lecture 7 protein foldingcmcd.hms.harvard.edu/activities/_media/bcmp201/lecture7.pdf · bonds are...
TRANSCRIPT

Lecture 7 Protein Folding
James Chou
BCMP201 Spring 2008

The problem of protein folding
How proteins fold?
How to predict protein folds?
Lecture Outline
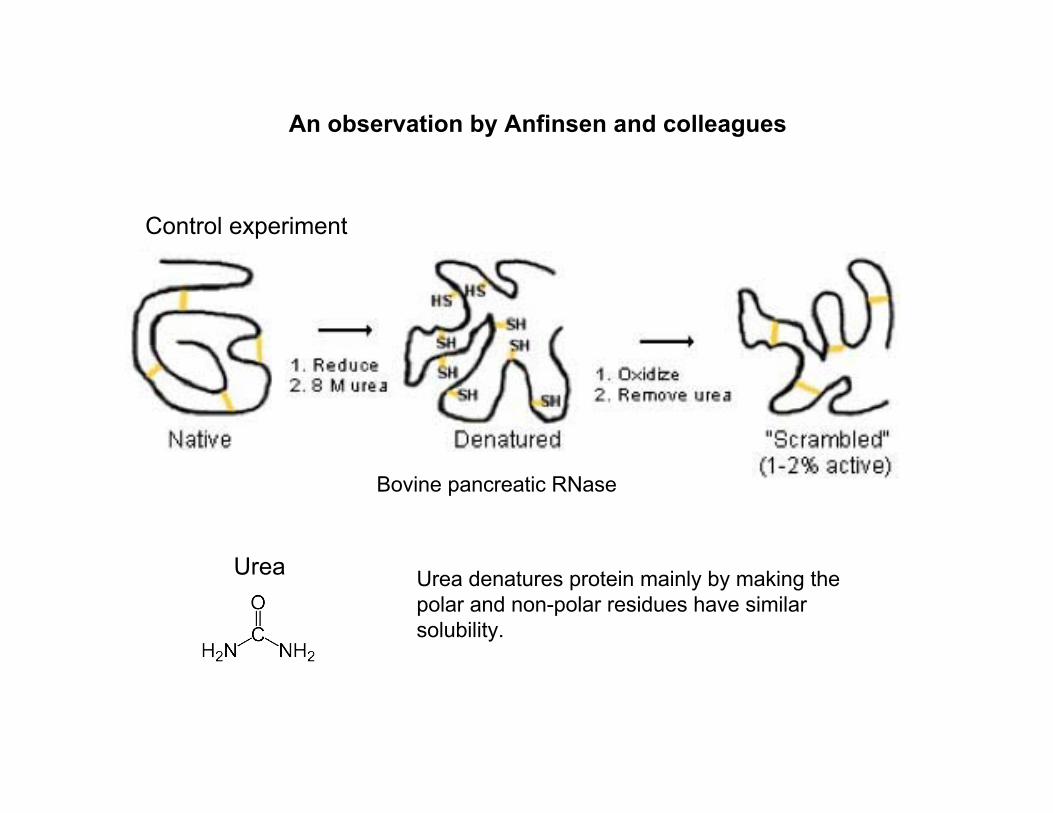
An observation by Anfinsen and colleagues
Urea Urea denatures protein mainly by making thepolar and non-polar residues have similarsolubility.
Control experiment
Bovine pancreatic RNase

Haber and Anfinsen. JBC 1961; 236(2):422-4
Refolding experiment

Anfinsen’s Dogma
Anfinsen et al., PNAS 1961; 47(9):1309

The problem of protein folding
Amino acidsequence
Tertiarystructure
?

Levinthal paradox
Zwanzig et al., PNAS 1992; 89:20-22
Assume each amino acid backbone can be in 3 conformational states,for 101 residues, there are 3100 = 5 x 1047 conformations.
Levinthal C. Extrait du Journal de Chimie Physique 1968; 65:44
If the protein can sample a new conformation at a rate of 1013 s-1, it willtake 1027 years to try them all. Longer than the age of the universe!
Therefore, proteins must fold in “pre-arranged pathways” and in acooperative manner.

