introduction to rna-seq & transcriptome analysis jessica kirkpatrick powerpoint by casey hanson...
TRANSCRIPT

RNA-Seq Lab | Jessica Kirkpatrick | 2015 1
Introduction to RNA-Seq & Transcriptome Analysis
Jessica Kirkpatrick
PowerPoint by Casey Hanson

RNA-Seq Lab | Jessica Kirkpatrick | 2015 2
Exercise
Use the Tuxedo Suite to:
1. Align RNA-Seq reads using TopHat (splice-aware
aligner).
2. Perform reference-based transcriptome assembly with
CuffLinks.
3. Obtain a new transcriptome using CuffLinks &
CuffMerge.
4. Use CuffDiff to obtain a list of differentially expressed
genes.
5. Report a list of significantly expressed genes.
RNA-Seq Lab | Jessica Kirkpatrick | 2015 3
Trapnell et al., Nature Protocols, March 2012
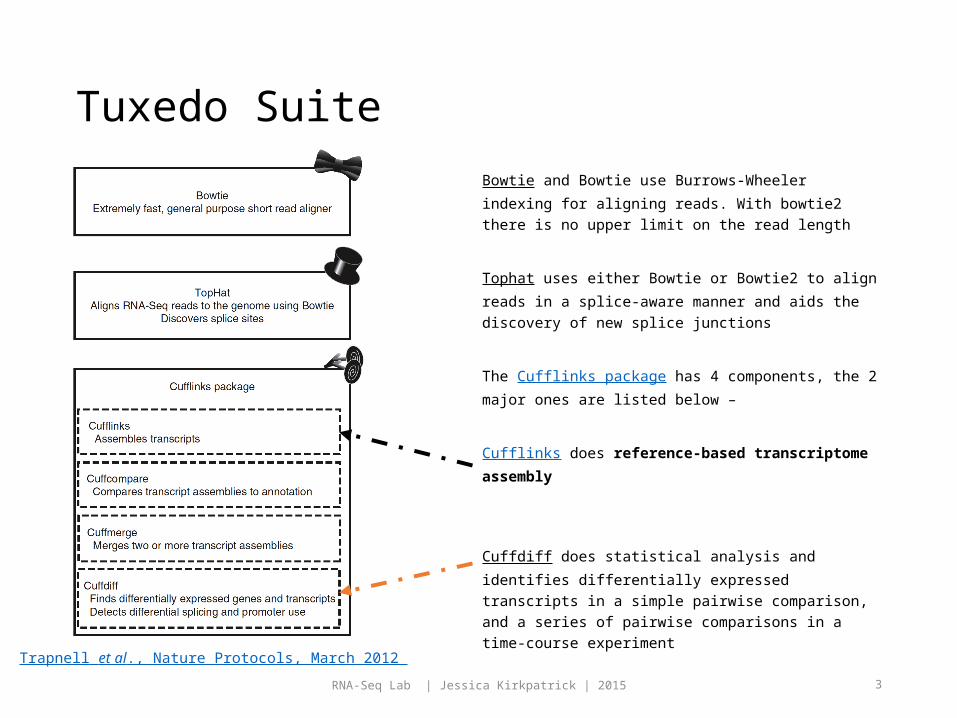
Tuxedo Suite
Bowtie and Bowtie use Burrows-Wheeler indexing for aligning reads. With bowtie2 there is no upper limit on the read length
Tophat uses either Bowtie or Bowtie2 to align reads in a splice-aware manner and aids the discovery of new splice junctions
The Cufflinks package has 4 components, the 2 major ones are listed below –
Cufflinks does reference-based transcriptome assembly
Cuffdiff does statistical analysis and identifies differentially expressed transcripts in a simple pairwise comparison, and a series of pairwise comparisons in a time-course experiment
RNA-Seq Lab | Jessica Kirkpatrick | 2015 4
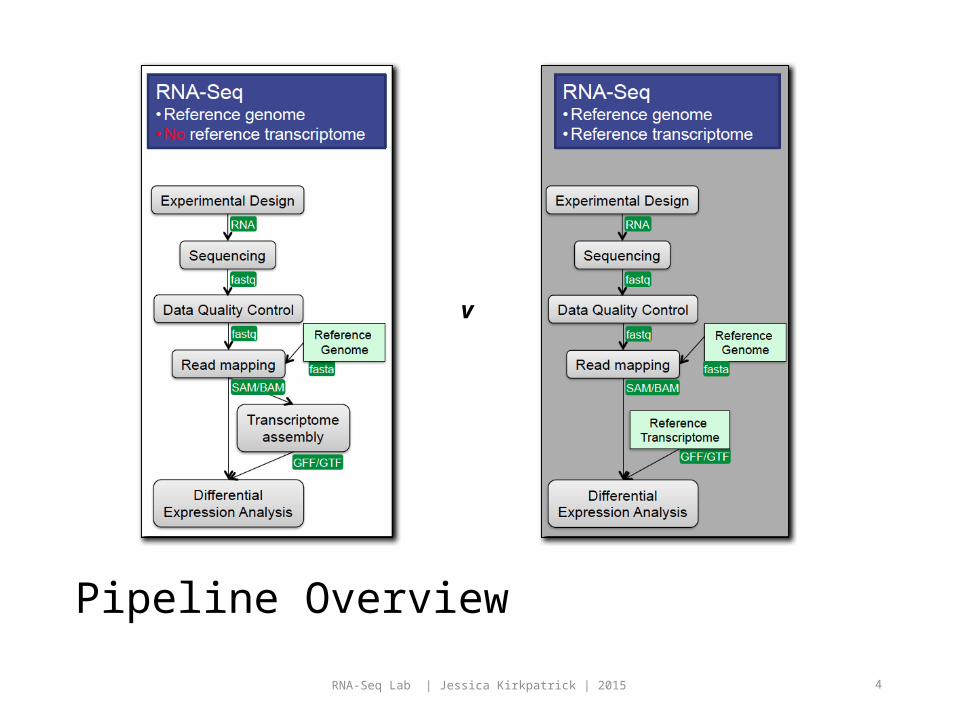
Pipeline Overview
v

