medicine 5th year, all lectures/pituitary (dr. sabir)
TRANSCRIPT

Endocrine diseasesEndocrine diseases
Dr. Sabir

Measurement of hormonesMeasurement of hormones
Circulating levels of most are very low and cannot be measured by simple chemical techniques.
Recent assays done by immunometric type which are automated

General principlesGeneral principles1) Timing of measurement: release of many hormones is
rhythmical ( e.g: pulsatile as in GH, circadian as in cortisol, or monthly as in sex steroids in premenopausal women). Basal levels are adequate for T3 & T4 (long half-lives)
2)Choice of dynamic biochemical tests:
A- if hormone deficiency is suspected choose a stimulation test
B- if hormone excess is suspected choose a suppression test
C- the more tests there are to choose from, the less likely it is that single test is infallible, so avoid interpreting one result in isolation.

General principlesGeneral principles
3) Imaging:
A- secretory cells also take up substrates, which can be labelled
B- most endocrine glands have a high prevalence of ‘incidentalomas’ so do not scan unless biochemistry confirms endocrine dysfunction or the primary problem is a tumor
4) Biopsy: many endocrine tumors are difficult to classify histologically ( e.g: adrenal ca and adenoma )

Classification of endocrine Classification of endocrine diseasedisease
1) Hormone excess: 1º gland overproduction
2º to excess trophic substance
2) Hormone deficiency: 1º gland failure
2º to deficient trophic substance
3) Hormone hypersensitivity: a- failure of inactivation of hormone b- target organ over-activity/hypersensitivity
4) Hormone resistance: a- failure of activation of hormone b- target organ resistance
5) Non-functioning tumors

EpidemiologyEpidemiology
Most common diseases (excluding DM): Thyroid disorders Subfertility: affects 5-10% of couples Menstrual disorders and hirsutism Osteoporosis 1º hyperparathyroidism Short stature & delayed puberty

Common presentationsCommon presentations
Body size & shape: short stature, tall stature, wt loss, wt gain
Metabolic: tiredness, weakness, ↑appetite, ↓ appetite, polydipsia/thirst, polyuria/nocturia, tremor, palpitation, anxiety
Local effects: swelling in neck, carpal tunnel syn, bone or muscle pain, protrusion of eyes, visual loss, headache
Reproductive: ↓ libido, impotence, oligo-/amenorrhea, subfertility, galactorrhea, gynecomastia, delayed puberty, precocious puberty
Skin: hirsutism, hair thinning, pigmentation, dry skin, excess sweating

Pituitary gland diseasesPituitary gland diseases

Pituitary GlandPituitary Gland
Weight 600 mg Is located within the sella turcica Anatomically and functionally distinct
anterior and posterior lobes


Pituitary DevelopmentPituitary Development
The pituitary originate from different sources.
The anterior pituitary from Rathke´s pouch (which is an embryonic invagination of the pharyngeal
epithelium). The posterior pituitary from an outgrow of
the hypothalamus.

HypothalamicHypothalamic
Hypothalamic neural cells synthesize specific releasing and inhibiting hormones that are secreted directly into the portal vessels of the pituitary stalk.
Hypothalamic-pituitary portal plexus provides the major blood source for the anterior pituitary.

Pituitary cells are exposed to sharp spikes of releasing factors and in turn release their hormones as discrete pulses.

Posterior lobe is directly innervated by hypothalamic neurons (supropticohypophyseal and tuberohypophyseal nerve tracts) via the pituitary stalk


Classification of diseasesClassification of diseases
1. Hormone excess: A- anterior pituitary: 1º)prolactinoma, acromegaly, Cushing’s, rare TSH-,LH-, & FSHomas 2º)disconnection hyperprolactinemia
B- hypothalamus and posterior pituitary: SIADH2. Hormone deficiency: A- ant pit: 1º) hypopituitarism. 2º)
Kallmann’s syndromeB- hypoth & post pit: cranial diabetes insipidus3. Hormone resistance: GH resistance (Laron dwarfism),
nephrogenic diab insip4. Non-functioning tumors: pit adenoma,
craniopharyngioma, metastatic tumors

Anterior PituitaryAnterior Pituitary
Is often referred to as the “MASTER GLAND” because, it orchestrates the complex regulatory functions of multiple other endocrine glands.

