence on gene expression. The expression can vary 15-20 fold depending on which codon that is in this position. Codons that start with the base A generally give a high gene expression (Stenström et al. 2001b), whereas codons rich on G give low gene expression if located in +2 position (Gonzalez de Valdivia and Isaksson 2004). These G-rich codons, (AGG, CGG, UGG, GGG) gave a similar low expression when placed in position +3 and +5 in the downstream region (Gonzalez de Valdivia and Isaksson 2004). The gene expression is also influenced by the localization of the SD-region. A strong SD sequence located eight bases upstream from the initiation codon showed a strong positive effect on gene expression, whereas a negative effect was shown when the strong SD was placed 15 bases downstream of the initiation codon (Jin et al. 2006). It has also been shown that the SD region

Methodology for high-throughput production of soluble recombinant proteins in E.coli - Katrin Markland
20
upstream of the initiation codon and the DR that follows the initiation codon act together to influence gene expression (Stenström et al. 2001a). Acetic acid formation Acetic acid is produced during oxygen limitation and during glucose excess in the presence of oxygen. With a high glucose concentration, the carbon flux into the metabolic pathways is larger then what is demanded for biosynthesis and the surplus of carbon is used to produce acetic acid. This may be due to an imbalance in the glycolysis, a saturation of the TCA-cycle or the electron transport chain (Lee 1996). The formation of acetic acid can be decreased by controlling the growth rate below 0.2 h-1 or 0.35 h-1 for complex or defined media respectively (Meyer et al. 1984) as well as using glycerol instead of glucose as the carbon source (Lee 1996). It has been shown that acetic acid has a negative impact on both growth and protein production. At acetic acid concentrations over 6 g/L, the growth was inhibited (Jensen and Carlsen 1990). However, the specific production rate of a human growth hormone was decreased already below an acetic acid concentration of 2.4 g/L (Jensen and Carlsen 1990). Since acetate has a negative impact on productivity it is likely to increase the productivity by reducing the accumulation of acetic acid. One pathway to acetate production is from acetylcoenzyme A by the acetate kinase-phosphotransacetylase pathway. Through mutations in the enzymatic genes acetate accumulation was seven to nine-fold less than in the wild type as well as an improved accumulation of the product, Interleukin-2 was received (Bauer et al. 1990). Acetic acid accumulation could also be decreased by inserting the als gene from Bacillus subtilis that converts pyruvate to acetoin which is a far less toxic compound to the cell than acetate. The specific activity for the product ß-galactosidase was increased with 60 % with the inserted gene (Aristidou et al. 1995). Summary There are many ways in which the specific productivity of a recombinant protein can be influenced. The choice of plasmid copy number and promoter as well as how the production is induced and to what levels, affects the specific productivity. Furthermore, it is also influenced by the translation initiation region and through by-product formation such as acetate, which can inhibit both growth and product formation.

Methodology for high-throughput production of soluble recombinant proteins in E.coli - Katrin Markland
21
2. PRESENT INVESTIGATION E.coli is a host with a large capacity to produce target proteins that are structurally and functionally diverse. However, we believe that this capacity have not been used to its full potential specifically concerning the production of more difficult proteins, i.e. proteins that are toxic to the cells or proteins with a low solubility. The aim with this thesis is to investigate and determine central parameters that can be used to increase the solubility, quality and productivity of such recombinant proteins. The first problem faced was the product quality. The definition here is seen as a soluble, active, full-length protein. The hypothesis is that folding and post-translational modifications are the rate limiting steps in production of difficult recombinant proteins. Therefore, focus has been to reduce the synthesis rate of the nascent polypeptide. This may benefit the production of recombinant proteins, since the produced proteins often are heterologous to E.coli. This means that the recombinant protein is produced in an unfamiliar environment and the chaperones in E.coli can therefore have problems recognizing the recombinant protein. If the protein is produced to high concentrations, the folding and post-translation machinery will be overloaded thus being the rate-limiting step. By decreasing the synthesis rate, the rate-limiting step can be shifted from folding and post-translational modifications to earlier steps in the process. Is it possible to increase the solubility and decrease the proteolysis through an altered synthesis rate as accomplished by different feed rates? The second problem refers to the nature of the protein. There will be problems with specific groups of proteins produced in the cytoplasm despite a reduced synthesis rate. The reason can be that they are toxic to the cell or need a more oxidizing environment than the cytoplasm can provide. The common solution is to secrete these proteins to the periplasm. The periplasmic space is more viscous, it is devoid of ATP and it has oxidizing conditions. For instance, proteins containing disulfide bridges have to be transported unfolded to the periplasm where the oxidizing conditions favour disulfide bond formation. As for folding and post-translational modifications we believe that the transport across the cytoplasmic membrane is the rate-limiting step. The transporters in the membrane have low affinity for these recombinant proteins and will have problems coping with the transport since these proteins are unknown and in a high concentration. With a reduced synthesis rate, the transportation rate across the cytoplasmic membrane would be lower thus giving the transporters more time. Therefore the transportation would be improved by a reduced synthesis rate. Can stress and degradation be affected by different feed rates for secreted proteins? In earlier work it has been observed that the feed rate of substrate has a strong and pronounced impact on the protein production concerning amounts and quality of the protein (Boström and Larsson 2004; Sandén et al. 2002). The substrate concentration determines the uptake rate of glucose. A high substrate concentration will result in a higher glucose uptake rate, which means that the cell will have more carbon and energy that can be used for growth. Furthermore, a high feed rate results in an increased amount of RNA-polymerase molecules as well as an increased amount of ribosomes. This affects transcription and translation rates respectively and thus theoretically the synthesis rate of the protein. From earlier work it was indeed observed that the uptake rate also affected the synthesis rate of a recombinant protein (Sandén et al. 2002). Given the central role of the feed, the strategy in this work was to select the feed rate to influence the quality and quantity of a recombinant protein.

Methodology for high-throughput production of soluble recombinant proteins in E.coli - Katrin Markland
22
Given the importance of the substrate uptake rate and taken into consideration the constraints of HTPP with regards to simplicity and low volume, can the substrate uptake rate and the total productivity be influenced without using the fed-batch technique?
2.1 Control of solubility and proteolysis in the cytoplasm (I) Is it possible to increase the solubility and decrease the proteolysis through an altered synthesis rate as accomplished by different feed rates? When producing recombinant proteins in E.coli, the protein is often heterologous. This can result in folding problems since chaperones and foldases may have problems recognizing these recombinant proteins. By reducing the synthesis rate of the recombinant protein, the chaperones and foldases can have a lower folding rate per polypeptide to recognize and aid the folding. Strategy We have postulated that proteins naturally subjected to proteolysis or inclusion body formation would benefit from being produced in E.coli provided that we can find the parameter to control this quality issue. We have used an isogenic strain producing the proteins MalE and MalE31 for this evaluation. MalE, represents an easy protein as well as being a control protein, it is a 41 kDa periplasmic receptor used to transport maltose and maltodextrin. It has been used as a fusion protein to enhance the solubility of the fused target protein as was discussed above. A mutated form of the maltose-binding protein, MalE31, was chosen as the difficult model protein. The mutations consist of two altered consecutive amino acids. Aspartic acid and proline is used in MalE31 instead of glycine and isoleucine in the wild type MalE. The amino acids are situated in positions 32 and 33 respectively. This double mutant seems to be less stable than the wild type, MalE, and it has been suggested that the mutant is more prone to aggregate (Betton and Hofnung 1996). The lacUV5-promoter is a well-known promoter that is insensitive to catabolite repression (Arditti et al. 1973). This promoter does not depend on cAMP i.e. the transcription rate is independent of the glucose concentration and thus independent of the feed rate. The lacUV5-promoter is induced by the natural substrate allolactose that can be converted from lactose or by the synthetic analogue isopropyl ß-D-thiogalactopyranoside (IPTG). A drawback of the lac-promoter and its’ derivatives is the induction kinetics. A plot of the specific production rate as a function of the IPTG concentration (qp = f(IPTG)), shows that the induction curve is very steep and small variations in IPTG concentration give dramatic effects on the production (Ruhdal Jensen and Hammer 1997). The reason for this is that IPTG also induces its’ own transport system, the permease LacY. This can be overcome and we have constructed a lacY mutant where IPTG is diffusing into the cell rather than being actively transported and this results in that the promoter is more precisely regulated (Ruhdal Jensen and Hammer 1997). The vectors used in this work have a low copy number and the two recombinant proteins are under the control of the lacUV5-promoter . They are fused to a purification tag that allows for an easy identification and quantification of the proteins through Western blot using antibodies against the tag. The tag used was Zbasic or Zb, which is a 7-kDa purification tag originating from the B-domain of Staphylococcal protein A. Positive charges have been added to the original domain thus making it positively charged even at high pH-values where it can be efficiently used in ion-exchange chromatography (Gräslund et al. 2000).
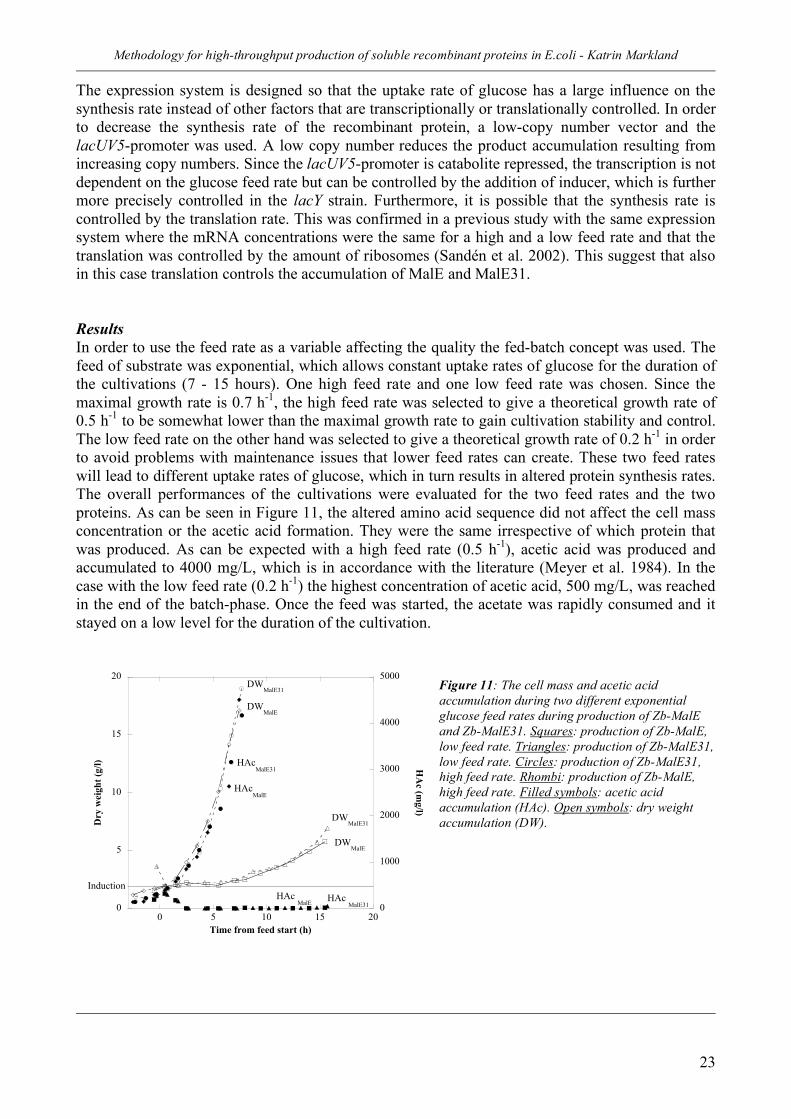
Methodology for high-throughput production of soluble recombinant proteins in E.coli - Katrin Markland
23
The expression system is designed so that the uptake rate of glucose has a large influence on the synthesis rate instead of other factors that are transcriptionally or translationally controlled. In order to decrease the synthesis rate of the recombinant protein, a low-copy number vector and the lacUV5-promoter was used. A low copy number reduces the product accumulation resulting from increasing copy numbers. Since the lacUV5-promoter is catabolite repressed, the transcription is not dependent on the glucose feed rate but can be controlled by the addition of inducer, which is further more precisely controlled in the lacY strain. Furthermore, it is possible that the synthesis rate is controlled by the translation rate. This was confirmed in a previous study with the same expression system where the mRNA concentrations were the same for a high and a low feed rate and that the translation was controlled by the amount of ribosomes (Sandén et al. 2002). This suggest that also in this case translation controls the accumulation of MalE and MalE31. Results In order to use the feed rate as a variable affecting the quality the fed-batch concept was used. The feed of substrate was exponential, which allows constant uptake rates of glucose for the duration of the cultivations (7 - 15 hours). One high feed rate and one low feed rate was chosen. Since the maximal growth rate is 0.7 h-1, the high feed rate was selected to give a theoretical growth rate of 0.5 h-1 to be somewhat lower than the maximal growth rate to gain cultivation stability and control. The low feed rate on the other hand was selected to give a theoretical growth rate of 0.2 h-1 in order to avoid problems with maintenance issues that lower feed rates can create. These two feed rates will lead to different uptake rates of glucose, which in turn results in altered protein synthesis rates. The overall performances of the cultivations were evaluated for the two feed rates and the two proteins. As can be seen in Figure 11, the altered amino acid sequence did not affect the cell mass concentration or the acetic acid formation. They were the same irrespective of which protein that was produced. As can be expected with a high feed rate (0.5 h-1), acetic acid was produced and accumulated to 4000 mg/L, which is in accordance with the literature (Meyer et al. 1984). In the case with the low feed rate (0.2 h-1) the highest concentration of acetic acid, 500 mg/L, was reached in the end of the batch-phase. Once the feed was started, the acetate was rapidly consumed and it stayed on a low level for the duration of the cultivation.
Figure 11: The cell mass and acetic acid accumulation during two different exponential glucose feed rates during production of Zb-MalE and Zb-MalE31. Squares: production of Zb-MalE, low feed rate. Triangles: production of Zb-MalE31, low feed rate. Circles: production of Zb-MalE31, high feed rate. Rhombi: production of Zb-MalE, high feed rate. Filled symbols: acetic acid accumulation (HAc). Open symbols: dry weight accumulation (DW).
0
5
10
15
20
0
1000
2000
3000
4000
5000
0 5 10 15 20
Dry
wei
gh
t (g
/l)
HA
c (mg
/l)
Time from feed start (h)
DWMalE31
DWMalE
DWMalE31
DWMalE
HAc MalE31
HAcMalE31
HAcMalE
HAc MalE
Induction

