iron_heme_proteins_peroxidases_review_eib2005 (2)
TRANSCRIPT

Iron: Heme Proteins,Peroxidases, Catalases &Catalase-peroxidases
B. Bhaskar, Latesh Lad & Thomas L. PoulosUniversity of California-Irvine, Irvine, CA, USA
Based in part on the article Iron: Heme Proteins, Peroxidases, &Catalases by Ann M. English which appeared in the Encyclopedia ofInorganic Chemistry, First Edition.
1 Introduction 12 Class I Peroxidases 23 Class II Peroxidases 74 Class III Peroxidase 85 Bacterial Di-heme Cytochrome C peroxidases
(BCcP) 106 Prostaglandin Endoperoxide H2 Synthases-1 and 2
(PGHS) 117 Mammalian Peroxidases 138 Catalases 169 References 17
Glossary
Compound I: two-electron oxidized intermediate of hemeperoxidases and catalasesCompound II: one-electron oxidized intermediate of hemeperoxidases and catalases
Abbreviations
CcP = Cytochrome c Peroxidase; BCCP = Bacterial Cyto-chrome c Peroxidases; APX = Soybean or pea orTobacco Ascorbate Peroxidase; MNP = Manganese Per-oxidase; LIP = Lignin Peroxidase; EPO = Eosinophil Per-oxidase; LPO = Lactoperoxidase; MPO = Myeloperoxidase;TPO = Thyroid Peroxidase; ARP = ArthomycesRamosus
Peroxidase; SPO = Salivary Peroxidase; PGHsynthase =Prostaglandin Hsynthase; KatG = Catalase-peroxidase;EPR = Electron Paramagnetic Resonance; RR = ResonanceRaman; BHA = Benzhydroxamic Acid; FA = FerulicAcid; DBNBS = 3,5-dibromo-4-nitrosobenzenesulfonic acid;ABTS = 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonicacid); MNPr = Methylnitrosopropane; INH = Isoniazid.
1 INTRODUCTION
The purpose of this article is to review structure–functionrelationships for heme peroxidases, catalases, and catalase-peroxidases for which detailed crystallographic informationis available. Peroxidases catalyze the peroxidation of varioussubstrates and catalases are similar except that a peroxide isboth an oxidant and a reductant.
The overall peroxidase cycle is
Fe3+R + H2O2
Resting State
Fe4+ − O R• + H2O
Compound I(1)
Fe4+ − O R• + S
Compound I
Fe4+ − O R + S•
Compound II(2)
Fe4+ − O R + S
Compound II
Fe3+R + H2O + S•
Resting State(3)
In the first step (equation 1), peroxide removes twoelectrons from the protein. One derives from iron and anotherfrom an organic group, R, giving the well-known compoundI. Compound I thus has two fewer electrons than the nativeenzyme. In most peroxidases, the oxidized organic group is theporphyrin leading to a porphyrin π -cation radical.1 However,in cytochrome c peroxidase (CcP), a tryptophan residue ratherthan the porphyrin is oxidized.2,3
Both compounds I and II have distinct spectroscopicsignatures and very often are relatively long-lived. This,together with the relative ease of obtaining these enzymes,has enabled peroxidases and catalases to occupy a pivotalposition in the history of enzymology. Peroxidases played animportant role in the development of modern enzymology asearly as the 1930s.4 The formation of distinct spectroscopicintermediates in the peroxidase reaction cycle helped shapethe development of rapid reaction kinetic methods pioneeredby Britton Chance in the 1940s.5 Since then, nearly everyconceivable sophisticated biophysical tool has been appliedto peroxidases, which has resulted in a vast and diverse rangeof studies.
The most well-known and thoroughly investigatedperoxidases are of nonmammalian origin. Establishmentof a detailed sequence database enabled Welinder6 todivide nonmammalian heme peroxidases into three distinctclasses. Class I comprises intracellular and/or peroxidases ofprokaryotic origin. These include yeast CcP, chloroplast andcytosolic ascorbate peroxidases (APX), and bacterial catalase-peroxidases (KatGs). Class II peroxidases are the secretedfungal peroxidases. Examples include lignin peroxidase(LIP) and manganese peroxidase (MnP) of Phanerochaetechrysosporium and the ink cap mushroom peroxidase,ARP from Coprinius cinerius (alternate name Arthomycesramosus). Chloroperoxidase from Caladariomyces fumago,while also a secreted fungal peroxidase, does not belongto class II owing to its specialized function and lack ofeither primary or tertiary structural homology with otherfungal peroxidases. Class III peroxidases are the classical

2 IRON: HEME PROTEINS, PEROXIDASES, CATALASES & CATALASE-PEROXIDASES
secretory plant peroxidases such as horseradish peroxidase(HRP), the most well-known and thoroughly investigatedclass III peroxidase.
2 CLASS I PEROXIDASES
2.1 Yeast Cytochrome c Peroxidase (CcP)
CcP is the most well-known and thoroughly studied class Iperoxidase and has been recently reviewed.7,8 CcP is a yeastmitochondrial enzyme that catalyzes the following reaction:
2cyt. c(Fe3+) + H2O2 + 2H+ cyt. c(Fe3+) + 2H2O (4)
Given the large number of studies centered on CcP, itis surprising that only recently have there been advances inunderstanding the biological function of CcP. Kwon et al.9
found that yeast CcP levels increase during oxidative stressindicating that the primary function of CcP is to serve asan antioxidant. In addition, yeast CcP may participate in asignaling process during oxidative stress.10
2.1.1 Yeast CcP Structure and Mechanism
CcP shares the same overall fold with other class I as wellas class II and III peroxidases (Figure 1(a)). CcP contains10 α-helices with a single β-sheet. The protein is dividedinto two distinct domains separated by heme viz. distal andproximal heme domains. Figure 1(b) shows the active sitearchitecture of CcP with the role of key residues in catalysis,which are typical of most peroxidases. On the distal side of theheme, Arg48, Trp51, and His52 form the catalytic triad anddistal heme pocket where exogenous ligands and substrateslike hydrogen peroxide bind. On the proximal side of theheme, His175 coordinates the heme iron while Trp191 formsa parallel stacking interaction with His175. Asp235 hydrogenbonds to both His175 and Trp191. The His175-Asp235 H-bond imparts anionic character to the proximal His175, whichis important for stabilization of the high oxidation state ofiron in compounds I and II.11,12 As outlined in Figure 1, His52operates as an acid-base catalyst in promoting heterolyticfission of the peroxide O–O bond while Arg48 helps tostabilize the developing negative charge on the leaving OHgroup. Mutagenesis studies clearly illustrate the critical role ofHis52 in CcP13 and other peroxidases,14,15 although the effectof changing Arg48 is less dramatic.16 Similarly mutagenesisof the distal His42 in HRP has demonstrated the critical roleof the distal His in peroxidase catalysis.14,15
Once compound I is formed, CcP reacts with ferrocy-tochrome c in the following multistep scheme:17
1. Fe4+ Trp•+ + cyt. c (Fe2+) Fe4+Trp•+/cyt. c (Fe2+)
2. Fe4+ Trp•+/cyt. c (Fe2+) Fe4+Trp/cyt. c (Fe3+)
3. Fe4+ Trp/cyt.c (Fe3+) Fe4+Trp + cyt. c (Fe3+)
4. Fe4+ Trp Fe3+Trp•+
In step 1, CcP compound I forms a complex withferrocytochrome c followed by electron transfer (step 2)and dissociation (step 3) of oxidized cytochrome c. Step 4represents the intramolecular electron transfer from Trp191 tothe iron giving Fe3+Trp•+, the intermediate required to oxidizethe second molecule of ferrocytochrome c. In this mechanism,formation of the Trp191 cation radical is essential for bothelectron transfer steps.
Recent 1.2-A and 1.3-A structures of resting state andcompound I CcP have provided new insights into the natureand stabilization of compound I.18 This was possible owingto the unexpected stability of compound I in the X-ray beam.Exposure of metalloprotein crystals to X-rays can often leadto reduction of the metal center,19 which can be problematicfor enzyme intermediates like compounds I and II where theredox potential of the oxyferryl centers are near 1000 mV.20
Spectroscopic studies of single crystals of resting state andcompound I showed that CcP compound I is remarkablystable to X-ray exposure. The oxyferryl center (Fe4+–O)is not affected by X-ray exposure as evidenced by bothabsorption and single-crystal EPR (electron paramagneticresonance) spectroscopies.18 In addition, the single-crystalEPR data indicated that by the end of X-ray diffractiondata collection, approximately 50% of the Trp191 π -cationicradical signal remains.
These higher resolution structures have provided insightinto small but functionally important conformational differ-ences between resting state CcP and compound I.18 There arethree such regions which occupy two discrete positions in theresting state structure but only one conformation in compoundI. The first of these is Arg48 in the distal heme pocket whichis considered to be a key residue involved in the formationof compound I.21,22 In resting state CcP, Arg48 occupies twopositions, one ‘out’ toward the heme propionate and the other‘in’ toward heme iron. In compound I, Arg48 occupies the‘in’ position which enables Arg48 to hydrogen bond withFe4+–O oxygen. The ‘in’ position is similar to the earliercompound I structures observed at lower resolution23,24 andin the CcP-fluoride complex.25 A second region is Met172 inthe proximal heme pocket which exists in two conformationsin resting state, but becomes more ordered in compound I.In compound I, Met172 occupies a position closer to theHis175 ligand (Figure 1). The third region that shows dis-crete differences between resting state and compound I is theproximal loop region involving residues 190–195 which playsan important role in complex formation with cytochrome c.Residues 193–195, including both side chains and the pep-tide backbone, adopt multiple conformations in the restingstate, but become more ordered in compound I. In the singleconformation that is observed in compound I, the side chain
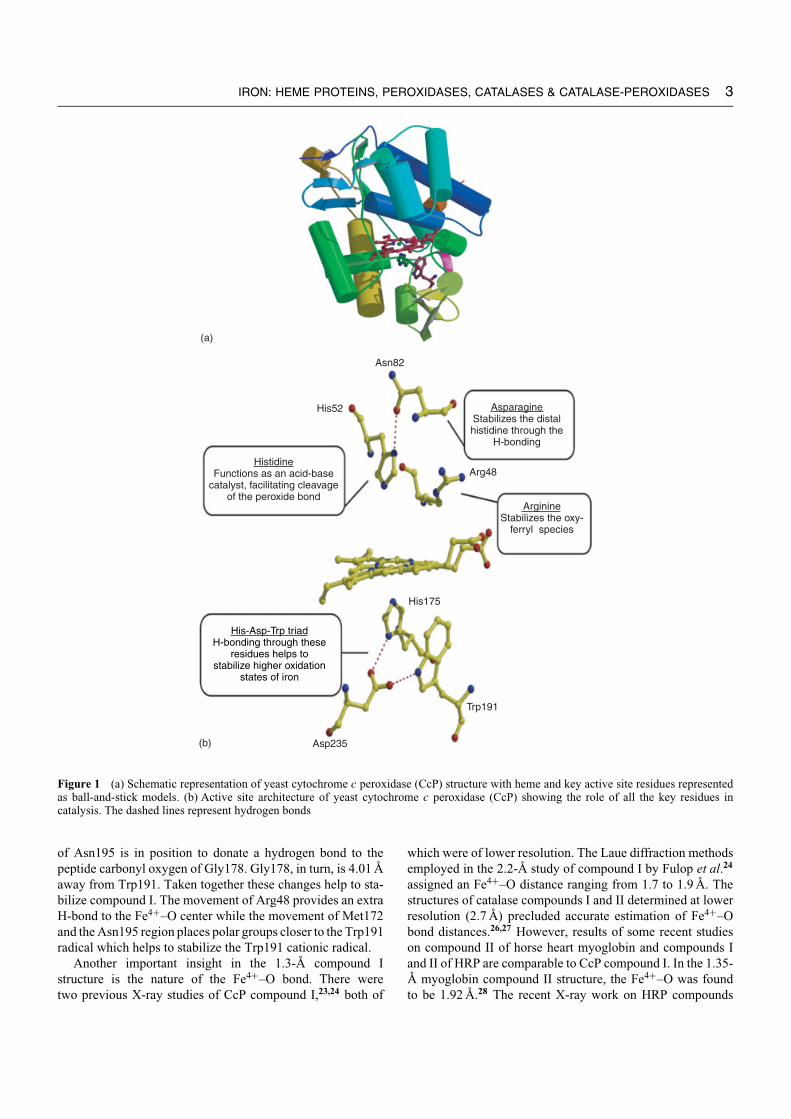
IRON: HEME PROTEINS, PEROXIDASES, CATALASES & CATALASE-PEROXIDASES 3
(a)
HistidineFunctions as an acid-base
catalyst, facilitating cleavageof the peroxide bond
AsparagineStabilizes the distalhistidine through the
H-bonding
ArginineStabilizes the oxy-
ferryl species
Asn82
His52
Arg48
His175
Trp191
Asp235(b)
His-Asp-Trp triadH-bonding through these
residues helps tostabilize higher oxidation
states of iron
Figure 1 (a) Schematic representation of yeast cytochrome c peroxidase (CcP) structure with heme and key active site residues representedas ball-and-stick models. (b) Active site architecture of yeast cytochrome c peroxidase (CcP) showing the role of all the key residues incatalysis. The dashed lines represent hydrogen bonds
of Asn195 is in position to donate a hydrogen bond to thepeptide carbonyl oxygen of Gly178. Gly178, in turn, is 4.01 Aaway from Trp191. Taken together these changes help to sta-bilize compound I. The movement of Arg48 provides an extraH-bond to the Fe4+–O center while the movement of Met172and the Asn195 region places polar groups closer to the Trp191radical which helps to stabilize the Trp191 cationic radical.
Another important insight in the 1.3-A compound Istructure is the nature of the Fe4+–O bond. There weretwo previous X-ray studies of CcP compound I,23,24 both of
which were of lower resolution. The Laue diffraction methodsemployed in the 2.2-A study of compound I by Fulop et al.24
assigned an Fe4+–O distance ranging from 1.7 to 1.9 A. Thestructures of catalase compounds I and II determined at lowerresolution (2.7 A) precluded accurate estimation of Fe4+–Obond distances.26,27 However, results of some recent studieson compound II of horse heart myoglobin and compounds Iand II of HRP are comparable to CcP compound I. In the 1.35-A myoglobin compound II structure, the Fe4+–O was foundto be 1.92 A.28 The recent X-ray work on HRP compounds

