metabolic engineering for the biosynthesis of … · metabolic engineering for the biosynthesis of...
TRANSCRIPT

Metabolic Engineering for the Biosynthesis of
Styrene and its Derivatives
by
Rebekah McKenna
A Dissertation Presented in Partial Fulfillment
of the Requirements for the Degree
Doctor of Philosophy
Approved March 2014 by the
Graduate Supervisory Committee:
David Nielsen, Chair
Cesar Torres
Laura Jarboe
Karmella Haynes
Michael Caplan
ARIZONA STATE UNIVERSITY
May 2014

i
ABSTRACT
Metabolic engineering is an extremely useful tool enabling the biosynthetic
production of commodity chemicals (typically derived from petroleum) from renewable
resources. In this work, a pathway for the biosynthesis of styrene (a plastics monomer)
has been engineered in Escherichia coli from glucose by utilizing the pathway for the
naturally occurring amino acid phenylalanine, the precursor to styrene. Styrene
production was accomplished using an E. coli phenylalanine overproducer, E. coli
NST74, and over-expression of PAL2 from Arabidopsis thaliana and FDC1 from
Saccharomyces cerevisiae. The styrene pathway was then extended by just one enzyme
to either (S)-styrene oxide (StyAB from Pseudomonas putida S12) or (R)-1,2-
phenylethanediol (NahAaAbAcAd from Pseudomonas sp. NCIB 9816-4) which are both
used in pharmaceutical production. Overall, these pathways suffered from limitations due
to product toxicity as well as limited precursor availability. In an effort to overcome the
toxicity threshold, the styrene pathway was transferred to a yeast host with a higher
toxicity limit. First, Saccharomyces cerevisiae BY4741 was engineered to overproduce
phenylalanine. Next, PAL2 (the only enzyme needed to complete the styrene pathway)
was then expressed in the BY4741 phenylalanine overproducer. Further strain
improvements included the deletion of the phenylpyruvate decarboxylase (ARO10) and
expression of a feedback-resistant choristmate mutase (ARO4K229L
). These works have
successfully demonstrated the possibility of utilizing microorganisms as cellular factories
for the production styrene, (S)-styrene oxide, and (R)-1,2-phenylethanediol.

ii
ACKNOWLEDGEMENTS
This work would not have been possible without the support of my advisor,
committee members, fellow labmates, friends, and collaborators. I would like to thank
my advisor Dr. David Nielsen for his support and guidance. I am very thankful to have
been given the opportunity to work in his lab. Also, I would like to thank my committee
members Dr. Michael Caplan, Dr. Karmella Haynes, Dr. Laura Jarboe, and Dr. Cesar
Torres for their assistance and agreeing to be on my committee. I would like to thank Dr.
Kristala Prather at MIT and Dr. Bradley Moore from UCSD for their donations of
number plasmids and strains that contributed greatly to these works. I would like to thank
Tom Colella in the GEL lab for his help with GC-MS. I would like to thank my lab
members, both former and current, as well as those from Dr. Sierks’ and Dr. Rege’s lab
for their help and assistance. I would like to thank Shawn Pugh, Warinsinee
Phusitkanchana, Luis Moya, Michael Wiehn, Vick Syradi, Ibrahim Halloum, and Thomas
Levario for making everyday an enjoyable day to go to lab. I am not sure how I would
have made it through this without our occasional racquetball games. I am also grateful for
the funding provided by ASU as well as the donors to the Ford, Barrett and Materials,
ARCS, and PEO Fellowships.

iii
TABLE OF CONTENTS
Page
LIST OF TABLES ................................................................................................................. vii
LIST OF FIGURES .............................................................................................................. viii
CHAPTER
1 INTRODUCTION .......................................................................................................... 1
1.1 Background and motivation ...................................................................................... 1
1.2 Metabolic pathways .................................................................................................. 5
1.3 Dissertation organization .......................................................................................... 9
2 BIOSYNTHESIS OF STYRENE BY ENGINEERED E. COLI.................................. 10
2.1 Introduction ............................................................................................................. 11
2.2 Materials and methods ............................................................................................ 15
2.2.1 Chemicals. ........................................................................................................ 15
2.2.2 Strains and media ............................................................................................. 16
2.2.3 Toxicity assays. ................................................................................................ 18
2.2.4 Cloning of candidate genes encoding PAL activity from A. variabilis, N.
punctiforme, and A. thaliana. .................................................................................... 18
2.2.5 Cloning of candidate genes encoding PADC activity from L. plantarum, B.
subtilis, and S. cerevisiae. ......................................................................................... 20
2.2.6 Assaying PAL activity in crude lysates of recombinant E. coli. ..................... 20
2.2.7 Assaying PAL activity in recombinant E. coli whole cells. ............................ 22
2.2.8 Assaying PADC activity in recombinant E. coli whole cells. ......................... 22

iv
Page
2.2.9 Co-expression of PAL and CADC isoenzymes in E. coli NST74 to convert L-
phenylalanine to styrene. .......................................................................................... 23
2.2.10 Co-expression of PAL and CADC isoenzymes in E. coli NST74 to convert
glucose to styrene in shake flask cultures. ................................................................ 23
2.2.11 Metabolite analysis by HPLC ........................................................................ 24
2.2.12 Confirmation of styrene biosynthesis by GC-MS .......................................... 25
2.3 Results and discussion ........................................................................................... 25
2.3.1 Assaying styrene toxicity ................................................................................. 25
2.3.2 Screening candidate PAL isoenzymes for activity in recombinant E. coli. ..... 27
2.3.3 Screening candidate PADC isoenzymes for tCA decarboxylase activity in
recombinant E. coli. .................................................................................................. 30
2.3.4 Biosynthesis of tCA and styrene from glucose in recombinant E. coli. .......... 33
2.4 Conclusion .............................................................................................................. 40
3 BIOSYNTHESIS OF (S)-STYRENE OXIDE AND (R)-1,2-PHENYLETHANEDIOL
FROM GLUCOSE ............................................................................................................ 42
3.1 Introduction ............................................................................................................. 43
3.2 Materials and methods ............................................................................................ 48
3.2.1 Strains and media ............................................................................................. 48
3.2.2 Toxicity assays ................................................................................................. 50
3.2.3 Cloning of PAL2 from A. thaliana, FDC1 from S. cerevisiae, and tktA and
aroB from E. coli ...................................................................................................... 50

v
Page
3.2.4 Construction of the plasmid pTrcColaK and the cloning of SMO- and SDO-
encoding genes .......................................................................................................... 51
3.2.5 Construction of tyrA and trpE deletion mutants in E. coli NST74 .................. 52
3.2.6 Construction of phenylalanine, (S)-styrene oxide, and (R)-1,2-
phenylethanediol producing strains .......................................................................... 52
3.2.7 Assaying SMO activity in recombinant E. coli whole resting cells ................ 52
3.2.8 HPLC analysis ................................................................................................. 53
3.3 Results and discussion ............................................................................................ 54
3.3.1 Assessing and investigating the mechanisms of product toxicity .................... 54
3.3.2 Screening and selecting pathway enzymes ...................................................... 56
3.3.3 Evaluating pathway function and performance ............................................... 58
3.3.4 Promoting availability of phenylalanine, the pathway precursor .................... 60
3.3.5 Improving production using more robust host strains ..................................... 62
3.4 Conclusion .............................................................................................................. 64
4 ENGINEERING THE STYRENE PATHWAY IN YEAST......................................... 65
4.1 Introduction ............................................................................................................. 65
4.2 Materials and methods ............................................................................................ 70
4.2.1 Strains and media ............................................................................................. 70
4.2.2 Toxicity assays. ................................................................................................ 71
4.2.3 Evolution of phenylalanine overproducing strains .......................................... 72
4.2.4 Transcriptional analysis of phenylalanine over-producing mutants. ............... 73
4.2.5 Investigating native expression and activity of FDC1 in S. cerevisiae............ 73

vi
Page
4.2.6 Cloning of PAL2 from A. thaliana. ................................................................. 74
4.2.7 Assaying the extracellular transport of trans-cinnamate. ................................ 75
4.2.8 Chromosomal disruption of ARO10 and integration of ARO4K229L
. .............. 75
4.2.9 Styrene production from glucose in S. cerevisiae shake flask cultures. .......... 75
4.2.10 Metabolite analysis ........................................................................................ 77
4.3 Results and discussion ............................................................................................ 77
4.3.1 Assaying styrene toxicity ................................................................................. 77
4.3.2 Evolving and engineering phenylalanine over-production in S. cerevisiae ..... 79
4.3.3 Investigating native FDC1 activity and factors influencing its expression ..... 85
4.3.4 Probing the styrene pathway via the exogenous addition of phenylalanine .... 86
4.3.5 Styrene production from glucose ..................................................................... 88
4.4 Conclusion .............................................................................................................. 91
5 DISCUSSION AND FUTURE WORK......................................................................... 93
5.1 Introduction ............................................................................................................. 93
5.2 Product removal as a method to overcome toxicity ................................................ 94
5.3 Improving precursor availability............................................................................. 98
5.4 Improving PAL Activity ....................................................................................... 101
5.5 Conclusion ............................................................................................................ 104
REFERENCES....... ............................................................................................................ 105

vii
LIST OF TABLES
Table Page
2.1. Strains, plasmids, and oligonucleotide primers…………………………………… 17
2.2. PAL specific sctivity………………………………………………………………. 28
2.3. Styrene fermentation titers………………………………………………………… 34
3.1. Strains and plasmids used in this study.……………………………………………49
4.1. List of strains and plasmids. ……………………………………………………….71
4.2. Common regulators of ARO1, ARO2, ARO3, and ARO8.………………………..83
4.3. Assaying the in vitro decarboxylase activity of FDC1 …………………………….86
5.1. Toxicity limit analysis of styrene and its derivatives………………………………94
5.2. Summary of titers and yields for styrene and its derivatives.……………….……100

viii
LIST OF FIGURES
Figure Page
1.1. Enzymatic pathway to convert glucose to styrene, (S)-styrene oxide and (R)-1,2-
phenylethanediol ................................................................................................................. 8
2.1. Styrene pathway. ...................................................................................................... 14
2.2. Toxicity of styrene and trans-cinnamate. ................................................................. 26
2.3. PAL activity. ............................................................................................................ 29
2.4. PADC activity. ......................................................................................................... 32
2.5. Mass spectra of styrene ............................................................................................ 35
2.6. Whole cell styrene production. ................................................................................ 37
3.1. (S)-styrene oxide and (R)-1,2-phenylethanediol pathway. ...................................... 46
3.2. (S)-styrene oxide and (R)-1,2-phenylethanediol toxicity. ....................................... 55
3.3. SMO activity. ........................................................................................................... 58
3.4. Comparing phenylalanine, (S)-styrene oxide, and (R)-1,2-phenylethanediol titers 60
3.5. Comparing phenylalanine titers ............................................................................... 62
4.1. Styrene biosynthesis by S. cerevisiae. ..................................................................... 69
4.2. Toxicity of exogenous styrene against S. cerevisiae BY4741 growing cells. ......... 78
4.3. Evolution of phenylalanine overproducing mutants of S. cerevisiae. ..................... 79

ix
Page
4.4. Transcriptional analysis of phenylalanine overproducing S. cerevisiae mutants. ... 82
4.5. Assessing the trans-membrane export of trans-cinnamate. ..................................... 88
4.6. Styrene biosynthesis from glucose by engineered S. cerevisiae. ............................. 91
5.1. Styrene accumulation in bioreactor ......................................................................... 97
5.2. In situ product removal of styrene via solvent extraction ........................................ 98

1
CHAPTER 1
INTRODUCTION
Abstract
This chapter describes background information on the field of metabolic
engineering and the motivation for my project on the construction of pathways from L-
phenylalanine. The metabolic pathway for styrene and its derivatives (S)-styrene oxide
and (R)-1,2-phenylethanediol will be provided. This chapter concludes with the
dissertation organization.
1.1 Background and motivation
A demand for commodity chemicals by renewable means rather than fossil fuels
has been increasing in recent years. Microorganisms are capable of naturally producing
various useful products including pharmaceuticals, fuels, and polymers; however, often at
too small of quantities for industrial economic viability. Metabolic engineering enables
efficient construction of both high yield pathways that already exist in nature and novel
pathways that may be constructed from the enzymatic parts of various microorganisms.
Utilizing a diversity of enzymes through the tools of metabolic engineering, researchers
have successfully catalyzed a wide array of biochemical reactions producing important
biofuels, pharmaceuticals, and other fine chemicals.
The most prominent application of metabolic engineering is currently in the
transportation fuel sector with a heavy focus on bio-ethanol and bio-butanol production.
Though these biofuels represent the highest volume of biologically produced chemicals,
they also have the lowest profit margin due to the discrepancy between the high cost of

2
processing suitable carbon substrates (sugars such as glucose) and the value of the final
product (Keasling 2010). While the biofuels sector has been of primary interest for the
last several decades, recent interest in utilizing metabolic engineering for the production
of pharmaceuticals and fine chemicals has developed. Not only does a biosynthetic route
offer a renewable means of producing commodity chemicals, it also gives us the ability to
circumvent the high energetic requirements of chemocatalytic production (as is the case
of styrene from ethylbenzene), control the stereospecificity of chiral compounds (a
necessity for pharmaceutical precursors), as well as manufacture compounds which can
only be produced naturally (such as amino acids, vitamins, flavors, and fragrances).
Several examples of fine chemicals synthesized from microbial platforms have recently
been reported, most notably monomers of polymers and co-polymers such as 1,5-
diaminopentane and 1,4-diaminopentane (Qian, Xia, and Lee 2011) (used to make
polyamids like Nylon) as well as isoprene (Lindberg, Park, and Melis 2010) (a monomer
for rubber and copolymer synthesis). Since these products are indistinguishable from
those of petrochemical origin, no change in industry infrastructure is necessary for their
incorporation into existing polymer and co-polymer synthesis methods. Thus, metabolic
engineering strategies provide new opportunities to develop products of commercial
interest by biosynthetic means thereby reducing our dependence on nonrenewable
resources and offering a ‘green chemistry’ approach to producing essential commodity
chemicals.
Monoaromatic compounds are an important and diverse class of fine chemicals
with applications ranging from use as solvents to monomers for polymer synthesis.
Several monoaromatic compounds have been microbially synthesized to date, but the

3
origin of these compounds have been mainly derived from p-coumarate via the L-tyrosine
pathway, making them phenolics(Boudet 2007). This approach simply replicates the
natural pathways commonly found in plants. In natural systems, phenolics play vital roles
in plant physiology and fitness. For example, plants produce phenolic polymers, like
lignin and suberin, as structural components of their cellular wall, as well as flavanoids
for pigmentation which is used to attract pollinators and as UV filtration(Boudet 2007).
With such an apparent necessity for phenolics in natural systems, there exists a plethora
of enzymes and enzymatic sources for phenolic based reactions. Examples of phenolics
which have been heterologously synthesized include phenol (a monomer for phenolic
resins)(Wierckx et al. 2005), p-hydroxybenzoate (a precursor to parabens)(Verhoef et al.
2007), caffeate (an antioxidant and antitumor agent)(Zhang and Stephanopoulos 2012),
tyrosol (an antioxidant)(Satoh et al. 2012), and p-hydroxystyrene (a useful
copolymer)(Qi et al. 2007; Verhoef et al. 2009). However, the number of biosynthesized
non-phenolic monoaromatics remains limited to-date. This, in part, is due to the fact that
non-phenolics are relatively uncommon in natural systems; therefore, pathway design is
more difficult and requires novel approaches. In addition, the pool of known enzymes
which express activity on non-phenolics is also greatly limited. Enzymes which
demonstrate activity on phenolics often utilize the para-hydroxyl group of the substrate
for stability serving as a hydrogen bond donor or acceptor(Brownlee et al. 2008; Serre et
al. 1999; Caruso et al. 2004; Rodríguez et al. 2010). Due to the necessity of the para-
hydroxyl group, these phenolic enzymes often possess limited, if any, substrate
promiscuity thereby limiting their potential use on non-phenolic homologues.

4
As previously mentioned, the number of non-phenolic monoaromatic compounds
engineered from renewable resources remains limited, and thus the focus of this work
will be on the biosynthetic production of monoaromatics derived via phenylalanine,
namely styrene and its derivatives, (S)-styrene oxide and (R)-1,2-phenylethanediol.
Styrene is a high-value monomer used in the production of polymers and copolymers,
most notably polystyrene, acrylonitrile-butadiene-styrene (ABS), styrene-acrylonitrile
(SAN), and styrene-butadiene rubber. As one of the most important monomers in the
plastics industry, its annual production exceeds 6 million metric tons per year
representing a $28 billion market(McKenna and Nielsen 2011). However, while its uses
and need by consumers is apparent, production of styrene via the dehydrogenation of
petroleum-derived ethylbenzene is also one of the most energy intensive processes
requiring 3 metric tons of steam per metric ton of styrene produced(McKenna and
Nielsen 2011). Not only is styrene an essential and valuable precursor to plastics, it is
also an important precursor to the fine chemicals (S)-styrene oxide and (R)-1,2-
phenylethanediol. These styrene derivatives are chiral building blocks used for the
production of pharmaceuticals as well as other compounds of interest. For example, (S)-
Styrene oxide is used as a precursor to the biocides levamisole and nematocide(Park, So,
et al. 2006) as well as the synthesis of cosmetics(Loprieno et al. 1976), surface coatings,
and agricultural(Loprieno et al. 1976) and biological(Panke et al. 2000; Han et al. 2006)
chemicals. (R)-1,2-Phenylethanediol is used in the synthesis of the pharmaceuticals (R)-
norfluoxetine and (R)-fluoxetine, which are used to treat psychiatric and metabolic
disorders, as well as β-lactam antibiotics(Cao et al. 2006; Kumar, Upadhyay, and Pandey
2004). Additionally, (R)-1,2-phenylethanediol is also used in the production of various

5
agrochemicals and pheromones(Gamenara and Dominguez de Maria 2009). In the face of
rising costs of oil, alternative, renewable production methods for these compounds will
secure the future accessibility to meet the needs of consumers.
1.2 Metabolic pathways
Phenylalanine, the necessary endogenous precursor to the production of styrenics,
is produced via the shikimic acid pathway in addition to tyrosine and tryptophan as seen
in Fig. 1.1. However, in E. coli, the aromatic amino acids are used only for the production
of proteins and not as precursors for secondary metabolites; therefore, the pathway is
tightly feedback-regulated. The shikimic acid pathway begins with the condensation of
phosphoenolpyruvate (PEP), a key intermediate in glycolysis, and erythrose-4-phosphate
(E-4P), a key intermediate in the pentose phosphate pathway, to yield the first major
precursor of the pathway, 3-deoxy-D-arbino-heptulosonate-7-phosphate (DAHP). This
reaction is achieved via the DAHP synthase enzymes AroF, AroG, and AroH which are
feedback sensitive to tyrosine, phenylalanine, and tryptophan, respectively. The
regulation of these enzymes is two-fold, including regulation at the transcriptional level
as well as at the protein level. Firstly, to control transcription, the aromatic amino acids
form a complex with their DNA-binding transcriptional regulator (TyrR for
phenylalanine and tyrosine, TrpR for tryptophan) which then binds to a palindromic
target sequence near the transcriptional promoter region of the genes aroF, aroG, and
aroH. The presence of the DNA-binding protein complex interferes with the proper
binding of RNA polymerase and thereby inhibits transcription from occurring. Secondly,
the proteins themselves are regulated via allosteric enzyme inhibition. Upon binding of
the aromatic amino acids to their respective feedback-sensitive DAHP synthase, a

6
conformational change of the enzyme occurs which represses its activity further. In
addition to feedback control of DAHP synthase activity, the shikimic acid pathway is also
tight regulated at the metabolite chorismate, which is a key branch point for the three
aromatic amino acids. To produce phenylalanine, chorismate is converted to prephenate
and subsequently to phenylpyruvate via the bifunctional chorismate mutase/prephenate
dehydratase enzyme PheA. However, unlike the DAHP synthases, PheA is only regulated
via allosteric enzyme inhibition and is not regulated transcriptionally(Keseler et al. 2005).
To achieve high flux through the shikimic acid pathway and obtain high titers of
phenylalanine, it is necessary to relieve the feedback regulation imposed on the key
enzymes in E. coli.
A feedback resistant strain of E. coli, NST74 (ATCC 31884), has been previously
engineered to overproduce phenylalanine(Tribe 1987). This was accomplished via NTG
(N-methyl-N'-nitro-N-nitrosoguanidine) mutagenesis and subsequent selection of mutants
in minimal media containing the phenylalanine anti-metabolites β-2-thienylalanine or p-
fluoro-DL-phenylalanine. In the presence of phenylalanine anti-metabolites, feedback
regulation of the shikimic acid pathway is activated and natural production of
phenylalanine ceases. In order to survive in the presence of the anti-metabolites, the
strain must evolve to de-regulate the feedback sensitive enzymes. This approach resulted
in the development of the strain E. coli NST74 with the relevant genotype aroH367(fbr),
tyrR366, tna-2, lacY5, aroF394(fbr), malT384, pheA101(fbr), pheO352, aroG397(fbr).
As described, E.coli NST74 possesses mutations which relieve the allosteric enzyme
inhibition of the DAHP synthase enzymes AroF, AroG, and AroH as well as the
chorismate mutase/prephenate dehydratase PheA. In addition, the DNA-binding

7
transcriptional regulator TyrR and the phenylalanine operator pheO has also been
mutated to alleviate repression. As a result of these key mutations, phenylalanine titers as
high as 1.98 g/L were achieved in 1 L bioreactor experiments(Tribe 1987). E. coli
NST74's ability to produce copious amounts of phenylalanine makes it an ideal host
platform for the biosynthetic production of the non-natural, monoaromatics styrene, (S)-
styrene oxide, and (R)-1,2-phenylethanediol.
The proposed biosynthesis of styrene and its derivatives (S)-styrene oxide and
(R)-1,2-phenylethanediol may be achieved from endogenously produced phenylalanine,
as illustrated in Fig. 1.1. Firstly, phenylalanine is deaminated to trans-cinnamate via the
expression of a suitable phenylalanine ammonia lyase (PAL). Secondly, trans-cinnamate
is decarboxylated to styrene via the expression of a suitable phenylacrylate decarboxylase
(PADC). To date, the only known PADC which demonstrates activity toward styrene is
the ferulate decarboxylase (FDC1) of Saccharomyces cerevisiae. Styrene may then be
oxidized to (S)-styrene oxide via the expression of a styrene monooxygenase, namely
StyAB from Pseudomonas putida S12, or (R)-1,2-phenylethanediol via the expression of
a naphthalene dioxygenase which demonstrates activity on styrene, namely
NahAaAbAcAd from Pseudomonas NCIB 9816. In each enzymatic step of the described
pathway, various enzymes were tested for their activity and specificity, which will be
discussed in further detail in the proceeding chapters.
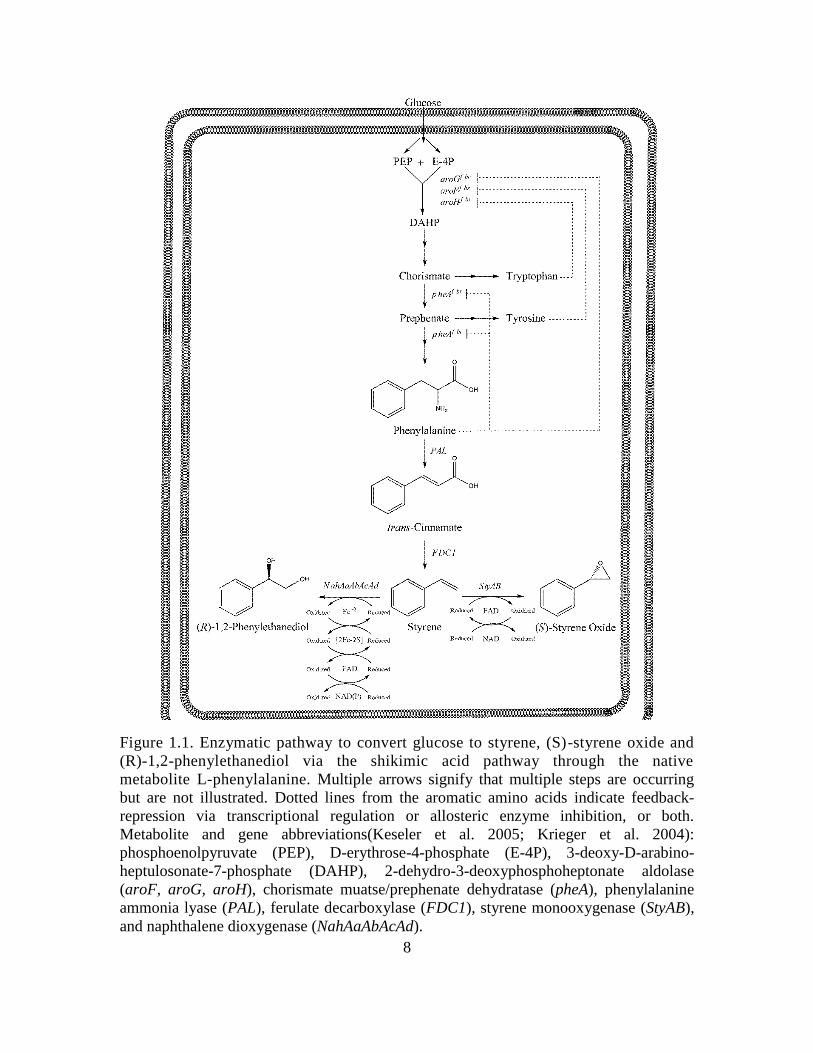
8
Figure 1.1. Enzymatic pathway to convert glucose to styrene, (S)-styrene oxide and
(R)-1,2-phenylethanediol via the shikimic acid pathway through the native
metabolite L-phenylalanine. Multiple arrows signify that multiple steps are occurring
but are not illustrated. Dotted lines from the aromatic amino acids indicate feedback-
repression via transcriptional regulation or allosteric enzyme inhibition, or both.
Metabolite and gene abbreviations(Keseler et al. 2005; Krieger et al. 2004):
phosphoenolpyruvate (PEP), D-erythrose-4-phosphate (E-4P), 3-deoxy-D-arabino-
heptulosonate-7-phosphate (DAHP), 2-dehydro-3-deoxyphosphoheptonate aldolase
(aroF, aroG, aroH), chorismate muatse/prephenate dehydratase (pheA), phenylalanine
ammonia lyase (PAL), ferulate decarboxylase (FDC1), styrene monooxygenase (StyAB),
and naphthalene dioxygenase (NahAaAbAcAd).

9
1.3 Dissertation organization
This dissertation is organized into five chapters. Chapter 1 discusses a background
into the field of metabolic engineering and the importance of determining methods to
create fine chemicals such as styrene, (S)-styrene oxide, and (R)-1,2-phenylethanediol
from renewable resources. This chapter also gives an outline of the metabolic pathways
constructed during this project as well as an overview of E. coli’s phenylalanine pathway.
Chapter 2 introduces us to pathway engineering through the development of a renewable
method for the biosynthesis of styrene in E. coli. This is the first example of a renewable
method for styrene production from renewable resources such as glucose. This chapter
will show that the styrene monomer is extremely toxic to the bacteria and severely limits
production titers. The styrene pathway is then extended to (S)-styrene oxide and (R)-1,2-
phenylethanediol in Chapter 3. When the endogenously produced styrene is converted to
these less toxic compounds, higher productivities are achieved. However, at this point the
pathway is limited not only by toxicity but by the ability to make ample amounts of the
endogenous precursor phenylalanine. Several methods to improve phenylalanine titers
will be discussed. Since the main difficulty in biosynthetically producing monoaromatics
in E. coli remains the toxicity threshold, Chapter 4, focuses on transferring the styrene
pathway to a new yeast host. The work encompasses using S. cerevisiae which will be
engineered to overproduce phenylalanine. The styrene pathway was incorporated into the
S. cerevisiae phenylalanine over-producer and tested for styrene production from glucose.
Chapter 5 will suggest future directions for the styrene pathway and its derivatives.

10
CHAPTER 2
BIOSYNTHESIS OF STYRENE BY ENGINEERED E. COLI
Abstract
Styrene is a large volume, commodity petrochemical with diverse commercial
applications, including as a monomer building-block for the synthesis of many useful
polymers. Here we demonstrate how, through the de novo design and development of a
novel metabolic pathway, styrene can alternatively be synthesized from renewable
substrates such as glucose. The conversion of endogenously-synthesized L-phenylalanine
to styrene was achieved by the co-expression of phenylalanine ammonia lyase and trans-
cinnamate decarboxylase. Candidate isoenzymes for each step were screened from
bacterial, yeast, and plant genetic sources. Finally, over-expression of PAL2 from A.
thaliana and FDC1 from S. cerevisiae (originally classified as ferulate decarboxylase) in
an L-phenylalanine over-producing E. coli host led to the accumulation of up to 260
mg/L in shake flask cultures. Achievable titers already approach the styrene toxicity
threshold (determined as ~300 mg/L). To the best of our knowledge, this is the first
report of microbial styrene production from sustainable feedstocks.
This work was published as:
McKenna, R. & Nielsen, D.R. Styrene biosynthesis from glucose by engineered E. coli.
Metab Eng 13, 544-554 (2011)

11
2.1 Introduction
Styrene is a versatile, large commodity chemical for which 60% of its global
annual consumption supports the production of numerous, industrially-important
polymers and co-polymers(SRI 2010). In 2006, over 6 million metric tons of styrene
were produced by U.S. manufacturers, a market that was valued at nearly $28 billion and
projected to grow by 4.3% per year through 2010(SRI 2010). Today, all commercially-
available styrene is derived from the world’s dwindling petroleum resources.
Conventional styrene synthesis is achieved through the chemocatalytic dehydrogenation
of petroleum-derived ethylbenzene(Wu, Koylinski, and Bozik 1981) which requires over
3 metric tons of steam per metric ton of styrene produced. This exorbitant requirement
renders styrene production as the most energy-intensive among commodity chemical
production routes, consuming nearly 200 trillion BTU of steam for its domestic annual
production alone (DoE 2002). With that being said, the goal of this study was to engineer
a biocatalyst capable of synthesizing styrene from renewable resources as a more
sustainable and greener source of styrene and styrene-derived polymers.
Beyond its petrochemical origins, styrene has been observed as a trace metabolite
in foods, in particular cheeses, where it acts as an aroma defect. For instance, the yeast
Penicillium camemberti has been reported to be capable of synthesizing low levels of
styrene from excess L-phenylalanine, but neither a defined pathway nor the requisite
genes have thus far been elucidated(Pagot et al. 2007). Styrene is also known to be
naturally synthesized by select plant species, including several trees in the Styracaceae
family (including several Styrax sp.). Here, styrene is also synthesized from excess L-
phenylalanine where it then subsequently accumulates as a minor constituent (<0.55% of

12
total dry weight) within benzoin resins (which are predominantly composed of benzoic
acid)(Fernandez et al. 2005). Again, however, neither the enzymes nor genes associated
with said pathway have been identified to date. Although it is possible to purify styrene
from plant resins via distillation or liquid-liquid extraction(Clark 1990), considering the
extremely low productivity, poor net yields, and low inherent value of styrene, its
potential, large-scale biological production by such a mechanism is rendered as
completely uneconomical and unsustainable. A more sustainable and inexpensive
approach, however, would involve the engineering of microorganisms that possess the
unique ability to synthesize styrene directly from renewable resources.
In recent years, a variety of additional, novel synthetic routes have been proposed
and engineered in microorganisms for the production (from renewable substrates such as
glucose) of a number of other useful, functionalized monoaromatic compounds with
structural similarity to styrene. For example, a biosynthetic pathway for the production of
p-hydroxystyrene (a monomer used in synthesis of photo-resist polymers) from
renewable sugars has been constructed using both Escherichia coli(Qi et al. 2007) and
Pseudomonas putida(Verhoef et al. 2009) as host platforms. Meanwhile, both phenol (a
precursor and monomer for phenolic resins)(Wierckx et al. 2005) and p-hydroxybenzoate
(a precursor to parabens, which are used as preservatives)(Verhoef et al. 2007) have also
been synthesized as individual products from glucose by engineered strains of P. putida.
Interestingly, each of the above non-natural metabolites were derived using L-tyrosine (or
its immediate precursor, 4-hydroxyphenylpyruvate) as a pathway precursor, thereby
making them each phenolics(Boudet 2007). To date, there remain few examples of
engineered biosynthetic pathways for the production of non-phenolic, monoaromatic

13
compounds using microbial biocatalysts. Moreover, there exist no previous reports
regarding the development of a styrene biosynthetic pathway or the engineering of
microbes capable of synthesizing styrene from renewable resources. With this in mind,
the present study describes the de novo design and development of a functional styrene
biosynthetic pathway and the engineering of E. coli strains capable of styrene
biosynthesis from glucose.
The proposed styrene biosynthesis pathway utilizes endogenously synthesized
(from glucose) L-phenylalanine as an intermediate precursor which is converted to
styrene by a series of two enzymatic steps, as in Fig. 2.1. First, endogenously-occurring
L-phenylalanine is converted to trans-cinnamic acid (tCA) through its deamination, as
catalyzed by phenylalanine ammonia lyase (PAL). Said activity and substrate specificity
has been previously reported for a number of PAL isoenzymes that have been identified
and characterized in yeast(Vannelli, Xue, et al. 2007; Qi et al. 2007; Gilbert and Tully
1982), plants(Young, Towers, and Neish 1966; Cochrane, Davin, and Lewis 2004), and
(although less prevalently) bacteria(Moffitt et al. 2007; Xiang and Moore 2005, 2006;
Young, Towers, and Neish 1966). Among previously characterized PAL isoenzymes,
considerable variability with respect to both substrate specificity and activity has been
reported. The most commonly studied PAL isoenzyme in recombinant systems, including
E. coli, remains the bifunctional PAL/TAL (TAL: tyrosine ammonia lyase, which also
catalyzes the deamination of L-tyrosine to p-coumaric acid) of the yeast Rhodotorula
sp.(Gilbert et al. 1985; Gilbert and Tully 1982; Gilbert, Stephenson, and Tully 1983; Cui,
Jia, and Sun 2008; Vannelli, Wei Qi, et al. 2007; Santos, Koffas, and Stephanopoulos
2011). Meanwhile, a number of PAL isoenzymes have been also been studied from plant

14
sources(Jones 1984) where, for example, Arabidopsis thaliana has been characterized as
possessing four distinct PALs (encoded by PAL1, PAL2, PAL3, and PAL4)(Cochrane,
Davin, and Lewis 2004). More recently, a number of prokaryotic PALs have been
isolated and characterized, beginning with that which is encoded by encP from
Streptomyces maritimus(Xiang and Moore 2002, 2005). Soon after, two additional PAL
isoenzymes were discovered in the cyanobacteria Nostoc puntiformes and Anabaena
variabilis(Moffitt et al. 2007; Xiang and Moore 2005, 2006). Interestingly, each of these
prokaryotic PALs was also found to be highly specific for L-phenylalanine, and thus do
not also display TAL activity that is so common among yeast PALs.
Figure 2.1 Styrene pathway. Enzymatic pathway to convert the precursor L-
phenylalanine to the product styrene via the intermediate trans-cinnamate. The two-step
pathway from L-phenylalanine is achieved by the co-expression of one or more genes
which encoded phenylalanine ammonia lyase (PAL) activity (A), and one or more genes
which encoded trans-cinnamic acid decarboxylase (CADC) activity (B).
The second step in the proposed styrene biosynthesis pathway involves the
subsequent decarboxylation of tCA by a phenylacrylate decarboxylase (PADC)
displaying trans-cinnamate decarboxylase activity to yield styrene as the final product.
Several genes encoding PADC activity have been characterized and reported in the
literature, including pdc from Lactobacillus plantarum and padC from Bacillus
subtilis(Tran et al. 2008). It currently remains unknown, however, if their enzyme

15
products could specifically decarboxylate tCA to produce styrene. Meanwhile, the yeast
Saccharomyces cerevisiae has demonstrated the ability to synthesize styrene when
supplied with exogenous tCA(Clausen et al. 1994); however, the underlying mechanism
and genetic basis for said activity presently remains unclear. For instance, previous
characterizations of PAD1 have shown that its over-expression in S. cerevisiae results in
increased resistance to tCA (as achieved by its conversion to styrene)(Larsson,
Nilvebrant, and Jonsson 2001). However, it has also been demonstrated that the
expression of PAD1 alone is insufficient for achieving PADC activity(Clausen et al.
1994), and it was later speculated that the expression of a second enzyme was necessary
to convert tCA to styrene(Jiang, Wood, and Morgan 2005). Most recently, it was reported
that tCA decarboxylase activity in S. cerevisiae maintains an essential dependence on the
co-expression of both PAD1 and FDC1, the latter a gene previously characterized as
encoding ferulic acid decarboxylase (note that ferulic acid is also a phenylacrylic
acid)(Mukai et al. 2010).
The present study describes the de novo design of a styrene biosynthetic pathway,
as supported through the comprehensive screening of composite pathways enzymes from
various genetic sources. The synthesis of styrene from glucose was ultimately achieved
through the co-expression of PAL and tCA decarboxylase enzymes in an L-phenylalanine
over-producing E. coli host platform.
2.2 Materials and Methods
2.2.1 Chemicals.