Methodology for high-throughput production of soluble recombinant proteins in E.coli - Katrin Markland
24
Soluble and insoluble fractions of the proteins were analyzed. The proteins were detected by Western blots using antibodies against protein A that will bind to the purification tag. As can be seen in Figure 12 the soluble fractions are generally higher than the insoluble fractions that consist of inclusion bodies. Both the soluble and the insoluble fractions increase with the inducer concentration. Using the highest amount of inducer does in most cases not result in more soluble protein but only results in an increased inclusion body formation. Furthermore, a low feed rate gives higher product concentrations than the high feed rate. And when comparing the two proteins, it is the native form (Zb-MalE) that is accumulated to the highest levels of both soluble protein and inclusion bodies. The lowest amount of soluble protein is achieved by the high feed rate in combination with the mutated form of the protein and the highest concentration of inducer. The reason for the higher ratio of insoluble proteins may come from harsh environmental conditions resulting in stressful conditions for the bacteria. These stressful conditions can render the holding chaperones unable to stabilize the protein thus causing it to aggregate.
Figure 12: The soluble and insoluble fractions of Zb-MalE and Zb-MalE31 as a function of feed rate and IPTG-concentration. All fractions are compared relative to a standard of Zb-MalE. The samples were taken at a time corresponding to five generations of production at exponential feed. In order to determine the reason for this lower solubility, the proteolytic stability of the proteins was investigated. This was done by withdrawing a sample from the bioreactor and adding chloramphenicol to stop the protein synthesis. After the addition of chloramphenicol, total protein samples were taken every five minutes and analyzed using Western blots. As can be seen in Figure 13a, the relative concentration of the native protein is around 100 % for the duration of the experiment. This was true for both low and high feed rates as well as for different concentrations of IPTG. On the western blots, some degradation bands in the sizes of 30, 33 and 38 kDa were visible but they could not be characterized further due to low concentrations. For the mutated form of MalE, the scenario looks completely different as can be seen in Figure 13b. During low feed rates, the relative concentration of the protein seems to be stable at first. But after 10-15 minutes, the protein is subjected to proteolysis and after 45 minutes, only 40-50 % remains of the soluble protein. The scenario for the high feed rate is even more severe. The product concentration is rapidly decreasing from the first minute to the end of the experiment. From initial values of 100 %, only 10 % soluble protein remains after 45 minutes. In both cases, the concentration of IPTG does not seem to be important for the proteolysis. As was the case for Zb-
Rel
ati
ve
pro
d c
on
c.
0
0,01
0,02
0,03
5 3010
030
0 5 3010
030
0 5 3010
030
0 3010
030
0
Soluble
Insoluble
IPTG conc. (µM)
Zb-MalE31Zb-MalE
High feed High feedLow feedLow feed
0.03
0.02
0.01
0
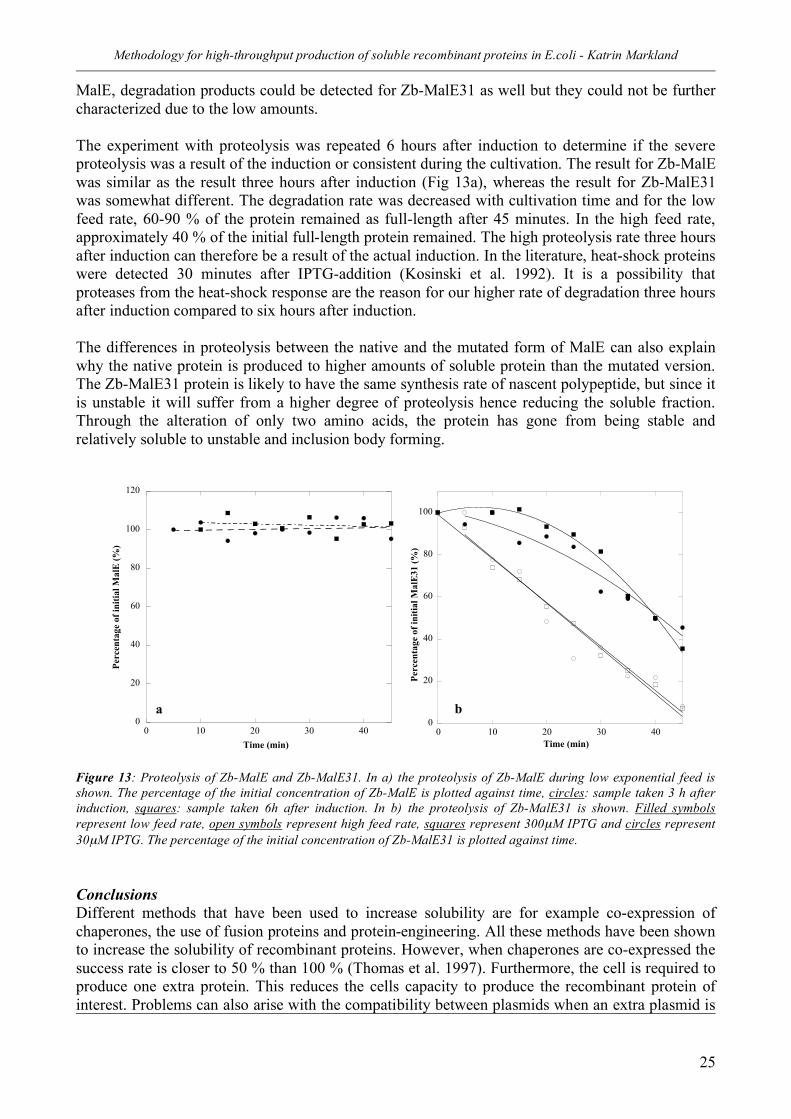
Methodology for high-throughput production of soluble recombinant proteins in E.coli - Katrin Markland
25
MalE, degradation products could be detected for Zb-MalE31 as well but they could not be further characterized due to the low amounts. The experiment with proteolysis was repeated 6 hours after induction to determine if the severe proteolysis was a result of the induction or consistent during the cultivation. The result for Zb-MalE was similar as the result three hours after induction (Fig 13a), whereas the result for Zb-MalE31 was somewhat different. The degradation rate was decreased with cultivation time and for the low feed rate, 60-90 % of the protein remained as full-length after 45 minutes. In the high feed rate, approximately 40 % of the initial full-length protein remained. The high proteolysis rate three hours after induction can therefore be a result of the actual induction. In the literature, heat-shock proteins were detected 30 minutes after IPTG-addition (Kosinski et al. 1992). It is a possibility that proteases from the heat-shock response are the reason for our higher rate of degradation three hours after induction compared to six hours after induction. The differences in proteolysis between the native and the mutated form of MalE can also explain why the native protein is produced to higher amounts of soluble protein than the mutated version. The Zb-MalE31 protein is likely to have the same synthesis rate of nascent polypeptide, but since it is unstable it will suffer from a higher degree of proteolysis hence reducing the soluble fraction. Through the alteration of only two amino acids, the protein has gone from being stable and relatively soluble to unstable and inclusion body forming.
Figure 13: Proteolysis of Zb-MalE and Zb-MalE31. In a) the proteolysis of Zb-MalE during low exponential feed is shown. The percentage of the initial concentration of Zb-MalE is plotted against time, circles: sample taken 3 h after induction, squares: sample taken 6h after induction. In b) the proteolysis of Zb-MalE31 is shown. Filled symbols represent low feed rate, open symbols represent high feed rate, squares represent 300µM IPTG and circles represent 30µM IPTG. The percentage of the initial concentration of Zb-MalE31 is plotted against time. Conclusions Different methods that have been used to increase solubility are for example co-expression of chaperones, the use of fusion proteins and protein-engineering. All these methods have been shown to increase the solubility of recombinant proteins. However, when chaperones are co-expressed the success rate is closer to 50 % than 100 % (Thomas et al. 1997). Furthermore, the cell is required to produce one extra protein. This reduces the cells capacity to produce the recombinant protein of interest. Problems can also arise with the compatibility between plasmids when an extra plasmid is
0
20
40
60
80
100
120
0 10 20 30 40
Per
cen
tag
e o
f in
itia
l M
alE
(%
)
Time (min)
a0
20
40
60
80
100
0 10 20 30 40
Per
cen
tag
e o
f in
itia
l M
alE
31
(%
)
Time (min)
b