4 IRON: HEME PROTEINS, PEROXIDASES, CATALASES & CATALASE-PEROXIDASES
I and II to 1.6 A resolution gives Fe4+–O distances of 1.7 Afor compound I and 1.8 A for compound II.29 The heme ofHRP compound II and the heme of CcP compound I areelectronically equivalent. Hence, the longer 1.8-A Fe4+–Odistance in HRP compound II is comparable to the 1.87-Adistance found in CcP compound I. These longer Fe4+–Odistances are at odds with some of the early EXAFS workon peroxidases. Chance et al.30 estimated a distance of 1.67 Ain CcP compound I while Penner–Hahn et al.31 estimateda distance of 1.6 A in HRP compounds I and II. However,Chance et al.30 estimated a Fe4+–O distance of 1.93 A inHRP compound II which is closer to the 1.8 A found inthe HRP crystal structure. Although EXAFS results differfrom that of the recent X-ray results, resonance Raman datamore closely match the more current results. Reczek et al.32
assigned a stretching frequency for the Fe4+–O stretch in CcPcompound I to be 753 cm−1, compared the 773–779 cm−1 forHRP32–35 indicating a weaker Fe4+–O bond in CcP compoundI. It was argued that the lower stretching frequency for theFe4+–O stretch was due to a strong hydrogen bond to theFe4+–O oxygen and greater single-bond character of theFe4+–O bond. This is consistent with the high-resolution CcPcompound I structure since both Arg48 and Trp51 formhydrogen bonds with ferryl oxygen and the long 1.87 AFe4+–O bond indicates a single bond. Overall, the resultsof the high-resolution structure of CcP coupled with a wealthof spectroscopic data strongly favor a single Fe4+–O bond. Asingle bond would also inevitably mean that the ferryl oxygenis protonated. In peroxidases, we know that the peroxideO–O bond undergoes heterolytic cleavage36 which leaves anoxygen atom with six valence electrons bound to the iron.If both electrons now reside primarily on the Fe-linked Oatom, this oxygen atom contains a full complement of eightvalence electrons, giving an Fe4+–O center which should bea strong base and exist primarily as a Fe4+–OH center if theFe4+–O bond is a single bond. The –OH group is stabilizedby the motion of Arg48 in the high-resolution structure ofCcP compound I.18 Therefore, the ferryl center of CcP maybe best described as a hydroxide ion with a single bond toFe4+ stabilized by hydrogen bonding interactions with Arg48and Trp51.
2.1.2 Stabilization of the Trp Cation Radical Yeast CcP
A fascinating aspect of CcP chemistry and biochemistry isthe formation and stabilization of the Trp191 cationic radicalin compound I while other peroxidases form a porphyrinπ -cationic radical. A wealth of biochemical, spectroscopic,and structural results are available on several mutants ofCcP which have emphasized the importance of Trp191 toenzyme catalysis.8 Other class I peroxidases like APX andKatG also have a proximal side Trp analogous to yeast CcPTrp191 but these peroxidase do not form a Trp radical. Itnow appears that the local environment surrounding Trp191
is designed to electrostatically stabilize the extra positivecharge on the Trp191 cationic radical. Indeed, the first clueto this puzzle came from several mutagenesis studies thatshowed that the Trp191Gly mutation resulted in a cavitythat binds cations such as potassium and imidazolium.37–39
These results and electrostatic calculations suggested thatthe local environment created a negative potential that canstabilize the Trp radical in CcP37,40,41 and these resultshave been further corroborated by computational studiesof Poulos and coworkers.42 Further structural comparisonsshowed that in APX and plant peroxidases, a cation-bindingsite [a K+ site for APX (see Cation-activated Enzymes) anda Ca2+ for most other peroxidases (see Calcium-bindingProteins)] is located ∼8 A away from the Trp/Phe residue,while only a water molecule is present at the same locationin CcP.18,40,43 Therefore, Patterson et al.44 proposed that thiscation-binding site may play a role in destabilizing the Trpradical in APX. This hypothesis was tested by engineeringa protein whose activity could be modulated by a designedcation site. On the basis of structural homology betweenAPX and CcP, the K+ binding site of APX was engineeredinto CcP.40,45 Crystal structures showed that K+ binds tothe engineered site in CcP exactly as in APX. Moreover,EPR experiments showed that the Trp191 radical becomesincreasingly less stable as a function of K+ concentrationwhile the steady state enzyme activity decreases. These resultsdemonstrated that long-range electrostatic effects can controlthe reactivity of a redox-active amino acid side chain. Buildingon this success, further metallo-engineering efforts enabled theselectivity of the engineered site to be switched from K+ toCa2+.46
2.1.3 CcP-cytochrome c Complex
CcP and cytochrome c were the first physiological redoxpartners where the crystal structure of each partner hadbeen determined. As such, the CcP-cytochrome c systemhas served as a paradigm for both protein–protein interactionsand interprotein electron transfer studies. The crystal structureof the CcP-cytochrome c complex47 provided a major advancein our understanding of how CcP works. The complex showsthat the cytochrome c heme is within ≈3.9 A from Ala194 inCcP. This provides a direct route of electron transfer from thecytochrome c heme to the Trp191 cationic radical. There arefew strong charge–charge interactions at the interface whichwas surprising since the complex is strongly dependent onionic strength.
Various biophysical probes indicate that CcP has two sitesof interaction with cytochrome c although it is less clear ifboth sites are involved in electron transfer. To probe thisquestion, CcP and cytochrome c have been site-specificallycross-linked using engineered disulfide bonds48 to generate acovalent complex that mimics the noncovalent complex. CcPin the covalent complex is unable to accept electrons from freeferrocytochrome c. A second cross-link was designed to block

the proposed second cytochrome c binding site49,50 but thiscomplex could still accept electrons from ferrocytochromec. Taken together, these results indicate that the covalentlyattached cytochrome c is blocking the one site used bycytochrome c for docking and transferring electrons and thatthis single competent electron transfer site is the same oneidentified in the crystal structure of the CCP-cytochrome c
complex.47
2.2 Ascorbate Peroxidase (APX)
The primary function of APX is to rid plant cells of toxicperoxides by using ascorbate as the reducing substrate. Peacytosolic APX is the most thoroughly studied APX and wasthe first APX crystal structure determined.43 This now hasbeen followed by the crystal structures of soybean cytosolic51
and tobacco stromal APX.52 The overall fold of APX is verysimilar to CcP except that APX is smaller and contains 250residues compared to 294 in CcP. The main difference is thatAPX lacks the β-sheet structure in CcP. APX also exists asa homodimer.
The key elements of the APX active site are the sameas in CcP including the homolog to CcP Trp191 (Trp179 inAPX) which, in CcP, forms the stable compound I cationradical. However, APX does not form a Trp179 radicalbut instead forms the more traditional porphyrin π -cationradical.44 In the absence of reducing substrates, compoundI decays to compound I∗. Recently Rodriguez–Lopez andcoworkers showed by low-temperature EPR and RP-HPLCthe existence of a transient Trp cation radical.53 As notedin Section 2.1.2, this difference has been attributed to thelocal electrostatic environment of CcP, which is designed tostabilize a Trp191 cation radical.40,41,43
Studies on APX provided some of the first evidencethat peroxidases can have different substrate binding sites.Early on, it was found that modification of the single Cysin pea APX eliminated enzyme activity but at that timethere was insufficient enzyme for more detailed studies. Thiswas followed by work on recombinant APX where boththe Cys modified form of APX and the Cys-to-Ser mutantcrystal structures led to the hypothesis that the substrate,ascorbate, binds near Cys32 (Figure 2).54 Moreover, Poulosand coworkers proposed that Arg172 in this region interactswith the ascorbate and helps to hold the substrate in placenear the heme propionates.55 As predicted, mutating Arg172to other residues blocked the oxidation of ascorbate but notthe oxidation of the small phenolic substrate, guaiacol. Thisled to the view that phenolic substrates interact with APX ata different site near the exposed heme edge. More recently,the crystal structure of the ascorbate-APX complex51 clearlyshows (Figure 2) that the ascorbate is positioned near thepropionate edge of the heme where it interacts with Arg172as predicted by the chemical modification and mutagenesisstudies.55
IRON: HEME PROTEINS, PEROXIDASES, CATALASES & CATALASE-PEROXIDASES 5
Heme C32L30
L-ascorbate
R172
Figure 2 Structure of active site of recombinant soybean ascorbateperoxidase (sAPX) showing the heme, Lys30, Cys32, and Arg172with the substrate ascorbate (This figure was generated fromcoordinates of 1OAF deposited in the Protein Data Bank)
2.3 Bacterial Catalase-peroxidases
2.3.1 Introduction
Over the last ten years, the catalase-peroxidases haveattracted significant interest in the fight against tuberculosis.Mycobacterium tuberculosis is a leading pathogen worldwide,infecting an estimated 2–3 million people each year.56 Oneof the primary drugs used to treat tuberculosis is isoniazid(INH), a prodrug that is oxidized by the KatG (gene encodingcatalase-peroxidase) catalase-peroxidase to an activated formthat inhibits cell wall synthesis.57,58 The real surge of interestin catalase-peroxidases started in 1992 when it was confirmedthat mutation of KatG in Mycobacterium tuberculosis confersisoniazid resistance.59 A putative reaction scheme of catalase-peroxidases is shown in Figure 3(a).
2.3.2 Structure and Mechanism
These are difficult enzymes to work with and onlyrecently have crystal structures become available for twocatalase-peroxidases: Haloarcula marismortui (HMCP)60 andBurkholderia pseudomallei (BpKatG).61 A typical subunit isapproximately 80 kDa in molecular mass, with a single heme b
prosthetic group. The primary structure of each subunit can bedivided into two distinct domains, N terminal and C terminal.The N-terminal domain contains the heme and active site,62
while the C-terminal domain does not contain a heme bindingmotif and its function remains unclear. The clear sequencesimilarity between the two domains suggests gene duplicationand fusion.63 Curiously, despite many years of study, the actualin vivo peroxidatic substrate of the catalase-peroxidases hasnot been identified.