16
Chemicals used in this study include L-phenylalanine (98.5%, VWR,
Westchester, PA), trans-cinnamic acid (99%, MP Biomedicals, Solon, OH), styrene
(99%, Alfa Aesar, Ward Hill, MA), methanol (99.8%, VWR, Westchester, PA), and
trifluoroacetic acid (99.5%, EMD, Darmstadt, Germany). All other chemicals used in this
study are from Sigma-Aldrich (St. Louis, MO).
2.2.2 Strains and Media.
All strains, plasmids, and oligonucleotide primers used in this study are listed in
Table 2.1. Custom oligonucleotide primers were synthesized by Integrated DNA
Technologies (Coralville, IA). cDNA of N. puntiformes and A. variabilis were gifts from
Prof. Bradley Moore (UCSD). Strains of B. subtilis, L. plantarum, and S. cerevisiae, as
well as the plasmids pSTV28 and pTrc99A were all gifts from Prof. Kristala Prather
(MIT). Genomic DNA was prepared from whole cells using the ZR Fungal/Bacterial
DNA MiniPrep (Zymo Research, Irvine, CA) according to vendor protocols. Strains were
routinely cultured in Luria-Bertani (LB) broth (supplemented with antibiotics as
necessary). Cultures were assayed for their ability to synthesize L-phenylalanine, trans-
cinnamate, and styrene by cultivation in phosphate-limited minimal media (herein
referred to as “MM1”) with glucose. MM1 was adapted from Qi et al. (2007), and is
composed of glucose (nominally 15 g/L), MgSO4∙7H2O (0.5 g/L), (NH4)2SO4 (4.0 g/L),
MOPS (24.7 g/L), KH2PO4 (0.3 g/L), K2HPO4 (0.7 g/L), and 5 mL/L ATCC Trace
Mineral Supplement (Catalog No. MD-TMS) (EDTA (0.5 g/L), MgSO4∙7H2O (3 g/L),
MnSO4∙7H2O (0.5 g/L), NaCl (1 g/L), FeSO4∙7H2O (0.1 g/L), Co(NO3)2∙6H2O (0.1 g/L),
CaCl2 (0.1 g/L), ZnSO4∙7H2O (0.1 g/L), CuSO4∙5H2O (0.01 g/L), AlK(SO4)2 (0.01 g/L),
17
H3BO3 (0.01 g/L), Na2MoO4∙2H2O (0.01 g/L), Na2SeO3 (0.001 g/L), Na2WO4∙2H2O
(0.10 g/L), and NiCl2∙6H2O (0.02 g/L)).
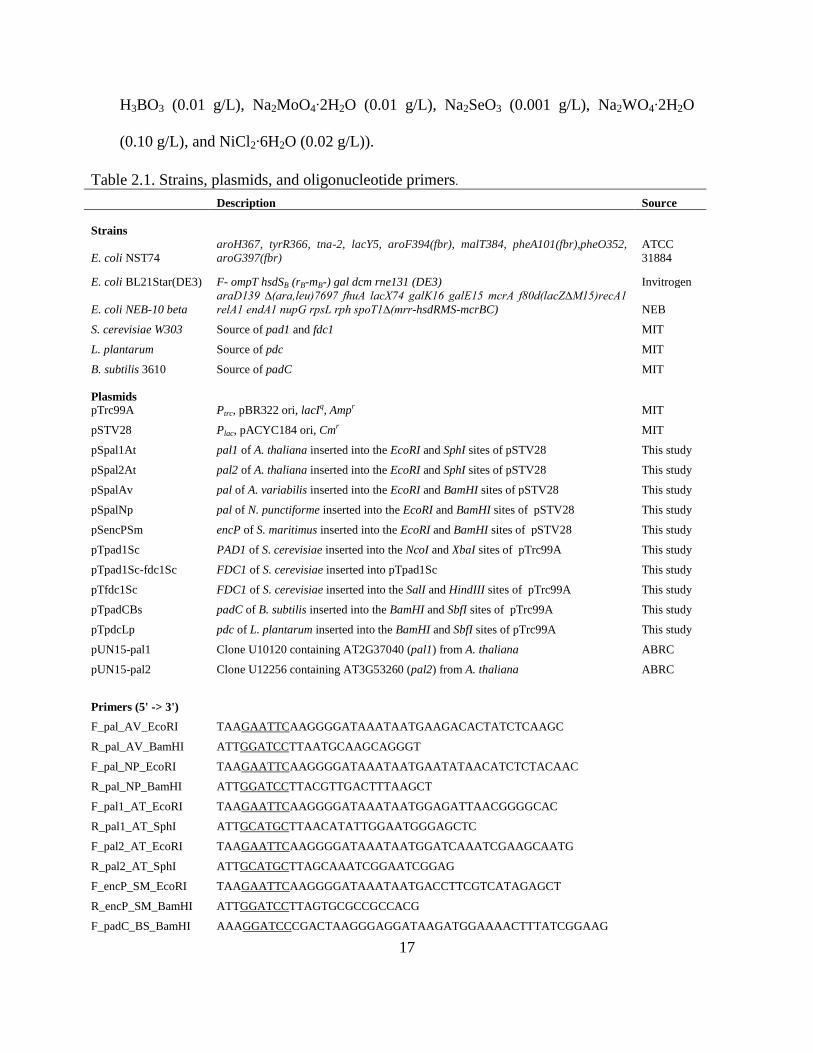
Table 2.1. Strains, plasmids, and oligonucleotide primers.
Description Source
Strains
E. coli NST74
aroH367, tyrR366, tna-2, lacY5, aroF394(fbr), malT384, pheA101(fbr),pheO352,
aroG397(fbr)
ATCC
31884
E. coli BL21Star(DE3) F- ompT hsdSB (rB-mB-) gal dcm rne131 (DE3) Invitrogen
E. coli NEB-10 beta
araD139 ∆(ara,leu)7697 fhuA lacX74 galK16 galE15 mcrA f80d(lacZ∆M15)recA1
relA1 endA1 nupG rpsL rph spoT1∆(mrr-hsdRMS-mcrBC) NEB
S. cerevisiae W303 Source of pad1 and fdc1 MIT
L. plantarum Source of pdc MIT
B. subtilis 3610 Source of padC MIT
Plasmids
pTrc99A Ptrc, pBR322 ori, lacIq, Ampr MIT
pSTV28 Plac, pACYC184 ori, Cmr MIT
pSpal1At pal1 of A. thaliana inserted into the EcoRI and SphI sites of pSTV28 This study
pSpal2At pal2 of A. thaliana inserted into the EcoRI and SphI sites of pSTV28 This study
pSpalAv pal of A. variabilis inserted into the EcoRI and BamHI sites of pSTV28 This study
pSpalNp pal of N. punctiforme inserted into the EcoRI and BamHI sites of pSTV28 This study
pSencPSm encP of S. maritimus inserted into the EcoRI and BamHI sites of pSTV28 This study
pTpad1Sc PAD1 of S. cerevisiae inserted into the NcoI and XbaI sites of pTrc99A This study
pTpad1Sc-fdc1Sc FDC1 of S. cerevisiae inserted into pTpad1Sc This study
pTfdc1Sc FDC1 of S. cerevisiae inserted into the SalI and HindIII sites of pTrc99A This study
pTpadCBs padC of B. subtilis inserted into the BamHI and SbfI sites of pTrc99A This study
pTpdcLp pdc of L. plantarum inserted into the BamHI and SbfI sites of pTrc99A This study
pUN15-pal1 Clone U10120 containing AT2G37040 (pal1) from A. thaliana ABRC
pUN15-pal2 Clone U12256 containing AT3G53260 (pal2) from A. thaliana ABRC
Primers (5' -> 3')
F_pal_AV_EcoRI TAAGAATTCAAGGGGATAAATAATGAAGACACTATCTCAAGC
R_pal_AV_BamHI ATTGGATCCTTAATGCAAGCAGGGT
F_pal_NP_EcoRI TAAGAATTCAAGGGGATAAATAATGAATATAACATCTCTACAAC
R_pal_NP_BamHI ATTGGATCCTTACGTTGACTTTAAGCT
F_pal1_AT_EcoRI TAAGAATTCAAGGGGATAAATAATGGAGATTAACGGGGCAC
R_pal1_AT_SphI ATTGCATGCTTAACATATTGGAATGGGAGCTC
F_pal2_AT_EcoRI TAAGAATTCAAGGGGATAAATAATGGATCAAATCGAAGCAATG
R_pal2_AT_SphI ATTGCATGCTTAGCAAATCGGAATCGGAG
F_encP_SM_EcoRI TAAGAATTCAAGGGGATAAATAATGACCTTCGTCATAGAGCT
R_encP_SM_BamHI ATTGGATCCTTAGTGCGCCGCCACG
F_padC_BS_BamHI AAAGGATCCCGACTAAGGGAGGATAAGATGGAAAACTTTATCGGAAG

18
R_padC_BS_SbfI ATACCTGCAGGATGTTTATTATAATCTTCCCGCG
F_pdc_LP_BamHI ATAGGATCCCTCTGGAGGCAGTTCTAATGACAAAAACTTTTAAAACACT
R_pdc_LP_SbfI ATACCTGCAGGCCAGAATGTTTCACGTGAA
F_pad1_SC_NcoI TTACCATGGAGGAACCTAGGCACACAATGGTCCTATTTCCAAGAAGAA
R_pad1_SC_XbaI ATTTCTAGATTACTTGCTTTTTATTCCTTCCC
F_fdc1_SC_SalI ATAGTCGACAGACATCAAAGGACGGTTCATGAGGAAGCTAAATCCAGCT
R_fdc1_SC_HindIII ATTAAGCTTTTATTTATATCCGTACCTTTTCCAAT
2.2.3 Toxicity assays.
To determine extent and effects of metabolite toxicity on E. coli, the impacts of
the exogenous addition of tCA and styrene (at increasing final concentrations) to growing
cultures was investigated. Seed cultures of E. coli NST74 were prepared in 5 mL of LB
broth and grown at 32oC overnight while shaking at 250 rpm. The seed culture (1 ml) was
then used to inoculate 50 ml of LB broth in a 250 mL shake flask. Cultures were grown
to an optical density (OD600) of either ~0.6 or ~2.0, at which time either tCA or styrene
was added to the flasks at an array of final concentrations between 0 to 1 g/L. Two
different initial OD600 levels were used to investigate the potential effects of product
toxicity as a function of cell density and/or growth stage. In all cases, culturing then
resumed at 32oC for another 6-8 h while cell growth, as determined by OD600
measurements, was periodically monitored using a UV/Vis spectrophotometer (Beckman
Coulter DU800, Brea, CA).
2.2.4 Cloning of candidate genes encoding PAL activity from A. variabilis, N.
punctiforme, and A. thaliana.
All genes used in this study were PCR amplified using a BioRad iCycler system
with Phusion DNA Polymerase (Finnzymes, Espoo, Finland) using custom
oligonucleotide primers. PCR cycling and reaction conditions were standardized

19
according to manufacturer instructions. Candidate PAL encoding genes were amplified
from the cDNA of A. variabilis and N. punctiforme. Candidate PAL encoding genes from
A. thaliana were derived from cDNA library plasmids containing the specific loci of
interest (Table 2.1) obtained from the ABRC (Ohio State University, Columbus, OH).
Whereas A. thaliana posses four distinct PALs, we focused on those encoded by PAL1
and PAL2 as these have displayed the greatest activity when expressed in recombinant E.
coli (noting also that PAL3 was found to be of ‘very low activity’)(Cochrane, Davin, and
Lewis 2004). Amplified linear DNA fragments were purified using the Zyppy Clean and
Concentrator kit (Zymo Research, Orange, CA). Purified fragments were treated by
endonuclease digestion using appropriate restriction enzymes (all from New England
Biolabs, Ipswich, MA). Amplified fragments containing the pal from both N. punctiforme
and A. variabilis were digested with BamHI and EcoRI whereas fragments containing
PAL1 and PAL2 from A. thaliana were digested with EcoRI and SphI. All digestions were
performed at 37oC according to manufacturer’s protocols. The expression vector pSTV28
was similarly digested with either BamHI and EcoRI or EcoRI and SphI. Digested
fragments were gel purified using the Zyppy Gel DNA recovery kit (Zymo Research,
Orange, CA). Linearized fragments were ligated using T4 DNA ligase (New England
Biolabs, Ipswich, MA) at 4oC overnight. Chemically competent E. coli NEB10-Beta
(New England Biolabs, Ipswich, MA) cells were used for all transformations.
Transformants were selected by plating on LB solid agar containing 34 mg/L
chloramphenicol and culturing at 37oC overnight. The transformant pool was screened
according to both colony PCR (employing the same primers as used in the initial
amplification) and restriction digest mapping of the resultant plasmids to identify those

20
clones harboring the successful construct. This approach resulted in construction of
plasmids pSpalAv, pSpalNp, pSpal1At, and pSpal2At, as listed in Table 2.1.
2.2.5 Cloning of candidate genes encoding PADC activity from L. plantarum, B. subtilis,
and S. cerevisiae.
Candidate PADC encoding genes, including pdc, padC, PAD1, and FDC1, were
amplified via PCR using genomic DNA templates derived from L. plantarum, B. subtilis,
and S. cerevisiae, respectively. PCR amplified DNA fragments were purified before
treatment by endonuclease digestion. Fragments containing padC and pdc were each
digested with BamHI and SbfI. The E. coli expression vector pTrc99A was similarly
digested with BamHI and SbfI. Meanwhile, the amplified fragment containing PAD1 was
digested with NcoI and XbaI whereas the FDC1 containing fragment was digested with
SalI and HindIII. The E. coli expression vector pTrc99A was similarly digested with
either NcoI and XbaI (for the insertion of PAD1) or SalI and HindIII (for the insertion of
FDC1). All digested fragments were gel purified then ligated at 4oC overnight before
their transformation into chemically competent E. coli NEB10-Beta. Selection was then
achieved by plating transformed cells on LB solid agar containing 100 mg/L ampicillin
and culturing at 37oC overnight. After confirmation of the correct transformant, these
works resulted in the generation of plasmids pTpadCBs, pTpdcLp, pTpad1Sc, and
pTfdc1Sc, as listed in Table 2.1. The SalI-HindIII digested FDC1 fragment was then also
cloned into the same sites in the newly generated plasmid pTpad1Sc by an analogous
protocol, resulting in the plasmid pTpad1Sc-fdc1Sc.
2.2.6 Assaying PAL activity in crude lysates of recombinant E. coli.

21
Each of the newly created PAL harboring plasmids (namely pSpalAv, pSpalNp,
pSpal1At, and pSpal2At) were individually transformed into E. coli BL21(DE3)
(Invitrogen, Carlsbad, CA). Seed cultures of each of the resultant strains were prepared
(in triplicate) in 5 mL LB broth supplemented with 34 mg/L chloramphenicol and
cultured at 32°C while shaking at 250 rpm overnight. 50 µl of each seed was then used to
again inoculate 5 ml LB broth. These cultures were grown until reaching an OD600 of
~0.6, at which point each was induced by the addition of isopropyl β-D-1-
thiogalactopyranoside (IPTG) at a final concentration of 0.2 mM. Induced cultures were
incubated for an additional 6 h after which an equal number of cells (determined by
OD600 measurement) were collected by centrifugation at 1400 x g for 4 min. The cell
pellet was re-suspended in 900 µL distilled water. Cell lysis was achieved using the
FastBreak Cell Lysis Reagent kit (Promega, Madison, WI) and the supernatant collected
after centrifugation at 11,000 x g for 2 min. PAL activity was analyzed at room
temperature in pH 7.5 50 mM Tris-HCl buffer containing 250 mM L-phenylalanine.
Activity assays were initiated by the addition of 5 µL of crude cell lysate. The production
of tCA was followed at 290 nm on a Beckman Coulter UV/Vis Spectrophotometer for a
total of 5 min at 20 sec intervals. A molar extinction coefficient of 9,000 M-1
cm-1
and a 1
cm path length were used to establish enzyme activity in terms of U mg-1
protein. The
PAL protein content in each crude lysate was determined via first separation by SDS-
PAGE using Mini-PROTEAN TGX 4-20% precast gels (Bio-Rad, Hercules, CA) and
standard protocols. Concentration was then analyzed using the ImageJ software package
(NIH, Bethesda, MD) and calibrated versus Precision Plus Unstained Protein Standards
(Bio-Rad, Hercules, CA).

22
2.2.7 Assaying PAL activity in recombinant E. coli whole cells.
Seed cultures of E. coli BL21(DE3) harboring one of pSpalAv, pSpalNp,
pSpal1At, or pSpal2At, were prepared in 5 mL LB broth and grown overnight. Shake
flasks (250 mL) containing 50 mL of LB were inoculated with 1 mL of each seed culture.
Cultures were grown until an OD600 of ~0.6 was reached, at which point the cultures were
induced by IPTG addition at a final concentration of 0.2 mM. Cultures were then
incubated for an additional 6 h (resulting in an OD600 of ~2) before an equal number of
cells were collected and centrifuged at 1400 x g for 5 min. The pellet was washed once
with PBS (phosphate buffered saline) at pH 7 before being re-suspended in 12 ml PBS
buffer. Finally, the appropriate substrate, L-phenylalanine or L-tyrosine at a final
concentration of 1 g/L, was added to the suspension. The suspensions were then shaken at
32°C for a total of three hours. Samples (1 mL) were taken every hour, centrifuged, and
750 µL of supernatant was collected for HPLC analysis to monitor the production of
either tCA or p-coumaric acid.
2.2.8 Assaying PADC activity in recombinant E. coli whole cells.
The plasmids pTpad1Sc-fdc1Sc, pTpad1Sc, pTfdc1Sc, pTpdcLp, and pTpadcBs
were each individually transformed into chemically competent E. coli BL21(DE3). A
seed culture of each strain was then grown in LB broth overnight. Shake flasks (250 mL)
containing 50 mL of LB were inoculated with 1 ml of seed culture. Cultures were grown
at 32°C until an OD600 of ~0.6, at which point they were induced by adding 0.2 mM
IPTG and then incubated for an additional 6 h. Cells were then collected and re-
suspended in 12 ml PBS buffer (as previously described) and the substrate (tCA or p-
coumaric acid) was added at a final concentration of 1 g/L. Samples (1 mL) were

23
removed from the culture at both the time of initiation as well as after 12 h of incubation
at 32oC and analyzed by HPLC using the methods described herein.
2.2.9 Co-expression of PAL and CADC isoenzymes in E. coli NST74 to convert L-
phenylalanine to styrene.
The L-phenylalanine over-producing strain E. coli NST74 (Table 2.1) was co-
transformed with the plasmids pSpal2At and pTfdc1Sc and selected for on LB agar
supplemented with 100 mg/L ampicillin and 34 mg/L chloramphenicol. The resultant
transformant was then grown overnight at 32°C in 5 mL LB broth. Shake flasks (250 mL)
containing 50 mL LB were inoculated with 1 mL of seed culture. The culture was then
grown at 32°C until an OD600 of ~0.6, at which time it was induced with 0.2 mM IPTG
and then incubated for an additional 8 h. Cells were then collected and re-suspended in 12
ml PBS buffer (as previously described) and L-phenylalanine added at a final
concentration of either 400 or 900 mg/L. Samples were taken periodically and analyzed
by HPLC to determine the content of L-phenylalanine, tCA, and styrene, according to the
methods presented below.
2.2.10 Co-expression of PAL and CADC isoenzymes in E. coli NST74 to convert glucose
to styrene in shake flask cultures.
The L-phenylalanine over-producing strain E. coli NST74 was co-transformed
with each of the following combinations of plasmids: pSpalAv and pTfdc1Sc, pSpalNp
and pTfdc1Sc, pSpal1At and pTfdc1Sc, and pSpal2At and pTfdc1Sc.Single colonies
were then selected from the resulting transformants and grown in 5 mL LB broth for 12 h
at 32°C while shaking at 250 rpm to prepare a seed culture. Each seed (1 mL) was then
used to inoculate 50 mL MM1. These cultures were performed in 100 mL serum bottles

24
outfitted with septa caps that were tightly sealed upon inoculation. A closed system was
used to avoid volatile product (i.e., styrene) losses. A large headspace was used to
preclude the depletion of oxygen from the bottle. Cultures were grown for 10 h prior to
being induced by the addition of IPTG at a final concentration of 0.2 mM. Culturing
continued for 29 to 48 h post induction while 1 ml samples were periodically taken from
each culture and analyzed for relevant metabolite contents via HPLC, using the methods
described herein.
2.2.11 Metabolite analysis by HPLC.
Samples were prepared by removing 1 mL of culture from a shake flask culture
and pelleting the cells at 11,000 x g for 2 min. The supernatant (0.75 mL) was then
transferred to a glass HPLC vial and sealed with a Teflon-lined cap. HPLC analysis was
carried out using a Hewlett Packard 1100 series HPLC system equipped with an auto
sampler, diode array (UV/Vis) detector, and reverse-phase Hypersil Gold SBC18 column
(4.6mm x 150 mm; Thermo Fisher, USA). Samples (5 µL) were injected for analysis at a
total constant flow rate of 1.0 ml/min and constant column temperature of 45oC. The
column was eluted with ‘solvent A’ (consisting of double-distilled water) and ‘solvent B’
(consisting of methanol (99.8% grade) plus 0.1% trifluoroacetic acid (TFA)). The eluent
began as a mixture of 95% solvent A and 5% solvent B before a linear gradient was
applied over 8 min to then reach a mixture of 20% solvent A and 80% solvent B. This
eluent composition was then held constant for 2 min before a second linear gradient was
then applied over the course of 4 min to achieve a final mixture of 95% solvent A and 5%
solvent B. The eluent was monitored at each of 215 nm for L-phenylalanine and 258 nm
for tCA, p-coumaric acid, hydroxystyrene and styrene. Under these conditions L-

25
phenylalanine, p-coumaric acid, tCA, p-hydroxystyrene, and styrene were eluted at 4.5,
6.7, 8.67, 8.78, and 10.4 min, respectively.
2.2.12 Confirmation of styrene biosynthesis by GC-MS.
Culture supernatant (1 ml) was added to hexane (1 ml) and vortexed for 20 min at
maximum speed. The biphasic mixture was centrifuged for 1 min at 11,000 x g to settle.
750 µl of the hexane layer was removed for analysis by GC-MS. GC-MS analysis was
performed on a Hewlett Packard 5890 Series II gas chromatograph with a flame ionizing
detector and Supelco MDN-5 S (30 m × 0.25 mm id) fused-silica capillary column using
helium as the carrier gas. The injector, column, and detector temperatures were initial set
at 280, 240, and 220 °C, respectively. The column temperature was then increased from
40 to 320 °C at 14 °C/min.
2.3 Results and discussion
2.3.1 Assaying styrene toxicity.
Under the studied conditions, maximal titers of L-phenylalanine produced by E.
coli NST74 after 48 h of culture in MM1 (with 1.5% glucose) reached about 700-1000
mg/L (results not shown). Whereas the theoretical yield of L-phenylalanine on glucose is
0.55 g/g(Baez-Viveros et al. 2004), achievable yields by E. coli NST74 under the present
conditions in shake flask cultures were only 0.052-0.074 g/g (or about 10% of
theoretical). Assuming complete conversion of all endogenously produced L-
phenylalanine to styrene were possible (if our engineered pathway could achieve a
particularly high flux), we would expect to be able to synthesize up to 440-630 mg/L of
styrene under the same culture conditions (corresponding to a glucose yield of 0.051-
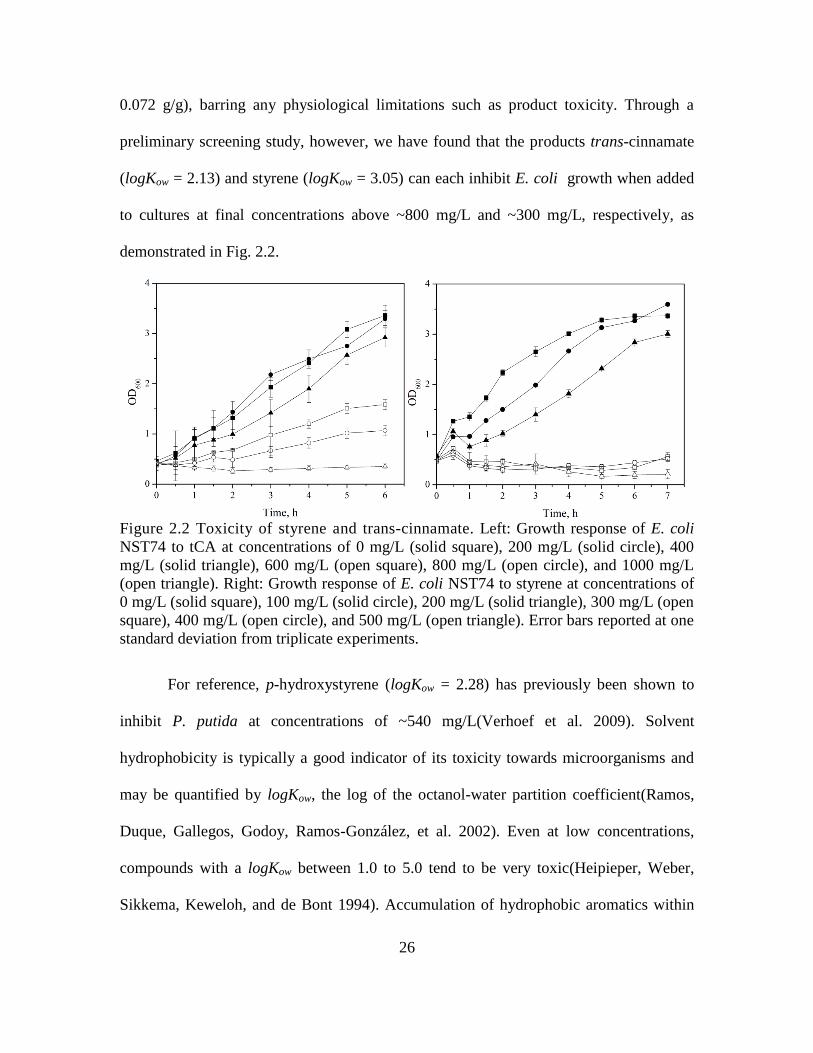
26
0.072 g/g), barring any physiological limitations such as product toxicity. Through a
preliminary screening study, however, we have found that the products trans-cinnamate
(logKow = 2.13) and styrene (logKow = 3.05) can each inhibit E. coli growth when added
to cultures at final concentrations above ~800 mg/L and ~300 mg/L, respectively, as
demonstrated in Fig. 2.2.
Figure 2.2 Toxicity of styrene and trans-cinnamate. Left: Growth response of E. coli
NST74 to tCA at concentrations of 0 mg/L (solid square), 200 mg/L (solid circle), 400
mg/L (solid triangle), 600 mg/L (open square), 800 mg/L (open circle), and 1000 mg/L
(open triangle). Right: Growth response of E. coli NST74 to styrene at concentrations of
0 mg/L (solid square), 100 mg/L (solid circle), 200 mg/L (solid triangle), 300 mg/L (open
square), 400 mg/L (open circle), and 500 mg/L (open triangle). Error bars reported at one
standard deviation from triplicate experiments.
For reference, p-hydroxystyrene (logKow = 2.28) has previously been shown to
inhibit P. putida at concentrations of ~540 mg/L(Verhoef et al. 2009). Solvent
hydrophobicity is typically a good indicator of its toxicity towards microorganisms and
may be quantified by logKow, the log of the octanol-water partition coefficient(Ramos,
Duque, Gallegos, Godoy, Ramos-González, et al. 2002). Even at low concentrations,
compounds with a logKow between 1.0 to 5.0 tend to be very toxic(Heipieper, Weber,
Sikkema, Keweloh, and de Bont 1994). Accumulation of hydrophobic aromatics within

27
the cytoplasmic membrane has been shown to disrupt its integrity, resulting in the
leakage of ions, metabolites, lipids, and proteins, as well as affecting the cells ability to
maintain its internal pH and an appropriate transmembrane proton gradient(Ramos,
Duque, Gallegos, Godoy, Ramos-González, et al. 2002; Weber et al. 1993). The
measured toxicity thresholds were found to be consistent at both low (OD600 0.6) and
high (OD600 2, data not shown) initial cell densities (reflecting early and late exponential
growth, respectively), indicating growth inhibition was independent of cell density and
growth stage. As a result of the inherent toxicity of styrene we anticipated that its
biosynthesis using an NST74 host platform would be limited to below its maximum
potential under our culture conditions. While no prior studies have specifically explored
either the effects of styrene toxicity on E. coli or strategies to improve its tolerance,
several Pseudomonas sp. have been shown to display enhanced styrene tolerance
characteristics(Weber et al. 1993). Whereas the present study is solely focused on the
prototyping of a novel pathway for styrene biosynthesis, it is clear from our preliminary
assays that styrene toxicity must eventually be overcome or effectively circumvented if
renewable styrene production is ever to become viable or sustainable.
2.3.2 Screening candidate PAL isoenzymes for activity in recombinant E. coli.
The activities of recombinant PALs from various genetic sources were analyzed
according to both in vitro (crude lysate) and in vivo (whole, resting cell) assays. All
recombinant PALs, expressed from plasmids pSencPSm, pSpalAv, pSpalNp, pSpal1At,
and pSpal2At (Table 2.1) in an E. coli BL21(DE3) background were recovered as crude
lysates. According to our in vitro assay results, all of the tested PALs showed comparable
levels of activity on L-phenylalanine as substrate (Table 2.2), with the exception of EncP

28
from S. maritimus whose activity was non-measureable. It has previously been shown
through kinetic studies on prokaryotic PALs that those derived from N. puntiformes and
A. variabilis possessed 500-1000 times greater activity than EncP from S. maritimus;
furthermore, the kcat/Km value of PAL from A. variabilis was found to be greater than that
of N. puntiformes (72.2 and 43.8 mM-1
s-1
, respectively)(Moffitt et al. 2007). Thus, it is
possible that EncP was in fact functionally expressed in our study; however, its activity
was too low to measure according to the protocols employed. The relative activities of
candidate PALs were then further explored through the use of whole-cell assays in
recombinant E. coli. Resting cells suspended in PBS buffer (pH 7) were supplemented
with L-phenylalanine or L-tyrosine and product (tCA or p-coumaric acid, respectively)
formation was monitored over the course of 3 h. Although the PALs from A. thaliana
were found to possess the greatest specific activity in crude lysates (Table 2.2), assays of
in vivo function provided a more stark contrast into the relative activities among all
candidate PALs, as shown in Fig. 2.3.
Table 2.2 PAL Specific Activity. Specific activity of PAL isoenzymes from A.
variabilis, N. punctiforme, and A. thaliana on L-phenylalanine and L-tyrosine when
expressed in recombinant E. coli BL21(DE3).
Strain Substrate Activity (U mg-1
protein)
BL21(DE3) L-phenylalanine n.d.
L-tyrosine n.d.
pSencPSm L-phenylalanine n.d.
L-tyrosine n.d.
pSpalAv L-phenylalanine 2.38 ± 0.64
L-tyrosine n.d.
pSpalNp L-phenylalanine 0.91 ± 0.32
L-tyrosine n.d.
pSpal1At L-phenylalanine 2.42 ± 1.07
L-tyrosine n.d.
pSpal2At L-phenylalanine 4.08 ± 0.11
L-tyrosine n.d.
n.d. – not detected
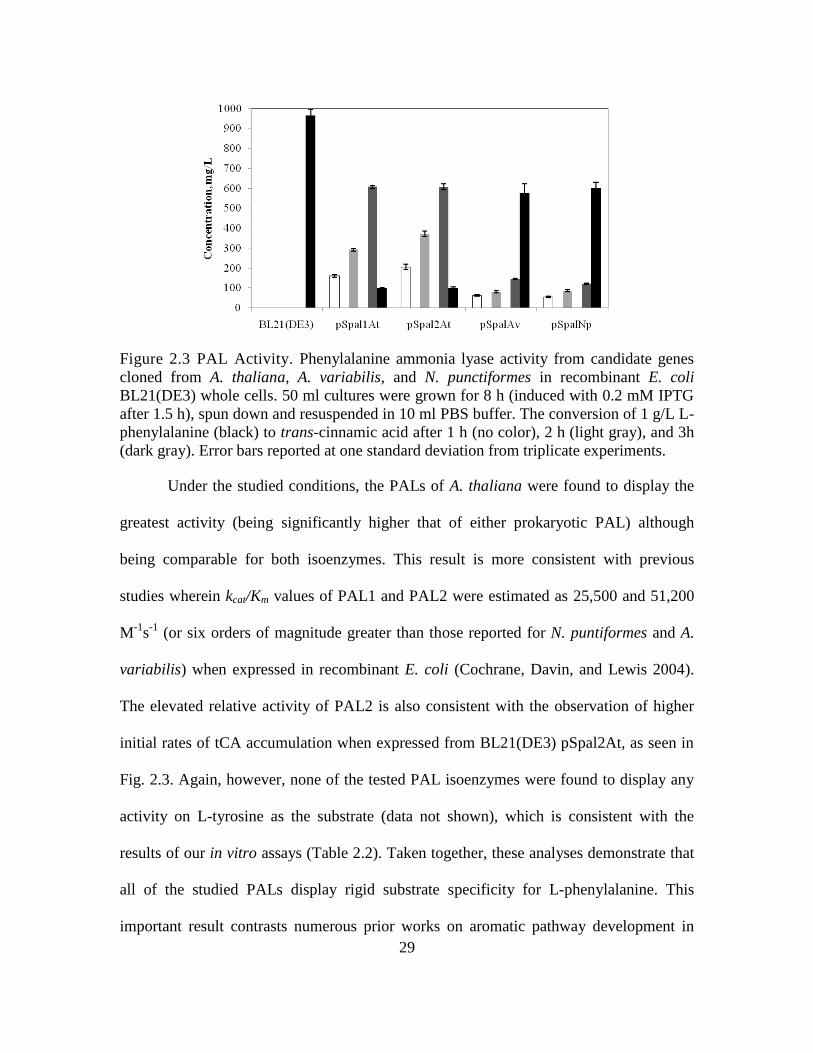
29
Figure 2.3 PAL Activity. Phenylalanine ammonia lyase activity from candidate genes
cloned from A. thaliana, A. variabilis, and N. punctiformes in recombinant E. coli
BL21(DE3) whole cells. 50 ml cultures were grown for 8 h (induced with 0.2 mM IPTG
after 1.5 h), spun down and resuspended in 10 ml PBS buffer. The conversion of 1 g/L L-
phenylalanine (black) to trans-cinnamic acid after 1 h (no color), 2 h (light gray), and 3h
(dark gray). Error bars reported at one standard deviation from triplicate experiments.
Under the studied conditions, the PALs of A. thaliana were found to display the
greatest activity (being significantly higher that of either prokaryotic PAL) although
being comparable for both isoenzymes. This result is more consistent with previous
studies wherein kcat/Km values of PAL1 and PAL2 were estimated as 25,500 and 51,200
M-1
s-1
(or six orders of magnitude greater than those reported for N. puntiformes and A.
variabilis) when expressed in recombinant E. coli (Cochrane, Davin, and Lewis 2004).
The elevated relative activity of PAL2 is also consistent with the observation of higher
initial rates of tCA accumulation when expressed from BL21(DE3) pSpal2At, as seen in
Fig. 2.3. Again, however, none of the tested PAL isoenzymes were found to display any
activity on L-tyrosine as the substrate (data not shown), which is consistent with the
results of our in vitro assays (Table 2.2). Taken together, these analyses demonstrate that
all of the studied PALs display rigid substrate specificity for L-phenylalanine. This
important result contrasts numerous prior works on aromatic pathway development in