Methodology for high-throughput production of soluble recombinant proteins in E.coli - Katrin Markland
26
used to express the chaperone. When fusion proteins are used, the cell is also required to produce one additional protein thereby consuming resources that could have been used for the recombinant protein. Additionally, the cost of cleaving the fusion protein from the recombinant product can be high and time consuming. In order to succeed with protein-engineering the structure of the protein needs to be known. Since the aim is to produce proteins for structure determination, this is not possible. Furthermore, protein engineering requires an additional step in the protein production process. This is a considerable drawback since production is to be as rapid as possible. Instead of using these methods we have shown that simply by using different feed rates of glucose, i.e. different glucose uptake rates, the solubility can be increased and the proteolysis can be reduced. A low feed rate results in the highest solubility and the lowest proteolysis. Therefore it is possible to use an altered synthesis rate as accomplished by different feed rates to increase the solubility and decrease the proteolysis without using extra and time-consuming steps in the protein production.
2.2 Control of stress and proteolysis in the periplasm (II) Can stress and degradation be affected by different feed rates for secreted proteins? Several proteins are not possible to produce to a high level in the cytoplasm, instead the periplasm is the natural alternative. For these proteins, we believe that the transport across the cytoplasmic membrane is the rate-limiting step in folding and post-translational modifications. We thus believe, that by reducing the synthesis rate of the protein these problems may be diminished. It is natural to decrease the synthesis as early as possible in the production chain due to carbon and energy considerations. In this work we have thus attempted to put the regulation under transcriptional control and we are using a promoter, PmalK, with different induction kinetics. Strategy We have investigated a new promoter for use in recombinant protein production, i.e. the promoter malK (Boström and Larsson 2002; Boström and Larsson 2004). MalK is a gene of the maltose operon and its’ promoter depends on both activation of MalT and formation of cAMP-CRP. This is achieved through three separate mechanisms and depending on how many mechanisms that are activated at the same time; the promoter is induced at different levels through different kinetics. The first mechanism consists of the formation of the inducer maltotriose from maltose taken up from the medium. The maltotriose leads to the transcriptional activation through activation of MalT. The second mechanism builds on the transcription from cAMP-CRP, which is formed when the glucose concentration in the medium is low and this activates the malK-promoter directly. Thirdly, maltotriose can also be formed from the degradation of internal glycogen and trehalose. See Figure 14 for the activation mechanisms. Figure 14: The activation of the malK-promoter (Boström and Larsson 2002)
Low glucose Maltose
cAMP-CRP formation
TRANSCRIPTION MALTOTRIOSE
Glycogen/trehalose
MalK MalT i MalK MalT a
medium
cytoplasm
degradation

Methodology for high-throughput production of soluble recombinant proteins in E.coli - Katrin Markland
27
Depending on how many cAMP-CRP and MalT binding sites that are occupied, the induction reaches different levels and kinetics from inducer exclusion and catabolite repression. It has been suggested that at least three out of four cAMP-CRP binding sites and all five MalT binding sites should be occupied for full induction. The lowest production rates came from batch cultivations on glucose (lack of maltotriose) or maltose (lack of cAMP) and to increase the production rate, a glucose-limited fed-batch is utilized where the formation of cAMP is accomplished. By using this fed-batch, glycogen is at the same time degraded to maltotriose. If the glucose-limited fed-batch is followed by an addition of maltose, more maltotriose is formed resulting in the highest production rates (Boström and Larsson 2004). The model protein in (I), Zb-MalE, is recognized as a periplasmic receptor protein and was chosen once again. To transport Zb-MalE to the periplasm, the signal peptide from OmpA was fused to the N-terminal of the protein. The expression vector was a low copy number plasmid to minimize the copy number effects. In this case the synthesis rate of the recombinant protein is controlled by the malK-promoter in combination with a low copy number plasmid which reduces the total synthesis rate. The malK-promoter does furthermore not have the same “on-off control” as the lacUV5-promoter, which makes it easier to control. It should therefore be possible to decrease the protein synthesis rate through the transcription rate thus allowing the control through medium uptake. Results The different feed rates were accomplished by using the fed-batch technique with glucose as the substrate. The feed rates were exactly the same as in work (I). The malK-promoter was induced by the addition of maltose after the glucose fed-batch phase. When glucose is replaced with maltose, the cell is forced to change its uptake system to use the new substrate. This has effects on the cell mass accumulation as can be seen in Figure 15. Independent of feed rate, both cytoplasmic production systems suffer from a 2-hour long lag-phase during the substrate change, which is well known and has been described in the literature since the 1940’s. However this lag-phase is drastically reduced when Zb-MalE is transported to the periplasm. This shows that the change of uptake system is influenced by the location of the recombinant product.
Figure 15: Cell mass accumulation during two different exponential feed rates of glucose followed by addition of maltose. Open symbols: cytoplasmic production, closed symbols: secretion to the periplasm. Squares: high feed rate, circles: low feed rate. Plus sign: cell accumulation without plasmid. Vertical lines indicate the addition of maltose.
0
5
10
15
20
25
30
0 5 10 15 20
Dry
wei
gh
t (g
/l)
Time from feed start (h)
maltose
addition
maltose
addition
glucose feed 0.5glucose feed 0.2

