jieli li 04/04/06. chief complaint progressive left sided weakness
TRANSCRIPT

Jieli Li
04/04/06

Chief Complaint
Progressive left sided weakness

HPI 63 y/o Caucasian male with no significant PMH presents
with progressive left leg weakness over 8 months. Per pt he now requires a walker and is only able to ambulate 20 feet.
Pt also reports left hand clumsiness, double vision (horizontal), difficulty swallowing and difficulty speaking.
He has also had falls x 2 due to weakness, not imbalance. Most recently he slumped over the walker onto dresser, no LOC, no head trauma. After this episode he called EMS. He is unable to cook, requires assistance to dress and bathe, and has not left his home in nearly one year.

History cont. PMH/PSH
None FH
mother – pancreatic cancer at age 85 father – Parkinsonism at age 85
SH Born in Bronx, completed 2 yrs of college, former
statistician, divorced, no children, no hx of STDs. Has only traveled to Canada, has had cats as pets
Habits: 10 pk yr smoking hx, quit 30 yrs ago. No etoh, no drugs

History cont. Meds
None
Allergies None
ROS Neg for f/c/night sweats, HA, hearing changes, sore
throat, CP, palpitations, cough, SOB, n/v, abdominal pain, diarrhea/constipation, dysuria

Physical Exam VS: 97, 130/72, 89, 16, 97% on RA Gen: cachectic male in NAD Skin: brown crusted lesions over feet, ears, scalp HEENT: NC/AT, TM intact and clear, nares clear,
oropharynx clear, MMM Neck: supple, no bruits/thyromegaly/LAD Heart: rrr, s1s2, no murmurs Lungs: decreased bibasilar breath sounds, R > L Abd: soft, nt/nd, na bs, no hsm Ext: no c/c/e

Physical Exam cont. Neuro: AAO x 4, speech spontaneous but slow and dysarthric,
comprehension intact, MS 30/30. Very labile affect, markedly depressed mood. Right eye deviated medially at rest, abduction of both eyes incomplete during lateral gaze. Mild left lower facial droop. Motor: bulk reduced, + pronator drift of left arm, strength 5/5 on right
except hip flexion is 2/5, strength is 4/5 on left except unable to flex hip or extend knee, ankle extension is 1/5.
Sensory: diffusely intact including proprioception DTR’s: 3+ diffusely except 4+ at ankle, bilateral up-going toes,
bilateral ankle clonus, sustained on left Coordination: dysmetria on FTN left worse than right, slow rapid
alternating movement on left Station/Gait: No truncal ataxia when sitting up. When pulled to
standing pt keeps balance but refuses to take a step.

Laboratory CBC:
9.6, 12.1/37.0, 311, MCV 78.7 (iron panel showed ACD pattern)
Anion gap panel: 136/4.0, 101/28, 14/0.8, 127 Ca 8.8, Mg 1.8, P 3.7
LFT’s: Albumin 2.1, total bili 0.4, AST 13, ALT11, alk phos 81, LDH
139 Coags:
INR 1.1, PT14, PTT 28.5

Labs cont. Vitamin B12 515 Folate 9.39 RPR NR HIV neg AFB neg ACE neg Serum brucellosis, proteus,
salmonella neg Serum histoplasma neg Serum crypto Ag neg Serum cocci neg Lyme screen neg
ESR 50, CRP 15 U. tox neg CEA 2.3 PSA 0.99 AFP 2.2 TSH 1.07 ANA neg, RF neg Anti-ds DNA neg C-ANCA/P-ANCA neg Cryo neg C3, C4 wnl

Lumbar Puncture 0 WBC, 0 RBC Gram stain no organism, no cells, no growth Glucose 58, Protein 71 (10-50), albumin 43.8 (13-24) India ink neg Fungal cx neg Crypto antigen neg VDRL neg AFB cx and smear neg TB PCR neg ACE 3 HSV neg Cytology neg

Brain MRI Mild hyperintensity in the belly of the pons that is
positive for contrast enhancement. There is evidence of mild to moderate cerebellar atrophy.
Repeat MRI later showed lesions in bilateral medial temporal lobes (brain biopsy of these lesions showed mild reactive gliosis which was nonspecific).