30
recombinant E. coli which have solely relied upon the use of bifunctional yeast
PAL/TALs(Gilbert et al. 1985; Gilbert and Tully 1982; Gilbert, Stephenson, and Tully
1983; Cui, Jia, and Sun 2008) and, which was specifically used to synthesize p-
hydroxystyrene(Qi et al. 2007). As these results illustrate that the first committed step in
our pathway is highly specific for the intended substrate (L-phenylalanine) alone, we
anticipate that this advantageous outcome will ultimately help to control product purity
(specifically styrene over p-hydroxystyrene or a mixture of products) while also
improving the activity and flux of our desired pathway. Meanwhile, the same cannot be
assured had the first committed step of our pathway been catalyzed by PAL/TAL, as was
the case for all engineered p-hydroxystyrene pathways reported to date(Qi et al. 2007;
Verhoef et al. 2009).
2.3.3 Screening candidate PADC isoenzymes for tCA decarboxylase activity in
recombinant E. coli.
Candidate PADC isoenzymes from L. plantarum, B. subtilis, and S. cerevisiae
were screened for their ability to decarboxylate tCA to produce styrene when expressed
in E. coli. Plasmids harboring the candidate PADC-encoding genes (Table 2.1) were
individually transformed into E. coli BL21(DE3), as described above. Whole, resting
cells were prepared in PBS buffer supplemented with 1g/L of tCA acid or p-coumaric
acid. The production of styrene or p-hydroxystyrene, respectively, was then followed
periodically, and the results after 12 h of culture are compared in Fig. 2.4. With the
exception of the strain expressing PAD1 from S. cerevisiae alone, all other strains
displayed decarboxylase activity on p-coumaric acid, leading to p-hydroxystyrene
biosynthesis. These results are consistent with previous reports on the functional

31
expression of pdc and padC in recombinant E. coli(Qi et al. 2007) to support p-
hydroxystyrene biosynthesis from glucose. However, our findings further and
importantly show that the enzymes from L. plantarum and B. subtilis cannot catalyze the
conversion of tCA to styrene (thus making them more specific for p-coumaric acid). In
the present study it was instead found that the sole expression of FDC1 from S. cerevisiae
(which was previously characterized to encode a ferulic acid decarboxylase) was
sufficient for achieving decarboxylase activity on either tCA or p-coumaric acid (with
perhaps a slight preference towards tCA, based on overall conversion). Though it was
previously reported that the co-expression of both FDC1 and PAD1 is necessary to
achieve tCA decarboxylase activity in the native S. cerevisiae (Mukai et al. 2010), we
now report that functional tCA decarboxylase activity in E. coli depends solely upon
FDC1 over-expression and is not dependent upon the co-expression of PAD1.
Furthermore, as can be seen in Fig. 2.4, comparable styrene titers were achieved when
either FDC1 was expressed alone or together with PAD1, indicating that the co-
expression of PAD1 does not increase (or otherwise alter) tCA decarboxylase activity in
E. coli, as has previously been suggested in the native S. cerevisiae (Larsson et al., 2001).
It is, however, plausible that the expression of ubix (which has been shown to be 50%
similar to PAD1(Mukai et al. 2010)) in our E. coli background could have served to
compensate for the absence of PAD1 expression, enabling tCA decarboxylase activity to
be achieved in E. coli expressing FDC1 alone. Future studies will explore the deletion of
ubix from the background of E. coli to further investigate the recombinant function of
FDC1. Most importantly, these results have demonstrated that FDC1 over-expression in
E. coli uniquely enables the decarboxylation of tCA to styrene.
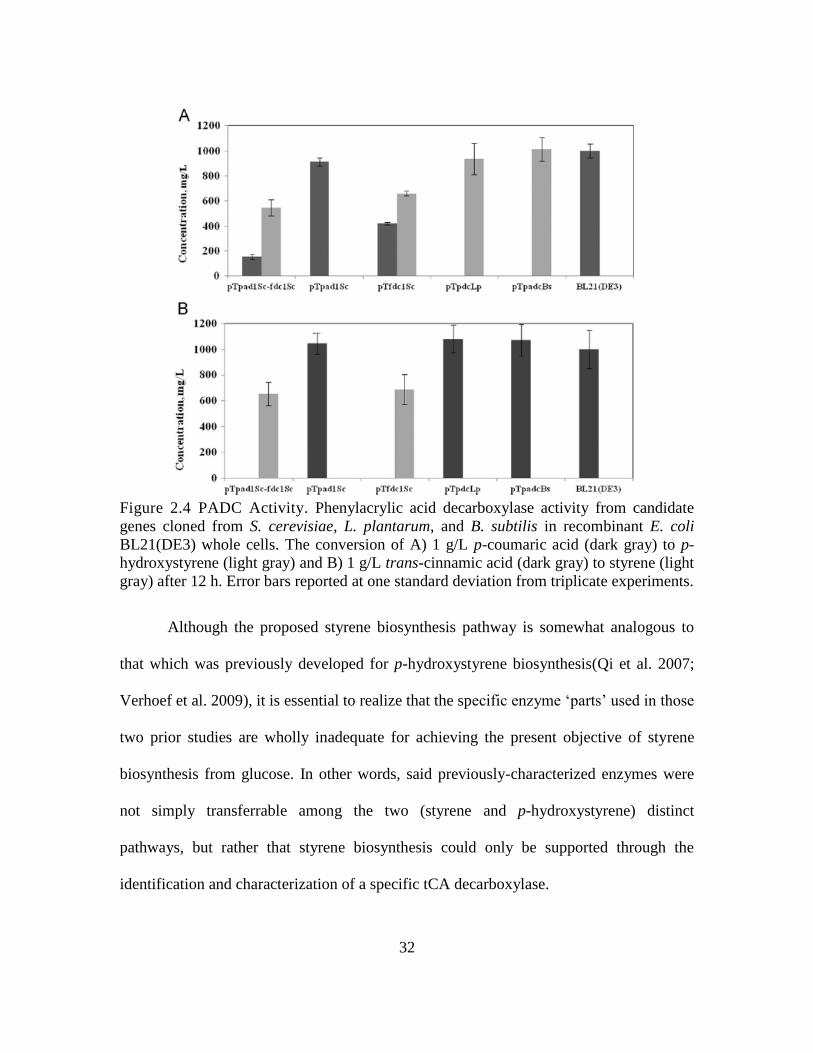
32
Figure 2.4 PADC Activity. Phenylacrylic acid decarboxylase activity from candidate
genes cloned from S. cerevisiae, L. plantarum, and B. subtilis in recombinant E. coli
BL21(DE3) whole cells. The conversion of A) 1 g/L p-coumaric acid (dark gray) to p-
hydroxystyrene (light gray) and B) 1 g/L trans-cinnamic acid (dark gray) to styrene (light
gray) after 12 h. Error bars reported at one standard deviation from triplicate experiments.
Although the proposed styrene biosynthesis pathway is somewhat analogous to
that which was previously developed for p-hydroxystyrene biosynthesis(Qi et al. 2007;
Verhoef et al. 2009), it is essential to realize that the specific enzyme ‘parts’ used in those
two prior studies are wholly inadequate for achieving the present objective of styrene
biosynthesis from glucose. In other words, said previously-characterized enzymes were
not simply transferrable among the two (styrene and p-hydroxystyrene) distinct
pathways, but rather that styrene biosynthesis could only be supported through the
identification and characterization of a specific tCA decarboxylase.

33
2.3.4 Biosynthesis of tCA and styrene from glucose in recombinant E. coli.
The biosynthesis of tCA from glucose was first investigated using strains of E.
coli NST74 that individually carried the plasmids pSpalAv, pSpalNp, pSpal1At, and
pSpal2At. Cultures were grown in MM1 media with 1.5% glucose. 48 h after induction
with 0.2 mM IPTG, the production of tCA was detected in each culture at final titers of
600 mg/L, 473 mg/L, 648 mg/L, and 918 mg/L, respectively. Neither styrene nor p-
coumaric acid were detected in any samples.
To test for styrene biosynthesis from glucose, FDC1 from S. cerevisiae was then
co-expressed with each of the PAL-encoding genes according to the following
constructed strains: NST74 pSpalAv pTfdc1Sc, NST74 pSpalNp pTfdc1Sc, NST74
pSpal1At pTfdc1Sc, and NST74 pSpal2At pTfdc1Sc. Conversion of glucose to styrene
was followed after IPTG induction and results for the strain NST74 pSpal2At pTfdc1Sc
are shown in Table 2.3 (other results not shown as this dataset was representative of the
results and trends observed with all other strains). Substantial L-phenylalanine
accumulation was not observed until 17 h post induction, after which time styrene titers
then also rose considerably, reaching a final titer of 260 mg/L in the culture medium 29 h
post-induction. The final styrene titers for the strains NST74 pSpalAv pTfdc1Sc, NST74
pSpalNp pTfdc1Sc, and NST74 pSpal1At pTfdc1Sc were found to be 210, 183, and 188
mg/L, respectively.

34
Table 2.3 Styrene Fermentation Titers. Biosynthesis of L-
phenylalanine, trans-cinnamic acid, and styrene by E. coli
NST74 pSpal2At pTfdc1Sc from glucose in MM1 media.
Errors reported at one standard deviation from triplicate
experiments.
time (h) L-phenylalanine
(mg/L)
trans-cinnamic
acid (mg/L)
styrene
(mg/L)
0 13.9 ± 0.03 0 0
13 56.4 ± 0.13 3.7 ± 0.01 7.3 ± 0.12
17 236.5 ± 0.57 6.3 ± 0.01 23 ± 0.38
21 167.4 ± 0.40 9.1 ± 0.02 205 ± 3.40
25 152.2 ± 0.36 13.5 ± 0.02 243 ± 4.03
29 179.5 ± 0.43 14.0 ± 0.03 260 ± 4.31
As styrene-producing strains were cultured, a strong ‘hydrocarbon’ aroma was
readily detected upon opening the sealed culture bottles. The high volatility of styrene
necessitated the use of sealed jars for culturing. To ensure that sufficient oxygen
remained available to the culture, a large headspace volume (200 mL headspace vs. 50
mL media) was used. Thus, cultures were maintained under aerobic conditions
throughout the culture (as confirmed by the inclusion of the indicator dye resazurin).
However, the use of such a large headspace volume also allows for the significant
accumulation of styrene vapor, which should also be accounted for when assessing
productivity. Under dilute and near-ambient conditions, the equilibrated headspace vapor
composition can be estimated by application of Henry’s Law. Using a dimensionless
Henry’s Law constant of 0.113(Yang 1992), it can be predicted that a headspace
equilibrated with an aqueous phase containing 260 mg/L styrene will contain an
additional 29 mg/L styrene, for a total of 18.9 mg in the 250 mL flask (note this would be
equivalent to an aqueous titer of 376 mg/L if no volatilization had occurred). In an
analogous manner, the final styrene production by strains NST74 pSpalAv pTfdc1Sc,

35
NST74 pSpalNp pTfdc1Sc, and NST74 pSpal1At pTfdc1Sc would be estimated as 15.2,
13.4, and 13.6 mg, respectively.
Following these fermentation studies, culture supernatants were extracted using
hexane, and the extracts were analyzed by GC-MS to confirm that it was in fact styrene
that was being synthesized by our cultures. Comparing the spectra of the dominant
metabolite peak which was recovered from the extract to the NIST08 spectral
database(Babushok et al. 2007), styrene was found to be the most probable compound.
As can be seen from Fig. 2.5, the mass spectra of the extracted sample and the library
reference provide an excellent match, providing confirmation that styrene was in fact
synthesized by our engineered strains of E. coli.
Figure 2.5 Mass spectra of styrene. Head to tail comparison of the standard mass
spectra showing the relative abundance of the mass-to-charge ratio of styrene from the
NIST08 library (lower) with that of the dominant metabolite peak obtained in hexane
extractions of the culture broth (upper).
Throughout the duration of culture, never was all of the L-phenylalanine observed
to be fully assimilated into the styrene pathway (as seen in Table 2.3). Furthermore, tCA
titers were observed to remain low throughout, indicating that almost all of the

36
synthesized tCA could be quickly converted to styrene. Taken together, these
observations suggest that low PAL activity presently remains as the flux limiting
condition in the engineered styrene biosynthesis pathway. As the first committed step in
the styrene pathway, high PAL activity is essential and must be improved in subsequent
generations of our strains. To start, the expression of PAL can likely be improved through
the use of codon optimized variants. This approach has shown to be useful, particularly in
cases for which pathways involve the expression of plant-derived genes in E. coli. For
example, codon optimization was applied to the amorphadiene oxidase from Artemisia
annua (a plant species) as part as a synthetic pathway to produce artemisinic
acid(Keasling 2008). Whereas the native protein originally showed neither in vivo nor in
vitro activity when expressed in E. coli, a codon optimized variant resulted in its
functional expression in E. coli, contributing to successful pathway development.
Whereas improvements in PAL expression can lead to enhancements in the
specific metabolite flux, the net flux can also be improved by increasing the pathway
‘driving force’. That is, by promoting the increased availability of the pathway’s
immediate precursor, L-phenylalanine. This notion was tested in resting cells assays
wherein E. coli NST74 pSpal2At pTfdc1Sc cell suspensions were supplemented with
exogenous L-phenylalanine at initial concentrations of either 400 or 950 mg/L. As shown
in Fig. 2.6, the conversion of 400 mg/L L-phenylalanine to styrene occurred rapidly and
completely (i.e., no residual L-phenylalanine or tCA was detectable), yielding a final
styrene titer of 250 mg/L after 30 h. Meanwhile, when 950 mg/L L-phenylalanine was
added to the resting cell cultures, nearly 500 mg/L styrene could be produced as all of the
L-phenylalanine was consumed. However, in this case, nearly an additional 250 mg/L
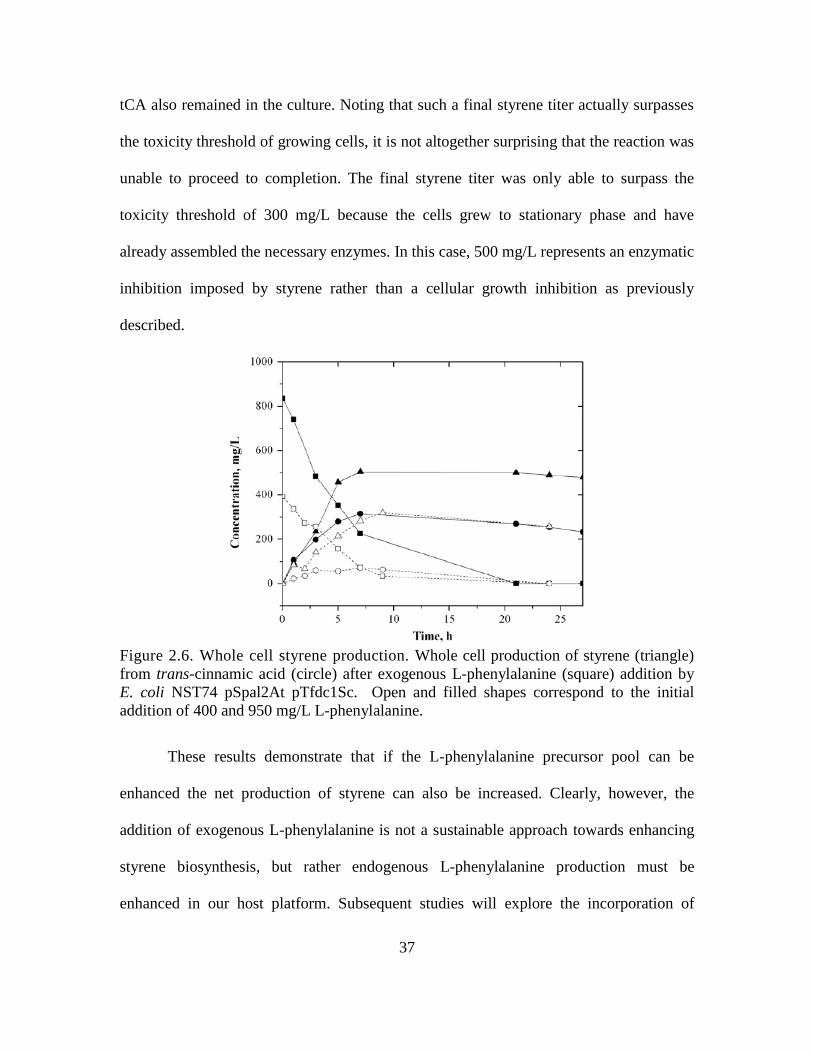
37
tCA also remained in the culture. Noting that such a final styrene titer actually surpasses
the toxicity threshold of growing cells, it is not altogether surprising that the reaction was
unable to proceed to completion. The final styrene titer was only able to surpass the
toxicity threshold of 300 mg/L because the cells grew to stationary phase and have
already assembled the necessary enzymes. In this case, 500 mg/L represents an enzymatic
inhibition imposed by styrene rather than a cellular growth inhibition as previously
described.
Figure 2.6. Whole cell styrene production. Whole cell production of styrene (triangle)
from trans-cinnamic acid (circle) after exogenous L-phenylalanine (square) addition by
E. coli NST74 pSpal2At pTfdc1Sc. Open and filled shapes correspond to the initial
addition of 400 and 950 mg/L L-phenylalanine.
These results demonstrate that if the L-phenylalanine precursor pool can be
enhanced the net production of styrene can also be increased. Clearly, however, the
addition of exogenous L-phenylalanine is not a sustainable approach towards enhancing
styrene biosynthesis, but rather endogenous L-phenylalanine production must be
enhanced in our host platform. Subsequent studies will explore the incorporation of

38
additional modifications into the genome of the E. coli NST74 host that have previously
been shown by other groups to result in L-phenylalanine yield enhancements. For
instance, it has been reported that the over-expression of endogenous transketolase I
(encoded by tktA) supports elevated L-phenylalanine biosynthesis(Gosset, Yong-Xiao,
and Berry 1996). TktA over-expression enables the enhanced biosynthesis of erythrose 4-
phosphate (E4P) which, when condensed with phosphoenolpyruvate (PEP), yields 3-
deoxy-D-arabino-heptulosonate 7-phosphate (DAHP), the first committed intermediate in
the aromatic amino acid biosynthesis pathway(Flores et al. 1996; Lütke-Eversloh and
Stephanopoulos 2008). Meanwhile, disruption of the global carbon storage regulatory
system of E. coli through deletion of csrA, increases PEP biosynthesis, leading to up to a
2-fold enhancement in L-phenylalanine yield(Tatarko and Romeo 2001).
For microbially-derived, renewable styrene to become an economically-viable
and sustainable alternative to petroleum-derived predecessor, titers and productivity must
ultimately be improved. Although the styrene titers achieved by our 1st generation strains
were modest, in comparison to the toxicity assays performed above it can be seen that
they are already approaching the inhibitory threshold. Thus, whereas increasing L-
phenylalanine yields could eventually translate into elevated styrene production, product
toxicity would soon become the subsequent limiting factor and must be addressed. As
complex phenotypes such as solvent tolerance are simply not monogenic in nature(Alper
et al. 2006), numerous specific mutations must applied in concert to achieve the
specifically desired result. Since little is presently known of styrene toxicity or tolerance,
rational approaches to engineering more tolerant strains may be less suitable than higher
throughput, combinatorial strategies. Although commonly employed, combinatorial

39
approaches towards enhancing desired phenotypes often rely upon the aggressive use of
chemomutagenesis, such techniques are less desirable as they can also result an
unforeseen and difficult to understand negative impacts on host fitness and/or
productivity(Bonomo et al. 2006), in addition to requiring laborious screening and
selection procedures. A more effective approach for the present application might involve
the use of an alternative host platform which inherently boasts greater tolerance(Fischer,
Klein-Marcuschamer, and Stephanopoulos 2008), a strategy which has been applied for
the production of biofuels, such as n-butanol(Nielsen et al. 2009). The bacterium P.
putida S12 has been engineered, for example, as a solvent tolerant platform for the
biosynthesis of both p-hydroxybenzoate and p-hydroxystyrene(Verhoef et al. 2007;
Verhoef et al. 2009), and might also make excellent starting point for styrene production.
Since our pathway is derived from the ubiquitous, proteinogenic amino acid L-
phenylalanine, its transference to an alternative host platform remains wholly compatible.
Alternatively, improved product tolerance can also be achieved through genome
evolution, as accomplished through serial adaptations to increasing styrene
concentrations. Such an approach has worked well for achieving tolerance to biofuels like
n-butanol and could also be coupled with genomic library screens to provide the first
comprehensive view of styrene inhibition and tolerance in E. coli(Reyes, Almario, and
Kao 2011). In an interesting example, prior works have shown that Pseudomonas sp. may
be adapted to styrene when also grown in the presence of acetate or similar carboxylic
acids as the sole carbon source(Weber et al. 1993). It has been suggested that this evolved
phenotype may be associated with genetic changes leading to reductions in membrane
fluidity, as well as through the activation of genes believed to be specifically associated

40
with enhanced tolerance to aromatic compounds. As an alternative strategy,
combinatorial procedures such as the global Transcription Machinery Engineering
(gTME)(Alper et al. 2006; Nicolaou, Gaida, and Papoutsakis 2010) may be implemented
to improve styrene tolerance in E. coli or alternative host platforms selected due to their
elevated tolerance baseline. The principals of gTME have been successfully employed to
enhance ethanol tolerance in both yeast(Alper et al. 2006) and E. coli(Alper and
Stephanopoulos 2007).
In addition to the development of styrene tolerant phenotypes, product toxicity
can also be enhanced through the use of in situ product recovery (ISPR). Common ISPR
approaches involve solvent extraction(Gyamerah and Glover 1996; Malinowski 2001;
Weilnhammer and Blass 1994), adsorption(Nielsen, Amarasiriwardena, and Prather
2010; Nielsen and Prather 2009), gas and vacuum stripping(Loser et al. 2005), and
membrane pervaporation(Vane 2005). Each of these approaches has been successfully
applied for the continuous recovery of biofuel compounds such as ethanol, for example,
and even aromatics like L-phenylacetylcarbinol(Khan and Daugulis 2010) and
benzaldehyde(Jain, Khan, and Daugulis 2010). The latter three approaches may be
particularly well-suited for styrene recovery given its volatile nature, as previously
discussed.
2.4 Conclusion
For the first time, the present study has demonstrated the biosynthesis of styrene
from renewable resources using an engineered microbial platform. Whereas low activity
of pathway enzymes and product toxicity remain as challenges which limit the

41
productivity of our styrene-producing strains, continued improvements will lead to the
development of robust biocatalysts for the sustainable production of this important, large
volume, commodity chemical.
Acknowledgements
We thank Prof. Bradley Moore (UCSD) and Prof. Kristala Prather (MIT) for their
kind gifts. We also thank Tom Colella (ASU) for his skillful technical assistance in GC-
MS analysis.

42
CHAPTER 3
BIOSYNTHESIS OF (S)-STYRENE OXIDE AND (R)-1,2-PHENYLETHANEDIOL
FROM GLUCOSE
Abstract
(S)-Styrene oxide and (R)-1,2-phenylethanediol are chiral aromatic molecular
building blocks used commonly as precursors to pharmaceuticals and other specialty
chemicals. Two pathways have been engineered in E. coli for their individual
biosynthesis directly from glucose. The novel pathways each constitute extensions of the
previously engineered styrene pathway, developed by co-expressing either styrene
monooxygenase (SMO) or styrene dioxygenase (SDO) to convert styrene to (S)-styrene
oxide and (R)-1,2-phenylethanediol, respectively. StyAB from Pseudomonas putida S12
was determined to be the most effective SMO. SDO activity was achieved using
NahAaAbAcAd of Pseudomonas sp. NCIB 9816-4, a naphthalene dioxygenase with
known broad substrate specificity. Production of phenylalanine was enhanced through a
number of mutations, most notably via deletion of tyrA and over-expression of tktA. As a
result, (R)-1,2-phenylethanediol reached titers as high as 1.23 g/L, and at 1.32 g/L (S)-
styrene oxide titers already approach their toxicity limit. This study additionally
demonstrates that greater flux through the styrene pathway can be achieved if its toxicity
is addressed, as achieved in this case by reacting styrene to less toxic products.
This work was published as: McKenna, R., Pugh, S., Thompson, B. & Nielsen, D.R.
Microbial production of the aromatic building-blocks (S)-styrene oxide and (R)-1,2-
phenylethanediol from renewable resources. Biotechnology Journal 8, 1465-1475 (2013).

43
3.1 Introduction
(S)-Styrene oxide and (R)-1,2-phenylethanediol (also referred to as (R)-styrene
glycol) are chiral aromatic molecules used in the synthesis of numerous high value
pharmaceuticals and specialty chemicals. For example, (S)-styrene oxide is used as a
precursor to the biocides levamisole and nematocide (Park, So, et al. 2006), as well as in
the synthesis of cosmetics (Loprieno et al. 1976), surface coatings, and numerous
agricultural and biological chemicals (Panke et al. 2000; Han et al. 2006). Meanwhile,
(R)-1,2-phenylethanediol is an optically active diol and precursor to the pharmaceuticals
(R)-norfluoxetine and (R)-fluoxetine (used to treat psychiatric and metabolic disorders),
as well as β-lactam antibiotics (Cao et al. 2006; Kumar, Upadhyay, and Pandey 2004).
Additionally, (R)-1,2-phenylethanediol is also used in the production of various
agrochemicals and pheromones (Gamenara and Dominguez de Maria 2009), as well as
chiral catalysts (King et al. 1979). Conventional production of (S)-styrene oxide occurs
by the partial oxidation of styrene, most often over a heavy metal catalyst. This
chemocatalytic process offers little control over product stereochemistry, and yields (S)-
styrene oxide at only 48 to 57% enantiomeric excess (Groves and Myers 1983; Zhang et
al. 1990) resulting in a mixture of isomers that must then be further processed to resolve
and isolate the desired (S)-enantiomer. As (R)-1,2-phenylethanediol is conventionally
produced from (S)-styrene oxide via a hydrolytic ring opening reaction, its enantiopurity
ultimately depends upon that of the substrate.
In contrast to heavy metal and other chemocatalysts, more stereoselective enzyme
biocatalysts can often provide greater control over product enantiopurity, while offering
additional benefits such as a reduced environmental footprint. For example, the

44
enzymatic conversion of styrene to (S)-styrene oxide via styrene monooxygenase (SMO)
has been extensively studied, with high conversion and enantiopurity (>99% e.e.)
routinely reported (Panke et al. 1998; Archelas and Furstoss 1997; Panke et al. 2000).
Two distinct biosynthetic routes to (R)-1,2-phenylethanediol, meanwhile, have been
previously described in the literature, each involving a single-step enzyme
biotransformation. For example, it has been shown that (S)-styrene oxide can be
transformed to (R)-1,2-phenylethanediol via the expression of an epoxide hydroxylase
(EH). Well-characterized EH homologs include those from bacteria such as Caulobacter
crescentus (Min and Lee 2012) and Agrobacterium radiobacter AD1 (Rui et al. 2005),
as well as potato (Solanum tuberosum) (Monterde et al. 2004). Alternatively, it has also
been shown that styrene itself can instead be directly dihydroxylated to (R)-1,2-
phenylethanediol at high conversion/yields and up to 74% e.e. via the expression of a
Pseudomonas sp. naphthalene dioxygenase that also displays broad substrate specificity
(Lee and Gibson 1996).
However, just as with conventional industrial production of (S)-styrene oxide and
(R)-1,2-phenylethanediol, such single-step enzymatic biotransformations are similarly
limited by the need to supply substrates (i.e., styrene or (S)-styrene oxide) derived from
non-renewable, petroleum feedstocks. Accordingly, previous enzymatic
biotransformation studies have yet to address concerns over renewability and
sustainability. In view of declining petroleum reserves and their ever-increasing costs, the
ability to produce chiral aromatic building blocks such as (S)-styrene oxide and (R)-1,2-
phenylethanediol directly from renewable resources using microbial biocatalysts would
represent a more sustainable approach. The engineering of a styrene biosynthesis

45
pathway in Escherichia coli (McKenna and Nielsen 2011) has recently, and for the first
time, enabled such a prospect. Building upon the styrene bioproduction platform, the
focus of this study was to systematically engineer novel enzyme pathways and microbial
biocatalysts to enable both (S)-styrene oxide and (R)-1,2-phenylethanediol to be
synthesized as individual products from renewable sugars alone.
As shown in Fig. 3.1, the proposed (S)-styrene oxide and (R)-1,2-
phenylethanediol pathways essentially represent extensions of the previously-engineered
styrene pathway, and thus similarly utilize L-phenylalanine as their immediate
endogenous precursor (McKenna and Nielsen 2011). As has been shown, phenylalanine
can be deaminated to trans-cinnamate by a phenylalanine ammonia lyase (PAL); for
example, PAL2 from Arabidopsis thaliana was previously found to display both high
recombinant activity in E. coli and strict substrate specificity for phenylalanine
(McKenna and Nielsen 2011). trans-Cinnamate can then be decarboxylated to styrene by
a phenylacrylate decarboxylase (PADC), a reaction that to date has only been proven to
be catalyzed by FDC1 (originally characterized as a ferulate decarboxylase) from
Saccharomyces cerevisiae (McKenna and Nielsen 2011). Once synthesized
endogenously, styrene could potentially then be oxidized to either (S)-styrene oxide or
(R)-1,2-phenylethanediol via co-expression of a third, appropriate oxidase. (S)-Styrene
oxide production, for instance, could be achieved by co-expressing a suitable styrene
monooxygenase (SMO), whereas (R)-1,2-phenylethanediol could be achieved by co-
expressing a suitable styrene dioxygenase (SDO).
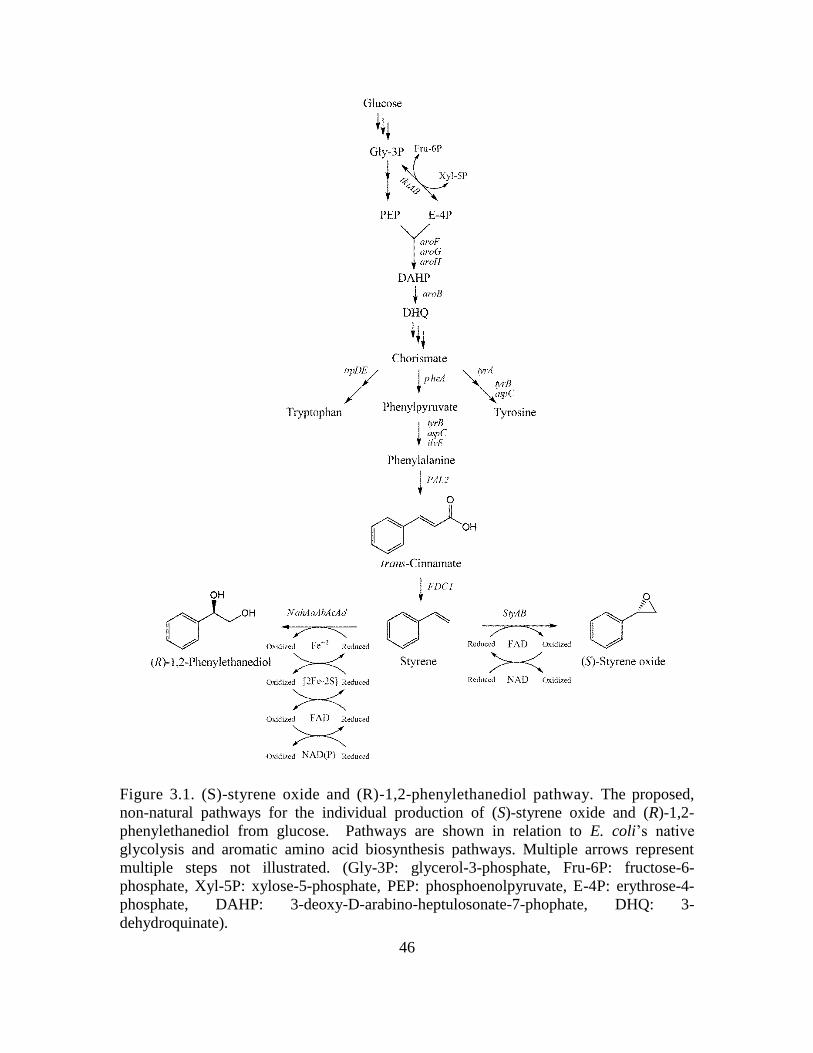
46
Figure 3.1. (S)-styrene oxide and (R)-1,2-phenylethanediol pathway. The proposed,
non-natural pathways for the individual production of (S)-styrene oxide and (R)-1,2-
phenylethanediol from glucose. Pathways are shown in relation to E. coli’s native
glycolysis and aromatic amino acid biosynthesis pathways. Multiple arrows represent
multiple steps not illustrated. (Gly-3P: glycerol-3-phosphate, Fru-6P: fructose-6-
phosphate, Xyl-5P: xylose-5-phosphate, PEP: phosphoenolpyruvate, E-4P: erythrose-4-
phosphate, DAHP: 3-deoxy-D-arabino-heptulosonate-7-phophate, DHQ: 3-
dehydroquinate).