Produces six major hormones:Produces six major hormones:
Prolactin (PRL) Growth hormone (GH) Adrenocorticotropin hormone (ACTH) Luteinizing hormone (LH) Follicle-stimulating hormone (FSH) Thyroid-stimulating hormone (TSH)


Anterior Pituitary InsufficiencyAnterior Pituitary Insufficiency

Reduced pituitary function can result from inherited disorders; more commonly, it is acquired and reflects the mass effects of tumors or the consequences of inflamation or vascular damage.

Causes of HypopituitarismCauses of Hypopituitarism
A. Developmental & Congenital– Kallmann Syndrome– Laurence-Moon-Biedl Syndrome– Prader-Willi Syndrome.

Causes of HypopituitarismCauses of Hypopituitarism
B. Acquired: 1. Trauma: accidental or neurosurgical 2.Vascular: apoplexy, Sheehan’s syn3. Neoplastic: pituitary or hypothalamic: adenomas,
craniopharyngiomas, or metastatic deposits, meningiomas, gliomas
4. Infective: basal meningitis(TB), AIDS, encephalitis5. Infiltrative : sarcoidosis, hemochromatosis.6. Functional: anorexia nervosa, starvation, emotional
deprivation7. Others: cranial irradiation, chemotherapy, empty sella
turcica

Causes of hypopituitarismCauses of hypopituitarism
Hypothalamic infiltration: sarcoidosis, histiocytosis X, amyloidosis, and
hemochromatosis. Frequently involve both hypothalamic and
pituitary. Diabetes insipidus occurs in half of patients with
these disorders. Growth retardation is seen if attenuated GH
secretion occurs before pubertal epiphyseal closure.
Hypogonadotropic hypogonadism and hyperprolactinemia are also common.

Causes of hypopituitarismCauses of hypopituitarism
Inflammatory causes: Pituitary damage can be seen with chronic
infections such as tuberculosis, opportunistic fungal infections associated with AIDS.
Other inflammatory processes, such as granulomas or sarcoidosis.

Causes of hypopituitarismCauses of hypopituitarism
Cranial irradiation: May result in Hypotalamic and pituitary
dysfunction, specially in children and adolescents who are more susceptible to damage following whole-brain or head and neck therapeutic irradiation.
Up to two-thirds of patients ultimately develop hormone insufficiency after a median dose of 50 Gy directed at the skull base.

Pituitary Apoplexy Pituitary Apoplexy
May occur spontaneously in a preexisting adenoma (usually nonfunctioning); postpartum (Sheehan´s syndrome); or in association with diabetes, hypertension, or acute shock. Hyperplasia of the pituitary during pregnancy increases the risk for hemorrhage and infarction.
Symtoms: headache with signs of meningeal irritation, vomiting, visual disturbance and cranial nerve palsy.

Empty sella turcicaEmpty sella turcica
Reported on pituitary imaging Causes: defect in diaphragm & extension of
subarachnoid space, or follow spontaneous infarction of a tumor
Sella appears empty but despite this pituitary function is usually normal
Pituitary tissue being eccentrically placed against floor or roof of the fossa
Hypopituitarism seen in <10% of cases

