kukkadapu krishna kishore - theses.fr
TRANSCRIPT

ANNÉE 2013
THÈSE / UNIVERSITÉ DE RENNES 1 sous le sceau de l’Université Européenne de Bretagne
pour le grade de
DOCTEUR DE L’UNIVERSITÉ DE RENNES 1
Mention : CHIMIE
Ecole doctorale Sciences de la Matière de Rennes
présentée par
Krishna Kishore Kukkadapu UMR 6510 CNRS
Chimie et Photonique Moléculaires
UFR Sciences et Propriétés de la Matière
Gamma-borylatedallylic acetates as 3 carbon functionalized units : synthesis and applications
Thèse soutenue à Rennes le Jeudi 6 juin 2013
devant le jury composé de :
Véronique BELLOSTAProfesseur –ESPCI / rapporteur
Stéphane PELLET-ROSTAING Chargé de recherche CNRS à l’ICSM-CEA / rapporteur
Florence MONGINProfesseur à l’Université de Renne1 / / examinateur
Mathieu PUCHEAULT Chargé de recherche CNRS / examinateur
Michel VAULTIERDirecteur de recherche CNRS// directeur de thèse
1

2
Table of contents :
Résumé de la thèse en français 5
Acknowledgements: 22
Abbreviations: 24
General Introduction: 27
PART A 30
Chapter I: Bibliography 30
I. 1. Synthesis & applications of borylated allylic electrophiles: 31
I. 1. i. Synthesis of borylated allylic electrophiles: 31
I. 1. ii. Applications of borylated allylic electrophiles: 33
I. 1. ii. a. In iridium catalysis: 33
I. 1. ii. b. In copper catalysis: 37
I. 1. ii. c. In palladium catalysis: 39
I. 1. ii. d. In Grignard reaction: 41
I. 1. ii. e. In Diels Alder reaction: 42
I. 1. ii. f. In Mitsunobu reaction: 43
I. 1. ii. g. In cyclopropane synthesis: 46
I. 2. Tsuji Trost Allylation: 48
I. 2. i. Stereochemistry in Tsuji Trost allylation: 51
I. 2. ii. Regioselectivity in Tsuji Trost allylation: 52
I. 2. iii. Asymmetric allylic alkylation (AAA): 54

I. 2. iv. Application in natural product synthesis: 58
I. 3. Selectivity issues in palladium catalyzed Tsuji Trost allylation of borylated allyl
acetates:
61
Objectives: 62
Chapter II: Palladium catalyzed Tsuji Trost allylation of borylated allyl acetates 64
II. 1. Synthesis of borylated allyl acetates: 65
II. 2. Reactivity of borylated allyl acetates under palladium catalysis: 67
II. 2. i. Regioselectivity with carbon nucleophiles: 69
II. 2. ii. One pot allylation followed by Suzuki Miyaura cross coupling: 72
II. 2. iii. Application of a one pot strategy: 74
II. 2. iv. Stereoselectivity: 74
II. 2. v. Regioselectivity with nitrogen nucleophiles: 79
II. 2. vi. One pot allylation followed by Suzuki Miyaura cross coupling: 83
II. 2. vii. Stereoselectivity: 84
II. 3. Some failure attempts in order to use borylated allylic derivatives: 87
Conclusion: 90
Chapter III: Chemo enzymatic resolution of borylated allylic alcohols in continuous
flow systems using ionic liquids & scCO2
91
Introduction: 92
III. 1. Ionic liquids as solvents in Green biocatalysis: 92
III.2. Green biocatalysis in super critical carbon dioxide (scCO2): 93
III. 3. Literature data on the mechanism of resolution using Candida Antartica Lipase: 94
3

III. 4. Kinetic resolution of borylated allylic alcohols in ionic liquids: 96
III. 5. Enzyme activity in Ionic liquids: 98
III. 6. Optimization of kinetic resolution: 100
III.7. Effect of water in kinetic resolution: 102
III.8. Recyclability of ionic liquids: 102
III. 9. Kinetic resolution using continuous flow systems: 103
III. 10. Results and discussion: 104
Conclusion: 107
PART B: Experimental part 108
Compounds synthesized 171
Conclusions and Perspectives 173
4

6 Juin 2013
Thèse présentée par Mr Krishna Kishore Kukkadapu
Pour l'obtention du grade de Docteur de l'Université de Rennes 1
Résumé de la thèse en français
Introduction générale:
Les boranes vinyliques, les acides boroniques vinyliques et les boronates vinyliques sont
des organoboranes où la différence d'électronégativité entre le carbone et le bore est très faible
[C (2.55)-B (2.04)] et la liaison entre ces deux atomes est donc peu polaire. Les propriétés
caractéristiques du bore permettent de réaliser une grande variété de réactions dans différentes
conditions. Beaucoup de groupes de recherche ont exploré les applications synthétiques des
organoboranes en synthèse organique. Par exemple les boranes vinyliques peuvent être
transformés en les alcènes correspondants par protonolyse,1 ils peuvent être facilement
oxydés avec H2O2 en présence de base (addition d'un groupe hydroxyle sur la double liaison)
pour donner des produits cis-anti Markovnikov.2 Ils peuvent aussi subir des réactions
d'addition pour donner des alcools allyliques,3 ou des cycloadditions [4+2] pour former deux
nouvelles liaisons carbone- carbone via des réactions de Diels-Alder.4 Les acides
1 Brown, H. C.; Zweifel, G. J. Am. Chem. Soc. 1961, 83, 3834. 2 Brown, H. C.; Liotta, R. J. Am. Chem. Soc., 1979, 101, 96.3 a) Jacob, P.; Brown, H. C. J. Am. Chem. Soc. 1976, 98, 7832.
b) Jacob, P.; Brown, H. C. J. Org. Chem. 1977, 42, 579.4 a) Matteson, D. S.; Waldbillig, J. O. J. Org. Chem. 1963, 28, 366.
b) Singleton, D. A.; Martinez, J. P. J. Am. Chem. Soc. 1990, 112, 7423.
c) Vaultier, M.; Truchet, F.; Carboni, B. Tetrahedron Lett. 1987, 28, 4169.
5

vinylboroniques peuvent être transformés en halogénures vinyliques via une halogénolyse,5
réagir via une réaction de cyclisation radicalaire utilisant la méthode catalytique de Corey en
présence d'un initiateur de réaction radicalaire pour obtenir des diols 1,3- ou 1,4.6 Ils peuvent
participer à des réactions de couplage au palladium de type Suzuki pour former de nouvelles
liaisons carbone-carbone.7 Ils peuvent réagir avec des anhydrides pour donner différentes
cétones -insaturatées via des catalyses au palladium8 ou au rhodium.
9 Les acides
vinylboroniques ont aussi été utilisés pour la formation de nouvelles liaisons carbone-azote,10
carbone-oxygène,11
carbone-fluor12
via des réactions catalysées au palladium ou au cuivre.
Les boronates vinyliques ont été employés pour former de nouvelles liaisons carbone-carbone
via des couplages de Suzuki- Miyaura. Ils sont employés dans la réaction multicomposant de
Petasis13
pour donner des hétérocycles azotés fonctionnalisés. Ils participent aux réactions de
métathèse croisée pour donner des boronates vinyliques hautement fonctionnalisés,14
ils
réagissent facilement avec des carbènes générés à partir de diazos pour donner des
cyclopropanes15
et ceci à travers des catalyses au palladium et au rhodium. Les boronates
vinyliques, en réaction avec des oxides de nitrile subissent des réactions de cycloaddition 1,3-
dipolaire pour donner des isoxazoles.16
5 Brown, H. C.; Campbell, J. B. J. Org. Chem. 1980, 45, 389.6 Batey, R. A.; Smil, D. V. J. Angew. Chem. Int. Ed. 1999, 38, 1798.7 Suzuki, A.; Miyaura, N. Chem. Rev. 1995, 95, 2457.8 Yamamoto, A.; Ryuki, K.; Shimizu, I. Helvetica Chimica Acta. 2001, 84, 2996.9 Frost, C. G.; Wadsworth, K. J. Chem. Commun. 2001, 2316.10 Tao, C-Z.; Xin, C.; Juan, L.; Guo, Q-X. Tetrahedron Letters. 2007, 48, 3525.11 Lam, P. Y. S.; Vincent, G.; Clark, C. G.; Deudon, S.; Jadhav, P. K. Tetrahedron Lett. 2001,
42, 3415.12 Takeru, F.; Tobias, R. Org. Lett. 2009, 11, 2860.13 a) Petasis, N. A.; Zavialov, I. A. J. Am. Chem. Soc. 1977, 119, 445.
b) Batey, R. A.; Mackay, D. B.; Santhakumar, V. J. Am. Chem. Soc. 1999, 121, 5075.14 a) Morril, C. ; Grubbs, R. H. J. Org. Chem. 2003, 68, 6031.
b) McNulty, L.; Wright. Z. J. Org. Chem. 2010, 75, 6001.15 a) Fontani, P.; Carboni, M.; Vaultier, M. Tetrahedron Lett. 1989, 30, 4815.
b) Toshiro, I.; Hiroshi, M.; Shinya, N. J. Org. Chem. 1990, 55, 4986.
c) Yasutaka, F.; Hideki, A. Org. Lett. 2008, 10, 769.
16 Bianchi, G.; Cogoli, A.; Grünanger, P. J. Organomet. Chem. 1966, 6, 598.
6

Les boronates vinyliques ont aussi été utilisés comme nucléophiles en réaction d'allylation17
avec catalyse au cuivre ou au palladium.
Les transformations précédentes des organoboranes fournissent des précurseurs
importants pour la synthèse totale de molécules bioactives complexes qui ont été utilisées
dans les domaines de la médecine, de l'agrochimie, des composés pharmaceutiques et de la
chimie fine. Les organoboranes peuvent être synthétisés facilement et ceci les rend
particulièrement précieux comme intermediaires clés en synthèse organique. Ils peuvent être
obtenus par hydroboration d'alcynes à partir d'alkylboranes.18
Les acides boroniques
vinyliques peuvent être synthétisés par une hydroboration d'alcynes avec des alkoxyboranes,
suivie d'hydrolyse.19
Les boronates vinyliques ont été obtenus via des réactifs
organométalliques par transmétallation avec le trimethylorthoborate,20
ou par hydroboration
d'alcynes avec des alkoxyboranes.
L'introduction d'une substitution en position allylique sur des boronates vinyliques leur
confère un degré élevé de flexibilité vis-à-vis des applications en synthèse organique. De tels
boronates vinyliques -substitués possèdent plusieurs sites réactionnels ce qui permet de les
considérer comme des substrats difficiles en ce qui concerne la sélectivité des réactions
(spécialement vis-à-vis des réactions catalysées par les métaux.21
Peu de groupes de recherche
ont exploré les applications de dérivés vinyl boroniques -substitués en synthèse organique
via des réactions de Grignard, Mitsunobu, Diels-Alder, ainsi que des cyclopropanations
asymétriques et des réactions catalysées par des métaux de transition.
17 a) Whittaker, A. M.; Richard, P. R.; Lalic, G. Org. Lett. 2010, 12, 3216.
b) Ortar, G. Tetrahedron Lett. 2003, 44, 4311.18 a) Brown, H. C.; Zweifel, G. J. Am. Chem. Soc. 1961, 83, 3834.
b) Brown, H. C.; Moerikofer, A. W. ibid, 1963, 85, 2063.19 Shyam, K. G.; Brown, H. C. ibid, 1975, 97, 5249.20 Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457.21
Carosi, L.; Hall, D. G. Angew. Chem. Int. Ed. 2007, 46, 5913.
7

Différentes méthodes
ont été employées pour leur préparation à partir de -céto
vinylboronates par réduction avec des hydrures ou des dérivés du zinc,22
par hydroboration
d'alcools propargyliques avec soit le pinacol borane ou le di-isopinocamphenyl borane suivie
par une refonctionnalisation dans ce dernier cas.23
Dans notre laboratoire, nous avons développé des réactions catalysées au palladium sur
des boronates vinyliques -substitués et nous nous sommes attachés à étudier en particulier les
chimio-, régio- and stéréoselectivités lors de la réaction d'allylation.
Cette thèse est divisée en trois chapitres. Le premier chapitre fait une brève revue de la
littérature sur la synthèse et la réactivité de dérivés allyliques -borylés. Dans le second
chapitre notre objectif est d'étudier la possibilité de générer des complexes -allyl palladium à
partir d'acétates allyliques et -borylés puis d'étudier leur réactivité vis-à-vis d'une variété de
réactifs nucléophiles (réaction de couplage de Tsuji-Trost24
) en mettant l'accent sur les
problèmes de chimio-, régio- et stéréo-selectivités. Le troisième chapitre décrit le
dédoublement chimio enzymatique d'alcools allyliques -borylés dans des systèmes à flux
continu utilisant des liquides ioniques et du CO2 supercritique.
Chapitre-I : Synthèse et applications de dérivés allyliques -borylés:
L’introduction d’un groupe fonctionnel en position allylique sur des boranes vinyliques est
très intéressante car elle permettra d’effectuer une grande variété de réactions, compte tenu de
la présence de multiples groupes fonctionnels sur ce synthon. Vaultier et al ont décrit la
synthèse d’électrophiles allyliques -borylés en partant d’alcools propargyliques (Schéma
1).25
22Jehanno, E.; Vaultier, M. Tetrahedron Lett. 1995, 36, 4439.
23 Fortineau, A.-D.; Robert,M.; Gueguan, J.-P.; Carrie, D.; Mortier, J.; Vaultier, M. C. R.
Acad. Sci. Serie IIc 1998, 1, 253.24
Trost, B. M.; Matthew, L. C. Chem. Rev. 2003, 2921.25 (a) Fortineau, A. D.; Robert, M.; Gueguan, J. P.; Carrie, D.; Mortier, J.; Vaultier, M. C. R.
Acad. Sci. Serie IIc 1998, 1, 253.
8

TMSCl, HMDS
0 oC to 50 oC,16 h
citric acid
MeOH, rt 1 h
R1
OH
R2 R1
OTMS
R2
pinacolborane
CH2Cl2, 0 oC to rt, 48 h
B OTMS
R2R1
O
O
A 70-96%
B 41-58% C 47-82%
B OH
R2R1
O
O
Entrée R1 R2 Rendement (%)A B C
a H H 96 41 47
b CH3 H 94 58 82
c Ph H 95 41 65
d CH3 CH3 70 50 74
Schéma 1: Alcools allyliques -borylés via une hydroboration avec le pinacolborane
Des alcools allyliques -borylés peuvent aussi être synthétisés par une séquence "one-pot" en
trois étapes via l’hydroboration de systèmes propargyliques protégés et en utilisant le
dicyclohexylborane.26
Peu d’applications des alcools allyliques -borylés ont été présentées dans la littérature.
Dennis Hall et al ont décrit la préparation d’allylboronates chiraux -substitutés, à partir de
dérivés allyliques -borylés, via des alkylations allyliques asymétriques par des catalyseurs à
l’iridium ou au cuivre portant des ligands chiraux.27, 28
Des boronates allyliques -substitués
ont été préparés avec de très hautes énantiosélectivités, jusqu’à 93%, et de bons rendements
(jusqu’à 87%). Walsh et al ont décrit une allylation chimiosélective catalysée au palladium sur
des réactifs bifonctionnels contenant à la fois un acétate allylique et un ester boronique
vinylique [Le groupe partant (acetate) est en du bore].29
Il a été montré que seuls des
(b) Berree, F.; Gernigon, N.; Hercouet, A.; Lin, C-H.; Carboni, B. Eur. J. Org. Chem. 2009,
329.26 Pietruszka, J.; Witt, A. J. Chem. Soc., Perkin Trans. 1 2000, 4293.27 Peng, F.; Hall, D. G. Tetrahedron Lett. 2007, 48, 3305.28 Carosi, L.; Hall, D. G. Angew. Chem. Int. Ed. 2007, 46, 5913.29 Hussain, M. M.; Walsh, P. J. Angew. Chem., Int. Ed. 2010, 49, 1834.
9

produits d’allylation sont obtenus avec une excellente chimioselectivité et des rendements
allant de 65 à 92%. Carboni et al ont décrit l’addition de réactifs organométalliques aux
boronates vinyliques possédant un group acetal en position , qui subit un réarrangement
allylique en présence d’acides de Lewis pour former des -alkoxy allyl boronates avec une
haute stéréoselectivité, l’isomère E étant très majoritaire.30
Par une simple oxydation, les alcools allyliques -borylés donnent des borono-3-acroleines
qui ont été employées pour préparer des allylboronates cycliques -chiraux via des
cycloadditions d’hétero Diels-Alder avec des éthers d’enol, catalysées par le complexe chiral
CrIII
de Jacobsen.31
Des réactions de Mitsunobu sur des alcools allyliques -borylés avec des nucléophiles tels
que l’acide benzoique, des phénols, des N-tosylamines en présence de triphenylphosphine
(PPh3) et de diethyl azodicarboxylate (DEAD) conduisent aux produits de substitution SN2.32
Par ailleurs, les alcenylboronates peuvent être employés pour la synthèse de derivés
cyclopropaniques optiquement purs en utilisant des auxiliaires chiraux.33
De plus ces
boronates peuvent être oxydés pour obtenir les alcools correspondants.
Chapitre II: Allylation de Tsuji-Trost catalysée au palladium sur des acétates
allyliques -borylés
Des acétates allyliques -borylés peuvent présenter des réactivités differenciées vis à vis
de complexes metalliques,34
compte tenu de la présence de plusieurs sites réactionnels dans
ces synthons et différents aspects de sélectivité sont donc à considérer dans leurs réactions
(Schéma 2).
30 Possémé, F.; Deligny, M.; Carreaux, F.; Carboni, B. J. Org. Chem. 2007, 72, 984.31 (a) Gao, X.; Hall, D. G.; Carreaux, F.; Carboni, B. Chem. Eur. J. 2006, 12, 3132.
(b) Favre, A.; Carreaux, F.; Carboni, B. Eur. J. Org. Chem. 2008, 4900. 32 Berree, F.; Gernigon, N.; Hercouet, A.; Carboni, B. Eur. J. Org. Chem. 2009, 329.33 Luithle, J. E. A.; Pietruszka, J. J. Org. Chem. 1999, 64, 8287. 34
Carosi, L.; Hall, D. G.; Angew. Chem. Int. Ed. 2007, 46, 5913.
10

(i) Chimioselectivité: réaction de Tsuji-Trost (a) versus couplage de Suzuki (a1).
(ii) Régioselectivité: attaque en position- (b) versus position- (b1) dans l’allylation de
Tsuji-Trost
(iii) Stéréoselectivité: stéreorétention (c) versus stéréoinversion (c1) lors de l’addition
nucléophile.
OAc
B
O
OB
O
O
Nu
B
O
O
Nu
B
O
O
OAc
PdL2 B
O
O
Nu
B
O
O
Nu
PdIIL2(a) (b) (c)
(a1) (b1) (c1)
Chemoselectivity Regioselectivity Stereoselectivity
Schéma 2: Problèmes de sélectivité dans les réactions catalysées au palladium sur des acetates -borylés
Dans ce chapitre notre objectif a été d’étudier la possibilité de générer des complexes
-allyles palladium à partir d’acétates allyliques -borylés et d’analyser leur réactivité vis-à-
vis d’un certain nombre de réactifs nucléophiles (réaction de couplage de Tsuji-Trost35
) en
incluant les aspects de chimio-, régio- et stéréo-selectivité.
Une allylation chimio-, régio- et stéréo-selective d’acetates allyliques -borylés a été
développée avec des nucléophiles carbonés, azotés et oxygénés. Une substitution ipso de
l’acétate a été obtenue, avec une rétention complète de configuration au niveau du centre
chiral, conduisant à des boronates vinyliques -fonctionnalisés. Ces réactions s’effectuent
avec de bons rendements et des excès énantiomériques supérieurs à 99% (Schéma 3).
35Trost, B. M.; Matthew, L. C. Chem. Rev. 2003, 2921.
11

Une réaction “one pot” de Tsuji-Trost, suivie par un couplage de Suzuki-Miyaura a été mise
au point, conduisant aux produits recherchés avec de bons rendements (Schéma 4).
Il est, en particulier, très difficile de contrôler la régioselectivité de l’allylation quand les deux
côtés du complexe portent des groupes aromatiques (Cas 1, Schéma 5). Cette méthode “one
pot” est donc particulièrement utile pour réaliser une allylation sélective sur la position
choisie et le boronate intermédiaire peut ensuite être transformé en le groupe aryle choisi (Cas
2, Schéma 5).
12

Cette procédure “one-pot” offre une alternative intéressante pour contrôler la régioselectivité.
La réaction tandem “one pot” Tsuji-Trost allylation / couplage de Suzuki-Miyaura entre
l'acétate allylique substitué par un phényle, le dimethyl malonate puis l'iodotoluène, donne le
produit désiré avec un rendement de 78%, rendement qui est supérieur à celui obtenu lors du
processus en deux étapes (45%). De plus, les boronates vinyliques peuvent être transformés
en d’autres groupes fonctionnels. Ils peuvent être activés par une catalyse au cuivre36
pour
obtenir des azides vinyliques avec de bons rendements (Schéma 6).
En utilisant des nucléophiles carbonés nous avons pu introduire de la chiralité par allylation
asymétrique de substrats racémiques et en utilisant différents ligands chiraux (Schéma 7).
36 Tao, C.-Z.; Guo, Q.-X. Tetrahedron Lett. 2007, 48, 3525.
13

Des dérivés -borylés et fonctionnalisés ont été obtenus avec de bons rendements (jusqu’à
80%) et des excès énantiomériques allant jusqu’à 78%. Les deux isomères ont été synthetisés
à partir de l’acetate racémique en utilisant les ligands chiraux appropriés. De la même
manière, l’allylation asymétrique d’acétates allyliques -borylés a été réalisée avec des
nucléophiles azotés. Une allylation de type Trost, suivie en “one pot” d’un couplage de
Suzuki-Miyaura, a donné les produits désirés avec des énantioselectivités jusqu’à 63% et des
rendements élevés (83-90%) (Schéma 8). Ces réactions s’avèrent complètement chimio-,
régio- et stéréo-sélectives.
Conclusion:
Une allylation chimio-, régio- et stereo-selective a été mise au point à partir d’acétates
allyliques -borylés et ceci en utilisant des nucléophiles carbonés,37
ou azotés. Au bilan, nous
avons donc réussi à employer un intermédiaire clé à trois atomes de carbone hautement
fonctionnalisé de manière chimio-, régio-, et stéréoselective. Les produits obtenus sont
37 Kukkadapu, K. K.; Ouach, A.; Lozano, P.; Vaultier, M.; Pucheault, M. Org. Lett. 2011, 13,
4132.
14

susceptibles d’être employés dans une grande gamme de transformations en utilisant le
potentiel de la chimie des boronates.
Chapitre III: Dédoublement chimio-enzymatique d'alcools allyliques -borylés
en système à flux continu, utilisant des liquides ioniques et du CO2
supercritique.
Les solvants jouent un rôle important pour obtenir de bons résultats dans les réactions de
chimie organique. Généralement ces solvants organiques sont volatiles et génèrent des résidus
organiques qui ne sont pas acceptables en termes environmentaux et doivent donc être évités.
Dans un contexte de chimie verte,38
le remplacement des solvants dangereux par des solvants
avec des effets bénins sur l'environnement est un défi très attractif. Ces problèmes ont conduit
les chercheurs à identifier des solvants alternatifs pour remplacer les solvants organiques,
comme les fluides supercritiques39
et les liquides ioniques40
qui paraissent comme les
meilleures alternatives.
Les liquides ioniques sont des sels d'oniums à bas points de fusion et composés seulement
d'anions et de cations. Ils sont liquides à, ou en dessous de, 100 °C. Les liquides ioniques ne
sont pas volatiles et présentent une tension de vapeur très faible. Ils sont très polaires,
recyclables et stables thermiquement jusqu'à 400 °C (donc utilisables à hautes températures).
Ils peuvent dissoudre des composés organiques et inorganiques. La synthèse de composés
énantioenrichis en utilisant les enzymes comme catalyseurs dans des conditions "sans solvant"
relève de la biocatalyse "verte". La grande efficacité catalytique des enzymes dans les liquides
ioniques est maintenant bien documentée.41
Cependant, des solvants organiques sont souvent
utilisés pour isoler les produits à partir des liquides ioniques, ce qui constitue un inconvénient
pour le développement de procédés verts. L'isolement de produits à partir de milieux de type
liquides ioniques par un autre solvant vert comme le CO2 supercritique (scCO2) est considéré
38 Collins, T. Science 2001, 291, 48.39 Noyori, R. Chem. Rev. 1999, 99, 353.40 Riisager, A.; Fehrmann, R.; Haumann, M.; Wasserscheid, P. Top. Catal. 2006, 40, 91.41 Lozano, P. Green Chem. 2010, 12, 555.
15

comme la stratégie la plus intéressante pour développer des procédés chimiques propres et
verts.
Ceci est dû à la capacité du scCO2 d'extraire, de dissoudre et de transporter des composés
chimiques en phase gazeuse, à savoir le gaz CO2 comprimé. Dans ce contexte, un système
hétérogène peut être utilisé avec succès pour des réactions dans scCO2. Des systèmes
biphasiques, basés sur des liquides ioniques et scCO2 représentent des alternatives
intéressantes aux solvants organiques pour le design de procédés propres utilisant des
biotransformations en environment non-aqueux et conduisant directement à des produits
purs.42
Les enzymes ne perdent pas leur activité quand elles sont supportées sur un milieu
liquide ionique, même à des hautes temperatures. Une telle stabilité des enzymes ainsi que
l'emploi du scCO2 sont les paramètres clés pour la mise en oeuvre d'un processus de
bioconversion vert en flux continu.
Dédoublement cinétique d'alcools allyliques -borylés dans les liquides
ioniques:
Andrade et al ont décrit la première application des enzymes comme catalyseurs pour la
synthèse énantiocontrôlée de composés contenant du bore par une acetylation énantiosélective
(via un dédoublement cinétique catalysé par une enzyme) dans du n-hexane solvant.43
42 Lozano, P.; Vaultier, M. Green Chem. 2007, 9, 780.43 Andrade, L. H.; Barcellos, T. Org. lett. 2009, 11, 3052.
16

Dans un tel dédoublement cinétique d'un composé racéemique, le rendement chimique du
procédé sera généralement limité à 50%. Différents types d'alcools secondaires (aromatiques,
allyliques, aliphatiques) contenant des boronates ont été acetylés en utilisant ce protocole et
de hautes sélectivités (> 98%) ont été obtenues. Dans notre laboratoire, nous nous sommes
intéressés à l'étude du dédoublement d'alcools allyliques -borylés dans des conditions sans
solvant (Schéma 9).
OH
B
OAc (3.0 eq), CAL-B
Ionic Liquid, 50 oC, t min
OH
B
OAc
B+ **
O
O
O
O
O
O
(S)-OH (R)-OAc
Schéma 9: Dédoublement cinetique d'alcools allyliques -borylés avec CAL-B sans solvant.
Le dédoublement chimioenzymatique d'alcools allyliques -borylés par acétylation sélective
avec l'acétate de vinyle dans les liquides ioniques a été développé en utilisant Candida
Antartica Lipase (CAL-B) comme enzyme à 50 oC. Différents liquides ioniques ont été
étudiés en fonction de:
i) Leur longueur de chaine (butyle, octyle, et dodécyle)
ii) Le motif cationique:
Ammonium: BTMA, TBMA
Imidazolium: BMIM
Pyrolidinium: BMPy
Piperidinium: BMPi
iii) La partie anionique (NTf2, BF4, PF6)
17
Nous nous sommes donc attachés à optimiser le système catalytique pour avoir des temps de
reaction minima, combinés à de bons rendements et de bonnes sélectivités. Les résultats ont
été pris sur la base de la formation du produit (R)-OAc. Différents liquides ioniques ont été
testés pour optimiser le dédoublement cinetique. Il faut noter que ces réactions n'ont pas été
effectuées sous atmosphère inerte. L'activité enzymatique (présentée en efficacité par
milligramme d'enzyme immobilisée utilisée) est un facteur clé pour obtenir un bon
dédoublement cinétique. Une activité enzymatique élevée a été trouvée dans le cas des

liquides ioniques contenant NTf2 par rapport à ceux contenant PF6 et BF4. Les rendements
faibles obtenus avec les autres liquides ioniques peuvent être dus à la nature hygroscopique de
ces derniers conduisant à une absorption d'humidité. La présence d'eau dans le milieu
réactionnel peut hydrolyser l'enzyme acetylée en donnant de l'acide acétique, ce qui arrête le
processus d'acétylation énantiosélectif. Nos résultats ont démontré une forte activité
enzymatique 7.6 (U/mg de IME) pour le liquide ionique [C12MIM][NTf2] (Entrée 3, Tableau
1). Le dédoublement cinétique utilisant les liquides ioniques est réalisé en seulement 2h, alors
qu'avec les solvants organiques tels que le n-hexane les temps de réaction sont de 12-14h.
Entrée liquide ionique Activite 'de l' % Rendementa % Rendementa
enzyme (%eea) (%eea)(U/mg IME) at 2 h at 6 h
1. [BMIM][NTf2] 2.7 45 (>99) 51 (89)
2. [OMIM][NTf2] 6.3 49 (>99) 50 (>99)
3. [C12MIM][NTf2] 7.6 50 (>99) 50 (>99)
4. [BTMA][NTf2] 2.2 39 (>99) 51b(>99)
5. [TBMA][NTf2] 2.4 41 (>99) 51b(>99)
6. [BMPy][NTf2] 2.8 45 (>99) 50 (90)
7. [BMPi][NTf2] 3.3 48 (>99) 49 (91)
8. [BMIM][PF6] 3.3 46 (88) 49 (74)
9. [OMIM][PF6] 1.8 32 (99) 48 (99)
10. [C12MIM][PF6] 1.8 40 (99) 49 (85)
11. [BMIM][BF4] 4.9 44 (99) 48 (99)
12. [C12MIM][BF4] 1.8 26 (99) 29 (99)
Tableau 1: Activité enzymatique dans les liquides ioniques
aCette conversion a été évaluée par analyse chromatographique en phase gazeuse sur phase chirale, en se
basant sur la formation de l'acétate (R) au cours de la réaction.bErreur possible sur l'intégration en chromatographie en phase gazeuse sur phase chirale
Dédoublement cinétique dans des systèmes à flux continu:
Un système à flux continu controlé avec un support hétérogène à 500 C a été testé initialement
18

en utilisant CAL-B et [BMIM][NTf2] comme support hétérogène (Schéma 10). Un tel procédé
ne doit pas générer de sous produit organique et les composés obtenus, après passage à travers
le support hétérogène, seront récupérés dans le collecteur. Le scCO2 gazeux comprimé sera
recyclé vers le cylindre par une condensation. Dans les expériences à l'échelle du laboratoire,
ce gaz comprimé sera simplement rejeté dans l'atmosphère.
OH
B
O
O
19
Résultats et discussion:
Les expériences initiales ont visé à l'optimisation du système réactionnel dans des conditions
de flux continu et en utilisant un support solide préparé avec CAL-B et [BMIM][NTf2]. La
vitesse de la phase mobile [0.1mL de substrat et 0.9mL de scCO2] est de 1mL / min à 100 bar
et ceci en maintenant le support hétérogène à 500
C. L'activité par gramme d'enzyme utilisée a
été trouvée à 13.3 mol/h/g (Tableau 2, entrée 1). Le dédoublement cinétique a été réalisé en
continu pendant 8h le premier jour, avec un taux de conversion de 40%. L'activité
enzymatique n'a pas changé quand le même support hétérogène a été utilisé une seconde fois
pendant une autre opération de 8h le jour suivant, et des résultats identiques ont été obtenus
(Tableau 2, entrée 2). Le troisième jour, nous avons changé la concentration à 12 mol/h tout
en conservant le même support hétérogène. Cette 3ème
opération a été réalisée pendant 8h. La
conversion est restée à 40% alors que l'activité enzymatique a doublé à 26.6 mol/h/g
(Tableau 2, entrée 3). Cependant, on n'a pas atteint une conversion totale dans ces conditions.
L'étude d'autres liquides ioniques comme [OMIM][NTf2] avec CAL-B comme support
hétérogène a par contre donné une conversion complète avec de très bons rendements et des
OAcCAL B / IL
scCO2
produit de départ 50oC

sélectivités élevées dans des conditions à flux continu (Tableau 2, entrées 4-6). L'activité
enzymatique reste la même pendant des temps d'opération longs (jusqu'à 8h) et elle a été
trouvée de 9.03 mol/h/g (Tableau 2, entrée 4).
Entrée CAL-B Concentration Débit % Conversion Activité durée desur liquid de l'enzyme réactionionique ( mol/ h) ( L/ min) %ee ( mol/ h)
1. [BMIM][NTf2] 6 0.1 40 (99.9) 13.3 8 h
2. [BMIM][NTf2] 6 0.1 40 (99.9) 13.3 8 h
3. [BMIM][NTf2] 12 0.1 40 (99.9) 26.6 8 h
4. [OMIM][NTf2] 3 0.05 50 (99.9) 9.03 8 h
5. [OMIM][NTf2] 3 0.05 50 (99.9) 9.03 8 h
6. [OMIM][NTf2] 6 0.1 50 (99.9) 18.07 8 h
Tableau 2: Dédoublement cinétique en flux continu avec un système scCO2/IL
Une seconde opération de 8h, à une concentration du substrat de 3 mol/h, a donné la même
activité enzymatique de 9.03 mol / h /g conduisant à un rendement de 50% et une selectivité
>99% (Tableau 2, entrée 5). Des études en changeant the flux de substrat de 0.05ml à 0.1ml
(ce qui accroit la concentration à 6 mol/h) ont montré que l'activité enzymatique a doublé à
18.07 mol/h/g avec un rendement de 50% et une sélectivité >99% (Tableau 2, entrée 6). En
conclusion, l'activité enzymatique reste inchangée après 3 jours d'opération en continu et en
changeant le flux et la concentration.
Conclusion:
Une acetylation énantioselective d'alcools allyliques -borylés racémiques par Candida
Antarctica Lipase B (CAL-B) et utilisant de l'acétate de vinyle comme donneur d'acyle a
permis de préparer des acétates et des alcools allyliques -borylés avec des rendements élevés
(> 99%) et des sélectivités élevées (ee’s > 99%) dans des conditions réactionnelles "sans
solvant". Ce dédoublement cinétique très efficace a été réalisé en réacteur à flux continu
pendant 3 jours dans un système biphasique liquides ioniques / scCO2 sans perte d'activité du
système enzymatique. Ceci constitue un exemple d'un procédé réellement "vert" et bénin pour
l'environnement.
20