PET scan
A moderately intense focus of activity in the region of the pons
Large areas of slightly heterogeneous moderate uptake are demonstrated in the thorax, in lobar or lobular distribution involving the right middle lobe and left lower lobe. Infiltrative vs. infectious etiology.

CT chest Right infrahilar irregular soft tissue abnormality
measuring 7 x 4.5 cm in size, with collapse and infiltration of right middle lobe and right lower lobe. + reactive right infrahilar adenopathy, measuring 1.4 cm in size.
Peribronchiolar soft tissue abnormality is also seen in the basilar segments of the left lower lobe.
Moderate sized right-sided pleural effusion, small left-sided pleural effusion. No pleural nodularity
Ill-defined sclerotic lesions in sternum, T10 vertebral body, proximal humeral head, suspicious for metastatic disease.

Lung and Pleural Biopsy Pulmonary and pleural tissue showing
marked fibroplasia, chronic inflammation, organizing pneumonia with sclerosis, aggregates of foamy histiocytes (postive for CD68), hyperplasia of mesothelial cells and type 2 pneumocytes.

Bone Scan
Diffuse, bilaterally symmetrical, prominently increased peripheral uptake of the radiotracer involving the clavicles, humeri, proximal forearms, femurs, and proximal/mid tibiae/fibulae is most suggestive of peripheral bone marrow expansion.

Bone Marrow Biopsy
Normocellular marrow with several small lymphoid aggregates, overall findings are non-diagnostic, but the possibility of a lymphoproliferative process cannot be ruled out.

Skeletal Survey
Sclerosis of the diaphyses and metaphyses of bilateral femurs and humeri, with sparing of epiphyses, consistent with the diagnosis of Erdheim-Chester Disease.

Clinical Course Briefly, pt was in house from 09/2005 to 01/2006 for workup
of his illness. Multiple subspecialty services were involved. While an inpatient he continued to have progressive neurologic deterioration with worsening dysarthria and development of bilateral exophthalmos.
After the diagnosis of Erdheim-Chester Syndrome, pt underwent a short steroid taper trial w/o improvement. Due to pt’s poor functional status, he was not felt to be a good candidate for interferon or chemo. Moreover, pt himself refused interferon. He agreed to continued tx with steroids (prednisone 1mg/kg/day) and was enrolled into a hospice program. He continues to be followed by neurology to assess response to tx (serial bone scan, skeletal films of long bones, neurologic assessment q2-3 months)


Overview Erdheim-Chester disease is a rare sporadic systemic
histiocytic disease of unknown etiology First identified by William Chester in 1930 Primarily affects middle-aged and older adults Vary from asymptomatic or minimally symptomatic
bone lesions to a severe multisystem disease Prognosis depends on the extent of extraosseous
disease 3-yr survival rate is 50% Mortality is usually from respiratory distress or cardiac
failure

Clinical Presentation Predominantly involves the long bones of the extremities. Bone pain is the most common presenting symptom 50% patients have disease involvement in other tissues
Skin - xanthomas Retroorbital and periorbital tissues - exophthalmos Pituitary-hypothalamic axis – can lead to diabetes insipidus Heart Kidney Retroperitoneum Skeletal muscle Lung – can lead to pulmonary fibrosis

Histiocytes & Histiocytosis Originate from pluripotent stem cells in the bone marrow Under the influence of various cytokines (e.g., GM-CSF,
TNF-a, IL–3, IL-4), these precursor cells can differentiate into specific groups of antigen-processing cells, some with phagocytic capabilities
These include tissue macrophages, monocytes, dendritic cells, interdigitating reticulum cells, and Langerhans cells
Each disease category in histiocytosis can be traced to reactive or neoplastic proliferation and disorder of cells in one of these groups

Pathology ECD used to be considered a variant of
Langerhans cell histiocytosis (LCH), in particular it was thought to be similar to Hand-Schuller Christian disease
Characterized by diffuse infiltration of the affected organs by lipid-laden histiocytes. They resemble Langerhans cells but are
immunohistologically different (CD68 +, CD1a -) Sometimes referred to as xanthogranulomatous
infiltration