Cooperativity in protein folding : How a globally optimal state canbe found without a global search?
Origin of cooperativity -- The probability of forming contact C2is much higher if C1 is formed than in the absence of C1.
Dill et al., PNAS 1993; 90:1942-6
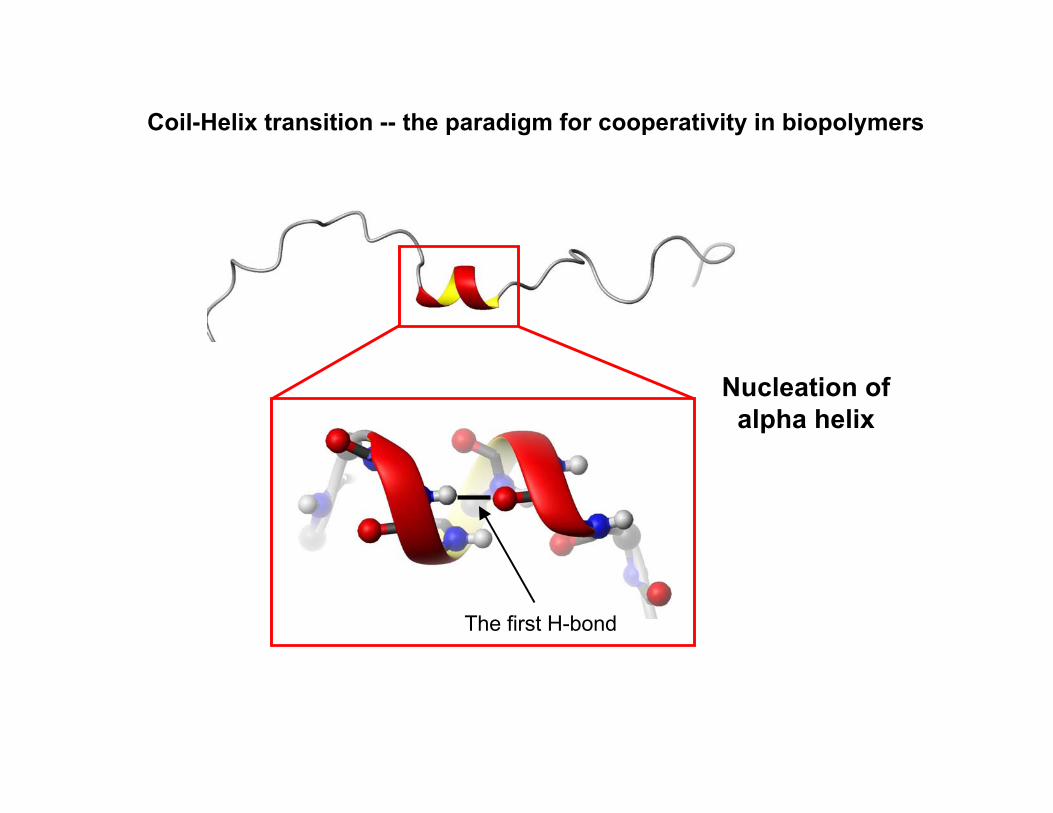
Nucleation ofalpha helix
The first H-bond
Coil-Helix transition -- the paradigm for cooperativity in biopolymers
�
! " #60
�
! " #40
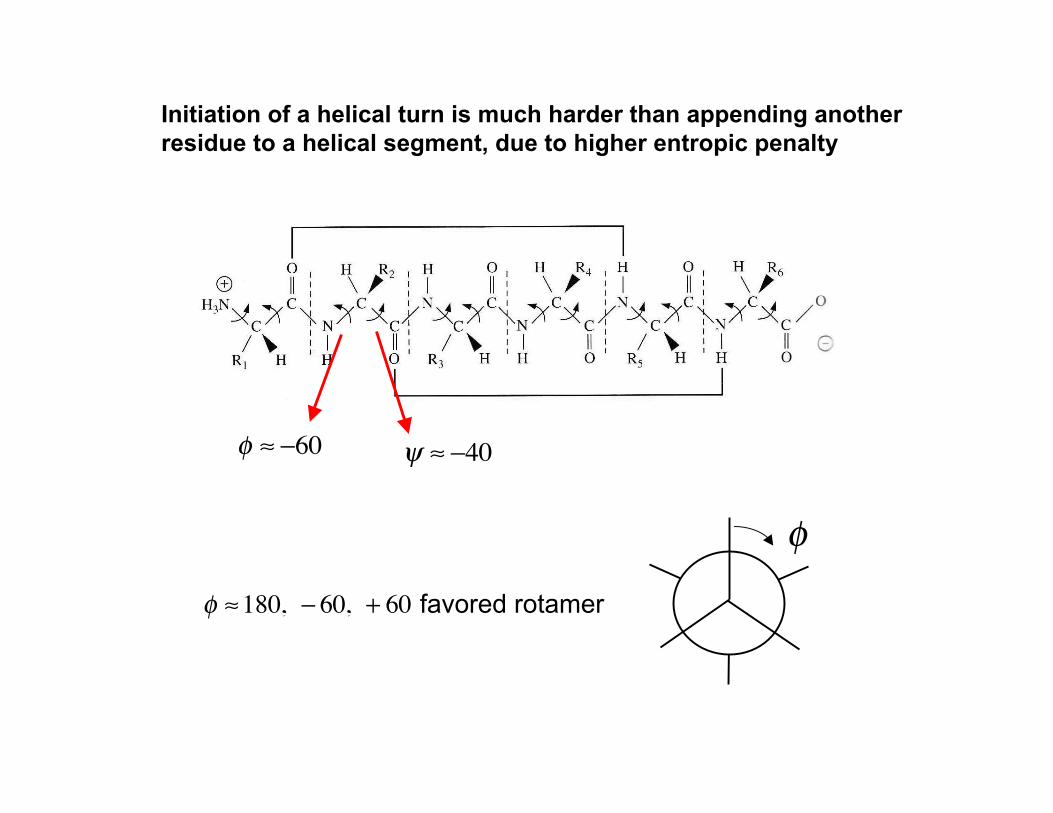
Initiation of a helical turn is much harder than appending anotherresidue to a helical segment, due to higher entropic penalty
�
!
�
! "180, # 60, + 60 favored rotamer

