manual para la evaluaciÓn de autoridades ... manual para la evaluaciÓn de autoridades reguladoras...
TRANSCRIPT

1
MANUAL PARA LA EVALUACIÓN DE AUTORIDADES REGULADORAS NACIONALES DE REFERENCIA
REGIONAL PARA MEDICAMENTOS Y PRODUCTOS BIOLOGICOS
Documento de trabajo adaptado conforme a requerimientos del Proyecto de "Autoridades Reguladoras Nacionales de Referencia Regional". Documentos base:
Practical Guidance for Conducting a Review (based on the WHO Data Collection Tool for the Review of Drug Regulatory Systems),
A WHO manual for assessment of the national regulatory system for vaccines,
Resolución CD 50.R9 "Fortalecimiento de las autoridades reguladoras nacionales de medicamentos y productos biológicos", 50.º Consejo Directivo, 62.a Sesión del Comité Regional de la Organización Mundial de la Salud para las Américas; 27 septiembre al 1 de octubre del 2010.
OPS 2011

2
Agradecimientos:
Este documento es el resultado de la colaboración entre diferentes instituciones y profesionales, especial reconocimiento para la Administración Nacional de Medicamentos, Alimentos y Tecnología Médica (ANMAT) de Argentina, Agencia Nacional de Vigilancia Sanitaria (ANVISA) de Brasil, Instituto de Salud Pública de Chile (ISP), Instituto Nacional de Vigilancia de Medicamentos y Alimentos (INVIMA) de Colombia, Centro para el Control Estatal de la Calidad de los Medicamentos (CECMED) de Cuba, Comisión Federal para la Protección contra Riesgos Sanitarios (COFEPRIS) de México y el Instituto Nacional de Higiene Rafael Rangel (INHRR) de Venezuela. Para los profesionales que han participado en la actualización y mejoramiento del instrumento de evaluación de Autoridades Reguladoras de Medicamentos: Patricia Aprea, Marina Rossi, María José Sánchez y Mónica Bobbi de ANMAT, Argentina; Celeste Sánchez y Biorkys Yáñez del CECMED, Cuba; Patricia Carmona e Isabel Sánchez del ISP de Chile; Renata Carvalho, Laura Castanheira, Patricia Andreotti y Tiago Rauber de ANVISA, Brasil ; Martha Rodríguez, Patricia León y Melissa Triana de INVIMA, Colombia; Ileana Herrera de la Dirección de Drogas de Costa Rica; Martha García de COFEPRIS, México y Ana Agaton del INHRR, Venezuela. El documento fue preparado bajo la coordinación y el apoyo de los profesionales del Proyecto de Medicamentos y Tecnologías Sanitarias de la OPS con las contribuciones de Maria Luz Pombo, Maria de los Ángeles Cortes, José María Parisi y José Luis Castro.
La versión final del documento fue editada por James Fitzgerald, Coordinador, Proyecto de Medicamentos y Tecnologías Sanitarias y José Peña Ruz, Asesor Regional de Asuntos Regulatorios. Las reuniones para el desarrollo y discusión del presente Manual fueron llevadas a cabo con el apoyo financiero brindado por la Canadian International Immunization Initiative Project CIII, Phase II, en su capitulo de Calidad de Vacunas, en el rubro de fortalecimiento de Agencias Reguladoras.

3
Tabla de contenidos:
Abreviaturas 4
Glosario 5
Introducción 6
Proceso de evaluación de la capacidad reguladora de medicamentos y productos
biológicos
11
Herramienta de recolección de datos 17
1. Información general - Módulo 1 20
2. Autoridad Reguladora Nacional - Módulo 2 21
3. Autorización de comercialización - Módulo 3 44
4. Licenciamiento de fabricantes - Módulo 4 57
5. Vigilancias y control posteriores a la comercialización - Módulo 5 69
6. Farmacovigilancia - Módulo 6 80
7. Ensayos clínicos - Módulo 7 88
8. Inspecciones regulatorias y actividades de fiscalización- Módulo 8 98
9. Laboratorio de control de calidad - Módulo 9 120
10. Liberación de lotes – Módulo 10 139
Anexos
Anexo 1: Términos de referencia para el equipo de evaluación 147
Anexo 2: Perfil de evaluadores regulador recomendado 148
Anexo 3: Ejemplo de un acuerdo de confidencialidad 149
Anexo 4: Ejemplo para evaluadores de una declaración de no conflicto de intereses 150
Anexo 5: Proyecto de programa de una evaluación reguladora 151
Anexo 6: Lista de instituciones a visitarse y miembros del personal a ser reunido 152
Anexo 7: Ejemplo del contenido de un formato de informe de evaluación 153
Anexo 8: Ejemplo de formato de un plan de acción correctiva 154
Anexo 9. Indicadores cuantitativos para propósitos reguladores 155
Anexo 10. Fuente principal de evidencia documentada 159

4
Abreviaturas
AC Autorización de comercialización ARN Autoridad Reguladora Nacional BPC Buenas Prácticas Clínicas BPD Buenas Prácticas de Distribución BPL Buenas Prácticas de Laboratorio BPM Buenas Prácticas de Manufactura CEI Comité de Ética Institucional CRO Compañía de Investigación por Contrato EC Estudio Clínico ESAVI Evento Supuestamente atribuido a la vacunación o inmunización ICH Conferencia Internacional de Armonización ISO Organización Internacional de Estandarización LOCM Laboratorio Oficial de Control de Medicamentos MS Ministerio de Salud OMS Organización Mundial de la Salud ONG Organización No Gubernamental OPS Organización Panamericana de la Salud PAI Programa Ampliado de Inmunizaciones PARF Red Panamericana para la Armonización de la Reglamentación Farmacéutica. PIC/S Pharmaceutical Inspection Cooperation Scheme POE Procedimiento Operativo Estandarizado RAM Reacciones Adversas a Medicamentos RS Registro Sanitario RCP Resumen de las Características del Producto SGC Sistema de gestión de Calidad

5
Glosario: Producto Farmacéutico: preparado que contiene uno o varios principios activos y excipientes, formulados en una forma farmacéutica o de dosificación. Medicamento: producto farmacéutico empleado para la prevención, diagnóstico, tratamiento, de una enfermedad o estado patológico, o para modificar sistemas fisiológicos en beneficio de la persona a la que le fue administrado. Esta definición incluye los medicamentos de origen biológico (productos biológicos) y de síntesis química1.
Productos biológicos: sustancias empleadas para fines de prevención (vacunas), del tratamiento (ejemplo: citoquinas y hormonas), o del diagnóstico de ciertas enfermedades (anticuerpos) y que son obtenidas a partir de organismos vivos, o sus derivados. Entre ellos se incluyen a los virus, sueros terapéuticos, toxinas, antitoxinas, vacunas, sangre, componentes o derivados de la sangre, productos alergénicos, hormonas, factores estimulantes de colonias, citoquinas, anticuerpos, etc., que son clasificados de acuerdo a su origen1.
Productos biotecnológicos: corresponden a los productos biológicos que han sido obtenidos mediante biotecnología, a partir de cultivos de células animales, así como proteínas obtenidas por la técnica del ADN recombinante expresadas en tejidos animales o en formas de vida microbianas, incluyendo a los anticuerpos monoclonales. En su mayoría estos productos son empleados en terapias de enfermedades crónicas1.
Tecnologías sanitarias: aplicación de conocimientos teóricos y prácticos estructurados en forma de dispositivos, medicamentos, vacunas, procedimientos y sistemas elaborados para resolver problemas sanitarios y mejorar la calidad de vida2. Licenciamiento de productores: hace referencia al permiso de instalación y funcionamiento de productores, importadores y distribuidores, en algunos países se designa a esta actividad como registro nacional de establecimientos o registro de empresas.
Introducción La función de la Organización Mundial de la Salud en la regulación de medicamentos esta basada en su mandato constitucional y varias resoluciones de la Asamblea Mundial de la Salud. Estos requieren a la OPS/OMS que apoye a los Estados Miembros en sus esfuerzos para implementar políticas de medicamentos y programas nacionales, para asegurar la equidad de acceso a medicamentos esenciales, seguridad y calidad, y el uso apropiado de medicamentos. Más específicamente, la OPS/OMS es requerida para proveer orientación y apoyo para establecer mecanismos reguladores eficaces de medicamentos nacionales, para asegurar que los medicamentos disponibles son de buena calidad y para luchar contra medicamentos falsificados. Resolución CD 50.R93.
1 Organización Panamericana de la Salud. Glosario de medicamentos: Desarrollo, evaluación y uso. Primera edición. Washington D.C.:
OPS; 1999. Disponible en
http://paho.publisher.ingentaconnect.com/content/paho/paho999/1999/00000001/00000001;jsessionid=3dv5qfulfl6lu.alice
2 Resolución WHA 60.29 “Tecnologías sanitarias”. OMS; 2007.
3 Organización Panamericana de la Salud. Resolución: CD50.R9 “Fortalecimiento de las autoridades reguladoras nacionales de medicamentos y
productos biológicos”. OPS; 2010. Disponible en:

6
El Consejo Directivo de la Organización Panamericana de la Salud aprobó en septiembre del 2010 la Resolución CD 50.R9 "Fortalecimiento de las autoridades reguladoras nacionales de medicamentos y productos biológicos". Dicha resolución está basada en el documento CD50/20 el cual describe una propuesta de fortalecimiento de la capacidad reguladora, cuyas finalidades principales son:
Garantizar la calidad, inocuidad y eficacia de los medicamentos y productos biológicos.
Designar autoridades regionales de referencia que cooperen con las instituciones de los países.
Como parte del fortalecimiento de las Autoridades Reguladoras los estados miembros y la OPS coordinaron el desarrollo e implementación de un procedimiento de calificación con el fin de calificar el desempeño de las autoridades nacionales respecto de las funciones básicas establecidas por la Organización Mundial de la Salud (OMS), y establecer su capacidad de liderazgo en las Américas. La designación de autoridades reguladoras de referencia regional permitirá reconocer las capacidades reguladoras instaladas en la Región, promover el intercambio de información entre los países en la regulación sanitaria de medicamentos y productos biológicos, además de contribuir al fortalecimiento de otras autoridades reguladoras nacionales. Por lo anteriormente señalado el Comité Directivo, Resolvió: 1. Instar a los Estados Miembros a que:
a) fortalezcan y evalúen su capacidad reguladora con respecto a las funciones propias de un organismo de regulación y fiscalización de medicamentos y productos biológicos, mediante un examen del cumplimiento de sus funciones esenciales;
b) utilicen los resultados de la calificación y la designación de la autoridad reguladora de referencia regional para fortalecer su desempeño en cuanto a la función rectora de la autoridad sanitaria;
c) apoyen a las autoridades reguladoras nacionales para que puedan beneficiarse de los procesos y la información de las autoridades reguladoras nacionales de referencia; d) promuevan la difusión de información sobre los resultados y procesos de regulación y fiscalización de medicamentos, productos biológicos y otras tecnologías sanitarias; e) promuevan el intercambio y la cooperación técnica entre países; f) participen activamente de la Red Panamericana para la Armonización de la Reglamentación Farmacéutica (Red PARF).
2. Solicitar a la Directora:
a) que apoye las iniciativas para el fortalecimiento y calificación de las autoridades reguladoras nacionales a fin de garantizar la calidad, inocuidad y eficacia de los medicamentos, biológicos y otras tecnologías sanitarias; b) que difunda ampliamente en los países de la Región de las Américas las herramientas y los procedimientos disponibles para la calificación de las competencias de las autoridades reguladoras
http://new.paho.org/hq/index.php?option=com_content&task=view&id=3149&Itemid=2401&lang=es

7
nacionales de medicamentos y productos biológicos y brinde apoyo para la elaboración del sistema de calificación de las autoridades reguladoras nacionales y su designación como autoridad reguladora de referencia regional; c) que mantenga y fortalezca la colaboración de la Organización Panamericana de la Salud con los Estados Miembros en materia de regulación en medicamentos y biológicos; d) que promueva la cooperación técnica entre las autoridades reguladoras de los países así como el reconocimiento de las capacidades instaladas en la Región;
e) que vele por que los procesos de compra de medicamentos y productos biológicos de la Organización Panamericana de la Salud se basen en la capacidad instalada de las autoridades reguladoras nacionales de referencia para garantizar la calidad, inocuidad y eficacia de dichos productos.

8
Objetivo:
El objetivo de este Manual es proporcionar a las Autoridades Reguladoras Nacionales una herramienta que les permita llevar a cabo la evaluación del cumplimiento de las funciones básicas que deben desempeñar como entes reguladores de medicamentos y productos biológicos. Alcance: Este Manual esta dirigido a la evaluación integral de las Autoridades Reguladoras Nacionales en sus capacidades de regulación de medicamentos y productos biológicos. Se incluye la evaluación de las funciones de regulación generales que son comunes a todos los medicamentos, considerando elementos que involucran fundamentalmente los medicamentos de síntesis química y la regulación de las vacunas. Se espera que
revisiones posteriores incluyan elementos de la regulación particular para otros productos biológicos de uso en la Región de América Latina y el Caribe, como lo son los derivados sanguíneos, sueros híper inmunes y productos biotecnológicos entre otros. Antecedentes La experiencia en países desarrollados ha mostrado que el desarrollo de las capacidades reguladoras ocurre en fases, durante un largo período de tiempo. Factores como el nivel de desarrollo del sector farmacéutico, la disponibilidad de recursos humanos capacitados, infraestructura, el tamaño y sofisticación de la autoridad reguladora y recursos financieros influyen en que las funciones de regulación puedan llevarse a cabo. Los países en desarrollo que dependen totalmente de productos importados y que cuentan con limitado personal calificado y otros recursos, necesitarán por lo tanto comenzar con actividades prioritarias limitadas, ampliándolas gradualmente conforme su sector farmacéutico se desarrolla y recursos empiezan a estar disponibles. Una regulación deficiente o inadecuada puede conducir a la disponibilidad de medicamentos con deficiencias en su calidad respecto de los estándares establecidos, nocivos e ineficaces en los mercados nacionales y en el comercio internacional. Esto puede resultar en un gran daño a la salud de los consumidores individuales e incluso a la salud de una población mayor. Por consiguiente, los países continuamente deben fortalecer responsabilidades claves de regulación de medicamentos para garantizar la seguridad, calidad y eficacia de medicamentos y la exactitud de la información sobre el producto. Los países deben evaluar su desempeño regulador de medicamentos usando indicadores que se enfocan en las estructuras, procesos y resultados. Deben identificar toda fortaleza o debilidad con motivo de considerar opciones reguladoras alternativas, eligiendo las más apropiadas y prácticas. La herramienta de recaudación de datos para la revisión de sistemas reguladores de medicamentos y el Manual para la evaluación del Sistema Regulatorio nacional para vacunas de la OMS han sido diseñados para ayudar a las autoridades reguladoras a desempeñar tal evaluación. En la Región de la Américas, esta herramienta ha sido adaptada a las necesidades del proyecto de Autoridades Reguladoras Nacionales de Referencia y se encuentra incluida en el presente documento. Principios de la regulación de medicamentos
Las estructuras de regulación de medicamentos que existen hoy (leyes, agencias reguladoras, juntas de evaluación, laboratorios nacionales de control de calidad (LOCM), centros de información de medicamentos, etc.) han evolucionado con el transcurso del tiempo. La regulación de medicamentos es una política pública en respuesta a los problemas o necesidades percibidas de la sociedad. Cada país establece su propio marco regulador y prioridades según los riesgos para la salud existentes o esperados y debe adaptarse continuamente a las cambiantes necesidades. En consecuencia, las leyes deben actualizarse para mantener el ritmo con los cambios en el ambiente en el cual se aplican.

9
Uno de los conceptos claves que crece con popularidad es la de combinar las actividades post-autorización y pre-autorización en el manejo y reducción de riesgos de medicamentos. Esto también debe ser considerado por los países cuando se elabora e implementa un marco regulador extenso. Leyes, normas y estructuras Las herramientas legales forman la base de regulación de medicamentos. Algunas leyes de medicamentos tradicionalmente omiten o exoneran ciertas áreas de la actividad farmacéutica como su alcance de control, dando lugar a una brecha reguladora. Para proteger al público de medicamentos y prácticas nocivas y dudosas, las leyes deben ser lo suficientemente extensas para cubrir todas las áreas de la actividad farmacéutica en el país. Mientras las leyes proporcionan las bases para la regulación de medicamentos, herramientas reguladoras tales como las normas y guías de orientación equipan a las autoridades reguladoras de medicamentos con el sentido práctico de implementar las leyes pertinentes. Normas y guías de orientación deben ser establecidas en forma escrita para todas las funciones de regulación. Estas herramientas luego deben ser usadas para guiar la práctica reguladora, así como también disponerlas públicamente al alcance de todas las partes involucradas para dar transparencia a los procesos reguladores. Estructuras de las autoridades reguladoras de medicamentos
La regulación de medicamentos abarca una variedad de funciones, tales como licenciamiento, inspección de establecimientos de fabricación y canales de distribución, controles de importación y exportación, evaluación y registro de productos, farmacovigilancia, control de calidad, liberación de lotes de vacunas y de productos derivados de sangre, control de promoción y publicidad y control de ensayos clínicos. Cada una de estas funciones se dirige a un diferente aspecto de la actividad farmacéutica y todos ellos deben actuar al unísono para la eficaz protección de los consumidores. En algunos países, todas las funciones relacionadas con la regulación de medicamentos están bajo la jurisdicción de un único organismo, el cual tiene autoridad total en la autorización y control de estas funciones, así como asume la responsabilidad de su eficacia. En otros casos, las funciones de regulación se les asignan a dos o más organismos, en los mismos o diferentes niveles del gobierno. Dos temas relevantes y de gran impacto se encuentran en el diseño estructural de las autoridades reguladoras de medicamentos que pueden presentar problemas en la eficacia reguladora: la fragmentación y la delegación no coordinada. Por lo tanto, las estructuras reguladoras deben estar diseñadas de tal manera que aseguren que un organismo coordinador central tenga responsabilidad y control general de todos los aspectos de la regulación de medicamentos para el país en su totalidad. Las autoridades reguladoras de medicamentos (ARNs), en algunos países cumplen funciones no reguladoras tales como la fabricación y la obtención y/o entrega de servicios. Conflictos de intereses en los mandatos y asignación de recursos pueden ocurrir entre estas múltiples funciones y deben ser manejadas adecuadamente. La sostenibilidad financiera de las ARNs es un factor fundamental en la continua implementación de las diversas funciones de regulación de los medicamentos. Los honorarios cobrados por los servicios reguladores y la subvención del gobierno son dos maneras diferentes de financiar a las autoridades reguladoras de medicamentos. El gobierno por lo tanto debe estar comprometido plenamente para asegurar la sostenibilidad financiera de la regulación de medicamentos. Uno de los mayores problemas que afectan a las ANRs es la escasez de personal capacitado. Pueden considerarse varias estrategias para aliviar la limitación de los recursos humanos: mejor planificación de recursos humanos, compartir recursos internacionales en la educación y capacitación, información y vigilancia de mercados, instituir incentivos, priorizar y simplificar los procesos de trabajo, así como también la ampliación y enriquecimiento de los trabajos. Implementando la regulación de medicamentos

10
Varias áreas de regulación de medicamentos tales como el sector informal, vigilancia post-comercialización y control de la información del medicamento, reciben relativamente pequeña atención en el proceso de implementación. Productos falsificados, de calidad dudosa, información defectuosa y reclamos exagerados de eficacia, suelen ser encontrados en el sector informal. Los fabricantes no autorizados, importadores, mayoristas, minoristas e incluso los individuos comprometidos en el negocio farmacéutico presentan retos mayores para la regulación de medicamentos. No importa cuán a fondo se realice la evaluación pre-comercialización, es sólo una de las funciones necesarias si la eficacia y lo que es más importante, la seguridad de medicamentos es garantizada. También funciones de vigilancia post-comercialización tales como la farmacovigilancia, las pruebas de control de calidad, la liberación de lotes y la re-evaluación de los productos registrados, deben representar áreas prioritarias en regulación de medicamentos. La información del medicamento es distribuida tan ampliamente como los propios medicamentos. Los sistemas de regulación de la información de medicamentos incluyen aprobaciones previas y autorregulación. Sin embargo, monitorear la exactitud y lo apropiado de la información es generalmente insuficiente y la efectividad del sistema regulador existente se desconoce. Monitoreo y evaluación
El proceso regulador debe ser rutinariamente y sistemáticamente monitoreado para identificar problemas en el proceso y determinar si esas actividades llevadas a cabo son consistentes con el procedimiento de acción propuesto. Varios enfoques tales como las auditorias internas, las revisiones de cuerpos supervisores y arbitrajes pueden usarse para evaluar el desempeño de la ANR. Estos enfoques pueden complementarse con la evaluación del desempeño e identificar áreas de posible mejoramiento. Proceso de evaluación de la capacidad reguladora de medicamentos y productos biológicos La evaluación reguladora debe ser comprendida como parte de un proceso global de mejoramiento. Es el paso preliminar para una autoridad, un país o una organización para identificar sus brechas y debilidades. Los resultados deben proporcionar la base para desarrollar estrategias y planes globales para el mejoramiento de su estructura, capacidades y eficiencia. La OPS/MS puede participar en este proceso al proporcionar pericia para realizar esta evaluación como también apoyar la implementación de medidas correctivas necesarias. Por lo tanto puede contribuir a un aumento global en el nivel de calidad de las organizaciones cuya responsabilidad son la regulación y fiscalización de los medicamentos y productos biológicos. El alcance de la evaluación de la capacidad reguladora puede variar con respecto a sus objetivos. Una evaluación global del sistema regulador revisará todas las organizaciones responsables para cada tipo de medicamento. Algunas evaluaciones se centrarán en funciones horizontales tales como las inspecciones, las cuales aplican a todas las categorías de productos o algunas funciones verticales tales como la revisión de la autorización, licenciamiento y funciones de vigilancia aplican para una categoría especial de producto. Así mismo, serán realizadas evaluaciones orientadas a la evaluación de la organización/estructura y el cumplimiento de todas las funciones desempeñadas por una autoridad específica. Cada evaluación de la capacidad reguladora debe organizarse paso a paso de la siguiente manera: planificación, preparación, sitio a visitar, informe, plan de acción y seguimiento. Estos pasos pueden variar dependiendo de la clase de evaluación realizada (autoevaluación o evaluación externa). El proceso descrito a continuación fue desarrollado por la OPS/OMS en colaboración con diferentes instituciones y profesionales de Autoridades Reguladoras Nacionales de la Región. Puede adaptarse para satisfacer las necesidades u organizaciones particulares. Expresión de una necesidad

11
Una evaluación siempre debe basarse en una necesidad expresada. Esta necesidad puede ser formal, por ejemplo, una solicitud oficial de un país a la OPS/OMS, o informal, en el caso de autoevaluación. Puede ser interno, por ejemplo, siguiendo un programa de gestión de calidad, o externo, si un Ministerio de Salud pide la evaluación de algún organismos que cumple funciones reguladoras delegadas por parte de la autoridad reguladora nacional. Esto es un gran paso porque tiene impacto sobre el resto del proceso: la definición del alcance, objetivos, etc. Después de esta solicitud, los términos iníciales de referencia y evaluación deben ser redactados (ver Anexo 1). Equipo de evaluación Un equipo de evaluación que comprende al menos seis evaluadores debe ser designado para emprender la evaluación. El criterio para su selección, en el caso de OPS, está descrito en el procedimiento de selección de expertos, el cual se basa principalmente en: formación académica, experiencia laboral y conocimientos en gestión (ver Anexo 2). Los evaluadores deben pertenecer a un grupo independiente externo a la organización evaluada y no estar involucrados en los procesos revisados. Es obligatorio que los expertos externos que participen firmen un acuerdo de confidencialidad (ver Anexo 3) y una declaración de no conflicto de intereses (ver Anexo 4). Los evaluadores deben seleccionarse considerando su formación académica y experiencia laboral en el área a ser evaluada. Si el equipo de evaluación está integrado por varios evaluadores, un líder del equipo debe nombrarse así como un "relator" a cargo de tomar notas y mantener la documentación recopilada durante la evaluación. Generalmente, un representante designado del país o de la organización evaluada acompaña al equipo de evaluación durante su evaluación. Si la organización que solicita la evaluación no es la organización evaluada, la composición del equipo final debe acordarse entre las dos organizaciones. Preparación Los términos de referencia de la evaluación deben ser discutidos, consolidados y estar de acuerdo entre el equipo de evaluación y el solicitante. La duración de la visita de revisión depende del tipo de evaluación y usualmente debe durar de cuatro a cinco días hábiles. Puede ser más largo según el alcance de la evaluación. Una fecha precisa de la evaluación debe planificarse en colaboración con todas las partes involucradas. El equipo de evaluación debe empezar recopilando información sobre la autoridad reguladora. Una solicitud para la documentación debe ser enviada al país/organización con antelación para preparar la evaluación. La información útil puede recopilarse de evaluaciones anteriores. El equipo de evaluación debe empezar revisando la información recopilada y completar la herramienta de recaudación de datos de la OPS/OMS. Es aconsejable usar herramientas electrónicas para recopilar información durante las fases de preparación, evaluación e informe. Basado en esta información, el líder del equipo debe preparar una agenda borrador mencionando las diferentes organizaciones, actividades a ser evaluadas (ver Anexo 5) y la persona responsable a ser visitada para cada actividad (ver Anexo 6). Visita Antes de iniciar la evaluación y establecer cualquier reunión, el equipo de evaluación debe hacer un resumen de la misión a los funcionarios de nivel superior de la organización, los cuales en la mayoría de los casos serán el Ministerio de Salud o la Dirección General de una Autoridad Reguladora Nacional. La sesión de apertura

12
Antes de la visita, una reunión de abertura debe ser organizada para definir los aspectos prácticos de la evaluación, revisar el plan, comprobar la disponibilidad de personal y para estar de acuerdo con el horario de evaluación. Realizando la evaluación Durante la visita, el equipo de evaluación debe recopilar la información requerida sobre la organización bajo evaluación en la secuencia elaborada en la herramienta de recaudación de datos de la OPS/OMS. En general los evaluadores deben pedir la documentación, revisar los registros, entrevistar al personal y formular observaciones. Los datos deben recopilarse y la información debe ser registrada directamente en la herramienta. Deben identificarse brechas y debilidades. Recomendaciones preliminares pueden ser discutidas con los miembros de la organización. En ciertos asuntos o temas, los evaluadores también pueden hacer entrevistas fuera de la organización de la ARN o de la institución evaluada, por ejemplo, los representantes de usuarios directos o actores claves (industria, sociedad civil, ONG, etc.), en orden de recoger opiniones o información sobre cómo la ARN se percibe u opera. Los evaluadores deben revisar muestras de registros para verificar: la existencia de regulación, su alcance, los procedimientos internos establecidos por la organización y los resultados de haber llevado a cabo dichos procedimientos. Al finalizar cada día de trabajo, el equipo de evaluación junto al líder deben ser convocados para organizar una sesión de rendición de cuentas sobre el resultado provisional de su evaluación, de manera de poder discutir los principales hallazgos. La sesión de clausura Al final de la visita, el equipo de evaluación debe preparar un informe preliminar detallando el alcance de la evaluación, fortalezas y debilidades identificadas y recomendaciones sobre las medidas correctivas que deben ser emprendidas. El equipo de evaluación luego debe organizar una sesión de clausura con el personal de la autoridad reguladora que ha sido evaluada. El tiempo estimado para esta sesión de clausura es la mitad de un día. Durante esta sesión de clausura, las principales brechas deben ser presentadas y las recomendaciones propuestas por el equipo de evaluación deben ser discutidas. El equipo de evaluación y el personal de la organización deben poder llegar a un acuerdo común sobre las recomendaciones. El informe de evaluación preliminar debe estar basado en la discusión con el personal de la autoridad reguladora. Después de esta discusión, un plan de acción preliminar debe ser elaborado por el equipo de evaluación y funcionarios superiores de la organización para especificar cómo deben ser implementadas las recomendaciones. Puede suceder que los evaluadores y el personal evaluado no estén de acuerdo en ciertos temas (brechas identificadas, medidas correctivas recomendadas, etc.). En tal evento, los evaluadores deben referirse a la evidencia documentada, que este menos sujeta a disputa y debe proporcionar un punto de referencia. Los evaluadores siempre deben tener presente que el contenido desarrollado del informe es distinto del plan de acción, el cual se basa en un compromiso entre el equipo de evaluación y la organización evaluada. Seguimiento Un borrador conteniendo los resultados, las brechas y recomendaciones debe ser hecho por el equipo de evaluación (ver Anexo 7) y ser presentado a la organización evaluada oportunamente y permitir comentarios. El plan de acción preliminar de las medidas correctivas (ver Anexo 8) debe anexarse al borrador.

13
Este borrador debe ser revisado por la organización evaluada que puede responder con comentarios técnicos o proponer algunas enmiendas al informe del equipo de evaluación. El informe debe ser enmendado si es necesario y finalizado por el equipo de evaluación. Luego el informe final debe ser presentado oficialmente a la organización evaluada y/o solicitante, si son diferentes. Siguiendo la evaluación reguladora Conforme al informe final, la organización evaluada debe preparar un plan de acción final de medidas correctivas. Este plan de acción debe tomar un formato operativo con la lista de medidas correctivas que se tomarán, fechas límite o plazos probables para su conclusión y un trabajo claro de responsabilidades para la conclusión de estas acciones. Aspectos financieros o recursos requeridos pueden agregarse a este plan o mencionarse por separado. Luego las acciones necesarias deben ser emprendidas por la organización con o sin el apoyo de intervenciones externas. La implementación del plan de acción debe ser revisado y documentado usando el formato de plan de implementación (ver Anexo 9). La reevaluación de la Autoridad no deberá ser programada para un plazo mayor a los 24 meses. Diferentes categorías de evaluaciones Siguiendo los diferentes pasos en el proceso de evaluación, diferentes evaluaciones reguladoras pueden realizarse durante un cierto período de tiempo. La evaluación preliminar Una evaluación preliminar puede ser organizada para tener un panorama global del sistema e identificar las brechas o deficiencias principales. La evaluación de seguimiento La evaluación de seguimiento puede ser organizada en una segunda etapa después que una evaluación preliminar y después que las medidas correctivas han sido implementadas. Esto puede ser usado para verificar la eficacia de las acciones emprendidas o revisar el nivel de implementación. Diferentes evaluaciones de capacidades reguladoras pueden ser realizadas basadas en su alcance (no aplica para ANR de Referencia), por ejemplo, una evaluación especializada puede centrarse en:
Funciones reguladoras específicas (licenciamiento, inspección, laboratorio, etc.) o en
Regulación de productos específicos (proceso de toma de decisiones para otorgar una autorización de mercado a un medicamento dado).
Duración de evaluación La duración de una evaluación de sistemas reguladores variará de un tipo de evaluación a otra, del nivel de madurez de la autoridad reguladora de medicamentos y también de las diversas funciones que tienen que ser evaluadas. El tiempo asignado debe basarse en factores como el tamaño de la organización y el número de ubicaciones. La duración de una evaluación dependerá también de la frecuencia de la auditoría. Metodología de evaluación La metodología de evaluación esta basada en varios conceptos que deben tenerse presente y ser seguidos.