RNA-Seq Lab | Jessica Kirkpatrick | 2015 5
Premise
1. Procedure:
Run 1A: Allow TopHat to select splice junctions de novo and
proceed through the steps without giving the software known
genes/gene models.
Run 1B: Force TopHat to use only known splice junctions (i.e. known
genes/gene models) and proceed through the steps making sure we
are doing our analysis in the context of these gene models.
2. Evaluation:
a. 2 metrics: # of mapped reads and # of significantly
different identified genes
b. Compare new transcriptome to known genes.
Question: Is there a difference in our results if the Tuxedo Suit is run two different ways?
RNA-Seq Lab | Jessica Kirkpatrick | 2015 6
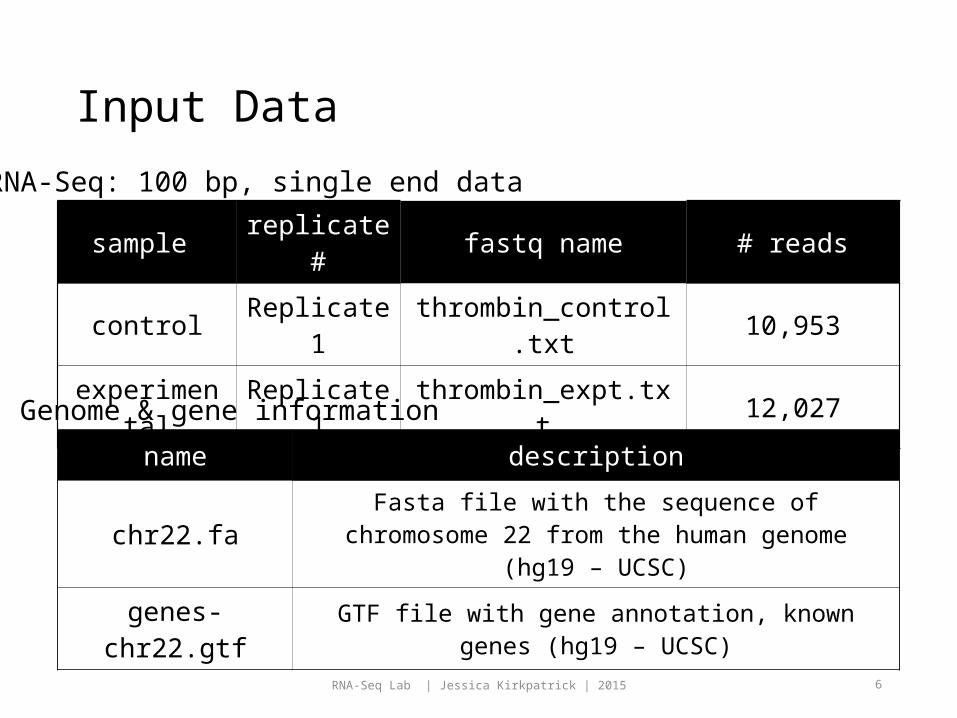
sample replicate
# fastq name # reads
controlReplicate
1thrombin_control.tx
t 10,953
experimental
Replicate 1 thrombin_expt.txt 12,027
name description
chr22.fa Fasta file with the sequence of chromosome 22 from the human genome (hg19 – UCSC)
genes-chr22.gtf GTF file with gene annotation, known genes (hg19 – UCSC)
RNA-Seq: 100 bp, single end data
Genome & gene information
Input Data
RNA-Seq Lab | Jessica Kirkpatrick | 2015 7
Accessing the IGB Biocluster
RNA-Seq Lab | Jessica Kirkpatrick | 2015 8
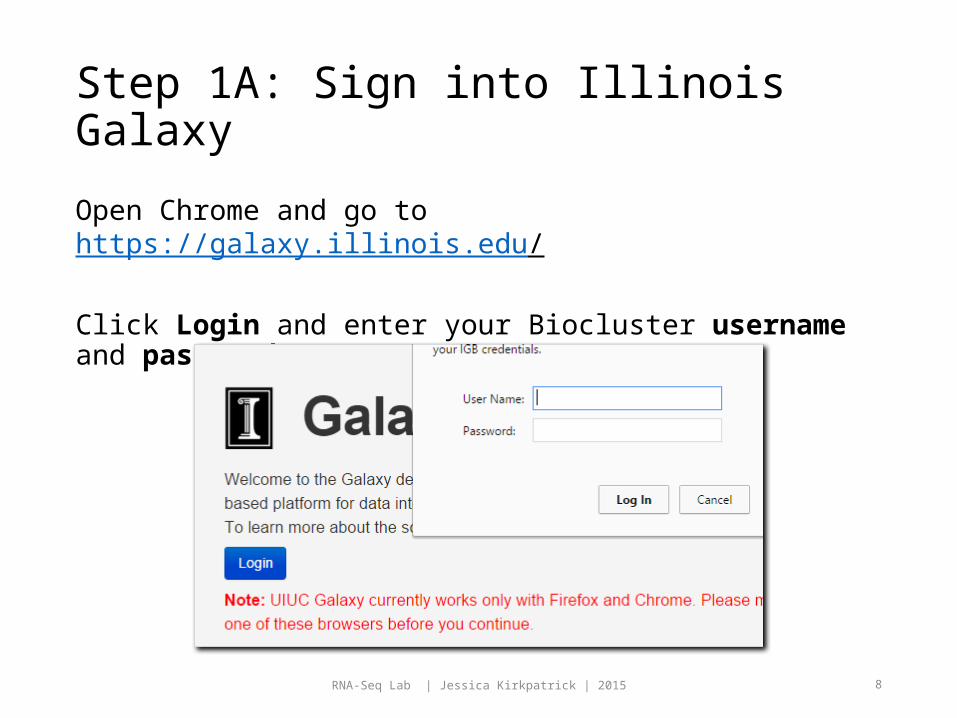
Step 1A: Sign into Illinois Galaxy
Open Chrome and go to https://galaxy.illinois.edu/
Click Login and enter your Biocluster username and password.
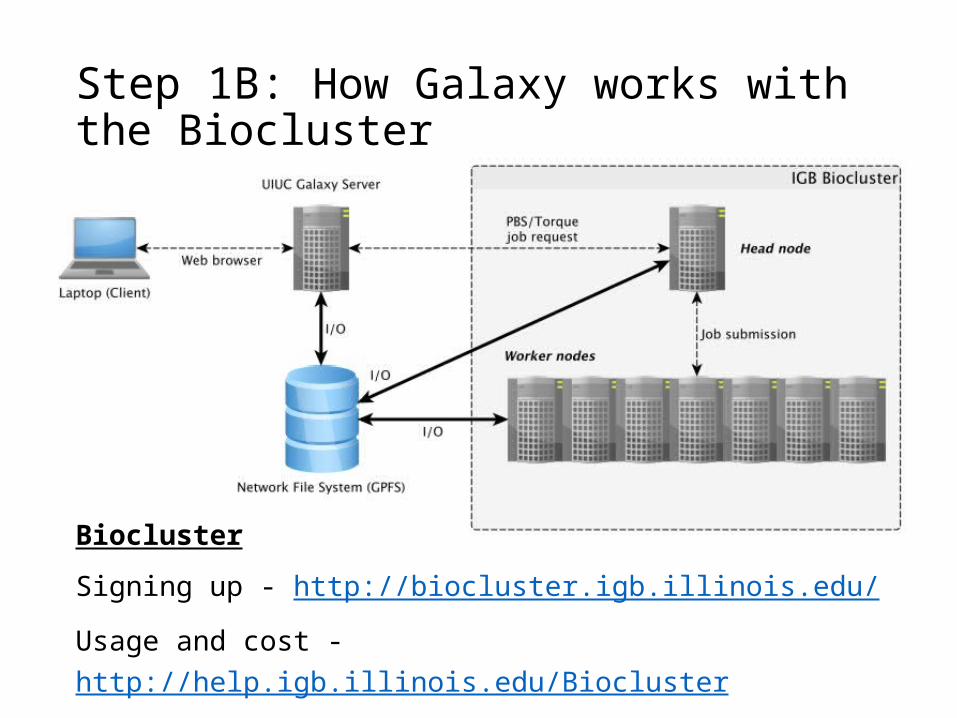
Step 1B: How Galaxy works with the Biocluster
Biocluster
Signing up - http://biocluster.igb.illinois.edu/
Usage and cost - http://help.igb.illinois.edu/Biocluster
RNA-Seq Lab | Jessica Kirkpatrick | 2015 10
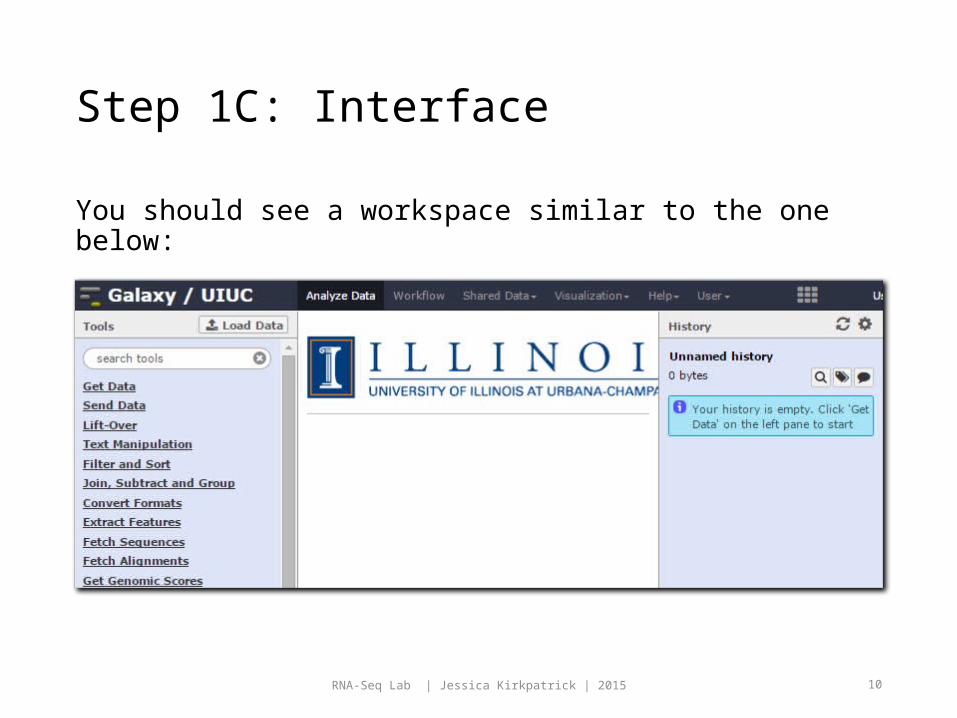
Step 1C: Interface
You should see a workspace similar to the one below:

RNA-Seq Lab | Jessica Kirkpatrick | 2015 11
Step 1B: Changing History Name
Click on Unnamed History in the History Pane on the left side :
Type RNA – Seq workshop and press Enter.

RNA-Seq Lab | Jessica Kirkpatrick | 2015 12
Step 2A: Accessing Input Files
At the top of the page, click Shared Data.
Then click Publish Histories.
RNA-Seq Lab | Jessica Kirkpatrick | 2015 13
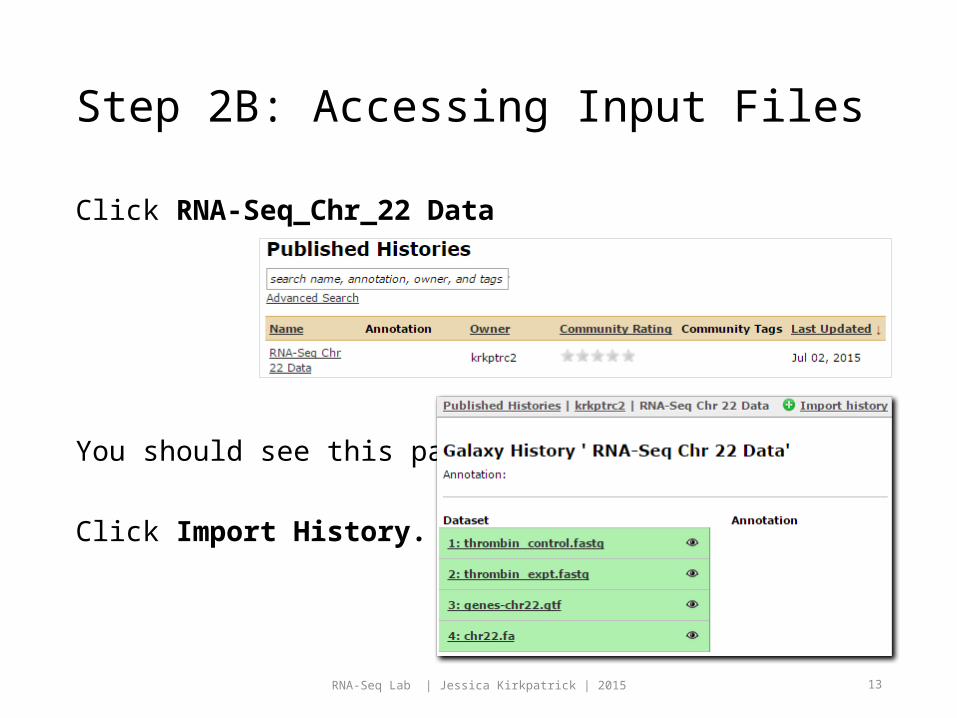
Step 2B: Accessing Input Files
Click RNA-Seq_Chr_22 Data
You should see this page.
Click Import History.