ECD vs. Hand-Schuller-Christian
ECD HSC
SkeletalMixed lytic/sclerotic lesions
Lytic lesions predominate
Age21-77 with average of 54
Children, adolescents, young adults
Immuno-
phenotypeCD1a neg, S-100 neg, CD68 pos
CD1a pos, S-100 pos, CD68 neg
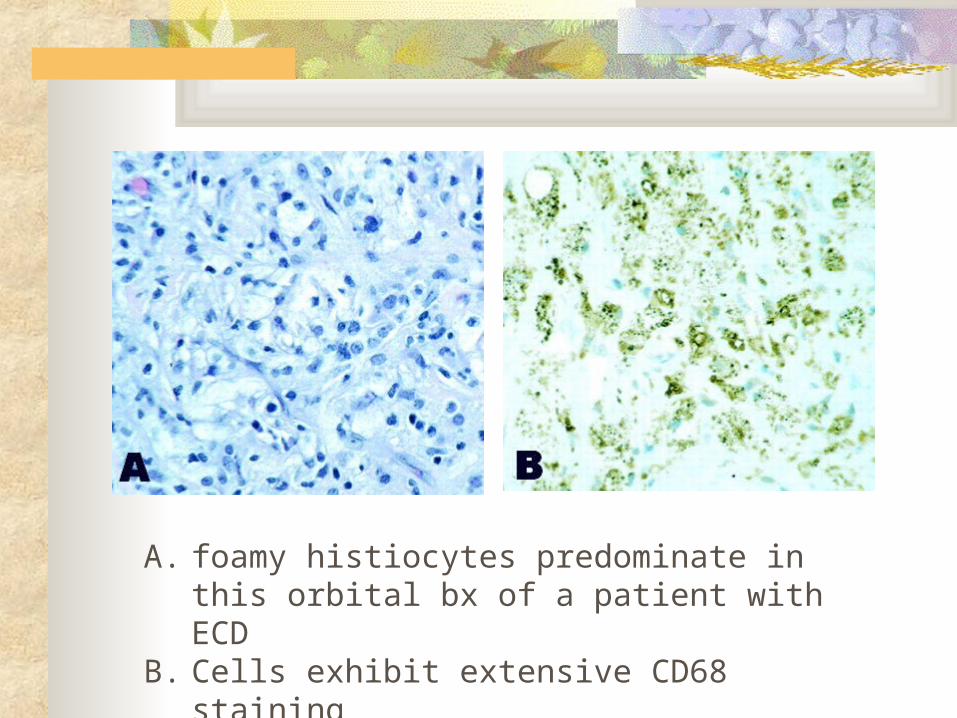
A. foamy histiocytes predominate in this orbital bx of a patient with ECD
B. Cells exhibit extensive CD68 staining