6 IRON: HEME PROTEINS, PEROXIDASES, CATALASES & CATALASE-PEROXIDASES
Por FeIII.......R
•+Por FeIV= O...R
Por FeIV= O...R
Por FeIII−O2+ −...R
Por FeII−O2 .....R
Por FeIV= O...R•+
Por FeIII.....R•+
Ferric enzyme
Compound I
Compound III
Compound II
M264 W111
Y238
Heme
OR
OR
OR
1 2 4
3
5
H2O2
H2O2 H2O2
H2O
H2OH2O + O2
AH2
AH2
•AH(+H2O)
•AH (+H2O)
(a) (b)
Figure 3 (a) Putative reaction scheme of catalase-peroxidases (KatG). (b) Schematic diagram showing the formation of a novel covalentadduct among side chain of residues Trp111 (Trp95), Tyr238 (Tyr218), and Met264 (Met244) of catalase-peroxidase of Burkholderiapseudomallei (BpKatG). The numbers in parenthesis represent the residues of Haloarcula marismortui (HMCP) which form a similar adduct.(This figure was generated from coordinates of 1MWV deposited in the Protein Data Bank)
As noted, the N-terminal domain of KatGs exhibits theclassic peroxidase fold. Superimposition of the N-terminalsegment of either BpKatG or HMCP onto CcP, APX, or HRPgive rms deviations of 0.97 A, 1.22 A, and 2.03 A, respectively,for BpKatG and 1.12 A, 1.31 A, and 1.99 A, respectively, forthe ten α-helical segments of HMCP. On the other hand,superimposition of the C-terminal region of either BpKatG orHMCP onto the same peroxidase proteins give rms deviationsof 3.62 A, 3.75 A, and 4.06 A, respectively, for BpKatG and3.51 A, 3.83 A, and 4.44 A, respectively, for HMCP.
The organization of the heme active site of both HMCPand BpKatG are almost identical to that of CcP and APX.On the distal side of the heme, Trp, Arg, and His form theperoxide-binding site. On the proximal side, the conservedAsp H-bonds with both the proximal His ligand and proximalside Trp just as in CcP and APX. However, comparisonof other residues surrounding the catalase-peroxidase groupreveals greater similarity to CcP with decreasing similarityto APX. Most notably, a potassium ion found in APX ina surface loop about 8 A from the proximal Trp is absent.This same loop forms the Ca2+ site in class III peroxidases.However, a Trp radical does not form in BpKatG as in yeastCcP compound I but instead a porphyrin π -cation radical hasbeen demonstrated for both EcKatG (catalase-peroxidase formEscherichia coli)64 and MtKatG (catalase-peroxidase from M.tuberculosis).65
One of the most surprising results from the crystal structuresof the KatGs was the identification of a novel Trp-Tyr-Met covalent cross-link adduct (Trp111-Tyr238-Met264 inBpKatG and Tyr218-Trp95-Met244 in HCMP) in the vicinityof the active site (Figure 3(b)). Although the mechanismleading to the formation of the adduct is unclear, a possibleexplanation for its formation involves hydrogen peroxideand the heme iron in an oxidative process. What is notclear is whether the adduct provides some functional role.Other unusual adducts have been observed in peroxidases
and in monofunctional catalases, including a hydroxy groupon the Cβ of Trp171 in lignin peroxidase,66 distal Trp51-Tyr52 covalent cross-link in a mutant of CcP,67 a methioninesulfoxide residue in the catalase from Proteus mirabilis,68
formation of a proximal covalent cross-link between the Nδ
of His392 and Cβ of Tyr415 in catalases HPII from E. coliconcomitant with the cyclization of heme b to form oxidizedheme d69 in Penicillium vitale.70 One rationale proposed forsome of these modifications is that they stabilize the enzyme byreducing the likelihood of other, more damaging oxidations.The Trp-Tyr-Met adduct may impart a similar protective rolein that it will prevent further modification of the oxygen-susceptible methionine residue. Another possibility is that theadduct forms a route for electron transfer from the cleft regionto the heme.61 Other metal-based protein modifications arediscussed in Metal-mediated Protein Modification.
One of the main goals for the structure determination ofthe catalase-peroxidases was to determine the INH bindingsite. The crystal structure of BpKatG revealed a region ofundefined electron density in a constricted region in the mainchannel leading to the distal heme cavity. The location ofthe density is over 10 A further away from the heme thanis the benzhydroxamic acid bound in HRP.71 However, thedensity is in close proximity to Ser324, the equivalent ofSer315 in MtKatG, which is thought to be involved in INHbinding. Mutation of this residue to a Thr leads to a loss ofINH activation with no loss of either peroxidase or catalaseactivity.72 Most of the residues in the pocket surroundingthe INH-like electron density are fully conserved betweenBpKatG and MtKatG, but HMCP and the other catalase-peroxidases of halo-archaebacterial origin have a one residueinsertion of Asp269 compared to BpKatG, which is locatedin a loop adjacent to the pocket. The insertion forces thering of the adjacent Pro270 into a position that would mostinterfere with the region of INH-like electron density. Thissuggests that HMCP should not bind INH in this region, and

thus explains why a similar region of electron density was notobserved in the HMCP electron density maps.
3 CLASS II PEROXIDASES
3.1 Lignin Peroxidase (LIP)
Lignin is a major component of woody plant cell wallsand lignin biodegradation plays a pivotal role in the earth’scarbon cycle. Lignin is not only an abundant renewablecarbon source but also protects cellulose against enzymaticdegradation. The metalloenzymes, LIP and laccase, secretedby filamentous white-rot fungi from Basidiomycetes, are theonly known sources capable of degrading the recalcitrantlignin. This rather heterogenous and complex biopolymerconsists of phenyl propanoid units linked by variousnonhydrolyzable C–C and C–O bonds.73 The best studiedwhite-rot fungus, Phaenerochaete chrysosporium, secretestwo heme peroxidases, LIP and a MnP. Both of theseperoxidases appear to be major components of the extracellulardegradative system of the fungus.74
The crystal structures of two fungal LIP isozymes havebeen characterized66,75,76 and more recently the recombinantenzyme.77 Fungal LIPs are globular and mostly helicalglycoproteins of about 40 kDa (including carbohydrate), with343–344 amino acids depending on the isozyme. LIP alsocontains the two Ca2+ sites found in the class III peroxidases(Figure 4). The overall fold of LIP is that of a typicalheme peroxidase, as in CcP,18,22 MnP,78 HRP,79 barley grainperoxidase80 and other nonligninolytic fungal peroxidases likethose from ARP81 and C. cinerius.82 The heme, which dividesthe protein into a proximal and a distal heme domain, is buriedin the interior of the protein with restricted access via a smallchannel. This channel, which is similar to substrate accesschannel found in ARP83 and HRP,71 has a significantly smallerentrance than other peroxidases. Attempts to determine crystalstructures of substrate bound form LIP have been unsuccessful.
LIP undergoes the typical peroxidase reaction cycle andforms compounds I and II. However, LIP compound I is notvery stable and readily converts to compound II, even inthe absence of reducing substrates. In the crystal-structuredetermination of LIP, a significant residual electron densitywas visible close to the surface residue Trp171. It wasinterpreted as a hydroxy group covalently bound to the Cβ
atom of Trp171. Subsequent chemical and EPR spectroscopicanalysis by spin trapping proved that Trp171 had, indeed, beenhyroxylated (Figure 4(b)).84 In view of the above findings, itwas proposed that Trp171 acts as an endogenous electrondonor of LIP compound I reduction, which implies theformation of a Trp radical may be part of the catalyticcycle. This assumption was tested by treating LIP withthe tryptophan-specific agent N -bromosuccinimide (NBS),which led to drastically reduced activity with respect to
IRON: HEME PROTEINS, PEROXIDASES, CATALASES & CATALASE-PEROXIDASES 7
b-OH Group
His47
His176
Trp171
(a) (b)
ca2+
Figure 4 (a) Schematic diagram showing the overall fold of therecombinant pristine lignin peroxidase (LIP) with key residuesinvolved in catalysis. Only one of the 2Ca2+ ions is shown.The second is hidden in this view. (b) Diagram showing theenvironment around β-hydroxy-Trp171. (This figure was generatedfrom coordinates of 1LLP deposited in the Protein Data Bank)
veratryl alcohol (VA) oxidation.85 Site-directed mutagenesisof Trp171 to Phe or Ser showed no residual activity towardVA.86 These experiments proved the involvement of Trp171in LIP catalysis. On the basis of this, a catalytic mechanismwas proposed for the hydroxylation of Trp171 starting withthe formation of Trp cation radical.85 Evidence for a transientTrp171 radical intermediate was obtained by using elegantspin-trapping experiments with methylnitrosopropane (MNPr)in combination with peptide mapping and crystallography.84
In the presence of H2O2, MNPr forms a covalent adductwith Trp171 which was confirmed by the crystal structure ofMNPr-modified LIP.84
In order to complement kinetic work, crystal structuresof recombinant LIP isozyme H8 expressed in E. coli inthe pristine state (lacking hydroxylation at Trp171), of theoxidatively processed enzyme (hydroxylated at Trp171), anda Trp171 Phe mutant,77 were solved. Recombinant enzymeswere not glycosylated and had to be refolded in vitro.87 Thestructure of the pristine state was virtually identical to thatof Cβ-hydroxylated form, except for the absence of the –OHgroup attached to Trp171. A water molecule was found inboth structures in a small cavity near the Cβ atom of Trp171,ideally positioned to attack the Cβ atom stereospecifically,leading to an S-configured Cβ atom only.77 The Trp171Phemutation exhibited no structural changes of the helix exceptin the side chain position of residue 171. Complete lack ofactivity of this mutant has been attributed to the absence ofthe redox-active indole side chain. Mutation experiments havenot only underlined the importance of Trp171 for the catalyticmechanism but also show that there are two distinct substrateinteraction sites in LIP.
3.2 Manganese Peroxidase (MnP)
Like LIP, MnP is produced by P . chrysosporium and thestructure78 closely resembles LIP. MnP, however, oxidizesMn(II) to Mn(III). Once formed, Mn(III) is bound to and

8 IRON: HEME PROTEINS, PEROXIDASES, CATALASES & CATALASE-PEROXIDASES
stabilized by a dicarboxylic acid and the Mn(III)-acid complexserves as a diffusible redox mediator that can oxidize varioussubstrates including lignin model compounds.88,89 The bindingsite for Mn(II) is shown in Figure 5 which was correctlypredicted by homology modeling.90 Note that one Mn(II)ligand is a heme propionate leading to the proposal that theheme propionate mediates electron transfer from Mn(II) tothe porphyrin radical of compound I. It is also interestingthat the specialized site for Mn(II) binding near one hemepropionate is also where ascorbate binds in APX. Thus, bothAPX and MnP use the same region of the protein, near oneheme propionate, for selective binding of the physiologicallyimportant substrate.
Using crystallographic methods, this binding site wasprobed by altering the amount of Mn2+ bound to the protein.Crystals grown in the absence of Mn(II), or in the presenceof EDTA, exhibited diminished electron density at this site.Crystals grown in excess Mn(II) exhibited increased electrondensity at the proposed binding site but nowhere else in theprotein. This suggested that there is only one major Mn(II)binding site in MnP. Crystal structures of a single mutant(D179N) and a double mutant (E35Q-D179N) at this sitewere determined.91 The mutant structures lack a cation at theMn(II) binding site. The structure of the Mn(II) binding siteis altered significantly in both mutants, resulting in increasedaccess to the solvent and substrate. Kinetic analyses of thesingle mutants, E35Q, E39Q, and D179N, yielded Km valuesfor the substrate Mn(II) that were ∼50-fold greater than the
Phe45
His46
Arg42
Asp39
Asp179
Fe3+
Mn2+
Asp35
Figure 5 Schematic diagram showing the heme environment andMn-binding site of manganese peroxidase (MnP)
corresponding Km value for the wild-type enzyme. Similarly,the kcat values for Mn(II) oxidation were ∼300-fold lowerthan that for the wild-type MNP.
Despite the presence of this unique Mn(II) binding site,the overall structure of MnP is very similar to all othernonmammalian heme peroxidases. The localized structuralalterations near the surface of the protein required to formthe Mn(II) site do not induce significant changes in thecore peroxidase structure. For example, the structure of P.chrysosporium LIP is very similar to that of MnP but lacksthe Mn(II) site. LIP has only one of the three acidic residues,Glu39 (Glu40 in LIP). In place of Glu35 and Asp179, LIP hasAla36 and Asn182, respectively.78 Although it is possible toaccommodate an aspartic acid in place of Asn182 in the LIPstructure, the space occupied by the side chain of Glu35 in MnPis filled by the backbone structure of the C terminus in LIP.MnP has a longer C terminus, which deviates considerably inits course from that of LIP. In addition, Arg177 pushes thepolypeptide chain out and away from the main body of theprotein to form the Mn(II) site in MnP. The correspondingresidue in LIP is an Ala180. Finally, MnP has an extra disulfidethat helps to force the polypeptide chain away from the body ofthe protein. These differences result in the formation of spacefor Glu35 near the cation-binding site. These comparisonssuggest that constructing a productive Mn(II) binding site inLIP by iterative protein engineering should be possible.
4 CLASS III PEROXIDASE
4.1 Horseradish Peroxidase (HRP) Structure
HRP is prepared from horseradish roots and over 30isozymes have been identified. Owing to its relative abundanceand ease of preparation, HRP is one of the most well-knownand thoroughly investigated peroxidases. For reviews seereferences.7,92
HRP consists of a single polypeptide chain of 308amino acids of Mr33 890 Da and with one noncovalentlybound protoporphyrin IX heme, four disulphide bridges,the two conserved Ca2+ binding sites, and contains ∼22%carbohydrate. HRP-C can also be overexpressed in E. colifrom a synthetic gene comprising 309 amino acids includingan N-terminal Met required for translation initiation.93 Theproduction and refolding of unglycosylated recombinantprotein paved the way for the long-awaited determinationof the crystal structure to 2.15 A.79
HRP exhibits the typical peroxidase fold and active sitestructure as shown in Figure 6(b). As with CcP, the nativeresting form of HRP-C contains a five-coordinate, high-spinFe3+ heme. Compound I of HRP contains two oxidizingequivalents, one as oxyferryl (Fe4+–O) and the other asporphyrin radical.94 A transient Trp π -cation radical hasbeen detected in the Phe221Trp mutant of HRP-C compound

