mammary cancer promotion by ovarian hormones involves igfr/akt/mtor signaling
TRANSCRIPT

Steroids 77 (2012) 791–797
Contents lists available at SciVerse ScienceDirect
Steroids
journal homepage: www.elsevier .com/locate /s teroids
Mammary cancer promotion by ovarian hormones involvesIGFR/AKT/mTOR signaling
Arunkumar Arumugam, Jacqueline Parada, Lakshmanaswamy Rajkumar ⇑Center of Excellence in Cancer Research, Department of Biomedical Sciences, Paul L. Foster School of Medicine, Texas Tech University Health Sciences Center,El Paso, TX 79905, United States
a r t i c l e i n f o a b s t r a c t
Article history:Received 27 December 2011Received in revised form 23 February 2012Accepted 16 March 2012Available online 29 March 2012
Keywords:EstrogenProgesteroneMammary lesionsMammary tumormTORIGFR
0039-128X/$ - see front matter � 2012 Elsevier Inc. Ahttp://dx.doi.org/10.1016/j.steroids.2012.03.009
Abbreviations: MNU, N-methyl-N-nitrosourea; ECOP, Copenhagen; IGFR, Insulin like growth factor rereceptor binding protein 2; SHC1, Src homolog and coof sevenless homolog 1; AKT, v-akt murine thymomERK1/2, Extracellular signal-regulated kinase 1/2; mrapamycin; p70S6K, ribosomal protein S6 kinase; 4initiation factor 4E binding protein 1; BCL2, B-cellBCL2-associated X protein; CASP 3, Caspase 3; CASP 8⇑ Corresponding author. Tel.: +1 915 783 5235; fax
E-mail address: rajkumar.lakshmanaswamy@ttuhs
In a previous study, we observed that N-methyl-N-nitrosourea (MNU)-induced mammary lesions are pro-moted to overt mammary cancers by exogenous administration of estradiol (E) and progesterone (P). Thepurpose of the present study was to identify the early molecular events occurring during the hormonalpromotion of mammary carcinogenesis and persistent activation of molecular pathways responsible fortumor growth. Seven-week-old female Copenhagen (COP) rats, which are resistant to MNU-inducedmammary carcinogenesis, were intraperitoneally administered a single dose of MNU (50 mg/kg bodyweight). Six weeks after carcinogen administration, the rats were treated with E + P, killed at 15th weekand 43rd week to obtain mammary lesions and tumor tissues and the molecular analysis were per-formed. Quantitative RT–PCR experiments showed increased mRNA expression of Igfr, Grb2, Sos1, andShc1 in mammary lesions and tumors. Immunoblot data also showed increased protein levels of IGFR,GRB2 and SHC1 in mammary lesions and tumors, which is in correlation with their respective RT–PCRdata. Activation of AKT and ERK1/2 were up regulated in E + P treated mammary lesions and tumors.Molecular analysis of mTOR pathway proteins revealed increased phosphorylation of p70S6K and4EBP1 in the hormone treated tumors indicating the activation of mTOR signaling. E + P treatmentreduced the protein expression of BAX and increased BCL2 expression along with down regulation ofactive caspase 3 and 8. Together, these data demonstrate that ovarian hormones promote the lesionsto mammary tumors by enhancing IGFR and Akt/mTOR signaling along with inhibition of apoptoticstimuli.
� 2012 Elsevier Inc. All rights reserved.
1. Introduction [4]. Hormone replacement therapy in postmenopausal women
The ovarian hormones estradiol (E) and progesterone (P) playan important role in the development of normal and neoplasticmammary glands [1]. Neoplastic growth of the breast is the mostcommon malignancy in women [2]. The risk of breast cancer ismainly influenced by age, genetics, reproductive history, exposureto long-term hormone treatment, radiation, socioeconomic status,and ethnicity [3]. The Women’s Health Initiative study has shownan increased risk of breast cancer in postmenopausal womenreceiving estrogen plus progestin hormone replacement therapy
ll rights reserved.
, estradiol; P, progesterone;ceptor; GRB2, Growth factorllagen homolog 1; SOS1, Son
a viral oncogene homolog 1;TOR, mammalian target of
EBP1, eukaryotic translationleukemia/lymphoma 2; BAX,, Caspase 8.
: +1 915 783 5222.c.edu (L. Rajkumar).
has also been shown to increase epithelial cell proliferation anddouble the incidence of lobular breast cancer [5,6]. These dataclearly illustrate the importance of E and P in increasing the riskof breast cancer and emphasize the significance of identifying theunderlying molecular changes induced by these hormones.
Experimental animal models for breast cancer are of greatimportance when studying its pathology and treatment strategies.Rat mammary cancers closely reflect human breast cancer at thephysiological, cellular, and molecular levels [7–9]. Copenhagen(COP) rats are extremely resistant to chemical and hormonalcarcinogenesis [7,10,11]. Korkola and Archer [11] reported thatadministration of a carcinogen causes preneoplastic lesions in themammary epithelial cells in both susceptible Wistar Furth andresistant COP rats. In COP rats, mammary lesions disappeared after50 days, but in Wistar Furth rats (the susceptible strain), they pro-gressed to palpable mammary tumors. Genetic analysis of the F1
generation of COP �Wistar Furth or Fisher F344 (another suscep-tible strain) crosses revealed three breast cancer-resistant locitermed mammary carcinoma suppressor (Mcs) 1, Mcs 2, and Mcs3 [7,12,13].