Methodology for high-throughput production of soluble recombinant proteins in E.coli - Katrin Markland
28
The recombinant protein suffers from degradation as can be seen in Figure 16. The soluble fraction of the protein was detected with Western blot using antibodies against protein A. Looking at the cytoplasmic production; the full-length protein is only visible at the low feed rate and not the high. This may be due to severe degradation or inclusion body formation before the protein is properly folded. Degradation bands are visible at both feed rates, the most abundant at 38- and 33-kDa. In the periplasmic system the high feed rate gave the highest amount of full-length protein but degradation bands are seen here as well. The reason for this degradation in both the cytoplasmic and periplasmic production systems may be stress responses in the cell. For the cytoplasmic production, the overproduction of recombinant proteins induces the heat-shock response whereas the envelope stress response is induced when proteins are misfolded in the periplasm. Both stress response systems produce among other proteins proteases, which can degrade the recombinant protein. The product accumulation was also investigated after the addition of maltose, since it has been shown to increase the production levels (Boström and Larsson 2004). The product pattern could not be distinguished from the production during the glucose-feed phase, however the sample taken in the maltose phase only covered the transition between glucose and maltose. Figure 16: The protein quality during cytoplasmic production and during secretion to the periplasm. Western blots showing the accumulation of soluble Zb-MalE and its degradation products during production at two selected glucose feed-profiles in fed-batch cultivation (g) and after the addition of maltose (m). M = molecular marker, R=purified Zb-MalE, Low = low feed rate, high = high feed rate. FL = full-length protein 48 kDa, 38 = a 38 kDa degradation product, 33 = a 33 kDa degradation product. In order to understand what caused the differences regarding degradation, the accumulation of acetic acid was investigated since acetate is known to influence productivity (Jensen and Carlsen 1990). Acetate accumulation is as stated in literature depending on the specific growth rate. When the cells have a growth rate below 0.35 h-1, there should be no accumulation of acetic acid. And as expected in the low feed systems the acetic acid concentration is less than 20 mg/L at the time of maltose addition, see Figure 17. After this time the concentration increases up to 70-90 mg/L, however there is only a slight difference between cytoplasmic production and when the protein is transported to the periplasm. Using the high feed rate, acetate accumulation would be expected. During cytoplasmic production at high feed rate, the concentration was as high as 6500 mg/L at the time of maltose addition. During the change in the substrate uptake system, the cells consumed some of the acetic acid and the final concentration was about 4000 mg/L. The secretion of the product showed an altogether different picture. The acetate concentration never reached above 800 mg /L.
52
35
28 21
M R g g g g m m m m
Low Low High High
cytoplasm periplasm
FL 38 33
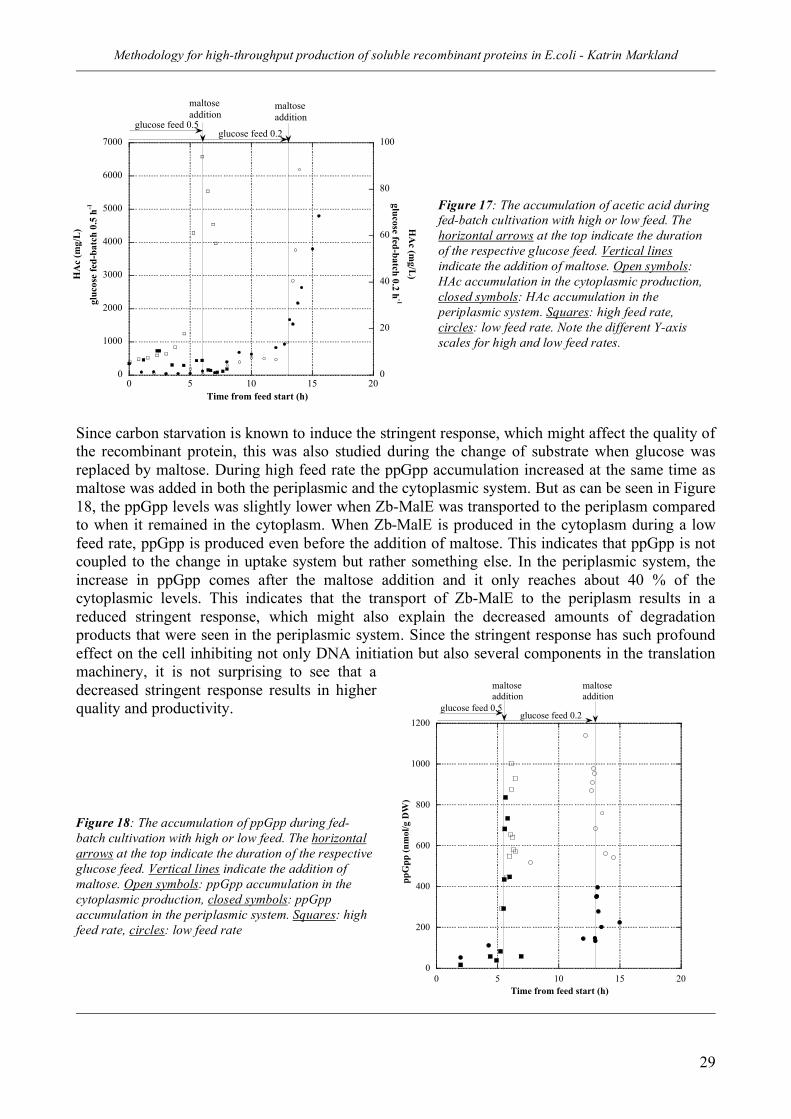
Methodology for high-throughput production of soluble recombinant proteins in E.coli - Katrin Markland
29
Figure 17: The accumulation of acetic acid during fed-batch cultivation with high or low feed. The horizontal arrows at the top indicate the duration of the respective glucose feed. Vertical lines indicate the addition of maltose. Open symbols: HAc accumulation in the cytoplasmic production, closed symbols: HAc accumulation in the periplasmic system. Squares: high feed rate, circles: low feed rate. Note the different Y-axis scales for high and low feed rates.
Since carbon starvation is known to induce the stringent response, which might affect the quality of the recombinant protein, this was also studied during the change of substrate when glucose was replaced by maltose. During high feed rate the ppGpp accumulation increased at the same time as maltose was added in both the periplasmic and the cytoplasmic system. But as can be seen in Figure 18, the ppGpp levels was slightly lower when Zb-MalE was transported to the periplasm compared to when it remained in the cytoplasm. When Zb-MalE is produced in the cytoplasm during a low feed rate, ppGpp is produced even before the addition of maltose. This indicates that ppGpp is not coupled to the change in uptake system but rather something else. In the periplasmic system, the increase in ppGpp comes after the maltose addition and it only reaches about 40 % of the cytoplasmic levels. This indicates that the transport of Zb-MalE to the periplasm results in a reduced stringent response, which might also explain the decreased amounts of degradation products that were seen in the periplasmic system. Since the stringent response has such profound effect on the cell inhibiting not only DNA initiation but also several components in the translation machinery, it is not surprising to see that a decreased stringent response results in higher quality and productivity. Figure 18: The accumulation of ppGpp during fed-batch cultivation with high or low feed. The horizontal arrows at the top indicate the duration of the respective glucose feed. Vertical lines indicate the addition of maltose. Open symbols: ppGpp accumulation in the cytoplasmic production, closed symbols: ppGpp accumulation in the periplasmic system. Squares: high feed rate, circles: low feed rate
0
1000
2000
3000
4000
5000
6000
7000
0
20
40
60
80
100
0 5 10 15 20
HA
c (m
g/L
)
glu
cose
fed
-batc
h 0
.5 h
-1
HA
c (mg/L
)
glu
cose fed
-batch
0.2
h-1
Time from feed start (h)
glucose feed 0.5glucose feed 0.2
maltose
additionmaltose
addition
0
200
400
600
800
1000
1200
0 5 10 15 20
pp
Gp
p (
nm
ol/
g D
W)
Time from feed start (h)
glucose feed 0.5glucose feed 0.2
maltose
addition
maltose
addition

Methodology for high-throughput production of soluble recombinant proteins in E.coli - Katrin Markland
30
Conclusions Several stress response systems are present in E.coli and they are activated to enable cell survival during harsh conditions. One of these systems is the stringent response which is activated during carbon and energy starvation as well as when charged tRNA’s are lacking. When the stringent response is induced the alarmone ppGpp is produced and effectively stops both transcription and translation (Magnusson et al. 2005). This has detrimental effects on the protein synthesis and increased levels of ppGpp should be avoided. A reduced stringent response has been shown to have a positive effect on protein production (Dedhia et al. 1996), however since growth is impaired a better approach is to use a strategy where the formation of ppGpp is avoided. Furthermore, acetic acid is another component with a negative effect on protein production (Jensen and Carlsen 1990). Methods (except from fed-batch cultivations) to decrease the accumulation of acetic acid include vector engineering where enzymatic genes have been modified or even incorporated from other bacteria (Bauer et al. 1990; Aristidou et al. 1995). Instead of using vector engineering we have shown that the feed rate can be used to decrease the accumulation of acetic acid for a secreted protein. For a high feed rate, the acetate concentration was 8-fold reduced if the protein was secreted to the periplasm. When the recombinant protein is transported to the periplasm both acetic acid accumulation and the stringent response is reduced. This can lead to a higher productivity and a high production rate can be used. This means that protein production is not limited to low growth rates but that also high growth rates can be used. It seems as if the cell utilizes the carbon and energy for the transport across the membrane instead of forming acetate. Therefore, stress and degradation for secreted proteins can be affected by different synthesis rates of the protein accomplished by the feed rates.
2.3 A cellular based system for substrate uptake rate control (III) Can the substrate uptake rate and the total productivity be influenced without using the fed-batch technique and without increasing the cultivation volume? Strategy The strategy of this work is based on control of the feed rate, i.e. the glucose uptake rate, to improve quality and quantity. This can be accomplished by mutations in the cells glucose uptake system. As described above, glucose is mainly transported into the cell through the glucose- and mannose- PTS systems. We have used strains with mutations in the substrate specific enzyme II in order to limit the substrate uptake rate. The first strain, PTSMan, has a mutation in the gene manX which codes for the enzyme IIABMan. The second strain, PTSGlc, has a mutation in the gene ptsG coding for the enzyme IICBGlc and the third strain, PTSGlcMan, has both mutations. Through these mutations, the cells are unable to maintain the same glucose uptake rates as wild type cells without mutations (Picon et al. 2004). For this work we needed a model protein, which was soluble and not subjected to proteolysis. The model protein chosen for this work was the protein ß-galactosidase (ß-gal). It is a stable protein and it has a high solubility even at high concentrations. ß-gal is a 116-kDa protein consisting of four domains that are 1023 amino acid residues long. Another advantage is the easy accessible activity analysis. ß-gal hydrolyze lactose into galactose and glucose, but also synthetic substrates exist and
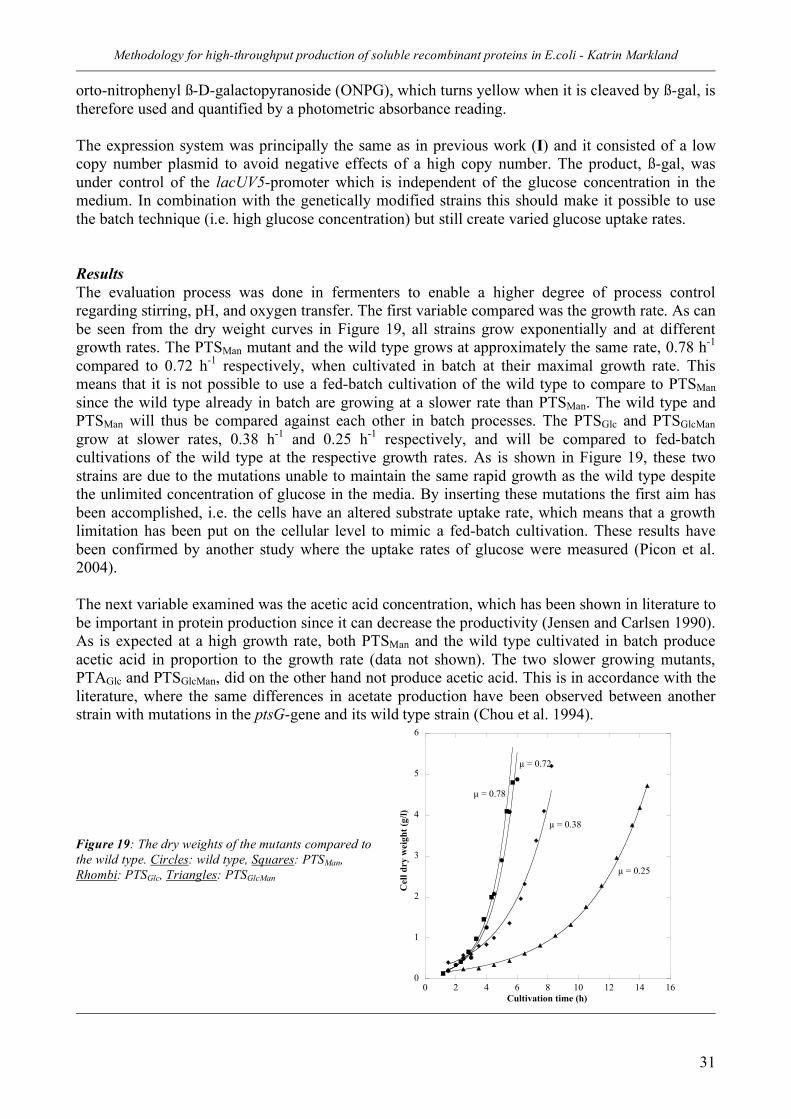
Methodology for high-throughput production of soluble recombinant proteins in E.coli - Katrin Markland
31
orto-nitrophenyl ß-D-galactopyranoside (ONPG), which turns yellow when it is cleaved by ß-gal, is therefore used and quantified by a photometric absorbance reading. The expression system was principally the same as in previous work (I) and it consisted of a low copy number plasmid to avoid negative effects of a high copy number. The product, ß-gal, was under control of the lacUV5-promoter which is independent of the glucose concentration in the medium. In combination with the genetically modified strains this should make it possible to use the batch technique (i.e. high glucose concentration) but still create varied glucose uptake rates. Results The evaluation process was done in fermenters to enable a higher degree of process control regarding stirring, pH, and oxygen transfer. The first variable compared was the growth rate. As can be seen from the dry weight curves in Figure 19, all strains grow exponentially and at different growth rates. The PTSMan mutant and the wild type grows at approximately the same rate, 0.78 h-1 compared to 0.72 h-1 respectively, when cultivated in batch at their maximal growth rate. This means that it is not possible to use a fed-batch cultivation of the wild type to compare to PTSMan since the wild type already in batch are growing at a slower rate than PTSMan. The wild type and PTSMan will thus be compared against each other in batch processes. The PTSGlc and PTSGlcMan grow at slower rates, 0.38 h-1 and 0.25 h-1 respectively, and will be compared to fed-batch cultivations of the wild type at the respective growth rates. As is shown in Figure 19, these two strains are due to the mutations unable to maintain the same rapid growth as the wild type despite the unlimited concentration of glucose in the media. By inserting these mutations the first aim has been accomplished, i.e. the cells have an altered substrate uptake rate, which means that a growth limitation has been put on the cellular level to mimic a fed-batch cultivation. These results have been confirmed by another study where the uptake rates of glucose were measured (Picon et al. 2004). The next variable examined was the acetic acid concentration, which has been shown in literature to be important in protein production since it can decrease the productivity (Jensen and Carlsen 1990). As is expected at a high growth rate, both PTSMan and the wild type cultivated in batch produce acetic acid in proportion to the growth rate (data not shown). The two slower growing mutants, PTAGlc and PTSGlcMan, did on the other hand not produce acetic acid. This is in accordance with the literature, where the same differences in acetate production have been observed between another strain with mutations in the ptsG-gene and its wild type strain (Chou et al. 1994). Figure 19: The dry weights of the mutants compared to the wild type. Circles: wild type, Squares: PTSMan, Rhombi: PTSGlc, Triangles: PTSGlcMan
0
1
2
3
4
5
6
0 2 4 6 8 10 12 14 16
Cel
l d
ry w
eigh
t (g
/l)
Cultivation time (h)
! = 0.78
! = 0.72
! = 0.38
! = 0.25