14
Una evaluación no debe basarse en impresiones, sentimientos o cualquier consideración subjetiva. Además, es importante para los evaluadores apoyar sus observaciones con evidencia objetiva. Leyes o regulaciones publicadas deben ser recolectadas y cualquier referencia a los procedimientos internos o instructivos/procedimientos operativos estándar deben ser citados. Estos documentos se usarán después como la base para las recomendaciones propuestas y ayudarán a demostrar lo adecuado de las proposiciones al abordar cualquier brecha identificada. La evidencia puede ser recolectada por diferentes medios tales como:
entrevista del personal,
lectura de documentos,
revisión de manuales,
estudio de registros,
lectura de informes,
visualización de archivos,
análisis de datos,
observación de actividades,
revisar condiciones. La evidencia recolectada mediante entrevistas debe, siempre que sea posible, ser confirmada por otros medios objetivos. Los indicios que apunten a posibles deficiencias o brechas deben investigarse a fondo. Los evaluadores deben revisar la evidencia recolectada y documentar las brechas identificadas. Los evaluadores no deben limitar sus actividades para verificar si un documento o ley está presente o ausente. Deben proseguir sus actividades para encontrar evidencia, ya sea en documentos, procedimientos, lineamientos o leyes que se han implementado. Por ejemplo, una ley podría haberse publicado sin que una regulación haya sido adoptada después o explicada adecuadamente a otros actores relevantes a través de procesos de difusión pública o documentos de orientación. La misma metodología se aplica a procedimientos administrativos: un procedimiento podría haberse establecido pero no implementado. En tales casos, los evaluadores deben tomar muestras de los registros para comprobar en qué medida el procedimiento se ha implementado. La evaluación reguladora es una manera de mejorar un sistema regulador y puede ser realizada dentro de la organización por personal interno, por un experto externo o por un tercero. Esto no debe entenderse como medios para hacer cumplir procedimientos específicos, normas o personas obligadas a actuar en una manera específica. Herramienta de recolección de datos y sistema de calificación
La herramienta para la recolección de datos, permite verificar el desempeño del sistema regulatorio y las funciones básicas de control que deben llevar a cabo las ARN. En el marco del lineamento estratégico de OMS/OPS que señala: "El fortalecimiento de las ARN permite garantizar el uso de productos médicos seguros, eficaces y de calidad", esta herramienta es eficaz, para el mejoramiento continuo de la autoridad reguladora. Las pautas de esta herramienta, han considerado las directrices de las Buenas Prácticas de Manufactura (BPM), de laboratorio (BPL), de distribución (BPD) y de investigación clínica (BPC) de la Organización Mundial de la Salud. Cada indicador tiene asignada una calificación, con la finalidad de que los resultados de su aplicación en las visitas a las ARNs respondan a criterios de evaluación objetivos y uniformes, los cuales se detallan a continuación: CRITICO: Abreviatura correspondiente: "C". Se ha otorgado a aquellos indicadores de la herramienta cuyo
incumplimiento puede afectar en grado crítico el sistema regulador y/o el adecuado desempeño de las funciones críticas de control. Debe ser cumplido de modo absoluto e incuestionable para obtener la evaluación positiva de la ARN. El no cumplimiento o cumplimiento parcial de uno de los factores que afectan en grado crítico implica la evaluación negativa para ese indicador y para el resultado general de la ARN. En

15
consecuencia, ello implicará la presentación de una nueva solicitud de evaluación en un plazo determinado, concebido en función del problema crítico identificado. NECESARIO: Abreviatura correspondiente: "N". Se ha asignado a aquellos indicadores de la herramienta cuyo
no cumplimiento afecta el desempeño del sistema regulador y/o la adecuada aplicación de las funciones críticas de control. Los indicadores deben ser cumplidos de modo absoluto e incuestionable para obtener la evaluación positiva. Por lo tanto se define por sí o por no. Su incumplimiento será calificado como negativo y requiere de su inclusión en el Plan de Desarrollo Institucional con un plazo definido para su solución. INFORMATIVO: Abreviatura correspondiente: "I". Se otorgó a aquellos indicadores de la herramienta que brindan información descriptiva y complementaria. Su incumplimiento o cumplimiento parcial no afecta el sistema regulador y/o la adecuada aplicación de las funciones críticas de control .Sin embargo, ella debe ser proporcionada por la Autoridad Nacional de Regulación al momento de la visita. Considerando el dinamismo y actualización permanente de la reglamentación farmacéutica conforme al desarrollo técnico científico, se ha establecido una escala de valoración en la implementación y cumplimiento de los indicadores: N.P.: No procede, la actividad o indicador no aplica conforme al sistema establecido en el país N.I.: No implementado, no hay evidencias de la actividad consultada E.I: En implementación (documentos / actividades en elaboración / implementación y/o existen evidencias
de resultados parciales) P.I.: Parcialmente implementado (documentos elaborados o actividades implementadas recientemente para
las cuales la ARN posee algunos resultados) I.: Implementado; existe evidencia de la actividad, procedimiento o lineamiento. La calificación de la ARN se determina de acuerdo a la implementación satisfactoria de los indicadores críticos de la herramienta de recolección de datos del sistema regulador y de las funciones de control del organismo regulador. La designación de ARN de referencia Regional corresponde a las autoridades reguladoras que alcancen el nivel IV de la tabla de calificación. Nivel IV: Autoridad Nacional Reguladora competente y eficiente en el desempeño de las funciones de regulación sanitaria recomendadas por la OPS/OMS para garantizar la eficacia, seguridad y calidad de los medicamentos. AUTORIDAD REGULADORA DE REFERENCIA REGIONAL. Nivel III: Autoridad Nacional Reguladora competente y eficiente que debe perfeccionar el desempeño de
determinadas funciones de regulación sanitaria recomendadas por la OPS/OMS para garantizar la eficacia, seguridad y calidad de los medicamentos. Nivel II: Estructuras u organizaciones con mandato de Autoridad Nacional Reguladora que cumplen determinadas funciones de regulación sanitaria recomendadas por la OPS/OMS para garantizar la eficacia, seguridad y calidad de los medicamentos. Nivel I: Dependencias de instituciones de salud que cumplen determinadas funciones de regulación sanitaria de medicamentos. Reevaluación: Las ARNs que hayan sido calificadas como Referencia Regional serán reevaluadas cada 3 (tres), años. Tabla de calificación:

16
Nivel
% de Implementación de indicadores críticos alcanzado
No implementado En implementación Parcialmente
implementado Implementado
I Hasta 50 % C 50-75 % C 0-24 % C
II Hasta 25 % C 25-50 % C 25-49 % C
III Hasta 15% C 25-50 % C 50-74 % C
IV 0 0-25 % 75-100 % C

17
1. Información general - Módulo 1
Objetivo: Este modulo debe ser completado para suministrar información general sobre el país en el cual la autoridad reguladora será evaluada.
Información sobre el país
La fuente de información (demográfico, socio económico, estadístico, etc.) puede ser encontrada para un país específico en los indicadores estadística/país de la OPS/OMS. Se recomienda que se recopile información en los siguientes temas: Indicadores generales:
Población total (en miles)
Tasa de crecimiento de población anual (%)
Población en zonas urbanas (%)
Ingreso nacional bruto per cápita (internacional $)
Población debajo del umbral de pobreza (% de la población que vive con menos de $1 al día)
Per cápita en dólares internacionales
Sistemas de Salud Indicadores de sistemas de atención de salud
Médicos (número) y Médicos (densidad por 1 000 habitantes)
Enfermeras (número) y Enfermeras (densidad por 1 000 habitantes)
Dentistas (número) y Dentistas (densidad por 1 000 habitantes)
Farmacéuticos (número) y Farmacéuticos (densidad por 1 000 habitantes)
Agentes Sanitarios de la Comunidad (número) y Agentes Sanitarios de la Comunidad (densidad por 1.000 habitantes)
Los evaluadores deben mantener presente que este conjunto de información básica debe ser de ayuda para ellos durante la evaluación y al elaborar el plan de acción para proporcionar recomendaciones adecuadas en relación a la situación del país.
Información sobre la evaluación
Este capítulo proporciona una comprensión clara del enfoque de la evaluación, en este se establece el tipo de evaluación a ser realizada: la finalidad, el alcance, los datos de la ARN y el equipo de evaluación que participará.
18
2. Autoridad Reguladora Nacional (ARN) - Módulo 2
Objetivo: Revisar la base legal, estructura organizacional existente, recursos asignados y buenas prácticas reguladoras ya implementadas.
Usando los resultados mencionados en el capítulo anterior, los evaluadores deben revisar a todas las autoridades/instituciones que realizan funciones reguladoras. En primera instancia, los evaluadores deben centrarse en la autoridad reguladora nacional, si existe. En algunos casos los evaluadores tendrán que evaluar a las autoridades regionales o locales. Debido a limitantes de tiempo, recursos humanos o financieros, el equipo de evaluación puede tener que decidir si muestrean un número limitado de autoridades regionales o locales. Donde estén siendo evaluadas múltiples autoridades reguladoras, este módulo debe completarse para cada autoridad reguladora. En todos los casos, si se involucra a más de una institución, los evaluadores deben verificar si la legislación proporciona una clara coordinación/nexo y evita superponerse de sus respectivas jurisdicciones como también debe buscar cualquier área potencial huérfana.
Indicador 2.1: Organización y Estructura
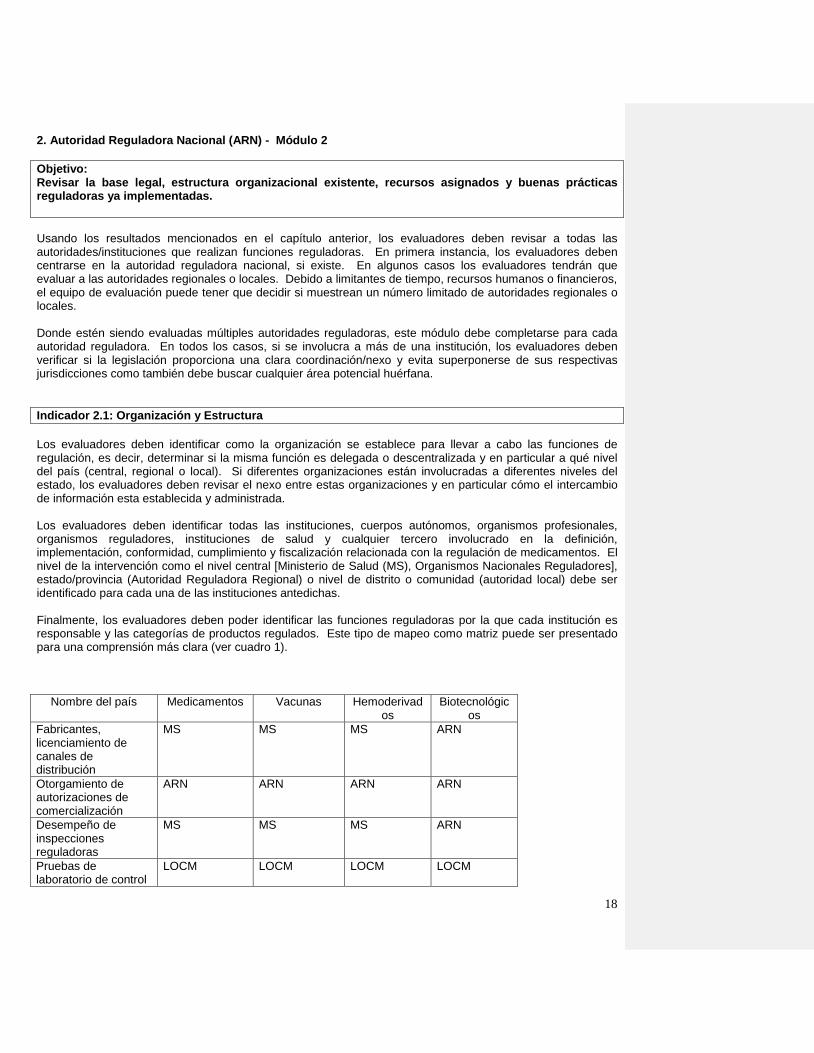
Los evaluadores deben identificar como la organización se establece para llevar a cabo las funciones de regulación, es decir, determinar si la misma función es delegada o descentralizada y en particular a qué nivel del país (central, regional o local). Si diferentes organizaciones están involucradas a diferentes niveles del estado, los evaluadores deben revisar el nexo entre estas organizaciones y en particular cómo el intercambio de información esta establecida y administrada. Los evaluadores deben identificar todas las instituciones, cuerpos autónomos, organismos profesionales, organismos reguladores, instituciones de salud y cualquier tercero involucrado en la definición, implementación, conformidad, cumplimiento y fiscalización relacionada con la regulación de medicamentos. El nivel de la intervención como el nivel central [Ministerio de Salud (MS), Organismos Nacionales Reguladores], estado/provincia (Autoridad Reguladora Regional) o nivel de distrito o comunidad (autoridad local) debe ser identificado para cada una de las instituciones antedichas. Finalmente, los evaluadores deben poder identificar las funciones reguladoras por la que cada institución es responsable y las categorías de productos regulados. Este tipo de mapeo como matriz puede ser presentado para una comprensión más clara (ver cuadro 1).
Nombre del país Medicamentos Vacunas Hemoderivados
Biotecnológicos
Fabricantes, licenciamiento de canales de distribución
MS MS MS ARN
Otorgamiento de autorizaciones de comercialización
ARN ARN ARN ARN
Desempeño de inspecciones reguladoras
MS MS MS ARN
Pruebas de laboratorio de control
LOCM LOCM LOCM LOCM

19
de calidad
Vigilancia de seguridad de productos comercializados
Centro de Vigilancia nacional
ARN
Controles de ensayo clínico
CEI MS
Cuadro 1: Ejemplo de participación institucional en el campo de medicamentos En esta etapa, los evaluadores deben empezar a identificar qué organización es responsable de coordinar todas las instituciones involucradas y comprender lo que significa que las actividades reguladoras estén siendo coordinadas.
Sub-Indicador 2.1.1: La regulación sanitaria de medicamentos se encuentra bajo la jurisdicción del Ministerio
de Salud y de otros organismos (instituciones, agencias, autoridades reguladoras) en los mismos o diferentes niveles de gobierno.
INFORMATIVO
Sub-Indicador 2.1.2: Las responsabilidades, funciones, organización, poderes y estructura del/de los
organismo/s responsables de la regulación farmacéutica se encuentran claramente definidos en los documentos constitutivos y otros complementarios, en particular en lo relacionado a las competencias y objetivos de la regulación farmacéutica que tiene bajo su control como las categorías de productos regulados y funciones reguladoras.
CRITICOI
Sub-Indicador 2.1.3: Todas las funciones de regulación se encuentran bajo la responsabilidad y supervisión
de al menos un organismo/ autoridad reguladora.
CRITICO
Sub-Indicador 2.1.4: La legislación define las instituciones involucradas en el sistema regulatorio de
medicamentos, sus competencias, funciones, roles, responsabilidades y poderes
CRITICO
Sub-Indicador 2.1.5: Cuando más de un organismo/autoridad reguladora se encuentra involucrado en
actividades reguladoras, la legislación define los canales de coordinación, y existe un mecanismo administrativo definido para ello.
NECESARIO
Sub-Indicador 2.1.6: Las actividades de Regulación son organizadas y desarrolladas a nivel central del país.
INFORMATIVO
Sub-Indicador 2.1.7: Si existen actividades descentralizadas a cargo de otras agencias/autoridades éstas
siguen los mismos estándares, lineamientos y procedimientos.
CRÍTICO
Sub-Indicador 2.1.8: En caso de descentralización, está establecido e implementado un mecanismo de intercambio de información de forma que el organismo descentralizado recibe los requisitos y/o directivas e informes de la autoridad central y le envía sus informes a la misma.
CRÍTICO
Sub-Indicador 2.1.9: Los mecanismos permiten una adecuada cooperación y colaboración entre las
organizaciones descentralizadas.
CRÍTICO

20
Indicador 2.2: Base Legal
El termino "legislación" se refiere a las leyes escritas, a menudo denominadas como Decretos o Reglamentos, que son promulgados por el Parlamento (el brazo legislativo del Gobierno). Las regulaciones son preparadas bajo la autoridad de un Decreto, denominado como el "Acto legislativo". Las regulaciones son promulgadas por el organismo a quien la autoridad a delegado para hacer regulaciones en el "acto legislativo", por ejemplo, consejo de administración, ministerio, etc. Directrices son documentos departamentales que son usados para interpretar la legislación y/o una regulación. Aunque pueden derivarse de la legislación, a menudo son usadas para asesorar sobre cómo cumplir con una regulación. La condición legal de estas directrices puede variar de un país a otro pero en todos los casos no tendrán el mismo nivel de empoderamiento como un acto legislativo. El país debe establecer un proceso para demostrar que los requisitos aplicables en cuanto a los medicamentos se han publicado oficialmente y están públicamente disponibles para los actores asociados que los implementarán. Es importante para los evaluadores comprender cómo las diferentes piezas de la legislación se redactan y qué organizaciones/instituciones se consultan durante este proceso, por ejemplo, público general, industria, organizaciones no gubernamentales (ONG) y otras partes interesadas. Los evaluadores deben identificar casos donde las disposiciones legales relevantes han sido definidas pero las regulaciones no han sido promulgadas y publicadas, que implican incertidumbre legal y potencial malentendido o mala interpretación. Evidencia documentada a ser estudiada
Organigrama del Ministerio de Salud
Reglamentos, leyes, decretos o circulares que establecen el sistema regulador. Los evaluadores deben revisar los requisitos legales vigentes y si es adecuado, las regulaciones promulgadas para mejorar la legislación. Los evaluadores deben establecer los términos de referencia, funciones, responsabilidades, poderes y estructura de la autoridad planteada en la legislación. Además debe establecer el alcance legal de las competencias asignadas a las autoridades reguladoras, y en particular si la ARN tiene cualquiera de los siguientes poderes reguladores:
autorizar, suspender, enmendar o retirar del mercado autorizaciones para medicamentos
autorizar u obligar el retiro de productos inseguros
controlar la importación y exportación de medicamentos
registrar los nombres de personal farmacéutico
licenciamiento de establecimientos y suspender o retirar estas licencias
inspeccionar los establecimientos donde tienen lugar actividades del medicamento regulado
tomar muestras de un medicamento y solicitar su análisis
autorizar, suspender o detener ensayos clínicos
participar en (o delegar responsabilidad de) vigilancia de seguridad de productos comercializados
controlar la promoción de medicamentos
monitorear el mercado y asegurar que los requerimientos del medicamento son implementados
facturar y cobrar honorarios para los servicios reguladores

21
Los evaluadores también deben identificar el rango de productos a ser monitoreados por la ARN: medicamentos humanos/veterinarios, dispositivos médicos, vacunas, otros productos biológicos, medicamentos herbarios y tradicionales, etc. Este alcance legal puede ser levemente diferente de las actividades ya conducidas por el ANR. Deben observarse diferencias significativas. Para cada una de estas actividades reguladoras vea el módulo a continuación antes de continuar la evaluación. Evidencia documentada a ser estudiada:
Reglamentos, leyes, decretos o circulares que establecen la Autoridad Reguladora Nacional,
Visión del ARN,
Misión del ARN. Referencia How to Develop and implement a national medicine policy, 2nd ed. Geneva, World Health Organization, 2001.
Sub-Indicador 2.2.1: La legislación define la creación de la ARN, su misión, términos de referencia, así como
también su alcance, funciones y responsabilidades.
CRÍTICO
Sub-Indicador 2.2.2: Si existe mas de un organismo/ autoridad involucrada en la regulación farmacéutica, la
legislación proporciona y define canales claros de coordinación entre los mismos y el respectivo empoderamiento.
CRÍTICO
Sub-Indicador 2.2.3: El desarrollo de las regulaciones involucra a la Autoridad Reguladora a cargo de su
implementación y cumplimiento.
CRÍTICO
Sub-Indicador 2.2.4: Durante el proceso de desarrollo de la legislación y de las regulaciones se involucra a
través de diferentes mecanismos a sectores de la sociedad civil tales como ONG's, representantes de la salud, industria, consumidores, pacientes y otras partes interesadas.
CRÍTICO
Sub-Indicador 2.2.5:. Las regulaciones han sido aprobadas y publicadas oficialmente.
CRÍTICO
Sub-Indicador 2.2.6: La legislación y regulaciones se encuentran públicamente disponibles para los actores
que deben cumplirla y se dispone de adecuados medios y canales de comunicación para su conocimiento
CRÍTICO
Sub-Indicador 2.2.7: Las bases legales específicas de la ARN le confieren la autoridad para decidir si un producto específico es un medicamento, un dispositivo médico u otra categoría de productos.
CRÍTICO

22
Sub-Indicador 2.2.8: Las bases legales específicas de la ARN le confieren la autoridad para realizar las
inspecciones en los establecimientos donde se realicen o pretendan realizar actividades relacionadas con los medicamentos regulados y para designar inspectores para conducirlas.
CRÍTICO
Sub-Indicador 2.2.9: Las bases legales específicas de la ARN ofrecen adecuado soporte, procedimientos y
responsabilidades para luchar contra la falsificación.
CRÍTICO
Sub-Indicador 2.2.10: La legislación confiere autoridad a la ANR para convocar expertos y comités, definir sus funciones y las circunstancias en las que debe ser convocado.
NECESARIO
Sub-Indicador 2.2.11: Las disposiciones legales establecen mecanismos de apelación para los productores
y/o titulares y otras figuras reguladas que han sido sancionadas, así como también mecanismos para el manejo de quejas y reclamaciones.
CRÍTICO
Indicador 2.3: Modelo de Administración
En orden a distribuir responsabilidades eficazmente, la ARN debe funcionar dentro de una organización administrativa que garantiza su independencia de acción. La gerencia del ARN debe ser organizada para considerar los siguientes tres componentes:
organización de trabajo diario
establecimiento de una estrategia de desarrollo
evolución del entorno científico
Las funciones, roles y responsabilidades de los diferentes órganos creados dentro de la ARN deben definirse claramente y establecerse los canales de comunicación. Evidencia documentada para ser estudiada:
Plan corporativo, plan estratégico o el plan gerencial
Sub-Indicador 2.3.1: Se encuentra definido en la organización de la ARN un Consejo Directivo, Dirección, Comité u Órgano administrativo responsable de establecer el Plan de desarrollo estratégico.
NECESARIO
Sub-Indicador 2.3.2: Las funciones de ese Consejo Directivo, Dirección, Comité u Órgano administrativo se
encuentran definidas, documentadas e implementadas en particular en lo relativo a asesorar sobre estrategias y orientaciones de la ARN.
NECESARIO
Sub-Indicador 2.3.3: Existen estructuras (dirección/unidad/departamento de planificación), dentro de la ARN
encargadas de manejar y organizar las actividades de trabajo.
NECESARIO
Sub-Indicador 2.3.4: Los roles y responsabilidades de tales estructuras se encuentran definidos,
documentados e implementados en particular sobre las decisiones de rutina, reportes e implementación de estrategias.
NECESARIO

23
Sub-Indicador 2.3.5: Están establecidos los canales de comunicación entre las estructuras dentro de la ARN
encargadas de manejar y organizar las actividades de trabajo, cuando existen varias y esta/s a su vez con los demás departamentos /unidades de la ARN.
NECESARIO
Sub-Indicador 2.3.6: Existe un Comité Científico para asesorar a la ARN sobre temas científicos de interés y orientaciones futuras.
NECESARIO
Indicador 2.4: Desarrollo Institucional
Basado en su declaración de misión y visión que ha sido creada de acuerdo con la política del gobierno, la ARN debe establecer una estrategia de desarrollo coherente que puede ser implementada y actualizarse regularmente. La ARN debe elaborar objetivos para las funciones reguladoras realizadas y monitorear el logro de tales objetivos usando indicadores apropiados que pueden ser cuantitativos o cualitativos, por ejemplo:
número de solicitudes recibidas (por año)
número de autorizaciones de comercialización concedidas (por año)
porcentaje de registros otorgados en un plazo de 6 meses y comparado con el número de solicitudes presentadas.
Evidencia documentada a ser estudiada:
Plan de desarrollo institucional de la ARN
Objetivos de la ARN
Indicadores principales de la ARN
Sub-Indicador 2.4.1: La ARN posee un plan de desarrollo institucional implementado y actualizado.
NECESARIO
Sub-Indicador 2.4.2: La ARN ha establecido y declarado su Visión y Misión.
CRÍTICO
Sub-Indicador 2.4.3: Están establecidos los objetivos generales de la ARN y han sido desglosados en objetivos específicos con plazos definidos para las diferentes funciones reguladoras.
CRÍTICO
Sub-Indicador 2.4.4: Existen planes de acción para alcanzar los objetivos establecidos.
CRÍTICO
Sub-Indicador 2.4.5: Existen indicadores establecidos para monitorear/evaluar el progreso de cumplimiento
con los objetivos/ plan de desarrollo institucional.
NECESARIO
Indicador 2.5: Sistema de Gestión de la Calidad
En esta sesión los evaluadores deben revisar el sistema de gestión de calidad de la ARN para todas las funciones reguladoras. Históricamente la inspectoría reguladora y el laboratorio de control de calidad son funciones que han sido sometidas a los procedimientos de garantía de la calidad antes de otras funciones.

24
Los evaluadores deben determinar si la puesta en práctica del sistema de gestión de calidad representa un reto global para el ARN en vez de construir puentes entre las "islas" del los sistemas de calidad parciales. Los evaluadores deben usar el cuestionario que se aplica a todas las actividades operacionales de la ARN. Preguntas más específicas se proporcionan en la herramienta en los siguientes capítulos sobre funciones de inspecciones reguladoras y en funciones de laboratorio de control de calidad. El número de personal empleado en las actividades del sistema de gestión de calidad también debe ser considerado por los evaluadores. Evidencia documentada a ser estudiada:
Designación de gerente de calidad y descripción del trabajo
Manual de calidad
Procedimiento para control de documentación (manual, procedimientos y registros)
Manejo de revisión de registros
Política de calidad y objetivos
Plan de calidad
Indicadores de calidad (plazo, notificación)
Lista de procedimientos y formularios internos
Procedimiento para planificación, implementación de auditoría interna y seguimiento con acciones correctivas
Informes de auditoría y seguimientos
Procedimientos para investigación de no-conformidad y registros.
Procedimientos para tratar quejas y registros.
Procedimiento para la iniciación, decisión e implementación de las medidas correctivas y preventivas y controlar eficiencia y registros.
Sub-Indicador 2.5.1: La ARN tiene implementado un sistema de gestión de calidad (SGC) para todos los
procesos reguladores.
NECESARIO
Sub-Indicador 2. 5.2: La máxima dirección de la ARN está comprometida con el desarrollo e implementación
de un sistema de gestión de calidad y en particular en proveer financiamiento y soporte organizacional para el mismo.
CRÍTICO
Sub-Indicador 2.5.3: El sistema de gestión de calidad está basado o reconoce estándares de referencia
(OMS, PIC/S, ISO, otros).
NECESARIO
Sub-Indicador 2.5.4: La política y objetivos de calidad están definidos y documentados.
NECESARIO
Sub-Indicador 2.5.5: La política y el plan de desarrollo han sido establecidos para la implementación del
sistema de gestión de calidad para todas las funciones regulatorias.
INFORMATIVO
Sub-Indicador 2.5.6: Está designada una persona calificada como responsable de desarrollar e implementar
el sistema de gestión de calidad.
CRÍTICO
Sub-Indicador 2.5.7: Se destinan y asignan recursos humanos para implementar el Sistema de Gestión de
Calidad.

25
INFORMATIVO
Sub-Indicador 2. 5.8: Está definido el sistema de documentación necesario para establecer, implementar y
mantener el SGC (Manual de calidad, registros, políticas, procedimientos de calidad, procedimientos operativos).
NECESARIO
Sub-Indicador 2.5.9: La documentación requerida por el sistema de gestión de calidad (SGC) se controla
siguiendo un procedimiento escrito.
NECESARIO
Sub-Indicador 2.5.10: Existe un procedimiento documentado para evaluar la organización y el desempeño del
Sistema de Gestión de Calidad (SGC) y los registros de los resultados.
NECESARIO
Sub-Indicador 2.5.11: Se realizan Auditorías internas del SGC como mínimo una vez cada 12 meses.
NECESARIO
Sub-Indicador 2.5.12: Las acciones correctivas y preventivas tomadas como resultado de auditorías o de no
conformidades son implementadas y documentadas y se verifica su eficacia.
CRÍTICO
Sub-Indicador 2.5.13: La Dirección de la ARN revisa la organización del SGC conforme a las bases
reguladoras al menos una vez cada 12 meses y están disponibles las evidencias documentadas.
NECESARIO
Indicador 2.6: Financiamiento de la ARN
Los evaluadores deben identificar si la ARN tiene una base de financiamiento sustentable. Deben determinar si la ARN es financiada por el gobierno, por los honorarios recaudados por los servicios provistos, por fuentes de donación o mediante una mezcla de fondos. La disponibilidad de un presupuesto adecuado es esencial para proporcionar sueldos para atraer personal con la capacitación y experiencia requerida, así como también para los establecimientos e infraestructura necesaria. Los evaluadores deben determinar si la ARN tiene la autoridad para facturar, recolectar y utilizar internamente los fondos que se generan para los servicios reguladores provistos. Evidencia documental a ser estudiada:
Lista de honorarios aplicables para licenciamiento, registro o autorización.
Presupuesto de la ARN.
Acuerdos de financiamiento.
Sub-Indicador 2.6.1: Están establecidas las fuentes de financiamiento de la ARN para desarrollar todas sus
funciones reguladoras.
CRÍTICO
Sub-Indicador 2.6.2: Están publicados los montos de las cuotas, tasas, tarifas o aranceles a pagar por los
servicios que presta la ANR.
CRÍTICO

26
Sub-Indicador 2.6.3: Existen disposiciones relacionadas con la reducción o exención de aranceles, tasas,
tarifas o cuotas, a fin de garantizar la disponibilidad de medicamentos esenciales para la sobrevida o los de un mercado muy limitado.
INFORMATIVO
Sub-Indicador 2.6.4: La ARN es financiada con fondos del presupuesto público.
INFORMATIVO
Sub-Indicador 2.6.5: El financiamiento es parcialmente proporcionado por donantes tales como la OMS,
Banco Mundial, etc.
INFORMATIVO
Sub-Indicador 2.6.6: La ARN tiene la autoridad para recaudar y utilizar internamente los fondos generados.
INFORMATIVO
Indicador 2.7: Gestión de Recursos Humanos
Los evaluadores deben revisar el organigrama de la ARN y revisar los deberes, funciones y responsabilidades del personal técnico y científico clave. Esto puede hacerse al estudiar las descripciones de puestos. Algunos ejemplos de las funciones claves para revisar y las personas a entrevistar se dan a continuación:
Director general/gerente general/jefe de la ARN.
Directores de departamento/unidad/sección involucrados en funciones reguladoras.
Gerente de calidad o representante.
Director de departamento/unidad/sección involucrados en funciones de apoyo (informática, logística, administración, finanzas, etc.).
Los evaluadores no deben limitar su investigación solo al personal clave: deben identificar y revisar la descripción del puesto de cada personal técnico o científico involucrado en cada función de reguladora. En los siguientes módulos los evaluadores tendrán la oportunidad de verificar si las funciones descritas en la descripción del puesto son aquellas en realidad realizadas. La entrada de carga de trabajo de la organización debe ser buscada cuando se hacen planes para el desarrollo y capacitación de recursos humanos. Especial atención debe darse para definir la educación mínima, experiencia y requisitos de capacitación para cada categoría del funcionario. Prioridad también debe darse a la organización de planificación de carreras y formación de equipos en el servicio gubernamental y evaluación de la necesidad de asistencia externa, pericia u otros tipos de servicios de apoyo. Los evaluadores deben seguir este enfoque general para la ARN al revisar y completar las secciones de recursos humanos de cada función reguladora, incluidas la educación, experiencia y capacitación del personal empleado para realizar las actividades reguladoras. En este contexto, la educación se refiere a grados, certificación y licenciamiento ganados como resultado de escolaridad formal o cursos de estudio en una institución de enseñanza superior (por ejemplo, magíster, doctorado, licenciatura, diplomado u otros). La capacitación se refiere en general a los programas cortos, centrados sobre temas específicos (por ejemplo, programa de capacitación de dos semanas sobre BPM). La experiencia incluye participación directa en las actividades que proporcionan pericia adicional en un área específica. Los siguientes indicadores pueden ser usados por los evaluadores para evaluar los procesos de recursos humanos y en particular el recambio y eficacia del proceso de contratación:
Número total de empleados de la ARN.
Número de personal científico.

27
Número de personal de apoyo.
Número de personal contratado.
Número de personal cuyos contratos concluyen en un período definido.
Número de personal contratado comparado con el número total de empleados.
Número de personal cuyos contratos concluyen comparado con número de personal contratado en un período definido.
Evidencia documental a ser estudiada:
Organigramas de la ARN.
Procedimientos internos para contratación, capacitación y calificación del personal y registros.
Procedimiento para evaluar el impacto de actividades de capacitación.
Procedimiento para evaluar las competencias del personal.
Código de conducta/código de ética.
Lista de personal con sus títulos.
Plan de capacitación.
Lista de capacitación realizada.
Descripciones de puestos de trabajo.
Curriculum vitae.
Plan de contratación.
Sub-Indicador 2.7.1: Existe un organigrama de la estructura de la ARN.
CRÍTICO
Sub-Indicador 2.7.2: Está identificado el personal clave de la ARN.
CRÍTICO
Sub-Indicador 2.7.3: Están establecidas las obligaciones, funciones y responsabilidades del personal clave en
la descripción de cargos.
CRÍTICO
Sub-Indicador 2.7.4: La ARN ha establecido las competencias necesarias (educación, entrenamiento,
capacidad y experiencia) para el personal clave a fin de realizar el trabajo asignado.
CRÍTICO
Sub-Indicador 2.7.5: La ARN posee la facultad para seleccionar y reclutar su propio personal siguiendo un
procedimiento documentado basado en criterios escritos propios (experiencia, educación mínima, entrenamiento de alto nivel, etc.).
NECESARIO
Sub-Indicador 2.7.6: Está establecido un periodo inicial de evaluación del personal, para revisar el desempeño, identificar necesidades de entrenamiento y establecer acuerdos sobre los objetivos a desarrollar.
NECESARIO
Sub-Indicador 2.7.7: Existe un programa de inducción del personal recientemente reclutado.
NECESARIO
Sub-Indicador 2.7.8: Está establecido un plan de entrenamiento para todo el personal para cumplir con las
necesidades identificadas.
NECESARIO

28
Sub-Indicador 2.7.9: La ARN realiza y mantiene registros de las actividades de entrenamiento del personal.
Está establecido un mecanismo para evaluar y demostrar la efectividad de las actividades de entrenamiento.
NECESARIO
Sub-Indicador 2.7.10: Existe fondo presupuestario para realizar entrenamiento del personal.
CRÍTICO
Indicador 2.8: Comités y Expertos Externos
Los evaluadores deben identificar si la ARN utiliza la pericia de expertos externos y/o si ha establecido comités de expertos para intervenir en una etapa específica en el proceso regulador. Los evaluadores también deben revisar la base legal para estas intervenciones. La ARN puede usar los servicios de expertos externos o quizá ya haya establecido uno o varios comités, por ejemplo, en autorizaciones de comercialización, farmacovigilancia, en control de promoción y publicidad de medicamentos o supervisión de ensayos clínicos. En caso de múltiples comités consultivos o técnicos, los temas detallados en este capítulo se aplican a cada comité consultivo identificado. Los temas principales están relacionados a procedimientos de selección y designación (publicidad para candidatos, revisión predefinida por un panel seleccionado, nombramientos y publicación de la decisión final así como exclusión de candidatos), la independencia de los diferentes expertos involucrados en los procesos reguladores y el manejo y prevención de potenciales conflictos de intereses. Evidencia documental a ser estudiada;
Procedimientos internos para selección y designación de expertos externos.
Procedimientos internos para la designación de miembros de los comités consultivos.
Lista de expertos externos.
Lista de los comités consultivos que intervienen en un proceso regulador.
Lista de miembros de los comités consultivos.
Composición de los diversos comités consultivos.
Términos de referencia de los diversos comités consultivos.
Procedimientos internos para administrar los comités consultivos.
Contrato modelo establecido entre la ARN y los expertos.
Sub-Indicador 2.8.1: La ARN ha establecido un Comité Asesor (en el que pueden participar especialistas
internos y expertos externos) involucrados en los procesos reguladores de la ARN.
INFORMATIVO
Sub-Indicador 2.8.2: La ARN utiliza expertos externos en sus procesos reguladores.
NECESARIO
Sub-Indicador 2.8.3: Existe un modelo de contrato entre la ARN y cada uno de los expertos externos
definiendo roles y con responsabilidades establecidas y firmadas por ambas partes.
NECESARIO
Sub-Indicador 2.8.4: Existe una política escrita/ procedimiento para la designación y captación de expertos
externos, definiendo la selección de los candidatos mediante un panel / jurado de selección y designación y haciendo pública la decisión final.
CRÍTICO

29
Sub-Indicador 2.8.5: Existe un procedimiento escrito para la gestión del Comité Asesor.
NECESARIO
Sub-Indicador 2.8.6: Existe una política general sobre potenciales conflictos de interés para expertos externos
ad hoc y miembros del Comité Asesor.
CRÍTICO
Sub-Indicador 2.8.7: Existe una política general sobre confidencialidad y código de conducta para expertos
externos “ad hoc” y miembros del Comité Asesor.
CRÍTICO
Sub-Indicador 2.8.8: Existe un mecanismo escrito para manejar potenciales conflictos de interés de expertos
internos o externos y miembros de comités, para recopilar declaraciones de interés y garantizar la actualización de estas declaraciones para todas las funciones de regulación.
NECESARIO
Sub-Indicador 2.8.9: La agenda del Comité Asesor es elaborada antes de la reunión y se registran las notas
de la discusión (acta).
NECESARIO
Sub-Indicador 2.8.10: La ARN participa en alguna red global con asociaciones científicas de prestigio y sociedades de profesionales.
INFORMATIVO
Indicador 2.9: Transparencia y Confidencialidad
Los evaluadores deben revisar las bases legales de la ARN, prácticas y políticas generales al tratar con temas de transparencia y confidencialidad en todos sus procedimientos y resultados. Sobre todo se deben aclarar los siguientes temas:
La definición, publicación y difusión de las directrices que requieren información para ser presentadas a la ARN en apoyo de diversos tipos de solicitudes;
La publicación del criterio usado por la ARN y los procedimientos seguidos cuando se decide si las solicitudes reúnen los requisitos;
La publicación de las decisiones de la ARN (listas completas de medicamentos registrados, renovados y retirados) y la información sobre la cual estas decisiones son basadas.
La documentación e implementación del mecanismo para apelar contra las decisiones de la ARN.
La consulta o participación de sectores selectos de sociedad civil (por ejemplo, ONG, representantes de profesionales de la salud, industria, consumidores y pacientes)
La organización de reuniones regulares programadas con actores claves y días abiertos para el público así como su representación en reuniones de actores relevantes.
Los evaluadores también deben evaluar las respuestas dadas a las preguntas sobre la disponibilidad de información al final de cada módulo sobre las funciones reguladoras. En este asunto los evaluadores pueden, con discreción, empezar las entrevistas fuera de los límites de la ARN; por ejemplo, con representantes de actores relevantes para recoger opiniones o información acerca de cómo la ARN se percibe u opera. Evidencia documental a ser estudiada:
Reglamentos, Leyes, Decretos o circulares aplicables a la ARN en esta área
Política de transparencia

30
Política de confidencialidad
Procedimientos para administrar confidencialidad
Procedimientos para administrar las reclamos y apelaciones
Disponibilidad pública de las disposiciones legales, orientación y procedimientos internos en el sitio web.
Sub-Indicador 2.9.1: La legislación provee los requerimientos para garantizar confidencialidad y transparencia
en el trabajo de la ARN.
CRÍTICO
Sub-Indicador 2.9.2: Se dispone de un mecanismo adecuado para la gestión del material/ información
confidencial.
NECESARIO
Sub-Indicador 2.9.3: Existe una política documentada sobre el acceso público a la información con
exenciones/excepciones.
NECESARIO
Sub-Indicador 2.9.4: La información sobre la legislación, regulación, procedimientos y lineamientos está
disponible públicamente en páginas Web u otros mecanismos que garanticen la adecuada disponibilidad de información y se mantiene debidamente actualizada.
CRÍTICO
Sub-Indicador 2.9.5: Se mantiene públicamente disponible un informe anual con el presupuesto asignado a la
ARN sobre un periodo base.
NECESARIO
Sub-Indicador 2.9.6: El proceso de toma de decisiones relacionado con la actividades reguladoras tales como
emisión o denegatoria de un registro; aplicación de medidas sanitarias de seguridad, liberación un lote; aprobación o denegatoria de un ensayo clínico, autorización de funcionamiento de un establecimiento elaborador, importador, distribuidor u otros está documentado y públicamente disponible.
NECESARIO
Sub-Indicador 2.9.7: La información sobre decisiones está públicamente disponible, incluyendo las decisiones
negativas en casos específicos (cuando la legislación lo permite) de manera oportuna.
NECESARIO
Sub-Indicador 2.9.8: La información sobre los resultados de la actuación de la ARN en todas sus funciones se
encuentra públicamente disponible y está permanentemente actualizada.
NECESARIO
Sub-Indicador 2.9.9:La información sobre sanciones, retiros y precauciones de salud pública está publicada y
disponible a todo el público.
CRÍTICO
Sub-Indicador 2.9.10: Están disponibles lineamientos sobre reclamos y apelaciones contra decisiones de la
ARN en cualquiera de sus funciones.
CRÍTICO

31
Sub-Indicador 2.9.11: Se dispone de mecanismos y procedimientos detallados para formular la apelación
contra decisiones de la ARN y para su manejo al interior de la ARN.
CRÍTICO
Sub-Indicador 2.9.12: La ARN consulta o involucra a sectores específicos de la sociedad civil (tales como
ONGs, representantes de profesionales de la salud, la industria, consumidores y pacientes) durante el desarrollo de guías o lineamientos.
NECESARIO
Sub-Indicador 2.9.13: La ARN ha establecido e implementado un procedimiento escrito para consultar, compilar y evaluar los comentarios recibidos durante el desarrollo de guías o lineamientos.
NECESARIO
Sub-Indicador 2.9.14: Las declaraciones de conflicto de interés de expertos internos y externos están
públicamente disponibles.
NECESARIO
Sub-Indicador 2.9.15: La industria, consumidores y representantes de los pacientes participan como observadores en reuniones técnicas/Comité de asesores.
NECESARIO
Sub-Indicador 2.9.16: Las agendas de los Comités técnicos/Asesores se circulan a sus participantes con
anticipación, las notas (actas) de las reuniones se registran y se brinda información pública de los tópicos discutidos de conformidad con las políticas nacionales vigentes
NECESARIO
Sub-Indicador 2.9.17: La ARN ha establecido una persona de contacto competente o una Unidad de
relaciones públicas, que es conocida por las partes interesadas.
NECESARIO
Sub-Indicador 2.9.18: La ARN organiza reuniones regularmente con los actores relevantes y brinda espacio
para las consultas al público en general, como por ejemplo con días abiertos al público.
NECESARIO
Sub-Indicador 2.9.19: La ARN participa cuando considera de su interés de reuniones organizadas por los
actores del sector (asociaciones de industriales, asociaciones de profesionales médicos, asociación de pacientes, etc.).
NECESARIO
Indicador 2.10: Independencia e Imparcialidad
Los evaluadores deben determinar si la ARN opera de una manera independiente e imparcial. Lo apropiado de los procedimientos que garantizan independencia e imparcialidad es que deben ser establecidos para expertos internos como externos. Dependiendo de la estructura de la ARN, los evaluadores pueden decidir enfrentar estos temas simultáneamente o por separado (la herramienta provee para ambas opciones). Especial atención debe prestárseles a los comités consultivos de la ARN con respecto a la implementación del procedimiento documentado para el manejo de tales comités.