792 A. Arumugam et al. / Steroids 77 (2012) 791–797
In our previous study, we demonstrated that administration of acarcinogen followed by long-term treatment with ovarian hor-mones can nullify the genetic resistance of COP rats [14]. Palpablemammary tumors were induced in COP rats by a single intraperi-toneal injection of N-methyl-N-nitrosourea (MNU) followed bylong-term E and P administration. Long-term hormone treatmentpromoted the growth of mammary lesions induced by the carcin-ogen to overt mammary cancers [14]. Identification of thepersistent molecular changes responsible for the promotion ofmammary lesions to overt mammary tumors may enrich ourknowledge of the role of ovarian hormones in the promotion ofbreast cancer. The pathways which are required to promote thetumor growth and identification of potential targets that are per-sistently activated during the tumor promotion and progressionwill have vast therapeutic value. So, the present study was aimedto identify the molecular targets that play a vital role in the ovarianhormone induced breast cancer promotion in genetically resistantCOP rats.
2. Experimental
2.1. Animals
Five-week-old virgin COP rats were purchased from HarlanSprague Dawley (Indianapolis, IN and San Diego, CA). The rats werehoused in a temperature-controlled room with a 12-h light and12-h dark cycle. They were provided food (Teklad, Madison, WI)and water ad libitum. All procedures followed the Animal Careand Use Committee guidelines of Texas Tech University HealthSciences Center.
2.2. Carcinogen and hormone treatments
A single intraperitoneal injection of MNU (Sigma, St. Louis, MO)at a dose of 50 mg/kg body weight was administered to seven-week-old female COP rats. MNU was dissolved in physiologicalsaline that had been adjusted to pH 5.0 [15].
Six weeks after administration of the carcinogen, the rats weredivided into the following groups (n = 30): (i) Control, (ii) E + P(30 mg each) (Fig. 1). Both hormones were purchased from Sigma(St. Louis, MO); they were packed in individual silastic capsules(size 0.078 in. ID � 0.125 in. OD, 2 cm in length; Dow Corning, Mid-land, MI). Control animals received empty silastic capsules. Allsilastic capsules were implanted dorsally and subcutaneously,and were primed before implantation by soaking in Medium 199(GIBCO-Invitrogen, Carlsbad, CA) overnight at 37 �C [15]. Eachtreatment was continued for 9 months. Silastic capsules were re-placed every 2 months.
2.3. Mammary tumorigenesis
Two weeks after hormone implantation, four rats from eachgroup were euthanized and the mammary lesions were identifiedby characteristic rich vascularization under a high-power dissect-ing microscope. The mammary lesions were then excised and apiece was fixed in 10% neutral buffered formalin for paraffin sec-tioning; the remaining sample was snap-frozen in liquid nitrogenfor molecular analysis. Mammary tumorigenesis in the remainingrats from each group was monitored by weekly palpation. The pal-pable mammary tumors were measured using a caliper. The twolargest measurements of the tumor were recorded once everyweek. The tumor volume was calculated using the formula4=3p � r2
1 � r2 where r1 is the minor radius and r2 is the major radius.Mammary tumors were surgically excised before they reached2.0 cm in diameter and processed similarly like the lesions asdescribed above.
2.4. RNA isolation, PI3K/AKT pathway focused PCR microarray and realtime RT–PCR
In brief, rat mammary lesions and tumor samples werehomogenized in Trizol reagent (Invitrogen, Carlsbad, CA). TotalRNA was isolated and treated using the Turbo DNA free kit(Ambion, Austin, TX) to remove any genomic DNA contamina-tion, as per the recommended protocol. The concentration andquality of RNA was determined using the NanoDrop 2000 spec-trophotometer (Thermo Scientific, Wilmington, DE). Initially,PI3K/AKT specific pathway focused microarray (SABiosciences,Frederick, MD) was performed using 1 lg of total RNA fromthe mammary lesions. The genes most differentially regulatedwere validated using real time RT–PCR. Real-time RT–PCR wasperformed with 200 ng of total RNA from rat mammary lesionsas well as from mammary tumors, using the QuantiTect ReverseTranscription Kit (Qiagen, Valencia, CA) according to the manu-facturer’s protocol. The single-step RT–PCR was run on the Ste-pone Plus Real Time PCR system (Applied Biosystems, FosterCity, CA). The real-time RT–PCR was run as follows: one cycleat 50 �C for 30 min, one cycle at 95 �C for 15 min, and 40 cyclesof denaturing at 95 �C for 15 s, 45 s at the specificannealing temperatures, followed by elongation at 72 �C for1 min. A dissociation protocol run was conducted to testthe melting temperature of the product. The DDCT method ofrelative quantification was used to determine the foldchange in gene expression after normalization with 18s RNAexpression.
PrimersIgfr forward: CTGGTCTCTCATCTTGGATGC, reverse: TCCAGCAG-
CGGTAGTTGTACT, Shc1 forward: GCGACGACGAGGAGAAAGTC, re-verse: TGATCGGCACTCCAGCAAA,
Grb2 forward: GGCTTCATCCCCAAGAATTACA, reverse: ATCAGG-AAGGCCCCGTCAT,
Sos1 forward: CAGAAGAAAGACAGTATCTACGGGAACT, reverse:CTTTTCAATTTCAGAAG ACTTGAACAA and 18s forward: GGGAGG-TAGTGACGAAAAATAACAAT, reverse: TTGCCCT CCAATGGATCCT.