Methodology for high-throughput production of soluble recombinant proteins in E.coli - Katrin Markland
32
The production of β-gal was compared in batch processes as is shown in Figure 20. The fastest growing mutant PTSMan shows both the highest activity as well as the highest specific production rate for the duration of the cultivation. It is higher than the wild type, which has less than half of both activity and specific productivity at the end of the cultivation. Concerning the other two mutants, PTSGlc and PTSGlcMan, they have very low activities in minimal medium and this applies for specific productivities as well. However, if the medium is supplemented with yeast extract it is possible to increase both activity and specific productivity. For PTSGlcMan the specific production rate is initially even higher than what it is for PTSMan in minimal medium. However, one drawback with the yeast supplement is that it also leads to an increase in growth rate and that is unwanted. Figure 20: a) ß-galactosidase activity and b) specific production rates of β-galactosidase during different growth rates. Production is constitutive. Open symbols: minimal medium, closed symbols: minimal medium supplemented with yeast extract. Circles: wild type, squares: PTSMan, rhombi: PTSGlc, triangles: PTSGlcMan. However, when production was induced by IPTG, the PTSGlc mutant grown in minimal medium showed initially the same production pattern as the wild type, see Figure 21. The wild type maintained production for six hours before it dropped whereas the production from PTSGlc started to drop after approximately three hours. During these conditions, production from PTSGlc was comparable to the wild type. The total ß-galactosidase accumulation at the end of the cultivation estimated to 33 % and 13% of the total protein, for the WT and PTSGlc respectively.
0
100
200
300
400
500
600
700
4 6 8 10 12
B-g
al
act
ivit
y (
U/m
l)
Cultivation time (h)
a
0
10
20
30
40
50
60
70
5 6 7 8 9 10 11 12 13
qp
(U
/mg,h
)
Cultivation time (h)
b

Methodology for high-throughput production of soluble recombinant proteins in E.coli - Katrin Markland
33
Figure 21: Cell dry weights and specific production rates of β-galactosidase when induction is induced by IPTG. Filled symbols: cell dry weight, open symbols: specific production rate, circles: AF1000, rhombi: PPA652. It was also vital to see how high cell densities that could be reached for the mutants compared to the wild type since a high cell density also results in a higher total productivity. The mutant PTSGlcMan was chosen in order to get a large difference in glucose uptake rate. At the end of the cultivation, the wild type had reached a cell density of 27 g/L dry weight whereas PTSGlcMan reached 34 g/L. The acetic acid concentration showed a huge difference between the two cultivations. The production of acetic acid has a major impact on the growth rate of the wild type. Already at 1.5 g/L, when the cell density is 9 g/L, the acetic acid starts to inhibit growth as can be seen in Figure 22. The growth rate drops gradually from 0.8 h-1 to 0.2 h-1 as the acetic acid concentration increases to 9 g/L. The oxygen consumption rate for the wild type and PTSGlcMan was also compared. It is important to see whether the mutations have resulted in a higher respiration that could possibly have a negative effect on cell mass accumulation and also the total protein production. The wild type reaches the highest oxygen consumption rate after 5 hours of cultivation when the cell dry weight is 5 g/L. For PTSGlcMan, it takes 23 hours and a cell dry weight of 23 g/L before the same oxygen consumption rate is reached. This is important in protein production since it allows for a longer protein production period before oxygen depletion limits the cultivation. To conclude, the cultivation with the wild type was ended due to growth limitations caused by acetate whereas PTSGlcMan was ended due to oxygen depletion.
0
2
4
6
8
10
-10
0
10
20
30
40
50
60
-4 -2 0 2 4 6 8 10 12
DW
(g
/l)
qp
(U/m
g,h
)
Time from induction (h)
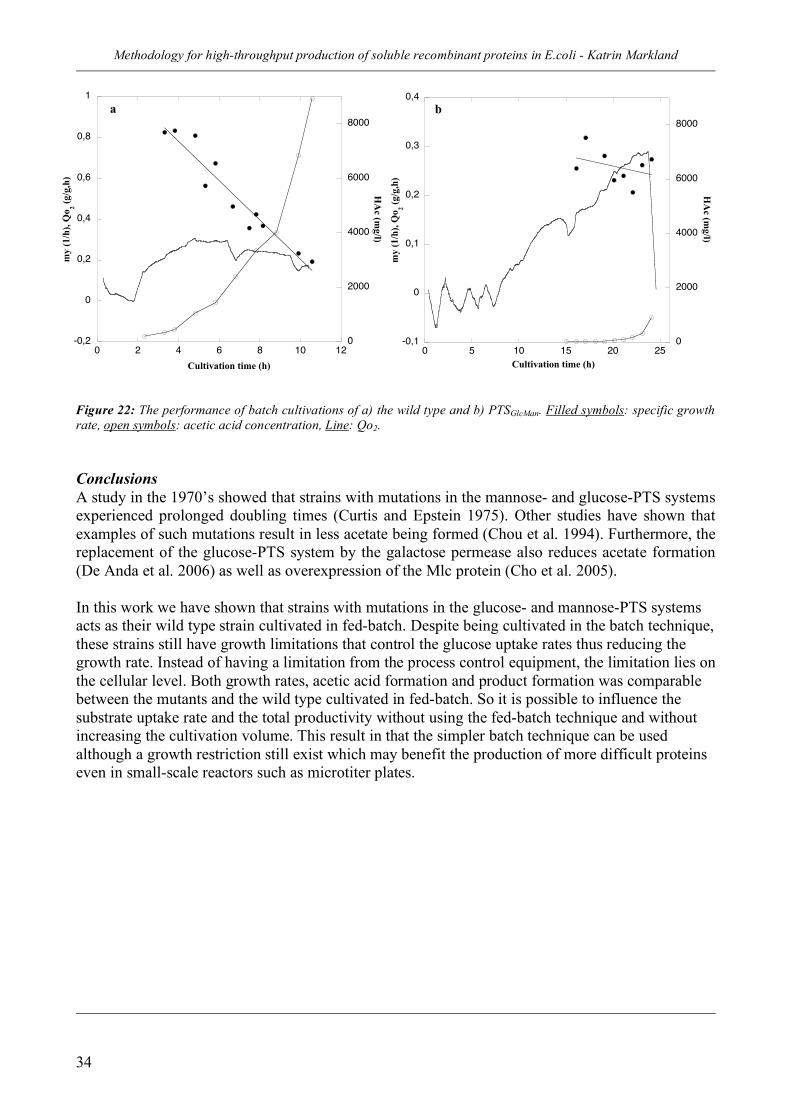
Methodology for high-throughput production of soluble recombinant proteins in E.coli - Katrin Markland
34
Figure 22: The performance of batch cultivations of a) the wild type and b) PTSGlcMan. Filled symbols: specific growth rate, open symbols: acetic acid concentration, Line: Qo2. Conclusions A study in the 1970’s showed that strains with mutations in the mannose- and glucose-PTS systems experienced prolonged doubling times (Curtis and Epstein 1975). Other studies have shown that examples of such mutations result in less acetate being formed (Chou et al. 1994). Furthermore, the replacement of the glucose-PTS system by the galactose permease also reduces acetate formation (De Anda et al. 2006) as well as overexpression of the Mlc protein (Cho et al. 2005). In this work we have shown that strains with mutations in the glucose- and mannose-PTS systems acts as their wild type strain cultivated in fed-batch. Despite being cultivated in the batch technique, these strains still have growth limitations that control the glucose uptake rates thus reducing the growth rate. Instead of having a limitation from the process control equipment, the limitation lies on the cellular level. Both growth rates, acetic acid formation and product formation was comparable between the mutants and the wild type cultivated in fed-batch. So it is possible to influence the substrate uptake rate and the total productivity without using the fed-batch technique and without increasing the cultivation volume. This result in that the simpler batch technique can be used although a growth restriction still exist which may benefit the production of more difficult proteins even in small-scale reactors such as microtiter plates.
-0,1
0
0,1
0,2
0,3
0,4
0
2000
4000
6000
8000
0 5 10 15 20 25
my
(1
/h),
Qo
2 (
g/g
,h)
HA
c (mg
/l)
Cultivation time (h)
b
-0,2
0
0,2
0,4
0,6
0,8
1
0
2000
4000
6000
8000
0 2 4 6 8 10 12
my
(1
/h),
Qo
2 (
g/g
,h)
HA
c (mg
/l)
Cultivation time (h)
a