32
Evidencia documental a ser estudiada:
Reglamentos, Leyes, Decretos o circulares aplicables a la ARN en esta área
Código de conducta para personal interno, expertos externos y miembros de los comités consultivos
Procedimientos internos para administrar el potencial conflicto de intereses
Lista de conflictos de intereses declarados
Procedimientos internos para administrar los comités consultivos
Minutas de comités consultivos.
Sub-Indicador 2.10.1: Existe un "Código de conducta" documentado para los miembros del personal
involucrado en las funciones de regulación.
CRÍTICO
Sub-Indicador 2.10.2: Existe una política interna / establecido un mecanismo sobre potenciales conflictos de
interés para los miembros del personal.
CRÍTICO
Sub-Indicador 2.10.3: Existe un modelo de formulario para realizar la declaración de conflicto de interés.
NECESARIO
Sub-Indicador 2.10.4: Existe una política escrita/procedimiento para evitar que coincidan responsabilidades
en un mismo miembro del personal de la ANR que comprometan la imparcialidad, que puedan considerarse incompatibles y conlleven a conflictos de intereses, como por ejemplo, responsable del registro y de suministros o responsable de registro y acuerdos de reembolso.
NECESARIO
Sub-Indicador 2.10.5: La ARN mantiene independencia respecto de investigadores, productores,
distribuidores, y mayoristas de medicamentos.
CRÍTICO
Sub-Indicador 2.10.6: La ARN mantiene independencia respecto del sistema/ instituciones proveedoras de
medicamentos.
CRÍTICO
Indicador 2.11: Infraestructura
Los evaluadores deben averiguar si y cómo la ARN proporciona edificios adecuados, espacio de trabajo, ambiente de trabajo y equipo para realizar las funciones reguladoras asignadas así como servicios de apoyo en términos de transporte y comunicación. Temas relacionados con el manejo de información no son discutidas bajo este título y se revisarán en el capítulo 3.13. Evidencia documental a ser estudiada:
Mapa o plan del establecimientos/ubicación
Contratos de mantenimiento
Contratos de servicios
Lista de equipo

33
Sub-Indicador 2.11.1: Los espacios, el ambiente de trabajo y el espacio disponible para archivar la
documentación son adecuados.
NECESARIO
Sub-Indicador 2.11.2: La ARN dispone del equipamiento necesario y adecuado para realizar las funciones
regulatorias
NECESARIO
Sub-Indicador 2.11.3: Los servicios de apoyo disponibles (por ejemplo en términos de transporte y
comunicación) son adecuados para el desarrollo de las actividades.
NECESARIO
Indicador 2.12: Monitoreo y Responsabilización
Los evaluadores deben revisar el recurso dispuesto por el Ministerio u otra organización para garantizar la supervisión o revisar cómo la ARN realiza sus actividades. Los evaluadores deben verificar si es una de las tareas de la ARN preparar informes generales y temáticos a intervalos regulares de cómo estas leyes están siendo implementadas. Estos informes deben, entre otros elementos, subrayar las deficiencias y debilidades en el sistema y proponer medidas correctivas. Evidencia documental a ser estudiada:
Política sobre informes
Informes anuales
Informes de autoevaluación
Sub-Indicador 2.12.1: La legislación establece requerimientos para que otras instituciones puedan
monitorear, supervisar y revisar el desempeño de la ARN en cuanto al cumplimiento de las funciones reguladoras que le fueron asignadas.
CRÍTICO
Sub-Indicador 2.12.2: Las funciones o procesos reguladores son monitoreados y revisados en forma regular y
sistemática para identificar problemas, brechas, debilidades e inconsistencias dentro de la ARN.
NECESARIO
Sub-Indicador 2.12.3: La ARN prepara informes generales y temáticos a intervalos regulares sobre las
funciones/procesos reguladores ejecutados, los que son enviados al/ a los organismo/s, institución/es o entidad/es que tengan a cargo la supervisión y revisión de la ARN.
NECESARIO
Indicador 2.13: Sistema de Gestión de la Información
La ARN es responsable de proteger la salud pública al asegurar que los medicamentos son eficaces para una finalidad propuesta y aceptablemente seguros, por consiguiente debe considerar la comunicación una misión fundamental. Los evaluadores deben revisar la estrategia de comunicaciones de la ARN y cómo ésta planea comunicar información exacta y oportuna acerca de los beneficios y riesgos de medicamentos a los actores claves incluyendo pacientes, publico, profesionales de la salud, investigadores e industria.

34
Diversos canales de comunicación pueden ser usados por la ARN para proporcionar el punto de contacto con diferentes actores dependiendo sus diferentes necesidades de información. Atención particular debe tomar la ARN con respecto al manejo de eventos adversos. Los evaluadores deben verificar si la ARN ha preparado un plan para tratar con los eventos graves o severos y adelantar información adecuada que podría necesitarse urgentemente para proteger al paciente. El éxito o eficacia de esta estrategia de comunicaciones debe ser evaluado periódicamente por la ARN. Los evaluadores deben revisar el sistema de gestión de información de la ARN y en particular cómo la información es recolectada, entrada en la base de datos y recuperada en caso de una consulta. Los evaluadores también deben determinar si la ARN ha establecido una red integrada de computadoras relacionadas a las funciones reguladoras. Procedimientos documentados deben establecerse para la recopilación y uso de aplicaciones de software o herramientas de búsqueda. La ARN debe tener desarrollado su propio sitio web o haber hecho un arreglo para usar el sitio web de otra organización/ministerio. Los evaluadores deben completar esta evaluación general al hacer preguntas más específicas antes de revisar la disponibilidad de información para cada función reguladora. El número de personal empleado en el sistema de gestión de información también debe ser considerado por los evaluadores. Evidencia documental a ser estudiada:
Número de páginas en el sitio web de la ARN o páginas web
Tiempo promedio dedicado al sitio web de la ARN o páginas web
Número de computadoras disponibles en la ARN
Número de personal de tecnología informática
Número de personas con acceso a internet
Lista de aplicaciones de software usadas Respecto de la comunicación:
Estrategia de comunicación
Páginas web del sitio de la ARN
Planes de crisis
Informe de la encuesta o estudio sobre conciencia del esquema de vigilancia de seguridad, o percepción pública de la ARN como fuente de información
Programa y minutos de reunión o conferencia
Sub-Indicador 2.13.1: La ARN usa sistemas informáticos para la automatización de actividades repetitivas y
para reportes de actividades.
NECESARIO
Sub-Indicador 2.13.2: La ARN usa sistemas informáticos para el eficiente manejo de datos de forma tal que la información sea recolectada, ingresada en una base de datos y recuperada en informes para su consulta.
NECESARIO
Sub-Indicador 2.13.3: La ARN usa sistemas informáticos para acceder a información técnica/científica.
NECESARIO
Sub-Indicador 2.13.4: La ARN ha desarrollado una red en la que se integran todas las computadoras
relacionadas con la información de las funciones reguladoras.
NECESARIO

35
Sub-Indicador 2.13.5: La ARN tiene su propia página Web o tiene algún acuerdo para utilizar la de otras
instituciones.
CRÍTICO
Sub-Indicador 2.13.6: La ARN emplea su propio personal técnico-informático o tiene garantizado el acceso a
servicios informáticos.
CRÍTICO
Sub-Indicador 2.13.7: Existen procedimientos para reunir datos, usar las aplicaciones o los programas y
herramientas de consultas.
NECESARIO
Referencias: Regulatory Situation of Herbal Medicines - A Worldwide Review (WHO/TRM; 1998; 49 pages) National medicine regulatory legislation: Guiding principles for small medicine regulatory authorities - Annex 8 in WHO Expert Committee on specifications for pharmaceutical preparation. 35 Report, WHO, 1999 ((WHO Technical Report Series, N°885) Effective Medicine Regulation: What can countries do? (WHO/HTP/EDM/MAC(11)/99.6) Effective medicine regulation - A multicountry study (WHO; 2002; 47 pages) WHO Policy Perspectives on Medicines N°7 - Effective medicines regulation: ensuring safety, efficacy and quality (November 2003, WHO Geneva) Example of a guideline on confidentiality - Appendix 2 in Annex 6 in WHO Expert Committee on specifications for pharmaceutical preparations. 40 Report, WHO, 2006 ((WHO Technical Report Series, N°937) Example of a Code of conduct - Appendix 1 in Annex 6 in WHO Expert Committee on specifications for pharmaceutical preparations. 40 Report, WHO, 2006 ((WHO Technical Report Series, N°937) Example of a guideline on conflict of interest - Appendix 3 in Annex 6 in WHO Expert Committee on specifications for pharmaceutical preparations. 40 Report, WHO, 2006 ((WHO Technical Report Series, N°937) Example of an invitation for expression of interest - Appendix 5 in Annex 6 in WHO Expert Committee on specifications for pharmaceutical preparations. 40 Report, WHO, 2006 ((WHO Technical Report Series, N°937) Model guidelines on conflict of interest and model proforma for a signed statement on conflict of interest (Annex 4 - Marketing authorization of pharmaceutical products with special reference to multisource (generic) products - A manual for medicine regulatory authorities) Measuring transparency in medicines registration, selection and procurement: Four country assessment studies (WHO/PSM/PAR/2006.7) Pharmaceuticals and the Internet Medicine Regulatory Authorities' Perspective - Joint NLN-WHI Workshop, 24-25 September 2001 (WHO; 2002; 80 pages) How to Implement Computer-Assisted Medicine Registration - A Practical Guide for Medicine Regulatory Authorities (MSH, WHO; 1998; 74 pages) Improving the quality and usefulness of medicine regulatory authority websites (WHO Medicine Information Vol. 15, No. 03 & 04, 2001 p.163 (WHO; 2000; 76 pages)

36
3. Autorización de comercialización (Registro) - Módulo 3
Objetivo: Los evaluadores deben revisar el marco jurídico dentro del cual las solicitudes de autorización de comercialización de los medicamentos son presentadas a la agencia reguladora, los procedimientos para la evaluación de estas solicitudes y el otorgamiento o rechazo de las autorizaciones de comercialización.
La solicitud de evaluaciones a la ARN debe estar basada en los criterios definidos para garantizar la seguridad, calidad y eficacia de los medicamentos. La legislación debe requerir al aspirante que suministre la información y datos necesarios para tal evaluación. Además el sistema de registro de medicamentos debe incluir la revisión y aprobación de la información provista con el producto como los folletos de información y etiquetas. Se puede utilizar diferentes procedimientos para la evaluación de diferentes categorías de medicamentos (por ejemplo, productos genéricos o nuevas entidades químicas). Para los productos obtenidos por síntesis química, indicados para usos estándares y conteniendo ingredientes ya conocidos, usualmente no hay necesidad de reevaluar la eficacia y seguridad de los principios activos. El énfasis debe ponerse en revisar otros factores, por ejemplo la presentación, intercambiabilidad (cuando es indicado), calidad del producto y la exactitud de la información acompañante. Categóricamente debe requerirse información más extensa para apoyar una solicitud de autorización de comercialización para una nueva sustancia activa, con el fin de proveer la garantía de calidad, eficacia y seguridad. Particularmente, deben requerirse datos detallados sobre el componente químico farmacéutico, propiedades farmacológicas, datos toxicológicos, estudios reproductivos y teratogénicos sobre animales, así como estudios clínicos. Los siguientes procedimientos de registro pueden establecerse dependiendo la disponibilidad de destreza técnica y de recursos:
La ARN registra un medicamento, pero no se hace ningún juicio de los productos registrados. El procedimiento no incluye la verificación de si el medicamento reúne la seguridad básica, eficacia y criterio de calidad, pero provee una base útil para desarrollar control adicional e ir mejorando el sistema de registro.
La ARN registra un medicamento siguiendo las decisiones puestas a disposición por las ARNs en otros países (una copia de una autorización, un certificado, etc.).
La ARN registra un medicamento después de los informes de evaluación o informes de inspección hechos por las ARNs en otros países como base para la toma de decisiones de las solicitudes.
La ARN registra un medicamento siguiendo su propia evaluación de calidad, seguridad y eficacia del producto conforme a la información presentada por el solicitante.
Los evaluadores deben revisar los métodos de la ARN para difundir información con respecto a la eficacia y seguridad de medicamentos nuevos en particular a pacientes o público en general. Tal información se distribuye usualmente a través de formularios, monografías farmacológicas y listas de medicamentos esenciales. Para asegurar la disponibilidad de medicamentos en una urgencia sanitaria pueden requerirse instrumentos jurídicos específicos. No siempre es factible autorizar este tipo de medicamentos usando el proceso de registro estándar, con frecuencia porque el fabricante tiene poco interés en el desarrollo y presentación de datos, particularmente en caso de un mercado muy pequeño.

37
También debe evaluarse la existencia de procedimientos específicos codificados para tales propósitos (por ejemplo, uso compasivo, autorización excepcional, autorización condicional, programas de tratamiento).
Indicador 3.1: Bases legales
Los evaluadores deben revisar los requisitos legales aplicables y determinar si las regulaciones apropiadas han sido promulgadas. La legislación debe proveer sanciones adecuadas y proporcionales, penalidades y proceso judicial conforme a las violaciones de la legislación vigente (vea inspección reguladora para ejecución y cumplimiento de actividades). Evidencia documental a ser evaluada:
Decretos, leyes, reglamentos, resoluciones, disposiciones legales, etc., destinados a la obligación de autorización de fabricantes y autorización de comercialización de medicamentos.
Sub-Indicador 3.1.1: El soporte legal obliga a que las compañías tengan registro sanitario antes de poner un
medicamento en el mercado.
CRÍTICO
Sub-Indicador 3.1.2: La legislación faculta a la ARN para conceder registro sanitario.
CRÍTICO
Sub-Indicador 3.1.3: La legislación faculta a la ARN para solicitar la suspensión, retiro o cancelación de
registro sanitario.
CRÍTICO
Sub-Indicador 3.1.4: Existe reglamentación para exigir que los solicitantes demuestren calidad, seguridad y
eficacia de los medicamentos sometidos a la solicitud de registro sanitario.
CRÍTICO
Sub-Indicador 3.1.5: Existe reglamentación para garantizar que el fabricante del medicamento cumple con las
Buenas Prácticas de Manufactura (BPM).
CRÍTICO
El evaluador debe verificar que existe reglamentación promulgada e implementada respecto a la obligación de que las licencias de los fabricantes de medicamentos (vacunas) sólo pueden ser garantizadas sobre la base del cumplimiento de las BPM. El evaluador debe identificar si la autoridad responsable de realizar la inspección ha verificado el cumplimiento de las BPM mediante inspecciones realizadas previo al otorgamiento de la licencia.
Sub-Indicador 3.1.6: Existe reglamentación relativa a la información que debe ser entregada con los
productos (envase, etiquetado, folletos, resumen de las características del producto, etc.)
CRÍTICO
Sub-Indicador 3.1.7: Existe requerimiento legal relativo al tiempo de validez del registro sanitario.
CRÍTICO
Sub-Indicador 3.1.8: Existe requerimiento legal para obligar a la actualización periódica del registro sanitario,
por ejemplo, al momento de la renovación o durante la aprobación de cambios post-registro.

38
CRÍTICO
Sub-Indicador 3.1.9: Existe reglamentación que exija la notificación a la ARN de cambios/variaciones
significativas del RS inicial que puedan afectar la calidad, seguridad y eficacia de los productos.
CRÍTICO
Sub-Indicador 3.1.10: La reglamentación prevé los casos que requieren demostración de equivalencia terapéutica.
CRÍTICO
Sub-Indicador 3.1.11: Las reglamentaciones prevén los casos de RS condicional o provisorio eximiendo de
requisitos específicos basados en criterios establecidos (como por ejemplo, para drogas huérfanas, interés de salud pública, presunto balance riesgo/beneficio positivo).
NECESARIO
Sub-Indicador 3.1.12: Está contemplada una exención de registro sanitario para medicamentos donados,
siguiendo criterios establecidos.
NECESARIO
Sub-Indicador 3.1.13: Las reglamentaciones especifican que el titular del RS es el responsable ante la
autoridad sanitaria de los medicamentos.
CRÍTICO
Indicador 3.2: Lineamientos y guías
Los evaluadores deben revisar los documentos de orientación publicados para diferentes tipos de actores claves y determinar si abarcan el alcance de la legislación y regulaciones vigentes. Debe comprobarse si hay coherencia con la orientación de la OPS/OMS y cualquier diferencia identificarla. Los evaluadores también deben considerar el uso interno de otra orientación relevante reconocida internacionalmente (ICH, PICS, etc). Evidencia documental a revisar:
Guías, procedimientos e, instructivos a los solicitantes, etc.
Guías, procedimientos, registros y formatos para la evaluación de las distintas partes de la solicitud.
Sub-Indicador 3.2.1: Existen guías que señalen los antecedentes a presentar para demostrar calidad,
seguridad y eficacia de los productos cuando se solicita registro sanitario (o licenciamiento de producto).
NECESARIO
Sub-Indicador 3.2.2: Existen lineamientos sobre el contenido de los folletos de información de productos, resumen de las características del producto, envase y etiquetado.
NECESARIO
Sub-Indicador 3.2.3: Existen lineamientos para la solicitud de RS en cuanto a requerimientos sobre validación
de métodos analíticos.
NECESARIO
Sub-Indicador 3.2.4: Existen lineamientos para la solicitud de RS en cuanto a requerimientos de estabilidad.
NECESARIO

39
Sub-Indicador 3.2.5: Existen lineamientos para optar a ciertas exenciones de requerimientos, como por
ejemplo, para solicitar bio-exenciones para estudios de bioequivalencia.
NECESARIO
Sub-Indicador 3.2.6: Existen lineamientos para los solicitantes (aplicantes) sobre el contenido, el formato y los
procedimientos a seguir para presentar una solicitud de registro sanitario.
NECESARIO
Sub-Indicador 3.2.7: Existen lineamientos para los solicitantes definiendo los tipos y objetivos de las
modificaciones, el formato y la documentación requerida y especificando que las modificaciones son sometidas a aprobación previa.
NECESARIO
Sub-Indicador 3.2.8: Existen lineamientos sobre la forma de proceder de la autoridad con los medicamentos
donados.
NECESARIO
El evaluador debe verificar si existen lineamientos respecto a autorizar la distribución de medicamentos donados, con o sin registro sanitario, en situaciones de salud pública bajo criterios establecidos, ya sea que la autorización se realice en el área de aprobación de registros sanitarios o en otra área de la ARN.
Indicador 3.3: Procedimientos de evaluación
Los evaluadores deben revisar los procedimientos de evaluación con respecto a:
resultados y logros esperados.
nivel de detalle.
son adecuados al entrenamiento recibido.
controles en procesos. Los evaluadores deben prestar atención especial al revisar si coinciden directrices vigentes, regulaciones y legislación. Los procedimientos documentados para evaluar las diferentes partes de la solicitud deben considerar los siguientes temas:
Datos de la caracterización farmacéutica, química y biológica cuando corresponda/parte del expediente sobre calidad
Farmacología animal y toxicología
Datos clínicos sobre seguridad y eficacia
Intercambiabilidad y bioequivalencia
Rotulación/envasado de los productos
Información sobre el producto o resumen de las características del producto. Los evaluadores deben evaluar en qué medida las inspecciones están involucradas en el proceso de registro, definiendo en particular qué clase de inspección está realizándose. Pueden usarse los siguientes indicadores para identificar el nivel de monitoreo por la ARN:
Número de establecimientos fabricantes inspeccionados para la inspección de pre-aprobación, para la autorización de la comercialización.

40
Los evaluadores también deben revisar cómo la información recopilada (farmacovigilancia) o presentada después de que la autorización de comercialización primaria es otorgada (modificaciones), es administrada para asegurar una corriente clara y continua del conocimiento acerca de los productos registrados.
Sub-Indicador 3.3.1: Existe un procedimiento escrito implementado que permita a los solicitantes reunirse con
la ARN antes de presentar la solicitud de RS.
INFORMATIVO
Sub-Indicador 3.3.2:. Existen procedimientos escritos /herramientas para la evaluación de las diferentes
partes de la solicitud.
CRÍTICO
El evaluador debe verificar que se dispone de instrucciones escritas para el personal y expertos internos y externos respecto a cómo evaluar las solicitudes en sus aspectos legales, de calidad y farmacéuticos.
Sub-Indicador 3.3.3: Existen procedimientos escritos /herramientas para la evaluación de requerimientos
específicos de ciertas categorías de productos (ej. genéricos, productos que contienen nuevas sustancias activas, nuevas concentraciones, productos innovadores, etc.).
CRÍTICO
Sub-Indicador 3.3.4: Existe un procedimiento para el intercambio de información entre el solicitante y la ARN
cuando es necesario.
NECESARIO
Sub-Indicador 3.3.5: Existen procedimientos para revisar periódicamente los RSs otorgados.
NECESARIO
Sub-Indicador 3.3.6: Existen procedimientos para la evaluación de solicitudes de modificación de RSs.
CRÍTICO
Sub-Indicador 3.3.7: Existe un formato modelo para el informe de evaluación y este informe entrega evidencia
para la toma de decisiones.
NECESARIO
Sub-Indicador 3.3.8: Se aplican los mismos criterios para la evaluación de solicitudes independiente del
origen o destino de uso de los productos (ej., nacional, importado, sector público/privado).
CRÍTICO
Sub-Indicador 3.3.9: La información del producto, resumen de las características del producto, folletos de
información al profesional y al paciente (o prospecto interno) y el etiquetado es aprobado por la ARN como parte del RS.
CRÍTICO
El evaluador debe verificar que la información relativa a: folleto de información al profesional, folleto de información al paciente, resumen de especificaciones del producto, rotulado gráfico de envases sea evaluada y aprobada como parte del dossier de registro, ya sea que una copia de esta información aprobada se entregue o no al solicitante una vez otorgado el registro sanitario.
Sub-Indicador 3.3.10: La información externa (fuentes de información y materiales de referencia) para la toma
de decisiones sobre las solicitudes presentadas están fácilmente disponibles.
NECESARIO

41
Sub-Indicador 3.3.11: Existe un control de calidad de la evaluación del dossier de registro y éste está
documentado.
NECESARIO
Sub-Indicador 3.3.12: En la evaluación de las solicitudes de RS, hay expertos externos involucrados.
INFORMATIVO
Sub-Indicador 3.3.13: En la revisión de solicitudes de RS, hay un Comité Asesor de expertos está
involucrado.
INFORMATIVO
Sub-Indicador 3.3.14: Existe un procedimiento para la toma de decisiones.
CRÍTICO
Sub-Indicador 3.3.15: El procedimiento de toma de decisiones considera un análisis de costo/beneficio.
INFORMATIVO
Sub-Indicador 3.3.16: Existe un procedimiento para emitir la autorización del RS y se realiza éste en un
formato estandarizado.
NECESARIO
Sub-Indicador 3.3.17: La ARN reconoce o usa decisiones reguladoras, informes o antecedentes de otras
ARNs u organismos internacionales para la toma de decisiones.
NECESARIO
Sub-Indicador 3.3.18: Si la ARN reconoce o usa decisiones reguladoras, informes o antecedentes de otras
ARNs u organismos internacionales para la toma de decisiones; existe criterios o procedimientos establecidos para este reconocimiento.
NECESARIO
Sub-Indicador 3.3.19: El procedimiento toma en cuenta la integración de las diferentes partes del dossier
incorporando un análisis de evaluación general riesgo/beneficio.
CRÍTICO
Sub-Indicador 3.3.20: Existe un procedimiento para asegurar la participación y comunicación entre los
evaluadores de la solicitud de registro y el personal del laboratorio de control de calidad para el cumplimiento del producto y/o de las inspecciones regulares para garantizar el cumplimiento de las buenas prácticas.
NECESARIO
Sub-Indicador 3.3.21: Existen plazo (s) para la evaluación de las solicitudes.
NECESARIO
Sub-Indicador 3.3.22: La ARN cumple los plazos establecidos reglamentariamente para la concesión de registro o de sus modificaciones.
NECESARIO
Sub-Indicador 3.3.23: Existe un sistema interno de seguimiento establecido para revisar los objetivos
programados.
NECESARIO

42
Sub-Indicador 3.3.24: Existe documentado un mecanismo acelerado con requisitos específicos de registro
para situaciones especiales (por ejemplo drogas huérfanas, interés de salud pública) con requisitos específicos, presunto balance riesgo/beneficio.
NECESARIO
Sub-Indicador 3.3.25: Los pasos/requerimientos para la evaluación de retiro/cancelación de RS están
documentados.
CRÍTICO
Sub-Indicador 3.3.26: Existe un formato modelo para la aprobación de una solicitud de registro sanitario.
NECESARIO
Sub-Indicador 3.3.27. Una resolución escrita del RS, firmada por una persona con delegación oficial es
comunicada al solicitante acompañada por la información aprobada del producto, incluyendo condiciones o restricciones de su aprobación.
CRÍTICO
Sub-Indicador 3.3.28. Cada medicamento recibe un número único de identificación que debe aparecer en el
envase/etiquetado e información del producto.
CRÍTICO
Indicador 3.4: Recursos humanos y otros recursos
La evaluación de recursos humanos debe centrarse en aspectos cuantitativos y cualitativos. Para los aspectos cuantitativos los evaluadores pueden decidir usar los siguientes indicadores para identificar si se están poniendo a disposición recursos humanos adecuados para llevar a cabo las actividades planificadas:
Carga de trabajo para las funciones realizadas con los siguientes indicadores: número de solicitudes recibidas de un medicamento nuevo, producto genérico, renovaciones y modificaciones, y si es aplicable incluir el número de revisiones periódicas basadas en porcentaje del total.
Número de personal científico involucrado.
Trabajo acumulado o atraso generado (carga de trabajo comparado con el número de decisiones tomadas).
Número promedio de días tomados por la ARN para la toma de decisiones en un medicamento nuevo, producto genérico, renovaciones y modificaciones.
Los evaluadores deben revisar si los evaluadores internos y los expertos externos son plenamente competentes basados en sus calificaciones en farmacia, farmacología clínica, medicina o una disciplina similar y tener experiencia práctica en al menos una de estas disciplinas así como en biofarmacia. Los evaluadores de la información sobre el producto y rotulación deben estar capacitados en farmacia, farmacología clínica, medicina o una disciplina similar, y tener experiencia práctica en al menos una de estas disciplinas. Pueden ser necesarias otras competencias para evaluar partes específicas de la aplicación, por ejemplo, en química, microbiología, etc. Si expertos externos o un comité asesor/técnico participan en este proceso, los evaluadores deben referirse a las preguntas aplicables en capítulo 3,8.

43
Evidencia documental a verificar:
Organigrama.
Perfiles de cargo.
Procedimientos de reclutamiento, entrenamiento y calificación del personal.
Lista del personal y su calificación.
Planes de capacitación.
Registro de capacitaciones realizadas.
Guía y procedimientos para la selección y uso de expertos externos en las actividades de autorización de comercialización.
Sub-Indicador 3.4.1: La descripción de cargos está definida para los siguientes miembros del personal: jefe
de registro (supervisor), jefe de unidad de registro por tipo de productos y asesores con sus áreas de evaluación (bioequivalencia, química, clínica, microbiológica, estadística, toxicológica, etc.).
NECESARIO
Sub-Indicador 3.4.2: Existe una adecuada pericia tanto interna como externa (educación, experiencia y
entrenamiento) para la evaluación de las diferentes partes de la solicitud de RS.
CRÍTICO
Idealmente el término de adecuado para cada campo de competencia está definido y se refleja en los planes de capacitación.
Sub-Indicador 3.4.3: Existe una planificación interna de recursos humanos para desarrollo y actualización y la
misma es periódicamente revisada.
NECESARIO
El evaluador debe revisar que existen planes de capacitación de expertos internos y externos desarrollados e implementados, reflejando el estatus actual de educación y experiencia.
Sub-Indicador 3.4.4: Existe monitoreo de las habilidades desarrolladas después de las actividades de
entrenamiento.
NECESARIO
El evaluador debe verificar que en el lugar hay un sistema de monitoreo de resultados y de implementación de las habilidades adquiridas en las actividades de entrenamiento y que se ha desarrollado indicadores para este proceso.
Sub-Indicador 3.4.5: Los productores o titulares de licencia nunca están involucrados en el trabajo de
evaluación de cualquier nivel/estado, incluyendo comités de expertos.
CRÍTICO
El evaluador debe verificar que exista procedimientos y criterios escritos destinados a cumplir con la confidencialidad, independencia de los diversos expertos involucrados en el proceso regulatorio y el manejo de potenciales conflictos de interés y para la evaluación continua del desempeño de los expertos externos.
Sub-Indicador 3.4.6: Existen líneas de autoridad que reflejan la independencia en la toma de decisiones en el
sistema de registro sanitario con relación a productores, proveedores o gobierno.
CRÍTICO
Sub-Indicador 3.4.7: Existe una oficina adecuada y espacio de almacenamiento para los archivos de RSs.

44
NECESARIO
El evaluador debe verificar que las dependencias del archivo tengan acceso controlado y espacio suficiente u necesario para una adecuado manejo y consulta de los dossier de RS.
Sub-Indicador 3.4.8: El recurso humano es adecuado en cantidad para desempeñar las funciones
involucradas con el RS en forma oportuna, de acuerdo a la demanda y a los plazos fijados para dar respuesta.
NECESARIO
La evaluación de lo adecuado del recurso humano en términos de cantidad debe realizarse en base a los parámetros cuantitativos señalados al inicio de este Indicador (4.4.)
Indicador 3.5: Registros y resultados
Los evaluadores deben revisar cómo toda la información recogida durante el proceso de registro es administrada y que clase de información es registrada y archivada por la organización. Mientras es revisada la implementación de procedimientos internos por la organización, los evaluadores deben muestrear los registros generados y comprobar su contenido. Los objetivos internos, la planificación y los plazos proyectados al futuro deben ser confirmados por la evidencia revisada. Los evaluadores también deben verificar si los resultados de este proceso (Autorización de comercialización, modificaciones, etc.) se usarán como insumo para los procesos relacionados como lo son la inspección reguladora, la vigilancia post-comercialización o la promoción de medicamentos.
Sub-Indicador 3.5.1: Existe un listado/ base de datos actualizado de todos los productos aprobados,
suspendidos o retirados y las solicitudes rechazadas.
CRÍTICO
Sub-Indicador 3.5.2: La ARN conserva un dossier de cada producto registrado así como también de las
modificaciones y renovaciones con la documentación de apoyo, incluyendo la solicitud, información aprobada del medicamento, informe de evaluación, evaluación riesgo/beneficio, etc. y todas las exenciones están documentadas en este dossier.
CRÍTICO
Sub-Indicador 3.5.3: La ARN conserva un registro (archivo o expediente) de cada aplicación rechazada.
NECESARIO
Indicador 3.6: Disponibilidad de la información
Los evaluadores deben revisar que tipo de información está públicamente disponible, si los medios usados (página web, boletín oficial u otro boletín de la ARN) son apropiados y si la información es mantenida y actualizada en forma regular. Evidencia documental a ser estudiada:
Reglamentos, leyes, decretos
Orientación publicada en el sitio web
Procedimientos internos, plantillas y registros
Formato del informe de evaluación e informes de evaluación
Minutas de comités consultivos o reuniones de la cadena de toma de decisiones
Formato de autorización de comercialización

45
Información sobre el producto y formato del resumen de características del producto
Base resumida para el formato de decisión
Lista de personal con sus calificaciones
Lista de expertos externos con sus calificaciones
Lista de productos/solicitud registrados autorizados
Lista de las solicitudes rechazadas o retiradas
Lista de solicitudes pendientes
Lista y planificación para la revisión periódica de las solicitudes
Lista de titulares de autorizaciones de comercialización
Sub-Indicador 3.6.1: Existe una lista actualizada, pública y disponible para todo público de todos los
productos aprobados.
CRÍTICO
Sub-Indicador 3.6.2: Se encuentra publicada y disponible para todo público, un resumen de las características
del producto y/o resumen del informe técnico de evaluación, como base para la toma de decisión.
NECESARIO
Sub-Indicador 3.6.3: Se encuentra publicada y disponible para todo público una lista/base de datos de todas
las licencias retiradas, suspendidas o rechazadas.
NECESARIO

46
4. Actividades de licenciamiento - Modulo 4:
Objetivo: Evaluar la estructura de la Organización en cuanto al otorgamiento del licenciamiento de fabricantes de medicamentos e ingredientes farmacéuticos activos.
Esta función es esencial para garantizar que los fabricantes de medicamentos y vacunas licenciados cumplen con los estándares de buenas prácticas de fabricación recomendados por la OMS, lo cual es fundamental para asegurar que todos los productos son de calidad, seguros y eficaces. Es necesario definir claramente las distintas categorías de licenciamiento, el contenido y el formato de las licencias; detalle de los criterios en que las solicitudes de licencia se evalúan y proporcionar orientación a los interesados sobre el contenido y el formato de solicitud de certificado y de las circunstancias en que se requiere una solicitud de renovación, ampliación o modificación de la licencia. El sistema debe estar asistido por el Sistema de Gestión de Calidad.
Indicador 4.1: Bases legales
Los evaluadores deben revisar los requisitos legales vigentes y determinar si han sido promulgadas regulaciones adecuadas Evidencia documental a estudiar:
Los actos, leyes, decretos y / o disposiciones relativas a la obligación de licenciamiento para los fabricantes de medicamentos y vacunas.
Guías, procedimientos, formularios, instrucciones para los solicitantes.
Guías, procedimientos, registros o formato para la evaluación de las diferentes partes del expediente de solicitud.
Sub-Indicador 4.1.1: La legislación exige que una compañía que produce o que planea producir un
medicamento y vacunas deba tener licencia o certificado de BPM que avale que el establecimiento cumple con las BPM recomendadas por la Organización Mundial de la Salud, acorde con el tipo de producto (biológico o no, inyectables, etc).
CRITICO
Sub-Indicador 4.1.2: La legislación faculta a la ARN para emitir la licencia pero también para suspender y
cerrar/detener actividades de producción.
CRITICO
El evaluador debe verificar la existencia de las normas legales en relación con la obligación de licenciar a los fabricantes (licencia de establecimiento) y que dichas disposiciones hayan sido promulgadas.
Sub-Indicador 4.1.3: Las disposiciones legales exigen a los productores cumplir con las BPM para solicitar el licenciamiento.
CRITICO
El evaluador debe verificar que las disposiciones legales existentes, son promulgadas y aplicadas con respecto a la obligación de que las licencias para la fabricación de vacunas y medicamentos, sólo podrán concederse en función del cumplimiento de las BPM. El evaluador debe verificar que la ARN haya comprobado el cumplimiento de las BPM a través de inspecciones antes de la expedición de la licencia.
Sub-Indicador 4.1.4: Las disposiciones legales exigen la notificación por parte del fabricante a la ARN, para
su aprobación, de cambios/variaciones significativas a las condiciones bajo las cuales fue expedida la licencia inicial que pudieran afectar la calidad, seguridad y eficacia de los productos.
Con formato: Resaltar

47
CRITICO
Sub-Indicador 4.1.5: Existen disposiciones legales que requieren al menos de una persona calificada
responsable de cada local de producción.
CRITICO
Sub-Indicador 4.1.6: En el caso de existir exenciones legales de requisitos para el licenciamiento o certificación de BPM, están definidos los criterios para su aplicación.
NECESARIO
Indicador 4.2: Lineamientos y guías para la presentación de solicitudes de licenciamiento de elaboradores
El procedimiento de presentación y los requerimientos de documentación/información a ser suministrada a la ARN para la licencia de manufactura debe estar en el lugar. Los procedimientos y la documentación permitirán a la ARN garantizar las evaluaciones y decisiones de alta calidad. Los evaluadores deben revisar los documentos de orientación publicados para todo tipo de fabricantes (de medicamentos, de vacunas, de productos en investigación y otros) y determinar si abarcan el alcance de legislación vigente y regulaciones. Evidencia documental a ser evaluada:
Instrucciones para los solicitantes
Sitio Web de la ARN
Requerimientos oficiales para la presentación de solicitudes de licenciamiento
Sub-Indicador 4.2.1: Un documento escrito provee información detallada sobre el cumplimiento de los
requerimientos de las BPM, incluyendo productos en investigación. El documento está basado principalmente en el modelo de la OMS y sus guías suplementarias.
CRITICO
El evaluador deberá verificar que el estándar mínimo de las BPM para el otorgamiento de las licencias responda a la Serie de Informes Técnicos (SIT) de la OMS más reciente, ó PIC/S - PE 009-X en adelante. En la actualidad la SIT No. 908, 2003 de la OMS es la vigente, el mismo es posterior a la emisión del Informe 32 (SIT No.823, 1992), y así mismo, revisiones de este documento son realizadas con cierta periodicidad por lo que su aplicación debe ser verificada por los evaluadores.
Sub-Indicador 4.2.2: Existen guías para los productores sobre el contenido de la solicitud, el formato y el procedimiento a seguir para presentar una solicitud de licencia de producción según corresponda
CRITICO
El evaluador debe revisar los requisitos oficiales, que deberán publicarse y estar disponibles. Debe verificar si están disponibles y cómo las instrucciones administrativas para todos los solicitantes de licencias de establecimiento y verificar que los requisitos están en consonancia con la OMS u otros estándares internacionales aceptados. Si procede, las diferencias deben ser identificadas. Las instrucciones son públicas y lo ideal es que puedan ser adquiridas a través del sitio Web de la ARN.
Sub-Indicador 4.2.3: En el caso de no aplicar requerimientos específicos para el licenciamiento existen
criterios definidos para su no aplicación (procedimientos / pasos).