Presentation and DiagnosisPresentation and Diagnosis
The clinical manifestations of hypopituitarism depend on which hormones are lost and the extent of the hormone deficiency.
GH (growth disorders) earliest to be lost, next FSH & LH are lost (menstrual disorders and infertility), then ACTH is lost, and lastly TSH (hypothyroidism)
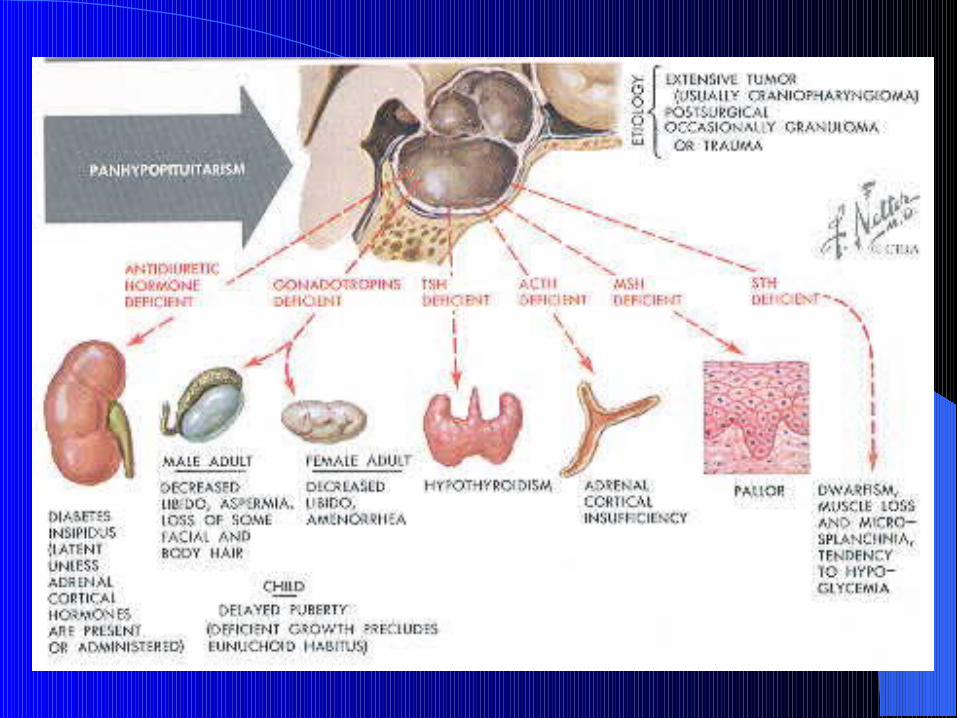

Clinical featuresClinical features
1) GH: in adults, lethargy, muscle weakness & ↑ fat mass( central obesity),atherosclerosis, wrinkly skin, osteoporosis
2) LH &FSH: in ♂ loss of libido & erectile dysfunction, gynecomastia and small testes, decreased shaving and in ♀ oligomenorrhea and amenorrhea. In both: sparse axillary and pubic hair, and ch anemia

C/F… cont.C/F… cont.
3) ACTH: cortisol insufficiency with retained aldosterone secretion hence normal K+ level, but postural hypotension and dilutional hyponatremia result from 3 causes:
A- failure of vasoconstriction and pooling of blood in legs on standing
B- ADH release is enhanced by hypotension & cortisol def.C- cortisol is required for normal water excretion by kidney.
Pallor ( unlike Addison’s) is due to lack of stimulation of melanocytes by ß-LPH in skin
4) TSH: secondary hypothyroidism, but frank myxoedema is rare bec thyroid gland retain some autonomous function

C/F…cont.C/F…cont.
PRL: deficiency is rare, except in Sheehan’s which is assoc. with failure of lactation
ADH deficiency is not reported with pituitary adenomas
Houssay phenomenon: amelioration of DM in hypopituitarism due to reduction in counter-regulatory hormones

Biochemical diagnosisBiochemical diagnosis
Is made by demostrating low ( or normal ) levels of trophic hormones in the setting of low target hormone levels.
Measurement of the pit hormone alone does not show that the level is inappropriately low and Dx may be missed

Basal TestsBasal Tests
LH&FSH:↓or↔ Testosterone or estradiol: ↓ TSH: ↓or ↔ T4: ↓ IGF-1: ↓ Cortisol: ↓ ( at 9 am) Prolactin: ↑

Dynamic testsDynamic tests
Short Synacthen test: to assess adrenal axis (but gives no measure of GH reserve, few false –ve bec ACTH def of <6wk may allow a normal response)
0 and 30 min values of cortisol are measured. Insulin tolerance test: insulin given to induce
hypoglycemia causing stress to↑ cortisol and GH Glucagon stimulation test: when ITT is
contraindicated

Investigation of the causeInvestigation of the cause
Pituitary imaging: MRI± contrast Occasionally biopsy is needed

TreatmentTreatment
Hormone replacement therapy, including glucocorticoids, thyroid hormone, sex steroids, growth hormone and vasopressin, is usually free of complications.
Glucocorticoid replacement require careful dose adjustments during stressful events.

RxRx
Cortisol replacement Hydrocortisone 15-30 mg/d. (10-20mg in morning and 5-10mg in early evening) OR prednisolone 3mg on waking and 2mg early evening ( total dose 5-7.5mg/d). Mineralocorticoid replacement is not required.
Thyroid hormone replacement: L-Thyroxine 100-150 μg /d. Unlike primary hypothyroidism, measuring TSH is not helpful in adjusting the replacement dose. The aim is to maintain serum T4 in the upper part of the reference range. It is dangerous to give thyroid replacement to patients with adrenal insufficiency without first giving glucocorticoid therapy, since this may precipitate adrenal crisis.