2.5. Western blotting
Mammary lesions and tumor samples were homogenized withradioimmunoassay precipitation buffer containing protease andphosphatase inhibitors (Roche Diagnostics, Indianapolis, IN). Thehomogenized lysates were centrifuged at 10,000g for 20 min at4 �C, and the supernatant fraction was collected. Protein concen-trations were determined using the BCA protein assay kit (Pierce,Rockford, IL). Proteins were resolved on Mini-Protean TGX gels(Bio-Rad, Hercules, CA) and transferred to Immobilon-P polyvinyl-idene fluoride membranes (Millipore, Bedford, MA). Antibodies forIGFR, Shc1, Grb2, BAX, BCL2, p70S6K, p4EBP1 total and phosphor-ylated AKT and ERK1/2 were obtained from Cell Signaling Technol-ogy, Boston, MA. CASP 8 and CASP 3 antibodies were purchasedfrom Abcam Inc., Cambridge, MA. After probing, the signal wasdeveloped using SuperSignal West Pico Chemiluminescent sub-strate detection solution (Pierce, Rockford, IL), and the membraneswere stripped and reprobed with b-actin antibody (Sigma, St.Louis, MO).
2.6. Statistical analysis
The data are expressed as mean ± SEM. The Mann–Whitneytest or Student’s t test was used to analyze differences betweenthe control and experimental groups using the GraphPad Prism5 software package version 5.03. P < 0.05 was consideredsignificant.

Fig. 1. (a) Seven-week-old COP rats were administered MNU (50 mg/kgb.w.). Six weeks post-carcinogen treatment empty silastic tubes or silastic tubes containing estradiol(30 mg) and progesterone (30 mg) were implanted subcutaneously. Two weeks after hormone implantation, four rats from each group were euthanized and the mammarylesions were excised. Treatments were continued for nine months and incidences of palpable tumors were recorded every week. The palpable mammary tumors weresurgically excised as needed. At the end of experimental period the rats were euthanized and the mammary glands and other tissues were removed and part of the tissueswere fixed in the 10% neutral buffered formalin and remaining were snap-frozen in the liquid nitrogen and stored in the �80 �C for molecular analysis. (b) Exogenous long-term treatment with E + P post-carcinogen treatment induced mammary tumors in 90% of rats. (c) Administration of hormones post-carcinogen treatment effectivelyincreased the growth of mammary tumors and also resulted in larger mammary tumors compared to the controls. Hormone treatment also decreased the latency period formammary tumor development. (d) Multiplicity of the tumors was also high in the E + P treatment group with average of 2.5 tumors per rat when compared to control ratshaving 0.2 tumor per rat. (e) Representative mammary lesions from control and E + P treated groups. E + P treated rats had larger and more mammary lesions, which isevident in the H and E staining. (f) Histology of mammary tumors from control and E + P treated animals. Histologically no significant difference was observed between themammary tumors from both the groups.
A. Arumugam et al. / Steroids 77 (2012) 791–797 793
3. Results
3.1. Effects of long-term exposure to estradiol plus progesterone onmammary tumorigenesis
It is well known that COP rats are resistant to mammary carci-nogenesis but develop mammary lesions on administration of car-cinogen but these lesions fail to progress further and disappearwithin 50 days [11]. In our previous study we reported that E + Ppromoted the growth of mammary lesions formed after MNUadministration and resulted in palpable overt mammary tumors[14]. Based on our data and others, our working hypothesis wasthat exogenous hormone treatment sustained the initial molecularalterations in the mammary lesions leading to the development ofpalpable overt mammary tumors. Hence, in our current investiga-tion, we studied the early molecular alterations and examined ifexogenous hormone treatment persistently sustained these altera-tions in the overt mammary tumors. To identify the early and per-sistent changes, we designed the experiments as detailed in Fig. 1a.Briefly, seven-week-old COP rats were treated with MNU. Sixweeks after MNU administration the rats were continuously
exposed to hormones E and P for 9 months. To identify the earlychanges, a set of rats were killed 2 weeks after the initiation of hor-mone treatments. Data on tumor incidence and tumor growth inthis experiment are in line with our previous studies, directly indi-cating the promotional effect of hormones on MNU-induced mam-mary lesions. A remarkable increase in the incidence of overtmammary tumor development was observed in E + P treatment(90%) than carcinogen alone treatment (10%) demonstrating theability of combined E + P treatment on tumor development(Fig. 1b). E + P treatment resulted in larger (730 ± 31 mm3) mam-mary tumors compared to controls (200 ± 26 mm3) (Fig. 1c). Overtmammary tumors were detected as early as 14 weeks after pelletimplantation in E + P group which shows that the latency periodfor the development of tumor was much lower in E + P treated ratsthan control (latency period of 26 weeks) (Fig. 1c). COP rats treatedwith E + P after carcinogen treatment developed more number oftumors per rat (average of 2.5) than compared to carcinogen alonetreatment group (average of 0.2) (Fig. 1d). Histopathological exam-ination revealed increased number of mammary lesions and com-paratively large sized lesions in E + P treated animals (Fig. 1e).Though the tumors induced by E + P treatment were larger in size
794 A. Arumugam et al. / Steroids 77 (2012) 791–797
and rapidly growing, there were no histological differences foundin the mammary tumors as well as mammary lesions of controland E + P treated rats (Fig. 1f).