48
NECESARIO
Sub-Indicador 4.2.4: Existen guías para los solicitantes sobre la definición de los tipos y objetivos de las
modificaciones, el formato y la documentación requerida.
NECESARIO
Sub-Indicador 4.2.5: Existe(n) disposición(es), sobre el contenido del Expediente Maestro del Sitio (Site Master File), o Planta de Fabricación, el cual corresponde como mínimo con lo recomendado por la OMS o PIC/S.
NECESARIO
Sub-Indicador 4.2.6: Existen documentos sobre la designación y calificación del profesional responsable de
cada local de producción.
NECESARIO
Indicador 4.3: Organización y estructura
Los evaluadores deben identificar la estructura de la organización para llevar a cabo esta función reguladora, si la función es delegada o descentralizada y en particular a qué nivel del país (central, regional o local). Si diferentes organizaciones están involucradas a diferentes niveles del estado, los evaluadores deben revisar el nexo entre estas organizaciones y en particular como el intercambio de información es establecido y administrado.
Sub-Indicador 4.3.1: Las actividades de licenciamiento son organizadas y desarrolladas a nivel central del país.
INFORMATIVO
Sub-Indicador 4.3.2: Actividades descentralizadas de otras agencias/autoridades deben seguir los mismos
estándares, guías y procedimientos.
CRITICO
Sub-Indicador 4.3.3: En caso de descentralización un mecanismo de intercambio de información debe ser
establecido e implementado para que la organización descentralizada reciba requisitos y/o directrices desde la autoridad central y se le reporte a ésta.
CRITICO
Sub-Indicador 4.3.4: El mecanismo debe permitir una apropiada cooperación y colaboración entre organismos
descentralizados.
CRITICO
Indicador 4.4: Sistema de aseguramiento de la calidad para las actividades de licenciamiento
El Sistema de aseguramiento de la calidad de la ARN debe estar implementado para las actividades de licenciamiento. El documento de la OMS (WHO TRS 902, Annex 8, 2002) debe ser usado como referencia. Evidencia documental a evaluar:
Organigrama.
Procedimientos para el manejo administrativo de las solicitudes ingresadas y documentos.
Archivos de documentación de las autorizaciones de licenciamiento.

49
Política y procedimientos para la trazabilidad de las solicitudes.
Reportes de auditorías internas y externas.
Sub-Indicador 4.4.1: Organigrama definido y responsabilidades para implementar el Sistema de
Aseguramiento de Calidad.
CRITICO
El evaluador debe identificar la organización creada en el lugar para: establecer, implementar o mantener el SGC. Las diferentes responsabilidades deben ser identificadas.
Sub-Indicador 4.4.2: Documentación escrita para la realización de actividades de licencia de fabricación.
CRITICO
El evaluador debe identificar la existencia de documentación escrita para la realización de licenciamiento de fabricantes. Los evaluadores deben chequear los procedimientos y registros para el manejo administrativo de las solicitudes ingresadas, documentos y archivos de documentación de autorizaciones de licenciamiento. Gerenciamiento del sistema para asegurar la trazabilidad de las acciones (expedientes de licencias de elaboración y actualizaciones, informes de evaluación, informes de reuniones, variaciones etc.). El SGC establecido debe asegurar que las actividades de licenciamiento de fabricantes estén identificadas, documentadas y archivadas a fin de asegurar que puedan ser trazables si fuera necesario por alguna razón, como en el caso de una apelación de una institución contra decisiones, a fin de comparar hallazgos anteriores y recientes, o para reconocer tendencias en las conclusiones referentes a instituciones o productos específicos.
Sub-Indicador 4.4.3: Auditoría del sistema implementado y documentado (interna y/o externa).
CRITICO
El evaluador debe verificar que un sistema interno y/o externo este establecido, el cual garantice auditar el sistema de licenciamiento. En el caso de que el sistema de calidad este certificado (ej. De acuerdo a estándares de ISO) los organismos de certificación/acreditación realizan auditorias al titular de la certificación con intervalos regulares a fin de renovar la certificación/acreditación.
Indicador 4.5: Procedimientos de evaluación de licenciamiento
La evaluación de las solicitudes para asegurar una buena práctica como un elemento clave en el proceso de licenciamiento debe proveer suficiente información científica que permitirán adoptar decisiones independientes y de alta calidad. Evidencia documental a evaluar:
Formatos y procedimientos para la evaluación de reportes.
Evaluación de reportes.
Evidencia documentada para la toma de decisiones basada en la evaluación de los reportes.
Políticas o acuerdos para el reconocimiento de licencias de manufactura de otras ARN. Los evaluadores deben identificar el nivel de participación de la inspectoría en el proceso de licenciamiento, definiendo en particular que clase de inspección se realiza. Pueden usarse los siguientes indicadores para identificar el nivel de monitoreo por la ARN:

50
Número de establecimientos inspeccionados comparados con el número de solicitudes recibidas.
Número de inspectores involucrados.
Número promedio de días ocupados en terreno. Para asegurar la mejor práctica de la evaluación de las solicitudes como un elemento clave en los procesos de autorización y licenciamiento, deberá proporcionar suficiente información científica que permita adoptar decisiones independientes y de alta calidad. Evaluación documental a ser evaluada:
Formato y procedimiento de los informes de evaluación.
Informes de evaluación.
Evidencia documentada para la toma de decisiones basada en la evaluación de informes.
Políticas o acuerdos para el reconocimiento de licencias de fabricación de otras ARN. El evaluador debe verificar que las instrucciones escritas y promulgadas están disponibles para el personal y los expertos internos/externos, respecto a como las solicitudes de licenciamiento de empresas tienen que ser evaluadas y como el Site Master File del elaborador y los resultados de la inspección de verificación de cumplimiento de BPM tienen que ser evaluados. La evaluación debe ser realizada para cada elaborador de cada vacuna en cuestión.
Sub-Indicador 4.5.1: Existe un procedimiento para reunirse con los solicitantes antes de que la solicitud sea
oficialmente presentada.
INFORMATIVO
Sub-Indicador 4.5.2: Existe un procedimiento/lista de chequeo para evaluar las solicitudes de licenciamiento.
NECESARIO
Sub-Indicador 4.5.3: Cuando proceda, existen criterios definidos para no aplicar ciertos requisitos o pasos
específicos.
NECESARIO
Verificar en los casos que proceda si están definidos por escrito criterios que contemplen los casos en que los procedimientos de licenciamiento de rutina no tengan que ser seguidos. Los criterios deben estar definidos y tienen que ser conocidos por los evaluadores. Las razones podrían ser por ejemplo, la escasez de un producto en el mercado y la necesidad de importar un producto no autorizado durante un tiempo, los acuerdos con las demás ARN que permiten abreviar o prescindir de la evaluación de la documentación o parte de ella, la decisión de aceptar una autorización expedida por otras ARN, etc. El evaluador debe identificar cuáles son los mecanismos establecidos para el procedimiento abreviado de concesión de licencias o reconocimiento de las decisiones.
Sub-Indicador 4.5.4: Existe un procedimiento para la evaluación de solicitudes de modificaciones de las
licencias otorgadas.
NECESARIO
Sub-Indicador 4.5.5: Existen y están documentadas las medidas de control de calidad al proceso de
evaluación de la solicitud.
NECESARIO

51
El personal también debe participar en las revisiones por pares de solicitudes de licenciamiento y las decisiones tomadas al respecto. El evaluador debe verificar la calidad del proceso de evaluación de la solicitud: doble revisión (doble chequeo) pudiendo ser por pares, comités, reuniones técnicas o según este definido por la ARN.
Sub-Indicador 4.5.6: Existe un procedimiento para la toma de decisiones en la emisión, renovación,
modificación o revocación de las licencias.
CRITICO
Sub-Indicador 4.5.7: La licencia se emite en un formato estandarizado (siguiendo un procedimiento u otro
documento que garantice la uniformidad en contenido y formato).
NECESARIO
Sub-Indicador 4.5.8: La ARN reconoce y/o usa decisiones reguladoras, reportes o información de otras ARNs
u Organismos internacionales para la toma de decisiones.
INFORMATIVO
Sub-Indicador 4.5.9: En caso de reconocer y/o usar decisiones reguladoras, reportes o información de otras
ARN u Organismos internacionales para la toma de decisiones, existen criterios establecidos y procedimientos escritos.
NECESARIO
Algunos países no tienen oportunidad de evaluar algunas o todas las solicitudes de licenciamiento con sus propios expertos y refieren sus decisiones a otras ARN. En este caso el país puede reconocer certificados, reportes o decisiones de Autoridades de ARN extranjeras. Ej. En el país de origen del producto. En ese caso, el evaluador debe verificar que las disposiciones y los procedimientos escritos establecidos que definen quien está autorizado a reconocer decisiones de otras Autoridades, reportes o certificados, lista de las ARN en cuestión.
Sub-Indicador 4.5.10: Existe un procedimiento operativo estándar (POE) en el lugar para garantizar la
participación y comunicación entre los evaluadores y los inspectores para el cumplimiento y aplicación de las BPM.
NECESARIO
Sub-Indicador 4.5.11: El mismo estándar de BPM es usado para el licenciamiento, independiente de la fuente
(Ej. nacional, extranjero, público o sector privado) de la producción.
CRITICO
Verificar que los mismos criterios y estándares sean usados para la evaluación de los procedimientos de licenciamiento. Se debe utilizar el mismo criterio básico para garantizar la misma calidad, independientemente del origen. Evidencia documental a ser evaluada:
Guías, procedimientos y protocolos para la evaluación de concesión de licencias.
Procedimientos, criterios, protocolos y registros de licenciamiento de establecimientos elaboradores de medicamentos y vacunas evaluadas por evaluadores internos y externos.
Guías y procedimientos de licencias especiales cuando proceda.
Registros de las licencias especiales otorgadas cuando proceda.
Sub-Indicador 4.5.12: Existen plazos definidos para la evaluación de las nuevas solicitudes, renovaciones y
modificaciones de las licencias, según corresponda.

52
NECESARIO
Sub-Indicador 4.5.13: Un sistema de seguimiento interno está establecido para verificar el cumplimiento de
los objetivos programados.
NECESARIO
El evaluador debe tener en cuenta que no necesariamente las licencias deben tener plazo de validez ni exigirse su renovación.
Sub-Indicador 4.5.14: Una licencia escrita, firmada por una persona con la adecuada delegación es enviada
al solicitante incluyendo condiciones o restricciones relacionadas con las licencias.
CRITICO
Sub-Indicador 4.5.15: Se encuentra establecido que las actualizaciones de los establecimientos y del
producto sean proporcionados por los productores a la ARN para mantener la información actualizada y de acuerdo con los términos de la licencia.
NECESARIO
El evaluador debe verificar por ejemplo procedimientos internos u otros documentos que establezcan dicha actualización.
Indicador 4.6: Recursos humanos y otros
Recursos humanos adecuados (internos y externos) deben ser designados con el fin de poder llevar a cabo las actividades licenciamiento de establecimientos elaboradores para medicamentos y vacunas. Se pueden utilizar como indicadores el número de autorizaciones y licencias expedidas, el tiempo promedio para la evaluación de las solicitudes, etc., relacionados con el personal disponible. Evidencia documental a ser evaluada:
Organigramas.
Descripciones de puestos de trabajo.
Procedimientos para la contratación, la formación y calificación del personal.
Lista de personal y su calificación.
Planes de capacitación.
Planes de capacitación continua.
Registros de los entrenamientos realizados.
Guías y procedimientos para la selección y el uso de expertos externos.
Política de confidencialidad y conflicto de intereses de la ARN.
Sub-Indicador 4.6.1: Existe personal calificado en cantidad suficiente para la evaluación de las solicitudes y la
emisión de licencias.
CRITICO
El evaluador debe identificar si los recursos humanos están a disposición para llevar a cabo actividades de licenciamiento. La evaluación de los recursos humanos debe centrarse en los aspectos cuantitativos y cualitativos. El evaluador debe revisar la adecuación de las competencias de los evaluadores internos y expertos externos en cuanto a sus calificaciones en microbiología, farmacia, farmacología clínica, medicina, inmunología o una disciplina similar y su experiencia práctica en al menos una de estas disciplinas, así como en biofarmacéuticos. Esto puede alcanzarse por la experiencia de trabajo, capacitación y otros.

53
Otras competencias pueden ser necesarias para la evaluación de partes específicas de la solicitud. Idealmente el término adecuado para cada área de competencia es definido y se refleja en los planes de capacitación. A juzgar por los recursos adecuados el evaluador puede utilizar indicadores como el número de solicitudes recibidas, el número de modificaciones recibidas, el número de personal científico involucrado, el número promedio de días empleados por la ANR para la toma de decisiones, el trabajo atrasado. Si los expertos externos o un comité asesor / técnico está implicado en este proceso, se refieren a los sub-indicadores
Sub-Indicador 4.6.2: Existen requisitos a cumplir para el personal que evalúa las solicitudes y la emisión de
licencias; dicho personal es evaluado periódicamente a fin de determinar si se encuentra capacitado para cumplir sus funciones.
NECESARIO
El evaluador debe verificar que existe un sistema para monitorear los resultados y la aplicación de las competencias adquiridas en actividades de capacitación y que los indicadores para este proceso han sido desarrollados.
Sub-Indicador 4.6.3: Existen procedimientos escritos para selección y uso de expertos externos.
CRITICO
El evaluador debe identificar los procedimientos escritos para la selección y el uso de expertos externos y el establecimiento y gestión de los comités y la elección de los miembros del comité. El evaluador debe verificar que los criterios y procedimientos escritos sobre la confidencialidad se aplican, la independencia de los diferentes expertos que participan en los procesos de regulación y el manejo de posibles conflictos de intereses, y para la evaluación continua del desempeño de los expertos externos. El evaluador debe revisar que los planes de capacitación de expertos internos y externos se desarrollan e implementan refleja el estado actual de la educación y la experiencia.
Sub-Indicador 4.6.4: Existe espacio para el almacenamiento de documentación y el equipamiento y demás
recursos materiales y de apoyo (computadoras, impresoras, fotocopiadoras, papelería, equipos de comunicación, movilidad).
NECESARIO
Sub-Indicador 4.6.5: Los servicios de apoyo están disponibles adecuadamente en términos de comunicación
e infraestructura.
NECESARIO
Indicador 4.7: Registros y resultados
Los evaluadores deben revisar cómo toda la información recogida durante el proceso de licenciamiento es administrada y qué clase de información es registrada y archivada por la organización. Los evaluadores deben muestrear los registros generados y comprobar su contenido. Los objetivos internos, la planificación a futuro y los plazos proyectados deben confirmarse con la evidencia revisada. Los evaluadores deben verificar si los resultados de este proceso (licencias, modificaciones) se usarán como entrada-insumo para procesos relacionados tal como la inspección reguladora.

54
Sub-Indicador 4.7.1: Existe una lista/base de datos de todos los productores licenciados.
CRITICO
El evaluador debe verificar que la lista o base de datos Incluya como información mínima por ejemplo: nombre del titular, establecimiento farmacéutico licenciado, operaciones farmacéuticas amparadas por la licencia, fecha de emisión y en la que expira cuando corresponda.
Sub-Indicador 4.7.2: La ARN retiene un Expediente Maestro del Sitio de Fabricación de cada sitio licenciado,
incluyendo los cambios aprobados con la documentación de soporte. Estos contienen a lo menos la siguiente información: empresa, personal, recintos, equipos, lista de medicamentos producidos (si es adecuado). Todas las exenciones deben estar documentadas en este expediente.
CRITICO
Indicador 4.8: Disponibilidad de la información
Los evaluadores deben revisar que clase de información está públicamente disponible, si los medios usados son apropiados como por ejemplo el portal Web de la ARN y/o en el Boletín/Diario Oficial de cada país y si la información es mantenida y actualizada en forma regular. Evidencia documental a ser evaluada:
Reglamentos, leyes y decretos.
Guías de orientación publicadas.
Buenas prácticas de fabricación adoptadas incluyendo la orientación complementaria vigente.
Procedimientos internos, formatos y registros para entregar licencias.
Archivos de sitios de fabricación del establecimiento.
Lista del personal con sus calificaciones.
Lista de sitios de fabricación autorizados, rechazados o retirados.
Lista de personas autorizadas responsables de las actividades farmacéuticas.
Sub-Indicador 4.8.1: La lista /base de datos de todos los productores licenciados es publicada y está públicamente disponible.
CRITICO
El evaluador debe verificar que la lista o base de datos incluya como información mínima por ejemplo: nombre del titular, establecimiento farmacéutico licenciado, operaciones farmacéuticas amparadas por la licencia, fecha de emisión y en la que expira cuando corresponda.
Sub-Indicador 4.8.2: La lista/base de datos de todas las licencias de los productores: retiradas, suspendidas o
rechazadas es pública y está públicamente disponible.
NECESARIO
Indicadores cuantitativos a efectos de regulación:
Concesión de licencias a los fabricantes.
Número de personal científico involucrado en el otorgamiento de licencias.
Número de plantas de fabricación con licencia.
Número de solicitudes recibidas para una nueva licencia en el año de referencia.
Número de modificaciones de una licencia inicial recibidas en el año de referencia.
Número de las decisiones adoptadas (positivas, negativas, suspensión, o retirada) en el año de referencia.
Número de solicitudes pendientes en el atraso.
Número promedio de días para emitir una decisión.

55

56
5.- Vigilancia y control posteriores a la comercialización - Controles de importación y exportación - Módulo 5.
Objetivo: Para ser plenamente eficaz, las actividades de vigilancia de mercado deben ser complementadas por los procedimientos administrativos encaminados a asegurar que los medicamentos y productos biológicos se importen solo si se han autorizado o recibido una licencia de importación antes de llegar el país.
Una consignación de medicamentos y productos biológicos importados debe cumplir con todos los detalles dados a conocer en la licencia pertinente de importación. Todos los productos que se importan deben haber sido registrados por la ARN o recibieron una autorización de comercialización. Licencias de importación así como controles aplicados a productos importados deben conferirse en relación a la autorización de mercado.
Indicador 5.1: Bases legales
Los evaluadores deben revisar los requisitos legales vigentes y determinar si las regulaciones adecuadas han sido promulgadas. La legislación debe proveer sanciones adecuadas y proporcionales, las penalidades y proceso judicial conforme a las violaciones de la legislación vigente (vea inspección reguladora para ejecución y cumplimiento de actividades).
Sub-indicador 5.1.1: La legislación requiere que una compañía que importa o intenta importar medicamentos
y productos biológicos medicamentos debe tener licencia o autorización sanitaria específica para ejercer su actividad emitida por la ARN.
CRÍTICO
Sub-indicador 5.1.2: La legislación requiere que una compañía que exporta o intenta exportar medicamentos
y productos biológicos medicamentos debe tener licencia o autorización sanitaria específica para ejercer su actividad, emitida por la ARN.
CRÍTICO
Sub-indicador 5.1.3: La legislación exige que una compañía que actúa como mayorista o distribuidor de
medicamentos deba tener licencia o autorización sanitaria específica para ejercer su actividad.
CRÍTICO
Sub-indicador 5.1.4 La legislación faculta a la ARN a suspender o restringir la importación, exportación,
distribución y dispensación de medicamentos.
CRÍTICO
Sub-indicador 5.1.5: Existen disposiciones legales concernientes a reenvasado / re-etiquetado.
CRÍTICO
Sub-indicador 5.1.6: Las disposiciones legales requieren que los importadores, exportadores, mayoristas y
distribuidores cumplan con la aplicación de Buenas Prácticas de Almacenamiento y de distribución para obtener su licencia o autorización sanitaria.
CRÍTICO
Sub-indicador 5.1.7: Las disposiciones legales requieren la notificación a la ARN de cambios/variaciones
significativas respecto de la licencia inicial que puedan afectar la calidad seguridad y eficacia de los productos.
CRÍTICO

57
Sub-indicador 5.1.8 Existe disposición legal que establezca que debe haber a lo menos una persona
responsable de la calidad para cada importador, exportador, mayoristas y distribuidores.
CRÍTICO
Sub-indicador 5.1.9: En caso de haber disposiciones legales para exenciones de requerimientos para el
licenciamiento o autorización sanitaria, están formalmente establecidos los criterios para su concesión.
CRÍTICO
Indicador 5.2: Controles de importación y exportación
Los evaluadores deben revisar los documentos de orientación publicados para diferentes tipos de “actores claves” y determinar si abarcan el alcance de la legislación y regulaciones vigentes. Debe comprobarse coherencia e identificar cualquier diferencia con la orientación de la OPS/OMS. La evaluación de recursos humanos debe centrarse en los aspectos cuantitativos y cualitativos. Con respecto a los aspectos cuantitativos, los evaluadores pueden decidir usar los siguientes indicadores para identificar si están siendo disponibles recursos humanos adecuados para llevar a cabo las actividades planificadas:
Carga de trabajo para las funciones realizadas con los siguientes indicadores: número de solicitudes recibidas, número de autorizaciones de importación entregadas, número de certificados de exportación emitidos.
Número de personal científico involucrado.
Número promedio de días tomados por la ARN para emitir una decisión. Los evaluadores deben revisar si el personal involucrado en los procedimientos de importación y exportación son plenamente competentes, basado en sus calificaciones en asuntos reguladores y procedimientos de importación y exportación. Los evaluadores deben identificar la estructura de la organización para llevar a cabo funciones reguladoras, si la función es delegada o descentralizada y en particular a qué nivel del país (central, regional o local). Si diferentes organizaciones están involucradas a diferentes niveles del estado los evaluadores deben revisar el nexo entre estas organizaciones y en particular cómo el intercambio de información está establecido y administrado. Los evaluadores deben revisar los procedimientos con relación a sus resultados esperados, su nivel de detalle con relación a la capacitación proporcionada y las medidas aplicadas para verificar las actividades descritas. Los evaluadores en particular deben revisar su coherencia con la orientación, regulaciones y legislación vigentes.
Sub-indicador 5.2.1: Las disposiciones legales exigen que los medicamentos que se importan cuenten con
registro sanitario previo al trámite de importación.
CRÍTICO
Sub-indicador 5.2.2: Las disposiciones legales exigen a los importadores que tengan una autorización para
cada acto de importación de un medicamento.
CRÍTICO
Sub-indicador 5.2.3: Las disposiciones legales exigen a los exportadores que tengan una autorización para
cada acto de exportación de un medicamento.
NECESARIO

58
Sub-indicador 5.2.4: Las disposiciones legales exigen a los importadores que registren la información sobre el
origen y el destino de los productos importados.
CRÍTICO
Sub-indicador 5.2.5:.Las disposiciones legales exigen a los exportadores que registren la información sobre el
origen y el destino de los productos exportados.
NECESARIO
Sub-indicador 5.2.6:. Existen guías para los importadores y exportadores sobre el formato y contenido de las
solicitudes y el procedimiento a seguir para la obtención de estas autorizaciones.
NECESARIO
Sub-indicador 5.2.7:. Las actividades se definen y organizan en el nivel central del país, y se desarrollan, bajo
su responsabilidad y control.
INFORMATIVO
Sub-indicador 5.2.8:. Las actividades descentralizadas a otras agencias o autoridades siguen los mismos
estándares, guías y procedimientos.
CRÍTICO
Sub-indicador 5.2.9: En el caso de descentralización, está establecido e implementado un mecanismo de intercambio de información para que la organización descentralizada reciba solicitudes y/o directivas de la autoridad central y a la vez pueda reportar a ésta.
CRÍTICO
Sub-indicador 5.2.10: Los mecanismos permiten la cooperación y la colaboración apropiada entre las
organizaciones descentralizadas.
CRÍTICO
Sub-indicador 5.2.11: Existen procedimientos documentados en la ARN para evaluar la solicitud de
importación o exportación de medicamentos.
NECESARIO
Sub-indicador 5.2.12: Existen procedimientos documentados para otorgar las autorizaciones de
importaciones y los certificados de exportación con formatos estandarizados.
CRÍTICO
Sub-indicador 5.2.13: La ARN mantiene registros o una base de datos sobre productos importados y
exportados.
NECESARIO
Sub-indicador 5.2.14: Existe colaboración o un convenio con la aduana u otra agencia de fiscalización en el
control de las importaciones y exportaciones.
NECESARIO
Mientras es revisada la implementación de procedimientos internos por la organización, los evaluadores deben muestrear los registros generados y comprobar su contenido. Los evaluadores deben verificar si los resultados de este proceso se usarán como entrada-insumo para procesos relacionados

59
Los evaluadores deben revisar que clase de información está públicamente disponible, si el medio usado (portal Web, boletín oficial u otro boletín de la ARN), son apropiados y si la información se mantiene y se actualiza en forma regular. Evidencia documental a ser estudiada:
Reglamentos, leyes, decretos.
Orientación publicada en controles de importación y exportación.
Procedimientos de control internos, plantillas y registros.
Lista del personal con sus calificaciones.
Lista de productos con autorización de importación y exportación. Referencia Guidelines on import procedures for pharmaceutical products - Annex 11 in WHO Expert Committee on specifications for pharmaceutical preparation. 35 Report, WHO, 1996 (WHO Technical Report Series, N°863)
Indicador 5.3: Control de Mercado
La aparición de productos falsificados y otros productos ilícitos dentro de los mercados nacionales e internacionales en los últimos años ha impuesto una dimensión extra en el trabajo de las autoridades reguladoras y los inspectores. También ha creado la necesidad de una mejor colaboración entre autoridades reguladoras, titulares de licencia, oficiales de aduana y autoridades encargadas de hacer cumplir la ley, así como la necesidad de mayor vigilancia por todas las personas involucradas en la fabricación, distribución y venta de medicamentos. Por consiguiente deben considerarse las disposiciones legales que facilitan el intercambio oportuno y eficaz de la información entre las partes interesadas, en el plano nacional e internacional, que puede, entre otros efectos, contrarrestar el comercio ilícito. A las ARNs se les recomienda vigilar la calidad y seguridad de los medicamentos para prevenir que los medicamentos nocivos, de calidad inferior a la norma y falsificados lleguen al público. Programas de vigilancia aseguran que muestras se colecten aleatoriamente del mercado y se prueben a intervalos programados. Deben ser desarrollados planes de control de calidad y estrategias de muestreo sobre la base de identificar o anticipar riesgos potenciales para la salud. Etiquetas del producto y prospectos deben verificarse con aquellos aprobados para el registro. Un sistema para recibir quejas acerca de la calidad del producto debe instituirse y todas las quejas deben ser investigadas y documentadas. La calidad del producto puede inspeccionarse al probar las muestras tomadas de los fabricantes y la cadena de distribución, aleatoriamente o cuando exista una base para sospechar que un producto quizá sea inferior al estándar o falsificado. Deben realizarse pruebas para asegurar que las muestras cumplan con requisitos del compendio (por ejemplo, Farmacopea británica, Farmacopea de EE.UU., Farmacopea Internacional, etc.) o que estén aprobadas las especificaciones del fabricante cuando sea apropiado. Los evaluadores deben revisar los requisitos legales vigentes y determinar si regulaciones adecuadas han sido promulgadas. La legislación debe autorizar a la ARN a muestrear productos en el mercado y aplicar sanciones adecuadas y proporcionales, las penalidades y proceso judicial conforme a las violaciones de la legislación vigente (vea inspección reguladora para ejecución y cumplimiento de actividades).

60
Los evaluadores deben revisar los procedimientos con relación a sus resultados esperados, su nivel de detalles y relevancia para la capacitación impartida y las medidas aplicadas para verificar las actividades descritas. Los evaluadores en particular deben revisar su coherencia con la orientación, regulaciones y legislación vigente.
Sub-indicador 5.3.1: Hay disposiciones legales con la definición de medicamentos falsificados y de un delito
farmacéutico.
CRÍTICO
Sub-indicador 5.3.2: Hay una disposición legal para el muestreo y análisis de muestras de los medicamentos comercializados.
CRÍTICO
Sub-indicador 5.3.3: Hay procedimientos documentados para muestrear y analizar los productos vendidos o
disponibles en el mercado.
NECESARIO
Sub-indicador 5.3.4: Los criterios para la recolección de muestras se basan en la evaluación de riesgos.
NECESARIO
Sub-indicador 5.3.5: Hay disposiciones y criterios para la compensación de las muestras recolectadas.
INFORMATIVO
Los evaluadores deben identificar como la organización establece para llevar a cabo esta función reguladora, si la función es delegada o descentralizada y en particular a qué nivel del país (central, regional o local). Si diferentes organizaciones están involucradas a diferentes niveles del estado, los evaluadores deben revisar el nexo entre estas organizaciones y en particular cómo el intercambio de información esta establecida y administrada. Mientras es revisada la implementación de procedimientos internos por la organización, los evaluadores deben muestrear los registros generados y verificar sus contenidos. Los siguientes indicadores pueden ser usados por los evaluadores para identificar el nivel de las actividades de control realizadas por el país:
Número de productos vigilados.
Número de productos detectados como no conforme.
Número de medicamentos probados por controles del mercado.
Número de denuncias judiciales y las sanciones legales. Los evaluadores deben revisar que la información está públicamente disponible, si el medio usado (portal Web, boletín oficial u otro boletín de la ARN) son apropiados y si la información es mantenida y actualizada de forma regular.
Sub-indicador 5.3.6: Las actividades se organizan y desarrollan en el nivel central del país.
INFORMATIVO
Sub-indicador 5.3.7: Las actividades descentralizadas a otras agencias o autoridades siguen los mismos estándares, guías y procedimientos.
CRÍTICO

61
Sub-indicador 5.3.8: En el caso de descentralización, está establecido e implementado un mecanismo de
intercambio de información para que la organización descentralizada reciba solicitudes y/o directivas de la autoridad central y a la vez pueda reportar a ésta.
CRÍTICO
Sub-indicador 5.3.9: Los mecanismos propician la apropiada colaboración entre las organizaciones
descentralizadas.
CRÍTICO
Sub-indicador 5.3.10: Hay procedimientos documentados para recopilar información, detectar y combatir la falsificación, los productos sub estándares, los productos sin registro y el comercio ilegal de medicamentos tal como la venta de medicamentos de origen desconocida o en establecimientos no licenciados/ autorizados.
CRÍTICO
Sub-indicador 5.3.11: Se ha establecido y se aplica una estrategia de vigilancia de mercados basada en
inspecciones, programas de análisis en laboratorio, y/o otras informaciones.
CRÍTICO
Sub-indicador 5.3.12: Se ha establecido un programa de vigilancia estructurado con tareas, procedimientos y
responsables para abarcar diversas formas farmacéuticas de diferentes drogas/medicamentos, basado en criterios de riesgo sanitario.
CRÍTICO
Sub-indicador 5.3.13. Existen procedimientos describiendo actividades de investigación de casos o
sospechas de productos con desvío de cualidad.
CRÍTICO
Sub-indicador 5.3.14:.El programa de vigilancia se examina periódicamente y se evalúa su eficacia.
NECESARIO
Sub-indicador 5.3.15: Hay un programa especial/específico para detectar y combatir la falsificación y el
comercio ilegal de medicamentos tal como la venta de medicamentos de origen desconocida o en establecimientos no licenciados/ autorizados.
CRÍTICO
Sub-indicador 5.3.16: Existen procedimientos describiendo actividades de investigación, retención y
procesamiento en casos o sospechas de productos falsificados.
CRÍTICO
Sub-indicador 5.3.17: La ARN mantiene los registros o información / base de datos de las muestras recogidas
(de productos falsificados y sub estándares y del comercio ilegal) y de los resultados de sus análisis.
NECESARIO
Sub-indicador 5.3.18: Se ha establecido e implementado un acuerdo de colaboración entre las aduanas u
otro organismo de fiscalización para las actividades de control de mercado.
NECESARIO
Sub-indicador 5.3.19. Existe una estructura reguladora de coordinación para combatir los medicamentos falsificados.
NECESARIO

62
Sub-indicador 5.3.20: Existe un procedimiento documentado para la comunicación con las autoridades de
salud y de la jurisdicción pertinente y para cooperar en las investigaciones y los incumplimientos de la reglamentación.
NECESARIO
Evidencia documental a ser estudiada:
Reglamentos, leyes, decretos.
Orientación publicada para el control de mercado.
Procedimientos internos, plantillas y registros.
Lista de personal con sus calificaciones.
Lista de programa de vigilancia, acciones implementadas, dentro de un período de referencia. Referencias Observations and recommendations on counterfeit medicines (Chap.8 - Quality assurance of pharmaceuticals - A compendium of guidelines and related materials - Volume1 - WHO; 1997; 238 pages) Guidelines for the development of measures to combat counterfeit medicines (WHO/EDM/QSM/99.1)
Indicador 5.4: Productos no conformes y procedimientos de retiro
La ARN debe establecer y administrar un sistema de notificación para que sea informado apropiadamente acerca de temas de calidad que afectan a los medicamentos ya en el mercado. El proceso para evaluar las quejas acerca de productos debe incluir la posibilidad de que la no-conformidad pueda deberse a productos falsificados. Provisiones adecuadas deben existir para manejar el retiro de medicamentos del mercado y su destrucción, requiriendo a los fabricantes que retiren productos que sean inseguros, defectuosos o inapropiadamente etiquetados y, cuando sea necesario, suspender la fabricación y comercialización donde los establecimientos u operaciones resultan ser de inferior calidad o a cesar actividades promocionales no-éticas. Ejemplos de posibles acciones que pueden ser tomadas incluyen suspender una autorización de comercialización, retirar lotes específicos, imprimir advertencias en boletines de medicamentos nacionales o colocar advertencias separadas a una lista de instituciones y de personas claves o medicamentos de prescripción. Los evaluadores deben revisar los requisitos legales vigentes y determinar si regulaciones adecuadas han sido promulgadas. Los evaluadores deben revisar los documentos de orientación publicadas para diferentes tipos de "actores claves" y determinar si cubren el alcance de la legislación y regulaciones aplicables. Debe verificarse coherencia con la orientación de la OPS/OMS e identificar cualquier diferencia.
Sub-indicador 5.4.1: La legislación le permite a la ARN exigir el retiro de medicamentos cuando se
encuentren en una situación de infracción respectos de las normas vigentes.
CRÍTICO
Sub-indicador 5.4.2: La legislación le permite a la ARN suspender o impedir la fabricación, importación,
exportación, distribución, venta y el uso de medicamentos.
CRÍTICO
Sub-indicador 5.4.3: La legislación le permite a la ARN suspender o impedir la comercialización o campañas
de promoción y detener la distribución de material promocional.