IRON: HEME PROTEINS, PEROXIDASES, CATALASES & CATALASE-PEROXIDASES 9
----- Fe(IV)-----
----- Fe(III)-----
----- Fe(III)---------- Fe(IV)-----
----- Fe(II)-----
O
++
+ +
−
Compound Ip-cation radical
Ferrous state
Compound IIICompound II
Ferric enzymeGround state
Peroxidasecycle
e−, H+
e−, H+
e−, H+
Oxidasecycle
O2
OO
H
HOHOO•
HOOH
HOOH
H2O
H2O
H2O
(a)
(b)
Phe41
His42
Arg38
Distal side
Proximal side
His170
Phe221
Asp247
F helix
B helix
H170
•H
Figure 6 (a) The five redox states of horseradish peroxidase (HRP) as determined from the high-resolution crystal structure of horseradishperoxidase (HRP). (b) Diagram showing the overall fold and active site environment containing key residues in catalysis of horseradishperoxidase (HRP)
I.95 Recently, Hajdu and coworkers29 worked out an effectivestrategy to obtain the 1.57–1.62 A crystal structures for high-valence redox intermediates and continuous snapshots of theX-ray driven catalytic reduction of a bound dioxygen species.These high-valence intermediates are readily susceptible toreduction by the X-ray beam and as such, the structureof compound I was obtained by merging data from 11crystals. Despite the problems with deconvoluting a mixtureof states, this tour de force study has provided the firstcrystallographic picture of all five HRP redox states as shownin Figure 6(a).
4.2 HRP-substrate Complexes
Crystal structures of HRP-C:substrate/inhibitor complexeshave also been solved. Of particular interest is the structureof HRP-C:benzhydroxamic acid (BHA) complex which wassolved by Gajhede and coworkers,71 the HRP-C:ferulicacid (FA) complex,96 and the HRP-C:FA:cyanide ternarycomplex.96 In all three complexes, changes in structure
are restricted to the substrate access channel and the hemesurroundings. The prevailing view is that enzyme-substratecomplexes are relatively nonspecific and weak in peroxidases.One possible reason is that tight binding of an oxidizedsubstrate could lead to enzyme modification. The products ofoxidation of substrates are phenoxyl radicals, very similarin structure to their respective substrate molecules, butare chemically reactive species. These products must leavethe active site immediately after formation, polymerize ordisproportionate. A prolonged retention of the radical in closeproximity to the enzyme active site would increase the risk ofoxidative damage. Loose association of the reducing substratewith the enzyme, as found in the structure of HRP-C: FAcomplex,96 is presumed to be an advantage.
These various structures together with the conserved struc-tures around the δ-edge heme suggests a common pattern ofbinding between class III peroxidases and aromatic substrates.Such common aromatic contacts have also been observed inthe complexes between class II peroxidase ARP and BHA,and salicylhydroxamic acid, respectively.83,97

10 IRON: HEME PROTEINS, PEROXIDASES, CATALASES & CATALASE-PEROXIDASES
5 BACTERIAL DI-HEME CYTOCHROMEC PEROXIDASES (BCcP)
5.1 Introduction
Bacterial CcPs have been isolated from anumber of organisms: Pseudomonas aeruginosa,98
Nitrosomonas europea,99 Paracoccus pantotropus,100
Paracoccus denitrificans,101 Rhodobacter capsulatus,102
Methylococcus capsulatus,103 Pseudomonas stutzeri,104 andPseudomonas nautica.105 The most well-studied andcharacterized BCcPs are those from P. aeruginosa and Par.denitrificans. BCcPs are homodimers with about 300 residuesper monomer. Each monomer contains two hemes whichare covalently tethered to the protein via thioether linksbetween the heme vinyl groups and Cys residues, exactlyas in many c-type cytochrome. One heme is high-potential(+270 mV), while the other is low-potential (between −190and −310 mV). Greenwood et al.106 proposed the followingmechanism:
Fe2+
high
Fe3+
low+ H2O2 Fe3+
high
Fe4+
low
=O + H2O (5)
Compound I
Fe3+
high
Fe4+
low
=O + e− + 2H+ Fe3+
high
Fe3+
low+ H2O (6)
Compound I Compound II
Fe3+
high
Fe3+
low+ e− Fe2+
high
Fe3+
low(7)
Compound II
The high-potential heme accepts an electron from thephysiological electron donor, which is then transferred tothe low-potential heme where peroxide reacts with the hemeto give compound I. In this case, the high-potential hemeprovides one of the two electrons required to activate peroxiderather than a Trp residue as in yeast CcP.
Most BCcPs require Ca2+. Addition of calcium chloride tothe reduced enzyme results in a transition of the low-potentialheme from low-spin to high-spin, which indicates a significantchange in the heme environment of the enzyme.107 It has beenproposed that calcium triggers a conformational change thatremoves the sixth ligand from the low-potential heme giving ahigh-spin heme and freeing up one axial coordination positionfor reaction with peroxide.105
5.2 Structure and Mechanism
The three-dimensional structure of the completely oxi-dized di-heme peroxidase (PAP) from P. aeruginosa wasdetermined at 2.4 A resolution.108 More recently, the three-dimensional structure of the completely oxidized di-hemeperoxidase from N. europea (NEP) was determined at 1.8 Aresolution.109 Preliminary X-ray diffraction analysis of di-heme peroxidases from R. capsulatus110 and P. nautica111
also have been reported. Unfortunately, the di-heme per-oxidases have proven extremely difficult to overexpress inhomologous/heterologous systems except in the case of thepurple phototrophic bacterium R. capsulatus.112
Figure 7 shows the overall structure of di-heme peroxidaseswith key residues involved in catalysis from P. aeruginosa andN. europea. Each monomer folds into two distinct domainswith the fold of each domain closely resembling that foundin class I c-type cytochromes. Each domain houses one hemewhich have been designated the high- and the low-potentialheme. Electrons enter from a redox partner to the high-potential heme followed by intramolecular electron transfer tothe low-potential heme. The closest approach of the 2 hemesis ≈10 A although the heme propionates are directly bridgedby the indole ring of Trp82. This interaction is the same as inTrp94 of PAP which is believed to act as an electron transferconduit between the two hemes.108 The two domains arerelated by a quasi two-fold axis, and the domain interface holdsa divalent cation, which is now known to be calcium. The likelyfunction of calcium is to maintain structural integrity and/orto modulate electron transfer between two heme domains.In both enzymes, calcium is not coordinated by negativelycharged carboxylates but by solvent and peptide carbonyloxygens. It is also known that calcium plays an important, ifas yet ill-defined, role in other peroxidases.
The high-potential heme in both enzymes has Met andHis as axial ligands. In the Pseudomonas enzyme, the low-potential heme has two His residues as axial heme ligands.108
Since the site of reaction with hydrogen peroxide is the low-potential heme, one His ligand must dissociate. Relevant tothis difference, the high-potential heme in PAP must first bereduced in order to activate the enzyme while NEP is fullyactive in the oxidized state. This implies that reduction ofthe high-potential heme in PAP leads to dissociation of theHis ligand in the PAP low-potential heme. The low-potentialheme of the Nitrosomonas enzyme is already in an activatedstate since the low-potential heme has only one His ligand andhence, has an open axial coordination site available for reactionwith peroxide.109 A comparison between the two enzymesillustrates the range of conformational changes required toactivate the Pseudomonas enzyme. This change involves thelarge repositioning of a loop containing the dissociable Hisheme ligand from the heme pocket to the molecular surfacewhere the His forms part of the dimer interface. In theNitrosomonas enzyme, the His ligand is already dissociatedso activation by reduction of the high-potential heme is notrequired. The mechanism by which a single electron caninduce such a large change remains a fascinating yet unsolvedproblem. Similar redox induced conformational changes andligand rearrangements have also been observed in cytochromecd1 nitrite reductase.113 This enzyme also has two hemesand upon reduction a Tyr ligand of one heme is displaced,which opens up a distal site for required nitrite reductionchemistry.

IRON: HEME PROTEINS, PEROXIDASES, CATALASES & CATALASE-PEROXIDASES 11
NEP
NEP
PAP
PAP
LP
HP
C-term
C-term
N-term
a5
a4a3
a2
a6
a7
a10
a1a8
a5
a6
a2a3
a4
a8
a9a11
α10
a1
b2
b1
b6
b5
b7b3
b4
a7
a9
N-term
Ca+2Ca+2
LP
HP
Loop out Loop in
H59
E102
E114H71
F93
Q104
F81
Q92
Figure 7 Schematic representation of the of di-heme peroxidases from Pseudomonas aeruginosa (PAP) and Nitrosomonas europea (NEP)showing key residues involved in catalysis. (This figure was generated from coordinates of 1EB7 and 1IQC deposited in the ProteinData Bank)
Little is known about the peroxide activation process inBCcPs. However, the NEP structure revealed a peroxide-binding site quite different from the more traditionalperoxidases. BCcPs do not have a distal His that servesas the catalytic acid-base required for heterolytic cleavage ofthe peroxide O–O bond. The NEP structure has a Glu residuedirectly adjacent to the peroxide-binding site that most likelyoperates as the acid-base catalyst. The only other peroxidaseknown to have a Glu in this position is chloroperoxidase.114
6 PROSTAGLANDIN ENDOPEROXIDE H2
SYNTHASES-1 AND 2 (PGHS)
6.1 Introduction
Prostaglandin endoperoxide H2 synthase (PGHS) [EC1.14.99.1] isoenzymes 1 and 2 catalyze the committed step inprostanoid synthesis115 via two sequential enzymatic reactions

