mechanisms of control of neuron survival by the endocannabinoid
TRANSCRIPT

Current Pharmaceutical Design, 2008, 14, 000-000 1
1381-6128/08 $55.00+.00 © 2008 Bentham Science Publishers Ltd.
Mechanisms of Control of Neuron Survival by the Endocannabinoid System
Ismael Galve-Roperh*, Tania Aguado, Javier Palazuelos and Manuel Guzmán*
Department of Biochemistry and Molecular Biology I, School of Biology, and Centro de Investigación Biomédica en Red
sobre Enfermedades Neurodegenerativas (CIBERNED), Complutense University, 28040 Madrid, Spain
Abstract: Endocannabinoids act as retrograde messengers that, by inhibiting neurotransmitter release via presynaptic CB1
cannabinoid receptors, regulate the functionality of many synapses. In addition, the endocannabinoid system participates
in the control of neuron survival. Thus, CB1 receptor activation has been shown to protect neurons from acute brain injury
as well as in neuroinflammatory conditions and neurodegenerative diseases. Nonetheless, some studies have reported that
cannabinoids can also exert neurotoxic actions. Cannabinoid neuroprotective activity relies on the inhibition of glutama-
tergic neurotransmission and on other various mechanisms, and is supported by the observation that the brain overpro-
duces endocannabinoids upon damage. Coupling of neuronal CB1 receptors to cell survival routes such as the phosphati-
dylinositol 3-kinase/Akt and extracellular signal-regulated kinase pathways may contribute to cannabinoid neuroprotec-
tive action. These pro-survival signals occur, at least in part, by the cross-talk between CB1 receptors and growth factor
tyrosine kinase receptors. Besides promoting neuroprotection, a role for the endocannabinoid system in the control of neu-
rogenesis from neural progenitors has been put forward. In addition, activation of CB2 cannabinoid receptors on glial cells
may also participate in neuroprotection by limiting the extent of neuroinflammation. Altogether, these findings support
that endocannabinoids constitute a new family of lipid mediators that act as instructive signals in the control of neuron
survival.
Key Words: Cannabinoid, endocannabinoid system, neuron, neuroprotection, neurodegeneration, neurogenesis, neuroinflam-mation.
INTRODUCTION
Preparations from Cannabis sativa L. (marijuana) have been used for many centuries both medicinally and recrea-tionally. However, the chemical structure of their active components - the cannabinoids - was not elucidated until the early 1960s. Among the approximately 70 cannabinoids pro-duced by marijuana,
9-tetrahydrocannabinol (THC) is the
most relevant owing to its high potency and abundance [1]. Since the early-mid 1990s it is widely accepted that THC acts in the organism by mimicking endogenous substances - the endocannabinoids (eCBs) anandamide (AEA, N-arachi- donoylethanolamine; [2]) and 2-arachidonoylglycerol (2AG; [3, 4]) that bind to and activate specific cell surface cannabi-noid receptors, two of which have been cloned and well characterized from mammalian tissues: CB1 [5] and CB2 [6]. eCB generation occurs by cleavage of plasma membrane lipid precursors, and is tightly controlled by neuronal activity (Fig. 1). Thus, engagement of various postsynaptic metabo- tropic and ionotropic neurotransmitter receptors triggers eCB production. Once generated, eCBs act retrogradely through presynaptic CB1 cannabinoid receptors, blunting membrane depolarization and inhibiting neurotransmitter release [7]. CB1 receptors are highly abundant in discrete areas of the brain, and their activation lowers the release of neurotrans-mitters such GABA and glutamate, thereby affecting pro-
*Address correspondence to these authors at the Department of Biochemis-
try and Molecular Biology I, School of Biology, Complutense University,
28040 Madrid, Spain; Tel: +34913944668; Fax: +34913944672;
E-mail: [email protected] and [email protected]
cesses such as motor activity, learning and memory [7, 8]. Besides this well established neuromodulatory role of eCBs, it is becoming evident that these compounds play an impor-tant role in the control of neuron survival. A large series of articles in this Special Issue provides details on the charac-teristics and therapeutic potential of eCB system-mediated neuroprotection in many particular neuropathologies related to acute brain injury, neurodegeneration and neuroinflamma-tion. Hence here we will focus on the general signalling mechanisms by which the eCB system can protect neurons from death.
NEUROPROTECTIVE ROLE OF THE ENDOCAN-NABINOID SYSTEM
The first reports on the ability of eCBs to protect neurons from death date more than one decade ago, when various cannabinoids were shown to exert neuroprotection upon ischemic/excitotoxic injury both in vitro [9, 10] and in vivo [11]. These observations founded a new area of research focused on the study of the eCB system as an endogenous neuroprotective system and the potential of cannabinoid compounds as new pharmacological tools for the manage-ment of various neuropathologies [12]. A number of can-nabinergic drugs, including synthetic and endogenous can-nabinoid receptor agonists and inhibitors of eCB transport and degradation, have been used in those studies. Just to mention a few examples, THC administration reduces neu-ronal loss and brain damage in excitotoxicity and ischemia models [13]. Likewise, AEA exerts a neuroprotective action
2 Current Pharmaceutical Design, 2008, Vol. 14, No. 00 Galve-Roperh et al.
in excitotoxicity [14, 15] and 2AG protects neurons in trau-matic brain injury [16]. In addition, administration of the synthetic cannabinoid receptor agonist WIN-55,212-2 con-fers neuroprotection in a model of neonatal hypoxic-ischemic encephalopathy [17].
A common approach to modulate eCB levels involves the use of inhibitors of their transport and degradation, which provides a means of elevating eCB levels and therefore of activating cannabinoid receptors in a more prolonged fashion and with higher sensitivity to physiological, on-demand regulation than acute administration of cannabinoid receptor agonists [18]. In agreement with the proposed neuroprotec-tive action of the latter agents, administration of inhibitors of eCB transport or degrading enzymes [fatty acid amide hy-drolase (FAAH) and monoacylglycerol lipase (MAGL)] can prevent behavioural alterations and memory impairment due to excitotoxic damage in a CB1 receptor-dependent manner [19, 20]. A similar strategy has proved useful in attenuating emotional symptoms of anxiety and depression [21].
In addition to the aforementioned pharmacological stud-ies, the use of CB1 receptor knock-out mice has provided further evidence for a neuroprotective role of the eCB sys-tem as those animals are more sensitive to various brain in-sults and neurodegenerative disorders that their correspond-ing wild-type controls. Thus, for example, increased excito-toxic injury is observed in CB1 receptor-deficient mice after brain stroke or kainic acid administration [22, 23]. CB1 re-ceptors can also favour hippocampal neuron survival in the hippocampus along life, as CB1 receptor-deficient mice show an enhanced loss of neurons in CA1 and CA3 with aging,
which is accompanied by a decrease in cognitive functions [24].