63
CRÍTICO
Sub-indicador 5.4.4: Existe un requisito legal de un sistema de registro en la cadena de distribución para
asegurar la trazabilidad de los lotes para facilitar un sistema eficaz de retiro.
CRÍTICO
Sub-indicador 5.4.5: Existe una disposición legal que faculte a la ARN para dictaminar la eliminación de productos defectuosos o que no cumplen con las normas.
CRÍTICO
Sub-indicador 5.4.6: Existe un requerimiento legal para los fabricantes/importadores de notificar a la ARN
sobre el inicio de un retiro.
CRÍTICO
Sub-indicador 5.4.7: Existe un requerimiento legal para los fabricantes/importadores de notificar a la ARN
sobre quejas acerca de la calidad de los productos.
CRÍTICO
Sub-indicador 5.4.8: Existen disposiciones legales que instruyan a los fabricantes, importadores y
exportadores acerca del procedimiento que deben usar para el retiro y la eliminación de productos defectuosos o que no cumplen con las normas.
CRÍTICO
Los evaluadores deben revisar los procedimientos con relación a los resultados esperados, su nivel de detalle en relación a la capacitación impartida y las medidas aplicadas para verificar las actividades descritas. Los evaluadores en particular deben revisar su coherencia con la orientación, regulaciones y legislación vigentes. Mientras es revisada la implementación de procedimientos internos por la organización, los evaluadores deben muestrear los registros generados y verificar sus contenidos. Los siguientes indicadores pueden ser usados por los evaluadores para identificar el nivel de las actividades de control realizadas por el país:
Número de quejas recibidas.
Número de medicamentos retirados.
Número de advertencias o avisos de cumplimiento expedidas por la ARN.
Los evaluadores deben revisar que la clase de información está públicamente disponible, si el medio usado (portal Web, boletín oficial u otro boletín de la ARN) son apropiados y si la información es mantenida y actualizada de forma regular.
Sub-indicador 5.4.9: Existen procedimientos documentados y implementados en la ARN para asegurar la
evaluación de la notificación de los retiros y de las quejas sobre los productos.
NECESARIO
Sub-indicador 5.4.10: Existen procedimientos documentados e implementados en la ARN para garantizar la
remoción de los productos defectuosos o que no cumplen con las normas; que permitan organizar un retiro efectivo y su posterior eliminación.
CRÍTICO

64
Sub-indicador 5.4.11: Existen criterios definidos para determinar los medios apropiados y el nivel de la
comunicación del retiro con un mecanismo de retroalimentación.
NECESARIO
Sub-indicador 5.4.12: La ARN utiliza mecanismos para evaluar la efectividad del retiro de un producto
defectuoso del mercado.
NECESARIO
Sub-indicador 5.4.13: La ARN aprueba la finalización del retiro y confirma la eliminación de los productos
defectuosos o que no cumplen con las normas.
NECESARIO
Sub-indicador 5.4.14: La ARN mantiene los registros o información/una base de datos sobre retiros y
productos defectuosos o que no cumplen normas que han sido desechados.
NECESARIO
Evidencia documental a ser estudiada:
Reglamentos, leyes, decretos.
Orientación para fabricantes, importadores-exportadores y distribuidores en como manejar/organizar un retiro, incluyendo la destrucción de productos defectuosos.
Procedimientos internos para manejar defectos de calidad (notificaciones de calidad sospechosa, defectos, retiros de lotes de compañías) y registros.
Lista del personal con sus calificaciones.
Procedimientos internos para organizar un retiro.
Lista de retiros realizados.

65
6. Farmacovigilancia - Módulo 6
Objetivo: Evaluar la estructura de la organización para recolectar información, evaluar y decidir sobre la seguridad de medicamentos.
El objetivo de este módulo es evaluar la vigilancia de las reacciones adversas a los medicamentos (RAM) y los reportes de ESAVis Debe establecerse un sistema de notificación para vigilar la seguridad de los medicamentos. A las ARNs se les recomienda recopilar, analizar y evaluar información sobre las RAM notificadas y tomar las decisiones apropiadas. El alcance y extensión de la farmacovigilancia deben definirse claramente en la legislación, regulaciones y orientación. Un comité consultivo debe establecerse dentro de la autoridad reguladora a cargo de vigilar quien puede participar en revisar los informes de las RAM. Los países deben considerar si tienen recursos adecuados para establecer su propio mecanismo de notificación de reacciones adversas y la capacidad reguladora de usar la información reunida. Inicialmente deben considerar promulgar los requisitos de informes que están en proporción con su estructura organizativa y considerar ampliar a mayor escala los requisitos de notificación hacia un procedimiento de registro formal, solo cuando esta estructura se convierte en una más sofisticada. Conexión con otros organismos internacionales y ARNs es un método lógico para adquisición, compartir e intercambiar información relevante sobre la seguridad de los medicamentos y para basar decisiones sobre que medida apropiada tomar. Programas de entrenamiento para promover farmacovigilancia entre los profesionales médicos y de la salud también deben ser llevados a cabo por la propia ARN u otra organización bajo su control.
Indicador 6.1: Bases legales
Los evaluadores deben revisar los requisitos legales vigentes y determinar si las regulaciones apropiadas han sido promulgadas. La legislación debe proveer sanciones adecuadas y proporcionales, penalidades y procesamiento judicial conforme a las violaciones de la legislación vigente (vea inspección reguladora para ejecución y cumplimiento de actividades).
Sub-indicador 6.1.1: Existe bases legales sobre la vigilancia post-comercialización de la seguridad de los
medicamentos
CRITICO
Sub-indicador 6.1.2: Las bases legales exigen a la ARN que implemente un sistema de vigilancia para
recopilar información útil en farmacovigilancia, para evaluar ésta información y tomar las decisiones
CRITICO
Sub-indicador 6.1.3: Existen bases legales para los Laboratorios farmacéuticos y titulares de RS, que
dispongan el registro, la recolección y mantenimiento de datos, la evaluación y el monitoreo de las reacciones/eventos adversos y para reportarlos a la ARN bajo los requisitos establecidos.
CRITICO
Sub-indicador 6.1.4: Las bases legales exigen a distribuidores e importadores que reporten las reacciones o
eventos adversos al titular de RS o a la ARN bajo los requisitos establecidos.
CRITICO

66
Sub-indicador 6.1.5: Las disposiciones legales exigen a los profesionales de la salud que reporten las
reacciones o eventos adversos a los titulares del RS o a la ARN u otra autoridad a cargo.
NECESARIO
Sub-indicador 6.1.6: Existen requisitos específicos para reportar temas de seguridad relacionados con
categorías específicas de los productos (vacunas, productos biológicos, biotecnológicos etc.).
NECESARIO
Sub-indicador 6.1.7: Existen requisitos específicos para Laboratorios Farmacéuticos y titulares del RS,
designar una persona capacitada a cargo del monitoreo de la seguridad post comercialización
NECESARIO
Sub-indicador 6.1.8: Existen bases legales para definir la terminología utilizada como por ejemplo evento
adverso, reacción adversa, eventos adversos serios, etc.
CRITICO
Sub-indicador 6.1.9: Las bases legales establecen la demora y/o la periodicidad para informar sobre eventos
adversos.
CRITICO
Sub-indicador 6.1.10: Existen requisitos específicos para las instituciones de salud (clínicas, hospitales, etc.) para designar a una persona encargada del monitoreo de la seguridad post comercialización.
NECESARIO
Indicador 6.2: Lineamientos y guías
Los evaluadores deben revisar los documentos de orientación publicados para diferentes tipos de “actores claves” y determinar si abarcan el alcance de la legislación y regulaciones vigentes. Si diferentes organizaciones toman parte, deben ser mencionados claramente los roles y responsabilidades de cada uno Debe comprobarse si hay coherencia con la orientación de la OPS/OMS y cualquier diferencia identificarla.
Sub-indicador 6.2.1: Existen guías sobre el monitoreo de la seguridad post - comercialización en relación con
el registro, el reporte y el formato que debe ser usado.
NECESARIO
Sub-indicador 6.2.2: Existen guías sobre la clasificación de eventos relacionados con la seguridad.
NECESARIO
Sub-indicador 6.2.3: La guía sobre reportes de seguridad aporta a la evaluación científica del balance entre
riesgo/beneficio de los medicamentos.
CRÍTICO
Sub-indicador 6.2.4: Existen pautas que definen los conocimientos científicos y el entrenamiento adecuado a
las personas calificadas y los puntos focales a cargo de la farmacovigilancia.
NECESARIO
Sub-indicador 6.2.5: Existen guías sobre el criterio para determinar los plazos y los medios para reportar los
eventos adversos (severos, esperados, etc.).
NECESARIO

67
Sub-indicador 6.2.6: Existen guías y regulaciones para la vigilancia pos comercialización que incluyen
monitoreo y manejo del evento adverso posterior a la inmunización.
CRÍTICO
Sub-indicador 6.2.7: Las Guías existentes para el seguimiento de eventos adversos con productos biológicos
se encuentran publicadas y distribuidas a los actores de la vigilancia.
CRÍTICO
Indicador 6.3: Organización y Estructura
Los evaluadores deben identificar la estructura de la organización para llevar a cabo esta función reguladora; si la función es delegada o descentralizada y en particular a qué nivel del país (central, regional o local). Si diferentes organizaciones se involucran a diferentes niveles del estado los evaluadores deben revisar el nexo entre estas organizaciones y en particular cómo el intercambio de información esta establecido y administrado. Los evaluadores pueden usar el número de puntos de contacto dentro del país, el número total de RAM y ESAVIs informadas para determinar el nivel de farmacovigilancia en relación con los indicadores de sistemas de atención de salud mencionados en el módulo 1.
Sub-indicador 6.3.1: Las actividades de vigilancia se organizan e implementan a nivel central del país.
INFORMATIVO
Sub-indicador 6.3.2: Las actividades descentralizadas a otras agencias o autoridades siguen los mismos
estándares, guías y procedimientos.
CRÍTICO
Sub-indicador 6.3.3: En el caso de descentralización está establecido o implementado un mecanismo de
intercambio de información para que la organización descentralizada reciba solicitudes y/o directivas de la autoridad central y a la vez pueda reportar a ésta.
CRÍTICO
Sub-indicador 6.3.4: Los mecanismos permiten la cooperación y la colaboración apropiada entre las
organizaciones descentralizadas.
CRÍTICO
Indicador 6.4: Procedimientos internos
Los evaluadores deben revisar los procedimientos con relación a los resultados esperados, su nivel de detalle y conveniencia en relación a la capacitación proporcionada y las medidas aplicadas para verificar las actividades descritas. Los evaluadores en particular deben revisar su coherencia con la orientación, regulaciones y legislación vigentes. En el contexto de farmacovigilancia, los evaluadores deben prestar atención especial a los retrasos tomados y los plazos aplicados por los fabricantes, así como los niveles administrativos, intermedios y centrales, para transmitir, investigar y evaluar información. Los evaluadores deben ver en que extensión la inspectoría está involucrada en controlar las prácticas de farmacovigilancia, en particular si las inspecciones son realizadas. Los siguientes indicadores pueden ser usados para identificar el nivel de vigilancia por la ARN:
Número de establecimientos inspeccionados para farmacovigilancia en el año de referencia.

68
Número promedio de días tomados por establecimiento para inspeccionar en terreno.
Sub-indicador 6.4.1: La información externa (fuentes de información y materiales de referencia) para la toma
de decisiones sobre reacciones adversas a medicamentos y el monitoreo de la seguridad está fácilmente disponible.
CRÍTICO
Sub-indicador 6.4.2: La información externa (fuentes de información y materiales de referencia) para la toma
de decisiones para los eventos sospechosos atribuidos a la inmunización y el monitoreo de la seguridad está fácilmente disponible.
CRÍTICO
Sub-indicador 6.4.3: Existen procedimientos documentados en la ARN para registrar y evaluar los reportes
diarios de reacciones adversas.
CRÍTICO
Sub-indicador 6.4.4: Existen procedimientos documentados en la ARN para analizar las tendencias de
seguridad para la detección de señales.
NECESARIO
Sub-indicador 6.4.5: Se ha establecido un sistema para la priorización de las señales de seguridad de
acuerdo al impacto de salud pública y para demostrar que los temas de alto riesgo se investigan inmediatamente, o en una primera instancia.
CRÍTICO
Sub-indicador 6.4.6: Se ha establecido un sistema de seguimiento interno (legal o no) para seguir los plazos
programados en el proceso.
NECESARIO
Sub-indicador 6.4.7: La falta de eficacia debida a medicamentos bajo sospecha de ser falsificados se
contempla durante el proceso de evaluación.
CRÍTICO
Sub-indicador 6.4.8: La falta de eficacia debida a productos biológicos bajo sospecha de ser falsificados se
contempla durante el proceso de evaluación.
CRÍTICO
Sub-indicador 6.4.9: Hay procedimientos documentados para la toma de decisiones y para definir las
acciones recomendadas a ser tomadas por la ARN y/o el fabricante u otros interesados directos.
CRÍTICO
Sub-indicador 6.4.10: La ARN organiza regularmente campañas para promover la adhesión a la vigilancia,
Incluyendo los actores del programa nacional de inmunización.
NECESARIO
Sub-indicador 6.4.11: Los consumidores están involucrados en el programa de monitoreo de la seguridad.
NECESARIO
Sub-indicador 6.4.12: La ARN tiene capacidad para detectar problemas relacionados con la seguridad de las
vacunas.
CRÍTICO

69
Indicador 6.5: Recursos Humanos y otros
La evaluación de recursos humanos debe centrarse en aspectos cuantitativos y cualitativos. Para los aspectos cuantitativos los evaluadores pueden decidir usar los siguientes indicadores para identificar si se están poniendo a disposición recursos humanos adecuados para llevar a cabo las actividades planificadas:
Carga de trabajo para las funciones realizadas con los siguientes indicadores: número de RAM informadas y reportes periódicos revisados.
Número de personal científico involucrado.
Trabajo acumulado o atraso generado (carga de trabajo comparado con el número de decisiones tomadas).
Número de investigaciones realizadas.
Número de cartas de advertencia/notificaciones de seguridad generadas.
Número promedio de días tomados por la ARN para tomar una decisión. Los evaluadores deben revisar si el personal involucrado en los procesos de vigilancia es plenamente competente en particular con respecto a las siguientes áreas:
Toxicología experimental.
Estudios en animales.
Prueba “in vitro”.
Farmacología clínica.
Fármaco-epidemiología.
Utilización de medicamentos.
Estadísticas y epidemiología. Si expertos externos o un comité asesor/técnico participan en este proceso regulador, los evaluadores deben referirse a las preguntas aplicables en el Capítulo 3.8.
Sub-indicador 6.5.1: Existe personal adecuado y pericia (educación, experiencia y capacitación) para las
actividades de monitoreo de seguridad de medicamentos y productos biológicos.
CRÍTICO
Sub-indicador 6.5.2: Se han implementado medidas de control de calidad documentadas como la revisión por
pares.
NECESARIO
Sub-indicador 6.5.3: Hay expertos externos participando en la evaluación de la información sobre seguridad
transmitida a través de la red de vigilancia.
NECESARIO
Sub-indicador 6.5.4: Existe un comité consultivo de expertos que participa en la revisión de la información
sobre seguridad transmitida a través de la red de vigilancia.
NECESARIO
Indicador 6.6: Registros y resultados
Los evaluadores deben revisar cómo toda la información recogida durante los procesos de registro y evaluación es administrada y que clase de información es registrada y archivada por la organización.

70
Mientras es revisada la implementación de procedimientos internos de la organización, los evaluadores deben muestrear los registros generados y comprobar su contenido. Los objetivos internos, la planificación a futuro y los plazos proyectados deben ser confirmados por la evidencia revisada. Los evaluadores deben verificar si los resultados de este proceso se usarán como entrada para procesos relacionados como la autorización de comercio o la inspección reguladora.
Sub-indicador 6.6.1: La información sobre seguridad recolectada se utiliza para tomar o modificar decisiones
sobre autorizaciones de comercialización originales para medicamentos como biológicos (agregando información, restringiendo el uso, retiro de productos, etc.).
CRÍTICO
Sub-indicador 6.6.2: La ARN mantiene la información/base de datos sobre eventos de seguridad reportados y
las acciones tomadas. La terminología utilizada es la recomendada por la OMS.
CRÍTICO
Sub-indicador 6.6.3: La ARN mantiene un archivo maestro de cada RAM, ESAVI con la documentación
comprobatoria.
CRÍTICO
Sub-indicador 6.6.4: La herramienta de consulta en los datos recolectados permite a la ARN evaluar e
interpretar las señales de seguridad (cálculo de tasa de incidencia, evaluación de causalidad).
NECESARIO
Sub-indicador 6.6.5: La información de ESAVIs serios es compartida con las otras autoridades nacionales
relacionadas con la inmunización como el programa nacional de inmunización.
CRÍTICO
Indicador 6.7: Disponibilidad de información
Los evaluadores deben revisar la información que está públicamente disponible, si los medios usados (portal Web, boletín oficial u otro boletín de la ARN) son apropiados y si la información es mantenida y actualizada en forma regular. Evidencia documental a ser estudiada:
Reglamentos, leyes, decretos.
Procedimientos internos y registros.
Formato RAM inicial y periódico para informe.
Formato para intercambio de información con otras ARNs y la OMS.
Lista de personal y sus calificaciones.
Sub-indicador 6.7.1: La información sobre las RAM, ESAVIs y sobre las medidas tomadas respecto del
monitoreo de la seguridad se comunican al público incluyendo el aviso sobre seguridad.
CRÍTICO
Sub-indicador 6.7.2: Existe un Sistema para retroalimentar a todos los niveles del sistema nacional de salud
sobre eventos asociados a la inmunización.
NECESARIO
Referencias: Safety of Medicines A guide to detecting and reporting adverse medicine reactions (WHO/EDM/QSM/2002.2)

71
Safety Monitoring of Medicinal Products: Guidelines for Setting Up and Running a Pharmacovigilance Centre (UMC; 2000; 28 pages) The Importance of Pharmacovigilance - Safety Monitoring of Medicinal Products (WHO; 2002; 52 pages) The safety of medicines in public health programs: Pharmacovigilance an essential tool (WHO, Geneva, 2006) Consumer reporting of adverse medicine reactions - General Policy Issues (WHO Medicine Information Vol. 14, No. 04, 2000) WHO guidelines on safety monitoring of herbal medicines in pharmacovigilance systems (WHO; 2004; 82 pages)

72
7. Ensayos Clínicos - Módulo 7
Objetivo: Revisar cómo la autoridad reguladora controla los ensayos clínicos (EC) conducidos en su territorio.
Una ARN debe definir si es necesaria una solicitud para conducir un ensayo clínico de un medicamento no registrado o un medicamento registrado con intención de nuevos usos. Para hacerlo, un sistema de registro debe incluir provisiones para la fabricación e importación de los materiales requeridos, sujeta a los controles apropiados. Tales pruebas deben tener lugar solo después que la acreditación formal se haya obtenido de la autoridad de registro competente. Por otro lado, evidencia escrita se requiere para verificar que el EC se esté conduciendo en conformidad con los principios declarados en la Declaración de la Asociación Médica Mundial de Helsinki y las directrices expedidas por el Consejo de Organizaciones Internacionales de las Ciencias Médicas. Tales pruebas deben llevarse a cabo siguiendo buenas prácticas clínicas y con la aprobación de un comité ético. Un comité consultivo puede establecerse dentro de la autoridad reguladora que puede participar al revisar las solicitudes de EC. Los países deben considerar si tienen recursos suficientes para establecer su propio mecanismo de registro de EC y si tienen la capacidad reguladora para evaluar la información suministrada y supervisar los estudios realizados en su territorio. Inicialmente deben considerar promulgar los requisitos de información que este en proporción con su estructura organizativa y considerar ampliar a mayor escala los requisitos hacia un procedimiento de registro formal, solo cuando esta estructura se convierte en una más sofisticada. Los evaluadores deben revisar los requisitos exigidos por la ARN para autorizar los diversos estudios clínicos y también verificar cómo la ARN organiza la vigilancia de los ensayos clínicos puestos en marcha para asegurar el cumplimiento con las condiciones originales de autorización (protocolo aprobado, curriculum de investigadores, etc.).
Indicador 7.1: Base legal
Los evaluadores deben revisar los requisitos legales vigentes y determinar si las regulaciones apropiadas han sido promulgadas. La legislación debe proveer sanciones adecuadas y proporcionales, penalidades y proceso judicial conforme a las violaciones de la legislación vigente (vea inspección reguladora para ejecución y cumplimiento de actividades).
Sub-indicador 7.1.1: Existe una base legal (leyes, decretos, resoluciones, normas u otros) para la
autorización de los ensayos clínicos así como también para su suspensión por la ARN o por otra autoridad delegada.
CRÍTICO
Sub-indicador 7.1.2: Existe una base legal (leyes, decretos, resoluciones, normas u otros) de la ARN o de
otra autoridad delegada que exige la conformación de Comité revisor/de ética.
CRÍTICO
Sub-indicador 7.1.3: Existe una base legal (leyes, decretos, resoluciones, normas u otros) de la ARN o de
otra autoridad delegada que exige el pronunciamiento ético científico del EC por parte del Comité revisor/de ética.
CRÍTICO
Sub-indicador 7.1.4: Las bases legales (leyes, decretos, resoluciones, normas u otros) requieren la
notificación a la ARN sobre cambios / variaciones (enmiendas) en el protocolo original del EC.

73
CRÍTICO
Sub-indicador 7.1.5: Las bases legales (leyes, decretos, resoluciones, normas u otros) exigen a los centros
de investigación, investigadores, patrocinadores, CRO y todos los involucrados en el ensayo clínico que cumplan con Buenas Prácticas Clínicas.
CRÍTICO
Sub-indicador 7.1.6: Las bases legales (leyes, decretos, resoluciones, normas u otros) exigen a los
laboratorios o centros de ensayo cumplir las Buenas Prácticas de Laboratorio.
CRÍTICO
Sub-indicador 7.1.7: Existen bases legales (leyes, decretos, resoluciones, normas u otros) sobre el
licenciamiento (autorización de funcionamiento) de productores e importadores de productos de investigación.
CRÍTICO
Sub-indicador 7.1.8: Existe una base legal (leyes, decretos, resoluciones, normas u otros) exigiendo que los
productos para investigación cumplan con BPM.
CRÍTICO
Sub-indicador 7.1.9: Existen bases legales (leyes, decretos, resoluciones, normas u otros) sobre los
requerimientos específicos del etiquetado para los establecimientos que producen productos de investigación.
CRÍTICO
Sub-indicador 7.1.10: Existen bases legales (leyes, decretos, resoluciones, normas u otros) requiriendo la
autorización de la importación y exportación de los productos de investigación.
CRÍTICO
Sub-indicador 7.1.11: Existen bases legales (leyes, decretos, resoluciones, normas u otros) exigiendo durante
el desarrollo del ensayo clínico, la notificación, recolección de datos, evaluación y monitoreo de eventos/reacciones adversas y el reporte a la ARN bajo condiciones específicas.
CRÍTICO
Sub-indicador 7.1.12: Existen bases legales (leyes, decretos, resoluciones, normas u otros) definiendo la
terminología utilizada para los fines del ensayo clínico como por ejemplo evento adverso, reacción adversa, evento adverso serio, inesperado, etc.
NECESARIO
Sub-indicador 7.1.13: Las bases legales (leyes, decretos, resoluciones, normas u otros) establecen los plazos
y/o la periodicidad para informar sobre los eventos adversos durante el ensayo clínico.
CRÍTICO
Sub-indicador 7.1.14: Existen bases legales (leyes, decretos, resoluciones, normas u otros) sobre
excepciones a los requisitos para la evaluación de los ensayos clínicos basados en criterios establecidos.
NECESARIO
Indicador 7.2. Guías (Directrices)
Los evaluadores deben revisar los documentos de orientación publicados para diferentes tipos de “actores claves” y si abarcan el alcance de la legislación y regulaciones vigentes. Si es aplicable, debe comprobarse si hay coherencia con la orientación de la OPS/OMS e identificar cualquier diferencia.

74
Sub-indicador 7.2.1: Hay guías para los solicitantes sobre el contenido, el formato y el procedimiento a seguir
para presentar una solicitud para la realización de los ensayos clínicos.
NECESARIO
Sub-indicador 7.2.2: Existen guías para los solicitantes sobre la documentación requerida, tipos, alcances y
condiciones adjuntas de las enmiendas / modificaciones de los ensayos clínicos.
NECESARIO
Sub-indicador 7.2.3: Existe una guía para los solicitantes sobre el monitoreo que dan a las notificaciones de
reacciones adversas y sobre el informe periódico de sus resultados a la ARN.
NECESARIO
Sub-indicador 7.2.4: Hay una guía para los solicitantes sobre los criterios para seleccionar el investigador
principal y sobre sus funciones y responsabilidades.
NECESARIO
Sub-indicador 7.2.5: Hay guías para autorizar a los productores y los importadores de los productos de
investigación.
NECESARIO
Sub-indicador 7.2.6: Hay guías para los fabricantes de los productos de investigación sobre el cumplimiento
de los requisitos de BPM.
NECESARIO
Sub-indicador 7.2.7: Se proporciona información detallada sobre los requisitos de cumplimiento con BPC en
una guía para los centros de investigación, investigadores, patrocinadores, CRO y todos los involucrados en el ensayo clínico. Ésta guía está de acuerdo con el modelo de OMS.
NECESARIO
Sub-indicador 7.2.8: Hay guías para el control de la importación, exportación o destrucción de los productos
de investigación.
NECESARIO
Indicador 7.3: Marco Ético (Supervisión Ética)
Los evaluadores deben poder evaluar el alcance de los controles aplicados por la ARN y revisar la supervisión ética aplicada a los ensayos clínicos. Los evaluadores deben prestar especial atención a la prevención de conflictos de intereses y asegurar que los temas de confidencialidad se tengan en cuenta. El equipo evaluador debe tener la oportunidad de entrevistar al presidente o miembros representativos del CEI así como visitar los establecimientos.
Sub-indicador 7.3.1: El marco ético que se ha establecido se basa en la Declaración de Helsinki y otras
normativas internacionales.
CRÍTICO
Sub-indicador 7.3.2: Hay requisitos en la composición, funciones y forma de operar del Comité de Ética.

75
NECESARIO
Sub-indicador 7.3.3: Existen procedimientos documentados para los Comités de Ética para examinar el
ensayo, especialmente los protocolos y enmiendas, la documentación del consentimiento informado y el procedimiento de reclutamiento.
NECESARIO
Sub-indicador 7.3.4: Existe una base legal (leyes, decretos, resoluciones, normas u otros) para el
establecimiento y funcionamiento de una autoridad nacional supervisora de los Comités de Ética.
CRÍTICO
Sub-indicador 7.3.5: Hay un procedimiento para la aprobación y supervisión de los Comités de Ética.
NECESARIO
Sub-indicador 7.3.6: El financiamiento del Comité de Ética se basa en los honorarios por los servicios
prestados.
INFORMATIVO
Sub-indicador 7.3.7: Existe claridad en cuanto al financiamiento del Comité de Ética y sus miembros.
CRÍTICO
Sub-indicador 7.3.8: Se ha establecido e implementado una política general sobre potenciales conflictos de
interés para los miembros del Comité de Ética.
CRÍTICO
Sub-indicador 7.3.9: Se ha establecido e implementado una política general sobre la confidencialidad y un
código de conducta para los miembros del Comité de Ética.
CRÍTICO
Sub-indicador 7.3.10: Se ha establecido e implementado una política general sobre potenciales conflictos de
interés para la autoridad nacional supervisora de los Comités de Ética.
CRÍTICO
Sub-indicador 7.3.11: Se ha establecido e implementado una política general sobre la confidencialidad y un
código de conducta para la autoridad nacional supervisora de los Comités de Ética.
CRÍTICO
Indicador 7.4: Organización y estructura
Los evaluadores deben identificar la estructura de la organización para llevar a cabo esta función reguladora; si la misma función es delegada o descentralizada y en particular a qué nivel del país (central, regional o local). Si diferentes organizaciones se involucran a diferentes niveles del país los evaluadores deben revisar el nexo entre estas organizaciones y en particular cómo el intercambio de información esta establecido y administrado.
Sub-indicador 7.4.1: Las actividades se organizan y desarrollan a nivel central del país.
INFORMATIVO
Sub-indicador 7.4.2: Las actividades centralizadas o descentralizadas a otras agencias o autoridades siguen
los mismos estándares, guías y procedimientos.
CRÍTICO

76
Sub-indicador 7.4.3: En el caso de descentralización, se ha establecido e implementado un mecanismo de
intercambio de información para que la organización descentralizada reciba solicitudes y/o directivas de la autoridad central y a la vez pueda reportar a ésta.
CRÍTICO
Sub-indicador 7.4.4: Los mecanismos deben permitir la armonización y la cooperación apropiada entre las organizaciones descentralizadas.
CRÍTICO
Indicador 7.5: Procedimientos de evaluación
Los evaluadores deben revisar los procedimientos con relación a sus resultados esperados, su nivel de detalle y conveniencia en relación a la capacitación proporcionada y las medidas aplicadas para verificar las actividades descritas. Los evaluadores en particular deben revisar su coherencia con la orientación, regulaciones y legislación vigente. Los evaluadores deben identificar el nivel en que la inspectoría esta involucrada en el control de ensayos clínicos, definiendo en particular que tipo de inspección esta siendo realizada. Los siguientes indicadores pueden ser usados para identificar el nivel de vigilancia por la ARN:
Número de ECs inspeccionados para BPM y BPC comparado al número de aplicaciones EC recibidas.
Número de inspectores involucrados.
Número promedio de días ocupados en terreno.
Sub-indicador 7.5.1: Existen procedimientos documentados para la evaluación del protocolo y toda la
documentación presentada con las solicitudes EC.
CRÍTICO
Sub-indicador 7.5.2: El manual del Investigador, el procedimiento para el consentimiento informado, el plan
de investigación y las herramientas de recolección de datos se evalúan y aprueban como parte de la aprobación del ensayo clínico.
CRÍTICO
Sub-indicador 7.5.3: Hay procedimientos documentados para la evaluación de la documentación presentada
de variación/enmienda de los protocolos de ensayos clínicos.
CRÍTICO
Sub-indicador 7.5.4: Expertos externos participan en la evaluación de la documentación presentada para la
aprobación de ensayos clínicos.
INFORMATIVO
Sub-indicador 7.5.5: Un comité consultivo de expertos participa en la evaluación de la documentación
presentada para la aprobación de ensayos clínicos.
INFORMATIVO
Sub-indicador 7.5.6: Hay procedimientos documentados para documentar la toma de decisiones sobre EC.
CRÍTICO
Sub-indicador 7.5.7: Hay un formato estándar para la elaboración del informe final de la evaluación del EC.

77
NECESARIO
Sub-indicador 7.5.8: Existen procedimientos documentados e implementados para asegurar la participación y
comunicación entre los asesores y los inspectores del área reguladora para el cumplimiento a las Buenas Prácticas Aplicables (BPM, BPC y BPL).
NECESARIO
Sub-indicador 7.5.9: Los Comités de Ética, investigadores, IRB/IEC y patrocinadores, son inspeccionados
periódicamente por el cuerpo de inspectores.
NECESARIO
Sub-indicador 7.5.10: Se emplean los mismos criterios para la evaluación de la documentación de EC
independientemente el origen de la solicitud (por ejemplo, sector nacional, extranjero, público/privado).
CRÍTICO
Sub-indicador 7.5.11: Existen límites de tiempo para la evaluación de la documentación de EC y la toma de
decisiones.
NECESARIO
Sub-indicador 7.5.12: Hay un mecanismo acelerado para la evaluación de la documentación y la toma de
decisiones de EC de valor particular en salud pública.
NECESARIO
Sub-indicador 7.5.13: Se ha establecido un sistema de seguimiento interno para seguir los plazos
proyectados al evaluar un EC.
NECESARIO
Sub-indicador 7.5.14: Existe un procedimiento documentado para otorgar la autorización de EC en un formato
estandarizado.
NECESARIO
Sub-indicador 7.5.15: Una autorización escrita, firmada por una persona con la delegación adecuada, que
incluye condiciones o restricciones adjuntas a la autorización es enviada al patrocinador.
NECESARIO
Indicador 7.6: Recursos humanos y otros
La evaluación de recursos humanos debe centrarse en aspectos cuantitativos y cualitativos. Para los aspectos cuantitativos los evaluadores pueden decidir usar los siguientes indicadores para identificar si se están poniendo a disposición recursos humanos adecuados para llevar a cabo las actividades planificadas:
Carga de trabajo para las funciones realizadas con los siguientes indicadores: número de ensayos clínicos y número de solicitudes de enmiendas recibidas.
Número de personal involucrado en el control de ensayos clínicos.
Trabajo acumulado o generado (carga de trabajo comparado con el número de decisiones en solicitudes de EC recibidas).
Número promedio de días tomados por la ARN para tomar una decisión y/o número promedio de días tomados por el CEI para tomar una decisión.
Los evaluadores deben revisar si el personal involucrado en los procesos son plenamente competentes en particular en las siguientes especialidades:

78
Bioestadístico,
Farmacólogo clínico,
Médico.
Los que evaluaban las aplicaciones de EC deben tener conocimiento apropiado de las leyes y regulaciones vigentes, un conocimiento amplio de principios y prácticas internacionalmente aceptadas para la conducción de investigación clínica en conformidad con la BPC, incluyendo los requisitos éticos para la protección de los sujetos humanos incluidos en la investigación. Si expertos externos o un comité asesor/técnico participan en este proceso regulador, los evaluadores deben referirse a las preguntas aplicables en el Capítulo 3.8.
Sub-indicador 7.6.1: Existe la experticia adecuada interna y externa (educación, experiencia y capacitación)
para la evaluación de la documentación de EC.
CRÍTICO
Sub-indicador 7.6.2: Los representantes del productor o del patrocinador o los investigadores nunca están
involucrados en la evaluación del EC (en cualquier nivel/etapa, incluidos los comités de expertos).
CRÍTICO
Sub-indicador 7.6.3: Hay líneas de la autoridad que reflejan la independencia en la toma de decisiones en el
sistema de aprobación de EC respecto de los patrocinadores, los investigadores o el gobierno.
CRÍTICO
Sub-indicador 7.6.4: Se cuenta con los servicios de apoyo adecuados en términos de comunicación, materiales, equipos e infraestructura.
NECESARIO
Indicador 7.7: Registros y resultados
Los evaluadores deben revisar cómo toda la información recogida durante el proceso de aprobación es administrada y que clase de información es registrada y archivada por la organización. Mientras es revisada la implementación de procedimientos internos de la organización, los evaluadores deben muestrear los registros generados y comprobar su contenido. Los objetivos internos, la planificación a futuro y los plazos proyectados deben ser confirmados por la evidencia revisada.
Sub-indicador 7.7.1: Hay una lista/base de datos interna de todos los EC aprobados y rechazados.
NECESARIO
Sub-indicador 7.7.2: La ARN mantiene un expediente de cada EC aprobado y rechazado, que incluye toda la
documentación presentada para la revisión del EC y los informes de evaluación.
CRÍTICO
Indicador 7.8: Disponibilidad de la información.
Los evaluadores deben revisar la información que está públicamente disponible, si los medios usados (portal Web, boletín oficial u otro boletín de la ARN) son apropiados y si la información es mantenida y actualizada en forma regular.
Sub-indicador 7.8.1: Los ensayos clínicos aprobados por la ANR están públicamente disponibles o
registrados en una base de datos nacional o internacional.