3.2. Long-term estradiol plus progesterone treatment induced thegrowth of mammary lesions by inducing IGFR/AKT/MAPK activation
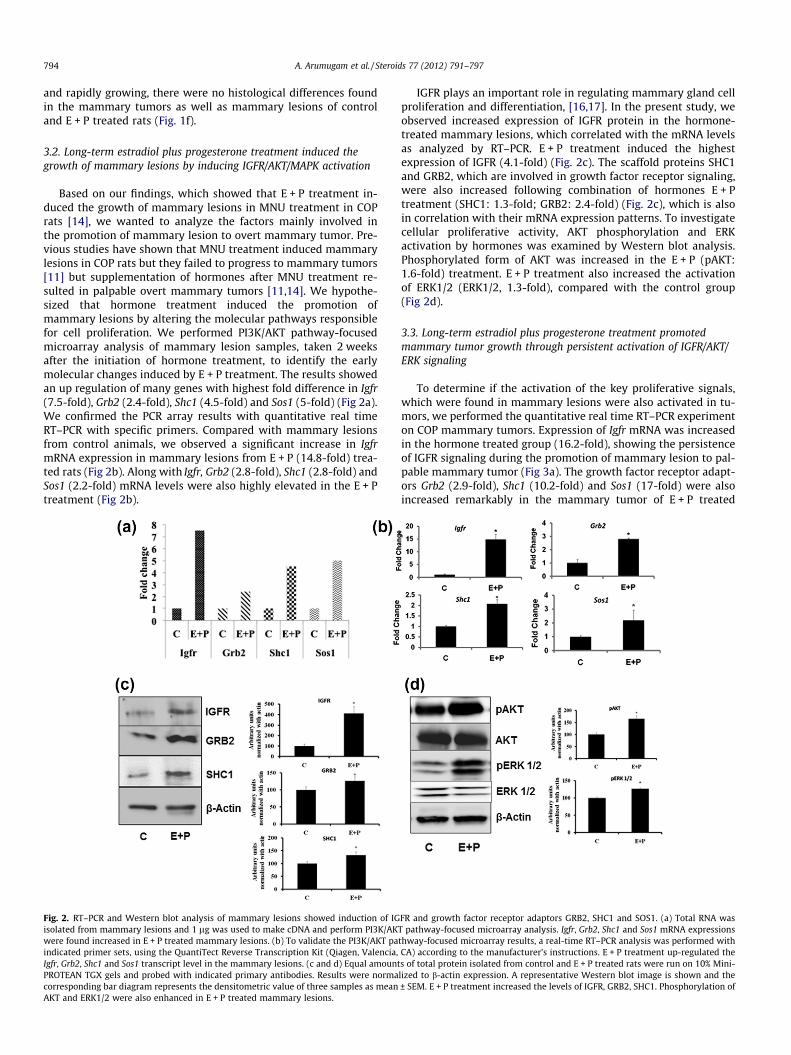
Based on our findings, which showed that E + P treatment in-duced the growth of mammary lesions in MNU treatment in COPrats [14], we wanted to analyze the factors mainly involved inthe promotion of mammary lesion to overt mammary tumor. Pre-vious studies have shown that MNU treatment induced mammarylesions in COP rats but they failed to progress to mammary tumors[11] but supplementation of hormones after MNU treatment re-sulted in palpable overt mammary tumors [11,14]. We hypothe-sized that hormone treatment induced the promotion ofmammary lesions by altering the molecular pathways responsiblefor cell proliferation. We performed PI3K/AKT pathway-focusedmicroarray analysis of mammary lesion samples, taken 2 weeksafter the initiation of hormone treatment, to identify the earlymolecular changes induced by E + P treatment. The results showedan up regulation of many genes with highest fold difference in Igfr(7.5-fold), Grb2 (2.4-fold), Shc1 (4.5-fold) and Sos1 (5-fold) (Fig 2a).We confirmed the PCR array results with quantitative real timeRT–PCR with specific primers. Compared with mammary lesionsfrom control animals, we observed a significant increase in IgfrmRNA expression in mammary lesions from E + P (14.8-fold) trea-ted rats (Fig 2b). Along with Igfr, Grb2 (2.8-fold), Shc1 (2.8-fold) andSos1 (2.2-fold) mRNA levels were also highly elevated in the E + Ptreatment (Fig 2b).
Fig. 2. RT–PCR and Western blot analysis of mammary lesions showed induction of IGisolated from mammary lesions and 1 lg was used to make cDNA and perform PI3K/AKwere found increased in E + P treated mammary lesions. (b) To validate the PI3K/AKT paindicated primer sets, using the QuantiTect Reverse Transcription Kit (Qiagen, Valencia,Igfr, Grb2, Shc1 and Sos1 transcript level in the mammary lesions. (c and d) Equal amountPROTEAN TGX gels and probed with indicated primary antibodies. Results were normacorresponding bar diagram represents the densitometric value of three samples as meanAKT and ERK1/2 were also enhanced in E + P treated mammary lesions.
IGFR plays an important role in regulating mammary gland cellproliferation and differentiation, [16,17]. In the present study, weobserved increased expression of IGFR protein in the hormone-treated mammary lesions, which correlated with the mRNA levelsas analyzed by RT–PCR. E + P treatment induced the highestexpression of IGFR (4.1-fold) (Fig. 2c). The scaffold proteins SHC1and GRB2, which are involved in growth factor receptor signaling,were also increased following combination of hormones E + Ptreatment (SHC1: 1.3-fold; GRB2: 2.4-fold) (Fig. 2c), which is alsoin correlation with their mRNA expression patterns. To investigatecellular proliferative activity, AKT phosphorylation and ERKactivation by hormones was examined by Western blot analysis.Phosphorylated form of AKT was increased in the E + P (pAKT:1.6-fold) treatment. E + P treatment also increased the activationof ERK1/2 (ERK1/2, 1.3-fold), compared with the control group(Fig 2d).