1st helical turn ...cccccccc...
! k f
kb
! "!!# !!! ...ccchcccc... ! << 1
2nd helical turn ...ccchcccc...
k f
kb
! "!!# !!! ...ccchhccc...
3rd helical turn ...ccchhccc...
k f
kb
! "!!# !!! ...ccchhhcc...
equilibrium const for each reaction s =k f
kb
Akf
kb
! "!!# !!! B dA
dt= !k f A[ ] + kb B[ ] = 0 Keq =
B[ ]A[ ]
=k f
kb

Fraction of residues in helix =
! k + 3( )k=1
N "3
# N " k " 2( )sk
N 1+! N " k " 2( )sk
k=1
N "3
#$
%&
'
()
,
where N is the number of residues in the polypeptide chain.
C
! k f
kb
! "!!# !!! H1
k f
kb
! "!!# !!! H2
k f
kb
! "!!# !!! $k f
kb
! "!!# !!! Hn"1
k f
kb
! "!!# !!! Hn
C! "!# !! Hn K
n=
Hn[ ]
C[ ]= ! s
n
The Zimm-Bragg-Lifson-Roig theory of coil-helix transition

f =
! k + 3( )k=1
N "3
# N " k " 2( )sk
N 1+! N " k " 2( )sk
k=1
N "3
#$
%&
'
()
The sharpness of the transition depends both on and N, being sharper at smaller values of and at larger N. The temperature dependence of s is given by
�
ln s∝−ΔGo kBT .
Melting curve of a coiled-coilprotein in water
Cooperative transition has a sigmoidal profile

Cooperativity results roughly in a two-state system
Luminescence decay kinetics ofCyt c measured at varioustimes after initial denaturation.
The process of unfoldingshows a bimodal distribution,one for largely unfolded stateand other includes nearlyfolded states.

The free energy of protein folding
From Alan Fersht. “Structure and Mechanism in Protein Science”
~50 kJ/mol for10 kDa protein

Hydrophobic interaction (entropic)
Pure H2O H2O around a hydrophobic molecule
Water molecules have moredegrees of freedom as the H-bonds are in the true tetrahedralarrangement.
Water molecules have lessdegrees of freedom in theclathrate cage arrangementsbecause some H-bondscannot point inside toward thehydrophobic sphere

Unfolded
More Hydrocarbon-Water Interfacial Area,More Water Ordered
Less Hydrocarbon-WaterInterfacial Area,Less Water Ordered
ΔS > 0
Formation of protein hydrophobic core in water
Folded

Hydrogen bonds (enthalpic)
Hydrogen bonds with H2O (hydration)Hydrogen bonds within protein
!H of H-bond = " 4 to 20( ) kJ mol"1

Other Enthalpic Interactions
Electrostatic or Coulomb interactions between ions
van der Waals interactions
Metal coordination
Disulfide bonds

Dissecting the free energy of protein folding
Unfolded !G! "!!# !!! Folded
!G = !H " T!S < 0, !G =~ "50 kJ/mol
!G ~ "50 kJ/mol
chain conformational
entropy ! T"S ! 0
~ 750 kJ/mol
H-bonds !H! 0
" "500 kJ/mol
VDW !H ~ " 50 kJ/mol
Electrostatic !H ! "50 kJ/mol
Hydrophobic effect ! T"S ! 0
~ !200 kJ/mol