Methodology for high-throughput production of soluble recombinant proteins in E.coli - Katrin Markland
35
3. CONCLUSIONS To increase the number of process settings, the aim of this work was to investigate and determine central parameters that can be used to control and increase the solubility, quality and productivity of recombinant proteins. These central parameters should be applicable under the constraints of high-throughput production i.e. the multi-parallel format at restricted volume. The central parameter in this work is the feed rate or the glucose uptake rate. The hypothesis was that if the synthesis rate could be reduced, solubility, quality and productivity could be increased. This reduced synthesis rate is accomplished by different feed rates of glucose. These different feed rates results in different uptake rates of glucose, which in turn affects the recombinant protein synthesis rate. We have shown that by lowering the feed rate, a higher solubility and a lower proteolysis can be achieved. However, it is not possible to use the fed-batch technique to control growth in the small-scale of high-throughput production. Therefore, mutant strains with altered glucose uptake rates were evaluated compared to the traditional fed-batch technique. These mutants enable fed-batch like growth conditions and receive the benefits from fed-batch even in the small-scale batch cultivations that is used in high-throughput production. Furthermore, a strong promoter such as the T7-promoter and a high inducer concentration are traditionally used in high-throughput production. However, this investigation has shown that by increasing the concentration of inducer, the ratio of soluble and insoluble proteins is shifted towards more insoluble proteins. Therefore, it is recommended to use primarily a weaker promoter but also lower inducer concentrations in order to reduce the metabolic burden on the cell thereby increasing the solubility of the recombinant protein. In order to control the recombinant protein production, a lacY strain can be used. This results in a more precise regulation of the promoter thus decreasing large production variations due to a small change in inducer concentration. Finally, if problems with solubility exist, an option is to transport the recombinant protein to the periplasm. This can benefit the quality of the recombinant protein as less degradation products have been formed and the stringent response has been reduced, which otherwise can have a negative impact on the productivity.

Methodology for high-throughput production of soluble recombinant proteins in E.coli - Katrin Markland
36
4. ABBREVIATIONS ATP adenosine triphosphate cAMP cyclic adenosine monophosphate CAT chloramphenicol-acetyl-transferase CRP catabolite repressor protein DNA deoxyribonucleic acid DOT dissolved oxygen tension DR downstream region Dsb thiol-disulfide oxidoreductase E.coli Escherichia coli F flow rate (L/h) GDP guanosine diphosphate GFP green fluorescent protein GTP guanosine triphosphate HSP heat shock proteins HTPP High-throughput protein production IPTG isopropyl-β-D-thiogalactopyranoside kDa kilo Dalton KLa oxygen transfer coefficient (h-1) MalE Maltose binding protein (=MBP) MalE31 Maltose binding protein with two altered amino acids mRNA messenger RNA OD optical density OMP outer membrane proteins ppGpp guanosine tetraphosphate pppGpp guanosine pentaphosphate PlacUV5 lacUV5 promoter PmalK malK promoter PphoA phosphatase promoter Ptac derivative of lac-promoter Ptrp tryptophane promoter PT7 T7-promoter PPIase peptidyl-prolyl cis/trans isomerase PPK polyP-kinase PPX exopolyphosphatase PTS phosphoenolpyruvate:phosphotransferase system qp specific productivity Q total productivity RNA ribonucleic acid RNAP RNA polymerase rRNA ribosomal RNA SD Shine Dalgarno sequence Si Substrate concentration of feed (g/L) TF trigger factor TIR translation initiation region tRNA transfer RNA V cultivation volume (L) X cell mass (g/L) YX/S Yield of biomass over substrate (g cells / g substrate) Zb Z-basic β-gal betagalactosidase µ specific growth rate σ32 = σH heat-shock sigma factor σ24 = σE extra-cytoplasmic stress sigma factor

Methodology for high-throughput production of soluble recombinant proteins in E.coli - Katrin Markland
37
5. ACKNOWLEDGEMENTS I would like to start with acknowledging The Swedish Centre for Bioprocess Technology (CBioPT) and the Royal Institute of Technology (KTH) for funding this project. Furthermore, I would like to thank all people that have been involved in the making of this thesis and first of all my supervisor, professor Gen Larsson for having me as a diploma worker and later on as a PhD-student. Big thanks for all your help making this thesis what it is today! Thank you Sophia Hober for spending time during your Christmas vacation to review this thesis. I greatly appreciated it! Thank you to Olle, for accepting me as a PhD-student and for having time to answer my questions when I popped my head into your office. Also to the other seniors at the department, Andres for never ending enthusiasm at the lab and around the coffee table, Lena for giving an superb overview of the stringent response in just about the right time! Thank you to Erik Holmgren at Biovitrum for taking part in my project and trying to teach me how to clone. It didn’t always go so well but I assure you that it was my fault and not yours! To Sofia Borg for contributions to paper III. To Anna Maria and Maria for coping with me during my diploma work, answering all my questions and also for the cultivations and analyses that followed both at Biovitrum and at KTH. Looking back at it, I had a lot of fun although it didn’t always feel like it when I was running western blot number one thousand…. I learned a lot from you guys! A huge thank you to Emma, for all the things that we have done together. Both PhD-courses in and out of Sweden, long cultivations from early morning to late evening, boring analyses and especially for all the chats and our gossip-moments! It hasn’t been easy every day, but it helped to have you by my side so thank you for being there! A big hug to former and present PhD-students for all beer nights, bowling, Bambi on ice (i.e. curling) and the fun crab- and Christmas parties that we have had and hopefully will continue to have! Thank you to Tommy for always fixing problems on the lab, to Ela for all the analyses, to Marita and Gunilla for fixing all the administrative stuff. A special thank you to my family for never ending support. You have been there from the very start always helping me out no matter what and I love you for it! To my wonderful fiancé Anders for always being there to listen to both complaints and fun stuff that had happened during the days and for trying to cheer me up when I had a bad day. Also for talking me into buying a house even if it took three years to get me there. And for putting a ring on my finger … I love you and I can’t wait for our next adventures!

Methodology for high-throughput production of soluble recombinant proteins in E.coli - Katrin Markland
38
6. REFERENCES Alba BM, Gross CA (2004) Regulation of the Escherichia coli σE-dependent envelope stress response. Molecular
Microbiology 52:613 - 619 Alba BM, Leeds JA, Onufryk C, Lu CZ, Gross CA (2002) DegS and YaeL participate sequentially in the cleavage of
RseA to activate the σE-dependent extracytoplasmic stress response. Genes and Development 16:2156 - 2168 Allen SP, Polazzi JO, Gierse JK, Easton AM (1992) Two novel heat shock genes encoding proteins produced in
response to heterologous protein expression in Escherichia coli. Journal of Bacteriology 174:6938-6947 Andersson L, Yang S, Neubauer P, Enfors S-O (1996) Impact of plasmid presence and induction on cellular responses
in fed-batch cultures of Escherichia coli. Journal of Biotechnology 46:255-263 Arditti R, Grodzicker T, Beckwith J (1973) Cyclic adenosine monophosphate-independent mutants of the lactose
operon of Escherichia coli. Journal of Bacteriology 114:652-655 Aristidou AA, San K-Y, Bennett GN (1995) Metabolic engineering of Escherichia coli to enhance recombinant protein
production through acetate reduction. Biotechnology Progress 11:475-478 Arsène F, Tomoyasu T, Bukau B (2000) The heat shock response of Escherichia coli. International Journal of Food
Microbiology 55:3 - 9 Balbás P, Soberón X, Merino E, Zurita M, Lomeli H, Valle F, Flores N, Bolivar F (1986) Plasmid vector pBR322 and
its special-purpose derivatives – a review. Gene 50:3-40 Baneyx F (1999) Recombinant protein expression in Escherichia coli. Current Opinion in Microbiology 10:411-421 Baneyx F (2004) Keeping up with protein folding. Microbial Cell Factories 3 Baneyx F, Mujacic M (2004) Recombinant protein folding and misfolding in Escherichia coli. Nature Biotechnology
22:1399-1408 Baneyx F, Palumbo JL (2003) Improving heterologous protein folding via molecular chaperone and foldase co-
expression. Methods in Molecular Biology 205:171-197 Bauer KA, Ben-Bassat A, Dawson M, de la Puente VT, Neway JO (1990) Improved expression of human Interleukin-2
in high-cell-density fermentor cultures of Escherichia coli K-12 by a phosphotransacetylase mutant. Applied and Environmental Microbiology 56:1296-1302
Bentley WE, Davis RH, Kompala DS (1991) Dynamics of induced CAT expression in E.coli. Biotechnology and Bioengineering 38:749-760
Berrow NS, Büssow K, Coutard B, Diprose J, Ekberg M, Folkers GE, Levy N, Lieu V, Owens RJ, Peleg Y, Pinaglia C, Quevillon-Cheruel S, Salim L, Scheich C, Vincentelli R, Busso D (2006) Recombinant protein expression and solubility screening in Escherichia coli: a comparative study. Acta Crystallographica section D 62:1218-1226
Betton J-M, Hofnung M (1996) Folding of a mutant maltose-binding protein of Escherichia coli which forms inclusion bodies. The Journal of Biological Chemistry 271:8046 - 8052
Betts JI, Baganz F (2006) Miniature bioreactors: current practices and future opportunities. Microbial Cell Factories 5:21
Booth I (1999) Adaptation to extreme environments. Biology of the prokaryotes, Edited by Joseph Lengeler, Gerhart Drews, Hans G. Schlegel, Blackwell Science: 652-673
Boström M, Larsson G (2002) Introduction of the carbohydrate-activated promoter PmalK for recombinant protein production. Applied Microbiology and Biotechnology 59:231-238
Boström M, Larsson G (2004) Process design for recombinant protein production based on the promoter, PmalK. Applied Microbiology and Biotechnology 66:200 - 208
Cashel M, Gentry DR, Hernandez VJ, Vinella D (1996) The Stringent Response. In Escherichia coli and Salmonella: Cellular and molecular biology (Neidhardt F.C., ed) ASM Press Vol 1:1458-1496
Chen Y, Song J, Sui S-f, Wang D-N (2003) DnaK and DnaJ facilitated the folding process and reduced inclusion body formation of magnesium transporter CorA overexpressed in Escherichia coli. Protein Expression and Purification 32:221-231
Cho S, Shin D, Ji GE, Heu S, Ryu S (2005) High-level recombinant protein production by overexpression of Mlc in Escherichia coli. J Biotechnol 119:197-203.
Chou CH, Bennett GN, San KY (1994) Effect of modified glucose uptake using genetic engineering techniques on high-level recombinant protein production in Escherichia coli dense cultures. Biotechnology and Bioengineering 44:952-960
Curtis SJ, Epstein W (1975) Phosphorylation of D-glucose in Escherichia coli mutants defective in glucosephosphotransferase, mannosephosphotransferase, and glucokinase. Journal of Bacteriology 122:1189-1199
Danese PN, Silhavy TJ (1998) CpxP, a stress-combative member of the Cpx regulon. Journal of Bacteriology 180:831 - 839
Dartigalongue C, Missiakas D, Raina S (2001) Characterization of the Escherichia coli σE regulon. The Journal of Biological Chemistry 276:20866 – 20875