Conclusions et Perspectives:
Dans la première partie de notre travail de recherche, nous avons mis en œuvre une réaction
d'allylation de Tsuji-Trost à partir d'intermédiaires clés hautement fonctionnalisés, à savoir
des acétates allyliques -borylés. Ces réactions ont été réalisées avec un excellent contrôle de
la chimio- régio- et stéréo-sélectivité. Nous avons aussi développé une stratégie "one-pot"
impliquant d'abord cette allylation de Tsuji-Trost suivie immédiatement de réactions de
Suzuki-Miyaura, et ceci à partir d'acétates allyliques -borylés. Ces composés ont, en outre,
été employés dans des réactions d'alkylation allylique asymétriques conduisant à des dérivés
allyliques -borylés énantioenrichis. Après allylation, tous les composés obtenus pourraient
être soumis à une grande variété de réactions mettant à profit la présence du groupe pinacol
boronique: par exemple, ils pourraient être employés dans des réactions d'addition 1,4
utilisant des catalyseurs au rhodium; ils pourraient aussi être transformés en dérivés halogénés
et ces composés halogénés vinyliques pourraient eux-même être des intermédiaires très utiles
pour différentes réactions notamment des couplages catalysés par des métaux de transition.
Un autre développement possible de ce travail serait d'étudier cette réaction d'allylation
d'acétates allyliques -borylés en milieu liquide ionique.
Dans la seconde partie de ma thèse nous avons dévéloppé avec succès un procédé de
dédoublement cinétique à partir d'un alcool allylique -borylé, en utilisant une enzyme
Candida Antartica Lipase (CAL-B) et des liquides ioniques. De plus nous avons démontré
qu'on pouvait réaliser ce dédoublement cinétique d'alcool allylique -borylé dans un système
en flux continu, en utilisant l'enzyme immobilisée sur le liquide ionique comme support et
avec du CO2 super critique. Comme développement ultérieur de ce travail, il serait intéressant
de l'étendre à un processus de dédoublement cinétique dynamique à partir de cet alcool
allylique -borylé et en y ajoutant, pour l'étape de racémisation, des composants tels que des
zéolithes ou des catalyseurs à base de métaux de transition par exemple. De tels procédés de
dédoublements cinétiques dynamiques en flux continu pourraient être étendus ensuite à
d'autres alcools allyliques -borylés. De telles méthodes s'inscrivent parfaitement dans le
contexte du développment d'une chimie plus respectueuse de l'environnement.
21

Acknowledgements:
With great admiration, respect and appreciation, I take this privilege to express my
sincere gratitude to my research supervisor Prof. Michel Vaultier, Director of Research,
CNRS for his constant encouragement, creative guidance, invaluable and stimulating
suggestions, which greatly enhanced my interest in the frontier areas of science. His
dedication and passion towards research in chemistry is a great inspiration to my career. It is a
great pleasure and privilege for me to work under his guidance for my Doctoral research. I am
most thankful for all his invaluable help professionally and personally for spending his
valuable time during my tenure.
I would like to thank Dr. Mathieu Pucheault for his support, encouragement and
interest throughout every aspect of my research work. I am highly indebted for his valuable
suggestions and pain taking efforts in teaching me several skills. Thanks to the group meetings
and Mechanistic classes arranged by him which helped me to enlighten my knowledge in
chemistry apart from my research work. I am thankful for his helping hand and ideas which
helped me to solve many of my research tasks throughout my research period.
I extend my sincere thanks to Dr. Mireille Blanchard-Desce, Director of UMR-6510
for giving me the opportunity to work in her group and to have access for the state of art
facilities during my research programme. I would also like to thank Prof. Pedro Lozano,
University of Murcia, Spain for his help during my three months research programme in his
lab, where i learnt very important process for biocatalysis under continuous flow operation.
Many thanks as well to the staff of UMR-6510 and CRMPO for their help and support
during my study. Many thanks as well to Dr. Emilie, Dr. Florence Mongin and to Dr. Floris
Chervallier for their fruitful suggestions during weekly joint group meetings.
I also thank the previous and present group members for giving friendly environment
in the lab especially Thomas, Nicolas, Katia, Emmanuelle, Aicha, Kevin, Sunitha, Venkat,
Shankar, Marina, Cedric, Vivek, Bilal, Jean-Marie and Anne-Claire for their countless
support and help during the lab time and greatly enjoyed the foot ball sessions with them
22

during summer. I also thank my other friends for the lighter moments we shared specially
with Ludovic, Yogesh, Eduardo, Sebastien, Elisa, Kassem, Dayaker and Tai. I would also like
to thank the students from Pedro laboratory in Spain namely Juana Mari, Berenice for their
professional and personal help during my stay in Spain. I would also like to thank other
friends Kalyan, Ravi, Deepthi, Kiran, Yalla reddy, Praveen, Pavan reddy, Kesav, Shyam and
Sreesailam who joined me for several occasions.
I extend my heartful thanks to my Industry supervisors Dr. Y. Krishna Reddy, Dr.
Srinu Guntha and Dr. Srinivasulu Bandaru and Dr. Rajesh Shenoy who helped me to gain
research knowledge while working at Albany Molecular Reseearch Inc., India after my
Master degree.
This thesis would not have seen the light of the day without the moral support of love
and affection from my beloved parents Anjaneyulu, Mahalakshmi and sisters Vani, Jayasri
and Jayanthi and brothers-in-laws Madhusudhan rao, Viswesawar rao and Mallikarjuna rao
for their incessant encouragement, constant support and understanding.
Financial assistance from UMR 6510 through Egide, France in the form of Fellowship
is greately acknowledged. Finally, I thank my Thesis Director Prof. Michel Vaultier for
allowing me to submit this work in the form of a thesis and helping me a lot in several
aspects. Once again I thank all named and unnamed who have been associated during my part
of research work.
Krishna Kishore. Kukkadapu
23

Abbreviations :
ACN
Acetonitrile
Ac2O
acetic anhydride
BF4
boron tetrafluoride
BMIM
1-butyl-3-methylimidazolium
BMPi
1-butyl-1-methylpiperidinium
BMPy
1-butyl-1-methylpyrrolidinium
Bn
Benzyl
(Boc)2O di-tert-butyl dicarbonate
BTMA
butyl-trimethyl-ammonium
Bz
Benzoyl
CAL-B
Candia antartica lipase – B
Cy2BH
Dicyclohexylborane
C12MIM
1-dodecyl-3-methylimidazolium
dba
dibenzylidene acetone
DCM
Dichloromethane
DEAD
Diethylazodicarboxylate
DIBAL-H
diisobutylaluminium hydride
DMAP
4-dimethylaminopyridine
DME Dimethoxyethane
DMF
Dimethylformamide
DMSO
dimethyl sulfoxide
Et2O diethyl ether
GC gas chromatography
24

HMDS
Hexamethyldisilazane
HPLC
high pressure liquid chromatography
IL
ionic liquid
IME
immobilized enzyme
Ipc2BH
Diisopinocampheylborane
[Ir(cod)Cl]2 iridium(I) chloride 1,5-cyclooctadiene complex
dimer
LiAlH4
lithium aluminum hydride
m-CPBA
3-chloroperbenzoic acid
MOM
methoxy methyl ether
m.s.
molecular sieves
NaH
sodium hydride
NMO
N-methylmorpholine-N-oxide
NMR
nuclear magnetic resonance
NTf2
Trifluoromethanesulfonimide
OMIM
1-octyl-3-methylimidazolium
PdCl2
palladium(II) chloride
Pd2(dba)3
tris(dibenzylideneacetone)dipalladium(0)
PF6
Hexafluorophoshpine
Pd(PPh3)4
tetrakis(triphenylphosphine)palladium(0)
Pd(OAc)2
palladium(II) acetate
PMBOH p-methoxybenzyl alcohol
scCO2
supercritical carbon dioxide
TBMA tributyl-methyl-ammonium
25

TBS
tert-butyldimethylsilyl chloride
THF
Tetrahydrofuran
TMS
Trimethylsilyl
TPSCl
Chlorotriphenylsilane
26

General Introduction:
Vinylboranes, vinylboronic acids and vinylboronates are organoboranes where the
electronegativity difference between carbon (2.55) and boron (2.04) is low and the bond
between them is less polar than usual carbon-metal bonds. The characteristic features of
borane allow performing wide range of reactions under different conditions. Several research
groups explored the synthetic applications of vinylboranes in organic synthesis. For example,
they can be transformed to their corresponding alkenes via protonolysis,44
can be easily
oxidized by hydrogen peroxide in presence of base (addition of hydroxy group at double
bond) to result in cis-, anti Markovnikov products.45
They also participate in addition
reactions to give allylic alcohols,46
they undergo [4+2] cycloaddition reactions to form two
new carbon- carbon bonds via Diels-Alder reaction.47
Vinylboronic acids can be transformed
to vinyl halides via halogenolysis,48
react via boron-tethered radical cyclisation using Corey’s
catalytic tributyl-stannane method in presence of radical initiator to afford 1,3- or 1,4-diols,49
participate in palladium-catalyzed Suzuki cross coupling reactions to give new carbon-carbon
bond,50
and react with anhydrides to result in various -unsaturated ketones via palladium51
and rhodium52
catalysis. Vinylboronic acids were also used for the synthesis of new carbon-
nitrogen,53
carbon-oxygen,54
carbon-fluorine55
bonds via palladium and copper catalysis.
44 Brown, H. C.; Zweifel, G. J. Am. Chem. Soc. 1961, 83, 3834.45 Brown, H. C.; Liotta, R. J. Am. Chem. Soc., 1979, 101, 96.46 a) Jacob, P.; Brown, H. C. J. Am. Chem. Soc. 1976, 98, 7832.
b) Jacob, P.; Brown, H. C. J. Org. Chem. 1977, 42, 579.47 a) Matteson, D. S.; Waldbillig, J. O. J. Org. Chem. 1963, 28, 366.
b) Singleton, D. A.; Martinez, J. P. J. Am. Chem. Soc. 1990, 112, 7423.
c) Vaultier, M.; Truchet, F.; Carboni, B. Tetrahedron Lett. 1987, 28, 4169.48 Brown, H. C.; James, B. C. J. Org. Chem. 1980, 45, 389.49 Batey, R.; Smil, D. V. J. Angew. Chem. Int. Ed. 1999, 38, 1798.50 Suzuki, A.; Miyaura, N. Chem. Rev. 1995, 95, 2457.51 Yamamoto, A.; Ryuki, K.; Shimizu, I. Helvetica Chimica Acta. 2001, 84, 2996.52 Frost, C. G.; Wadsworth, K. J. Chem. Commun. 2001, 2316.53 Tao, C-Z.; Xin, C.; Juan, L.; Guo, Q-X. Tetrahedron Letters. 2007, 48, 3525.54 Lam, P. Y. S.; Vincent, G.; Clark, C. G.; Deudon, S.; Jadhav, P. K. Tetrahedron Lett. 2001,
42, 3415.55 Takeru, F.; Tobias, R. Org. Lett. 2009, 11, 2860.
27

Vinylboronates were used to synthesize new carbon-carbon bonds via Suzuki-Miyaura cross-
coupling reaction under palladium catalysis, participate in Petasis (modified Mannich)
multicomponent reaction56
to give functionalized nitrogen based heterocycles, they undergo
olefin cross-metathesis to afford highly functionalized vinylboronate derivatives,57
readily
react with carbene generated from diazo compounds to afford cyclopropane derivatives58
under palladium and rhodium catalysis. Vinylboronates on treatment with arylnitrile oxides
undergo 1,3-dipolar cycloaddition reaction to give isoxazole derivatives;59
vinylboronates
were also used as nucleophiles in allylation60
with copper and palladium catalysis.
The above transformations of organoboranes provide important precursors for building
complex bioactive molecules which were developed as medicine, agrochemicals,
pharmaceuticals and fine chemicals. Organoboranes can be easily synthesized and this easy
access made them useful key intermediates for organic synthesis. Vinylboranes can be
synthesized via hydroboration of alkynes with alkylboranes;61
vinylboronic acids can be
synthesized via hydroboration of alkynes with alkoxyboranes followed by hydrolysis62
whereas vinylboronates were synthesized from organometallic reagents by transmetallation
with trimethylorthoborate,63
also prepared from hydroboration of alkynes with alkoxyboranes.
Grafting a substitution in the allylic position of vinyl boronates confers to these units a
high degree of versatility with regard to their use in organic synthesis. -substitued
56 a) Petasis, N. A.; Zavialov, I. A. J. Am. Chem. Soc. 1977, 119, 445.
b) Batey, R. A.; Mackay, D. B.; Santhakumar, V. J. Am. Chem. Soc. 1999, 121, 5075.57 a) Morril, C. ; Grubbs, R. H. J. Org. Chem. 2003, 68, 6031.
b) McNulty, L.; Wright. Z. J. Org. Chem. 2010, 75, 6001.58 a) Fontani, P.; Carboni, M.; Vaultier, M. Tetrahedron Lett. 1989, 30, 4815.
b) Toshiro, I.; Hiroshi, M.; Shinya, N. J. Org. Chem. 1990, 55, 4986.
c) Yasutaka, F.; Hideki, A. Org. Lett. 2008, 10, 769.59 Bianchi, G.; Cogoli, A.; Grünanger, P. J. Organomet. Chem. 1966, 6, 598.60 a) Whittaker, A. M.; Richard, P. R.; Lalic, G. Org. Lett. 2010, 12, 3216.
b) Ortar, G. Tetrahedron Lett. 2003, 44, 4311.61 a) Brown, H. C.; Zweifel, G. J. Am. Chem. Soc. 1961, 83, 3834.
b) Brown, H. C.; Moerikofer, A. W. ibid, 1963, 85, 2063.62 Shyam, K. G.; Brown, H. C. ibid, 1975, 97, 5249.63 Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457.
28

vinylboronate derivatives contain multiple reaction sites which make them challenging
substrates to obtain selectivity (especially for metal-catalyzed reactions64
). Few research
groups explored the applications of -substitued vinylboron derivatives for organic synthesis
via Grignard, Mitsunobu, Diels-Alder, asymmetric cyclopropanation and transition metal-
catalyzed reactions.
Various methods
have been developed for their preparation either from -keto
vinylboronates by reduction with hydride or zinc derivatives,65
or from hydroboration of
propargylic alcohols with either pinacol borane66
or diisopinocampheylborane followed by a
refunctionalization in this last case.
In our lab we developed palladium-catalyzed reaction on -substitued vinylboronates
where we investigated chemo-, regio- and stereoselectivity during allylation reaction. This
thesis was divided into 3 chapters.
1. The first chapter describes a brief literature survey on the synthesis and reactivity of -
borylated allylic derivatives.
2. In the second chapter our goal is to study the possibility of generating palladium -
allyl complexes from -borylated allylic acetates and study their reactivity towards a
variety of nucleophilic reagents (Tsuji-Trost coupling reaction67
) including chemo-,
regio- and stereo-selectivity.
3. The third chapter describes the chemoenzymatic resolution of -borylated allylic
alcohols in continuous flow systems using ionic liquids & scCO2.
64Carosi, L.; Hall, D. G. Angew. Chem. Int. Ed. 2007, 46, 5913.
65Jehanno, E.; Vaultier, M. Tetrahedron Lett. 1995, 36, 4439.
66 Fortineau, A.-D.; Robert, M.; Gueguan, J.-P.; Carrie, D.; Mortier, J.; Vaultier, M. C. R.
Acad. Sci. Serie IIc 1998, 1, 253.67
Trost, B. M.; Matthew, L. C. Chem. Rev. 2003, 2921.
29

PART A
Chapter I: Bibliography
I.1: Synthesis and applications of
-borylated allylic electrophiles
I.2: Tsuji-Trost allylation
I.3: Selectivity issues in palladium-catalyzed
Tsuji-Trost allylation of -borylated allyl
acetates
30

I. 1. Synthesis & applications of -borylated allylic electrophiles:
I. 1. i. Synthesis of -borylated allylic electrophiles:
Vinylboronates -substituted with leaving group such as acetate has attracted much interest.
This highly functionalized three carbon building block bearing boronate is an electron-
deficient olefin, which offers synthetic potential for various functional group transformations.
This chapter describes the synthesis and applications of -borylated allylic systems in organic
synthesis. Vaultier et al reported the synthesis of -borylated allylic eletrophiles starting from
propargylic alcohol systems (Scheme 11).68
Protection of propargylic alcohols as
trimethylsilyl derivatives affords 1 in 70-96% yield.
68 (a) Fortineau, A. D.; Robert, M.; Gueguan, J. P.; Carrie, D.; Mortier, J.; Vaultier, M. C. R.
Acad. Sci. Serie IIc 1998, 1, 253.
(b) Jehanno, E.; Vaultier, M. Tetrahedron Lett. 1995, 36, 4439.
(c) Berree, F.; Gernigon, N.; Hercouet, A.; Lin, C-H.; Carboni, B. Eur. J. Org. Chem. 2009,
329.
31

Hydroboration of 1 with pinacolborane results in the formation of TMS protected -borylated
allylic alcohols 2 in 41-58% yield. Deprotection of 2 with citric acid in methanol affords
borylated allylic alcohols 3 in 47-82% yield (Scheme 11). borylated allylic electrophiles can
be synthesized from 3 via acetylation.
-borylated allylic alcohols can also be synthesized by a three step one-pot sequence via
hydroboration of protected propargylic systems using dicyclohexylborane,69
followed by
oxidation with trimethylamine oxide, leading to alkenylboronic esters 4. Transesterification of
4 with diols results in the formation of -substituted pinacolboronate derivatives 5 in 38-60%
yields (Scheme 12).
PG1O
OB
O
OMe
PhPh
OMe
PhPh
PG1O
Cy2BH, DME
0 oC to rt, 2 h BCy2
PG1O Me3NO
B(OCy)2
PG1O
HO
HO
OMe
PhPh
OMe
PhPh
PG1 = Bn 47%
PG1 = MOM 38%
PG1 = Bz 60%
4
5
2 h, rt
Scheme 12: Protected -borylated allylic alcohols via hydroboration with dicyclohexylborane
rt, 2 h
Alternatively, -borylated allylic alcohols were synthesized via direct hydroboration of silyl-
or benzyl-protected alkynes 6 with dioxaborolane 7 to give protected -borylated allylic
alcohol derivatives 8 in yields ranging from 30 to 91%. Silyl and benzyl protecting groups did
not interfere in the synthesis of corresponding alkenylboronic esters whereas ether, ester and
acetal protecting groups failed to give alkenylboronic ester 8 (Scheme 13).
69 Pietruszka, J.; Witt, A. J. Chem. Soc., Perkin Trans. 1 2000, 4293.
32

I. 1. ii. Applications of -borylated allylic electrophiles:
Introduction: Organic chemists explored the interest to use vinylboranes in organic
synthesis, and many strategies and applications were developed during these studies on
vinylboranes. Major contribution on vinylboranes involves the formation of characteristic new
C-C bond. Introducing a functional group at allylic position for vinylboranes brings the
interest to perform a variety of reactions because of the multiple functional groups present in
this type of molecule.
I. 1. ii. a. In iridium catalysis:
Dennis Hall et al reported a transition metal (TM)-catalyzed enantioselective allylation
method for the preparation of chiral -substituted allylboronates from achiral starting
materials (Scheme 14). 70
70 Peng, F.; Hall, D. G. Tetrahedron Lett. 2007, 48, 3305.
33
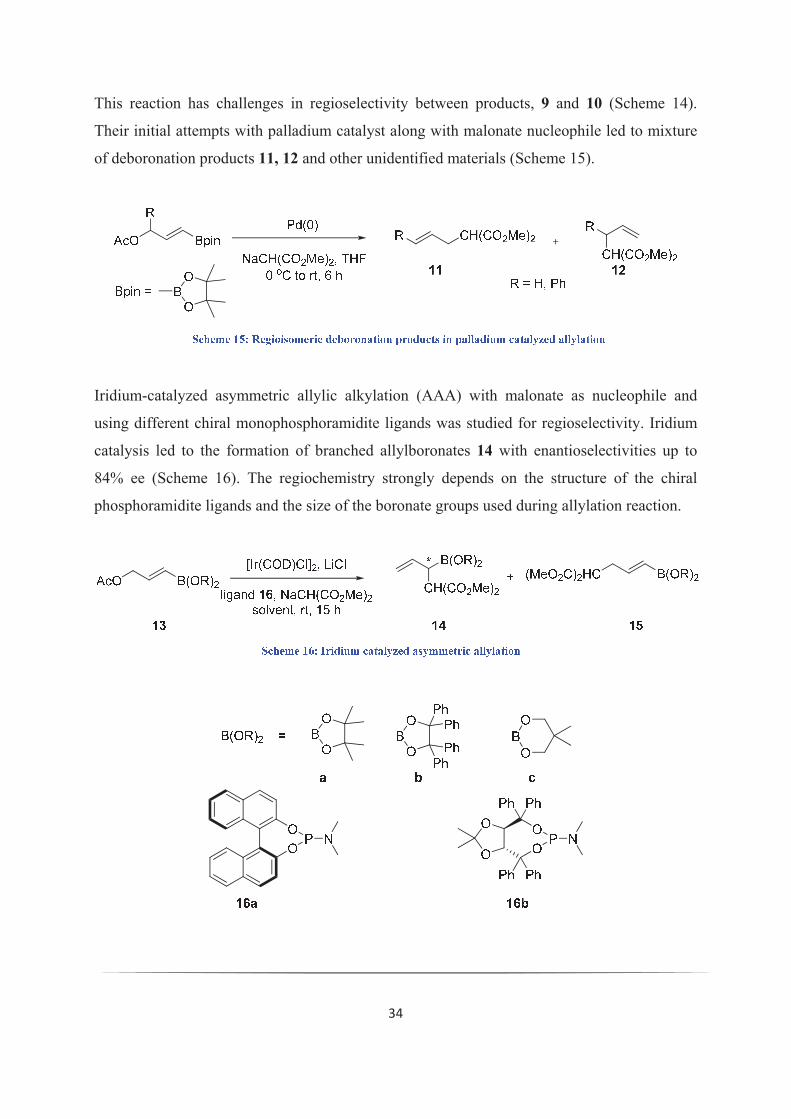
This reaction has challenges in regioselectivity between products, 9 and 10 (Scheme 14).
Their initial attempts with palladium catalyst along with malonate nucleophile led to mixture
of deboronation products 11, 12 and other unidentified materials (Scheme 15).
Iridium-catalyzed asymmetric allylic alkylation (AAA) with malonate as nucleophile and
using different chiral monophosphoramidite ligands was studied for regioselectivity. Iridium
catalysis led to the formation of branched allylboronates 14 with enantioselectivities up to
84% ee (Scheme 16). The regiochemistry strongly depends on the structure of the chiral
phosphoramidite ligands and the size of the boronate groups used during allylation reaction.
34

Iridium-catalyzed asymmetric allylic alkylations with different ligands in different solvent
combinations were studied to optimize the reaction conditions as shown in Table 3. Of all the
above mentioned ligands from Table 3, only allylation with 16d in THF solvent was found to
give branched type products 14a (Table 3, entry 4) whereas other ligands resulted in the
formation of linear products 15a (Table 3, entries 1-3, 5) during asymmetric allylation.
Allylation failed with other solvents like ether, dichloromethane and toluene. Use of more
polar solvents like DMF, Dioxane and DMSO gave linear products in majority (Table 3,
35

entries 9-11). Increasing the substitution on the boronate ring resulted in the formation of
linear product 15b (Table 3, entry 12) whereas changing the boronate cyclic system to six
membered ring gave branched type product as major compound (Table 3, entry 13). Also, it
was observed that these products were unstable during isolation, therefore they were readily
treated with aldehydes under Lewis acid catalysis to give homoallylic alcohol derivatives 17
with chirality transfer in one-pot. This type of addition between allylboron/crotylboron
derivatives to aldehydes is a popular method for stereoselective C-C bond formation (Scheme
17).
The allylboronation proceeds via six-membered chair-like transition state (Mechanism 1). The
addition of aldehydes to -substituted allylboronates of type 18 proceeds with near perfect
transfer of chirality to give two diastereomeric products 21 and 22. These Z and E allylic
alcohol products are stereoisomers, and their proportion is highly dependent on the nature of
the -substituent (R1) and the structure of the boronic ester.
71 The selectivity between 21 and
22 can be explained in terms of steric and dipolar effects on the two competing Zimmerman-
Traxler type transition state structures 19 and 20. With a non-polar alkyl substituent R1, steric
interactions play a dominant role. Transition structure 19 can be destabilized by steric
interactions between a large boronic ester and the pseudo-equatorial -substituent R1. On the
other hand, chair-like transition structure 20 features unfavorable allylic interactions due to
the pseudo-axial position of the R1 substituent.
71 (a) Hoffmann, R. W. Pure Appl. Chem. 1988, 60, 123.
36
(b) Hoffmann, R. W.; Neil, G.; Schlapbach, A. Pure Appl.Chem. 1990, 62, 1993.