79
NECESARIO
Sub-indicador 7.8.2: La lista de solicitudes de EC rechazadas, incluidos los informes de evaluación resumidos
está públicamente disponible.
NECESARIO
Evidencia documental a ser estudiada:
Reglamentos, leyes, decretos.
Directrices.
Buenas prácticas clínicas.
Buenas prácticas de laboratorio.
Lista del comité ético.
Lista de los miembros del comité ético.
Procedimientos internos.
Procedimientos para seleccionar y nombrar a los miembros de los comités éticos.
Procedimientos para revisar un aplicación/protocolo de EC por el CEI
Procedimientos internos, plantillas y registros.
Procedimientos internos para tratar las reacciones adversas y los registros de la droga y/o el medicamento.
Lista de personal con sus calificaciones.
Lista de los ensayos clínicos autorizados, rechazados o retirados (por año).

80
8. Inspecciones Reguladoras y actividades de fiscalización - Módulo 8
Objetivo: Evaluar la estructura de la organización para inspeccionar y vigilar el cumplimiento con los requerimientos vigentes y tomar las acciones necesarias para su cumplimiento.
Indicador 8.1: Bases legales
Existen disposiciones legales para inspeccionar a los establecimientos, a fin de verificar el cumplimiento de normativas, estándares y buenas prácticas de manufactura. Los evaluadores deben revisar los requisitos legales aplicables y determinar si las regulaciones apropiadas han sido promulgadas. Debe haber disposiciones legales para inspeccionar las instalaciones y/o los establecimientos de titulares autorizados para comercialización, fabricantes, instituciones de investigación, agentes importadores y exportadores, distribuidores, mayoristas, minoristas y otros canales de distribución (farmacias, clínicas, consultorios, etc.) basados en los estándares aplicables. Debe haber disposiciones legales para inspeccionar las actividades antedichas para todas las categorías de los medicamentos. Debe haber un requisito legal para el cumplimiento de buenas prácticas (por ejemplo, buenas prácticas de fabricación, buenas prácticas clínicas, buenas prácticas de laboratorio, buenas prácticas de distribución, buenas prácticas de vigilancia, buenas prácticas de dispensación, etc.) Los evaluadores deben poder determinar si el alcance de la inspección y las actividades de fiscalización abarca todos los requisitos que tienen que satisfacerse por los "actores claves". Evidencia a ser estudiada:
Leyes, Reglamentos, Decretos, Acuerdo, u otras Disposiciones legales mandatorias en inspecciones de Buenas Prácticas y en otras actividades de fiscalización.
Normas y Guías nacionales en BPM. Se tiene establecida en el país una organización para que realice la inspección, monitoreo del cumplimiento de los requisitos aplicables y realice los actos de autoridad necesarios. El asesor deberá investigar como ha establecido la ARN sus estrategias de aplicación de la ley para promover el cumplimiento de la regulación de vacunas. Deberán tener procedimientos internos apropiados, establecidos e instrumentados. El asesor deberá identificar la unidad de la ARN o el departamento con la atribución para realizar las inspecciones y los actos de autoridad. La unidad inspectora de BPM deberá tener un organigrama que muestre claramente las responsabilidades y la estructura de reporte de la misma, así como la relación entre las funciones de inspección y autorización (licenciamiento). Deberá contarse con la descripción de la relación entre la unidad de inspección de BPM y otros departamentos de la ARN así como con otras agencias gubernamentales, en caso de que operen como organismos separados. La ARN deberá mantener colaboración cercana con otras agencias de aplicación de la Ley tales como la policía y las cortes judiciales que puedan intervenir. El asesor deberá verificar que se puede evitar cualquier conflicto de interés y que se puede demostrar la independencia en la toma de decisiones. El personal del servicio de inspección, incluyendo el personal subcontratado y los expertos, deberá estar libre de cualquier presión comercial, financiera o de otra índole, que pueda afectar su juicio y libertad de acción. La unidad de inspección deberá asegurar que las personas u organizaciones externas a la organización de inspección no pueden influir en el resultado de las inspecciones.

81
El asesor deberá revisar los requisitos legales aplicables y averiguar si se ha aplicado la regulación adecuada. Los servicios nacionales de inspección son responsables de asegurar que se cumplen los requisitos establecidos en la legislación nacional relevante. Deberá haber disposiciones legales para inspeccionar los establecimientos de los titulares de autorizaciones de comercialización, fabricantes, importadores y exportadores, distribuidores, mayoristas, minoristas e instituciones involucradas en el manejo o distribución de los productos. Deberá haber disposiciones legales para inspeccionar las actividades antes mencionadas para todas las categorías de medicamentos. Deberá haber requisitos legales para el cumplimiento de las buenas prácticas actuales (p.ej. BPM, BPC, BPL, BPD, etc.).
Sub-idicador 8.1.1: Existen disposiciones legales para inspeccionar a los establecimientos, a fin de verificar el
cumplimiento de normativas, estándares y buenas prácticas.
CRÍTICO
Los evaluadores deben revisar la guía o documento oficial, el cuál deberá ser publicado y encontrarse disponible a los usuarios, dicho documento deberá ser coherente con recomendaciones de la OMS u otras guías internacionalmente aceptadas, tales como TRS 902 11, TRS 908, TRS 822 u otro documento equivalente actual.
Sub-indicador 8.1.2: Existe disposición legal que especifique la periodicidad de inspecciones del
cumplimiento de la regulación.
CRíTICO
Los evaluadores deberán verificar que ha sido emitida una licencia de manufactura basada en el cumplimiento del requerimiento legal. Que las inspecciones de BPM son realizadas antes de emitir la licencia y tener establecidos intervalos de tiempo definidos y apropiados para verificar cumplimiento. Deberá identificar los requisitos o criterios para las inspecciones post-licencia.
Sub-indicador 8.1.3: Existe disposición legal que permita la inspección de plantas/ sitios o productores
extranjeros.
CRÍTICO
Los evaluadores deben verificar que la ARN tenga establecido que podrá inspeccionar las instalaciones de los productores extranjeros, con la finalidad de asegurar que cumplen con la reglamentación y requisitos de BPM en las operaciones de fabricación y de control; además de asegurarse que los productos importados se autorizan en el país de origen y en su caso, que han sido inspeccionados para el cumplimiento de las BPM.
Sub-indicador 8.1.4: Existe disposición legal para la designación de inspectores y sus facultades.
CRÍTICO
Sub-indicador 8.1.5: Los inspectores tienen las facultades y autoridad adecuadas.
CRÍTICO
Los inspectores tienen las facultades y autoridad para inhibir la producción, intervenir partidas, intervenir la producción y el producto existente en el establecimiento inspeccionado, decomisar, clausurar preventivamente, dependiendo de las características del producto y el riesgo sanitario constatado. La ley debe asignar poderes y autoridad adecuada a los inspectores en la descarga de sus responsabilidades, permitiéndoles en particular:

82
Inspeccionar en cualquier momento razonable cualquier sitio donde productos regulados son fabricados, envasados, almacenados, distribuidos, probados o vendidos.
Tomar muestras.
Hacer copias de documentos.
Tomar fotografías de las instalaciones y equipamientos.
Incautar o detener productos regulados cuando se cree que hay una violación.
Abrir y revisar cualquier receptáculo o paquete que contiene artículos sujetos a la legislación pertinente.
Recibir cooperación total del personal en las instalaciones inspeccionadas quienes no deben dificultar las investigaciones que están siendo emprendidas.
El asesor deberá identificar si las atribuciones legales de los inspectores les otorgan el poder y autoridad adecuados para suspender o detener la producción. Este puede ser el caso por ejemplo cuando hay violaciones a los requisitos legales o con el fin de garantizar la calidad de los productos que serán comercializados, en el caso de que se sospeche la existencia de defectos de calidad o en caso de que haya defectos de calidad confirmados que ameriten una alerta rápida.
Sub-indicador 8.1.6: Autoridad para tener acceso a cualquier sitio de producción y documentos que son
relevantes para la inspección.
CRÍTICO
El asesor deberá verificar que la ley otorga el poder y autoridad adecuados a los inspectores para inspeccionar en cualquier momento que sea razonable, cualquier sitio de producción en el que se manejen vacunas o medicamentos, tener acceso a toda la documentación, copiar los documentos o tomar fotografías del sitio y de los equipos.
Sub-indicador 8.1.7: En el caso de existir exenciones de los requerimientos a inspeccionar, están definidos los criterios para su aplicación.
CRÍTICO
La ARN debe identificar y establecer el requisito legal para permitir excepciones para cada situación, establecer criterios y capacitar al personal para evaluar y aprobar dichas excepciones, así como informar a la comunidad sobre el ítem.
Sub-indicador 8.1.8: La legislación contempla sanciones adecuadas y proporcionales, multas y
procesamiento penal ante la convicción de infracciones a la legislación vigente.
CRÍTICO
La legislación debe proveer sanciones adecuadas y proporcionales, penalidades y proceso judicial conforme a las violaciones de la legislación vigente.
Sub-indicador 8.1.9: Atribución para inspeccionar y tomar muestras en cualquier establecimiento en el país,
en el que se produzcan medicamentos y productos biológicos.
CRÍTICO
El asesor deberá verificar que la ley otorga poder y autoridad adecuados a los inspectores para inspeccionar en cualquier momento que sea razonable cualquier sitio dentro del país en el que se produzcan vacunas y tomar muestras.
Indicador 8. 2: Guías y lineamientos
Sub-indicador 8.2.1: Existe un documento que describa los criterios para la aplicación de sanciones de
acuerdo a la naturaleza de la infracción.

83
NECESARIO
La legislación debe proveer sanciones adecuadas y proporcionales, penalidades y proceso judicial conforme a las violaciones de la legislación vigente.
Sub-indicador 8.2.2: Existe un plazo para el cumplimiento de BPM, BPC, BPL y BPD en todas las
instalaciones nacionales (en caso de que las disposiciones legales hayan sido adoptadas recientemente).
CRÍTICO
Los evaluadores deberán revisar los lineamientos escritos donde se establezcan los plazos para el cumplimiento a los requisitos, dicha información deberá encontrarse disponible para guiar a los fabricantes, importadores, distribuidores y comercializadores.
Sub-indicador 8.2.3: Existen directrices para conducir inspecciones pre y post licenciamiento.
CRÍTICO
Los evaluadores deben revisar los documentos de orientación publicados para diferentes tipos de “actores claves” y determinar si abarcan el alcance de la legislación y regulaciones vigentes. Debe comprobarse si hay coherencia con la orientación de la OPS/OMS y cualquier diferencia identificarla. Los evaluadores también deben considerar el uso interno de otra orientación relevante reconocida internacionalmente (ICH, PICS, etc.). Los evaluadores deben revisar la guía o documento oficial, el cuál deberá ser publicado y encontrarse disponible a los usuarios, dicho documento deberá ser coherente con recomendaciones de la OMS u otras guías internacionalmente aceptadas, tales como TRS 902 11, TRS 908, TRS 822 u otro documento equivalente actual.
Sub-indicador 8.2.4: El código de BPM nacional está publicado y es equivalente a las BPM de la OMS (TRS 902 11, TRS 908, TRS 822 o cualquier actualización) o algún otro estándar reconocido.
CRÍTICO
El asesor deberá revisar las guías de BPM, mismas que deberán estar publicadas y disponibles para todos los interesados y verificar que las guías están alineadas con las guías de la OMS o alguna otra internacionalmente aceptada. Deberán identificarse aquellas diferencias de relevancia.
Sub-indicador 8.2.5: Disposiciones (o criterios escritos) para el reconocimiento de
certificados/reportes/decisiones de otras ARN (de ser procedente).
NECESARIO
El evaluador deberá identificar si existen disposiciones o criterios escritos para el reconocimiento de certificados/reportes/decisiones de otras ARN. El evaluador deberá identificar la cooperación internacional existente (ej. OMS. EDQM, PICS, FDA, etc.) que permita el intercambio de información. Por ejemplo, en caso de la importación de productos fabricados en un país extranjero, el país importador reconoce los certificados, reportes o decisiones de la autoridad competente del país de origen, de ser el caso, las disposiciones deberán estar por escrito. Estas disposiciones deberán indicar cuál es la instancia autorizada para reconocer las decisiones de otras autoridades, el listado de los países o autoridades reconocidas y el procedimiento o criterios de reconocimiento.
Indicador 8.3: Organización y Estructura
Sub-indicador 8.3.1: Las actividades de inspección son organizadas y desarrolladas a nivel central del país.

84
INFORMATIVO.
Los evaluadores deben identificar la estructura de la organización para llevar a cabo esta función reguladora; si la función es delegada o descentralizada y en particular a qué nivel del país (central, regional o local).
Sub-indicador 8.3.2: En caso de actividades descentralizadas de otras agencias/autoridades siguen los
mismos estándares, directrices y procedimientos.
CRÍTICO
Si diferentes organizaciones se involucran a diferentes niveles del estado los evaluadores deben revisar el nexo entre estas organizaciones y en particular cómo el intercambio de información está establecido y administrado.
Sub-indicador 8.3.3: En caso de descentralización, un mecanismo de intercambio de información está
establecido e implementado para que los organismos descentralizados reciban los requerimientos y/o directivas desde la autoridad central y reporten a ésta.
CRÍTICO
Si diferentes organizaciones se involucran a diferentes niveles del estado los evaluadores deben revisar el nexo entre estas organizaciones y en particular cómo el intercambio de información está establecido y administrado.
Sub-indicador 8.3.4: Los mecanismos permiten colaboración y cooperación apropiada entre los organismos
descentralizados.
CRÍTICO
Si diferentes organizaciones se involucran a diferentes niveles del estado los evaluadores deben revisar el nexo entre estas organizaciones y en particular cómo el intercambio de información está establecido y administrado.
Indicador 8.4: Sistema de gestión de la calidad
Sub-indicador 8.4.1: Está implementado un sistema de gestión de calidad para las inspecciones.
NECESARIO
Los evaluadores deben revisar el sistema de gestión de calidad implementado dentro de la inspectoría y el cumplimiento de los requisitos de referencia en los sistemas de gestión de calidad, así como tambien si existen lineamientos definidos, tales como: WHO Technical Report Series, N° 902, 2002 annex 8 y Quality Systems Requirements for National Good Manufacturing Practice Inspectorates o PIC/s - PI 002-x - Recommendation on Quality System Requirements for Pharmaceutical Inspectorates. Deberá haber un compromiso formal de cumplir con los principios recomendados de calidad asegurando que la política de calidad de la unidad de inspección está documentada, es relevante para los objetivos y está instrumentada. El sistema de calidad deberá incluir todas las actividades involucradas en la inspección. Deberá describirse en el manual de calidad la estructura administrativa, membresía, operación y estatus legal de la unidad de inspección de BPM. El asesor deberá revisar el sistema de gestión de calidad instrumentado en la unidad de inspección y el cumplimiento de los requisitos de los sistemas de gestión de calidad que debieran tenerse en cuenta como referencia. Podría utilizarse como referencia la Guía de la OMS (en el anexo 8, WHO TRS 902) o algún estándar internacional.

85
El sistema de gestión de calidad deberá ser tan amplio que permita llevar a cabo todos los indicadores de la función. Evidencia documental a ser evaluada:
Organigrama.
Manual de inspección.
Procedimientos de planeación, preparación, conducción y reporte de la inspección.
Listas de control y reportes de inspección.
Política y procedimientos de rastreabilidad de las actividades de inspección.
Reportes de auditorías internas y externas.
Sub-indicador 8.4.2: La gestión de la unidad de inspección está comprometida con el desarrollo e
implementación del sistema de gestión de calidad.
CRÍTICO
Sub-indicador 8.4.3: El sistema de gestión de calidad está basado en el reconocimiento de estándares de
referencia (OMS, PIC/S, ISO, otros).
NECESARIO
Los evaluadores deberán comprobar que existe un documento de orientación de la OPS/OMS o un estándar internacional en los sistemas de gestión de calidad puede usarse como referencia. Cualquiera que sea el caso, el cuestionario incluido en este módulo debe abordar los temas principales con respecto a un sistema de gestión de calidad.
Sub-indicador 8.4.4: Una persona calificada está designada como responsable en la Unidad de Inspección
para el desarrollo e implementación de un sistema de gestión de calidad.
CRÍTICO
La unidad de inspección de BPM deberá tener una organización que le permita mantener la capacidad de llevar a cabo sus funciones satisfactoriamente. Deberá designarse a una persona o personas para que desempeñen la función de aseguramiento de la calidad incluyendo la instrumentación y mantenimiento del sistema de calidad. El asesor deberá verificar y revisar la organización desarrollada para establecer, instrumentar o mantener el Sistema de Gestión de Calidad. Deberán estar identificadas las diferentes responsabilidades. En general este indicador deberá evaluarse de acuerdo con las guías de la OMS sobre requisitos de gestión de calidad para la unidad nacional de inspección de BPM (Anexo 8 en TRS 902 15 o cualquier actualización).
Sub-indicador 8.4.5: Está definida la documentación necesaria para establecer, implementar y mantener el
SGC (manual de calidad, registros y procedimientos).
NECESARIO
Los evaluadores deberán comprobar el funcionamiento del sistema documental; considerando que lo que no ha sido registrado, no se ha hecho, y por lo tanto no existe. Los procesos y POE tienen que hacer referencia al documento de registro pertinente, el cual debe contener los siguientes campos: identificación (organización, numeración, paginado y fecha), título; autoría, aprobación y actualizaciones con sus respectivas fechas.
Sub-indicador 8.4.6: La documentación requerida por el SGC es controlada siguiendo un procedimiento
escrito.
NECESARIO

86
Los evaluadores deben verificar que el sistema de calidad contempla establecer y mantener una organización estructurada de la documentación, que vincule las políticas, los procesos y los procedimientos, así como un formato y un contenido definidos y específicos para los POE. Además debe contener los procesos necesarios para generar nuevos documentos y formularios; controlar la aprobación, distribución y archivo de documentos y registros; controlar los cambios en los documentos; y controlar y archivar los documentos que sean obsoletos.
Sub-indicador 8.4.7: Existe un plan documentado para evaluar la organización y el desarrollo del SGC y los
registros de resultados.
NECESARIO
Dado el conjunto de etapas y requisitos que componen la implementación de un sistema de calidad, deben establecerse plazos de ejecución, que debe tener en cuenta aspectos tales como el tamaño del laboratorio, el número y grado de complejidad de sus procedimientos y sus prioridades, estado actual con respecto a la calidad, disponibilidad de personas que se puedan asignar al desarrollo del SGC, tanto de tiempo completo como de tiempo parcial, etc.; estableciendo con ello un plan de acción que incluya las políticas del sistema de calidad, sus objetivos y resultados esperados; la responsabilidad de la dirección en la implementación del sistema de calidad, y la planificación necesaria para implementarlo.
Sub-indicador 8.4.8: Documentación escrita para llevar a cabo la inspección y actividades relacionadas.
CRÍTICO
El evaluador deberá identificar que existe la documentación escrita tal como el manual de inspección (calidad), guías, POE, listas de control, formatos de reporte, etc. El evaluador deberá verificar si existen POE relacionados con la planeación, preparación, conducción y reporte de la inspección (incluyendo aseguramiento de la calidad de los reportes de inspección). EL evaluador deberá identificar si existen POE relacionados con la gestión del sistema de calidad, del sistema de licenciamiento y las relaciones entre las inspecciones y otras actividades tales como evaluación y control de calidad. El manual deberá incluir una muestra en blanco de las diversas listas de control, certificados y reportes utilizados durante el proceso de inspección y describir la forma en la que se procesan, almacenan y archivan, y/o desechan. La unidad de inspección deberá tener procedimientos documentados para manejar quejas sobre sus actividades.
Sub-indicador 8.4.9: Sistema de auditoría documentado e instrumentado (externo o interno).
CRÍTICO
El evaluador deberá identificar si está establecido un sistema interno y/o externo que garantice la auditoría del propio sistema de gestión de calidad. En caso de que el sistema de gestión de calidad esté certificado (por ejemplo de acuerdo con normas ISO) los cuerpos de acreditación generalmente auditan al titular de la certificación en intervalos regulares a fin de renovar la certificación/acreditación. La gerencia de la unidad de inspección deberá revisar periódicamente el sistema de gestión de calidad para asegurar su efectividad y aplicabilidad. El evaluador deberá revisar el sistema instrumentado y que estén documentados los procedimientos de revisión periódica interna. Deberá haber procedimientos para acciones correctivas y preventivas en caso de que se detecten fallas en el sistema de gestión de calidad, o en la conducción de las inspecciones y en la actuación general del servicio de inspección. Los inspectores deberán ser evaluados antes de permitírseles realizar inspecciones. Deberán realizarse revisiones periódicas para examinar la actuación de los inspectores de manera individual a fin de

87
asegurar la consistencia entre ellos, y en las operaciones y procedimientos de la unidad de inspección de BPM. Deberá mantenerse un registro de todas las auditorías y revisiones, mismo que deberá incluir los hallazgos, conclusiones, recomendaciones y acciones de seguimiento. Este registro deberá guardarse por uN periodo de tiempo específico.
Sub-indicador 8.4.10: Son realizadas auditorías al SGC de la Unidad de inspección al menos una vez cada
12 meses.
NECESARIO
Los evaluadores deberán verificar que cuentan con un sistema de auditorias como parte de la gestión de los procesos y es un examen planificado sistemático, independiente y documentado, realizado con una frecuencia definida y adecuada, con el fin de evaluar la eficacia de los sistemas de aseguramiento de la calidad. Específicamente se busca evaluar:
Si los procedimientos son eficaces y adecuados para lograr los objetivos definidos.
Si los procedimientos se siguen de forma efectiva.
Si las actividades y sus resultados cumplen con los requisitos, procesos y procedimientos establecidos.
Sub-indicador 8.4.11: Las acciones correctivas o preventivas tomadas como resultado de auditorías o de
otras no conformidades son implementadas y documentadas y su eficacia es verificada.
CRÍTICO
Los evaluadores deben verificar que existe un sistema de gestión de no conformidades como un proceso organizado para detectar, analizar, documentar e informar las no conformidades, con el objeto de identificar sus causas y tomar las acciones correctivas necesarias, contemplando: detección y reconocimiento, documentación e informe, Investigación y análisis, tratamiento de las no conformidades, acciones correctivas y preventivas, planes de contingencia, seguimiento y monitoreo; así como iniciativas para el mejoramiento.
Sub-indicador 8.4.12: Instrumentación del Sistema de Gestión de Calidad para asegurar la trazabilidad de las
actividades.
CRÍTICO
El Sistema de Gestión de Calidad deberá asegurar que las actividades de inspección estén documentadas y archivadas a fin de estar en posibilidad de rastrearlas si por alguna razón así resultara necesario, como por ejemplo en caso de una apelación por parte de una determinada institución a las decisiones tomadas por la autoridad, lo que permita a esta última comparar los hallazgos recientes y anteriores o identificar tendencias en los hallazgos relacionados con dichas instituciones o productos específicos. Los registros deberán demostrar que todos los procedimientos se han seguido durante el desarrollo de cada inspección de Buenas Prácticas de Manufactura, incluyendo la inspección inicial, la recomendación para la emisión de una autorización de mercado, inspecciones de rutina y acciones correctivas/preventivas. Todos los registros deberán resguardarse por un periodo de tiempo apropiado y conservarse bajo condiciones que garanticen su seguridad y confidencialidad, a menos que de otro modo sea requerido por la legislación nacional.
Indicador 8.5: Planificación interna y procedimientos

88
Los evaluadores deben revisar la inspección y los procedimientos operativos con relación a los resultados esperados, su nivel de detalle en relación a la capacitación proporcionada y las medidas aplicadas para verificar las actividades descritas. Los evaluadores en particular deben revisar su coherencia con la orientación, regulaciones y legislación vigentes. Los siguientes indicadores pueden ayudar a los evaluadores a determinar el nivel de las actividades de fiscalización que están siendo realizadas:
Número de medidas administrativas expedidas.
Número de licencias retiradas o suspendidas.
Número de procesamientos judiciales presentados.
Número de sanciones legales aplicadas.
Número promedio de días para tomar una medida administrativa. El evaluador deberá identificar la existencia y aplicación de un Manual de Procedimiento Normalizado de Operación (PNO) (manual, listas de verificación) para inspectores internos y para expertos externos con atribuciones para realizar inspecciones o para participar en éstas con respecto a los procesos técnicos (ej. cómo preparar, anunciar, desarrollar y documentar las inspecciones). El evaluador deberá revisar el manual de inspecciones/Manual de Procedimiento Normalizado de Operación relacionado a la planeación, preparación, conducción y reporte de inspecciones, así como copias en blanco de las listas de verificación, certificados y reportes utilizados durante el proceso de inspección. El evaluador deberá revisar los Procedimientos Normalizados de Operación relacionados con el manejo de los retiros de los productos del mercado, el Sistema de Gestión de Calidad, el sistema de licenciamiento y la relación entre las inspecciones y otras actividades como la evaluación y el control de calidad.
Sub-indicador 8.5.1: Las inspecciones son establecidas conforme a un programa de monitoreo basado en actividades que serán evaluadas (BPM, BPD, BPC, Farmacias, distribuidoras, etc).
NECESARIO
Sub-indicador 8.5.2: Existe un procedimiento para la designación de inspectores.
NECESARIO
Sub-indicador 8.5.3: Existen procedimientos /herramientas para la planificación de actividades de inspección
señalando en particular la duración y frecuencia de las inspecciones.
NECESARIO
Sub-indicador 8.5.4: La planificación interna para las actividades de inspección está establecida y actualizada
para cubrir todas las actividades de regulación.
NECESARIO
El evaluador deberá verificar que existan planes para las inspecciones pre-autorización, inspecciones en proceso, actividades relacionadas a la vigilancia post-comercialización y muestreo de productos para todo tipo de inspecciones (inspecciones de rutina, inspecciones específicas, inspecciones de seguimiento y las inspecciones concisas). La planeación para el desarrollo de las inspecciones deberá basarse en elementos de manejo de riesgos (ej. identificación de hallazgos de manera previa a las inspecciones, reconocimiento o sospecha de violaciones, problemas de calidad, reporte de ESAVIs para productos específicos, renovación del producto o de la licencia de un establecimiento, etc.).

89
Los evaluadores deberán revisar que la periodicidad de las inspecciones esté definida e incluida en la planeación. La periodicidad deberá basarse en el motivo de la inspección, con un enfoque en el manejo de riesgos (ej. es recomendable que la periodicidad de las inspecciones a los fabricantes no sea por debajo de una vez cada dos años, ya que la falta de continuidad puede dar lugar a un desconocimiento de la situación actual de las BPM o a permitir el desarrollo de deficiencias significativas, además de que los titulares de nuevas autorizaciones deberán ser inspeccionados más frecuentemente hasta que los inspectores tengan la certeza de que los fabricantes están cumpliendo con los lineamientos de las BPM.
Sub-indicador 8.5.5: Existen procedimientos/herramientas para preparar las actividades previas a la inspección y la elaboración de planes de inspección.
NECESARIO
Sub-indicador 8.5.6: Existen procedimientos /herramientas para desarrollar las inspecciones en BPM, y en
particular sobre la evaluación de pasos críticos de los procesos y validación.
NECESARIO
Los evaluadores deberán confirmar que el enfoque de las inspecciones en BPM abarca los ítems de la guía de verificación e incluye actividades críticas de los sistemas, procesos y otras unidades de la organización que tengan un impacto directo sobre la calidad de los productos.
Sub-indicador 8.5.7: Existen procedimientos /herramientas para verificar el cumplimiento con las BPM, BPD,
BPC, BPL, Buenas Prácticas de Vigilancia y Buenas Prácticas de dispensación.
CRÍTICO
Sub-indicador 8.5.8: Reportes disponibles para todos los tipos de inspecciones de BPM.
CRÍTICO
El evaluador deberá verificar que los reportes estén disponibles para todos los tipos de inspecciones de BPM e identificar que los formatos importantes (copias en blanco) sean utilizados. El evaluador deberá revisar la manera en que la información es compilada durante el proceso de inspección, y cómo los reportes son registrados y archivados por la organización. El evaluador deberá revisar ejemplos de reportes para los diferentes tipos de inspecciones de BPM. El evaluador deberá revisar que los reportes estén elaborados en formatos aprobados, y con las firmas y fechas correspondientes.
Sub-indicador 8.5.9: Las inspecciones están basadas en requerimientos específicos para categorías
especificas de productos o instalaciones (biológicos, productos estériles, radiofármacos, etc).
NECESARIO
Sub-indicador 8.5.10: Está definido un formato estándar para reportar deficiencias.
NECESARIO
Sub-indicador 8.5.11: Existen registros de los reportes acordes al procedimiento.
NECESARIO
Sub-indicador 8.5.12: Las observaciones de no cumplimiento son clasificados /categorizados de acuerdo al
riesgo (crítico, mayor y menor).
NECESARIO

90
Los evaluadores deberán revisar ejemplos de los reportes de inspección, con especial atención a inspecciones con hallazgos y observaciones relevantes, lista de deficiencias (criticas, mayores y otras), recomendaciones, resumen y conclusiones.
Sub-indicador 8.5.13: Hay mecanismos y medidas de control de calidad documentadas para verificar el
cumplimiento del procedimiento de inspección establecido.
NECESARIO
Los reportes de las inspecciones deberán ser analizados de conformidad con los requisitos (formato, resultados, conclusiones y acciones tomadas por los inspectores).
Sub-indicador 8.5.14: Están involucrados en los procesos de inspección los expertos externos.
INFORMATIVO
El evaluador deberá verificar que los criterios y los procedimientos estén por escrito y sean aplicados con base en la confidencialidad, la independencia (ej. de los fabricantes) de los diferentes expertos involucrados en las actividades de inspección y el manejo de potenciales conflictos de interés.
Sub-indicador 8.5.15: En los casos que proceda, un Comité de expertos internos de la ARN está involucrada
en la inspección, evaluación de informes de la inspección y toma de decisiones.
NECESARIO
Para los diferentes tipos de productos y sitios de fabricación a ser inspeccionados, deberá considerarse contar con un equipo de expertos interno durante las inspecciones. Por ejemplo, expertos en pruebas de laboratorios de vacunas, expertos en eventos adversos, responsables de la evaluación científica de la autorización de mercado de un producto específico, etc.
Sub-indicador 8.5.16: Existen procedimientos para la toma de decisiones.
CRÍTICO
Sub-indicador 8.5.17: Existen procedimientos para seguimiento de deficiencias / infracciones exigiendo su
cumplimiento (incluyendo plazos) y emitir advertencias sobre las mismas.
CRÍTICO
El evaluador deberá revisar si a los resultados de las inspecciones se les brinda el seguimiento correspondiente. Se debe contar con lineamientos escritos sobre cómo dar seguimiento a las deficiencias. El evaluador deberá verificar la evidencia documental de que las acciones se han instrumentado. El evaluador deberá identificar si se cuenta con un Procedimiento Normalizado de Operación (POE) para el seguimiento de las deficiencias y monitoreo de las acciones correctivas. El evaluador deberá revisar ejemplos de reportes de inspección, con especial atención a los resultados y observaciones realizadas durante la inspección, lista de deficiencias detectadas, recomendaciones, resumen y conclusiones. El evaluador deberá detectar los resultados de las acciones tomadas, si es aplicable.
Sub-indicador 8.5.18: Existen procedimientos o mecanismos internos para asegurar una adecuada
integración y comunicación entre los inspectores, Laboratorio de CC y personal de licenciamiento o registro.
NECESARIO
Sub-Indicador 8.5.19: El mismo criterio es usado para realizar las inspecciones; nacionales, extranjeras,
públicas o privadas.
CRÍTICO

91
Sub-indicador 8.5.20: Existen tiempos límites para la presentación y entrega de informes de inspección.
NECESARIO
Sub-indicador 8.5.21: Está establecido un sistema interno de seguimiento para revisar los objetivos
programados.
NECESARIO
Indicador 8.6: Inspección a canales de distribución
La distribución de los medicamentos y en particular las vacunas es una actividad crítica relacionada a la calidad de estos productos. Las instancias involucradas en la distribución de las vacunas deberán haber sido autorizadas e inspeccionadas antes de llevar a cabo sus actividades. Evidencia documental a ser estudiada:
Disposiciones legales para el seguimiento a los canales de distribución y para regular las acciones y sanciones.
Código Nacional de Buenas Prácticas de Distribución equivalente con el de la OMS.
Sub-indicador 8.6.1: Disposición para el monitoreo subsecuente de la distribución, según proceda.
NECESARIO
El evaluador deberá revisar que las disposiciones estén establecidas y la autoridad tenga el mandato para monitorear todos los canales de distribución en el país.
Sub-indicador 8.6.2: El Código Nacional de Buenas Prácticas de Distribución deberá ser equivalente con el
de la OMS (en TRS 9377 o cualquier actualización) y deberá estar publicado.
NECESARIO
El evaluador deberá verificar que exista una Guía oficial de Buenas Prácticas de Distribución, la cual deberá estar publicada y disponible para las partes interesadas. La Guía deberá estar alineada con las disposiciones de la OMS u otras guías internacionales en la materia. De ser necesario, las diferencias deberán ser identificadas.
Sub-indicador 8.6.3: Solamente los productos autorizados se distribuyen.
CRÍTICO
El evaluador deberá verificar que existen las disposiciones legales para que solamente las vacunas autorizadas sean distribuidas en el país. Las disposiciones deberán también regular las acciones y sanciones de la autoridad si productos no autorizados son encontrados en el mercado. La ARN deberá contar con planes de acción para detectar productos no autorizados que sean circulados a nivel mundial y trabajar en conjunto con autoridades e instancias extranjeras.
Sub-indicador 8.6.4: Mandato para inspeccionar y recolectar muestras en cualquier punto de la cadena de
distribución de vacunas en el país.
CRÍTICO
El evaluador deberá verificar que la legislación atribuya a los inspectores el mandato y autoridad necesarios para inspeccionar en cualquier momento los lugares en el país en donde las vacunas son manejadas para su distribución y para la toma de muestras.
indicador 8. 7: Recursos humanos y otros

92
La evaluación de recursos humanos debe centrarse en aspectos cuantitativos y cualitativos. Para los aspectos cuantitativos los evaluadores pueden decidir usar los siguientes indicadores para identificar si se están poniendo a disposición recursos humanos adecuados para llevar a cabo las actividades planificadas:
Número de inspectores involucrados por categoría de inspección.
Número de establecimientos inspeccionados por categoría de inspección comparado con el número de establecimientos autorizados por categoría de inspección.
Número promedio de días ocupados en terreno por categoría de inspección. Estos indicadores deben permitir a los evaluadores determinar el alcance de las actividades de la inspectoría y el nivel de fiscalización realizada a los "actores claves". Como resultado debe poder proveer información sobre el cumplimiento de estos "actores claves" con la regulación en cuestión. Los evaluadores también deben determinar si los inspectores tienen el soporte logístico necesario para realizar su misión y completar las actividades planificadas.
Sub indicador 8.7.1: Administración de los recursos humanos
CRÍTICO
El evaluador deberá identificar que los recursos humanos estén debidamente designados (número de personal, estructura organizacional, descripción de los puestos, designaciones oficiales) a fin de que estén en posibilidad de desempeñar sus funciones de inspección. El número de empresas inspeccionadas, el promedio de días utilizados para una determinada inspección, pueden considerarse como indicadores.
Evidencia documental a ser revisada:
Estructuras organizacionales de la Autoridad Nacional Reguladora.
Procedimientos internos para el reclutamiento, capacitación y calificación del personal y registros.
Listado del personal con nivel de estudios.
Plan de capacitación.
Listados de cursos de capacitación atendidos.
Descripción de las funciones desempeñadas por el personal.
Currícula Vitae.
Plan de contratación.
Sub-indicador 8.7.2: La descripción de cargo/funciones están establecidos para los siguientes miembros del
personal: jefe de la unidad de inspección (supervisor), jefe de la unidad de inspección por productos, líder del equipo de inspección e inspector.
CRÍTICO
Sub-indicador 8.7.3: Está definida la experticia requerida para ser designado como experto externo o interno en base a educación, experiencia y entrenamiento.
CRÍTICO
Los evaluadores deben revisar si los inspectores son plenamente competentes para la inspección de la BPM y debe determinar en particular si tienen las siguientes aptitudes:
Académicamente capacitado en una disciplina reconocida científica/tecnológica relacionada con ciencias farmacéuticas;
Experiencia personal en la fabricación o control farmacéutico;
Haber completado satisfactoriamente un curso reconocido de capacitación sobre auditoría de los sistema de gestión de calidad;

93
Haber experimentado al menos 10 días de capacitación de por año (por ejemplo, cursos, simposios, conferencias, etc.);
Tener conocimiento práctico competente de las directrices aplicables BPM a los medicamentos y/o a los procedimientos de inspección BPM de la ARN;
Haber recibido una capacitación apropiada en los procedimientos y técnicas actuales para inspecciones BPM antes de que se conduzca una inspección independiente;
Tener cualidades personales como la integridad, la discreción y el carácter requerido de un inspector BPM.
Sub-indicador 8.7.4: Personal debidamente calificado (estudios, capacitación, habilidades y experiencia) para
llevar a cabo inspecciones y actividades relacionadas.
CRÍTICO
El evaluador deberá identificar que los recursos humanos estén debidamente calificados para el desempeño de las actividades de inspección. La evaluación de los recursos humanos deberá enfocarse tanto en elementos cuantitativos como cualitativos. El evaluador deberá revisar los criterios de reclutamiento del personal y solicitar el listado del personal, sus niveles de estudio y experiencia. El evaluador deberá revisar que los inspectores cuenten con las debidas aptitudes y asegurarse de que cumplan con las siguientes:
Académicamente calificados en una disciplina científica relacionada a la farmacéutica.
Experiencia en fabricación o control farmacéutico.
Capacitación en auditorías del sistema de gestión de calidad.
Cubrir al menos 10 días de capacitación por año (cursos, simposios, conferencias, etc.).
Conocimiento de los lineamientos de BPM en vacunas y otros medicamentos.
Cualidades de integridad, tacto y carácter para desempeñar las funciones de inspector. El evaluador también deberá identificar que los inspectores tengan el apoyo logístico necesario para el desarrollo y logros de sus actividades. Las actividades de inspección requerirán de un buen entrenamiento y debida remuneración al personal.
Sub-indicador 8.7.5: Plan de capacitación del personal desarrollado e instrumentado.
NECESARIO
La autoridad competente de las inspecciones de BPM deberá tener establecido un sistema documentado del reclutamiento y capacitación de su personal. La capacitación recibida y requerimientos de capacitación de su personal deberán ser regularmente revisados y mantener un registro de la capacitación individual. El evaluador deberá revisar que los planes de capacitación para inspectores estén desarrollados e instrumentados, reflejando el estatus actual de educación y experiencia. Antes de conducir inspecciones, el inspector deberá cubrir con los procedimientos y técnicas de BPM. El evaluador deberá revisar que se cuente con una plan de capacitación anual y los registros de no menos un año, así como identificar si se proporciona capacitación en técnicas de inspección, sistemas computarizados, monitoreo, etc
Sub-indicador 8.7.6: Monitoreo del desarrollo de habilidades posterior a las actividades de capacitación.
NECESARIO
El evaluador deberá identificar si el monitoreo del desarrollo de las habilidades adquiridas en una determinada capacitación está establecido, por ejemplo, como parte del sistema de gestión de calidad, aplicación de una evaluación a través de indicadores específicos.