Rx… cont.Rx… cont.
Sex hormone replacement : in men testosterone im, orally, as patch or implant and in women : cyclical estrogen/progestogen orally or as patch/implant.
Fertility: HCG + FSH, and pulsatile LHRH also used.

Rx…cont.Rx…cont.
Growth hormone (GH) is administered, by daily subcutaneous self-injection, to young patients with GH deficiency, renal failure or Turner's syndrome to assist them in attaining their growth potential. Growth hormone therapy may also help young adults to achieve a higher peak bone mineral density. Side-effect is sodium retention, manifest as peripheral oedema or carpal tunnel syndrome. For this reason, GH replacement is started at a low dose, with monitoring of the response by measurement of serum insulin-like growth factor-1 (IGF-1) levels

Diabetes insipidusDiabetes insipidus
Persistent excretion of excessive quantities of dilute urine, and thirst.
2) cranial: def. of ADH production by hypothalamus
3) nephrogenic:: renal tubules unresponsive to ADH: a- genetic, b- metabolic (hypokalemia, hypercalcemia),c- drugs: lithium, demeclocycline, d- poisoning: by heavy metals, e-: ch kidney dis: polycystic kid dis, sickle cell anemia

C/FC/F
Polyuria and polydipsia
Patient may pass 5-20 litres of urine/24 hr of low specific gravity and osmolality.
DDx: diabetes mellitus and primary polydipsia

DxDx
Elevated plasma osmolality (> 300 mOsm/kg) and urine is not maximally concentrated ( <600 mOsm/kg)

RxRx
Cranial DI: DDAVP ( desmopressin) analogue of ADH as nasal spray, also present as tablets and im injection, dose determined by measuring plasma Na conc &/or osmolality

Pituitary TumorsPituitary Tumors
Almost always benign adenomas Are the most common cause of pituitary
hormone hypersecretion and hyposecretion syndromes in adults.
Account for 10% of intracranial tumors Divided by size: microadenoma <1cm,
macroadenoma >1cm


Histological typesHistological types
1. Chromophobe: 70%,some are non-secretory, but cause hypopituitarism. Half produce PRL, a few produce GH or ACTH, local pressure effect in 30%
2. Acidophil: 15%, secrete GH or PRL, local effect in 10%
3. Basophil: 15%, secrete ACTH, local effect rare

Clasification of pituitary Clasification of pituitary adenomas.adenomas.
Rare thyrotoxicosis, macro-orchidism in men
1%LH/FSH/TSH
Chushing;s disease7%ACTH
Acromegaly/ galactorrhea7%GH &PRL
Acromegaly/Gigantism20%GH only
30%No obvious hormone
Hypogonadism, galactorrea35%PRL only
Syndrome%Hormone

Features of local pressureFeatures of local pressure
Headache Visual field defect ( bitemporal
hemianopia) Cranial nerve palsies( III, IV, VI) Diabetes insipidus Sleep and appetite disturbance ( from
hypothalamic pressure) CSF rhinorrhea: from erosion of floor of
sella

Other Sellar MassesOther Sellar Masses
Craniopharyngiomas Sella Chordomas Meningiomas Histiocytosis X Pituitary metastases Hypothalamic hamartomas and
gangliocytomas

ImagingImaging
Pituitary MRI defines intra- and supra-sellar extension

THERAPEUTIC MODALITIES FOR HYPOTHALAMIC THERAPEUTIC MODALITIES FOR HYPOTHALAMIC
AND PITUITARY TUMOURSAND PITUITARY TUMOURS commentMedicalRadiotherapySurgery
2nd line
2nd line
Dopamine agonists cause macroadenomas to shrink
1st lineDopamine agonists
2nd lineProlactinoma
-1st lineNon-functioning macroadenoma

-2nd line1st lineCraniopharyngioma
Radiotherapy is used in children and to prevent Nelson’s syndrome
-2nd line1st lineCushing’s disease
Medical Rx does not cause macroadenomas to shrink
2nd line: somatostatin analogues, Dopamine agonists, GH-receptor antagonists
2nd line1st lineAcromegaly

If there is evidence of pressure on visual pathways, then urgent treatment is required. The chances of recovery of a visual field defect are proportional to the duration of symptoms; full recovery is unlikely if the defect has been present for longer than 4 months. It is crucial that serum prolactin is measured before emergency surgery is performed. If the prolactin is > 5000 mU/l, then the lesion may be a macroprolactinoma and a therapeutic trial of a dopamine agonist for just a few days may successfully shrink the lesion and make surgery unnecessary. Pituitary function should be retested 4-6 weeks following surgery to detect the development of any new hormone deficits following surgery; rarely, the surgical treatment of a sellar lesion can result in recovery of hormone secretion that was deficient pre-operatively.