RNA-Seq Lab | Jessica Kirkpatrick | 2015 14
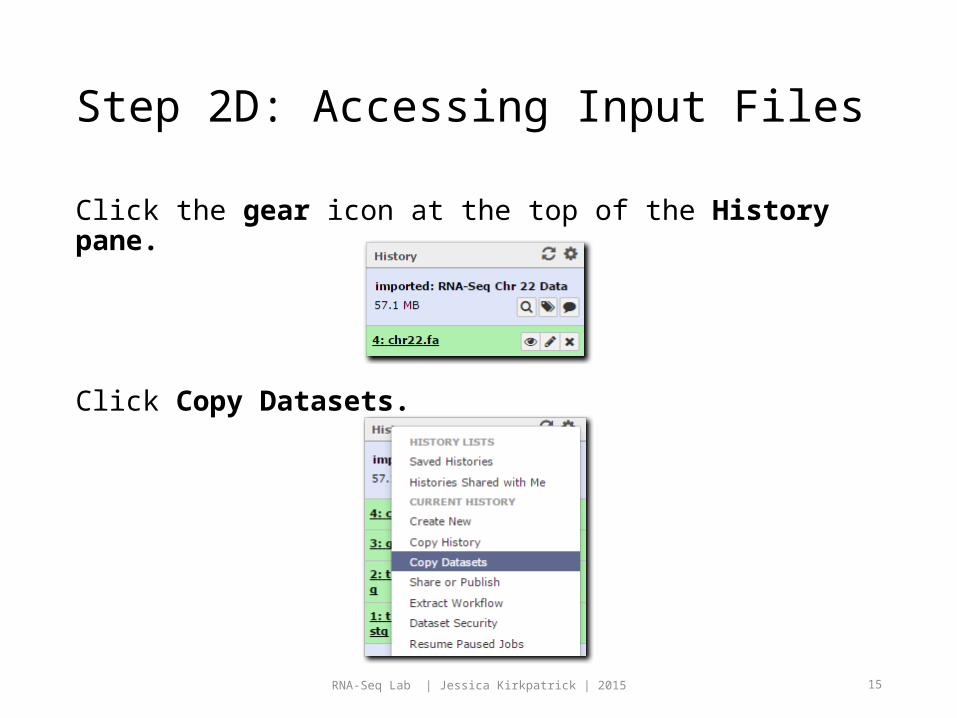
Step 2C: Accessing Input Files
Click start_using_this_history
You should see an imported history like the following.
RNA-Seq Lab | Jessica Kirkpatrick | 2015 15
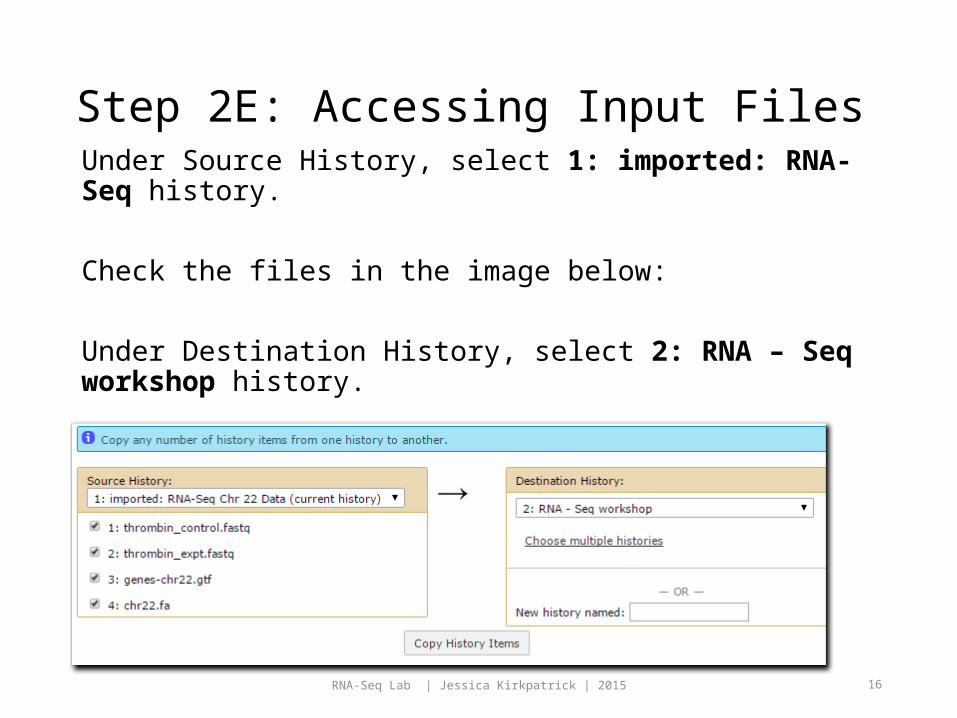
Step 2D: Accessing Input Files
Click the gear icon at the top of the History pane.
Click Copy Datasets.
RNA-Seq Lab | Jessica Kirkpatrick | 2015 16
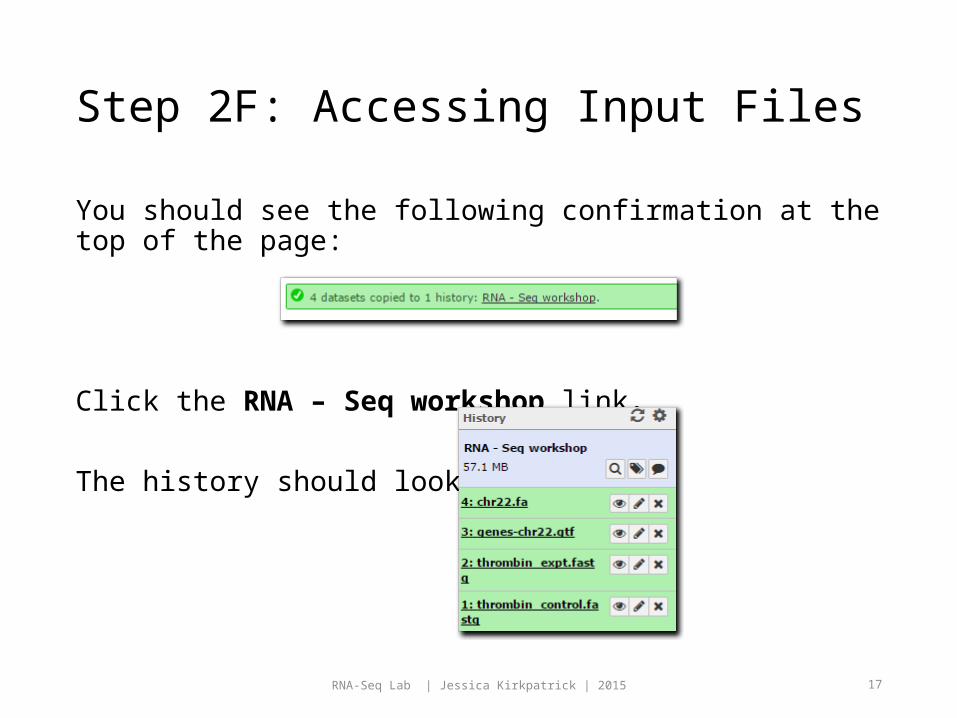
Step 2E: Accessing Input Files Under Source History, select 1: imported: RNA-Seq history.
Check the files in the image below:
Under Destination History, select 2: RNA – Seq workshop history.
Click the Copy History Items button.
RNA-Seq Lab | Jessica Kirkpatrick | 2015 17
Step 2F: Accessing Input Files
You should see the following confirmation at the top of the page:
Click the RNA – Seq workshop link.
The history should look like this :

RNA-Seq Lab | Jessica Kirkpatrick | 2015 18
In this exercise, we will be aligning RNA-Seq reads to a reference genome in
the absence of gene models. Splice junctions will be found de novo.
Remember, we are not going to provide any genic structure information.
.
Run 1A: de novo Alignment

RNA-Seq Lab | Jessica Kirkpatrick | 2015 19
Step 3A: Align Reads de novo Using TopHat2
At the top right of the page, click the search box :
Type TopHat2
Select TopHat2 under NGS: RNA Analysis

RNA-Seq Lab | Jessica Kirkpatrick | 2015 20
Step 3B: Align Reads de novo Using TopHat2You should a page similar to the one below. We will run TopHat2 first on the thrombin experimental data.
Make sure your inputs match the screenshot below:

RNA-Seq Lab | Jessica Kirkpatrick | 2015 21
Step 3C: Align Reads de novo Using TopHat2
The rest of the page contains parameters.
We will change the following parameters:
1. Library Type: FR Unstranded
2. Minimum Intron Length: 70
3. Maximum Intron Length: 500000
4. Maximum number of alignment to be allowed: 20

RNA-Seq Lab | Jessica Kirkpatrick | 2015 22
Step 3C: Align Reads de novo Using TopHat2
The rest of the page contains parameters.
We will change the following parameters:
5. Number of mismatches allowed in each segment alignments for reads mapped independently : 2
6. Use Own Junctions: No
7. Use Coverage Search: Yes
8. Maximum intron length that may be found during coverage search: 500000

RNA-Seq Lab | Jessica Kirkpatrick | 2015 23
Step 3E: Align Reads de novo Using TopHat2
The rest of the page contains parameters.
We will change the following parameters:
9. Use Microexon Search: No 10.Do Fusion Search: No11.Set Bowtie2 settings: No12.Specify read group: No
Click Execute when you have set the parameters.

RNA-Seq Lab | Jessica Kirkpatrick | 2015 24
Step 3F: Align Reads de novo Using TopHat2
You will see confirmation in the Main Pane denoting which tracks have been added to run.
You should see the tracks at the top of the History Pane
A gray track means the job isn't running.A yellow track means the job is running.A green track means the job is finished.

RNA-Seq Lab | Jessica Kirkpatrick | 2015 25
Step 3G: Align Reads de novo Using TopHat2
You will see confirmation in the Main Pane denoting which tracks have been added to run.
You should see the tracks at the top of the History Pane
A gray track means the job isn't running.A yellow track means the job is running.A green track means the job is finished.

RNA-Seq Lab | Jessica Kirkpatrick | 2015 26
Step 3H: Align Reads de novo Using TopHat2We want to run TopHat2 for the control dataset now.
Navigate to the TopHat2 page again.
This time use 1: thrombin_control.fastq for RNA-Seq FASTQ file.