12 IRON: HEME PROTEINS, PEROXIDASES, CATALASES & CATALASE-PEROXIDASES
Arachidonic acid
Ty385
Heme
His207
Gin203
Tyr385
His388Arachidonic acid
(a) (b)
COOH
Phospholipid
Arachidonic acid
COOH
HO
O •
•
O2
COOH
O
O
•
O O ••
COOHO
O
OOHPGG2
COOHO
O
OH
2e−
PGH2
Prostaglandins
Thromboxane A2Prostacyclin
Synthases
Phospholipase A2
Cyclooxygenase
Peroxidase
O2
Figure 8 (a) Putative reaction scheme of prostaglandin endoperoxide H2 synthases (PGHS). (b) Structure of PGHS-1 with key residues incatalysis in complex with substrate arachidonic acid (AA). (This figure was generated from coordinates of 1DIY deposited in the ProteinData Bank)
(Figure 8(a)): (1) the bis-oxygenation of arachidonic acid(the cyclooxygenase activity or COX) forms PGG2; and(2) reduction of 15-hydroperoxide of PGG2 in the peroxidase(POX) active site to form PGH2.
PGHS enzymes are targets of nonsteroidal anti-inflammatory (NSAI) drugs, which include over the counterdrugs such as aspirin and ibuprofen (Advil/Motrin). More-over, PGHS isozymes play essential roles in pathologiesthat include, for PGHS-1, thrombosis and for PGHS-2,
inflammation, pain, fever, various cancers, and neurologi-cal disorders like Alzheimer’s and Parkinson’s diseases.116
PGHS-1 (cox1 gene) is thought to be a housekeeping enzymepresent in all tissues. The isolated protein is usually a homod-imer of Mr140 kDa, which includes 3.5% carbohydrate. Asecond more recently discovered PGHS (PGHS-2, cox2 gene)is induced in a few specific tissues by mitogens, growthfactors, and cytokines.117 Despite the 60% sequence iden-tity between the 2 PGHSs, specific inhibitors of PGHS-1 are

available and selective inhibitors of PGHS-2 are already onhand.118,119 Inhibition of PGHS-1 may be associated withgastric and renal toxicity of the NSAI agents and specificinhibitors of PGHS-2 can alleviate unwanted side effects ofanti-inflammatory reagents.
6.2 Structure and Mechanism
Garavito and coworkers solved the first structure of aPGHS at 3.4 A resolution.120 Crystal structures of otherPGHS isoforms are structurally homologous121,122 as might beexpected from high sequence identity. PGHS is a homodimericmembrane protein with one heme per subunit. Each monomerconsists of three structural domains: a N-terminal epidermalgrowth factor (EGF) domain, a membrane-binding domain ofabout 48 amino acids in length, and a large C-terminal globularcatalytic domain with heme binding site facing the solvent andcontaining both cyclooxygenase and peroxidase active sites.The structure of the C-terminal segments beyond Pro583 (17amino acids in PGHS-1 and 35 amino acids in PGHS-2)have not been resolved crystallographically. Nonetheless, thisregion is of great interest as PGHS-1 and PGHS-2 contain C-terminal Lys-Asp-Glu-Leu (KDEL) like sequences that maytarget PGHSs to the endoplasmic reticulum and the associatednuclear envelope.115 The structure supports the proposedmodel from biochemical studies that cyclooxygenase andperoxidase active sites are distinct but share a common hemecofactor. Despite the low sequence homology between PGHSsand other peroxidases,123 the secondary and tertiary structureof PGHS is similar to the structure of myeloperoxidase123 andCcP.22 However, unlike other systems, the heme in PGHS ismore exposed to solvent.
The structure of PGHS-1 in a complex with arachidonicacid is shown in Figure 8(b). The peroxidase active site is verysimilar to other peroxidases. Histidine serves as an axial hemeligand (Figure 8(b)), while in the distal pocket, His207 servesas the acid-base catalytic group required for heterolytic fissionof the peroxide O–O bond. However, PGHS-1 has Gln203in place of the distal Arg found in other peroxidases. Thissame type of His/Gln distal site pocket is found in anothermammalian peroxidase, myeloperoxidase.
The mechanism shown below requires that Tyr385 firstbe oxidized to a radical. This reaction is achieved via thetraditional peroxidase mechanism.
(1) Fe3+PTyr385 + ROOHFe4+ − OP
•Tyr385 + ROH
Compound I(2) Fe4+ − OP
•Tyr385 Fe4+ − OPTyr385
•
(3) Fe4+ − OPTyr385• + AA Fe4+ − OPTyr385
Compound II + AA•
(4) Fe4+ − OPTyr385 + AA• + 2O2
Fe4+ − OPTyr385 + O2AAOO•
(5) Fe4+ − OPTyr385 + O2AAOO•
Fe4+ − OPTyr385• + O2AAOOH
IRON: HEME PROTEINS, PEROXIDASES, CATALASES & CATALASE-PEROXIDASES 13
In step 1, the usual compound I is formed via reaction witha hydroperoxide. Tyr385 is less than 5 A from the heme, sothe oxidation of Tyr385 to a radical in step 2 by the porphyrinradical can readily occur. As shown in Figure 8(b), C13 ofarachidonic acid (AA) directly contacts Tyr385 thus enablingregio- and stereoselective abstraction of the pro-S hydrogento give the arachidonic acid radical, AA•. After the twooxygenation reactions in steps 4 and 5, the AA peroxy radicalin step 5 oxidizes Tyr385 to regenerate the Try385 radical.Details of the cyclooxygenase reaction in steps 4 and 5 areoutlined in Figure 8(a). The product dissociates and anotherarachidonic acid can bind to compound I, without regeneratingthe resting protein. Thus the oxyferryl center appears to bean innocent bystander. Reduction of O2AAOOH to O2AAOHproceeds via the traditional peroxidase mechanism to givecompound I.
7 MAMMALIAN PEROXIDASES
7.1 Myeloperoxidase (MPO)
MPO (Myeloperoxidase) [EC 1.11.1.7] catalyzes thehydrogen peroxide–dependent two-electron oxidation ofhalides (CI−, I−, Br−) and thiocyanate to the correspondinghypohalous acids and hypothiocyanate which are cytotoxic toinvading pathogens. In addition, MPO is capable of the single-electron oxidation of a wide variety of aromatic alcohols andamines.124
MPO occurs in two types of mammalian phagocytic whiteblood cells: monocytes and neutrophils. MPO is localizedin azurophil granules, which comprise 1–5% of dry weightof neutrophils. Following microbial ingestion, degranulationleads to rapid release of MPO into the phagosome.125
Microbial ingestion or phagocytosis is accompanied by a‘respiratory burst’ giving rise to the superoxide anion O2
−which, in turn, dismutates to molecular oxygen and hydrogenperoxide. MPO is able to oxidize chloride ions to HOCl, aneffective agent in killing bacteria, fungi, and viruses.
MPO is a covalently linked dimeric glycoprotein of Mr
140 kDa comprised of 745 amino acids. MPO consists ofthree different isoenzyme forms termed a, b, and c.126 Thenature of the chemical differences between these isoformsis not fully understood, and only isoform c has beencrystallized.127 Catalytically active recombinant human MPOhas been expressed in Chinese hamster ovary cells. However,incomplete posttranslational processing of the recombinantenzyme yields a monomeric form of the enzyme consistingof a single polypeptide chain of Mr 84 kDa with alteredcarbohydrate content.128
The heme in ferric MPO exhibits a relatively strongabsorption band near 680 nm, which is responsible forthe characteristic green color. The heme Soret band isconsiderably red-shifted compared to other heme-containing

14 IRON: HEME PROTEINS, PEROXIDASES, CATALASES & CATALASE-PEROXIDASES
proteins including its close relatives, EPO (415 nm) and LPO(412 nm).124 The origin of the red-shift has been attributedto the effects of heme-protein covalent linkages in distortingthe planarity of the porphyrin ring and to possible electron-withdrawing effects of the unique sulfonium ion linkage toMet243.129,130 MPO exhibits a characteristic EPR spectrum ofhigh-spin ferric heme in an environment of rhombic symmetrywith gx = 6.90, gy = 5.07.124 Compound I of MPO exhibitsan EPR spectrum characteristic of a hexa-coordinated low-spin heme iron similar to other heme peroxidases.124 However,complete formation of compound I requires a large excess ofperoxide and binding of H2O2 to the ferric (Fe3+) enzyme andthis appears to be reversible.
7.1.1 Structure of MPO
The crystal structure of MPO was first determined in1987,131 and more recently, the data have been extended to1.8 A (Figure 9)132 which has enabled the nature of the hemeto be unambiguously established. The heme is covalentlyattached to the protein via two ester linkages between thecarboxyl groups of Glu242 and Asp94 and modified methylgroups on pyrrole rings A and C of the heme as well as asulfonium ion linkage between the sulfur atom of Met243 andthe beta-carbon of the vinyl group on pyrrole ring A.
MPO is a covalently linked dimer which is ellipsoidalin shape with overall dimensions of 110 × 60 × 50 A3. Thedimer can be cleaved by reduction of a disulfide bond into twoidentical halves. Each half of the dimer termed hemi-MPO hasthe same optical and catalytic properties of the dimer. Hemi-MPO consists of two polypeptides of 466 and 108 amino acidresidues, and a heme prosthetic group covalently bound to thelarge polypeptide. Like CcP and LIP, MPO is largely a helicalbundle protein with very little β-sheet structure. The bulk ofthe large polypeptide folds into five separate domains and one
Ca2+
Sulfate
Sugar
Heme
Figure 9 Structure of myeloperoxidase (MPO) along with the hemeenvironment as observed in the high-resolution crystal structure.(This figure was generated from coordinates of 1CXP deposited inthe Protein Data Bank)
open loop that surround this core. The small polypeptide wrapsaround the surface of the molecule with its C-terminal helixpenetrating the interior core. There are four intrachain disulfidebonds in the large polypeptide and a single disulfide bridgein the small polypeptide. Four potential asparagine-linkedglycosylation sites have been identified in the larger subunit.Sugar residues near the Asn317 glycosylation site close tothe dimer interface play a role in dimer–dimer interaction, inaddition to the lone disulfide bridge.
Crystallography revealed a bound chloride ion at theamino terminus of the helix containing the proximal His336which can be replaced with bromide.132 Crystal structures ofthe human MPO-cyanide, MPO-cyanide-bromide, and MPO-cyanide-thiocyanate have also been determined by Fenna andcoworkers to 1.9 A.133 These results support a model for asingle common binding site for halides and thiocyanate assubstrates or as inhibitors near the δ-meso carbon of theporphyrin ring in myeloperoxidase.
7.2 Lactoperoxidase (LPO)
The mucosal surfaces of the body provide protectionagainst potential infections. The exocrine glands and theirsecretions provide both specific immunological and lessspecific nonimmunological protection to the mucosal surfacesof eye, nose, mouth, trachiobronchial tree, and intestinal tract.The antibacterial system includes lysozyme, lactoferrin, andperoxidase. Peroxidase also provides an antibacterial systemin bovine and human milk. LPO (lactoperoxidase) is a heme-containing, calcium-containing glycoprotein of Mr 78 kDawith one heme group per molecule. In bovine milk, LPO is,next to xanthine oxidase (see Molybdenum: MPT-containingEnzymes), the most abundant enzyme. Its concentrationis around 30 mg/L, constituting 0.5% of whey proteins.134
Variations in enzyme levels were reported to depend on thesexual cycle of the cow, season, feeding regime, and breed.135
The properties and biological function of LPO have recentlybeen reviewed.7,136 This secretory peroxidase is isolated fromcow’s milk. Crystalline LPO from bovine milk was obtained in1943 and shown to be green colored like MPO. Its isolation andpurification was simplified by precipitation of casein by rennet.The enzyme with a single polypeptide chain has been knownto exist as several isoenzyme forms. The Soret maxima ofLPO of 412 nm with molar absorptivity of 112 mM−1 cm−1 istypical of most peroxidases. It recently has been demonstratedthat the heme in LPO is attached to the protein by ester bondsbetween the heme 1- and 5-methyl groups, and Glu375 andAsp275, respectively.137
The heme environment of MPO, both in the proximaland distal heme pockets, is conserved in LPO.123 Aswith other peroxidases, LPO reacts with H2O2 to givecompound I. In the absence of any electron-donor substrate,compound I decomposes to compound I∗ species which isspectroscopically different from compound I. Compound IIis formed spontaneously upon addition of one equivalent of