The use of a hindered ester, such as pinacolate, aggravates interactions between R1 and the
dioxaborolane unit in structure 19, and tends to encourage transition structure 20 leading to
mixtures of products 21 and 22 in modest selectivities.72
I. 1. ii. b. In copper catalysis:
Dennis Hall et al reported copper-catalyzed asymmetric allylic alkylation on -borylated
allylic chloride derivatives using Grignard reagent and a chiral ligand. Enantioenriched -
substituted allylboronates with high level of selectivities (up to 93%) and yields up to 87%
were obtained (Scheme 18).73
AAA using copper catalyst was developed using
dichloromethane as solvent with slow addition of Grignard reagent and various
phosphoramidite ligands, and various cyclic boronate groups were investigated. The
combination of ligand 16e with boronic ester 23d affords optically active -substituted
allylboronate 24d (Scheme 18, entry 5) in 93% ee.
72 Hoffmann, R. W.; Weidmann, U. J. Organomet. Chem. 1980, 195, 137.73 Carosi, L.; Hall, D. G. Angew. Chem. Int. Ed. 2007, 46, 5913.
37

Cl B(OR)2+ EtMgBrS
COOCu
(CuTC)
ligand 16, CH2Cl2, -78 oC
Et
B(OR)2
upto 87% yield23 24
B(OR)2 =O
BO
OB
O
PhPh
PhPh
OB
O
a b c d
in
23
OB
O
Entry product ligand %ee
1. 24a 16d 87
2. 24b 16d 52
3. 24c 16d 86
4. 24d 16d 91
5. 24d 16e 93
, 4 h
Scheme 18: Asymmetric allylic alkylation with copper catalyst
The resulting -substituted allylic boronate reacts with aldehydes in presence of Lewis acid
catalyst at low temperature via stereoselective allylation, to give homoallylic alcohol
derivative 25 with chirality transfer in 75% yield and 92% selectivity (Scheme 19).
Also, chiral -substituted allylic trifluoroborate salts were prepared from -substituted allylic
38

boronates which have significant potential in carbonyl allylation chemistry.74
Allylic
trifluoroborate salts react with ketones via allylboration to give the homoallylic alcohol 26
containing a quaternary center, in 95% yield with 85% selectivity (Scheme 19).
I. 1. ii. c. In palladium catalysis:
Walsh et al reported palladium-catalyzed chemoselective allylation on bifunctional reagents
that contain both allylic acetate and vinylboronate ester groups (Scheme 20).75
Where the
leaving group (acetate) is -to the boron, this type of substrates were considered to be
bifunctional reagents as palladium can catalyze both functional groups i.e., allylic acetate via
Tsuji allylation and vinylboronate ester groups via transmetallation. Competitive reactions
between Tsuji-Trost and Suzuki could occur for these substrates, but it was observed that only
allylation products 27 were formed with excellent chemoselectivity and yields ranging from
65 to 92%.
Nucleophiles like malonates, primary amines and secondary amines successfully underwent
chemoselective Tsuji-Trost allylation (Scheme 20).
74 Batey, R. A.; Thadani, A. N.; Smil, D. V. Tetrahedron Lett. 1999, 40, 4289.75 Hussain, M. M.; Walsh, P. J. Angew. Chem., Int. Ed. 2010, 49, 1834.
39

Allylation between allylic acetate systems that contain pinacolborane substitution (Bpin) and
allylic systems without pinacolborane substitution were investigated for regioselectivity.
Interestingly, allylation occurred with high regioselectivity at benzylic position affording 28
(Table 4, entries 2 and 3) for allylic systems that contain pinacolborane. Whereas, allylation at
the other position was observed affording 29 (Table 4, entry 1) for the allylic system which
doesn’t have pinacolborane substituent (regioselectivity 1:9). Therefore, regioselectivity in
allylation was quite opposite for the systems which have boron-substitution in -allyl
palladium complex.
Allylations were performed using palladium complex without interference of pinacolborane
moiety. Also, since palladium complex catalyzes both allylation and Suzuki reaction, a one-
pot tandem allylation followed by Suzuki cross-coupling reaction strategy was developed, to
give a variety of 2-arylated allylic amines 30 with yields ranging from 65 to 70% (Schme 21).
40

Also, allylic substitution followed by oxidation in one-pot provides enol ethers which undergo
keto-enol tautomerization to provide -substituted ketones 31 in 82 to 85 % yields. This type
of products were not easy to synthesize by Tsuji-Trost allylation alone (Scheme 22).
I. 1. ii. d. In Grignard reaction:
Carboni et al reported the addition of organometallic reagents to vinylboronates possessing an
acetal group in the -position, which undergo allylic rearrangement in presence of Lewis acid
to form -alkoxy allyl boronates 32 (Scheme 23) with high stereoselectivity and E-isomer as
major.76
The reaction was independent on the nature of the metal and the size of the entering
group. Organometallic reagents like n-BuLi, PhLi, BuMgCl react with -boryl allyllic
derivatives at -78 oC to give -alkoxy allyl boronates in 50-65% yield.
OEt
OEt
Bpin RMX, BF3.Et2O
THF, -78 oC, 25 minBpin
R
OEt
E/Z > 99:1 50-65%
RMX = n-BuLi, s-BuLi, i-PrMgCl
32
Scheme 23: Grignard reaction on -boryl alkoxy derivatives
This type of products ( -substituted -ethoxy-allylboronates) 32 were difficult to purify,
hence they are readily treated with aldehydes via allylboration to result in the formation of
homoallylic alcohols 33 in one-pot with 75% yield (Scheme 24).
76 Possémé, F.; Deligny, M.; Carreaux, F.; Carboni, B. J. Org. Chem. 2007, 72, 984.
41

Typical reation mechanism (Mechanism 2) involves the attack of Grignard reagent directly on
the boronate moiety to give a tetravalent intermediate, which, on further rearrangement, forms
the -substituted allylic boronate derivative as shown below (1,2-anionotropic shift).
I. 1. ii. d. In Diels Alder reaction:
-boryl allylic alcohols on simple oxidation provide 3-boronoacrolein which was used to
synthesize cyclic -chiral allylboronate 34 via hetero-Diels-Alder cycloaddition between 3-
boronoacrolein and enol ethers, catalyzed by Jacobsen’s chiral chromium (III) catalyst
(Scheme 25).77
O
Bpin
OEt
+
O
Bpin
OEt
34(85%, 96% de)
NCr
O
O
CH3
Cl
Chromium catalyst
4 Å m.s., rt, 4 h
Chromium catalyst
Scheme 25: Hetero- [4+2]-cycloaddition of 3-boronoacrolein
77 (a) Gao, X.; Hall, D. G.; Carreaux, F.; Carboni, B. Chem. Eur. J. 2006, 12, 3132.
(b) Favre, A.; Carreaux, F.; Carboni, B. Eur. J. Org. Chem. 2008, 4900.
42

Stereoselective total synthesis of several styryllactones were achieved efficiently from
common intermediate 34. Further, this intermediate can be oxidized by hydrogen peroxide to
give corresponding alcohol 35, which can be readily converted to corresponding acetate 36
which is a useful intermediate in allylic substitution chemistry (Scheme 26).
The cyclic -chiral allylboronate 34 adds to a variety of aldehydes to give diastereomerically
pure products. A three component hetero- [4+2]-cycloaddition between 3-boronoacrolein,
enol ethers and aldehydes, catalyzed by Jacobsen’s chiral catalyst, was developed to give -
hydroxy alkyl pyrans 37 in yields ranging from 73 to 92% (Scheme 27). This -hydroxy alkyl
pyran unit shows a broad range of biological properties like antibiotic and anticancer activity.
I. 1. ii. e. In Mitsunobu reaction:
Mitsunobu reaction of -borylated allylic alcohols with nucleophiles like benzoic acid,
phenols, N-tosylamines in presence of triphenylphosphine (PPh3) and diethyl
azodicarboxylate (DEAD) leads to SN2 substitution products (Scheme 28).78
78 Berree, F.; Gernigon, N.; Hercouet, A.; Carboni, B. Eur. J. Org. Chem. 2009, 329.
43

The typical mechanism involves the reaction of triphenylphosphine with DEAD to generate a
phosphonium intermediate that converts the allylic alcohol oxygen atom to a leaving group 39
as in classical Mitsunobu reactions (Mechanism 3). Addition of the nucleophile to the boron
atom in 39 leads to the borate 40 that rearranges by an anionotropic 1,2-shift to afford -
substituted allylboronates 41 in SN2 manner, anti to the leaving group which is similar to
Grignard reaction on -borylated allylic derivatives.
The resulting Mitsunobu product 38 was used as allylating reagent. A three component one-
pot reaction was developed via Mitsunobu followed by allylboration sequences to give (Z)-
homoallylic alcohols 42 (Scheme 29). Different boronates (Scheme 29, entries 1,3,4,5)
substituted with alkyl, aryl and allyl were treated with various nucleophiles like benzoic acid,
phenols, tosylamides and aldehydes in presence of triphenylphosphine and di-tert-butyl
azodicarboxylate to obtain 42. Substituted enamides or enol benzoates were synthesized in
one-pot sequence with a high diastereoselectivity, up to >99%.
44

Trans-whisky lactone 44 was synthesized using this one-pot strategy by treating -borylated
allylic alcohol with benzoic acid under Mitsunobu conditions followed by allylboration
sequence to give intermediate 43. Compound 43, on treatment with NaOMe followed by
oxidation in presence of BF3.Et2O, afforded trans-whisky lactone 44 in 57% yield (Scheme
30).
45

Ruthenium-catalyzed cycloisomerization reaction of enyne derivative 45 was developed by
treating a -borylated allylic alcohol with N-tosyl propargylamine under Mitsunobu
conditions to give compound 45 in 69% yield. Ring closing metathesis of 45 with Grubb’s
catalyst readily converts 45 to a cyclic diene which, on allylboration with aldehydes, afforded
homoallylic alcohol 46 in 36% yield. This protocol was useful to synthesize pyrrolidines with
quaternary stereogenic centers of defined stereochemistry (Scheme 31).
I. 1. ii. f. In cyclopropane synthesis:
Cyclopropane rings were useful intermediates in organic synthesis79
and this strained ring was
observed in naturally occurring terpenes, steroids, amino acids, fatty acids, alkaloids, and
nucleic acid derivatives.80
Many cyclopropane-containing non-natural compounds also have
important biological activities. Enantiopure cyclopropane81
derivatives show important
biological activity, for example FR-900848 is a potent antibiotic against filamentous fungi,
and U-106305 is an inhibitor of cholesteryl ester transfer protein (CETP). Alkenylboronates
can be employed for the synthesis of optically pure cyclopropane derivatives 47 using chiral
79 Patai, S.; Rappoport, Z., Eds. The Chemistry of the Cyclopropyl Group; Wiley: New York.
1987, 1.80 Faust, R.; Angew. Chem. Int. Ed. 2001, 40, 2251.81 Barrett, A.; Kasdorf, K. Chem. Commun. 1996, 325.
46

auxiliaries (Scheme 32),82
further this boronate can be oxidized to get corresponding alcohol
derivatives.
CH2I2, Pd(OAc)2
Et2O, 0 oC, 1 h
BR
BR OR1*
OR1*
OMe
OMeHO
HO
HO
HO
PhPh
PhPh
PhHO
PhHO
Ph HO
HO CO2Pri
CO2Pri
OR1*
OR1*
OR1* =
47 85-96%dr upto 93:7
Scheme 32: Cyclopropanation of chiral alkenylboronates
Chiral -borylated allylic alcohols were subjected to cyclopropanation via Pd(OAc)2-
catalyzed decomposition of diazomethane afforded diastereomers 48 and 49 in 98% yield
(Scheme 33).82
On the other hand, enantiopure cyclopropylboronic ester 49 was obtained by
cyclopropanation of -borylated allylic alcohol using bis(iodomethyl)zinc as reagent and bis-
methanesulfonamide as catalyst.83
Belactosin A is a Streptomyces metabolite that inhibits the cell cycle progression of human
tumour cells, Belactosin A was synthesized using asymmetric cyclopropylamine as a key
intermediate (Scheme 34).84
This cyclopropylamine was synthesized from pure benzoate 50
which was converted into enantiomerically pure trifluoroborate 51 in 90% yield. This was
followed by amination, via the dichloroborane, with benzyl azide leading to 52 in 73% yield.
82 Luithle, J. E. A.; Pietruszka, J. J. Org. Chem. 1999, 64, 8287. 83 Denmark, S. E.; O’Connor, S. P. J. Org. Chem. 1997, 62, 3390.84 Pietruszka, J; Solduga, G. Eur. J. Org. Chem. 2009, 5998.
47

Boc protection of 52 followed by hydrogenolysis gave intermediate 53 in 86% yield. Boc
protection of 53 followed by saponification afforded enantiomerically pure building block 54
in 92% yield. It is the key intermediate for the total synthesis of Belactosin A.
BzO
NHBn
1. (Boc)2O, Et3NMeOH, 24 h, rt
2. Pd/C, H2, 3 d
BzO
NHBoc
1. DMAP, (Boc)2O,ACN, rt, 15 h
2. NaOH, MeOH, 30 min
HO
NBoc2H2N
NH
O COOHHN
O
OO
Belactosin A
53 86%
8 steps
BzO
BF3K
BzO
BO
O
MeO
Ph
Ph
Ph
OMe
Ph
51 90%
KHF2, MeOH
80 oC, 2 d
1. SiCl4, Toluene / ACNrt, 2 h
2. BnN3, 5 h
52 73%
54 92%
50
Scheme 34: Application in the synthesis of Belactosin A
I. 2. Tsuji-Trost allylation:
Allylation reactions catalyzed by transition-metal complexes bring a lot of interest and they
are used as very powerful tool in organic synthesis for C-C and C-heteroatom bond formation
(Scheme 35).85 Allylation process involves activation of the allylic position by the formation
of a -allyl palladium complex followed by reaction of this ambident electrophile with an
anion to result in allyl substituted derivatives.86
85 Tsuji, J. Tetrahedron Lett. 1965, 4387. 86 Trost, B. M.; Fullerton, T. J. J. Am. Chem. Soc. 1973, 95, 292.
48

The reaction mechanism (Mechanism 4) involves in the catalytic cycle first olefin
complexation (coordination) with palladium to give a -complex. The next step is oxidative
addition in which the leaving group is expelled to give a -allyl complex. In the case of soft
nucleophiles, nucleophile attacks at proximus or distal carbon atom of the allyl group
generating another -complex by reductive elimination. The palladium detaches from the
alkene via dissociation in completion of reaction and can start again the catalytic cycle.
The typical geometry in -allyl complex for mono-substituted unsymmetrical olefin is shown
below (Scheme 36). Between the syn and anti isomers of monosubstituted olefin, syn isomer
49

is the favoured the geometry because of the less steric hindrance between the R group and
ligand (L) in -complex.
R
RPd PdLL LL
syn(favourable)
anti(non-favourable)
Scheme 36: -allyl complex for mono substituted unsymmetrical olefin
Similarly, in case of disubstituted -allyl complex the syn-syn isomer geometry is favoured
when compared to anti-anti isomer due to steric factor. However, in some cases, anti
geometry is favoured because of steric hindrance between the substituent in ligand and R
group of -allyl complex (Scheme 37).
The most used leaving groups in allylation reaction are acetates, halides and carbonates at
allylic position. When allylic systems substituted with carbonates are subjected to allylation
reaction, the alkoxide ion generated during the -complex formation itself acts as nucleophile
during allylation. No base is required and the reaction can be carried without adding base
(Scheme 38).87
Tsuji allylation of 55 with enol carbonate produces 56 wih a quaternary
stereogenic center in 96% yield and 88% ee, when chiral ligand (S)-t-Bu-PHOX used as
ligand, 56 is a useful building block for synthetic chemistry (Scheme 38).
87 Behenna, D. C. ; Stoltz, B. M. J. Am. Chem. Soc. 2004, 126, 15044.
50

Many other leaving groups were employed in allylation such as carbamates, sulfones, halides,
phosphates and epoxides (Scheme 39).
OAc OCO2R OCONHRO
OP(O)(OR)2 Cl NO2 SO2R
Acetates Carbonates Carbamates Oxiranes
Phosphates Halides Nitro Sulfones
Scheme 39: Allylic systems used inTsuji-Trost allylation
I. 2. i. Stereochemistry in Tsuji-Trost allylation:
The stereochemistry of this allylation depends on the type of nucleophile used. The -allyl
complex 57 in Tsuji-Trost allylation is formed by SN2 type inversion, and subsequent attack
of nucleophile, i.e. either soft or hard nucleophiles, determines the configuration of the
product. Soft nucleophiles are those derived from conjugate acids whose pKa<25, like bases
generated from dialkyl malonate, -ketoester, enamine and -diketone which attack directly
on allyl moiety via SN2 reaction to give product 58 with inversion of configuration at this
step. Allylation with soft nucleophiles involves a double inversion mechanism which leads to
overall retention of product 58a (Scheme 40).
51

Whereas, reaction with hard nucleophiles follows a different mechanism. Hard nucleophiles
are those derived from conjugate acids whose pKa > 20, such as organometallic reagents like
Grignard reagent, organozirconium, organozinc and organotin reagents which first attack the
metal center in -complex 57 via transmetallation followed by reductive elimination to give
the allylation product 58b with overall inversion of configuration (Scheme 41).
CO2Me
OAc
CO2Me
Me
(overall inversion)
Pd(PPh3)4 / PPh3
MeMgBr, THF, 0 oC to rt, 8 h
90%
Scheme 41: Stereochemistry of allylation with hard nucleophiles
52

I. 2. ii. Regioselectivity in Tsuji-Trost allylation:
Symmetrical allylic systems during palladium catalysis do not generate regioselectivity issues,
whereas unsymmetrical allylic systems during palladium catalysis have regioselectivity
issues. Allylation occurs at less substituted carbon in majority, according to steric effect
(Scheme 42).88
Soft nucleophiles like malonate and morpholine attack the unsymmetrical -allyl complex at
the less substituted carbon in majority to result in allylation products according to steric
factor. But hard nucleophiles, like PhZnCl, attack unsymmetrical -allyl complex at more
substituted carbon, and this is due to the fact that hard nucleophile first attacks on palladium
in the -allyl complex via transmetallation. Then the ligand and phenyl group orient for a
more stable -allyl complex (Shown below). After this stable -complex formation, the
phenyl group attacks at adjacent carbon to give the allylated product.
iBuMe
PdPh3P Ph
iBuMe
PdPh PPh3
-Complex in hard nucleophiles
88 Trost, B. M.; Hung, M. H. J. Am. Chem. Soc. 1984, 106, 6837.
53

The stereochemical version of allylation in unsymmetrical -allyl complex is shown below.
Soft nucleophiles attack at less substituted carbon with stereoretention in 97% yield as major
product, however 3% of the other isomer with stereoretention was formed as minor product in
allylation (Scheme 43).
I. 2. iii. Asymmetric allylic alkylation (AAA):
Introducing enantioselectivity89
in allylation reactions starting from a racemic substrate
represents a new dimension to their use in organic synthesis. Ligands play important roles for
developing enantioselectivity in allylation reactions, and the chiral information on the ligand
is directly responsible for the enantioselectivity. The ability to transform achiral, prochiral, or
chiral material to enantiopure material in allylation is termed as asymmetric allylic alkylation
(AAA, Scheme 44).
RIR
OAc
PdL*n RIR
PdL*n
R1R R1R
CH(CO2R'')2CH(CO2R'')2
* *CH2(CO2RII)2
-OAc+
Scheme 44: Asymmetric allylic alkylation
Trost et al synthesized different chiral ligands for allylation reaction.90
Most of the chiral
ligands are commercially available for various synthetic needs. The most extensively studied
example to demonstrate the efficiency of ligand is 1,3-diphenylprop-2-enyl acetate 59.
89 Trost, B. M.; Strege, P. E. J. Am. Chem. Soc. 1977, 99, 1649.90 Trost, B. M.; Vranken, D. L. Chem. Rev. 1996, 96, 395.
54

However, the results from this system do not necessarily translate into high enantioselectivity
for other substrates. Chiral ligands based on nitrogen and phosphines were extensively used
for allylation reactions because of the strong binding nature of these ligands to palladium
catalyst. A model asymmetric allylic allylation reaction of 1,3-diphenylallyl acetate (59)
under palladium catalysis with malonate nucleophile, under different chiral ligands was
studied for enantioselectivities in the product 60 (Scheme 45, Table 5).
Ph
OAc
Ph
MeOOC COOMe Pd(allyl2Cl2), ligand*
solvent, reflux Ph PhNa+
COOMeMeOOC
*
59 60
Scheme 45: Asymmetric allylic alkylation on 1,3-diphenylallyl acetate
Entry ligand % yield % ee
1. L1 98 91
2. L2 83 95
3. L3 86 90
4. L4 68 85
5. L5 86 77
6. L6 89 81
7. L7 85 85
8. L8 97 88
9. L9 99 99
10. L10 56 92
11. L11 92 96
12. L12 80 34
13. L13 81 95
14. L14 89 99
Table 5: Enantioselectivity studies in allylation
55

Various Chiral ligands used in Tsuji-Trost allylation:
56

A wide variety of bidentate ligands ranging from bisphosphines91
(Table 5, entries 3, 4, 12)
and bisamines92
(Table 5, entries 1, 2, 13, 14) are capable of inducing enantioselectivity to
give 60 with good yields. Oxazoline ligands93
during allylation gave 60 with high
enantioselectivities up to 99% and yields up to 99% (Table 5, entries 7, 8, 9 and 11). In case
of allylation with sodium dimethylmalonate using ligand (S)-BINAP in THF, a selectivity was
observed as low as 34% (Table 5, entry 12). It was improved to 94% when the solvent system
changed to dichloromethane.94
Allylation reaction conditions need to be optimized for each
new ligand/substrate/nucleophile/solvent combination in order to find the best efficiency for
the reaction.
However, ligands not only introduce chirality into the products but they also influence the
regioselectivity during allylation reaction. Simple allylation of optically pure 1-phenyl-p-
tolyl-disubstituted allyl acetate 61 with dimethylmalonate affords the products 62 and 63 in
1:1 ratio when triphenylphosphine is used as ligand. The formation of regioisomers can be
greatly influenced by the ligands used in the reaction.95
For instance, using chiral ligands
derived from phosphino-dihydrooxazoles (R)-L9 and (S)-L9 each of the regioisomers 62 and
63 could be obtained in high yield and high enantioselectivity (Scheme 46).
91 Yamazaki, A.; Morimoto, T.; Achiwa, K. Tetrahedron: Asymmetry 1993, 4, 2287.92 Gamez, P.; Dunjic, B.; Fache, F.; Lemaire, M. J. Chem. Soc.,Chem. Commun. 1994, 1417.93 Vonmatt, P.; Pfaltz, A. Angew. Chem., Int. Ed. Engl. 1993, 32,566.94 Yamaguchi, M.; Shima, T.; Yamagishi, T.; Hida, M. TetrahedronLett. 1990, 31, 5049.95 Vonmatt, P.; Lloyd-Jones, G. C.; Pregosin, P. S. Helv.Chim.Acta. 1995, 78, 265.
57

This shows that ligands can dictate regiochemistry in allylation, however this is applicable
only to this substrate, and the results from this system can not be generalized to other
substrates. The selectivity is not well documented for the Trost allylation when similar aryl
groups were present on both the sides of -complex, and it is very difficult to control the
regioselectivity in allylation when chemically equivalent groups are present on both sides.
The Trost allylation products are directly used for the synthesis of many natural products.
I. 2. iv. Application in natural product synthesis:
Helmchen et al reported the synthesis of enantiomerically pure (-)-wine lactone based on a
palladium-catalyzed enantioselective allylic substitution with the lithium anion of malonate
(Scheme 47).96
Apart from malonate nucleophiles, -ketoesters were also used as nucleophiles in palladium-
catalyzed allylation by using chiral Trost ligand in the synthesis of (-)-nitramine (Scheme
48).97
96 Bergner, E. J.; Helmchen, G. Eur. J. Org. Chem. 2000, 419.97 Trost, B. M.; Radinov, R.; Grenzer, E. M. J. Am. Chem. Soc. 1997, 119, 7879.
58

Trost et al reported the use of primary alcohols as nucleophiles in palladium-catalyzed
allylation by using chiral Trost ligand in the synthesis of (-)-malyngolide (Scheme 49).98
(+)-Cyclophellitol, an HIV virus inhibitor, was synthesized via palladium-based allylation
using carboxylate nucleophile.99
Pivalic acid was used as oxygen nucleophile in palladium-
catalysed allylation using chiral Trost ligand to result in the adduct in 44% yield with 97% ee.
This compound was a key intermediate for the synthesis of (+)-Cyclophellitol (Scheme 50).
OAc
OAc
OAc
OAc
Pd2dba3-CHCl3, CH2Cl2, L15
Pivalic acid, H2O, NaOH, rt, 24 h OAc
OAc
OAc
OCOtBuO
OH
OH
OHHO
44%, 97%ee(+)-Cyclophellitol
7 steps
Scheme 50: Tsuji-Trost allylation in the synthesis of (+)-Cyclophellitol
Azides are interesting nucleophiles in allylation for the C-N bond formation, (-)-Epibatidine
was synthesized via palladium-catalyzed stereoselective allylation using azide as nucleophile
(Scheme 51).100
98 Trost, B. M.; Tang, W.; Schulte, J. L. Org. Lett. 2000, 2, 4013.99 Trost, B. M.; Hembre, E. J. Tetrahedron Letters. 1999, 40, 219.100 Trost, B. M.; Cook, G. C. Tetrahedron Lett. 1996, 37, 7485.
59
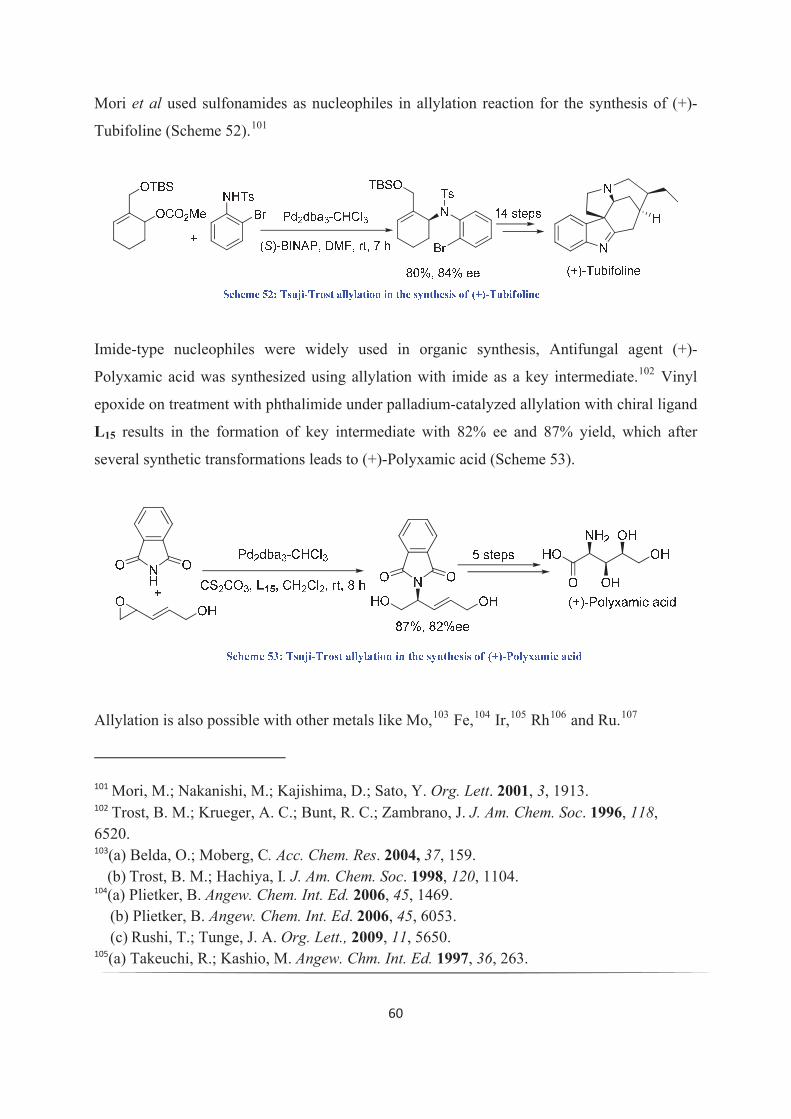
Mori et al used sulfonamides as nucleophiles in allylation reaction for the synthesis of (+)-
Tubifoline (Scheme 52).101
Imide-type nucleophiles were widely used in organic synthesis, Antifungal agent (+)-
Polyxamic acid was synthesized using allylation with imide as a key intermediate.102
Vinyl
epoxide on treatment with phthalimide under palladium-catalyzed allylation with chiral ligand
L15 results in the formation of key intermediate with 82% ee and 87% yield, which after
several synthetic transformations leads to (+)-Polyxamic acid (Scheme 53).
Allylation is also possible with other metals like Mo,103
Fe,104
Ir,105
Rh106
and Ru.107
101 Mori, M.; Nakanishi, M.; Kajishima, D.; Sato, Y. Org. Lett. 2001, 3, 1913.102 Trost, B. M.; Krueger, A. C.; Bunt, R. C.; Zambrano, J. J. Am. Chem. Soc. 1996, 118,
6520.103(a) Belda, O.; Moberg, C. Acc. Chem. Res. 2004, 37, 159.
(b) Trost, B. M.; Hachiya, I. J. Am. Chem. Soc. 1998, 120, 1104.104(a) Plietker, B. Angew. Chem. Int. Ed. 2006, 45, 1469.
(b) Plietker, B. Angew. Chem. Int. Ed. 2006, 45, 6053.
(c) Rushi, T.; Tunge, J. A. Org. Lett., 2009, 11, 5650.105(a) Takeuchi, R.; Kashio, M. Angew. Chm. Int. Ed. 1997, 36, 263.
60

I. 3. Selectivity issues in palladium-catalyzed Tsuji-Trost allylation of
-borylated allyl acetates:
-borylated allylic acetates contain many reactive centres, especially when this substrate will
be subjected to palladium catalysis (Scheme 54). Palladium can activate the allylic system, as
well as the boronate present in the substrate. The activation of allylic system by replacing the
acetate functional group, with palladium catalyst to give a -allyl complex, followed by attack
of nucleophile is called the Tsuji-Trost allylation reaction.
(b) Takeuchi, R.; Kashio, M. J. Am. Chem. Soc. 1998, 120, 8647.
(c) Bartels, B.; Helmchen, G. Chem. Commun. 1999, 741.
(d) Bartels, B.; Garcõ´a-Yebra, C.; Rominger, F.; Helmchen, G. Eur. J. Inorg. Chem. 2002,
2569.
(e) Kanayama, T.; Yoshida, K.; Takemoto, Y. Angew. Chem., Int. Ed. 2003, 42, 2054.
(f) Graening, T.; Hartwig, J. F. J. Am. Chem. Soc. 2005, 127, 17192.
(g) Weihofen, R.; Tverskoy, O.; Helmchen, G. Angew. Chem., Int. Ed. 2006, 45, 5546.106 (a) Evans, P. A.; Nelson, J. D. Tetrahedron Letters, 1998, 39, 1725.
(b) Evans, P. A.; Nelson, J. D. J. Am. Chem. Soc. 1998, 120, 5581.
(c) Hayashi, T.; Okada, A.; Suzuka, T.; Kawatsura, M. Org. Lett. 2003, 5, 1713.
(d) Evans, P. A.; Leahy, D. K. J. Am. Chem. Soc. 2000, 122, 5012.
(e) Evans, P. A.; Robinson, J. E.; Nelson, J. D. J. Am. Chem. Soc. 1999, 121, 6761.107 (a) Trost, B. M.; Fraisse, P. L.; Ball, Z. T. Angew. Chem. Int. Ed. 2002, 41, 1059.
(b) Morisaki, Y.; Kondo, T.; Take-aki, M. Organometallics, 1999, 18, 4742.
61