RNA-Seq Lab | Jessica Kirkpatrick | 2015 27
Step 3I: Align Reads de novo Using TopHat2
Configure the parameters as before (below) and click execute:
1. Library Type: FR Unstranded2. Minimum Intron Length: 703. Maximum Intron Length: 5000004. Maximum number of alignment to be allowed: 205. Number of mismatches allowed in each segment
alignments for reads mapped independently : 26. Use Own Junctions: No7. Use Coverage Search: Yes 8. Maximum intron length that may be found during
coverage search: 5000009. Use Microexon Search: No 10. Do Fusion Search: No11. Set Bowtie2 settings: No12. Specify read group: No
RNA-Seq Lab | Jessica Kirkpatrick | 2015 28
Step 4A: Renaming Files
In galaxy, it is important to rename output files to something meaningful.
For example, to rename 9: Tophat2_on_data2_and data4:accepted_hits
Click the pencil icon
RNA-Seq Lab | Jessica Kirkpatrick | 2015 29
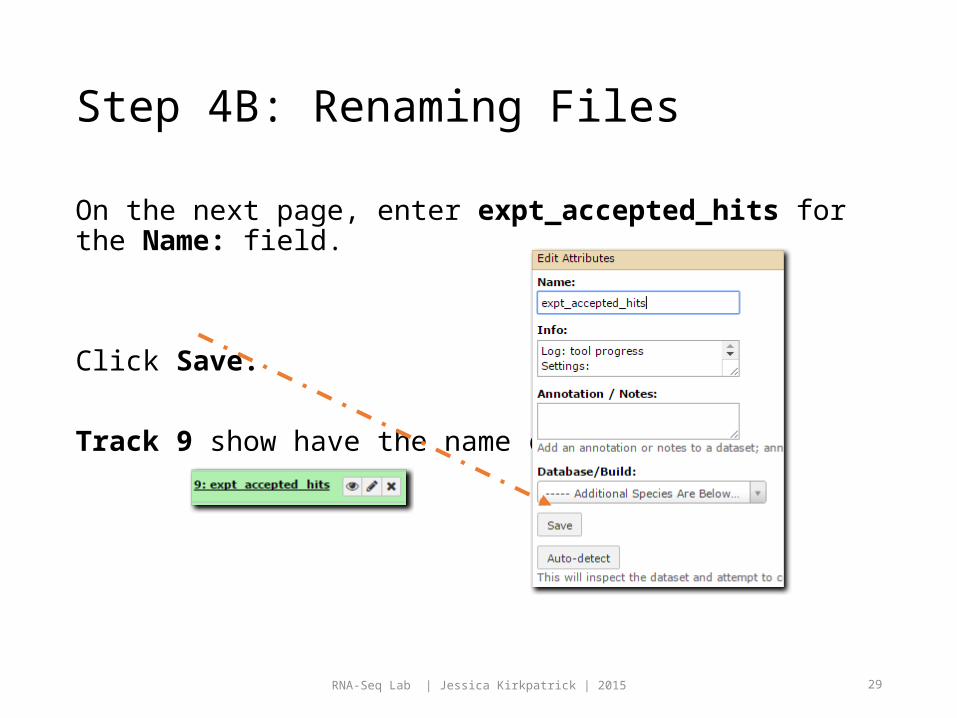
Step 4B: Renaming Files
On the next page, enter expt_accepted_hits for the Name: field.
Click Save.
Track 9 show have the name change:

RNA-Seq Lab | Jessica Kirkpatrick | 2015 30
Step 4C: Renaming Files
In this manner, rename the following tracks with the respective names:
5. expt_align_summary6. expt_insertions7. expt_deletions8. expt_splice_junctions
10.ctrl_align_summary11.ctrl_insertions12.ctrl_deletions13.ctrl_splice_junctions14.ctrl_accepted_hits

RNA-Seq Lab | Jessica Kirkpatrick | 2015 31
Step 5A: Evaluating de novo Alignment
Click the eye icon 5: expt_align_summary
You should see the results on the screen, like below :
In the experimental group, 148 reads were not aligned.

RNA-Seq Lab | Jessica Kirkpatrick | 2015 32
Step 5B: Evaluating de novo Alignment
Click the eye icon 10: ctrl_align_summary
You should see the results on the screen, like below :
In the control group, 101 reads were not aligned.

RNA-Seq Lab | Jessica Kirkpatrick | 2015 33
In this exercise, we will be aligning RNA-Seq reads to a reference
genome in the presence of gene information. This obviates the
need for TopHat to find splice junctions de novo.
.
Run 1B: Informed Alignment

RNA-Seq Lab | Jessica Kirkpatrick | 2015 34
Step 6A: Informed Align Reads Using TopHat2
We want to re-run the analysis for the experimental group, but using a gene-model annotation this time.
Instead of repeating the previous steps, we can save some time by clicking on the update icon on track 9: expt_accepted_hits.
Click on track 9.
Click the update icon.

RNA-Seq Lab | Jessica Kirkpatrick | 2015 35
Step 6B: Informed Align Reads Using TopHat2
Keep the same parameters as before, but change the following:
1. Use Own Junctions: Yes 2. Use Gene Annotation Model: Yes3. Gene Model Annotations: 3: genes-chr22.gtf4. Use Raw Junctions: No5. Only look for supplied junctions: No
Click Execute.

RNA-Seq Lab | Jessica Kirkpatrick | 2015 36
Step 6C: Informed Align Reads Using TopHat2
This should generate tracks 15 through 19.
Rename the tracks the following:
15.expt-genes_align_summary16.expt-genes_insertions17.expt-genes_deletions18.expt-genes_splice_junctions19.expt-genes_accepted_hits

RNA-Seq Lab | Jessica Kirkpatrick | 2015 37
Step 6D: Informed Align Reads Using TopHat2
We want to re-run the analysis for the control group, but using a gene-model annotation this time.
Instead of repeating the previous steps, we can save some time by clicking on the update icon on track 14: ctrl_accepted_hits.
Click on track 14.
Click the update icon.

RNA-Seq Lab | Jessica Kirkpatrick | 2015 38
Step 6E: Informed Align Reads Using TopHat2
Keep the same parameters as before, but change the following:
1. Use Own Junctions: Yes 2. Use Gene Annotation Model: Yes3. Gene Model Annotations: 3: genes-chr22.gtf4. Use Raw Junctions: No5. Only look for supplied junctions: No
Click Execute.

RNA-Seq Lab | Jessica Kirkpatrick | 2015 39
Step 6F: Informed Align Reads Using TopHat2
This should generate tracks 15 through 19.
Rename the tracks the following:
20.ctrl-genes_align_summary21.ctrl-genes_insertions22.ctrl-genes_deletions23.ctrl-genes_splice_junctions24.ctrl-genes_accepted_hits

RNA-Seq Lab | Jessica Kirkpatrick | 2015 40
Step 7A: Evaluating Informed Alignment
Click the eye icon 15: expt-genes_align_summary
You should see the results on the screen, like below :
In the experimental group, 39 reads were not aligned.

RNA-Seq Lab | Jessica Kirkpatrick | 2015 41
Step 7B: Evaluating Informed Alignment
Click the eye icon 20: ctrl-genes_align_summary
You should see the results on the screen, like below :
In the control group, 27 reads were not aligned.