47
Enzymatic oxidation is typically the first step associated with the aerobic
biodegradation of styrene (as well as other aromatics), a process that numerous
Pseudomonas sp. have been naturally evolved to perform and for which they particularly
are well renowned (Smith 1990; Stanier, Palleroni, and Doudoroff 1966). Accordingly,
pseudomonads represent a useful genetic repository for SMO and SDO encoding genes.
To date, several isoenzymes displaying SMO activity have been identified and
characterized in Pseudomonas sp., including, for example, StyAB from P. putida S12,
XylMA (originally characterized as xylene monooxygenase) from P. putida mt-2, and
CymA1A2 (originally characterized as cymene monooxygenase) from P. putida F1
(Nishio et al. 2001; Wubbolts, Reuvekamp, and Witholt 1994; Panke et al. 1998). All
three are two-component monooxygenases consisting of an FAD-dependent hydroxylase
(StyA, XylM, CymA1, respectively) and an NADH-FAD oxidoreductase (StyB, XylA,
CymA2, respectively) (Nishio et al. 2001; Wubbolts et al. 1994; Wubbolts, Reuvekamp,
and Witholt 1994; Otto et al. 2004). The epoxidation of styrene to (S)-styrene oxide
accordingly requires FADH2, which is in turn regenerated through NADH oxidation (Otto
et al. 2004). On the other hand, whereas no specific SDOs have been reported in the
literature to date, as a result of its very relaxed substrate specificity, naphthalene
dioxygenase of P. putida NCIB 9816-4 (encoded by the nahAaAbAcAd operon) has been
shown capable of utilizing styrene as an alternative substrate, oxidizing it directly to (R)-
1,2-phenylethanediol (Lee and Gibson 1996). NahAaAbAcAd is also a multi-component
enzyme, containing an iron-sulfur flavoprotein reductase, an iron-sulfur ferredoxin, and a
two-subunit oxygenase. Similar to SMO, oxidized cofactors are ultimately regenerated by
the consumption of NAD(P)H.

48
3.2 Materials and Methods
3.2.1 Strains and media
All strains and plasmids used in this study are listed in Table 1. Custom
oligonucleotides were synthesized by Integrated DNA Technologies (IDT, Coralville,
IA). Genomic DNA (gDNA) templates were prepared from whole cells using the Zyppy
Fungal/Bacterial DNA MiniPrep kit (Zymo Research, Irvine, CA) according to vendor
protocols. Strains were routinely cultured in Luria-Bertani (LB) broth supplemented with
appropriate antibiotics, as necessary, at the following final concentrations: 100 mg/L
ampicillin, 34 mg/L chloramphenicol, and 50 mg/L kanamycin. As required, auxotrophic
strains were additionally supplemented with tyrosine and tryptophan, each initially at 50
mg/L. All cultures were grown aerobically at 32oC with shaking at 250 rpm, unless
otherwise noted. Seed cultures were prepared in 5 mL of LB broth with appropriate
antibiotics and grown overnight. A 1%v./v. seed was then used to inoculate all flask
cultures. To test for production of aromatic species, strains were cultivated in shake
flasks (250 mL) containing 50 mL phosphate-limited minimal media (MM1) with
1.5%w./v. glucose, as previously described (McKenna and Nielsen 2011). Cultures were
grown until an OD600 of ~0.6, at which point they were induced by 0.2 mM IPTG
addition and incubated an additional 72 h. Samples were taken at 24 h intervals and
analyzed by HPLC to determine concentrations of phenylalanine, trans-cinnamate,
styrene, (S)-styrene oxide, and (R)-1,2-phenylethanediol.
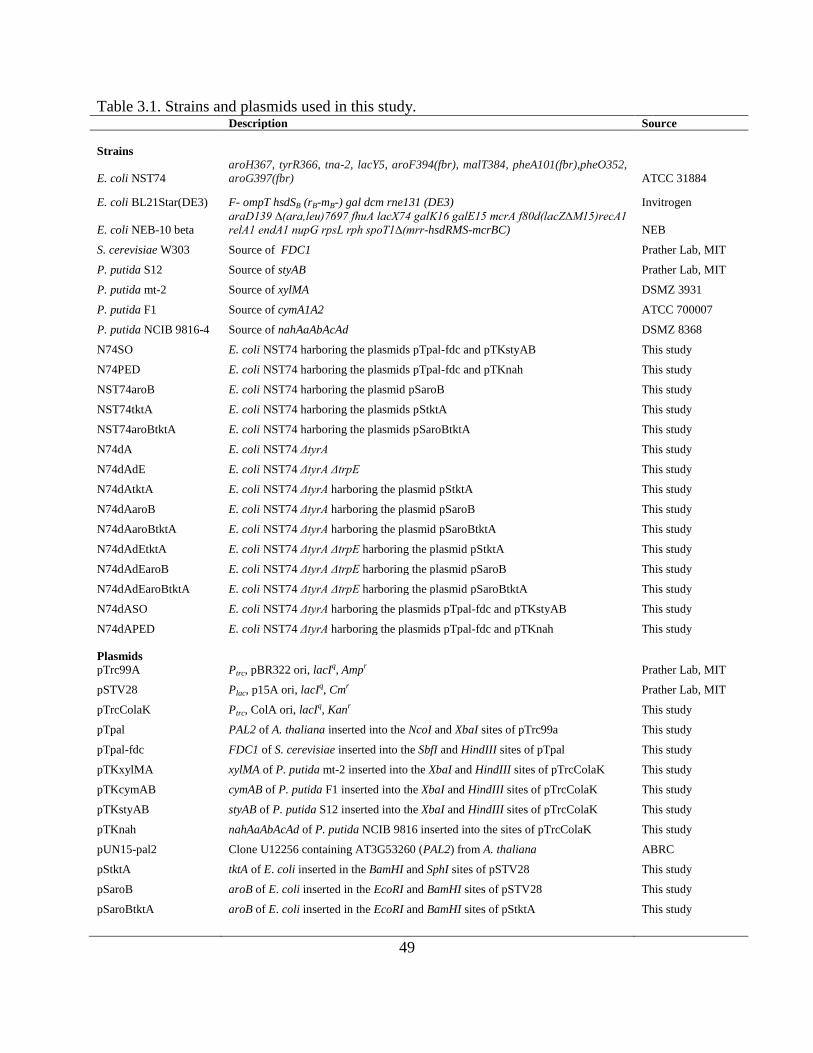
49
Table 3.1. Strains and plasmids used in this study. Description Source
Strains
E. coli NST74
aroH367, tyrR366, tna-2, lacY5, aroF394(fbr), malT384, pheA101(fbr),pheO352,
aroG397(fbr) ATCC 31884
E. coli BL21Star(DE3) F- ompT hsdSB (rB-mB-) gal dcm rne131 (DE3) Invitrogen
E. coli NEB-10 beta
araD139 ∆(ara,leu)7697 fhuA lacX74 galK16 galE15 mcrA f80d(lacZ∆M15)recA1
relA1 endA1 nupG rpsL rph spoT1∆(mrr-hsdRMS-mcrBC) NEB
S. cerevisiae W303 Source of FDC1 Prather Lab, MIT
P. putida S12 Source of styAB Prather Lab, MIT
P. putida mt-2 Source of xylMA DSMZ 3931
P. putida F1 Source of cymA1A2 ATCC 700007
P. putida NCIB 9816-4 Source of nahAaAbAcAd DSMZ 8368
N74SO E. coli NST74 harboring the plasmids pTpal-fdc and pTKstyAB This study
N74PED E. coli NST74 harboring the plasmids pTpal-fdc and pTKnah This study
NST74aroB E. coli NST74 harboring the plasmid pSaroB This study
NST74tktA E. coli NST74 harboring the plasmids pStktA This study
NST74aroBtktA E. coli NST74 harboring the plasmids pSaroBtktA This study
N74dA E. coli NST74 ΔtyrA This study
N74dAdE E. coli NST74 ΔtyrA ΔtrpE This study
N74dAtktA E. coli NST74 ΔtyrA harboring the plasmid pStktA This study
N74dAaroB E. coli NST74 ΔtyrA harboring the plasmid pSaroB This study
N74dAaroBtktA E. coli NST74 ΔtyrA harboring the plasmid pSaroBtktA This study
N74dAdEtktA E. coli NST74 ΔtyrA ΔtrpE harboring the plasmid pStktA This study
N74dAdEaroB E. coli NST74 ΔtyrA ΔtrpE harboring the plasmid pSaroB This study
N74dAdEaroBtktA E. coli NST74 ΔtyrA ΔtrpE harboring the plasmid pSaroBtktA This study
N74dASO E. coli NST74 ΔtyrA harboring the plasmids pTpal-fdc and pTKstyAB This study
N74dAPED E. coli NST74 ΔtyrA harboring the plasmids pTpal-fdc and pTKnah This study
Plasmids
pTrc99A Ptrc, pBR322 ori, lacIq, Ampr Prather Lab, MIT
pSTV28 Plac, p15A ori, lacIq, Cmr Prather Lab, MIT
pTrcColaK Ptrc, ColA ori, lacIq, Kanr This study
pTpal PAL2 of A. thaliana inserted into the NcoI and XbaI sites of pTrc99a This study
pTpal-fdc FDC1 of S. cerevisiae inserted into the SbfI and HindIII sites of pTpal This study
pTKxylMA xylMA of P. putida mt-2 inserted into the XbaI and HindIII sites of pTrcColaK This study
pTKcymAB cymAB of P. putida F1 inserted into the XbaI and HindIII sites of pTrcColaK This study
pTKstyAB styAB of P. putida S12 inserted into the XbaI and HindIII sites of pTrcColaK This study
pTKnah nahAaAbAcAd of P. putida NCIB 9816 inserted into the sites of pTrcColaK This study
pUN15-pal2 Clone U12256 containing AT3G53260 (PAL2) from A. thaliana ABRC
pStktA tktA of E. coli inserted in the BamHI and SphI sites of pSTV28 This study
pSaroB aroB of E. coli inserted in the EcoRI and BamHI sites of pSTV28 This study
pSaroBtktA aroB of E. coli inserted in the EcoRI and BamHI sites of pStktA This study

50
3.2.2 Toxicity assays
The toxicity effects of (S)-styrene oxide and (R)-1,2-phenylethanediol on E. coli
were determined by monitoring the impacts of the exogenous addition of increasing final
concentrations of (S)-styrene oxide or (R)-1,2-phenylethanediol to growing cultures.
Approximately 1 ml of an E. coli NST74 seed was used to inoculate 50 ml of LB broth in
a 250 mL shake flask. At the time when the cultures reached an optical density (OD600) of
~0.6, (S)-styrene oxide and (R)-1,2-phenylethanediol were added to the flasks at an array
of final concentrations between 0 to 1.8 g/L and 0 to 9 g/L, respectively. Culturing then
resumed for another 6 h while cell growth, as determined by OD600 measurements, was
periodically monitored using a UV/Vis spectrophotometer (Beckman Coulter DU800,
Brea, CA).
3.2.3 Cloning of PAL2 from A. thaliana, FDC1 from S. cerevisiae, and tktA and aroB
from E. coli
All genes were PCR amplified using a BioRad iCycler system, Phusion DNA
Polymerase (New England Biolabs, Ipswich, MA), and custom oligonucleotide primers
(see Supplementary Information; note: primer names indicate restriction sites used for
cloning). PCR cycling and reaction conditions were standardized according to
manufacturer instructions. All PCR amplified DNA fragments were purified using the
Zyppy DNA Clean and Concentrator kit (Zymo Research, Irvine, CA). Gene fragments
and plasmids were treated by endonuclease digestion according to manufacturer’s
protocols. All digested fragments were first gel purified using the Zyppy DNA
purification kit (Zymo Research, Irvine, CA) and then ligated with T4 DNA Ligase (New
England Biolabs, Ipswich, MA) at 4oC overnight before the mixture was then

51
transformed into chemically competent E. coli NEB10-Beta. Transformants were selected
on LB solid agar with appropriate antibiotics and cultured at 37oC overnight.
Transformant pools were screened using colony PCR with final confirmation by gene
sequencing. PAL2 was amplified from cDNA of clone U12256 from the Arabidopsis
Biological Resource Center (ABRC, Columbus, OH) and cloned into pTrc99a, resulting
in construction of the plasmid pTpal. FDC1 was amplified from gDNA of S. cerevisiae
and cloned into pTpal, resulting in construction of the plasmid pTpal-fdc (Table 3.1).
Using methods analogous to those described above, tktA and aroB were each PCR
amplified from gDNA template prepared from E. coli NST74 and cloned into pSTV28.
These works resulted in the generation of the plasmids pStktA and pSaroB (Table 3.1).
3.2.4 Construction of the plasmid pTrcColaK and the cloning of SMO- and SDO-
encoding genes
The plasmid pTrcColaK was constructed by ligating a PCR amplified expression
cassette from pTrc99a (containing the trc promoter, laqIq repressor, and multi-cloning
site) together with a PCR amplified fragment from pCOLADuet-1 (EMD Millipore,
Billerica, MA) containing the ColA origin of replication and kanamycin resistance
marker. Using methods analogous to those described above, xylAM, cymA1A2, styAB,
and nahAaAbAcAd were PCR amplified from gDNA templates from P. putida mt-2, F1,
S12, and NCIB 9816-4, respectively, and individually cloned into pTrcColaK. These
works resulted in the generation of the plasmids pTKstyAB, pTKcymAB, pTKxylMA,
and pTKnah (Table 3.1).

52
3.2.5 Construction of tyrA and trpE deletion mutants in E. coli NST74
Chromosomal in-frame gene deletion was accomplished using an approach
adapted from the methods of Datsenko and Wanner (Datsenko and Wanner 2000).
Deletion cassettes harboring the kanamycin resistance gene flanked by the FLP
recognition target sites were PCR amplified from the gDNA of the Keio collection
mutants E. coli JW2581-1 and JW1256-1 (Baba et al. 2006) for the chromosomal
integration of tyrA::FRT-Kan-FRT and trpE::FRT-Kan-FRT, respectively. This resulted
in the construction of the phenylalanine over-producing strains N74dA (NST74 ΔtyrA)
and N74dAdE (NST74 ΔtyrA ΔtrpE).
3.2.6 Construction of phenylalanine, (S)-styrene oxide, and (R)-1,2-phenylethanediol
producing strains
The previously engineered phenylalanine over-producing strain E. coli NST74
was individually transformed with pStktA, pSaroB, and pSaroBtktA to generate strains
NST74tktA, NST74aroB, and NST74aroBtktA, respectively. E. coli NST74 and N74dA
were also co-transformed with the plasmids pTpal-fdc and pTKsty or pTKnah resulting in
strains N74SO, N74PED, N74dASO, and N74dAPED, respectively. Analogous methods
were used to construct the strains N74dAtktA, N74dAaroB, N74dAaroBtktA,
N74dAdEtktA, N74dAdEaroB, and N74dAdEaroBtktA (Table 3.1).
3.2.7 Assaying SMO activity in recombinant E. coli whole resting cells
The three SMO harboring plasmids were individually transformed into E. coli
BL21(DE3) to generate strains E. coli BL21:pTKstyAB, E. coli BL21:pTKcymAB, and
E. coli BL21:pTKxylMA. Shake flasks (250 mL) containing 50 mL of LB with 1%w./v.

53
glucose were inoculated with 1 mL of overnight seed culture. Cultures were grown until
an OD600 of ~0.6, induced by addition of 0.2 mM IPTG, and then incubated for 6 h before
an equal number of cells (~0.5 g/L dry cell weight) were collected from each by
centrifuging at 2000 x g for 5 min. The pellet was washed once with pH 7 PBS
(phosphate buffered saline) before being re-suspended in 25 ml PBS buffer with 1%w./v.
glucose (for cofactor regeneration) in a 250 ml shake flask. Finally, 0.22 g/L styrene was
added to the suspension before sealing the flask. Cultures were shaken at 32°C for 3 h
and sampled every hour to monitor both styrene depletion and (S)-styrene oxide
production via HPLC.
3.2.8 HPLC analysis
Samples (1 mL) were centrifuged at 11,000 x g for 2 min to pellet cells and
supernatants (0.75 mL) were then transferred to glass HPLC vials with Teflon-lined caps.
All HPLC analysis was carried out using a Hewlett Packard 1100 series HPLC system.
For aromatics analysis, separation was performed on a reverse-phase Hypersil Gold
SBC18 column (4.6mm x 150 mm; Thermo Fisher, USA) operated at 45oC. The column
was eluted at a total constant flow rate of 1.0 ml/min using ‘solvent A’ (consisting of
double-distilled water) and ‘solvent B’ (consisting of methanol (99.8% grade) plus
0.1%v./v. trifluoroacetic acid (TFA)). The eluent began as a mixture of 95% solvent A
and 5% solvent B before a linear gradient was applied over 8 min to then reach a mixture
of 20% solvent A and 80% solvent B. This eluent composition was then held constant
for 2 min before a second linear gradient was then applied over the course of 4 min to
achieve a final mixture of 95% solvent A and 5% solvent B. The eluent was monitored
using a diode array detector set at 215 nm for L-phenylalanine, (S)-styrene oxide, and

54
(R)-1,2-phenylethanediol and 258 nm for trans-cinnamate and styrene. Under these
conditions L-phenylalanine, trans-cinnamate, styrene, (S)-styrene oxide, and (R)-1,2-
phenylethanediol were eluted at 4.3, 9.5, 11.1, 8.2 and 6.0 min, respectively. Glucose
analysis was performed on the same HPLC system, however now using an RID detector
and an anion exchange column (Aminex HPX-87H; BioRAD, Hercules, CA) operated at
35oC. The column was eluted with 0.005 M H2SO4 at a constant flow rate of 0.8 ml/min.
External calibrations were used to determine concentrations for each analyte.
3.3 Results and Discussion
3.3.1 Assessing and investigating the mechanisms of product toxicity
Prior to investigating their production from glucose, preliminary toxicity screens
were first performed for both (S)-styrene oxide and (R)-1,2-phenylethanediol to better
anticipate the effects of their accumulation on E. coli growth. By screening for changes in
E. coli growth rate and yield under the stress of the added chemicals, toxicity thresholds
of ~1.6 and ~8 g/L were approximated for (S)-styrene oxide and (R)-1,2-
phenylethanediol, respectively, as shown in Fig. 2A and 2B. While it is certainly
plausible that exogenous chemical addition does not offer a complete view of cell
response to their presence relative to when the same compounds are instead synthesized
endogenously within the cell, experience indicates that, at least in the case of aromatic
solvents, said approach can provide a reasonable first approximation (McKenna and
Nielsen 2011). By this same approach, toxicity limits for the pathway intermediates
styrene and trans-cinnamate against E. coli were previously approximated as 300 and 800
mg/L, respectively (McKenna and Nielsen 2011). For many organic solvents, the octanol-
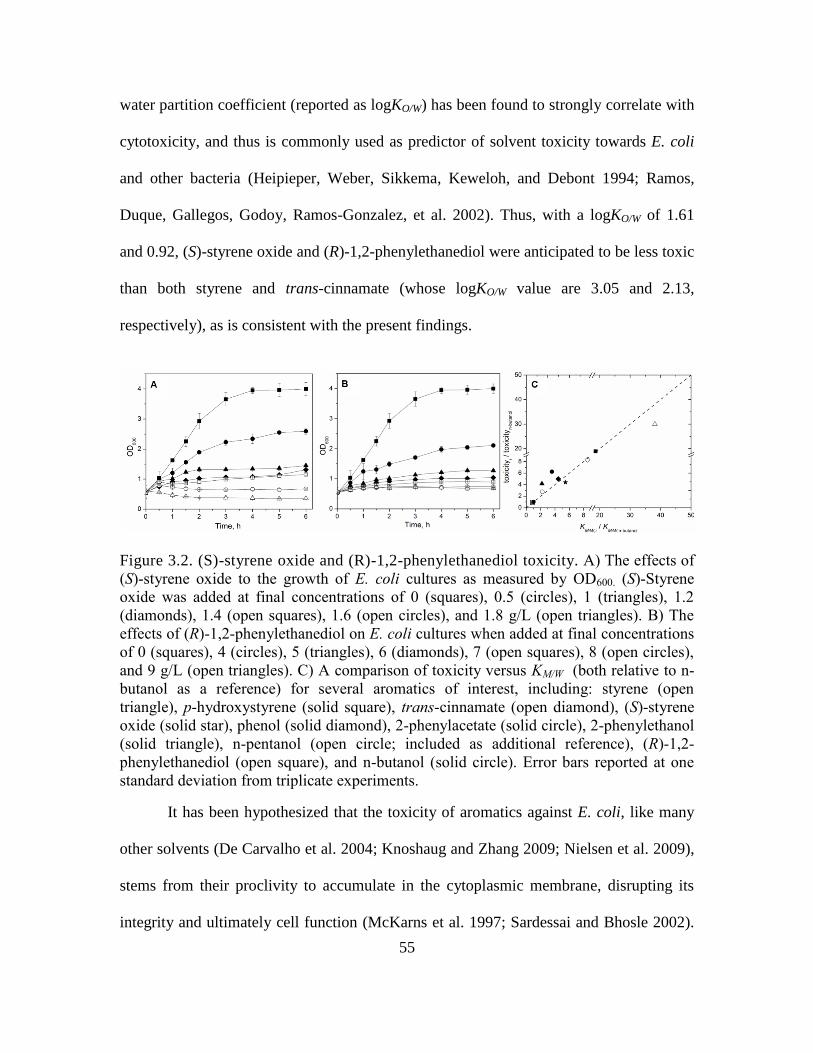
55
water partition coefficient (reported as logKO/W) has been found to strongly correlate with
cytotoxicity, and thus is commonly used as predictor of solvent toxicity towards E. coli
and other bacteria (Heipieper, Weber, Sikkema, Keweloh, and Debont 1994; Ramos,
Duque, Gallegos, Godoy, Ramos-Gonzalez, et al. 2002). Thus, with a logKO/W of 1.61
and 0.92, (S)-styrene oxide and (R)-1,2-phenylethanediol were anticipated to be less toxic
than both styrene and trans-cinnamate (whose logKO/W value are 3.05 and 2.13,
respectively), as is consistent with the present findings.
Figure 3.2. (S)-styrene oxide and (R)-1,2-phenylethanediol toxicity. A) The effects of
(S)-styrene oxide to the growth of E. coli cultures as measured by OD600. (S)-Styrene
oxide was added at final concentrations of 0 (squares), 0.5 (circles), 1 (triangles), 1.2
(diamonds), 1.4 (open squares), 1.6 (open circles), and 1.8 g/L (open triangles). B) The
effects of (R)-1,2-phenylethanediol on E. coli cultures when added at final concentrations
of 0 (squares), 4 (circles), 5 (triangles), 6 (diamonds), 7 (open squares), 8 (open circles),
and 9 g/L (open triangles). C) A comparison of toxicity versus KM/W (both relative to n-
butanol as a reference) for several aromatics of interest, including: styrene (open
triangle), p-hydroxystyrene (solid square), trans-cinnamate (open diamond), (S)-styrene
oxide (solid star), phenol (solid diamond), 2-phenylacetate (solid circle), 2-phenylethanol
(solid triangle), n-pentanol (open circle; included as additional reference), (R)-1,2-
phenylethanediol (open square), and n-butanol (solid circle). Error bars reported at one
standard deviation from triplicate experiments.
It has been hypothesized that the toxicity of aromatics against E. coli, like many
other solvents (De Carvalho et al. 2004; Knoshaug and Zhang 2009; Nielsen et al. 2009),
stems from their proclivity to accumulate in the cytoplasmic membrane, disrupting its
integrity and ultimately cell function (McKarns et al. 1997; Sardessai and Bhosle 2002).

56
Testing this hypothesis, membrane-water partitioning coefficients (KM/W) were predicted
for (S)-styrene oxide and (R)-1,2-phenylethanediol, as well as other aromatics that have
been examined by our group (unpublished data) or for which prior literature reports were
available (Keweloh, Heipieper, and Rehm 1989; Park, Buehler, et al. 2006), according to
the model of Sikkema et al. (Sikkema, de Bont, and Poolman 1994):
Predictions of KM/W for each species, as well as their toxicity thresholds for E. coli, are
also compared in Fig. 2C. To provide a frame of reference, however, the results are
compared relative to the predicted KM/W and previously report toxicity of n-butanol (note:
E. coli’s n-butanol toxicity limit is ~1%w./v.) (Nielsen et al. 2009; Winkler, Rehmann,
and Kao 2010), as it is generally accepted that n-butanol’s toxicity arises due to its
membrane accumulation (Bowles and Ellefson 1985; Osborne et al. 1990). The observed
strong linear correlation existing between the relative toxicity of aromatics versus their
predicted relative tendency to accumulate in the membrane provides direct support for the
hypothesis that membrane accumulation/disruption is the predominant toxicity
mechanism of aromatics against E. coli.
3.3.2 Screening and selecting pathway enzymes
In addition to providing preliminary targets for maximal end-product
accumulation, these findings further suggest that a high turnover of trans-cinnamate, and
especially styrene, must be achieved to avoid the inhibitory effects of their intermediate
accumulation on (S)-styrene oxide and (R)-1,2-phenylethanediol producing strains.
Furthermore, if this condition can be achieved, it may then also be possible to achieve

57
higher final titers of (S)-styrene oxide and (R)-1,2-phenylethanediol than have been
previously reported for either styrene or trans-cinnamate under otherwise equivalent
conditions (McKenna and Nielsen 2011). To achieve this, however, high metabolite flux
through the entire pathway must be realized, including at the final pathway steps (i.e.,
SMO and SDO). Thus, identifying a SMO and SDO with the greatest inherent activity in
E. coli was an important initial aim. Based on previous reports, SMO candidates encoded
by styAB from P. putida S12, xylMA from P. putida mt-2, and cymA1A2 from P. putida
F1 were selected for screening and analysis in E. coli. Recombinant SMO activity was
screened via whole resting cell assays for each of E. coli BL21(DE3) pTKstyAB, E. coli
BL21(DE3) pTKxylMA, and E. coli BL21(DE3) pTKcymAB. The results are compared
in Fig. 3.3, where (S)-styrene oxide production via the biotransformation of exogenously
supplied styrene was monitored for a period of 3 h. As can be seen, E. coli BL21(DE3)
pTKstyAB displayed the highest rates of styrene degradation and (S)-styrene oxide
accumulation, suggesting that StyAB possessed the greatest recombinant activity;
followed by XylMA from mt-2 then CymA1A2 from F1. These findings are consistent
with previous reports which found the specific activity of StyAB for styrene to be 180
U/g (Panke et al. 1998), whereas XylMA (22 U/g (Wubbolts et al. 1994)) and CymA1A2
(2.30 U/g (Nishio et al. 2001)) were much less active. Furthermore, this result is not
altogether surprising as styrene is the natural substrate of StyAB, whereas XylMA and
CymA1A2 only incidentally display SMO activity as a result of their relaxed substrate
specificities. StyAB was accordingly selected for use in the proposed pathway and in all
subsequent studies. Meanwhile, as only a single SDO candidate was identified, the
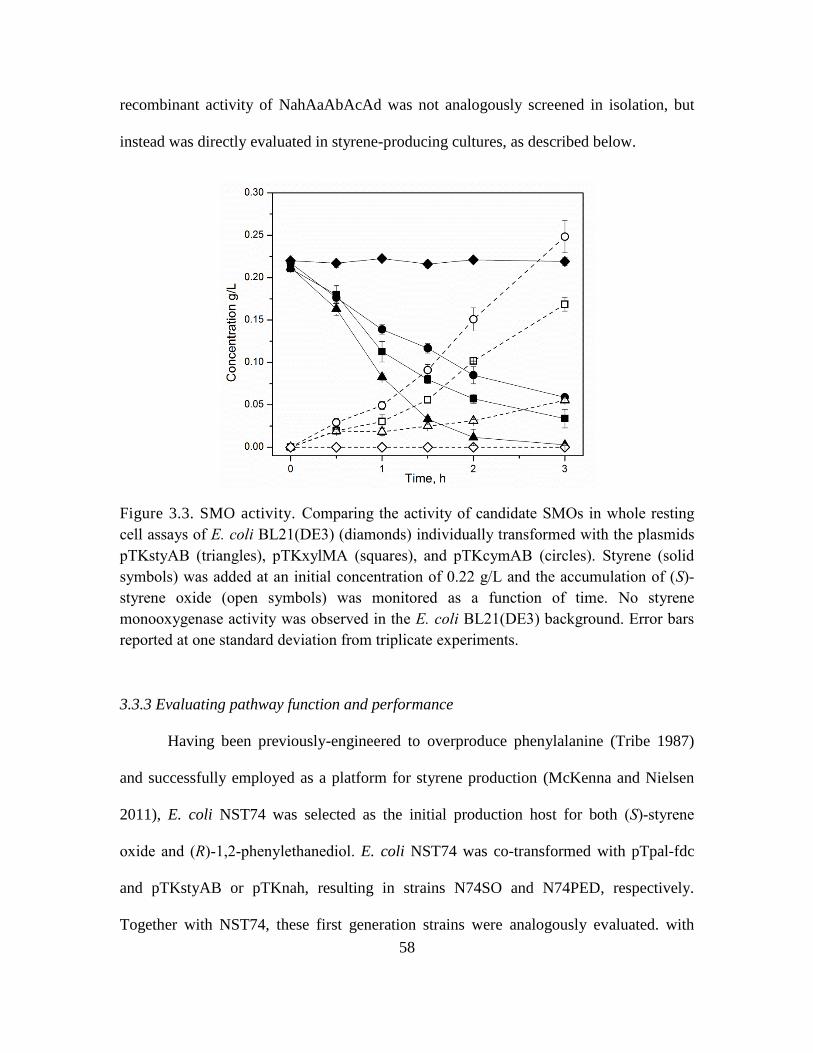
58
recombinant activity of NahAaAbAcAd was not analogously screened in isolation, but
instead was directly evaluated in styrene-producing cultures, as described below.
Figure 3.3. SMO activity. Comparing the activity of candidate SMOs in whole resting
cell assays of E. coli BL21(DE3) (diamonds) individually transformed with the plasmids
pTKstyAB (triangles), pTKxylMA (squares), and pTKcymAB (circles). Styrene (solid
symbols) was added at an initial concentration of 0.22 g/L and the accumulation of (S)-
styrene oxide (open symbols) was monitored as a function of time. No styrene
monooxygenase activity was observed in the E. coli BL21(DE3) background. Error bars
reported at one standard deviation from triplicate experiments.
3.3.3 Evaluating pathway function and performance
Having been previously-engineered to overproduce phenylalanine (Tribe 1987)
and successfully employed as a platform for styrene production (McKenna and Nielsen
2011), E. coli NST74 was selected as the initial production host for both (S)-styrene
oxide and (R)-1,2-phenylethanediol. E. coli NST74 was co-transformed with pTpal-fdc
and pTKstyAB or pTKnah, resulting in strains N74SO and N74PED, respectively.
Together with NST74, these first generation strains were analogously evaluated. with

59
accumulation of the respective end-products being monitored in each case. As seen in
Fig. 3.4, after 72 h, up to 0.97±0.03 g/L (8.08±0.25 mM) of (S)-styrene oxide was
produced by N74SO at a yield of 0.083±0.005 g/g (0.125±0.008 mol/mol). N74PED,
meanwhile, produced 0.41±0.02 g/L (2.95±0.14 mM) (R)-1,2-phenylethanediol at
0.035±0.002 g/g yield (0.046±0.003 mol/mol). It should be noted that phenylalanine,
trans-cinnamate, and styrene were undetectable in these strains at all times, indicating
rapid turnover of intermediates and efficient flux through the pathway. In comparison,
phenylalanine production by NST74 reached up to 1.1±0.03 g/L (6.66±0.18 mM) at a
yield of 0.095±0.005 g/g (0.104±0.008 mol/mol), or 25% of theoretical. Relative to
phenylalanine, it is hypothesized that higher molar titers and yields of (S)-styrene oxide
resulted due to the rapid turnover of phenylalanine by StyAB, which in turn increased
flux through the amino acid biosynthesis pathway. By this logic, the relatively lower
titers and yields of (R)-1,2-phenylethanediol by N74PED could perhaps have stemmed
from low SDO activity displayed by NahAaAbAcAd. If this is the case, identifying or
engineering for more active SDO isoenzymes could result in future improvements. A
BlastP search based on NahAaAbAcAd reveals several additional, four-subunit candidate
dioxygenases from Pseudomonas sp. (including P. fluorescens, P. stutzeri, P.
chlororaphis, P. xanthomarina, P. monteilii, P. balearica), as well as one from Rhanella
sp. LCY15. Whereas all are similarly classified as naphthalene dioxygenases, their future
screening could lead to the identification of more robust enzyme ‘parts’ for the (R)-1,2-
phenylethanediol pathway.
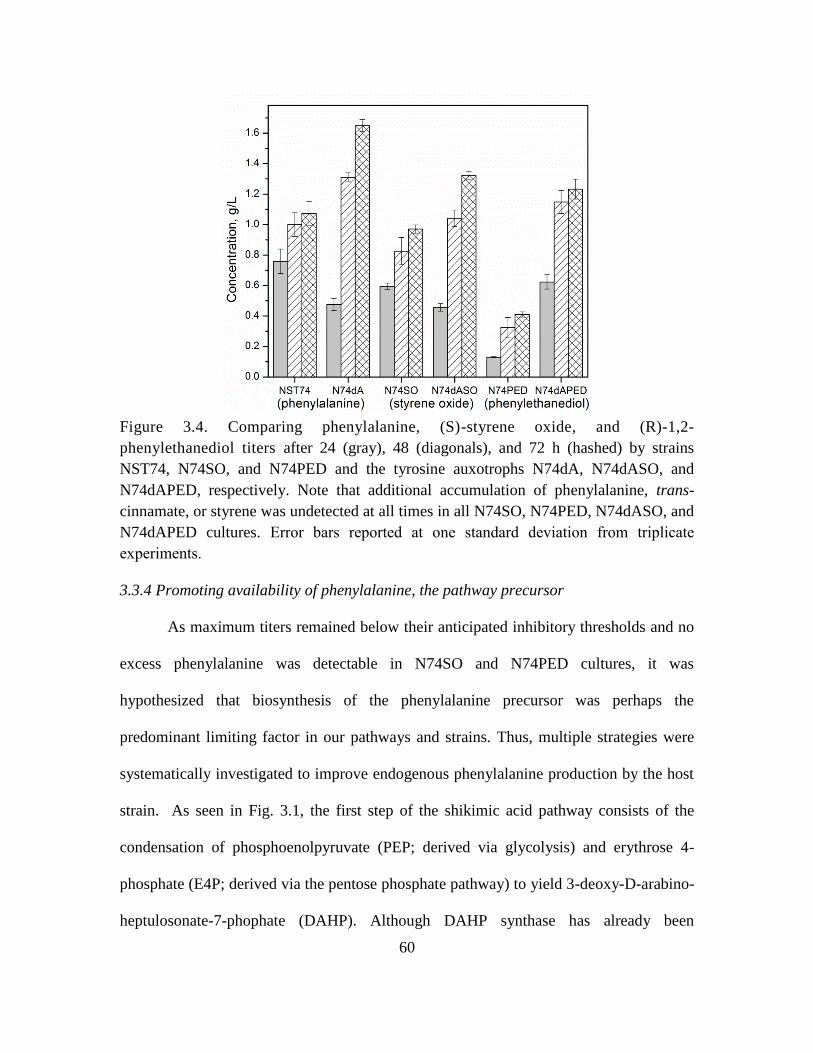
60
Figure 3.4. Comparing phenylalanine, (S)-styrene oxide, and (R)-1,2-
phenylethanediol titers after 24 (gray), 48 (diagonals), and 72 h (hashed) by strains
NST74, N74SO, and N74PED and the tyrosine auxotrophs N74dA, N74dASO, and
N74dAPED, respectively. Note that additional accumulation of phenylalanine, trans-
cinnamate, or styrene was undetected at all times in all N74SO, N74PED, N74dASO, and
N74dAPED cultures. Error bars reported at one standard deviation from triplicate
experiments.
3.3.4 Promoting availability of phenylalanine, the pathway precursor
As maximum titers remained below their anticipated inhibitory thresholds and no
excess phenylalanine was detectable in N74SO and N74PED cultures, it was
hypothesized that biosynthesis of the phenylalanine precursor was perhaps the
predominant limiting factor in our pathways and strains. Thus, multiple strategies were
systematically investigated to improve endogenous phenylalanine production by the host
strain. As seen in Fig. 3.1, the first step of the shikimic acid pathway consists of the
condensation of phosphoenolpyruvate (PEP; derived via glycolysis) and erythrose 4-
phosphate (E4P; derived via the pentose phosphate pathway) to yield 3-deoxy-D-arabino-
heptulosonate-7-phophate (DAHP). Although DAHP synthase has already been