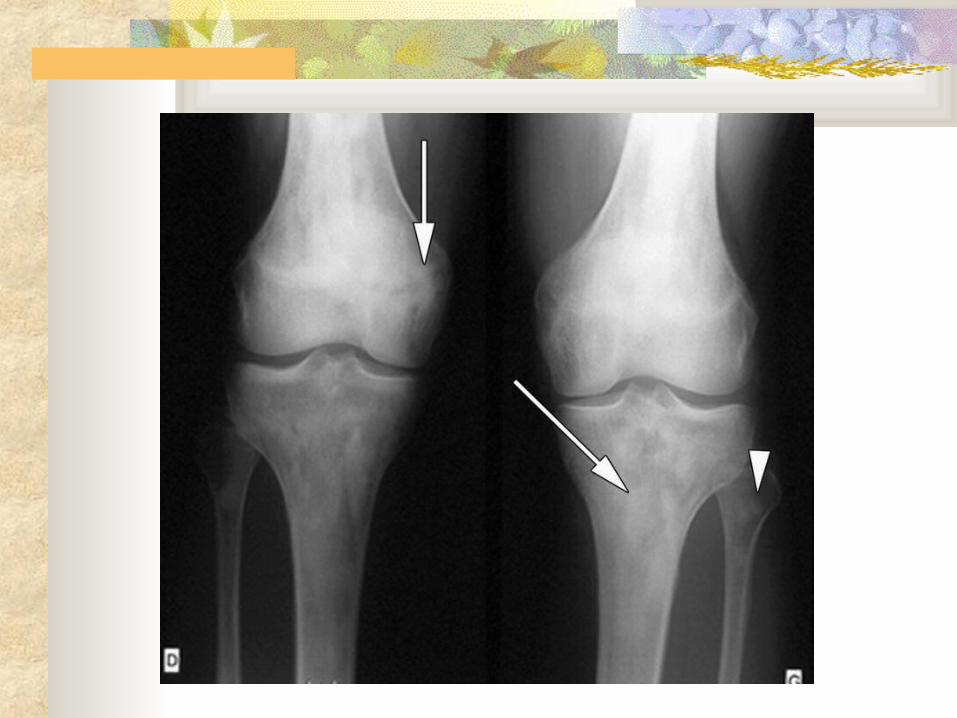
Radiological Diagnosis Skeletal Survey
Bilateral, symmetric, patchy, sclerotic lesions of the metaphyses and diaphyses of long bones, with epiphyseal sparing
These changes are considered virtually pathoneomonic
Bone Scan Usually showing increased tracer uptake in areas that
are abnormal on routine radiographs

Pulmonary Involvement About 20% of Erdheim-Chester Disease patients have
involvement of the lungs Dyspnea is a frequent presenting feature Most have diffuse interstitial infiltrates and pleural and/or
interlobar septal thickening on imaging Histopathology
Accumulation of histiocytes with variable amounts of fibrosis and variable lymphoplasmacytic infiltrate
Immunostains CD68 +, CD1a -

CNS Involvement Diabetes insipidus
Most frequent CNS sign Associated with the presence of orbital involvement
in some cases ? Extension from the orbit along the optic nerves and
chiasm to the hypothalamic-pituitary axis Cerebellar symptoms and signs
Gait ataxia Multiple Sclerosis Spinal epidural and extradural masses

CNS Involvement The evolution of the neurological symptoms and signs is
usually slowly progressive Neurological problems may arise in isolation or in
association with symptoms and signs of systemic disease. CSF analysis is usually normal or shows small increases
of protein MRI often shows intense gadolinium enhancement, which
may persist for a prolonged period Extra-axial lesions – often involve the dura over the convexities
or along the falx Intra-axial lesions – often involve the cerebellum and pons,
rarely pituitary gland

Treatments Various therapies have been tried including
Corticosteroids – usually results in only transient improvement
Chemo Radiation therapy Interferon cyclosporin

References
Allen TC. Pulmonary and Ophthalmic Involvement with Erdheim-Chester Disease: A Case Report and Review of the Literature. Arch Pathol Lab Med 2004; 128: 1428-1431
Johnson MD, Aulino JP, JagasiaM, Mawn, LA. Erdheim-Chester Disease Mimicking Multiple Meningiomas Syndrome. Am J Neuroradiol 2004; 25: 134-137
Kenn W, Eck M, Allolio B, Jakob F, Illg A, Marx A, Mueller-Hermelink HK, Hahn D. Erdheim-Chester disease: evidence of a disease entity different from Langerhans cell histiocytosis? Three cases with detailed radiological and immunohistochemical analysis. Hum Pathol 2000; 31(6):734-9

References cont.
Veyssier-Belot C, Cacoub P, Capparros-Lefebvre D, Wechsler J, Brun B, Remy M, Wallaert B, Petit H, Grimaldi A, Wechsler B, Godeau P. Erdheim-Chester disease. Clinical and radiologic characteristics of 59 Cases. Medicine (Baltimore) 1996; 75(3): 157-69.
Wright RA, Hermann RC, Parisi JE. Neurological manifestations of Erdheim-Chester disease. J Neurol Neurosurg Psychiatry 1999; 66: 72-75