This allylation results in the formation of either branched or linear products, depending on the
catalyst/ligand used. For example, palladium majorly gives linear products whereas
molybdenum, iron and rhodium give branched products. In the case of iridium-catalyzed
allylation, branched-type allylic substrates give branched products whereas linear-type allylic
substrates tend to give mixtures. The main challenge in Tsuji-Trost allylation is selectivity.
-borylated allylic electrophiles can display different reactivities towards metal complexes
because of the multiple reaction sites present in these synthons and many selectivity issues
can arise from their reaction. Therefore, -borylated allylic acetates are interesting substrates
to study selectivity issues in palladium catalyzed allylation.
Achieving selective palladium-catalyzed allylic substitution on -borylated allylic derivatives
with mild nucleophiles is a much greater challenge and leads “to mixtures of regioisomeric
deboronation products and other unidentified materials” as noticed by Hall et al.108
The issue
of chemoselectivity was not well documented for the substrates having many reactive sites.
Palladium-catalyzed reaction of -borylated allylic acetates generate several selectivity issues
(Scheme 54).
1. Issue of chemoselectivity between Tsuji-Trost (a) and Suzuki (a1) reactions
2. Issue of regioselectivity during allylation of unsymmetrical -allyl complex between
-position (b) and -position (b1).
3. Issue of stereoselectivity between stereoretention (c) and stereoinversion (c1) during
allylation.
Objectives:
Walsh et al reported palladium-catalyzed chemoselective allylation on -borylated allylic
acetates, where the leaving group (acetate) is -to the boron.109
However, the -borylated
allylic acetates offer challenge to perform allylation because of the multiple reactive sites
108 Peng, F.; Hall, D. G. Tetrahedron Lett. 2007, 48, 3305.109 Hussain, M. M.; Walsh, P. J. Angew. Chem. Int. Ed. 2010, 49, 1834.
62

present in it. Palladium-catalyzed reaction of -borylated allylic acetates generate chemo,
regio and stereoselectivity issues shown in Scheme 54. Therefore, in the second chapter we
will be interested in generating a -complex from -borylated allylic acetates inorder to study
its reactivity in Tsuji-Trost allylation, as well as the chemo, regio and stereoselectivity of the
reaction with various nucleophiles.
63

Chapter II: Palladium-catalyzed Tsuji-Trost
allylation of -borylated allyl acetates
II.1: Synthesis of -borylated allyl acetates
II.2: Palladium-catalyzed Tsuji-Trost allylation
of -borylated allyl acetates
64

II. 1. Synthesis of -borylated allyl acetates:
II. 1. i. From hydroboration of propargylic acetate systems:
According to previous reports,25
-borylated allylic acetates can be synthesized via
hydroboration of TMS-protected propargylic systems with pinacolborane. Deprotection of
TMS-protected -borylated derivatives with citric acid affords stable -borylated allylic
alcohols in 70-76% yield. Subsequent acetylation with acetic anhydride leads to the final -
borylated allyl acetates in 75-80% yield. In this protocol removal of excess pinacolborane by
silica gel chromatography was problematic. Hence, we moved to diisopinocampheylborane
(readily generated from -pinene by hydroboration with borane-dimethylsulfide complex).
Hydroboration was performed on propargylic acetates instead of TMS-protected propargylic
alcohols (Scheme 55).
2
(Ipc)2BH
-35 oC to rt,16 h OAcR
2 Me2S.BH3+THF BH
OAc
0 oC to rt, 4 h
B
AcOR
O
O
1. CH3CHO / 40 oC,16 h
2. Pinacol / rt, 6 h
R'R R'
R'
B(Ipc)2
Entry R R' 64 Yield (%)
a. H H 64a 75
b. CH3 H 64b 80
c. Ph H 64c --
d. CH3 CH3 64d --
64
Scheme 55: Hydroboration of propargylic acetates
65

Hydroboration of propargylic acetates with diisopinocamphenylborane was smoothly carried
out from -35 oC to room temperature for 16 h. Refunctionalization of camphenyl derivative
with acetaldehyde by refluxing for 16 h resulted in the diethylboronate which was
transesterified with pinacol in one pot to give final -borylated allylic acetates 64a and 64b in
75 and 80% yield respectively. This method was not successful when the R, R’ substituents at
allylic position were Ph, H and Me, Me (Scheme 55, entries c and d). Indeed, in this case
unseperable complex mixture was obtained, which didn’t show the required product by crude
1HNMR.
Another attempt by direct hydroboration of propargylic acetates with pinacolborane to give -
borylated allyl acetate derivative failed and no product was observed (Scheme 56). In this
case starting material was not consumed.
Therefore, a modified method was developed for the preparation of -borylated allyl acetate
derivatives.
II. 1. ii. From hydroboration of propargylic alcohol systems:
Contrary to previous results with propargylic acetates, direct hydroboration of propargylic
alcohols to give -borylated allyl alcohol derivatives was successful with the four substrates.
This protocol was more efficient in terms of yield (Scheme 57). Hydroboration of propargylic
alcohols with diisopinocampheylborane, followed by refunctionalization with acetaldehyde,
and transesterification with pinacol, afforded -borylated allyl alcohol derivatives 65 in yields
ranging from 75 to 80% (Scheme 57, entries a, b, c, d). The -borylated allyl alcohol
66

derivatives were acylated to get the final -borylated allyl acetate derivatives 66 in 85%
yields. This protocol was more efficient than previous approaches.
2(Ipc)2BH
-35 oC to rt,16 h OHR
2 Me2S.BH3+THF BH
OH
0 oC to rt, 4 h
B
HOR
O
O
1.CH3CHO / 40 oC,16 h
2.Pinacol / rt, 6 h
R'R R'
R'
B(Ipc)2
Ac2O, Et3N
DMAP, CH2Cl20 oC to rt, 2 h
B
AcOR
O
OR'
Entry R R' 65 Yield (%) 66 Yield (%)
a. H H 65a 80 66a 85
b. CH3 H 65b 76 66b 85
c. Ph H 65c 75 66c 85
d. CH3 CH3 65d 79 66d 85
65 66
Scheme 57: Hydroboration of propargyl alcohols and synthesis of target molecules 66
II. 2. Reactivity of -borylated allyl acetates under palladium catalysis:
As discussed earlier in Scheme 54, palladium-catalyzed reaction of -borylated allylic acetates
generate many selectivity issues, and our initial attempts on these substrates with palladium
catalyst gave interesting results. The allylated branched-type product 67 (Table 6) was
observed in good yield, and high regioselectivity under many reaction conditions. Product 68
was not observed although theoretically possible (Table 6). A high yield was observed for 1%
Pd(OAc)2 and 3% PPh3 catalytic system (77%, entry 1) when malonate was used as
nucleophile. Increase in the ligand amount from 3% to 4% (entry 5) didn’t change the yield.
Reactions failed with less ligand loading, i.e. less than 3% (entry 10). Investigation by using
N-heterocyclic carbene ligands during allylation was not successful and even failed in
combination with different bases like K2CO3, KOtBu and triethylamine (entry 9). Pyridyl-type
67

ligands gave lower conversion, this might be due to the problem of coardination beween
pyridyl ligand and palladium metal (entry 11).
OAc
B(pin) B(pin)
1.1mol% [Pd] , n% ligand,THF
CO2MeMeO2CNa
2.
3. rt to reflux, 4 h
MeO2C CO2Me
B(pin)(or)
67 68
CO2MeMeO2C
(1.1 eq)
Entry Catalyst Ligand Yield 67(%)
1. Pd(OAc)2 PPh3(3%) 77
2. PdCl2 PPh3(3%) 72
3. [Pd(allyl)Cl]2 PPh3(3%) 76
4. Pd(OAc)2 ---- 0
5. Pd(OAc)2 PPh3(4%) 77
6. Pd2(dba)3.CHCl3 PPh3(2%) 70
7. Pd(dba)2 PPh3(2%) 70
8. Pd(PPh3)4 ---- 75
9. Pd(OAc)2 NHC-carbene(3%) 0
10. Pd(OAc)2 PPh3(1%) 0
11. [Pd(allyl)Cl]2 Pyridyl Trost(3%) 25
NN
NHC-Carbene
NH HNOO
N N
(R,R)-DACH- pyridyl Trost ligand
Table 6: Optimization of catalytic system for regioselective allylation
As expected, the reaction with Pd(II) alone without ligand wasn’t successful [Pd(OAc)2, Entry
4] and the use of hindered phosphine ligand is required for this reaction. Other catalytic
systems like Pd2(dba)3 and PdCl2 along with PPh3 ligand reacted smoothly to give almost
similar yields (~70 %, entries 2 and 7). Catalysts like Pd(PPh3)4, [Pd(allyl)Cl]2 gave almost
68

equal yields to that of palladium(0) generated by reduction of Pd(OAc)2 with PPh3 (~75 %,
entries 3 and 8). Catalyst loading of 1% is enough to carry out the reaction in good yields.
Increased amount of nucleophile to 2 equivalents didn’t change the yield. A typical ratio of
catalyst (1%) / ligand (3%) is required for the reaction to be successful. No products of direct
transmetallation between boron and palladium were observed.
II. 2. i. Regioselectivity with carbon nucleophiles:
Keeping in view of easy handling, 1% Pd(OAc)2/3% PPh3 in THF was selected as catalytic
system for allylation reaction using different nucleophiles. Using these optimized conditions,
a variety of nucleophiles were tested for their chemo-, and regio-selective allylation and
initially the attempts were carried out with enolate-type nucleophiles (Scheme 58).
Firstly, sodium salt of dimethylmalonate (generated by treatment of dimethylmalonate with
NaH) was used as nucleophilic source in the optimized catalytic system. The allylation on -
allyl complex with sterically less crowded substrates (when R, R1 = H) gave 74% yield with
ipso substitution (Table 7, entry 1). A little increase in the steric hindrance from hydrogen to
methyl (when R= Me, R1 =H) didn’t change the position of allylation and gave 77% yield
(Table 7, entry 2). Further increase in crowding from methyl to dimethyl group (when R, R1 =
Me) also resulted in the same type of allylation products in 80% yield (Table 7, entry 3).
Presence of the boron atom in the -allyl complex drives allylation to -position irrespective
of the nuclophile used. In case of phenyl substituent, a lower 61% yield was obtained (Table
7, entry 4). This is unexpected, considering the traditional outcome of the Tsuji-Trost reaction
in the presence of palladium complexes. However, another product 69d’was obtained in 15%
yield by direct coupling of boronate moiety with malonate.
69

-ketoester (3-oxobutyric acid methyl ester) was tested under these optimized conditions
using NaH as base (Table 8). The unsubstituted -borylated allylic acetate reacted smoothly to
give a regioselective allylation product with substitution at -position in 76% yield (Table 8,
entry 1). Methyl and dimethyl substituted -borylated allylic acetate derivatives underwent
allylation reaction with -ketoester nucleophile, in 80% and 83% yield respectively (Table 8,
entries 2 and 3).
1,3-diketones (pentane-2,4-dione) underwent allylation with -borylated allylic acetate
derivatives to give regioselective allylation products at -position (Table 9). Methyl-
70

susbstituted derivative in this allylation gave the product in 80% yield (Table 9, entry 1)
whereas the dimethyl substituted derivative was obtained in 82% yield (Table 9, entry 2).
Cyanoacetates (methyl 2-cyanoacetate) also reacted efficiently under these allylation
conditions, gave regioselective products with substitution at -position in high yields (Table
10). In case of unsubstituted -borylated allylic acetate derivatives, a 77% yield (Table 10,
entry 1) was obtained. The methyl substituted derivative gave 80% yield (Table 10, entry 2)
and the dimethyl substrate gave 79% yield (Table 10, entry 3).
Sterically hindered nucleophiles, such as 2-oxocyclopentanecarboxylic acid methyl ester,
successfully underwent allylation reaction to give products with quaternary centers in very
good yields. Unsubstituted (when R = H) -borylated allylic acetate derivatives, in this
reaction, gave the allylation product in 79% yield (Table 11, entry 1), whereas methyl
71

substituted (when R = Me) derivative gave 75% yield of the allylated product (Table 11, entry
2).
Allylation on -borylated allylic acetate derivatives with aqueous NaCN resulted in -
borylated allylic alcohol derivatives instead of cyano group substitution at -position. Another
nucleophile generated from acetophenone (using NaH and t-BuOK) was not successful in this
allylation.
II. 2. ii. One-pot allylation followed by Suzuki-Miyaura cross coupling:
The boronate moiety, present in allylation products after chemo-, and regio-selective
allylation of -borylated allylic acetates, can further be transformed via Suzuki-Miyaura cross
coupling for new carbon-carbon bond formation (Scheme 59).
Starting from 70 the cross coupling was performed with phenyl iodide in THF along with 1%
Pd(OAc)2 and 3% PPh3 as catalytic system and using aqueous K2CO3. In the case of methyl
substituent, sequential allylation (70a, 77% yield), followed by Suzuki cross coupling (71a,
91% yield), led to final product with an overall yield of 70% (Scheme 59, entry 1). Similarly,
in the case of phenyl substituent, sequential allylation (70b, 61% yield), followed by Suzuki
cross-coupling (71b, 75% yield), led to final product with an overall yield of 45% (Scheme
59, entry 2).
72
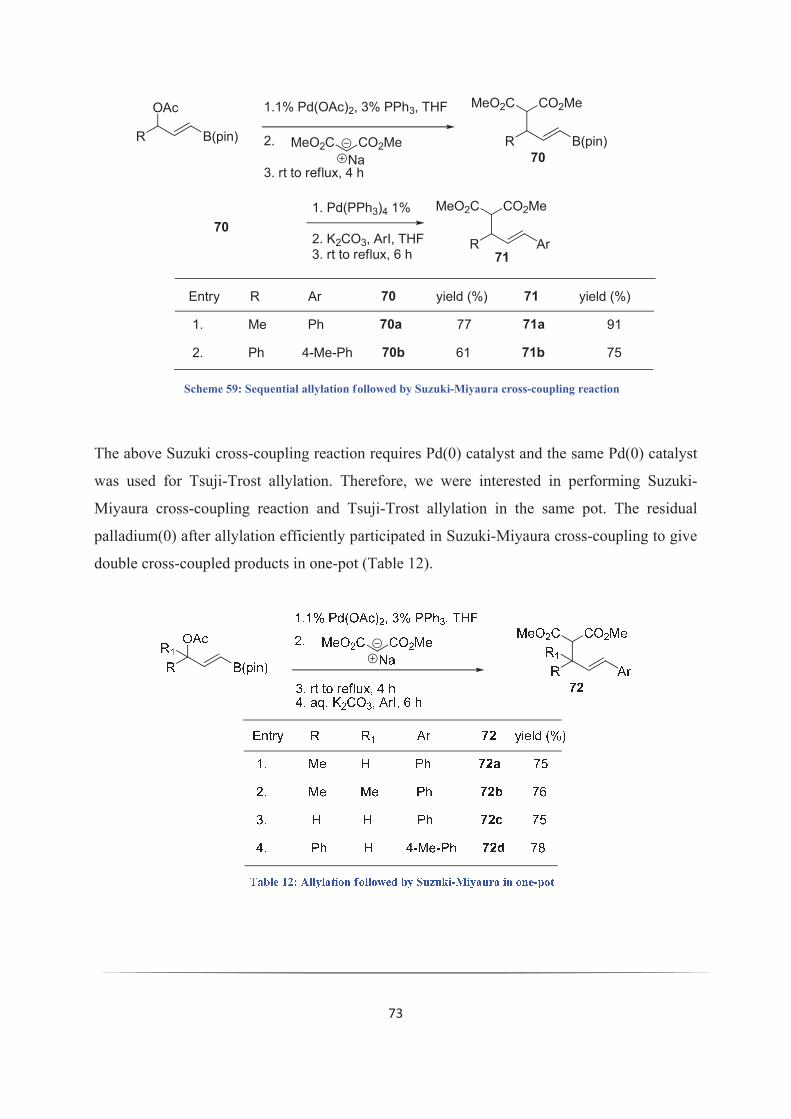
1. Pd(PPh3)4 1%
2. K2CO3, ArI, THF3. rt to reflux, 6 h
R Ar
MeO2C CO2Me
Entry R Ar 70 yield (%) 71 yield (%)
1. Me Ph 70a 77 71a 91
2. Ph 4-Me-Ph 70b 61 71b 75
1.1% Pd(OAc)2, 3% PPh3, THF
CO2MeMeO2CNa
2.
3. rt to reflux, 4 h
R
MeO2C CO2Me
B(pin)R
OAc
B(pin)
70
71
70
Scheme 59: Sequential allylation followed by Suzuki-Miyaura cross-coupling reaction
The above Suzuki cross-coupling reaction requires Pd(0) catalyst and the same Pd(0) catalyst
was used for Tsuji-Trost allylation. Therefore, we were interested in performing Suzuki-
Miyaura cross-coupling reaction and Tsuji-Trost allylation in the same pot. The residual
palladium(0) after allylation efficiently participated in Suzuki-Miyaura cross-coupling to give
double cross-coupled products in one-pot (Table 12).
73

It was observed that one-pot reaction gave good yields when compared with sequential cross-
couplings. In the case of methyl substituent, the yield of one-pot strategy to obtain double
cross-coupled product was 75% (Table 12, entry 1). This was more than the yield of
sequential cross coupling, 70%. Similarly, dimethyl-substituted -borylated allylic acetate (R,
R1 = Me) gave the one pot product in 76% yield (Table 12, entry 2) and for unsubstituted -
borylated allylic acetate (R, R1 = H) this one-pot strategy gave 75% yield (Table 12, entry 3).
In case of phenyl substituent the yield of one-pot sequence was 78% (Table 12, entry 4) which
was superior to stepwise process where the yield was 45%.
II. 2. iii. Application of the one-pot strategy:
In particular, it is very difficult to control the regioselectivity in allylation when both sides of
the -complex is flagged by similar aromatic groups (Case 1).110
This one-pot method is
useful especially to carry out selective allylation at desired position and the resulting boronate
can be transformed to the required aryl group (Case 2) using Suzuki-Miyaura cross-coupling
reaction (Scheme 60) in high yields.
74
110 Vonmatt, P.; Lloyd-Jones, G. C.; Pregosin, P. S. Helv.Chim.Acta. 1995, 78, 265.

After solving the issue of chemo and regioselectivity we focused our studies on the
development of stereochemistry and asymmetric allylation (which transforms the racemic
material to enantiopure material). Preliminary investigation was carried with allylation of
enantiopure -borylated allylic acetate substrates. The stereochemistry of the products after
allylation was studied to establish the absolute configuration.
II. 2. iv. Stereoselectivity:
The (S)- -borylated allylic acetate (ee> 99% by Chiral GC) was synthesized in the laboratory
by the same route, but starting from commercially available optically pure propargylic
alcohol. Allylation was performed on this (S)-enantiomer using the same optimized
conditions, i.e. 1 mol% Pd(OAc)2 and 3 mol% PPh3 with 1.1 equivalent of nucleophile
(generated from freshly distilled dimethylmalonate on treatment with NaH). The allylation
product 73 was obtained in 76% yield with 88% ee (Scheme 61). However, the absolute
configuration can’t be assigned directly from this product at this stage. Hence, we were
interested to convert 73 to an already existing product in order to establish the absolute
configuration. The ambiguity between inversion or retention was solved when >99% ee
compound (Table 13, entry 1) was cross-coupled with phenyl iodide using Suzuki reaction in
order to measure the specific rotation of 74 (Scheme 61).
B
OAc1% Pd(OAc)2, 3% PPh3, THF
, 4 h, reflux
B
MeO2C CO2Me
88%ee, 76%yield
(S)CO2MeMeO2C
NaO
O
O
O
73
Scheme 61: Stereochemistry of allylation
B
MeO2C CO2MePd(0), aq. Na2CO3
PhI, THF, reflux, 6 h Ph
MeO2C CO2Me
D
(S)O
O
>99%ee(Table 13, entry 1)
73 74
(Table 13, entry 4)
75
It was observed that 74 has specific rotation [ ]D = -70 (c 1.8, CHCl3) which can be compared
with existing known compound. From literature, for (S) product, the specific rotation for 74

was observed [ ]D = -51.2 (c 1.8, CHCl3), with 80% ee.111
Hence, the allylation product was
assigned with absolute configuration (S). Therefore, allylation of -borylated allylic acetate
proceeds with retention of configuration which is in agreement with the Tsuji-Trost allylation
mechanism. According to Tsuji-Trost allylation, the -allyl complex was formed by SN2
inversion followed by nucleophilic attack in SN2 inversion manner to result in double
inversion product with overall retention of configuration. The high level of selectivity was
observed with Trost ligands in allylation. These ligands are sterically crowded and readily
form -complex when treated with allylic acetates.
B
OAc 1% [Pd(allyl)Cl]2 0.5%, L 2%,THF
, 4 h, reflux
B
MeO2C CO2Me
(S) CO2MeMeO2C
NaO
O
O
O(S)
Entry Substrate Ligand % yield % ee
1. (S)-OAc (S,S)-DACH phenyl 80 >99 (S)Trost ligand (L15)
2. (S)-OAc (R,R)-DACH phenyl 80 >97(S)Trost ligand (L15)
3. (S)-OAc (R,R) + (S,S)-DACH 80 >98 (S)phenyl Trost ligand(L15)
4. (S)-OAc PPh3 76 88 (S)
Table 13: Stereochemical influence of ligands in allylation
Matched pair in allylation, i.e. (S)- -borylated allylic acetate on allylation using (S,S)-DACH
phenyl Trost ligand, resulted in (S)-product in 80% yield with ee >99% (Table 13, entry 1,
HPLC A), whereas the same (S)-OAc on allylation with mismatched pair (R,R)-DACH
phenyl Trost ligand resulted in the same (S)-product in 80% yield with 97% ee (Table 13,
entry 2). It was quite surprising that both the Trost ligands resulted in the same configuration
in the product. Therefore, we tested the (S)-OAc on allylation with racemic mixture of Trost
ligands, and the product was obtained with 80% yield and ee > 98% (Table 13, entry 3).
111 Plietker, B. Angew. Chem. Int. Ed. 2006, 45, 1469.
76
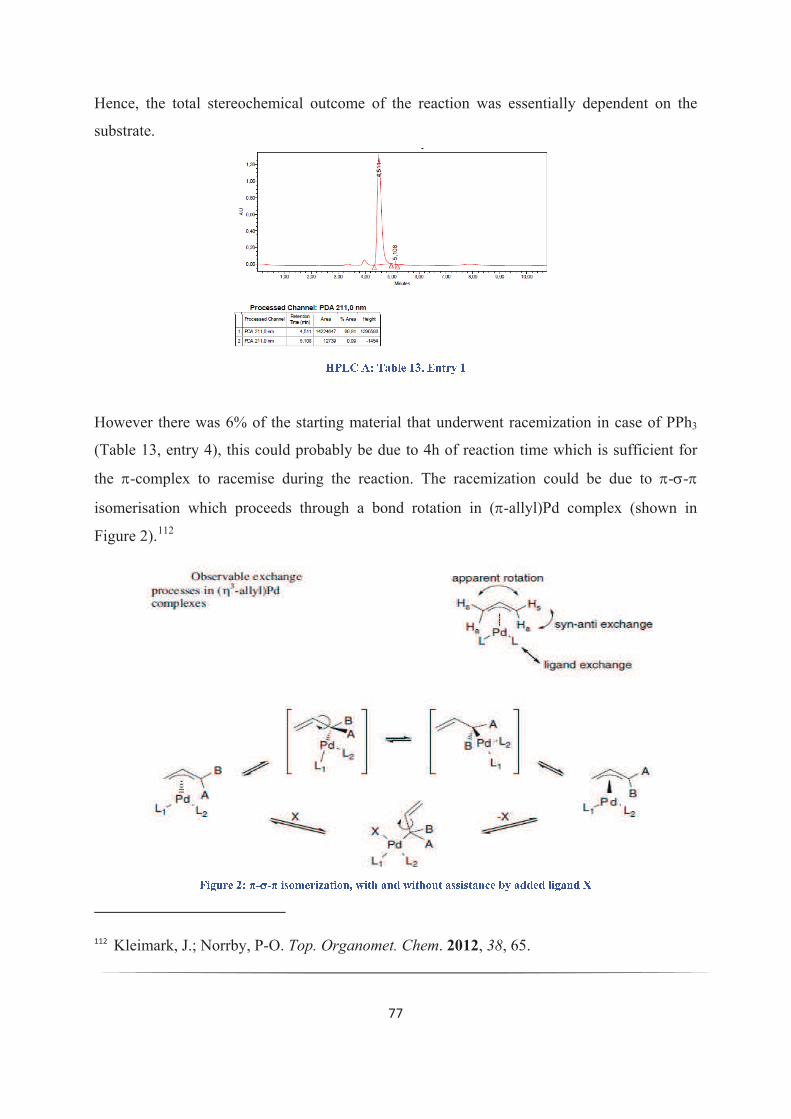
Hence, the total stereochemical outcome of the reaction was essentially dependent on the
substrate.
However there was 6% of the starting material that underwent racemization in case of PPh3
(Table 13, entry 4), this could probably be due to 4h of reaction time which is sufficient for
the -complex to racemise during the reaction. The racemization could be due to - -
isomerisation which proceeds through a bond rotation in ( -allyl)Pd complex (shown in
Figure 2).112
112 Kleimark, J.; Norrby, P-O. Top. Organomet. Chem. 2012, 38, 65.
77

Asymmetric allylic alkylation (AAA) where the nonchiral or prochiral material was converted
to chiral material during allylation was studied using -borylated allylic acetates. Allylation
with malonate as nucleophile along with several Trost ligands were investigated and the
enantioselectivity was studied, starting from racemic -borylated allylic acetates. The chiral
ligands used in AAA of -borylated allylic acetates are shown below:
Asymetric allylic alkylation with (S,S)-DACH phenyl Trost ligand instead of
triphenylphosphine resulted 80% yield with 78% ee (Table 14, entry 1) of (S)-product,
whereas the (R,R)-DACH phenyl Trost ligand gave 80% yield with 72% ee of other
enantiomer (R)-product (Table 14, entry 2). Increase in ligand steric crowding from phenyl to
naphthyl group, i.e. (R,R)-DACH naphthyl Trost ligand in allylation, increased the yield up to
84% but resulted in a drastic decrease in enantioselectivity, 54% (Table 14, entry 3). Further
increase in steric crowding from naphthyl to a modified diamine chiral ligand (R,R)-ANDEN
phenyl Trost ligand gave the other enantiomer (S) with very good yields up to 85% but with
tremendous decrease in selectivity, 30% (Table 14, entry 4) was obtained. Overall, increase in
the steric hindrance of ligand decreased the enantioselectivity, and the simplest ligand turned
out to give the best selectivities. The nitrogen-based pyridyl ligand, i.e. (R,R)-DACH pyridyl
78

Trost ligand in this allylation, gave a low 25% yield and a very poor selectivity of 11% ee
(Table 14, entry 5).
The boronate moiety obtained after allylation obtained can be transformed to other functional
groups. For instance, Chan-Lam-Evans coupling of vinylboronates on treatment with sodium
azide under copper catalyst affords efficient C-N bond formation to result in the -
functionalized vinyl azide 75 in 78% yield (Scheme 62).
In short, a chemo-, regio-, and stereo-selective allylation on -borylated allylic acetates was
achieved with carbon nucleophiles. A further study of allylation was studied on -borylated
79

allylic acetates using nitrogen and oxygen based nucleophiles as the products can be useful
bulding blocks.
II. 2. v. Regioselectivity with nitrogen nucleophiles:
Palladium-catalyzed reaction of -borylated allylic acetates generate chemo-, regio- and
stereo-selective issues. Allylation with nitrogen nucleophiles was investigated to confirm a
general strategy for selective substitutions in -borylated allylic acetates (Table 15).
Preliminary experiments were carried out on the optimization of catalytic system using aniline
as nucleophile. The branched-type products (Table 15, product 76) were obtained with high
regioselectivity in good yields, and the other product 77 was not observed although
theoretically possible. 1% [Pd(allyl)Cl]2 and 3% PPh3 system was found to give high 76%
yield (Table 15, entry 3).
1.1mol% [Pd] , n% ligand,THF
2. PhNH2 (1.1eq), rt to reflux,THF, 4 h
B(pin)
OAc
B(pin)
NHPh
B(pin)
NHPh
(OR)
76 77
Entry Pd source Ligand (n%) Yield 76 (%)
1. Pd(OAc)2 PPh3 (3%) 70
2. PdCl2 PPh3 (3%) 65
3. [Pd(allyl)Cl]2 PPh3 (3%) 76
4. Pd(OAc)2 ---- 0
5. Pd(OAc)2 PPh3 (4%) 70
6. Pd2(dba)3.CHCl3 PPh3 (2%) 70
7. Pd(dba)2 PPh3 (2%) 70
8. Pd(PPh3)4 ---- 70
Table 15: Optimization of the catalytic system for nitrogen nucleophiles
Carbene and nitrogen-based ligands were not efficient in this catalytic system, in the case of
80

carbon nucleophiles. Hence, those ligands were not tested in allylation with nitrogen
nucleophiles. The most efficient catalytic system in allylation with malonate nucleophiles was
found to be 1% Pd(OAc)2 and 3% PPh3 but in aniline allylation it resulted in a yield of 70%
(Table 15, entry 1). Further increase in the ligand amount up to 4% didn’t change the yield
(Table 15, entry 5). As expected, palladium(II) catalyst alone i.e., Pd(OAc)2, failed in this
allylation (Table 15, entry 4). A low yield of 65% in this allylation was observed when the
reaction was catalyzed by 1% PdCl2 and 3% PPh3 (Table 15, entry 2). Allylation with other
catalytic systems like Pd(dba)2 and Pd(PPh3)4 gave yields similar to that of Pd(OAc)2 (Table
15, entries 6 and 7). The optimized conditions in allylation with aniline on -borylated allylic
acetates was found to be 1% [Pd(allyl)Cl]2 and 3% PPh3. Therefore, this optimized catalytic
system was used for extension studies. Only THF was used as solvent in all these allylations
(Scheme 63).
Aniline was used as nucleophile with other substituted -borylated allylic acetates derivatives.
Studies were done by increasing the steric hindrance at -position with different alkyl groups.
The dimethyl-substituted derivative gave a high yield, 77% (Table 16, entry 3) on allylation.
81

The unsubstituted derivative reacted equally well to give a yield of 75% (Table 16, entry 1),
whereas the methyl-substituted derivative resulted in a yield of 76% (Table 16, entry 2).
Aqueous sodium azide was successfully employed as nucleophile in allylation with -
borylated allylic acetates, using the optimized condition of 1% [Pd(allyl)Cl]2 and 3% PPh3.
The unsubstituted derivative resulted in an excellent yield of 85% (Table 17, entry 1), while
the methyl-substituted derivative gave a yield of 81% (Table 17, entry 2) and further increase
in steric hindrance from methyl to dimethyl resulted in a yield of 80% (Table 17, entry 3).
R
HN
B(pin)
R1
O
Entry R R1 78 yield(%)
1. H H 78g 76
2. Me H 78h 73
3. Me Me 78i 78
R
OAc
B(pin) +
1. [Pd(allyl)Cl]2 1%, PPh3 3%
2. THF, rt to reflux, 4h
R1
p-Anisidine
78
Table 18: p -Anisidine as nucleophile
p-Anisidine was also found to be a good nucleophile in this allylation, and the unsubstituted
derivative gave a yield of 76% (Table 18, entry 1,). A little increase in steric hindrance by
82

methyl substitution resulted in a yield of 73% (Table 18, entry 2), whereas the disubstituted
derivative gave a yield of 78% (Table 18, entry 3).
Several nitrogen nucleophiles such as pyrrolidine, aq. NH4OH, allyl amine, succinimide,
phthalimide, TMSN3, TsNH2, Bn2NH, 4-nitroaniline, benzamide, tert-butyl carbamate, benzyl
carbamate and heterocyclic bases such as imidazole, pyrrole and purine were not reactive,
even in presence of added bases like NaH, t-BuOK under this catalytic system. Investigation
of the allylation for a -borylated allylic acetate, where phenyl group was presented at the
allylic position, with aqueous sodium azide as nucleophile resulted in a direct coupling of
nucleophile with boronate (Scheme 64).
Thus, a chemo-, and regio-selective allylation was obtained with nitrogen nucleophiles on -
borylated allylic acetates. The boron moiety presented in the products of allylation can be
conveniently converted to other functional groups, like Suzuki-Miyaura, which involves a
new C-C bond formation.
II. 2. vi. One- pot allylation followed by Suzuki-Miyaura cross coupling:
Palladium(0) presented after allylation was effectively catalyzing the Suzuki-Miyaura cross-
coupling in a one-pot sequence (Table 19) to give double cross-coupled products. The
dimethyl substrate resulted in an yield of 72% (Table 19, entry 2), whereas the methyl
substrate resulted in 77% yield in a one pot reaction (Table 19, entry 1).
83