hydrogen peroxide to native LPO and is stable for 20 min. Athreefold excess of peroxide can also be used to obtain purecompound II, and addition of 117-fold excess peroxide to thesame solution produces pure compound III.138
Recent studies have shown that LPO reacts with H2O2 tosequentially give two compound I intermediates: the first withan oxyferryl (Fe4+–O) and a porphyrin radical cation, and thesecond with the same ferryl species and a presumed proteinradical.139,140 Using spin-trapping experiments followed byEPR spectroscopy, LPO was found to react with the spin-trap3,5-dibromo-4-nitrosobenzenesulfonic acid (DBNBS) to givea protein-bound radical. Furthermore, LPO was shown toundergo H2O2-dependent formation of dimers and trimer. Thedimer involves a cross-link between Tyr289 in each of twoLPO molecules and retains full catalytic activity.139
A recent study by Harvey and Bruck followed theoxidation of mitoxantrone, an anthraquinone type anticancerdrug, spectrophotometrically under turnover conditions andsingle turnover conditions using stopped-flow methods. Withcompound I and compound II, mitoxantrone formed bindingcomplexes that were deactivated with increasing substrateconcentration.141 Under turnover conditions, compound IIwas the steady state intermediate, but with increasingH2O2, compound II reacted with H2O2 to form thecatalytically inactive intermediate compound III. Nitrite
IRON: HEME PROTEINS, PEROXIDASES, CATALASES & CATALASE-PEROXIDASES 15
prevented formation of compound III by reducing compoundII to the native state.
The physiological role of LPO is to use H2O2 togenerate OSCN− which inhibits microbial growth. H2O2 isgenerated endogenously, for example, by polymorphonuclearleukocytes (PMN), in the process of phagocytosis. In addition,under aerobic conditions many lactobacilli, lactococci, andstreptococci produce sufficient H2O2 to activate LPO.Figure 10 outlines the proposed LPO reaction mechanism.134
The first step in the enzymatic mechanism is the initiationreaction of the resting ferric (Fe3+) LPO to its groundstate, using H2O2, according to Fe3+ + H2O2 Fe2+ + HO
•2,
followed by propagation reactions, as illustrated in Figure 10.The propagation reactions include conversion of LPO fromground state to compound I by reaction with H2O2. At lowSCN− (<3 µM) and halide concentrations, compound I reactswith one-electron donors that are present (proteins, peptides)to form compound II that is continuously reduced to groundstate at a low rate. At an excess of [H2O2] (>0.5 mM),compound II may react to form compound III, leading to aferrylperoxidase adduct and irreversible inactivation of LPO.The agent that oxidizes SCN− or halides is compound I. Muchof the interest in the LPO system stems from its potential forgenerating natural biopreservatives in food, feed specialities,cosmetics, and related products. Some of the applications of
N N
NN
Fe2+
CH3
O O OO
HIS CH3PROT-S
CH3
−2e
N N
NN
Fe3+
CH3
O O OO
HIS CH3PROT-S
CH3 O •
+ H2O2 + H2O
2SCN−
(SCN)2
AH+ A• + Compound II
+1e
+1e
+2e
Compound IIILPO-Fe4+
+
Ground state Compound I
•
+ excess H2O2
+HO2
AH+
Figure 10 Proposed pathways in the lactoperoxidase-catalyzed reaction mechanism (LPO system)

16 IRON: HEME PROTEINS, PEROXIDASES, CATALASES & CATALASE-PEROXIDASES
LPO and their functionality have been reviewed by de Witand van Hooijdonk.134
8 CATALASES
8.1 Introduction
Catalases are ubiquitous metalloenzymes present in almostall aerobic prokaryotic and eukaryotic organisms. Althoughthe full range of biological functions of catalases remainsuncertain, the primary role is the removal of hydrogen peroxidefrom aerobic organisms by catalyzing its dismutation to waterand oxygen (equation 8)
2H2O2 2H2O + O2 (8)
This type of antioxidant function is essential for allorganisms that are exposed to dioxygen, which readily formsH2O2 both enzymatically, through the action of oxidases,and nonenzymatically, as a side product of respiration orauto-oxidation of cell components. Hydrogen peroxide exertsits toxic effects in a variety of ways, including transitionmetal activation of peroxides via Fenton chemistry thatgenerates highly reactive superoxide or hydroxy radicals thatcan lead to DNA, protein, and lipid damage. Catalase deficient(acatalasemic) cells lead to an accumulation of oxidativeagents resulting in increased susceptibility to thermal injury142
or HIV proliferation,143 high rates of DNA mutations,144
and inflammation.145 Besides this primary protective role,the catalase system is also believed to have physiologicalsignificance in various biological oxidations, such as themetabolization of methanol and ethanol.146,147
8.2 Functional Classes
Four groups of catalases have been identified. The first,most fully characterized group contains the monofunctional‘classic’ heme-containing catalases, which are homotetramerswith one heme per subunit and are present in both prokaryotesand eukaryotes.69,148 The subunit sizes within this groupvary, consisting of approximately 500 residues (small-subunitcatalases) or 700 residues (large-subunit catalases). Thesecond group is composed of the nonheme manganese-containing catalases, homohexameric enzymes with twomanganese ions in the active site, that have so far onlybeen reported in prokaryotes.149–151 The third group, heme-containing catalase-peroxidases, were described in Section 2.The final group is composed of miscellaneous heme-containing enzymes with secondary reactions in the formof low levels of catalase activity. Heme-containing enzymesthat fall in this group include methemoglobin, metmyoglobinand chloroperoxidase from Caldariomyces fumago.
8.3 ‘Classic’ Monofunctional Heme-containingCatalases
Catalases of Penicillium vitale (PVC)152 and beef livercatalase (BLC)153,154 were the first catalases crystallographi-cally characterized. At present, the crystal structures of sevencatalases from this group are known: beef liver,155 Penicil-lium vitale,70,156 Micrococcus lysodeikticus (MLC),157 E. colihydroperoxidase (HPII),158 Proteus mirabilus (PMC PR),159
Saccharomyces cerevisiae (SCC-A),160 and human erythro-cyte (HEC).161 There is considerable structural similarityamong the catalases in this subgroup. The conformationdisplayed by the homologous molecular regions appears tobe exclusive of ‘classic’ monofunctional heme catalases, andhence is now typically referred to as the ‘catalase fold’. Atypical catalase fold consists of four consecutive regions alongthe polypeptide chain (Figure 11(a)), which entail an amino
NADPH Heme
H61
N133
Y343
(b)(a)
Figure 11 (a) Ribbon drawing of the beef liver catalase structure (PDB code 7CAT) showing the location of the heme (red) and NADPH(yellow). The four regions of a typical catalase fold are represented with different colors: amino acid terminal arm (purple); anti-paralleleight-stranded β-barrel domain (green); wrapping domain (blue), and α-helical domain (cyan). (b) Diagram showing the heme environment ina monofunctional heme-containing MLC catalase. (This figure was generated from coordinates of 1GWE deposited in the Protein Data Bank)

acid terminal arm, an anti-parallel 8-stranded β-barrel domain,an extended structural part which is referred to as the ‘wrap-ping’ domain, and finally an α-helical domain. Of the variousregions in monofunctional heme catalases, the anti-paralleldomain appears to be the central feature of a typical catalasefold since the first half of the β-barrel domain contains mostof the residues that define the heme distal side, whereas thesecond half of the β-barrel contributes to the NADPH bindingpocket in those catalases that bind this cofactor.162
Each subunit of a heme catalase binds a single molecule ofheme and some mammalian catalases also possess a secondcofactor, NADPH. The binding of NADPH in catalases wasat first totally unexpected,162,163 but has since been a frequentfeature of small-subunit catalases from both prokaryoticand eukaryotic organisms. However, the actual biochemicalfunction of NADPH in these catalases is still not fullyunderstood. One possible role is protection of the enzymeagainst inactivation by its own substrate, especially underconditions of low-peroxide concentrations.164 The NADPHbinding pocket is located on the molecular surface, just abovean entrance channel with the nicotinamide active carbonsituated approximately 20 A from the closest heme atom(Figure 11(a)).162
The heme group characterized in the active site of variouscatalases varies from either being a heme d (a cis-hydroxyγ -spirolactone) as observed in the active site of HPII andPVC70,165 or the conventional heme b as seen in all othercatalase structures. Catalase HPII and presumably PVC,initially bind heme b and then catalyze its conversion to hemed utilizing H2O2.166 This heme conversion is concomitantwith formation of a unique covalent bond between Cβ of theessential Tyr415 and the Nδ of His392, and a mechanismrelating to its formation to heme modification has beenproposed.69 Although different heme variant groups existamong the catalases, the location and environment of the hemegroup is very similar among the catalase structures. In general,the heme groups are deeply buried in the catalase tetramerwith iron atoms situated approximately 20 A from the nearestmolecular surface. The deep burial of the heme groups resultsin catalases requiring channels for efficient communicationbetween the active center and the surface molecule. Themost significant and best-conserved channel reaches the distalside of the heme pocket, whereas a second, less conservedchannel is observed at a location that corresponds to theNADPH binding pocket in catalases that bind the dinucleotide.These channels narrow as the heme is approached and thenarrow entrance of these channels is strongly believed to bea ‘discrimination gate’ between substrates of various sizesand thus accounts for the reduced activity of certain catalasestoward larger substrates such as aliphatic alcohols.167 This isin good agreement with the evidence that MLC, which hasone of the narrowest entrances, is slower at catalyzing largersubstrates and faster for small substrates relative to othercatalases.168
IRON: HEME PROTEINS, PEROXIDASES, CATALASES & CATALASE-PEROXIDASES 17
The heme pocket in these catalases differs considerablyfrom that of a typical heme peroxidase (Figure 11(b)). Themost obvious difference between the two sets of enzymes isthe coordination of the proximal iron ligand. In peroxidases,the proximal ligand is a histidine, while in the classicmonofunctional heme catalases, the imidazole nitrogen isreplaced by an oxygen of the phenolic hydroxy group of atyrosine. This oxygen, which also hydrogen bonds with aconserved arginine, is most likely deprotonated possessinga localized negative charge that can contribute to thestabilization of the Fe4+ oxidation state required in the catalyticcycle.162 On the distal side of the heme, a His and Asn residueform the peroxide pocket and are considered essential forcatalysis. The His is generally considered to operate as anacid-base catalytic group as in peroxidases,7,21 while the Asnserves the function of the distal side Arg in peroxidases.
8.4 Catalytic Mechanisms
The catalytic mechanism for the formation of compound Iin the catalase-peroxidases is believed to occur in a mannervery similar to that of the heme peroxidases.60,61 The maindifference in the enzymatic mechanism between catalases andperoxidases is compound I reduction. In the catalase cycle,a second peroxide molecule is also used as a reducing agentfor compound I rather than a small organic molecule asin most other peroxidases. Crystal structures of the variouscatalytic intermediate states (compound I and compoundII) have been determined for classic monofunctional heme-containing catalases.27,160,169 Only minor local rearrangementsare observed relative to the native enzyme.27 In bothcompounds I and II, the iron appears slightly displaced abovethe heme ring toward the bound oxygen. In compound I, butnot in compound II, the presence of extra electron density,assigned as weakly bound anion, is situated on the hemeproximal side ∼18 A from the iron. Interestingly, in the higherresolution compound I structure of PVC at 1.8 A, no extradensity was observed, similar to that reported in PMC PR.159
In both the PMC PR and the PVC compound I structures,the coordinated oxygen is situated between 1.7 and 1.8 Aabove the iron atom which is similar to that found in theHRP compound I structure (1.7 A).29 In both the PMC PRand the PVC compound I structures, this coordinated solventmolecule is hydrogen bonded to the Nε and Nδ atoms of theessential His and Asn residues.
9 REFERENCES
1. D. Dolphin, A. Forman, D. C. Borg, J. Fajer, and R. H. Felton,Proc. Natl. Acad. Sci. U.S.A., 1971, 68, 614.
2. A. F. Coulson and T. Yonetani, Biochem. Biophys. Res.Commun., 1972, 49, 391.