3.3. Long-term estradiol plus progesterone treatment promotedmammary tumor growth through persistent activation of IGFR/AKT/ERK signaling
To determine if the activation of the key proliferative signals,which were found in mammary lesions were also activated in tu-mors, we performed the quantitative real time RT–PCR experimenton COP mammary tumors. Expression of Igfr mRNA was increasedin the hormone treated group (16.2-fold), showing the persistenceof IGFR signaling during the promotion of mammary lesion to pal-pable mammary tumor (Fig 3a). The growth factor receptor adapt-ors Grb2 (2.9-fold), Shc1 (10.2-fold) and Sos1 (17-fold) were alsoincreased remarkably in the mammary tumor of E + P treated
FR and growth factor receptor adaptors GRB2, SHC1 and SOS1. (a) Total RNA wasT pathway-focused microarray analysis. Igfr, Grb2, Shc1 and Sos1 mRNA expressionsthway-focused microarray results, a real-time RT–PCR analysis was performed withCA) according to the manufacturer’s instructions. E + P treatment up-regulated thes of total protein isolated from control and E + P treated rats were run on 10% Mini-lized to b-actin expression. A representative Western blot image is shown and the± SEM. E + P treatment increased the levels of IGFR, GRB2, SHC1. Phosphorylation of

A. Arumugam et al. / Steroids 77 (2012) 791–797 795
animals (Fig 3a). Immunoblot analysis of mammary tumor samplesrevealed that IGFR levels were increased by 4.3-fold in the E + Ptreated animals (Fig 3b). GRB 2 protein levels were up regulatedby 2.9-fold in the E + P combination treatment, similarly exposureto E + P up regulated SHC1 protein levels by 3.4-fold when com-pared to control mammary tumors, demonstrating the activationof IGFR pathway by E and P right from the beginning of mammarytumorigenesis (Fig 3b). To analyze the effect of hormones on AKT/ERK activation we analyzed the total and active forms of AKT andERK1/2 in the tumor tissues of E + P treatment. Phosphorylationof AKT and ERK1/2 were highly up regulated in E + P treated mam-mary tumors emphasizing the proliferative effect of hormonesresulting in a tumor promotional environment (Fig 3c).
3.4. Hormone treatment induced mTOR activation and inhibitedapoptosis
Since the growth of mammary tumor and activation of AKT washigh in E + P treatment, we investigated the downstream pathwaysregulating tumor cell growth and cell death. Our data shows thatactivation of mTOR was remarkably up regulated by E + P (2.2-fold)treatment (Fig 4a). Immunoblotting data revealed that phosphory-lated (activated) p70S6K and p4EBP1 downstream targets of mTORand p70S6K respectively, were increased in the mammary tumorsof hormone treated rats (p-p70S6K in E + P 5.4-fold; p4EBP1
Fig. 3. Mammary tumors induced by hormones E + P treatment showed persistent exprRNA was isolated from mammary tumors and 200 ng was used to perform Real-time R(Qiagen, Valencia, CA) according to the manufacturer’s instructions. E + P treatment up-amounts of total protein isolated from control and E + P treated rats were run on 10% Mnormalized to b-actin expression. A representative Western blot image was shown and thmean ± SEM. (b) Expression of IGFR, GRB2, and SHC1 proteins was increased in E + P treatactivated in the hormone treated mammary tumors when compared to carcinogen alon
6.2-fold) showing the presence of active mTOR signaling in thesetumors (Fig 4a). Next, we investigated the effect of E + P treatmenton the cellular balance of pro and antiapoptotic signals. We foundthat BAX was notably down regulated (to 0.56-fold) and BCL2 lev-els were increased (3.6-fold) in mammary tumors of E + P treatedrats (Fig 4b). Analysis of effectors of apoptosis caspase 3 and 8showed a reduction in the levels of active caspase 3 (to 0.72-fold)and 8 (to 0.69-fold) in E + P treated mammary tumors when com-pared to control tumors (Fig 4b). Taken together, it is clear thatE + P treatment inhibits apoptosis by affecting BCL2 family proteinsand caspases.
4. Discussion
Tumor promotion and progression requires persistent changes inthe tumor microenvironment to facilitate transformed cell prolifer-ation. We recently demonstrated the significance of hormonal pro-motion of mammary carcinogenesis by long-term ovarian hormoneadministration in MNU-treated COP rats [14]. Hormone treatmentresulted in a high incidence of overt, palpable mammary tumors to-gether with a reduced latency period in COP rats, which signifies thepromotional role of hormones in mammary carcinogenesis [14]. Inthe present study, we attempted to identify the early molecularevents occurring during hormonal promotion of mammary lesionsto palpable mammary tumors. We also investigated the persistence
ession of IGFR and growth factor receptor adaptors GRB2, SHC1 and SOS1. (a) TotalT–PCR with indicated primer sets, using the QuantiTect Reverse Transcription Kit
regulated the Igfr, Grb2, Shc1 and Sos1 transcript levels compared to controls. Equalini-PROTEAN TGX gels and probed with indicated primary antibodies. Results weree corresponding bar diagram represents the densitometric value of three samples asment, in correlation with RT–PCR data. (c) AKT and downstream MAPK, ERK1/2 weree treatment.