RNA-Seq Lab | Jessica Kirkpatrick | 2015 42
sample # fastq name # readsUnmapped Reads
de novo Informed
control thrombin_control.txt 10,953 101 27
experimental thrombin_expt.txt 12,027 163 39
Step 8: Comparison of Alignments
There are fewer unmapped reads with the informed alignment, or Run 1B
(i.e. when we use the known genes, and known splice sites)!
TopHat’s prediction of splice junctions de novo is not working very well for
this dataset. (This is likely due to the low number of reads in our dataset.)
Conclusions

RNA-Seq Lab | Jessica Kirkpatrick | 2015 43
Next, we will utilize our RNA-Seq alignments to assembly gene
transcripts, thereby permitting us to get relative gene abundances
between the two samples (control and experimental).
Finding Differentially Expressed Genes

RNA-Seq Lab | Jessica Kirkpatrick | 2015 44Trapnell et al., Nature Protocols, March 2012
Reminder: Cufflinks
The Cufflinks package has 4 components, the 2 major ones are listed below –
Cufflinks does reference-based transcriptome assembly
Cuffdiff does statistical analysis and identifies differentially expressed transcripts in a simple pairwise comparison, and a series of pairwise comparisons in a time-course experiment
RNA-Seq Lab | Jessica Kirkpatrick | 2015 45
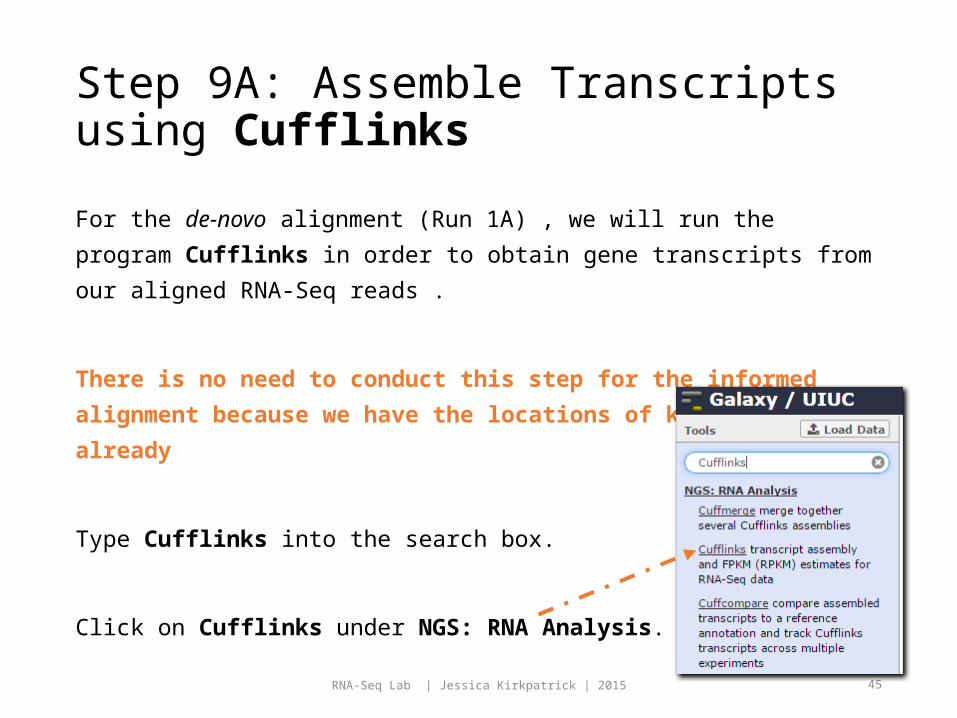
Step 9A: Assemble Transcripts using Cufflinks
For the de-novo alignment (Run 1A) , we will run the program
Cufflinks in order to obtain gene transcripts from our aligned
RNA-Seq reads .
There is no need to conduct this step for the informed
alignment because we have the locations of known genes
already
Type Cufflinks into the search box.
Click on Cufflinks under NGS: RNA Analysis.

RNA-Seq Lab | Jessica Kirkpatrick | 2015 46
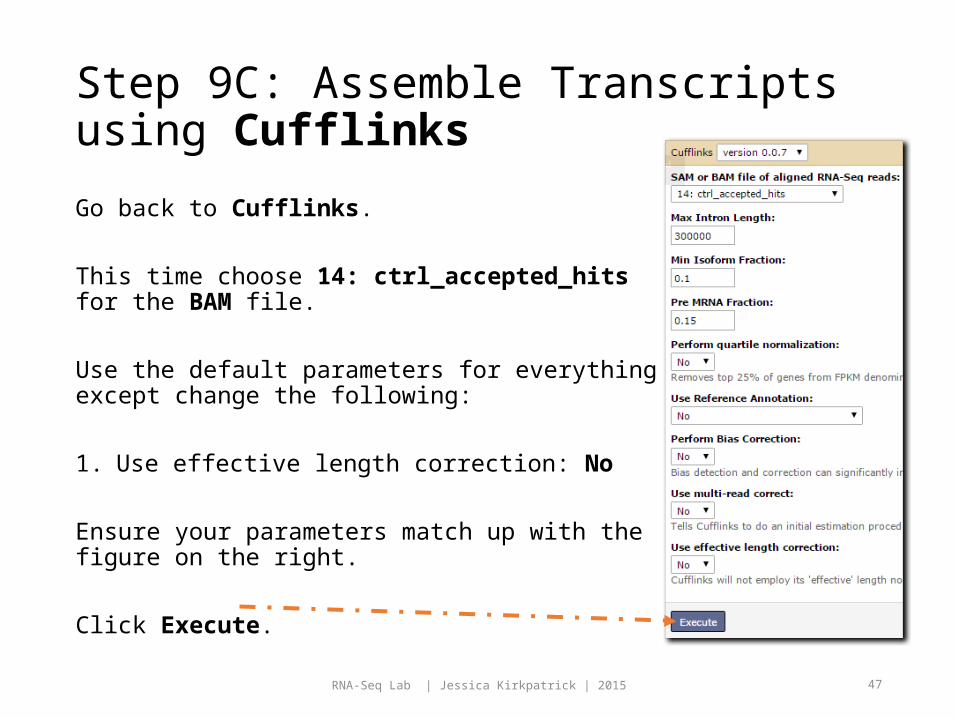
Step 9B: Assemble Transcripts using Cufflinks
Choose 9: expt_accepted_hits for the BAM file.
Use the default parameters for everything except change the following:
1. Use effective length correction: No
Ensure your parameters match up with the figure on the right.
Click Execute.
RNA-Seq Lab | Jessica Kirkpatrick | 2015 47
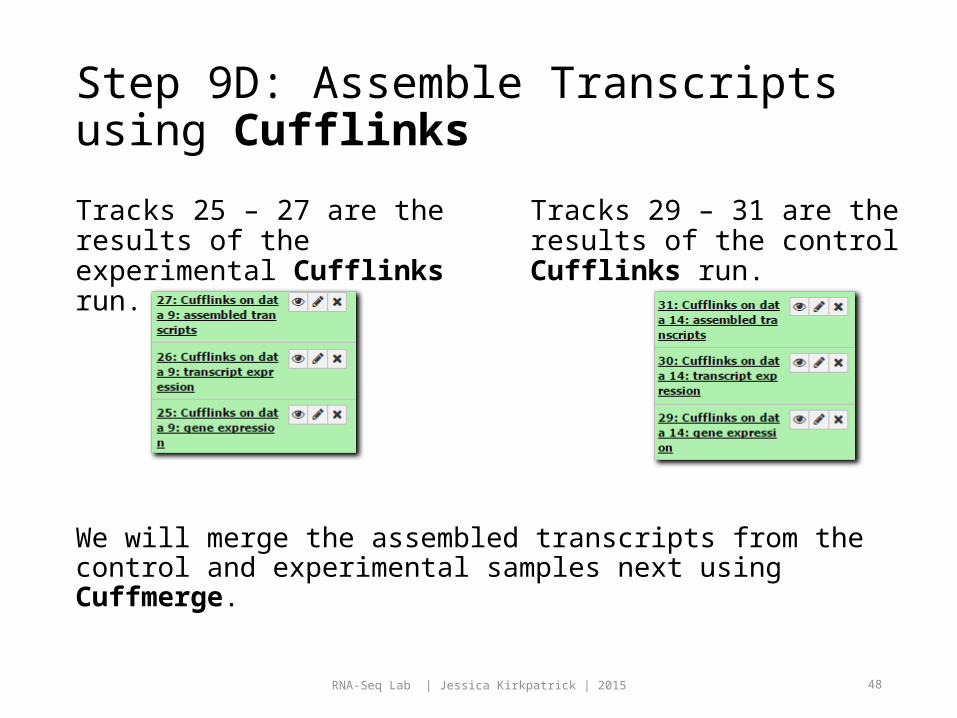
Step 9C: Assemble Transcripts using Cufflinks
Go back to Cufflinks.
This time choose 14: ctrl_accepted_hits for the BAM file.
Use the default parameters for everything except change the following:
1. Use effective length correction: No
Ensure your parameters match up with the figure on the right.
Click Execute.
RNA-Seq Lab | Jessica Kirkpatrick | 2015 48
Step 9D: Assemble Transcripts using Cufflinks
Tracks 25 – 27 are the results of the experimental Cufflinks run.
Tracks 29 – 31 are the results of the control Cufflinks run.
We will merge the assembled transcripts from the control and experimental samples next using Cuffmerge.