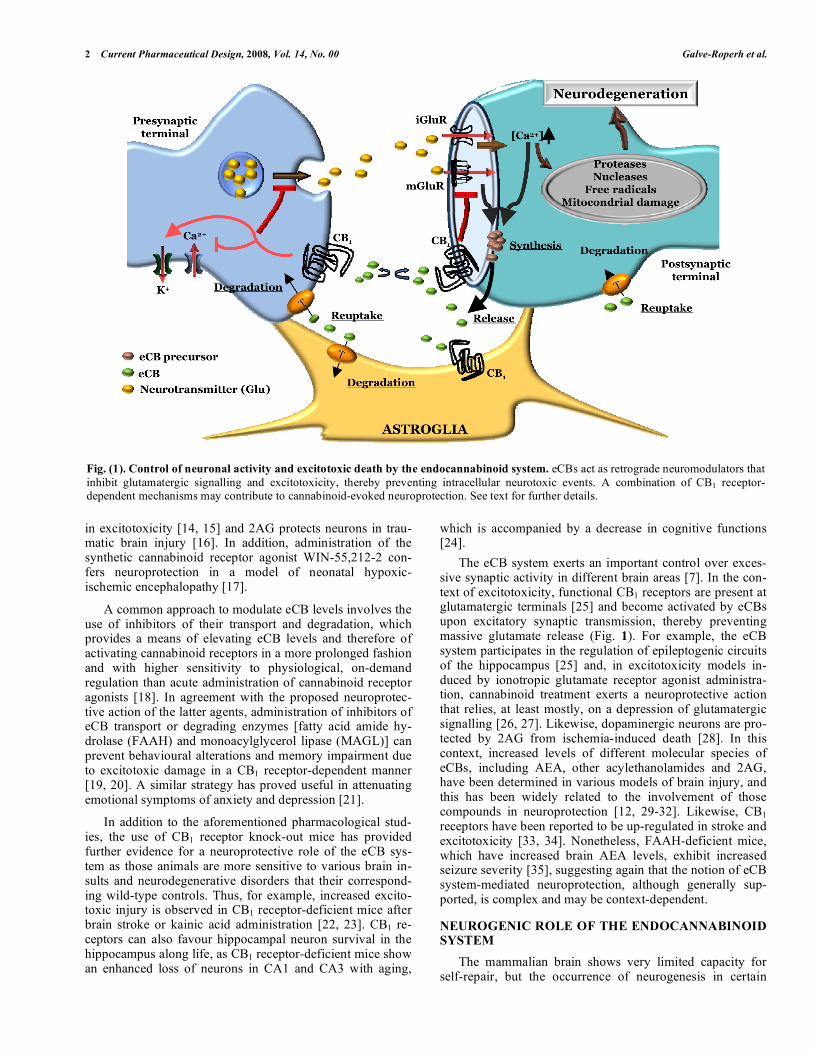
The eCB system exerts an important control over exces-sive synaptic activity in different brain areas [7]. In the con-text of excitotoxicity, functional CB1 receptors are present at glutamatergic terminals [25] and become activated by eCBs upon excitatory synaptic transmission, thereby preventing massive glutamate release (Fig. 1). For example, the eCB system participates in the regulation of epileptogenic circuits of the hippocampus [25] and, in excitotoxicity models in-duced by ionotropic glutamate receptor agonist administra-tion, cannabinoid treatment exerts a neuroprotective action that relies, at least mostly, on a depression of glutamatergic signalling [26, 27]. Likewise, dopaminergic neurons are pro-tected by 2AG from ischemia-induced death [28]. In this context, increased levels of different molecular species of eCBs, including AEA, other acylethanolamides and 2AG, have been determined in various models of brain injury, and this has been widely related to the involvement of those compounds in neuroprotection [12, 29-32]. Likewise, CB1 receptors have been reported to be up-regulated in stroke and excitotoxicity [33, 34]. Nonetheless, FAAH-deficient mice, which have increased brain AEA levels, exhibit increased seizure severity [35], suggesting again that the notion of eCB system-mediated neuroprotection, although generally sup-ported, is complex and may be context-dependent.
NEUROGENIC ROLE OF THE ENDOCANNABINOID SYSTEM
The mammalian brain shows very limited capacity for self-repair, but the occurrence of neurogenesis in certain
Fig. (1). Control of neuronal activity and excitotoxic death by the endocannabinoid system. eCBs act as retrograde neuromodulators that
inhibit glutamatergic signalling and excitotoxicity, thereby preventing intracellular neurotoxic events. A combination of CB1 receptor-dependent mechanisms may contribute to cannabinoid-evoked neuroprotection. See text for further details.

Mechanisms of Control of Neuron Survival Current Pharmaceutical Design, 2008, Vol. 14, No. 00 3
areas of the adult brain suggests that neural progenitor cells can participate in structural and functional neurorepair. Un-der normal conditions, neurogenesis in the adult brain ap-pears to be restricted to discrete germinal areas - the subven-tricular zone and the hippocampal dentate gyrus [36]. While some reports indicate that neurogenesis in the adult central nervous system may be more widespread than previously thought, improved characterization of these observations is required [37]. Newly generated cells mature into functional neurons in the adult mammalian brain; in the hippocampus they can participate in memory processing [38], while in the olfactory bulb they can contribute to the control of olfactory inputs [36].
Within the many signalling systems involved in the in-struction of neuronal generation, e.g. trophic factors and cy-tokines [39], the eCB system has been recently proposed to cooperate in the regulation of neural cell development and neurogenesis [40, 41]. Cannabinoid receptors are not only expressed in differentiated neurons of many brain areas [7, 42], but they are also present in neuroblasts and neural pro-genitors of the adult brain [40, 41], in which they modulate cell proliferation [43-45] and differentiation [46, 47]. Re-garding proliferation, CB1 receptor-deficient mice show de-creased progenitor proliferation in the hippocampus and the subventricular zone [43, 44]. Regarding differentiation, the situation seems more complex as, for example, in certain pharmacological paradigms cannabinoid administration in-hibits synaptogenesis via CB1 receptors [48], while in others it does not affect the absolute number of newly born hippo-campal neurons [49] and in others it promotes neurogenesis and exerts anxiolytic and antidepressant effects [46].
Some studies on neurogenesis have also been conducted in genetically-modified animals. Thus, in CB1
-/- and FAAH
-/-
adult mice no alterations are evident in hippocampal neuro-genesis under basal conditions, though hippocampal astro-gliogenesis is depressed in the former and enhanced in the latter animals [47]. In contrast, upon excitotoxic damage CB1 receptor-deficient mice show a strongly depressed neuro-genic response [34]. In that situation, activation of CB1 re-ceptors in neural progenitors induce cell proliferation and contribute to the generation of new neurons by promoting the production of growth factors such as fibroblast growth factor-2 (FGF-2) [34]. CB2 receptor activation can also stimulate adult neural progenitor proliferation [45, 50] and, accordingly, CB2
-/- mice show lower progenitor proliferation
after excitotoxicity than their wild-type controls [45, 50]. Thus, the eCB system may participate in the control of brain structural plasticity and repair by modulating neural cell pro-liferation and commitment, although the precise role of CB1 and CB2 receptors in neuronal specification and differentia-tion remains to be elucidated.
CB1 CANNABINOID RECEPTOR-DEPENDENT
NEUROPROTECTION: SIGNAL TRANSDUCTION
MECHANISMS
Besides the various non-cell autonomous processes in-volved in cannabinoid receptor-mediated neuroprotection, such as the CB1 receptor-dependent reduction of glutamater-gic secretion (see above) and the CB2 receptor-dependent inhibition of proinflammatory-mediator release from acti-
vated microglia (see below), it is likely that neuron-cell in-trinsic, CB1 receptor-triggered intracellular signal transduc-tion events also contribute to cannabinoid neuroprotective activity. The precise nature of those mechanisms is not fully understood, but there is accruing evidence that CB1 receptor activation can couple to at least two important cell survival signalling routes: the phosphatidylinositol 3-kinase (PI3K)/ Akt pathway and the extracellular signal-regulated (ERK) pathway (Fig. 2).