18 IRON: HEME PROTEINS, PEROXIDASES, CATALASES & CATALASE-PEROXIDASES
3. M. Sivaraja, D. B. Goodin, M. Smith, and B. M. Hoffman,Science, 1989, 245, 738.
4. H. B. Dunford, Horseradish Peroxidase: Structure and KineticProperties, in ‘Peroxidases in Chemistry and Biology’, eds.J. Everse, E. Everse, and M. B. Grisham, CRC Press, BocaRaton, FL, Vol. 2, 1991, p. 1.
5. B. Chance, J. Biol. Chem., 1943, 151, 553.
6. K. G. Welinder, Curr. Opin. Struct. Biol., 1992, 2, 388.
7. H. B. Dunford, ‘Heme Peroxidases’, John Wiley & Sons,Danvers, MA, 1999.
8. J. E. Erman and L. B. Vitello, Biochim. Biophys. Acta, 2002,1597, 193.
9. M. Kwon, S. Chong, S. Han, and K. Kim, Biochim. Biophys.Acta, 2003, 1623, 1.
10. C. Charizanis, H. Juhnke, B. Krems, and K. D. Entian, Mol.Gen. Genet., 1999, 262, 437.
11. D. B. Goodin and D. E. McRee, Biochemistry, 1993, 32, 3313.
12. K. Choudhury, M. Sundaramoorthy, J. M. Mauro, andT. L. Poulos, J. Biol. Chem., 1992, 267, 25656.
13. J. E. Erman, L. B. Vitello, M. A. Miller, A. Shaw, K. A.Brown, and J. Kraut, Biochemistry, 1993, 32, 9798.
14. S. L. Newmyer and P. R. Ortiz de Montellano, J. Biol. Chem.,1995, 270, 19430.
15. S. L. Newmyer and P. R. Ortiz de Montellano, J. Biol. Chem.,1996, 271, 14891.
16. L. B. Vitello, J. E. Erman, M. A. Miller, J. Wang, andJ. Kraut, Biochemistry, 1993, 32, 9807.
17. M. A. Miller, R. Q. Liu, S. Hahm, L. Geren, S. Hibdon,J. Kraut, B. Durham, and F. Millett, Biochemistry, 1994, 33,8686.
18. C. A. Bonagura, B. Bhaskar, H. Shimizu, H. Li, M. Sundara-moorthy, D. E. McRee, D. B. Goodin, and T. L. Poulos,Biochemistry, 2003, 42, 5600.
19. I. Schlichting, J. Berendzen, K. Chu, A. M. Stock, S. A. Maves,D. E. Benson, R. M. Sweet, D. Ringe, G. A. Petsko, and S. G.Sligar, Science, 2000, 287, 1615.
20. W. L. Purcell and J. E. Erman, J. Am. Chem. Soc., 1976, 98,7033.
21. T. L. Poulos and J. Kraut, J. Biol. Chem., 1980, 255, 8199.
22. B. C. Finzel, T. L. Poulos, and J. Kraut, J. Biol. Chem., 1984,259, 13027.
23. S. L. Edwards, H. X. Nguyen, R. C. Hamlin, and J. Kraut,Biochemistry, 1987, 26, 1503.
24. V. Fulop, R. P. Phizackerley, S. M. Soltis, I. J. Clifton,S. Wakatsuki, J. Erman, J. Hajdu, and S. L. Edwards,Structure, 1994, 2, 201.
25. S. L. Edwards, T. L. Poulos, and J. Kraut, J. Biol. Chem.,1984, 259, 12984.
26. H. M. Jouve, P. Andreoletti, P. Gouet, J. Hajdu, andJ. Gagnon, Biochimie, 1997, 79, 667.
27. P. Gouet, H. M. Jouve, P. A. Williams, I. Andersson,P. Andreoletti, L. Nussaume, and J. Hajdu, Nat. Struct. Biol.,1996, 3, 951.
28. H. P. Hersleth, B. Dalhus, C. H. Gorbitz, and K. K. Andersson,J. Biol. Inorg. Chem., 2002, 7, 299.
29. G. I. Berglund, G. H. Carlsson, A. T. Smith, H. Szoke,A. Henriksen, and J. Hajdu, Nature, 2002, 417, 463.
30. B. Chance, L. Powers, Y. Ching, T. Poulos, G. R. Schonbaum,I. Yamazaki, and K. G. Paul, Arch. Biochem. Biophys., 1984,235, 596.
31. J. E. Penner-Hahn, T. J. McMurry, M. Renner, L. Latos-Grazynsky, K. S. Eble, I. M. Davis, A. L. Balch, J. T. Groves,J. H. Dawson, and K. O. Hodgson, J. Biol. Chem., 1983, 258,12761.
32. C. M. Reczek, A. J. Sitter, and J. Terner, J. Mol. Struct., 1989,214, 27.
33. S. Hashimoto, J. Teraoka, T. Inubushi, T. Yonetani, andT. Kitagawa, J Biol Chem., 1986, 261, 11110.
34. A. J. Sitter, C. M. Reczek, and J. Terner, J. Biol. Chem., 1985,260, 7515.
35. R. Makino, T. Uno, Y. Nishimura, T. Iizuka, M. Tsuboi, andY. Y. Ishimura, J. Biol. Chem., 1986, 261, 8376.
36. G. R. Schonbaum and S. Lo, J. Biol. Chem., 1972, 247, 3353.
37. M. A. Miller, G. W. Han, and J. Kraut, Proc. Natl. Acad. Sci.U.S.A., 1994, 91, 11118.
38. M. M. Fitzgerald, M. J. Churchill, D. E. McRee, andD. B. Goodin, Biochemistry, 1994, 33, 3807.
39. Y. Cao, R. A. Musah, S. K. Wilcox, D. B. Goodin, andD. E. McRee, Protein Sci., 1998, 7, 72.
40. C. A. Bonagura, M. Sundaramoorthy, H. S. Pappa, W. R.Patterson, and T. L. Poulos, Biochemistry, 1996, 35, 6107.
41. G. M. Jensen, S. W. Bunte, A. Warshel, and D. B. Goodin, J.Phys. Chem. B, 1998, 102, 8221.
42. T. L. Poulos, T. P. Barrows, B. Bhaskar, C. A. Bonagura, andH. Li, Int. J. Quantum Chem., 2002, 88, 211.
43. W. R. Patterson and T. L. Poulos, Biochemistry, 1995, 34,4331.
44. W. R. Patterson, T. L. Poulos, and D. B. Goodin, Biochem-istry, 1995, 34, 4342.
45. C. A. Bonagura, M. Sundaramoorthy, B. Bhaskar, andT. L. Poulos, Biochemistry, 1999, 38, 5538.
46. C. A. Bonagura, B. Bhaskar, M. Sundaramoorthy, andT. L. Poulos, J. Biol. Chem., 1999, 274, 37827.
47. H. Pelletier and J. Kraut, Science, 1992, 258, 1748.
48. H. S. Pappa and T. L. Poulos, Biochemistry, 1995, 34, 6573.
49. S. H. Northrup, J. O. Boles, and J. C. Reynolds, Science, 1988,241, 67.
50. H. S. Pappa, S. Tajbaksh, A. J. Saunders, G. J. Pielak, andT. L. Poulos, Biochemistry, 1996, 35, 4837.
51. K. H. Sharp, M. Mewies, P. C. Moody, and E. L. Raven, Nat.Struct. Biol., 2003, 10, 303.

52. K. Wada, T. Tada, Y. Nakamura, T. Ishikawa, Y. Yabuta,K. Yoshimura, S. Shigeoka, and K. Nishimura, J. Biochem.(Tokyo), 2003, 134, 239.
53. A. N. Hiner, J. I. Martinez, M. B. Arnao, M. Acosta,D. D. Turner, E. Lloyd Raven, and J. N. Rodriguez-Lopez,Eur. J. Biochem., 2001, 268, 3091.
54. D. Mandelman, J. Jamal, and T. L. Poulos, Protein Sci., 1998,37, 17610.
55. E. H. Bursey and T. L. Poulos, Biochemistry, 2000, 39, 7374.
56. C. Dye, S. Scheele, P. Dolin, V. Pathania, and M. C.Raviglione, JAMA, 1999, 282, 677.
57. Y. Zhang, S. Dhandayuthapani, and V. Deretic, Proc. Natl.Acad. Sci. (USA), 1996, 93, 13212.
58. R. A. Slayden and C. E. Barry III, Microbes Infect., 2000, 2,659.
59. Y. Zhang, B. Heym, B. Allen, D. Young, and S. Cole, Nature,1992, 358, 591.
60. Y. Yamada, T. Fujiwara, T. Sato, N. Igarashi, and N. Tanaka,Nat. Struc. Biol., 2002, 9, 691.
61. X. Carpena, S. Loprasert, S. Mongkolsuk, J. Switala,P. C. Loewen, and I. Fita, J. Mol. Biol., 2003, 327, 475.
62. M. Zamocky, G. Regelsberger, C. Jakopitsch, and C. Obinger,FEBS Lett., 2001, 492, 177.
63. K. G. Welinder, Biochim. Biophys. Acta, 1991, 1080, 215.
64. A. Hillar, B. Peters, R. Pauls, A. Loboda, H. Zhang,A. G. Mauk, and P. C. Loewen, Biochemistry, 2000, 39, 5868.
65. S. Chouchane, I. Lippai, and R. S. Magliozzo, Biochemistry,2000, 39, 9975.
66. T. Choinowski, W. Blodig, K. H. Winterhalter, and K. Piontek,J. Mol. Biol., 1999, 286, 809.
67. B. Bhaskar, C. E. Immoos, H. Shimizu, F. Sulc, P. J. Farmer,and T. L. Poulos, J. Mol. Biol., 2003, 328, 157.
68. A. Buzy, V. Bracchi, R. Sterjiades, J. Chroboczek, P. Thibault,J. Gagnon, H. M. Jouve, and G. Hudry-Clergeon, J. Protein.Chem., 1995, 14, 59.
69. J. Bravo, I. Fita, J. C. Ferrer, W. Ens, A. Hillar, J. Switala, andP. C. Loewen, Protein Sci., 1997, 6, 1016.
70. G. N. Murshudov, A. I. Grebenko, V. V. Barynin, Z. Dauter,K. S. Wilson, B. K. Vainshtein, W. R. Melik-Adamyan,J. Bravo, J. M. Ferran, J. C. Ferrer, J. Switala, P. C. Loewen,and I. Fita, J. Biol. Chem., 1996, 271, 8863.
71. A. Henriksen, D. J. Schuller, K. Meno, K. G. Welinder,A. T. Smith, and M. Gajhede, Biochemistry, 1998, 37,8054.
72. N. L. Wengenack, J. R. Uhl, A. L. St Amand, A. J. Tomlinson,L. M. Benson, S. Naylor, B. C. Kline, F. Rr. Cockerill, andF. Rusnak, J. Infect. Dis., 1997, 176, 722.
73. K. Piontek, A. T. Smith, and W. Blodig, Biochem. Soc. Trans.,2001, 29, 111.
74. P. J. Harvey, R. Floris, T. Lundell, J. M. Palmer, H. E. Schoe-maker, and R. Wever, Biochem. Soc. Trans., 1992, 20,345.
IRON: HEME PROTEINS, PEROXIDASES, CATALASES & CATALASE-PEROXIDASES 19
75. K. Piontek, T. Glumoff, and K. Winterhalter, FEBS Lett.,1993, 315, 119.
76. T. L. Poulos, S. L. Edwards, H. Wariishi, and M. H. Gold, J.Biol. Chem., 1993, 268, 4429.
77. W. Blodig, A. T. Smith, W. A. Doyle, and K. Piontek, J. Mol.Biol., 2001, 305, 851.
78. M. Sundaramoorthy, K. Kishi, M. H. Gold, and T. L. Poulos,J. Biol. Chem., 1994, 269, 32759.
79. M. Gajhede, D. J. Schuller, A. Henriksen, A. T. Smith, andT. L. Poulos, Nat. Struct. Biol., 1997, 4, 1032.
80. A. Henriksen, K. G. Welinder, and M. Gajhede, J. Biol.Chem., 1998, 273, 2241.
81. N. Kunishima, K. Fukuyama, H. Matsubara, H. Hatanaka,Y. Shibano, and T. Amachi, J. Mol. Biol., 1994, 235, 331.
82. J. F. W. Petersen, A. Kadziola, and S. Larsen, FEBS Lett.,1994, 339, 291.
83. H. Itakura, Y. Oda, and K. Fukuyama, FEBS Lett., 1997, 412,107.
84. W. Blodig, A. T. Smith, K. Winterhalter, and K. Piontek,Arch. Biochem. Biophys., 1999, 370, 86.
85. W. Blodig, W. A. Doyle, A. T. Smith, K. Winterhalter,T. Choinowski, and K. Piontek, Biochemistry, 1998, 37,8832.
86. W. A. Doyle, W. Blodig, N. C. Veitch, K. Piontek, andA. T. Smith, Biochemistry, 1998, 37, 15097.
87. W. A. Doyle and A. T. Smith, Biochem. J., 1996, 315, 15.
88. U. Tuor, H. Wariishi, H. E. Schoemaker, and M. H. Gold,Biochemistry, 1992, 31, 4986.
89. H. Wariishi, K. Valli, and M. H. Gold, Biochem. Biophys. Res.Commun., 1991, 176, 269.
90. F. Johnson, G. H. Loew, and P. Du, in ‘Plant Peroxidases:Biochemistry and Physiology’, eds. K. G. Welinder, S. K. R.,C. Penel, and H. Greppin, Geneva, Switzerland, 1993.
91. M. Sundaramoorthy, K. Kishi, M. H. Gold, and T. L. Poulos,J. Biol. Chem., 1997, 272, 17574.
92. M. Gajhede, Horseradish Peroxidase, in ‘Handbookof Metalloproteins’, eds. A. Messerschmidt, R. Huber,T. L. Poulos, and K. Wieghardt, John Wiley & Sons,Chichester, 2001, Vols. I, 2, p. 195.
93. A. T. Smith, N. Santama, S. Dacey, M. Edwards, R. C. Bray,R. N. Thorneley, and J. F. Burke, J. Biol. Chem., 1990, 265,13335.
94. J. E. Penner-Hahn, K. S. Eble, T. J. McMurry, M. Renner,A. L. Balch, J. T. Groves, J. H. Dawson, and K. O. Hodgson,J. Am. Chem. Soc., 1986, 108, 7819.
95. A. Morimoto, M. Tanaka, S. Takahashi, K. Ishimori, H. Hori,and I. Morishima, J. Biol. Chem., 1998, 273, 14753.
96. A. Henriksen, A. T. Smith, and M. Gajhede, J. Biol. Chem.,1999, 274, 35005.
97. K. Tsukamoto, H. Itakura, K. Sato, K. Fukuyama, S. Miura,S. Takahashi, H. Ikezawa, and T. Hosoya, Biochemistry, 1999,38, 12558.