RNA-Seq Lab | Jessica Kirkpatrick | 2015 49
Step 10A: Merge Transcripts Using CuffMerge
In the search box, type Cuffmerge
Click Cuffmerge under NGS: RNA Analysis.
RNA-Seq Lab | Jessica Kirkpatrick | 2015 50
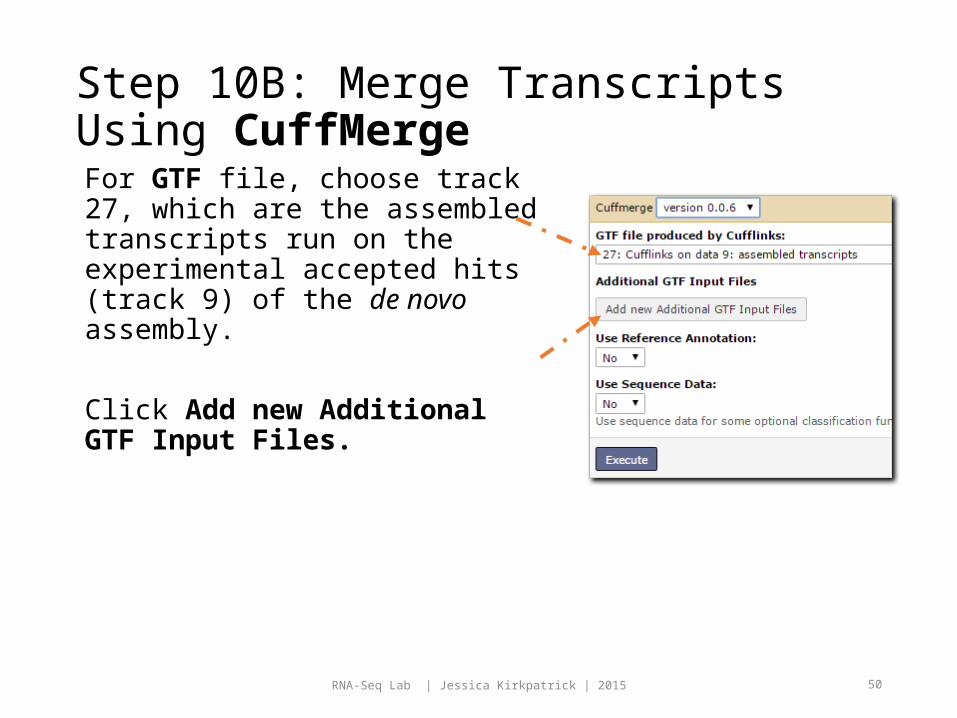
Step 10B: Merge Transcripts Using CuffMergeFor GTF file, choose track 27, which are the assembled transcripts run on the experimental accepted hits (track 9) of the de novo assembly.
Click Add new Additional GTF Input Files.
RNA-Seq Lab | Jessica Kirkpatrick | 2015 51
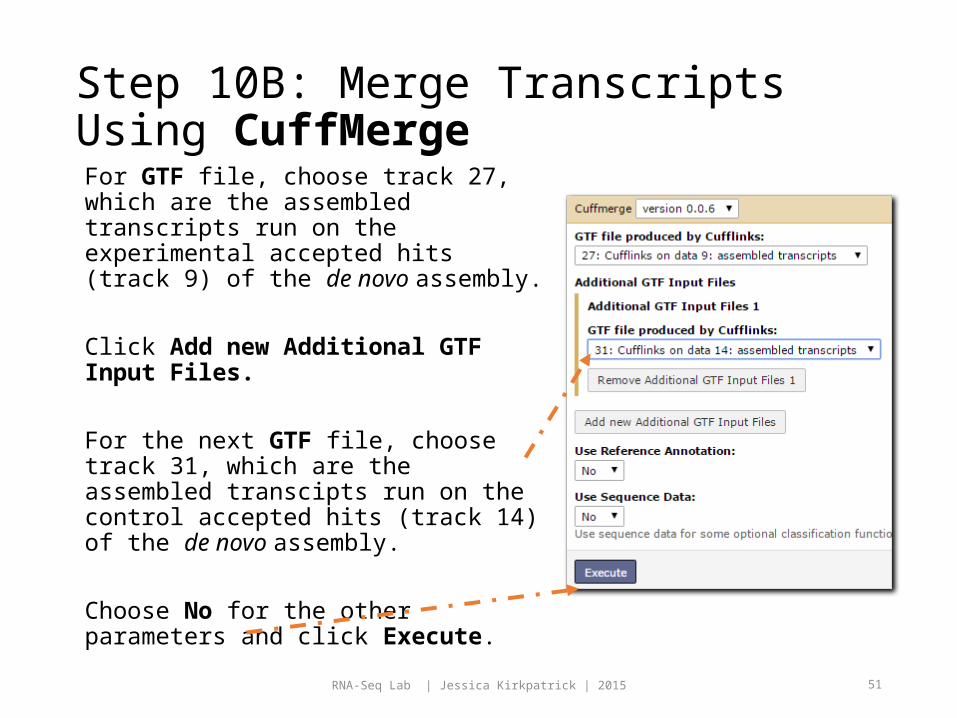
Step 10B: Merge Transcripts Using CuffMergeFor GTF file, choose track 27, which are the assembled transcripts run on the experimental accepted hits (track 9) of the de novo assembly.
Click Add new Additional GTF Input Files.
For the next GTF file, choose track 31, which are the assembled transcipts run on the control accepted hits (track 14) of the de novo assembly.
Choose No for the other parameters and click Execute.
RNA-Seq Lab | Jessica Kirkpatrick | 2015 52
Step 11A: Differential Gene Expression
For the de novo assembly, lets find out how many differentially expressed (DE) genes are present. We will use Cuffdiff to do this.
To do this, we need a GTF file and a BAM file for both the control and experimental assemblies.
We could use Cuffdiff on the informed alignments, as well, but we normally recommend using htseqcount and edgeR instead.
Type Cuffdiff into the search and click its link:
RNA-Seq Lab | Jessica Kirkpatrick | 2015 53
Step 11B: Differential Gene Expression
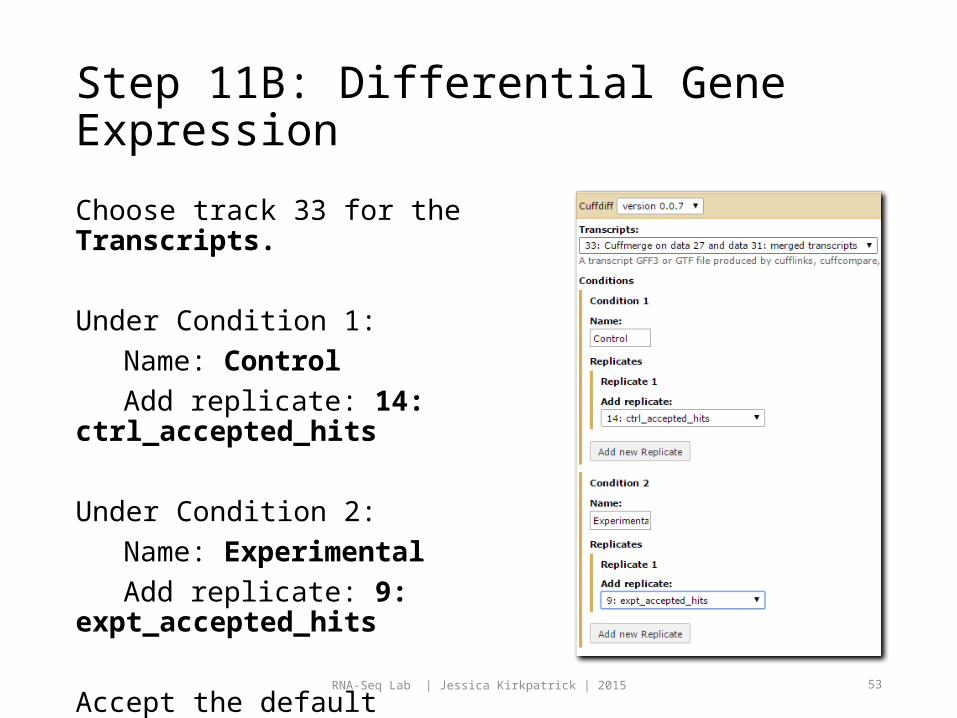
Choose track 33 for the Transcripts.
Under Condition 1:Name: ControlAdd replicate: 14:
ctrl_accepted_hits
Under Condition 2:Name: ExperimentalAdd replicate: 9:
expt_accepted_hits
Accept the default parameters and click Execute.