CB1 receptor activation evokes PI3K/Akt stimulation in primary cortical neurons [51] and in the mouse hippocam-pus, striatum and cerebellum in vivo [52], and this event has been related with cannabinoid-mediated neuroprotection. CB1-mediated PI3K/Akt activation is also observed in cells heterologously expressing the receptor [53] and in primary glial cells [54, 55] and astrocytoma cells lines [56, 57], and is dependent on Gi/o protein dissociation [53]. The down-stream targets by which CB1 receptors may signal neuropro-tection via Akt are as yet unclear, but one of them could be glycogen synthase kinase-3, a potentially neurotoxic protein that is phosphorylated and inactivated by Akt [52, 53].
It is well documented that cannabinoid administration leads to CB1 receptor-mediated ERK pathway activation in the brain in vivo, for example in the hippocampus [58], the striatum [59], the frontal cortex [60] and the cerebellum [61]. This process is also observed in cells heterologously express-ing the receptor [62] as well as in primary glial cells [63] and astrocytoma cells lines [56, 57]. CB1 receptor-dependent ERK activation relies on Gi/o protein dissociation [62] and may be dependent, at least in part, on the inhibition of the adenylyl cyclase/protein kinase A/cAMP pathway [58, 64, 65]. In line with the notion that ERK actions are highly pro-miscuous, CB1 receptor coupling to this pathway most likely signals via different context-dependent effectors. For exam-ple, induction of various transcription factors such as the early-response genes c-fos [66, 67] and Krox-24 [58, 68] and phosphorylation/activation of transcription factors such as Elk-1 [59] or other targets such as p90 ribosomal S6 kinase [54] have been implicated in CB1 receptor-evoked ERK ef-fects in neural cells.
Gi/o protein-coupled receptors can conceptually activate the PI3K/Akt and ERK pro-survival pathways through G protein subunit release both “directly” - via activation of targets such as class IB PI3K isoforms - and “indirectly” - via transactivation of growth factor tyrosine kinase receptors and subsequent stimulation of the downstream “canonical” class IA PI3K/Akt and ERK pathways [69] (Fig. 2). Although for cannabinoid receptors the former process cannot be ex-cluded, and indeed has received some experimental support [56, 70], the latter possibility has focused much more atten-tion. The phylogenetically ancient eCB system must have evolved simultaneously with other signalling systems and in the process established multiple levels of interactions with cell surface and intracellular proteins at the ligand, receptor and post-receptor levels. Thus, accumulating evidence sug-gests that receptor cross-talk may be involved in coordinat-ing the coincident actions of cannabinoids and growth fac-tors in neural cell survival. For example, CB1 receptor acti-vation at low signal inputs promotes the survival of various cell lines expressing epidermal growth factor (EGF) recep-

4 Current Pharmaceutical Design, 2008, Vol. 14, No. 00 Galve-Roperh et al.
tors [57, 67, 71]. This activates in turn tumor necrosis factor -converting enzyme (TACE/ADAM17), a member of the
disintegrin-metalloprotease family, through cytoplasmic Src family member tyrosine kinases [57]. Proteolytic ectodomain shedding of EGF-like precursors by TACE would liberate active ligands at the cell surface, thereby inducing rapid EGF receptor tyrosine phosphorylation and downstream activation of mitogenic pathways. Other studies in cell lines indicate that CB1 receptors may also transactivate vascular endothe-lial growth factor receptors [72].
Another connection of cannabinoids with cell survival mediators is that of neuronal CB1 receptors with the brain-derived neurotrophic factor (BDNF) signalling system. Thus, CB1 receptors are involved in BDNF production, and this process plays a pivotal role in the neuroprotective response elicited by endocannabinoids upon excitotoxic damage in brain areas such as the hippocampus and the striatum [23, 73, 74]. In addition, studies on the cooperativity of cannabi-noid- and BDNF-induced migration of cortical GABAergic interneurons have revealed that CB1 receptors promote neu-ronal differentiation through transactivation of BDNF TrkB receptors in a Src kinase-dependent fashion [75]. Likewise, CB1 receptors might cross-talk to the bFGF signalling sys-tem. Thus, for example, increased production of bFGF may be involved CB1 receptor-mediated hippocampal neurore-
generative response after excitotoxic injury [34], and the CB1 receptor antagonist rimonabant inhibits bFGF-stimulated axonal growth [76]. Taken together, these data support that promotion of signalling through transactivation of tyrosine kinase receptors may constitute a common mechanism in-volved in CB1 receptor-mediated neuroprotection.
CB2 CANNABINOID RECEPTOR-DEPENDENT NEU-
ROPROTECTION: TARGETING OF MICROGLIAL
CELLS
Cannabinoids are known to improve the symptoms of several models of neuroinflammatory disorders such as ex-perimental autoimmune encephalomyelitis (EAE) at least in part by their ability to modulate microglial cell activation [77, 78]. Here we will focus on this topic, as the ability of cannabinoids to regulate other aspects of glial cell biology in various neuropathologies is discussed elsewhere in this Spe-cial Issue. Amelioration of EAE motor symptoms can occur at different levels and involves both CB1 and CB2 receptors [79-81]. Genetic and pharmacological studies support that neuronal CB1 receptors are majorly involved in preventing inflammation-induced neuron death [79]. Nonetheless, the absence of CB2 receptors significantly exacerbates EAE pa-thology, and the administration of CB2 receptor-selective agonists exerts beneficial symptomatic effects [80, 82]. In
Fig. (2). CB1 receptor-dependent signalling mechanisms involved in neuroprotection. CB1 receptor activation can couple to at least two
important cell survival signalling routes: the phosphatidylinositol 3-kinase (PI3K)/Akt pathway and the extracellular signal-regulated (ERK)
pathway. This is reliant on heterotrimeric G protein dissociation, and can occur both “directly” and “indirectly” - via transactivation of
growth factor tyrosine kinase receptors. See text for further details.

Mechanisms of Control of Neuron Survival Current Pharmaceutical Design, 2008, Vol. 14, No. 00 5
this context, CB2 receptor activation decreases leukocyte rolling, leukocyte/endothelial interactions and leukocyte in-filtration into the nervous system [83], contributes to the attenuation of excitotoxic damage during demyelination and ischemic injury [84, 85] and inhibits microglial activation [86]. Microglial cells express CB2 receptors (Fig. 3), which are strongly up-regulated during neuroinflammation [87] and in plaques of multiple sclerosis [88] and Alzheimer’s disease patients [89]. Microglia also synthesize and degrade eCBs such as 2AG, the levels of which are negatively controlled by the IFN- released by primed T-cells invading the central nervous system during EAE, thus indicating that impaired 2AG production may be associated with neurodegeneration in this disorder [90]. In addition, cannabinoids down-regu- late the production of proinflammatory cytokines as well as nitrogen and oxygen reactive species by microglial cells and autoreactive T cells [86]. Overall, this attenuation of micro-glial activation is considered to participate in cannabinoid-induced neuroprotection under neuroinflammatory condi-tions and in excitotoxicity [86, 91]. Additionally, brain mi-croglia can be replenished from bone marrow-derived pro-genitors during inflammation and brain injury [92, 93], and myeloid progenitors mobilize through the bloodstream and target the inflamed central nervous system in a process that is under the negative control of CB2 receptors [82]. In sum-mary, eCBs can contribute to neuron survival by their CB2 receptor-dependent central and peripheral immunomodula-tory actions (Fig. 3).