The boronate obtained after allylation can also be transformed to other functional groups like
azide 81 via Chan-Lam-Evans cross-coupling with copper catalysis. This involves the
treatment of vinylboronates with sodium azide in presence of copper catalyst like CuSO4 in
MeOH to yield -functionalized vinyl azide in 80% yield (Scheme 65).
II. 2. vii. Stereoselectivity:
Palladium-catalyzed asymmetric allylation was studied, using nitrogen-based nucleophiles, on
-borylated allylic acetate. Stereochemistry at -position in allylation was assigned in
comparison with already reported material.113
Here, we performed allylation, followed by
Suzuki-Miyaura cross-coupling in one-pot in order to establish the enantioselectivitiy (Table
20).
113 Plietker, B. Angew. Chem. Int. Ed. 2006, 45, 6053.
84
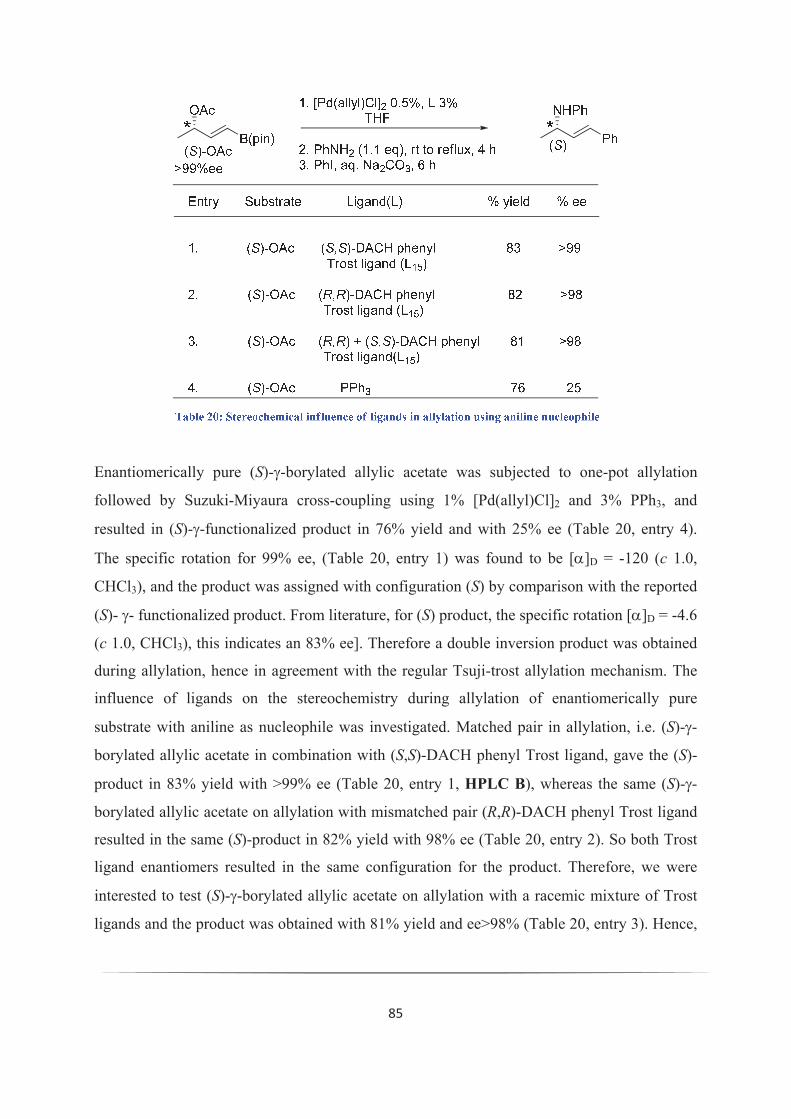
Enantiomerically pure (S)- -borylated allylic acetate was subjected to one-pot allylation
followed by Suzuki-Miyaura cross-coupling using 1% [Pd(allyl)Cl]2 and 3% PPh3, and
resulted in (S)- -functionalized product in 76% yield and with 25% ee (Table 20, entry 4).
The specific rotation for 99% ee, (Table 20, entry 1) was found to be [ ]D = -120 (c 1.0,
CHCl3), and the product was assigned with configuration (S) by comparison with the reported
(S)- - functionalized product. From literature, for (S) product, the specific rotation [ ]D = -4.6
(c 1.0, CHCl3), this indicates an 83% ee]. Therefore a double inversion product was obtained
during allylation, hence in agreement with the regular Tsuji-trost allylation mechanism. The
influence of ligands on the stereochemistry during allylation of enantiomerically pure
substrate with aniline as nucleophile was investigated. Matched pair in allylation, i.e. (S)- -
borylated allylic acetate in combination with (S,S)-DACH phenyl Trost ligand, gave the (S)-
product in 83% yield with >99% ee (Table 20, entry 1, HPLC B), whereas the same (S)- -
borylated allylic acetate on allylation with mismatched pair (R,R)-DACH phenyl Trost ligand
resulted in the same (S)-product in 82% yield with 98% ee (Table 20, entry 2). So both Trost
ligand enantiomers resulted in the same configuration for the product. Therefore, we were
interested to test (S)- -borylated allylic acetate on allylation with a racemic mixture of Trost
ligands and the product was obtained with 81% yield and ee>98% (Table 20, entry 3). Hence,
85

the total stereochemical outcome of the product in allylation with aniline is dependent on the
substrate.
The higher level of selectivity in allylation was observed with Trost ligands. These ligands are
sterically crowded and readily form stable -complex when treated with allylic acetates. In
case of PPh3 ligand the starting material underwent racemization (Table 20, entry 4). The
racemization in case of PPh3 ligand could be due to bond rotation in ( -allyl)Pd complex
via - - isomerisation (shown in Figure 2) considering 4 h of reaction time.
Racemic -borylated allylic acetate was converted via asymmetric allylation to chiral -
borylated allylic synthons. Asymmetric allylation of -borylated allylic acetates with (S,S)-
DACH phenyl Trost ligand resulted in 83% yield with 30% ee (Table 21, entry 1) of (S)-
product, whereas (R,R)-DACH phenyl Trost ligand gave 83% yield with 63% ee of the other
enantiomer (R)-product (Table 21, entry 2). Increase in the ligand steric hindrance from
phenyl to naphthyl group, using (R,R)-DACH naphthyl Trost ligand, increased the yield up to
87% but the enantioselectivity was dropped to 5% (Table 21, entry 3). Further increase in
steric hindrance, from naphthyl to a modified diamine chiral ligand, (R,R)-ANDEN phenyl
Trost ligand, gave the products in excellent yield of 90% but the enantioselectivity was very
low 8% (Table 21, entry 4).
86

OAc
B(pin)
NHPh
Ph
1. [Pd(allyl)Cl]2 0.5%, L 3%THF
2. PhNH2 (1.1 eq), rt to reflux, 4 h3. PhI, aq. Na2CO3, 6 h
*rac-OAc
Entry Substrate Ligand % yield % ee
1. rac-OAc (S,S)-DACH phenyl 83 30 (S)Trost ligand (L15)
2. rac-OAc (R,R)-DACH phenyl 83 63 (R)Trost ligand (L15)
3. rac-OAc (R,R)-DACH naphthyl 87 5 (R)Trost ligand (L16)
4. rac-OAc (R,R)-ANDEN phenyl 90 8 (R)Trost ligand (L17)
5. rac-OAc (R,R)-DACH pyridyl 0 0Trost ligand (L18)
Table 21: Asymmetric allylation with aniline, followed by Suzuki Miyaura in one pot
Nitrogen-based pyridyl ligand, i.e. (R,R)-DACH pyridyl Trost ligand, failed to give products
(Table 21, entry 5). Sterically crowded ligands gave very poor selectivities, although excellent
yields. The simple ligands gave good selectivities.
Chemo- and regio-selective allylation was also successful with -borylated allylic acetates
using oxygen nucleophiles and the products were obtained in 74% yield using 1mol%
Pd(OAc)2 and 3mol% PPh3 as catalytic system (Scheme 66).
II. 3. Some failure attempts of -borylated allylic derivatives:
Here we indicate some reactions that we attempted but failed to give the desired product.
87

1. Trost allylation on trifluoroborate salts using aqueous sodium azide as nucleophile, under
palladium (0) catalyst (Scheme 67).
This reaction was carried out with THF as the solvent and it was observed that the reaction
has solubility problem and no product obtained. Hence we studied other solvents like acetone,
DMF and THF/H2O. We also investigated the addition of base K2CO3 but all these attempts
failed to give the desired product.
2. Haibo et al reported a palladium-catalyzed decarboxylative cross-coupling of aryl
potassium aryltrifluoroborates with -oxocarboxylic acids in the presence of K2S2O8,
resulting in the formation of aryl ketones as shown below (Scheme 68).114
A similar reaction was attempted with -substituted vinyl trifluoroborates on treatment with
with -oxocarboxylic acids in presence of K2S2O8, but this reaction wasn’t successful to give
the -substituted , -unsaturated systems (Scheme 69).
114 Mingzong, L.; Cong, W.; Haibo, G. Org. Lett. 2011, 13, 2062.
88

3. Meike et al reported a Friedel-Crafts alkylation at room temperature with calcium and
lithium salts (Lewis acid) as catalysts. Allylic alcohol on treatment with resorcinol dimethyl
ether, under lithium or calcium Lewis acid catalyst, results in the formation of alkylated
product (Scheme 70).115
This type of Friedel-Crafts alkylation was attempted on -borylated allylic alcohol but no
alkylated product was observed even after 48 h of reaction time, and the starting material was
completely unreactive for this catalytic reaction (Scheme 71).
4. Grubb’s et al reported a 1,3-isomerization of allylic alcohols via rhenium oxo catalysis
using O3ReOSiPh3 as catalyst, under very mild conditions in 30 min (Scheme 72).116
115 Meike, N.; Matthias. J. M. Angew. Chem., Int. Ed. 2010, 49, 3684.
116 Morrill, C.; Grubbs, R. H. J. Am. Chem. Soc. 2005, 127, 2842.
89

A similar 1,3-isomerization of allylic alcohol was studied using -borylated allylic alcohol
under same conditions as mentioned, but it was observed that only the starting material was
present after 30 min reaction time. The prolonged reaction times like 1h, 2h, 4h, 8h and 24h
resulted in the same starting material only, and no isomerized product was isolated (Scheme
73).
Conclusion:
A chemo-, regio-, and stereo-selective allylation was achieved on -borylated allylic acetates
using carbon,117
and nitrogen nucleophiles. Overall, we have managed to use a highly
functionalized three-carbon building block in a chemo-, regio-, and stereoselective manner.
The resulting products could be used in a large variety of transformations taking advantage of
further reactions of the pinacol boronate moiety.
117 Kukkadapu, K. K.; Ouach, A.; Lozano, P.; Vaultier, M.; Pucheault, M. Org. Lett. 2011, 13,
4132.
90

Chapter III: Chemo enzymatic resolution of -borylated
allylic alcohols in continuous flow systems
using ionic liquids & scCO2
91

Introduction:
Organic solvents play an important role in organic chemistry to get a successful chemical
reaction. In majority these organic solvents are volatile and generate organic waste which
were not environmentally acceptable and should be avoided. In green chemistry118
replacing
hazardous solvents with environmentally benign solvents is highly attractive. These problems
led to identify alternative solvents like supercritical fluids119
and ionic liquids
120 which are
considered as best alternatives for organic solvents.
III. 1. Ionic liquids as solvents in green biocatalysis:
Ionic liquids attracted great attention as green solvent and were used in large number of
chemical transformations.121
Ionic liquids are low-melting onium salts composed solely of
anions and cations that are liquids, at or below 100 °C. The combination of bulky organic
cations and inorganic or organic anions counterparts lowers the lattice energy thereby melting
point is diminished for the resulting salts.122
Ionic liquids differs from molten salts like
sodium chloride (which are high-melting salts). Ionic liquids are non-volatile, exhibit very
low vapor pressure. They are highly polar, recyclable and thermally stable up to 400 °C (safe
to use at high temperatures) and can dissolve organic and inorganic materials. Many reactions
have been reported using ionic liquid media like Friedel-Crafts reaction,123
olefin
metathesis,124
hydrogenation,125
hydroformylation,126
etc. Ionic liquids are green solvents and
very good alternatives for organic solvents. Synthesis of enantioenriched products using
enzyme catalysts under organic solvent free media is called green biocatalysis. Green
biocatalysis in ionic liquids attracted the interest of scientists to perform different reactions for
118 Collins, T. Science 2001, 291, 48.119 Nayori, R. Chem. Rev. 1999, 99, 353.120 Riisager, A.; Fehrmann, R.; Haumann, M.; Wasserscheid, P. Top. Catal. 2006, 40, 91.121 Wasserscheid, P. Ionic Liquid in Synthesis : Wiley VCH, 2007.122 Hamaguchi, H-O.; Ozawa, R. Adv. Chem. Phys. 2005, 131, 85.123
Ross, J.; Xiao, J. Green Chem. 2002, 4, 129. 124
Yao, Q.; Zhang, Y. Angew. Chem., Int. Ed. 2003, 42, 3395. 125
Obert, K.; Roth, D.; Ehrig, M.; Schoenweiz, A.; Assenbaum, D.; Lange, H.; Wasserscheid,
P.; Appl. Catal., A 2009, 356, 43.
92
126 Hamza, K. ; Blum, J. ; Eur. J. Org. Chem. 2007, 4706.

green chemistry development. It is a highly effective approach for pollution prevention.
Minimising the formation of side products and the design of new methodologies for obtaining
pure products are becoming challenging problems. Enzyme catalysis in ionic liquids can solve
this up to certain extent. The high catalytic efficiency of enzymes in ionic liquids is now well
documented.127
However, organic solvent was often used to isolate products from ionic liquids, which is a
drawback for green process development. Alternative strategies were reported in literature
like membrane technology developed for isolating (S)-ibuprofen from (rac)-ibuprofen.128
Isolation of products from ionic liquid media by another green solvent such as scCO2 is
considered to be the most interesting strategy for developing a clean & green chemical
process.
III.2. Green biocatalysis in supercritical carbon dioxide (scCO2):
Supercritical carbon dioxide (scCO2) brings the attention of scientists regarding its use as
green solvent in continuous flow systems. This is due to its ability to extract, dissolve and
transport the chemicals in gas phase. It is a compressed CO2 gas. Hence, a heterogeneous
system can be successfully employed for reactions in scCO2. Biphasic systems based on ionic
liquids and supercritical carbon dioxide (scCO2) represent interesting alternatives to organic
solvents for designing continuous clean bio transformations in non-aqueous environment that
directly provide pure products.129
The reaction with scCO2 under heterogeneous medium was
successfully carried out for many synthetic transformations like kinetic resolution,130
dynamic
kinetic resolution131
and other synthetic reactions.132
The enzyme immobilized on ionic liquid
support (IME) was used as solid support and the substrate along with scCO2 used as mobile
127 Lozano, P. Green Chem. 2010, 12, 555.128 Branco, L. B.; Crespo, J. G.; Afonso, C. A. M. Chem. Eur. J. 2002, 8, 3865.129 Lozano, P.; Vaultier, M. Green Chem. 2007, 9, 780.130 Tomoko, M.; Kazunori, W.; Tadao, H.; Kaoru, N.; Yoshitaka, A.; Yukihiro, Misumi.;
Shinichiro, I.; Takao, I. Chem. Commun., 2004, 2286. 131 Lozano, P.; Diego, T. D.; Mira, C.; Montage, K.; Vaultier, M.; Iborra, J. L. Green Chem.
2009, 11, 538.132 (a) Huabin, X.; Tao, W.; Youyuan. D.; J. Supercrit. Fluids, 2009, 49, 52.
93
(b) Firas, Z.; Lasse, G.; Peter, S. S.; Alexei, L.; Walter, L. Chem. Commun., 2008, 79.

phase. The reactor was filled with IME, known concentration of substrate was pumped
through the reactor using controlled flow of scCO2. The reaction occurs on solid support with
very less residence time, the products after passing through the heterogeneous support will be
collected at the collection chamber and the compressed scCO2 gas is recycled back to the
cylinder by condensation process using back pressure (Picture 1). Enzymes tend to lose their
activity when heated because of denaturation. But enzymes on ionic liquid support don’t lose
their activity even at high temperatures. The stability of enzyme on ionic liquid support along
with scCO2 even at high temperatures are key parameters for carrying out integral green
bioprocess in continuous operation.
III. 3. Literature data on the mechanism of resolution using Candida
Antartica Lipase (CAL-B or Novozyme-435):
The enantioselectivity in acetylation of enzyme (CAL-B or Novozyme-435) is due to the
oxyanion active site (Picture 2). It’s a tetrahedral coordinate geometry obtained by the
hydrogen bonding interactions of Ser-His-Asp triad.133
The spatial arrangement of hydrogen-
bond donors in the active site lowers the free energy of the transition state. The oxyanion is
stabilized by two backbone amide hydrogen atoms and the side-chain hydroxyl group of
133 Anders, M.; Kar, H.; Mats, H. J. Am. Chem. Soc. 2001, 123, 4354-4355.
94

Thr40. The transition state of trans-esterification proceeds through an oxyanion and this active
site introduces the enantioselectively in acetylation.
A typical enantioselective acetylation (Mechanism 4) involves the interaction of acylating
agent to the active site of Ser-His-Asp protein, A, and a tetrahedral intermediate, B, is formed.
The alcohol part of the ester leaves and an acyl enzyme is formed, C. A second tetrahedral
intermediate, D, is formed after nucleophilic attack by a second alcohol. The newly formed
ester leaves, completing the catalytic cycle.
Mechanism 4: Reaction mechanism of t rans-acetylation
Lozano et al reported an efficient kinetic resolution of racemic 1-phenylethanol in continuous
flow process by selective acetylation of benzylic alcohols on treatment with CAL-B (Scheme
74), affording the products in equal yields with high selectivity.134
The racemic 1-
134 Lozano, P.; Diego, T. D.; Carrié, D.; Vaultier, M. Chem. Comm. 2002, 692.
95

phenylethanol reacts with CAL-B and only the (R)-OH converts to (R)-OAc whereas (S)-OH
remains unreacted for this catalytic system.
III. 4. Kinetic resolution of -borylated allylic alcohols in ionic liquids:
Andrade et al135
reported the first application of enzymes as catalysts for synthesizing
enantiopure boron compounds via enantioselective acetylation (Enzyme-catalyzed kinetic
resolution) in n-hexane as solvent. Kinetic resolution being used for separating the two
enantiomers of a racemic mixture, the chemical yield of the process will be limited to 50%.
Various types of aromatic, allylic and aliphatic secondary alcohols containing boronates were
acetylated using this protocol. High enantioselectivities, more than 98%, were obtained.
In our laboratory we were interested to investigate the kinetic resolution of -borylated allylic
alcohols under solvent free media (Scheme 75). Furthermore, we wanted to apply this
135 Andrade, L. H.; Barcellos, T. Org. lett. 2009, 11, 3052.
96

knowledge to continuous flow reactor with scCO2 in order to develop continuous kinetic
resolution of -borylated allylic alcohols.
OH
B
OAc (3.0 eq), CAL-B
Ionic Liquid, 50 oC, t min
OH
B
OAc
B+ **
O
O
O
O
O
O
(S)-OH (R)-OAc(+,-)-rac OH
50% 50%
Scheme 75: Kinetic resolution of -borylated allylic alcohols under solvent free media with CAL-B
Chemoenzymatic resolution of -borylated allylic alcohols by selective acetylation with vinyl
acetate in ionic liquids was developed using CAL-B as enzyme at 50 oC. Preliminary
experiments were focused on optimising the catalytic system in different ionic liquids.
Various ionic liquids were screened based on their chain length (butyl, octyl, and dodecyl),
anionic counterpart (NTf2, BF4, PF6), and cationic counterpart: Ammonium (BTMA &
TBMA), Imidazolium (BMIM), Pyrrolidinium (BMPy), Piperidinium (BMPi).
The reaction mixture samples were injected into chiral GC to study the reaction profile at
different reaction times. The relative conversion of racemic alcohol with respect to time to
obtain enantiopure products was plotted in graph to find enzyme activity in ionic liquids. The
ionic liquids used in kinetic resolution are shown below:
97

III. 5. Enzyme activity in Ionic liquids:
In kinetic resolution (Scheme 75), enzyme acetylates only (R)-OH to (R)-OAc. The only
product formed is (R)-OAc whereas the (S)-OH present in the racemic mixture remains
unreactive, hence products obtained in this reaction were (S)-OH and (R)-OAc. But after
prolonged reaction times under enzyme catalysis we observed that (S)-OH can also be
acetylated to (S)-OAc in minor yields (~3-5%). Therefore, we focused to optimize the
catalytic system with less reaction time, high yield and good selectivity. Here the results were
taken based on the (R)-OAc product formation. Various ionic liquids were screened for
optimisation of kinetic resolution. It is to be noted that these reactions were performed under
non-inert conditions. Enzyme activity (efficiency per milligram quantity of immobilized
enzyme used) is the key factor to obtain kinetic resolution. Enzyme activity is the rate at
which resolution occurs, the more enzyme activity results more efficient catalytic system.
A chemoenzymatic enantioselective acetylation was performed on -borylated allylic alcohol
(0.01g, 0.05mmol) with vinyl acetae (0.015 mL, 0.15mmol) in 0.485 mL of ionic liquid using
0.01 g of CAL-B enzyme at 50 oC. This reaction was monitored using different ionic liquids
and the results were plotted in graph between time and rate of conversion and the reaction was
monitored at regular intervals of time (15 min, 30 min, 1h, 2h, 4h, 6h, 8h, 24h).
98

Calculation of enzyme activity for [BTMA][NTf2] ionic liquid:
The enzyme activity was calculated from [BTMA][NTf2] ionic liquid reaction profile (kinetic
resolution) by plotting the reaction progress with respect to time in minutes (Graph 1).
The enzyme activity was found by multiplying the slope of Graph 1 with the concentration
( mol) of the substrate per mg of enzyme used. In case of [BTMA][NTf2] ionic liquid the
slope from this graph was found to be 0.44. The concentration of the substrate used was 50.51
mol per 10 mg of enzyme.
Enzyme activity =Slope x mol of substrate
mg of enzyme used
99

Enzyme activity in[BTMA][NTf2]
0.44 x 50.51
10= = 2.2 U/mg of IME
The enzyme activity in [BTMA][NTf2] was found to be 2.2 U/mg of IME. Similarly, enzyme
activity was calculated for other ionic liquid reactions to optimise the reaction conditions.
III. 6. Optimisation of kinetic resolution:
A high enzyme activity was found in case of NTf2-based ionic liquids, compared to PF6 and
BF4 ionic liquids. Kinetic resolution in NTf2-based ionic liquids having ammonium as
cationic counterpart like [BTMA] and [TBMA] ions showed similar enzyme activity of 2.2
and 2.4 respectively with conversion upto 40% at 2 h and 51% at 6 h of reaction time with a
selectivity >99% (Table 22, entries 4 and 5). Changing the cationic counter ion of the ionic
liquid from ammonium to imidazolium by using [BMIM][NTf2] showed an increased
enzymatic activity to 2.7, with conversion upto 45% with 99% selectivity were obtained at 2
h, and 51% conversion at 6 h with selectivity of 89% were obtained (Table 22, entry 1). By
increasing the chain length of imidazolium ionic liquid from butyl to octyl by using
[OMIM][NTf2], enzyme activity increased to 6.3 with 49% conversion at 2 h and 50%
conversion at 6 h with selectivity of 99% (Table 22, entry 2). Further increase in chain length
from octyl to dodecyl by using [C12MIM][NTf2] gave a high enzymatic activity of 7.6 with
50% conversion and selectivity >99% (Table 22, entry 3).
Other cationic counter ions based on pyrrolidinium [BMPy] and piperidinium [BMPi] showed
low enzymatic activities of 2.8 and 3.3 respectively with moderate conversion of 45% and
48% at 2 h with 99% selectivity, the selectivity was further decreased to 90% at 6 h (Table 22,
entries 6 and 7).
Ionic liquid based on BF4 anionic counterpart, [BMIM][BF4], showed enzyme activity of 4.9
with a conversion of 44% at 2 h and 48% conversion at 6 h with 99% selectivity (Table 22,
entry 11). Increasing chain length from butyl to dodecyl by using [C12MIM][BF4] reduced the
enzyme activity to 1.8 and the conversion was very poor, 26% at 2 h and 29% at 6 h (Table
22, entry 12).
100

Entry Ionic liquid Enzyme activity % Conversiona %Conversiona
(U/mg IME) (%ee) at 2 h (%ee) at 6 h
1. [BMIM][NTf2] 2.7 45 (>99) 51 (89)
2. [OMIM][NTf2] 6.3 49 (>99) 50 (>99)
3. [C12MIM][NTf2] 7.6 50 (>99) 50 (>99)
4. [BTMA][NTf2] 2.2 39 (>99) 51b(>99)
5. [TBMA][NTf2] 2.4 41 (>99) 51b(>99)
6. [BMPy][NTf2] 2.8 45 (>99) 50 (90)
7. [BMPi][NTf2] 3.3 48 (>99) 49 (91)
8. [BMIM][PF6] 3.3 46 (88) 49 (74)
9. [OMIM][PF6] 1.8 32 (99) 48 (99)
10. [C12MIM][PF6] 1.8 40 (99) 49 (85)
11. [BMIM][BF4] 4.9 44 (99) 48 (99)
12. [C12MIM][BF4] 1.8 26 (99) 29 (99)
aThis conversion was evaluated from chiral GC based on the (R)-OAc formation in the reactionb Possible integration error in chiral GC
Table 22: Enzyme activity in ionic liquids
Ionic liquids based on PF6 anionic counter part gave a poor conversion. In case of
[BMIM][PF6], the enzyme activity was found to be 3.3 with 46% conversion and poor
selectivity (88%) at 2 h whereas the conversion was increased to 49% but tremendous drop in
selectivity was observed, 74% at 6 h (Table 22, entry 8). Increasing the chain length from
butyl to octyl decreased the enzyme activity from 3.3 to 1.8 with a conversion of 32% at 2 h,
whereas it is 48% at 6 h with selectivitiy up to 99% (Table 22, entry 9). Using further
increased chain lengths from octyl to dodecyl resulted in enzyme activity of 1.8 with a
conversion of 40% at 2 h, and 49% at 6 h with selectivity of 85% (Table 22, entry 10). The
low yields may be due to the hygroscopic nature of these ionic liquids which tend to absorb
101

moisture and the presence of water in reaction medium might hydrolyze the acetylated
enzyme to acetic acid and thereby enantioselective acetylation process was arrested.
From the above results, high enzymatic activity of 7.6 (U/mg of IME) was found for
[C12MIM][NTf2] ionic liquid. Kinetic resolution using ionic liquids occurs in 2 h, faster than
with organic solvents like n-hexane for which the reaction time was 12-14 h.
III.7. Effect of water in kinetic resolution:
A study has been conducted to know the effect of added water on the reaction profile. The
ionic liquid [OMIM][NTf2] was tested under the reaction conditions of Scheme 75 and it was
observed that increased amount of water decreased the product formation (From Table 23).
The conversion, when no water was added at 30 min, was 36% which was decreased to 25%
when 2 L of water was added, and the conversion did not reach 50% even at 8 h of reaction.
Amount of water 0 L 2 L 4 L 6 L 8 Ladded
% of (R)-OAc 36% 25% 15% 10% 8%formed at 30 min
% of (R)-OAc 50% 42% 36% 25% 20%formed at 8 h
Table 23: Reaction profile by the addition of water
A further amount of added water to 8 L gave less conversion (only 8% conversion was
observed at 30 min which reached to 20% after 8 h). This could be due to hydrolysis of
acetylated enzyme which stops the chemoenzymatic kinetic resolution. Therefore it was
necessary to perform the reaction in ionic liquids under water free conditions.
III.8. Recyclability of ionic liquids:
After solving the issue of low conversion we were interested to study the recyclability of the
catalytic system. Recyclability test was studied using [C12MIM][NTf2] ionic liquid as in
102

Scheme 32. After 1st reaction cycle the products were extracted from the reaction media using
n-hexane or ethyl acetate (3 times each) and the same media (which contains the ionic liquid
and enzyme) was used for the second reaction cycle. It was observed that the second reaction
cycle showed the same productivity (Table 24) in 50% yield and selectivity >99% after 2 h
and 6 h. The enzyme activity remains unchanged for two consecutive reactions, therefore we
were interested to make use of this catalytic system as a heterogeneous solid support for
continuous flow systems.
III. 9. Kinetic resolution using continuous flow systems:
Enzymes can be immobilized136
(IME) on solid supports while keeping their activity and
stability. Then, fixed-bed reactors can be used for heterogeneous enzymatic catalysts using
ionic liquid/scCO2 mixtures as solvent in continuous flow systems allowing for the synthesis
of products in very good yields and selectivities. The main advantages of scCO2 are its ability
to extract, dissolve and transport chemicals. Enzyme behaviour in scCO2 and ionic liquids, as
well as the phase behaviour of ionic liquids/scCO2, are key parameters for carrying out
integral green bioprocess in continuous operation.
136 González-Sabõn, J.; Gotor, V.; Rebolledo, F. Tetrahedron Asymmetry 2002, 13, 1315.
103