61
effectively feedback-deregulated in E. coli NST74, it has been shown that this initial
condensation step is typically limited by low E4P availability. As such, overexpression of
transketolase I (TktA) promotes increased E4P production (Gosset, Yong-Xiao, and Berry
1996; Kambourakis, Draths, and Frost 2000; Draths et al. 1992). Also reported, flux through
the shikimic acid pathway is improved by preventing DAHP accumulation, as achieved
by overexpressing 3-dehydroquinate synthase (AroB) (Kambourakis, Draths, and Frost 2000;
Keseler et al. 2005). Meanwhile, in addition to serving as a precursor to phenylalanine,
chorismate is also the key branch point metabolite from which all other aromatic amino
acids, namely tryptophan (via anthranilate synthase; TrpED) and tyrosine (following its
conversion to prephenate via chorismate dehydratase; TyrA). While trpE and tyrA
deletion will lead to tryptophan and tyrosine auxotrophies (necessitating their
supplementation to cultures), said mutations will enable an assessment of maximum
achievable production levels by our strains. The individual and combined effects of these
strategies on phenylalanine production were next investigated with the goal of developing
a more robust host. As seen in Fig. 3.5, relative to NST74, deletion of tyrA (N74dA) led
to a 1.5-fold increase in phenylalanine production after 72 h at a similar yield, whereas
the additional deletion of trpE (N74dAdE) actually negated this improvement. Then, by
combining tyrA deletion with tktA overexpression (N74dAtktA), phenylalanine titer and
yield were each improved by >50%, reaching 37% of the theoretical yield. However, tktA
overexpression also imposed a marked negative effect on cell growth, causing an initial
lag period that lasted nearly 16 h (data not shown). The impact on phenylalanine
production too is seen in Fig. 3.5, where titers at 24 h are lowest for tktA overexpressing
strains. In contrast, tyrA deletion rendered no impact on cell growth (data not shown).
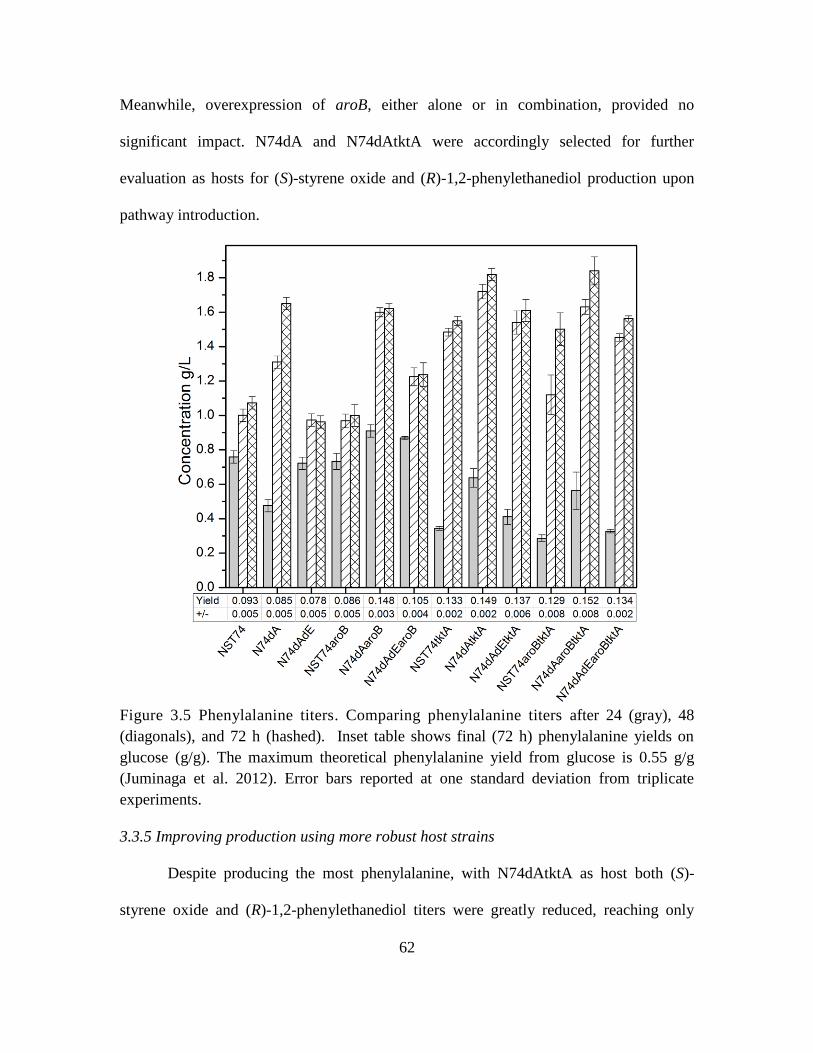
62
Meanwhile, overexpression of aroB, either alone or in combination, provided no
significant impact. N74dA and N74dAtktA were accordingly selected for further
evaluation as hosts for (S)-styrene oxide and (R)-1,2-phenylethanediol production upon
pathway introduction.
Figure 3.5 Phenylalanine titers. Comparing phenylalanine titers after 24 (gray), 48
(diagonals), and 72 h (hashed). Inset table shows final (72 h) phenylalanine yields on
glucose (g/g). The maximum theoretical phenylalanine yield from glucose is 0.55 g/g
(Juminaga et al. 2012). Error bars reported at one standard deviation from triplicate
experiments.
3.3.5 Improving production using more robust host strains
Despite producing the most phenylalanine, with N74dAtktA as host both (S)-
styrene oxide and (R)-1,2-phenylethanediol titers were greatly reduced, reaching only

63
0.62±0.07 g/L and 0.32±0.02 g/L, respectively. This was likely due to the compromised
fitness of this strain, as indicated by its poor growth phenotype (noted above), which may
have been further exacerbated by the increased burden associated with introducing the
pathway plasmids. In contrast, with N74dA as host (S)-styrene oxide and (R)-1,2-
phenylethanediol production was greatly improved (Fig. 3.4). Relative to N74SO, (S)-
styrene oxide production by N74dASO was improved by ~30%, to a final titer of
1.32±0.03 g/L (11.0±0.25 mM) at a yield of 0.115±0.005 g/g (0.172±0.007 mol/mol) or
39% of theoretical, and now approaches its estimated toxicity limit (~1.6 g/L). However,
with a final titer of 1.23±0.07 g/L (8.9±0.51 mM) and a yield of 0.109±0.002 g/g
(0.142±0.003 mol/mol) or 32% of theoretical, a more striking difference, was the ~3-fold
increase in (R)-1,2-phenylethanediol production by N74dAPED (relative to N74PED).
Another important outcome of this study is the demonstration that enhanced flux
through the styrene pathway can be realized if styrene’s toxic effects can be addressed or
overcome, as was achieved in this case by its in vivo transformation to a less toxic
product, namely (S)-styrene oxide or (R)-1,2-phenylethanediol. For example, under
analogous conditions, prior works have shown that E. coli can produce up to 260±4.31
mg/L (2.5±0.04 mM) styrene before reaching its toxicity limit, representing a yield of
0.07 g/g (0.12 mol/mol) or 27% of its theoretical maximum (McKenna and Nielsen
2011). However, by simply extending the styrene pathway by one step to efficiently
convert styrene to (S)-styrene oxide, for example, net flux through the pathway was
increased by 4.4-fold while the yield was improved by over 40%. Though slightly less
dramatic, the same is also true of (R)-1,2-phenylethanediol production. Thus, as styrene is
the precursor to both (S)-styrene oxide and (R)-1,2-phenylethanediol, these findings also

64
imply that it is possible for the molar flux to styrene in E. coli to reach at least these
levels. This realization provides important motivation to the continued development of
the styrene pathway, and the development of styrene tolerant microbes as platforms to
produce this important monomer at high levels from renewable resources.
3.4 Conclusion
A novel method for the direct production of the chiral aromatic building blocks
(S)-styrene oxide and (R)-1,2-phenylethanediol from renewable glucose has been
established. While initial experiments demonstrated that low availability of precursor
phenylalanine limited production of both compounds, a systematic approach towards
enhancing flux through the phenylalanine biosynthesis pathway ultimately resulted in the
improved bioproduction of both (S)-styrene oxide and (R)-1,2-phenylethanediol. Toxicity
of both compounds was strongly correlated with a model of membrane accumulation and
disruption, and was likely a limiting factor in the case of (S)-styrene oxide. These works
demonstrate the versatility of the styrene pathway as a platform for producing other
useful and valuable aromatic fine chemicals, and show that greater flux through the
styrene pathway is possible if styrene’s toxicity can first be effectively addressed.

65
CHAPTER 4
ENGINEERING THE STYRENE PATHWAY IN YEAST
Abstract
Here we report the first heterologous production of styrene in yeast from glucose.
This was achieved from the native aromatic amino acid phenylalanine in a two-step
biosynthetic pathway by expressing the phenylalanine ammonia lyase PAL2 from
Arabidopsis thaliana and the ferulate decarboxylase FDC1, which is native to
Saccharomyces cerevisiae. A phenylalanine overproducing strain of S. cerevisiae was
first engineered via EMS mutagenesis and selection on antimetabolites resulting in a
strain capable of producing 357±32.5 mg/L phenylalanine. This strain, 22A74D, was
further engineered to knockout the Ehrlich pathway (ΔARO10) and to incorporate the
feedback-resistant DAHP synthase ARO4K229L
. After expressing PAL2 and relying on
native expression of FDC1, styrene titers reached 28.8±2.1 mg/L.
4.1 Introduction
Like most monomers used in conventional plastics production, at present, all
commercially-available styrene is solely derived from non-renewable petroleum
feedstocks. More specifically, the predominant means by which styrene is synthesized is
the chemocatalytic dehydrogenation of petroleum-derived ethylbenzene (Wu, Koylinski,
and Bozik 1981; Mimura and Saito 2000). With the global annual demand of styrene to
surpass 41 million tons by 2020 (James and Castor 2011) (a >$28 billion U.S.
market(SRI 2010)), the net energy requirements for just this single transformation step
amount to over 200 trillion BTU of steam each year (DoE 2002). However, driven by
concerns over depleting feedstock availability and deleterious environmental impacts,

66
there is growing interest in the development of ‘green’ processes for the production of
biorenewable replacements to conventional petroleum products, including numerous
monomers and plastics.
Advances in metabolic and pathway engineering have been paramount to the
continuously expanding range of conventional monomer compounds that can now be
synthesized from renewable biomass feedstocks (Adkins et al. 2012; Curran and Alper
2012; Erickson, Nelson, and Winters 2012; Lee et al. 2011). Along these lines, in prior
works a novel and non-natural pathway for styrene biosynthesis from biomass-derived
glucose was recently engineered in the bacterium Escherichia coli (McKenna and Nielsen
2011). The previously-engineered pathway, depicted in Fig. 4.1, utilizes phenylalanine, a
naturally occurring proteinogenic amino acid, as its precursor (McKenna and Nielsen
2011). Phenylalanine is first deaminated to trans-cinnamate by phenylalanine ammonia
lyase (PAL). It was found that PAL2 from Arabidopsis thaliana displays both the greatest
activity and highest substrate specificity (i.e., for phenylalanine) when recombinantly
expressed in E. coli (McKenna and Nielsen 2011). Next, trans-cinnamate is
decarboxylated to styrene via the expression of the phenylacrylate decarboxylase FDC1
(originally characterized as a ferulate decarboxylase) from Saccharomyces cerevisiae
(McKenna and Nielsen 2011; Mukai et al. 2010). When PAL2 and FDC1 were
subsequently co-expressed in a previously-engineered L-phenylalanine over-producing E.
coli background grown in glucose minimal media, the resultant styrene titers reached
~260 mg/L in 29 h at a yield of about 0.030 g/g (McKenna and Nielsen 2011). As E. coli
was found to possess a low toxicity threshold for styrene (between only 200 to 300
mg/L), this suggested that end-product toxicity (likely caused by membrane

67
accumulation/disruption (McKenna et al. 2013)) was the principal limiting factor
(McKenna and Nielsen 2011).
As sensitivity to solvent-like products – including, for example, n-
butanol(Atsumi, Hanai, and Liao 2008) and (S)-styrene oxide(McKenna et al. 2013), and
2-pentanone(Lan et al. 2013),– commonly arises as a productivity-limiting factor in E.
coli, engineering more robust hosts for renewable chemical production is an important
aim in industrial biotechnology. Relative to E. coli, the yeast biosynthetic platforms can
offer several inherent advantages of importance to robust and large–scale renewable
chemicals production. The most significant of these attributes often includes: faster
growth rates, elevated solvent tolerance, and the ability to withstand low temperatures
and pH (Demain and Vaishnav 2009; Sudbery 1996; Curran et al. 2013; Nevoigt 2008;
Ostergaard, Olsson, and Nielsen 2000). Among yeast, S. cerevisiae is particularly
attractive for metabolic engineering studies due to its well characterized genetics,
physiology, and metabolism, as well as the plethora of diverse genetic toolkits for the
stable expression and introduction of heterologous enzymes and pathways (Alberti,
Gitler, and Lindquist 2007).
With an eye towards evaluating and ultimately improving the prospects of
industrial scale renewable styrene production, the objective of the present study was to
engineer the yeast S. cerevisiae to synthesize styrene from glucose. In addition to the
promising phenotypic traits previously mentioned, several additional and complementary
factors were further responsible for motivating this specific aim. First, past studies have
shown that S. cerevisiae is a suitable host for the engineering of other, non-native
aromatic biosynthesis pathways, including for the production of protocatechuate,

68
catechol, vanillin, naringenin, and 2-phenylethanol, among others (Curran et al. 2013;
Hansen et al. 2009; Jiang, Wood, and Morgan 2005; Stark et al. 2002). Second, in
addition to displaying general solvent tolerance (Matsui et al. 2008; Kawamoto, Kanda,
and Tanaka 2001), S. cerevisiae has specifically been shown to display elevated tolerance
to aromatics such as 2-phenylethanol (Etschmann et al. 2002). Third, it was hypothesized
that improved function of styrene pathway enzymes might be achieved in S. cerevisiae
since: i) PAL2 is of eukaryotic origin, and ii) FDC1 is native to S. cerevisiae. Contingent
upon the native regulation of FDC1 (to be addressed later in this study) this suggests that
a functional pathway could be in fact constructed via the expression of a single
heterologous enzyme, thus minimizing the prospects of metabolic burden. Lastly, S.
cerevisiae does not naturally possess the transporter required for phenylalanine efflux.
For example, in previous works it has been shown that although intracellular
concentrations of phenylalanine in S. cerevisiae exceeded 60 mM, its content in the
associated supernatant was undetectable (Luttik et al. 2008). Retaining phenylalanine
intracellularly may enhance its availability to the styrene pathway.
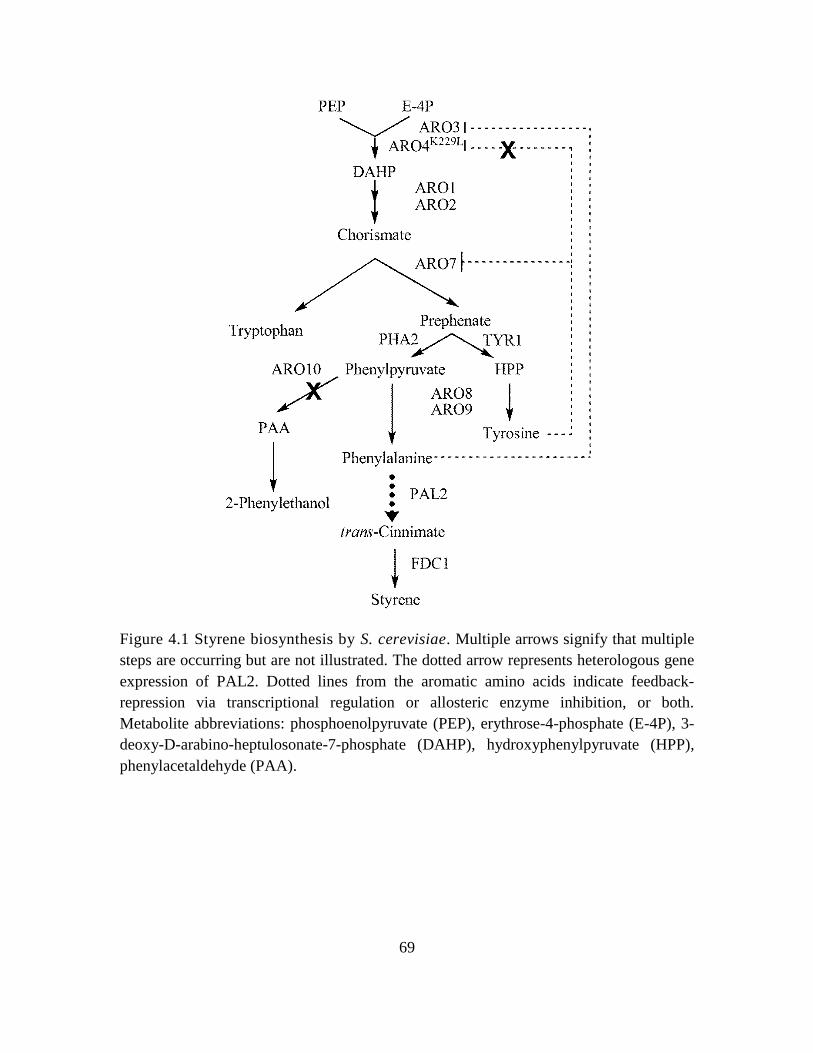
69
Figure 4.1 Styrene biosynthesis by S. cerevisiae. Multiple arrows signify that multiple
steps are occurring but are not illustrated. The dotted arrow represents heterologous gene
expression of PAL2. Dotted lines from the aromatic amino acids indicate feedback-
repression via transcriptional regulation or allosteric enzyme inhibition, or both.
Metabolite abbreviations: phosphoenolpyruvate (PEP), erythrose-4-phosphate (E-4P), 3-
deoxy-D-arabino-heptulosonate-7-phosphate (DAHP), hydroxyphenylpyruvate (HPP),
phenylacetaldehyde (PAA).

70
4.2 Materials and Methods
4.2.1 Strains and media.
All strains and plasmids used in this study are listed in Table 4.1. Custom
oligonucleotide primers were synthesized by Integrated DNA Technologies (Coralville,
IA) and presented in the supplementary material. All S. cerevisiae strains were purchased
from Thermo Scientific (Waltham, MA). Yeast plasmids used were derived from the
Gateway vector collection and purchased from AddGene (Cambridge, MA). Genomic
DNA was prepared from whole cells using the ZR Fungal/Bacterial DNA MiniPrep
(Zymo Research, Irvine, CA) according to vendor protocols. E. coli strain NEB10β
(New England Biolabs, Ipswich, MA) was used for all cloning and plasmid propagation,
except for pDONR221 which was propagated in One Shot ccdB Survival 2 T1 E. coli
(Life Technologies, Grand Island, NY). E. coli strains were routinely cultured at 37ºC in
Luria-Bertani (LB) broth (supplemented with appropriate antibiotics, as necessary). Yeast
strains were routinely cultured at 32ºC in Yeast Extract Peptone Dextrose (YPD)
medium, yeast synthetic dextrose (SD) medium, or yeast synthetic minimal (SD-Leu)
medium. YPD medium was composed of of 10 g/L yeast extract, 20 g/L peptone, and 20
g/L glucose. SD medium was composed of 6.7 g/L yeast nitrogen base, 20 g/L glucose,
and 20 mg/L of each Uracil, Histidine, Leucine, and Methionine. SD-Leu medium was
composed of 6.7 g/L yeast nitrogen base, 20 g/L glucose, 20 mg/L each Uracil, Histidine,
and Methionine.

71
Table 4.1. List of strains and plasmids.
Strain Genotype Source
E. coli NEB-10 beta
araD139 ∆(ara,leu)7697 fhuA lacX74 galK16 galE15 mcrA
f80d(lacZ∆M15)recA1 relA1 endA1 nupG rpsL rph
spoT1∆(mrr-hsdRMS-mcrBC)
New England
Biolabs
One Shot ccdB
Survival 2 T1 E. coli
F-mcrA Δ(mrr-hsdRMS-mcrBC) Φ80lacZΔM15
ΔlacX74 recA1 araΔ139 Δ(ara-leu)7697galU galK rpsL
(StrR) endA1 nupG fhuA::IS2
Life Technologies
BY4741 MATa his3Δ0 leu2Δ0 met15Δ0 ura3Δ0 Thermo Scientific
BY4741ΔFDC1
(YDR539W) MATa his3Δ0 leu2Δ0 met15Δ0 ura3Δ0 fdc1Δ Thermo Scientific
BY4741-PAL MATa his3Δ0 leu2Δ0 met15Δ0 ura3Δ0-425GPDPAL This Study
22A75D BY4741 phenylalanine overproducer This Study
22A75D-PAL 22A75D-425GPDPAL This Study
22A75D10-PAL 22A75D aro10Δ-425GPDPAL This Study
22A75D104-PAL 22A75D aro10Δ::aro4K229L
-425GPDPAL This Study
Plasmids
pTrc99A Ptrc, pBR322 ori, lacIq, Amp
R Prather Lab, MIT
pFA6-KanMX KanR2, pBR322 ori, AmpR AddGene
pDONR221 attP1-ccdB-CmR-attP2 cassette, pUC ori, Kan
R Life Technologies
pDONR-PAL PAL2 from A. thaliana inserted into pDONR221 This study
425GPD
PGPD, attR1-ccdB-CmR-attR2 cassette, pBR322 ori, LEU2,
AmpR AddGene
425GPDPAL PAL2 from A. thaliana inserted into 425GPD This study
pACYCDuet-1 PT7, p15A ori, lacIq, Cm
R Novagen
pACYC- ARO4K229L
-
KanMX pACYC with the integration cassette aro4K229L
-KanMX This Study
pUN15-PAL2
Clone U12256 containing AT3G53260 (PAL2) from A.
thaliana ABRC
4.2.2 Toxicity assays.
To determine impact of styrene on S. cerevisiae growth, the effect of its
exogenous addition to growing cultures at increasing final concentrations was
investigated. Seed cultures of S. cerevisiae BY4741 were prepared in 5 mL of YPD broth
and grown at 32oC overnight while shaking at 250 rpm. The seed culture (1 ml) was then
used to inoculate 50 ml of YPD broth in a 250 mL shake flask. Cultures were grown to an

72
optical density at 600 nm (OD600) of ~0.6, at which time styrene was added to the flasks
at an array of final concentrations ranging between 0 to 1 g/L. Culturing resumed at 32oC
for an additional 6-8 h while cell growth, as determined by OD600 measurements, was
periodically monitored using a UV/Vis spectrophotometer (DU800, Beckman Coulter,
Brea, CA).
4.2.3 Evolution of phenylalanine overproducing strains.
Evolution of a phenylalanine over-producing phenotype in S. cerevisiae was
achieved through random mutagenesis and high-throughput selection using the
phenylalanine anti-metabolite m-fluoro-DL-phenylalanine to provide selective pressure.
S. cerevisiae BY4741 was first treated with ethylmethanesulphonate (EMS) according to
standard protocols(Winston 2001) before then being plated on minimal media
supplemented with m-fluoro-DL-phenylalanine. In the first round of mutagenesis,
selection occurred on SD media plates supplemented with either 18 or 22 mg/L m-fluoro-
DL-phenylalanine. Note that the BY4741’s minimum inhibitory concentration of m-
fluoro-DL-phenylalanine was previously found to be approximately 15 mg/L (data not
shown). Two mutants (designated 18A and 22A) were subsequently isolated from the
first round of mutagenesis before then being cultured in SD media for 48 h at 32oC.
Supernatants were analyzed by high performance liquid chromatography (HPLC; as
described below) to test for their comparative ability to produce 2-phenylethanol. Note
that 2-phenylethanol, which is endogenously produced from phenylpyruvate via ARO10,
was used as a surrogate to indicate phenylalanine over-production because phenylalanine
is not exported to the extracellular media(Luttik et al. 2008). Strains 18A and 22A were
then subjected to a second round of mutagenesis as preformed above, however, in this

73
case the resultant mutants were selected for on SD media plates supplemented with 25,
50, or 75 mg/L m-fluoro-DL-phenylalanine. From this, 18 mutants were then isolated
before then being similarly cultured and characterized with respect to their 2-
phenylethanol production abilities. The key regulatory genes (as well as 500 bp upstream
of the start codon) of the phenylalanine pathway ARO3, ARO4, ARO7, ARO8, GCN4, and
PHA2 were sequenced in the strains BY4741, 22A, and 22A75D.
4.2.4 Transcriptional analysis of phenylalanine over-producing mutants.
Relative transcription levels of each of ARO1, ARO2, ARO3, ARO4, ARO7,
ARO8, ARO9, and PHA2 were quantified at mid-log phase in the strains BY4741, 22A,
and 22A75D. Approximately 1.5×108
cells of each strain were collected by centrifugation
at 17,000 x g for 1 min. The supernatant was discarded and RNA was extracted from the
cell pellet using the YeaStar RNA Extraction Kit (Zymo Research, Irvine, CA). cDNA
was synthesized using the SuperScript VILO cDNA Synthesis Kit (Life Technologies)
and RT-qPCR was performed using SYBR Green (Life Technologies) based quantitative
PCR according to manufacturer’s protocols. Custom oligonucleotide primers for RT-
qPCR experiments, including those for the reference housekeeping gene 26S (Martorell,
Querol, and Fernández-Espinar 2005), were designed and synthesized, the sequences of
which are provided in Table S1 (see Supplementary Information). RT-qPCR was
performed on an Applied Biosystems StepOne Real-Time PCR (Applied Biosystems)
using a 60ºC annealing temperature. Data analysis was performed using StepOne
software and relative transcriptional levels were calculated using the 2-ΔΔCt
method(Livak
and Schmittgen 2001).
4.2.5 Investigating native expression and activity of FDC1 in S. cerevisiae.

74
S. cerevisiae BY4741 seed cultures were prepared (in triplicate) in 5 mL YPD
broth and cultured at 32°C while shaking at 250 rpm overnight. 1 ml of each seed was
then used to again inoculate 50 ml SD media. These cultures were grown until reaching
an OD600 of ~0.6, at which point each was induced by either trans-cinnamate, ferulic
acid, p-coumaric acid, or phenylalanine at a final concentration of 0.2 mM. Induced
cultures were incubated for an additional 12 h after which an equal number of cells
(OD600 = 4) were collected by centrifugation at 2800 x g for 4 min. Cells were lysed using
Zymolyase (Zymo Research) and the supernatant collected after centrifugation at 11,000
x g for 2 min. FDC1 activity was assayed at room temperature in pH 7.5 50 mM Tris-HCl
buffer containing 250 mM trans-cinnamate, and initiated by the addition of 5 µL of crude
cell lysate. The accumulation of styrene was then followed at 247 nm on a UV/Vis
Spectrophotometer for a total of 5 min at 20 sec intervals. A molar extinction coefficient
of 10,000 M-1
cm-1
and a 1 cm path length were used to establish enzyme activity in
terms of U mg-1
total protein. The total protein content in each lysate sample was
determined by Bradford Assay using bovine serum albumin (BSA) as an external
standard.
4.2.6 Cloning of PAL2 from A. thaliana.
The PAL2 encoding gene from A. thaliana was derived from cDNA library
plasmids containing the specific loci of interest (Table 4.1) obtained from the
Arabidopsis Biological Research Center (ABRC; Ohio State University, Columbus, OH).
PAL2 was PCR amplified using Phusion DNA Polymerase (Finnzymes, Espoo, Finland)
using custom oligonucleotide primers (supplementary material). Using Gateway Cloning
Technology (Alberti, Gitler, and Lindquist 2007), amplified linear DNA fragments

75
flanked with attB sequences were purified using the Zyppy Clean and Concentrator kit
(Zymo Research). The BP reaction between the DNA fragment and pDONR221 (Life
Technologies) was created using Gateway BP Clonase II Enzyme Mix (Life
Technologies) following manufacturer’s protocols. Transformants were selected by
plating on LB solid agar containing kanamycin and culturing at 37oC overnight. The
resultant donor plasmid, pDONR-PAL, was mixed with the desired destination plasmid,
425-GPD, using the Gateway LR Clonase II Enzyme Mix (Life Technologies).
Transformants were selected by plating on LB solid agar containing ampicillin and
confirmed using colony PCR. This approach resulted in construction of the constitutively
induced, high copy number (2µ) plasmid 425GPDPAL, as listed in Table 4.1.
4.2.7 Assaying the extracellular transport of trans-cinnamate.
S. cerevisiae BY4741 and the FDC1 knockout mutant BY4741ΔFDC1
(YDR539W, clone 5834 (Thermo Scientific, Waltham, MA)) were each transformed with
plasmid 425GPDPAL. Cultures grown in SD-Leu media supplemented with 200 mg/L
phenylalanine while the extracellular accumulation of trans-cinnamate, styrene, and 2-
phenylethanol were periodically monitored via HPLC over the course of 24 h.
4.2.8 Chromosomal disruption of ARO10 and integration of ARO4K229L
.
Targeted chromosomal disruption of ARO10 in strain 22A75D was performed via
homologous recombination. Gene disruption cassettes were generated via PCR to contain
40 base pairs of homology on both sides of the targeted integration site (i.e., ARO10) in
addition to the KanMX selectable marker (as obtained from pFA6-KanMX4). Following
transformation, colonies were selected on YPD solid agar plates containing 200 mg/L
G418. Clones carrying the successful ARO10 disruption cassette were further confirmed

76
by PCR. This resulted in the strain 22A75D10. In addition, a copy of the feedback
resistant mutant ARO4K229L
, whose expression was driven by the native ARO4 promoter,
was likewise integrated into 22A75D at the ARO10 locus, thereby also and
simultaneously resulting in its chromosomal disruption. In this case, however, the gene
disruption cassette was first constructed in the E. coli expression vector pACYCDuet-1.
The ARO4 point mutation (K229L) was generated (and confirmed by sequencing) via
overlap extension using the primers listed in Table S1 and inserted into the BamHI and
EcoRI sites of pACYCDuet-1. Subsequently, the KanMX selectable marker together with
its promoter was inserted downstream of ARO4K229L
to generate the plasmid pACYC-
ARO4K229L
-KanMX. The entire cassette was then PCR amplified using primers whose
overhangs each contained 40 base pairs of homology to ARO10. The resultant fragment
was then transformed into 22A74D as described above, resulting in the strain
22A75D104.
4.2.9 Styrene production from glucose in S. cerevisiae shake flask cultures.
Each of the S. cerevisiae strains BY4741, 22A75D, 22A75D10, and 22A75D104
were individually transformed with the plasmid 425GPDPAL. The transformants were
each grown in 5 mL SD-Leu broth for 12 h at 32°C while shaking at 250 rpm to prepare
seed cultures. Each seed (1 mL) was then used to inoculate 50 mL SD-Leu media in 250
ml shake flask fitted with a glass cap. A closed system with large headspace was used to
avoid volatile product (i.e., styrene) losses while also precluding the exhaustion of
oxygen. Culturing continued for 48 h while 1 ml samples were periodically taken for
analysis of cell growth and metabolite production.