Fig. 4. Activation of mTOR pathway and anti-apoptotic pathway was evident in the mammary tumors induced by hormone treatments. Equal amounts of total proteinisolated from control and E + P treated rats were run on 10% Mini-PROTEAN TGX gels and probed with indicated primary antibodies. Results were normalized to b-actinexpression. A representative Western blot image was shown and the corresponding bar diagram represents the densitometric value of 3 samples as mean ± SEM. (a)Phosphorylation of mTOR and its downstream target protein, p70S6K and 4E-BP1 were elevated in the hormone treatments. (b) E + P increased the expression of BCL2 andreduced the levels of BAX. E + P treatment also down regulated the active forms of caspases 8 and 3.
796 A. Arumugam et al. / Steroids 77 (2012) 791–797
of the proliferative signals involved in promotion and progression ofmammary tumors. The results demonstrate that the ovarian hor-mones E and P, have a significant influence on the early tumormicroenvironment, and converge the proliferative and anti-apopto-tic signaling pathways to promote the tumor growth.
The tumor microenvironment consists of epithelium and thesurrounding stromal compartment, including adipocytes, extracel-lular matrix, endothelial cells and a few neuronal cells [18]. Astrong epithelial–stromal interaction occurs during the process ofmammary gland development and tumorigenesis [18]. Growth fac-tors produced in the mammary stromal compartment interact withtheir receptors on the epithelial cell surface to promote cell prolif-eration [18]. Increase in the levels of IGFR in both mammary le-sions and mammary tumor identifies the persistent change in theIGF signaling axis to favor the transformed cell proliferation inthe genetically resistant COP rats. Exogenous hormone treatmentsincrease the levels of Igfr in mammary lesions, thus increasing sen-sitivity to IGF1. IGF1 signaling is strongly associated with mito-genic activity in the mammary gland and is required for itsmorphological development [19]. The importance of IGF1 signalingin the mammary gland has been shown using IGF1-knockouttransgenic mice [19–21]. Impaired mammary duct developmentis observed with IGF1 knockout, and treatment with E and IGF1 in-duces ductal morphogenesis of the mammary gland [20]. IGF1 andits receptor levels are strongly associated with the risk of breastcancer, and interactions between IGFR and ERa have been reportedin mammary gland tumorigenesis [21,22]. Blockage of IGF-IRexpression by using antisense oligodeoxynucleotides to IGF-IRmRNA, inhibited progestin stimulated growth of primary C4HD(progestin responsive) cells [23–25]. These findings support theidea that the observed increase in the IGFR level in E + P treatedmammary lesions and tumors could have contributed to the pro-motion of mammary lesions to palpable mammary tumors.
The ovarian hormones E and P, promote mammary lesions toovert mammary tumors. Increased SHC1 and GRB2 protein expres-sion in the hormone treatment group indicates induction of growthfactor receptor/PI3K/AKT signaling. The transforming potential ofSHC1 and GRB2 scaffold proteins is well known [26,27]. SHC1and GRB2 together with SOS1 are key mediators of the IGF1 signal-ing pathway [17,28,29]. SOS1 is another adaptor protein that func-tions in growth factor receptor signaling. These results indicatethat ovarian hormones increase the activation of IGFR by alteringthese scaffold proteins and thus induce sustained growth of mam-mary lesions.
The increased activation of AKT and ERK1/2 in the hormone-treated mammary lesions show that growth factor receptor/PI3Ksignaling induces cell proliferation via the AKT and ERK1/2 path-way. A recent study has shown that induction of IRS-2 by IGFRcaused the activation of ERK1/2 resulted in cell proliferation [30].Treatment of antisense oligonucleotide against IGFR mRNA re-duced breast tumor growth along with inactivation of AKT andERK1/2 [31]. Our data, along with these studies, suggests that acti-vation of ERK1/2 by E + P treatment could be responsible for pro-motion and progression of mammary tumor in COP rats.
From our data it is evident that E + P treatment activated AKT,which was found to be increased in both mammary lesions and tu-mors. It is well known that PI3K/AKT pathway leads to the activa-tion of mTOR signaling [32], our data on mammary tumorscorrelate with this concept. The 40S ribosomal protein S6 kinase(S6K) is a downstream target of mTOR [33]. Strong induction ofphospho 4E-BP1 (elF4E binding protein) shows the aggressive nat-ure of tumors induced by hormones in E + P treated animals. 4EBP1activation along with mTOR activation has been reported in hor-mone induced breast cancer proliferation [34]. Many studies dem-onstrate concurrent activation of PI3K/AKT and ERK pathways intumor progression [35–37]. It has been shown that activations of

A. Arumugam et al. / Steroids 77 (2012) 791–797 797
p70S6K and 4E-BP1 are required for the induction and mainte-nance of transformed phenotype [38–40]. Our data is consistentwith these findings, and we speculate that induction of p70S6Kand 4E-BP1 by E + P treatment could have played a role in inducingand sustaining growth and survival of transformed cells. A clinicalstudy also reports that irrespective of the upstream oncogenicstimuli, hyper phosphorylation of 4E-BP1 is associated with poorprognosis [41].