Firstly, the enzyme was immobilised on ionic liquid using acetonitrile as solvent and the
acetonitrile was removed by evaporation. This solid support was used in a continuous flow
reactor (Picture 4). This continuous flow reactor was operated using scCO2 as solvent, which
is a compressed gas, and the flow was controlled using a pressure regulator. The substrate was
diluted in hexane (for a typical lab-scale experiment) connected with pump to control the flow
rate of substrate. A controlled flow system having heterogeneous support at 50 oC was
experimented initially using CAL-B and [BMIM][NTf2] as heterogeneous support (Scheme
76). This total operation will not result in any organic waste, the products after passing
through the heterogeneous support will be collected at the collection chamber and the
compressed scCO2 gas will be recycled back to the cylinder by condensation process using
back pressure. In normal lab-scale experiment, this compressed gas after collecting the
product was left to the atmosphere.
CAL B / ILOAc
scCO2
OH
B
OAc
B+**
O
O
O
O
(S)-OH (R)-OAcproducts50oC
III. 10. Results and discussion:
Initial experiments were focused to optimize the reaction system under continuous flow
systems using solid support made of CAL-B and [BMIM][NTf2], with a total flow rate of
1mL/min of mobile phase (0.1 mL substrate and 0.9mL of scCO2) at 100 bars while
maintaining the heterogeneous support at 50 OC. The enzyme activity under continuous flow
104

systems was calculated by multiplying concentration with percentage of conversion of the
product per gram of enzyme used.
For example, in the first continuous flow reaction, the substrate concentration was 6 mol/h,
whose conversion rate was found to be 40% using 0.18 g of enzyme on solid stationary phase.
The enzyme activity per gram of enzyme used was found to be 13.3 mol/h/g (Table 25, entry
1). The kinetic resolution was done continuously for 8 h in a day and a 40% conversion was
observed. Enzyme activity didn’t change when the same heterogeneous support was used for
the second time of another 8 h operation (Table 25, entry 2). However, the full conversion
was not reached but we observed reproducibility. Changing the concentration to 12 mol/h,
while keeping same heterogeneous support for 3rd
time operation of 8 h, was done. Here, the
concentration was doubled but still the conversion remains 40% whereas the enzyme activity
was doubled to 26.6 mol/h/g (Table 25, entry 3). However, the full conversion was not
reached.
Therefore, to increase the conversion rate, another ionic liquid where the enzyme activity was
better than with [BMIMNTf2] ionic liquid was studied (from Table 25). Investigation by other
ionic liquid [OMIM][NTf2] along with CAL-B as heterogeneous support resulted in very
good yields with high selectivity under continuous flow operation. The total flow rate of
mobile phase is 1 mL/min (0.05 mL of substrate and 0.95 mL of scCO2) at 100 bars pressure
and heterogeneous support was maintained at 50 oC. The continuous flow operation using 3
mol/h concentration gave the products in good conversion of 50% with high selectivity of
>99% (Graph 2) after 8 h of continuous operation.
105

In the continuous flow reaction using [OMIM][NTf2], the substrate concentration was 3
mol/h, whose conversion rate was found to be 50% using 0.16 g of enzyme on solid
stationary phase.
The enzyme activity remains the same for very long operation times. Upto 8 h, it was found to
be 9.03 mol/h/g (Table 25, entry 4). Another day of operation for 8 h with 3 mol/h
concentration of the substrate gave the same enzymatic activity of 9.03 mol/h/g with 50%
conversion and >99% selectivity (Table 25, entry 5). By changing the flow rate from 0.05
mL to 0.1 mL of substrate (which increases the concentration to 6 mol/h) it was observed
that the enzymatic activity was doubled to 18.07 mol/h/g with a conversion of 50% and
selectivity of >99% (Table 25, entry 6). Therefore, enzyme activity remains the same after 3
days of continuous operation by changing flow rate and concentration.
106
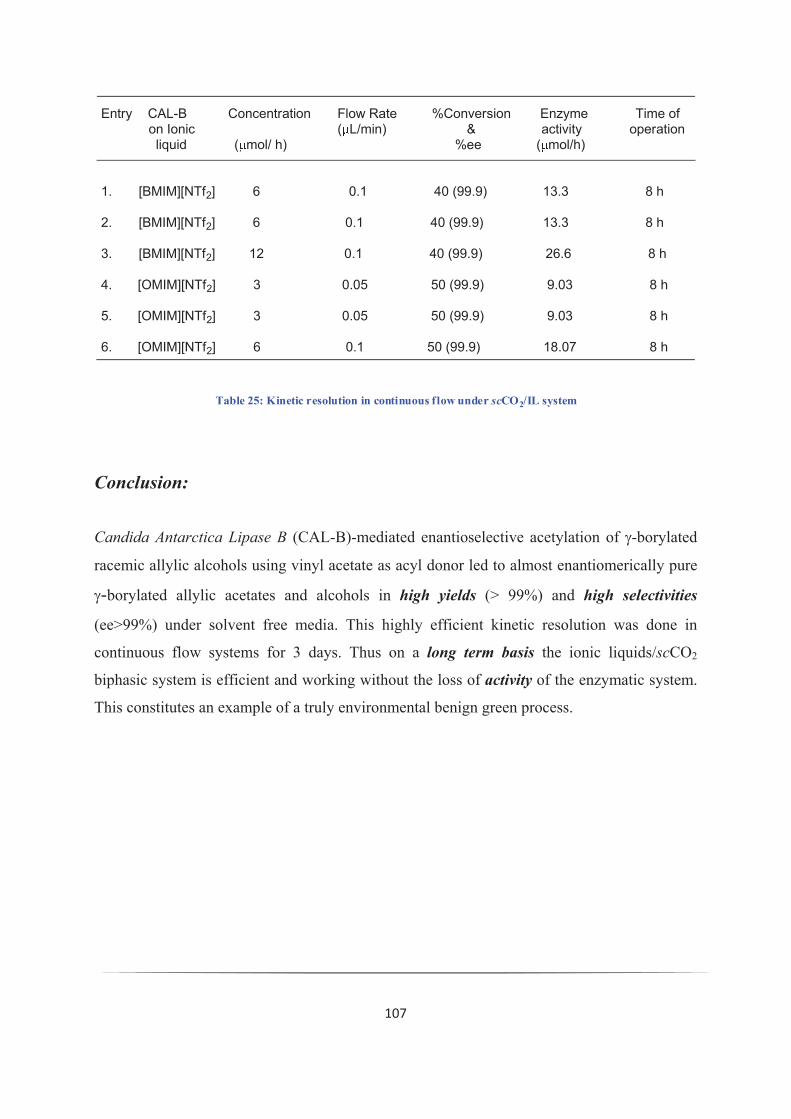
Entry CAL-B Concentration Flow Rate %Conversion Enzyme Time ofon Ionic ( L/min) & activity operationliquid ( mol/ h) %ee ( mol/h)
1. [BMIM][NTf2] 6 0.1 40 (99.9) 13.3 8 h
2. [BMIM][NTf2] 6 0.1 40 (99.9) 13.3 8 h
3. [BMIM][NTf2] 12 0.1 40 (99.9) 26.6 8 h
4. [OMIM][NTf2] 3 0.05 50 (99.9) 9.03 8 h
5. [OMIM][NTf2] 3 0.05 50 (99.9) 9.03 8 h
6. [OMIM][NTf2] 6 0.1 50 (99.9) 18.07 8 h
Table 25: Kinetic resolution in continuous flow under scCO2/IL system
Conclusion:
Candida Antarctica Lipase B (CAL-B)-mediated enantioselective acetylation of -borylated
racemic allylic alcohols using vinyl acetate as acyl donor led to almost enantiomerically pure
-borylated allylic acetates and alcohols in high yields (> 99%) and high selectivities
(ee>99%) under solvent free media. This highly efficient kinetic resolution was done in
continuous flow systems for 3 days. Thus on a long term basis the ionic liquids/scCO2
biphasic system is efficient and working without the loss of activity of the enzymatic system.
This constitutes an example of a truly environmental benign green process.
107

PART-B
Experimental
108

General Procedures. All reactions were carried out using oven-dried glassware under Argon
atmosphere or unless specified. Ether, THF, hexanes, pentane, and toluene, distilled from Na /
Benzophenone; DMF, benzene, CH2Cl2, and CHCl3, distilled from CaH2; Ethyl acetate,
Heptane and acetone, simple distillation; stored over molecular sieves. All reagents were
purchased from Sigma-Aldrich, Acros chemicals or Alfa Aesar and used without further
purification unless specified. Analytical thin layer chromatography (TLC) was carried out
using 0.25 mm silica plates purchased from Merck. Eluted plates were visualized using
KMnO4 stain or anisaldehyde stain. Silica gel chromatography was performed using 230–400
mesh silica gel purchased from Merck.
NMR spectra were recorded on standard 300 MHz FT spectrometers instrument Bruker FT
NMR (AVANCE 300) which referenced to the residual solvent signals (1H: CDCl3, 7.26 ppm;
acetone-D5, 2.05 ppm, CD3OD, 3.31ppm, D2O, 4.79 ppm, CD3CN, 1.94 ppm, DMSO, 2.25
ppm; 13
C: CDCl3, 77.0 ppm; acetone-D6, 29.9 ppm, CD3OD, 49 ppm, CD3CN, 1.32 ppm and
118.26 ppm, DMSO, 39.52 ppm) and recorded at 20-250C on a Bruker FT NMR instrument
(AVANCE 300). NMR spectra are reported as chemical shifts in values in ppm relative to
calibrated CDCl3. Splitting patterns are abbreviated as follows: singlet (s), doublet (d), triplet
(t), quartet (q), multiplet (m), doublet of doublet (dd), triplet of doublet (td), doublet of triplet
(dt). Determination of enantiomeric excesses was carried out using Waters HPLC 600
controller and pumps, equipped with a 2996 Photodiode Array Detector. Unless specified,
chromatographic conditions used for enantiomers separation were:
- Chiralpak AS-H 250mm column and Chiralpak AD-H 250mm columns
- 90/10 n-hexane / iPrOH mixture as mobile phase at 1mL/ min flow rate.
High-resolution mass spectra (HRMS) were recorded using a Waters-MicroMass analytical
LCT (ESI) spectrometer and obtained from the CRMPO analysis center at the University of
Rennes1.
Kinetic resolution was determined by GC using -DEX 110 Cyclodextrin Supelco chiral
column.
109
Optical rotations were measured by using a Perkin- Elmer model 141 polarimeter. Solution of
compounds was prepared in spectroscopic grade solvent.

Chapter-II Experimental:
II. 1. i. Synthesis of -borylated allyl acetates from hydroboration of propargylic
acetates (Scheme 55):
In a dried schlenk 26 mmol of freshly distilled -pinene was added to 26 mmol of
BH3.THF in 20 mL dry THF at 0 oC slowly for a period of 10 min and slowly warmed to rt
for 4 h. A white suspension of diisopinocampheylborane observed which was cooled to -35
oC, 26 mmol of propargylic acetate derivative was slowly added for a period of 30min
allowed to warm to rt, stirred for 5 h at rt and 260 mmol of freshly distilled acetaldehyde was
added at 0 oC and heated the reaction at 45
oC for 12 h. Distilled off the excess acetaldehyde
and 26 mmol of pinacol was added at rt and stirred for another 5 h. The solvent was removed
and the residue was purified on silicagel column chromatography.
Acetic acid 3-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-allyl ester (64a):
B
O
OAcO
Yield : 4.40 g (75% )
1H NMR (300 MHz, CDCl3) 6.61 (td, 1H, J = 4.67 Hz, J = 18.1 Hz), 5.66 (td,1H, J = 1.8
Hz, J = 18.1 Hz), 4.65 (dd, 2H, J = 1.81 Hz, J = 4.67 Hz), 2.05 (s, 3H), 1.23 (s, 12H);
13C NMR (75 MHZ, CDCl3)
11BNMR (CDCl3) .
110

Acetic acid 1-methyl-3-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-allyl ester (64b):
B
OAc
O
O
Yield : 4.98 g (80%)
Rf 0.60 (Heptane/Ethyl acetate (5:1)).
1H NMR (300 MHz, CDCl3) 6.42(dd, 1H, J = 4.7Hz, J = 18.1Hz), 5.45 (dd,1H, J=18.1 Hz,
J=1.6 Hz), 5.25-5.35 (m, 1H), 1.94 (s, 3H), 1.19 (d, 3H, J = 6.6 Hz), 1.15 (s,12H);
13C NMR (75 MHz,CDCl3) 170.1, 151.1, 83.0, 71.2, 24.7, 21.1, 19.5;
11BNMR (CDCl3) .
II. 1. ii. Hydroboration of propargylic alcohols and synthesis of target molecule 66
(Scheme 57):
In a dried schlenk 26 mmol of freshly distilled -Pinene was added to 26 mmol of BH3.THF
in 20 mL dry THF at 0 oC slowly for a period of 10 min and slowly warmed to rt for 4h. A
white suspension of diisopinocampheylborane observed which was cooled to -35 oC, 26 mmol
of propargylic alcohol derivative was slowly added for a period of 30 min allowed to warm to
rt, stirred for 5 h at rt and 260 mmol of freshly distilled acetaldehyde was added at 0 oC and
heated the reaction at 45 oC for 12 h. Distilled off the excess acetaldehyde and 26 mmol of
pinacol was added at rt and stirred for another 5 h. The solvent was removed and the residue
was purified on silicagel column chromatography to give -borylated allylic alcohol
derivatives.
(E)-3-(4,4,5,5-Tetramethyl-[1,3,2]dioxaborolan-2-yl)-prop-2-en-1-ol (65a):
111

HO B
O
O
Yield: 3.82 g (80%)
Rf 0.45 (Heptane/Ethyl acetate (1:1)).
1H NMR(300 MHz, CDCl3) 6.61 (dd, 1H, J = 18.1 Hz, J = 4.9 Hz), 5.58 (dd, 1H, J = 1.5
Hz, J = 18.1 Hz), 4.30 (dd, J = 4.0 Hz, J = 1.8 Hz, 2H), 1.27 (s, 12H);
13C NMR (75 MHz, CDCl3)
11B NMR (96 MHz, CDCl3) 29.0.
(E)-4-(4,4,5,5-Tetramethyl-[1,3,2]dioxaborolan-2-yl)-but-3-en-2-ol (65b):
B
OH
O
O
Yield: 3.90 g (76%)
Rf 0.45 (Heptane/Ethyl acetate (1:1)).
1H NMR (300 MHz, CDCl3) 6.61 (dd, 1H, J = 18.1 Hz, J = 4.9 Hz), 5.58 (dd, 1H, J = 1.5
Hz, J = 18.1 Hz), 4.30-4.40 (m, 1H), 2.29 (br, 1H), 1.28 (s, 12H), 1.24 (d, J = 5.6 Hz, 3H);
13C NMR (75 MHz, CDCl3) 156.46, 83.48, 69.68, 24.89, 22.77;
11B NMR (96 MHz, CDCl3) 29.9.
(E)-1-Phenyl-3-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-prop-2-en-1-ol (65c):
Ph B
OH
O
O
112

Yield: 5.33g (75%)
Rf 0.45 (Heptane/Ethyl acetate (1:1)).
1H NMR(300 MHz, CDCl3) 7.40-7.29 (m, 5H), 6.79 (dd, 1Hz, J = 18.0 Hz, J = 5.2 Hz),
5.78 (dd, 1Hz, J = 18.0 Hz, J = 1.5 Hz), 5.28 (dd, 1Hz, J = 5.2 Hz, J = 1.5 Hz), 2.12 (s, 1H),
1.28 (s, 12H);
13C NMR (75 MHz, CDCl3)
11B NMR (96 MHz, CDCl3) 29.0.
(E)-2-Methyl-4-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-but-3-en-2-ol (65d):
B
OH
O
O
Yield: 4.13 g (79%)
Rf 0.45 (Heptane/Ethyl acetate (1:1)).
1H NMR(300 MHz, CDCl3) 6.75 (d, 1H, J = 18.3 Hz), 5.65 (d, 1H, J = 18.3 Hz), 1.65 (s,
1H), 1.32 (s, 6H), 1.29 (s, 12H);
13C NMR (75 MHz, CDCl3) 160.5, 83.1, 72.0, 26.6, 24.4;
11B NMR (96 MHz, CDCl3) 29.0.
Acetylation of -borylated allylic alcohol derivatives (Scheme 57):
In a dried schlenk introduced 0.182 mol of DMAP, 1.82 mmol of -borylated allylic alcohol
and 3.01 mmol of triethyl amine in 3mL of dry THF at 0 oC and stirred for 45 min then added
1.99 mmol of Ac2O slowly for a period of 5 min at 0 oC, stirred at rt for 2 h. Diluted the
reaction mass with diethyl ether (50 mL) washed with 1N HCl (50 mL x 3 times) followed by
sat. NaHCO3 (50 mL x 3 times) dried over MgSO4 and the residue was purified by silica gel
column chromatography to give the -borylated allylic acetate derivative in 80% yield.
(E)-Acetic acid 3-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-allyl ester (66a):
113

AcO B
O
O
Yield: 0.35 g (85%)
Rf 0.45 (Heptane/Ethyl acetate (1:1)).
1H NMR (300 MHz, CDCl3) 6.61 (td, 1H, J = 4.67 Hz, J = 18.1 Hz), 5.66 (td,1H, J = 1.8
Hz, J = 18.1 Hz), 4.65 (dd, 2H, J = 1.81 Hz, J = 4.67 Hz), 2.05 (s, 3H), 1.23 (s, 12H);
13C NMR (75 MHz, CDCl3)
11BNMR (CDCl3) .
(E)-Acetic acid 1-methyl-3-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-allyl ester
(66b):
B
OAc
O
O
Yield: 0.37 g (85 %)
Rf 0.60 (Heptane/Ethyl acetate (5:1)).
1H NMR (300 MHz, CDCl3) 6.42(dd, 1H, J = 4.7Hz, J = 18.1Hz), 5.45 (dd,1H, J=18.1 Hz,
J=1.6 Hz), 5.25-5.35 (m, 1H), 1.94 (s, 3H), 1.19 (d, 3H, J = 6.6 Hz), 1.15 (s,12H);
13C NMR (75 MHz,CDCl3) 170.1, 151.1, 83.0, 71.2, 24.7, 21.1, 19.5;
11BNMR (CDCl3) .
(E)-Acetic acid 1-phenyl-3-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-allyl ester
(66c):
Ph B
OAc
O
O
114

Yield: 0.46 g (85%)
Rf 0.45 (Heptane/Ethyl acetate (1:1)).
1H NMR(300 MHz, CDCl3) 7.31-7.36 (m, 5H), 6.70 (dd, 1H, J = 4.8 Hz, J = 18.0 Hz), 6.31
(d, 1H, J = 4.8 Hz), 5.67 (dd, 1H, J = 18.0 Hz, J = 1.7 Hz), 2.13 (s, 12H), 1.27 (s, 12H);
13C NMR(75 MHz, CDCl3) 169.8, 149.3, 138.3, 128.5, 128.4, 128.2, 127.3, 83.4, 24.8, 24.7,
21.1;
11B NMR (96 MHz, CDCl3) 29.2.
(E)-Acetic acid 1,1-dimethyl-3-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-allyl ester
(66d):
B
OAc
O
O
Yield: 0.39 g (85%)
Rf 0.45 (Heptane/Ethyl acetate (1:1)).
1H NMR(300 MHz, CDCl3) 6.52 (d, 1H, J = 18.0 Hz), 5.51 (d, 1H, J = 18.0Hz), 2.01 (s,
3H), 1.61 (s, 6H), 1.22 (s, 12H);
11B NMR (96 MHz, CDCl3) 29.0.
Typical experimental for Tsuji-Trost allylation using carbon nucleophiles (Scheme 58):
In a dried schlenk reactor, were dissolved the boronate (1eq), Pd(OAc)2 (1 mol %) and PPh3
(3 mol%) in 2 mL of anhydrous THF. In another schlenk reactor, to a solution of NaH (60%
suspension in oil, 1.1 eq) washed with 2 mL dry ether was added freshly distilled dimethyl
malonate at 0 oC (1.1 eq). After 1h at room temperature, the malonate salt was added to the
palladium-boronate mixture at RT. After 4 h under reflux, the reaction mixture was
concentrated, dissolved in CH2Cl2 (20 mL/mmol). This organic solution was washed with
water (10 mL/mmol), brine (2 x 10mL/mmol), dried over MgSO4, and concentrated under
reduced pressure. The residual oil was purified by silica gel column flash chromatography.
115

(E)-2-[3-(4,4,5,5-Tetramethyl-[1,3,2]dioxaborolan-2-yl)-allyl]-malonic acid dimethyl
ester (69a):
B
O
O
O
OO
O
Yield : 97 mg (74%), colorless oil
Rf 0.52 (Heptane/Ethyl acetate (1:1)).
1H NMR (300 MHz, CDCl3) 6.56 (m, 1H), 5.52 (td, 1H, J = 1.5 Hz, J = 17.9 Hz), 3.72 (s,
6H), 3.53 (t, 1H, J = 7.5 Hz), 2.76 (dt, 2H, J = 1.5 Hz, J = 6.3 Hz), 1.24 (s, 12H);
13C NMR (75 MHz, CDCl3) 169.1, 148.5, 83.1, 52.5, 50.5, 34.4, 24.6;
11B NMR (96 MHz, CDCl3) 29.4;
HRMS (ESI) [M + Na+]/z calcd. 321.1485, found 321.1487.
(E)-2-[1-Methyl-3-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-allyl]-malonic acid
dimethyl ester (69b):
B
O
O
O
OO
O
Yield: 100 mg (77%), colorless liquid
Rf 0.52 (Heptane/Ethyl acetate (1:1)).
1H NMR (300 MHz, CDCl3) 6.54 (dd, 1H, J = 7.4 Hz, J = 17.9 Hz), 5.50 (d,1H, J = 17.9
Hz), 3.72 (s, 3H), 3.68 (s, 3H), 3.35 (d, 1H, J = 9.1 Hz), 3.05-2.97 (m, 1H), 1.24 (s, 12H),
1.09 (d, 3H, J = 6.8 Hz);
13C NMR (75 MHz, CDCl3) 168.5, 168.4, 153.9, 83.1, 56.8, 52.3, 52.2, 39.3, 24.7,
17.4;
11B NMR (96 MHz, CDCl3) 28.8;
HRMS (ESI) [M + Na+]/z calcd. 335.1641, found 335.1644.
116

(E)-2-[1,1-Dimethyl-3-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-allyl]-malonic acid
dimethyl ester (69c):
B
O
O
O
OO
O
Yield: 102 mg (80%), white amorphous solid
Rf 0.45 (Heptane/Ethyl acetate (1:1)).
1H NMR (300 MHz, CDCl3) 6.72 (d, 1H, J = 18.2 Hz), 5.45 (d, 1H, J = 18.2 Hz), 3.67 (s,
6H), 3.41 (s, 1H), 1.25 (s, 12H), 1.22 (s, 6H);
13C NMR (75 MHz, CDCl3) 168.1, 159.0, 83.1, 59.9, 52.0, 40.1, 24.7, 24.5;
11B NMR (96 MHz, CDCl3) 28.6;
HRMS (ESI) [M + Na+]/z calcd. 349.1798, found 349.1799.
(E)-2-[1-Phenyl-3-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-allyl]-malonic acid
dimethyl ester (69d):
COOMeMeOOC
B
O
O
Yield: 95mg (61%), colorless oil
Rf 0.49 (Heptane/Ethyl acetate (1:1)).
1H NMR(300 MHz, CDCl3) 7.28-7.19 (m, 5H), 6.71 (dd, 1H, J = 17.8 Hz, J = 7.4 Hz), 5.50
(dd, 1H, J = 1.2 Hz, J = 17.8 Hz), 4.21 (ddd, 1H, J = 7.4 Hz, J = 1.0 Hz, J = 11.3 Hz), 3.92 (d,
1H, J = 11.3 Hz), 3.73 (s, 3H), 1.22 (s,12H) ;
13C NMR (75 MHz, CDCl3) 168.0, 167.1, 151.5, 139.0, 128.6, 128.2, 127.1, 83.2, 56.7,
52.6, 52.3, 51.1, 24.7, 24.7;
11B NMR (96 MHz, CDCl3) 28.6;
117

HRMS (ESI) [M + Na+]/z calcd. 397.1798, found 397.1799.
(E)-2-Acetyl-5-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-pent-4-enoic acid methyl
ester (69e):
B
O
O
OO
O
Yield: 95mg (76%), colorless oil
Rf 0.52 (Heptane/Ethyl acetate (1:1)).
1H NMR (300 MHz, CDCl3) 6.53 (m, 1H), 5.51 (td, 1H, J = 1.5 Hz, J = 17.9 Hz), 3.73 (s,
3H), 3.62 (t, 1H, J = 7.3 Hz), 2.71 (dt, 2H, J = 1.5 Hz, J = 7.6 Hz), 2.23 (s, 3H), 1.24 (s, 12H);
13C NMR (75 MHz, CDCl3) 202.1, 169.5, 148.8, 83.2, 58.2, 52.5, 33.7, 29.2, 24.7;
11B NMR (96 MHz, CDCl3) 28.6;
HRMS (ESI) [M + Na+]/z calcd. 305.1536, found 305.1537.
(E)-2-Acetyl-3-methyl-5-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-pent-4-enoic acid
methyl ester (69f):
B
O
O
OO
O
Yield: 99 mg (80%), colorless oil
Rf 0.50 (Heptane/Ethyl acetate (1:1)).
1H NMR (300 MHz, CDCl3) 6.53 (dq, 1H, J = 7.4 Hz, J = 17.9 Hz, J = 2.3 Hz, J = 15.6
Hz), 5.49 (dd, 1H, J = 3.1 Hz, J = 17.9 Hz), 3.72 (s, 1.5H), 3.67 (s, 1.5H), 3.44 (dd, 1H, J =
2.3 Hz, J = 9.7 Hz), 3.08-3.00 (m, 1H), 2.23 (s, 1.5H), 2.18 (s, 1.5H), 1.24 (d, 12H, J = 1.3
Hz), 1.07(dd, 3H, J = 3.6 Hz, J = 6.7 Hz );
13C NMR (75 MHz, CDCl3) 202.3, 168.9, 154.1, 83.2, 65.1, 52.3, 39.2, 29.6, 24.7, 17.6;
118

11B NMR (96 MHz, CDCl3) 28.9;
HRMS (ESI) [M + Na+]/z calcd. 319.16927, found 319.1695.
(E)-2-Acetyl-3,3-dimethyl-5-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-pent-4-enoic
acid methyl ester (69g):
B
O
O
OO
O
Yield: 102 mg (83%), white crystals
Rf 0.51 (Heptane/Ethyl acetate (1:1)).
1H NMR (300 MHz, CDCl3) 6.75 (d, 1H, J = 18.2 Hz), 5.45 (d, 1H, J = 18.2 Hz), 3.68 (s,
3H), 3.51 (s, 1H), 2.19 (s, 3H), 1.26 (s, 12H), 1.22 (s, 3H), 1.19 (s, 3H);
13C NMR (75 MHz, CDCl3) 202.4, 168.8, 159.1, 83.1, 67.1, 51.9, 40.5, 31.5, 25.0, 24.7,
24.2;
11B NMR (96 MHz, CDCl3) 28.6;
HRMS (ESI) [M + Na+]/z calcd. 333.1849, found 333.1848.
(E)-3-[1-Methyl-3-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-allyl]-pentane-2,4-dione
(69h):
B
O
O
O
O
Yield: 93 mg (80%), colorless liquid
Rf 0.48 (Heptane/Ethyl acetate (1:1)).
1H NMR (300 MHz, CDCl3) 6.43 (dd, 1H, J = 7.6 Hz, J = 17.9 Hz), 5.47 (dd, 1H, J = 1.0
Hz, J = 17.9 Hz), 3.64 (d, 1H, J = 10.4 Hz), 3.14-3.06 (m, 1H), 2.18 (s, 3H), 2.11 (s, 3H), 1.23
(s, 12H), 0.99 (d, 1H, J = 6.6 Hz);
119

13C NMR (75 MHz, CDCl3) 203.4, 153.8, 83.2, 74.8, 39.6, 29.9, 24.7, 17.7;
11B NMR (96 MHz, CDCl3) 29.5;
HRMS (ESI) [M + Na+]/z calcd. 303.1743, found 303.1747.
(E)-3-[1,1-Dimethyl-3-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-allyl]-pentane-2,4-
dione (69i):
B
O
O
O
O
Yield: 95 mg (82%), white crystals
Rf 0.51 (Heptane/Ethyl acetate (1:1)).
1H NMR (300 MHz, CDCl3) 6.75 (d, 1H, J = 18.2 Hz), 5.43 (d, 1H, J = 18.2 Hz), 3.75 (s,
1H), 2.16 (s, 6H), 1.27 (s, 12H), 1.16 (s, 6H);
13C NMR (75 MHz, CDCl3) 203.6, 159.1, 83.2, 75.3, 41.4, 32.4, 24.7, 24.6;
11B NMR (96 MHz, CDCl3) 29.2;
HRMS (ESI) [M + Na+]/z calcd. 317.1900, found 317.1901.
(E)-2-Cyano-5-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-pent-4-enoic acid methyl
ester (69j):
BNC
OOO
O
Yield: 90 mg (77%), colorless liquid
Rf 0.50 (Heptane/Ethyl acetate (1:1)).
1H NMR (300 MHz, CDCl3) 6.59 (m, 1H), 5.68 (m, 1H), 3.83 (s, 3H), 3.64 (m, 1H), 3.09
(m, 0.6H), 2.80(m, 1.4H), 1.28(s, 12H);
120

13C NMR (75 MHz, CDCl3) 166.1, 145.8, 116.0, 83.3, 53.4, 37.4, 31.6, 24.7;
11B NMR (96 MHz, CDCl3) 29.5;
HRMS (ESI) [M + Na+]/z calcd. 288.1383, found 288.1385.
(E)-2-Cyano-3-methyl-5-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-pent-4-enoic acid
methyl ester (69k):
BNC
OOO
O
Yield: 93 mg (80%), colorless liquid
Rf 0.50 (Heptane/Ethyl acetate (1:1)).
1H NMR (300 MHz, CDCl3) 6.55 (dd, 1H, J = 6.7 Hz, J = 17.9 Hz), 5.62 (d, 1H, J = 17.9
Hz), 3.81 (s, 1.5H), 3.79 (s, 1.5H), 3.59 (dd, 1H, J = 5.6 Hz, J = 21.8 Hz), 3.06-2.97 (m, 1H),
1.27 (d, 12H, J = 1.0 Hz), 1.23(d, 3H, J = 6.7 Hz);
13C NMR (75 MHz, CDCl3) 165.8, 151.3, 115.0, 83.4, 53.4, 43.5, 40.1, 24.7, 17.8;
11B NMR (96 MHz, CDCl3) 29.1;
HRMS (ESI) [M + Na+]/z calcd. 302.1539, found 302.1542.
(E)-2-Cyano-3,3-dimethyl-5-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-pent-4-enoic
acid methyl ester (69l):
BNC
OOO
O
Yield: 91 mg (79%), white crystals
Rf 0.51 (Heptane/Ethyl acetate (1:1)).
1H NMR (300 MHz, CDCl3) 6.61 (d, 1H, J = 18.1 Hz), 5.56 (d, 1H, J = 18.1 Hz), 3.76 (s,
3H), 3.43 (s, 1H), 1.29 (s, 3H), 1.27 (s, 12H), 1.26 (s, 3H);
121