77
4.2.10 Metabolite analysis.
Samples were prepared for metabolite analysis via HPLC by first removing 1 mL
of culture from a shake flask culture and pelleting the cells at 11,000 x g for 2 min. The
supernatant (0.75 mL) was then transferred to a glass HPLC vial and sealed with a
Teflon-lined cap. Analysis was carried out using a Hewlett Packard 1100 series HPLC
system (Palo Alto, CA) equipped with an auto sampler, diode array (UV/Vis) detector,
and reverse-phase Hypersil Gold SBC18 column (4.6mm x 150 mm; Thermo Fisher,
USA). Samples (5 µL) were injected for analysis according to the methods of McKenna
and Nielsen(McKenna and Nielsen 2011). The eluent was monitored at 215 nm for L-
phenylalanine and 2-phenylethanol and 258 nm for trans-cinnamate and styrene. Under
these conditions L-phenylalanine, 2-phenylethanol, trans-cinnamate, and styrene were
eluted at 4.5, 7.1, 8.67, and 10.4 min, respectively.
4.3 Results
4.3.1 Assaying styrene toxicity
To assess if S. cerevisiae BY4741 would indeed be a suitable host for styrene
production, a cursory evaluation of its ability to tolerate exogenous styrene at increasing
concentrations was first performed. Due to its hydrophobic nature, styrene readily
accumulates within the hydrophobic core of the membrane lipid-bilayer(Juan L. Ramos
2002; Heipieper, Weber, Sikkema, Keweloh, and de Bont 1994; Isken and de Bont 1998;
McKenna et al. 2013).
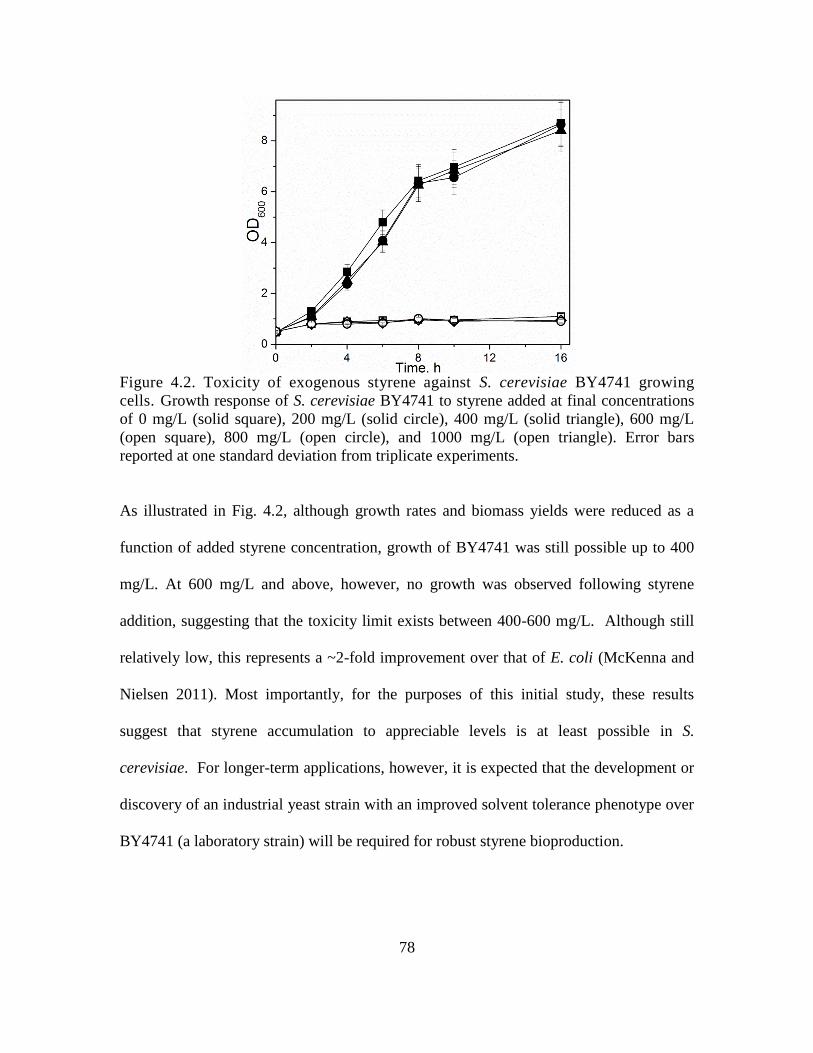
78
Figure 4.2. Toxicity of exogenous styrene against S. cerevisiae BY4741 growing
cells. Growth response of S. cerevisiae BY4741 to styrene added at final concentrations
of 0 mg/L (solid square), 200 mg/L (solid circle), 400 mg/L (solid triangle), 600 mg/L
(open square), 800 mg/L (open circle), and 1000 mg/L (open triangle). Error bars
reported at one standard deviation from triplicate experiments.
As illustrated in Fig. 4.2, although growth rates and biomass yields were reduced as a
function of added styrene concentration, growth of BY4741 was still possible up to 400
mg/L. At 600 mg/L and above, however, no growth was observed following styrene
addition, suggesting that the toxicity limit exists between 400-600 mg/L. Although still
relatively low, this represents a ~2-fold improvement over that of E. coli (McKenna and
Nielsen 2011). Most importantly, for the purposes of this initial study, these results
suggest that styrene accumulation to appreciable levels is at least possible in S.
cerevisiae. For longer-term applications, however, it is expected that the development or
discovery of an industrial yeast strain with an improved solvent tolerance phenotype over
BY4741 (a laboratory strain) will be required for robust styrene bioproduction.
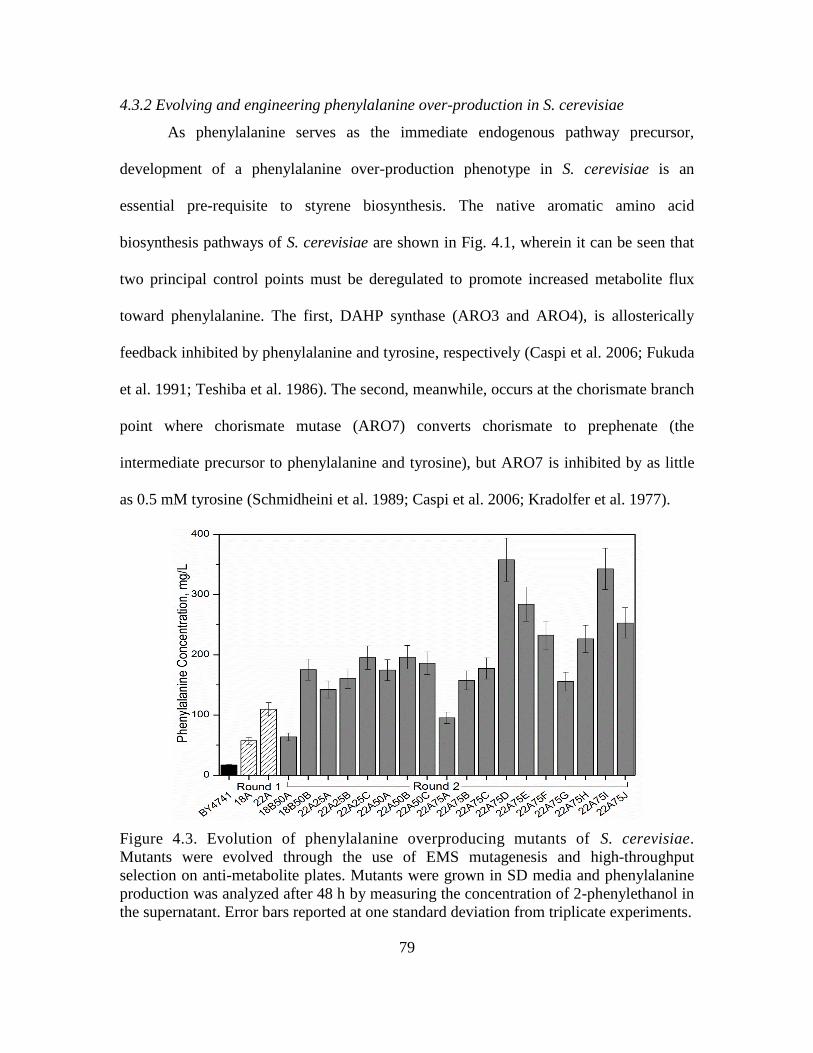
79
4.3.2 Evolving and engineering phenylalanine over-production in S. cerevisiae
As phenylalanine serves as the immediate endogenous pathway precursor,
development of a phenylalanine over-production phenotype in S. cerevisiae is an
essential pre-requisite to styrene biosynthesis. The native aromatic amino acid
biosynthesis pathways of S. cerevisiae are shown in Fig. 4.1, wherein it can be seen that
two principal control points must be deregulated to promote increased metabolite flux
toward phenylalanine. The first, DAHP synthase (ARO3 and ARO4), is allosterically
feedback inhibited by phenylalanine and tyrosine, respectively (Caspi et al. 2006; Fukuda
et al. 1991; Teshiba et al. 1986). The second, meanwhile, occurs at the chorismate branch
point where chorismate mutase (ARO7) converts chorismate to prephenate (the
intermediate precursor to phenylalanine and tyrosine), but ARO7 is inhibited by as little
as 0.5 mM tyrosine (Schmidheini et al. 1989; Caspi et al. 2006; Kradolfer et al. 1977).
Figure 4.3. Evolution of phenylalanine overproducing mutants of S. cerevisiae. Mutants were evolved through the use of EMS mutagenesis and high-throughput
selection on anti-metabolite plates. Mutants were grown in SD media and phenylalanine
production was analyzed after 48 h by measuring the concentration of 2-phenylethanol in
the supernatant. Error bars reported at one standard deviation from triplicate experiments.

80
To overcome feedback resistance in the phenylalanine biosynthesis pathways, the
use of antimetabolites, in this case m-fluoro-DL-phenylalanine, in parallel with EMS
mutagenesis provided the needed selection pressure for evolving phenylalanine over-
producing mutants(Drake 1976). Two mutants were isolated from the first round of
selection on SD plates containing 18 mg/L (strain 18A) and 22 mg/L (strain 22A) m-
fluoro-DL-phenylalanine. Both mutants were subsequently tested for their respective
phenylalanine production capacities in shake flask cultures (Fig.4.3). As phenylalanine is
not exported from S. cerevisiae, the extracellular accumulation of 2-phenylethanol, which
is naturally and readily produced as a degradation product of phenylpyruvate (the
precursor to phenylalanine; Fig. 4.1), was used as a measure of increased flux through the
phenylalanine biosynthesis pathway. 18A and 22A showed 3.3- and 6.4-fold
improvements in phenylalanine production potential (57.24±5.20 and 109.56±9.24
mg/L), respectively, relative to the parent control (BY4741).
A second round of mutagenesis and selection was performed with the objective of
further deregulating phenylalanine biosynthesis. In this case, the isolated mutants 18A
and 22A were themselves subjected to further mutagenesis where the selective pressure
was elevated by application of m-fluoro-DL-phenylalanine at increased concentrations
(namely 25 mg/L, 50 mg/L, or 75 mg/L). In contrast to the first round, this second round
of mutagenesis netted numerous colonies (over 50) at all three selection pressures. Thus,
to screen for the best performers, only the fastest growing mutants were selected (in this
case, a total of 18) and subsequently tested for their ability to overproduce phenylalanine.
Amongst the pools, the top performing mutant, named 22A75D, was able to produce an
estimated 357±32.5 mg/L of phenylalanine in 48 h, representing a ~21-fold improvement

81
over BY4741. The performance of mutant 22A75D was followed closely by 22A75I,
which was able to produce up to 342±28.9 mg/L phenylalanine.
In an effort to better understand the genetic bases responsible for the evolved
phenylalanine over-production phenotype in the most productive mutant (22A75D), key
genes (as well as 500 bp upstream of the start codon) involved in the phenylalanine
biosynthesis pathway were sequenced and compared with those of its parent (22A) and
the control (BY4741). The subset of genes of interest included those associated with the
known bottleneck enzymes DAHP synthase (ARO3 and ARO4) and chorismate mutase
(ARO7), as well as phenylalanine prephenate dehydratase (PHA2), aromatic
aminotransferase (ARO8), and the activator protein GCN4, a transcriptional activator
involved in the expression of all steps in the phenylalanine biosynthesis pathway (Caspi
et al. 2006). Interestingly, however, no mutations were observed in any of the genes
investigated, or in their associated upstream sequences. It is possible, perhaps, that a
global transcription factor was the cause of the increase rather than single point mutations
in the individual phenylalanine pathways genes themselves. As no changes in any open
reading frame tested were detected, this would imply that the evolved phenotype was not
accrued as a result of relieving allosteric inhibition.
Accordingly, attention was next directed towards understanding if it were instead
transcriptional changes that were responsible for the observed phenotypic changes.
Changes in the transcription level of each of the genes of interest were measured with the
use of RT-qPCR, and quantified relative to that of the wild-type control (BY4741). As
illustrated in Fig. 4.4, in 22A75D up-regulation of ARO8 was found to be most
significant (a 9.3-fold increase), followed by ARO1 (6.8-fold), ARO2 (5.8-fold), and
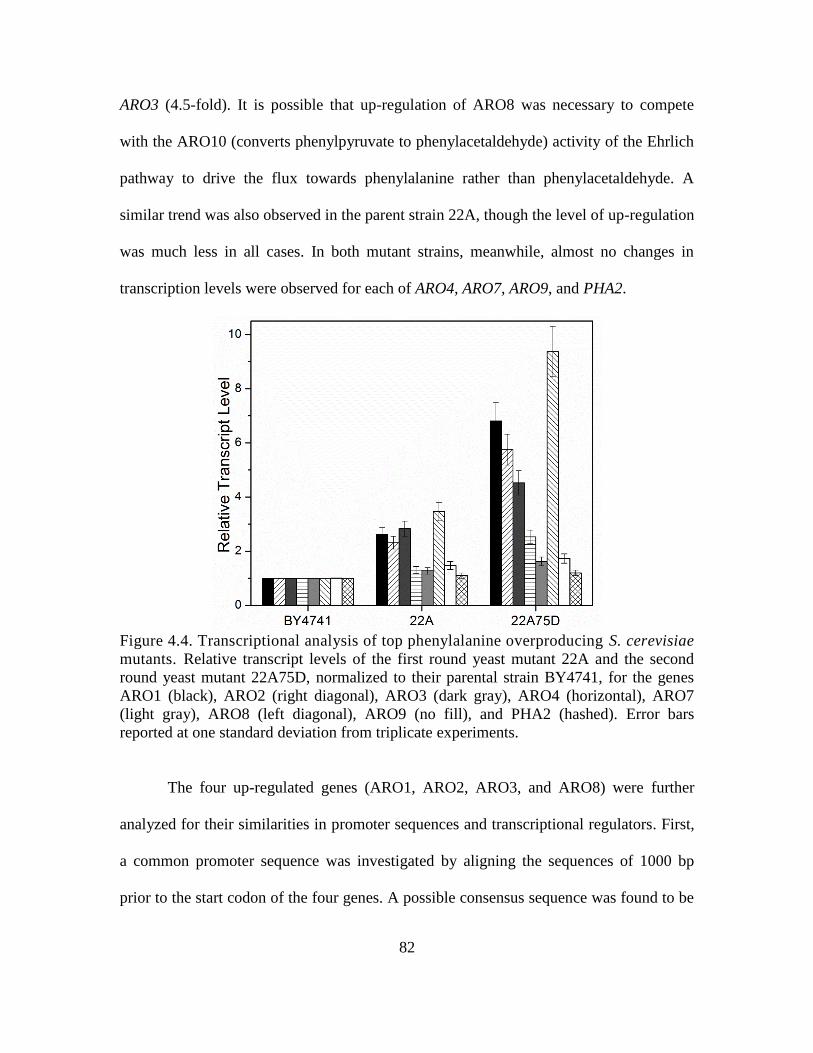
82
ARO3 (4.5-fold). It is possible that up-regulation of ARO8 was necessary to compete
with the ARO10 (converts phenylpyruvate to phenylacetaldehyde) activity of the Ehrlich
pathway to drive the flux towards phenylalanine rather than phenylacetaldehyde. A
similar trend was also observed in the parent strain 22A, though the level of up-regulation
was much less in all cases. In both mutant strains, meanwhile, almost no changes in
transcription levels were observed for each of ARO4, ARO7, ARO9, and PHA2.
Figure 4.4. Transcriptional analysis of top phenylalanine overproducing S. cerevisiae
mutants. Relative transcript levels of the first round yeast mutant 22A and the second
round yeast mutant 22A75D, normalized to their parental strain BY4741, for the genes
ARO1 (black), ARO2 (right diagonal), ARO3 (dark gray), ARO4 (horizontal), ARO7
(light gray), ARO8 (left diagonal), ARO9 (no fill), and PHA2 (hashed). Error bars
reported at one standard deviation from triplicate experiments.
The four up-regulated genes (ARO1, ARO2, ARO3, and ARO8) were further
analyzed for their similarities in promoter sequences and transcriptional regulators. First,
a common promoter sequence was investigated by aligning the sequences of 1000 bp
prior to the start codon of the four genes. A possible consensus sequence was found to be
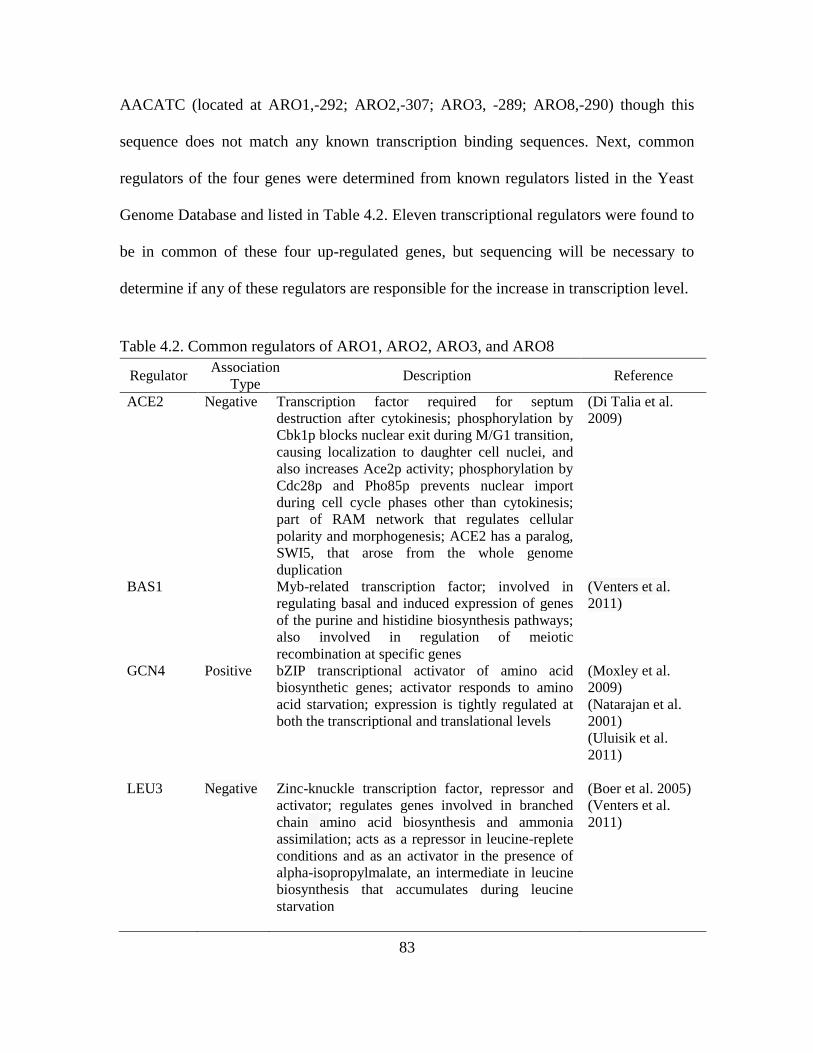
83
AACATC (located at ARO1,-292; ARO2,-307; ARO3, -289; ARO8,-290) though this
sequence does not match any known transcription binding sequences. Next, common
regulators of the four genes were determined from known regulators listed in the Yeast
Genome Database and listed in Table 4.2. Eleven transcriptional regulators were found to
be in common of these four up-regulated genes, but sequencing will be necessary to
determine if any of these regulators are responsible for the increase in transcription level.
Table 4.2. Common regulators of ARO1, ARO2, ARO3, and ARO8
Regulator Association
Type Description Reference
ACE2 Negative Transcription factor required for septum
destruction after cytokinesis; phosphorylation by
Cbk1p blocks nuclear exit during M/G1 transition,
causing localization to daughter cell nuclei, and
also increases Ace2p activity; phosphorylation by
Cdc28p and Pho85p prevents nuclear import
during cell cycle phases other than cytokinesis;
part of RAM network that regulates cellular
polarity and morphogenesis; ACE2 has a paralog,
SWI5, that arose from the whole genome
duplication
(Di Talia et al.
2009)
BAS1 Myb-related transcription factor; involved in
regulating basal and induced expression of genes
of the purine and histidine biosynthesis pathways;
also involved in regulation of meiotic
recombination at specific genes
(Venters et al.
2011)
GCN4 Positive bZIP transcriptional activator of amino acid
biosynthetic genes; activator responds to amino
acid starvation; expression is tightly regulated at
both the transcriptional and translational levels
(Moxley et al.
2009)
(Natarajan et al.
2001)
(Uluisik et al.
2011)
LEU3 Negative Zinc-knuckle transcription factor, repressor and
activator; regulates genes involved in branched
chain amino acid biosynthesis and ammonia
assimilation; acts as a repressor in leucine-replete
conditions and as an activator in the presence of
alpha-isopropylmalate, an intermediate in leucine
biosynthesis that accumulates during leucine
starvation
(Boer et al. 2005)
(Venters et al.
2011)
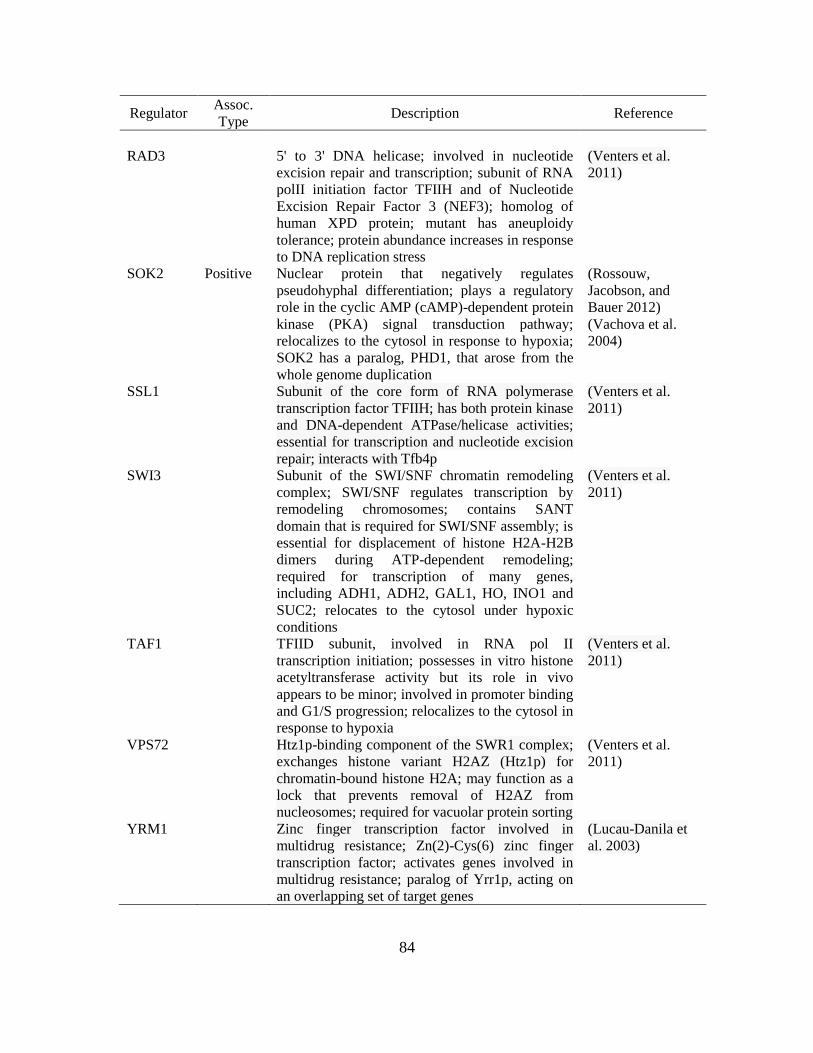
84
Regulator Assoc.
Type Description Reference
RAD3
5' to 3' DNA helicase; involved in nucleotide
excision repair and transcription; subunit of RNA
polII initiation factor TFIIH and of Nucleotide
Excision Repair Factor 3 (NEF3); homolog of
human XPD protein; mutant has aneuploidy
tolerance; protein abundance increases in response
to DNA replication stress
(Venters et al.
2011)
SOK2 Positive Nuclear protein that negatively regulates
pseudohyphal differentiation; plays a regulatory
role in the cyclic AMP (cAMP)-dependent protein
kinase (PKA) signal transduction pathway;
relocalizes to the cytosol in response to hypoxia;
SOK2 has a paralog, PHD1, that arose from the
whole genome duplication
(Rossouw,
Jacobson, and
Bauer 2012)
(Vachova et al.
2004)
SSL1 Subunit of the core form of RNA polymerase
transcription factor TFIIH; has both protein kinase
and DNA-dependent ATPase/helicase activities;
essential for transcription and nucleotide excision
repair; interacts with Tfb4p
(Venters et al.
2011)
SWI3 Subunit of the SWI/SNF chromatin remodeling
complex; SWI/SNF regulates transcription by
remodeling chromosomes; contains SANT
domain that is required for SWI/SNF assembly; is
essential for displacement of histone H2A-H2B
dimers during ATP-dependent remodeling;
required for transcription of many genes,
including ADH1, ADH2, GAL1, HO, INO1 and
SUC2; relocates to the cytosol under hypoxic
conditions
(Venters et al.
2011)
TAF1 TFIID subunit, involved in RNA pol II
transcription initiation; possesses in vitro histone
acetyltransferase activity but its role in vivo
appears to be minor; involved in promoter binding
and G1/S progression; relocalizes to the cytosol in
response to hypoxia
(Venters et al.
2011)
VPS72 Htz1p-binding component of the SWR1 complex;
exchanges histone variant H2AZ (Htz1p) for
chromatin-bound histone H2A; may function as a
lock that prevents removal of H2AZ from
nucleosomes; required for vacuolar protein sorting
(Venters et al.
2011)
YRM1 Zinc finger transcription factor involved in
multidrug resistance; Zn(2)-Cys(6) zinc finger
transcription factor; activates genes involved in
multidrug resistance; paralog of Yrr1p, acting on
an overlapping set of target genes
(Lucau-Danila et
al. 2003)

85
While this level of analysis offers some clues as to changes associated with the
evolved trait, elucidating the entire picture will likely only be possible through whole-
genome sequencing. However, given that the achievable phenylalanine titers are still
quite modest (only up to ~350 mg/L), such an undertaking does not seem warranted at
this time. For now, however, and for the purpose of this study, a phenylalanine over-
producing host has been developed that can now serve as a test platform for engineering
styrene biosynthesis.
4.3.3 Investigating native FDC1 activity and factors influencing its expression
The fact that FDC1 – the second enzyme in the established styrene pathway (Fig.
4.1) – is native to S. cerevisiae was an important factor motivating its selection as a
potential host platform for styrene biosynthesis. However, as factors controlling its native
regulation have not yet been fully resolved, it was initially unclear if: i) background
expression of FDC1 would occur in the specific strain and under the culture conditions of
interest, or ii) expression levels would be sufficient for supporting ample flux through the
last step of the pathway. With regards to the first point, it has previously been shown that
PAD1 expression was induced in the presence of ferulic acid, coumaric acid, and trans-
cinnamate, when said species were added to growing cultures(Larsson, Nilvebrant, and
Jonsson 2001). The potential role of trans-cinnamate (a styrene pathway intermediate and
known substrate of FDC1) as an inducer of FDC1 expression, however, was not tested
and has not before been reported. Thus, to better understand native expression of FDC1,
particularly within the context of the styrene pathway, an investigation of factors
influencing its induction was performed.

86
Potential inducers of interest included both phenylalanine and trans-cinnamate, as
well as coumaric acid and ferulic acid as both structural homologs to trans-cinnamate.
BY4741 was cultured in minimal media supplemented with 200 mg/L of each species for
12 h before collecting the cells by pelleting and preparing crude cell lysate. Cellular
extracts were then each tested for in vitro decarboxylase activity on trans-cinnamate, as
measured spectrophotometrically. As seen in Table 4.3, a positive result was observed for
cells cultured in the presence of each of ferulic acid, trans-cinnamate, or coumaric acid,
indicating that all three are suitable inducers of FDC1 expression. Interestingly, lysates
prepared from cells grown in the presence of trans-cinnamate displayed the greatest
specific trans-cinnamate decarboxylase activity. In contrast, phenylalanine was not an
inducer of FDC1, nor was FDC1 activity detected in the control. This implies that FDC1
expression is not constitutive. Furthermore, its expression in the final strain will be
contingent upon PAL2 expression, which could be of benefit with respect to minimizing
metabolic burden. Further experiments, however, are certainly necessary before the
mechanism of transcription initiation can be fully understood.
Table 4.3. Assaying the in vitro decarboxylase activity of FDC1 against a pool of
structurally-related, phenylacrylic acid substrates.
Compound Induced activity mU/mg total protein
trans-cinnamic acid Positive 0.46±0.02
p-coumaric acid Positive 0.39±0.02
ferulic acid Positive 0.21±0.03
phenylalanine Negative Not Detected
control Negative Not Detected
4.3.4 Probing the styrene pathway via the exogenous addition of phenylalanine
Preliminary studies were next performed to begin probing the functionality of the
styrene pathway, with the particular objectives of determining: i) if styrene can be

87
produced from exogenously supplied phenylalanine, and ii) if native expression levels of
FDC1 are sufficient for supporting flux through the pathway. To test this, S. cerevisiae
BY4741 was first transformed with 425GPDPAL (a high copy (2μ) number, constitutive
plasmid) to enable the heterologous expression of PAL2 from A. thaliana. Cultures
grown in SD-Leu minimal media were then supplied phenylalanine (200 mg/L) while the
extracellular accumulation of key pathway metabolites trans-cinnamate and styrene, as
well as the natural degradation product 2-phenylethanol, were monitored via HPLC for a
period of 24 h. In BY4741, although only less than half of the phenylalanine was
consumed, as shown in Fig. 4.5, styrene and 2-phenylethanol constituted the major
accumulated end-products (20.4±0.8 and 43±0.45 mg/L, respectively). Importantly, no
trans-cinnamate was detected in the culture medium at any time. This finding suggested
that sufficient FDC1 activity was present so as to swiftly convert all trans-cinnamate to
styrene as it was produced (i.e., implying that native levels of FDC1 expression are
sufficient for circumventing the creation of a flux bottleneck).
However, the question still remained as to whether the reason for not detecting
trans-cinnamate in the culture medium was alternatively due to its potential inability to
be exported by the cells. Whereas it has previously been shown that trans-cinnamate can
be imported by S. cerevisiae (Mukai et al. 2010), its natural ability to be exported has
never been explicitly reported. To test this, the control strain BY4741ΔFDC1 was
similarly transformed with 425GPDPAL and analogously assayed. In this case, as seen in
Fig. 4.5, 2-phenylethanol was the major product (98±2.98 mg/L) and no styrene
production was observed. More importantly, however, in this case extracellular
accumulation of trans-cinnamate (26.2±1.6 mg/L) was in fact observed, implying that it
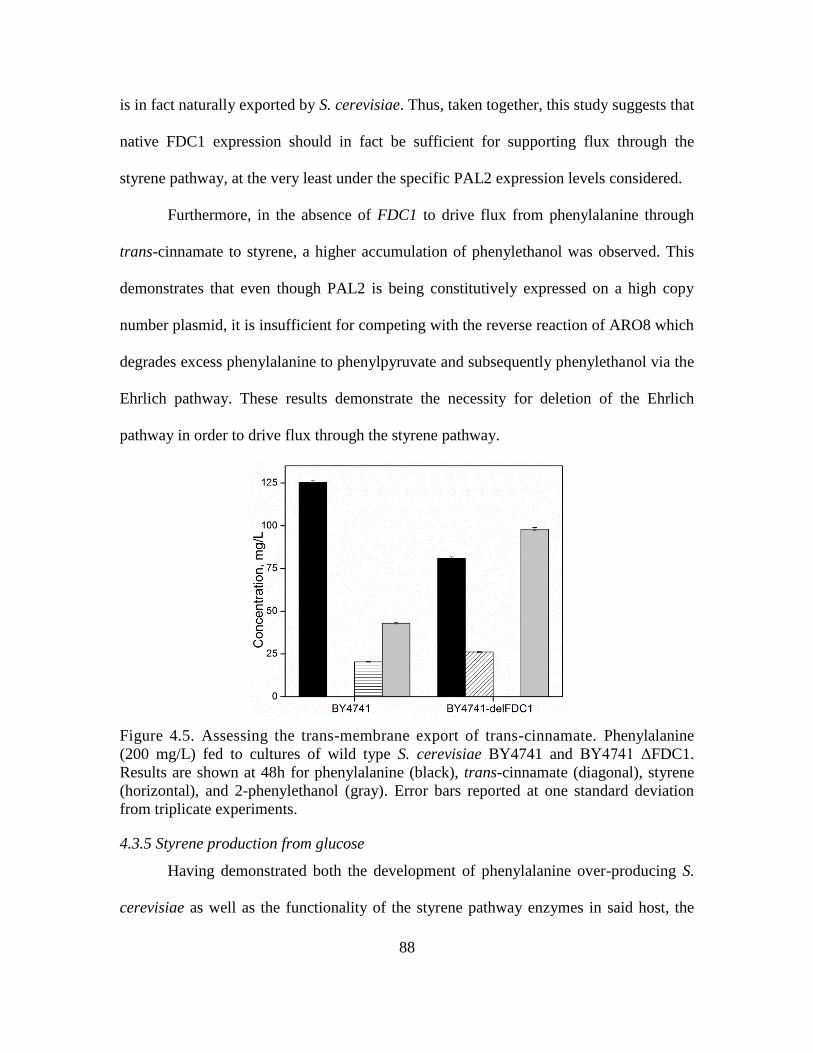
88
is in fact naturally exported by S. cerevisiae. Thus, taken together, this study suggests that
native FDC1 expression should in fact be sufficient for supporting flux through the
styrene pathway, at the very least under the specific PAL2 expression levels considered.
Furthermore, in the absence of FDC1 to drive flux from phenylalanine through
trans-cinnamate to styrene, a higher accumulation of phenylethanol was observed. This
demonstrates that even though PAL2 is being constitutively expressed on a high copy
number plasmid, it is insufficient for competing with the reverse reaction of ARO8 which
degrades excess phenylalanine to phenylpyruvate and subsequently phenylethanol via the
Ehrlich pathway. These results demonstrate the necessity for deletion of the Ehrlich
pathway in order to drive flux through the styrene pathway.
Figure 4.5. Assessing the trans-membrane export of trans-cinnamate. Phenylalanine
(200 mg/L) fed to cultures of wild type S. cerevisiae BY4741 and BY4741 ΔFDC1.
Results are shown at 48h for phenylalanine (black), trans-cinnamate (diagonal), styrene
(horizontal), and 2-phenylethanol (gray). Error bars reported at one standard deviation
from triplicate experiments.
4.3.5 Styrene production from glucose
Having demonstrated both the development of phenylalanine over-producing S.
cerevisiae as well as the functionality of the styrene pathway enzymes in said host, the

89
final objective of this study was to demonstrate that styrene could by produced directly
from glucose by S. cerevisiae. When the plasmid 425GPDPAL was transformed into
BY4741 (strain BY4741-PAL) and cultured for 48 h in SD-Leu media for production of
styrene, minimal amounts (less than 5 mg/L, Fig. 4.6) of styrene were detected. This was
not unexpected since the concentration of phenylalanine was minimal in BY4741. Once
the plasmid 425GPDPAL was transformed into the phenylalanine overproducer 22A75D
(to generate strain 22A75D-PAL) and cultured for styrene production, styrene
biosynthesis levels increased to 18.2±1.5 mg/L with 54.4±4.6 mg/L co-production of
phenylethanol. The co-production of phenylethanol demonstrates that even though PAL
is being over-expressed, it cannot compete with native metabolism and the natural
regulation of phenylalanine production. In S. cerevisiae, the production of phenylalanine
is not only regulated via feedback-inhibition but also via the Ehrlich pathway which
converts the phenylalanine to phenylpyruvate in order to utilize the nitrogen from the
amino acid (Hazelwood et al. 2008; Dickinson, Salgado, and Hewlins 2003). At this
point, further strain improvements were necessary to increase styrene titers.
Due to the endogenous Ehrlich pathway present in yeast, phenylpyruvate is
subsequently converted to phenylacetaldehyde and phenylethanol rather than styrene by
means of ARO10(Caspi et al. 2006; Hazelwood et al. 2008). To prevent degradation of
phenylpyruvate, ARO10 was subsequently deleted from strain 22A75D to create strain
22A75D10. The ARO10 knockout strain hosting the 425GPDPAL plasmid, strain
22A75D10-PAL, had a modest improvement in styrene synthesis achieving a final titer of
22.7±1.9 mg/L. Since trans-cinnamate was not observed in the culture media, it was

90
believed that the styrene titers suffered from a lack of precursor availability, and to
improve titers, an increase in phenylalanine was needed.
To enhance flux through the phenylalanine pathway, it has been previously
reported that the tyrosine sensitive DAHP synthase mutant ARO4K229L
alleviates
feedback inhibition from tyrosine and can increase flux through the shikimic acid
pathway by as much as 4.5 fold(Curran et al. 2013; Luttik et al. 2008). In an attempt to
relieve the feed-back regulation, we incorporated the tyrosine-sensitive feedback resistant
DAHP synthase mutant ARO4K229L
onto the genome while simultaneously deleting
ARO10. The strain 22A75D104 was then transformed with the plasmid 425GPDPAL to
achieve the strain 22A75D104-PAL (expressing both ARO4K229L
and PAL2).
22A75D104-PAL achieved a 25% increase (Fig. 4.6) in styrene production to 28.8±2.1
mg/L with a glucose yield of 0.00144±0.00011 g/g. It is important to note, however, that
ARO4 is feed-back sensitive to tyrosine (not phenylalanine) (Caspi et al. 2006);
therefore, it is not surprising that it did not have a more impactful result. To date, a
feedback-resistant mutant of the phenylalanine sensitive DAHP synthase homolog ARO3
has not been discovered. While this point-mutation demonstrates a modest increase in
flux through the shikimic acid pathway, achievable phenylalanine yields are still quite
low in yeast suggesting that the pathway is tightly regulated beyond the DAHP synthase.
In order for higher titers to be realized, further de-regulation of the phenylalanine
biosynthesis pathway in S. cerevisiae must be achieved.
With only a modest improvement from its predecessor, we believe that the
bottleneck is a result of poor PAL activity. Although flux through the phenylalanine
pathway greatly improved in mutant 22A75D, we believe that the activity of PAL is not
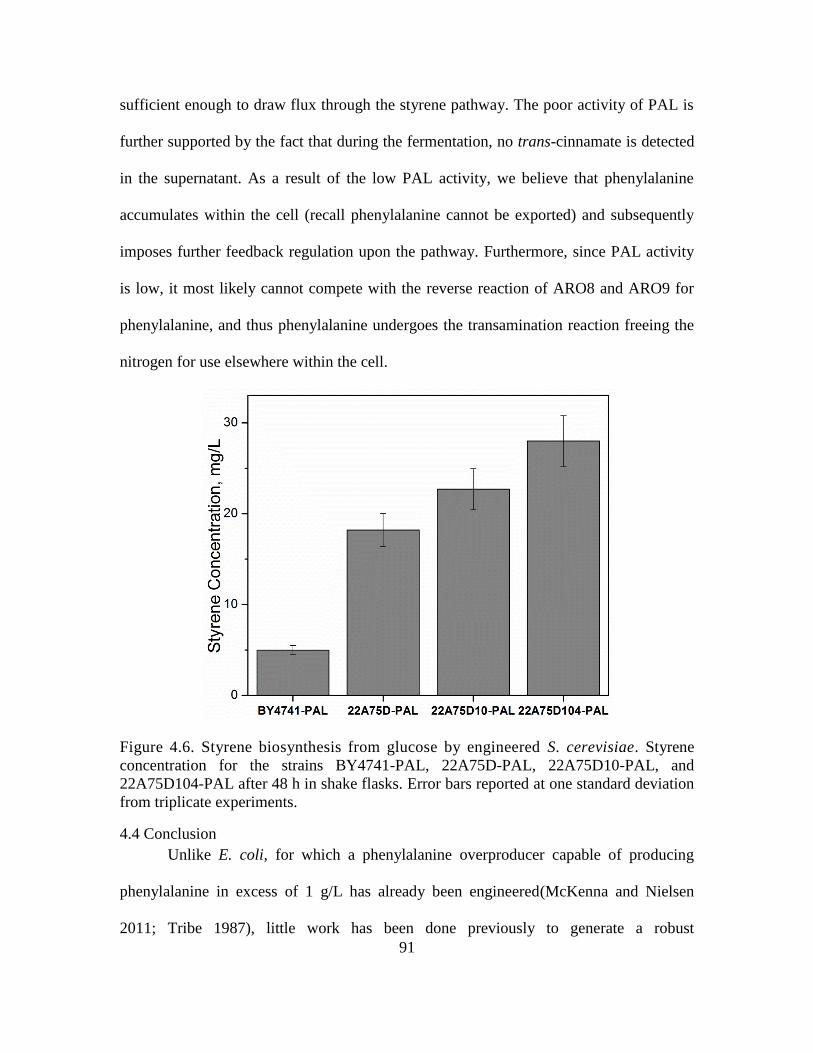
91
sufficient enough to draw flux through the styrene pathway. The poor activity of PAL is
further supported by the fact that during the fermentation, no trans-cinnamate is detected
in the supernatant. As a result of the low PAL activity, we believe that phenylalanine
accumulates within the cell (recall phenylalanine cannot be exported) and subsequently
imposes further feedback regulation upon the pathway. Furthermore, since PAL activity
is low, it most likely cannot compete with the reverse reaction of ARO8 and ARO9 for
phenylalanine, and thus phenylalanine undergoes the transamination reaction freeing the
nitrogen for use elsewhere within the cell.
Figure 4.6. Styrene biosynthesis from glucose by engineered S. cerevisiae. Styrene
concentration for the strains BY4741-PAL, 22A75D-PAL, 22A75D10-PAL, and
22A75D104-PAL after 48 h in shake flasks. Error bars reported at one standard deviation
from triplicate experiments.
4.4 Conclusion
Unlike E. coli, for which a phenylalanine overproducer capable of producing
phenylalanine in excess of 1 g/L has already been engineered(McKenna and Nielsen
2011; Tribe 1987), little work has been done previously to generate a robust

92
phenylalanine overproducer in S. cerevisiae. Through systematic strain and pathway
engineering, the biosynthetic production of styrene from glucose by S. cerevisiae has
been demonstrated for the first time. Although limited PAL activity as well as precursor
availability of the pathway precursor phenylalanine limits the overall productivity and
remains a challenge, continued improvements on engineering a more robust
phenylalanine overproducer as well as enhancing PAL activity will lead to greater yields
and a more sustainable approach for styrene biosynthesis.