Antiapoptotic role of sex steroids are well known and the regu-lation of E on BCL2 family of proteins during induction of chemo-resistance has been reported earlier [42,43]. By increasing thelevel of anti-apoptotic protein BCL2 and reducing pro-apoptoticprotein BAX, and further by reducing the active form of caspase 3and 8, E + P treatment could have reduced the apoptotic cell deathfavouring tumor progression. In conclusion, increased activation ofAKT and ERK signaling together with differential growth factorreceptor signaling, predominantly IGFR, are early molecular altera-tions in mammary lesions that occur in response to the administra-tion of ovarian hormones. These hormone-influenced alterations inthe tumor microenvironment, which is rich in proliferative signal-ing proteins, have tumor-promoting capabilities that compete withthe genetic resistance of COP rats during the early phases of tumorgrowth. The convergence of IGFR and phosphorylation of AKTresulting in activation of ERK1/2 could lead to progression of mam-mary lesions to palpable tumors. Hormone induced expression ofp70S6K, 4E-BP1 and anti-apoptotic proteins, in our findings sug-gest that targeting these proteins or proteins that regulate thesetargets could be a useful approach for enhancing and prolongingthe effectiveness of endocrine therapies. Overall, our results indi-cate that evaluation of proliferative signals and associated path-ways in early-stage breast cancer could serve as a biomarker forbreast cancer prevention and therapy.
References
[1] Pike MC, Spicer DV, Dahmoush L, Press MF. Estrogens, progestogens, normalbreast cell proliferation, and breast cancer risk. Epidemiol Rev 1993;15:17–35.
[2] Coughlin SS, Ekwueme DU. Breast cancer as a global health concern. CancerEpidemiol 2009;33:315–8.
[3] McPherson K, Steel CM, Dixon JM. ABC of breast diseases. Breast cancer-epidemiology, risk factors, and genetics. BMJ 2000;321:624–8.
[4] Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML,et al. Risks and benefits of estrogen plus progestin in healthy postmenopausalwomen: principal results from the women’s health initiative randomizedcontrolled trial. JAMA 2002;288:321–33.
[5] Hofseth LJ, Raafat AM, Osuch JR, Pathak DR, Slomski CA, Haslam SZ. Hormonereplacement therapy with estrogen or estrogen plus medroxyprogesteroneacetate is associated with increased epithelial proliferation in the normalpostmenopausal breast. J Clin Endocrinol Metab 1999;84:4559–65.
[6] Ravdin PM. Hormone replacement therapy and the increase in the incidence ofinvasive lobular cancer. Breast Dis 2009;30:3–8.
[7] Isaacs JT. Genetic control of resistance to chemically induced mammaryadenocarcinogenesis in the rat. Cancer Res 1986;46:3958–63.
[8] Shepel LA, Gould MN. The genetic components of susceptibility to breastcancer in the rat. Prog Exp Tumor Res 1999;35:158–69.
[9] Thompson HJ, Singh M. Rat models of premalignant breast disease. J MammaryGland Biol Neoplasia 2000;5:409–20.
[10] Lu SJ, Archer MC. Ha-ras oncogene activation in mammary glands of N-methyl-N-nitrosourea-treated rats genetically resistant to mammaryadenocarcinogenesis. Proc Natl Acad Sci USA 1992;89:1001–5.
[11] Korkola JE, Archer MC. Resistance to mammary tumorigenesis in Copenhagenrats is associated with the loss of preneoplastic lesions. Carcinogenesis1999;20:221–7.
[12] Gould MN, Zhang R. Genetic regulation of mammary carcinogenesis in the ratby susceptibility and suppressor genes. Environ Health Perspect1991;93:161–7.
[13] Shepel LA, Lan H, Haag JD, Brasic GM, Gheen ME, Simon JS, et al. Geneticidentification of multiple loci that control breast cancer susceptibility in therat. Genetics 1998;149:289–99.
[14] Rajkumar L, Arumugam A, Elsayed A, Schecter S, Kotkowski E, Castillo R, et al.Long-term hormonal promotion overcomes genetic resistance to mammarycancer. Steroids 2010;76:31–7.
[15] Rajkumar L, Canada A, Esparza D, Collins K, Moreno E, Duong H. Decreasinghormonal promotion is key to breast cancer prevention. Endocrine2009;35:220–6.
[16] Sachdev D, Yee D. The IGF system and breast cancer. Endocr Relat Cancer2001;8:197–209.
[17] Laban C, Bustin SA, Jenkins PJ. The GH-IGF-I axis and breast cancer. TrendsEndocrinol Metab 2003;14:28–34.
[18] Imagawa W, Pedchenko VK, Helber J, Zhang H. Hormone/growth factorinteractions mediating epithelial/stromal communication in mammary glanddevelopment and carcinogenesis. J Steroid Biochem Mol Biol 2002;80:213–30.
[19] Kleinberg DL, Feldman M, Ruan W. IGF-I: an essential factor in terminal endbud formation and ductal morphogenesis. J Mammary Gland Biol Neoplasia2000;5:7–17.
[20] Ruan W, Kleinberg DL. Insulin-like growth factor I is essential for terminal endbud formation and ductal morphogenesis during mammary development.Endocrinology 1999;140:5075–81.
[21] Lanzino M, Morelli C, Garofalo C, Panno ML, Mauro L, Ando S, et al. Interactionbetween estrogen receptor alpha and insulin/IGF signaling in breast cancer.Curr Cancer Drug Targets 2008;8:597–610.
[22] Fox EM, Andrade J, Shupnik MA. Novel actions of estrogen to promoteproliferation: integration of cytoplasmic and nuclear pathways. Steroids2009;74:622–7.