RadiotherapyRadiotherapy
Following surgery, imaging is repeated usually after a few months and, if there is any residual mass and the histology confirms a radiosensitive tumor, external radiotherapy may be given to reduce the risk of recurrence. Radiotherapy is not useful in patients requiring urgent therapy because it takes many months or years to be effective and there is a risk of acute swelling of the mass. Radiotherapy carries a life-long risk of hypopituitarism (50-70% in the first 10 years) and annual pituitary function tests are obligatory.

HYPERPROLACTINAEMIA HYPERPROLACTINAEMIA
The cardinal features are galactorrhoea and hypogonadism. Prolactin stimulates milk secretion but not breast development, so that galactorrhoea almost never occurs in men, and is only possible in those in whom gynaecomastia is induced by hypogonadism.

Causes of hyperprolactinemiaCauses of hyperprolactinemia
Physiological Stress (e.g. post-seizure), Pregnancy, Lactation, Nipple
stimulation, Sleep, Coitus, Exercise, Baby crying Drugs: A)Dopamine antagonists Antipsychotics (phenothiazines and butyrophenones) Antidepressants Antiemetics (e.g. metoclopramide, domperidone) B) Dopamine-depleting drugs Reserpine, Methyldopa C) Oestrogens Oral contraceptive pill

Causes…. Cont.Causes…. Cont.
Pathological Common Disconnection hyperprolactinaemia (e.g.
non-functioning pituitary macroadenoma) Prolactinoma (usually microadenoma) Primary hypothyroidism Polycystic ovarian syndrome Macroprolactinaemia

Causes…cont.Causes…cont.
Uncommon Hypothalamic disease Pituitary tumour secreting prolactin and
growth hormone Renal failure Rare Chest wall reflex (e.g. post-herpes zoster) Ectopic source

Clinical assessment Clinical assessment
In women, in addition to galactorrhoea, the hypogonadism associated with hyperprolactinaemia causes secondary amenorrhoea and anovulation with infertility. In men there is decreased libido, reduced shaving frequency and lethargy

InvestigationInvestigation
The upper limit of normal for many assays of s.prolactin is 500 mU∼ /l ( 14 ng∼ /ml). In non-pregnant and non-lactating pts, prolactin concentrations of 500-1000 mU/l are likely to be induced by stress or drugs, and a repeat measurement is indicated. Levels between 1000 and 5000 mU/l are likely to be due to either drugs, a microprolactinoma or 'disconnection' hyperprolactinaemia. Levels > 5000 mU/l are highly suggestive of a macroprolactinoma, and the higher the level, the bigger the tumour. Some macroprolactinomas cause levels over 100 000 mU/l.

TreatmentTreatment
Dopamine agonist drugs are first-line therapy. However, it is possible to withdraw dopamine agonist therapy without recurrence of hyperprolactinaemia after 5-10 years of treatment in some pts with a microadenoma. Also, after the menopause, suppression of prolactin is only required in microadenomas if galactorrhoea is troublesome, since hypogonadism is then physiological and tumour growth unlikely. In pts with macroadenomas, drugs can only be withdrawn after curative surgery or radiotherapy and under close supervision.

DrugsDrugs
Bromocriptine : 2.5-15 mg/day8-12-hourly orally. Useful in treating infertilityProven long-term efficacy. S/E: Ergotamine-like (nausea, headache, postural hypotension, constipation)It needs frequent dosing so poor compliance

AcromegalyAcromegaly
Acromegaly is an insidious disorder caused by a pituitary GH-secreting adenoma resulting in high circulating levels of GH and IGF-I.
Annual incidence : 3–4 cases/million. Prevalence : 40-90 cases/million.
Due to its insidious nature, diagnosis of acromegaly may be considerably delayed.
Mortality rate for acromegalics is 2–3 times that of the general population, but with effective treatment survival can be improved to that of the
age-matched population.

morbidity and mortalitymorbidity and mortality
Most frequent causes of death are cardiovascular
and respiratory complications. Sleep apnea is a significant cause of morbidity. Cardiovascular disease and diabetes increase
mortality. Patients with acromegaly may also be at increased
risk for cardiac hypertrophy, hypertension,
arthritis, sleep apnea, and development of other neoplastic lesions, particularly in the colon.