Denaturation by Heat -- break H-bonds and other enthalpicallyfavorable interactions
!G ~ "50 kJ/mol
chain conformational
entropy ! T"S ! 0
~ 750 kJ/mol
H-bonds
VDW
Electrostatic
Hydrophobic effect ! T"S ! 0
~ !200 kJ/mol
Heating makes ΔHless negative
!G = !H " T!S < 0

Denaturation by Cold -- reduce the contribution fromhydrophobic effect
!G ~ "50 kJ/mol
chain conformational
entropy ! T"S ! 0
~ 750 kJ/mol
H-bonds
VDW
Electrostatic
!G = !H " T!S < 0
Near freezing T, entropyof H2O around non-polarresidues is less differentfrom those around polarresidues
Hydrophobic effect

Pathways of protein folding
From Alan Fersht. “Structure and Mechanism in Protein Science”

Folding pathway of Barnase
Bond et al., PNAS 1997; 94:13409-13
Barnase is a bacterial protein that consists of 110 amino acids and hasribonuclease activity.

Arcus et al., JMB 1995; 254(2):305-21
NMR studies of denatured Barnase reveal residual 2nd structures
1H (ppm)

Chaperone assisted protein folding
Two most important types of chaperones, Hsp60 and Hsp70
Hsp60 - In bacteria, provide a folding chamber
Hsp70 - In all living organisms, mainly to block aggregation

Hsp60 - GroEL-GroES chaperone

Hsp60 - GroEL-GroES chaperone
Fenton and Horwich, Quarterly Rev of Biophysics 2003; 36(2):229-56

substrate
DnaK binds unfolded proteins by recognizing an extended regionof the polypeptide chain that is rich in hydrophobic residues

E J
J
free unfolded protein
Bukau and Horwich, Cell 1998; 92:351-66

Computational Protein Folding
Amino acidsequence
Tertiarystructure

The free energy landscape of protein folding
Dill and Chan, Nature Struct Biol 1997; 4:10-19
Ideal funnel Reality funnel

The basic idea is to solve Newton’s equation of motion for every atom in the system
�
!"qi V = mi
d2ri
dt2 ,
ri – position of the ith atom mi – mass of the ith atom V – total potential energy of the system
A brief note on molecular dynamics simulation
The total potential energy is a function of the atomic positions (3N) of all the atoms in thesystem. Due to the complicated nature of this function, there is no analytical solution tothe equations of motion above; they must be solved numerically.

Example of computer simulation using the Verlet algorithm
All the integration algorithms assume the positions, velocities and accelerationscan be approximated by a Taylor series expansion:
�
r t + !t( ) = r t( ) + v t( )!t +1
2a t( )!t 2
r t "!t( ) = r t( ) " v t( )!t +1
2a t( )!t 2
Summing the above two equations, we get
�
r t + !t( ) = 2r t( ) " r t "!t( ) + a t( )!t 2
The Verlet algorithm uses positions and accelerations at time t and the positionsfrom time t-dt to calculate new positions at time t+dt.
�
!"qi V = mi
d2ri
dt2

MD simulation of membrane, water and water channel

http://www.stanford.edu/group/pandegroup/folding/villin/
Game - Folding@home by the Pande group at Stanford

Jumping out of the false minima
Temperature annealing
Melt the system at 2000 C andslowly cool down to 20 C.Perform MD simulation at each Tstep.
Metropolis Monte Carlo
accept the move if x < exp!"GkBT
#
$%&
'(
and reject otherwise.
1. Make a move (alter the conf.)
2. Calculate ΔG for the move
3. Generate a random number, x, between 0 and 1

Statistical methods for predicting protein structures
On Feb 12, 2008, there are 48891Structures in PDB. Pure statistical ormachine-learning methods are becomingmore and more powerful due to the rapidlyexpanding data base.

The Chou-Fasman Method
v = p1, p2 ,…, p20[ ], pi = % residues of AA type i
αβ α/β V
coil
Chou PY, Fasman GD. (1974). Prediction ofprotein conformation. Biochemistry.13(2):222-45.
Generate similar vectors for known classes offolds, e.g., α, β, α/β, … etc, from the PDB.
Early predictions of protein structural properties use aminoacid composition
Prediction of Protein Cellular AttributesUsing Pseudo- Amino Acid Composition.Proteins 2001; 43:246-55.

Knowledge-based methods for predicting protein structures
Artificial Intelligence - Neural network
Pattern Recognition - Evidence theory, Support Vector Machine
Homology Modeling - Multiple AA sequence alignment PSI-BLAST (take into account sequence evolution information)
Combining All You’ve Got - Rosetta from the Baker Lab