CANNABINOID RECEPTOR-INDEPENDENT NEU-
ROPROTECTION
In addition to the CB receptor-dependent promotion of neuron survival, some cannabinoids can also exert neuropro-tective actions by receptor-independent mechanisms. On the one hand, the intrinsic antioxidant properties of phenol-containing cannabinoids as cannabidiol and THC are well known [94, 95] and, for example, cannabidiol administration protects neurons from ischemic damage [96] and in a Parkin-son’s disease model [97] independently of CB receptors. On the other hand, eCBs are actively metabolized in the brain, and the resulting products constitute a variety of bioactive lipids that may control neuron survival. For example, cy-clooxygenase-2-mediated metabolism of eCB substrates [98] generates different neuroactive prostaglandins [99] and prostamides [100], and AEA hydrolysis by FAAH yields ethanolamine, that is protective for neuroblastoma cells [101]. During brain injury, alterations in eCBs levels are not restricted to AEA and 2AG species, and other fatty acid ethanolamides – including various putative eCBs - are also affected [12]. Among them, palmitoylethanolamide exerts anti-inflammatory effects that are most likely mediated by peroxisome proliferator-activated receptor- [102]. Micro-glial cells produce palmitoylethanolamide and their motility is increased after focal cerebral ischemia, a situation in which palmitoylethanolamide levels raise [31]. Hence, under certain circumstances cannabinoid-mediated neuroprotection
Fig. (3). CB2 receptor-dependent mechanisms involved in neuroprotection. Cannabinoids can contribute to neuron survival by their CB2
receptor-dependent central and peripheral immunomodulatory actions. These include for example inhibition of microglial activation, leuko-cyte infiltration and myeloid progenitor-derived microglial replenishment. See text for further details.

6 Current Pharmaceutical Design, 2008, Vol. 14, No. 00 Galve-Roperh et al.
comprises various components that include CB receptor-dependent and independent actions. Nonetheless, it is plausi-ble that at least some of those receptor-independent actions will be ascribed in the future to membrane receptors such as GPR55 - recently proposed as a new metabotropic cannabi-noid receptor [103] - or intracellular targets such as perox-isome proliferator-activated receptors and [104].
CANNABINOID-INDUCED NEUROTOXICITY
In contrast to the large number of reports showing can-nabinoid-mediated neuroprotection, some other studies have shown that high eCB levels can be deleterious to neurons. Thus, for example, AEA can induce brain damage by a mechanism that depends, at least in part, on the activation of TRPV1 vanilloid receptors [105, 106]. It is therefore con-ceivable that, besides the intrinsic differences in the pharma-cokinetics of synthetic, plant-derived and endogenous can-nabinoid agonists, an important factor that determines the balance of protective versus toxic actions of these com-pounds is their distinct ability to engage (neuroprotective) metabotropic CB1 receptors or (neurotoxic) ionotropic TRPV1 receptors [105, 107]. Nonetheless, the situation may be more complex as, under certain experimental conditions, administration of the CB1 receptor antagonist SR141716 (rimonabant) has been shown to exert neuroprotection from excitotoxic/ischemic damage [108-110]. Various cell signal-ling mechanisms have been proposed for cannabinoid-
evoked neuron killing, including prostanoid synthesis and generation of free radicals by cyclooxygenase [111], stimula-tion of the pro-apoptotic c-Jun N-terminal kinase [112] and p38 mitogen-activated protein kinase [113] cascades, activa-tion of calpains [106, 114] and increase of p53-dependent lysosomal permeability [115]. Finally, different cannabinoid agonists are known to induce cyclooxygenase-2 expression [116, 117], which may also contribute to the final balance between neuronal protection and death.
CONCLUDING REMARKS
Research conducted during the last decade and summa-rized here provides support for a neuroprotective action of cannabinoids in which various cells types and both CB1 and CB2 receptors are most likely involved (Fig. 4). However, while in several animal models cannabinoids have shown promising symptomatic relief together with decreased neu-ron damage, data from human studies are still very scarce, except perhaps for the alleviation of multiple sclerosis-associated symptoms such as spasticity, tremor, neuropathic pain and nocturia. Besides exerting neuroprotection by act-ing on differentiated neural cells, the functionality of the eCB system in neurogenic areas prompts the study of its po-tential contribution to endogenous processes of neurorepair. This may be of importance not only in the adult brain but perhaps more in the younger, immature brain, which pos-sesses higher plasticity and thus potential of repair. Much
Fig. (4). Summary of cannabinoid receptor-dependent processes involved in neuroprotection. Cannabinoid receptors and their endoge-
nous ligands are present in undifferentiated and differentiated neural cells. Neurons can be protected by cannabinoids through a combination
of cell-autonomous and indirect actions. Cannabinoids also promote astrocyte and oligodendrocyte survival and inhibit microglial activation.
In addition, the eCB system controls neural progenitor cell proliferation and lineage commitment. See text for further details.

Mechanisms of Control of Neuron Survival Current Pharmaceutical Design, 2008, Vol. 14, No. 00 7
more research is nonetheless required to define in detail the molecular mechanisms of the control of neuron generation and survival by the eCB system and the pathophysiological consequences of cannabinoid-evoked neuroprotection.
ACKNOWLEDGEMENTS
T.A. and J.P. are supported by Comunidad Autónoma de Madrid (Spain) and Ministerio de Educación y Ciencia (FPI Program; Spain), respectively. Research in our laboratory is financially supported by Comunidad de Madrid (S-SAL-2006/261 and 950344), Picasso Program (HF2005-0017), Fundación de Investigación Médica Mutua Madrileña Auto-movilística and Ministerio de Educación y Ciencia (SAF2006-00918).
ABBREVIATIONS
AEA = N-arachidonoylethanolamine (anandamide)
2AG = 2-Arachidonoylglycerol
BDNF = Brain-derived neurotrophic factor
FGF-2 = Fibroblast growth factor-2
eCB = Endocannabinoid
EAE = Experimental autoimmune encephalomyelitis
EGF = Epidermal growth factor
ERK = Extracellular signal-regulated kinase
FAAH = Fatty acid amide hydrolase
MAGL = Monoacylglycerol lipase
PI3K = Phosphatidylinositol 3-kinase
THC = 9-Tetrahydrocannabinol
REFERENCES
[1] Gaoni Y, Mechoulam R. Isolation, structure and partial synthesis of an active constituent of hashish. J Am Chem Soc 1964; 86: 1646-7.
[2] Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA,
Griffin G, et al. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 1992; 258: 1946-9.
[3] Mechoulam R, Ben Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR, et al.. Identification of an endogenous 2-
monoglyceride, present in canine gut, that binds to cannabinoid re-ceptors. Biochem Pharmacol 1995; 50: 83-90.