Methodology for high-throughput production of soluble recombinant proteins in E.coli - Katrin Markland
39
De Anda R, Lara AR, Hernández V, Hernández-Montalvo V, Gosset G, Bolivar F, Ramirez OT (2006) Replacement of
the glucose phosphotransferase transport system by galactose permease reduces acetate accumulation and improves process performance of Escherichia coli for recombinant protein production without impairment of growth rate. Metabolic Eng 8:281-290
De Las Penas A, Connolly L, Gross CA (1997) The σE-mediated response to extracytoplasmic stress in Escherichia coli is transduced by RseA and RseB, two negative regulators of σE. Molecular Microbiology 24:373 - 385
Death A, Notley L, Ferenci T (1993) Derepression of LamB protein facilitates outer membrane permeation of carbohydrates into Escherichia coli under conditions of nutrient stress. Journal of Bacteriology 175:1475-1483
Dedhia N, Richins R, Mesina A, Chen W (1996) Improvement in recombinant protein production in ppGpp-deficient Escherichia coli. Biotechnology and Bioengineering 53:379-386
Deuerling E, Patzelt H, Vorderwülbecke S, Rauch T, Kramer G, Schaffitzel E, Mogk A, Schulze-Specking A, Langen H, Bukau B (2003) Trigger Factor and DnaK possess overlapping substrate pools and binding specificities. Molecular Microbiology 47:1317-1328
Doig SD, Diep A, Baganz F (2005) Characterisation of a novel miniaturized bubble column bioreactor for high throughput cell cultivation. Biochemical Engineering Journal 23:97-105
Dong J, Iuchi S, Kwan H-S, Lu Z, Lin EC (1993) The deduced amino-acid sequence of the cloned cpxR gene suggests the protein is the cognate regulator for the membrane sensor, CpxA, in a two-component signal transduction system of Escherichia coli. Gene 136:227-230
Duetz WA, Rüedi L, Hermann R, O'Connor K, Büchs J, Witholt B (2000) Methods for intense aeration, growth, storage, and replication of bacterial strains in microtiter plates. Applied and Environmental Microbiology 66:2641-2646
Edelmann P, Gallant J (1977) Mistranslation in E.coli. Cell 10:131-137 Erickson JW, Gross CA (1989) Identification of the σE subunit of Escherichia coli RNA polymerase: a second
alternative σ factor involved in high-temperature gene expression. Genes and Development 3:1462 - 1471 Fahnert B, Lilie H, Neubauer P (2004) Inclusion bodies: Formation and utilization. Advances in Biochemical
Engineering/Biotechnology 89:93-142 Fuchs C, Köster D, Wiebusch S, Mahr K, Eisbrenner G, Märkl H (2002) Scale-up of dialysis fermentation for high cell
density cultivation of Escherichia coli. Journal of Biotechnology 93:243-251 Gellerfors P, Pavlu B, Axelsson K, Nyhlen C, Johansson S (1990) Separation and identification of growth hormone
variants with high performance liquid chromatography techniques. Acta paediatrica Scandinavica 370:93-100 Georgiou G (1988) Optimizing the production of recombinant proteins in microorganisms. AIChE Journal 34:1233-
1248 Georgiou G, Shuler M, Wilson D (1988) Release of periplasmic enzymes and other physiological effects of ß-lactamase
overproduction in Escherichia coli. Biotechnology and Bioengineering 32:741-748 Georgiou G, Valax P (1996) Expression of correctly folded proteins in Escherichia coli. Current Opinion in
Biotechnology 7:190 - 197 Glick BR (1995) Metabolic load and heterologous gene expression. Biotechnology Advances 13:247-261 Gonzalez de Valdivia EI, Isaksson LA (2004) A codon window in mRNA downstream of the initiation codon where
NGG codons give strongly reduced gene expression in Escherichia coli. Nucleic Acid Research 32:5198-5205 Grabherr R, Nilsson E, Striedner G, Bayer K (2001) Stabilizing plasmid copy number to improve recombinant protein
production. Biotechnology and Bioengineering 77:142 - 147 Gross CA (1996) Function and regulation of the heat shock proteins. In Escherichia coli and Salmonella: Cellular and
molecular biology (Neidhardt F.C., ed) ASM Press 1:1382-1399 Grossman AD, Erickson JW, Gross CA (1984) The htpR gene product of E.coli is a sigma factor for heat-shock
promoters. Cell 38:383-390 Grossman AD, Taylor WE, Burton ZF, Burgess RR, Gross CA (1985) Stringent response in Escherichia coli induces
expression of heat shock proteins. Journal of Molecular Biology 186:357-365 Gräslund T, Lundin G, Uhlén M, Nygren P-Å, Hober S (2000) Charge engineering of a protein domain to allow
efficient ion-exchange recovery. Protein Engineering 13:703-709 Gualerzi CO, Pon CL (1990) Initiation of mRNA translation in prokaryotes. Biochemistry 29:5881-5889 Hammarström M, Hellgren N, van den Berg S, Berglund H, Härd T (2002) Rapid screening for improved solubility of
small human proteins produced as fusion proteins in Escherichia coli. Protein Science 11:313-321 Hedrén M, Ballagi A, Mörtsell L, Rajkai G, Stenmark P, Sturesson C, Nordlund P (2006) GRETA, a new
multifermenter system for structural genomics and process optimization. Acta Crystallographica section D 62:1227-1231
Hellebust H, Murby M, Abrahmsén L, Uhlén M, Enfors S-O (1989) Different approaches to stabilize a recombinant fusion protein. BIO/Technology 7:165-168
Hermann R, Lehmann M, Büchs J (2002) Characterization of gas-liquid mass transfer phenomena in micro-titer plates. Biotechnology and Bioengineering 81:178-186

Methodology for high-throughput production of soluble recombinant proteins in E.coli - Katrin Markland
40
Horn U, Krug M, Sawistowski J (1990) Effect of high cell density cultivation on plasmid copy number in recombinant Escherichia coli cells. Biotechnology Letters 12:191-196
Jensen EB, Carlsen S (1990) Production of recombinant human growth hormone in Escherichia coli: expression of different precursors and physiological effects of glucose, acetate and salts. Biotechnology and Bioengineering 36:1-11
Jin H, Zhao Q, Gonzalez de Valdivia EI, Arcell DH, Stenström M, Isaksson LA (2006) Influences on gene expression in vivo by a Shine Dalgarno sequence. Molecular Microbiology 60:480-492
John GT, Goelling D, Kliman I, Schneider I, Heinzle E (2003a) pH-sensing 96-well microtitre plates for the characterization of acid production by dairy starter cultures. Journal of Dairy Research 70:327-333
John GT, Kliman I, Wittman C, Heinzle E (2003b) Integrated optical sensing of dissolved oxygen in microtitre plates: a novel tool for microbial cultivation. Biotechnology and Bioengineering 81:829-836
Jones CH, Danese PN, Pinkner JS, Silhavy TJ, Hultgren SJ (1997) The chaperone-assisted membrane release and folding pathway is sensed by two signal transduction systems. The EMBO Journal 16:6394-6406
Jones KL, Kim S-W, Keasling JD (2000) Low-copy plasmids can perform as well as or better than high-copy plasmids for metabolic engineering of bacteria. Metabolic Engineering 2:328-338
Jones SA, Melling J (1984) Persistance of pBR322-related plasmids in Escherichia coli grown in chemostat culture. FEMS Microbiology Letters 22:239-243
Kanehara K, Ito K, Akiyama Y (2002) YaeL (EcfE) activates the σE pathway of stress response through a site-2 cleavage of anti-σE, RseA. Genes and Development 16:2147 - 2155
Kapust RB, Waugh DS (1999) Escherichia coli maltose-binding protein is uncommonly effective at promoting the solubility of polypeptides to which it is fused. Protein Science 8:1668-1674
Kimata K, Inada T, Tagami H, Aiba H (1998) A global repressor (Mlc) is involved in glucose induction of the ptsG gene encoding major glucose transporter in Escherichia coli. Molecular Microbiology 29:1509-1519
Kimata K, Takahashi H, Inada T, Postma P, Aiba H (1997) cAMP receptor protein-cAMP plays a crucial role in glucose-lactose diauxie by activating the major glucose transporter gene in Escherichia coli. Proceedings of the National Academy of Sciences USA 94:12914-12919
Kosinski MJ, Rinas U, Bailey JE (1992) Isopropyl-β-D-thiogalacto-pyranoside influences the metabolism of Escherichia coli. Applied Microbiology and Biotechnology 36:782-784
Kumar S, Wittman C, Heinzle E (2004) Minibioreactors. Biotechnology Letters 26:1-10 Kuroda A (2006) A polyphosphate-Lon protease complex in the adaptation of Escherichia coli to amino acid starvation.
Bioscience, Biotechnology and Biochemistry 70:325-331 Langer T, Lu C, Echols H, Flanagan J, Hayer MK, Hartl FU (1992) Successive action of DnaK, DnaJ and GroEL along
the pathway of chaperone-mediated protein folding. Nature 356:683-689 Laskowska E, Kucynska-Wisnik D, Bak M, Lipinska B (2003) Trimethoprim induces heat shock proteins and protein
aggregation in E.coli cells. Current Microbiology 47:286-289 Lee SY (1996) High-cell density culture of Escherichia coli. Trends in Biotechnology 14:98-105 Lengeler JW, Postma PW (1999) Global regulatory networks and signal transduction pathways. Biology of the
prokaryotes, Edited by Joseph Lengeler, Gerhart Drews, Hans G. Schlegel, Blackwell Science: 491-523 Lilie H, Schwarz E, Rudolph R (1998) Advances in refolding of proteins produced in E.coli. Current Opinion in
Microbiology 9:497-501 Lipinska B, Fayet O, Baird L, Georgopoulos C (1989) Identification, characterization, and mapping of the Escherichia
coli htrA gene, whose product is essential for bacterial growth only at elevated temperatures. Journal of Bacteriology 171:1574-1584
Magnusson LU, Farewell A, Nyström T (2005) ppGpp: a global regulator in Escherichia coli. TRENDS in Microbiology 13:236-242
Mar Carrio M, Villaverde A (2001) Protein aggregation as bacterial inclusion bodies is reversible. FEBS Letters 489:29-33
Mecsas J, Rouviere PE, Erickson JW, Donohue TJ, Gross CA (1993) The activity of σE, an Escherichia coli heat-inducible σ-factor, is modulated by expression of outer membrane proteins. Genes and Development 7:2618 - 2628
Mere L, Bennett T, Coassin P, England P, Hamman B, Rink T, Zimmerman S, Negulescu P (1999) Miniaturized FRET assays and microfluidics: key components for ultra-high-throughput screening. DDT 4:363-369
Meyer H-P, Leist C, Fiechter A (1984) Acetate formation in continuous culture of Escherichia coli K12 D1 on defined and complex media. Journal of Biotechnology 1:355-358
Mileykovskaya E, Dowhan W (1996) The Cpx two-component signal transduction pathway is activated in Escherichia coli mutant strains lacking Phosphatidylethanolamine. Journal of Bacteriology 179:1029 - 1034
Miroux B, Walker JE (1996) Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. Journal of Molecular Biology 260:289-298