94
Sub-indicador 8.7.7: Está definida la calificación mínima, experiencia y entrenamiento para desarrollar todos
los tipos de inspecciones.
CRÍTICO
Sub-indicador 8.7.8: Los inspectores tienen conocimiento de los procedimientos judiciales locales si se
supone que inicien acciones en las cortes de justicia.
NECESARIO
Sub-indicador 8.7.9: Existe un procedimiento para calificar, supervisar y/o confirmar la competencia requerida
para inspectores principiantes.
NECESARIO
Sub-indicador 8.7.10: Los inspectores son entrenados para áreas específicas (BPM, BPD, BPC,
BPL).Siguiendo un programa de entrenamiento. Las actividades de entrenamiento son registradas y tales registros se actualizan.
NECESARIO
Sub-indicador 8.7.11: Los productores o representantes de los titulares de licencia nunca son convocados para conducir las inspecciones.
CRÍTICO
Sub-indicador 8.7.12: La lista de inspectores que están calificados en sus respectivas áreas de competencia
está disponible y se mantiene actualizada.
NECESARIO
Sub-indicador 8.7.13: Existe una oficina adecuada y espacio de almacenamiento suficientes para las
actividades de inspectorado.
NECESARIO
Sub-indicador 8.7.14: Adecuados servicios de apoyo son provistos en términos de transporte y comunicación.
NECESARIO
Indicador 8. 8: Registros y resultados
Sub-indicador 8.8.1: Existe una lista/base de datos actualizada de todas las instalaciones inspeccionadas.
CRÍTICO
Los evaluadores deberán revisar que exista una lista o base de datos de los establecimientos inspeccionados que contenga como mínimo: Nombre o razón social del establecimiento, domicilio, líneas de fabricación, distribución y/o comercialización autorizadas y fecha de la última inspección aplicada.
Sub-indicador 8.8.2: La ARN retiene un archivo de cada inspección incluyendo la información sobre la misión,
el informe de inspección, los comentarios de la empresa inspeccionada y la decisión tomada finalmente con la documentación que la avala, y cada exención está documentada en este archivo.
CRÍTICO
Sub-indicador 8.8.3: La información recopilada de las compañías es usada para tomar o corregir decisiones
reguladoras sobre la licencia inicial o RS (recogida o retiro de productos, etc.).
CRÍTICO

95
Indicador 8.9: Disponibilidad de la información
Sub-indicador 8.9.1: Existe un documento de notificación a las compañías que indique los inspectores
asignados para realizar la inspección.
NECESARIO
Los evaluadores deberán revisar que exista un documento de los inspectores, dicho documento puede tratarse de credencial, carta de presentación, oficio de visita, etc.
Sub-indicador 8.9.2: La lista/base de datos actualizada de todas las instalaciones inspeccionadas es pública y
está disponible públicamente.
NECESARIO
Los evaluadores deberán comprobar que la lista o base de datos se encuentra actualizada y esta se encuentra disponible a través de un medio de comunicación de acceso al público en general, como es diario oficial, página web, etc.
Sub-indicador 8.9.3: Los datos de la última inspección están públicamente disponible.
NECESARIO
Sub-indicador 8.9.4: La información actualizada relacionada con actividades de fiscalización, tales como
advertencias de conformidad, multas, procesamientos, etc., son públicas y están públicamente disponibles.
NECESARIO
Los evaluadores deben revisar que tipo de información está públicamente disponible, si los medios usados (portal Web, boletín oficial u otro boletín de la ARN) son apropiados y si la información es mantenida y actualizada en forma regular.
Evidencia documental a ser estudiada.
Reglamentos, leyes, decretos.
Publicaciones, orientación.
Política de calidad y sus objetivos.
Designación de gerentes de calidad y descripción del puesto.
Manual de inspección de calidad.
Minutas de revisión de la dirección.
Procedimiento para el de control de documentación.
Planes de inspección.
Lista de equipo (logística, computadora, etc.).
Procedimiento para planificación, implementación de auditoría interna y seguimiento de las medidas correctivas.
Informes de auditoría y seguimientos.
Indicadores de calidad (plazo, notificación).
Procedimiento para la investigación de no-conformidades y registros.
Procedimiento para la iniciación, decisión e implementación de medidas correctivas y preventivas y verificar la eficacia y los registros.
Procedimiento para tratar quejas y devoluciones.
Procedimiento para la inspección (preparación, planificación, realización y seguimiento).
Procedimiento para muestreo y registros.
Formato del informe de inspección.
Informes de inspección.
Planificación de inspecciones para un año específico.
Procedimiento para entregar o retirar un certificado de BPM.

96
Procedimiento para aprobar o cerrar instalaciones, entregando cartas y/o avisos de advertencia de no-conformidades.
Procedimiento para reclutamiento, capacitación y calificación de inspectores con recalificación periódica y registros.
Lista de inspectores con sus alcances de intervención.
Lista de establecimientos inspeccionados.
Lista de cartas y/o notificaciones de advertencia sobre no-conformidades entregadas.
Formato/plantilla del certificado.

97
9. Laboratorio Oficial de Control de Medicamentos (LOCM) - Módulo 9
Objetivo: Evaluar la estructura de la organización para analizar productos farmacéuticos.
La función tiene por objeto garantizar que la ARN es capaz de verificar, mediante los análisis apropiados, si los medicamentos, incluyendo los productos biológicos son de la calidad requerida antes y durante su comercialización. Los recursos y la capacidad técnica para llevar a cabo estas actividades a nivel de país varían enormemente pero cada ARN debe tener acceso a un laboratorio de control de calidad el cual también desempeñará un papel importante en el proceso de registro y la vigilancia post-comercialización de la calidad del producto. Se recomienda en general que todos los países deban tener acceso al menos a un laboratorio pequeño donde puedan realizarse pruebas básicas y que tales establecimientos básicos gradualmente se amplíen. Las pruebas podrían realizarse adecuadamente y más eficazmente en costos en una institución ya existente, como en un laboratorio universitario de farmacia o laboratorio independiente. En el caso particular del LOCM de vacunas, un laboratorio funcional para la realización de ensayos críticos es un recurso importante para el Sistema Regulatorio Nacional. Un equipo de expertos en vacunología puede colaborar en la toma de decisiones regulatorias tales como la evaluación de solicitudes de autorización de comercialización, revisión de datos de ensayos clínicos, entre otras.
Indicador 9.1: Bases legales
El evaluador debe revisar los requisitos legales vigentes y determinar si las regulaciones adecuadas han sido promulgadas. Para el caso especial de las vacunas se ha establecido a nivel mundial un procedimiento denominado “liberación de lote”; para lo cual es esencial que los LOCM que realizan la liberación de lotes cumplan con los requisitos del módulo 11 de este manual. Pruebas documentadas a estudiar:
Leyes, Decretos, Reglamentos y Disposiciones legales que establezcan la existencia de una autoridad nacional responsable del control de medicamentos y vacunas o acuerdos con un laboratorio externo independiente.
Procedimientos y registros.
Sub-indicador 9.1.1: Existen bases legales para el establecimiento de un Laboratorio Oficial de Control de Medicamentos (LOCM) o acuerdos operacionales para usar un Laboratorio de Control Externo.
CRÍTICO
El evaluador debe verificar que existe un LOCM responsable de controlar medicamentos y vacunas dentro de la estructura de la ARN o de otra autoridad nacional con incumbencia en medicamentos o que el país tiene acuerdos operativos con un laboratorio externo independiente.
Sub-indicador 9.1.2: El LOCM es parte de la ARN.
INFORMATIVO
Sub-indicador 9.1.3: En caso de que el LOCM no forme parte de la ARN, existe una base legal que
establezca los mecanismos de relación entre ambos.
CRÍTICO

98
Sub-indicador 9.1.4: La legislación faculta al LOCM para desarrollar pruebas de control de calidad de
medicamentos y vacunas y emitir resultados oficiales.
CRÍTICO
Indicador 9.2: Directrices
Los evaluadores deben revisar los documentos de orientación publicados para diferentes tipos de "actores claves" y si abarcan el alcance de la legislación y regulaciones aplicables. Debe comprobarse si hay coherencia con la orientación de la OPS/OMS y cualquier diferencia debe ser identificada.
Sub-indicador 9.2.1: Existen lineamientos establecidos para el cumplimiento de Buenas Prácticas de
Laboratorio en el LOCM.
CRÍTICO
Sub-indicador 9.2.2: Están definidas claramente las responsabilidades para el aporte científico-técnico del
LOCM en el período pre-comercialización y post-comercialización.
CRÍTICO
El evaluador debe verificar que la documentación defina por escrito las responsabilidades del LOCM y el rol técnico y/o científico que desempeña en el período pre-comercialización y post-comercialización de los productos. Teniendo en cuenta el enfoque de equipo para la realización de las inspecciones, deben existir lineamientos que regulen la participación de personal científico del LOCM como expertos en las inspecciones.
Sub-indicador 9.2.3: El LOCM está involucrado en la definición de métodos de control y especificaciones
durante la evaluación de la documentación de registro de producto.
NECESARIO
El evaluador debe verificar que la ARN tiene un sistema para garantizar que durante la evaluación de la documentación de registro del producto sometido a autorización de comercialización y de las modificaciones presentadas, el personal científico de LOCM está involucrado en la definición de las especificaciones del producto y los métodos de laboratorio utilizados para asegurar la calidad de los productos durante el ciclo de vida del producto.
Sub-indicador 9.2.4: Existen lineamientos generales sobre toma de muestras.
CRÍTICO
Sub-indicador 9.2.5: Existen lineamientos establecidos sobre los estándares aplicados por el LOCM. Si
existen diferencias con los establecidos por OPS/OMS, cuentan con adecuada justificación.
CRÍTICO
Sub-indicador 9.2.6: Existen lineamientos establecidos para tratar con medicamentos en que se sospecha
falsificación.
INFORMATIVO
Sub-indicador 9.2.7: Existen lineamientos establecidos sobre manejo de quejas y reclamos contra las
actividades del LOCM.
NECESARIO
Sub-indicador 9.2.8: Existen lineamientos establecidos sobre manejo de apelaciones contra las decisiones
del LOCM.

99
NECESARIO
Indicador 9.3: Organización y estructura
Los evaluadores deben identificar la estructura de la organización para llevar a cabo las actividades de LOCM; si la función es delegada o descentralizada y en este último caso a qué nivel del país (central, regional o local). Si diferentes organizaciones se involucran en diferentes niveles del estado los evaluadores deben revisar el nexo entre estas organizaciones y en particular cómo el intercambio de información esta establecido y administrado.
Sub-indicador 9.3.1: Las actividades de ensayos son organizadas y desarrolladas a nivel central del país.
INFORMATIVO
Sub-indicador 9.3.2: Las actividades descentralizadas de otras agencias/autoridades siguen los mismos
estándares, guías y procedimientos. (No aplica en caso que las actividades del LOCM estén centralizadas).
CRÍTICO
Sub-indicador 9.3.3: En caso de descentralización, un mecanismo de intercambio de información está
establecido e implementado para que la organización descentralizada reciba requerimientos y/o directivas de la autoridad central y reportar a ésta. (No aplica en caso que las actividades del LOCM estén centralizadas).
CRÍTICO
Sub-indicador 9.3.4: Los mecanismos permiten apropiada colaboración entre los organismos
descentralizados. (No aplica en caso que las actividades del LOCM estén centralizadas).
CRÍTICO
Indicador 9.4: Sistema de Gestión de la Calidad (SGC)
Los evaluadores deben revisar cómo la organización satisface los requisitos establecidos para los sistemas de gestión de calidad adoptados como referencias. Estos quizá sean un documento de orientación de la OPS/OMS o un estándar internacional en sistemas de gestión de calidad. Cualquiera que sea el caso, el cuestionario incluido en este módulo debe abordar los temas principales con respecto a un sistema de gestión de calidad (vea también capítulo 3.5). Pruebas documentadas a estudiar:
Organigrama del SGC.
Listado de procedimientos operativos.
Listas de verificación (chequeo), Formatos de reportes.
Políticas y procedimientos para trazabilidad de los registros de ensayos.
Informes de las auditorías internas y externas.
Sub-indicador 9.4.1: Existe un Sistema de Gestión de Calidad establecido, implementado y mantenido por el LOCM y está orientado a demostrar la competencia técnica del mismo.
CRÍTICO
El sistema de gestión de la calidad del LOCM debe estar implementado tanto para las actividades pre-comercialización como para las post-comercialización de los medicamentos, incluyendo a las vacunas.
Sub-indicador 9.4.2: La gestión del LOCM está comprometida con el desarrollo e implementación del sistema
de gestión de calidad.
CRÍTICO

100
Sub-indicador 9.4.3: El SGC está basado o reconoce estándares de referencia (OMS, PIC/S, ISO, otros).
NECESARIO
Pueden usarse como referencia documentos de orientación de la OMS o una norma internacional sobre sistema de gestión de la calidad. Se recomienda utilizar como guía las Buenas Practicas de Laboratorio de la OMS y complementariamente la norma ISO 17025 que representa el estándar internacional para la acreditación de laboratorios oficiales designados a través de organismos nacionales de acreditación.
Sub-indicador 9.4.4: Está definido el organigrama y las responsabilidades para la implementación del SGC.
CRÍTICO
El evaluador debe identificar la organización creada para establecer, implementar y mantener el sistema de gestión de calidad. Deben identificarse las diferentes responsabilidades.
Sub-indicador 9.4.5: Una persona calificada está designada como responsable en el LOCM para el
desarrollo, implementación y ejecución del SGC.
NECESARIO
Sub-indicador 9.4.6: Está definida la documentación necesaria para establecer, implementar y mantener el
SGC (Manual de Calidad, Registros, Procedimientos, etc.).
NECESARIO
Sub-Indicador 9. 4.7: Toda la documentación que forma parte del SGC está controlada y revisada
siguiendo un procedimiento documentado.
NECESARIO
Sub-indicador 9.4.8: Existe documentación que establezca el mecanismo de evaluación periódica de la
organización y el desarrollo del SGC, los registros y resultados.
NECESARIO
Sub-indicador 9.4.9: Existe un sistema de gestión para garantizar la trazabilidad de las actividades.
CRÍTICO
El evaluador debe comprobar si el SGC implementado garantiza que las actividades de ensayo están documentadas (resultados del muestreo y de los ensayos, resultados fuera de especificación, validación de métodos) y que los documentos son archivados de manera trazable para, si es necesario, obtener datos (apelaciones de una institución contra las decisiones del LOCM, defectos de calidad detectados en los productos en el mercado, comparar hallazgos antiguos y recientes, o reconocer tendencias referidas a consistencia de la calidad de los productos autorizados).
Sub-indicador 9.4.10: Las acciones correctivas, preventivas y de mejora definidas para tratar los hallazgos
evidenciados como resultado de auditorías (no conformidades u observaciones) o de otras actividades (autocontrol, revisiones por la dirección) son implementadas y documentadas y su eficacia es verificada.
NECESARIO
Sub-indicador 9.4.11: Existe un sistema documentado de auditorías internas y externas.
NECESARIO

101
El evaluador debe verificar que el LOCM realiza auditorías internas periódicamente y que existen POE para la realización de las mismas. En caso de que el SGC esté certificado (por ejemplo, según normas ISO 17025), tener en cuenta que los organismos de acreditación auditan al titular de la certificación en intervalos regulares con el fin de renovar la certificación/acreditación.
Sub-indicador 9.4.12: Existen evidencias de que El LOCM participa en esquemas internacionales de estudios
de desempeño, estudios colaborativos y / o comparaciones interlaboratorios.
NECESARIO
Se recomienda la participación del LOCM en esquemas internacionales de pro-eficiencia o estudios organizados por la OMS, EDQM, NIBSC u otras instituciones. La participación permite comparar el rendimiento propio con un punto de referencia internacional. El evaluador deberá verificar la existencia de registros de participación en los esquemas estudios de desempeño internacionales y estudios colaborativos y comprobar las fechas de participación, el alcance, los productos, la institución coordinadora.
Sub-indicador 9.4.13: Los resultados indican un rendimiento adecuado en esquemas de evaluación de
desempeño, estudios colaborativos y/o comparaciones interlaboratorios.
NECESARIO
El evaluador debe verificar si se ha logrado un rendimiento adecuado y chequear para las distintas pruebas el número de resultados adecuada frente al número de resultados inadecuados.
Indicador 9.5: Procedimientos de control de calidad
Los evaluadores deben revisar los procedimientos con relación a los resultados esperados, su nivel de detalle en relación a la capacitación proporcionada y los controles aplicados para verificar las actividades descritas. Deben revisar en particular su coherencia con la orientación, regulaciones y legislación vigentes. El desempeño consistente de los ensayos es esencial para poder comparar los resultados del LOCM con las pruebas realizadas en el lugar de fabricación o en otros LOCM. Sólo resultados consistentes permitirá al LOCM detectar tendencias relacionadas a la consistencia lote a lote de cada producto. Pruebas documentadas a estudiar:
Procedimientos para cada ensayo.
Procedimientos para la manipulación de la muestra.
Procedimientos para el manejo de resultados fuera de especificación.
Sub-indicador 9.5.1: Existen procedimientos o mecanismos de comunicación efectiva con usuarios y partes
interesadas.
NECESARIO
Sub-indicador 9.5.2: Existen procedimientos /herramientas para la preparación de estándares, muestras, etc.
CRÍTICO
Sub-indicador 9.5.3: Existen procedimientos escritos para desarrollar los ensayos.
CRÍTICO

102
El evaluador debe identificar la existencia de POE escritos y ejecutados para cada prueba, incluyendo la descripción exacta del procedimiento de ensayo, los materiales y equipos utilizados (incluyendo los estándares), el cálculo de resultados de las pruebas y los intervalos de confianza.
Sub-indicador 9.5.4: Existe una planificación adecuada de ensayos y coordinación de actividades.
NECESARIO
Sub-indicador 9.5.5: Existen procedimientos para el manejo y retención de muestras.
NECESARIO
Sub-indicador 9.5.6: Existen procedimientos para ensayos de categorías de productos que requieran
consideraciones especiales.
NECESARIO
Sub-indicador 9.5.7: Están definidos las especificaciones y criterios de validez para cada ensayo.
CRÍTICO
El evaluador debe verificar que estén claramente definidas las especificaciones y criterios de validez para todas las pruebas realizadas sobre medicamentos y vacunas en la fase pre y post-comercialización en el LOCM.
Sub-indicador 9.5.8: Existen estrategias, programas y procedimientos para la implementación y validación de
nuevos/mejoras de ensayos.
NECESARIO
Sub-indicador 9.5.9: Existe/n procedimiento/s para la evaluación de los resultados de los ensayos y toma de
decisiones.
CRÍTICO
Sub-indicador 9.5.10: Existe/n procedimiento/s para el manejo de resultados fuera de especificaciones que
incluyan la política de reensayo.
CRÍTICO
El evaluador debe verificar la existencia de procedimientos escritos sobre cómo manejar los resultados fuera de especificaciones para cada sistema de ensayo utilizado. Se deberá identificar la política de reensayo para cada prueba. La repetición de las pruebas a veces se rige por los requisitos de Farmacopea que deben ser respetados en los procedimientos internos.
Sub-indicador 9.5.11: Existe un formato estándar de hoja de trabajo analítico o como se le denomine, para registrar información sobre la muestra, los procedimientos de ensayo, los datos primarios obtenidos en el análisis, cálculos (cuando aplique) y resultados del análisis.
NECESARIO
Sub-indicador 9.5.12: Los criterios adoptados por el LOCM para la evaluación de un producto son
seleccionados de acuerdo al principio activo y los tipos de muestras a evaluar, independientemente del origen del producto.
CRÍTICO
Indicador 9.6: Recursos humanos

103
Los recursos humanos adecuados y competentes (número de personal, organigramas, descripción del puesto, funciones, nombramiento oficial) deben estar designados con el fin de poder llevar a cabo todo tipo de pruebas de laboratorio. La evaluación de recursos humanos debe centrarse en aspectos cuantitativos y cualitativos. Para los aspectos cuantitativos los evaluadores pueden decidir usar los siguientes indicadores para identificar si se están poniendo a disposición recursos humanos adecuados para llevar a cabo las actividades planificadas:
Número de personal científico involucrado en las actividades de análisis.
Número de productos analizados y certificados emitidos.
Número de productos analizados y encontrados como no-conformes. Los siguientes indicadores deben permitirles a los evaluadores determinar en qué medida las actividades de laboratorio de control de calidad están involucradas en las actividades de conformidad a lo establecido:
Número de productos analizados por categoría de producto comparado con el número de autorizaciones concedidas por categoría.
Número de medicamentos retirados en base a resultados del LOCM.
Número de registros suspendidos o retirados en base a resultados del LOCM. Los evaluadores deben revisar si el personal involucrado en los procesos de control es plenamente competente: los analistas deben ser graduados en farmacia, química analítica, microbiología u otra área relevante y con capacitación apropiada impartida en disciplinas o técnicas específicas (espectrofotometría, instrumental, etc.) y también debe tener conocimiento del sistema de gestión de calidad del laboratorio. Los evaluadores deben revisar los registros de capacitación del personal de LOCM para asegurar la adecuación de las competencias del personal de laboratorio. El LOCM debe tener un sistema que permita mantener la experticia en la realización de los ensayos. El evaluador también deberá establecer si el personal cuenta con el apoyo logístico necesario para llevar a cabo sus actividades. Pruebas documentadas a estudiar:
Organigrama.
Procedimientos de designación, formación y calificación del personal y sus registros.
Lista de personal con su respectiva calificación.
Plan de Capacitación.
Lista de los entrenamientos realizados.
Descripción de tareas.
Currícula Vitae.
Sub-indicador 9.6.1: Existe un mecanismo para la designación de analistas y la definición de sus
responsabilidades y facultades en el LOCM.
CRÍTICO
Sub-indicador 9.6.2: Las descripciones de cargo están establecidas para directivos, técnicos, y personal
involucrado en los ensayos, y/o calibraciones, validaciones y verificaciones.
CRÍTICO
Sub-indicador 9.6.3: Está definida la experticia requerida para ser designado como analista en base a
educación, experiencia y entrenamiento.
CRÍTICO
Sub-indicador 9.6.4: Existe un plan de capacitación del personal desarrollado e implementado.

104
NECESARIO
El evaluador debe revisar que los planes de capacitación del personal de laboratorio se han desarrollado e implementado reflejando el estado actual de educación y experiencia en las distintas metodologías de ensayo.
Sub-indicador 9.6.5: Se realiza el monitoreo del desarrollo de las competencias adquiridas después de las
actividades de entrenamiento.
NECESARIO
El evaluador debe comprobar si se monitorea y registra después del entrenamiento la competencia adquirida.
Sub-indicador 9.6.6: Existe un mecanismo para manejar conflictos de interés garantizando la actualización de la declaración de conflictos de interés.
CRÍTICO
Sub-Indicador 9.6.7: Existe un mecanismo para garantizar los requerimientos de confidencialidad, por
ejemplo la observancia de un código de conducta por los analistas.
CRÍTICO
Sub-indicador 9.6.8: Existe un número de analistas adecuado para implementar el plan de trabajo.
INFORMATIVO
El evaluador debe determinar si son adecuados los recursos humanos disponibles para implementar y ejecutar las actividades planificadas.
Sub-indicador 9.6.9: El LOCM participa de manera efectiva y suficiente en las actividades de
fiscalización/vigilancia conforme a lo establecido.
INFORMATIVO
Indicador 9.7: Infraestructura y equipamiento
Los evaluadores deben revisar la disponibilidad y la adecuación del equipo para realizar las pruebas, la adecuación de las instalaciones, ambiente de trabajo y espacio de trabajo. Los siguientes indicadores pueden ser usados por los evaluadores:
Número de personal científico involucrado en las actividades de ensayos o pruebas analíticas.
Área de superficie del LOCM.
Número de personal científico comparado con el área de superficie.
Número de análisis no realizados debido a falta de equipo adecuado. Debe verificarse el cumplimiento de requisitos específicos para los sectores en los que se lleven a cabo ensayos tales como pruebas microbiológicas, pruebas con tecnología PCR o valoraciones con organismos vivos utilizando sistemas de cultivo celular. Pruebas documentadas a estudiar:
Manuales de Operación de equipos, POE y libros/cuadernos de registro.
Planes de Calibración y Mantenimiento.
Protocolos e Informes de Calificación.

105
Sub-indicador 9.7.1: Existe un espacio de trabajo, ambiente y almacenamiento adecuados para las
actividades del laboratorio.
CRÍTICO
El evaluador deberá verificar la adecuación de los locales, el ambiente y espacio de trabajo.
Sub-indicador 9.7.2: Existe un adecuado equipamiento e instrumentos para realizar los ensayos.
CRÍTICO
Sub-indicador 9.7.3: Existen protocolos de calificación y sus respectivos registros.
CRÍTICO
El evaluador debe verificar la existencia de protocolos de calificación e informes para todos los equipos y comprobar si los informes reflejan los protocolos. El evaluador debe verificar si los resultados han sido evaluados, analizados y comparados con los criterios de aceptación pre-determinados
Sub-indicador 9.7.4: Hay manuales de operación, procedimientos operativos y sus correspondientes registros
(uso y mantenimiento).
CRÍTICO
El evaluador debe identificar para los diferentes equipos e instrumentos la existencia de manuales o POE detallando las operaciones y registros que demuestren el correcto uso y mantenimiento.
Sub-indicador 9.7.5: Existe un plan de calibración y mantenimiento de los equipos.
CRÍTICO
El evaluador debe verificar la existencia de planes de calibración y mantenimiento para todos los equipos existentes.
Sub-indicador 9.7.6: Adecuados servicios de apoyo son suministrados en términos de transporte y
comunicación.
NECESARIO
Sub-indicador 9.7.7: La relación de analistas/área de laboratorio permite que las actividades de LOCM se
lleven a cabo de manera eficiente.
INFORMATIVO
Sub-indicador 9.7.8: La capacidad instalada y analítica del LOCM es suficiente para dar respuesta a todas las
solicitudes de análisis.
INFORMATIVO
Indicador 9.8: Política de validación
La validación de los ensayos, procedimientos y calificación de equipos son la base para obtener resultados confiables. Pruebas documentadas a estudiar:
Plan Maestro de Validación.
Protocolos e informes de validación de métodos.

106
Sub-indicador 9.8.1: Existe un Plan Maestro de Validación, se han validados los métodos no compendiados y
verificados los métodos compendiados.
CRÍTICO
El evaluador debe verificar si existe un Plan Maestro de Validación. Las pruebas codificadas en Farmacopeas deben ser verificadas en el laboratorio para obtener los mismos resultados según lo estipulado en los procedimientos de la Farmacopea. Las pruebas no codificadas y cualquier otra "in house" deben ser validadas para demostrar resultados cualitativos y cuantitativos consistentes.
Sub-indicador 9.8.2: Existen procedimientos para la transferencia de métodos validados (por ejemplo, desde
el fabricante).
CRÍTICO
El evaluador debe verificar si existen métodos validados transferidos desde el fabricante o cualquier otro laboratorio al LOCM y la existencia de un procedimiento de transferencia que incluya la revalidación de los sistemas de prueba.
Indicador 9.9: Estándares de referencia, materiales y reactivos
Los evaluadores deben evaluar los procedimientos existentes para la adquisición, preparación, manejo y prueba de estándares/materiales de referencia y reactivos. Para los ensayos biológicos, en vista de la probable comparación entre los resultados de ensayos y en el plano inter-laboratorios, el uso de estándares y materiales de referencia es de importancia crítica. Pruebas documentadas a estudiar:
Políticas, procedimientos y registros para establecer y calificar materiales de referencia nacionales.
Sub-indicador 9.9.1: En caso de contar con estándares/materiales de referencia nacionales de sustancias
químicas, existe un sistema para establecer y calificar dichos materiales.
CRÍTICO
El evaluador debe verificar que exista un sistema adecuado para uso y calificación de estándares nacionales de referencia tal como se recomienda en el Anexo 2, la OMS TRS 932, 2006 o sus actualizaciones.
Sub-indicador 9.9.2: Existen criterios definidos para la selección de estándares/materiales de referencia.
CRÍTICO
Sub-indicador 9.9.3: Existe un sistema regular de suministro que asegure la disponibilidad de
estándares/materiales de referencia?
NECESARIO
Sub-indicador 9.9.4: Existe/n procedimiento/s para el adecuado manejo, uso y verificación de
estándares/materiales de referencia.
CRÍTICO
El evaluador debe verificar que los estándares/materiales de referencia o de trabajo se preparan, manipulan, almacenan y utilizan correctamente. El LOCM no debería utilizar estándares internacionales en las pruebas de rutina.
Sub-indicador 9.9.5: Existe/n procedimiento/s para recepción, almacenamiento y uso de reactivos de calidad
garantizada.

107
CRÍTICO
El evaluador debe verificar que todos los reactivos utilizados en el LOCM son de calidad garantizada y están etiquetados según lo establecido (fecha de elaboración, fecha de caducidad, especificaciones, pruebas para las que se utiliza, condiciones de almacenamiento, etc.).
Sub-indicador 9.9.6: Existe/n procedimiento/s para la preparación de reactivos en el laboratorio.
NECESARIO
Sub-indicador 9.9.7: Existen registros de la preparación y estandarización de soluciones volumétricas.
NECESARIO
Sub-indicador 9.9.8: Existe un sistema regular de abastecimiento de estándares o materiales de referencia.
CRÍTICO
El evaluador debe verificar que el LOCM haya establecido un sistema para el suministro regular de estándares de referencia. Cabe señalar que los estándares de referencia internacionales sólo están disponibles en cantidades limitadas.
Indicador 9.10: Monitoreo, análisis y manejo regulatorio de los resultados del LOCM de vacunas
El LOCM debe monitorear y analizar los resultados de laboratorio. Es esencial el seguimiento de la calidad de las vacunas para cada producto específico y cada fabricante, para detectar las tendencias de incumplimiento o resultados que no cumplen con los límites de confianza especificados.
Pruebas documentadas a estudiar:
Análisis de tendencias de los resultados de LOCM y del fabricante.
Procedimientos escritos o lineamientos para acciones correctivas en caso de desvíos.
Sub-indicador 9.10.1: Se lleva a cabo la verificación (el chequeo) sistemática(o) del cumplimiento de las
especificaciones.
CRÍTICO
El evaluador debe verificar que todos los resultados son verificados (chequeados) sistemáticamente contra las especificaciones definidas.
Sub-indicador 9.10.2: Son tomadas acciones correctivas en caso de desvíos.
CRÍTICO
El evaluador debe verificar (chequear) lotes fuera de especificación y el análisis de tendencias con el fin de definir si el LOCM lleva a cabo una acción correctiva apropiada en caso de desvíos en los resultados de los ensayos respecto a las especificaciones establecidas. Deben existir procedimientos escritos o lineamientos sobre acciones correctivas a implementar en caso de desvíos.
Sub-indicador 9.10.3: Se adoptan acciones regulatorias en caso de incumplimiento.
CRÍTICO
El evaluador debe comprobar si se ejecutan acciones regulatorias en caso de incumplimiento de las especificaciones en los resultados de los ensayos y verificar que acciones regulatorias se han tomado y si las mismas han sido apropiadas.

108
Sub-indicador 9.10.4: Se lleva a cabo el seguimiento y análisis de tendencias de datos de los resultados de
laboratorio.
CRÍTICO
El evaluador debe verificar si se realiza el análisis sistemático de tendencia de los resultados de todos o un grupo definido de ensayos y para productos y fabricantes específicos.
Sub-indicador 9.10.5: Se lleva a cabo el seguimiento y análisis de tendencia de los datos obtenidos de
materiales de referencia.
CRÍTICO
El evaluador debe comprobar si se realiza sistemáticamente el análisis de tendencia de los datos de los materiales de referencia.
Sub-indicador 9.10.6: Se lleva a cabo la comparación de los resultados obtenidos con los del fabricante.
CRÍTICO
El evaluador debe comprobar si los resultados obtenidos en los ensayos de laboratorio en el LOCM son comparados de manera rutinaria con los resultados obtenidos en los laboratorios de control de calidad de los diferentes productores y para diferentes productos. En caso de detectar desvíos o modificaciones en la tendencia de los datos debe evaluarse la razón, si es necesario, en colaboración con el laboratorio de control de calidad del fabricante.
Indicador 9.11: Programa de seguridad
Los evaluadores deben verificar la disponibilidad del programa de seguridad y cualquier medida para comprobar su eficacia. El trabajo de laboratorio está vinculado a riesgos específicos por el uso de materiales químicos, radiactivos y biológicos (organismos vivos tales como virus, etc.). Los temas de seguridad para proteger al personal y el medio ambiente son de gran importancia. Pruebas documentadas a estudiar:
Programa de seguridad.
Lista de sustancias peligrosas.
Procedimiento para el almacenamiento, manipulación y eliminación de sustancias peligrosas.
Sub-indicador 9.11.1: Las listas de sustancias peligrosas están disponibles.
NECESARIO
El evaluador debe comprobar si están disponibles las listas de sustancias peligrosas utilizadas.
Sub-indicador 9.11.2: Está designado un responsable del equipo para la gestión del programa de seguridad.
NECESARIO
El evaluador debe confirmar si se ha designado una persona o grupo de personas como responsable de todos los aspectos del programa de seguridad del LOCM.
Sub-indicador 9.11.3: Existe un procedimiento para el almacenamiento, manejo y disposición de sustancias
peligrosas.
CRÍTICO

109
El evaluador debe comprobar si están disponibles y en ejecución los procedimientos para el almacenamiento, manipulación y eliminación de sustancias peligrosas
Sub-indicador 9.11.4: Están definidos los requerimientos de inmunización del personal y los mismos se han
cumplimentado.
NECESARIO
El evaluador debe comprobar si existe un programa de inmunización para el personal de laboratorio y si el mismo se ha cumplimentado. Se debería prestar especial atención a la protección de mujeres embarazadas que trabajan con materiales microbiológicos como los virus vivos.
Indicador 9.12: Sub contratación
Los evaluadores deben revisar cómo el laboratorio de control de calidad supervisa los análisis que se subcontratan a otro laboratorio/institución. Se sugiere la revisión de los contratos entre partes para verificar que se describan las obligaciones de cada parte y la existencia de mecanismos de control del LOCM sobre el laboratorio contratado.
Sub-indicador 9.12.1: Está establecida la lista de todos los ensayos, que el LOCM contrata a otros
laboratorios u organizaciones.
CRÍTICO
Sub-indicador 9.12.2: Existe un acuerdo/contrato formal definiendo el alcance y responsabilidades de cada
parte.
CRÍTICO
Sub-indicador 9.12.3: El acuerdo tiene en consideración los puntos relacionados a conflictos de interés y confidencialidad del laboratorio contratado.
CRÍTICO
Sub-indicador 9.12.4: El acuerdo/contrato formal asegura que los requerimientos establecidos por el LOCM
son aplicados de la misma manera por la organización sub-contratada?
CRÍTICO
Sub-indicador 9.12.5: Existe un mecanismo o procedimiento documentado que asegura la competencia técnica del laboratorio sub-contratista.
NECESARIO
Indicador 9.13: Registros y resultados
Los evaluadores deben revisar cómo toda la información recogida durante el proceso de análisis es administrada y que clase de información es registrada y archivada por la organización. Mientras es revisada la implementación de procedimientos internos de la organización, los evaluadores deben muestrear los registros generados y comprobar su contenido. Los evaluadores deben verificar si los resultados de este proceso se usaran como entrada-insumo para procesos relacionados tales como la autorización de comercialización.