[23] Elizalde PV, Lanari C, Molinolo AA, Guerra FK, Balana ME, Simian M, et al.Involvement of insulin-like growth factors-I and -II and their receptors inmedroxyprogesterone acetate-induced growth of mouse mammaryadenocarcinomas. J Steroid Biochem Mol Biol 1998;67:305–17.
[24] Balana ME, Labriola L, Salatino M, Movsichoff F, Peters G, Charreau EH, et al.Activation of ErbB-2 via a hierarchical interaction between ErbB-2 and type Iinsulin-like growth factor receptor in mammary tumor cells. Oncogene2001;20:34–47.
[25] Carnevale RP, Proietti CJ, Salatino M, Urtreger A, Peluffo G, Edwards DP, et al.Progestin effects on breast cancer cell proliferation, proteases activation, andin vivo development of metastatic phenotype all depend on progesteronereceptor capacity to activate cytoplasmic signaling pathways. Mol Endocrinol2007;21:1335–58.
[26] Alam SM, Rajendran M, Ouyang S, Veeramani S, Zhang L, Lin MF. A novel role ofShc adaptor proteins in steroid hormone-regulated cancers. Endocr RelatCancer 2009;16:1–16.
[27] Dharmawardana PG, Peruzzi B, Giubellino A, Burke Jr TR, Bottaro DP.Molecular targeting of growth factor receptor-bound 2 (Grb2) as an anti-cancer strategy. Anticancer Drugs 2006;17:13–20.
[28] Surmacz E. Function of the IGF-I receptor in breast cancer. J Mammary GlandBiol Neoplasia 2000;5:95–105.
[29] Ravichandran KS. Signaling via Shc family adapter proteins. Oncogene2001;20:6322–30.
[30] Wang J, Kuiatse I, Lee AV, Pan J, Giuliano A, Cui X. Sustained c-Jun-NH2-kinaseactivity promotes epithelial–mesenchymal transition, invasion, and survival ofbreast cancer cells by regulating extracellular signal-regulated kinaseactivation. Mol Cancer Res 2010;8:266–77.
[31] Salatino M, Schillaci R, Proietti CJ, Carnevale R, Frahm I, Molinolo AA, et al.Inhibition of in vivo breast cancer growth by antisense oligodeoxynucleotidesto type I insulin-like growth factor receptor mRNA involves inactivation ofErbBs, PI-3K/Akt and p42/p44 MAPK signaling pathways but not modulation ofprogesterone receptor activity. Oncogene 2004;23:5161–74.
[32] Morgensztern D, McLeod HL. PI3K/Akt/mTOR pathway as a target for cancertherapy. Anticancer Drugs 2005;16:797–803.
[33] Jacinto E, Lorberg A. TOR regulation of AGC kinases in yeast and mammals.Biochem J 2008;410:19–37.
[34] Boulay A, Rudloff J, Ye J, Zumstein-Mecker S, O’Reilly T, Evans DB, et al. Dualinhibition of mTOR and estrogen receptor signaling in vitro induces cell deathin models of breast cancer. Clin Cancer Res 2005;11:5319–28.
[35] Liu Z, Hou P, Ji M, Guan H, Studeman K, Jensen K, et al. Highly prevalent geneticalterations in receptor tyrosine kinases and phosphatidylinositol 3-kinase/aktand mitogen-activated protein kinase pathways in anaplastic and follicularthyroid cancers. J Clin Endocrinol Metab 2008;93:3106–16.
[36] Simi L, Pratesi N, Vignoli M, Sestini R, Cianchi F, Valanzano R, et al. High-resolution melting analysis for rapid detection of KRAS, BRAF, and PIK3CA genemutations in colorectal cancer. Am J Clin Pathol 2008;130:247–53.
[37] Tsao H, Goel V, Wu H, Yang G, Haluska FG. Genetic interaction between NRASand BRAF mutations and PTEN/MMAC1 inactivation in melanoma. J InvestDermatol 2004;122:337–41.
[38] She QB, Halilovic E, Ye Q, Zhen W, Shirasawa S, Sasazuki T, et al. 4E-BP1 is a keyeffector of the oncogenic activation of the AKT and ERK signaling pathwaysthat integrates their function in tumors. Cancer Cell 2010;18:39–51.
[39] Mamane Y, Petroulakis E, Rong L, Yoshida K, Ler LW, Sonenberg N. eIF4E – fromtranslation to transformation. Oncogene 2004;23:3172–9.
[40] Polunovsky VA, Bitterman PB. The cap-dependent translation apparatusintegrates and amplifies cancer pathways. RNA Biol 2006;3:10–7.
[41] Armengol G, Rojo F, Castellvi J, Iglesias C, Cuatrecasas M, Pons B, et al. 4E-binding protein 1: a key molecular ‘‘funnel factor’’ in human cancer withclinical implications. Cancer Res 2007;67:7551–5.
[42] Teixeira C, Reed JC, Pratt MA. Estrogen promotes chemotherapeutic drugresistance by a mechanism involving Bcl-2 proto-oncogene expression inhuman breast cancer cells. Cancer Res 1995;55:3902–7.
[43] Huang Y, Ray S, Reed JC, Ibrado AM, Tang C, Nawabi A, et al. Estrogen increasesintracellular p26Bcl-2 to p21Bax ratios and inhibits taxol-induced apoptosis ofhuman breast cancer MCF-7 cells. Breast Cancer Res Treat 1997;42:73–81.