93
CHAPTER 5
DISCUSSION AND FUTURE WORK
Abstract
The styrene pathway has successfully been engineered in both E. coli and S.
cerevisiae. Furthermore, the styrene pathway has been extended for production of the
styrene derivatives (S)-styrene oxide and (R)-1,2-phenylethanediol in engineered E. coli.
Currently, however, product toxicity, poor precursor availability, and poor PAL activity
remain a challenge. This chapter discusses those weaknesses of the current processes and
suggests potential future directions.
5.1 Introduction
These works have demonstrated the possibility of using microorganisms for the
production of styrene, a plastics monomer commercially produced from petroleum
derivatives. This renewable approach has created a ‘green’ process for not only styrene
but has also opened the door for the biosynthetic production of the styrene derivatives
(S)-styrene oxide and (R)-1,2-phenylethanediol by extension of the styrene pathway. In
our endeavors to maximize both titers and yields of styrene and its derivatives, we have
pushed the boundaries of our E. coli host and are limited by product toxicity and/or
precursor availability. Though we have demonstrated that the yeast S. cerevisiae may be
a more robust, solvent tolerant host, precursor availability remains a limiting factor. Thus,
in order to further improve upon the achievable titers and yields as well as establish a
robust, economical process for styrene production we must continue to explore routes for

94
process improvements, including i) relieving or circumventing product toxicity, ii)
enhancing precursor availability, and iii) improving recombinant PAL activity.
5.2 In situ product removal as a method to overcome toxicity
Due to the hydrophobic nature of styrene and its derivatives, it is believed that
product accumulation occurs within the lipid bi-layer of the cell membrane resulting in
reduced membrane fluidity and integrity. According to the model of Sikkema et al., there
exists a linear relationship between a compounds hydrophobicity, as measured by its
octanol-water partition coefficient (log KOW), and its propensity to accumulate in the
membrane lipid bi-layer of an organism, as measured by the membrane-water partition
coefficient (log KMW)(Sikkema, de Bont, and Poolman 1994). In order to understand the
toxic effects of a particular solvent, we have previously demonstrated that the toxicity
limit may be estimated by adding the compound directly to the culture medium and
monitoring the growth response. When the concentration of a product reaches a threshold
at which the cells can no longer proliferate, we designate this threshold as the toxicity
limit. When we compared the toxicity limit of styrene and its derivatives with their
predicted membrane-water partition coefficient (KM/W), we found that there was a strong
linear correlation (Fig 3.2) providing further evidence to support the hypothesis that the
intra-membrane accumulation of hydrophobic entities is the cause of reduced host fitness.
Table 5.1: Toxicity Limit Analysis of Styrene and its Derivatives
Product (Log KOW)
Host Highest Titer Maximum
Toxicity
Threshold
% of Toxicity
Limit
Achieved
Styrene (3.05)
E. coli 260 ± 4.3mg/L 300 mg/L 87%
S. cerevisiae 28.8 ± 2.1 mg/L 600 mg/L 5%
(S)-Styrene Oxide (1.61)
E. coli 1.32 ± 0.03 g/L 1.6 g/L 83%
(R)-1,2-Phenylethanediol (0.92)
E. coli 1.23 ± 0.07 g/L 8 g/L 15%

95
Product toxicity remains the greatest hindrance in achieving a high titer
biosynthetic process for styrene production. In E. coli, styrene titers are currently limited
to 260±4.3 mg/L, approximately 87% of the estimated toxicity limit of ~300 mg/L. It is
important to note however, that it is unlikely that a product could ever truly reach the
toxicity limit, rather in actuality, there must exist a balance between host fitness and the
metabolic burden associated with expressing a heterologous pathway. Similar to styrene,
the (S)-styrene oxide titers also approach the toxicity limit achieving 1.32 ± 0.03 g/L,
approximately 83% of the estimated toxicity limit of 1.6 g/L. In the case of (S)-styrene
oxide however, precursor availability of phenylalanine was also a limiting factor. In an
attempt to overcome our toxicity obstacle, a new host, S. cerevisiae, was evaluated for its
potential to produce styrene. Though the toxicity limit of S. cerevisiae was twice higher
(~600 mg/L), other factors including poor precursor availability and low PAL activity
limited the final titer to 28.8 mg/L, 5% of the toxicity limit. While finding a more
solvent-tolerant host and improving its ability to produce styrene remains a goal, a more
attractive option to addressing product toxicity may be in situ product removal (ISPR).
Not only would ISPR circumvent the toxicity issue, it would also, by nature,
recover the compound of interest. Some examples of ISPR approaches include liquid-
liquid extraction (or solvent extraction)(Gyamerah and Glover 1996; Malinowski 2001;
Weilnhammer and Blass 1994), adsorption(Nielsen, Amarasiriwardena, and Prather
2010; Nielsen and Prather 2009), gas and vacuum stripping(Loser et al. 2005), and
membrane pervaporation(Vane 2005). These approaches have been successfully applied
for the continuous recovery of many chemical compounds including ethanol as well as
the aromatics L-phenylacetylcarbinol(Khan and Daugulis 2010) and benzaldehyde(Jain,

96
Khan, and Daugulis 2010). Although the hydrophobic and volatile nature of styrene
proved to negatively limit titers due to the associated toxicity, these qualities conversely
make styrene amenable to ISPR.
Taking advantage of styrene’s volatility, we first attempted to utilize gas stripping
coupled to a cold trap for retention of styrene in a bioreactor model. The outlet gas of the
bioreactor was monitored for the concentration of stripped styrene by taking 100 µL gas
samples from the effluent using a Hamilton gas tight syringe (Hamilton, Reno NV) for
subsequent analysis by gas chromatography (GC). GC analysis of styrene gas was
performed on a Hewlett Packard 5890 Series II gas chromatograph with a flame ionizing
detector and Agilent DB-5 (30 m × 0.25 mm id) fused-silica capillary column using
helium as the carrier gas. The injector, column, and detector temperatures were set at 250
°C.
The accumulated concentration of styrene that was produced by the culture after
42 h was determined from the gas samples to be ~464 mg/L, refer to Fig. 5.1 for time
course production of styrene. While gas stripping proved effective for removing styrene
from the culture medium and increasing the yield by almost two-fold, our lab-scale cold-
trap was ill-equipped for retaining stripped styrene. Due to the quick rate of loss of
styrene during gas stripping, other approaches such as solvent extraction (via overlays) or
pervaporation are most likely the best option for styrene removal and recovery.
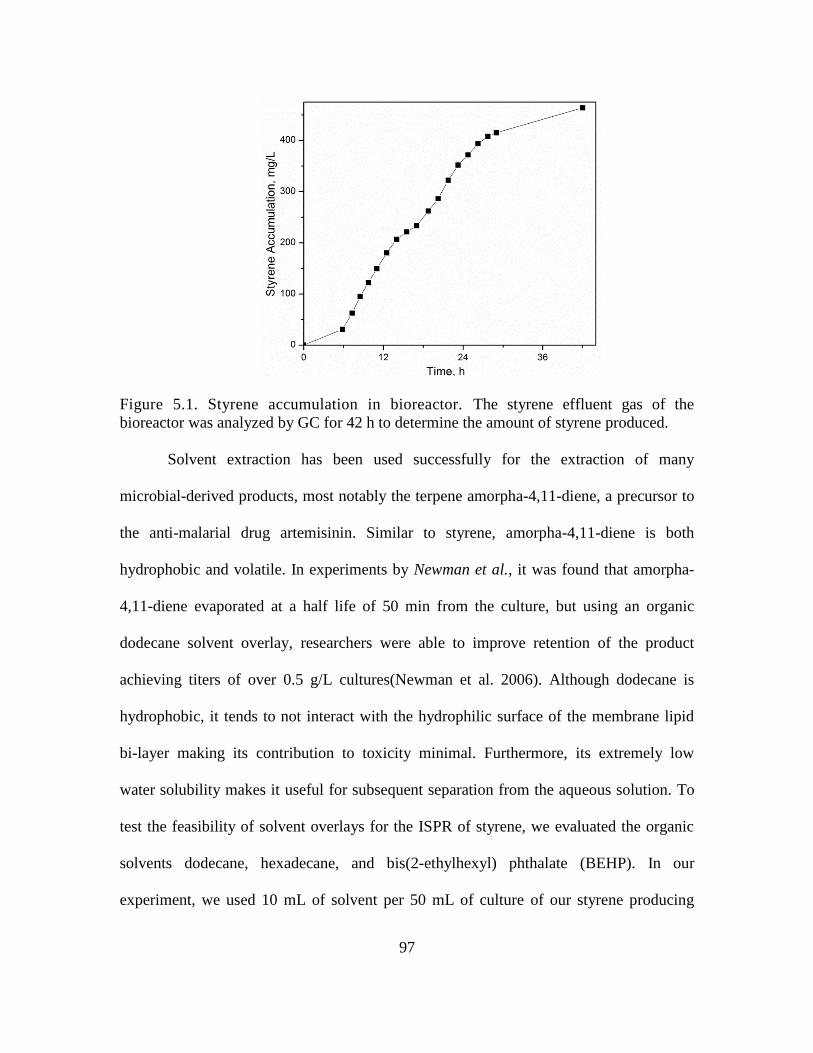
97
Figure 5.1. Styrene accumulation in bioreactor. The styrene effluent gas of the
bioreactor was analyzed by GC for 42 h to determine the amount of styrene produced.
Solvent extraction has been used successfully for the extraction of many
microbial-derived products, most notably the terpene amorpha-4,11-diene, a precursor to
the anti-malarial drug artemisinin. Similar to styrene, amorpha-4,11-diene is both
hydrophobic and volatile. In experiments by Newman et al., it was found that amorpha-
4,11-diene evaporated at a half life of 50 min from the culture, but using an organic
dodecane solvent overlay, researchers were able to improve retention of the product
achieving titers of over 0.5 g/L cultures(Newman et al. 2006). Although dodecane is
hydrophobic, it tends to not interact with the hydrophilic surface of the membrane lipid
bi-layer making its contribution to toxicity minimal. Furthermore, its extremely low
water solubility makes it useful for subsequent separation from the aqueous solution. To
test the feasibility of solvent overlays for the ISPR of styrene, we evaluated the organic
solvents dodecane, hexadecane, and bis(2-ethylhexyl) phthalate (BEHP). In our
experiment, we used 10 mL of solvent per 50 mL of culture of our styrene producing
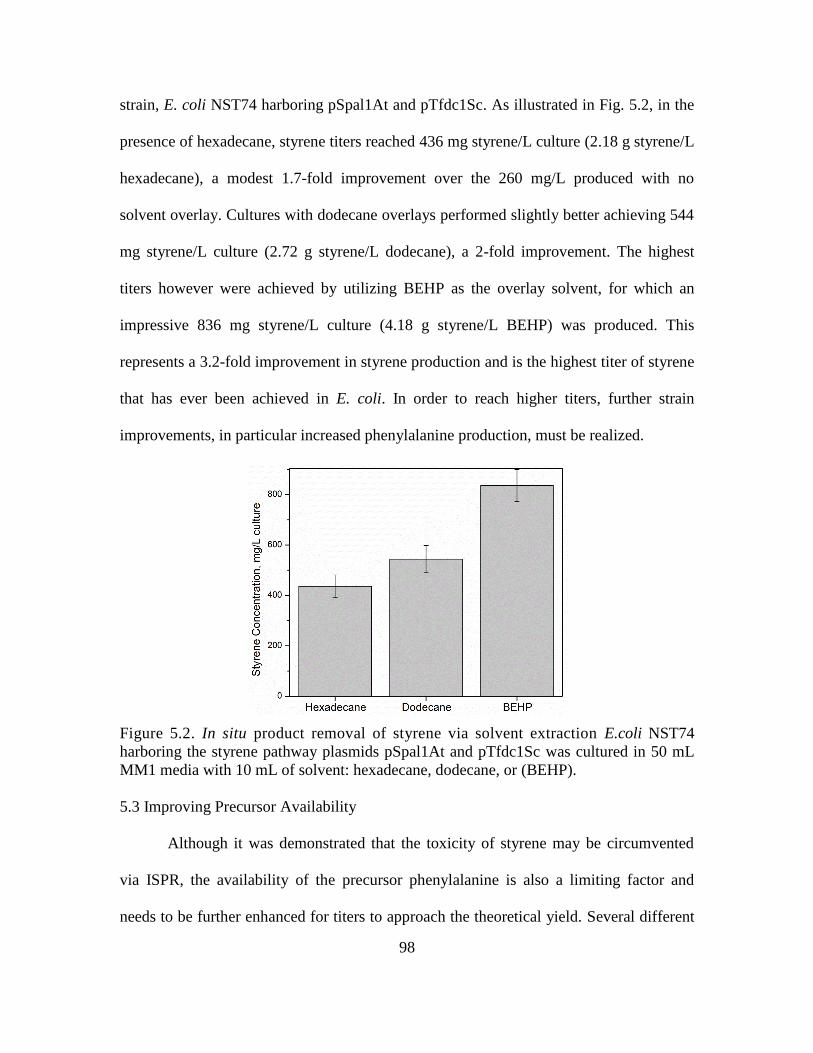
98
strain, E. coli NST74 harboring pSpal1At and pTfdc1Sc. As illustrated in Fig. 5.2, in the
presence of hexadecane, styrene titers reached 436 mg styrene/L culture (2.18 g styrene/L
hexadecane), a modest 1.7-fold improvement over the 260 mg/L produced with no
solvent overlay. Cultures with dodecane overlays performed slightly better achieving 544
mg styrene/L culture (2.72 g styrene/L dodecane), a 2-fold improvement. The highest
titers however were achieved by utilizing BEHP as the overlay solvent, for which an
impressive 836 mg styrene/L culture (4.18 g styrene/L BEHP) was produced. This
represents a 3.2-fold improvement in styrene production and is the highest titer of styrene
that has ever been achieved in E. coli. In order to reach higher titers, further strain
improvements, in particular increased phenylalanine production, must be realized.
Figure 5.2. In situ product removal of styrene via solvent extraction E.coli NST74
harboring the styrene pathway plasmids pSpal1At and pTfdc1Sc was cultured in 50 mL
MM1 media with 10 mL of solvent: hexadecane, dodecane, or (BEHP).
5.3 Improving Precursor Availability
Although it was demonstrated that the toxicity of styrene may be circumvented
via ISPR, the availability of the precursor phenylalanine is also a limiting factor and
needs to be further enhanced for titers to approach the theoretical yield. Several different

99
theoretical yields have been previously calculated from known stoichiometry of
phenylalanine biosynthesis from glucose based on different metabolic scenarios(Baez-
Viveros et al. 2004; Takagi et al. 1996; Patnaik and Liao 1994; Förberg, Eliaeson, and
Häggström 1988). In wild type E. coli, the phosphotransferase system (PTS) imports
glucose and converts one mole of phosphoenolpyruvate (PEP) to one mole of pyruvate
(PYR). If the competing aromatic amino acid pathways for tyrosine and tryptophan are
knocked out and there is no feed-back inhibition imposed upon the phenylalanine
pathway, the maximum theoretical yield of phenylalanine is 0.275 g/g (assuming carbon
dioxide is produced at a rate of 3.33 mol/mol glucose)(Förberg, Eliaeson, and Häggström
1988). If, however, E. coli is engineered to recycle PYR to PEP, resulting in less carbon
being lost as carbon dioxide (2.62 mol/mol glucose), the yield is increased to 0.34 g/g
(Förberg, Eliaeson, and Häggström 1988). Recycling PYR requires the expression of the
PEP synthetase (PpsA) which is not typically induced by growth on glucose. The
maximum PYR able to be recycled was determined by the break point when the TCA
(citric acid) cycle balanced NADPH. The maximum theoretical yield possible would be if
all PYR is recycled to PEP or the PTS was inactivated resulting in a yield of 0.55
g/g(Förberg, Eliaeson, and Häggström 1988). This theoretical yield is highly improbable
to ever experimentally reach though since it assumes no carbon enters the TCA cycle
which would severely hinder cell viability. Current experimental yields from this project
have yet to reach the most probable theoretical yield of 0.275 g/g, though some strain
improvements have proven effective in working toward that goal.

100
Table 5.2. Summary of titers and yields for styrene and its derivatives
Product Host Highest Titer Max Yield
(styrene/glucose)
% of
Theoretical
Yield1
Styrene
E. coli
NST74 260 ± 4.3mg/L, 25.0 ± 0.04 mM
0.03 ± 0.005 g/g, 0.05 ± 0.009 mol/mol
17%
S. cerevisiae
22A75D 28.8 ± 2.1 mg/L, 0.28 ± 0.02 mM
0.0014 ± .0001 g/g 0.0024 ± .0001 mol/mol
-
(S)-Styrene
Oxide E. coli
NST74ΔtyrA 1.32 ± 0.03 g/L, 11.0 ± 0.25 mM
0.12 ± 0.005 g/g, 0.17 ± 0.01 mol/mol
60%
(R)-1,2-
Phenylethanediol E. coli
NST74ΔtyrA 1.23 ± 0.07 g/L, 8.9 ± 0.51 mM
0.11 ± 0.002 g/g, 0.14 ± 0.01 mol/mol
47%
1 Assumes a maximum yield of 0.275 g-phenylalanine/g-glucose
Through strain development, we have been able to achieve as high as 60% of the
theoretical yield. In Chapter 3, it was shown that (S)-styrene oxide titers were raised in E.
coli NST74 from 1.1 g/L to 1.32 g/L (resulting in a yield of 0.12 g/g, Table 5.2) by
deleting the competing tyrosine pathway. While effective, the drawback of this approach
is that tyrosine must now be supplemented to the culture medium. In addition to deleting
the competing tyrosine pathway, it was also shown that enhancing the production of the
phenylalanine precursor erythrose-4-phosphate, via overexpression of the transketolase
tktA, had a significant impact on improving phenylalanine titers achieving ~1.55 g/L, a
40% increase. However, when tktA was overexpressed in combination with the
heterologous pathway, an improvement in product titers was not observed, presumable
due to increased metabolic burden. Single gene mutations, deletions, or overexpression of
pathway genes, in efforts to drive more flux through the pathway, are not only time
consuming but often result in minimal yield improvements.
In order to have a more global impact on the phenylalanine pathway, methods
beyond single gene alterations needs to be considered. For example, other approaches
may include media optimization or modifying the sugar transport system. For example,

101
lycopene titers were increased 7-fold in E. coli by i) removing the PTS to increase PEP
availability, ii) feeding glycerol, a non-PTS sugar, and iii) optimizing media components
via statistical design of experiments(Zhang et al. 2013). While modifying the PTS has
shown to be effective in E.coli, yeast rely on facilitated diffusion for glucose import so
conservation of PEP is not a concern(Lagunas 1993).
As an alternative approach, procedures such as global Transcription Machinery
Engineering (gTME)(Alper et al. 2006; Nicolaou, Gaida, and Papoutsakis 2010) may be
implemented to increase phenylalanine titers as well as perhaps improve styrene
tolerance in our hosts E. coli and S. cerevisiae. The principals of gTME have been
successfully employed to enhance ethanol tolerance in both yeast(Alper et al. 2006) and
E. coli (Alper and Stephanopoulos 2007). Furthermore, gTME was also applied in S.
cerevisiae to increase ethanol tolerance through mutagenesis of the Spt15p transcription
factor encoding the TATA-binding protein resulting in a 15% increase in ethanol
yield(Alper et al. 2006). Furthermore, in E. coli gTME was used to mutate RpoD,
encoding sigma factor σ70
, to increase ethanol tolerance as well as increase lycopene
production in parallel(Alper and Stephanopoulos 2007). In theory, gTME could be
applied to RpoD as well as the sigma factor responsible stationary phase activity, σ38
encoded by RpoS, in hope that phenylalanine titers would increase during both the
exponential and stationary phases (when synthesis is typically stagnant) given a high-
throughput selection method was used.
Investigating strategies for yeast specifically, there are several strategies that can
be made to the aromatic amino acid pathway in S. cerevisiae to further increase
phenylalanine titers. First, the transketolase TKL1 may be overexpressed to increase

102
production of E-4P. This may be especially advantageous since TKL1 is believed to be
feedback regulated by hydroxyphenylpyruvate, the precursor to tyrosine. Second, the
glucose-6-phosphate dehydrogenase, ZWF1, may be knocked out to force entry into the
pentose phosphate pathway via E-4P. When these two modifications were combined for
muconic acid production, their synergistic effect resulted in a two-fold increase in titers
(Curran et al. 2013). In addition to increasing E-4P precursor availability, further
deregulation of the phenylalanine pathway would likely increase flux. For example, the
tyrosine sensitive chorismate mutase ARO7 may be de-regulated via single point
mutation ARO7G141S
(Curran et al. 2013); however, this is likely to increase titers of both
phenylalanine as well as tyrosine.
It is possible based on the results of increasing phenylalanine in E. coli presented
in Chapter 3 (Fig. 3.5) that deletion of TYR1 to create a tyrosine auxotroph may direct
flux to phenylalanine. This approach has not yet been investigated in S. cerevisiae to
enhance phenylalanine concentrations, but the TYR1 knockout is available from Thermo
Scientific (Strain 852464). However, since this strategy would require supplementation of
exogenous tyrosine to the culture media at relatively high concentrations, it is likely that
tyrosine would impose feedback regulation upstream in the pathway. Alternatively, it
may be more optimal to use a gene knock-down approach which has been previously
shown to be possible in yeast with as little as 21 bp hairpin structured and silence genes
through translational repression which has been shown to be capable of reducing activity
up to 60%(Elbashir, Lendeckel, and Tuschl 2001; McManus et al. 2002).

103
5.4 Improving PAL Activity
PAL activity was shown to be the rate limiting step for the styrene pathway in
both hosts, E. coli and S. cerevisiae. Strategies to improve enzyme expression include
tuning the ribosomal binding site (RBS) and translation initiation region, using a stronger
promoter (such as trc or T7), increasing the copy number of the protein expression
system, changing the transcriptional promoter (constitutive versus inducible), or changing
the concentration of the inducer. Previous attempts at improving PAL activity in E. coli
involved using a stronger promoter (trc versus lac) and a plasmid with a higher copy
number (20 verses 10 copies/cell); although, this resulted in minimal increase of activity
(data not shown). With the exception of tuning the RBS, the aforementioned strategies
specifically address transcriptional improvements, as opposed to translational
improvements. One of the key problems associated with expressing heterologous
enzymes in engineered hosts is that the codon usage is foreign and generally not optimal
for high level expression. Not only could improper codon usage create a potential
bottleneck, it may also unnecessarily increase the metabolic burden on the host. As a
result of the redundancy of the genetic code, each organism demonstrates preferences for
certain codon usage which is dependent on the relative abundance and diversity of tRNA
within the host(Jana and Deb 2005). Codon optimization offers the chance to alter the
codon usage of a protein without altering the protein's sequence. Not only can codon
optimization tune the tRNA usage and improve protein translation, it can also be used to
optimize the GC content as well as remove unwanted RNA secondary
structures(Gustafsson, Govindarajan, and Minshull 2004; Wu et al. 2004). Codon
optimization has proven useful for increasing product titers, for example mevalonate in E.

104
coli(Anthony et al. 2009) and muconic acid in S. cerevisiae(Curran et al. 2013). While
PAL activity remains an obstacle, particularly in S. cerevisiae, codon optimization, as
well as other previously mentioned techniques, may prove effective in improving titers.
While the previously suggested enzyme activity improvement techniques rely on
improving PAL2 from A. thaliana, it is possible that this specific isoenzyme may not
have the highest activity in all hosts. While it was previously demonstrated that PAL2
had the highest recombinant activity in E. coli, the pool of candidate enzymes was small
consisting of only four isozymes (Chapter 2). In future works, it is suggested that
alternative PAL isoenzymes be tested for their possible greater PAL activity. Examples
of other PAL plant sources include Nicotiana tabacum(O'Neal and Keller 1970;
Fukasawa-Akada, Kung, and Watson 1996), Bambusa oldhamii(Hsieh et al. 2011; Hsieh
et al. 2010), Lycoris radiata(Jiang et al. 2011), Salvia Miltiorrhiza(HU et al. 2009),
Cynara cardunculus(De Paolis et al. 2008; Pandino et al. 2011), and Daucus carota(De
Lorenzo et al. 1987), to name a few.
5.5 Conclusion
These works constitute a 'proof of concept' that it is possible to engineer
microorganisms with the pathways necessary to replace chemical products of
petrochemical origin with those biosynthesized from renewable resources. Though these
processes are not currently at an economical scale, there is immense opportunity for
future improvement. Through in situ product removal, enhanced precursor availability,
and increased enzyme activity there is potential for increased titers that may one day be
able to compete with the petroleum industry.

105
REFERENCES
Adkins, J., S. Pugh, R. McKenna, and D. R. Nielsen. 2012. Engineering microbial
chemical factories to produce renewable "biomonomers". Front Microbiol 3:313.
Alberti, Simon, Aaron D. Gitler, and Susan Lindquist. 2007. A suite of Gateway®
cloning vectors for high-throughput genetic analysis in Saccharomyces cerevisiae.
Yeast 24 (10):913-919.
Alper, H., J. Moxley, E. Nevoigt, G. R. Fink, and G. Stephanopoulos. 2006. Engineering
yeast transcription machinery for improved ethanol tolerance and production.
Science 314 (5805):1565-1568.
Alper, H., and G. Stephanopoulos. 2007. Global transcription machinery engineering: A
new approach for improving cellular phenotype. Metabolic Engineering 9
(3):258-267.
Anthony, Jennifer R, Larry C Anthony, Farnaz Nowroozi, Gina Kwon, Jack D Newman,
and Jay D Keasling. 2009. Optimization of the mevalonate-based isoprenoid
biosynthetic pathway in Escherichia coli for production of the anti-malarial drug
precursor amorpha-4, 11-diene. Metabolic Engineering 11 (1):13-19.
Archelas, A., and R. Furstoss. 1997. Synthesis of enantiopure epoxides through
biocatalytic approaches. Annual Review of Microbiology 51:491-525.
Atsumi, S., T. Hanai, and J. C. Liao. 2008. Non-fermentative pathways for synthesis of
branched-chain higher alcohols as biofuels. Nature 451 (7174):86-9.
Baba, T., T. Ara, M. Hasegawa, Y. Takai, Y. Okumura, M. Baba, K. A. Datsenko, M.
Tomita, B. L. Wanner, and H. Mori. 2006. Construction of Escherichia coli K-12
in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2:2006
0008.
Babushok, V. I., P. J. Linstrom, J. J. Reed, I. G. Zenkevich, R. L. Brown, W. G. Mallard,
and S. E. Stein. 2007. Development of a database of gas chromatographic
retention properties of organic compounds. Journal of Chromatography A 1157
(1-2):414-421.
Baez-Viveros, J. L., J. Osuna, G. Hernandez-Chavez, X. Soberon, F. Bolivar, and G.
Gosset. 2004. Metabolic engineering and protein directed evolution increase the
yield of L-phenylalanine synthesized from glucose in Escherichia coli.
Biotechnology and Bioengineering 87 (4):516-524.

106
Bonomo, J., T. Warnecke, P. Hume, A. Marizcurrena, and R. T. Gill. 2006. A
comparative study of metabolic engineering anti-metabolite tolerance in
Escherichia coli. Metabolic Engineering 8 (3):227-239.
Boudet, A. M. 2007. Evolution and current status of research in phenolic compounds.
Phytochemistry 68 (22-24):2722-2735.
Boudet, Alain-Michel. 2007. Evolution and current status of research in phenolic
compounds. Phytochemistry 68 (22–24):2722-2735.
Bowles, L. K., and W. L. Ellefson. 1985. Effects of Butanol on Clostridium-
Acetobutylicum. Applied and Environmental Microbiology 50 (5):1165-1170.
Brownlee, June, Panqing He, Graham R. Moran, and David H. T. Harrison. 2008. Two
Roads Diverged: The Structure of Hydroxymandelate Synthase from
Amycolatopsis orientalis in Complex with 4-Hydroxymandelate†,‡. Biochemistry
47 (7):2002-2013.
Cao, L., J. T. Lee, W. Chen, and T. K. Wood. 2006. Enantioconvergent production of
(R)-1-phenyl-1,2-ethanediol from styrene oxide by combining the Solanum
tuberosum and an evolved Agrobacterium radiobacter AD1 epoxide hydrolases.
Biotechnol Bioeng 94 (3):522-529.
Caruso, Francesco, Joseph Tanski, Adriel Villegas-Estrada, and Miriam Rossi. 2004.
Structural Basis for Antioxidant Activity of trans-Resveratrol: Ab Initio
Calculations and Crystal and Molecular Structure. Journal of Agricultural and
Food Chemistry 52 (24):7279-7285.
Caspi, Ron, Hartmut Foerster, Carol A. Fulcher, Rebecca Hopkinson, John Ingraham,
Pallavi Kaipa, Markus Krummenacker, Suzanne Paley, John Pick, Seung Y. Rhee,
Christophe Tissier, Peifen Zhang, and Peter D. Karp. 2006. MetaCyc: a
multiorganism database of metabolic pathways and enzymes. Nucleic Acids
Research 34 (suppl 1):D511-D516.
Clark, GS. 1990. Phenethyl alcohol. Perfum. Flavor 15:37-44.
Clausen, Monika, Christopher J. Lamb, Roland Megnet, and Peter W. Doerner. 1994.
PAD1 encodes phenylacrylic acid decarboxylase which confers resistance to
cinnamic acid in Saccharomyces cerevisiae. Gene 142 (1):107-112.
Cochrane, Fiona C., Laurence B. Davin, and Norman G. Lewis. 2004. The Arabidopsis
phenylalanine ammonia lyase gene family: kinetic characterization of the four
PAL isoforms. Phytochemistry 65 (11):1557-1564.

107
Cui, J. D., S. R. Jia, and A. Y. Sun. 2008. Influence of amino acids, organic solvents and
surfactants for phenylalanine ammonia lyase activity in recombinant Escherichia
coli. Letters in Applied Microbiology 46 (6):631-635.
Curran, K. A., and H. S. Alper. 2012. Expanding the chemical palate of cells by
combining systems biology and metabolic engineering. Metab Eng 14 (4):289-97.
Curran, Kathleen A., John M. Leavitt, Ashty S. Karim, and Hal S. Alper. 2013. Metabolic
engineering of muconic acid production in Saccharomyces cerevisiae. Metabolic
Engineering 15 (0):55-66.
Datsenko, K. A., and B. L. Wanner. 2000. One-step inactivation of chromosomal genes
in Escherichia coli K-12 using PCR products. Proceedings of the National
Academy of Sciences of the United States of America 97 (12):6640-6645.
De Carvalho, C. C. C. R., A. A. R. L. Da Cruz, M. N. Pons, H. M. R. V. Pinheiro, J. M.
S. Cabral, M. M. R. Da Fonseca, B. S. Ferreira, and P. Fernandes. 2004.
Mycobacterium sp., Rhodococcus erythropolis, and Pseudomonas putida behavior
in the presence of organic solvents. Microscopy Research and Technique 64
(3):215-222.
De Lorenzo, Giulia, Alessandra Ranucci, Daniela Bellincampi, Giovanni Salvi, and
Felice Cervone. 1987. Elicitation of phenylalanine ammonia-lyase in Daucus
carota by oligogalacturonides released from sodium polypectate by homogeneous
polygalacturonase. Plant Science 51 (2):147-150.
De Paolis, Angelo, Domenico Pignone, Anita Morgese, and Gabriella Sonnante. 2008.
Characterization and differential expression analysis of artichoke phenylalanine
ammonia‐lyase‐coding sequences. Physiologia plantarum 132 (1):33-43.
Demain, Arnold L., and Preeti Vaishnav. 2009. Production of recombinant proteins by
microbes and higher organisms. Biotechnology Advances 27 (3):297-306.
Dickinson, J. Richard, L. Eshantha J. Salgado, and Michael J. E. Hewlins. 2003. The
Catabolism of Amino Acids to Long Chain and Complex Alcohols in
Saccharomyces cerevisiae. Journal of Biological Chemistry 278 (10):8028-8034.
DoE. 2002. Steam System Opportunity Assessment for the Pulp and Paper, Chemical
Manufacturing, and Petroleum Refining Industries. edited by O. o. E. E. a. R. E.
U. S. D. o. Energy.
Drake, J.W. and Baltz, R.H. 1976. The Biochemistry of Mutagenesis. Annual Review of
Microbiology 45:11-37.