[4] Sugiura T, Kondo S, Sukagawa A, Nakane S, Shinoda A, Itoh K, et al.. 2-Arachidonoylglycerol: a possible endogenous cannabinoid
receptor ligand in brain. Biochem Biophys Res Commun 1995; 215: 89-97.
[5] Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI.
Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 1990; 346: 561-4.
[6] Munro S, Thomas KL, Abu Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature 1993; 365: 61-5.
[7] Freund TF, Katona I, Piomelli D. Role of Endogenous Cannabi-noids in Synaptic Signaling. Physiol Rev 2003; 83: 1017-66.
[8] Chevaleyre V, Takahashi KA, Castillo PE. Endocannabinoid-
mediated synaptic plasticity in the CNS. Annu Rev Neurosci 2006; 29: 37-76.
[9] Skaper SD, Buriani A, Dal Toso R, Petrelli L, Romanello S, Facci L, et al. The ALIAmide palmitoylethanolamide and cannabinoids,
but not anandamide, are protective in a delayed postglutamate paradigm of excitotoxic death in cerebellar granule neurons. Proc
Natl Acad Sci U S A 1996; 93: 3984-9.
[10] Shen M, Thayer SA. Cannabinoid receptor agonists protect cul-
tured rat hippocampal neurons from excitotoxicity. Mol Pharmacol 1998; 54: 459-62.
[11] Nagayama T, Sinor AD, Simon RP, Chen J, Graham SH, Jin K, et al. Cannabinoids and neuroprotection in global and focal cerebral
ischemia and in neuronal cultures. J Neurosci 1999; 19: 2987-95.
[12] Mechoulam R, Shohami E. Endocannabinoids and traumatic brain injury. Mol Neurobiol 2007; 36: 68-74.
[13] van der Stelt M, Veldhuis WB, Bar PR, Veldink GA, Vliegenthart JF, Nicolay K. Neuroprotection by Delta9-tetrahydrocannabinol,
the main active compound in marijuana, against ouabain-induced in vivo excitotoxicity. J Neurosci 2001; 21: 6475-9.
[14] van der Stelt M, Veldhuis WB, van Haaften GW, Fezza F, Bisogno T, Bar PR, Veldink GA, Vliegenthart JF, Di Marzo V, Nicolay K.
Exogenous anandamide protects rat brain against acute neuronal in-jury in vivo. J Neurosci 2001; 21: 8765-71.
[15] Veldhuis WB, van der Stelt M, Wadman MW, van Zadelhoff G,
Maccarrone M, Fezza F, Veldink GA, Vliegenthart JF, Bar PR, Nicolay K, Di Marzo V. Neuroprotection by the endogenous can-
nabinoid anandamide and arvanil against in vivo excitotoxicity in the rat: role of vanilloid receptors and lipoxygenases. J Neurosci
2003; 23: 4127-33.
[16] Panikashvili D, Simeonidou C, Ben-Shabat S, Hanus L, Breuer A,
Mechoulam R, Shohami E. An endogenous cannabinoid (2-AG) is neuroprotective after brain injury. Nature 2001; 413: 527-31.
[17] Fernandez-Lopez D, Martinez-Orgado J, Nunez E, Romero J, Lorenzo P, Moro MA, et al. Characterization of the neuroprotective
effect of the cannabinoid agonist WIN-55212 in an in vitro model of hypoxic-ischemic brain damage in newborn rats. Pediatr Res
2006; 60: 169-73.
[18] Mangieri RA, Piomelli D. Enhancement of endocannabinoid sig-naling and the pharmacotherapy of depression. Pharmacol Res
2007; 56: 360-6.
[19] Karanian DA, Brown QB, Makriyannis A, Kosten TA, Bahr BA.
Dual modulation of endocannabinoid transport and fatty acid amide hydrolase protects against excitotoxicity. J Neurosci 2005; 25:
7813-20.
[20] Zani A, Braida D, Capurro V, Sala M. Delta9-tetrahydrocannabinol
(THC) and AM 404 protect against cerebral ischaemia in gerbils through a mechanism involving cannabinoid and opioid receptors.
Br J Pharmacol 2007; 152: 1301-11.
[21] Kathuria S, Gaetani S, Fegley D, Valino F, Duranti A, Tontini A, et al. Modulation of anxiety through blockade of anandamide hy-
drolysis. Nat Med 2003; 9: 76-81.
[22] Parmentier-Batteur S, Jin K, Mao XO, Xie L, Greenberg DA. In-
creased severity of stroke in CB1 cannabinoid receptor knock-out mice. J Neurosci 2002; 22: 9771-5.
[23] Marsicano G, Goodenough S, Monory K, Hermann H, Eder M, Cannich A, et al. CB1 cannabinoid receptors and on-demand de-
fense against excitotoxicity. Science 2003; 302: 84-8.
[24] Bilkei-Gorzo A, Racz I, Valverde O, Otto M, Michel K, Sastre M, et al. Early age-related cognitive impairment in mice lacking can-
nabinoid CB1 receptors. Proc Natl Acad Sci U S A 2005; 102: 15670-5.
[25] Monory K, Massa F, Egertova M, Eder M, Blaudzun H, Westen-broek R, et al. The endocannabinoid system controls key epilepto-
genic circuits in the hippocampus. Neuron 2006; 51: 455-66.
[26] Pintor A, Tebano MT, Martire A, Grieco R, Galluzzo M, Scattoni
ML, et al. The cannabinoid receptor agonist WIN 55,212-2 attenu-ates the effects induced by quinolinic acid in the rat striatum. Neu-
ropharmacology 2006; 51: 1004-12.
[27] Shouman B, Fontaine RH, Baud O, Schwendimann L, Keller M, Spedding M, et al. Endocannabinoids potently protect the newborn
brain against AMPA-kainate receptor-mediated excitotoxic dam-age. Br J Pharmacol 2006; 148: 442-51.
[28] Melis M, Pillolla G, Bisogno T, Minassi A, Petrosino S, Perra S, et al. Protective activation of the endocannabinoid system during
ischemia in dopamine neurons. Neurobiol Dis 2006; 24: 15-27.
[29] Guan XL, He X, Ong WY, Yeo WK, Shui G, Wenk MR. Non-
targeted profiling of lipids during kainate-induced neuronal injury. Faseb J 2006; 20: 1152-61.
[30] Hansen H, Moesgaard B, Petersen G. Putative neuroprotective
actions of N-acyl-ethanolamines. Pharmacol Ther 2002; 95: 119.

8 Current Pharmaceutical Design, 2008, Vol. 14, No. 00 Galve-Roperh et al.
[31] Franklin A, Parmentier-Batteur S, Walter L, Greenberg DA, Stella
N. Palmitoylethanolamide increases after focal cerebral ischemia and potentiates microglial cell motility. J Neurosci 2003; 23: 7767-
75.
[32] Berger C, Schmid PC, Schabitz WR, Wolf M, Schwab S, Schmid
HH. Massive accumulation of N-acylethanolamines after stroke. Cell signalling in acute cerebral ischemia? J Neurochem 2004; 88:
1159-67.
[33] Jin KL, Mao XO, Goldsmith PC, Greenberg DA. CB1 cannabinoid receptor induction in experimental stroke. Ann Neurol 2000; 48:
257-61.