Presentation and DiagnosisPresentation and Diagnosis
Manifestations of GH hypersecretion are indolent and often are not clinically diagnosed.

C/FC/F
Acral bony overgrowth results in frontal bossing, increased hand and foot size, mandibular enlargement prognathism, and widened space between the lower incisor teeth. Patients with acromegaly may also be at increased risk for cardiac hypertrophy, hypertension, arthritis, sleep apnea, and development of other neoplastic lesions, particularly in the colon.

Acromegaly

GigantismGigantism
In children and adolescents, initiation of GH secretion prior to epiphyseal long bone closure is associated with the development of pituitary gigantism.

Clinical featuresClinical features
Increased heel pad thickness, shoe or glove size, ring tightening, and a large fleshy nose.
Hyperhidrosis, deep sounding voice, oily skin, arthropathy, kyphosis, carpal tunnel syndrome proximal muscle weakness and skin tags.
Generalized visceromegaly occurs, including cardiomegaly, and macroglossia.

Acromegaly/gigantism

DiagnosisDiagnosis
Diagnosis of acromegaly is based on clinical findings, elevated serum IGF-I levels, and the inability to suppress serum GH during an OGTT.
A single measurement of GH provides inadequate information regarding GH elevation (pulsatile manner) .
During an OGTT in normal patients, serum GH should suppress to less than 2 ng/mL if the regular GH RIA is used.

Diagnosis…contDiagnosis…cont
If GH and IGF-I levels suggest acromegaly, the presence of a pituitary adenoma should be confirmed using MRI.
In rare cases, a pituitary mass may not be identified. Although this may be due to an occult pituitary microadenoma or a partially empty sella, an ectopic tumor secreting GH or GHRH may rarely be present.
Circulating GHRH blood levels or chest and abdominal imaging confirm peripheral ectopic GHRH secretion.

TreatmentTreatment
Primary goal of treatment is to normalize GH levels Surgical tumor excision ( trans-sphenoidal) is indicated for most patients with small, well localized microadenomas unless there is a contraindication to surgery.
This procedure generally results in a rapid and substantial reduction of serum GH levels
immediately postoperatively and normalization of IGF-I levels in the weeks following surgery.
Only approximately 70–80% of patients with microadenomas and less than 50% of patients with macroadenomas achieve a circulating GH level less than 5 ng/mL after surgery.

Rx…cont.Rx…cont.
Fatalities, meningitis, and visual impairment are major drawbacks of surgery but are rare ( <2% )
CSF leak, permanent anterior lobe deficits, diabetes insipidus, and local nasal complications occur in approximately 5% of patients

Nonsurgical Treatment Nonsurgical Treatment Options for Acromegaly Options for Acromegaly
Nonsurgical treatment options for acromegaly include medical therapy with somatostatin analogs or dopamine agonists and radiotherapy.
These therapies have been most effective when used in conjunction with surgery.

TreatmentTreatment
Radiotherapy: External radiotherapy ( 45-50 Gy total dose) is usually employed as second-line treatment if acromegaly persists after surgery, to stop tumour growth and lower GH levels. However, GH levels fall slowly (over many years) and there is a risk of hypopituitarism

TreatmentTreatment
Medical: In patients with persisting acromegaly after surgery, most centres employ medical therapy to lower GH levels to < 5 mU/l. Medical therapy may be discontinued after several years in patients who have received radiotherapy. Somatostatin analogues (e.g. octreotide or lanreotide) can be administered as slow-release injections every few weeks.

TreatmentTreatment
Octreotide reduces GH hypersecretion in approximately 95% of acromegalic patients within 30–60 min of injection and ameliorates clinical and metabolic anomalies associated with the disease.

Side-Effect of OctreotideSide-Effect of Octreotide
Asymptomatic cholesterol gallstone development, in up to 25% of patients.
Short term side-effects that often resolve with continued treatment include abdominal
pain, diarrhea, fat malabsorption, nausea, and flatulence.
bradycardia in 25% of patients