RNA-Seq Lab | Jessica Kirkpatrick | 2015 54
Step 11C: Differential Gene Expression
When done, click the eye icon on track 47:
You should see output like the following:
Count the number of "yes" answers in the significant column as you scroll down.
There should be 3. These are the DE genes.

55
Conclusion
We did the following today
Use the Tuxedo Suite to:
1. Align RNA-Seq reads using TopHat (splice-aware aligner).
2. Perform reference-based transcriptome assembly with
CuffLinks.
3. Obtain a new transcriptome using CuffLinks &
CuffMerge.
4. Use CuffDiff to obtain a list of differentially expressed
genes.
5. Report a list of significantly expressed genes.RNA-Seq Lab | Jessica Kirkpatrick | 2015

RNA-Seq Lab | Jessica Kirkpatrick | 2015 56
Useful linksOnline resources for RNA-Seq analysis questions –
http://www.biostars.org/ - Biostar (Bioinformatics explained)
http://seqanswers.com/ - SEQanswers (the next generation sequencing
community)
Most tools have a dedicated lists
Information about the various parts of the Tuxedo suite is available here -
http://ccb.jhu.edu/software.shtml
Genome Browsers tutorials –
http://www.broadinstitute.org/igv/QuickStart/ - IGV tutorials
http://www.openhelix.com/ucsc/ - UCSC browser tutorials
(openhelix is a great place for tutorials, UIUC has a campus-wide subscription)
Contact us at:

RNA-Seq Lab | Jessica Kirkpatrick | 2015 57
Extra MaterialIGV

RNA-Seq Lab | Jessica Kirkpatrick | 2015 58
The Integrative Genomics Viewer (IGV) is a tool that supports the visualization
of mapped reads to a reference genome, among other functionalities. We will use
it to observe where hits were called for the de-novo alignment (Run 1A) for the two
samples (control and experimental), the new transcriptome generated by
CuffMerge, and the differentially expressed genes.
.
Visualization Using IGV

RNA-Seq Lab | Jessica Kirkpatrick | 2015 59
In this step, we will start IGV and load the chr22.fa file, the known genes
file
(genes-chr22.gtf), the hits for both sample groups, and the merged
transcriptome. These files are located in
[course_directory]/05_Transcriptomics/results
Step 9: Start IGV
Graphical Instruction: Load Genome
1. Within IGV, click the ‘Genomes’ tab on the menu bar.
2. Click the the ‘Load Genome from File’ option.
3. In the browser window, select chr22.fa (genome).
Graphical Instruction: Load Other Files
1. Within IGV, click the FILE tab on the menu bar.
2. Click the ‘Load from File’ option.
3. Select the genes-chr22.gtf file (known genes file).
4. Perform Steps 1-3 for the files to the right.
Files to Load
genes-chr22.f
ctrl_accepted_hits.b
am
expt_accepted_hits.
bam
merged.gtf
RNA-Seq Lab | Jessica Kirkpatrick | 2015 60
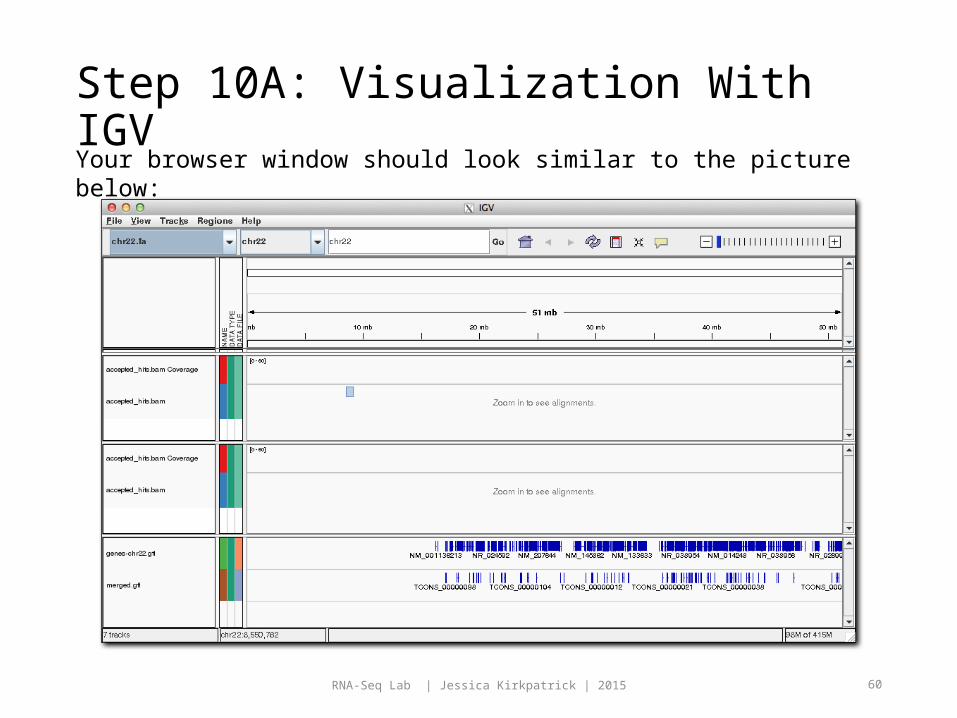
Step 10A: Visualization With IGVYour browser window should look similar to the picture below:

RNA-Seq Lab | Jessica Kirkpatrick | 2015 61
Step 10B: Visualization With IGVClick here and type the following location of a differentially expressed gene:
chr22:19960675-19963235
Move to the left and right of the gene. What do you see?

RNA-Seq Lab | Jessica Kirkpatrick | 2015 62
Step 10C: Visualization with IGV
Looks like the new transcriptome (merged.gtf) compares
poorly to the known gene models. This is very likely due to
the very low number of reads in our dataset.
We can see that there are many more reads for one dataset
compared to the other. Hence, it makes sense that the gene
was called as being differentially expressed.
Note the intron spanning reads.