[34] Aguado T, Romero E, Monory K, Palazuelos J, Sendtner M, Mar-
sicano G, et al. The CB1 cannabinoid receptor mediates excitotox-icity-induced neural progenitor proliferation and neurogenesis. J
Biol Chem 2007; 282: 23892-8.
[35] Clement AB, Hawkins EG, Lichtman AH, Cravatt BF. Increased
seizure susceptibility and proconvulsant activity of anandamide in mice lacking fatty acid amide hydrolase. J Neurosci 2003; 23:
3916-23.
[36] Zhao C, Deng W, Gage FH. Mechanisms and functional implica-tions of adult neurogenesis. Cell 2008; 132: 645-60.
[37] Gould E. How widespread is adult neurogenesis in mammals? Nat Rev Neurosci 2007; 8: 481-8.
[38] Shors TJ, Miesegaes G, Beylin A, Zhao M, Rydel T, Gould E. Neurogenesis in the adult is involved in the formation of trace
memories. Nature 2001; 410: 372-6.
[39] Guillemot F. Cell fate specification in the mammalian telencepha-lon. Prog Neurobiol 2007; 83: 37-52.
[40] Harkany T, Guzman M, Galve-Roperh I, Berghuis P, Devi LA, Mackie K. The emerging functions of endocannabinoid signaling
during CNS development. Trends Pharmacol Sci 2007; 28: 83-92.
[41] Galve-Roperh I, Aguado T, Palazuelos J, Guzman M. The endo-
cannabinoid system and neurogenesis in health and disease. Neuro-scientist 2007; 13: 109-14.
[42] Pacher P, Batkai S, Kunos G. The endocannabinoid system as an
emerging target of pharmacotherapy. Pharmacol Rev 2006; 58: 389-462.
[43] Jin K, Xie L, Kim SH, Parmentier-Batteur S, Sun Y, Mao XO, et al. Defective adult neurogenesis in CB1 cannabinoid receptor
knockout mice. Mol Pharmacol 2004; 66: 204-8.
[44] Aguado T, Monory K, Palazuelos J, Stella N, Cravatt B, Lutz B, et
al. The endocannabinoid system drives neural progenitor prolifera-tion. FASEB J 2005; 19: 1704-6.
[45] Palazuelos J, Aguado T, Egia A, Mechoulam R, Guzman M,
Galve-Roperh I. Non-psychoactive CB2 cannabinoid agonists stimulate neural progenitor proliferation. FASEB J 2006; 20: 2405-
7.
[46] Jiang W, Zhang Y, Xiao L, Van Cleemput J, Ji SP, Bai G, et al.
Cannabinoids promote embryonic and adult hippocampus neuro-genesis and produce anxiolytic- and antidepressant-like effects. J
Clin Invest 2005; 115: 3104-16.
[47] Aguado T, Palazuelos J, Monory K, Stella N, Cravatt B, Lutz B, et
al. The endocannabinoid system promotes astroglial differentiation by acting on neural progenitor cells. J Neurosci 2006; 26: 1551-61.
[48] Kim D, Thayer SA. Cannabinoids inhibit the formation of new
synapses between hippocampal neurons in culture. J Neurosci 2001; 21: RC146.
[49] Rueda D, Navarro B, Martinez-Serrano A, Guzman M, Galve-Roperh I. The endocannabinoid anandamide inhibits neuronal pro-
genitor cell differentiation through attenuation of the RAP1/B-RAF/ERK pathway. J Biol Chem 2002; 16: 16.
[50] Molina-Holgado F, Rubio-Araiz A, Garcia-Ovejero D, Williams RJ, Moore JD, Arevalo-Martin A, et al. CB2 cannabinoid receptors
promote mouse neural stem cell proliferation. Eur J Neurosci 2007; 25: 629-34.
[51] Molina-Holgado F, Pinteaux E, Heenan L, Moore JD, Rothwell NJ,
Gibson RM. Neuroprotective effects of the synthetic cannabinoid HU-210 in primary cortical neurons are mediated by phosphatidy-
linositol 3-kinase/AKT signaling. Mol Cell Neurosci 2005; 28: 189-94.
[52] Ozaita A, Puighermanal E, Maldonado R. Regulation of PI3K/Akt/GSK-3 pathway by cannabinoids in the brain. J Neuro-
chem 2007; 102: 1105-14.
[53] Gomez del Pulgar T, Velasco G, Guzman M. The CB1 cannabinoid
receptor is coupled to the activation of protein kinase B/Akt. Bio-chem J 2000; 347: 369-73.
[54] Gomez Del Pulgar T, De Ceballos ML, Guzman M, Velasco G. Cannabinoids protect astrocytes from ceramide-induced apoptosis
through the phosphatidylinositol 3-kinase/protein kinase B path-way. J Biol Chem 2002; 277: 36527-33.
[55] Molina-Holgado E, Vela JM, Arevalo-Martin A, Almazan G,
Molina-Holgado F, Borrell J, et al. Cannabinoids promote oli-godendrocyte progenitor survival: involvement of cannabinoid re-
ceptors and phosphatidylinositol-3 kinase/Akt signaling. J Neurosci 2002; 22: 9742-53.
[56] Galve-Roperh I, Rueda D, Gomez Del Pulgar T, Velasco G, Guz-man M. Mechanism of extracellular signal-regulated kinase activa-
tion by the CB1 cannabinoid receptor. Mol Pharmacol 2002; 62: 1385-92.
[57] Hart S, Fischer OM, Ullrich A. Cannabinoids induce cancer cell proliferation via tumor necrosis factor alpha-converting enzyme
(TACE/ADAM17)-mediated transactivation of the epidermal growth factor receptor. Cancer Res 2004; 64: 1943-50.
[58] Derkinderen P, Valjent E, Toutant M, Corvol JC, Enslen H, Ledent
C, et al. Regulation of extracellular signal-regulated kinase by can-nabinoids in hippocampus. J Neurosci 2003; 23: 2371-82.
[59] Valjent E, Pages C, Rogard M, Besson MJ, Maldonado R, Caboche J. Delta 9-tetrahydrocannabinol-induced MAPK/ERK and Elk-1
activation in vivo depends on dopaminergic transmission. Eur J Neurosci 2001; 14: 342-52.
[60] Moranta D, Esteban S, Garcia-Sevilla JA. Acute, chronic and with-drawal effects of the cannabinoid receptor agonist WIN55212-2 on
the sequential activation of MAPK/Raf-MEK-ERK signaling in the rat cerebral frontal cortex: short-term regulation by intrinsic and
extrinsic pathways. J Neurosci Res 2007; 85: 656-67.
[61] Tonini R, Ciardo S, Cerovic M, Rubino T, Parolaro D, Mazzanti M, et al. ERK-dependent modulation of cerebellar synaptic plastic-
ity after chronic Delta9-tetrahydrocannabinol exposure. J Neurosci 2006; 26: 5810-8.
[62] Bouaboula M, Poinot-Chazel C, Bourrie B, Canat X, Calandra B, Rinaldi-Carmona M, et al. Activation of mitogen-activated protein
kinases by stimulation of the central cannabinoid receptor CB1. Biochem J 1995; 312: 637-41.