13C NMR (75 MHz, CDCl3) 165.1, 155.5, 115.3, 83.4, 52.9, 48.0, 41.1, 24.9, 24.7,
24.1;
11B NMR (96 MHz, CDCl3) 29.0;
HRMS (ESI) [M + Na+]/z calcd. 316.1696, found 316.1698.
(E)-2-Oxo-1-[3-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-allyl]-
cyclopentanecarboxylic acid ethyl ester (69m):
B O
OO OO
Yield: 106 mg (79%), colorless liquid
Rf 0.51(Heptane/Ethyl acetate (1:1)).
1H NMR (300 MHz, CDCl3) 6.46 (m, 1H), 5.51 (td, 1H, J = 1.3 Hz, J = 17.7 Hz ), 4.17 (q,
2H, J = 7.1 Hz), 2.82 (dddd, 1H, J = 1.3 Hz, J = 6.8 Hz, J = 14.1 Hz), 2.45 (m, 4H), 1.99 (m,
3H), 1.24 (m, 15H) ;
13C NMR (75 MHz, CDCl3) 214.2, 170.5, 147.7, 83.1, 61.4, 59.6, 39.6, 37.8, 32.0, 24.6,
19.4,14.0 ;
11B NMR (96 MHz, CDCl3) 29.4;
HRMS (ESI) [M + Na+]/z calcd. 345.1849, found 345.1848.
(E)-1-[1-Methyl-3-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-allyl]-2-oxo-
cyclopentanecarboxylic acid ethyl ester (69n):
OOEt
O
B O
O
Yield: 105 mg (75%), colorless liquid
122

Rf 0.51(Heptane/Ethyl acetate (1:1)).
1H NMR (300 MHz, CDCl3) 6.45 (dddd, 1H, J = 6.9 Hz , J = 1.6 Hz, J = 17.9 Hz), 5.50
(dddd, 1H, J = 1.3 Hz, J = 3.1 Hz, J = 17.9 Hz), 4.20 (m, 2H), 3.24 (m, 1H), 2.50 (m, 2H),
2.17 (m, 1H), 1.95 (m, 3H), 1.28 (m, 15H), 1.02 (dd, 3H, j = 6.8 Hz, j = 14.7 Hz) ;
13C NMR (75 MHz, CDCl3) 214.2, 169.7, 153.3, 83.1, 64.7, 61.5, 43.0, 39.0, 28.3, 24.7,
19.7, 14.8, 14.0;
11B NMR (96 MHz, CDCl3) 28.8;
HRMS (ESI) [M + Na+]/z calcd. 359.20057, found 359.2005.
Typical one-pot reaction experimental procedure (Table 12):
To a dried argon filled Schlenk 0.416 mmol of gamma-borylated allylic acetate, 2.1 mol of
Pd(OAc)2 and 6.3 mol of PPh3 were dissolved in 2mL of anhydrous THF and stirred for 1 h
at RT. In another Schlenk freshly distilled 50 L (0.457 mmol) of dimethyl malonate were
added at 0 oC to a solution of 18 mg of NaH (0.458 mmol, 60% in oil washed with 2 mL of
anhydrous Et2O). After 1 h at RT, the solution was added at RT to the boronate-palladium
complex mixture. After 4 h under refluxing THF, 0.63mmol of aryliodide and a degassed
saturated aqueous solution of K2CO3 (0.63 mmol) were added to the reaction mixture at room
temperature. After 6 h under refluxing conditions, the reaction mixture was concentrated
under reduced pressure. The residue was dissolved in CH2Cl2 (10 mL), washed with water (5
mL), brine (2 x 5 mL). Organic phases were dried over MgSO4 and purified by silica gel
column flash chromatography.
(E)-dimethyl 2-(4-phenylbut-3-en-2-yl)malonate(72a):
Yield: 87 mg (75%), colorless liquid
Rf 0.65(Heptane/Ethyl acetate (1:1))
1H NMR (300 MHz, CDCl3) 7.34 (m, 5H), 6.48 (s, 1H, J = 15.8 Hz), 6.16 (dd, 1H, J = 18.2
Hz), 3.75 (s, 3H), 3.67 (s, 3H), 3.42 (d, 1H, J = 8.9 Hz), 3.16 (m, 1H), 1.20 (d, 3H, J = 6.7
Hz);
123

13C NMR (75 MHz, CDCl3) 168.6, 137.0, 131.1, 130.7, 128.4, 127.3, 126.2, 57.7, 52.4,
37.7,18.4;
HRMS (ESI) [M + Na+]/z calcd. 285.1102, found 285.1105.
(E)-2-(1,1-Dimethyl-3-phenyl-allyl)-malonic acid dimethyl ester (72b)
O
O
O
O
Yield: 87 mg (76%), colorless liquid
Rf 0.51(Heptane/Ethyl acetate (1:1)).
1H NMR (300 MHz, CDCl3) 7.37-7.20 (m, 5H), 6.47 (d, 1H, J = 16.2Hz), 6.39 (d, 1H, J =
16.2Hz), 3.69 (s, 6H), 3.45 (s, 1H), 1.34 (s, 6H) ;
13C NMR (75 MHz, CDCl3) 168.2, 137.4, 136.5, 128.4, 127.4, 127.1 126.2, 60.9, 52.0, 38.6,
25.5.;
HRMS (ESI) [M + Na+]/z calcd. 299.1259, found 299.1256.
(E)-2-(3-Phenyl-allyl)-malonic acid dimethyl ester (72c):
O
O
O O
Yield: 77 mg (75%), colorless liquid
Rf 0.51(Heptane/Ethyl acetate (1:1)).
1H NMR (300 MHz, CDCl3) 7.34-7.21 (m, 5H), 6.50 (d, 1H, J = 15.7Hz), 6.18-6.08 (m,
1H), 3.74 (s, 6H), 3.55 (t, 1H, J = 7.5Hz), 2.83-2.78 (m, 2H) ;
13C NMR (75 MHz, CDCl3) 169.2, 136.9, 132.9, 128.4, 127.3, 126.1, 125.3, 52.5, 51.7,
32.2.
124

(E)-2-(1-Phenyl-3-p-tolyl-allyl)-malonic acid dimethyl ester (72d):
MeOOC COOMe
Yield: 109 mg (78%), colorless liquid
Rf 0.51 (Heptane/Ethyl acetate (1:1)).
1H NMR (300 MHz, CDCl3) 7.31-7.0 (m, 9H), 6.47 (d, 1H, J = 15.7 Hz), 6.30 (dd, 1H, J =
8.5 Hz, J = 15.7 Hz), 4.27 (dd, 1H, J = 10.8 Hz, J = 8.6 Hz), 3.95 (d, 1H, J = 10.9 Hz), 3.69
(s, 3H), 3.51 (s, 3H), 2.30 (s, 3H);
13C NMR (75 MHz, CDCl3) 168.3, 167.9, 140.4, 137.5, 134.1, 131.8, 129.3, 128.8, 128.1,
128.0,127.2, 126.4, 57.8, 52.7, 52.6, 49.3, 21.3.
(S,E)-2-[1-Methyl-3- (4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-allyl]-malonic
acid dimethyl ester (73):
B
O
O
MeOOC COOMe
Yield: 100 mg (77%)
Rf 0.52 (Heptane/Ethyl acetate (1:1)).
1H NMR (300MHz, CDCl3) 6.54 (dd, 1H, J = 7.4 Hz, J = 17.9 Hz), 5.50 (d,1H, J = 17.9
Hz), 3.72 (s, 3H), 3.68 (s, 3H), 3.35 (d, 1H, J = 9.1 Hz), 3.05-2.97 (m, 1H), 1.24 (s, 12H),
1.09 (d, 3H, J = 6.8 Hz);
13C NMR (75 MHz, CDCl3) 168.5, 168.4, 153.9, 83.1, 56.8, 52.3, 52.2, 39.3, 24.7,
17.4;
125

11B NMR (96 MHz, CDCl3) 28.8;
HRMS (ESI) [M + Na+]/z calcd. 335.1641, found 335.1644.
(S,E)-2-(1-Methyl-3-phenyl-allyl)-malonic acid dimethyl ester (74):
Ph
COOMeMeOOC
Yield: 77 mg (75%), pale yellow solid
Rf 0.65(Heptane/Ethyl acetate (1:1))
1H NMR (300 MHz, CDCl3) 7.34 (m, 5H), 6.48 (s, 1H, J = 15.8 Hz), 6.16 (dd, 1H, J = 18.2
Hz), 3.75 (s, 3H), 3.67 (s, 3H), 3.42 (d, 1H, J = 8.9 Hz), 3.16 (m, 1H), 1.20 (d, 3H, J = 6.7
Hz);
13C NMR (75 MHz, CDCl3) 168.6, 137.0, 131.1, 130.7, 128.4, 127.3, 126.2, 57.7, 52.4,
37.7,18.4;
HRMS (ESI) [M + Na+]/z calcd. 285.1102, found 285.1105.
(E)-2-(3-Azido-1-methyl-allyl)-malonic acid dimethyl ester 75 (Scheme 62):
NaN3 (32 mg, 0.48 mmol) and CuSO4 (0.2 mmol) were placed in an oven-dried
roundbottomed flask. Subsequently methanol (3mL) and (E)-2-[1-Methyl-3- (4,4,5,5-
tetramethyl-[1,3,2]dioxaborolan-2-yl)-allyl]-malonic acid dimethyl ester (0.1 g, 0.32 mmol)
were added. After 4 h at room temperature, the reaction mixture was concentrated under
reduced pressure. The residue was dissolved in CH2Cl2 (10 mL), washed with water (5 mL),
brine (2 x 5 mL). Organic phases were dried over MgSO4 and purified by silica gel column
flash chromatography affording 75 as colorless liquid.
126

N3
COOMeMeOOC
Yield : 58 mg (78%), colorless liquid
Rf 0.51( Heptane/Ethyl acetate (1:1)).
1H NMR (300 MHz, CDCl3) 6.00 (d, 1H, J = 13.9 Hz), 5.30 (dd, 1H, J = 13.4 Hz, J = 9.1
Hz), 3.72 (s, 3H), 3.70 (s, 3H), 3.29 (d, 1H, J = 8.7 Hz), 3.00 (m, 1H), 1.10 (d, J = 6.8 Hz,
3H);
13C NMR (75 MHz, CDCl3) 168.5, 168.4, 128.2, 121.0, 57.7, 52.6, 52.5, 34.8, 18.7;
HRMS (ESI) [M + Na+]/z calcd. 250.08038, found 250.0805.
Typical experimental for allylic substitution reaction using nitrogen nucleophiles
(Scheme 63):
To a dried argon filled shlenk introduced the boronate (1 eq), Pd(allyl)Cl]2 (0.5 mol%)
or Pd(OAc)2 (1 mol%) and PPh3 (3 mol %) were added in 2 mL dry THF and stirred for 1 h at
rt, Nucleophile (1.1 eq) was added to the boronate palladium complex mixture at rt and
refluxed for 4 h, reaction compiles and the crude was concentrated and separated between (10
mL) DCM and (5 mL) water, washed the organic layer with brine (2 x 5 mL), dried over
MgSO4, purified by silica gel column flash chromatography to get the respective yields which
were described below.
(E)-N-(3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)allyl)aniline (78a):
H B
O
O
NHH
Yield: 86 mg (75%), colorless oil
127

Rf 0.58 (Heptane / Ethyl acetate (1:1)).
1H NMR (300 MHz, CDCl3) 7.18-7.13 (m, 2H), 6.72-6.58 (m, 4H), 5.72 (d, 1H, J = 18.0
Hz), 3.87 (dd, 2H, J = 1.6 Hz, J = 4.6 Hz), 1.26 (s, 12H);
13C NMR (75 MHz, CDCl3) 150.0, 147.8, 129.1, 117.4, 112.8, 83.2, 47.5, 24.7;
11B NMR (96 MHz, CDCl3) 28.4;
HRMS (ESI) [M + Na+]/z calcd. 282.16413, found 282.1643.
(E)-N-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)but-3-en-2-yl)aniline (78b):
B
O
O
NH
Yield: 80 mg (76%), colorless liquid
Rf 0.65 (Heptane / Ethyl acetate (1:1)).
1H NMR (300 MHz, CDCl3) 7.16 (m, 2H), 6.68-6.54 (m, 4H), 5.66 (d, 1H, J = 18.0 Hz),
4.05-4.00 (m, 1H), 1.32 (d, 3H, J = 6.7 Hz), 1.25 (s, 12H);
13C NMR (75 MHz, CDCl3) 155.4, 147.2, 129.1, 117.1, 113.1, 83.2, 51.9, 24.7, 21.2;
11B NMR (96 MHz, CDCl3) 28.9;
HRMS (ESI) [M + Na+]/z calcd. 296.17978, found 296.1797.
(E)-N-(2-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)but-3-en-2-yl)aniline (78c):
B
O
O
NH
Yield: 87 mg (77%), white amorphous solid
Rf 0.61(Heptane / Ethyl acetate (1:1)).
1H NMR (300MHz, CDCl3) 7.11-7.06 (m, 2H), 6.78- 6.60 (m, 4H), 5.64 (d, 1H, J = 18.3
Hz), 1.38 (s, 6H), 1.26 (s, 12H);
128

13C NMR (75 MHz, CDCl3) 160.2, 146.3, 128.6, 117.1, 115.4, 83.1, 55.4, 27.9, 24.7;
11B
NMR (96 MHz, CDCl3) 28.9;
HRMS (ESI) [M + Na+]/z calcd. 310.19543, found 310.1955.
(E)-2-(3-Azido-propenyl)-4,4,5,5-tetramethyl-[1,3,2]dioxaborolane (78d):
H B
O
O
N3H
Yield: 79 mg (85%), colorless oil
Rf 0.58 (Heptane /Ethyl acetate (1:1)).
1H NMR (300MHz, CDCl3) 6.60-6.50 (m, 1H), 5.74 (td, 1H, J = 1.6 Hz, J = 17.9 Hz), 3.86
(dd, 2H, J = 1.4 Hz, J = 5.3 Hz), 1.26 (s, 12H);
13C NMR (75 MHz, CDCl3) 144.9, 83.4, 54.2, 24.7;
11B NMR (96 MHz, CDCl3) 29.1.
HRMS (ESI) [M + Na+]/z calcd. 232.12281, found 232.1232.
(E)-2-(3-Azido-but-1-enyl)-4,4,5,5-tetramethyl-[1,3,2]dioxaborolane (78e):
B
O
O
N3
Yield: 76 mg (81%), colorless oil
Rf 0.60 (Heptane / Ethyl acetate (1:1)).
1H NMR (300 MHz, CDCl3) 6.53 (dd, 1H, J = 6.2 Hz, J = 17.9 Hz), 5.68 (d, 1H, J = 17.9
Hz), 4.08-3.99 (m, 1H), 1.32 (d, 3H, J = 6.8 Hz), 1.29 (s, 12H);
13C NMR (75 MHz, CDCl3) 150.2, 83.5, 60.4, 24.7, 19.2;
11B NMR (96 MHz, CDCl3) 29.7;
129

HRMS (ESI) [M + Na+]/z calcd. 246.13898, found 246.1392.
(E)-2-(3-Azido-3-methyl-but-1-enyl)-4,4,5,5-tetramethyl-[1,3,2]dioxaborolane (78f)
B
O
O
N3
Yield: 75 mg (80%), white amorphous solid
Rf 0.63 (Heptane / Ethyl acetate (1:1)).
1H NMR (300 MHz, CDCl3) 6.56 (d, 1H, J = 18.1 Hz), 5.64 (d, 1H, J = 18.1 Hz), 1.34 (s,
6H), 1.28 (s, 12H);
13C NMR (75 MHz, CDCl3) 154.2, 83.4, 62.7, 25.8, 24.7;
11B NMR (96 MHz, CDCl3) 29.7;
HRMS (ESI) [M + Na+]/z calcd. 260.15463, found 260.1548.
(E)- (4-Methoxy-phenyl)-[3-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-allyl]-amine
(78g):
H B
O
O
NHH
O
Yield: 97 mg (76%), colorless oil
Rf 0.62 (Heptane / Ethyl acetate (1:1)).
1H NMR (300MHz, CDCl3) 6.77-6.73 (m, 3H), 6.57 (dd, 2H, J = 9.0 Hz, J = 6.6 Hz), 5.71
(td, 1H, J = 1.8 Hz, J = 18.0 Hz), 3.82 (dd, 2H, J = 1.8 Hz, J = 4.7 Hz), 3.73 (s, 3H), 1.26 (s,
12H);
13C NMR (75 MHz, CDCl3) 152.0, 150.3, 142.0, 114.8, 114.0, 83.2, 55.7, 48.4, 24.7;
11B NMR (96 MHz, CDCl3) 28.9;
130

HRMS (ESI) [M + Na+]/z calcd. 312.17469, found 312.1748.
(E)- (4-Methoxy-phenyl)-[1-methyl-3-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-
allyl]-amine (78h):
B
O
O
NH
O
Yield: 93 mg (73%), colorless liquid
Rf 0.59 (Heptane / Ethyl acetate (1:1)).
1H NMR (300MHz, CDCl3) 6.77–6.72 (m, 2H), 6.63–6.53 (m, 3H), 5.64 (dd, 1H, J = 18.0
Hz, J = 1.4 Hz), 3.98-3.93 (m, 1H), 3.74 (s, 3H), 1.31 (d, 3H, J = 6.7 Hz), 1.26 (s, 12H);
13C NMR (75 MHz, CDCl3) 155.8, 151.9, 141.4, 114.6, 83.1, 55.7, 52.9, 24.7, 21.2;
11B NMR (96 MHz, CDCl3) 28.9;
HRMS (ESI) [M + Na+]/z calcd. 326.19034, found 326.1905.
(E)- [1,1-Dimethyl-3-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-allyl]-(4-methoxy-
phenyl)-amine (78i):
B
O
O
NH
O
Yield: 97 mg (78%), white amorphous solid
Rf 0.59 (Heptane / Ethyl acetate (1:1)).
1H NMR(300MHz, CDCl3) 6.77-6.61 (m, 5H), 5.59 (d, 1H, J = 18.3 Hz), 3.73 (s, 3H), 1.32
(s, 6H), 1.27 (s, 12H);
131

13C NMR (75 MHz, CDCl3) 160.6, 152.7, 140.0, 118.7, 114.1, 83.1, 55.9, 55.6, 27.8, 24.7;
11B NMR (96 MHz, CDCl3) 28.4;
HRMS (ESI) [M + Na+]/z calcd. 340.20599, found 340.2062.
(E)- (3-Azido-propenyl)-benzene (79)
N3
Yield: 44 mg (83%), colorless oil
Rf 0.68 (Heptane / Ethyl acetate (1:1)).
1H NMR (300MHz, CDCl3) 7.42-7.26 (m, 5H), 6.68 (d, 1H, J = 15.7 Hz), 6.29-6.20 (m,
1H), 3.96 (d, 2H, J = 7.3 Hz);
13C NMR (75 MHz, CDCl3) 135.9, 134.5, 128.6, 128.1, 126.6, 122.3, 53.0.
Typical experimental procedure for one-pot reaction (Table 19):
To a dried argon filled Schlenk 0.416 mmol of gamma-borylated allylic acetate, 2.1 mol of
Pd(OAc)2 and 6.3 mol of PPh3 were dissolved in 2mL of anhydrous THF and stirred for 1 h
at RT. Nucleophile (1.1 eq) was added to the boronate palladium complex mixture at rt and
refluxed for 4 h. After 4 h under refluxing THF, 0.63 mmol of aryliodide and a degassed
saturated aqueous solution of K2CO3 (0.63 mmol) were added to the reaction mixture at room
temperature. After 6 h under refluxing conditions, the reaction mixture was concentrated
under reduced pressure. The residue was dissolved in CH2Cl2 (10 mL), washed with water (5
mL), brine (2 x 5 mL). Organic phases were dried over MgSO4 and purified by silica gel
column flash chromatography.
(E)-(1-Methyl-3-phenyl-allyl)-phenyl-amine (80a):
NH
132

Yield: 72 mg (77%), colorless liquid
Rf 0.59 (Heptane / Ethyl acetate (1:1)).
1H NMR (300MHz, CDCl3) 7.26-7.03 (m, 6H), 6.62-6.44 (m, 4H), 6.14 (dd, 1H, J = 5.8 Hz,
J = 15.9 Hz), 4.05 (m, 1H), 1.30 (d, 3H, J = 6.6 Hz),
13C NMR (75 MHz, CDCl3) 147.2, 136.9, 133.0, 129.2, 129.1, 128.4, 127.2, 126.2, 117.2,
113.3, 50.7, 21.9.
(E)-(1,1-Dimethyl-3-phenyl-allyl)-phenyl-amine (80b):
NH
Yield: 66 mg (72%), white amorphous solid
Rf 0.59 (Heptane / Ethyl acetate (1:1)).
1H NMR (300MHz, CDCl3) 7.41-7.23 (m, 5H), 7.13- 7.08 (m, 2H), 6.75 (dd, 3H, J = 7.8
Hz, J = 16.5 Hz), 6.57 (d, 1H, J = 16.2 Hz), 6.44 (d, 1H, J = 16.2 Hz), 1.49 (s, 6H) ;
13C NMR (75 MHz, CDCl3) 137.9, 137.2, 128.8, 128.5, 127.9, 127.2, 126.3, 117.7, 115.8,
100.0, 68.0, 54.6, 28.7.
(E)-(3-Azido-1-methyl-allyl)-phenyl-amine 81(Scheme 65):
N3
NH
NaN3 (17 mg, 0.28 mmol) and CuSO4 (5 mg, 0.1 mmol) were placed in an oven-dried round
bottomed flask. Subsequently methanol (3 mL) and (E)- [1-Methyl-3-(4,4,5,5-tetramethyl-
[1,3,2]dioxaborolan-2-yl)-allyl]-phenyl-amine (0.05 g, 0.18 mmol) were added. After 4h at
room temperature, the reaction mixture was concentrated under reduced pressure. The residue
was dissolved in CH2Cl2 (10 mL), washed with water (5 mL), brine (2 x 5 mL). Organic
133

phases were dried over MgSO4 and purified by silica gel column flash chromatography
affording 30 mg of 81 as a colorless liquid.
Yield: 30 mg (80%), colorless liquid.
Rf 0.69 (Heptane / Ethyl acetate (1:1)).
1H NMR (300MHz, CDCl3) 7.22 (t, 2H, J = 8.5 Hz), 6.76 (t, 1H, J = 7.3 Hz), 6.61 (d, 2H, J
= 7.6 Hz), 6.13 (dd, 1H, J = 13.4 Hz, J = 1.0 Hz), 5.41 (dd, 1H, J = 13.5 Hz), 4.07 (m, 1H),
1.35 (d, 3H, J = 6.6 Hz);
13C NMR (75 MHz, CDCl3) 146.8, 129.2, 127.3, 122.6, 117.6, 113.3, 48.4, 22.2.
HRMS (ESI) [M + Na+]/z calcd. 211.1044, found 211.1102.
(E)-4,4,5,5-Tetramethyl-2-(3-phenoxy-but-1-enyl)-[1,3,2]dioxaborolane (82):
B
O
O
O
Yield: 85 mg, (74%), colorless liquid
Rf 0.65 (Heptane / Ethyl acetate (1:1)).
1H NMR (300MHz, CDCl3) 7.24-7.21 (m, 1H), 6.92-6.85 (m, 3H), 6.69 (dd, 1H, J = 4.9 Hz,
J = 18.2 Hz), 5.71 (dd, 1H, J = 1.4 Hz, J = 18.2 Hz), 4.85-4.81 (m, 1H), 1.44 (d, 3H, , J = 6.5
Hz), 1.25 (s, 12H); );
13C NMR (75 MHz, CDCl3) 157.9, 152.9, 129.3, 120.5, 115.6, 83.3, 74.8, 24.7, 20.8;
11B NMR (96 MHz, CDCl3) 28.8.
HRMS (ESI) [M + Na+]/z calcd. 297.1632, found 297.1631.
Chiral ligands used in allylation:
134

NH HNOO
PPh2 Ph2P
NH HNOO
PPh2 Ph2P
(R,R)-DACH-Naphthyl Trost(R,R)-L2
(R,R)-DACH- Phenyl Trost Ligand(R,R)-L1
HNHN
O O
PPh2
(R,R)-ANDEN- Phenyl Trost(R,R)- L3
Ph2P
NH HNOO
N N
(R,R)-DACH- PyridylTrost Ligand (R,R)-L4
135

HPLC-1(69b)
136

HPLC-2 (Table 13, entry 4)
137

HPLC-3 (Table 13, entry 1)
138
HPLC-4 (Table 13, entry 2)
139

HPLC-5 (Table 13, enry 3)
140

HPLC-6 (Table 14, enry 1)
141

HPLC-7 (Table 14, entry 2)
142

HPLC-8 (Table 14, entry 3)
143

HPLC-9 (Table 14, entry 4)
144
HPLC-10 (Table 14, entry 5)
145
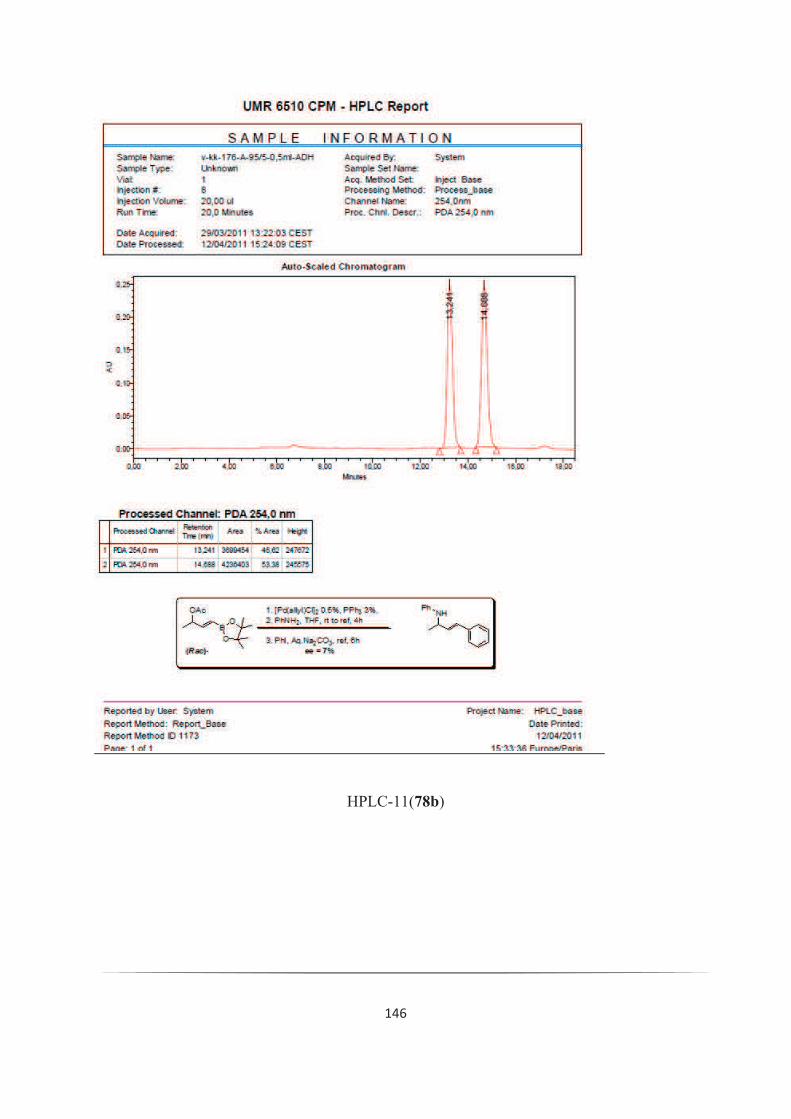
HPLC-11(78b)
146

HPLC-12 (Table 20, entry 1)
147

HPLC-13 (Table 20, entry 2)
148

HPLC-14 (Table 20, entry 3)
149

HPLC-15 (Table 21, entry 1)
150
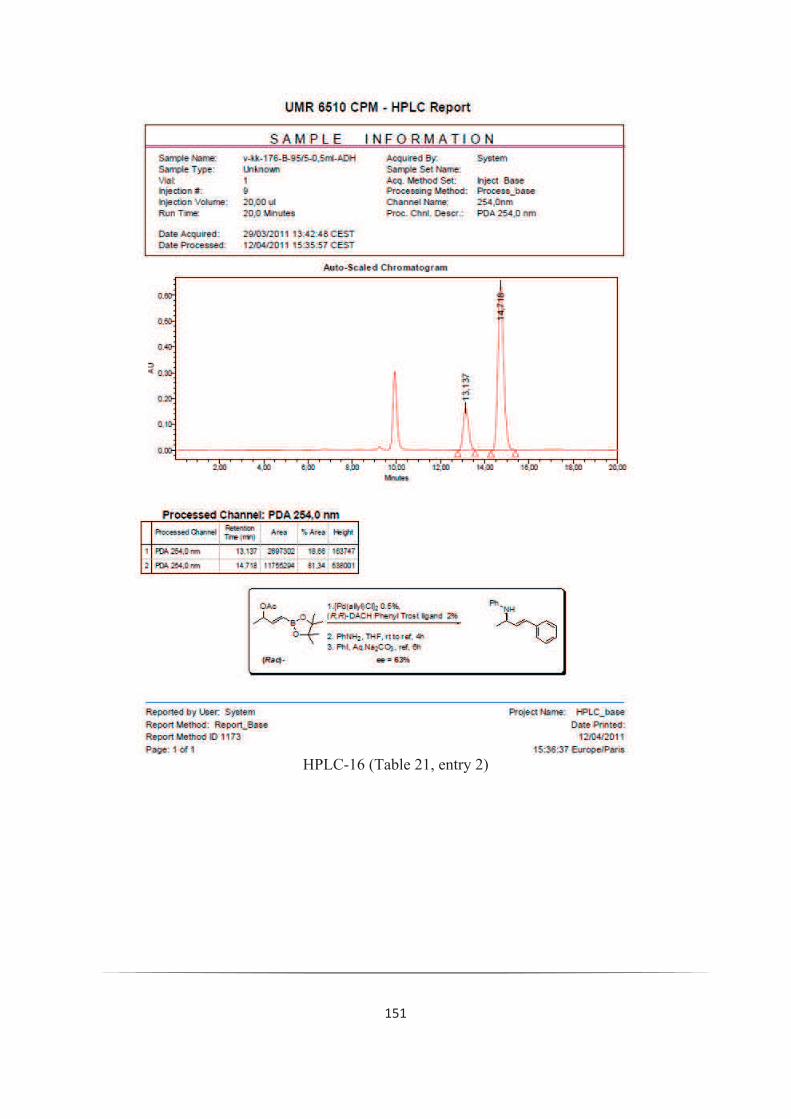
HPLC-16 (Table 21, entry 2)
151