20 IRON: HEME PROTEINS, PEROXIDASES, CATALASES & CATALASE-PEROXIDASES
98. N. Ellfolk and R. Soininen, Acta Chem. Scand., 1970, 24,2126.
99. D. M. Arciero and A. B. Hooper, J. Biol. Chem., 1994, 269,11878.
100. C. F. Goodhew, I. B. Wilson, D. J. Hunter, and G. W.Pettigrew, Biochem J , 1990, 271, 707.
101. G. W. Pettigrew, Biochim. Biophys. Acta, 1991, 1058, 25.
102. S. P. Hanlon, R. A. Holt, and A. G. McEwan, FEMSMicrobiol. Lett., 1992, 97, 283.
103. J. A. Zahn, D. M. Arciero, A. B. Hooper, J. R. Coats, andA. A. DiSpirito, Arch. Microbiol., 1997, 168, 362.
104. J. Villalain, I. Moura, M. C. Liu, W. J. Payne, J. LeGall,A. V. Xavier, and J. J. Moura, Eur. J. Biochem., 1984, 141,305.
105. T. Alves, S. Besson, L. C. Duarte, G. W. Pettigrew, F. M.Girio, B. Devreese, I. Vandenberghe, J. Van Beeumen, G.Fauque, and I. Moura, Biochim. Biophys. Acta, 1999, 1434,248.
106. C. Greenwood, N. Foote, J. Peterson, and A. J. Thomson,Biochem. J , 1984, 223, 379.
107. N. Foote, J. Peterson, P. M. Gadsby, C. Greenwood, andA. J. Thomson, Biochem. J., 1985, 230, 227.
108. V. Fulop, C. J. Ridout, C. Greenwood, and J. Hajdu, Structure,1995, 3, 1225.
109. H. Shimizu, D. J. Schuller, W. N. Lanzilotta, M. Sundara-moorthy, D. M. Arciero, A. B. Hooper, and T. L. Poulos,Biochemistry, 2001, 40, 13483.
110. L. De Smet, D. Leys, and J. J. Van Beeumen, ActaCrystallogr., Sect. D Biol. Crystallogr., 2002, 58, 522.
111. J. M. Diaz, C. Bonifacio, T. Alves, J. J. Moura, I. Moura, andM. J. Romao, Acta Crystallogr., Sect. D Biol. Crystallogr.,2002, 58, 697.
112. L. De Smet, G. W. Pettigrew, and J. J. Van Beeumen, Eur. J.Biochem., 2001, 268, 6559.
113. P. A. Williams, V. Fulop, E. F. Garman, N. F. Saunders,S. J. Ferguson, and J. Hajdu, Nature, 1997, 389, 406.
114. M. Sundaramoorthy, J. Terner, and T. L. Poulos, Chem. Biol.,1998, 5, 461.
115. W. L. Smith, D. L. DeWitt, and R. M. Garavito, Annu. Rev.Biochem., 2000, 69, 145.
116. R. M. Garavito, Prostaglandin Endoperoxide H2 Synthases-1and -2, in ‘Handbook of Metalloproteins’, eds. A. Messer-schmidt, R. Huber, T. L. Poulos, and K. Wieghardt, JohnWiley & Sons, Chichester, 2001, Vols. 1, 2, p. 245.
117. J. Stubbe and W. A. van der Donk, Chem. Rev., 1998, 98, 705.
118. W. L. Smith and L. J. Marnett, in ‘Metals in BiologicalSystems’, eds. H. Sigel and A. Sigel, Marcel Dekker, NewYork, 1994, Vol. 30.
119. J. Hayllar and I. Bjarnason, Lancet, 1995, 346, 521.
120. D. Picot, P. J. Loll, and R. M. Garavito, Nature, 1994, 367,243.
121. C. Luong, A. Miller, J. Barnett, J. Chow, C. Ramesha, andM. F. Browner, Nat. Struct. Biol., 1996, 3, 927.
122. R. G. Kurumbail, A. M. Stevens, J. K. Gierse, J. J. McDonald,R. A. Stegeman, J. Y. Pak, D. Gildehaus, J. M. Miyashiro,T. D. Penning, K. Seibert, P. C. Isakson, and W. C. Stallings,Nature, 1996, 384, 644.
123. J. Zeng and R. E. Fenna, J. Mol. Biol., 1992, 226, 185.
124. R. E. Fenna, Myeloperoxidase, in ‘Handbook of Metallopro-teins’, eds. A. Messerschmidt, R. Huber, T. L. Poulos, andK. Wieghardt, John Wiley & Sons, Chichester, 2001, Vols. 1,2, p. 211.
125. S. J. Klebanoff, Peroxidases in Chemistry and Biology, in‘Peroxidases in Chemistry and Biology’, eds. J. Everse,E. Everse, and M. B. Grisham, CRC Press, Boca Raton, FL,1991, Vol. 1, p. 1.
126. S. O. Pember, R. Shapiro, and J. M. J. Kinkade, Arch.Biochem. Biophys., 1983, 221, 391.
127. B. J. Sutton, C. Little, R. L. Olsen, and N. P. Willassen, J.Mol. Biol., 1988, 199, 395.
128. N. Moguilevsky, L. Garcia-Quintana, A. Jacquet, C. Tournay,L. Fabry, L. Pierard, and A. Bollen, Eur. J. Biochem., 1991,197, 605.
129. R. E. Fenna, J. Zeng, and C. Davey, Arch. Biochem. Biophys.,1995, 316, 653.
130. I. M. Kooter, N. Moguilevsky, A. Bollen, L. A. van der Veen,C. Otto, H. L. Dekker, and R. Wever, J. Biol. Chem., 1999,274, 26794.
131. R. E. Fenna, J. Biol. Chem., 1987, 196, 919.
132. T. J. Fiedler, C. A. Davey, and R. E. Fenna, J. Biol. Chem.,2000, 275, 11964.
133. M. Blair-Johnson, T. Fiedler, and R. E. Fenna, J. Biol. Chem.,2001, 40, 13990.
134. J. N. de Wit and A. C. M. van Hooijdonk, Neth. Milk Dairy J.,1996, 50, 227.
135. B. Reiter and J. P. Perraudin, Lactoperoxidase: Biologicalfunctions, in ‘Peroxidases in Chemistry and Biology’, eds.J. Everse, E. Everse, and M. B. Grisham, CRC Press, BocaRaton, FL, 1991, Vol. I, p. 143.
136. K. D. Kussendrager and A. C. van Hooijdonk, Br. J. Nutr.,2000, 84, S19.
137. C. Colas, J. M. Kuo, and P. R. Ortiz de Montellano, J. Biol.Chem., 2001, 277, 7191.
138. H. Kohler, A. Taurog, and H. B. Dunford, Arch. Biochem.Biophys., 1988, 264, 438.
139. O. M. Lardinois, K. F. Medzihradszky, and P. R. Ortiz deMontellano, J. Biol. Chem., 1999, 274, 35441.
140. O. M. Lardinois and P. R. Ortiz de Montellano, Biochem.Biophys. Res. Commun., 2000, 270, 199.
141. T. B. Bruck and P. J. Harvey, Biochem. Biophys. Acta, 2003,1649, 154.
142. J. A. Leff, L. K. Burton, E. M. Berger, B. O. Anderson,C. P. Wilke, and J. E. Repine, Inflammation, 1993, 17, 199.

143. J. A. Leff, M. A. Oppegard, T. J. Curiel, K. S. Brown,R. T. Schooley, and J. E. Repine, Free Radic. Biol. Med.,1992, 13, 143.
144. B. Halliwell and O. I. Aruoma, FEBS Lett., 1991, 281, 9.
145. B. Halliwell and J. M. Gutteridge, Methods Enzymol., 1990,186, 1.
146. C. J. Masters and R. S. Holmes, TIBS, 1979, 4, 233.
147. N. Oshino, R. Oshino, and B. Chance, Biochem. J., 1973, 131,555.
148. M. Zamocky and F. Koller, Prog. Biophys. Mol. Biol., 1999,72, 19.
149. M. W. Fraaije, H. P. Roubroeks, W. R. Hagen, and W. J. VanBerkel, Eur. J. Biochem., 1996, 235, 192.
150. S. V. Antonyuk and W. R. Melik-Adamyan, Crystallogr.Rep., 2000, 45, 105.
151. V. V. Barynin, M. M. Whittaker, S. V. Antonyuk, V. S.Lamzin, P. M. Harrison, P. J. Artymiuk, and J. W. Whittaker,Structure, 2001, 9, 725.
152. B. K. Vainshtein, W. R. Melik-Adamyan, V. V. Barynin,A. A. Vagin, and A. I. Grebenko, Nature, 1981, 293,411.
153. M. R. N. Murthy, T. J. Reid III, A. Sicignano, N. Tanaka, andM. G. Rossmann, J. Mol. Biol., 1981, 152, 465.
154. T. J. Reid III, M. R. N. Murthy, A. Sicignano, N. Tanaka,W. D. Musick, and M. G. Rossmann, Proc. Natl. Acad. Sci.U.S.A., 1981, 78, 4767.
155. I. Fita and A. M. Silva, Acta Crystallogr., 1986, B42,497.
156. B. K. Vainshtein, W. R. Melik-Adamyan, V. V. Barynin,A. A. Vagin, A. I. Grebenko, V. V. Borisov, K. S. Bartels,I. Fita, and M. G. Rossmann, J. Mol. Biol., 1986, 188, 49.
IRON: HEME PROTEINS, PEROXIDASES, CATALASES & CATALASE-PEROXIDASES 21
157. G. N. Murshudov, W. R. Melik-Adamyan, A. I. Grebenko,V. V. Barynin, A. A. Vagin, B. K. Vainshtein, Z. Dauter, andK. S. Wilson, FEBS Lett., 1992, 312, 127.
158. J. Bravo, N. Verdaguer, J. Tormo, C. Betzel, J. Switala,P. C. Loewen, and I. Fita, Structure, 1995, 3, 491.
159. P. Gouet, H. M. Jouve, and O. Y. Dideberg, J. Mol. Biol.,1995, 249, 933.
160. M. J. Mate, M. Zamocky, L. M. Nykyri, C. Herzog, P. M.Alzari, C. Betzel, F. Koller, and I. Fita, J. Mol. Biol., 1999,286, 135.
161. C. D. Putnam, A. S. Arvai, Y. Bourne, and J. A. Tainer, J.Mol. Biol., 2000, 296, 295.
162. I. Fita and M. G. Rossmann, Proc. Natl. Acad. Sci. U.S.A.,1985, 82, 1604.
163. H. N. Kirkman and G. F. Y. Gaetani, Proc. Natl. Acad. Sci.U.S.A., 1984, 81, 43434.
164. D. C. De Luca, R. Dennis, and W. G. Smith, Arch. Biochem.Biophys., 1995, 320, 129.
165. J. Bravo, M. J. Mate, T. Schneider, J. Switala, K. Wilson,P. C. Loewen, and I. Fita, Proteins, 1999, 34, 155.
166. P. C. Loewen, J. Switala, I. von Ossowski, A. Hillar,A. Christie, B. Tattrie, and P. Nicholls, Biochemistry, 1993,32, 10159.
167. P. R. Ortiz de Montellano, Annu. Rev. Pharmacol. Toxicol.,1992, 32, 89.
168. P. Jones and D. N. Middlemiss, Biochem. J., 1972, 130,411.
169. G. N. Murshudov, A. I. Grebenko, J. A. Brannigan, A. A. Ant-son, V. V. Barynin, G. G. Dodson, Z. Dauter, K. S. Wilson,and W. R. Melik-Adamyan, Acta Crystallogr., Sect. D Biol.Crystallogr., 2002, 58, 1972.