108
Draths, K. M., D. L. Pompliano, D. L. Conley, J. W. Frost, A. Berry, G. L. Disbrow, R. J.
Staversky, and J. C. Lievense. 1992. Biocatalytic synthesis of aromatics from D-
glucose: the role of transketolase. Journal of the American Chemical Society 114
(10):3956-3962.
Erickson, B., Nelson, and P. Winters. 2012. Perspective on opportunities in industrial
biotechnology in renewable chemicals. Biotechnol J 7 (2):176-85.
Etschmann, M., W. Bluemke, D. Sell, and J. Schrader. 2002. Biotechnological production
of 2-phenylethanol. Applied Microbiology and Biotechnology 59 (1):1-8.
Fernandez, X., L. Lizzani-Cuvelier, A. M. Loiseau, C. Perichet, C. Delbecque, and J. F.
Arnaudo. 2005. Chemical composition of the essential oils from Turkish and
Honduras Styrax. Flavour and Fragrance Journal 20 (1):70-73.
Fischer, C. R., D. Klein-Marcuschamer, and G. Stephanopoulos. 2008. Selection and
optimization of microbial hosts for biofuels production. Metabolic Engineering 10
(6):295-304.
Flores, N., J. Xiao, A. Berry, F. Bolivar, and F. Valle. 1996. Pathway engineering for the
production of aromatic compounds in Escherichia coli. Nat Biotechnol 14
(5):620-3.
Förberg, Cecilia, Thomas Eliaeson, and Lena Häggström. 1988. Correlation of theoretical
and experimental yields of phenylalanine from non-growing cells of a rec
Escherichia coli strain. Journal of Biotechnology 7 (4):319-331.
Fukasawa-Akada, Tomoko, Shain-dow Kung, and John C Watson. 1996. Phenylalanine
ammonia-lyase gene structure, expression, and evolution in Nicotiana. Plant
molecular biology 30 (4):711-722.
Fukuda, Kazuro, Makoto Watanabe, Kozo Asano, Kozo Ouchi, and Seigo Takasawa.
1991. A mutated ARO4 gene for feedback-resistant DAHP synthase which causes
both o-fluoro-dl-phenylalamine resistance and β-phenethyl-alcohol
overproduction in Saccharomyces cerevisiae. Current Genetics 20 (6):453-456.
Gamenara, D., and P. Dominguez de Maria. 2009. Candida spp. redox machineries: an
ample biocatalytic platform for practical applications and academic insights.
Biotechnol Adv 27 (3):278-85.
Gilbert, H. J., I. N. Clarke, R. K. Gibson, J. R. Stephenson, and M. Tully. 1985.
Molecular-Cloning of the Phenylalanine Ammonia-Lyase Gene from
Rhodosporidium-Toruloides in Escherichia-Coli K-12. Journal of Bacteriology
161 (1):314-320.

109
Gilbert, H. J., J. R. Stephenson, and M. Tully. 1983. Control of Synthesis of Functional
Messenger-Rna Coding for Phenylalanine Ammonia-Lyase from
Rhodosporidium-Toruloides. Journal of Bacteriology 153 (3):1147-1154.
Gilbert, H. J., and M. Tully. 1982. Synthesis and Degradation of Phenylalanine
Ammonia-Lyase of Rhodosporidium-Toruloides. Journal of Bacteriology 150
(2):498-505.
Gosset, G., J. Yong-Xiao, and A. Berry. 1996. A direct comparison of approaches for
increasing carbon flow to aromatic biosynthesis in Escherichia coli. J Ind
Microbiol 17 (1):47-52.
Groves, J. T., and R. S. Myers. 1983. Catalytic Asymmetric Epoxidations with Chiral
Iron Porphyrins. Journal of the American Chemical Society 105 (18):5791-5796.
Gustafsson, Claes, Sridhar Govindarajan, and Jeremy Minshull. 2004. Codon bias and
heterologous protein expression. Trends in Biotechnology 22 (7):346-353.
Gyamerah, M., and J. Glover. 1996. Production of ethanol by continuous fermentation
and liquid-liquid extraction. Journal of Chemical Technology and Biotechnology
66 (2):145-152.
Han, J. H., M. S. Park, J. W. Bae, E. Y. Lee, Y. J. Yoon, S. G. Lee, and S. Park. 2006.
Production of (S)-styrene oxide using styrene oxide isomerase negative mutant of
Pseudomonas putida SN1. Enzyme and Microbial Technology 39 (6):1264-1269.
Hansen, Esben H., Birger Lindberg Møller, Gertrud R. Kock, Camilla M. Bünner,
Charlotte Kristensen, Ole R. Jensen, Finn T. Okkels, Carl E. Olsen, Mohammed
S. Motawia, and Jørgen Hansen. 2009. De Novo Biosynthesis of Vanillin in
Fission Yeast (Schizosaccharomyces pombe) and Baker's Yeast (Saccharomyces
cerevisiae). Applied and Environmental Microbiology 75 (9):2765-2774.
Hazelwood, Lucie A., Jean-Marc Daran, Antonius J. A. van Maris, Jack T. Pronk, and J.
Richard Dickinson. 2008. The Ehrlich Pathway for Fusel Alcohol Production: a
Century of Research on Saccharomyces cerevisiae Metabolism. Applied and
Environmental Microbiology 74 (8):2259-2266.
Heipieper, H. J., F. J. Weber, J. Sikkema, H. Keweloh, and J. A. M. Debont. 1994.
Mechanisms of Resistance of Whole Cells to Toxic Organic-Solvents. Trends in
Biotechnology 12 (10):409-415.
Heipieper, Hermann J., Frans J. Weber, Jan Sikkema, Heribert Keweloh, and Jan A. M.
de Bont. 1994. Mechanisms of resistance of whole cells to toxic organic solvents.
Trends in Biotechnology 12 (10):409-415.

110
Hsieh, Lu-Sheng, Yi-Lin Hsieh, Chuan-Shan Yeh, Chieh-Yang Cheng, Chien-Chih
Yang, and Ping-Du Lee. 2011. Molecular characterization of a phenylalanine
ammonia-lyase gene (BoPAL1) from Bambusa oldhamii. Molecular biology
reports 38 (1):283-290.
Hsieh, Lu-Sheng, Guo-Jhang Ma, Chien-Chih Yang, and Ping-Du Lee. 2010. Cloning,
expression, site-directed mutagenesis and immunolocalization of phenylalanine
ammonia-lyase in Bambusa oldhamii. Phytochemistry 71 (17):1999-2009.
HU, Yong-Sheng, Lei ZHANG, Peng DI, and Wan-Sheng CHEN. 2009. Cloning and
Induction of Phenylalanine Ammonia-lyase Gene from Salvia miltiorrhiza and Its
Effect on Hydrophilic Phenolic Acids Levels. Chinese Journal of Natural
Medicines 7 (6):449-457.
Isken, S., and Jan A. M. de Bont. 1998. Bacteria tolerant to organic solvents.
Extremophiles 2 (3):229-238.
Jain, A. N., T. R. Khan, and A. J. Daugulis. 2010. Bioproduction of benzaldehyde in a
solid-liquid two-phase partitioning bioreactor using Pichia pastoris. Biotechnology
Letters 32 (11):1649-1654.
James, D.H., and W.M. Castor. 2011. Styrene. In Ullmann's Encyclopedia of Industrial
Chemistry: Wiley-VCH.
Jana, S, and JK Deb. 2005. Strategies for efficient production of heterologous proteins in
Escherichia coli. Applied Microbiology and Biotechnology 67 (3):289-298.
Jiang, Hanxiao, Karl V. Wood, and John A. Morgan. 2005. Metabolic Engineering of the
Phenylpropanoid Pathway in Saccharomyces cerevisiae. Applied and
Environmental Microbiology 71 (6):2962-2969.
Jiang, Hanxiao, Karl V. Wood, and John A. Morgan. 2005. Metabolic Engineering of the
Phenylpropanoid Pathway in Saccharomyces cerevisiae. Appl. Environ.
Microbiol. 71 (6):2962-2969.
Jiang, Yumei, Nan Xia, Xiaodan Li, Wenbiao Shen, Lijian Liang, Chunyan Wang, Ren
Wang, Feng Peng, and Bing Xia. 2011. Molecular cloning and characterization of
a phenylalanine ammonia-lyase gene (LrPAL) from Lycoris radiata. Molecular
biology reports 38 (3):1935-1940.
Jones, D. Hugh. 1984. Phenylalanine ammonia-lyase: Regulation of its induction, and its
role in plant development. Phytochemistry 23 (7):1349-1359.
Juan L. Ramos, Estrella Duque, María-Trinidad Gallegos, Patricia Godoy, María Isabel
Ramos-González, Antonia Rojas, Wilson Terán, and Ana Segura. 2002.

111
Mechanisms of solvent tolerance in gram-negative bacteria. Annual Review of
Microbiology 56:743-768.
Juminaga, D., E. E. K. Baidoo, A. M. Redding-Johanson, T. S. Batth, H. Burd, A.
Mukhopadhyay, C. J. Petzold, and J. D. Keasling. 2012. Modular Engineering of
L-Tyrosine Production in Escherichia coli. Applied and Environmental
Microbiology 78 (1):89-98.
Kambourakis, Spiros, K. M. Draths, and J. W. Frost. 2000. Synthesis of Gallic Acid and
Pyrogallol from Glucose: Replacing Natural Product Isolation with Microbial
Catalysis. Journal of the American Chemical Society 122 (37):9042-9043.
Kawamoto, T., T. Kanda, and A. Tanaka. 2001. Preparation of an organic solvent-tolerant
strain from baker's yeast. Applied Microbiology and Biotechnology 55 (4):476-
479.
Keasling, Jay D. 2008. Synthetic Biology for Synthetic Chemistry. ACS Chemical
Biology 3 (1):64-76.
Keasling, Jay D. 2010. Manufacturing Molecules Through Metabolic Engineering.
Science 330 (6009):1355-1358.
Keseler, Ingrid M., Julio Collado-Vides, Socorro Gama-Castro, John Ingraham, Suzanne
Paley, Ian T. Paulsen, Martín Peralta-Gil, and Peter D. Karp. 2005. EcoCyc: a
comprehensive database resource for Escherichia coli. Nucleic Acids Research 33
(suppl 1):D334-D337.
Keweloh, H., H. J. Heipieper, and H. J. Rehm. 1989. Protection of Bacteria against
Toxicity of Phenol by Immobilization in Calcium Alginate. Applied Microbiology
and Biotechnology 31 (4):383-389.
Khan, T. R., and A. J. Daugulis. 2010. Application of Solid-Liquid TPPBs to the
Production of L-Phenylacetylcarbinol From Benzaldehyde Using Candida utilis.
Biotechnology and Bioengineering 107 (4):633-641.
King, R. B., J. Bakos, C.D. Hoff, and L. Marko. 1979. 1,2-Bis(diphenylphosphino)-1-
phenylethane: a chiral ditertiary phosphine derived from mandelic acid used as a
ligand in asymmetric homogenous hydrogenation catalysts. Journal of Organic
Chemistry 44:1729-1731.
Knoshaug, E. P., and M. Zhang. 2009. Butanol Tolerance in a Selection of
Microorganisms. Applied Biochemistry and Biotechnology 153 (1-2):13-20.

112
Kradolfer, P., J. Zeyer, G. Miozzari, and R. Huetter. 1977. Dominant regulatory mutants
in chorismate mutase of Saccharomyces cerevisiae. FEMS Microbiology Letters 2
(4):211-216.
Krieger, Cynthia J., Peifen Zhang, Lukas A. Mueller, Alfred Wang, Suzanne Paley,
Martha Arnaud, John Pick, Seung Y. Rhee, and Peter D. Karp. 2004. MetaCyc: a
multiorganism database of metabolic pathways and enzymes. Nucleic Acids
Research 32 (suppl 1):D438-D442.
Kumar, P., R. K. Upadhyay, and R. K. Pandey. 2004. Asymmetric dihydroxylation route
to (R)-isoprenaline, (R)-norfluoxetine and (R)-fluoxetine. Tetrahedron-
Asymmetry 15 (24):3955-3959.
Lagunas, Rosario. 1993. Sugar transport in Saccharomyces cerevisiae. FEMS
Microbiology Letters 104 (3):229-242.
Lan, Ethan I., Yasumasa Dekishima, Derrick S. Chuang, and James C. Liao. 2013.
Metabolic engineering of 2-pentanone synthesis in Escherichia coli. AIChE
Journal 59 (9):3167-3175.
Larsson, S., N. O. Nilvebrant, and L. J. Jonsson. 2001. Effect of overexpression of
Saccharomyces cerevisiae Pad1p on the resistance to phenylacrylic acids and
lignocellulose hydrolysates under aerobic and oxygen-limited conditions. Applied
Microbiology and Biotechnology 57 (1-2):167-74.
Lee, J. W., H. U. Kim, S. Choi, J. Yi, and S. Y. Lee. 2011. Microbial production of
building block chemicals and polymers. Curr Opin Biotechnol 22 (6):758-67.
Lee, K., and D. T. Gibson. 1996. Stereospecific dihydroxylation of the styrene vinyl
group by purified naphthalene dioxygenase from Pseudomonas sp. strain NCIB
9816-4. J Bacteriol 178 (11):3353-6.
Lee, K., and D. T. Gibson. 1996. Toluene and ethylbenzene oxidation by purified
naphthalene dioxygenase from Pseudomonas sp strain NCIB 9816-4. Applied and
Environmental Microbiology 62 (9):3101-3106.
Lindberg, Pia, Sungsoon Park, and Anastasios Melis. 2010. Engineering a platform for
photosynthetic isoprene production in cyanobacteria, using Synechocystis as the
model organism. Metabolic Engineering 12 (1):70-79.
Loprieno, N., A. Abbondandolo, R. Barale, S. Baroncelli, S. Bonatti, G. Bronzetti, A.
Cammellini, C. Corsi, G. Corti, D. Frezza, C. Leporini, A. Mazzaccaro, R. Nieri,
D. Rosellini, and A. M. Rossi. 1976. Mutagenicity of Industrial Compounds -
Styrene and Its Possible Metabolite Styrene Oxide. Mutation Research 40
(4):317-324.

113
Loser, C., A. Schroder, S. Deponte, and T. Bley. 2005. Balancing the ethanol formation
in continuous bioreactors with ethanol stripping. Engineering in Life Sciences 5
(4):325-332.
Lütke-Eversloh, Tina, and Gregory Stephanopoulos. 2008. Combinatorial pathway
analysis for improved L-tyrosine production in Escherichia coli: Identification of
enzymatic bottlenecks by systematic gene overexpression. Metabolic Engineering
10 (2):69-77.
Luttik, M. A. H., Z. Vuralhan, E. Suir, G. H. Braus, J. T. Pronk, and J. M. Daran. 2008.
Alleviation of feedback inhibition in Saccharomyces cerevisiae aromatic amino
acid biosynthesis: Quantification of metabolic impact. Metabolic Engineering 10
(3–4):141-153.
Malinowski, J. J. 2001. Two-phase partitioning bioreactors in fermentation technology.
Biotechnol Adv 19 (7):525-38.
Martorell, P., A. Querol, and M. T. Fernández-Espinar. 2005. Rapid Identification and
Enumeration of Saccharomyces cerevisiae Cells in Wine by Real-Time PCR.
Applied and Environmental Microbiology 71 (11):6823-6830.
Matsui, Ken, Shinya Teranishi, Shohei Kamon, Kouichi Kuroda, and Mitsuyoshi Ueda.
2008. Discovery of a Modified Transcription Factor Endowing Yeasts with
Organic-Solvent Tolerance and Reconstruction of an Organic-Solvent-Tolerant
Saccharomyces cerevisiae Strain. Applied and Environmental Microbiology 74
(13):4222-4225.
McKarns, S. C., C. Hansch, W. S. Caldwell, W. T. Morgan, S. K. Moore, and D. J.
Doolittle. 1997. Correlation between hydrophobicity of short-chain aliphatic
alcohols and their ability to alter plasma membrane integrity. Fundamental and
Applied Toxicology 36 (1):62-70.
McKenna, R., and D. R. Nielsen. 2011. Styrene biosynthesis from glucose by engineered
E. coli. Metabolic Engineering 13 (5):544-554.
McKenna, Rebekah, Shawn Pugh, Brian Thompson, and David R. Nielsen. 2013.
Microbial production of the aromatic building-blocks (S)-styrene oxide and (R)-
1,2-phenylethanediol from renewable resources. Biotechnology Journal 8
(12):1465-1475.
Mimura, Naoki, and Masahiro Saito. 2000. Dehydrogenation of ethylbenzene to styrene
over Fe2O3/Al2O3 catalysts in the presence of carbon dioxide. Catalysis Today
55 (1–2):173-178.

114
Min, J. Y., and E. Y. Lee. 2012. Biosynthesis of (R)-1,2-phenylethanediol and (R)-4-
chloro-1,2-phenylethanediol by using two recombinant cells expressing
enantiocomplementary epoxide hydrolases. Journal of Industrial and Engineering
Chemistry 18 (1):160-164.
Moffitt, M. C., G. V. Louie, M. E. Bowman, J. Pence, J. P. Noel, and B. S. Moore. 2007.
Discovery of two cyanobacterial phenylalanine ammonia lyases: Kinetic and
structural characterization. Biochemistry 46 (4):1004-1012.
Moffitt, Michelle C., Gordon V. Louie, Marianne E. Bowman, Janelle Pence, Joseph P.
Noel, and Bradley S. Moore. 2007. Discovery of Two Cyanobacterial
Phenylalanine Ammonia Lyases: Kinetic and Structural Characterization†,‡.
Biochemistry 46 (4):1004-1012.
Monterde, M. I., M. Lombard, A. Archelas, A. Cronin, M. Arand, and R. Furstoss. 2004.
Enzymatic transformations. Part 58: Enantioconvergent biohydrolysis of styrene
oxide derivatives catalysed by the Solanum tuberosum epoxide hydrolase.
Tetrahedron-Asymmetry 15 (18):2801-2805.
Mukai, N., K. Masaki, T. Fujii, M. Kawamukai, and H. Iefuji. 2010. PAD1 and FDC1 are
essential for the decarboxylation of phenylacrylic acids in Saccharomyces
cerevisiae. Journal of Bioscience and Bioengineering 109 (6):564-569.
Nevoigt, Elke. 2008. Progress in Metabolic Engineering of Saccharomyces cerevisiae.
Microbiology and Molecular Biology Reviews 72 (3):379-412.
Newman, Jack D., Jessica Marshall, Michelle Chang, Farnaz Nowroozi, Eric Paradise,
Douglas Pitera, Karyn L. Newman, and Jay D. Keasling. 2006. High-level
production of amorpha-4,11-diene in a two-phase partitioning bioreactor of
metabolically engineered Escherichia coli. Biotechnology and Bioengineering 95
(4):684-691.
Nicolaou, Sergios A., Stefan M. Gaida, and Eleftherios T. Papoutsakis. 2010. A
comparative view of metabolite and substrate stress and tolerance in microbial
bioprocessing: From biofuels and chemicals, to biocatalysis and bioremediation.
Metabolic Engineering 12 (4):307-331.
Nielsen, D. R., G. S. Amarasiriwardena, and K. L. J. Prather. 2010. Predicting the
adsorption of second generation biofuels by polymeric resins with applications for
in situ product recovery (ISPR). Bioresource Technology 101 (8):2762-2769.
Nielsen, D. R., E. Leonard, S. H. Yoon, H. C. Tseng, C. Yuan, and K. L. Prather. 2009.
Engineering alternative butanol production platforms in heterologous bacteria.
Metab Eng.

115
Nielsen, D. R., and K. J. Prather. 2009. In Situ Product Recovery of n-Butanol Using
Polymeric Resins. Biotechnology and Bioengineering 102 (3):811-821.
Nishio, T., A. Patel, Y. Wang, and P. C. K. Lau. 2001. Biotransformations catalyzed by
cloned p-cymene monooxygenase from Pseudomonas putida F1. Applied
Microbiology and Biotechnology 55 (3):321-325.
O'Neal, Denny, and CJ Keller. 1970. Partial purification and some properties of
phenylalanine ammonia-lyase of tobacco (Nicotiana tabacum). Phytochemistry 9
(7):1373-1383.
Osborne, S. J., J. Leaver, M. K. Turner, and P. Dunnill. 1990. Correlation of biocatalytic
activity in an organic-aqueous two-liquid phase system with solvent concentration
in the cell membrane. Enzyme Microb Technol 12 (4):281-91.
Ostergaard, Simon, Lisbeth Olsson, and Jens Nielsen. 2000. Metabolic Engineering of
Saccharomyces cerevisiae. Microbiology and Molecular Biology Reviews 64
(1):34-50.
Otto, K., K. Hofstetter, M. Rothlisberger, B. Witholt, and A. Schmid. 2004. Biochemical
characterization of StyAB from Pseudomonas sp strain VLB120 as a two-
component flavin-diffusible monooxygenase. Journal of Bacteriology 186
(16):5292-5302.
Pagot, Y., J. M. Belin, F. Husson, and H. E. Spinnler. 2007. Metabolism of phenylalanine
and biosynthesis of styrene in Penicillium camemberti. Journal of Dairy Research
74 (2):180-185.
Pandino, Gaetano, Sara Lombardo, Giovanni Mauromicale, and Gary Williamson. 2011.
Phenolic acids and flavonoids in leaf and floral stem of cultivated and wild
Cynara cardunculus L. genotypes. Food chemistry 126 (2):417-422.
Panke, S., B. Witholt, A. Schmid, and M. G. Wubbolts. 1998. Towards a biocatalyst for
(S)-styrene oxide production: Characterization of the styrene degradation pathway
of Pseudomonas sp. strain VLB120. Applied and Environmental Microbiology 64
(6):2032-2043.
Panke, S., M. G. Wubbolts, A. Schmid, and B. Witholt. 2000. Production of enantiopure
styrene oxide by recombinant Escherichia coli synthesizing a two-component
styrene monooxygenase. Biotechnology and Bioengineering 69 (1):91-100.
Panke, S., M. G. Wubbolts, A. Schmid, and B. Witholt. 2000. Production of enantiopure
styrene oxide by recombinant Escherichia coli synthesizing a two-component
styrene monooxygenase. Biotechnol Bioeng 69 (1):91-100.

116
Park, M. So, J. W. Bae, J. H. Han, E. Y. Lee, S. G. Lee, and S. Park. 2006.
Characterization of styrene catabolic genes of Pseudomonas putida SN1 and
construction of a recombinant Escherichia coli containing styrene monooxygenase
gene for the production of (S)-styrene oxide. Journal of Microbiology and
Biotechnology 16 (7):1032-1040.
Park, J. B., B. Buehler, T. Habicher, B. Hauer, S. Panke, B. Witholt, and A. Schmid.
2006. The efficiency of recombinant Escherichia coli as biocatalyst for
stereospecific epoxidation. Biotechnology and Bioengineering 95 (3):501-512.
Patnaik, R., and J. C. Liao. 1994. Engineering of Escherichia coli central metabolism for
aromatic metabolite production with near theoretical yield. Appl Environ
Microbiol 60 (11):3903-8.
Qi, W. W., T. Vannelli, S. Breinig, A. Ben-Bassat, A. A. Gatenby, S. L. Haynie, and F. S.
Sariaslani. 2007. Functional expression of prokaryotic and eukaryotic genes in
Escherichia coli for conversion of glucose to p-hydroxystyrene. Metab Eng 9
(3):268-76.
Qi, Wei Wei, Todd Vannelli, Sabine Breinig, Arie Ben-Bassat, Anthony A. Gatenby,
Sharon L. Haynie, and F. Sima Sariaslani. 2007. Functional expression of
prokaryotic and eukaryotic genes in Escherichia coli for conversion of glucose to
-hydroxystyrene. Metabolic Engineering 9 (3):268-276.
Qian, Zhi-Gang, Xiao-Xia Xia, and Sang Yup Lee. 2011. Metabolic engineering of
Escherichia coli for the production of cadaverine: A five carbon diamine.
Biotechnology and Bioengineering 108 (1):93-103.
Ramos, Juan L., Estrella Duque, María-Trinidad Gallegos, Patricia Godoy, María Isabel
Ramos-González, Antonia Rojas, Wilson Terán, and Ana Segura. 2002.
Mechanisms of solvent tolerance in gram-negative bacteria. Annual Review of
Microbiology 56 (1):743-768.
Reyes, Luis H., Maria P. Almario, and Katy C. Kao. 2011. Genomic Library Screens for
Genes Involved in n-Butanol Tolerance in Escherichia coli. PLoS ONE 6
(3):e17678.
Rodríguez, Héctor, Iván Angulo, Blanca de las Rivas, Nuria Campillo, Juan A. Páez,
Rosario Muñoz, and José M. Mancheño. 2010. p-Coumaric acid decarboxylase
from Lactobacillus plantarum: Structural insights into the active site and
decarboxylation catalytic mechanism. Proteins: Structure, Function, and
Bioinformatics 78 (7):1662-1676.
Rui, L., L. Cao, W. Chen, K. F. Reardon, and T. K. Wood. 2005. Protein engineering of
epoxide hydrolase from Agrobacterium radiobacter AD1 for enhanced activity

117
and enantioselective production of (R)-1-phenylethane-1,2-diol. Appl Environ
Microbiol 71 (7):3995-4003.
Santos, Christine Nicole S., Mattheos Koffas, and Gregory Stephanopoulos. 2011.
Optimization of a heterologous pathway for the production of flavonoids from
glucose. Metabolic Engineering In Press, Corrected Proof.
Sardessai, Y., and S. Bhosle. 2002. Tolerance of bacteria to organic solvents. Research in
Microbiology 153 (5):263-268.
Satoh, Yasuharu, Kenji Tajima, Masanobu Munekata, Jay D. Keasling, and Taek Soon
Lee. 2012. Engineering of a Tyrosol-Producing Pathway, Utilizing Simple Sugar
and the Central Metabolic Tyrosine, in Escherichia coli. Journal of Agricultural
and Food Chemistry 60 (4):979-984.
Schmidheini, T, P Sperisen, G Paravicini, R Hütter, and G Braus. 1989. A single point
mutation results in a constitutively activated and feedback-resistant chorismate
mutase of Saccharomyces cerevisiae. Journal of Bacteriology 171 (3):1245-1253.
Serre, Laurence, Alain Sailland, Denise Sy, Philippe Boudec, Anne Rolland, Eva Pebay-
Peyroula, and Claudine Cohen-Addad. 1999. Crystal structure of Pseudomonas
fluorescens 4-hydroxyphenylpyruvate dioxygenase: an enzyme involved in the
tyrosine degradation pathway. Structure 7 (8):977-988.
Sikkema, J., J. A. de Bont, and B. Poolman. 1994. Interactions of cyclic hydrocarbons
with biological membranes. J Biol Chem 269 (11):8022-8.
Smith, M. R. 1990. The biodegradation of aromatic hydrocarbons by bacteria.
Biodegradation 1 (2-3):191-206.
SRI. 2010. Styrene. Access Intelligence LLC Inc.
Stanier, R. Y., N. J. Palleroni, and M. Doudoroff. 1966. The aerobic pseudomonads: a
taxonomic study. J Gen Microbiol 43 (2):159-271.
Stark, D., T. Münch, B. Sonnleitner, I. W. Marison, and U. von Stockar. 2002. Extractive
Bioconversion of 2-Phenylethanol from l-Phenylalanine by
Saccharomycescerevisiae. Biotechnology Progress 18 (3):514-523.
Sudbery, Peter E. 1996. The expression of recombinant proteins in yeasts. Current
Opinion in Biotechnology 7 (5):517-524.
Takagi, Mutsumi, Yoshinori Nishio, Gyuseop Oh, and Toshiomi Yoshida. 1996. Control
of L-phenylalanine production by dual feeding of glucose and L-tyrosine.
Biotechnology and Bioengineering 52 (6):653-660.

118
Tatarko, M., and T. Romeo. 2001. Disruption of a global regulatory gene to enhance
central carbon flux into phenylalanine biosynthesis in Escherichia coli. Curr
Microbiol 43 (1):26-32.
Teshiba, Sadao, Rolf Furter, Peter Niederberger, Gerhard Braus, Gerhard Paravicini, and
Ralf Hütter. 1986. Cloning of the ARO3 gene of Saccharomyces cerevisiae and its
regulation. Molecular and General Genetics MGG 205 (2):353-357.
Tran, Ngoc Phuong, Jerome Gury, Veronique Dartois, Thi Kim Chi Nguyen, Helene
Seraut, Lise Barthelmebs, Patrick Gervais, and Jean-Francois Cavin. 2008.
Phenolic Acid-Mediated Regulation of the padC Gene, Encoding the Phenolic
Acid Decarboxylase of Bacillus subtilis. J. Bacteriol. 190 (9):3213-3224.
Tribe, D.E. 1987. Novel microorganism and method. US Patent 4,681,852.
Vane, L. M. 2005. A review of pervaporation for product recovery from biomass
fermentation processes. Journal of Chemical Technology and Biotechnology 80
(6):603-629.
Vannelli, T., Z. X. Xue, S. Breinig, W. W. Qi, and F. S. Sariaslani. 2007. Functional
expression in Escherichia coli of the tyrosine-inducible tyrosine ammonia-lyase
enzyme from yeast Trichosporon cutaneum for production of p-hydroxycinnamic
acid. Enzyme and Microbial Technology 41 (4):413-422.
Vannelli, Todd, Wei Wei Qi, James Sweigard, Anthony A. Gatenby, and F. Sima
Sariaslani. 2007. Production of p-hydroxycinnamic acid from glucose in
Saccharomyces cerevisiae and Escherichia coli by expression of heterologous
genes from plants and fungi. Metabolic Engineering 9 (2):142-151.
Verhoef, S., H. J. Ruijssenaars, J. A. de Bont, and J. Wery. 2007. Bioproduction of p-
hydroxybenzoate from renewable feedstock by solvent-tolerant Pseudomonas
putida S12. J Biotechnol 132 (1):49-56.
Verhoef, S., N. Wierckx, R. G. Westerhof, J. H. de Winde, and H. J. Ruijssenaars. 2009.
Bioproduction of p-hydroxystyrene from glucose by the solvent-tolerant
bacterium Pseudomonas putida S12 in a two-phase water-decanol fermentation.
Appl Environ Microbiol 75 (4):931-6.
Verhoef, Suzanne, Harald J. Ruijssenaars, Jan A. M. de Bont, and Jan Wery. 2007.
Bioproduction of p-hydroxybenzoate from renewable feedstock by solvent-
tolerant Pseudomonas putida S12. Journal of Biotechnology 132 (1):49-56.
Verhoef, Suzanne, Nick Wierckx, R. G. Maaike Westerhof, Johannes H. de Winde, and
Harald J. Ruijssenaars. 2009. Bioproduction of p-Hydroxystyrene from Glucose
by the Solvent-Tolerant Bacterium Pseudomonas putida S12 in a Two-Phase

119
Water-Decanol Fermentation. Applied and Environmental Microbiology 75
(4):931-936.
Weber, F J, L P Ooijkaas, R M Schemen, S Hartmans, and J A de Bont. 1993. Adaptation
of Pseudomonas putida S12 to high concentrations of styrene and other organic
solvents. Appl. Environ. Microbiol. 59 (10):3502-3504.
Weilnhammer, C., and E. Blass. 1994. Continuous Fermentation with Product Recovery
by in-Situ Extraction. Chemical Engineering & Technology 17 (6):365-373.
Wierckx, Nick J. P., Hendrik Ballerstedt, Jan A. M. de Bont, and Jan Wery. 2005.
Engineering of Solvent-Tolerant Pseudomonas putida S12 for Bioproduction of
Phenol from Glucose. Applied and Environmental Microbiology 71 (12):8221-
8227.
Winkler, J., M. Rehmann, and K. C. Kao. 2010. Novel Escherichia coli hybrids with
enhanced butanol tolerance. Biotechnol Lett 32 (7):915-20.
Winston, Fred. 2001. EMS and UV Mutagenesis in Yeast. In Current Protocols in
Molecular Biology: John Wiley & Sons, Inc.
Wu, CY, TP Koylinski, and JE Bozik. 1981. Preparation of styrene from ethylbenzene
US Patent 4,255,599.
Wu, Xiaoqiu, Hans Jörnvall, Kurt D Berndt, and Udo Oppermann. 2004. Codon
optimization reveals critical factors for high level expression of two rare codon
genes in Escherichia coli: RNA stability and secondary structure but not tRNA
abundance. Biochemical and Biophysical Research Communications 313 (1):89-
96.
Wubbolts, M. G., J. Hoven, B. Melgert, and B. Witholt. 1994. Efficient Production of
Optically-Active Styrene Epoxides in 2-Liquid Phase Cultures. Enzyme and
Microbial Technology 16 (10):887-894.
Wubbolts, M. G., P. Reuvekamp, and B. Witholt. 1994. Tol Plasmid-Specified Xylene
Oxygenase Is a Wide Substrate Range Monooxygenase Capable of Olefin
Epoxidation. Enzyme and Microbial Technology 16 (7):608-615.
Xiang, L. K., and B. S. Moore. 2002. Inactivation, complementation, and heterologous
expression of encP, a novel bacterial phenylalanine ammonia-lyase gene. Journal
of Biological Chemistry 277 (36):32505-32509.
Xiang, L. K., and B. S. Moore. 2005. Biochemical characterization of a prokaryotic
phenylalanine ammonia lyase. Journal of Bacteriology 187 (12):4286-4289.

120
Xiang, L. K., and B. S. Moore. 2006. Biochemical characterization of a prokaryotic
phenylalanine ammonia lyase (vol 187, pg 4286, 2005). J Bacteriol 188
(14):5331-5331.
Yang, C. L. Yaws and H. C. 1992. Henry's law constant for compound in water. Edited
by C. L. Yaws, Thermodynamic and Physical Property Data. Houston: Gulf
Publishing Company.
Young, M. R., G. H. N. Towers, and A. C. Neish. 1966. Taxonomic Distribution of
Ammonia-Lyases for L-Phenylalanine and L-Tyrosine in Relation to
Lignification. Canadian Journal of Botany 44 (3):341-&.
Zhang, Congqiang, Xixian Chen, Ruiyang Zou, Kang Zhou, Gregory Stephanopoulos,
and Heng-Phon Too. 2013. Combining Genotype Improvement and Statistical
Media Optimization for Isoprenoid Production in E. coli. PLoS ONE 8
(10):e75164.
Zhang, Haoran, and Gregory Stephanopoulos. 2012. Engineering E. coli for caffeic acid
biosynthesis from renewable sugars. Applied Microbiology and Biotechnology:1-
9.
Zhang, W., J. L. Loebach, S. R. Wilson, and E. N. Jacobsen. 1990. Enantioselective
Epoxidation of Unfunctionalized Olefins Catalyzed by (Salen)Manganese
Complexes. Journal of the American Chemical Society 112 (7):2801-2803.