HPLC-17 (Table 21, entry 3)
152
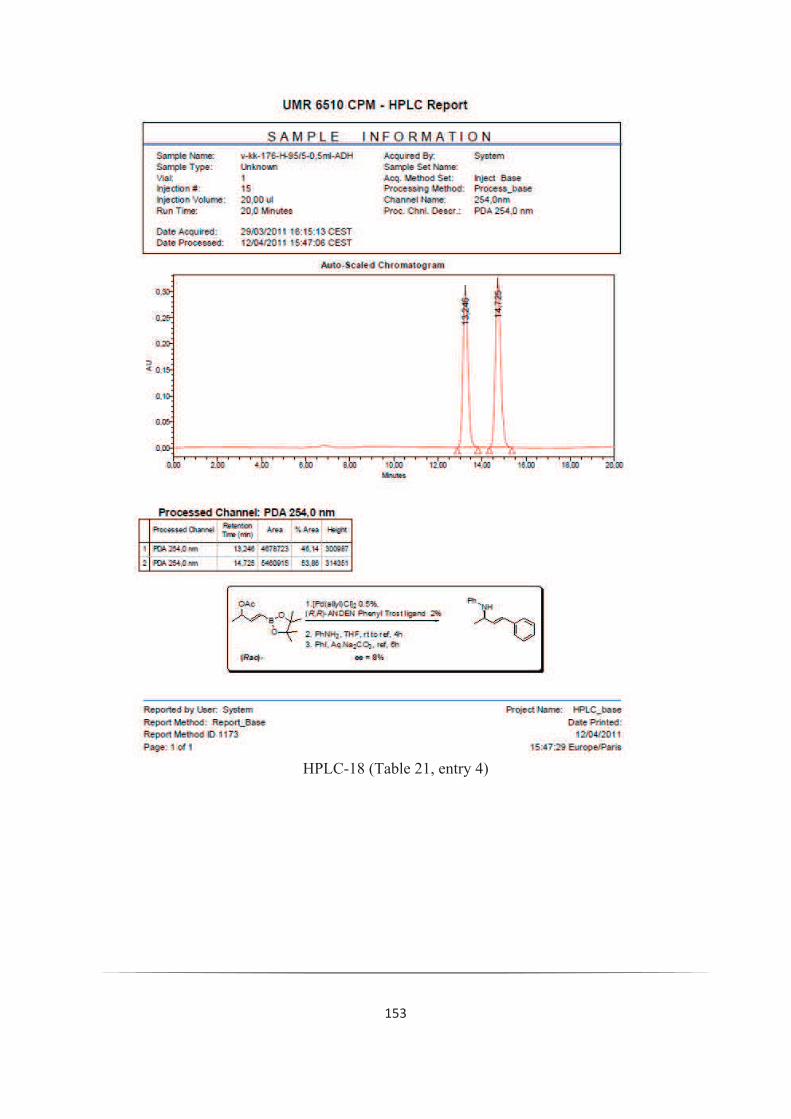
HPLC-18 (Table 21, entry 4)
153

HPLC-19 (Table 20, entry 4)
154

Chapter-III Experimental:
Typical experimental procedure for kinetic resolution in ionic liquids (Scheme 75):
To -borylated alcohol (0.01 g, 0.05 mmol) were added vinylacetate (0.015 mL, 0.15
mmol) and 10mg of CAL-B in ionic liquid (0.485 mL) at rt and heated the reaction at 50 oC.
The reaction was monitored at regular intervals of time (15 min, 30 min, 1 h, 2 h, 4 h, 6 h, 8 h,
24 h) with different ionic liquids and the results were plotted in graph with rate of conversion
vs Time of reaction.
OH
B
OAc (3.0 eq), CAL-B
Ionic Liquid, 50 oC, t min
OH
B
OAc
B+ **
O
O
O
O
O
O
(S)-OH (R)-OAc(+,-)-rac OH
50% 50%
Scheme 75: Kinetic resolution of -borylated allylic alcohols under solvent free media with CAL-B
[rac-OH]
= racemic alcohol (starting material)
[IS]
= Internal standard (Butyl Buterate)
[R-Product]
= Final product- [R-OAc]
[R-OH]
= [R-OH] (which was not formed)
[S-OH]
= Final product-[S-OH]
[S-Product] = [S-OAc] (which was not formed)
[VA]
= vinyl acetate
[Vr]
= Total volume of the reaction
IME
= Immobilized enzyme
(S)-OH [ ]D24
= +11.8 (c 1.0, MeOH)
(R)-OAc [ ]D24
= +42.5 (c 1.0, MeOH)
155

Reaction profile in [BTMA][NTf2] ionic liquid:
The progress of the reaction was monitored by Chiral-GC. The racemic -borylated allylic
acetate was separated in chiral GC with the retention time of isomers tS = 25.3; tR = 25.5
(Picture 5).
The reaction was monitered by comparing with authentic (S)-OAc (synthesized in laboratory)
retention time whose absolute configuration was already known, tS = 25.3 (Picture 6).
The chemoenzymatic resolution was successfully carried on -Borylated allylic alcohol in
ionic liquids. A typical reaction profile was shown below (Picture 7) where the only
compound formed during the reaction was (R)-OAc whose retention time is tR = 25.5 and no
peaks were observed at 25.3.
156
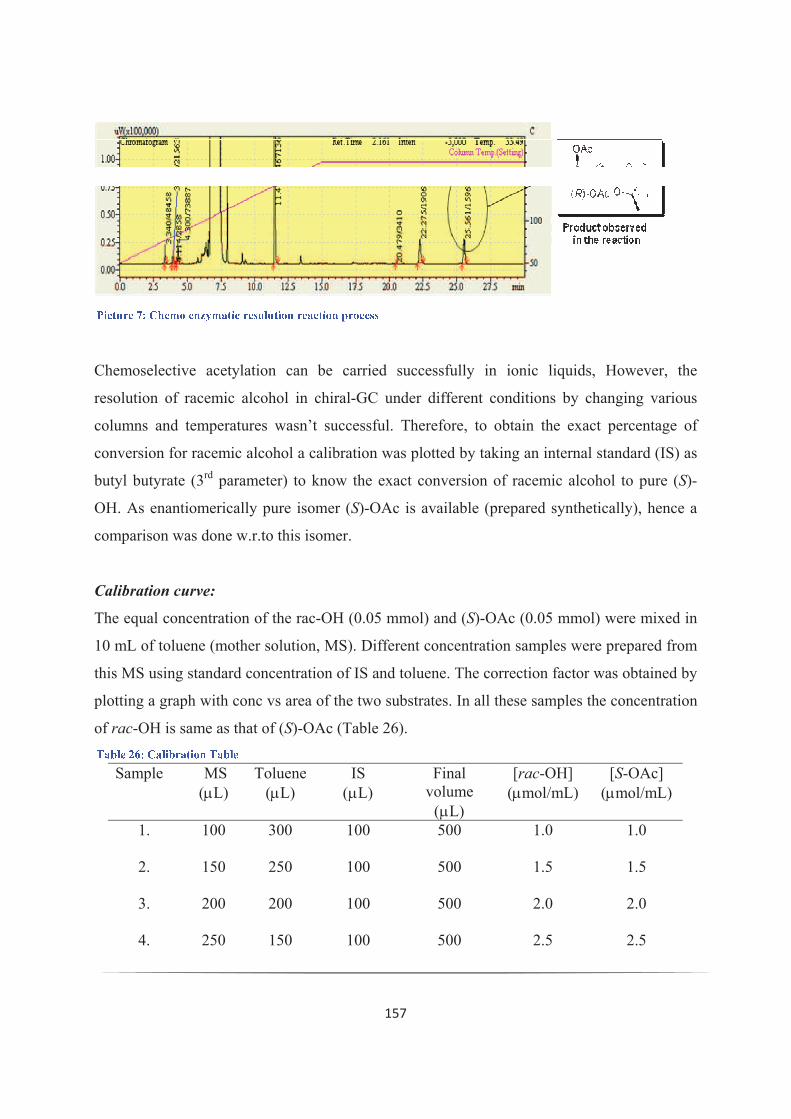
Chemoselective acetylation can be carried successfully in ionic liquids, However, the
resolution of racemic alcohol in chiral-GC under different conditions by changing various
columns and temperatures wasn’t successful. Therefore, to obtain the exact percentage of
conversion for racemic alcohol a calibration was plotted by taking an internal standard (IS) as
butyl butyrate (3rd
parameter) to know the exact conversion of racemic alcohol to pure (S)-
OH. As enantiomerically pure isomer (S)-OAc is available (prepared synthetically), hence a
comparison was done w.r.to this isomer.
Calibration curve:
The equal concentration of the rac-OH (0.05 mmol) and (S)-OAc (0.05 mmol) were mixed in
10 mL of toluene (mother solution, MS). Different concentration samples were prepared from
this MS using standard concentration of IS and toluene. The correction factor was obtained by
plotting a graph with conc vs area of the two substrates. In all these samples the concentration
of rac-OH is same as that of (S)-OAc (Table 26).
Sample MS
( L)
Toluene
( L)
IS
L)
Final
volume
L)
[rac-OH]
( mol/mL)
[S-OAc]
( mol/mL)
1. 100 300 100 500
1.0 1.0
2. 150 250 100 500
1.5 1.5
3. 200 200 100 500
2.0 2.0
4. 250 150 100 500
2.5 2.5
157

5. 300 100 100 500
3.0 3.0
6. 350 50 100 500
3.5 3.5
7. 400 0 100 500 4.0 4.0
These 7 samples were injected in GC (same conditions used for the reaction monitoring).
The chiral GC areas observed for same internal standard concentration shown in Table 27.
Sample A-OH A-S-OAc A-IS
[IS]
( mol/mL)
[S-OAc]
( mol/mL)
[Rac-OH]
( mol/mL)
1. 121565 177941 1636903 15 1.0 1.0
2. 189332 261633 1608050 15 1.5 1.5
3. 259268 353723 1608770 15 2.0 2.0
4. 337548 453942 1661121 15 2.5 2.5
5. 414080 550208 1667355 15 3.0 3.0
6. 465811 624893 1628294 15 3.5 3.5
7. 561627 735411 1841803 15 4.0 4.0
A calibration was done with internal standard to the concentrations of rac-OH and S-OAc, and
also to the areas from GC. (The concentrations and areas were divided with respective
concentration of Internal standard and areas from GC, Table 28)
A-(S)-OAc
Sample / A-IS A rac-OH
/ A-IS [S-OAc] /
[IS][rac-OH]
/ [IS]
1 0.108 0.074 0.066 0.066
2 0.162 0.117 0.1 0.1
3 0.219 0.161 0.133 0.133
4 0.273 0.203 0.166 0.166
5 0.329 0.248 0.2 0.2
6 0.383 0.286 0.233 0.233
7 0.399 0.304 0.266 0.266
From this calibrated values a plot of racemic alcohol concentration vs its area with respect to
internal standard gives the exact correction factor for the concentration of rac-OH.
158
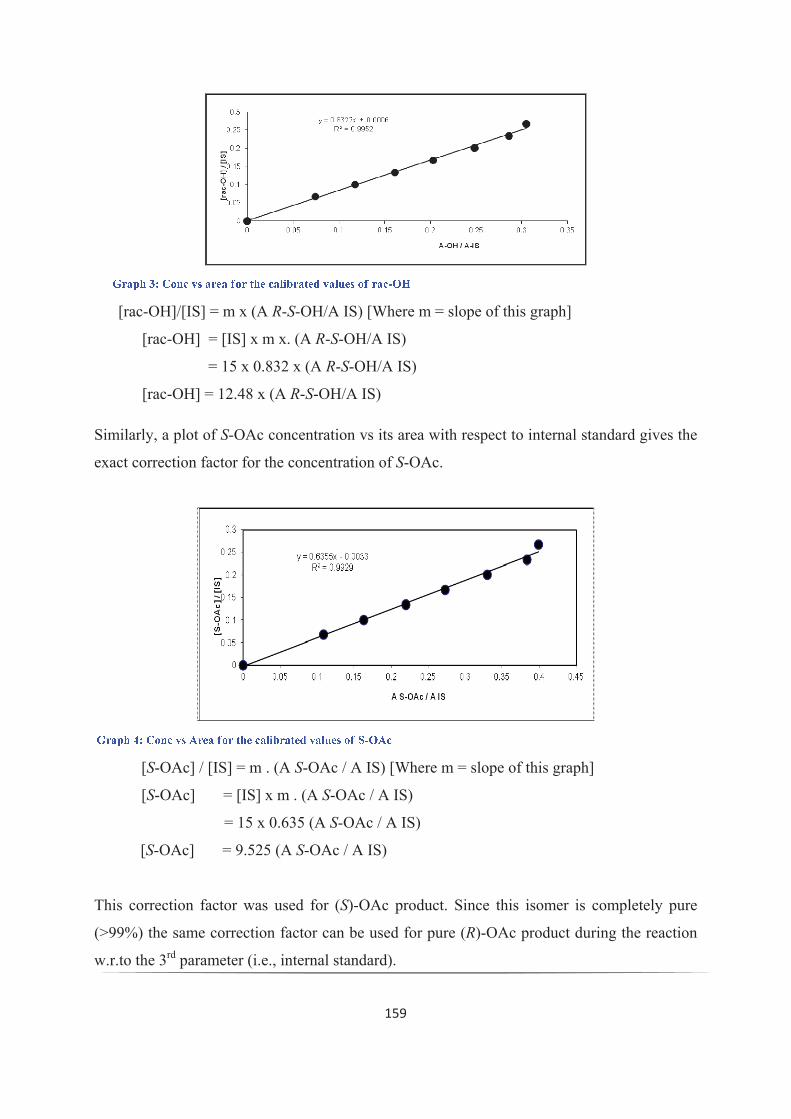
[rac-OH]/[IS] = m x (A R-S-OH/A IS) [Where m = slope of this graph]
[rac-OH] = [IS] x m x. (A R-S-OH/A IS)
= 15 x 0.832 x (A R-S-OH/A IS)
[rac-OH] = 12.48 x (A R-S-OH/A IS)
Similarly, a plot of S-OAc concentration vs its area with respect to internal standard gives the
exact correction factor for the concentration of S-OAc.
[S-OAc] / [IS] = m . (A S-OAc / A IS) [Where m = slope of this graph]
[S-OAc] = [IS] x m . (A S-OAc / A IS)
= 15 x 0.635 (A S-OAc / A IS)
[S-OAc] = 9.525 (A S-OAc / A IS)
159
This correction factor was used for (S)-OAc product. Since this isomer is completely pure
(>99%) the same correction factor can be used for pure (R)-OAc product during the reaction
w.r.to the 3rd
parameter (i.e., internal standard).
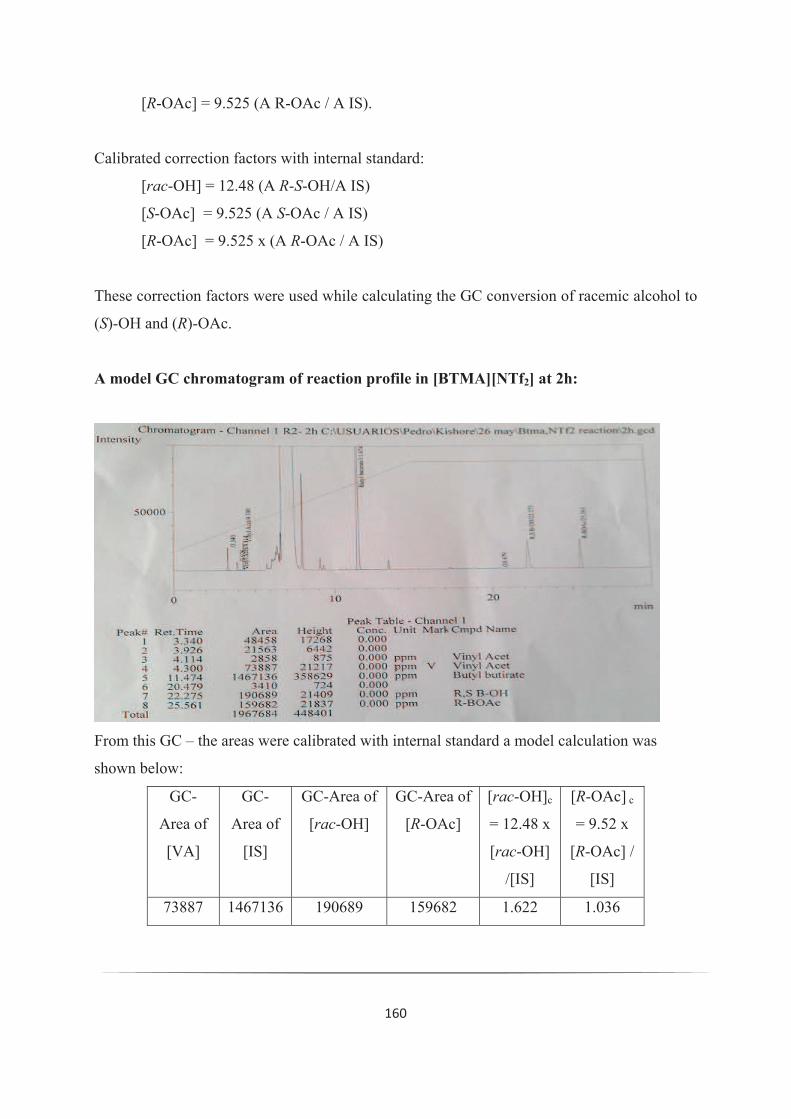
[R-OAc] = 9.525 (A R-OAc / A IS).
Calibrated correction factors with internal standard:
[rac-OH] = 12.48 (A R-S-OH/A IS)
[S-OAc] = 9.525 (A S-OAc / A IS)
[R-OAc] = 9.525 x (A R-OAc / A IS)
These correction factors were used while calculating the GC conversion of racemic alcohol to
(S)-OH and (R)-OAc.
A model GC chromatogram of reaction profile in [BTMA][NTf2] at 2h:
From this GC – the areas were calibrated with internal standard a model calculation was
shown below:
GC-
Area of
[VA]
GC-
Area of
[IS]
GC-Area of
[rac-OH]
GC-Area of
[R-OAc]
[rac-OH]c
= 12.48 x
[rac-OH]
/[IS]
[R-OAc] c
= 9.52 x
[R-OAc] /
[IS]
73887 1467136 190689 159682 1.622 1.036
160

Example : At 2 h reaction time in [BTMA][NTf2] the GC-area were calibrated.
[rac-OH]C = calibrated area with respect to internal standard = 12.48 x [rac-OH] / [IS] =
1.622
[R-OAc]c = calibrated area w.r.to internal standard = 9.525 x [R-OAc] / [IS] = 1.036
Therefore, Correction in the area of [rac-OH] = 1.622 / (1.622 + 1.036) = 61%
Therefore, Correction in the area of [R-OAc] = 1.036 / (1.622 + 1.036) = 39%
Since the initial percentage of [R-OH] and [S-OH] in Racemic mixture is 50/50.
Therefore the area of [R-OH] remaining = 50-[R-OAc] = 50 – 39 = 11%
Area of [S-OH] = 50-[S-OAc] = 50-0 = 50%.
In this reaction profile
[R-OH] [S-OH] [R-OAc] [S-OAc]
11% 50% 39% 0%
Same calibration was done for each chromatogram using correction factor to know the exact
% of conversion and were shown in graph.
Kinetic resolution in ionic liquids using CAL-B: (Scheme 75)
Typical reaction conditions:
Ionic liquid volume [IL] = 0.485 mL
Vinyl acetate volume [VA] = 0.15 mL
Racemic alcohol substrate [rac-OH] = 10 mg (50.51 mol)
CAL-B enzyme = 10 mg (IME)
The reaction was monitored with Chiral GC and the reaction profiles in ionic liquids were
shown in graphical representation at each interval of time for ex: 15 min, 30 min, 1h, 2h, 4h,
6h, 8h, and 24h respectively. From this graph the enzyme activity was calculated.
161

1. Reaction profile in [BMIM][NTf2]: (Table 22, entry 1)
Enzyme activity in [BMIM][NTf2] = slope x mol of [rac-OH] / mg of IME
= (0.544 x 50.51) / 10 = 2.74 U / mg of
Reaction profile in [OMIM][NTf2] : (Table 22, entry 2)
Enzyme activity in [OMIM][NTf2] = (1.255 x 50.51) / 10 = 6.33 U / mg of IME
Reaction profile in [C12MIM][NTf2] : (Table 22, entry 3)
162

Enzyme activity in [C12MIM][NTf2] = (1.51 x 50.51) / 10 = 7.63 U/mg of IME
Reaction profile in [BTMA][NTf2] : (Table 22, entry 4)
Enzyme activity in [BTMA][NTf2] = (0.44 x 50.51) /10 = 2.26 U/mg of IME
5. Reaction profile in [TBMA][NTf2] : (Table 22, entry 5)
Enzyme activity in [TBMA][NTf2] = (0.48 x 50.51) /10 = 2.45 U/mg of IME
6. Reaction profile in [BMPy][NTf2] : (Table 22, entry 6)
163
Enzyme activity in [BMPy][NTf2] = (0.56 x 50.51) /10 = 2.83 U/mg of IME
7. Reaction profile in [BMPi][NTf2] : (Table 22, entry 7)
Enzyme activity in [BMPi][NTf2] = (0.65 x 50.51) /10 = 3.31 U/mg of IME
8. Reaction profile in [BMIM][PF6] : (Table 22, entry 8)
Enzyme activity in [BMIM][PF6] = (0.69 x 50.51) /10 = 3.34 U/mg of IME
9. Reaction profile in [OMIM][PF6] : (Table 22, entry 9)
164

Enzyme activity in [OMIM][PF6] = (0.37 x 50.51) /10 = 1.87 U/mg of IME
10. Reaction profile in [C12MIM][PF6] : (Table 22, entry 10)
Enzyme activity in [C12MIM][PF6] = (0.36 x 50.51) /10 = 1.86 U/mg of IME
11. Reaction profile in [BMIM][BF4] : (Table 22, entry 11)
Enzyme activity in [BMIM][BF4] = (0.98 x 50.51) /10 = 4.97 U/mg of IME
165
12. Reaction profile in [C12MIM][BF4] : (Table 22, entry 12)
Enzyme activity in [C12MIM][BF4] = (0.36 x 50.51) /10 = 1.82 U/mg of IME
Enzymatic Resolution in Continuous flow reactors: (Scheme 76)
Experimental procedure: Vinyl acetate (3 mmol) and rac-OH (1 mmol) were dissolved in 50
mL of hexane in a flask and pumped under controlled flow (0.1 mL / min) through the
stationary phase which was filled with CAL-B on ionic liquid as a heterogeneous support. A
controlled flow (0.9 mL / min) with 100 bar pressure of scCO2 was used as mobile phase.
Heterogeneous support (stationary phase) preparation for CAL-B / [BMIM][NTf2]:
(Table 25, entry 1)
100 mg of [BMIM][NTf2] and 200 mg of CAL-B were mixed in 2 mL ACN solvent and the
solvent was evaporated to get the enzyme coated with ionic liquid of 300 mg mixture. The
stationary phase was prepared by 270 mg of this mixture.
The amount of enzyme present in the stationary phase = (270 / 300) x 200 = 180 mg = 0.18 g.
1st Cycle: [rac-OH] = 1 mmol, [VA] = 3 mmol were mixed in 50mL of hexane
Total flow rate = 1 mL /min (scCO2 = 0.9 mL and Substrate = 0.1 mL).
Concentration of [rac-OH] = 1 mmol = 1 x 10-3
mol / Lt,
Amount of Substrate = 0.1 mL / min = 0.1 x 10-3
Lt / min,
Substrate flow rate per min during reaction = Concentration x Amount of Substrate
= 1 x 10-3
x 0.1 x 10-3
mol / min = 0.1 mol/min
166

Substrate flow rate per hour = 60 x 0.1 mol/min = 6 mol/min
The conversion from GC was plotted in graph below:
The % of conversion observed from graph to form (R)-OAc = 40% (from graph)
The overall productivity per hour = 6 x 0.4 = 2.4 mol/ h.
Note: 0.18g of enzyme was presented in the stationary phase.
The enzyme activity per gram of CAL-B in [BMIM][NTf2] in continuous flow system
= 2.4/0.18 = 13.33 mol/h/g of enzyme.
2nd
Cycle (Table 25, entry 2): [rac-OH] = 1 mmol, [VA] = 3 mmol were mixed in 50mL of
hexane, the same stationary phase and same flow rate 0.1 mL /min was used for the second
cycle for another 8h. It was observed the same percentage of conversion (40%) and the
enzyme activity didn’t changed remains same 13.33 mol/ h/ g of enzyme.
3rd
Cycle (Table 25, entry 3): [rac-OH] = 2 mmol, [VA] = 6 mmol were mixed in 50mL of
hexane
The same stationary phase was used but the concentration was doubled.
Total flow rate = 1 mL/min (scCO2 = 0.9 mL and Substrate = 0.1 mL).
Concentration of [rac-OH] = 2 mmol = 2 x 10-3
mol/Lt,
Amount of Substrate = 0.1 mL/min = 0.1 x 10-3
Lt/min,
Substrate flow rate per min during reaction = Concentration x Amount of Substrate L/min
= 2 x 10-3
x 0.1 x 10-3
mol/min = 0.2 mol/min
Substrate flow rate per hour = 60 x 0.2 mol/min = 12 mol/min
167

The conversion from GC was plotted in graph below:
The % of conversion observed from graph to form (R)-OAc = 40% (from graph)
The overall productivity per hour = 12 x 0.4 = 4.8 mol/h.
Note: 0.18g of enzyme was presented in the stationary phase.
The enzyme activity per gram of CAL-B in [BMIM][NTf2] in continuous flow system
= 4.8/0.18 = 26.66 mol/h/g of enzyme.
Heterogeneous support (stationary phase) preparation for CAL-B / [OMIM][NTf2]:
(Table 25, entry 4)
100mg of [OMIM][NTf2] and 200 mg of CAL-B were mixed in 2 mL ACN solvent and the
solvent was evaporated to get the enzyme coated with ionic liquid of 300 mg mixture. The
stationary phase was prepared by 250 mg of this mixture.
The amount of enzyme present in the stationary phase = (250/300) x 200 = 166 mg = 0.166 g.
1st Cycle: [rac-OH] = 1 mmol, [VA] = 3 mmol were mixed in 50 mL of hexane
Total flow rate = 1 mL /min (scCO2 = 0.95 mL and Substrate = 0.05 mL).
Concentration of [rac-OH] = 1 mmol = 1 x 10-3
mol/Lt,
Amount of Substrate = 0.05 mL /min = 0.05 x 10-3
Lt/min,
Substrate flow rate per min during reaction = Concentration x Amount of Substrate
= 1 x 10-3
x 0.05 x 10-3
mol/min = 0.05 mol/min
Substrate flow rate per hour = 60 x 0.05 mol/min = 3 mol/min
The conversion from GC was plotted in graph below:
168

The % of conversion observed from graph to form (R)-OAc = 50% (from graph)
The overall productivity per hour = 3 x 0.5 = 1.5 mol/h.
Note: 0.166 g of enzyme was presented in the stationary phase.
The enzyme activity per gram of CAL-B in [OMIM][NTf2] in continuous flow system
= 1.5/0.16 = 9.03 mol/h/g of enzyme.
2nd
Cycle (Table 25, entry 5): [rac-OH] = 1 mmol, [VA] = 3 mmol were mixed in 50 mL of
hexane, the same stationary phase and same flow rate 0.05 mL was used for the second cycle
for another 8 h. It was observed the same percentage of conversion (50%) and the enzyme
activity didn’t changed, it remains same 9.03 mol/h/g of enzyme.
3rd
Cycle (Table 25, entry 6): [rac-OH] = 1 mmol, [VA] = 3 mmol were mixed in 50 mL of
hexane, the same stationary phase was used but the substrate flow was doubled.
169

Total flow rate = 1 mL /min (scCO2 = 0.9 mL and Substrate = 0.1 mL).
Concentration of [rac-OH] = 1 mmol = 1 x 10-3
mol/Lt,
Amount of Substrate = 0.1 mL /min = 0.1 x 10-3
Lt/min,
Substrate flow rate per min during reaction = Concentration x Amount of Substrate L/min
= 1 x 10-3
x 0.1 x 10-3
mol/min = 0.1 mol/min
Substrate flow rate per hour = 60 x 0.1 mol/min = 6 mol/min
The conversion from GC was plotted in graph below:
The % of conversion observed from graph to form (R)-OAc = 50% (from graph)
The overall productivity per hour = 6 x 0.5 = 3 mol/h.
Note: 0.166g of enzyme was presented in the stationary phase.
The enzyme activity per gram of CAL-B in [OMIM][NTf2] in continuous flow system
= 3/0.16 = 18.07 mol/h/g of enzyme.
170

Compounds Synthesized:
171

172

173

Conclusion & Perspectives:
In the first part of our research we have developed a chemo, regio-, and stereo-
selective Tsuji-Trost allylation reaction starting with highly functionalized building
blocks, the -borylated allylic acetates. We also developed a one-pot strategy of Tsuji-
Trost allylation, followed by Suzuki-Miyaura reactions, using -borylated allylic
acetates. Further, -borylated allylic acetates were employed for asymmetric allylic
alkylation to give enantioenriched -borylated allyl derivatives. The resulting
products, after allylation, could be subjected to a wide range of reactions using the
pinacol boronate moiety: for example it could be employed in 1,4-addition reactions
using rhodium catalysts, it could be subjected to halogenolysis since the resulting
vinyl halide derivatives could be, as well, useful key intermediates for various
synthetic transformations and transition metal catalyzed cross couplings. As an
extension to this work, it would be interesting to test such a Tsuji-Trost allylation
reaction of -borylated allylic acetates in ionic liquids.137
In the second part we successfully developed a kinetic resolution process for a -
borylated allylic alcohol, by using an enzyme, Candida Antartica Lipase (CAL-B),
along with ionic liquids. Further, we developed this kinetic resolution of a -borylated
allylic alcohol in continuous flow systems using immobilized enzyme (CAL-B) on
ionic liquid support, along with scCO2. As an extension to this work, it would be
interesting to perform a dynamic kinetic resolution process in continuous flow systems
in combination with components for the racemization step such as zeolites or
transition metal catalysts, for instance. Further, such enzyme-mediated kinetic
dynamic resolution process in continuous flow systems could be extended to other -
borylated allylic alcohols. Such new technologies are perfectly in line with a
development of a sustainable chemistry.
137 Liao, M-C, Duan, X-H, Liang, Y-M. Tetrahedron Lett. 2005, 46, 3469.
174

ANNEXE 2 (Modèle dernière page de thèse)
VU : VU :
Le Directeur de Thèse Le Responsable de l'École Doctorale
(Nom et Prénom)
VU pour autorisation de soutenance
Rennes, le
Le Président de l'Université de Rennes 1
Guy CATHELINEAU
VU après soutenance pour autorisation de publication :
Le Président de Jury,
(Nom et Prénom)
175

176