Methodology for high-throughput production of soluble recombinant proteins in E.coli - Katrin Markland
41
Missiakas D, Mayer MP, Lemaire M, Georgopoulos C, Raina S (1997) Modulation of the Escherichia coli σE (RpoE) heat-shock transcription-factor activity by the RseA, RseB and RseC proteins. Molecular Microbiology 24:355 - 371
Moat AG, Foster JW, Spector MP (2002) Microbial Physiology. Wiley-Liss: 162 Mosavi LK, Peng Z (2003) Structure-based substitutions for increased solubility of a designed protein. Protein
engineering 16:739-745 Murby M, Samuelsson E, Nguyen TN, Mignard L, Power U, Binz H, Uhlén M, Ståhl S (1995) Hydrophobicity
engineering to increase solubility and stability of a recombinant protein from respiratory syncytial virus. European Journal of Biochemistry 230:38-44
Murby M, Uhlén M, Ståhl S (1996) Upstream strategies to minimize proteolytic degradation upon recombinant production in Escherichia coli. Protein Expression and Purification 7:129-136
Märkl H, Zenneck C, Dubach ACH, Ogbonna JC (1993) Cultivation of Escherichia coli cells to high cell densities in a dialysis reactor. Applied Microbiology and Biotechnology 39:48-52
Nakano K, Rischke M, Sato S, Märkl H (1997) Influence of acetic acid on the growth of Escherichia coli K12 during high-cell-density cultivation in a dialysis reactor. Applied Microbiology and Biotechnology 48:597-601
Neidhardt FC, Ingraham JL, Schaechter M (1990) Physiology of the bacterial cell. Sinauer Associates, Inc: 361-367 Nikaido H, Vaara M (1985) Molecular basis of bacterial outer membrane permeability. Microbiological Reviews 49:1-
32 Parker J (1989) Errors and alternatives in reading the universal code. Microbiological Reviews 53:273-298 Parker J, Pollard JW, Friesen JD, Stanners CP (1978) Stuttering: High-level mistranslation in animal and bacterial cells.
Proceedings of the National Academy of Sciences USA 75:1091-1095 Picon A, Teixeira de Mattos MJ, Postma PW (2004) Reducing the glucose uptake rate in Escherichia coli affects
growth rate but not protein production. Biotechnology and Bioengineering 90:191 - 200 Plumbridge J (1998) Control of the expression of the manXYZ operon in Escherichia coli: Mlc is a negative regulator of
the mannose PTS. Molecular Microbiology 27:369 - 380 Plumbridge J (1999) Expression of the phosphotransferase system both mediates and is mediated by Mlc regulation in
Escherichia coli. Molecular Microbiology 33:260-273 Plumbridge J (2002) Regulation of gene expression in the PTS in Escherichia coli: the role and interactions of Mlc.
Current Opinion in Microbiology 5:187 - 193 Puskeiler R, Kaufmann K, Weuster-Botz D (2005) Development, parallelization, and automation of a gas-inducing
milliliter-scale bioreactor for high-throughput bioprocess design (HTBD). Biotechnology and Bioengineering 89:512-523
Raivio TL, Popkin DL, Silhavy TJ (1999) The Cpx envelope stress response is controlled by amplification and feedback inhibition. Journal of Bacteriology 181:5263-5272
Raivio TL, Silhavy TJ (2001) Periplasmic stress and ECF sigma factors. Annual Review of Microbiology. 55:591 - 624 Risenberg D (1991) High-cell density cultivation of Escherichia coli. Current Opinion in Biotechnology 2:380-384 Rose RE (1988) The nucleotide sequence of pACYC184. Nucleic Acids Research 16:355 Rouvière PE, De Las Penas A, Mecsas J, Lu CZ, Rudd KE, Gross CA (1995) rpoE, the gene encoding the second heat-
shock sigma factor, σE, in Escherichia coli. The EMBO Journal 14:1032-1042 Ruhdal Jensen P, Hammer K (1997) Artificial promoters for metabolic optimization. Biotechnology and
Bioengineering 58:191-195 Sandén AM, Prytz I, Tubulekas I, Förberg C, Le H, Hektor A, Neubauer P, Pragai Z, Harwood C, Ward A, Picon A,
Teixeira de Mattos J, Postma P, Farewell A, Nyström T, Reeh S, Pedersen S, Larsson G (2002) Limiting factors in Escherichia coli fed-batch production of recombinant proteins. Biotechnology and Bioengineering 81:158-166
Schein CH (1990) Solubility as a function of protein structure and solvent components. Biotechnology 8:308-317 Schmidt FR (2004) Recombinant expression systems in the pharmaceutical industry. Applied Microbiology and
Biotechnology 65:363-372 Shibui T, Nagahari K (1992) Secretion of a functional Fab fragment in Escherichia coli and the influence of culture
conditions. Applied Microbiology and Biotechnology 37:352-357 Snyder WB, Davis LJB, Danese PN, Cosma CL, Silhavy TJ (1995) Overproduction of NlpE, a new outer membrane
lipoprotein, suppresses the toxicity of periplasmic LacZ by activation of the Cpx signal transduction pathway. Journal of Bacteriology 177:4216-4223
Sorensen HP, Mortensen KK (2005) Soluble expression of recombinant proteins in the cytoplasm of Escherichia coli. Microbial Cell Factories 4:1
Stenström CM, Holmgren E, Isaksson LA (2001a) Cooperative effects by the initiation codon and its flanking regions on translation initiation. Gene 273:259-265
Stenström CM, Jin H, Major LL, Tate WP, Isaksson LA (2001b) Codon bias at the 3’-side of the initiation codon is correlated with translation initiation efficiency in Escherichia coli. Gene 263:273-284

Methodology for high-throughput production of soluble recombinant proteins in E.coli - Katrin Markland
42
Straus DB, Walter WA, Gross CA (1987) The heat shock response of E.coli is regulated by changes in the concentration of σ32. Nature 329:348-351
Terpe K (2003) Overview of tag protein fusions: from molecular and biochemical fundamentals to commercial systems. Applied Microbiology and Biotechnology 60:523-533
Thomas JG, Ayling A, Baneyx F (1997) Molecular chaperones, folding catalysts, and the recovery of active recombinant proteins from E.coli. Applied Biochemistry and Biotechnology 66:197-238
Thomas JG, Baneyx F (1997) Divergent effects of chaperone overexpression and ethanol supplementation on inclusion body formation in recombinant Escherichia coli. Protein Expression and Purification 11:289-296
Tsai LB, Lu HS, Kenny WC, Curless CC, Klein ML, Lai P-H, Fenton DM, Altrock BW, Mann MB (1988) Control of misincorporation of de novo synthesized norleucine into recombinant Interleukin-2 in E.coli. Biochemical and Biophysical Research Communications 156:733-739
Uhlén M, Forsberg G, Moks T, Hartmanis M, Nilsson B (1992) Fusion proteins in biotechnology. Current Opinion in Biotechnology 3:363-369
Waldo GS, Standish BM, Berendzen J, Terwilliger TC (1999) Rapid protein-folding assay using green fluorescent protein. Nature Biotechnology 17:691-695
Wang Q, Kaguni JM (1989) A novel sigma factor is involved in expression of the rpoH gene of Escherichia coli. Journal of Bacteriology 171:4248 - 4253
Weber RF, Silverman PM (1988) The Cpx proteins of Escherichia coli K12: structure of the CpxA polypeptide as an inner membrane component. Journal of Molecular Biology 203:467-478
Veinger L, Diamant S, Buchner J, Goloubinoff P (1998) The small heat-shock protein IbpB from Escherichia coli stabilizes stress-denatured proteins for subsequent refolding by a multichaperone network. The Journal of Biological Chemistry 273:11032-11037
Wendrich TM, Blaha G, Wilson DN, Marahiel MA, Nierhaus KH (2002) Dissection of the mechanism for the stringent factor RelA. Molecular Cell 10:779-788
Weuster-Botz D, Puskeiler R, Kusterer A, Kaufmann K, John GT, Arnold M (2005) Methods and milliliter scale devices for high-throughput bioprocess design. Bioprocess and Biosystems Engineering 28:109-119
Wickner S, Maurizi MR, Gottesman S (1999) Posttranslational quality control: folding, refolding and degrading proteins. Science 286:1888-1893
Woestenenk EA, Hammarström M, van den Berg S, Härd T, Berglund H (2004) His tag effect on solubility of human proteins produced in Escherichia coli: a comparison between four expression vectors. Journal of Structural and Functional Genomics 5:217-229
Yanisch-Perron C, Vieira J, Messing J (1985) Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 33:103-119
Yansura DG, Henner DJ (1990) Use of Escherichia coli trp promoter for direct expression of proteins. Methods in Enzymology 185:54-60
Zhang Z, Li Z-H, Wang F, Fang M, Yin C-C, Zhou Z-Y, Lin Q, Huang H-L (2002) Overexpression of DsbC and DsbG markedly improves soluble and functional expression of single-chain Fv antibodies in Escherichia coli. Protein Expression and Purification 26:218-228