[63] Sanchez C, Galve-Roperh I, Rueda D, Guzman M. Involvement of sphingomyelin hydrolysis and the mitogen-activated protein kinase
cascade in the Delta9-tetrahydrocannabinol-induced stimulation of glucose metabolism in primary astrocytes. Mol Pharmacol 1998a;
54: 834-43.
[64] Davis MI, Ronesi J, Lovinger DM. A predominant role for inhibi-tion of the adenylate cyclase/protein kinase A pathway in ERK ac-
tivation by cannabinoid receptor 1 in N1E-115 neuroblastoma cells. J Biol Chem 2003; 278: 48973-80.
[65] Kim SH, Won SJ, Mao XO, Jin K, Greenberg DA. Molecular mechanisms of cannabinoid protection from neuronal excitotoxic-
ity. Mol Pharmacol 2005;
[66] Porcella A, Gessa GL, Pani L. Delta9-tetrahydrocannabinol in-
creases sequence-specific AP-1 DNA-binding activity and Fos-related antigens in the rat brain. Eur J Neurosci 1998; 10: 1743-51.
[67] Zhao Q, He Z, Chen N, Cho YY, Zhu F, Lu C, et al. 2-
Arachidonoylglycerol stimulates activator protein-1-dependent transcriptional activity and enhances epidermal growth factor-
induced cell transformation in JB6 P+ cells. J Biol Chem 2005; 280: 26735-42.
[68] Bouaboula M, Bourrie B, Rinaldi Carmona M, Shire D, Le Fur G, Casellas P. Stimulation of cannabinoid receptor CB1 induces krox-
24 expression in human astrocytoma cells. J Biol Chem 1995b; 270: 13973-80.
[69] Dorsam RT, Gutkind JS. G-protein-coupled receptors and cancer. Nat Rev Cancer 2007; 7: 79-94.
[70] Jordan JD, He JC, Eungdamrong NJ, Gomes I, Ali W, Nguyen T, et
al. Cannabinoid receptor-induced neurite outgrowth is mediated by Rap1 activation through G(alpha)o/i-triggered proteasomal degra-
dation of Rap1GAPII. J Biol Chem 2005; 280: 11413-21.
[71] Preet A, Ganju RK, Groopman JE. Delta9-Tetrahydrocannabinol
inhibits epithelial growth factor-induced lung cancer cell migration in vitro as well as its growth and metastasis in vivo. Oncogene
2008; 27: 339-46.

Mechanisms of Control of Neuron Survival Current Pharmaceutical Design, 2008, Vol. 14, No. 00 9
[72] Rubovitch V, Gafni M, Sarne Y. The involvement of VEGF recep-
tors and MAPK in the cannabinoid potentiation of Ca2+ flux into N18TG2 neuroblastoma cells. Brain Res Mol Brain Res 2004; 120:
138-44.
[73] Khaspekov LG, Brenz Verca MS, Frumkina LE, Hermann H, Mar-
sicano G, Lutz B. Involvement of brain-derived neurotrophic factor in cannabinoid receptor-dependent protection against excitotoxic-
ity. Eur J Neurosci 2004; 19: 1691-8.
[74] De March Z, Zuccato C, Giampa C, Patassini S, Bari M, Gasperi V, et al. Cortical expression of brain derived neurotrophic factor and
type-1 cannabinoid receptor after striatal excitotoxic lesions. Neu-roscience 2008; 152: 734-40.
[75] Berghuis P, Dobszay MB, Wang X, Spano S, Ledda F, Sousa KM, et al. Endocannabinoids regulate interneuron migration and
morphogenesis by transactivating the TrkB receptor. Proc Natl Acad Sci U S A 2005; 102: 19115-20.
[76] Williams EJ, Walsh FS, Doherty P. The FGF receptor uses the endocannabinoid signaling system to couple to an axonal growth
response. J Cell Biol 2003; 160: 481-6.
[77] Pryce G, Baker D. Emerging properties of cannabinoid medicines in management of multiple sclerosis. Trends Neurosci 2005; 28:
272-6.
[78] Pertwee RG. Cannabinoids and multiple sclerosis. Mol Neurobiol
2007; 36: 45-59.
[79] Pryce G, Ahmed Z, Hankey DJ, Jackson SJ, Croxford JL, Pocock
JM, et al. Cannabinoids inhibit neurodegeneration in models of multiple sclerosis. Brain 2003; 126: 2191-202.
[80] Maresz K, Pryce G, Ponomarev ED, Marsicano G, Croxford JL,
Shriver LP, et al. Direct suppression of CNS autoimmune inflam-mation via the cannabinoid receptor CB1 on neurons and CB2 on
autoreactive T cells. Nat Med 2007; 13: 492-7.
[81] Arevalo-Martin A, Vela JM, Molina-Holgado E, Borrell J, Guaza
C. Therapeutic action of cannabinoids in a murine model of multi-ple sclerosis. J Neurosci 2003; 23: 2511-6.
[82] Palazuelos J, Davoust N, Julien B, Hatterer E, Aguado T, Mechou-lam R, et al. The CB2 cannabinoid receptor controls myeloid pro-
genitor trafficking: involvement in the pathogenesis of an animal model of multiple sclerosis. J Biol Chem 2008; 283: 13320-9.
[83] Ni X, Geller EB, Eppihimer MJ, Eisenstein TK, Adler MW, Tuma
RF. Win 55212-2, a cannabinoid receptor agonist, attenuates leu-kocyte/endothelial interactions in an experimental autoimmune en-
cephalomyelitis model. Mult Scler 2004; 10: 158-64.
[84] Zhang M, Martin BR, Adler MW, Razdan RK, Jallo JI, Tuma RF.
Cannabinoid CB2 receptor activation decreases cerebral infarction in a mouse focal ischemia/reperfusion model. J Cereb Blood Flow
Metab 2007; 27: 1387-96.
[85] Docagne F, Muneton V, Clemente D, Ali C, Loria F, Correa F, et
al. Excitotoxicity in a chronic model of multiple sclerosis: Neuro-protective effects of cannabinoids through CB1 and CB2 receptor
activation. Mol Cell Neurosci 2007; 34: 551-61.
[86] Fernandez-Ruiz J, Romero J, Velasco G, Tolon RM, Ramos JA, Guzman M. Cannabinoid CB2 receptor: a new target for control-
ling neural cell survival? Trends Pharmacol Sci 2007; 28: 39-45.
[87] Maresz K, Carrier EJ, Ponomarev ED, Hillard CJ, Dittel BN.
Modulation of the cannabinoid CB2 receptor in microglial cells in response to inflammatory stimuli. J Neurochem 2005; 95: 437-45.
[88] Benito C, Romero JP, Tolon RM, Clemente D, Docagne F, Hillard CJ, et al. Cannabinoid CB1 and CB2 receptors and fatty acid amide
hydrolase are specific markers of plaque cell subtypes in human multiple sclerosis. J Neurosci 2007; 27: 2396-402.
[89] Ramirez BG, Blazquez C, Gomez del Pulgar T, Guzman M, de
Ceballos ML. Prevention of Alzheimer's disease pathology by can-nabinoids: neuroprotection mediated by blockade of microglial ac-
tivation. J Neurosci 2005; 25: 1904-13.
[90] Witting A, Chen L, Cudaback E, Straiker A, Walter L, Rickman B,
et al. Experimental autoimmune encephalomyelitis disrupts endo-cannabinoid-mediated neuroprotection. Proc Natl Acad Sci U S A
2006; 103: 6362-7.