110
Sub-indicador 9.13.1: Existe una lista /base de datos de todos los productos ensayados.
CRÍTICO
Sub-indicador 9.13.2: Los registros de ensayos se mantienen fácilmente disponibles en el LOCM.
CRÍTICO
Sub-indicador 9.13.3: Existe un formato estándar de informe de resultados de ensayo emitido por el
laboratorio, con la información necesaria para la correcta identificación del solicitante, la muestra, el tipo de ensayo realizado, los resultados y las conclusiones del análisis de una muestra.
CRÍTICO
Evidencia documental a ser estudiada:
Reglamentos, leyes, decretos.
Archivos de información del laboratorio.
Política de calidad y objetivos.
Designación de gerentes de calidad y descripción del puesto.
Manual de calidad.
Registros de revisión de administración del LOCM.
Procedimientos para control de documentación.
Plan de prueba o programa.
Lista de equipos.
Procedimientos para planificación, implementación de auditoría interna y seguimiento de medidas correctivas.
Informes de auditoría y seguimientos.
Indicadores de calidad (plazos, notificación).
Procedimientos para investigaciones de no-conformidades y registros.
Procedimientos para tratar quejas y registros.
Procedimientos para iniciación, selección e implementación de acciones correctivas y preventivas y verificar eficiencia y registros.
Procedimientos internos para gestión de muestras.
Procedimientos internos para la puesta a prueba, validación analítica, calificación de equipos y mantenimiento; calibración y registros.
Planificación de mantenimiento de equipos.
Política de repetición de pruebas.
Procedimientos internos para la manipulación de resultados fuera de especificación.
Procedimientos internos para la manipulación de materiales de referencia y reactivos.
Organigrama.
Plan de capacitación e implementación de seguimiento.
Lista del personal con sus calificaciones.
Lista de equipos.
Lista de actividades de mantenimiento y calibración.
Lista de estudios de preeficiencia en los cuales el LOCM ha participado.
Registros de libros.
Mapa del laboratorio.
Directrices de seguridad.
Contratos de actividades subcontratadas.
Lista de análisis subcontratado.

111
10. Liberación de lotes de productos biológicos (vacunas)- Modulo 10
Objetivo: La función tiene por objeto asegurar que las vacunas y otros biológicos como los derivados sanguíneos comercializados estén sujetos al escrutinio de las Autoridades Reguladoras Nacionales, basado en el proceso de liberación lote a lote, debido al riesgo inherente al origen biológico de estos productos.
La evaluación inicial de lotes de vacuna se realiza durante el proceso de concesión de licencias cuando los métodos de fabricación y control y la consistencia de producción de varios lotes son evaluados. Una vez registradas, las vacunas deben ser liberadas lote a lote por la ARN, basado como mínimo en la revisión del protocolo resumen de fabricación y control del lote, el cual incluye los pasos de fabricación y los resultados de control efectuados durante toda su manufactura, desde la obtención del principio activo, controles en proceso y hasta la obtención del producto terminado.
Indicador 10.1: Sistema de liberación de lote
El sistema de liberación de lotes (también llamado en inglés “batch release”) es un sistema único establecido mundialmente para la liberación reglamentaria de cada producción de vacunas. Esto es realizado para garantizar la consistencia en la calidad y seguridad de todos los lotes de vacunas comercializados. Evidencia documental que debe ser estudiada:
Leyes, decretos, Reglamentos o disposiciones que establecen mandato de la ARN para desarrollar la liberación de lotes.
Organigrama de la ARN/LOCM.
Certificados de liberación de lotes.
Política, procedimientos de reconocimiento de decisiones de otras ARNs, informe o certificados incluyendo lista de ARNs/LOCM.
Política de ensayo.
Sub-indicador 10.1.1: Disposición para la liberación de lotes, incluido el funcionario responsable para firmar el
certificado de liberación de lotes.
CRÍTICO
El evaluador debe comprobar que existe la base legal para realizar liberación de lotes. La ARN/LOCM debe tener la autoridad para firmar los certificados de liberación de lotes oficiales.
Sub-indicador 10.1.2: La liberación de lotes se aplica a todas las vacunas.
CRÍTICO
El evaluador debe verificar que la base legal para realizar liberación de lotes aplica para todas las vacunas comercializadas en el país, tanto las producidas localmente, como las importadas, las empleadas en el PAI, como en consulta médica privada.
Sub-indicador 10.1.3: Basado como mínimo en la revisión del protocolo resumen de fabricación y control del
lote.
CRÍTICO
El evaluador debe verificar que la ARN/LOCM expide el certificado de liberación de lotes basado como mínimo en la revisión del protocolo resumen de fabricación y control del lote expedido por el fabricante. La evaluación del protocolo deber ser desarrollada apropiadamente al comparar los datos críticos de cada lote incluidos los datos de ensayos con las especificaciones autorizadas de los productos.

112
Sub-indicador 10.1.4: Es obligatorio el protocolo resumen de fabricación y control del lote como parte de los
requisitos de concesión del registro de las vacunas.
CRÍTICO
Sub-indicador 10.1.5: Existe Política/criterios de aceptación de la liberación de lotes realizada por otras ARN
(ej. el certificado de liberación de lotes del país de origen).
NECESARIO
El evaluador debe revisar si el país no tiene la capacidad para realizar la liberación de lotes por si mismo, y si el país por el contrario reconoce los certificados de la liberación de lotes emitidos por las autoridades extranjeras ARN/LOCM, por ejemplo, en el país de origen del producto o por otras ARN/LOCM competentes. Las provisiones escritas deben estar establecidas y abarcar lo que es permitido en cuanto al reconocimiento de decisiones de otras autoridades, informes o certificados, lista de los ARN/LOCM y procedimientos definidos.
Sub-indicador 10.1.6: Política de ensayo y frecuencia con que se realizan.
CRÍTICO
El evaluador debe confirmar que el LOCM ha definido parámetros apropiados de ensayos y frecuencia de comprobación de liberación de lotes de cada producto en procedimientos escritos, aprobados e implementados. En general, los ensayos de muestras es requerido para la liberación de lotes de los productos locales además de la revisión de los protocolos resumidos de producción de lotes.
Indicador 10.2: Sistema de Gestión de calidad para liberación de lotes
El sistema de gestión de calidad de la ARN/LOCM debe estar implementado para liberación de lotes. Documento de orientación de la OMS (como el anexo 8 de la Serie de Informes Técnicos, TRS 902 usado para inspectorías) o una norma internacional de Sistema de Gestión de Calidad podría usarse como una referencia. El SGC deberá ser evidenciado en todos los sub-indicadores de la función. Evidencia documental a ser evaluada:
Organigrama.
Directrices, la lista de los procedimientos para realizar las actividades de liberación de lotes.
Política y procedimientos para trazabilidad de actividades de liberación de lotes.
Informes de auditorías internas y externas.
Sub-indicador 10.2.1: Organigrama y responsabilidades definidas para implementar el Sistema de Gestión de
Calidad.
CRÍTICO
El evaluador debe identificar la organización asignada para establecer, implementar y mantener el SGC. Deben identificarse las diferentes responsabilidades.
Sub-indicador 10.2.2: Existe disponible documentación escrita para el desarrollo de actividades de liberación
de lotes.
CRÍTICO
El evaluador debe identificar la documentación escrita existente para realizar actividades de liberación de lotes incluidas lineamientos, procedimientos operativos estandarizados, listas de verificación y formatos de certificado de liberación de lotes.
Sub-indicador 10.2.3: Existe un Sistema de Gestión para garantizar trazabilidad de las actividades de
liberación de lotes.

113
CRÍTICO
El evaluador debe comprobar el SGC establecido asegura que las actividades de liberación de lotes están documentadas (muestreo y resultados de ensayos, datos de liberación de lotes, resultados fuera de-especificación). Los documentos deben archivarse en orden de disponer de trazabilidad, como en el caso de una apelación de una institución contra las decisiones, en el caso de los defectos de calidad detectados en los productos en el mercado, a fin de comparar resultados históricos y recientes, o a reconocer tendencias en los resultados con respecto a consistencia de calidad de los productos autorizados. El evaluador debe comprobar que los registros, los informes y los certificados de todas las actividades de liberación de lotes para todos los productos están disponibles y son almacenados por un tiempo definido.
Sub-indicador 10.2.4: Existe un Sistema de auditoria documentado e implementado (interno o externo).
CRÍTICO
El evaluador debe comprobar que un sistema de auditoría interna o externa se ha establecido y cubre todas las actividades de liberación de lotes.
Indicador 10.3: Gestión de Recursos Humanos
La Gerencia de la ARN/LOCM debe establecer el apoyo logístico necesario al personal designado para poder realizar las actividades de liberación de lotes eficientemente. Deben ser evaluados: el plan de desarrollo institucional (PDI), y los registros de capacitación del personal. Evidencia documental a ser evaluada:
Organigramas de la ARN/LOCM.
Procedimientos internos para reclutamiento, capacitación y calificación del personal y sus registros.
Lista del personal con su calificación.
Descripciones de puestos.
Curricula vitae.
Plan de capacitación.
Lista de las capacitaciones, entrenamientos desarrolladas.
Sub-indicador 10.3.1: Personal adecuadamente calificado (educación, entrenamiento, aptitudes y
experiencia), para desarrollar la liberación de lotes.
CRÍTICO
El evaluador debe identificar que los recursos humanos designados son adecuados y competentes (número y antecedentes de personal, organigramas, descripción del puesto, responsabilidades, nombramiento oficial), para poder realizar eficientemente la liberación de lotes. El evaluador debe examinar las competencias adecuadas del personal de laboratorio, si realizan ensayos o la competencia de los evaluadores de documentación y debe verificar en particular si tienen las siguientes competencias:
Líder o responsable, académicamente calificado.
Personal con educación y experiencia en técnicas de ensayos químicas, físicas y microbiológicas, según corresponda.
Conocimiento competente de los sistemas de gestión de calidad para laboratorios como ISO17025, según corresponda.
Pueden usarse como indicadores: el número de liberación de lotes basada en la evaluación de la documentación o pruebas de ensayo, pruebas realizadas y las técnicas usadas (titulación de virus contenido en vacunas víricas vivas, pruebas inmunológicas como determinación de antígenos víricos y bacterianos, pruebas microbiológicas, prueba usando tecnología de PCR etc.).

114
Sub-indicador 10.3.2: Plan de capacitación del personal desarrollado e implementado.
CRÍTICO
El evaluador debe revisar los planes de capacitación para el personal que esta realizando liberación de lotes basado en la revisión documental y verificar que la capacitación permite comprender e interpretar correctamente la documentación de datos analíticos del productor.
Sub-indicador 10.3.3: Existe monitoreo de las actitudes desarrolladas después de actividades de
entrenamiento.
CRÍTICO
El evaluador debe identificar si un sistema esta establecido para monitorear el desarrollo de aptitudes del personal para conducir liberación de lotes y pruebas de ensayo después que recibir capacitación.
Sub-indicador 10.3.4: Existe un número suficiente de personal para implementar el plan de trabajo.
CRÍTICO
El evaluador debe identificar si se ponen a disposición recursos humanos adecuados para ejecutar y realizar las actividades planificadas.
Indicador 10. 4: Gestión de procesos de liberación de lotes
Para conseguir resultados fiables y comparables el proceso de liberación lote tiene que administrarse de una manera uniforme. Evidencia documental a ser evaluada:
POE para revisar el protocolo resumido del lote.
Procedimientos para liberación de lotes incluidos criterios de aceptación.
Registros, informes y certificados de liberación de lote.
Provisiones para exención de liberación de lotes.
Sub-indicador 10.4.1: POE desarrollados y usados para la revisión del protocolo resumen de fabricación y
control del lote.
CRÍTICO
El evaluador debe identificar la existencia de POE escritos e implementados para examinar protocolos de liberación de lotes para cada producto.
Sub-indicador 10.4.2: Criterios definidos de aceptación para la liberación de lotes.
CRÍTICO
El evaluador debe identificar la existencia de criterios de aceptación escritos y hechos cumplir a lotes de liberación de productos específicos.
Sub-indicador 10.4.3: Registros de liberación de lotes, informes y certificados disponibles.
CRÍTICO
El evaluador debe comprobar que registros, informes y certificados de todas las actividades de liberación de lotes para todos los productos están disponibles y almacenado por tiempo definido.
Sub-indicador 10.4.4: Procedimientos para exención de liberación de lote.
CRÍTICO

115
El evaluador debe verificar si se han definido los criterios cuando los procedimientos corrientes para liberación de lotes no pueden seguirse. Las razones de la exención de la liberación de lotes podrían ser por ejemplo, una escasez de un producto en el mercado y la necesidad de importar un producto no autorizado por un tiempo definido.
Indicador 10.5: Acceso a la documentación relacionada de productos para guiar las áreas particulares del escrutinio en la liberación de lotes.
Realizar de manera confiable la liberación de lote conforme al trabajo del laboratorio requiere el cumplimiento de todos los parámetros específicos del producto. Intercambio de información entre la autoridad responsable de Registro y el LNC es obligatorio a fin de lograr el completo control de calidad de los productos en el mercado. Evidencia documental a ser estudiada:
Información sobre el producto contenida en el dossier de registro sanitario.
Sub-indicador 10.5.1: Componentes relevantes del dossier de Registro Sanitario aprobado están actualizadas
y disponibles para el personal involucrado en la liberación de lotes (Ej. Modificaciones al registro sanitario otorgado).
CRÍTICO
El evaluador debe comprobar si la autoridad responsable para liberación de lotes tiene acceso a la documentación de los productos. Es necesario contar con la documentación completa y actualizada, los archivos del registro deben disponibles y deben compartirse entre las autoridades responsables de la autorización y liberación de lotes.
Sub-indicador 10.5.2: Acceso al resumen de características del producto (RCP) e informes cuando sea
necesario (incluida inspección de BPM, datos de laboratorio, defectos de calidad, desempeño de vacuna y ESAVIs).
CRÍTICO
El evaluador debe comprobar si la autoridad responsable para liberación de lotes tiene acceso al RCP, la documentación de los productos y otra información relevante para demostrar la seguridad y calidad de productos incluidos informes de inspección, datos de laboratorio, información de fallas a la calidad, retiros y reportes de eventos adversos, etc.
Indicador 10. 6: Vigilancia y análisis de datos para liberación de lotes
Disponer de un mecanismo apropiado de vigilancia y análisis de datos es esencial para conseguir resultados consistentes e interpretación a fin de conducir las acciones adecuadas en caso de no conformidad. Evidencia documental a ser evaluada:
Tendencia de datos.
Revocación de las autorizaciones.
Retiros.
Inspecciones específicas de productos.
Sub-indicador 10.6.1: Análisis de la consistencia de lotes.
CRÍTICO

116
El evaluador debe comprobar que la consistencia de lotes se analiza adecuadamente y es documentada por la ARN/LOCM de forma regular.
Sub-indicador 10. 6.2: Medidas correctivas tomadas en el caso de desviación
CRÍTICO
El evaluador debe revisar que acciones correctivas apropiadas se toman en caso de desviaciones, tales como investigaciones específicas, inspecciones específicas de productos etc.
Sub-indicador 10.6.3: Medidas reglamentarias son tomadas en el caso de incumplimiento.
CRÍTICO
El evaluador debe examinar que acciones reglamentarias apropiadas se toman en caso de incumplimiento, como revocación de autorización, retiros, suspensión de importación, etc.
Sub-indicador 10.6.4: Seguimiento de la comunicación sobre datos de la calidad con los actores involucrados
incluyendo los productores.
NECESARIO
El evaluador debe comprobar que la ARN/LOCM dispone de un mecanismo adecuado para dar seguimiento y mantener la comunicación sobre datos de calidad con todas las partes involucradas tales como productores, importadores, distribuidores o los usuarios de los productos. Indicadores cuantitativos para las finalidades reglamentarias de liberación de lote:
Número del personal científico involucrado en actividades de liberación de lotes.
Número de lotes analizados y liberados en el año de referencia.
Número de lotes liberados en base a revisión de documentos.
Número de lotes importados en el año de referencia.
Número de lotes analizados (ensayos), durante actividades posteriores a la comercialización en el año de referencia.
Número de vacunas/lotes retirados en el año de referencia.

117
Anexo 1: Términos de Referencia para el equipo evaluador: Fecha y lugar: Propósito de la misión:
Revisar, en cooperación con los funcionarios de la (nombre de ARN) de (nombre del país), la situación reglamentaria de medicamentos y preparar un informe sobre los hallazgos de la evaluación.
Discutir el informe con los “interesados directos” pertinentes para lograr consenso en su contenido.
Desarrollar un plan de acción nacional en cooperación con los funcionarios de la (nombre de ARN) de (nombre del país).
Método de trabajo:
Entrevista con funcionarios de la (nombre de ARN) de (nombre del país) y otros actores relevantes.
Evaluar los documentos pertinentes, los informes, procedimientos, etc.
Reunirse y discutir con personas claves e “interesados directos”. Actividades:
Visita a la (nombre de ARN) de (nombre del país) y otras instituciones relevantes.
Entrevistas a directivos y equipo técnico usando la herramienta de recolección de datos OPS/OMS y otros documentos pertinentes.
Entrevista, si es necesario, a asociaciones empresariales farmacéuticas y asociaciones profesionales pertinentes como colegios de farmacia, médicos y asociaciones de la salud, etc.
Revisar los documentos, los informes del país y estudios ya existentes.
Escribir un proyecto de informe de evaluación que indique las brechas principales en la reglamentación de medicamentos, así como también las posibles recomendaciones para su mejoramiento.
Discutir el proyecto de informe de evaluación con el personal clave de la ARN y finalizar el reporte.
Elaborar un plan de acción nacional en cooperación con los funcionarios de (nombre de ARN) para mejorar la reglamentación de medicamentos en (nombre del país).
Resultados esperados:
Informe de evaluación del Sistema Regulador de Medicamentos
Herramienta completada de recopilación de datos de la OPS/OMS
Plan de acciones correctivas

118
Anexo 2: Procedimiento de selección de expertos
Para la selección de expertos que participan en las evaluaciones de las ARNs, en el marco de Autoridades Reguladoras de Medicamentos de Referencia de la Organización Panamericana de la Salud, se usa el Procedimiento ad-hoc.

119
Anexo 3: El ejemplo de compromiso de confidencialidad
CONVENIO DE CONFIDENCIALIDAD DE LOS MIEMBROS DE EQUIPO DE PARTICIPANTES QUE EVALUARAN EL SISTEMA NACIONAL DE REGULACIÓN DE MEDICAMENTOS Y PRODUCTOS BIOLÓGICOS. Por medio del presente declaro que mientras este visitando y evaluando los Estados Miembros de OPS como un asesor experto, usted tendrá acceso a cierta información perteneciente a las instituciones evaluadas. Tal información en adelante denominado "la Información" debe considerarse y manejarse como confidencial y es de propiedad de las partes mencionadas. En este sentido, estoy de acuerdo en: 1. No usar la Información para otras finalidades diferentes de aquellos solicitadas por esta misión; y 2. No revelar o suministrarle la Información a cualquier persona que no sea parte del equipo evaluador y
está unido mediante las obligaciones similares de la confidencialidad y la no utilización como figura en este documento.
Sin embargo, no estaré obligado por cualquier convenio de confidencialidad y la no utilización en la medida en que sea claramente capaz de demostrar esa parte de la Información: 1. Fue conocida por mí antes de participar en la evaluación o mas allá de las instituciones evaluadas; o 2. Fue de dominio público al momento de ser parte del equipo evaluador mas allá de las instituciones
evaluadas; o 3. Podría pasar a formar parte del dominio público mediante otras fuentes ajenas a mí. Además, me comprometo a no comunicar mis resultados o aquellos de los del equipo expertos con los cuales participaré, así como también las recomendaciones sugeridas o decisiones a cualquier tercero, salvo si explícitamente es solicitado por escrito. Confirmo que la información revelada por mí en la Declaración de los Intereses es correcta y que ninguna situación de conflicto de intereses real, potencial o evidente se conocida por mí. Me comprometo a advertir con prontitud de cualquier cambio en las circunstancias anteriores, incluido si un tema surge durante el curso de mi trabajo para OPS. Por la presente acepto y estoy de acuerdo con las condiciones y provisiones contenidas en este documento. Nombre ________________ Firma _____________________ Institución ________________Lugar__________________ Fecha_____________________________

120
Anexo 4: Ejemplo de una declaración de no conflicto de intereses para evaluadores
DECLARACIÓN DE NO CONFLICTOS DE INTERESES PARA LOS MIEMBROS DEL EQUIPO QUE PARTICIPA EN LA EVALUACIÓN DEL SISTEMA NACIONAL DE REGULACIÓN. Por medio del presente declaro que mientras este visitando y evaluando los Estados Miembros de OPS como un asesor experto usted tendrá acceso a cierta información perteneciente a las instituciones evaluadas. Tal información en adelante denominado "la Información" debe considerarse y manejarse como confidencial y es de propiedad de las partes mencionadas. En este sentido, declaro que:
Que yo cumpliré mis funciones exclusivamente como un asesor;
No tengo ninguna situación de conflicto de intereses real, potencial o evidente, incluyendo ningún interés financiero o de otro tipo en, y/u otra relación con un tercer, que:
o puede tener un interés comercial atribuido en obtener el acceso a cualquier información confidencial obtenida en el curso de la visita de evaluación, o
o puede tener un interés personal en el resultado de la visita de evaluación incluidos, pero no limitado a terceros tales como los fabricantes de medicamentos o vacunas.
Me comprometo a advertir con prontitud de cualquier cambio en las circunstancias anteriores, incluido si un tema surge durante el curso de mi trabajo para OPS. Por la presente acepto y estoy de acuerdo con condiciones y provisiones contenidas en este documento. Nombre ________________ _______ Firma ____________________ Institución_______________________ Lugar__________________ Fecha_____________________________

121
Anexo 5: Propuesta de Programa de Evaluación al Sistema Regulador
Horario Actividades Experto involucrado
Domingo
Llegada al país Equipo
20:00 hrs. Reunión, equipo de expertos, revisión general de la misión.
Equipo
Lunes
8:30 - 9:00 Apertura de la Misión OPS y ARN
9:00 - 9:30 Objetivos y alcances Líder del equipo
9:30 -10:15 Estructura, Misión, Visión y Objetivos de la ANR
Director ARN
Café
10:30 -13:00 Funciones del Organismo Regulador Directores y equipo
13:00 -14:00 Almuerzo
14:00 – 17:30 Funciones del Organismo Regulador Directores y equipo
19:00 – 20:00 Reunión de equipo evaluador Equipo Martes
8:30 – 17:30 Evaluación del Sistema Regulador y ARN Equipo 1
8:30 – 17:30 Evaluación de Registro y autorización de mercado
Equipo 2
8:30 – 17:30 Fiscalización y licenciamiento de productores Equipo 3
8:30 – 17:30 Laboratorio Nacional de Control Equipo 4
19:00 – 20:00 Reunión de equipo evaluador Equipo
Miércoles
8:30 – 17:30 Evaluación de Farmacovigilancia Equipo 1
8:30 – 17:30 Evaluación de Autorización de Ensayos Clínicos
Equipo 2
8:30 – 17:30 Visita a laboratorio productor Equipo 3
8:30 – 17:30 Vigilancia de Mercado Equipo 4
19:00 – 20:00 Reunión de equipo evaluador Equipo
Jueves
8:30 – 16:00 Informe y propuesta de plan de acción Equipo
16:00 – 17:00 Reunión con Directivos de la ARN Equipo y ARN
17:00 – 19:00 Preparar presentación Equipo
19:00 – 20:00 Reunión de equipo evaluador Equipo Viernes
9:00 – 11:00 Presentación de los resultados de la Evaluación
Equipo y ARN
11:00 – 13:00 Discusión del Informe con Directivos de la ARN
Equipo y ARN
13:00 – 14:00 Almuerzo
14:00 -15:30 Planificación de acciones correctivas Equipo
15:30 – 17:00 Finalización de Informe Equipo

122
Anexo 6: Lista de instituciones que se sugiere sean visitadas y miembros del personal de la ARN a ser entrevistados. Durante la visita de evaluación deben planificarse reuniones con los siguientes actores:
Ministerio de salud/Ministerio de industria,
Representantes de la autoridad reguladora y otras organizaciones involucradas en funciones de reglamentación,
Personal de autoridad reguladora u organización,
Representantes de la Asociación de Industria de fabricantes, distribuidores, importadores y exportadores,
Representantes de la Asociación Profesional de médicos generales, enfermeras y farmacéuticos,
Colegios de profesionales (colegio de médicos, colegio de farmacéuticos),
Representantes de asociaciones de consumidor,
Periodistas,
Asociaciones/organizaciones no gubernamentales, Organismos nacionales de medicamentos,
Organizaciones de investigación de salud,
Presidente o representantes de Comités consultivos
Presidente o representantes de IRB/CEI,
Representantes de asociaciones universitarias,
Otros.

123
Anexo 7: Ejemplo del contenido de un formato de informe:
Informe de evaluación de autoridades reguladoras de Medicamentos. Tabla de contenidos: Resumen Ejecutivo y recomendaciones principales: 1. Información general sobre la evaluación: 1.1. Propósito de la evaluación 1.2. El lugar y la fecha de la evaluación 1.3. El método de trabajo 1.4. Los miembros de la misión. 2. Información general sobre el país 3. Resultados y recomendaciones 3.1. Organización del Sistema Nacional de Regulación de Medicamentos. 3.2. La legislación y regulaciones. 3.3. La Autoridad Reguladora:
3.3.1 Recursos humanos. 3.3.2 Herramientas reglamentarias. 3.3.3 Infraestructura. 3.3.4 Transparencia y Confidencialidad. 3.3.5 Tecnología de la información.
3.4 Funciones Regulatorias (bases legales, estructura, procedimientos y resultados y recomendaciones):
3.4.1 Registro de productos. 3.4.2 Inspecciones y fiscalización. 3.4.3 Concesión de licencias de los productores. 3.4.4 Ensayos clínicos. 3.4.5 Vigilancia del Mercado. 3.4.6 Farmacovigilancia. 3.4.7 Laboratorio de Control de calidad.
4. Conclusiones y recomendaciones 5. Agradecimientos Anexos: 1. Herramienta de recopilación de datos OPS/OMS completada 2. Lista de Organizaciones visitadas 3. Lista de personas entrevistadas 4. Lista de documentos entregados 5. Organigrama de la ARN 6. Lista de personas que participaron en la discusión del reporte 7. Lista de personas que participaron en la preparación del plan de acción 8. Plan de acción

124
Anexo 8: Ejemplo de formato de Plan de acciones correctivas Fecha: País: Nombre de la ARN: Preparado por: (nombre de los evaluadores, nombre de los funcionarios de la ARN) Periodo cubierto:
Referencia a los hallazgos
Recomendaciones Objetivo a ser alcanzado
Principales tareas/actividades a ser desarrolladas
Plazo Persona responsable
Recursos necesarios

125
Anexo 9: Indicadores Cuantitativos de los procesos regulatorios.
Modulo 3: Autoridad Reguladora Nacional 3.1 Número total de empleados de la ARN (Ej. 2008) 3.2 Número de personal científico 3.3 Número de personal de apoyo 3.4 Número de personal contratado en sistemas de informática. 3.5 Número de personal dedicado a evaluación de calidad. 3.6 Número de personal reclutado en el año de referencia (Ej. 2008). 3.7 Número de personal cuyos contratos concluyen comparado con número de personal contratado en un
período definido. Modulo 4: Autorización de Mercado-Registro. 4.1 Número de personal científico involucrado 4.2 Número de productos con Registro vigente 4.3 Número de solicitudes recibidas en un año de referencia ( Ej. 2008) 4.4 Número de solicitudes recibidas de un medicamento nuevo 4.5 Número de solicitudes recibidas de un producto genérico 4.6 Número de solicitudes recibidas de modificaciones 4.7 Número de solicitudes recibidas de renovaciones 4.8 Número de decisiones tomadas (aprobaciones, denegaciones, suspensiones) en un año de referencia. 4.9 Número de solicitudes pendientes. 4.10 Número promedio de días tomados por la ARN para la toma de decisiones en un medicamento nuevo. 4.11 Número promedio de días tomados por la ARN para la toma de decisiones en un producto genérico. 4.12 Número promedio de días tomados por la ARN para la toma de decisiones de modificaciones. 4.13 Número promedio de días tomados por la ARN para la toma de decisiones en las renovaciones. Modulo 5: Licenciamiento de productores. 5.1 Número de personal científico involucrado en concesión de licencias de establecimiento 5.2 Número de plantas de fabricación licenciadas. 5.3 Número de plantas de fabricación de `principios activos licenciadas 5.4 Número de solicitudes recibidas para nuevas instalaciones en el año de referencia (Ej. 2008) 5.5 Número de modificaciones de una licencia inicial recibido en el año de referencia 5.6 Número de decisiones adoptadas (positivo, negativo, suspensión o retiros) en el año de referencia 5.7 Número de solicitudes pendientes. 5.8 Número promedio de días para la toma de decisión. Modulo 6: Vigilancia del Mercado. Control de importación y exportación 6.1 Número de solicitudes recibidas (importación y exportación) 6.2 Número de autorizaciones para importación concedida en el año de referencia o número de productos
autorizados para importación. 6.3 Número de certificados para exportación expedido en el año de referencia 6.4 Número promedio de días para expedir estos documentos administrativos. Control del Mercado 6.5 Número de productos monitoreados 6.6 Número de productos detectados no conforme o de la calidad deficiente en el año de referencia. Productos deficientes/Procedimientos de Retiro 6.7 Número de quejas de los medicamentos recibido en el año de referencia.

126
6.8 Número de productos/lotes retirados en el año de referencia Modulo 7: Farmacovigilancia 7.1 Número de profesionales de ARN involucrados en la evaluación y manejo de RAM. 7.2 Número de puntos de contacto (jurídica o persona natural que han notificado una RAM) 7.3 Número de RAM informadas por Titulares de registro, productores, importadores o distribuidores y
evaluados en el año de referencia 7.4 Número de RAM informadas por profesionales de la salud y evaluados en el año de referencia 7.5 Número de RAM informadas por pacientes y evaluadas en el año de referencia 7.6 Número de informe periódicos recibidos y evaluados en el año de referencia. 7.7 Número de decisiones adoptadas (retiro de productos, cartas a los médicos, avisos para usuarios,
alertas, etc.) en el año de referencia 7.8 Número de RAM pendiente-atraso en la evaluación. 7.9 Número promedio de días para toma de decisiones en temas de farmacovigilancia. Módulo 8: Ensayos Clínicos 8.1 Número de personal involucrado en el control de ensayos clínicos 8.2 Número de solicitudes de ensayos clínicos recibido en el año de referencia 8.3 Número de solicitudes para enmiendas de ensayos clínicos recibido en el año de referencia 8.4 Número de decisiones adoptadas (aprobaciones, denegaciones, suspensiones) en solicitudes de
estudios clínicos en el año de referencia. 8.5 Número promedio de días para toma de decisiones de la ARN 8.6 Número promedio de días para toma de decisiones del CEI 8.7 Número de personal involucrado en la inspección de ensayos clínicos 8.8 Número de ensayos clínicos inspeccionados en el año de referencia. 8.9 Número promedio de días en el sitio de inspección. Modulo 9: Inspección y actividades de fiscalización. 9.1 Número total de inspectores para el sector de medicamentos. 9.2 Número total de inspecciones llevadas a cabo en el año de referencia 9.3 Número de inspectores de fabricación 9.4 Número de establecimientos de producción inspeccionados incluidos fabricantes extranjeros en el año
de referencia. 9.4.1 Número de establecimientos de producción visitados para inspección para aprobaciones previas a la autorización de comercialización.
9.5 Número de establecimientos de producción de principios activos inspeccionados en el año de referencia 9.6 Número de establecimientos de producción de principios activos visitados para inspección para
aprobaciones previas a la autorización de comercialización. 9.7 Número promedio de días en el sitio de inspección 9.8 Número de inspectores para importadores, exportadores, mayoristas y distribuidores. 9.9 Número de establecimientos de intermediarios/importación/exportación inspeccionados en el año de
referencia. 9.10 Número de establecimientos de intermediarios/importación/exportación de principios activos
inspeccionados en el año de referencia. 9.11 Número de medidas administrativas (aviso de incumplimiento o notas de advertencia) expedidas en
cada uno de los tres últimos años. 9.12 Número promedio de días para tomar una medida administrativa como incumplimiento o nota de
advertencia. 9.13 Número de licencias retiradas o suspendidas en cada uno de los tres últimos años por temas de
incumplimiento. 9.14 Número de acciones penales presentadas a tribunal o sanciones penales solicitadas en cada uno de los
tres últimos años.

127
9.15 Número de sanciones legales impuestas por el poder judicial en cada uno de los tres últimos años. Modulo 10: Control de Calidad 10.1 Número de medicamentos analizados en el marco de una solicitud de Registro en el año de referencia 10.2 Número de ingredientes farmacéuticos activos analizados en el marco de una solicitud de registro en el
año de referencia. 10.3 Número de medicamentos analizados para control de importaciones en el año de referencia. 10.4 Número de medicamentos analizados para control del mercado en el año de referencia. 10.5 Número de medicamentos analizados/certificados emitidos en el año de referencia 10.6 Número de medicamentos analizados y encontrados no conforme 10.7 Número de medicamentos retirados basados en los resultados expedido por el LOCM en el año de
referencia. 10.8 Número de Registros suspendidos o retirados basado en los resultados expedido por LOCM en el año
de referencia 10.9 Número de personal científico en el laboratorio de control de calidad. 10.10 Superficie del laboratorio de control de calidad 10.11 Número de análisis no realizados debido a falta de equipo adecuado

128
Anexo 10. Fuente principal de evidencia documental
Sistema Regulador. Lista de Códigos, ley, decreto o la circular que establece el Sistema Regulador. Autoridad Reguladora:
Código, Reglamento, ley, decreto o circular estableciendo la autoridad reguladora.
Plan institucional, estratégico y gerencial de la ARN.
Misión, la visión, objetivos y los indicadores del ARN.
Manual de calidad.
Lista de los procedimientos internos.
Lista de formularios y plantillas internas.
Lista de los aranceles aplicables para licenciamiento o autorización de registro.
Organigrama.
Código de conducta/código deontológico.
Lista de personal con sus calificaciones.
Lista de los expertos externos.
Informe anual, informe de autoevaluación. Funciones Reguladoras: Registro, concesión de licencias de fabricantes, inspección, control de ensayos clínicos, farmacovigilancia, control de mercados, laboratorio de control de calidad:
Disposiciones legales, ley, el decreto o circular para cada una de las funciones de regulación.
Orientación publicada en este del campo.
Procedimientos internos.
Las listas del equipo. NOTA: Una solicitud de documentación debe enviársele con antelación a la organización siendo visitada para prepararse para la evaluación. La solicitud debe ser compatible con finalidad y alcance de la evaluación.