[91] Eljaschewitsch E, Witting A, Mawrin C, Lee T, Schmidt PM, Wolf
S, et al. The endocannabinoid anandamide protects neurons during CNS inflammation by induction of MKP-1 in microglial cells. Neu-
ron 2006; 49: 67-79.
[92] Hanisch UK, Kettenmann H. Microglia: active sensor and versatile
effector cells in the normal and pathologic brain. Nat Neurosci 2007; 10: 1387-94.
[93] Davoust N, Vuaillat C, Cavillon G, Domenget C, Hatterer E, Ber-nard A, et al. Bone marrow CD34+/B220+ progenitors target the
inflamed brain and display in vitro differentiation potential toward microglia. FASEB J 2006; 20: 2081-92.
[94] Hampson AJ, Grimaldi M, Axelrod J, Wink D. Cannabidiol and (-
)Delta9-tetrahydrocannabinol are neuroprotective antioxidants. Proc Natl Acad Sci U S A 1998; 95: 8268-73.
[95] Marsicano G, Moosmann B, Hermann H, Lutz B, Behlt C. Neuro-protective properties of cannabinoids against oxidative stress: role
of the cannabinoid receptor CB1. J Neurochem 2002; 80: 448-56.
[96] Hayakawa K, Mishima K, Nozako M, Hazekawa M, Irie K, Fuji-
oka M, et al. Delayed treatment with cannabidiol has a cerebropro-tective action via a cannabinoid receptor-independent myeloper-
oxidase-inhibiting mechanism. J Neurochem 2007; 102: 1488-96.
[97] Lastres-Becker I, Molina-Holgado F, Ramos JA, Mechoulam R, Fernandez-Ruiz J. Cannabinoids provide neuroprotection against 6-
hydroxydopamine toxicity in vivo and in vitro: Relevance to Park-inson's disease. Neurobiol Dis 2005; 19: 96-107.
[98] Kozak KR, Crews BC, Morrow JD, Wang LH, Ma YH, Weinander R, et al. Metabolism of the endocannabinoids, 2-arachidonylglyce-
rol and anandamide, into prostaglandin, thromboxane, and prosta-cyclin glycerol esters and ethanolamides. J Biol Chem 2002; 277:
44877-85.
[99] Sang N, Zhang J, Chen C. PGE2 glycerol ester, a COX-2 oxidative
metabolite of 2-arachidonoyl glycerol, modulates inhibitory synap-tic transmission in mouse hippocampal neurons. J Physiol 2006;
572: 735-45.
[100] Correa F, Docagne F, Clemente D, Mestre L, Becker C, Guaza C. Anandamide inhibits IL-12p40 production by acting on the pro-
moter repressor element GA-12: possible involvement of the COX-2 metabolite prostamide E2. Biochem J 2008; 409: 761-70.
[101] Matas D, Juknat A, Pietr M, Klin Y, Vogel Z. Anandamide protects from low serum-induced apoptosis via its degradation to ethanola-
mine. J Biol Chem 2007; 282: 7885-92.
[102] Lo Verme J, Fu J, Astarita G, La Rana G, Russo R, Calignano A, et
al. The nuclear receptor peroxisome proliferator-activated receptor-alpha mediates the anti-inflammatory actions of palmitoylethano-
lamide. Mol Pharmacol 2005; 67: 15-9.
[103] Ryberg E, Larsson N, Sjogren S, Hjorth S, Hermansson NO, Leonova J, et al. The orphan receptor GPR55 is a novel cannabi-
noid receptor. Br J Pharmacol 2007; 152: 1092-101.
[104] Sun Y, Alexander SP, Garle MJ, Gibson CL, Hewitt K, Murphy
SP, et al. Cannabinoid activation of PPAR alpha; a novel neuropro-tective mechanism. Br J Pharmacol 2007; 152: 734-43.
[105] Maccarrone M, Finazzi-Agro A. The endocannabinoid system, anandamide and the regulation of mammalian cell apoptosis. Cell
Death Differ 2003; 10: 946-55.
[106] Cernak I, Vink R, Natale J, Stoica B, Lea PMt, Movsesyan V, et al. The "dark side" of endocannabinoids: a neurotoxic role for anan-
damide. J Cereb Blood Flow Metab 2004; 24: 564-78.
[107] Bari M, Battista N, Fezza F, Finazzi-Agro A, Maccarrone M. Lipid
rafts control signaling of type-1 cannabinoid receptors in neuronal cells. Implications for anandamide-induced apoptosis. J Biol Chem
2005; 280: 12212-20.
[108] Hansen HH, Azcoitia I, Pons S, Romero J, Garcia-Segura LM,
Ramos JA, et al. Blockade of cannabinoid CB1 receptor function protects against in vivo disseminating brain damage following
NMDA-induced excitotoxicity. J Neurochem 2002; 82: 154-8.
[109] Pegorini S, Zani A, Braida D, Guerini-Rocco C, Sala M. Vanilloid VR1 receptor is involved in rimonabant-induced neuroprotection.
Br J Pharmacol 2006; 147: 552-9.
[110] Muthian S, Rademacher DJ, Roelke CT, Gross GJ, Hillard CJ.
Anandamide content is increased and CB1 cannabinoid receptor blockade is protective during transient, focal cerebral ischemia.
Neuroscience 2004; 129: 743-50.
[111] Chan GC, Hinds TR, Impey S, Storm DR. Hippocampal neurotox-
icity of Delta9-tetrahydrocannabinol. J Neurosci 1998; 18: 5322-32.
[112] Downer EJ, Fogarty MP, Campbell VA. Tetrahydrocannabinol-
induced neurotoxicity depends on CB1 receptor-mediated c-Jun N-

10 Current Pharmaceutical Design, 2008, Vol. 14, No. 00 Galve-Roperh et al.
terminal kinase activation in cultured cortical neurons. Br J Phar-
macol 2003; 140: 547-57.
[113] Derkinderen P, Ledent C, Parmentier M, Girault JA. Cannabinoids
activate p38 mitogen-activated protein kinases through CB1 recep-tors in hippocampus. J Neurochem 2001; 77: 957-60.
[114] Movsesyan VA, Stoica BA, Yakovlev AG, Knoblach SM, Lea PM,
Cernak I, et al. Anandamide-induced cell death in primary neuronal cultures: role of calpain and caspase pathways. Cell Death Differ
2004; 11: 1121-32.
[115] Gowran A, Campbell VA. A role for p53 in the regulation of
lysosomal permeability by delta9-tetrahydrocannabinol in rat corti-
cal neurones: implications for neurodegeneration. J Neurochem
2008; 105: 1513-24.
[116] Mestre L, Correa F, Docagne F, Clemente D, Guaza C. The syn-
thetic cannabinoid WIN 55,212-2 increases COX-2 expression and PGE2 release in murine brain-derived endothelial cells following
Theiler's virus infection. Biochem Pharmacol 2006; 72: 869-80.
[117] Ramer R, Weinzierl U, Schwind B, Brune K, Hinz B. Ceramide is involved in r(+)-methanandamide-induced cyclooxygenase-2 ex-
pression in human neuroglioma cells. Mol Pharmacol 2003; 64: 1189-98.